
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of specific and active surface areas in optimizing hard carbon irreversible capacity loss in sodium ion batteries†
Adrian
Beda
abc,
Cyril
Vaulot
ab,
François
Rabuel
cd,
Matthieu
Morcrette
 cd and
Camélia
Matei Ghimbeu
*abc
cd and
Camélia
Matei Ghimbeu
*abc
aUniversité de Haute-Alsace, CNRS, Institut de Science des Matériaux de Mulhouse (IS2M) UMR 7361, F-68100 Mulhouse, France. E-mail: camelia.ghimbeu@uha.fr
bUniversité de Strasbourg, F-67081 Strasbourg, France
cRéseau sur le Stockage Electrochimique de l’Energie (RS2E), HUB de l’Energie, FR CNRS 3459, 80039 Amiens Cedex, France
dUniversité de Picardie Jules Verne, Laboratoire de Réactivité et Chimie des Solides (LRCS), UMR 7314 CNRS, HUB de l’Energie, 80039 Amiens, France
First published on 7th March 2022
Abstract
Oxygen physi- and chemisorption were used to determine the hard carbon total specific and active surface areas, respectively, which have been correlated with irreversible capacity loss during the first charge/discharge cycle in Na-ion half-cells. Lowering the specific and active surface areas allow to reduce the irreversible capacity loss to 8%. Carbon basal planes and edge planes/defects contributions are discussed.
Na-ion batteries (NIBs) have been studied intensively during the last decade and have become a credible alternative for current lithium-ion battery (LIB) technology.1 The low price and worldwide abundance of sodium and the possible utilization of sustainable and renewable electrode materials are valuable advantages of NIBs. The performance of NIBs is approaching that of LIBs; however, there are still many challenges to be addressed, such as the high initial irreversibility and the low stability over cycling. Among a large variety of anodic carbon materials used in NIBs, hard carbon materials deliver the highest specific capacity, i.e., ∼300 mA h g−1,2vs. only 31 mA h g−1 for graphite.3 Instead, a high irreversible capacity is generally observed during the first charge/discharge cycle, and, in some cases, capacity fading during long-term cycling. The reason for this capacity loss is widely associated to the decomposition (reduction) of electrolyte at the carbon surface which results in the formation of the so-called solid electrolyte interphase (SEI) layer.4 The composition, thickness, morphology and structure of the SEI dictate the Coulombic efficiency (CE), stability and rate capability of the HC anode.
The main factors impacting SEI formation on hard carbon in NIBs have been barely studied compared to LIBs. Dedryvere et al.5 showed that the electrolyte formulation (i.e., salt, solvent and/or additive used) strongly impacts the SEI chemical composition and long-term cycling. Komaba et al. highlighted the importance of the electrode binder type6 and electrolyte additive presence7 on SEI formation and irreversible capacity. The electrochemical cycling conditions (charging rates) were shown to contribute to SEI build-up, leading to different thicknesses and morphologies.8 Bommier and Ji4 reviewed in more detail the non-electrode factors (i.e., electrolytes, additives and solvents) inducing SEI and irreversible capacity.
In addition, the properties of hard carbon are well known to have a strong influence on the SEI layer. It is often mentioned that the HC specific surface area, functional groups, defects and impurities contribute to electrolyte decomposition and SEI formation. For anodic carbon-based materials in LIBs, this topic has been extensively investigated, and insightful correlations have been found,9,10 whereas studies on hard carbons for NIBs are rare.
The specific surface area is considered a main parameter that leads to electrolyte decomposition and an increase in irreversible capacity. Bommier et al.11 reported a decreasing tendency of irreversible capacity with the surface area, assessed by N2 adsorption. However, assessment via N2 adsorption fails to analyse narrow pores (<2 nm), which are known to exist in HC. Later, our group12 proposed the method of CO2 adsorption for surface area determination, and a similar trend was found for the irreversible capacity. In addition, we recently proposed the use of other gases, such as O2 and H2, to evaluate the complex hard carbon porosity,13 which could help in better understanding the impact of porosity on electrochemical performance. Other factors, such as edge defects, can contribute to the irreversible capacity14 due to the strong binding energy of Na ions at these specific sites. The main difficulty, however, is the evaluation of carbon defects, which is customarily done in a qualitative way, using the ID/IG ratio deduced from Raman spectra. Alternatively, we proposed12 the quantitative determination of defects/active sites via the assessment of active surface area (ASA) through the oxygen chemisorption followed by temperature programmed desorption coupled with mass spectrometry (TPD-MS). The active surface comprises the contribution of different carbon edge planes and defects (i.e., vacancies, dislocations, point defects, Stone–Wales defects).15,16 This quantification is of great interest since it may allow us to discriminate between the effect of edge planes/defects and basal planes on the irreversible capacity. Notably, the total specific surface area (TSA) determined by gas adsorption, includes both the basal planes surface and the active surface area.
