
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring multivalent carbohydrate–protein interactions by NMR
Jon I.
Quintana†
 a,
Unai
Atxabal†
a,
Luca
Unione
*ab,
Ana
Ardá
*ab and
Jesús
Jiménez-Barbero
*abcd
a,
Unai
Atxabal†
a,
Luca
Unione
*ab,
Ana
Ardá
*ab and
Jesús
Jiménez-Barbero
*abcd
aCICbioGUNE, Basque Research & Technology Alliance (BRTA), Bizkaia Technology Park, Building 800, 48160 Derio, Bizkaia, Spain. E-mail: aarda@cicbiogune.es; jjbarbero@cicbiogune.es
bIkerbasque, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Bizkaia, Spain
cDepartment of Organic Chemistry, II Faculty of Science and Technology, EHU-UPV, 48940 Leioa, Spain
dCentro de Investigación Biomédica En Red de Enfermedades Respiratorias, Madrid, Spain
First published on 8th February 2023
Abstract
Nuclear Magnetic Resonance (NMR) has been widely employed to assess diverse features of glycan–protein molecular recognition events. Different types of qualitative and quantitative information at different degrees of resolution and complexity can be extracted from the proper application of the available NMR-techniques. In fact, affinity, structural, kinetic, conformational, and dynamic characteristics of the binding process are available. Nevertheless, except in particular cases, the affinity of lectin-sugar interactions is weak, mostly at the low mM range. This feature is overcome in biological processes by using multivalency, thus augmenting the strength of the binding. However, the application of NMR methods to monitor multivalent lectin–glycan interactions is intrinsically challenging. It is well known that when large macromolecular complexes are formed, the NMR signals disappear from the NMR spectrum, due to the existence of fast transverse relaxation, related to the large size and exchange features. Indeed, at the heart of the molecular recognition event, the associated free-bound chemical exchange process for both partners takes place in a particular timescale. Thus, these factors have to be considered and overcome. In this review article, we have distinguished, in a subjective manner, the existence of multivalent presentations in the glycan or in the lectin. From the glycan perspective, we have also considered whether multiple epitopes of a given ligand are presented in the same linear chain of a saccharide (i.e., poly-LacNAc oligosaccharides) or decorating different arms of a multiantennae scaffold, either natural (as in multiantennae N-glycans) or synthetic (of dendrimer or polymer nature). From the lectin perspective, the presence of an individual binding site at every monomer of a multimeric lectin may also have key consequences for the binding event at different levels of complexity.
 Jon Imanol Quintana | Jon I. Quintana studied chemistry at the University of the Basque Country. In 2017, he joined the chemical glycobiology lab at CIC bioGUNE, where he employed different NMR techniques to characterize the binding of a monoclonal antibody to different oligosaccharides. In 2018, he started his PhD under the supervision of Prof. Jesús Jiménez-Barbero and Dr Ana Ardá, studying molecular recognition events between lectins and sugar by means of NMR, ITC, and MD simulations. In 2021, he performed a secondment with Dr Martina Delbianco (MPIKI, Potsdam), where he worked on the synthesis of glycans using a solid phase synthesizer. He obtained his PhD in 2022 and his research focuses on understanding host–pathogen interactions mediated through glycans. |
 Unai Atxabal | Unai Atxabal studied Biochemistry at the University of the Basque Country (Spain) and completed a MSc in Medicinal Chemistry focusing in drug development and organic synthesis at the University of Copenhagen (Denmark). He is currently working on his PhD within the Marie-Skłodowska-Curie Innovative Training Network “BactiVax” under the supervision of Prof. Jesús Jiménez-Barbero and Dr June Ereño-Orbea. The PhD project is focused on studying molecular recognition processes of antigens by key receptors using state-of-the-art NMR techniques combined with molecular dynamics and other biophysical techniques. The goal is to understand these processes to design and synthesize novel drug-like compounds. |
 Luca Unione | Luca Unione studied Chemistry at the University of Napoli Federico II (Italy). He received the PhD in 2016 in Madrid (Spain), in the framework of a ITN-Marie Skłodowska-Curie Action, working in the field of structural characterization of carbohydrates, glycomimetics, and their interactions with glycan binding proteins. After carrying out a postdoctoral stay in the company Atlas molecular Pharma in Biscay (Spain, he moved to Utrecht University (The Netherlands) as postdoctoral fellow (2019–2021) under the International Human Frontier Science Program (HFSP). Since 2022 he is Ikerbasque Research Fellow (tenure track) at CIC bioGUNE. His research interest is focused on the understanding of the role of glycans as regulators in biomedical processes, with a special focus on chemo-enzymatic synthesis of glycans and the study of glycan–protein interactions through a multidisciplinary approach. |
 Ana Ardá | Ana Ardá studied Chemistry at the University of A Coruña, from where she received her doctorate in Chemistry in 2006. Between 2006 and 2008 she carried out a postdoctoral stay in at Utrecht University, with Prof. Hans Kamerling where she started to work with carbohydrates. In 2008 she moved to Centro de Investigaciones Biológicas (CIB-CSIC) in Madrid to join Prof. Jiménez-Barbero's group. In 2014 she moved to CIC bioGUNE where she is Ikerbasque Research Associate and Associated Principal Investigator. Her research interests are focused on the study of glycan–protein interactions through a multidisciplinary approach, with a special focus on NMR techniques. |
 Jesús Jiménez-Barbero | Jesús Jiménez-Barbero is Ikerbasque Research Professor and Scientific Director of CIC bioGUNE since 2014. He received his PhD in 1987 at Madrid. After postdoctoral stays at Zürich, Mill Hill, and Pittsburgh, he returned to Madrid (CSIC) and started to work on protein–carbohydrate interactions. In 2002 he was promoted to CSIC Research Professor at CIB-CSIC, where he developed his scientific activity until he moved to Bilbao. His scientific interest is focused in the field of molecular recognition and Chemical Glycobiology, employing a multidisciplinary approach that combines synthesis, biochemistry, molecular biology, molecular modeling, and especially NMR, enjoying a wide network of scientific collaborations worldwide. |
Introduction
Carbohydrates are essential biomolecules that are found ubiquitously in every living species. The roles of carbohydrates, also known as sugars, glycans, or saccharides are multiple; they are the main energy source for humans and one of the major components of the cell wall of plants. However, despite these renowned functions, carbohydrates also have a role in recognition events related to health and disease that are of paramount relevance.The interactions mediated through carbohydrates occur in multiple ways and with different kinds of entities. Glycans can interact between themselves, for instance, in cell–cell recognition.1 However, the most studied and relevant systems involve protein–carbohydrate interactions. These interactions are essential for cell adhesion,2 signalling events,3 host-pathogen interactions,4 cancer development,5 and many more.
Carbohydrates are also present in pathogens like virus, bacteria, parasites, or fungi. The glycans on the surface of these entities are often the first interface with the host cell. As a result, targeting those carbohydrates can be useful to avoid infection. As many viruses display a dense coat of glycans, another approach that has been proposed to battle pathogens is the use of glycan binding proteins.6,7 Fittingly, several glycan binding proteins have shown the ability of neutralising various viruses, including HIV, and therefore, these proteins are in the pipeline to be used to treat and prevent infections.
A relevant feature to take into consideration is the conformation of the carbohydrates. As stated before, glycans are rather flexible molecules. This feature can be, on many occasions, detrimental for the binding, due to high entropic penalties. Indeed, in certain systems, the glycan might need a specific conformation and presentation for the binding event to take place, which can result in high entropic penalties in flexible molecules. Thus, the design of synthetic molecules (glycomimetics) which already are preorganized for the binding may be a proper strategy to consider to target a biologically relevant sugar-binding protein.8
Among the entities that interact with glycans, lectins are sugar binding proteins with no catalytic function (they are not enzymes) and do not provide a direct immune response (they are not antibodies). There are fourteen different types or group of lectins in the animal kingdom.9 Three of the most relevant lectin families found in humans are C-type, I-type and S-type lectins. C-type (Ca2+-dependent) lectins are found both as transmembrane and as soluble proteins. Some of the lectins of this family, like DC-SIGN, langerin, and MGL have key roles in pathogen recognition and have become targets in the field of drug discovery.10
Within I-type lectins, the study of sialic acid binding immunoglobulin-type lectins (Siglecs) is nowadays a topic of major interest.11 The Siglec family of transmembrane lectins is comprised by 15 members, which contain an N-terminal domain that recognizes sialic acids.12 All siglecs (except for Siglec-4 and 6) are expressed in immune cells and help our immune system to distinguishing between self and non-self signals.
Galectins (earlier dubbed as S-type lectins) are β-galactoside binding proteins. This family is formed by 16 members, which are found ubiquitously in the human body.13 These lectins are expressed in the cytoplasm and then secreted through a non-classical pathway or transported to the nucleus. Through their ability of binding β-galactosides, galectins participate in cell–cell interactions, and they are also involved in immune responses, inflammation and signalling events, among many others. Due to their involvement in several diseases, galectins have been targeted for inhibitors development, for which different approaches have been used. Glycomimetics have been employed,14 ranging from monovalent molecules with multiple chemical decorations to relatively simple molecules displayed along multivalent scaffolds. One of the main drawbacks in developing mimetics for galectins is the difficulty of designing a molecule that is specific for just one galectin. However, there are various promising molecules that are fairly specific for Gal-1,15 Gal-3,16 and Gal-8.17
Given all these structural and dynamic features, the affinity of most individual protein–carbohydrate interactions are rather weak (KD values in the mM–μM range).18 In biological systems, however, this low-affinity binding is usually overcome through the engagement of simultaneous synergic interactions between the receptor and the ligand, a phenomenon known as multivalency.19 Therefore, multivalency is commonly used by nature to enhance the innately weak interactions occurring during carbohydrate–lectin molecular recognition.
There are diverse ways in which multivalent presentations can enhance affinity: chelation, subsite binding, statistical rebinding, steric stabilization and clustering effects.20–22 However, in the development of multivalent ligands there are various factors that should be taken into consideration. Firstly, the nature of the scaffold. Rigidity is a crucial feature to consider, since it is directly related to entropy. Flexible linkers may display a large entropic penalty upon binding. However, flexibility can also be advantageous as it might adopt the proper conformation for favourable interactions to take place.23,24 The chemical nature of the linker is also relevant, since it might establish additional interactions with secondary binding sites and therefore, improve the affinity.25 Undoubtedly, the choice of the ligand is a key factor in the outcome, as well as its effective concentration within the scaffold. Usually, the higher the effective concentration of the ligand is, the higher the affinity. However, at high concentrations of the ligand, steric clashes may take place and the effectiveness of the approach decreases. Usually, the higher the concentration of the ligand is, the higher the affinity. However, at high concentration of the ligand, steric clashes may take place and the effectivity of the approach decreases.
Methods to assess molecular recognition
There are various techniques that can be applied to characterize protein–carbohydrate interactions. X-Ray crystallography has been for decades the most employed biophysical technique for unravelling the structure of lectin–sugar complexes, as atomic resolution information of the complex can be obtained.26 However, the process of obtaining crystals can be very tedious and the proteins are not always crystallisable. Additionally, due to the intrinsic flexibility of glycans, crystallographic structures usually have erroneous conformations, linkages, or even wrong residues.27–29 Cryo electron microscopy (CryoEM) is also becoming an important structural biology technique.99 Indeed, there are already different complexes that have been evaluated through cryoEM.30,31Other biophysical techniques, as surface plasmon resonance (SPR) or more recently, biolayer interferometry (BLI) have also been widely employed to monitor protein–carbohydrate interactions.32,33 These techniques may yield the thermodynamics, kinetics and binding energy of the interaction and can be performed without any type of labelling. Another powerful label-free technique is isothermal titration calorimetry (ITC), which enables obtaining the thermodynamic profile of the binding event.34 These methods, which are extremely useful, provide key energy and thermodynamic data, but no direct information on the epitope and the paratope of the binding is obtained.
NMR to the rescue
Nuclear Magnetic Resonance (NMR) is extensively used to study protein–carbohydrate interactions. Through NMR, the binding affinities and information on the epitopes can be obtained. Moreover, the 3D structure of the protein and/or the glycan ligand can also be deduced.35 Additionally, due to the intrinsic chemical properties of the glycans, NMR has been the technique of choice of many research groups to analyse the structure, conformation, and dynamics of carbohydrates, as well as their interactions with biomolecular receptors, including proteins.36,37The NMR methods employed to analyse interactions are classified into two groups. In ligand-based methods, changes in the NMR signals of the ligand (the glycan, herein) are observed, whereas in receptor-based methods changes in the NMR signals of the macromolecule (the lectin, herein) are monitored.38
Ligand-based NMR methods
Ligand-based NMR methods to characterize protein–carbohydrate interactions rely on the changes on the NMR properties of the glycan.39 These variations can be observed through two strategies: (i) taking advantage of the dramatic changes on the molecular motion of the ligand upon binding to the macromolecule or (ii) through transfer of magnetization from the NMR signals of the macromolecule to those of the glycan.For a small or medium size glycans, as depicted in Fig. 1, their motional properties (fast Brownian motion, slow relaxation, fast diffusion, and positive NOE values) are different to those of the receptor (slow Brownian motion, fast relaxation, slow diffusion, and negative NOE values). However, when the glycan binds to the protein, its rotational motion properties change, and are similar to those of the large macromolecule.
