
Marine waste upcycling—recovery of nylon monomers from fishing net waste using seashell waste-derived catalysts in a CO2-mediated thermocatalytic process†
Soosan
Kim‡
a,
Yong Tae
Kim
 b,
Lee Seul
Oh
b,
Hyung Ju
Kim
*b and
Jechan
Lee
*c
b,
Lee Seul
Oh
b,
Hyung Ju
Kim
*b and
Jechan
Lee
*c
aDepartment of Environmental Engineering, Ajou University, Suwon 16499, Republic of Korea
bChemical & Process Technology Division, Korea Research Institute of Chemical Technology, Daejeon 34114, Republic of Korea. E-mail: hjkim@krict.re.kr
cSchool of Civil, Architectural Engineering, and Landscape Architecture & Department of Global Smart City, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea. E-mail: jechanlee@skku.edu
First published on 2nd August 2022
Abstract
A lot of fishing nets have been abandoned, lost, or discarded at sea. Herein, we aimed at applying seashell waste-derived catalytic materials to a thermocatalytic process to recover a valuable commodity chemical (e.g., caprolactam) from fishing net waste (FNW) made of polyamide 6. For catalyst synthesis, seashell waste was carbonized in N2 and CO2 environments (denoted as SSWC-N2 and SSWC-CO2, respectively); here, the basicity of SSWC-CO2 was two-fold more than that of SSWC-N2. The thermocatalytic conversion of FNW was also conducted under N2 and CO2 atmospheres. Using SSWC-CO2 in the thermocatalytic conversion conducted under a CO2 atmosphere maximized the caprolactam recovery (80 wt% of FNW feedstock, the highest yield reported to date) possibly because the base-catalyzed decomposition of polyamide 6 was enhanced by more reactive cleavage of the amide linkage in CO2. SSWC-CO2 was reused for at least three cycles. In conclusion, SSWC-CO2 is a promising alternative catalyst to recover caprolactam from FNW. The findings from this study offer insights into developing a new thermocatalytic upcycling process for marine waste such as FNW and seashell waste. This aids in reducing microplastic pollution and increasing economic potential for marine waste valorization.
 Jechan Lee | Dr Jechan Lee received his PhD in chemical engineering from the University of Wisconsin–Madison in 2015 and M.S. in environmental engineering from Columbia University in 2010. After his PhD, he worked as a postdoctoral researcher at the Catalysis Center for Energy Innovation at the University of Delaware (2015–2016) and Sejong University (2016–2018). He was an assistant professor in the Department of Environmental and Safety Engineering (2018–2021) and as associate professor in the Department of Environmental and Safety Engineering/Department of Energy Systems research (2021–2022) at Ajou University. He has currently joined the faculty at Sungkyunkwan University (SKKU). His research interests are in the areas of waste upcycling, waste-to-energy, biorefinery, and CO2 utilization. He has currently authored more than 210 peer-reviewed SCI(E) papers. |
1. Introduction
Microplastics are a major problem in the fish-eat-fish world of sea and ocean environments. These prolific plastic particles are consumed by small fish, which are then eaten by larger fish, thereby transferring this pollutant up the food chain eventually to humans.1 Microplastics originate from the colossal use of modern day plastics; these plastics deteriorate into smaller particles that may be consumed by marine fauna. Fishing nets are a major source of microplastics that has not previously been considered and are typically made of non-biodegradable nylon. Approximately 6.4 × 105 t of fishing nets were dumped, abandoned, or lost directly at sea, accounting for more than half the debris discarded in oceans.2 The fishing net waste (FNW) often becomes entangled with each other and is almost impossible to separate. Entangled FNW not only poses a risk to marine mammals such as sea lions, large whales, and seals that may become entangled in the nets,3 but they also block sunlight to plankton and algae, thereby disturbing primary production processes in marine ecosystems.4 The management of FNW (e.g., collecting and recycling) has recently emerged as a means to address this problem.5Conventional disposal and recycling methods for FNW (e.g., landfilling, mechanical methods, and incineration), are no longer sustainable. These methods are considered downcycling, which is characterized by inefficient heat energy recovery and yields lower-value products than the corresponding original commodities.6 Recently, upcycling approaches that use waste as feedstock to produce renewable chemicals have gained considerable attention. Among different waste materials, plastic waste is an emerging feedstock to produce energy and high-value chemicals and to synthesize useful carbonaceous materials.7–10
The thermocatalytic plastic upcycling process involves the catalytic conversion of plastics to high-value chemicals (e.g., monomers and intermediates), at high temperatures between 300 and 700 °C. This process is attractive because it addresses plastic pollution by sustainably producing potentially value-added products.11 It has high potential for the establishment of thermocatalytic plastic upcycling; hence, they have certainly shown great promise.12 For example, styrene (a monomer of polystyrene) has been successfully recovered from expanded polystyrene waste with yields of up to 62.9% in the presence of Al2O3-supported metal catalysts (e.g., Zn/Al2O3 and Mg/Al2O3) at 450 °C.13,14 Polyethylene terephthalate monomers have been recovered from abandoned beverage bottles via thermocatalytic glycolysis (with ethylene glycol),15 or aminolysis (with ethanolamine)16 in the presence of guanidine catalysts such as triazabicyclodecene. A Pt/SrTiO3 catalyst prepared by atomic layer deposition was used to make linear hydrocarbons from disposable polyethylene bags via thermocatalytic hydrogenolysis.17 Another study achieved a >99% yield of linear hydrocarbons at 300 °C under 1.2 MPa H2 without any solvent; the non-catalytic thermal treatment of disposable polyethylene bags did not yield any linear hydrocarbons.17
Recently, CO2 has been suggested as a reaction medium for the thermocatalytic conversion of plastic waste, as a strategy for CO2 utilization. For instance, using CO2 and a HZSM-11 catalyst to thermocatalytically treat teabag waste at 500 °C improved nylon monomer recovery compared to that using typical thermal treatment.18 Jung et al. investigated the thermocatalytic production of H2 from polymeric waste with a Ni/SiO2 catalyst in a CO2 environment.19 They found that utilizing CO2 enhanced H2 production from waste fishing nets, reaching >1090 mmol gcat−1 h−1; this was nine fold higher than the production rate obtained without CO2.19
The preparation of catalysts from waste materials is another waste-upcycling method. Utilizing waste materials to prepare and use a catalyst to generate renewable target products from other waste materials creates an eco-friendlier and more cost effective waste upcycling process.20–22 Seashell waste is a potential waste material that is highly abundant; it is generated worldwide and approximately 35–70 wt% of seashells become waste.23 In China, approximately 10 million t of seashell waste are landfilled annually.24 Although a small fraction of seashell waste is re-used for handicrafts and fertilizers, its re-use for such purposes is limited due to its cost ineffectiveness and environmental problems such as soil solidification.25 Improperly treated seashell waste (e.g., illegal dumping) pollutes reclaimed land and public water and generates foul odors.26 The preparation of solid base catalysts from seashell waste has been widely investigated for transesterification reactions27–30 and catalytic pyrolysis of polymeric waste.31 However, no studies have been performed on the thermocatalytic upcycling of plastic waste using catalysts made from natural seashell waste under different atmospheres.
This study investigated the recovery of caprolactam from FNW via thermocatalytic conversion under a CO2 atmosphere using seashell waste-derived catalysts (SSWCs); as such, this demonstrated a strategy to concurrently upcycle marine waste while utilizing CO2. To the best of our knowledge, this is the first ever study on synergistical CO2 utilization for waste valorization and catalytic material preparation. Fig. 1 schematically describes the concept of this study. Two different catalysts were prepared from seashell waste via thermal treatment under N2 and CO2 atmospheres, and their catalytic performances were compared to maximize CO2 utilization during waste upcycling. This work aims to offer an environmentally friendly treatment and concomitant production of value-added products from marine waste. This proposed strategy will also contribute to solving regional/global environmental problems, such as microplastic pollution.