Oxygen-based functional groups might favour an irreversible capacity due to its interactions with the electrolyte. However, it must be considered that the functional groups are usually bonded on carbon edges (i.e., defects/active sites); therefore, the active sites have a greater impact than the functional groups. This has been evidenced by performing reductive treatments under H2, which led to the removal of oxygen-based functional groups while preserving the active sites and observing no significant impact on the irreversible capacity.12,17 Metal-based inorganic impurities are inherently present in some natural precursors and have been proven to induce high irreversible capacity and low Coulombic efficiency.18,19 Moreover, non-SEI factors such as Na-ion trapping in defects and porosity have been proposed to contribute to irreversibility.20 In summary, among the abovementioned parameters that influence the irreversible capacity, the specific surface area and the active sites appear to be the most significant ones for pure HCs.
Herein, the total surface area and the active surface area were determined by O2 physisorption and chemisorption, respectively, and correlated with the irreversible capacity loss during the first charge/discharge cycle of the Na-ion half-cell.
A series of ten hard carbon materials were prepared using phenolic and carbohydrate precursors, by polymerization and hydrothermal carbonisation, respectively, followed by pyrolysis of the recovered solid product at temperatures ranging from 1300 to 1600 °C. A brief description of the experimental synthesis procedure is provided in the ESI,† and an exhaustive description can be found in our previous work.21
First, the total surface area of carbon powders was determined by O2 adsorption. The choice of this gas was motivated by our recent findings, which illustrate the ability of O2 molecule to detect both ultramicropores (<0.7 nm) and supermicropores (0.7 nm < d < 2.0 nm). In addition, its quadrupole moment is much lower than that of N2 and CO2, limiting the interactions with the HC