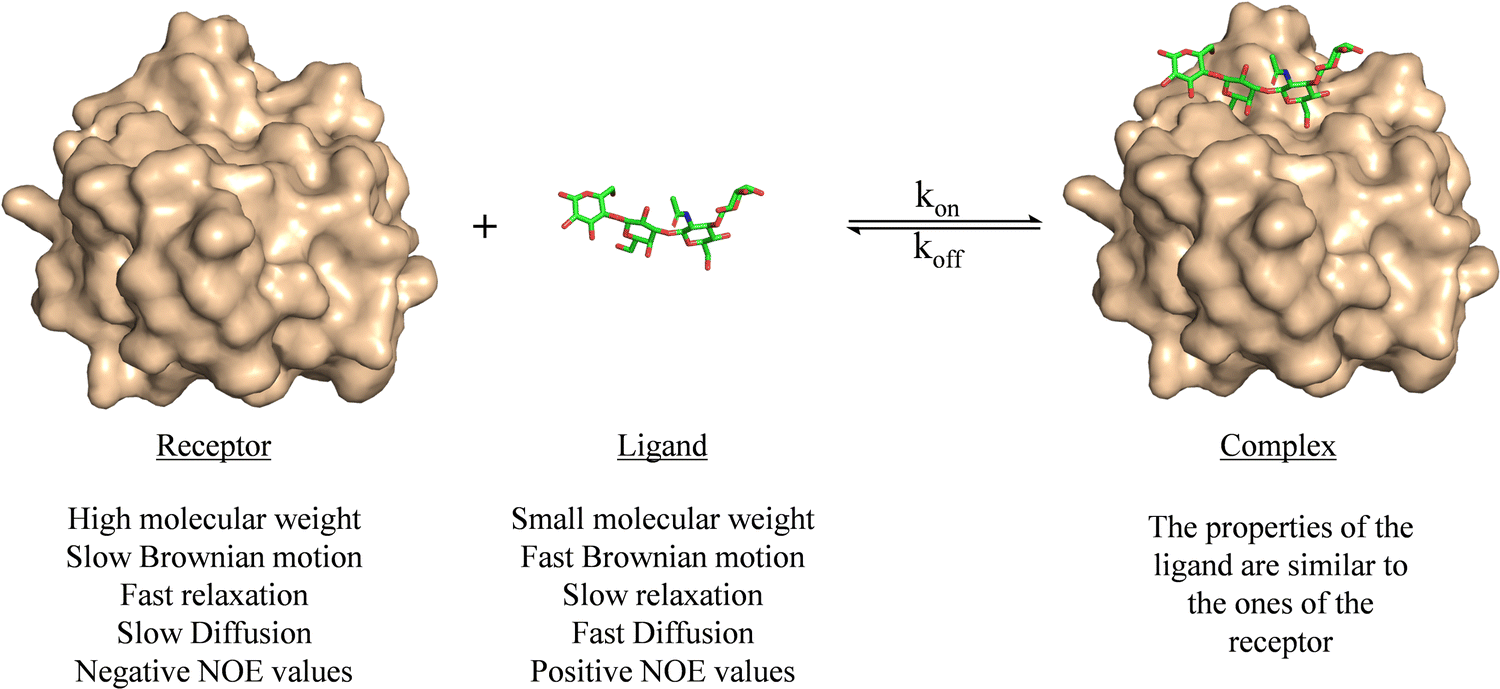 | ||
| Fig. 1 Motional properties of a receptor, a ligand, and the corresponding binary complex. The structures are taken from the PDB code 4YM0. | ||
The transferred NOE
NOEs developed on the fraction of the bound ligand but observed following dissociation from the complex are known as transferred NOEs (trNOESY). This experiment has found large use in identify and characterize glycan–protein interactions by NMR. In fact, one of the most significant NMR-related changes that carbohydrates may undergo upon protein binding is the change in the sign of the NOE. The NOE intensity and sign depends on the tumbling rate, which is related to the molecular size. For the free small sugars, the NOEs are positive or close to zero. However, when bound to the protein (using usually ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:20 protein:glycan ratio) the rotational properties of the glycan are similar to those of the macromolecule, and the corresponding NOEs are negative (Fig. 2). Therefore, the change in sign of the NOE can be exploited to characterize protein–sugar interactions.40,41
:5 to 1:20 protein:glycan ratio) the rotational properties of the glycan are similar to those of the macromolecule, and the corresponding NOEs are negative (Fig. 2). Therefore, the change in sign of the NOE can be exploited to characterize protein–sugar interactions.40,41
 | ||
| Fig. 2 Top: A ligand and its receptor in equilibrium. Bottom: (A) NOESY spectrum of the free ligand (B) NOESY spectrum of the ligand bound to the protein (trNOESY). (C) ROESY spectrum of the ligand in the presence of the protein (trROESY). | ||
Selection of the mixing time of the NOESY is also very relevant, as exemplified by Weimar and Peters when studying the interaction of α-Fuc-(1-6)-β-GlcNAc-OMe with Aleuria aurantia agglutinin.42 Whereas for the free disaccharide, NOEs are positive and small, for the complex, NOEs are negative and ca. ten times higher in absolute values (See Fig. 3). Transferred NOESY (trNOESY) experiments are usually performed with short mixing times (ca. 100 ms).43 Under these conditions, the contribution of the free ligand is almost negligible, meaning that the obtained information basically describes the conformation of the bound ligand.
 | ||
| Fig. 3 Dependence of the NOE on the mixing time and the diverse molecular motion regimes for the bound and free states. NOEs for the free ligand are displayed as blue dots. Transferred NOEs are displayed as orange dots. Adapted from ref. 42. | ||
One of the drawbacks of trNOESY experiments is the existence of spin-diffusion. Nevertheless, this effect can be quantitatively taken into account through full relaxation matrix calculations44 and moreover, can also be distinguished in ROESY experiments, which give rise to positive cross peaks for directly related proton pairs, the 3-spin-diffusion mediated signals are negative (Fig. 2).45 Additionally, the existence of chemical exchange events between the free and bound species can also be assessed through ROESY experiments, since chemical exchange peaks are always negative (Fig. 2).46
Transverse relaxation-based experiments
The large difference in the time scale of molecular motions between small and large ligands is the also the base of T2-based relaxation methods to screen and monitor ligand binding to proteins.47 Transverse relaxation is very fast for large molecules that tumble in the ns time scale. In contrast, it is slow, for free small molecules. When the small ligand binds to the macromolecule, its motion changes, as well as its relaxation properties. Therefore, T2-relaxation times of free and bound ligands are dramatically different and these differences can be easily monitored through a particular type of NMR experiments, which use the so-called CPMG filter.48 In these experiments, which were initially applied to 1H-NMR, but have found its particular niche in 19F-NMR,49 the filter is able to reduce or eliminate the protein and the bound ligand signals without significantly affecting the signal intensities of the free ligands. In fact, as alternative to 1H-NMR, the most employed nucleus that has been used in NMR to characterize carbohydrate structure and interactions is 19F.50 Generally, the hydroxyl group is substituted by fluorine atom, although on some occasions hydrogen atoms in methyl groups have also been substituted by 19F.51 The use of 19F is very appropriate since its intrinsic sensitivity is very high. Additionally, this nucleus has a broad spectral dispersion and it is not present in any natural biomolecule, avoiding non-desired signals and providing simple NMR spectra, with no or minor crowding. However, depending on the position that this chemical modification is being introduced, it can alter the conformational behavior and physical properties of the sugar, as well as the interactions with a lectin.52 The role and applicability of 19F in NMR studies will not be further discussed herein, as it has been extensively reviewed.53,54 From the NMR perspective, the use glycomimetics decorated with other NMR active nuclei is indeed relevant. For instance, 77Se is an NMR relatively abundant (7.60%) active isotope that displays a high chemical shift dispersion. Indirect 77Se detection has proved to be useful to characterize the binding of galectins to various disaccharides through 2D 1H,77Se CPMG-HSQMBC experiments.55–57 Illyés et al. have synthesized thio and selenogalactosides presenting clusters bearing four galactose epitopes and showed their efficacy targeting the bacterial lectin PA-IL from Pseudomonas aeruginosa.58 Although in this approach the binding was characterized through common 1H-STD-NMR experiments, the presence of selenium in glycomimetics is a promising alternative to analyse protein–carbohydrate interactions.Saturation transfer difference-NMR
Saturation Transfer Difference (STD-NMR) is probably the most employed NMR technique to study protein-ligand interactions.59 Through STD-NMR experiments, a clear picture of the “binding epitope” of the ligand towards the target receptor can be deduced. The binding epitope defines the region (protons, in this context) of the ligand that is spatially close to the protein.In STD-NMR, two different spectra are recorded. In the first one, the reference, a selective saturation is applied in a region devoid of any NMR signal, usually 100 ppm. A second spectrum is recorded, in which only protons of the protein are selectively saturated (on-resonance spectrum). As the protein is saturated, the magnetization is rapidly spread throughout the polypeptide chain protons. Fittingly, if a given ligand binds to the saturated protein, the ligand will also receive this magnetization (Fig. 4). As a result, the signals of those protons that are closer in space to the protein will suffer a decrease in their intensities. The subtraction of the on-resonance spectrum to the off-resonance one will result in a spectrum in which only the signals of those protons that are close to the protein will be present. This difference spectrum, defined as STD-NMR spectrum, contains the binding epitope of the ligand.
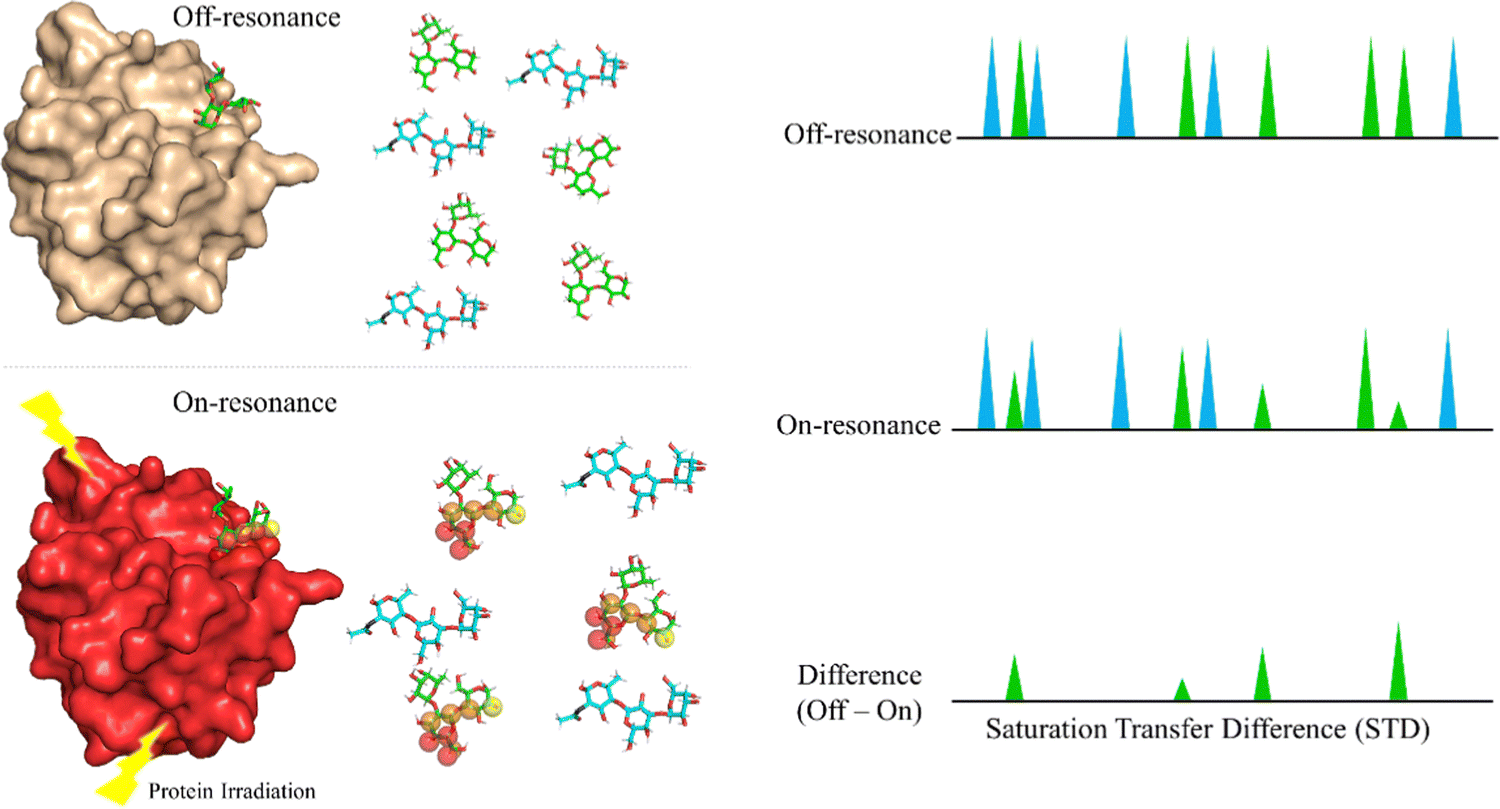 | ||
| Fig. 4 The STD NMR experiment for a mixture of ligands A (blue) and B (green). In the off-resonance spectrum, the protein is not saturated. In the on-resonance spectrum, the protein is saturated and the magnetization is transferred to protons of the bound ligand, B. As a result, some of the protons of B suffer a decrease in intensity. The signals of A display the same intensity in both spectra. Thus, the STD NMR spectrum shows that only ligand B is bound and defines its binding epitope to the target receptor. | ||
The difference between the off-resonance (I0) and on-resonance (Ion) intensities is the STD (ISTD) intensity. Usually, the intensity for each proton (ISTD,i) is shown as the relative value compared to the proton that displays the most intense STD (ISTD,max).

One of the drawbacks of STD-NMR is the overlap of numerous signals, especially for carbohydrates. Most of their 1H-NMR signals appear between 4.5–3.5 ppm. In this narrow chemical shift range, many signals overlap, which hinders the precise analysis of the binding epitope. 2-D STD-NMR experiments have been developed in which the second dimension provides significant enhancement in the spectral dispersion. For instance, the synthesis of 13C labelled carbohydrates or carbohydrates with fluorine tags has enabled performing STD-HSQC and STD-TOCSY experiments.61,62
Diffusion ordered spectroscopy
Diffusion Ordered Spectroscopy (DOSY) experiments can also be used to monitor protein–carbohydrate interactions.63 DOSY experiments are pseudo-2D experiments, which relate the diffusion coefficient, in the Y axis, to the regular 1H NMR spectrum (chemical shifts) in the X axis. Since the diffusion properties of a molecule depends on its molecular weight, size and shape, its diffusion coefficient will be different in the free and protein-bound states (Fig. 5).64 Thus, through DOSY, binding of ligands to proteins can be easily monitored.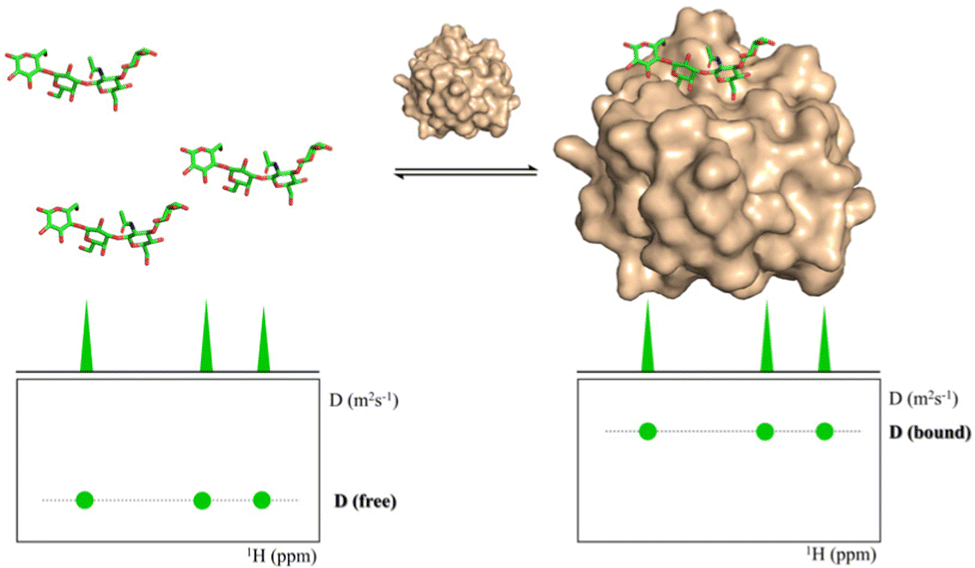 | ||
| Fig. 5 DOSY spectra of a free ligand (left) and of the ligand in the presence of its receptor (right). | ||
Receptor-based NMR methods
In receptor-based methods, changes on the chemical shift of the protein are observed. Usually, labelling with NMR active heteronuclei is necessary to perform the required 1H,15N-HSQC-based titrations.Indeed, the use of 13C (usually 13C-labelled glucose) or 15N (usually 15NH4Cl) containing precursors allows the introduction of 13C and/or 15N atoms into recombinant proteins. The presence of 15N nuclei in proteins permits obtaining 1H–15N correlations in which every proton attached to a 15N provides a cross peak.65
Two different types of experiments can be performed: 1H,15N-HSQC (Heteronuclear Single Quantum Coherence) for small single domain proteins and 1H,15N-TROSY (Transverse relaxation optimized spectroscopy) variant for the larger proteins.66
Since these spectra reflect the unique structure of each protein, they are considered a fingerprint, and any modification on the protein, such as ligand binding, can be monitored in the spectrum.