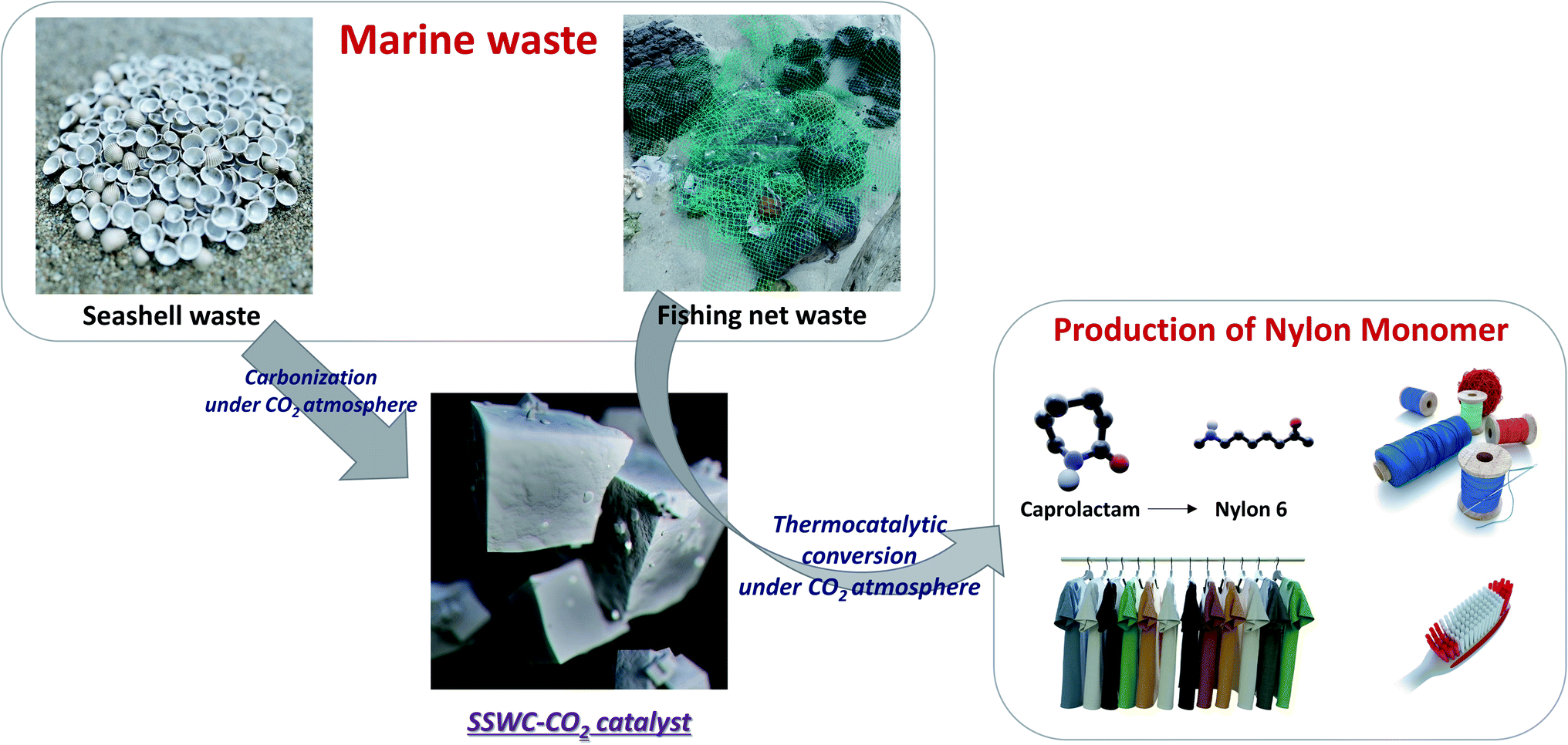 | ||
| Fig. 1 Conceptual diagram of marine waste upcycling proposed in this study. | ||
2. Materials and methods
2.1. Feedstocks and catalyst preparation
FNW and seashell waste were collected from a beach near Buan County, North Jeolla Province, Republic of Korea. Collected FNW was cut to a length of 20–25 mm (Fig. S1†), while collected seashell waste was thoroughly washed, dried in an oven at 60 °C for 24 h, and pulverized to fine powder between 0.15 and 0.6 mm. To prepare two different SSWCs, seashell waste powder (100 g) was first carbonized at 400 °C (2.5 °C min−1) under flowing ultra-high purity (UHP) N2 or CO2 gas at 300 mL min−1 for 2 h. The resultant char was mixed with a 6 M potassium hydroxide (KOH) solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, w/w) and maintained at 60 °C for 24 h. Then, the material was heated to 400 °C at 10 °C min−1, under flowing UHP N2 or UHP CO2 at 300 mL min−1 and maintained for 30 min. This was followed by heating to 700 °C at 3 °C min−1, after which the sample was maintained for 1 h under flowing UHP N2 or UHP CO2 at 300 mL min−1. The resultant catalyst was treated with a 2 M hydrochloric acid (HCl) solution at 90 °C for 1 h, followed by washing with deionized water. Finally, the sample was dried at 60 °C for 24 h. The samples prepared under N2 and CO2 atmospheres were denoted as SSWC-N2 and SSWC-CO2, respectively.
:3, w/w) and maintained at 60 °C for 24 h. Then, the material was heated to 400 °C at 10 °C min−1, under flowing UHP N2 or UHP CO2 at 300 mL min−1 and maintained for 30 min. This was followed by heating to 700 °C at 3 °C min−1, after which the sample was maintained for 1 h under flowing UHP N2 or UHP CO2 at 300 mL min−1. The resultant catalyst was treated with a 2 M hydrochloric acid (HCl) solution at 90 °C for 1 h, followed by washing with deionized water. Finally, the sample was dried at 60 °C for 24 h. The samples prepared under N2 and CO2 atmospheres were denoted as SSWC-N2 and SSWC-CO2, respectively.
2.2. Feedstock characterization
FNW was analyzed with Fourier-transform infrared spectroscopy (FTIR) using a Nicolet iS50 FTIR spectrometer (Thermo Fisher Scientific; Waltham, MA, USA) to identify its chemical composition.The American Society for Testing and Materials (ASTM) D6980-17, ASTM D1203-21, and ASTM D5630-94 standard test methods were used to determine the moisture, volatile matter, and ash content in FNW, respectively. The difference between the initial sample mass and the total moisture, volatile matter, and ash content was considered as the fixed matter content in FNW. The elemental composition of FNW was determined using a FlashSmart 2000 elemental analyzer (Thermo Fisher Scientific; Waltham, MA, USA). Thermogravimetric analysis (TGA) of a 30 mg sample of FNW, which was heated at 10 °C min−1, was conducted using an STA449 F3 thermal analyzer (NETZSCH; Selb, Germany).
2.3. Catalyst characterization
The composition of the SSWC-N2 and SSWC-CO2 catalysts was determined using an inductively coupled plasma-optical emission spectrometer (ICP-OES; iCAP™ PRO XP DUO, Thermo Fisher Scientific; Waltham, MA, USA).A JSM-7900F scanning electron microscope (SEM) (JEOL; Akishima, Tokyo, Japan) was used to characterize the morphology of the SSWC-N2 and SSWC-CO2 catalysts. X-ray diffraction (XRD) was conducted using an X-ray diffractometer (D/MAX-2200 Ultima, Rigaku Corp.; Tokyo, Japan); the XRD used Cu Kα radiation (λ = 0.154 nm) and operated at 40 kV and 40 mA. The crystalline phase of the sample was identified using the International Center for Diffraction Data database.
Physisorption measurements were performed using an ASAP 2020 system (Micromeritics; Norcross, GA, USA) using N2 to assess the adsorption–desorption isotherms at −196 °C. Prior to measurement, the samples were degassed at 90 °C for 30 min, followed by heating at 150 °C for 12 h in a vacuum.
A temperature programmed desorption of NH3 (NH3-TPD) was performed to analyze surface acidity using an AutoChem II 2920 system (Micromeritics; Norcross, GA, USA). The samples were pretreated in the presence of He for 1 h at 500 °C and 0.5 h at 150 °C. Then, the samples were saturated with 15 vol% NH3 in He. Changes in thermal conductivity detector (TCD) signals were recorded between 150 and 500 °C at a ramp rate of 5 °C min−1, in which the samples were maintained for 1 h in the presence of flowing He at 50 mL min−1.
Chemisorption measurements of samples were conducted using an ASAP2010 system (Micromeritics; Norcross, GA, USA), using 99.999 vol% CO2 as analysis gas. The samples were pretreated with He for 2 h at 500 °C; then, they were evacuated for 1 h at 35 °C such that the pressure was below 0.67 kPa. Adsorption isotherms were recorded at 35 °C to measure the CO2 uptake by the samples.
A sequence of temperature-programmed reduction of H2 (H2-TPR), temperature-programmed oxidation of O2 (O2-TPO), and a repeat of H2-TPR was carried out to probe reducibility using an AutoChem II 2920 (Micromeritics; Norcross, GA, USA). The samples were pretreated in the presence of Ar at 500 °C for 1 h and then cooled to 50 °C. During H2-TPR, changes in the TCD signal were recorded between 50 and 500 °C at a ramp rate of 10 °C min−1, in which the samples were maintained for 1 h in the presence of 10 vol% H2 in Ar at 50 mL min−1. A liquid trap was maintained at −70 °C to remove moisture from the gas prior to entering the TCD. After the sample was cooled to 50 °C in the presence of Ar, O2-TPO was conducted within 40–500 °C at a ramp rate of 10 °C min−1, where the samples were maintained for 1 h in the presence of 5 vol% O2/Ar at 50 mL min−1. After cooling the samples to 50 °C in the presence of Ar, H2-TPR was repeated.