surface. Moreover, the N2 and Ar gases were found unsuitable to determine the narrow porosity of hard carbons.13 Thus, the O2 adsorption/desorption isotherms were recorded at 77 K, as detailed in the ESI† and presented in Fig. 1. For the sake of clarity, the samples were divided into two categories (high and low adsorbed O2 volumes). Type I isotherms are seen for some HCs (Fig. 1a) and are characterized by a steep increase in the O2 adsorbed volume at low relative pressure, P/P0, followed by a plateau. This isotherm shape is specific to microporous carbons with narrow pore size distributions.
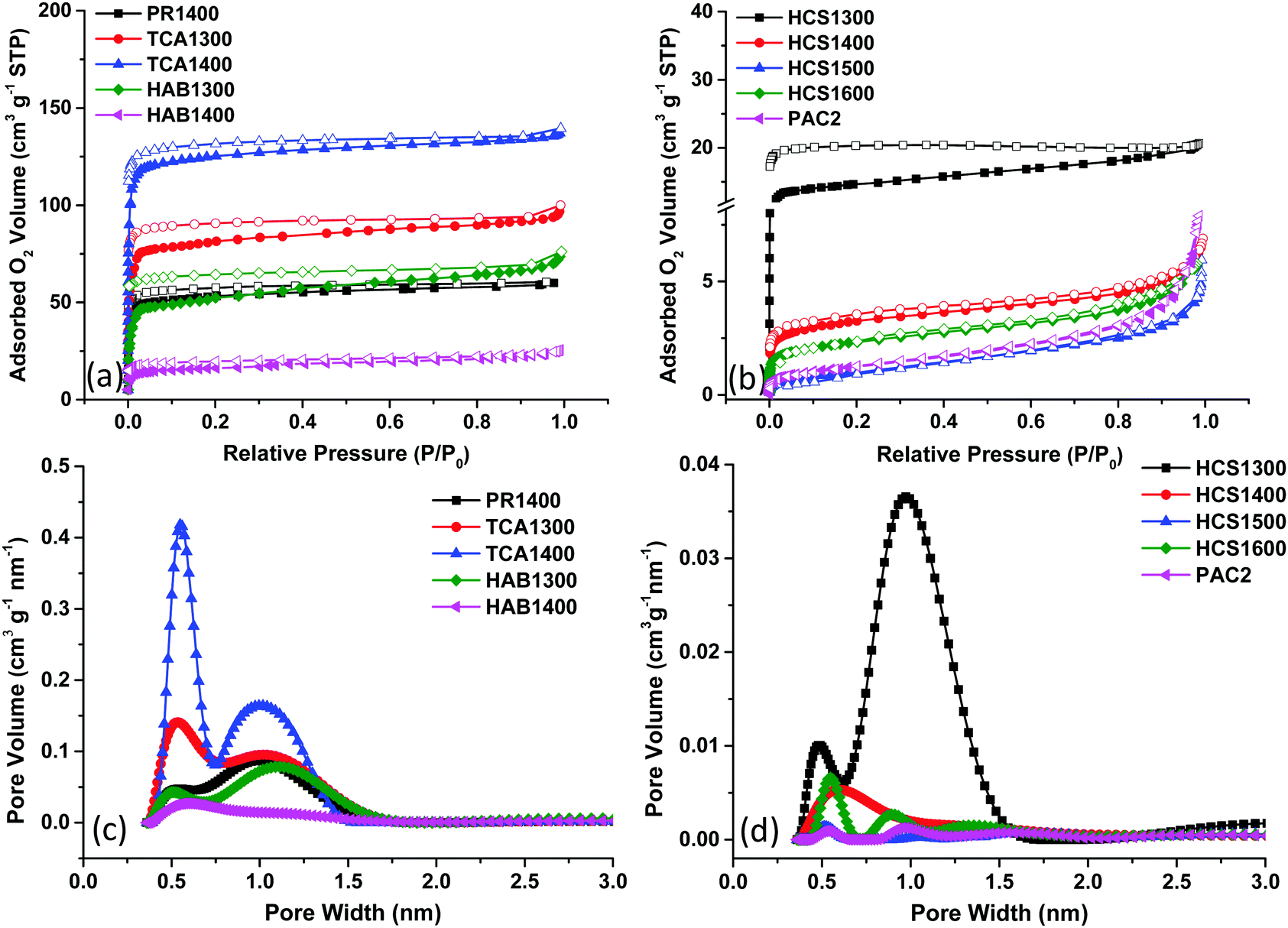 | ||
| Fig. 1 Hard carbon O2 adsorption/desorption isotherms (a and b) and the corresponding two-dimensional nonlocal density functional theory (2D-NLDFT) pore size distribution (c and d). | ||
Both ultramicropores (<0.7 nm) and supermicropores (∼1.1 nm) are seen (Fig. 1c). The average pore size, L0, ranges between 0.77 and 5.02 nm (Table 1). The total surface area (TSA) assessed by the BET model varies from 52 to 421 m2 g−1 for the HAB, PR and TCA materials. Notably, the BET surface obtained with N2 was below 25 m2 g−1 for all materials except TCA100 (104 m2 g−1).21 Thus, O2 adsorption offers much better insights into HC porosity and allows for better discrimination of a possible porosity effect on electrochemical performance than adsorption methods involving other gases. The second series of HCs (Fig. 1b) shows type II isotherms (except HCS1300, type I), which are associated with rather nonporous materials. For some isotherms (eg. HCS1300), the desorption branch is not closing and this might be related to HC narrow pores and in particular their ink-bottle neck shape,22 contributing to the O2 trapping and partial irreversible desorption. The total surface area was much lower (<50 m2 g−1) than that of the previous series of samples (Table 1). The pore volume is much lower as well (Fig. 1d), with most pores sizes below 2.0 nm. Some larger pores (>10 nm) are observed only on the low surface area samples (Fig. S1, ESI†).