This sensitivity of the chemical shift towards changes in the chemical environment of the nuclei can be exploited to monitor binding events.67 When a ligand binds to a protein, the nearby protein nuclei suffer changes in their chemical shifts, defined as Chemical Shift Perturbations (CSP), which can be applied to monitor the binding events (Fig. 6). The equilibrium of the system, i.e. the concentration of the protein (P), ligand (L), and protein–ligand complex (PL) is defined by the following equations:

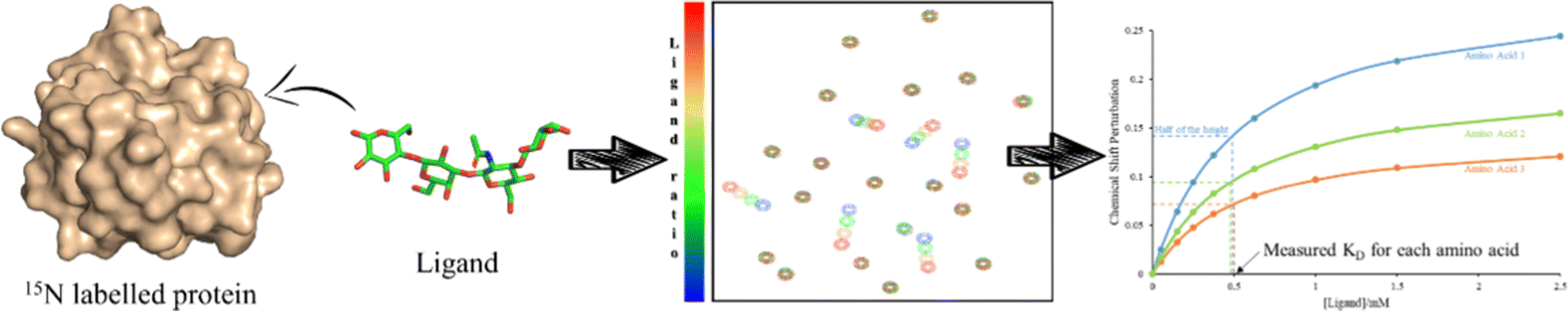 | ||
| Fig. 6 A 1H,15N-HSQC-based titration protocol. To a 15N-labeled protein, different quantities of a certain ligand are added. After each addition a 1H,15N-HSQC spectrum is recorded. From the titration data, the KD can be obtained. | ||
Having explained these NMR methods, it is worth mentioning that their application to study multivalent effects is intrinsically challenging. It is well known that when large macromolecular complexes are formed, the NMR signals disappear from the NMR spectrum, due to the existence of fast transverse relaxation, related to the large size and exchange features. Indeed, at the heart of the molecular recognition event, the associated free-bound chemical exchange process for both partners takes place in a particular timescale. Depending on the time frame of the exchange, lines may be sharp, broaden or even disappear due to the existence of a fast transverse relaxation. This fact is also usually associated to the binding affinity. For tight binding, the exchange rate will be slow, and provided that the generated supramolecule is very large, the NMR peaks will be very broad and will not be detected. Therefore, other approaches different than the direct detection should be employed. Usually, competition experiments with small (labelled) and well-defined binders are employed to unravel details of the binding event. Moreover, precipitation of the formed complexes in solution may also take place. Therefore, under these circumstances, only partial information can be usually extracted, for the individual components or for specific cases. Therefore, experiments with the components of the multivalent entity (either glycan or protein monomer) are also performed to try later to extend the findings achieved with this reductionistic approach to the whole system. Obviously, this has pros and cons. Moreover, in case that the NMR approach provides some information, it is evident that within any multivalent architecture, there are several “monomers” that are repeated. Given the features of NMR spectroscopy, these monomers cannot be directly distinguished, since their chemical environment is identical and will provide identical chemical shifts. Some methodologies to circumvent this initial problem are given below (specific isotope labelling, paramagnetic NMR).
Specific examples
When considering multivalent interactions from the NMR perspective, in this review article, we have distinguished, in a subjective manner, the existence of multivalent presentations in the glycan or in the lectin. From the glycan perspective, we have also considered whether multiple epitopes of a given ligand are presented in the same linear chain of a saccharide (i.e., poly-LacNAc oligosaccharides) or decorating different arms of a multiantennae scaffold, either natural (as in multiantennae N-glycans) or synthetic (of dendrimer or polymer nature). From the lectin perspective, the presence of an individual binding site at every monomer of a multimeric lectin may also have key consequences for the binding event at different levels of complexity.Targeting lectins with multivalent natural or synthetic ligands
Using a natural polysaccharide backbone to provide the multivalent presentation of the interacting epitopes,69 a dextran skeleton decorated with LacNAc (Fig. 7) moieties has been employed to target human galectin-3 (hGal3). The multivalent presentation of the epitopes in the dextran backbone was achieved through propargylation and bioconjugation with lactose, and using maltose and mannobiose as controls.70 Binding studies of the multivalent conjugates were performed from the receptor's perspective by using 1H,15N-HSQC based titrations. Despite the multivalent presentation, KDs in the medium–high μM range were measured, as those described for free lactose.71,72 The lack of affinity enhancement suggests that the interactions do not cooperatively provide added value to the interaction. Nevertheless, chemical-shift perturbations were also observed for cross peaks belonging to residues that are not located at the canonical galactose binding site of hGal3, but at the opposite face. Curiously, this locus had been previously reported to interact with β-mannans, as described below.72
 | ||
| Fig. 7 Chemical structure of lactose substituted propargylated dextran molecules employed for targeting hGal3. | ||
Alternatively, a series of HPMA-based glycopolymers (Fig. 8) bearing different LacNAc contents were designed and tested through ELISA assays.73
 | ||
| Fig. 8 Glycopolymers designed for targeting hGal-1 and hGal-3.73 For each scaffold 5 polymers were synthesized with different carbohydrate contents. From left to right polymers 7a, 7b, and 7c. | ||
The selectivity of the designed multimeric compounds for hGal-1 versus hGal-3 was remarkable. Herein, we should mention that Gal-1 is a non-covalent homodimer74 while the CRD of Gal-3 is a monomer.75 The phenomenon has also been investigated by NMR,76 from both the ligand and receptor's perspectives. STD-NMR experiments highlighted that the recognized epitope for the monomer presentations of di- and tri-LacNAc moieties is that of the LacNAc (ligand 1) moiety.
Then, the binding events were followed from the protein perspective by 1H,15N-HSQC titrations, with 15N-labeled Gal-1 and Gal-3 (Fig. 9). The observed chemical shift perturbations (CSP) generated by the di- and tri-LacNAc monomers again matched those induced by simple LacNAc, although a clear decrease in the cross-peak intensities in the HSQC spectra were observed, which did not occur when LacNAc was used.
 | ||
| Fig. 9 Schematic representation of 1H,15N-HSQC spectra of human galectin-1 (on the top) and galectin-3 (below) with the monomer of ligands 7b (left) and ligand 7c (right) adapted from Bertuzzi et al.72 The 1H,15N-HSQC spectra of both apo Gal-1 (top) and Gal-3 (below) are colored in blue. Top: Upon the addition of the ligands to the Gal-1 (red spectra), a significant reduction of signal intensities is observed, especially in the presence of the monomer of ligand 7c. Bottom: Signal reductions re observed when ligands were added to Gal-3 (green spectra), being more notorious at the right-hand side. | ||
It is tempting to guess that statistical rebinding events taking place when the di- or tri-valent ligands are employed, producing the increase of the transverse relaxation rate of the protein nuclei, further enhanced by the free-bound exchange process. Thus, the HSQC cross-peak intensities are decreased. Fittingly, the decrease was much larger in the presence of Gal-1 than with Gal-3. They were also larger for the tri-LacNAc than for the di-LacNAc analogue.
Receptor-based 1H,15N-HSQC experiments were employed to monitor the binding of the glycopolymers to both galectins. However, the addition of just small amounts of the polymers triggered the disappearance of the lectins’ cross-peaks. However, the addition of several equivalents of LacNAc to the NMR tubes containing the lectin/polymer mixtures permitted the recovery of the HSQC signals of the 15N-labeled lectin. Thus, the molar equivalents of LacNAc required to recover the cross peaks were a marker of the relative binding affinities for the different partners. The analysis of the results permitted concluding that the glycopolymer with just one LacNAc entity per monomer showed the largest potency versus Gal-1 per active LacNAc moiety, while no selectivity among the three glycopolymers was deduced for Gal-3. Dynamic light scattering and cryo-electron microscopy experiments allowed deducing the existence of supramolecular and cross-linked entities, further supporting the NMR results.76
The interaction of natural ligands versus galectins has also been studied by NMR. Typical multivalent presentations can be found in polysaccharides, multiantennae N-glycans, or polyLacNAc chains. Regarding polysaccharides, it is obvious that they may display multiple repeating units of the same oligosaccharide unit, which may provide numerous contact points to the partner lectins. As example of these interactions, the interaction of Davanat (Fig. 10), a galactomannan (GM), composed of β1–4-linked D-mannopyranosyl units periodically decorated with Galα1–6 moieties (59 kDa average molecular weight) to the Gal-1 homodimer has been studied.77,78 The use of 1H,15N-HSQC experiments allowed to distinguish an alternative binding site for long galactomannans, other than the canonical β-galactoside-binding region. This report evidenced the possibility of the existence of simulataneous binding sites for galectins. The existence of a non-canonical binding site was demonstrated by the fact that simple lactose is indeed able to bind the preformed Gal-1/Davanat complex. Moreover, DOSY experiments also assessed that Gal-1 binding alters Davanat conformation, likely perturbing the putative glycan–glycan interactions that take place between the Davanat saccharide chains. Alternatively, a detailed characterization of the recognition phenomena at the canonical and alternative sites was achieved by using two small galactomannans as models.
 | ||
| Fig. 10 Chemical structure of the repeating unit of the polysaccharide Davanat. | ||
In a publication from the same research group, the binding of GMs, with diverse Gal/Man molar ratios, to Gal-3 was also scrutinized. In fact, following the same methodology, it was observed that the intensities of the HSQC cross peaks from the carbohydrate recognition domain (CRD) and N-terminal domain (NTD) of galectin-3 were differentially affected, showing diverse degrees of broadening.72
Addition of simple lactose to the NMR tube containing the mixture of Gal-3 bound to GM partially recovered the intensities, strongly suggesting that the binding of lactose at the β-Gal site competes with Gal-3 binding to GM, but that there are additional binding events. Indeed, a fraction of GM still interacts with Gal-3 as shown by the still observable cross peak broadening. The effective KD was estimated in the low micromolar range (per Gal-3 binding site), one order of magnitude stronger than that for LacNAc, thus assessing the existence of multiple binding events at different sites, but also suggesting the existence of statistical rebinding processes. Intriguingly, although the effective binding affinity depended on the Gal/Man ratio, no clear structural explanation at the supramolecular level could be deduced.
Nature also provides spectacular multiantennae N-glycan structures that can interact with their receptor lectins. The experimental demonstration of the type of multivalent effects that these molecules may display, mediated by clustering, cross-linking, or statistical rebinding processes remains a challenge. NMR has been also applied to try to approach this scientific problem. In particular, one alternative approach to monitor the interaction of multiantennae glycans to lectins is the use of paramagnetic nuclei.79 It is well known that the presence of a paramagnetic lanthanide nucleus (Fig. 11A and B) attached to a lanthanide-bind tag linked to the reducing end of N-glycans provides80 pseudocontact shifts (PCS) that are proportional to the distance between the sugar nuclei and the metal (1/r3). In this manner, it is possible to distinguish the NMR resonance signals of the nuclei belonging to sugars at equivalent positions in the different arms, to estimate their specific distances to the lanthanide, and therefore to decipher the conformational features of the glycan.81 Thus, this methodology makes possible to distinguish between signals that in the presence of a diamagnetic metal are overlapped, such as those belonging to the same residues in the different arms of multiantennae N-glycans.80–82 Moreover, since cross peaks for every particular monosaccharide moiety are identified, 1H–13C HSQC experiments of the N-glycan, decorated with the lanthanide-binding tag, recorded in the presence and in the absence of Ricinus communis agglutinin and Datura stramonium lectins (Fig. 11C and D) allowed observing differential signal intensity decrease for each anomeric peak of the diverse Gal and GlcNAc units, thus revealing the preferences of these lectins for each arm of the N-glycan.81
 | ||
| Fig. 11 (A) Overlapped HSQC of the tetra-antennary glycan in the presence of diamagnetic metal. (B) HSQC of the tetra-antennary glycan with a paramagnetic metal. (C) HSQC of the previous molecule in the presence of a lectin. (D) Structure of the tetra-antennary N-glycan and the name of each branch. (E) Plot of the difference in intensity between spectra (C and D). Adapted from Canales et al.81 | ||
A similar strategy has been applied to study the interaction between a bivalent sialylated N-glycan and the hemagglutinin from the strain HK/68 of the influenza virus.83 The Neu5Acα2-6Gal units are recognized by the hemagglutinin on the surface of the virus, enabling the attachment of the virus to the host cell through binding to this epitope. Interestingly, microarray and infection studies have postulated that H3N2 human influenza viruses have evolved to recognize Neu5Acα2-6Gal at the non-reducing end of long polyLacNAc structures, whereas the early viruses preferred these terminal epitopes with shorter LacNAc structures.84 In this context, the paramagnetic NMR approach was applied to the study of the interaction of two biantennae N-glycans with either one or two LacNAc units capped with α2,6 linked sialic acids. First, the conformational features of the glycans were determined by the PCS analysis, while the combination of the observation and analysis of the PCS, together with STD-NMR in the presence of HK-68 allowed to deduce the existence of interaction of both sialic acids at the two arms of the N-glycan located far away from the Dy3+ (over 30 Å) present PCS. Intriguingly, in the STD-NMR experiments with Dy3+, STD signals arising from both sialic acids are present, showing that both participate in the binding, a feature that had not been proved before.
Despite the proven efficacy of this methodology, it involves chemically modifying the ligand, meaning that a non-natural modification needs to be introduced, which could affect to the properties of the binding. An alternative to the inclusion of a paramagnetic nucleus in the epitope is the labelling with 13C fragments of the molecule. In the context of protein–carbohydrate interactions, Moure et al. shed light into the interaction between several galectins (β-galactoside binding lectins) and polylactosamine molecules.61 The molecules analysed in this work consist on hexasaccharides containing three repeating units of LacNAc (tri-LacNAc), for which the galactose units were labelled with 13C. The use of 13C enables using STD-HSQC experiments with high sensitivity and no protein background, which profits a higher signal dispersion compared to traditional STD.85 This approach enables having a broad chemical shift dispersion as in the one with the paramagnetic tag, however, the advantage of 13C labelling is that the modification is minimal, and the ligand is identical to the natural structure.