2.4. Thermocatalytic conversion of FNW
Fig. S2† presents a schematic of the reactor setup used for the thermocatalytic conversion of FNW. For non-catalytic experiments, 1 g of FNW was loaded into the reactor, which was purged by flowing UHP N2 or UHP CO2 at 100 mL min−1 for 10 min at room temperature (∼23 °C) to remove all oxygen within the reactor. Then, the FNW feedstock was heated to 500 °C at 10 °C min−1 under flowing N2 or CO2 (the same gas used for purging) at 100 mL min−1; the feedstock was maintained at 500 °C for 1 min. For in situ thermocatalytic conversion, a FNW and catalyst mixture (FNW/catalyst = 20, mass basis) was loaded into the reactor (Fig. S2a†). For ex situ thermocatalytic conversion, a catalyst bed was established after the feedstock to allow vaporized species from the FNW migrate through the catalyst bed (Fig. S2b†). The subsequent steps in this process were the same as those performed during the non-catalytic experiments.2.5. Product analysis
A fusion gas analyzer micro-gas chromatograph (Micro GC) system (INFICON; Bad Ragaz, Switzerland) (see Fig. S2†) was used to identify and quantify the non-condensable compounds (i.e., gaseous products) created during the thermocatalytic conversion of FNW; this system was directly connected to the reactor outlet. Condensable compounds (i.e., caprolactam and other liquid by-products) were collected in the condenser (Fig. S2†) and quantified using an 8890 GC equipped with a 5977B mass spectrometer (MS) (Agilent Technologies; Santa Clara, CA, USA). The sample analysis conditions that were applied to the Micro GC and GC-MS systems are detailed in Tables S1 and S2,† respectively. Condensable compounds were identified by comparing their GC-MS spectrograms with those present in the National Institute of Standards and Technology mass spectral library. Pure caprolactam (product no. C2204, Sigma Aldrich; St Louis, MO, USA) was used to confirm GC-MS peaks corresponding to caprolactam by matching the mass spectrum with the retention time. The actual amount of caprolactam was quantified with an external standard method using a GC equipped with a flame ionization detector; the calibration curve with this method is shown in Fig. S3.† The caprolactam yield was calculated using eqn (1):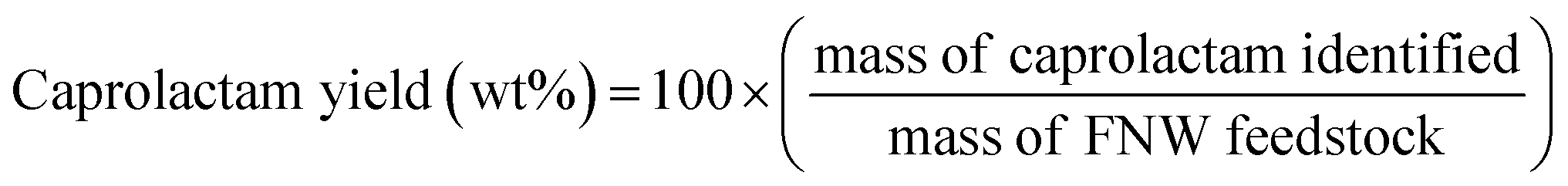 | (1) |
3. Results and discussion
3.1. Feedstock characterization
To identify the major repeating units of FNW, FTIR was utilized to characterize the key functional groups of FNW. The resultant FTIR spectra are shown in Fig. 2a; the FNW sample exhibited various peaks that corresponded to the characteristic peaks of polyamide 6,32 indicating the composition of FNW.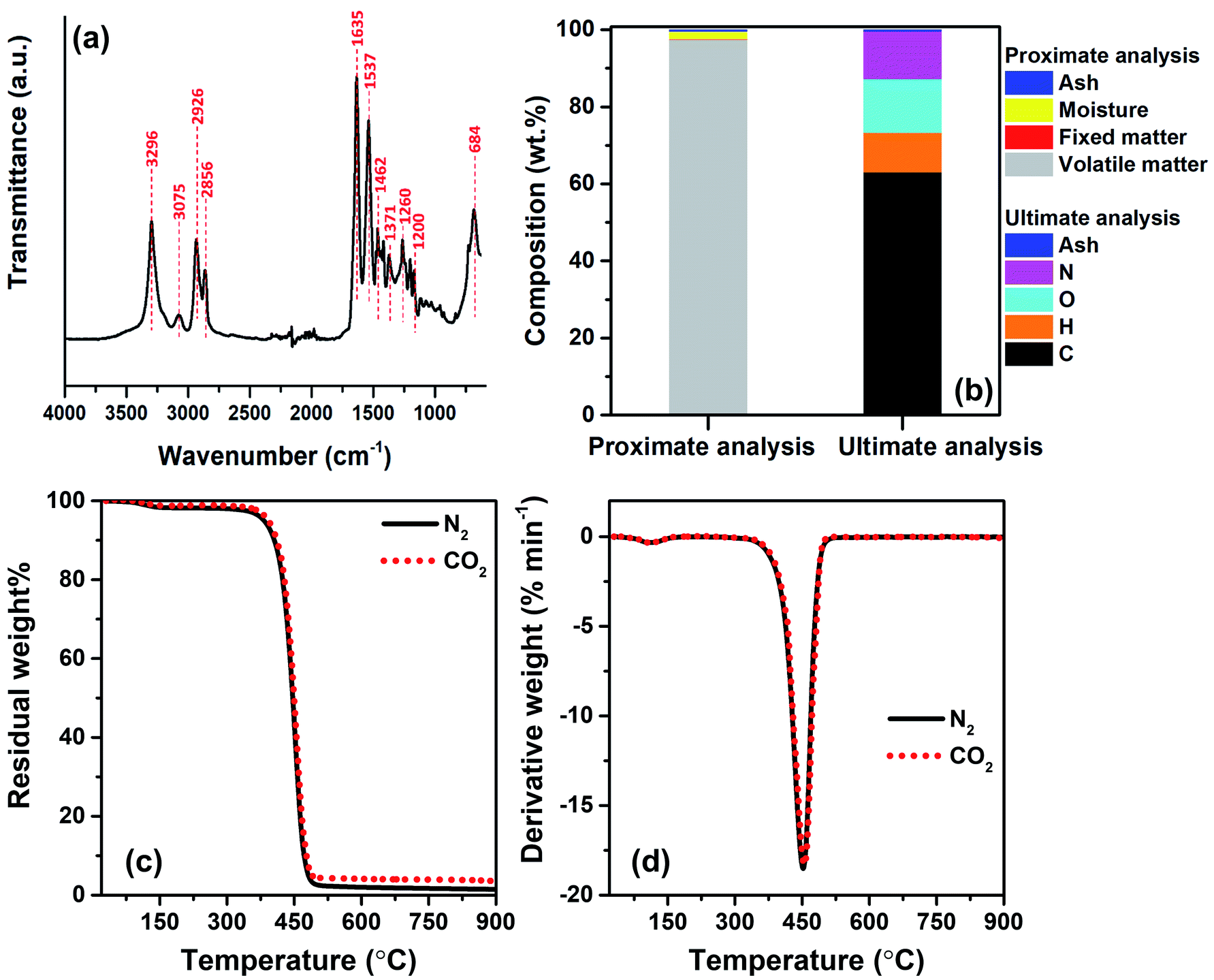 | ||
| Fig. 2 (a) FTIR spectrum of FNW; (b) proximate and ultimate analyses results of FNW; (c) residual mass change in terms of temperature for the TGA of FNW under N2 and CO2 environments; and (d) derivative weight variation in terms of temperature for the TGA of FNW under N2 and CO2 environments. | ||
Proximate analysis of FNW showed that it is composed of 97.48 wt% volatile matter, 0.01 wt% fixed carbon, 2.01 wt% moisture, and 0.50 wt% ash. Ultimate analysis revealed that FNW was composed of 63.06 wt% carbon, 10.28 wt% hydrogen, 13.89 wt% oxygen, 12.27 wt% nitrogen, and 0.50 wt% ash; the composition of FNW is summarized in Fig. 2b.
Fig. 2c and d illustrate the apparent thermal decomposition patterns of FNW under N2 and CO2 atmospheres, respectively, as recorded by TGA. An approximate 2 wt% mass loss was observed from 100 to 350 °C, corresponding to moisture evaporation (Fig. 2c), while a distinct decomposition zone between 350 and 500 °C occurred during the thermal degradation of FNW. Approximately 98 wt% of FNW was decomposed within this zone, which was consistent with its volatile matter content (Fig. 2b). Approximately 1 wt% of FNW remained after TGA; this residual mass was consistent with its fixed carbon and ash content (Fig. 2b). The mass change (Fig. 2c) and mass degradation rate (Fig. 2d) of FNW under a N2 environment were difficult to differentiate from those obtained under a CO2 environment. This means that CO2 had no apparent effect on the thermal degradation of FNW (i.e., the overall loss of thermal mass from FNW); however, CO2 may affect the chemical composition and yield of products derived from FNW.