| Sample | TSA O2 m2 g−1 | V T cm3 g−1 | L 0 nm | ASA21 m2 g-1 | ASA/(TSA–ASA) | 1st CD mA h g−1 | 1st CC mA h g−1 | 1st Cirrev mA h g−1 | ICL (%) |
|---|---|---|---|---|---|---|---|---|---|
| HCS1300 | 48 | 0.0245 | 1.23 | 11.9 | 0.329 | 342 | 305 | 37 | 11 |
| HCS1400 | 10 | 0.0082 | 1.47 | 3.3 | 0.492 | 345 | 308 | 37 | 11 |
| HCS1500 | 2.2 | 0.0071 | 4.28 | 1.2 | 1.20 | 322 | 297 | 25 | 8 |
| HCS1600 | 6.8 | 0.0069 | 1.84 | 1.3 | 0.236 | 336 | 309 | 27 | 8 |
| PR1400 | 179 | 0.1130 | 1.14 | 3.5 | 0.019 | 349 | 293 | 56 | 16 |
| TCA1300 | 277 | 0.1189 | 0.89 | 11.8 | 0.054 | 423 | 296 | 127 | 30 |
| TCA1400 | 421 | 0.1658 | 0.77 | 21.8 | 0.044 | 349 | 239 | 110 | 32 |
| HAB1300 | 173 | 0.0905 | 1.22 | 16.1 | 0.102 | 307 | 240 | 67 | 23 |
| HAB1400 | 52 | 0.0304 | 1.14 | 9.9 | 0.235 | 319 | 271 | 48 | 15 |
| PAC2 | 4.0 | 0.0093 | 5.02 | 6.4 | −2.904 | 325 | 281 | 44 | 14 |
Next, the active surface area of hard carbon powders was determined as explained in the ESI.† In the first stage, the carbon surface was treated under vacuum (up to 950 °C) and then exposed to O2 chemisorption at 300 °C. The active sites (edge planes/defects) react with oxygen to form complex C–O groups, which are subsequently thermally decomposed by heating to 950 °C and analysed by TPD-MS. The desorption rates of CO and CO2 gases are shown in Fig. 2. CO gas evolution (Fig. 2a and b) is predominant compared to CO2 (Fig. 2c and d) and occurs in the temperature range of 400–900 °C, while CO2 gas evolution is limited to ∼ 700 °C. This indicates the presence of stable oxygen functional groups, such as phenols, ethers, quinones, and, to a lesser extent, anhydrides.23 The gas desorption profiles are similar among the samples, except for PAC2. However, the intensity of the peaks is different, reflecting different amounts of released gases. The integration of these peaks allows us to obtain the desorbed quantities, as presented in Fig. S2 (ESI†). The HAB and TCA series exhibit the largest ASA values of 9.9 to 21.8 m2 g−1. Worth to note that the same series showed the highest total surface area, as well. In addition, HCS1300 revealed a high ASA value (11.9 m2 g−1) despite its lower total surface area (48 m2 g−1), while PR1400 had a low ASA (3.5 m2 g−1), although its total surface area was much higher (179 m2 g−1). These results indicate that the active surface area does not always follow the same trend as the total surface area. When the HCS material was heat treated at a higher temperature, i.e., 1500 °C instead of 1300 °C (HCS1500), both ASA and TSA become the smallest (1.2 and 2.2 m2 g−1) among the materials. Therefore, both temperature and precursors impact the active surface area, as reported before.24 For all materials, TSA is higher than ASA, with one exception: PAC2, for which ASA is higher than TSA (6.4 vs. 4.0 m2 g−1). Thus, for PAC2, the surface area observed is mainly due to edges/defects rather than basal planes.
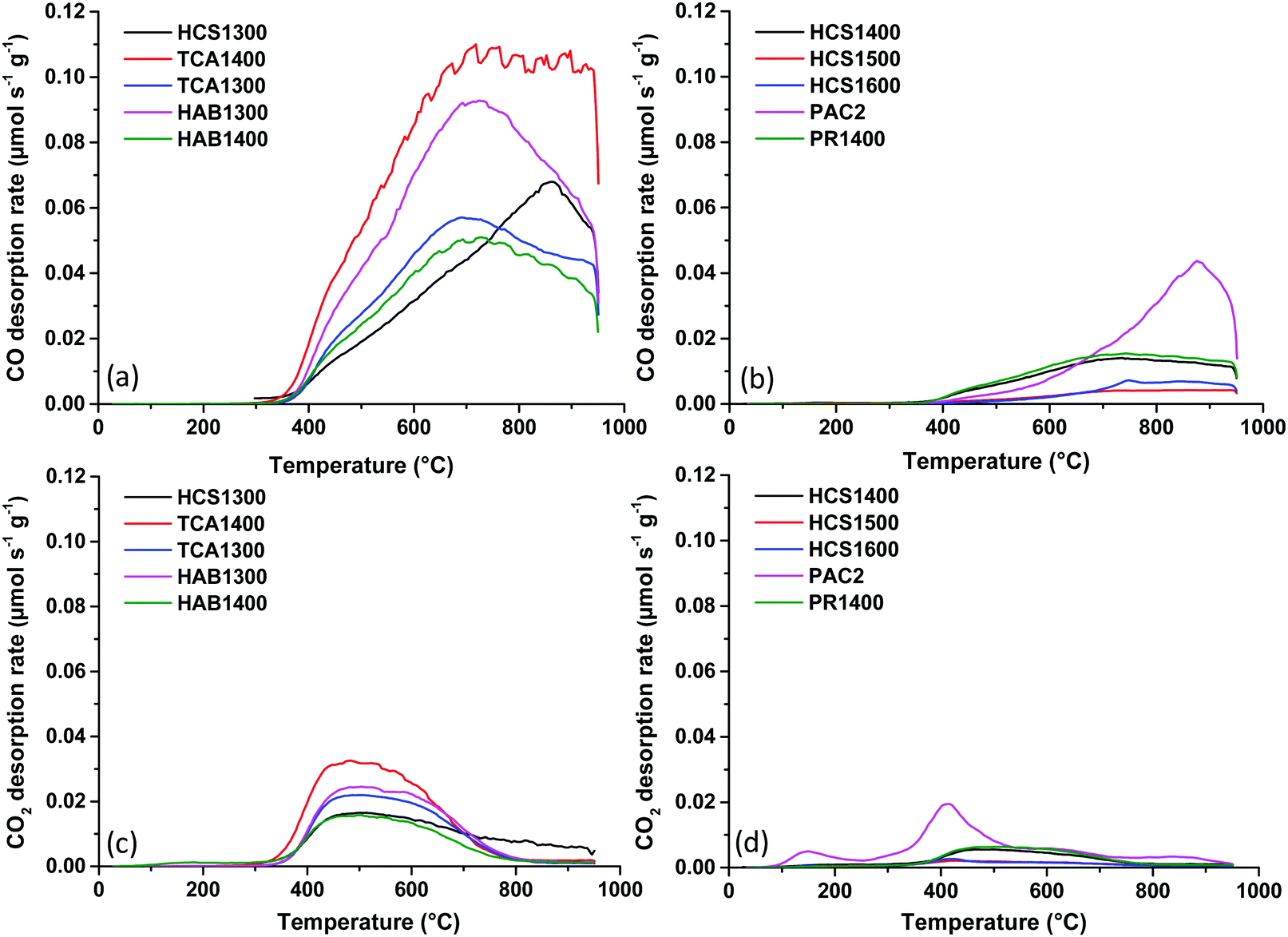 | ||
| Fig. 2 CO (a and b) and CO2 (c and d) desorption profiles of hard carbon materials obtained by TPD-MS after the O2 chemisorption step. | ||