Despite having a broader signal dispersion due to the capability of working in 2D, spectral overlap is still a bottleneck on many occasions. In the case of the tri-LacNAc, the internal galactose and the one in the reducing-end are isochronous, and thus cannot be differentiated. A solution to this issue is that of synthesizing molecules with the same structure but in which the labelled residue is different (Fig. 12). This approach was previously applied to decipher the conformational features of a series of selectively labelled linear β1–6 linked glucose hexasaccharides.86 The design of selective labelling of galactoses proved to be very useful to characterize the binding to galectins. The interaction with five different galectins was analysed, each with each own preferences towards the three epitopes. The case of galectin-7 is very representative of the usefulness of the selective labelling. STD-HSQC experiments with the tri-LacNAc 1, which is labelled in the three galactoses showed STD intensities mainly for terminal galactose, although the internal and reducing end units also showed STD. In order to differentiate between these galactose units, STD-HSQC experiments were performed with molecules 2 and 3, in which the galactoses are selectively labelled. In the STD with molecule 3 no STD effects are detected, whereas for triLacNAc 2 clear STD signals arise. These experiments showed clearly how Gal-7 is exclusively recognizing the terminal and internal LacNAc and not the one in the reducing-end.61
 | ||
| Fig. 12 Left: Structure of the polyLacNAc and the naming of each LacNAc epitope. Right: Molecules synthesized by Moure et al.61 | ||
More on multiple binding modes
Uniformly labelled 13C-labelled glycans had already been used to describe the multivalent binding of chitin oligosaccharides to hevein domains.87 Indeed, the specific interaction of hevein with a variety of GlcNAc-oligomers (Fig. 13) was assed by NMR, and assisted by ITC and analytical ultracentrifugation. Through this work, the existence of statistical rebinding was elegantly demonstrated. Hevein recognises short glycans, from di- to tetra-saccharides with 1:1 stoichiometry, and mM affinity. Nevertheless, the existence of multiple binding poses was evident already for the trisaccharide.88 Fittingly, for the penta-saccharide a dramatic increase in binding affinity (from millimolar to low micromolar) was measured. Fittingly, the NMR studies on the complex formed by a 13C-labeled pentasaccharide molecule and the lectin demonstrated the existence of an extended binding site that allowed for the existence of multiple binding modes in a highly dynamic process. Moreover, the ITC titrations could not be fitted by a simple 1:1 stoichiometry, suggesting the presence of higher-order complexes, which were also deduced by DOSY NMR and analytical ultracentrifugation data. Nevertheless, titration experiments at different glycan/hevein ratios demonstrated that the higher order complexes were less stable than the dynamic 1:1 alternative, in which the pentasaccharide is extended along the lectin site, on top of the statistical rebinding events.
 | ||
| Fig. 13 Scheme of a poly GlcNAc oligosaccharide. | ||
The existence of multiple binding modes for particular ligands has also been demonstrated in the investigations of age-related macular degeneration, paying attention to the structural basis of the interaction of diverse modules of complement factor H with sulfated glycosaminoglycans (GAG).89 A receptor-based NMR approach, including site directed mutagenesis allowed demonstrating that the GAG interacting site is occupies the centre of an extended binding groove, with multivalent recognition of the sulfated GAGs.
The AB5 toxins
The interaction of shiga-like toxins and related families with the cell surface oligosaccharides represent the paradigm of multivalent interactions in the glycan field and beyond. In fact, the gain in avidity using pentavalent inhibitors of these bacterial toxins that share a AB5 architecture is spectacular.90 Using model bivalent ligands (a tethered trisaccharide dimer, Fig. 14) and receptor-based NMR methods,91 it has been shown that sites 1 and 2 within a single B subunit are simultaneously occupied by the ligand. Indeed, using the 15N-enriched Shiga-like toxin B5 subunit, the analysis of the chemical shift perturbations of the lectin cross peaks strongly suggested that the bivalent ligand simultaneously binds to sites 1 and 2, through a cluster effect.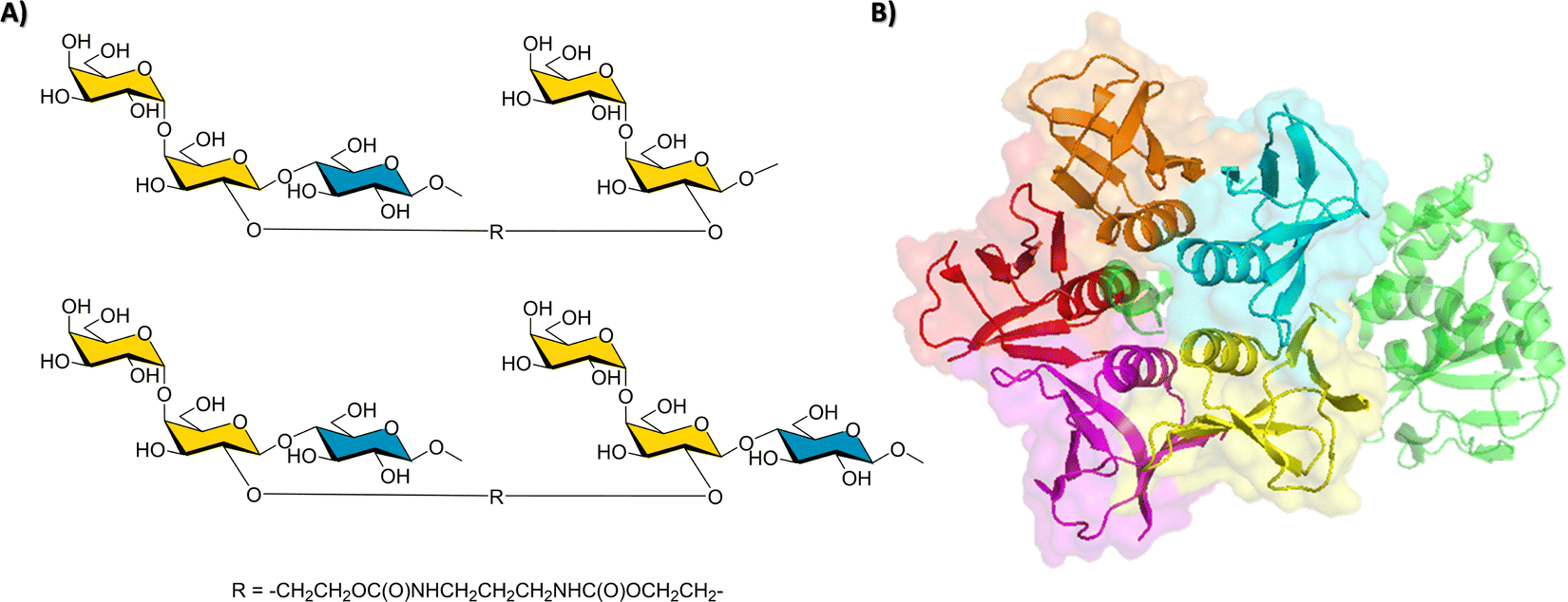 | ||
| Fig. 14 (A) The key employed bivalent molecules used to target Shiga-like toxin.91 (B) Structure of a Shiga-like toxin (PDB 4M1U), the A subunit is represented in green, whereas the B subunits are depicted with its surface in red, orange, blue, yellow, and purple. | ||
A different NMR approach, measuring residual dipolar couplings (RDC), allowed to describe the binding mode of the saccharide fragment of globotriaosylceramide and the B-subunit homopentamer of verotoxin 1 (VTB).92 The analysis of the RDC for the free and bound saccharide showed that the oligosaccharide binds in a single binding locus per monomer (site II). Fittingly, this is one of the three possible sites deduced by X-ray crystallography for the same molecular complex. No NMR experimental evidences were found for binding at the other two possible sites, which are likely low affinity sites. Interestingly, the paradigmatic STARFISH inhibitor invented by Bundle and co-workers90 was designed to bridge sites 1 and 2, although it exclusively binds to site 2 in the two adjacent molecules of VTB in the crystal structure. Nevertheless, it cannot be discarded that the low affinity sites I and III may contribute to the molecular recognition events in physiological conditions.
In a similar context, the synergic application of inter-ligand NOE and STD NMR experiments, using the DEEP protocol, permitted demonstrating the presence of a cryptic binding subsite on the ganglioside recognition site of cholera toxin-B.53 The combination of the experiments with computing data acquired through Hamiltonian replica exchange molecular dynamics (HREMD) revealed that, although the subsite could not be deduced by inspection of the X-ray crystal structure of the GM1/CTB complex, the MD simulations predicted that it can be easily generated by simple rearrangements of the orientation of Lys138 and Ile59, close to the known Neu5NAc and Gal binding subsites.
In the same subject, the binding of the histo blood group antigens (HBGA) to the CTB-pentamers and the El Tor variant has been investigated by STD-NMR and trNOESY experiments.93 Interestingly, no significant differences were observed, and similar binding affinities were deduced for both toxin genotypes. However, the HBGA antigens interact at a binding site distinct from that of GM1, the canonical binder. Indeed, the blood group H tetrasaccharide and the GM1-oligosaccharide simultaneously bind to the classical CTB.
Noroviruses
An NMR-based fragment-based drug discovery (FBDD) protocol has also been employed for developing multivalent inhibitors versus noroviruses.94 This well-known protocol tries to identify several fragment hits for adjacent binding sites. Then, the fragments can be chemically merged or linked together to try to increase the association. Alternatively, displaying these molecules into a polymer backbone might also yield high avidity multivalent ligands. NMR perfectly combines with this medicinal chemistry protocol since it provides the structural perspective for the rational design.It has been described that that the HBGA are related to the life cycle of the virus. More specifically, it had been previously demonstrated that α-L-fucose (the common part of A, B and H blood groups antigens) is necessary for binding.95 The authors employed STD-NMR spectroscopy combined with T2-filtering experiments to screen a commercial small-compound library (Maybidge Ro5 500 fragment library) versus norovirus virus like particles (VLP).
The protocol is fairly robust: The initial screening process by STD-NMR and spin-lock filtered experiments, (VLP:ligand ratio, 1:10), led to a very high hit rate. Then, these hits were subjected to a competition STD-NMR experiment to identify those that indeed bind to the Fuc subsite of the HBGA binding locus, using an excess of Fuc as competitor. The observed decrease in the STD intensity of the putative ligands indicated those that were competitors. Using small mixtures of just 9 molecules with a small molar excess of Fuc, the hits were ranked according to their relative binding ability.
Since the VP1 proteins of VLP are dimers, (25 Å distance between binding sites, and the dimers placed at ca. 75 Å from each other), a multivalent polymer was synthetized, placing one Fuc moiety and one identified ligand every 30 propionylamide units. It was guessed that this geometry would allow simultaneous binding to the HBGA binding clefts at the dimer and also at the vicinal dimers.96 Fittingly, an outstanding 1000-fold gain of potency over Fuc was obtained. The binding mode and bioactive conformation of the heterobifunctional moiety of the polymer was deduced (Fig. 15) by a combined STD-NMR and trNOE approach.97,98
 | ||
| Fig. 15 Left: Binding epitope map of the heterobifunctional part of the polymer determined by STD-NMR in the presence of the Norovirus VLPs. Right: Scheme of the polymer to which the ligand is attached. | ||
Indeed, the interaction of different ligands with noroviruses has been extensively studied by NMR. Recently, the comparison of the results obtained through NMR have been compared to those obtained by other techniques, highlighting the pros and cons of the diverse experimental approaches, and providing explanations to the observed reasons.99 The authors conclude that the combination of Mass Spectrometry techniques and NMR experiments provides the best insights for understanding the HBGA binding events by norovirus capsid proteins, providing reliable and reproducible binding affinities.
The molecular details of the recognition of the HBGA by a Human Norovirus had been previously determined by STD-NMR. The binding specificity was obtained as well as details on the bioactive conformation of the glycans.100
Moreover, the binding of HBGAs and sialoglycans to a variety of human and murine norovirus capsid proteins has been extensively studied by NMR experiments.101 Interestingly, on top of the usually employed STD-NMR experiments, the use of chemical shift perturbation NMR experiments allowed redefining the glycan recognition code for noroviruses. In particular, the norovirus P-domains from both species did not bind to the sialyl-containg glycans. Moreover, the murine P-domains did not bind to the HBGA either, while the infection through MNV-1 of cells deficient in sialoglycans did not present any difference to other cells that were expressing the corresponding glycans.
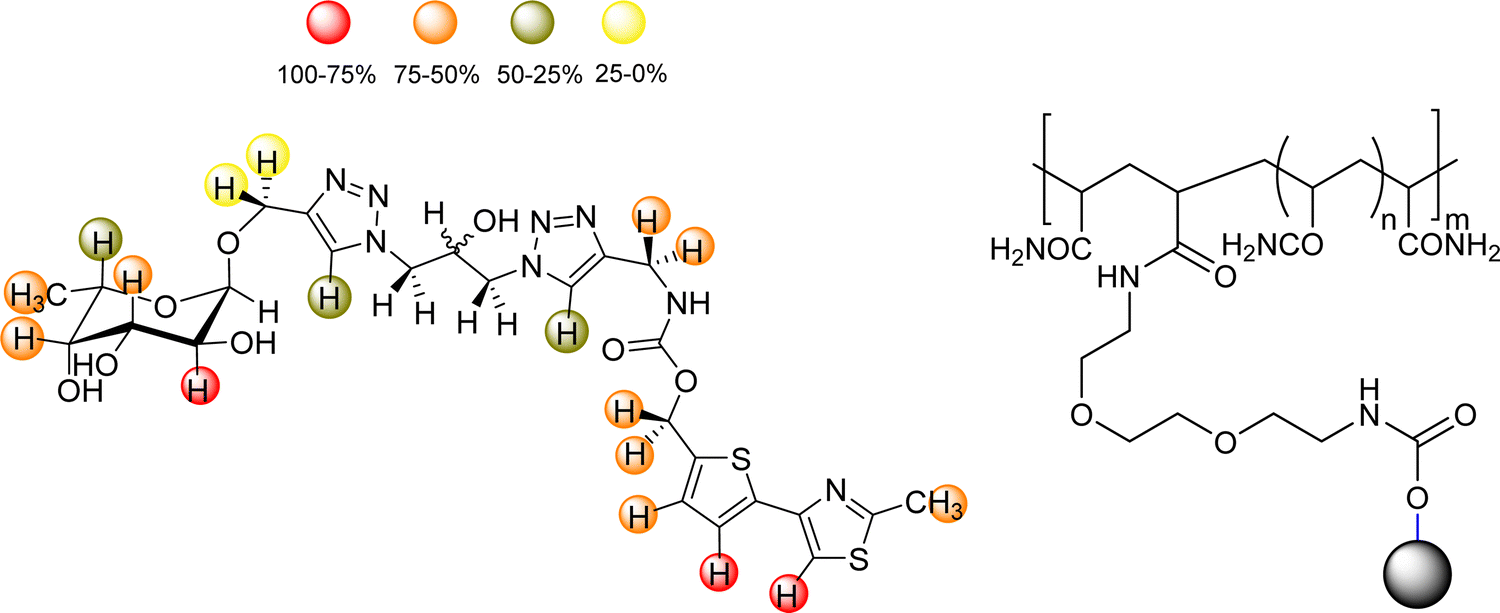
Additionally, glycomacromolecules functionalized with Fuc moieties (Fig. 16) have been developed to Targeting the Human Norovirus Capsid Protein in a precise manner.102 The design was based on the fact that the P domain dimer (P-dimer) contains two distinct HBGA binding loci, although two additional sites have been recently found between the two outer canonical binding sites The distances between the different sites were assessed through X-ray crystallography, being 11 Å (between Fuc sites 1 and 3), 17 Å (Fuc sites 1 and 4) and 27 Å (Fuc sites 1 and 2), as can be seen in Fig. 17.