3.2. Non-catalytic thermal conversion of FNW
Fig. 3a shows that the non-catalytic thermal conversion of FNW produces gas, caprolactam, and liquid; no solids remained as full thermal degradation was achieved at 500 °C (Fig. 2c and d). Gaseous products from non-catalytic thermal conversion were composed of non-condensable gases such as H2, CO, CO2, and C1–C4 hydrocarbons; liquid products included a mixture of by-products in addition to caprolactam. Fig. S4† shows a representative GC-MS chromatogram of the liquid products yielded from the thermocatalytic conversion of FNW at 500 °C. The liquid by-products identified were classified as azepines (7-butyl-3,4,5,6(2H)-tetrahydroazepine, (E)-N-(azepan-2-ylidene)octan-1-amine, and 1-(3,4,5,6-tetrahydro-2H-azepin-7-yl)-2-azepanone), imines (2-methyl-6-tridecyl-6-piperidene, (Z)-octadec-9-en-1-imine, and 3-(pyrrolidin-1-yl)propan-1-imine), and cyclic dimers (1,8-diazacyclotetradecane-2,9-dione and 1,6,14-triazacyclohenicosane-2,7,15-trione) (see Table S3†).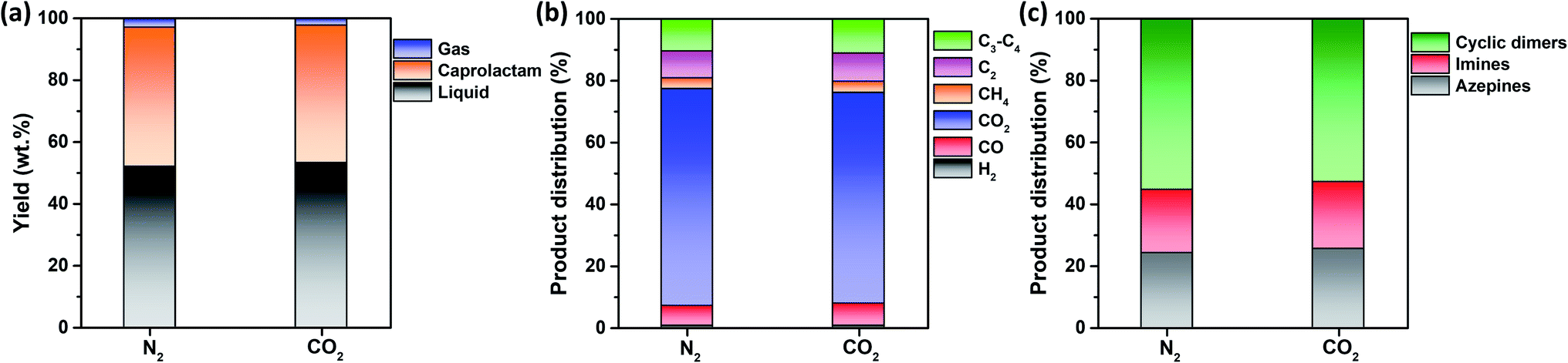 | ||
| Fig. 3 (a) Yields of gaseous and liquid products, and caprolactam produced from the non-catalytic thermal conversion of FNW at 500 °C under N2 and CO2 environments; (b) distribution of gaseous products; (c) distribution of liquid products. | ||
In Fig. S5,† the change in the yield of caprolactam produced through the non-catalytic thermal conversion of FNW under N2 and CO2 atmospheres from 500 to 900 °C is presented. The caprolactam yield clearly decreased with increasing temperature from 500 to 900 °C. This is most likely due to thermal degradation of caprolactam itself at temperatures higher than 500 °C. The caprolactam yield obtained under N2 and CO2 was not significantly different at 500 and 600 °C; however, using the CO2 medium in non-catalytic thermal conversion of FNW lowered the caprolactam yield at >600 °C (Fig. S5†). The use of CO2 as a thermal conversion medium enhances the thermal cracking of volatile species evolved from feedstock (biomass or plastics),33 which is favored at temperatures exceeding 700 °C.34 This results in higher yields of gaseous products and lower yields of liquid and solid products under a CO2 atmosphere at >600 °C than those obtained using a typical inert medium such as N2.33 As such, at temperatures lower than 700 °C, the caprolactam yield achieved in N2 and CO2 was not significantly different. The temperature of 500 °C led to the highest caprolactam yield (Fig. S5†), and the full thermal degradation of FNW occurred at ∼500 °C under both N2 and CO2 atmospheres (Fig. 2c and d). Therefore, 500 °C was used in further investigations on the thermocatalytic conversion of FNW.
Fig. 3a shows that the product yields obtained from the non-catalytic thermal conversion of FNW under N2 and CO2 atmospheres did not substantially differ. Enhanced thermal cracking of volatiles by CO2 is favored at temperatures exceeding 700 °C;34 as such, for the non-catalytic thermal conversion of FNW at 500 °C, CO2 was an inert agent (e.g., N2). The distribution of gaseous and liquid products derived from FNW under a CO2 atmosphere was similar to that under an N2 atmosphere (Fig. 3b and c). The non-catalytic thermal conversion of FNW yielded approximately 45 wt% of caprolactam (Fig. 3a).
3.3. Catalyst characterization
The seashell waste used as the feedstock for the preparation of two SSWC catalysts was made mainly of polymorph of CaCO3, such as aragonite, as proven by the XRD analysis (Fig. S6†). We prepared the SSWC-N2 and SSWC-CO2 catalysts via carbonization of seashell waste and sequential KOH-activation. Table 1 and Fig. 4 show the porosity of the SSWC catalysts through N2-physisorption; both catalysts had low specific surface areas with type III isotherms, indicating low adsorbent–adsorbent interactions. SSWC-N2 had a Brunauer, Emmett, and Teller specific surface area of 3.05 m2 g−1; this was 1.82-fold higher than that of SSWC-CO2. Both catalysts consist of bimodal pores, but the broad large peak above 10 nm indicates that the pores are irregular (Fig. 4). The low geometric surface areas indicate that the catalysts went through a molten state during the preparation to form the fused catalyst, mostly composed of surfaces.| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameterd (nm) | Basicitye (mmol g−1) | |||
|---|---|---|---|---|---|---|---|
| BET | Externalb | Microporeb | Mesoporec | Totald | |||
| a Seashell waste-derived catalysts under N2: SSWC-N2, seashell waste-derived catalysts under CO2: SSWC-CO2, and BET: Brunauer, Emmett, and Teller. b Determined by the t-plot method. c Calculated by subtracting micropore volume from total pore volume. d Determined by the Barrett–Joyner–Halenda (BJH) desorption method at a relative pressure (P/P0) of 0.99. e Measured using the CO2 chemisorption method at a temperature of 35 °C. | |||||||
| SSWC-N2 | 3.05 | 1.88 | 0.0005 | 0.1695 | 0.017 | 26.5 | 3.34 |
| SSWC-CO2 | 1.68 | 1.13 | 0.0002 | 0.0088 | 0.009 | 23.0 | 8.10 |
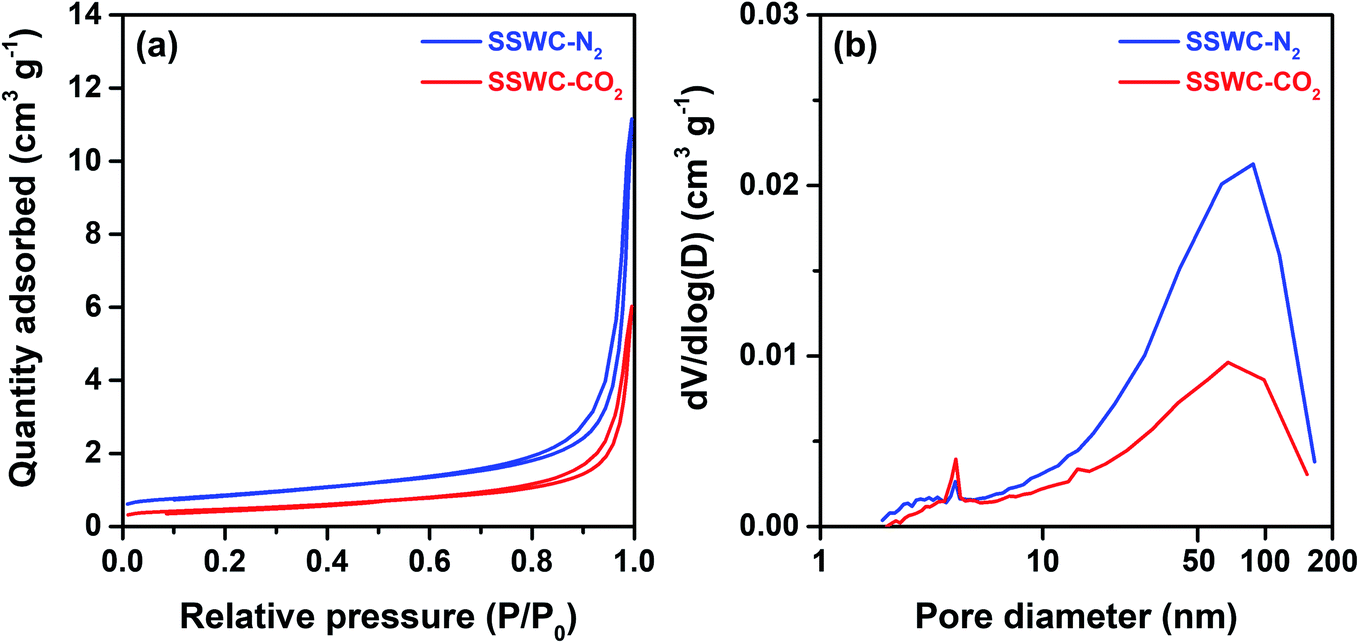 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherm; (b) pore size distribution under N2 physisorption for SSWC-N2 and SSWC-CO2. | ||
Fig. 5a shows the SEM images of the SSWC-N2 and SSWC-CO2 catalysts. It is clearly shown that SSWC-CO2 is more crystalline than SSWC-N2, having a typical cubic shape morphology. As listed in Table S4,† the two catalysts were composed of various inorganic chemicals, with CaCO3 being predominant. The carbonization of seashell waste under a CO2 atmosphere increased the CaCO3 content while decreasing the K content in the catalyst compared with that during carbonization under a N2 atmosphere. The XRD patterns of SSWC-N2 and SSWC-CO2 are shown in Fig. 5b; the diffraction peaks of both catalysts were almost identical, representing CaCO3 as per the ICDD database (JCPDS no. 01-072-1937), consistent with the ICP-OES results (Table S4†). This also indicates that a polymorphic transition of aragonite occurred during the carbonization of the seashell waste (Fig. S6† and 5b). Moreover, SSWC-CO2 showed a higher XRD intensity than SSWC-N2, indicating that the former has a higher degree of crystallinity than the latter; these XRD results were supported by the SEM results (Fig. 5a).