The first galvanostatic discharge/charge cycle was measured for all materials vs. Na in half-cells (Fig. S3, ESI†). Similar conditions, i.e., electrode formulation/processing, electrolyte type and testing parameters, were used to focus on how the carbon-related factors (total surface area and active surface area) impact the irreversible capacity loss (ICL).
The specific electrochemical signature of HC can be observed, i.e., a sloping region down to ∼0.1 V, followed by a potential plateau near 0 V. The presence of irreversible reactions is observed based on the difference between discharge and charge profiles. An ideal redox system would lead to identical discharge/charge capacities, but, in practice, due to SEI build-up during the 1st cycle and Na+ trapping, the capacity recovered during charge (dashed line Fig. S3, ESI†) is always lower than during discharge.
The obtained irreversible capacities loss are listed in Table1. They vary from 32% for TCA1400 to 8% for HCS1500 and HCS1600, which correspond to the initial Coulombic efficiencies (iCEs) of 78% and 92%, respectively. An effect of the pyrolysis temperature on the ICL is seen for all materials, i.e., it decreases with increasing temperature. However, this effect is less obvious for the TCA series. Interestingly, we observed that when the same pyrolysis temperature (1400 °C) is used with different precursors, the ICL values vary greatly: 32% for TCA1400, 16% for PR1400, 15% for HAB1400 and 11% for HCS1400.
The irreversible capacity loss was plotted vs. the total surface area determined by O2 adsorption (Fig. 3a). An increase in irreversible capacity is noticed with an increase in the surface area. Similar results were reported for graphite and hard carbon materials in LIBs10 and NIBs11 by using the N2 adsorption method for BET surface determination. However, in the present case, the TSA determined by N2 adsorption does not allow any correlation with the ICL (Fig. S4a, ESI†) since many narrow pores cannot be detected with this gas.13 If CO2 adsorption is used for TSA determination, then the obtained results follow similar trend as seen for the O2 adsorption. Moreover, when the total pore volume VT is plotted against the ICL (Fig. S4b, ESI†), an analogous correlation is obtained as for the total specific surface area. Such increase of hard carbon irreversible capacity with the pore volume was also observed in LIBs by Guerin et al.25 In addition, the impact of the average pore size, L0, was investigated (Fig. S4c, ESI†), and very interestingly, it can be observed that the irreversible capacity loss gradually decreases as the pore size increases. Thus, the highest ICL is seen for very small pores, and can be associated with both Na-ion trapping in narrow spaces20 and electrolyte decomposition with SEI formation. Smaller pores (<2 nm) contribute to an increase in the surface area and thus induce undesirable reactions with the electrolyte. Therefore, the high surface area and pore volume along with the narrow pores favour irreversible capacity loss.