 | ||
| Fig. 16 Left: Scheme of the histoblood group antigens (HBGA 0, A and B). Right: The glycomacromolecules synthesized by Bücher et al., adapted from ref. 98. | ||
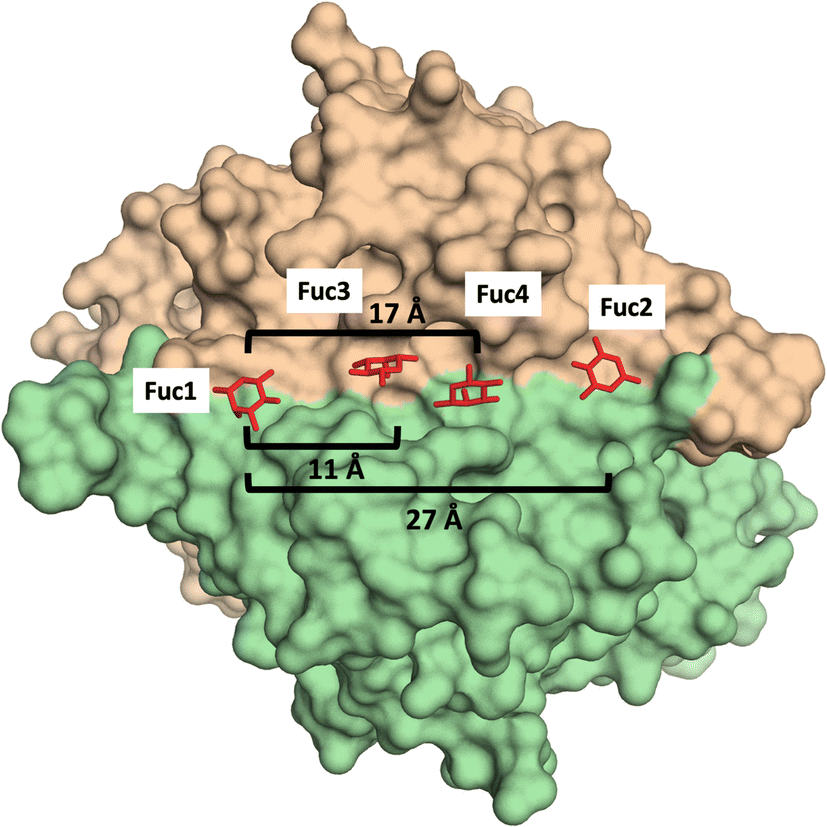 | ||
| Fig. 17 X-Ray crystal structure of the human norovirus capsid protein dimer in complex with four L-Fuc molecules (red). Binding to the four binding sites is a dose-dependent and stepwise process, where followed order is indicated according to the Fuc numbering in the image. Distances between different Fuc pockets are indicated. PDB ID: 4Z4R. | ||
Ligand and receptor-based NMR experiments were employed to disentangle the challenging multivalent interaction. The multivalent nature of the presented Fuc moieties at the polymer lead to precipitation of the ligands in the presence of the protein dimers. Nevertheless, the obtained NMR spectra were still useful to provide the binding epitope on the glycomacromolecules: the Fuc moieties were in close contact with the protein while the polymer backbone was not. Nevertheless, no information on the number of units at the individual glycomacromolecule binding to the protein could be extracted.
Although precipitation problems were also observed during the TROSY-based chemical shift perturbation (CSP) experiments, information on the binding mode of one particular glycomacromolecule to the GII.4 P-dimers could be extracted, showing that the CSP were basically identical to those observed when simple Fuc was added. This fact also supports that that the scaffold does not provide major interactions. Nevertheless, additional CSPs were observed at remote positions of the binding site, which were interpreted in terms of allosteric effects.
C-Type lectins: DC-SIGN and langerin
DC-SIGN represents a paradigmatic case of lectin oligomers. Indeed, four CRDs are displayed at the extracellular domain (ECD) of this lectin present in dendritic cells,103,104 where modulates the interplay of the adaptive and innate immune responses. Interestingly, both DC-SIGN and langerin belong to the family of C-type lectins expressed on the surface of dendritic cells (DC).105–110 Although both recognize Man-containing saccharides, HIV uses DC-SIGN in immature DC cells to ease the trans-infection of T-cells, opposing to langerin, which contributes to the virus elimination.110 Therefore, the development of ligands that selectively interact with DC-SIGN and not (or weakly) with langerin is of paramount importance. This tetrameric architecture of DC-SIGN opens the door for enhancing the interaction strength via avidity. Hence, numerous attempts of multivalent display of modified glycomimetics have been reported in the quest of achieving binding avidities in the nM range.111,112 A wise approach has used the natural sugar structure as starting scaffold, which is then decorated with diverse fragments containing a variety of functional groups in a FBDD approach.113–115The quest for allosteric regulation of DC-SIGN and its selective recognition versus langerin has also led to the design of heteromultivalent molecules.116 In this context, Rademacher and coworkers have described an elegant and multidisciplinary approach to discriminate DC-SIGN and langerin, continuously increasing the complexity of the employed molecules in different publications along the years. Initially, a library of mannosides derivatized at C1 and C6 was screened for langerin using a 19F NMR reporter displacement assay. A ligand with micromolar affinity (KI = 0.23 ± 0.03 mM and KD = 0.5 ± 0.2 mM) was then discovered.
This ligand was conjugated to DSPE-PEG2kDa lipids to be displayed on liposomes. Fittingly, no meaningful binding was observed for langerin+ cell, while the interaction with DC-SIGN+ cells occurred, especially when the hetero-multivalent liposomes were employed.117 Based on this evidence, it was hypothesized that the glycomimetic bearing liposome might target a secondary binding pocket on DC-SIGN. Then, the binding of glycomimetic 48 was scrutinized by different NMR techniques to provide a structural perspective of the findings (Fig. 18). The dissociation constant was estimated independently by 19F-based CSP and 19F R2 filtered experiments,117 assisted by 1H,15N-HSQC based titrations, yielding almost identical values within the micromolar range (KD ∼ 0.46 mM). Moreover, 19F R2 filtered experiments were carried out under inhibitory conditions, observing that neither high Man concentrations nor EDTA addition completely abrogated DC-SIGN binding to the glycomimetic, while the same experiments with langerin resulted in complete inhibition. In fact, in the STD-NMR experiments, the signals arising from the Man residue were substantially reduced in presence of EDTA, while those of the biphenyl aglycone highly increased (Fig. 18). The addition of deuterated Man further enhanced the saturation received by protons at the aromatic system. Thus, the data demonstrated suggest the presence of a Ca2+-independent, secondary binding site for DC-SIGN, which displays specific interactions with the glycomimetic, driven by the biphenyl system.91H,15N-HSQC based titrations showed the characteristic CSPs at the carbohydrate binding domain, (N344, N365, E358, N366, N367, S360 and F313) together with others at residues located far away. Interestingly, these contacts had previously been identified by Aretz et al.118 Additional experiments carried out under Ca2+-free buffer and competition experiments with high Man concentrations demonstrated the increment of the CSP of remote residues, while those at the sugar binding site were abolished in the absence of Ca2+. Fittingly, it was finally demonstrated that targeting the putative allosteric binding pocket potentiated glycan recognition and (Fig. 19) allosteric activation, selectively for DC-SIGN over langerin. Therefore, although formally no NMR methods were applied to the monitor the interaction of the multivalent system with the lectins, they were instrumental to provide the rational for the observed functionality. This investigation provided ground-breaking information on the differentiation of ligands targeting DC-SIGN versus langerin, as continuation of previous investigations of the research group on those systems using similar NMR protocols.
 | ||
| Fig. 18 STD NMR epitope mapping of the glycomimetic with DC-SIGN. Left: Epitope map under Ca2+ containing buffer conditions, where the Ca2+-dependent binding mode of the glycomimetic was determined. Right: Epitope map in the presence of EDTA to determine the Ca2+ independent binding mode. Image adapted from Wawrzinek et al.112 | ||
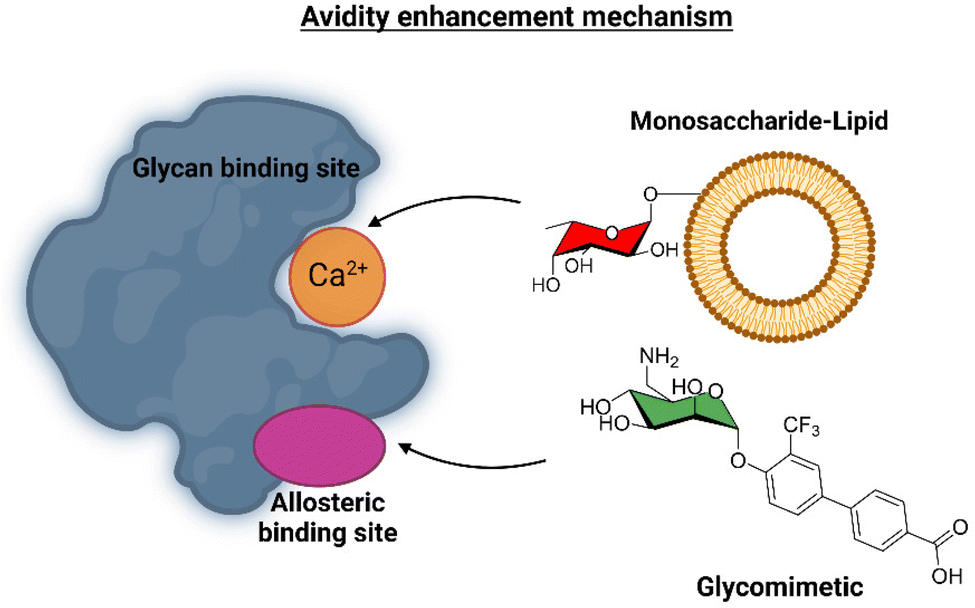 | ||
| Fig. 19 Scheme of the avidity enhancement mechanism for DC-SIGN in the presence of the glycomimetic and Fuc-bearing liposome particles. The binding of the glycomimetic to the DC-SIGN allosteric binding site causes structural rearrangements that potentiate the binding at the canonical sugar binding site resulting in a cooperative avidity enhancement.112 | ||
Langerin has also been used as target for glycosaminoglycans. In particular, the combination of experimental data obtained through STD-NMR and trNOESY experiments allowed deducing that while small heparin-like oligosaccharides bind to langerin in a Ca2+-dependent way in the canonical site, a long hexasaccharide, with an extra O-sulfate moiety at the non-reducing end, interacts with the lectin in a previously identified Ca2+-independent binding site. Indeed, the extra sulfate abolishes the interaction at the Ca2+ locus. Curiously, HEP-like oligosaccharides can also bind to the Ca2+-dependent binding site, in contrast to large heparin (6 kDa) that is bound at the multimerization interface between langerin monomers.119
Indeed, further NMR investigations combining glycan array screening with NMR spectroscopy allowed deducing that the interaction of heparin hexasaccharides to the elusive secondary site did not require the presence of Ca2+ ions, while activated an intradomain allosteric network of langerin that had previously been identified, although it was just linked to the affinity and release of Ca2+ ions.120
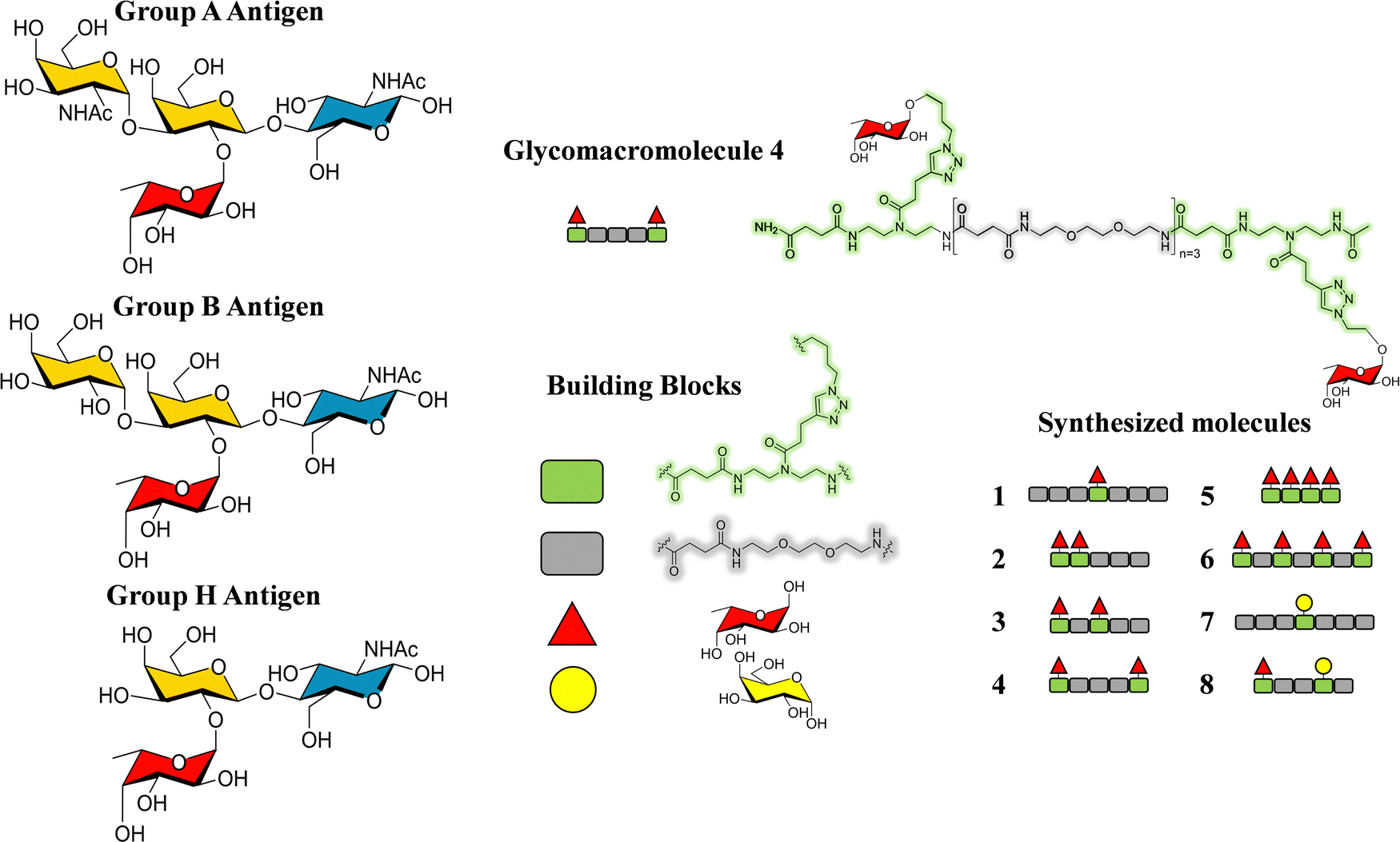
As a landmark in the field, combining multivalent presentation with NMR, an elegant series of asymmetrically branched precision glycooligomers have been built using chemical synthesis to study multivalent lectin–sugar interactions, such as in the Fig. 20.121
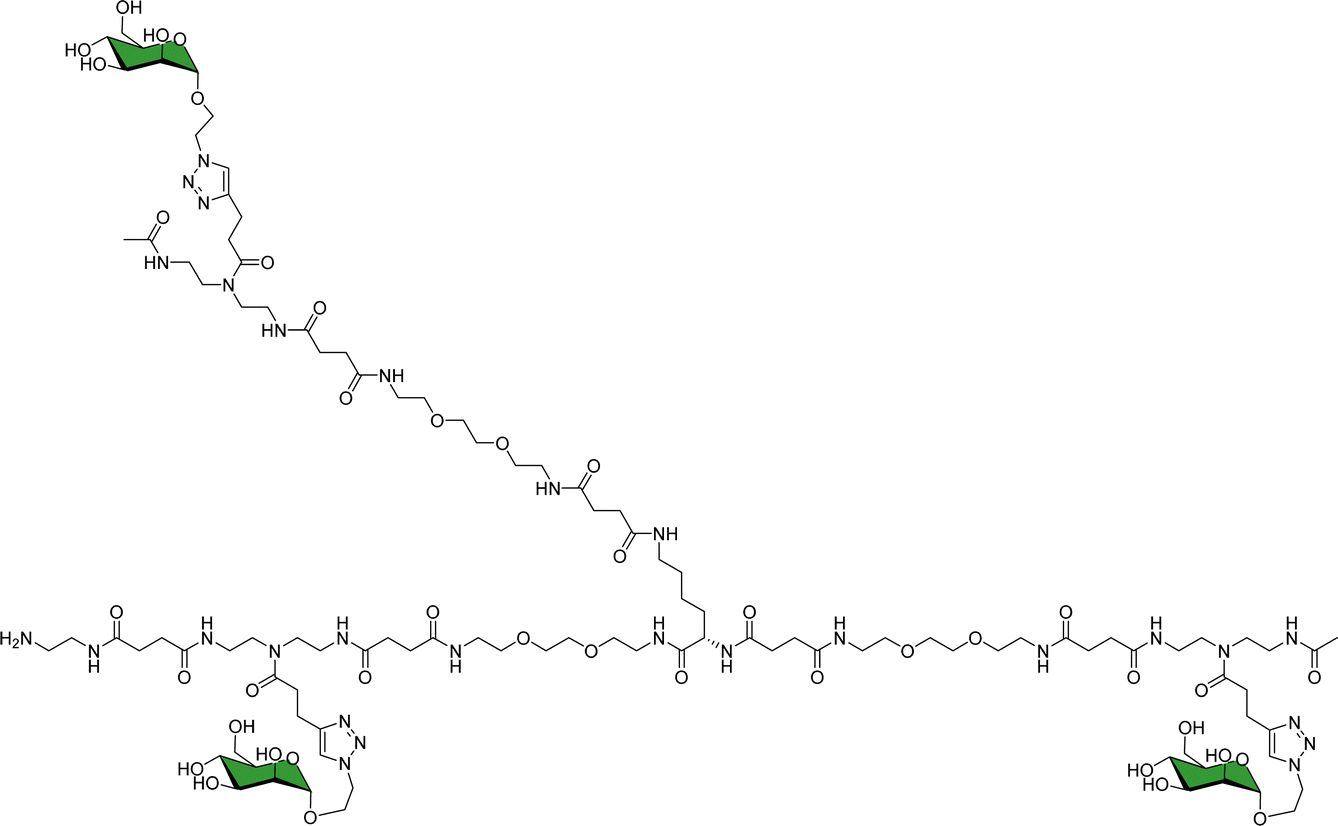 | ||
| Fig. 20 Schematic view of one of the key glycooligomers employed to target multivalent lectin–glycan interactions.121 | ||
The binding features of the glycomacromolecules to langerin were monitored via19F-NMR based T2-filter competition assay, using the basic monovalent N-acetylmannosamine analogue decorated with 19F as spy molecule. The 19F-NMR based strategy revealed a clear correlation between the glycooligomers architecture and the resulting binding affinity. Again, this work showed how the combined use of NMR and multivalent presentation provides structural evidences on the way towards tuning and modulation of key glycan–lectin interactions. Further elaboration of this concept has allowed the generation of a specific glycomimetic ligand for langerin that is able to specifically target human Langerhans cells in the human skin, when conjugated to liposomes. In this case, the ligand was designed and built based on previous knowledge acquired for the interaction of heparin oligosaccharides with langerin.122
The use of 19F-NMR-based experiments in fragment-based screening has been employed also in the search for druggable pockets in multimeric lectins, as in β-propeller lectins.123 The hits identified by 19F-NMR were further validated by orthogonal methods, such as SPR and TROSY NMR experiments. In that sense, the NMR approach identified druggable pockets in a bacterial b-propeller lectin, which could be used in the design of allosteric inhibitors.