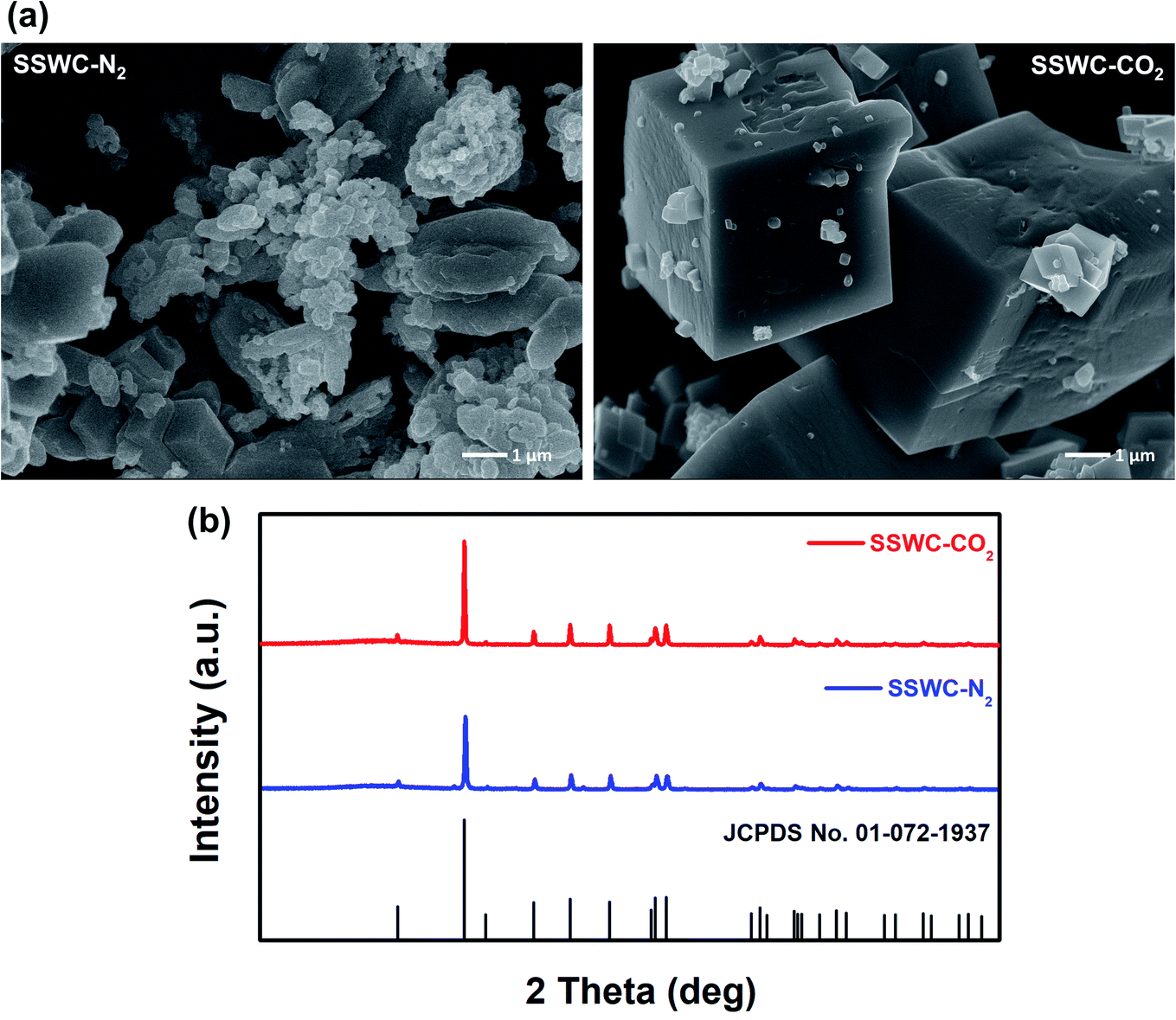 | ||
| Fig. 5 (a) Scanning electron microscopy (SEM) images of SSWC-N2 and SSWC-CO2 (SEM images with a magnification of 5 μm are given in Fig. S7†); (b) XRD patterns of SSWC-N2 and SSWC-CO2. | ||
Table 1 demonstrates that the increase in the CaCO3 concentration of SSWC-CO2 elevated CO2 uptake, which was 2.43-fold higher than that of SSWC-N2. Both catalysts exhibited negligible reducibility through the H2-TPR/O2-TPO/H2-TPR sequence (Fig. S8†). This means that most impurities in SSWC, which occurred as various metals as shown in Table S4,† were not exposed to the surface. Similarly, nearly zero acid sites were observed on the surface of both catalysts as a result of NH3-TPD (Fig. S9†). The characterization results indicate that the atmospheric variations, where seashell waste carbonization occurred (e.g., N2 or CO2), may alter the physicochemical properties of the SSWC, affecting its basicity.
3.4. Thermocatalytic conversion of FNW using the SSWC catalysts
This study hypothesized that catalyst configuration during thermocatalytic processing may affect caprolactam recovery from FNW. Thus, two different catalyst loading configurations were tested: in situ and ex situ. For in situ configuration, the thermocatalytic conversion of FNW occurred within a reactor where FNW and the catalyst were mixed together to enable direct contact between the two (Fig. S2a†).35,36Ex situ configuration involved no direct contact between FNW and the catalyst; here, nets were first thermally cracked to release high molecular weight volatile species. Then, volatile species were passed through a catalyst bed in which they were further transformed into lower molecular weight species (Fig. S2b†).35,36Fig. 6a shows yields for gaseous and liquid products, and caprolactam obtained via the thermocatalytic conversion of FNW under a N2 atmosphere with and without the SSWC-N2 catalyst, using in situ and ex situ configurations. The ex situ configuration did not significantly affect the caprolactam yield compared with that of the non-catalytic process; however, it increased the gaseous product yield while decreasing the liquid product yield compared with those from non-catalytic thermal conversion. This indicates that the catalyst improved the thermal cracking of high molecular weight volatiles released from FNW via thermal degradation. The use of the SSWC-N2 catalyst in the in situ configuration increased the caprolactam yield to 63.6 wt%; thus, the thermal depolymerization of FNW to caprolactam was more selective using the in situ configuration than that using ex situ configuration. This may be attributed to the intimate contact of the catalyst with high molecular weight volatile species generated via thermal depolymerization.37,38 The high diffusion efficiency of polymer-derived (e.g., FNW-derived) heavy volatiles enables their diffusion into catalyst pores; this facilitates direct contact between volatiles and catalytic sites using in situ configuration.39,40 This may amplify the catalytic effect for promoting the thermal depolymerization of FNW, which may potentially contribute to higher caprolactam yields than those derived from using ex situ configuration. The distributions of gaseous and liquid products in the three cases were similar (Fig. 6b and c). As the in situ configuration yielded the highest quantity of caprolactam, further experiments were conducted using this configuration.
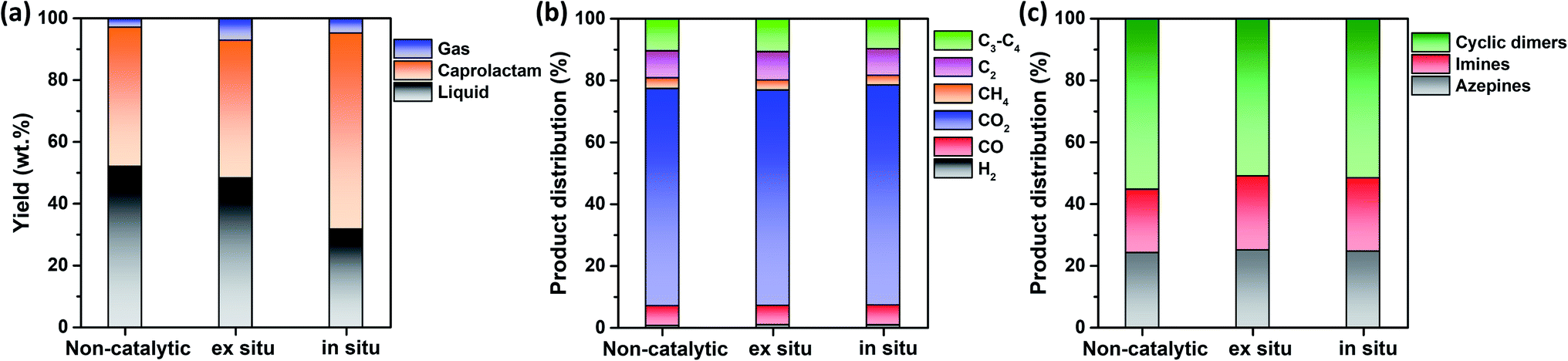 | ||
| Fig. 6 (a) Yields of gaseous and liquid products, and caprolactam produced from the thermocatalytic conversion of FNW at 500 °C under an N2 environment using SSWC-N2 in different catalyst configurations; (b) distribution of gaseous products; (c) distribution of liquid products. | ||
The base-catalyzed decomposition of polyamide 6 proceeds through an anionic chain mechanism, which occurs more rapidly than non-catalytic decomposition.41 As shown in Fig. 7, this mechanism involves the base-catalyzed deprotonation of amide groups and the cleavage of the amide link that occurs on the polyamide 6 chain, followed by intramolecular cyclization that produces lactam units (i.e., caprolactam). As the FNW feedstock used in this study is largely made up of polyamide 6 (Fig. 2a), the thermocatalytic conversion of these nets into caprolactam may follow a similar pathway.
 | ||
| Fig. 7 Anionic chain reaction occurring in thermal degradation of polyamide 6 in the presence of a base catalyst. | ||
Fig. 8 shows that the SSWC-CO2 catalyst yields more caprolactam than the SSWC-N2 catalyst under both N2 and CO2 atmospheres. For example, under an N2 environment, the SSWC-CO2 catalyst yielded more caprolactam (70.3 wt%) than the SSWC-N2 catalyst (63.6 wt%). It is likely that this higher yield using the SSWC-CO2 catalyst was due to the higher basicity of this catalyst compared with that of the SSWC-N2 catalyst (Table 1). The use of CO2 as a thermal conversion medium further enhanced the effectiveness of the SSWC catalyst in yielding caprolactam from FNW; for example, the yield reached 80 wt% when using the SSWC-CO2 catalyst under a CO2 environment. CO2 molecules have been observed to interact with polar functional groups, modifying their reactivity on the catalyst surface.42,43 Thus, the synergistic effect of the CO2 reaction environment and the SSWC catalyst is most likely because the molecular interaction between CO2 and the polar functional groups of FNW (e.g., amides) causes the more reactive base-catalyzed deprotonation of amide groups and the cleavage of amide linkages.
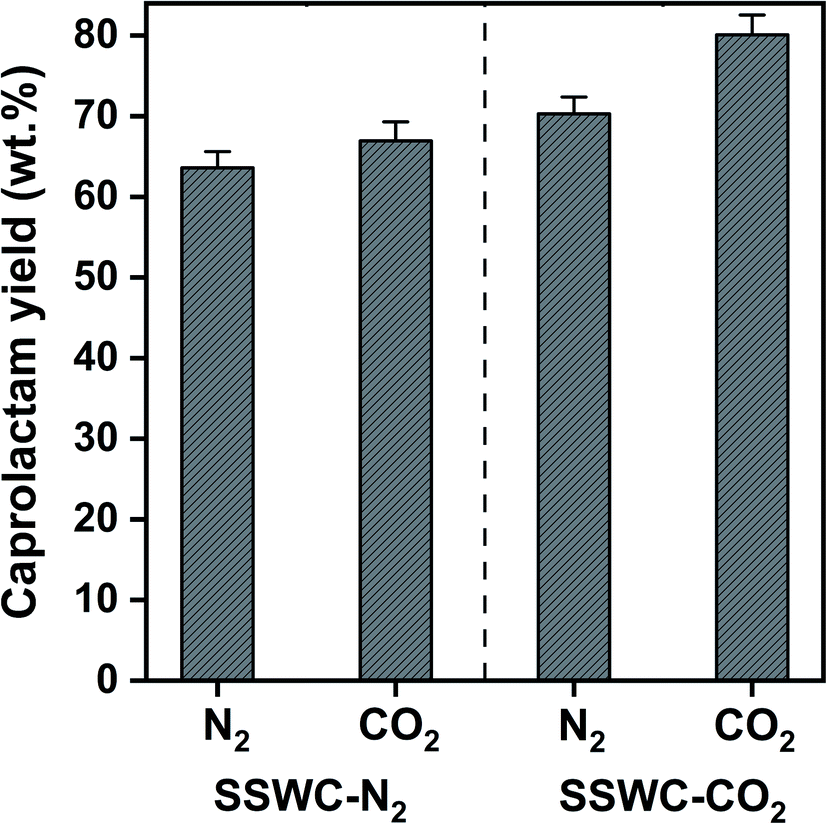 | ||
| Fig. 8 Caprolactam yield from the thermocatalytic conversion of FNW at 500 °C under N2 and CO2 environments using SSWC-N2 and SSWC-CO2 in an in situ catalyst configuration. | ||
Although there have been a limited number of literature reports, our results are compared with the most recent studies on thermocatalytic treatment of commercial nylon 6 and the feedstock similar to FNW, as summarized in Table S5.† The study done by Eimontas et al. has claimed 83% caprolactam yield (based on GC-MS area% without further quantification) with a ZSM-5 zeolite catalyst,44 but the GC-MS area %-based yield cannot reflect the actual caprolactam yield. In another recent study that quantified the caprolactam yield using an external standard method (a similar method to ours), 70 wt% caprolactam yield was obtained from commercial nylon 6 with a scallop shell catalyst calcined under air.31 In comparison, our yield (80 wt%; corresponding to ∼91 GC-MS area %, as shown in Fig. S4†) is the highest among those that have been reported.
To assess the reusability of the SSWC-CO2 catalyst, the spent SSWC-CO2 catalyst was reused at 500 °C under a CO2 atmosphere for three cycles; the results are shown in Fig. 9. A slight loss in the caprolactam yield was observed after each cycle. This is most likely due to coke deposition on the catalyst surface, as indicated by TGA analysis of the spent catalyst in air (Fig. S10†). The peaks ranging from 400 to 800 °C were attributed to the oxidation of coke.45 In Fig. S10b,† the coke oxidation peak moved to higher temperature with increasing the number of reuse cycles, which indicates that harder oxidized coke was formed. Fig. S11† shows a SEM image of the spent SSWC-CO2 catalyst after the 3rd reuse. In addition, the XRD patterns of fresh and spent catalysts are compared in Fig. S12.† The fresh catalysts exhibited higher XRD intensities than the spent catalysts, indicating that the spent ones have a lower degree of crystallinity than the fresh ones. Despite the yield loss, there was no significant reduction in the caprolactam yield as per the Student's t-test at the 95% confidence limit. Fig. 9 also shows that an increase in the SSWC-CO2 catalyst loading did not influence the caprolactam yield; this demonstrates that the catalyst is reusable at an elevated catalyst loading.
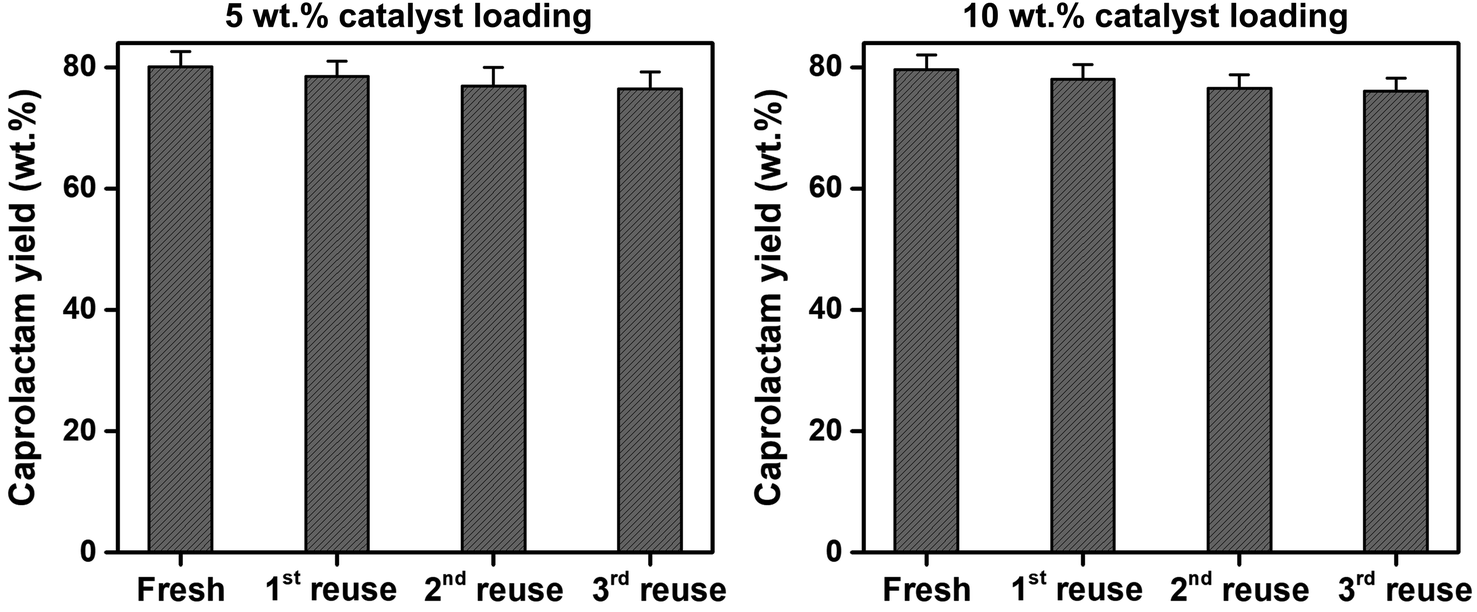 | ||
| Fig. 9 Reusability of the spent SSWC-CO2 catalyst with different catalyst loadings for the thermocatalytic conversion of FNW at 500 °C under a CO2 environment. | ||
Fig. 10 describes the marine waste upcycling process proposed in this study. As previously discussed, the thermocatalytic processing of FNW using the SSWC-CO2 catalyst produced gases including CO2, combustible gases (e.g., H2, CO, and C1–C4 hydrocarbons), and caprolactam-rich liquid products. Gaseous products were fed into a CO2 separation system (e.g., a monoethanolamine-based CO2 capture process46); the separated CO2 was recycled into the thermocatalytic FNW treatment or seashell waste carbonization. Combustible gases may be used as fuel to produce the heat and power required for thermocatalytic treatment and catalyst preparation. Caprolactam (80% wt yield based on the feedstock) was separated from the caprolactam-rich liquid product via caprolactam purification using toluene or acetone as a solvent, and introducing a CO2 flow.47 This entire process potentially helps develop an energy-independent marine waste upcycling system that effectively utilizes CO2.48
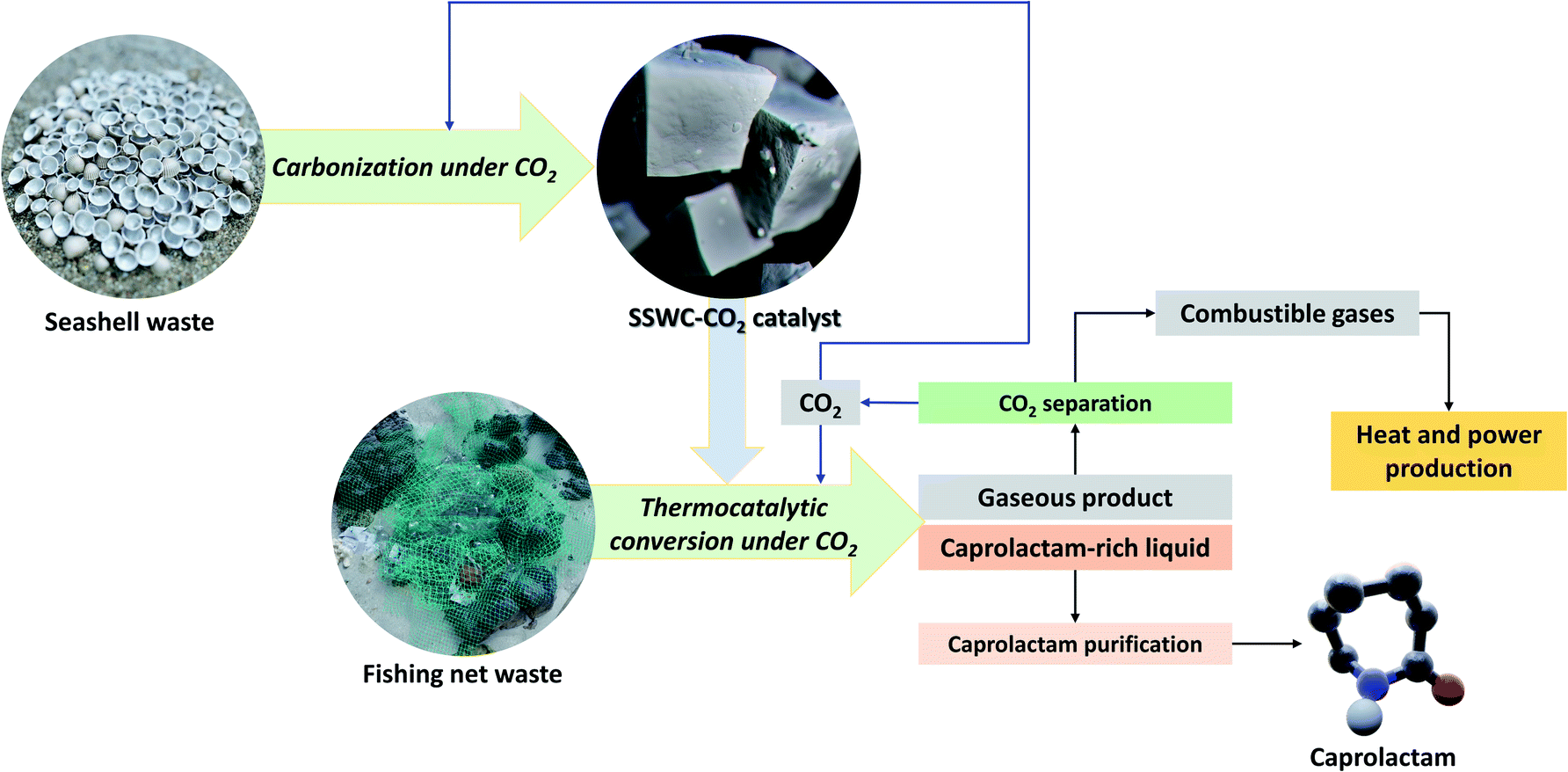 | ||
| Fig. 10 Concept underpinning the thermocatalytic upcycling of marine waste (FNW and seashell waste) to monomer (caprolactam) under a CO2 environment with the recycling of by-products. | ||
Globally, 640000 t of fishing nets are abandoned in oceans every year,5 contributing to the release of 7.1 × 1015 microplastic pieces (this estimate is based on data in Table S6†).49 The thermocatalytic process depicted in Fig. 10 is able to convert 80% wt of FNW into caprolactam, valued at roughly one billion USD (Table S6†). This implies that the proposed marine waste upcycling process may potentially realize the reduction of microplastic pollution and increase the economic potential for marine waste valorization, such as with FNW and seashell waste.
4. Conclusions
This study performed the thermocatalytic treatment of FNW with catalysts prepared by carbonizing seashell waste under N2 and CO2 atmospheres (SSWC-N2 and SSWC-CO2, respectively). This method reclaimed value from different marine waste materials (e.g., FNW and seashell waste) simultaneously. FNW used as feedstock in this study was made of polyamide 6, as confirmed by FTIR analysis. The carbonization environment materially affected the properties of the catalyst, such as its CaCO3 content and basicity. The SSWC-CO2 catalyst had a higher basicity and CaCO3 content than SSWC-N2. The SSWC catalysts enhanced the caprolactam yield (a value-added chemical used as the monomer of nylon 6), compared with the non-catalytic thermal conversion of FNW because the selective depolymerization of the polyamide bond to caprolactam involved base-catalyzed reactions. For the thermocatalytic conversion of FNW using a SSWC catalyst, the in situ catalyst configuration provided a higher caprolactam yield than that from the ex situ configuration. Base-catalyzed caprolactam production using the SSWC-CO2 catalyst (in situ configuration) was further promoted in the thermocatalytic conversion of FNW carried out under a CO2 atmosphere; the caprolactam yield reached 80 wt% of the FNW feedstock (a higher reported yield than previous studies), and the SSWC-CO2 catalyst was stable after three consecutive cycles. There is no single simple solution for concurrently achieving marine waste upcycling with CO2 utilization. Although this industry is still in its infancy, continuous research and development should help advance this industry to become economically viable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Government of Republic of Korea (Ministry of Science and ICT) (No. 2020R1C1C1003225).References
- D. Kindy, With Ropes and Nets, Fishing Fleets Contribute Significantly to Microplastic Pollution, Smithsonian Magazine, 2021 Search PubMed.
- Sea Around Us, https://www.seaaroundus.org, accessed January 13, 2022.
- One Green Planet, https://www.onegreenplanet.org, accessed January 13, 2022.
- S. Orasutthikul, D. Unno and H. Yokota, Constr. Build. Mater., 2017, 146, 594–602 CrossRef.
- M. Charter, R. Carruthers and S. F. Jensen, Products from Waste Fishing Nets, Circular Ocean led by the Environmental Research Institute, Scotland, 2018 Search PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- J. Gong, X. Chen and T. Tang, Prog. Polym. Sci., 2019, 94, 1–32 CrossRef CAS.
- B. Zhang, C. Song, C. Liu, J. Min, J. Azadmanjiri, Y. Ni, R. Niu, J. Gong, Q. Zhao and T. Tang, J. Mater. Chem. A, 2019, 7, 22912–22923 RSC.
- N. Liu, Z. Hu, L. Hao, H. Bai, P. He, R. Niu and J. Gong, J. Environ. Chem. Eng., 2022, 10, 106959 CrossRef CAS.
- F. Zhang, F. Wang, X. Wei, Y. Yang, S. Xu, D. Deng and Y.-Z. Wang, J. Energy Chem., 2022, 69, 369–388 CrossRef.
- L. O. Mark, M. C. Cendejas and I. Hermans, ChemSusChem, 2020, 13, 5808–5836 CrossRef CAS PubMed.
- J. Lee, E. E. Kwon, S. S. Lam, W.-H. Chen, J. Rinklebe and Y.-K. Park, J. Cleaner Prod., 2021, 321, 128989 CrossRef CAS.
- J. Shah, M. R. Jan and Adnan, J. Ind. Eng. Chem., 2014, 20, 3604–3611 CrossRef CAS.
- J. Shah, M. R. Jan and Adnan, J. Anal. Appl. Pyrolysis, 2015, 114, 163–171 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- S. Kim, N. Lee, S. W. Lee, Y. T. Kim and J. Lee, Chem. Eng. J., 2021, 412, 128626 CrossRef CAS.
- S. Jung, D. Choi, Y.-K. Park, Y. F. Tsang, N. B. Klinghoffer, K.-H. Kim and E. E. Kwon, Chem. Eng. J., 2020, 399, 125889 CrossRef CAS.
- J. Gong, J. Liu, X. Chen, Z. Jiang, X. Wen, E. Mijowska and T. Tang, J. Mater. Chem. A, 2015, 3, 341–351 RSC.
- J. A. Bennett, K. Wilson and A. F. Lee, J. Mater. Chem. A, 2016, 4, 3617–3637 RSC.
- D. Li, W. Chen, J. Wu, C. Q. Jia and X. Jiang, J. Mater. Chem. A, 2020, 8, 24977–24995 RSC.
- Z. Yao, M. Xia, H. Li, T. Chen, Y. Ye and H. Zheng, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2502–2530 CrossRef CAS.
- K. H. Mo, U. J. Alengaram, M. Z. Jumaat, S. C. Lee, W. I. Goh and C. W. Yuen, Constr. Build. Mater., 2018, 162, 751–764 CrossRef CAS.
- E.-I. Yang, S.-T. Yi and Y.-M. Leem, Cem. Concr. Res., 2005, 35, 2175–2182 CrossRef CAS.
- G.-L. Yoon, B.-T. Kim, B.-O. Kim and S.-H. Han, Waste Manag., 2003, 23, 825–834 CrossRef CAS PubMed.
- N. Viriya-empikul, P. Krasae, B. Puttasawat, B. Yoosuk, N. Chollacoop and K. Faungnawakij, Bioresour. Technol., 2010, 101, 3765–3767 CrossRef CAS PubMed.
- S. Hu, Y. Wang and H. Han, Biomass Bioenergy, 2011, 35, 3627–3635 CrossRef CAS.
- W. Jindapon, S. Jaiyen and C. Ngamcharussrivichai, Energy Convers. Manage., 2016, 122, 535–543 CrossRef CAS.
- A. Perea, T. Kelly and Y. Hangun-Balkir, Green Chem. Lett. Rev., 2016, 9, 27–32 CrossRef CAS.
- N. Chaihad, I. Kurnia, A. Yoshida, C. Watanabe, K. Tei, P. Reubroycharoen, Y. Kasai, A. Abudula and G. Guan, J. Anal. Appl. Pyrolysis, 2020, 146, 104750 CrossRef CAS.
- K.-H. Lee, K.-W. Kim, A. Pesapane, H.-Y. Kim and J. F. Rabolt, Macromolecules, 2008, 41, 1494–1498 CrossRef CAS.
- N. Lee, S. Kim and J. Lee, J. CO2 Util., 2021, 44, 101414 CrossRef CAS.
- C. Park, S. Lee and J. Lee, Chem. Eng. J., 2022, 427, 131459 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- E. Butler, G. Devlin, D. Meier and K. McDonnell, Renewable Sustainable Energy Rev., 2011, 15, 4171–4186 CrossRef CAS.
- K. Wang, P. A. Johnston and R. C. Brown, Bioresour. Technol., 2014, 173, 124–131 CrossRef CAS PubMed.
- Y. Shirazi, S. Viamajala and S. Varanasi, Front. Chem., 2020, 8, 786 CrossRef CAS PubMed.
- Y.-K. Park, J. S. Jung, J. Jae, S. B. Hong, A. Watanabe and Y.-M. Kim, Chem. Eng. J., 2019, 377, 119742 CrossRef CAS.
- H. Shafaghat, H. W. Lee, Y. F. Tsang, D. Oh, J. Jae, S.-C. Jung, C. H. Ko, S. S. Lam and Y.-K. Park, Chem. Eng. J., 2019, 366, 330–338 CrossRef CAS.
- H. Bockhorn, A. Hornung, U. Hornung and J. Weichmann, Thermochim. Acta, 1999, 337, 97–110 CrossRef CAS.
- S.-i. Fujita, Y. Onodera, H. Yoshida and M. Arai, Green Chem., 2016, 18, 4934–4940 RSC.
- C. Park and J. Lee, Green Chem., 2020, 22, 2628–2642 RSC.
- J. Eimontas, S. Yousef, N. Striūgas and M. A. Abdelnaby, Renewable Energy, 2021, 179, 1385–1403 CrossRef CAS.
- H. Zhang, Y. Wang, S. Shao and R. Xiao, Sci. Rep., 2016, 6, 37513 CrossRef CAS PubMed.
- K. Li, A. Cousins, H. Yu, P. Feron, M. Tade, W. Luo and J. Chen, Energy Sci. Eng., 2016, 4, 23–39 CrossRef CAS.
- L. Moens, US Pat., US5919927A, 1999 Search PubMed.
- S. Kim, J. Byun, H. Park, N. Lee, J. Han and J. Lee, Energy, 2022, 241, 122876 CrossRef CAS.
- L. S. Wright, I. E. Napper and R. C. Thompson, Mar. Pollut. Bull., 2021, 173, 113115 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02060b |
| ‡ Current affiliation: Department of Earth and Environmental Engineering, Columbia University, New York, NY 10027, USA. |
| This journal is © The Royal Society of Chemistry 2022 |