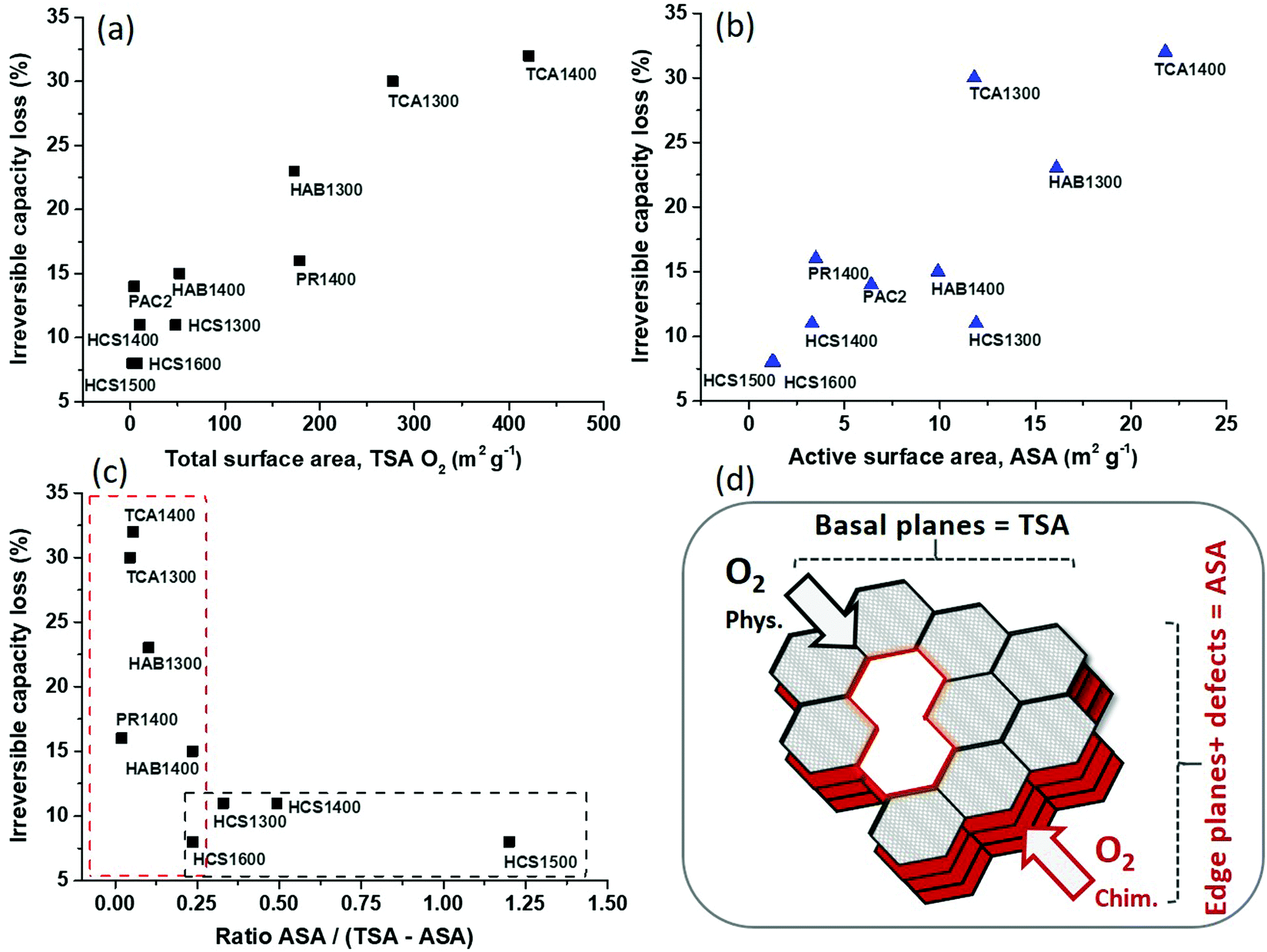 | ||
| Fig. 3 Relationship between the 1st irreversible capacity loss and (a) total surface area (TSA O2), (b) active surface area (ASA), (c) ratio ASA/(TSA O2-ASA). (d) Representation of hard carbon structure containing the active surface (edge planes and defects) and the basal planes surface (TSA-ASA). | ||
Additionally, the active surface area plays a key role in the irreversible capacity (Fig. 3b). An increase behaviour of the irreversible capacity loss with the active surface area is found. It can be noticed that three points are deviating from this correlation which correspond to PR1400, HCS1300 and TCA1300. These materials, as mentioned, exhibit either high TSA compared to ASA (PR1400) or high ASA compared to TSA (HCS1300). In the first case (PR1400), the correlation point is superior to the trend, while for the second case, the point is inferior to the trend. This might suggest that materials with lower TSA give lower irreversible capacity. Moreover, the data point for TCA1300 deviates the most from the correlation. This material has both high ASA and TSA O2 (11.8 and 277 m2 g−1), however, its counterpart, treated at 1400 °C, has even higher values (21.8 and 421 m2 g−1), but still follows the trend. One possible explanation can be related to the TSA found via N2 adsorption, which is the highest for TCA1300 among all materials (104 m2 g−1),21 suggesting that this material has a tendency for additional irreversible reactions. Moreover, the TCA series presented heteroatoms (N and S) in their structure (0.5 at% S for TCA1300, Table S1 ESI†), in contrast to the other HCs showing only O. The removal of these heteroatoms during pyrolysis could be responsible for the high ASA values found for these materials. In fact, the impact of N and S functional groups on the irreversible capacity of hard carbon in NIBs is scarcely documented. For instance, Agrawal et al. showed slightly higher iCEs for N-doped hard carbon spheres,26 while Lu et al. improved the iCE by S doping.27 We also found that O-functional groups have less impact on the initial Coulombic efficiency than the specific or active surface area do.21 Indeed, the O content in the materials prepared at 1300 °C is quite similar, i.e., 2.22 to 3.15 at%, and, notably, TCA1300 has the lowest O content (2.2 at%) and the highest C content (97.22 at%), which leads to the lowest O/C ratio (Table S1 ESI†). Therefore, the surface chemistry cannot explain the high irreversible capacity of TCA1300; it is most likely induced by its high specific surface area and variety of pores detected by both N2 and O2 gas adsorption. Worth to remark that no strict correlation between irreversible capacity and the defect ratios ID/IG was observed, altough an increase trend is noticed (Fig. S4d, ESI†).