Given its relevance in modulate immune response, DC-SIGN has been one of the key targets for drug discovery campaigns. It has been shown that DC-SIGN binds Man and Fuc-containing glycans from viral proteins, including the gp120 glycoprotein from the HIV envelope. Different NMR studies have demonstrated that the DC-SIGN Man/Fuc binding site shows a large plasticity and can indeed bind these sugar residues in different modes. For instance, the interaction of this ligand DC-SIGN with a glycomimetic pseudotrisaccharide (Fig. 21) deduced by a combined ligand-based NMR approach, using STD-NMR and trNOESY experiments.120 The use of molecular modelling protocols together with CORCEMA-ST calculations, assessed that the experimental data can only be explained by using an ensemble of binding poses, which can account for the large inhibition provided by the glycomimetic. Indeed, it was also demonstrated that the pseudomannotrioside is also able to promote clustering without any multivalent presentation.124
 | ||
| Fig. 21 Scheme of the pseudomannotrioside employed to target DC-SIGN.124 | ||
The existence of multiple binding modes has also been assessed for DC-SIGNR (also dubbed L-SIGN), a C-type lectin highly related to DC-SIGN.125 In particular, the interaction of Man9GlcNAc with the carbohydrate-recognition-domain of the lectin was investigated by receptor-based NMR techniques. Interestingly the lectin displays micro- to millisecond dynamics in the presence of the Man9 glycan, with extensive line broadening. The data strongly suggest the existence of multiple binding modes, which can interconvert over a range of time scales.125
Other receptors
DC-SIGN is linked to multiple host-pathogen interactions. Indeed, it is related to HIV infection through the binding to the gp120 glycoprotein, as stayed above. In this context, the study of the binding of glycans to the key broadly neutralizing 2G12 antibody developed against gp120 has also been tackled by NMR methods. Indeed, 2G12 targets clusters of high-Man glycans at the gp120 surface. As a matter of fact, these high-Man glycans have been foreseen as targets structures for developing glycomimetics that can elicit antibodies resembling 2G12-like. Indeed, STD-NMR and trNOESY experiments have been used to demonstrate that 2G12 binds to branched Man glycans beyond the pentasaccharide, using different binding modes as a way to enhance affinity.126As a further step, diverse oligomannosides were used to prepare glyconanoparticles targeting 2G12.127 The use of STD-NMR methods allowed demonstrating that the Man glycans, when clustered onto gold nanoparticles, are able to interact with 2G12 with high affinity, and even inhibited the binding between 2G12 and the gp120 glycoprotein. The observed affinity was dependent on the particular Man-glycan and the density on the gold surface, as also demonstrated by SPR. Moreover, some glyconanoparticles were able to restrict the interaction of 2G12 with a recombinant virus.
Also related to the battle against HIV, the interaction of cyanovirin-N (CV-N) with Man glycans has been extensively studied by NMR, specially using receptor-based methods,128 since it is relatively small and provides very well defined 1H–15N spectra.129 CV-N is a potent antiviral lectin with two binding sites.130 However, the fine details of the interaction of the glycan and the lectin within the complex, including the involvement of the saccharide hydroxyl groups has been deciphered by employing a 13C-labelled oligosaccharide. 13C-based methods had previously been used to study the interaction between cyanovirin-N (CV-N) and a linear mannose trisaccharide Manα(1-2)Manα(1-2)ManαOMe (Man3).131 In this system, the interaction between the trimannoside and CV-N is too strong, resulting in very low quality STD-NMR experiments, since the off-rate is too low. However, this dynamic regime enables observing the bound state of the 13C-labelled glycan in the 1H–13C HSQC spectrum in the presence of equimolar amounts of the lectin (Fig. 22), directly observing the chemical shift perturbation of the signals of the ligand generated by the lectin. As a result, changes in the linewidth and intensity of the peaks are also observed, yielding information on the dynamics changes of the sugar between the free and bound states.
 | ||
| Fig. 22 (A) Scheme of Man3. (B) In black 1H–13C HSQC of the 13C labelled Man3 and in blue in the presence of CV-N. The shift of the signal is indicated with arrows (C) sugar region of the 13C filtered NOESY-HSQC (D) side chain region of the 13C filtered NOESY-HSQC. NOEs between the glycan and CV-N are indicated. Adapted from ref. 131. | ||
The use of 13C labelled carbohydrates and the study of its complex with a 15N or 13C/15N labelled lectin opens up the possibility of using a large variety of NMR experiments to characterize the binding events. The main advantage is that the complexity of the spectra can be significantly simplified through filtering the magnetization through 13C or 15N, yielding information on the 1H nuclei attached to those labels. For instance, CNH-NOESY experiments can be performed, observing direct intermolecular NOEs between protons attached to 13C labelled nuclei (sugar) and those attached to 15N labelled nuclei (lectin). Another possibility that labelling generates is detecting intermolecular NOEs between protons attached to 13C labelled nuclei of the sugar and other protons of protein (Fig. 20C and D). Additionally, interresidual NOEs within the sugar can be detected, reducing the spectral complexity. Finally, through 2D 1H–13C-HSQC-NOESY experiments, the conformation of the free and bound ligand could be elucidated. In this case, slight differences in the conformation between the two states were encountered.
The frontiers
NMR methods are continuously advancing thanks to the great developments in experiments, hardware, and access to ultra-high field magnets at the GHz scale. Nevertheless, besides the intrinsic challenges of multivalent recognition events, monitoring dynamic processes in the seconds timescale or below also defy the limits of the technique. From a personal perspective, we herein cite two topics in which NMR will make important contributions in the years to come: Monitoring glycosylation events and on-cell NMR.Monitoring O-glycosylation events
Mucins are highly O-glycosylated glycoproteins displayed at the cell surface of mammals. Indeed, modifications in the glycosylation of mucins in humans have been linked with cancer development. Mucin-1, as key example, displays multiple repetitions of the same 20-mer polypeptide. Therefore, the presentation of the putative Ser and Thr residues to be glycosylated also constitute an example of multivalent presentation for the GalNAc transferases in charge of the O-glycosylation event. In this context, using a 15N-labelled 80 amino acids MUC-1 construct (with four tandem repeat domains), NMR methods have been employed to disentangle the mechanism and sequence of the O-glycosylation process (Fig. 23), the key role of the neighbouring tandem repeats for further glycosylation at the multiple acceptor sites, and the existence of local conformational motions at the polypeptide moiety.132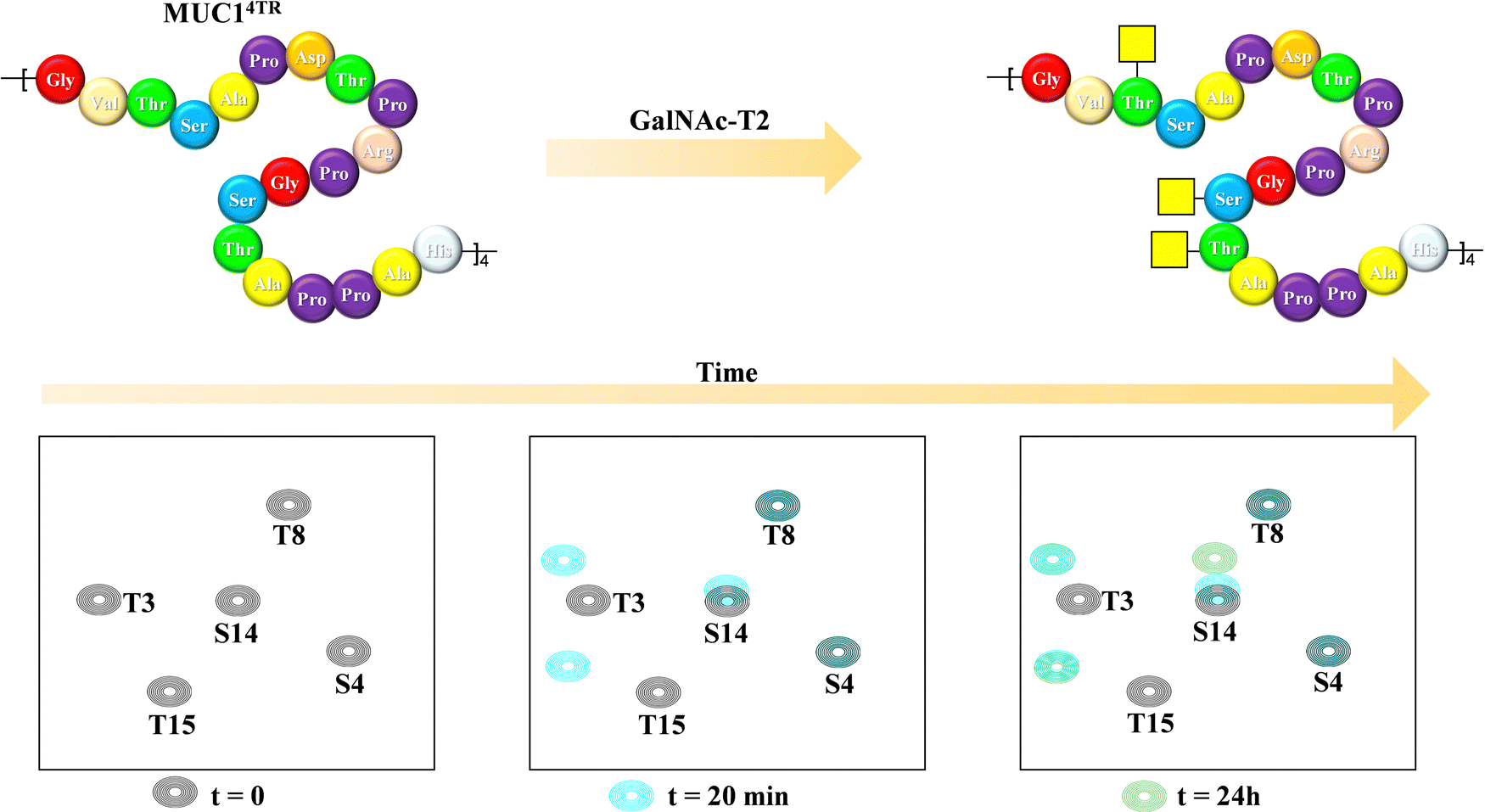 | ||
| Fig. 23 Glycosylation of the MUC1 antigen by GalNAc-T2 analysed through 1H,15N-HSQC of a 20-amino acid mucin tetramer, adapted from ref. 132. | ||
On-cell NMR
Glycans and lectins are mostly found on the cell surface. It is obvious that the characterization of its molecular recognition features using systems close to the natural environment may be of great interest to understand the interactions with the aim of modulating them. Therefore, on-cell NMR133 is a valuable approach that is starting to be realistic and has shown applications in the glycan-recognition field. It is likely that, given the density of the glycans or lectins on the cell surface, the conditions are of a multivalent character.Generally speaking, on-cell NMR methods (Fig. 24) could use either ligand-based or receptor-based NMR approaches. The motional properties and the kinetics of the free-bound chemical exchange process are essential to define the approach. In the glycan–protein interaction field, ligand-based approaches have been usually employed. In particular, STD-NMR has been widely applied to probe the interaction of membrane-associated lectins to a variety of glycans. In this case, it is absolutely essential that blank experiments are carried out in the absence of the ligand and in the absence the membrane receptor to be able to disregard possible STD signals that may arise from non-specific binding between the ligands and the cell. Therefore, the experiment with the same cells should be repeated but without the expression of the target protein. In case that some signals are still observed, the difference between the STD NMR spectrum acquired in the presence of the receptor and that obtained in the absence of it should be subtracted to give the STDD spectrum, which should only show the STD signals arising from specific interactions.
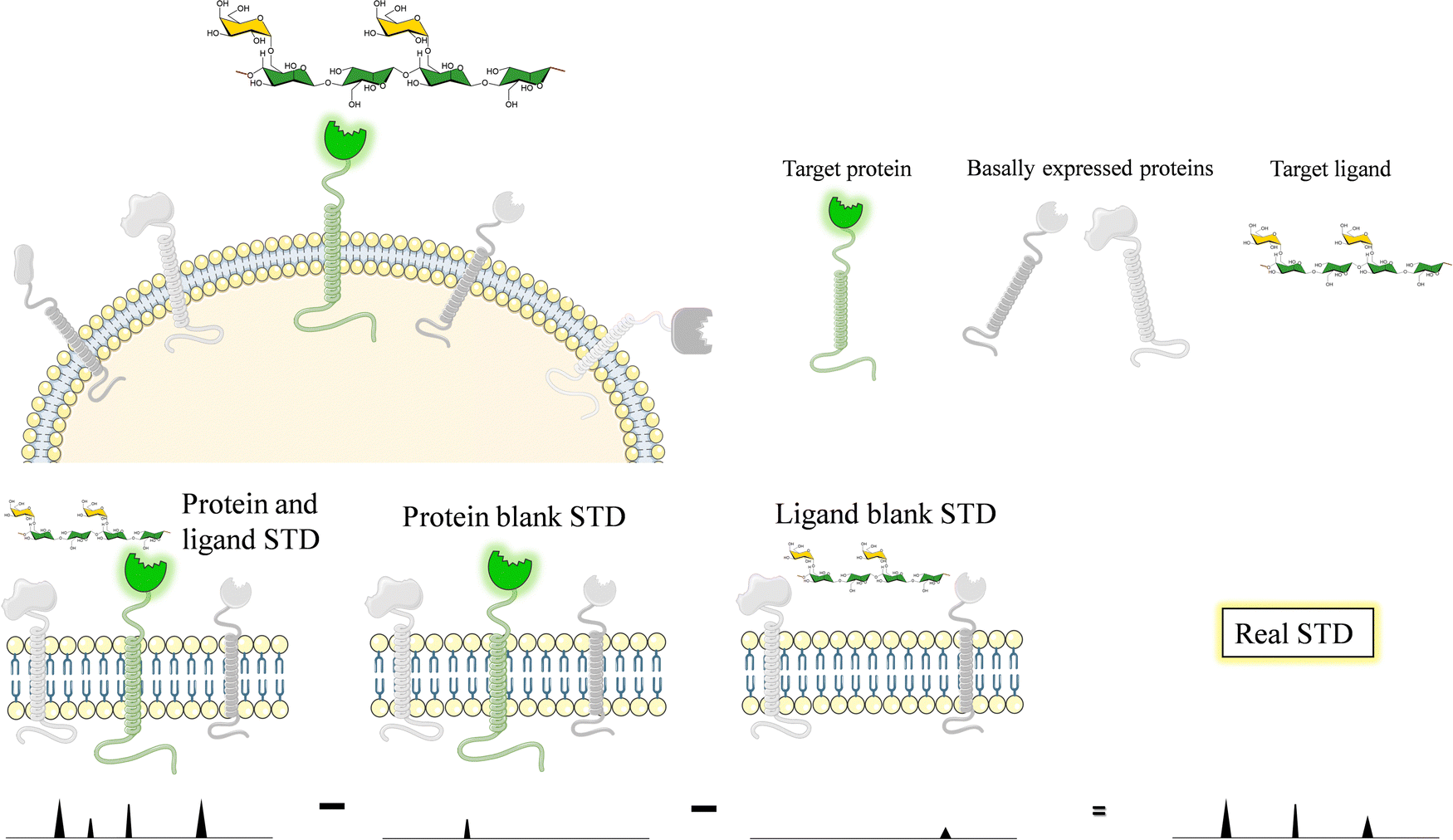 | ||
| Fig. 24 Scheme of the protocol to carry out on-cell NMR experiments. | ||
This approach was first applied to investigate the interaction of membrane bound DC-SIGN with a mannan polysaccharide.134 The quality of the STD-NMR spectrum was very high, displaying signals only when the employed K562 cells were transfected with DC-SIGN, showing the high specificity of the interaction.
The methodology has also been applied to disentangle the interaction of the Neu5Ac-α-(2,6)-Gal-β-(1–4)-GlcNAc trisaccharide with H1 and H5 influenza hemagglutinins from human and avian strains. The trimeric lectins were transfected on the surface of HEK 293T human cells. Interestingly, under these conditions, the HAs keep their native trimeric geometry and binding features.135 The authors demonstrated, through STD-NMR methods that the glycan epitopes recognized by the two HA variants were different.
On-cell STD NMR methods have also been applied as a fast and reliable method to screen diverse ligands136 targeting the FimH lectin, a mannose-binding bacterial adhesin that is a virulence factor and therefore, a therapeutic target for treating infections of the urinary tract. In this case, the binding epitopes of a series of dendrimers decorated with Man were deduced, while the ability of the multivalent molecules to prevent FimH-mediated yeast agglutination was also determined (Fig. 25).
 | ||
| Fig. 25 The multivalent molecules employed to target FimH.136 | ||
Conclusions and outlook
The application of NMR to study molecular recognition events involving multivalent entities remains challenging. However, as described in the precedent paragraphs, different NMR-based methods, usually combined with other techniques can be successfully used to access to key information on the interaction. Without being exhaustive, we have herein selected a variety of examples that cover a wide range of applications, using different concepts, approaches and methodologies. The latest developments with ultra-high field magnets, beyond the GHz, novel technologies, such as dynamic nuclear polarization, as well as the extension of on-cell methods will certainly provide new avenues to disentangle these multivalent events and approaching the results of the in vitro experiments performed in the lab to those actually taking place in the biological environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank generous funding by the European Research Council (RECGLYCANMR, Advanced Grant No. 788143), the Agencia Estatal de Investigación (Spain) for grant PDI2021-1237810B-C21, and CIBERES, an initiative of Instituto de Salud Carlos III (ISCIII), Madrid, Spain. We also thank Marie-Skłodowska-Curie actions (TN BactiVax, under grant agreement No. 860325).References
- I. Bucior and M. M. Burger, Curr. Opin. Struct. Biol., 2004, 14, 631–637 CrossRef CAS PubMed
.
- S. B. Oppenheimer, Integr. Comp. Biol., 1978, 18, 13–23 CAS
- K. Furukawa, Y. Ohkawa, Y. Yamauchi, K. Hamamura, Y. Ohmi and K. Furukawa, J. Biochem., 2012, 151, 573–578 CrossRef CAS PubMed
- K. A. Karlsson, Adv. Exp. Med. Biol., 2001, 491, 431–443 CrossRef CAS PubMed
- A. F. F. R. Nardy, L. Freire-de-Lima, C. G. Freire-de-Lima and A. Morrot, Front. Oncol., 2016, 6, 1–7 CrossRef PubMed
- M. Mazalovska and J. C. Kouokam, BioMed Res. Int., 2018, 2018, 1–12 CrossRef PubMed
- Y. Li, D. Liu, Y. Wang, W. Su, G. Liu and W. Dong, Front. Immunol., 2021, 12, 1–12 Search PubMed
- R. Claveria-Gimeno, S. Vega, O. Abian and A. Velazquez-Campoy, Expert Opin. Drug Discovery, 2017, 12, 363–377 CrossRef CAS PubMed
- K. de Schutter and E. J. M. van Damme, Molecules, 2015, 20, 9029–9053 CrossRef CAS PubMed
- P. Valverde, J. D. Martínez, F. J. Cañada, A. Ardá and J. Jiménez-Barbero, ChemBioChem, 2020, 21, 2999–3025 CrossRef CAS PubMed
- M. P. Lenza, U. Atxabal, I. Oyenarte, J. Jiménez-Barbero and J. Ereño-Orbea, Cells, 2020, 9, 1–19 CrossRef PubMed
- C. Büll, R. Nason, L. Sun, J. van Coillie, D. M. Sørensen, S. J. Moons, Z. Yang, S. Arbitman, S. M. Fernandes, S. Furukawa, R. McBride, C. M. Nycholat, G. J. Adema, J. C. Paulson, R. L. Schnaar, T. J. Boltje, H. Clausen and Y. Narimatsu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–12 CrossRef PubMed
- L. Johannes, R. Jacob and H. Leffler, J. Cell Sci., 2018, 131, 1–9 CrossRef PubMed
- S. Bertuzzi, J. I. Quintana, A. Ardá, A. Gimeno and J. Jiménez-Barbero, Front. Chem., 2020, 8, 1–17 CrossRef PubMed
- A. Dahlqvist, H. Leffler and U. J. Nilsson, ACS Omega, 2019, 4, 7047–7053 CrossRef CAS
- P. Bojarová, M. R. Tavares, D. Laaf, L. Bumba, L. Petrásková, R. Konefał, M. Bláhová, H. Pelantová, L. Elling, T. Etrych, P. Chytil and V. Křen, J. Nanobiotechnol., 2018, 16, 1–16 CrossRef PubMed
- M. Hassan, F. Baussière, S. Guzelj, A. P. Sundin, M. Håkansson, R. Kovačič, H. Leffler, T. Tomašič, M. Anderluh, Ž. Jakopin and U. J. Nilsson, ACS Med. Chem. Lett., 2021, 12, 1745–1752 CrossRef CAS PubMed
- J. Hirabayashi, J. Biochem., 2008, 144, 139–147 CrossRef CAS PubMed
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. de Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC
- K. S. Bücher, P. B. Konietzny, N. L. Snyder and L. Hartmann, Chem. – Eur. J., 2019, 3301–3309 Search PubMed
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed
- R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef CAS PubMed
- T. A. Shewmake, F. J. Solis, R. J. Gillies and M. R. Caplan, Biomacromolecules, 2008, 9, 3057–3064 CrossRef CAS PubMed
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, 1800425 CrossRef PubMed
-
J. M. Rondeau and H. Schreuder, Protein Crystallography and Drug Discovery, Elsevier Ltd, 2015 Search PubMed
- T. Lütteke, M. Frank and C. W. von der Lieth, Carbohydr. Res., 2004, 339, 1015–1020 CrossRef PubMed
- J. Agirre, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 171–186 CrossRef CAS PubMed
- J. Agirre, G. J. Davies, K. S. Wilson and K. D. Cowtan, Curr. Opin. Struct. Biol., 2017, 44, 39–47 CrossRef CAS PubMed
- J. H. Lee, G. Ozorowski and A. B. Ward, Science, 2016, 351, 1043–1048 CrossRef CAS PubMed
- M. Atanasova, H. Bagdonas and J. Agirre, Curr. Opin. Struct. Biol., 2020, 62, 70–78 CrossRef CAS PubMed
- A. Penezic, G. Deokar, D. Vignaud, E. Pichonat, H. Happy, P. Subramanian, B. Gasparović, R. Boukherroub and S. Szunerits, Plasmonics, 2014, 9, 677–683 CrossRef CAS
- Y. Ji and R. J. Woods, Adv. Exp. Med. Biol., 2018, 1104, 259–273 CrossRef CAS PubMed
- B. N. Murthy, S. Sinha, A. Surolia, S. S. Indi and N. Jayaraman, Glycoconjugate J., 2008, 25, 313–321 CrossRef CAS PubMed
- A. Ardá and J. Jiménez-Barbero, Chem. Commun., 2018, 54, 4761–4769 RSC
- R. Marchetti, S. Perez, A. Arda, A. Imberty, J. Jimenez-Barbero, A. Silipo and A. Molinaro, ChemistryOpen, 2016, 5, 274–296 CrossRef CAS PubMed
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618–13630 CrossRef CAS PubMed
-
U. Atxabal, A. Gimeno and J. Jiménez-Barbero, Comprehensive Glycoscience, 2nd edn, 2021, pp. 329–345 Search PubMed
- L. Unione, S. Galante, D. Díaz, F. J. Cañada and J. Jiménez-Barbero, MedChemComm, 2014, 5, 1280–1289 RSC
- F. Ni and H. A. Scheraga, Acc. Chem. Res., 1994, 27, 257–264 CrossRef CAS
-
T. Peters, Carbohydrates in Chemistry and Biology, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008, pp. 1003–1023 Search PubMed
- T. Weimar and T. Peters, Angew. Chem., Int. Ed. Engl., 1994, 33, 88–91 CrossRef
- P. M. Nieto, Front. Mol. Biosci., 2018, 5, 1–7 CrossRef PubMed
- V. Jayalakshmi and N. R. Krishna, J. Magn. Reson., 2002, 155, 106–118 CrossRef CAS PubMed
- J. L. Asensio, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Biochem., 1995, 233, 618–630 CrossRef CAS PubMed
- D. Jeannerat and J. Furrer, Comb. Chem. High Throughput Screening, 2011, 15, 15–35 CrossRef PubMed
- P. J. Hajduk, E. T. Olejniczak and S. W. Fesik, J. Am. Chem. Soc., 1997, 119, 12257–12261 CrossRef CAS
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS
-
(a) J. D. Martínez, P. Valverde, S. Delgado, C. Romanò, B. Linclau, N. C. Reichardt, S. Oscarson, A. Ardá, J. Jiménez-Barbero and F. J. Cañada, Molecules, 2019, 24, 2337 CrossRef PubMed
- G. Fittolani, E. Shanina, M. Guberman, P. H. Seeberger, C. Rademacher and M. Delbianco, Angew. Chem., Int. Ed., 2021, 60, 13302–13309 CrossRef CAS PubMed
- L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2017, 23, 3957–3965 CrossRef CAS PubMed
-
(a) B. Xu, L. Unione, J. Sardinha, S. Wu, M. Ethève-Quelquejeu, A. Pilar Rauter, Y. Blériot, Y. Zhang, S. Martín-Santamaría, D. Díaz, J. Jiménez-Barbero and M. Sollogoub, Angew. Chem., Int. Ed., 2014, 53, 9597–9602 CrossRef CAS PubMed
- J. Jiménez-Barbero, B. Linclau, A. Ardá, N. C. Reichardt, M. Sollogoub, L. Unione and S. P. Vincent, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC
- R. Hevey, Chem. – Eur. J., 2021, 27, 2240–2253 CrossRef CAS PubMed
- M. Raics, I. Timári, T. Diercks, L. Szilágyi, H. Gabius and K. E. Kövér, ChemBioChem, 2019, 20, 1688–1692 CrossRef CAS PubMed
- T. Diercks, F. J. Medrano, F. G. FitzGerald, D. Beckwith, M. J. Pedersen, M. Reihill, A. Ludwig, A. Romero, S. Oscarson, M. Cudic and H. Gabius, Chem. – Eur. J., 2021, 27, 316–325 CrossRef CAS PubMed
- I. Timári, S. Balla, K. Fehér, K. E. Kövér and L. Szilágyi, Pharmaceutics, 2022, 14, 201 CrossRef PubMed
- T. Z. Illyés, L. Malinovská, E. Rőth, B. Tóth, B. Farkas, M. Korsák, M. Wimmerová, K. E. Kövér and M. Csávás, Molecules, 2021, 26, 542 CrossRef PubMed
-
(a) M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS
-
(a) S. Monaco, L. E. Tailford, N. Juge and J. Angulo, Angew. Chem., Int. Ed., 2017, 56, 15289–15293 CrossRef CAS PubMed
- M. J. Moure, A. Gimeno, S. Delgado, T. Diercks, G. Boons, J. Jiménez-Barbero and A. Ardá, Angew. Chem., Int. Ed., 2021, 133, 18925–18930 CrossRef
- A. Ardá, P. Blasco, D. Varón Silva, V. Schubert, S. André, M. Bruix, F. J. Cañada, H.-J. Gabius, C. Unverzagt and J. Jiménez-Barbero, J. Am. Chem. Soc., 2013, 135, 2667–2675 CrossRef PubMed
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1992, 114, 3139–3141 CrossRef CAS
- P. Groves, M. O. Rasmussen, M. D. Molero, E. Samain, F. J. Cañada, H. Driguez and J. Jiménez-Barbero, Glycobiology, 2004, 14, 451–456 CrossRef CAS PubMed
- G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185–189 CrossRef CAS
- K. Pervushin, R. Riek, G. Wider and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12366–12371 CrossRef CAS PubMed
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Science, 1996, 274, 1531–1534 CrossRef CAS PubMed
- S. Bertuzzi, A. Gimeno, R. Núñez-Franco, G. Bernardo-Seisdedos, S. Delgado, G. Jiménez-Osés, O. Millet, J. Jiménez-Barbero and A. Ardá, Chem. – Eur. J., 2020, 26, 15643–15653 CrossRef CAS PubMed