Thus, the obtained results illustrate a strong correlation between irreversible capacity and both total surface area and active surface area. To discriminate between the effect of each surface, a different strategy was approached. We subtracted the edge plane contribution from the total surface area to obtain the carbon basal planes area (TSA-ASA). Then, the ratio between the edge surface/defects (ASA) and basal surface (TSA-ASA) was determined (Table 1). The obtained correlation between the irreversible capacity and the edge/basal surface ratio is depicted in Fig. 3c. Two behaviour regimes can be distinguished. In the first regime, a drastic decrease of irreversible capacity, down to ∼10%, occurs with the increase of edge/basal planes ratio from ∼0.05 to ∼0.25. Then, in the second regime, a quasi-plateau is observed, up to a ratio of ∼1.2. The lowest irreversible capacity (8%) is attained for the largest ratio edge/basal planes (1.2) and corresponds to the HCS1500 material, which possesses the lowest TSA and ASA surface areas (2.2 and 1.2 m2 g−1). Notably, for one of the materials, PAC2, negative ratio edge/basal planes have been obtained (Table 1, and not included in Fig. 3c) because the TSA is smaller than the ASA; thus, the edge planes surface is higher than the basal planes surface. This type of carbon structure, with a predominant edge structure, seems to not be favourable since the irreversible capacity is rather high, i.e., 14%. If the TSA obtained by CO2 adsorption is used,21 then only the first regime is observed (Fig. S5, ESI†). This behaviour can be explained by the fact that CO2 adsorption is capable of analysing very narrow pores (ultramicropores), while O2 adsorption can detect both ultramicropores and supermicropores, as highlighted by our recent work.13 Therefore, to limit the irreversible capacity, hard carbon materials with low specific and active surface areas are recommanded. Very narrow pores (<2 nm) contribute as well to irreversible capacity, and they must be avoided. In this direction, H2 gas might be more accurate than O2 to assess the pores smaller than 0.5 nm13 and to evaluate more precisely their impact. Heteroatoms (N and S) appears to have a certain influence on the irreversibility, however, deeper investigations are required to understand their precise role. Nevertheless, the use of pure carbon materials, exempt of inorganic impurities (like herein), is another important criterion, since such impurities found in natural-derived hard carbons might also lead to irreversible capacity increase.19
In summary, it was found that the hard carbon irreversible capacity diminishes with both the total surface area and active surface area. Therefore, the materials possessing low basal and edge plane surfaces provide the smallest irreversible capacity (8%), corresponding to a high Coulombic efficiency (92%).
AB: resources, investigation, validation, data curation, writing – review and editing; CV: investigation, methodology, validation, review and editing; FR: investigation, methodology, validation, review and editing; MM: supervision, review and editing; CMG: conceptualization, funding acquisition, project administration, data curation, writing – original draft.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Institut de Sciences des Matériaux de Mulhouse (UMR7361 CNRS – UHA) via the Emergent Project, is acknowledged. This work was also partially financially supported by the European Union’s Horizon 2020 Program (project NAIMA, call: LC-BAT-02, Contract no. 875629). It was realized in the frame of the RS2E network (French research network on electrochemical energy storage) and the laboratory of excellency for electrochemical energy storage (STORE-EX).Notes and references
- E. Goikolea, V. Palomares, S. J. Wang, I. R. de Larramendi, X. Guo, G. X. Wang and T. Rojo, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- B. Xiao, T. Rojo and X. Li, ChemSusChem, 2019, 12, 133 CrossRef CAS PubMed.
- D. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS.
- C. Bommier and X. Ji, Small, 2018, 14, 1703576 CrossRef PubMed.
- J. Fondard, E. Irisarri, C. Courreges, M. R. Palacin, A. Ponrouch and R. Dedryvere, J. Electrochem. Soc., 2020, 167, 070526 CrossRef CAS.
- M. Dahbi, T. Nakano, N. Yabuuchi, T. Ishikawa, K. Kubota, M. Fukunishi, S. Shibahara, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Electrochem. Commun., 2014, 44, 66 CrossRef CAS.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165 CrossRef CAS PubMed.
- Y. Rangom, R. R. Gaddam, T. T. Duignan and X. S. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 34796 CrossRef CAS PubMed.
- F. Béguin, F. Chevallier, C. Vix-Guterl, S. Saadallah, V. Bertagna, J. N. Rouzaud and E. Frackowiak, Carbon, 2005, 43, 2160 CrossRef.
- M. Winter, P. Novák and A. Monnier, J. Electrochem. Soc., 1998, 145, 428 CrossRef CAS.
- C. Bommier, W. Luo, W.-Y. Gao, A. Greaney, S. Ma and X. Ji, Carbon, 2014, 76, 165 CrossRef CAS.
- C. Matei Ghimbeu, J. Górka, V. Simone, L. Simonin, S. Martinet and C. Vix-Guterl, Nano Energy, 2018, 44, 327 CrossRef CAS.
- A. Beda, C. Vaulot and C. M. Ghimbeu, J. Mater. Chem. A, 2021, 9, 937 RSC.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- N. R. Laine, F. J. Vastola and J. P. L. Walker, J. Phys. Chem., 1963, 67, 2030 CrossRef CAS.
- P.-C. Tsai, S.-C. Chung, S.-K. Lin and A. Yamada, J. Mater. Chem. A, 2015, 3, 9763 RSC.
- H. Wang, F. Sun, Z. Qu, K. Wang, L. Wang, X. Pi, J. Gao and G. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 18554 CrossRef CAS.
- C. del Mar Saavedra Rios, L. Simonin, A. de Geyer, C. Matei Ghimbeu and C. Dupont, Energies, 2020, 13(14), 3513 CrossRef CAS.
- A. Beda, J. M. Le Meins, P. L. Taberna, P. Simon and C. M. Ghimbeu, Sustainable Mater. Technol., 2020, 26, e00227 CrossRef CAS.
- E. Memarzadeh Lotfabad, P. Kalisvaart, A. Kohandehghan, D. Karpuzov and D. Mitlin, J. Mater. Chem. A, 2014, 2, 19685 RSC.
- A. Beda, F. Rabuel, M. Morcrette, S. Knopf, P. L. Taberna, P. Simon and C. Matei Ghimbeu, J. Mater. Chem. A, 2021, 9, 1743 RSC.
- E. R. Buiel, A. E. George and J. R. Dahn, Carbon, 1999, 37, 1399 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379 CrossRef CAS.
- C. del Mar Saavedra Rios, A. Beda, L. Simonin and C. Matei Ghimbeu, Hard Carbon for Na-ion Batteries: From Synthesis to Performance and Storage Mechanism, in Na-ion Batteries, ed. L. Monconduit and L. Croguennec, ISTE Ltd 2020. Published by ISTE Ltd and John Wiley & Sons, Inc., 2021 Search PubMed.
- K. Guerin, A. Fevrier-Bouvier, S. Flandrois, B. Simon and P. Biensan, Electrochim. Acta, 2000, 45, 1607 CrossRef CAS.
- A. Agrawal, S. Janakiraman, K. Biswas, A. Venimadhav, S. K. Srivastava and S. Ghosh, Electrochim. Acta, 2019, 317, 164 CrossRef CAS.
- M. Lu, W. Yu, J. Shi, W. Liu, S. Chen, X. Wang and H. Wang, Electrochim. Acta, 2017, 251, 396 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ya00004k |
| This journal is © The Royal Society of Chemistry 2022 |