- J. Rahkila, F. S. Ekholm, A. Ardá, S. Delgado, J. Savolainen, J. Jiménez-Barbero and R. Leino, ChemBioChem, 2019, 20, 203–209 CrossRef CAS PubMed
- L. A. Grischenko, L. N. Parshina, L. V. Kanitskaya, L. I. Larina, L. N. Novikova and B. A. Trofimov, Carbohydr. Res., 2013, 376, 7–14 CrossRef CAS PubMed
- C. Diehl, O. Engström, T. Delaine, M. Håkansson, S. Genheden, K. Modig, H. Leffler, U. Ryde, U. J. Nilsson and M. Akke, J. Am. Chem. Soc., 2010, 132, 14577–14589 CrossRef CAS PubMed
- M. C. Miller, H. Ippel, D. Suylen, A. A. Klyosov, P. G. Traber, T. Hackeng and K. H. Mayo, Glycobiology, 2016, 26, 88–99 CrossRef CAS PubMed
- M. R. Tavares, M. Bláhová, L. Sedláková, L. Elling, H. Pelantová, R. Konefał, T. Etrych, V. Křen, P. Bojarová and P. Chytil, Biomacromolecules, 2020, 21, 641–652 CrossRef CAS PubMed
- M. F. López-Lucendo, D. Solís, S. André, J. Hirabayashi, K. Kasai, H. Kaltner, H.-J. Gabius and A. Romero, J. Mol. Biol., 2004, 343, 957–970 CrossRef PubMed
- J. Seetharaman, A. Kanigsberg, R. Slaaby, H. Leffler, S. H. Barondes and J. M. Rini, J. Biol. Chem., 1998, 273, 13047–13052 CrossRef CAS PubMed
- S. Bertuzzi, A. Gimeno, A. Martinez-Castillo, M. G. Lete, S. Delgado, C. Airoldi, M. Rodrigues Tavares, M. Bláhová, P. Chytil, V. Křen, N. G. A. Abrescia, A. Ardá, P. Bojarová and J. Jiménez-Barbero, Int. J. Mol. Sci., 2021, 22, 6000 CrossRef CAS PubMed
- M. C. Miller, A. Klyosov and K. H. Mayo, Glycobiology, 2009, 19, 1034–1045 CrossRef CAS PubMed
- M. C. Miller, I. V. Nesmelova, D. Platt, A. Klyosov and K. H. Mayo, Biochem. J., 2009, 421, 211–221 CrossRef CAS PubMed
- G. Pintacuda, M. John, X. C. Su and G. Otting, Acc. Chem. Res., 2007, 40, 206–212 CrossRef CAS PubMed
- A. Canales, A. Mallagaray, J. Pérez-Castells, I. Boos, C. Unverzagt, S. André, H. J. Gabius, F. J. Cañada and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2013, 52, 13789–13793 CrossRef CAS PubMed
- A. Canales, I. Boos, L. Perkams, L. Karst, T. Luber, T. Karagiannis, G. Domínguez, F. J. Cañada, J. Pérez-Castells, D. Häussinger, C. Unverzagt and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2017, 56, 14987–14991 CrossRef CAS PubMed
- K. Kato and T. Yamaguchi, Glycoconjugate J., 2015, 32, 505–513 CrossRef CAS PubMed
- B. Fernández de Toro, W. Peng, A. J. Thompson, G. Domínguez, F. J. Cañada, J. Pérez-Castells, J. C. Paulson, J. Jiménez-Barbero and Á. Canales, Angew. Chem., Int. Ed., 2018, 57, 15051–15055 CrossRef PubMed
- W. Peng, R. P. de Vries, O. C. Grant, A. J. Thompson, R. McBride, B. Tsogtbaatar, P. S. Lee, N. Razi, I. A. Wilson, R. J. Woods and J. C. Paulson, Cell Host Microbe, 2017, 21, 23–34 CrossRef CAS PubMed
- L. Unione, M. J. Moure, M. P. Lenza, I. Oyenarte, J. Ereño-Orbea, A. Ardá and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2022, 61, e202201432 CrossRef CAS PubMed
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed
- J. L. Asensio, F. J. Cañada, H.-C. Siebert, J. Laynez, A. Poveda, P. M. Nieto, U. Soedjanaamadja, H.-J. Gabius and J. Jiménez-Barbero, Chem. Biol., 2000, 7, 529–543 CrossRef CAS PubMed
- J. Jiménez-Barbero, F. Javier Cañada, J. L. Asensio, N. Aboitiz, P. Vidal, A. Canales, P. Groves, H.-J. Gabius and H.-C. Siebert, Adv. Carbohydr. Chem. Biochem., 2006, 60, 303–354 CrossRef PubMed
- B. E. Prosser, S. Johnson, P. Roversi, A. P. Herbert, B. S. Blaum, J. Tyrrell, T. A. Jowitt, S. J. Clark, E. Tarelli, D. Uhrín, P. N. Barlow, R. B. Sim, A. J. Day and S. M. Lea, J. Exp. Med., 2007, 204, 2277–2283 CrossRef CAS PubMed
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS PubMed
- P. I. Kitov, H. Shimizu, S. W. Homans and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 3284–3294 CrossRef CAS PubMed
- G. S. Thompson, H. Shimizu, S. W. Homans and A. Donohue-Rolfe, Biochemistry, 2000, 39, 13153–13156 CrossRef CAS PubMed
- F. Vasile, J. J. Reina, D. Potenza, J. E. Heggelund, A. Mackenzie, U. Krengel and A. Bernardi, Glycobiology, 2014, 24, 766–778 CrossRef CAS PubMed
- C. Rademacher, J. Guiard, P. I. Kitov, B. Fiege, K. P. Dalton, F. Parra, D. R. Bundle and T. Peters, Chem. – Eur. J., 2011, 17, 7442–7453 CrossRef CAS PubMed
- B. Fiege, M. Leuthold, F. Parra, K. P. Dalton, P. J. Meloncelli, T. L. Lowary and T. Peters, Glycoconjugate J., 2017, 34, 679–689 CrossRef CAS PubMed
- J. Guiard, B. Fiege, P. I. Kitov, T. Peters and D. R. Bundle, Chem. – Eur. J., 2011, 17, 7438–7441 CrossRef CAS PubMed
- A. Poveda and J. Jiménez-Barbero, Chem. Soc. Rev., 1998, 27, 133 RSC
- T. Peters and B. M. Pinto, Curr. Opin. Struct. Biol., 1996, 6, 710–720 CrossRef CAS PubMed
- T. Peters, R. Creutznacher, T. Maass, A. Mallagaray, P. Ogrissek, S. Taube, L. Thiede and C. Uetrecht, Biochem. Soc. Trans., 2022, 50, 347–359 CrossRef CAS PubMed
- B. Fiege, C. Rademacher, J. Cartmell, P. I. Kitov, F. Parra and T. Peters, Angew. Chem., Int. Ed., 2012, 51, 928–932 CrossRef CAS PubMed
- R. Creutznacher, T. Maass, P. Ogrissek, G. Wallmann, C. Feldmann, H. Peters, M. Lingemann, S. Taube, T. Peters and A. Mallagaray, Viruses, 2021, 13, 416 CrossRef CAS PubMed
- K. S. Bücher, H. Yan, R. Creutznacher, K. Ruoff, A. Mallagaray, A. Grafmüller, J. S. Dirks, T. Kilic, S. Weickert, A. Rubailo, M. Drescher, S. Schmidt, G. Hansman, T. Peters, C. Uetrecht and L. Hartmann, Biomacromolecules, 2018, 19, 3714–3724 CrossRef PubMed
- M. Taouai, V. Porkolab, K. Chakroun, C. Cheneau, J. Luczkowiak, R. Abidi, D. Lesur, P. J. Cragg, F. Halary, R. Delgado, F. Fieschi and M. Benazza, Bioconjugate Chem., 2019, 30, 1114–1126 CrossRef CAS PubMed
- N. Varga, I. Sutkeviciute, R. Ribeiro-Viana, A. Berzi, R. Ramdasi, A. Daghetti, G. Vettoretti, A. Amara, M. Clerici, J. Rojo, F. Fieschi and A. Bernardi, Biomaterials, 2014, 35, 4175–4184 CrossRef CAS PubMed
- T. B. H. Geijtenbeek, A. Engering and Y. van Kooyk, J. Leukoc. Biol., 2002, 71, 921–931 CrossRef CAS PubMed
- T. B. H. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. Van Vliet, G. C. F. Van Duijnhoven, J. Middel, I. L. M. H. A. Cornelissen, H. S. L. M. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. Van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed
- P. Stoitzner and N. Romani, Eur. J. Immunol., 2011, 41, 2526–2529 CrossRef CAS PubMed
- J. Valladeau, O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, C. Caux, S. Lebecque and S. Saeland, Immunity, 2000, 12, 71–81 CrossRef CAS PubMed
- T. Doebel, B. Voisin and K. Nagao, Trends Immunol., 2017, 38, 817–828 CrossRef CAS PubMed
- L. De Witte, A. Nabatov, M. Pion, D. Fluitsma, M. A. W. P. De Jong, T. De Gruijl, V. Piguet, Y. Van Kooyk and T. B. H. Geijtenbeek, Nat. Med., 2007, 13, 367–371 CrossRef CAS PubMed
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704–8719 CrossRef CAS PubMed
- R. Wawrzinek, E.-C. Wamhoff, J. Lefebre, M. Rentzsch, G. Bachem, G. Domeniconi, J. Schulze, F. F. Fuchsberger, H. Zhang, C. Modenutti, L. Schnirch, M. A. Marti, O. Schwardt, M. Bräutigam, M. Guberman, D. Hauck, P. H. Seeberger, O. Seitz, A. Titz, B. Ernst and C. Rademacher, J. Am. Chem. Soc., 2021, 143, 18977–18988 CrossRef CAS PubMed
- M. Singh, B. Tam and B. Akabayov, Molecules, 2018, 23, 233 CrossRef PubMed
- M. J. Harner, A. O. Frank and S. W. Fesik, J. Biomol. NMR, 2013, 56, 65–75 CrossRef CAS PubMed
- J. B. Jordan, D. A. Whittington, M. D. Bartberger, E. A. Sickmier, K. Chen, Y. Cheng and T. Judd, J. Med. Chem., 2016, 59, 3732–3749 CrossRef CAS PubMed
- C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 4, 243–271 CrossRef
- E. C. Wamhoff, J. Hanske, L. Schnirch, J. Aretz, M. Grube, D. Varón Silva and C. Rademacher, ACS Chem. Biol., 2016, 11, 2407–2413 CrossRef CAS PubMed
- J. Aretz, H. Baukmann, E. Shanina, J. Hanske, R. Wawrzinek, V. A. Zapol'skii, P. H. Seeberger, D. E. Kaufmann and C. Rademacher, Angew. Chem., Int. Ed., 2017, 56, 7292–7296 CrossRef CAS PubMed
- J. C. Muñoz-García, E. Chabrol, R. R. Vivès, A. Thomas, J. L. de Paz, J. Rojo, A. Imberty, F. Fieschi, P. M. Nieto and J. Angulo, J. Am. Chem. Soc., 2015, 137, 4100–4110 CrossRef PubMed
- N. Bertleff-Zieschang, J. Bechold, C. Grimm, M. Reutlinger, P. Schneider, G. Schneider and J. Seibel, ChemBioChem, 2017, 18, 1477–1481 CrossRef CAS PubMed
- K. Neuhaus, E.-C. Wamhoff, T. Freichel, A. Grafmüller, C. Rademacher and L. Hartmann, Biomacromolecules, 2019, 20, 4088–4095 CrossRef CAS PubMed
- E. C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. del Frari, R. van Dalen, D. Hartmann, N. M. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CrossRef CAS PubMed
- E. Shanina, S. Kuhaudomlarp, K. Lal, P. H. Seeberger, A. Imberty and C. Rademacher, Angew. Chem., Int. Ed., 2022, e202109339 CAS
-
(a) C. Guzzi, P. Alfarano, I. Sutkeviciute, S. Sattin, R. Ribeiro-Viana, F. Fieschi, A. Bernardi, J. Weiser, J. Rojo, J. Angulo and P. M. Nieto, Org. Biomol. Chem., 2016, 14, 335–344 RSC
- F. Probert, S. B.-M. Whittaker, M. Crispin, D. A. Mitchell and A. M. Dixon, J. Biol. Chem., 2013, 288, 22745–22757 CrossRef CAS PubMed
- P. M. Enríquez-Navas, F. Chiodo, M. Marradi, J. Angulo and S. Penadés, ChemBioChem, 2012, 13, 1357–1365 CrossRef PubMed
- P. M. Enríquez-Navas, M. Marradi, D. Padro, J. Angulo and S. Penadés, Chem. – Eur. J., 2011, 17, 1547–1560 CrossRef PubMed
- C. A. Bewley, K. R. Gustafson, M. R. Boyd, D. G. Covell, A. Bax, G. M. Clore and A. M. Gronenborn, Nat. Struct. Biol., 1998, 5, 571–578 CrossRef CAS PubMed
- C. A. Bewley, S. Kiyonaka and I. Hamachi, J. Mol. Biol., 2002, 322, 881–889 CrossRef CAS PubMed
- S. R. Shenoy, L. G. Barrientos, D. M. Ratner, B. R. O’Keefe, P. H. Seeberger, A. M. Gronenborn and M. R. Boyd, Chem. Biol., 2002, 9, 1109–1118 CrossRef CAS PubMed
- G. Nestor, T. Anderson, S. Oscarson and A. M. Gronenborn, J. Am. Chem. Soc., 2017, 139, 6210–6216 CrossRef CAS PubMed
- H. Coelho, M. de las Rivas, A. S. Grosso, A. Diniz, C. O. Soares, R. A. Francisco, J. S. Dias, I. Compañon, L. Sun, Y. Narimatsu, S. Y. Vakhrushev, H. Clausen, E. J. Cabrita, J. Jiménez-Barbero, F. Corzana, R. Hurtado-Guerrero and F. Marcelo, JACS Au, 2022, 2, 631–645 CrossRef CAS PubMed
- T. Höfurthner, B. Mateos and R. Konrat, ChemPlusChem, 2021, 86, 938–945 CrossRef PubMed
- S. Mari, D. Serrano-Gómez, F. J. Cañada, A. L. Corbí and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2005, 44, 296–298 CrossRef CAS PubMed
- F. Vasile, F. Gubinelli, M. Panigada, E. Soprana, A. Siccardi and D. Potenza, Glycobiology, 2018, 28, 42–49 CrossRef CAS PubMed
- A. Palmioli, P. Sperandeo, S. Bertuzzi, A. Polissi and C. Airoldi, Bioorg. Chem., 2021, 112, 104876 CrossRef CAS PubMed
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |