
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe quest for organo-alkali metal monomers: unscrambling the structure–reactivity relationship
Nathan
Davison
* and
Erli
Lu
 *
*
Chemistry – School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: erli.lu@newcastle.ac.uk; n.davison1@newcastle.ac.uk
First published on 30th May 2023
Abstract
Organo-alkali metal reagents are essential tools in synthetic chemistry. Alkali metal organometallics aggregate in solution and solid-state forming clusters and polymers. The structure of these aggregates and their structure–reactivity relationship have been of great interest for many decades. This Perspective will look at the strategies that have been employed to isolate low aggregates and, in particular, monomeric complexes of the most common alkali metal alkyls (M = Li–Cs, R = methyl, trimethylsilylmethyl, bis/tris(trimethylsilylmethyl), butyls and benzyl) and the relationship between level of aggregation, structure and reactivity.
 Nathan Davison (left) and Erli Lu (right) | Nathan Davison is a 4th-year PhD student at Newcastle University. Nathan started his PhD study in January 2020 in Dr Erli Lu's group, working on Group-1 metal chemistry. Nathan has published six research articles to date. Dr Erli Lu is a Lecturer in Inorganic Chemistry at Newcastle University, and a Newcastle University Academic Track (NUAcT) Fellow. Erli was awarded a PhD degree in rare-earth metal chemistry from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in January 2012, under the supervision of Prof. Yaofeng Chen (currently at South China University of Technology). After the PhD, Erli moved to the UK in June 2012, initially sponsored by an EU Marie Curie International Incoming Fellowship, working in actinide chemistry as a postdoctoral fellow for 7 years (2012–19) in the Steve Liddle group at Universities of Nottingham (2012–15) and Manchester (2015–19). In September 2019, Erli started his independent career at Newcastle, moving his research focus from the heaviest metals (actinide) to the lightest ones (Group-1). Erli has published 44 research articles and reviews (nine as an independent academic) and three book chapters. |
1. Introduction
The Jekyll and Hyde of the periodic table – there are two very different sides to alkali metal (Group 1) chemistry. On the one hand predictable, stable, relatively unreactive compounds such as table salt (sodium chloride), yet on the other highly reactive, often pyrophoric, alkali metal organometallic reagents. Both sides play an essential and widespread role in society.1,2Within the organo-alkali metal complexes, and organometallic complexes in general, alkyllithium compounds dominate, being some of the most widely used reagents in both academia and industry.1,3,4 First synthesised in 1917 by Wilhelm Schlenk,5 they are reagents that at a first glance may appear simple and predictable, however on further investigation quickly reveal a much more complicated picture.
A key part of this picture is aggregation. Alkali metal organometallic compounds aggregate in both solution and solid-state, which stabilises the polar and reactive M–C bond.4,6 The preferred coordination number and coordination sphere of the metal, the size and bulkiness of the carbanion substituent, and the strength of the M–C bond interaction all influence the aggregation level. For the alkali metals, M–C bonds are predominately ionic and polarised and therefore the metal and organic substituent can be usually thought of as a cation and anion respectively. When the MR units aggregate, each metal forms interactions with multiple R− moieties and each R− forms interactions with multiple metal centres. This stabilisation from aggregating apparently means that, the [MR]n aggregates are not as reactive as the hypothetical MR ‘naked’ monomers.4
Coordinating solvent or Lewis basic ligands, such as ethers or amines break down the oligomers into smaller aggregates, which coordinate to the metal through dative bonds via the N/O lone pair of electrons.4 Some of the most common ligands are shown in Fig. 1.
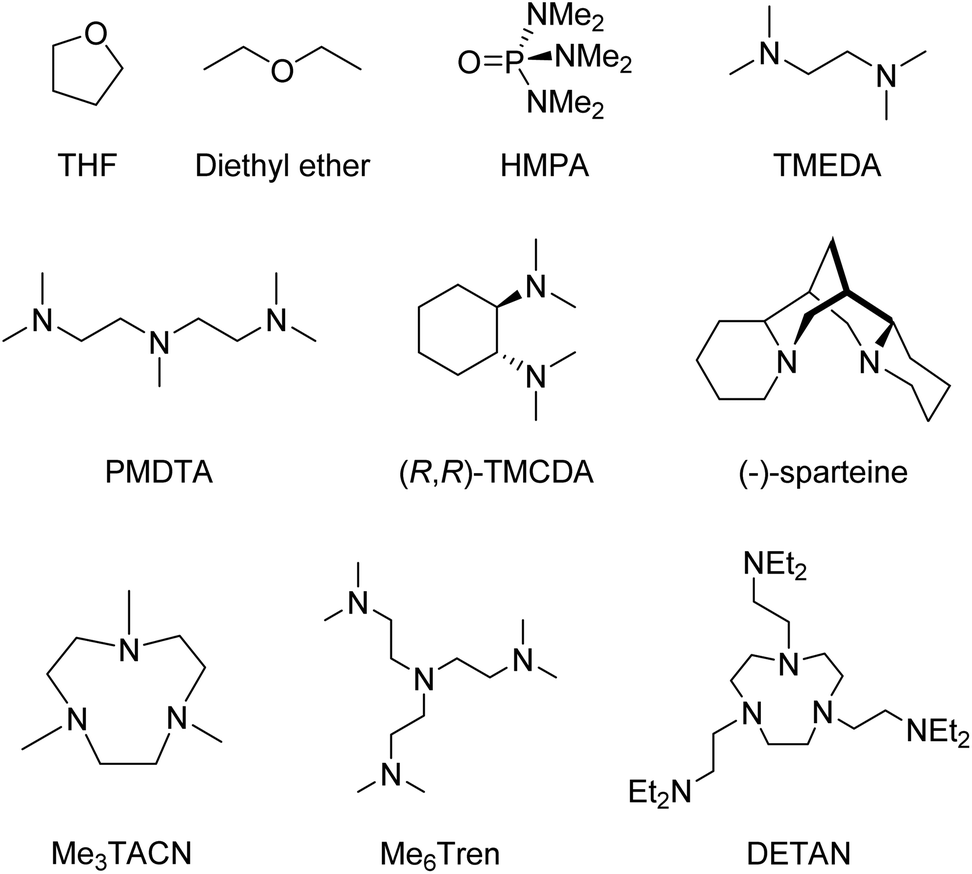 | ||
| Fig. 1 A selection of common neutral N/O-ligands in organo-alkali metal chemistry. | ||
There are two main reasons we want to break organo-alkali metal aggregates. Firstly, deaggregation to a monomeric species is often postulated to be a key step in alkali metal organometallic mediated reactions and therefore structural, mechanistic and computational studies of these isolated monomers are considered of high value for establishing mechanistic understanding.3,6 Secondly, it is hoped that by breaking the alkali metal organometallics into smaller aggregates, the reactivity can be increased, and/or unlocking novel reactivity patterns (Fig. 2).6 In general, for organolithiums, calculations have revealed the Li–C bond to be more ionic with decreasing aggregation size and hence, presumably, monomers should be the most ionic and most reactive.7 Nevertheless, readers will soon find out that this is a moot point in Fig. 2: lower aggregates do not necessarily lead to higher reactivity. On the other hand, a very recent report from our group did prove that lower aggregates (specifically an organosodium monomer) can unlock new reaction patterns cf. their parent polymeric complexes.8
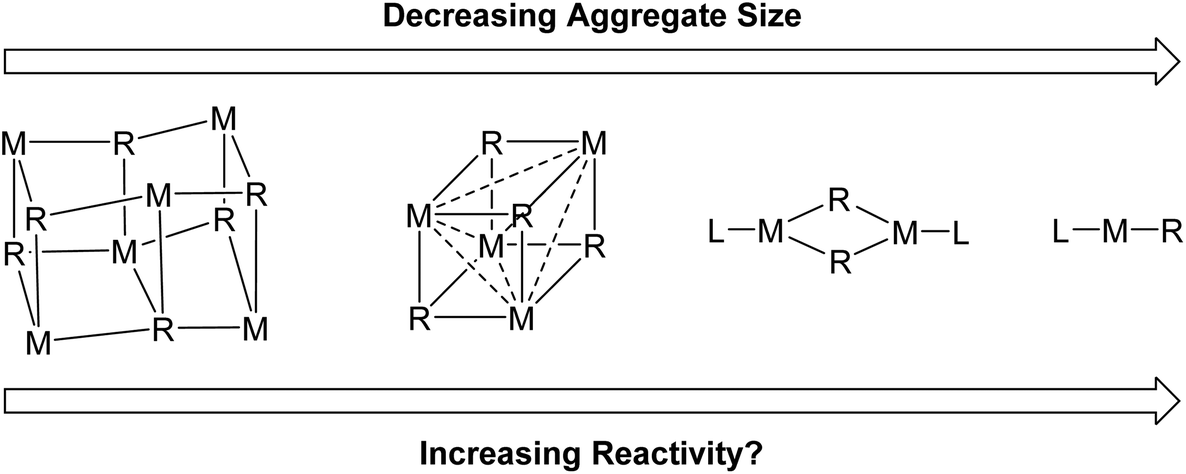 | ||
| Fig. 2 The presumed aggregate size–reactivity relationship. | ||
The metal, carbanion substituent and ligands all influence both the structure and reactivity. The influence of ligands is two-fold. On the one hand, they coordinate to the metal centre and deaggregate the clusters into, hypothetically more reactive, lower aggregate species (Fig. 2). On the other hand, sterically bulky ligands can also reduce the metal centre's accessibility for incoming substrates, i.e., shielding the reactive site, and hence reduce the reactivity. As one descends the Group 1 metal series, the metals’ ionic radii get larger and thus, can accommodate a larger coordination number. From a bonding perspective, the M–C bond gets more polar (can be superficially understood as the metals’ electronegativity gets smaller) and ionic and hence, more reactive.1,9,10 The steric and electronic properties of the carbanion substituent affects the level of aggregation as well. Electron donating groups destabilise the carbanion, whereas electron withdrawing groups help to stabilise it.11 An example of this is the greater reactivity and instability of tert-butyllithium (tBuLi) over n-butyllithium (nBuLi).
Aggregation and the aggregate size–reactivity relationship are some of the most important topics (if not the most important ones) in organo-alkali metal chemistry, or organometallic chemistry in general. Given the unique position of monomers in the aggregate size spectrum, an overview of the state-of-the-art would be of significant scientific value. However, despite a number of reviews focusing on aggregation in organo-alkali metal chemistry,3,4,6 none of these specifically focuses on the monomers. This status quo, along with our own recent work in this direction, motivates us to summarise the community's achievements to date towards synthesising organo-alkali metal monomers and their reactivity studies, along with our perspectives in this regard. It is important to note that this Perspective is not a comprehensive review: we focus on the most used alkyls, namely methyl (Me), trimethylsilylmethyl (CH2SiMe3), bis/tris(trimethylsilyl)methyl (CH(SiMe3)2 and C(SiMe3)3), butyls (nBu, secBu, tBu) and benzyl (Bn). There are also many other examples of monomeric and low (<4) aggregate organo-alkali metal complexes with less common alkyls (e.g., isopropyl;12 CHPh2;13,14 C(SiMe3)2Ph;15 CPh3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16), aryl,17–21 or hetero-atom substituted and stabilised alkyl:22 these cases are not included in this Perspective.
16), aryl,17–21 or hetero-atom substituted and stabilised alkyl:22 these cases are not included in this Perspective.
To help our readers follow the discussion in this Perspective, for each alkyl group, a table is provided to summarise SCXRD structural parameters and references of the monomers and key aggregates (Tables 1–6). While we believe that the tables provide a comprehensive coverage of the monomers, they should not be treated as comprehensive summarisations of all the aggregates (dimers and higher): only the ones which are relevant to our discussion are included.
| Metal | Formula | Aggregate size | M–CH3 bond length (Å) | Ref. |
|---|---|---|---|---|
| Li | [(MeLi)4]∞ | Tetrameric polymer | 2.31(5) (within tetramer), 2.36(5) (between tetramers). [[CD3Li]4]∞ at = 1.5 K = 2.256(6) (within tetramer), 2.356(6) (between tetramers) | 5, 52, 71 and 72 |
| [MeLi(THF)]4 | Tetramer | 2.230(5), 2.248(4), 2.228(5), 2.242(5), 2.241(5), and 2.253(5) | 53 | |
| [{(MeLi)4(μ-κ1-N-TMEDA)2}∞] | Tetrameric polymer | 2.274(6), 2.234(6), 2.279(6) | 59 | |
| [{(MeLi)4(μ-κ1-O-DEM)1.5}∞] (DEM: diethoxymethane) | Tetrameric polymer | 2.240(6), 2.271(4), 2.273(5) | 65 | |
| [Li(μ-CH3)((R,R)-TMCDA)]2 (1) | Dimer | 2.252(7), 2.277(7), 2.214(7), 2.219(7) | 60 | |
| [Li(μ-CH3)(κ2-N,N′-PMDTA)]2[MeLi]4 (2) | Dimer | 2.188(4), 2.195(5), 2.21(3), 2.21(2) | 61 | |
| [Li(CH3)(DETAN)] (3) | Monomer | 2.099(5) | 62 | |
| [Li(CH3)(Me3TACN)] (4) | Monomer | 2.1076(18) | 64 | |
| Na | [(MeNa)4]∞ | Tetrameric polymer, connected through discrete Na/methyl ions | [(CD3Na)4]∞ at 1.5 K = 2.528(4), 2.569(2), 2.834(4), 2.911(3), 264.4(6), 2.679(5), 2.569(4), 2.589(5) | 67 and 73 |
| K | [MeK]∞ | Polymer | 3.22 | 68–70 |
| Rb | [MeRb]∞ | Polymer | 3.36 | 70 |
| Cs | [MeCs]∞ | Polymer | 3.53 | 70 |
| Metal | Formula | Aggregate size | M–C bond lengths (Å) | Ref. |
|---|---|---|---|---|
| Li | [(DABCO)7(LiCH2SiMe3)8] | Octamer | 2.152(2), 2.152(2), 2.191(2), 2.195(2), 2.193(2), 2.163(2), 2.271(2), 2.230(2) | 86 |
| [LiCH2SiMe3]6 | Hexamer | 2.28(2), 2.22(2), 2.20(1), 2.17(4), 2.28(2), 2.20(1), 2.15(2), 2.24(2), 2.17(2), 2.24(1), 2.15(2), 2.29(1), 2.17(2), 2.29(2), 2.20(1), 2.15(1), 2.27(2), 2.24(1) | 76 | |
| [(Et2O)2(LiCH2SiMe3)4] | Tetramer | 2.287(3), 2.292(3), 2.340(3), 2.266(3), 2.291(3), 2.298(3), 2.222(3), 2.244(3), 2.247(3), 2.203(3), 2.214(3), 2.255(3) | 79 | |
| [(tBuOMe)2(LiCH2SiMe3)4] | Tetramer | 2.342(9), 2.381(9), 2.393(11), 2.293(8), 2.345(12), 2.455(8), 2.158(8), 2.204(7), 2.274(8), 2.133(8), 2.187(8), 2.307(11) | 79 | |
| [(DME)(LiCH2SiMe3)]2 | Dimer | 2.196(2), 2.226(2), 2.261(2), 2.213(2) | 79 | |
| [(SC)-LiCH2SiMe3]2, SC = cyclohexyldimethoxysilane | Dimer | 2.365(4), 2.365(4) | 80 | |
| [(TMEDA)LiCH2SiMe3]2 | Dimer | 2.317(4), 2.253(4), 2.183(4), 2.306(4) | 81 | |
| [{(−)-Sparteine}LiCH2SiMe3]2 | Dimer | 2.147(3), 2.661(3) | 81 | |
| [(Qu)n(LiCH2SiMe3)2], n = 2 or 3; Qu = quinuclidine | Dimer | n = 2: 2.185(2), n = 3: 2.169(4), 2.280(4), 2.181(4), 2.284(4) | 83 | |
| [Li(CH2SiMe3)(PMDTA)] (7) | Monomer | 2.113(2) | 81 | |
| [Li(CH2SiMe3)(R,R-TMCDA)] (8) | Monomer | 2.069(3) | 83 | |
| [Li(CH2SiMe3)(Me6Tren)] (9) | Monomer | 2.122(5) | 48 | |
| Na | [NaCH2SiMe3]∞ | Polymer | 2.642(2), 2.577(2), 2.825(2), 2.601(2) | 87 |
| [(TMEDA)NaCH2SiMe3]∞ | Polymer | 2.523(9)–2.530(8) | 88 | |
| [(PMDETA)2Na3(CH2SiMe3)3] | Trimer | 2.4672(77)–2.7322(74) | 84 | |
| [Na(CH2SiMe3)(Me6Tren)] (10) | Monomer | 2.5054(14) | 8 and 84 | |
| K | [(TMEDA)3(KCH2SiMe3)4] | Tetramer | 2.922(3)–3.247(3) | 88 |
| [(PMDETA)KCH2SiMe3]∞ | Polymer | 2.9151(19), 3.0358(19) | 88 |
| Metal | Formula | Aggregate size | M–C bond lengths (Å) | Ref. |
|---|---|---|---|---|
| Li | [LiCH(SiMe3)2]∞ | Polymer | 2.14(3), 2.21(3), 2.22(3), 2.18(3), 2.27(2), 2.13(3) | 89 |
| [Li(CH(SiMe3)2)(THF)]2 | Dimer | 2.204(2), 2.274(3) | 91 | |
| [Li(CH(SiMe3)2)(TMEDA)] (12) | Monomer | 2.070(3)/2.083(3) | 91 | |
| [Li(CH(SiMe3)2)(PMDTA)] (11) | Monomer | 2.13(5) | 92 | |
| Na | [NaCH(SiMe3)2]∞ | Polymer | 2.555(10) (average) | 90 |
| [Na(CH(SiMe3)2)(THF)]∞ | Polymer | 2.778(4), 2.657(4) | 91 | |
| [NaCH(SiMe3)2]2(TMEDA)3 | Dimer | 2.520(2) | 91 | |
| K | [K(CH(SiMe3)2)(THF)]∞ | Polymer | 2.956(4), 3.012(4) | 93 |
| [K(CH(SiMe3)2)(tBuOMe)]∞ | Polymer | 2.988(8), 3.012(8) | 94 | |
| [{KCH(SiMe3)3}2(PMDTA)]2 | Open tetramer | 3.095(5), 3.040(4), 3.017(4), 3.059(5) | 94 | |
| Rb | [Rb(CH(SiMe3)2)(PMDTA)]2 | Dimer | 3.361(9) and 3.485(8) | 90 |
| Cs | [Cs(CH(SiMe3)2)(TMEDA)]∞ | Polymer | 3.425(2) | 94 |
| Metal | Formula | Aggregate size | M–C bond lengths (Å) | Ref. |
|---|---|---|---|---|
| Li | [LiC(SiMe3)3]2 | Dimer | 2.291(6), 2.303(6) | 95 |
| [Li(THF)4][Li{C(SiMe3)3}2] | Ate complex | 2.16(1), 2.20(1) | 99 | |
| [Li(THF)3][Li{C(SiMe3)3}2] | Ate complex | 2.172(8), 2.164(8) | 100 | |
| [Li(TMEDA)2][Li{C(SiMe3)3}2] | Ate complex | 2.213(5) | 101 | |
| [Li{C(SiMe3)3}2][Cl{Li(PMDTA)}2] | Ate complex | 2.171(13) | 103 | |
| Na | [Na(TMEDA)2(Et2O)] [Na{C(SiMe3)3}2] | Ate complex | 2.479(6) | 102 |
| K | [KC(SiMe3)3]∞ | Polymer | 3.090(11), 3.104(11) | 96 |
| [KC(SiMe3)3(TMEDA)]∞ | Polymer | 2.92(1) | 98 | |
| Rb | [RbC(SiMe3)3]∞ | Polymer | 3.291(7), 3.287(7) | 97 |
| Cs | [Cs{C(SiMe3)3}(C6H6)3]·0.5C6H6 (13) | Monomer | 3.325(12) | 97 |
| Butyl | Formula | Aggregate size | Li–C bond lengths (Å) | Ref. |
|---|---|---|---|---|
| n Bu | [n-BuLi]6 | Hexamer | 2.280(3)–2.295(3) | 110 |
| [(n-BuLi)4(TMEDA)]∞ | Polymeric tetramer | 2.145(11)–2.326(8) | 120 and 125 | |
| [Li(n-Bu)(TMEDA)]2 | Dimer | 2.215(11)–2.227(9) | 120 | |
| [Li(n-Bu)((−)-sparteine)]2 | Dimer | 2.234(5), 2.291(5), 2.321(5), 2.230(5) | 126 | |
| [Li(n-Bu)((R,R)-TMCDA)]2 | Dimer | 2.279(6); 2.233(7); 2.214(6); 2.261(6) | 127 | |
| [Li(n-BuLi)(DPE-6)]2 | Dimer | 2.212(2) to 2.235(2) | 128 | |
| [(n-BuLi)2(TtBuTAC)]2 | Open tetramer | 2.274(3), 2.378(3), 2.131(3), 2.184(3), 2.199(3), 2.455(3) | 133 | |
| [(n-BuLi)2(PMDTA)]2 | Open tetramer | 2.207(5), 2.225(6), 2.121(6), 2.146(5) | 50 | |
| s Bu | [Li(sec-Bu)((R,R)-TMCDA)] (14) | Monomer | 2.104(11) Å | 60 |
| t Bu | [tert-BuLi]4 | Tetramer | 2.147(17)–2.370(14) | 109 |
| [(tert-BuLi)(Et2O)]2 | Dimer | 2.174(6), 2.194(6) | 109 | |
| [Li(tert-Bu)((−)-sparteine)] (15) | Monomer | 2.114(4) | 132 | |
| [Li(tert-Bu)((R,R)-TMCDA)] (16) | Monomer | 2.064(15) | 109 | |
| [Li(tert-Bu)(TEEDA)] (17) | Monomer | 2.101(3) | 49 | |
| [Li(tert-Bu)(TtBuTAC)] (18) | Monomer | 2.083(2) | 133 |
| Metal | Formula | Aggregate size | M–C bond lengths (Å) | Ref. |
|---|---|---|---|---|
| Li | [Li(CH2Ph)(Et2O)]∞ | Polymer | 2.189(8)–2.229(8) | 144 |
| [Li(CH2Ph)(THF)2]∞ | Polymer | 2.36 and 2.31 | 143 | |
| [Li(CH2Ph)(TMEDA)(THF)] (19) | Monomer | 2.210(5) | 149 | |
| [Li(CH2Ph)(Me3TACN)] (20) | Monomer | 2.18(2)/2.15(3) | 107 | |
| [Li(CH2Ph)(PMDTA)] (21) | Monomer | 2.144(5) | 13 | |
| [Li(CH2Ph)(Me6Tren)] (22) | Monomer | 2.352(3) | 153 | |
| Na | [Na(CH2Ph)(THF)2]∞ | Polymer | 2.543(2), 3.113(2) | 144 |
| [Na(CH2Ph)(TMEDA)]4 | Eight-membered ring tetramer | 2.64 (average) (Cα), 2.76 (average) (Cipso) | 149 | |
| [Na(CH2Ph)(PMDTA)]∞ | Polymer | 2.75, 2.79 | 13 | |
| [Na(CH2Ph)(Me6Tren)] (23) | Monomer | 2.556(1) (Cα), 3.183(1) (Cipso) | 153 | |
| K | [(KCH2Ph)3(THF)4]∞, [(KCH2Ph)2(THF)]∞ | Polymer | 2.965–3.485, 2.92 | 146 and 147 |
| [K(CH2Ph)(PMDTA)0.5(PhCH3)]∞ | Polymer | 3.171(2)–3.297(2) | 151 | |
| [K(CH2Ph)(Me6Tren)] (24) | Monomer | 3.097(4)–3.250(4) (to aromatic ring) | 153 | |
| Rb | [Rb(CH2Ph)(THF)2]∞ | Polymer | 3.105(6), 3.269(6), 3.420(6), 3.230(6) | 145 |
| [Rb(CH2Ph)(PMDTA)]∞ | Polymer | 3.26(2)–3.30(2) | 151 | |
| Cs | [Cs(CH2Ph)(THF)0.5]∞ | Polymer | 3.267(2), 3.432(2), 3.372(2) | 145 |
2. The challenges
Before starting surveying the M–R monomer complexes (M: Group-1 metals; R: –CH3, –CH2SiMe3, –CH(SiMe3)2, –C(SiMe3)3, nBu, secBu, tBu, –CH2Ph), we would like to bring to our readers’ awareness a few challenges in Group-1 metal organometallic chemistry faced by the community, and how the community's effort tackling these challenges has reshaped, and is still reshaping, the landscape of organometallic chemistry.2.1 Challenge 1: beyond SCXRD – what is going on in solution?
Among all the five Group-1 metals (Li, Na, K, Rb, Cs), by far the most studied are organolithiums and hence, most of the discussion in the following sections are dominated by organolithium complexes. Synthetic organometallic chemists have devoted decades of effort to synthesising organolithium monomers and studying their structures and reactivity, but such monomeric complexes still present grand scientific challenges: (1) their comprehensive characterisation in both solution and solid-state is challenging, especially regarding the relationship between the solid-state structures (SCXRD) and solution-state structures; (2) their structure–reactivity relationship is still unclear, despite decades of effort.3,6 Almost all the organolithium-mediated reactions take place in solution, and it is now well-accepted that the SCXRD structures do not necessarily reflect the solution-state structures, therefore a cautious approach must be adopted when using the plethora of SCXRD structures as a base to deduce the structure–reactivity relationship in solution.Unlike the intuitive SCXRD, characterising solution-state structures requires sophisticated and specialised techniques. NMR is the most useful probe.3 Diffusion-ordered NMR spectroscopy (DOSY) separates NMR signals via their diffusion coefficients and allow the estimation of molecular weight of the solution-phase species.23–27 Other than DOSY, hetero-nuclear 1J coupling constant can provide direct information regarding the surrounding environment of the metal centre, e.g., the number of metal–carbon bonds. For organolithium compounds, 13C–6/7Li 1JLi–C-coupling has been used to determine the number of Li–C interactions and hence the aggregation state.28 But this technique usually requires isotope (e.g., 13C and 6Li) enriched samples,29–32 which causes an extra barrier. 7Li residual quadrupolar couplings have themselves also been used to determine aggregation state.33,34 Heteronuclear Overhauser effect spectroscopy (HOESY)23 and exchange spectroscopy (EXSY)26 have also been utilised to study aggregation and exchange. Early research often used cryoscopic measurements (freezing point depression), but the calculated value shows only a statistic average of the degrees of aggregation in solution.35,36 Computational calculations can further add information such as structural, geometry and energy calculations, bonding insights and reaction pathways.37
2.2 Challenge 2: metal scope – synthesising organo-heavy alkali metal monomers
Another challenge is synthesising monomeric organo-heavy alkali metal (Na, K, Rb, Cs) complexes. Organosodium and organopotassium compounds were among the very first organometallic complexes, which were synthesised as early as 1858 by James Alfred Wanklyn.38 Despite being reported nearly 60 years earlier than the first organolithium complexes in 1917,5 the organosodium/potassium complexes have received much less interest compared with their lithium counterparts. This is partly due to the presumed poorer thermostability of these heavier Group-1 metals complexes. However, we have to point out here that, the presumed low thermostability of these heavy alkali metal complexes is not necessarily true: recently, we proved that [NaCH2SiMe3]∞ is at least as stable as its Li congener in C6D6 solution.8Another result of the larger metal ionic radii is their low solubility:39 the larger metal cations could facilitate the formation of polymeric structures, which would be less soluble than lower aggregates (e.g., hexamer, tetramer, dimer). Moreover, as we understand, there had been little motivation for the community to develop the heavy alkali metal chemistry: the paradigm is that they follow similar, if not identical, reactivity patterns with their Li counterparts. In other words, historically speaking, the heavier alkali metal alkyl complexes did not exhibit unique enough reactivity for the synthetic chemistry community to pursue, beyond the ubiquitous, easier-to-operate, and commercially available organolithium reagents. Bringing together these factors, organo-heavy alkali metal complexes remain largely unexplored nowadays.
Nonetheless, the status quo started changing since the late 2010s. There is a current trend of renaissance of organosodium chemistry,8,40–47 largely driven by the sustainable and economic merits of sodium (higher abundance Na 2.36% vs. Li 0.002%, lower cost/toxicity). Among these recent advances, we reported the first “Na-exclusive” reactivity (vide infra),8 which highlighted the grand potential of organo-heavy alkali metal complexes as a “pasture-new” of alkali metal chemistry.
2.3 Challenge 3: ligand design
Given the strong aggregating tendency and presumed high reactivity, supporting ligands are of vital importance to isolate organo-alkali metal monomers. The most successful strategy to break the aggregates and isolate monomers is to employ neutral multidentate amines (see Fig. 1 for examples), which coordinate to the metal and kinetically stabilise the monomers.4 These ligands must be multidentate enough to break down the parent aggregates and saturate the coordination sphere to prevent re-assembling. Ligands should also be as chemically robust as possible, containing no reactive C–O or N–H bonds, to avoid quick decomposition, although ligand metalation is still observed in cases of highly reactive monomeric species.48–503. Organo-alkali metal monomers: the state-of-the-art
In the following sections, we will cover the synthesised and SCXRD-characterised organo-alkali metal monomers, including their structural features and reactivity. We will especially focus on their structural and reactivity differences in comparison with their higher aggregates congeners. This section will be categorised into five parts according to the alkyls: (3.1) R = Methyl; (3.2) R = CH2SiMe3; (3.3) R = CH(SiMe3)2 and C(SiMe3)3; (3.4) R = nBu, secBu or tBu; (3.5) R = Bn.3.1 R = Methyl
Methyllithium (MeLi) was first synthesised by Schlenk in 19175 and is the parent archetypal member of the organolithium, or generally speaking, organo-alkali metal family. MeLi has been widely used as a methyl synthon, a nucleophilic reagent and a strong Brønsted base.51 The unsolvated SCXRD structure of MeLi was first reported by Weiss and co-workers as early as 1964, which is tetrameric with strong interaction between the tetrameric units, forming a “polymer of tetramer” structure.52 A discrete MeLi tetramer SCXRD structure was reported in 1993 as a tetrahydrofuran (THF) adduct.53 Other than the solid-state SCXRD structures, 1H, 6/7Li, 13C and 13C–7Li coupling NMR spectroscopic methods54,55 and the differential vapour-pressure technique56 were used to probe the aggregation of MeLi in THF, diethyl ether (Et2O) and triethylamine (TEA) solutions. These solution-state studies all indicated that MeLi exists as tetramers in such Lewis basic solvents, where solvent-coordination likely occurs.
Ligand-free MeLi monomers have been observed in inert gas matrix and as a short-lived species in sub-millimeter spectroscopy.57,58 In the regime of synthetic coordination chemistry, the mainstream, and so far, the most successful strategy to isolate “bottleable” MeLi monomers is utilising neutral amine ligands. This is also the prevailing strategy for synthesising other monomeric Group-1 metal alkyl complexes (LMR; L: neutral ligand; M: Group-1 metal; R: alkyl) throughout this Perspective.
Since monodentate ligands such as the abovementioned THF, Et2O and TEA could not break MeLi's tetrameric structures any further, Weiss and co-workers turned to bidentate N,N,N′N′-tetramethylethylenediamine (TMEDA):59 upon treating with the bidentate TMEDA, the tetramer–polymer structure is deaggregated into an infinite network of discrete TMEDA-capped tetramer units, where each Li is capped with only one N atom of TMEDA, and the TMEDA acts as a bridging ligand. The inability of THF and TMEDA to deaggregate the tetrameric units highlights the strength of the [MeLi]4 tetrameric unit, and subsequent difficulty of its deaggregation.
Dimeric MeLi complexes using bidentate ligand (R,R)-N,N,N′,N′-tetramethyl-1,2-diaminocyclohexane [(R,R)-TMCDA] (1) and tridentate ligand N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) (2) were reported by Strohmann and co-workers in 200760 and 2020,61 respectively. The MeLi dimers 1 and 2 (Fig. 3 & 4) both feature bridging methyl groups, and the TMCDA and PMDTA both act as bidentate ligand. Unusually, the tridentate PMDTA coordinates to Li via its two, instead of three, N atoms. We rationalise this is due to the flexibility of PMDTA: a rigid ligand backbone may be essential to facilitate higher coordination number and therefore, unlock MeLi monomers.
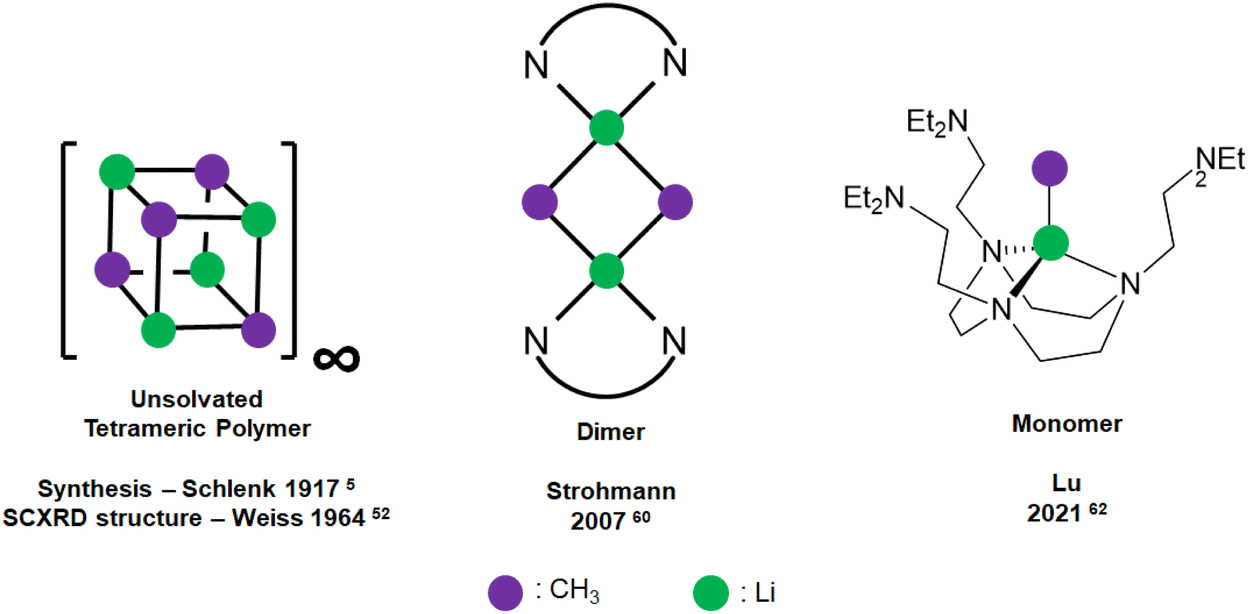 | ||
| Fig. 3 Schematic representation of MeLi SCXRD structures: unsolvated tetrameric polymer (left); dimeric (middle); monomeric (right). | ||
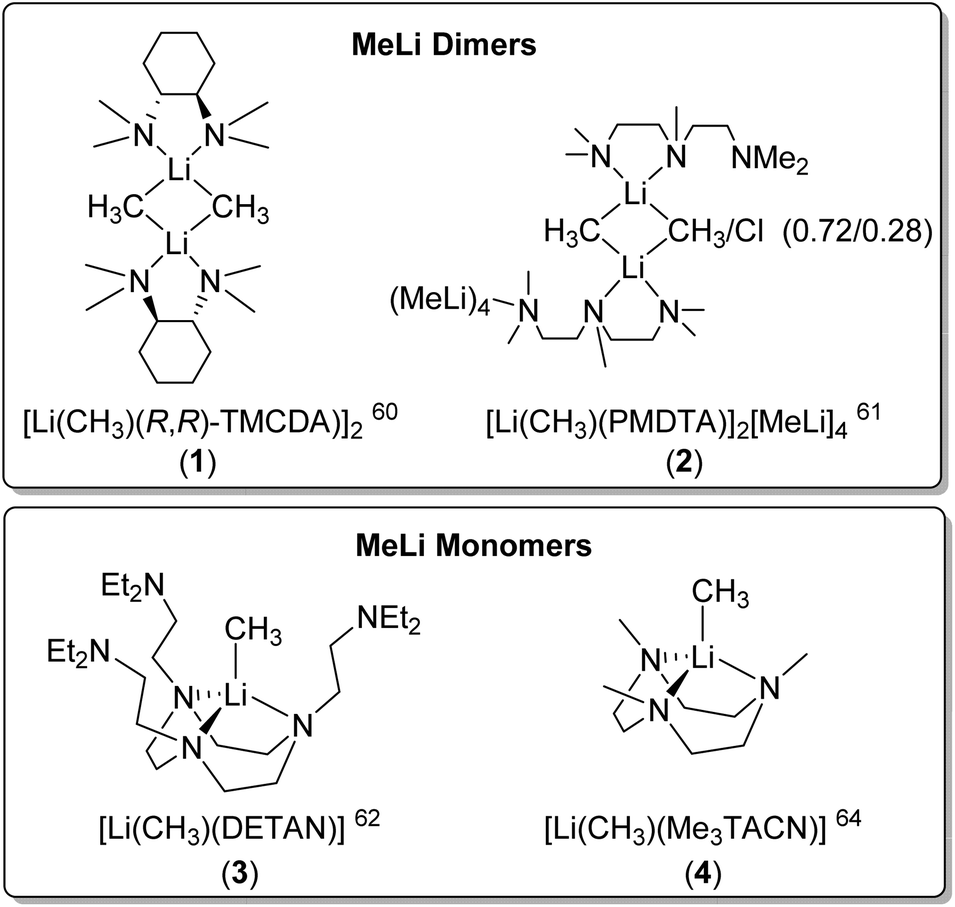 | ||
| Fig. 4 The reported SCXRD-characterised MeLi dimers and monomers. | ||
Following this line, in 2021, we designed a hexadentate amine ligand, namely DETAN (N,N′,N′′-tris-(2-N-diethylaminoethyl)-1,4,7-triaza-cyclononane) (Fig. 1 & 4). The DETAN ligand features a rigid TACN macrocyclic backbone, combined with three flexible sidearms. With the DETAN ligand, we synthesised the first MeLi monomer: [Li(CH3)(κ3-N,N′,N′′-DETAN)] (3) (Fig. 4).62 The SCXRD structure of 3 is exhibited in Fig. 5. Complex 3 features a short terminal Li–C bond (2.099(5) Å), which in the computational studies was found to be predominantly ionic and polarised but with a non-negligible orbital overlap-driven covalency. In 3's SCXRD structure (Fig. 5), the macrocyclic backbone in the DETAN ligand provides structural rigidity and the three coordination-free pendant arms form a protective cavity around the monomeric MeLi unit.623 is stable at −20 °C indefinitely, but decompose via ligand C–H and C–N bond cleavage to produce N,N-diethylethenamine and de-coordination at room temperature. This decomposition itself is evidence of enhanced reactivity of MeLi by forming a monomer: the [(MeLi)4]∞ is stable even in solutions of diethyl ether.1,63
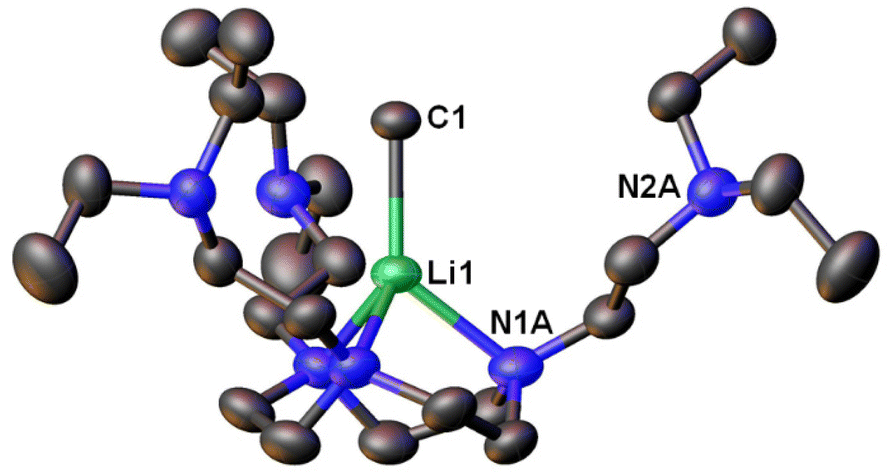 | ||
| Fig. 5 SCXRD structure of the first MeLi monomer [Li(CH3)(κ3-N,N′,N′′-DETAN)] (3).62 | ||
Very recently, Manßen, Sirsch, Anwander and co-workers reported the second ligand-supported MeLi monomer using the 1,3,7-trimethyl-1,3,7-triazacyclononane (Me3TACN) ligand, namely [Li(CH3)(Me3TACN)] (4) (Fig. 4).64 The SCXRD structure of 4 is exhibited in Fig. 6. The Li–CH3 bond lengths in 4 is 2.1076(18) Å, which is very close to that in 3 (2.099(5) Å).
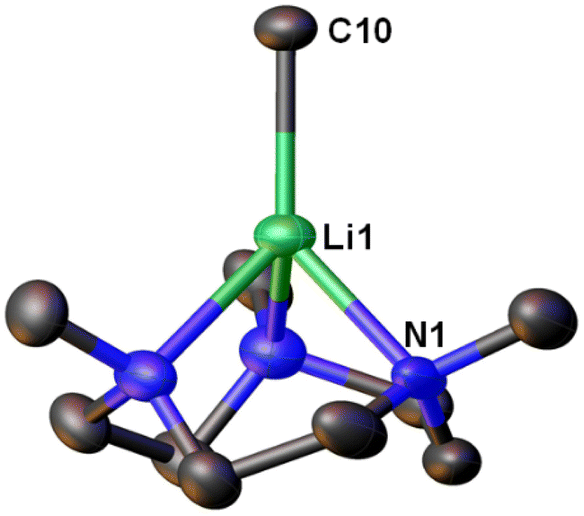 | ||
| Fig. 6 SCXRD structure of the second MeLi monomer [Li(CH3)(κ3-N,N′,N′′-Me3TACN)] (4).64 | ||
The synthesis of 4 is a part of the Anwander group's effort to pursue high-purity MeLi.64 While commercial solutions of MeLi are contaminated with lithium chloride, the authors (Anwander and co-workers) designed a route to achieve high-purity MeLi by utilising potassium bis(trimethylsilyl)amide (KN(SiMe3)2, i.e., KHMDS) as a halide-trapping reagent. Both the pure and commercial MeLi solutions reacted with Me3TACN to form 4, but the compound from the commercial solution resulted in a single-crystal structure with a chloride site occupancy of 9% (at the C10 position in Fig. 6), whereas the pure MeLi resulted in a chloride-free structure. Like 3, 4 was found to decompose readily at room temperature. Reactivity studies showed enchanted deprotonation reactivity of 4 towards toluene, which is not deprotonated by the unsolvated MeLi aggregate [(MeLi)4]∞.
Other than using Lewis basic amine/ether ligands, another strategy to deaggregate MeLi is via multimetallic chelation of the methyl carbanion. In 2001, Stalke and co-workers reported a reaction of dilithium triazasulfite with [(MeLi)4]∞ to form the Li3(μ-Me) complex [(THF)3Li3(μ-Me){(NtBu)3S}] (5) (Fig. 7, left).65 In 2022, Carmona, Jover and co-workers reported a complex [Mo2{(μ-H)Li(THF)(μ-CH3)}2(AdDipp2)2] (AdDipp2 = HC(NDipp)2; Dipp = 2,6-diisopropylphenyl) (6) with one MeLi unit coordinated to Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo bond, forming a H–Mo–Mo–C–Li framework (Fig. 7, right).66 In both these complexes, the methyl carbanion (CH3−), although monomeric, acts as a bridging ligand shared by multiple metal sites (Fig. 7). Since their significant structural differences compared with all other complexes we discuss herein, the two metal-cluster-stabilised monomeric methyl anion complexes 5 and 6 are not included in Table 1 (vide supra).
Mo bond, forming a H–Mo–Mo–C–Li framework (Fig. 7, right).66 In both these complexes, the methyl carbanion (CH3−), although monomeric, acts as a bridging ligand shared by multiple metal sites (Fig. 7). Since their significant structural differences compared with all other complexes we discuss herein, the two metal-cluster-stabilised monomeric methyl anion complexes 5 and 6 are not included in Table 1 (vide supra).
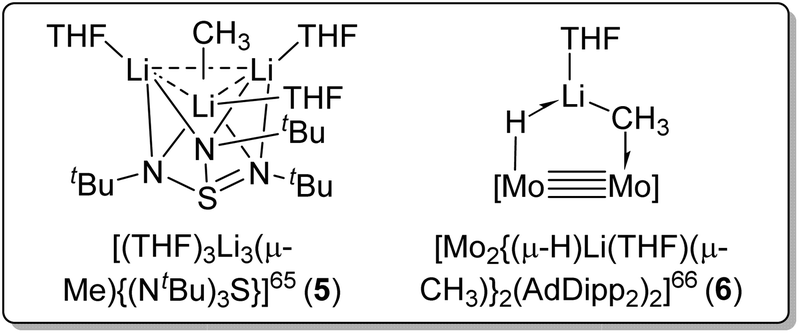 | ||
| Fig. 7 Multimetallic chelation strategy to isolate monomeric methyl anion [CH3]−. | ||
While the structures of polymeric methylsodium (MeNa), methylpotasium (MeK), methylrubidium (MeRb) and methylcesium (MeCs) have been identified by powder X-ray/neutron diffraction (Table 1), to date, no ligand-supported complexes of these species have been reported.63,67–70
3.2 R = CH2SiMe3
Trimethylsilylmethyl lithium (LiCH2SiMe3) is a widely used and commercially available organolithium and organosilicon compound. The CH2SiMe3 alkyl group is popular in organometallic chemistry for a number of reasons: (1) compared with ethyl, isopropyl and butyls, CH2SiMe3 does not have β-hydrogen atoms, hence it is immune to β-hydrogen elimination; (2) the trimethylsilyl group is believed to enhance solubility of the organometallic complexes; (3) more profoundly, compared with its carbon analogue, i.e., neo-pentyl, the M–C bond in MCH2SiMe3 complexes enjoys extra stabilisation via partial overlap between the M–C σ bonding orbital and the C–Si σ* antibonding orbital, i.e., known as the α-Si effect.74,75 The α-Si effect originates from the favourable C–Si σ* antibonding orbital energy level.LiCH2SiMe3 is the starting material for almost all the MCH2SiMe3 complexes via salt elimination, with reports known for where M is a d-block, s-block, f-block or p-block metal. LiCH2SiMe3 is commercially available as pentane solutions. Given the underpinning infrastructural roles of LiCH2SiMe3, research into its own structure and reactivity is relatively limited, partially due to a prejudice that there is little, if any, new chemistry remaining to be discovered in such an infrastructural complex.
The unsolvated solid-state structure of LiCH2SiMe3 is a hexamer, namely [LiCH2SiMe3]6, which was characterised by Oliver and co-workers in 1986.76 The SCXRD structure features a cage structure, which comprises two-deck six-membered rings. A [LiCH2SiMe3]6 polymorph was reported in 2020, which only have trivial structural differences compared with the original 1986 report.77,78 It remains hexameric in solution in cyclohexane, however in benzene, it was observed that at lower concentration it is likely mainly tetrameric; whereas when the concentration increases, a move towards a hexameric species was seen.35
The [LiCH2SiMe3]6 can be further deaggregated into tetramers and dimers using ether or amine ligands. In 2010, the Stalke group reported an Et2O-solvated tetramer [(Et2O)2(LiCH2SiMe3)4], where two out of the four Li centres are coordinated by a Et2O molecule.79 A bidentate ether, dimethoxy ethane (DME) can further deaggregate LiCH2SiMe3 into a dimer [(DME)(LiCH2SiMe3)]2.79 Similar dimeric structures were also reported with other bidentate ligands, such as O,O′-cyclohexyldimethoxysilane (SC)-coordinated [(SC)-LiCH2SiMe3]2,80N,N′-TMEDA-coordinated [(TMEDA)(LiCH2SiMe3)]2,81N,N′-(−)-sparteine-coordinated [{(−)-sparteine}(LiCH2SiMe3)]2,81 and N,O-Me2N(CH2)2OMe-coordinated [(Me2N(CH2)2OMe)(LiCH2SiMe3)]2.82 Monodentate N-donor quinuclidine (Qu) was also reported to form two different dimers,83 the symmetrical [(Qu)(LiCH2SiMe3)]2 and the asymmetrical [{(Qu)(LiCH2SiMe3)}{(Qu)2(LiCH2SiMe3)}].
There are three reports of LiCH2SiMe3 monomers. The first example, [Li(CH2SiMe3)(PMDTA)] (7), was reported in 2008 by the Stalke group (Fig. 8).81 Compared with the bidentate-ligand-coordinated dimers reported in the same paper, the tridentate PMDTA facilitate the formation of monomer. While, no reactivity studies were reported in the paper, it was noted that the monomeric complex showed increased sensitivity towards air, compared to the donor-free hexamer and increased reactivity relative to that of the dimeric aggregates synthesised, because of improved accessibility to the methyl carbanion and lithium-centre. In 2023, Hevia and co-workers reported 1H DOSY NMR on 7, which showed that it is monomeric in C6D12 solution.84
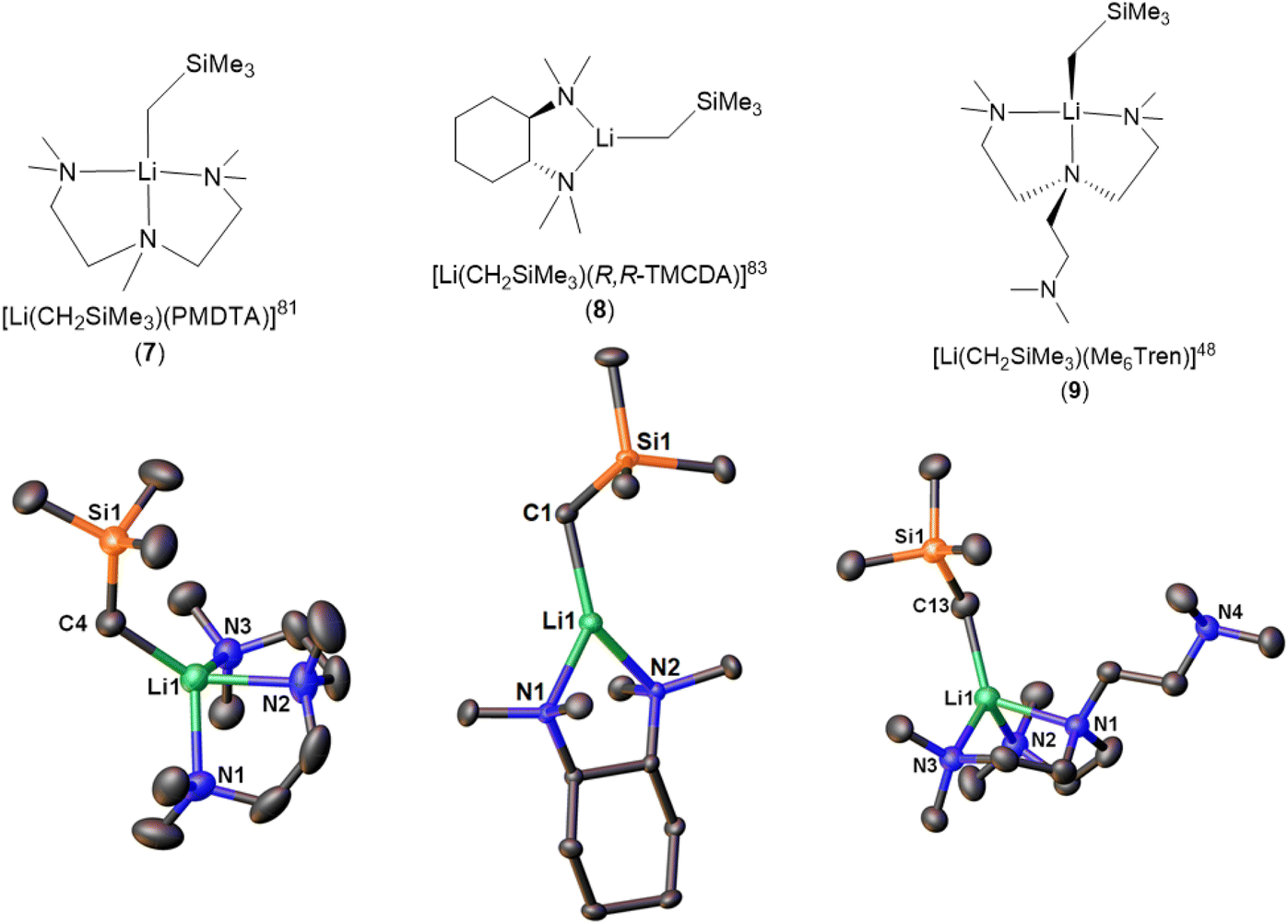 | ||
| Fig. 8 The LiCH2SiMe3 monomers (7–9) and their SCXRD structures. | ||
The second LiCH2SiMe3 monomer, namely [Li(CH2SiMe3)(R,R-TMCDA)] (8), was reported by the Strohmann group in 2019 (Fig. 8).83 Compared to the abovementioned Qu-coordinated dimers from the same paper, the rigid bidentate R,R-TMCDA ligand enabled the isolation of a monomer. Interestingly, the authors (Strohmann and co-workers) found that the monomer 8 was less reactive than the Qu-coordinated dimers for the deprotonation of N,N-dimethylbenzylamine. The authors attributed the unexpected low reactivity of the monomer 8 to its rigid TMCDA ligand, which kinetically prevents incoming substrate from approaching the Li–C reactive centre. This observation challenged the popular perception that a monomeric organolithium complex will be more reactive than its higher aggregates and emphasised the versatility of organolithium chemistry.
The third and the latest LiCH2SiMe3 monomer, [Li(CH2SiMe3)(Me6Tren)] (9) (Fig. 8), was reported by us in 2022.48 Notably, in the SCXRD structure of 9, only two of the three Me6Tren side arms coordinate to the Li centre, while the other is coordination-free (Fig. 8). Variable-temperature NMR (VT-NMR) studies proved that 9's partially coordinated geometry retains in C6D6 solution: lowering the temperature allowed us to observe the fast coordination-dissociation equilibrium in solution.48
Compared with the relatively inert 8, 9 exhibited higher C–H activation reactivity towards toluene, surpassing the [LiCH2SiMe3]6 hexamer.48 While the [LiCH2SiMe3]6 hexamer does not react with toluene for at least several hours at room temperature, 9 deprotonates neat toluene within seconds at room temperature. 9's high reactivity was also reflected by its relative low stability. While 9 is stable as a crystalline solid at −35 °C for several weeks, it decomposes via C–H and N–C bond cleavage of one (likely the coordination-free) sidearm of the Me6Tren ligand, to afford N,N-dimethylethenamine, tetramethylsilane and a lithium amide complex [Li{N(CH2CH2NMe2)2}] in C6D6 or n-hexane solution at room temperature for 3 days. We conducted thorough kinetic studies of 9's decomposition and confirmed that it is a two-stage process.48
A comparison between the reactivities of the LiCH2SiMe3 monomers 8 and 9 is intriguing. Since 8 is quite inert but 9 is rather reactive (both benchmarked towards toluene Csp3–H activation), it is obvious that the perception, i.e., “the smaller the better”83 (the term ‘better’ means more reactive), has to be taken with caution, if not being treated as a groundless “folklore”.85 The perception was also challenged on the other end of the aggregate size spectrum, i.e., the higher aggregates are not necessarily lower in reactivity. The Stalke group reported a DABCO-coordinated LiCH2SiMe3 octamer, namely [(DABCO)7(LiCH2SiMe3)8] (DABCO: 1,4-diazabicyclo[2.2.2]-octane),86 which can conduct toluene C–H activation at room temperature, hence is more reactive than the lower aggregate hexamer [LiCH2SiMe3]6.
Heavy alkali metal alkyl MCH2SiMe3 (M: Na, K, Rb, Cs) complexes are much less studied compared with LiCH2SiMe3. In 2010, Klett and co-workers reported the unsolvated crystal structure of [NaCH2SiMe3]∞, which is a polymer chain comprised of tetrameric subunits.87 Very recently, we performed 1H DOSY NMR on the [NaCH2SiMe3]∞, the result revealed that it exists as a tetramer in C6D6 solution.8 In 2011, Klett also reported the reactions of Na/KCH2SiMe3 with bi- and tridentate ligands TMEDA and PMDTA, to form helical chain polymers [(TMEDA)Na(CH2SiMe3)]∞ and [(PMDTA)K(CH2SiMe3)]∞, and a tetramer [(TMEDA)3(KCH2SiMe3)4], respectively.88
The first and so far the only NaCH2SiMe3 monomer, [Na(CH2SiMe3)(Me6Tren)] (10) (Fig. 9), was reported simultaneously by the Hevia group84 and us8 in 2023, but from rather different perspectives especially regarding reactivity standing points. Hevia and co-workers employed Me6Tren, among other ligands (e.g., PMDTA), to increase the solubility of [NaCH2SiMe3]∞ and to deliver benzylic C–H activation of toluene and substituted toluenes, which were subsequently treated with Weinreb amide to afford the nucleophilic substitution products.84 The core of NaCH2SiMe3 reactivity in the Hevia work is Brønsted basicity, which was found to be enhanced by the formation of the monomer 10. In comparison with the toluene C–H activation of 10, Hevia and co-workers found that treating [LiCH2SiMe3]6 with Me6Tren cannot deliver toluene C–H activation.84 We are particularly interested in such an observation, since it is apparently contradict with our previous report that the isolated [Li(CH2SiMe3)(Me6Tren)] (9) can activate toluene C–H bond.48 A closer examination revealed that the 9 mediated toluene C–H activation is concentration dependent: since we conducted the reaction in neat toluene,48 Hevia and co-workers used stoichiometric toluene in hexane solution.84 This delicate but crucial difference leads to totally different, and apparently contradictory conclusions, once again emphasising the importance of details in organo-alkali metal chemistry.
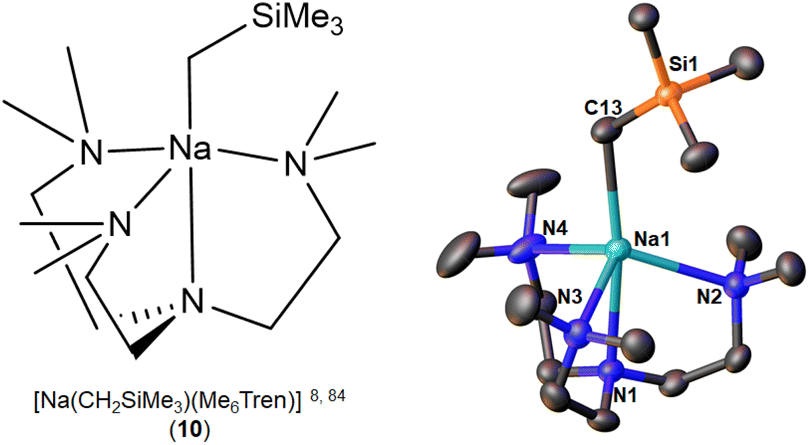 | ||
| Fig. 9 The only NaCH2SiMe3 monomer [Na(CH2SiMe3)(Me6Tren)] (10) and its SCXRD structure. | ||
We compared the reactivity of [NaCH2SiMe3]∞, [Na(CH2SiMe3)(Me6Tren)] (10) and [Li(CH2SiMe3)(Me6Tren)] (9) towards benzophenone.8 As expected, we found that both the [NaCH2SiMe3]∞ polymer and [Li(CH2SiMe3)(Me6Tren)] (9) resulted in the nucleophilic addiction product [MO{C(Ph)2(CH2SiMe3)}]4 (M: Li, Na). In contrast, methylation resulted using [Na(CH2SiMe3)(Me6Tren)] (10), which produced 1,1-diphenyl ethylene, free Me6Tren and [Na9(OSiMe3)10]−[Na(C6H6)(Me6Tren)]+. We expanded the substrate scope of this C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O methylenation, to produce terminal alkenes using 10 to include a variety of ketones, aldehydes and amides. We then further developed a ligand-catalysis strategy to conduct ketone/aldehyde methylenations, using [NaCH2SiMe3]∞ as the CH2 feedstock and Me6Tren as the catalyst (5 mol%). It was postulated that [NaCH2SiMe3]∞ rapidly reacts with the carbonyls in a nucleophilic addition manner to form a cluster alkoxide intermediate, which is subsequently slowly dissembled by the catalytic amount of Me6Tren into lower aggregate(s) intermediate(s), which then undergoes methylenation, eliminates NaOSiMe3 species and regenerates the ligand catalyst Me6Tren. The most striking feature of this work is the metal identity-dependent distinct reaction patterns between 9 and 10 (Fig. 10), which was analysed using DFT calculations and revealed to be metal identity dependent.8
O methylenation, to produce terminal alkenes using 10 to include a variety of ketones, aldehydes and amides. We then further developed a ligand-catalysis strategy to conduct ketone/aldehyde methylenations, using [NaCH2SiMe3]∞ as the CH2 feedstock and Me6Tren as the catalyst (5 mol%). It was postulated that [NaCH2SiMe3]∞ rapidly reacts with the carbonyls in a nucleophilic addition manner to form a cluster alkoxide intermediate, which is subsequently slowly dissembled by the catalytic amount of Me6Tren into lower aggregate(s) intermediate(s), which then undergoes methylenation, eliminates NaOSiMe3 species and regenerates the ligand catalyst Me6Tren. The most striking feature of this work is the metal identity-dependent distinct reaction patterns between 9 and 10 (Fig. 10), which was analysed using DFT calculations and revealed to be metal identity dependent.8
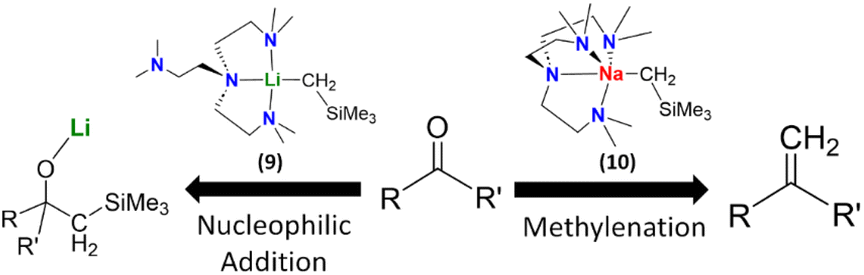 | ||
| Fig. 10 Distinct reaction patterns between 9 (nucleophilic addition) and 10 (methylenation).8 | ||
All the reported MCH2SiMe3 (M: Li, Na, K) complexes, monomers and higher aggregates, and their M–C bond lengths are summarised in Table 2. It should be noted that there are no Rb/CsCH2SiMe3 complexes to date.
3.3 R = Bis/tris(trimethylsilyl)methyl (CH(SiMe3)2 and C(SiMe3)3)
The CH(SiMe3)2 and C(SiMe3)3 alkyl groups have allowed the synthesis of a plethora of complexes with interesting aggregation and structural features, including unusual ate complexes. In addition to an increase in steric bulkiness, replacing protons with trimethylsilyl groups results in additional electronic stabilisation of the carbanionic centre, via the aforementioned α-Si effect. Furthermore, the extra trimethylsilyl groups improve solubility in hydrocarbon and aromatic solvents – an issue with many Na–Cs organometallics. These factors mean that the crystals structures of a number of heavier alkali metal bis/tris(trimethylsilyl)methyl complexes have been reported.The crystal structure of unsolvated bis(trimethylsilyl)methyllithium and bis(trimethylsilyl)methylsodium were found to be linear 1D polymers.89,90 The corresponding heavier alkali metal bis(trimethylsilyl)methyl complexes have been unable to be isolated as an unsolvated structure, due to their insolubility in hydrocarbon solvents. The solution aggregation behaviour was also investigated using DOSY NMR and cryoscopy measurements. Bis(trimethylsilyl)methyllithium was found to be dimeric in solution in cyclohexane. Bis(trimethylsilyl)methylsodium showed concentration-dependent aggregation behaviour, with dilute cyclohexane solutions resulting in tetramers, whereas hexamers were observed in more concentrated solutions.91
Bis(trimethylsilyl)methyllithium forms the THF-solvated dimer [Li(CH(SiMe3)2)(THF)]2. The coordination of TMEDA or PMDTA results in monomeric complexes [Li(CH(SiMe3)2)(PMDTA)] (11) and [Li(CH(SiMe3)2)(TMEDA)] (12) (Fig. 11), both in solid-state and cyclohexane solution.91,92 In contrast, bis(trimethylsilyl)methylsodium retains a 1D polymeric chain structure when solvated with THF, with one THF molecule coordinated to each sodium. The addition of TMEDA resulted in crystals of the dimeric complex [NaCH(SiMe3)2]2(TMEDA)3, in which two TMEDA coordinated ‘monomers’ are bridged through an additional TMEDA molecule.91
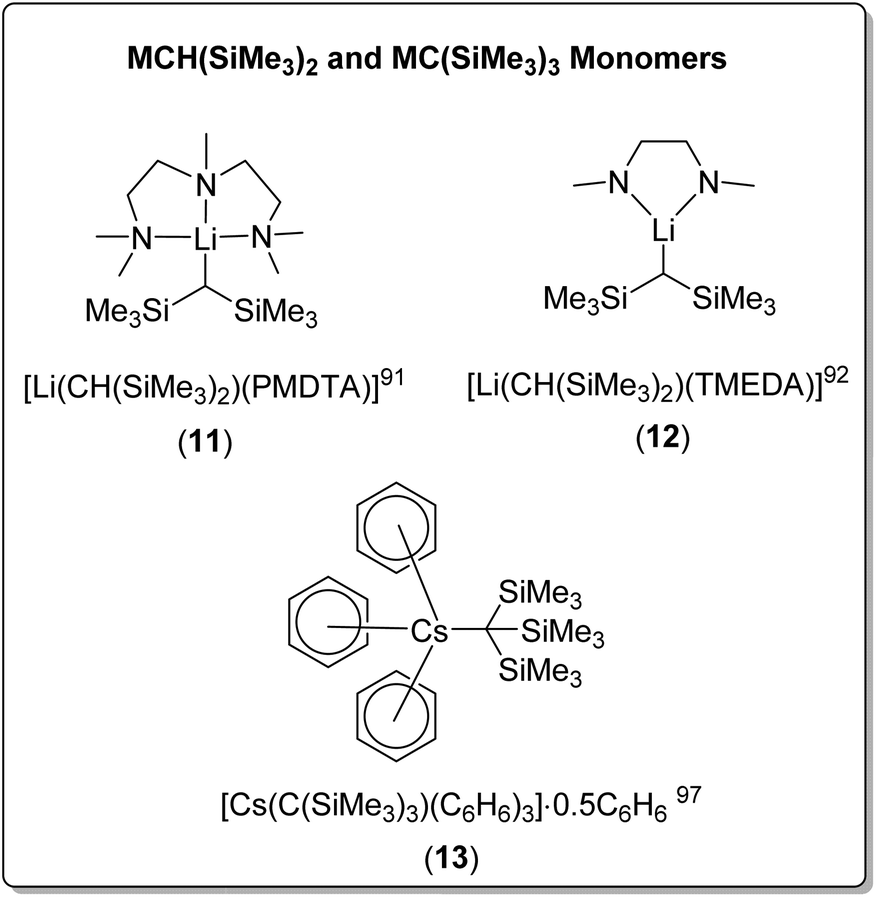 | ||
| Fig. 11 The reported SCXRD-characterised bis/tris(trimethylsilyl)methyllithium monomeric complexes. | ||
It is interesting to note that, in contrast to many organolithium and sodium compounds, neither bis(trimethylsilyl)methyllithium or sodium were reported to undergo decomposition in THF, highlighting the unusual stability of these bis(trimethylsilyl)methyl compounds.
Single-crystal structures have been reported for bis(trimethylsilyl)methylpotasium complexes with THF, tert-butyl methyl ether (tBuOMe), and PMDTA. Both the THF and tBuOMe form mono-solvated 1D linear chain polymers. The single-crystal structure of the PMDTA complex revealed the open tetramer [{KCH(SiMe3)3}2(PMDTA)]2.93,94 Single-crystal structures of organorubidium and organocaesium complexes are uncommon, however a dimeric bis(trimethylsilyl)methylrubidium complex has been reported using the PMDTA ligand, [Rb(CH(SiMe3)2)(PMDTA)]2, and a 1D linear chain polymer complex of bis(trimethylsilyl)methylcaesium was reported using TMEDA, [Cs(CH(SiMe3)2)(TMEDA)]∞.90,94
The even bulkier tris(trimethylsilyl)methyllithium was found to be dimeric when unsolvated in the solid-state, [LiC(SiMe3)3]2, however cryoscopic molecular mass determination suggested that complex was monomeric in benzene in solution.95 Surprisingly, a search of the CSD revealed that the unsolvated sodium analogue has not been reported, whereas the unsolvated K and Rb complexes are known. Due to their high reactivity and low solubility in hydrocarbon and aromatic solvent, unsolvated crystal structures for heavier alkali metal organometallics are very rare. Both tris(trimethylsilyl)methylpotasium and tris(trimethylsilyl)methylrubidium were found to have a 1D polymeric structure, however in the rubidium complex, the rubidium cation was found to interact additionally with the eight methyl groups from the trimethylsilyl component, in addition to the carbanionic carbon-centre.96,97
Both the tris(trimethylsilyl)methyl-potassium and -rubidium complexes involved benzene as a solvent in the crystallisation conditions, but no coordination to benzene was observed in the crystal structures. In contrast, the crystallisation of tris(trimethylsilyl)methylcaesium from benzene resulted in a remarkable benzene-solvated monomeric organocaesium complex, [Cs{C(SiMe3)3}(C6H6)3]·0.5C6H6 (13) (Fig. 11), with each caesium cation coordinated to the central anionic carbon and three benzene molecules. Uncoordinated benzene was also found in the crystal.97 The coordination of TMEDA to tris(trimethylsilyl)methylpotasium results in a polymeric chain structure, linked via coordination of the potassium cation, through one of methyl groups of the trimethylsilyl moiety.98
Compared to the structures of the other alkyl groups discussed in this perspective, the THF adduct of tris(trimethylsilyl)methyllithium features a unique structural category: solvated lithate complexes [LiR2]−[Li(solv)n]+. Some representatives of this ate complex family are presented in Fig. 12. It should be noted that, though the examples in Fig. 12 feature mono-metallic organolithium fragments, they are different from the other neutral organo-alkali metal monomers in this Perspective, hence are not numbered.
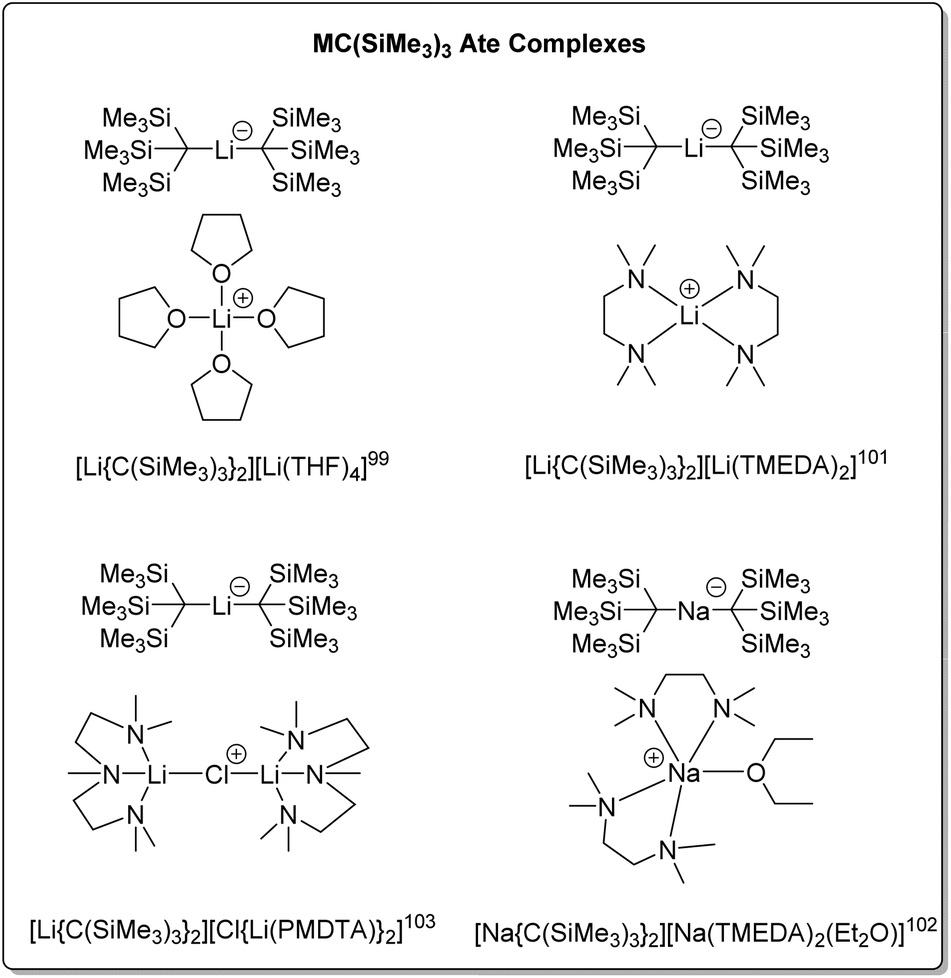 | ||
| Fig. 12 The reported SCXRD-characterised tris(trimethylsilyl)methyllithium/sodium ate complexes. | ||
In 1983, Eaborn, Smith and co-workers reported the crystal structure of the product from the metalation of tris(trimethylsilyl)methane and methyllithium in THF – the first structurally characterised lithate complex [Li(THF)4][Li{C(SiMe3)3}2].99 The anion exhibits a linear C–Li–C linkage. It was postulated that the steric hindrance of the bulky tris(trimethylsilyl)methyl group disfavours the normally observed contact ion pair structure [Li–R(L)x]n, as well as stabilisation of the negative charge on the carbon atoms of the anion by the silicon atoms, enabling the isolation of the lithate complex. The related crystal structure, [Li(THF)3][Li{C(SiMe3)3}2] has also been reported.100
The addition of TMEDA to [Li(THF4)][Li{C(SiMe3)3}2] resulted in the ate complex [Li(TMEDA)2][Li{C(SiMe3)3}2].101 Both complexes were subject to solution and solid-state NMR studies. The solution NMR studies confirmed the presence of the ate complexes in THF and toluene solutions, together with another species, with exchange between the species observed at NMR time-scale at higher temperatures. The sodium ate complex [Na(TMEDA)2(Et2O)][Na{C(SiMe3)3}2] has also been prepared via the metalation of tris(trimethylsilyl)methane using methylsodium with TMEDA in diethyl ether. The anion of this complex features a linear C–Na–C linkage with the sodium atom two-coordinate.102
The reaction of tris(trimethylsilyl)methane with MeLi in the presence of PMDTA, resulted in the complex [(PMDTA)Li(μ-CI)Li(PMDTA)][Li{C(SiMe3)3}], with the cation featuring a linear Li–Cl–Li linkage.103 As previously mentioned in Section 3.1, MeLi is often contaminated with LiCl as a result of its synthetic procedure, which leads to this structure.
What affect does this ate structure type have on reactivity? Reich and co-workers studied the structure and reactivity of tris(trimethylsilyl)methyllithium complexes in solution (Fig. 13). Tris(trimethylsilyl)methyllithium was found to exists as a mixture of three species in THF/ether solution. A separated ion pair (SIP), a contact ion pair (CIP), and an ate complex, referred to as a triple ion pair (TIP). These species were found to slowly exchange at NMR time-scale. It was observed that at around −70 °C, the NMR signals corresponding to the CIP and SIP coalesced. Above 50 °C, the signals of TIP also coalesced with the CIP and SIP into an averaged signal. In diethyl ether solution, only the CIP was detected. The addition of hexamethylphosphoramide (HMPA) completely converted tris(trimethylsilyl)methyllithium to a HMPA-solvated SIP species.104–106
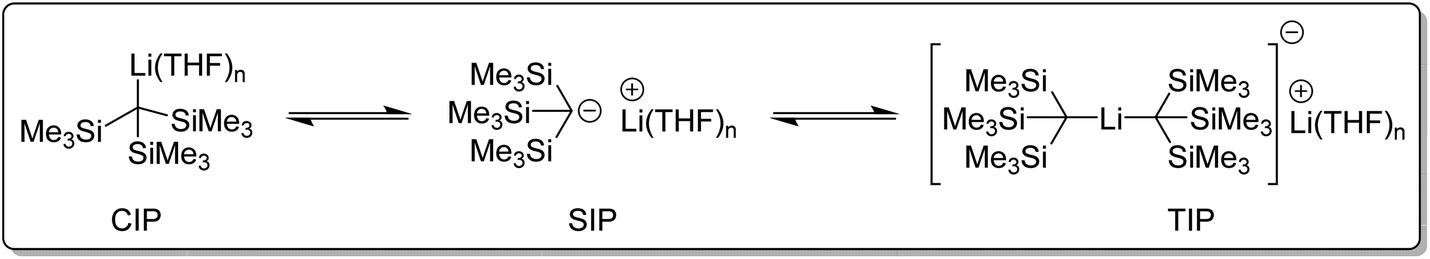 | ||
| Fig. 13 The three tris(trimethylsilylmethyl)lithium (CIP, SIP and TIP) species in THF/ether solution.104–106 | ||
The reactivity of the three species (CIP, TIP and SIP) was investigated and compared using low-temperature rapid-injection NMR (RINMR). At −132 °C The THF-solvated SIP was over fifty times more reactive towards methyl iodide (MeI) than the monomeric CIP and over 5 × 1010 times as reactive as the TIP, i.e., the reactivity sequence is SIP ≫ CIP ⋙ TIP. Monitoring of the reaction between MeI and tris(trimethylsilyl)methyllithium in 3:2 THF/ether at −85 °C, showed that the NMR signals corresponding to the SIP and CIP species disappeared in under one second to form tris(trimethylsilyl)ethane. The signals corresponding to the TIP remained, which reacted slowly over a period of 1–2 hours. Kinetic experiments revealed that, at these low temperatures, the TIP and CIP do not react directly with MeI, but must first dissociate to the SIP, which then reacts rapidly. This is particularly interesting to note since it highlights that the structure of the isolated solid-state crystal structure, i.e. [Li(THF)3/4][Li{C(SiMe3)3}2] for the THF adduct of tris(trimethylsilyl)methyllithium, is not necessarily the same as the reactive species in solution.
3.4 R = Butyls (n-, sec-, tert-)
Despite being perhaps the most well-known, widely used and most studied organometallic reagent,4 no isolated monomeric complexes of nBuLi have been reported. An often encountered problem with trying to isolate n-/tert-/sec-BuLi monomers, is the tendency of the lower aggregate species to lithiate the ligands used.49,50,107–109 However, a number of studies have taken place to investigate the structure–reactivity relationship of butyllithium reagents.
n
BuLi crystallises from pentane as an unsolvated hexamer.110 In solution, nBuLi mainly exists as a hexamer in hydrocarbon solvents at room temperature, but is primary an octamer at low temperature.32,35,111 It is tetrameric in diethyl ether,35,110 and a tetramer–dimer mixture in THF.25,110,112 This intriguing mixture of tetramer and dimer in THF has enabled insightful NMR studies to take place. Reich and co-workers reported rapid injection NMR (RINMR) studies of the relative reactivity of the nBuLi–THF dimer and tetramer towards a number of substrates, with astonishing findings.113,114 They found that for the metalation of trimethylsilylacetylene at −130 °C, the nBuLi dimer reacts in under two seconds, whereas the tetramer reacts over several hours. However, the rate of reaction of the tetramer is zero order to trimethylsilylacetylene and is consistent with the rate of tetramer to dimer dissociation, i.e., essentially, the tetramer itself does not react (or very slow) with trimethylsilylacetylene at −130 °C, but has to deaggregate to the dimer (Fig. 14). It was estimated that towards this substrate, the dimer least 40000 times as reactive as the tetramer.113 Further experiments reviled that nBuLi dimer is 320000000 times as reactive as the tetramer toward phenylthioacetylene at −130 °C.112,114
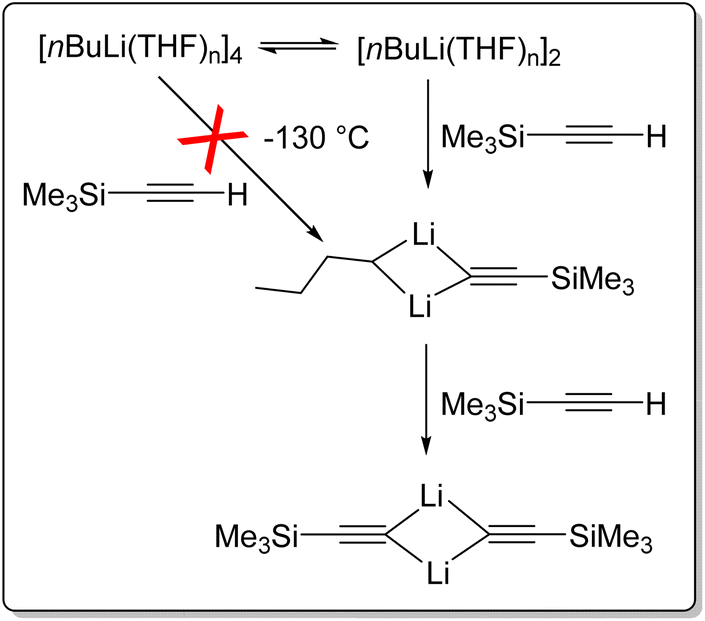 | ||
| Fig. 14 Rapid injection NMR (RINMR) studies of reactions between nBuLi (dimer & tetramer) and trimethylsilylacetylene at −130 °C.113 | ||
It is well-known that the reactivity of nBuLi is enhanced via the addition of TMEDA,115–119 which forms dimers in solution and solid-state when 1:1120–124 (polymeric tetramers has been reported in solid-state when 0.25 equivalents of TMEDA were added124,125). This enables the deprotonation of toluene, with no reaction taking place in the absence of TMEDA.115 Additionally, the nBuLi aggregates react with ferrocene to produce monolithioferrocene in only very low yield, while the addition of TMEDA enables the synthesis of 1,1′-dilithioferrocene in high yield.116–119
Dimeric nBuLi complexes were also isolated by the coordination of ligands such as (−)-sparteine,126 (R,R)-TMCDA127 and 1,2-dipiperidinoethane (DPE-6).128 A dimer [Li(nBu)(R,R-TMCDA)]2 was found to be able to deprotonate benzene, which the unsolvated [nBuLi]6 hexamer is stable towards.127
There is evidence from on 7Li quadrupolar couplings that nBuLi forms a monomer with PMDTA in solution in d8-toluene.33 However to date, the only product that has been crystallised with PMDTA has been the open tetramer, [(nBuLi)2(PMDTA)]2.50
sec-Butyllithium (sBuLi) and tert-butyllithium (tBuLi) are more sterically bulky than nBuLi and form lower aggregates in solution and solid-state.4 Solution-state NMR studies have revealed that unsolvated sBuLi is a hexamer–tetramer mixture in hydrocarbon solvents,129 a dimer–monomer mixture in THF130 and monomeric when PMDTA was added to the THF solution,130 however this has not been crystallographically characterised.
Only one crystallographically characterised monomer of sBuLi has been reported to date. In 2007, Strohmann and co-workers reported a sBuLi monomer using the (R,R)-TMCDA ligand, namely, [Li(sec-Bu)(R,R-TMCDA)] (14) (Fig. 15).60 Monomer 14 was found to be extremely reactive and able to deprotonate benzene and toluene using sub-stoichiometric amounts of (R,R)-TMCDA. The crystal structure of the sBuLi monomer 14 revealed an open Li centre, barely shielded by the ligand and hence offering a free and accessible coordination site for potential substrates. This is in sharp contrast with the inert [Li(CH2SiMe3)(R,R-TMCDA)] monomer (9), which was also reported by the Strohmann group.83
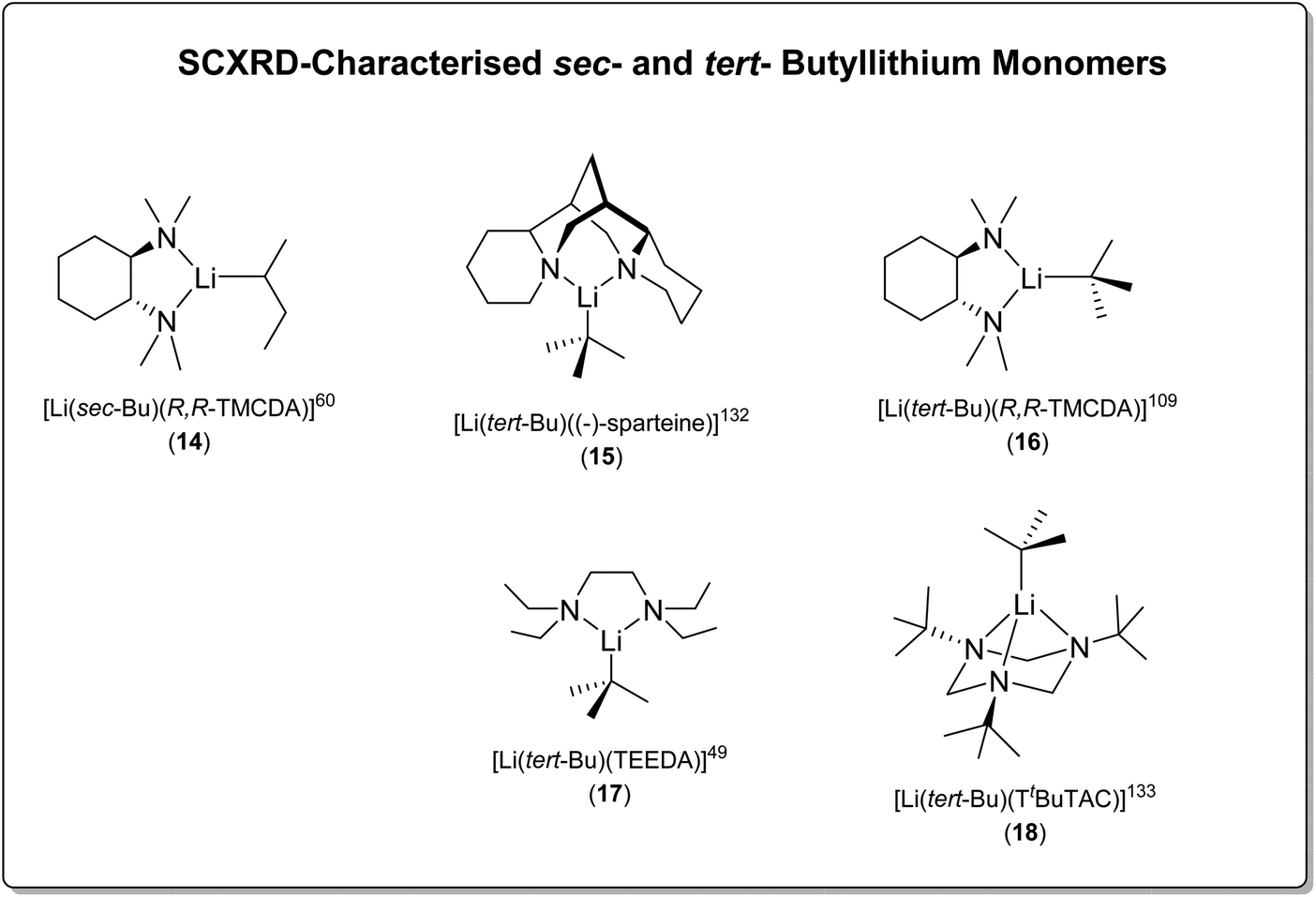 | ||
| Fig. 15 The SCXRD-characterised sBuLi (14) and tBuLi (15–18) monomers. | ||
t BuLi, the bulkiest butyllithium, is an unsolvated tetramer when crystallised from pentane,110 and is tetrameric in solution in hydrocarbon solvents.131 From diethyl ether, a bis-solvated dimer results in solid-state110 and is also dimeric in diethyl ether solution.130
t BuLi is monomeric in THF solution.130 Interestingly, it was observed that when TMEDA was added to tBuLi–THF solution,130 the TMEDA stayed as largely free ligand and was poor at competing with THF for coordination. This observation, among others, raised the question that whether TMEDA, despite its widespread use, is actually a good ligand for lithium in the 1990s,85 which still remains largely unanswered nowadays.
In 2003, the Strohmann group reported the first SCXRD-characterised tBuLi monomer, supported by the chiral amine ligand (−)-sparteine, namely [Li(tert-Bu)((−)-sparteine)] (15) (Fig. 15).132 This was followed by reports from the same group in 2007 and 2008 of [Li(tert-Bu)(R,R-TMCDA)] (16)109 and [Li(tert-Bu)(TEEDA)] (17)49 (TEEDA is TMEDA's ethyl-substituted analogue; N,N,N′,N′-tetraethylethylenediamine), respectively. In 2014, the Mitzel group employed a tridentate macrocyclic ligand 1,3,5-tri-tert-butyl-1,3,5-triazacyclohexane (TtBuTAC) to stabilise a tBuLi monomer [Li(tert-Bu)(TtBuTAC)] (18)133 (Fig. 15).
From a reactivity perspective, as observed in the previous sections, ligand plays underpinning roles for the tBuLi monomers’ reactivity. The (−)-sparteine-coordinated tBuLi monomer 15 is inert towards toluene/benzene C–H activation at room temperature,132 which is less reactive than THF-solvated tBuLi dimer.113 In contrast, the R,R-TMCDA-, TEEDA- and TtBuTAC-coordinated tBuLi monomers 16–18 all exhibited high reactivity towards toluene/benzene C–H activation and/or ligand lithiation.49,109,133 A particularly interesting case is the ligand (TEEDA) lithiation of 17. Instead of α-lithiation (deprotonating N–CH2–CH3), 17 was found to undergo β-lithiation (deprotonating N–CH2–CH3) of the TEEDA ligand at room temperature.49 The preference for α- over β-lithiation was explained by the distance and arrangement of the α- and β-hydrogen atoms toward the carbanionic tBu group, which is an example of the structure–reactivity relationship per se.49,109 This type of structure–reactivity relationship is known as the complex-induced proximity effect (CIPE).134
Organolithium-mediated deprotonation reactions can proceed via two possible reaction pathways. Firstly, a ‘direct’ deprotonation route, where pre-coordination of the lithium-centre to a functional group does not occur. Deprotonation takes place via a bimolecular Brønsted acid–base reaction between RLi and the substrate through the most stable transition state. In these cases, selectivity can be determined via steric factors or electrostatic interactions.134 The second possibility is the aforementioned CIPE, which proceeds via the pre-coordination of functional group to the lithium-centre. In this intermediate complex, the reactive groups (i.e., the carbanionic centre and acidic protons) are in close proximity to each other. The spatial proximity of these groups results in a decreased reaction barrier and hence the observed selectivity.134
The CIPE explains the difference in the observed regioselective (α vs. β deprotonation) in the decomposition reactions of the aforementioned tert-butyllithium monomers (Fig. 16, top). In the reaction between (R,R)-TMCDA and tert-BuLi, the isolated monomeric complex [Li(tert-Bu)(R,R-TMCDA)] (16) is the intermediate pre-coordinated complex in the CIPE. The structure of this complex shows the α-proton position in close spatial proximity to the carbanionic centre, which results in α-lithiated (R,R)-TMCDA. α-lithiation is challenging due to the repulsion between the nitrogen lone pair of elections and carbanion centre but are sought after targets due to them being important building blocks in synthesis.109
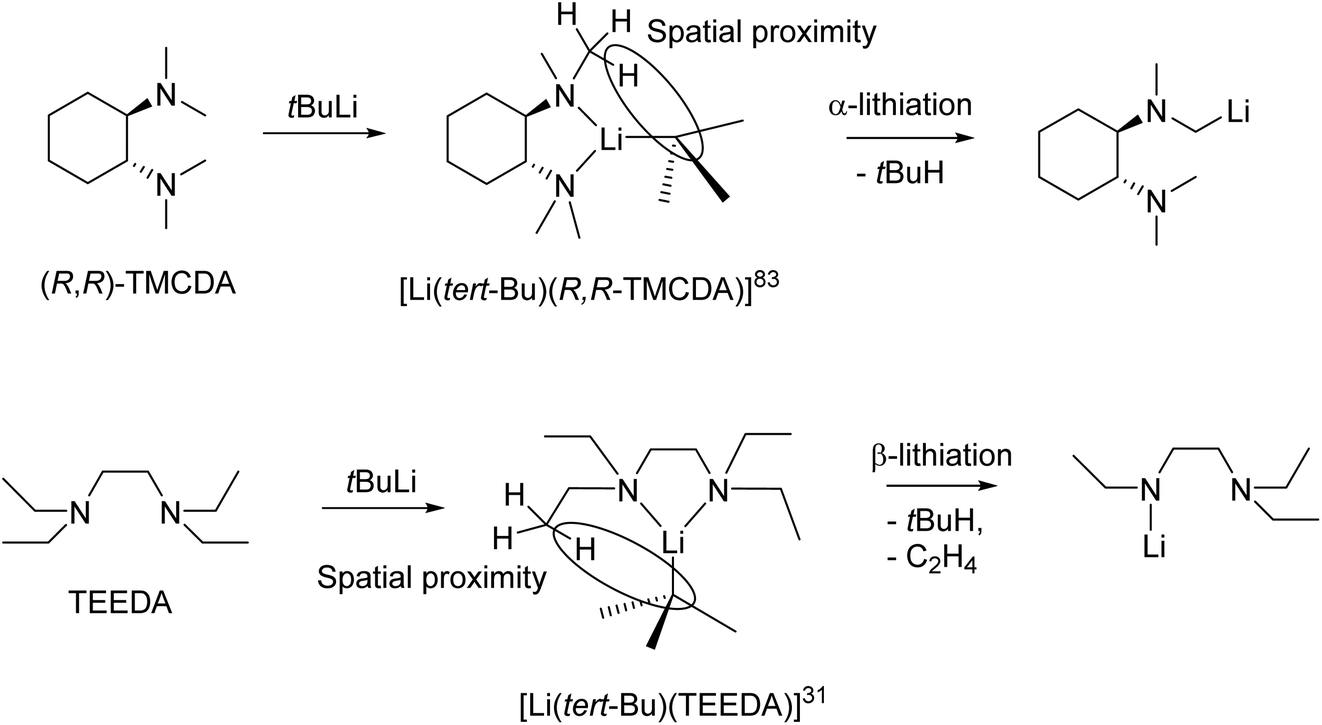 | ||
| Fig. 16 Regioselective deprotonation of (R,R)-TMCDA/tert-BuLi and TEEDA/tert-BuLi due to the CIPE. | ||
Likewise, the monomeric complex [Li(tert-Bu)(TEEDA)] (17), is the pre-coordinated complex in a reaction between TEEDA and tert-butyllithium. The structure reveals close spatial proximity between the β-proton position and the carbanionic centre (Fig. 16, bottom). This closer proximity of the β-protons compared to the α-protons to the carbanionic centre, results in a lower energy barrier for β-deprotonation (92 kJ mol−1) compared to α-deprotonation (119 kJ mol−1).49 Therefore deprotonation occurs at the β-position, followed by elimination of ethene, resulting in the observed product. After initial deprotonation of the Me6Tren side-arm, a similar elimination of N,N-dimethylethenamine, occurred following the decomposition of [Li(CH2SiMe3)(Me6Tren)] (9).48
Other ligands have also been studied regarding their CIPE and its effect in ligand lithiation, with TMEDA/tert-BuLi found to undergo α-lithiation.49 1,3,5-Trimethyl-1,3,5-triazacyclohexane/tert-BuLi was found to deprotonate at the methylene bridge of the triazacyclohexane, rather than the expected methyl group, which again was a result of closer proximity of the methylene bridge to the carbanionic centre, compared to the methyl group, in the pre-coordinate complex.135,136
The CIPE is not a new concept, though. It has been investigated for decades, due to its importance in understanding organolithium selectivity and control and improving our knowledge of the structure–reactivity relationship. An early interesting example was observed in 1983 by Julia, who showed that the selectivity of the reaction between (Z)-thioenol and sec-BuLi, could be altered via the addition of HMPA. It was postulated that, in the absence of HMPA, the lithium-centre coordinates to the methoxyl group, resulting in deprotonation of the nearest proton. Therefore, lithiation occurred β- to sulphur. In contrast, in the presence of HMPA, the lithium-cation is likely saturated with HMPA ligands, preventing pre-coordination of the substrate to the lithium-centre. This results in the most acidic proton being deprotonated, with lithiation occurring α- to sulphur.137 The CIPE effect has been reviewed more widely, and in more detail, in a number of reviews and book chapters.134,138–140
A further case of ligands altering the selectivity of organolithium reactions was observed by Clayden. It was found that the addition of six equivalents of HMPA to the reaction between n-, sec-, or tert-BuLi and THF, followed by quenching with phenyl chlorothioformate, resulted in a product formed via lithium but-3-en-1-olate, suggesting possible β-deprotonation, i.e., overriding the usual directing effect of the oxygen. In contrast, when the reactions were performed in the absence of HMPA, a different product formed via α-deprotonation.141
Very recently, Thomas and co-workers conducted 6Li/13C rapid-injection NMR (RINMR) studies on 6Li enriched tBuLi, in the presence of a variety of Lewis basic ligands.142 The RINMR studies on 6Li enriched tBuLi at −125 °C showed that, tBuLi was monomeric in 3:2 d8-THF:Et2O and also monomeric when one equivalent of HMPA was added. However, when two or three equivalents of HMPA were added, NMR signals consistent with a triple ion pair (TIP) [{(tBu)2Li}−·{Li(HMPA)4}+] were observed. They propose that this is likely in equilibrium with small quantities of a highly reactive separated ion pair (SIP) species [(L4Li)+·tBu−].142 The authors (Thomas and co-workers) claimed that the formation of TIP/SIP plays essential roles in delivering the observed abnormal deprotonation site selectivity of a number of O-heterocycles (Fig. 17). The pre-coordination of the lithium cation to the heteroatom prior to deprotonation, is responsible for the selectivity of lithiation. In the SIP, the lithium-centre is saturated, preventing the coordination of the lithium cation to the heterocycle and therefore, the resulting directing effect and ‘normal’ selectivity, can be overridden. The TIP [{(tBu)2Li}−·{Li(HMPA)4}+] was found to be inert for deprotonation, while the SIP [(L4Li)+·tBu−] was very reactive. To our understanding, this raises a question about deprotonation: what is the real active species for deprotonation? We, as the authors of this Perspective, understand that deprotonation is a Brønsted acid–base reaction by definition: a Brønsted base centre is the essential reactive centre. In this regard, a naked tBu− carbon anion (such as in the SIP) is a superb Brønsted base centre, and can deprotonate wherever it is thermodynamically favourable. In contrast, the {(tBu)2Li}− anion in the TIP is not a good Brønsted base centre.
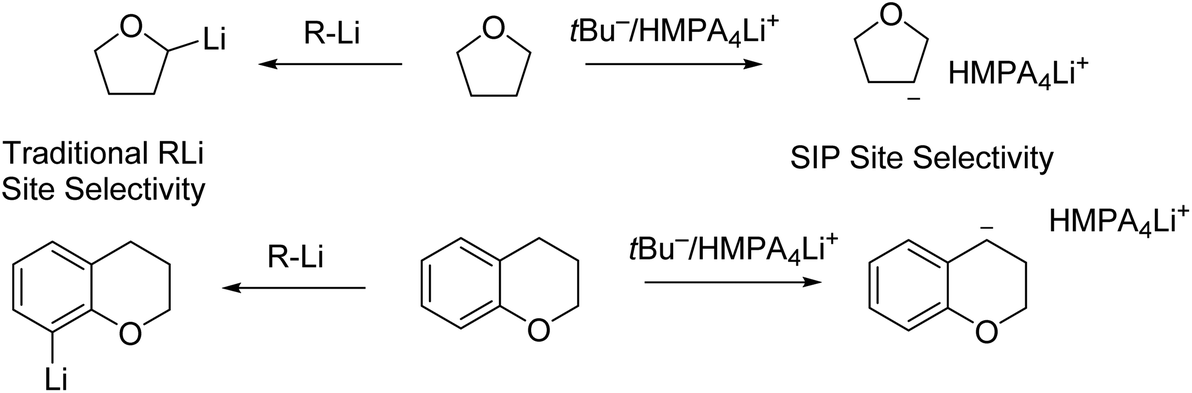 | ||
| Fig. 17 Contrasting site selectivity towards oxygen heterocycles between traditional organolithium reagents and tBu−/(HMPA)4Li+ separation ion pair (SIP) species. | ||
Ligand is not the only factor influencing the reactivity: the alkyl group also have a profound effect. It is particularly intriguing to compare toluene C–H activation reactivity, of the R,R-TMCDA-coordinated monomers of LiCH2SiMe3, sBuLi and tBuLi (Fig. 18). The authors (Strohmann and co-workers) employed DFT calculations to plot Connolly surface, as a probe to gauge the different kinetic shielding effects of CH2SiMe3 and sBu over the Li–C (the presumed reactivity centre).60,83 The striking reactivity difference in the series of 8, 14 and 16 once again highlights the delicate nature of organolithium, or organo-alkali metal chemistry in general.
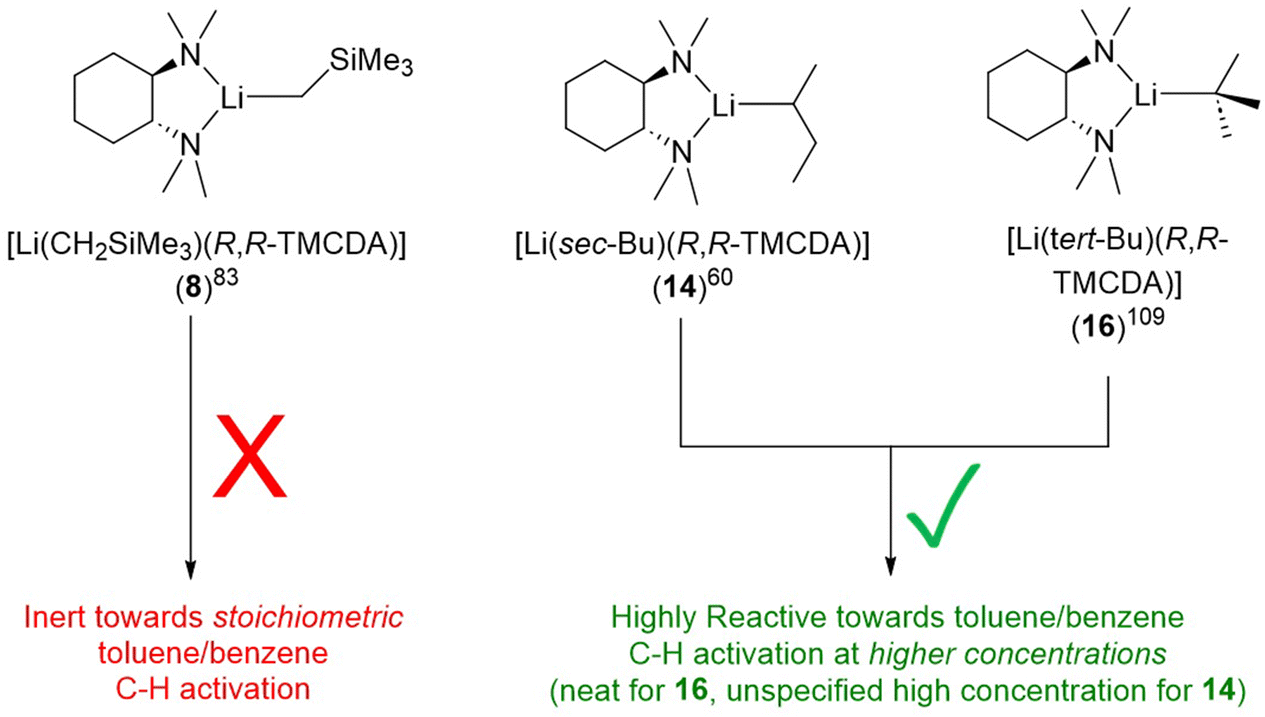 | ||
| Fig. 18 The alkyl's effect on reactivity: different reactivity of the R,R-TMCDA-coordinated LiCH2SiMe3, sBuLi and tBuLi monomers. | ||
3.5 R = CH2Ph (benyl: Bn)
Like R = bis/tris(trimethylsilyl)methyl, R = benzyl has single crystal structures reported for all the Group-1 metals (Li–Cs), therefore important information can be gained by not just comparing the ligand/alkyl effects, but also the effects of metal identity. It is intuitive that the heavier alkali metal cations feature larger ionic radii, hence could accommodate higher coordination numbers, and as a result, tend to form larger aggregates. Regarding the alkyl group, benzyl is generally more stable than the saturated alkyls for two reasons: (1) the sp2-hybrdized Cipso is slightly more electronegative than a sp3-hybrdized C (by approximate 0.2 Pauling electronegativity units), hence acting as an electron-withdrawing group to stabilise the negatively charged carbon centre; (2) the [PhCH2]− can delocalise and allow the negative charge to be shared through the whole benzyl fragment.14In solution, benzyllithium (BnLi) exists a monomer in THF.55,56 The THF solvated adducts and their single-crystal structures, are known for all benzyl-Li–Cs, which all exist as polymeric structures (vide infra). It is interesting to note, however, the change in how the metal-centre coordinates to the benzyl group as the group is descended.
The 1D BnLi polymer [Li(CH2Ph)(THF)2]∞ crystallises with each lithium coordinated to two benzyl groups and two THF molecules.143 The lithium-centre forms almost exclusively η1-Li–C σ-interactions with two –CH2 moieties of the benzyl group (Cα). In diethyl ether, a similar polymeric structure results, namely [Li(CH2Ph)(Et2O)]∞, but with only one diethyl ether molecule coordinated to each lithium.144
Benzylsodium (BnNa) also forms a 1D polymer with two benzyl groups and two THF ligands coordinated to the sodium, [Na(CH2Ph)(THF)2]∞.145 In this complex, η2-coordination of the sodium cation to the Cα and Cipso carbons of the benzyl anion, is observed in the single-crystal structure.
Two benzylpotassium (BnK) THF solvates have been reported: the sheet-like 2D polymer [(KCH2Ph)3(THF)4]∞ and the chain-like polymer [(KCH2Ph)2(THF)]∞, with only one THF coordinated potassium per dimer unit.146,147 The benzylrubidium (BnRb) THF solvate also forms a 1D polymer [Rb(CH2Ph)(THF)2]∞, whereas the benzylcaesium (BnCs) THF solvate is a 2D polymer [Cs(CH2Ph)(THF)0.5]∞.145
In the BnK THF solvated polymers, contacts between the potassium cations to both the Cα and phenyl ring carbons are observed, whereas in the rubidium compound, the interaction of the rubidium cation is exclusively with the π-system of the phenyl ring, resulting in a coordination-free CH2 site. For the BnCs analogue, each caesium cation is coordinated to one THF ligand and two η1, one η3 and one η6 interactions are observed to four benzyl anions.143–147
The bidentate amine ligand TMEDA, forms a monomeric TMEDA/THF adduct complex with BnLi [Li(CH2Ph)(TMEDA)(THF)] (19) (Fig. 19) and an eight-membered ring tetramer with BnNa.148,149 The tridentate ligands, Me3TACN and PMDTA, form monomers with BnLi too, namely [Li(CH2Ph)(Me3TACN)] (20)107 and [Li(CH2Ph)(PMDTA)] (21),150 respectively (Fig. 19). In contrast, PMDTA forms polymeric chains with BnNa, BnK and BnRb.13,151
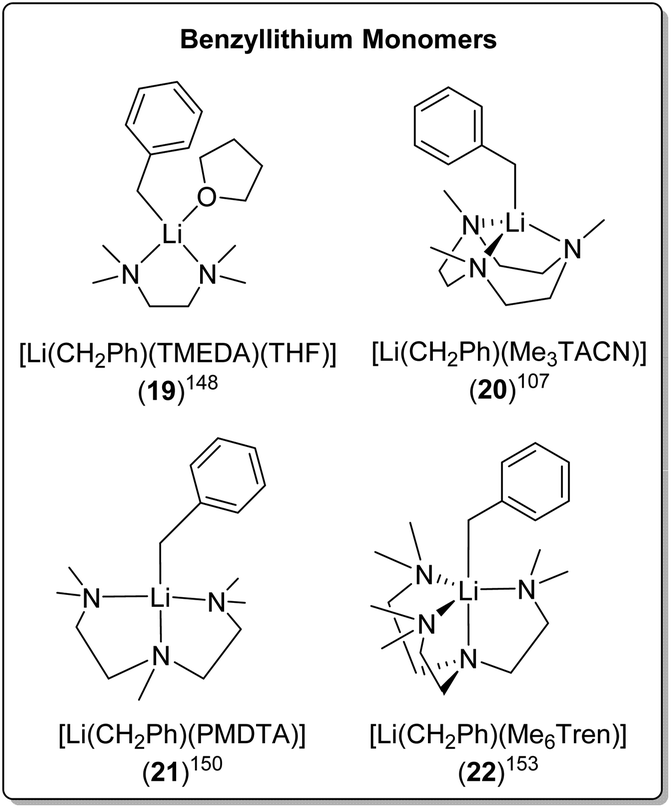 | ||
| Fig. 19 SCXRD-characterised BnLi monomers. | ||
The deprotonation of toluene using nBuLi/nBuNa and TMEDA produced the mixed lithium–sodium benzyl complex [(PhCH2)4Li2−xNa2+x(TMEDA)4], which was found to possess an octagonal ring structure.152 The structure is similar to that of [Na(CH2Ph)(TMEDA)]4.149 Both metal cations are in the same coordination environment and are interchangeable in the bulk crystal.152
Davidson, Mulvey, Robertson and co-workers reported a series of monomeric benzyl-lithium, -sodium and -potassium complexes, utilising the tetratentate Me6Tren ligand (22 for Li, 23 for Na & 24 for K, see Fig. 19 & 20).153 They were the first examples of crystallographically characterised monomeric ligand-supported benzylsodium and benzylpotassium compounds. In all three cases of 22–24, η4 coordination of the Me6Tren ligand to the metal centre is observed in the SCXRD structures.
The series of [M(CH2Ph)(Me6Tren)] {M: Li (22), Na (23), K (24)} provide a unique platform to establish metal identity–structure relationship. As elucidated in Fig. 20, the lithium cation in 22 is exclusively coordinated via σ-bonds to the CH2 moiety (Cα) of the benzyl anion. In 23, the sodium cation exhibits an interaction with both the Cα and Cipso carbons of the benzyl group, with the sodium cation moving towards the phenyl ring. In contrast, in 24, the potassium cation is exclusively coordinated to the benzyl group through the delocalised π-system of the phenyl ring, resulting in a planar naked CH2, with no interaction to the potassium cation. NMR analysis of the complexes suggested that the solid-state structures persist in solution-state.153 A similar trend was observed with alkali metal 3,5-dimethylbenzy complexes,154 and alkali metal diaryl methyl and fluorenyl complexes.14
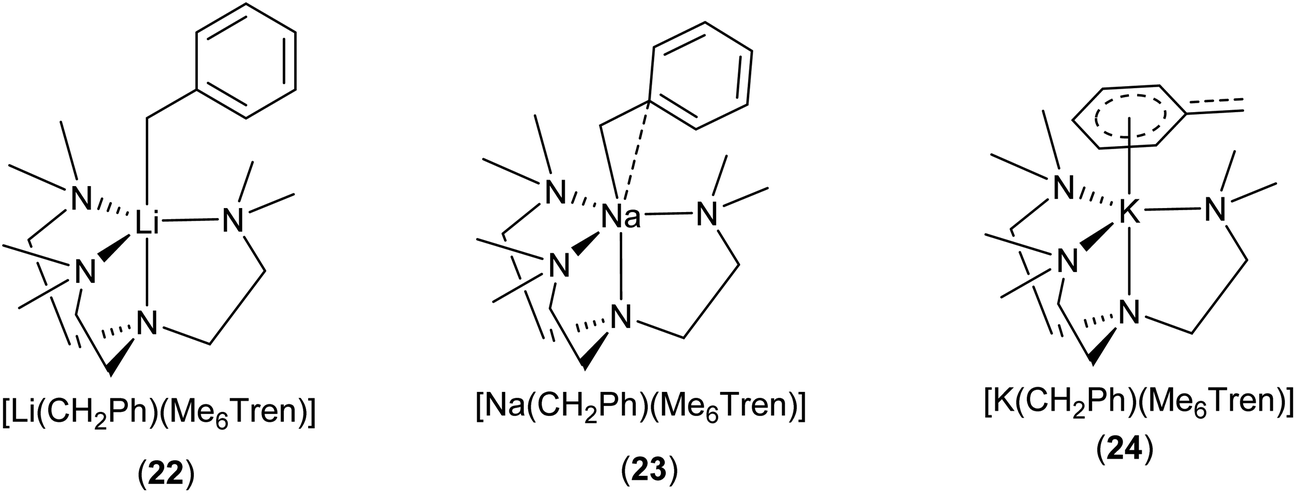 | ||
| Fig. 20 The Me6Tren coordinated benzyl-lithium/-sodium/-potassium monomers 22–24 and their different coordination modes with benzyl.153 | ||
Both the THF and Me6Tren complexes show a clear trend of the preference of the more polarisable, larger and softer alkali metals to coordinate to the aromatic π-system of the benzyl group, whereas the more polarising, smaller and harder alkali metals prefer coordination at the anionic CH2− site.14,145,153,154 The change in coordination environment can be explained by the stronger charge delocalisation on the benzyl anion with descending Group 1 metals.
4. Conclusions and outlook
Is the quest for organo-alkali metal monomers complete? Much has been learned over the past two decades in particular, but many knowledge gaps remain.While there are numerous reports of different structures, there are much fewer reports of comprehensive reactivity studies of monomeric organo-alkali metal complexes. It is extremely desirable for us to have an understanding of how reactivity is influenced by varying factors such as the aggregation size, alkali metal, ligand and the alkyl group. Importantly, we also need to understand why we may observe different reactivity in order to improve our understanding of the structure–reactivity relationship, which would be greatly beneficial in reaction design and help us unlock novel reactivity. A comprehensive understanding of both the solution and solid-state structure, together with computational calculations, are crucial to help us understand this.
One primary motivation for pursuing the monomers, is enhancing the reactivity, as the perception is “the smaller, the better”. However, it is quite clear now that a monomer is not necessarily more reactive than its higher aggregates. This is also true from the other way round: a higher aggregate is not necessarily less reactive than its lower congeners. A clear and generally applicable aggregate size–reactivity relationship is still out-of-reach at the moment. Actually, given the vast structural versatility of organo-alkali metal complexes, and the very delicate nature of their chemistry, it is likely that such a general aggregate size–reactivity relationship would be very little more than oversimplified “folklore”. A case-by-case approach, taking even seemingly trivial details into account, may be more appropriate. A potential universal approach to estimate reactivity and gauge aggregate size–reactivity relationship is DFT calculations, to probe the steric shielding around the reactive M–C bond. This approach was firstly implemented by the Strohmann group for their TMCDA-coordinated organolithium monomers.60,83
Nevertheless, the reactions do not necessarily involve both the metal cation and the carbon anion, i.e., the M–C bond does not necessarily act as the reactive centre. If a multidentate ligand saturates the coordination sphere of the metal cation, decreasing the Lewis acidity of the metal, should the reactivity then be dominated by the carbanion? Yet, there are a number of examples of poorly reactive monomers where the carbon anion is still fairly exposed, but with a shielded metal centre. In traditional contact ion pairs (CIP), the strong interaction between the metal and carbanion means the metal plays a greater role in reactivity, whereas in separated ion pairs (SIP), reactivity is dominated by the carbanion.142 The recent work from the Thomas group, especially the comparison between their TIP and SIP (see the end of Section 3.4), is very intriguing in this regard.142
Another open question is the definition of “reactivity”. What are we talking about when we are saying “more/less reactive”? In almost all the cases to date, “more reactive” means faster and easier deprotonation, usually on the methyl of toluene, and in less common cases, deprotonation of benzene or ligands. The reactivity landscape is quite homogeneous in this regard. It would be very tempting to be able to tune different reaction patterns for different aggregate sizes, e.g., monomer conducts nucleophilic addition, polymer conducts deprotonation, and so on. This new concept will unlock brand new chemical space. Very recent, we reported the first clear-cut case of such aggregate size-dependent reaction patterns. A NaCH2SiMe3 monomer [Na(CH2SiMe3)(Me6Tren)] (10) conducted CO bond methylenation, while [NaCH2SiMe3]∞ polymer underwent nucleophilic addition towards the CO bond.8
Exploring different metal identities would potentially unlock another totally unexplored chemical space. Very recent works from the Hevia group84 and us,8 started challenging the paradigm that all Group-1 metal alkyl complexes following the same reaction patterns. We proved that, by changing metal identity (e.g., from Li to Na), they can be tuned to exhibit totally different reaction patterns.8
One motivation of employing the ligands in organo-alkali metal chemistry is increasing their solubility. Some polymeric complexes, such as [NaCH2SiMe3]∞, are poorly soluble hence difficult to use. Ligand-coordinated lower aggregates are generally more soluble than their polymeric parent complexes. Nevertheless, the other potential way to circumvent the solubility issue is “removing the solvent altogether”, i.e., solvent-free solid-state synthesis. Since 2022, a few groups, including us, started introducing mechanochemical ball milling155–157 into alkali metal chemistry. The early effort has already led to exciting discoveries, such as new room-temperature stable alkali metal electride158 and solvent-free Birch reductions.158,159
The more pieces of the ‘jigsaw’ we have, the better our understanding and to be comprehensive both the solid- and solution-state structure (aggregation state and dynamic behaviour), computational studies and reactivity studies are desired. All the factors that affect the structure and reactivity, highlight the complexity and beauty of organo-alkali metal chemistry. It is clear the degree of aggregation can have a dramatic impact on reactivity, but there are still many mysteries and exciting discoveries awaiting when it comes to alkali metal mediated reaction mechanisms and the structure–reactivity relationship of Group 1 metal organometallics.
Author contributions
E. L. and N. D. conceptualised the central idea of the Perspective. N. D. wrote the manuscript under the supervision of E. L.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Newcastle University Chemistry Technical Support Team (Dr Laura McCorkindale, Dr Amy Roberts and Mr Niall Straughan) for supporting our research. E. L. thanks the Newcastle University Academic Track (NUAcT) Fellowship Scheme for financial support. N. D. thanks Newcastle University for a NUAcT PhD studentship and the Royal Society of Chemistry Research Enablement Grants (E22-3348740748).References
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194–204, DOI:10.1002/zaac.201700394
.
-
F. Totter and P. Rittmeyer, Organolithium Compounds – Industrial Applications and Handling, in Organometallics in Synthesis, A Manual, ed. M. Schlosser, Wiley, New York, 1994, ch. 2, pp. 167–194 Search PubMed
- V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204–14210, 10.1039/C4DT01004C
- H. J. Reich, Chem. Rev., 2013, 113, 7130–7178, DOI:10.1021/cr400187u
- W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1917, 50, 262–274, DOI:10.1002/cber.19170500142
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320–3334, DOI:10.1002/chem.200900041
- A. Münch, L. Knauer, H. Ott, C. Sindlinger, R. Herbst-Irmer, C. Strohmann and D. Stalke, J. Am. Chem. Soc., 2020, 142, 15897–15906, DOI:10.1021/jacs.0c06035
- N. Davison, C. L. McMullin, L. Zhang, S.-X. Hu, P. G. Waddell, C. Wills, C. Dixon and E. Lu, J. Am. Chem. Soc., 2023, 145, 6562–6576, DOI:10.1021/jacs.3c01033
-
M. J. Harvey, Alkali Metals: Organometallic Chemistry, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2014, pp. 1–13. DOI:10.1002/9781119951438.eibc0004.pub2
-
J. David Smith, Organometallic Compounds of the Heavier Alkali Metals, in Advances in Organometallic Chemistry, ed. R. West and A. F. Hill, Academic Press, 1999, vol. 43, pp. 267–348 Search PubMed
-
R. J. Ouellette and J. D. Rawn, in Organic Chemistry, ed. R. J. Ouellette and J. D. Rawn, Academic Press, 2nd edn, 2018, pp. 51–86. DOI:10.1016/B978-0-12-812838-1.50003-7
- C. Strohmann, V. H. Gessner and A. Damme, Chem. Commun., 2008, 3381–3383, 10.1039/B804855J
- S. Corbelin, N. P. Lorenzen, J. Kopf and E. Weiss, J. Organomet. Chem., 1991, 415, 293–313, DOI:10.1016/0022-328X(91)80130-C
- A. Rae, K. M. Byrne, S. A. Brown, A. R. Kennedy, T. Krämer, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2022, 28, e202104260, DOI:10.1002/chem.202104260
- F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015, DOI:10.1021/om0006209
- J. J. Brooks and G. D. Stucky, J. Am. Chem. Soc., 1972, 94, 7333–7338, DOI:10.1021/ja00776a012
- U. Schümann, J. Kopf and E. Weiss, Angew. Chem., Int. Ed. Engl., 1985, 24, 215–216, DOI:10.1002/anie.198502151
- T. Maetzke and D. Seebach, Helv. Chim. Acta, 1989, 72, 624–630, DOI:10.1002/hlca.19890720321
- G. S. Girolami, M. E. Riehl, K. S. Suslick and S. R. Wilson, Organometallics, 1992, 11, 3907–3910, DOI:10.1021/om00059a067
- R. C. Crittendon, B. C. Beck, J. Su, X.-W. Li and G. H. Robinson, Organometallics, 1999, 18, 156–160, DOI:10.1021/om980753i
- N. J. Hardmann, B. Twamley, M. Stender, R. Baldwin, S. Hino, B. Schiemenz, S. M. Kauzlarich and P. P. Power, J. Organomet. Chem., 2002, 643–644, 461–467, DOI:10.1016/S0022-328X(01)01443-7
- H. H. Karsch, K. Zellner, P. Mikulcik, J. Lachmann and G. Müller, Organometallics, 1990, 9, 190–194, DOI:10.1021/om00115a029
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364, 10.1039/C5SC00670H
- S. Bachmann, B. Gernert and D. Stalke, Chem. Commun., 2016, 52, 12861–12864, 10.1039/C6CC07273A
- I. Keresztes and P. G. Williard, J. Am. Chem. Soc., 2000, 122, 10228–10229, DOI:10.1021/ja002278x
- M. Granitzka, A.-C. Pöppler, E. K. Schwarze, D. Stern, T. Schulz, M. John, R. Herbst-Irmer, S. K. Pandey and D. Stalke, J. Am. Chem. Soc., 2012, 134, 1344–1351, DOI:10.1021/ja210382c
- A.-C. Pöppler, M. M. Meinholz, H. Faßhuber, A. Lange, M. John and D. Stalke, Organometallics, 2012, 31, 42–45, DOI:10.1021/om2010639
- L. D. McKeever, R. Waack, M. A. Doran and E. B. Baker, J. Am. Chem. Soc., 1968, 90, 3244–3244, DOI:10.1021/ja01014a051
- G. Fraenkel, A. M. Fraenkel, M. J. Geckle and F. Schloss, J. Am. Chem. Soc., 1979, 101, 4745–4747, DOI:10.1021/ja00510a060
- D. Seebach, R. Hässig and J. Gabriel, Helv. Chim. Acta, 1983, 66, 308–337, DOI:10.1002/hlca.19830660128
- H. J. Reich, D. P. Green, M. A. Medina, W. S. Goldenberg, B. Ö. Gudmundsson, R. R. Dykstra and N. H. Phillips, J. Am. Chem. Soc., 1998, 120, 7201–7210, DOI:10.1021/ja980684z
- R. D. Thomas, R. M. Jensen and T. C. Young, Organometallics, 1987, 6, 565–571, DOI:10.1021/om00146a022
- A.-C. Pöppler, H. Keil, D. Stalke and M. John, Angew. Chem., Int. Ed., 2012, 51, 7843–7846, DOI:10.1002/anie.201202116
- F. Rüttger, T. Patten, J. Kretsch, A. Krawczuk, D. Stalke and M. John, Chem. – Eur. J., 2023, 29, e202203995, DOI:10.1002/chem.202203995
- H. L. Lewis and T. L. Brown, J. Am. Chem. Soc., 1970, 92, 4664–4670, DOI:10.1021/ja00718a032
- W. Bauer and D. Seebach, Helv. Chim. Acta, 1984, 67, 1972–1988, DOI:10.1002/hlca.19840670736
-
S. O. Nilsson Lill, Computational Perspectives on Organolithiums, in Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley, 2014, ch. 2, pp. 33–52 Search PubMed
- J. A. Wanklyn, Proc. R. Soc. London, 1858, 9, 341–345, DOI:10.1098/rspl.1857.0084
- D. Seyferth, Organometallics, 2006, 25, 2–24, DOI:10.1021/om058054a
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262, DOI:10.1002/anie.202010963
- S. Asako, H. Nakajima and K. Takai, Nat. Catal., 2019, 2, 297–303, DOI:10.1038/s41929-019-0250-6
- H. N. C. Wong, Nat. Catal., 2019, 2, 282–283, DOI:10.1038/s41929-019-0259-x
- S. Asako, I. Takahashi, H. Nakajima, L. Ilies and K. Takai, Commun. Chem., 2021, 4, 76, DOI:10.1038/s42004-021-00513-2
- S. G. Davey, Nat. Rev. Chem., 2021, 5, 368, DOI:10.1038/s41570-021-00294-1
- J. H. Harenberg, R. R. Annapureddy, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2022, 61, e202203807, DOI:10.1002/anie.202203807
- A. Tortajada, D. E. Anderson and E. Hevia, Helv. Chim. Acta, 2022, 105, e202200060, DOI:10.1002/hlca.202200060
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2022, 62, e202218498, DOI:10.1002/anie.202218498
- N. Davison, P. G. Waddell, C. Dixon, C. Wills, T. J. Penfold and E. Lu, Dalton Trans., 2022, 51, 10707–10713, 10.1039/D1DT03532K
- V. H. Gessner and C. Strohmann, J. Am. Chem. Soc., 2008, 130, 14412–14413, DOI:10.1021/ja8058205
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 4566–4569, DOI:10.1002/anie.200605105
-
V. Bhatt, Essentials of Coordination Chemistry, Academic Press, 2016, ch. 7, pp. 173–190 Search PubMed
- E. Weiss and E. A. C. Lucken, J. Organomet. Chem., 1964, 2, 197, DOI:10.1016/S0022-328X(00)80512-4
- C. A. Ogle, B. K. Huckabee, H. C. I. V. Johnson, P. F. Sims, S. D. Winslow and A. A. Pinkerton, Organometallics, 1993, 12, 1960–1963, DOI:10.1021/om00029a061
- L. D. McKeever, R. Waack, M. A. Doran and E. B. Baker, J. Am. Chem. Soc., 1969, 91, 1057–1061, DOI:10.1021/ja01033a003
- T. Fox, H. Hausmann and H. Günther, Magn. Reson. Chem., 2004, 42, 788–794, DOI:10.1002/mrc.1443
- P. West and R. Waack, J. Am. Chem. Soc., 1967, 89, 4395–4399, DOI:10.1021/ja00993a025
- L. Andrews, J. Chem. Phys., 1967, 47, 4834–4842, DOI:10.1063/1.1701708
- D. B. Grotjahn, T. C. Pesch, J. Xin and L. M. Ziurys, J. Am. Chem. Soc., 1997, 119, 12368–12369, DOI:10.1021/ja972613q
- H. Köster, D. Thoennes and E. Weiss, J. Organomet. Chem., 1978, 160, 1–5, DOI:10.1016/S0022-328X(00)91191-4
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2007, 129, 8592–8593, DOI:10.1021/ja072970t
- L. Knauer and C. Strohmann, Chem. Commun., 2020, 56, 13543–13546, 10.1039/D0CC05547F
- N. Davison, E. Falbo, P. G. Waddell, T. J. Penfold and E. Lu, Chem. Commun., 2021, 57, 6205–6208, 10.1039/D1CC01420J
- E. Weiss, Angew. Chem., Int. Ed. Engl., 1993, 32, 1501–1523, DOI:10.1002/anie.199315013
- J. Lebon, A. Mortis, C. Maichle-Mössmer, M. Manßen, P. Sirsch and R. Anwander, Angew. Chem., Int. Ed., 2022, 62, e202214599, DOI:10.1002/anie.202214599
- B. Walfort, L. Lameyer, W. Weiss, R. Herbst-Irmer, R. Bertermann, J. Rocha and D. Stalke, Chem. – Eur. J., 2001, 7, 1417–1423, DOI:10.1002/1521-3765(20010401)7:7<1417::AID-CHEM1417>3.0.CO;2-Q
- M. Pérez-Jiménez, J. Campos, J. Jover, S. Álvarez and E. Carmona, Angew. Chem., Int. Ed., 2022, 61, e202116009, DOI:10.1002/anie.202116009
- E. Weiss, S. Corbelin, J. K. Cockcroft and A. N. Fitch, Angew. Chem., Int. Ed. Engl., 1990, 29, 650–652, DOI:10.1002/anie.199006501
- E. Weiss and G. Sauermann, Angew. Chem., Int. Ed. Engl., 1968, 7, 133–134, DOI:10.1002/anie.196801331
- E. Weiss, T. Lambertsen, B. Schubert and J. K. Cockcroft, J. Organomet. Chem., 1988, 358, 1–14, DOI:10.1016/0022-328X(88)87066-9
- E. Weiss and H. Köster, Chem. Ber., 1977, 110, 717–720, DOI:10.1002/cber.19771100234
- E. Weiss and G. Hencken, J. Organomet. Chem., 1970, 21, 265–268, DOI:10.1016/S0022-328X(00)83621-9
- E. Weiss, T. Lambertsen, B. Schubert, J. K. Cockeroft and A. Wiedenmann, Chem. Ber., 1990, 123, 79–81, DOI:10.1002/cber.19901230116
- E. Weiss, S. Corbelin, J. K. Cockcroft and A. N. Fitch, Chem. Ber., 1990, 123, 1629–1634, DOI:10.1002/cber.19901230807
- F. C. Whitmore and L. H. Sommer, J. Am. Chem. Soc., 1946, 68, 481–484, DOI:10.1021/ja01207a036
- L. H. Sommer and F. C. Whitmore, J. Am. Chem. Soc., 1946, 68, 485–487, DOI:10.1021/ja01207a037
- B. Tecle, A. F. M. Maqsudur Rahman and J. P. Oliver, J. Organomet. Chem., 1986, 317, 267–275, DOI:10.1016/0022-328X(86)80537-X
- A. E. Sedykh, R. Bissert, D. G. Kurth and K. Müller-Buschbaum, Z. Kristallogr. – Cryst. Mater., 2020, 235, 353, DOI:10.1515/zkri-2020-0053
- J. O. Bauer, Z. Kristallogr. – New Cryst. Struct., 2020, 235, 353, DOI:10.1515/ncrs-2019-0662
- T. Tatic, K. Meindl, J. Henn, S. K. Pandey and D. Stalke, Chem. Commun., 2010, 46, 4562–4564, 10.1039/c002504f
- J. O. Bauer and C. Strohmann, J. Am. Chem. Soc., 2015, 137, 4304–4307, DOI:10.1021/jacs.5b00861
- T. Tatic, H. Ott and D. Stalke, Eur. J. Inorg. Chem., 2008, 3765–3768, DOI:10.1002/ejic.200800610
- T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 6666–6669, DOI:10.1002/anie.201102068
- L. Knauer, J. Wattenberg, U. Kroesen and C. Strohmann, Dalton Trans., 2019, 48, 11285–11291, 10.1039/C9DT02182E
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498, DOI:10.1002/anie.202218498
- D. B. Collum, Acc. Chem. Res., 1992, 25, 448–454, DOI:10.1021/ar00022a003
- T. Tatić, S. Hermann and D. Stalke, Organometallics, 2012, 31, 5615–5621, DOI:10.1021/om3005806
- S. E. Baillie, W. Clegg, P. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and L. Russo, Chem. Commun., 2011, 47, 388–390, 10.1039/C0CC02164D
- W. Clegg, B. Conway, A. R. Kennedy, J. Klett, R. E. Mulvey and L. Russo, Eur. J. Inorg. Chem., 2011, 721–726, DOI:10.1002/ejic.201000983
- J. L. Atwood, T. Fjeldberg, M. F. Lappert, N. T. Luong-Thi, R. Shakir and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1984, 1163–1165, 10.1039/C39840001163
- P. B. Hitchcock, M. F. Lappert, W.-P. Leung, L. Diansheng and T. Shun, J. Chem. Soc., Chem. Commun., 1993, 1386–1387, 10.1039/C39930001386
- M. Von Pilgrim, M. Mondeshki and J. Klett, Inorganics, 2017, 5, 39, DOI:10.3390/inorganics5020039
- M. F. Lappert, L. M. Engelhardt, C. L. Raston and A. H. White, J. Chem. Soc., Chem. Commun., 1982, 1323–1324, 10.1039/C39820001323
- P. B. Hitchcock, A. V. Khvostov and M. F. Lappert, J. Organomet. Chem., 2002, 663, 263–268, DOI:10.1016/S0022-328X(02)01766-7
- W. M. Boesveld, P. B. Hitchcock, M. F. Lappert, D.-S. Liu and S. Tian, Organometallics, 2000, 19, 4030–4035, DOI:10.1021/om0004287
- W. Hiller, M. Layh and W. Uhl, Angew. Chem., Int. Ed. Engl., 1991, 30, 324–326, DOI:10.1002/anie.199103241
- C. Eaborn, P. B. Hitchcock, K. Izod, A. J. Jaggar and J. D. Smith, Organometallics, 1994, 13, 753–754, DOI:10.1021/om00015a004
- C. Eaborn, P. B. Hitchcock, K. Izod and J. D. Smith, Angew. Chem., Int. Ed. Engl., 1995, 34, 687–688, DOI:10.1002/anie.199506871
- C. Eaborn, W. Clegg, P. B. Hitchcock, M. Hopman, K. Izod, P. N. O'Shaughnessy and J. D. Smith, Organometallics, 1997, 16, 4728–4736, DOI:10.1021/om970469b
- C. Eaborn, P. B. Hitchcock, J. D. Smith and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1983, 827–828, 10.1039/C39830000827
- T. Viefhaus, A. Walz, M. Niemeyer, W. Schwarz and J. Weidlein, Z. Anorg. Allg. Chem., 2000, 626, 2040–2042, DOI:10.1002/1521-3749(200010)626:10<2040::AID-ZAAC2040>3.0.CO;2-M
- A. G. Avent, C. Eaborn, P. B. Hitchcock, G. A. Lawless, P. D. Lickiss, M. Mallien, J. D. Smith, A. D. Webb and B. Wrackmeyer, J. Chem. Soc., Dalton Trans., 1993, 3259–3264, 10.1039/DT9930003259
- S. S. Al-Juaid, C. Eaborn, P. B. Hitchcock, K. Izod, M. Mallien and J. D. Smith, Angew. Chem., Int. Ed. Engl., 1994, 33, 1268–1270, DOI:10.1002/anie.199412681
- N. H. Buttrus, C. Eaborn, P. B. Hitchcock, J. D. Smith, J. G. Stamper and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1986, 969–970, 10.1039/C39860000969
- H. J. Reich, W. H. Sikorski, J. L. Thompson, A. W. Sanders and A. C. Jones, Org. Lett., 2006, 8, 4003–4006, DOI:10.1021/ol061489p
- A. C. Jones, A. W. Sanders, W. H. Sikorski, K. L. Jansen and H. J. Reich, J. Am. Chem. Soc., 2008, 130, 6060–6061, DOI:10.1021/ja8003528
- H. J. Reich, W. H. Sikorski, A. W. Sanders, A. C. Jones and K. N. Plessel, J. Org. Chem., 2009, 74, 719–729, DOI:10.1021/jo802032d
- J. Arnold, V. Knapp, J. A. R. Schmidt and A. Shafir, J. Chem. Soc., Dalton Trans., 2002, 3273–3274, 10.1039/B205792C
- D. Bojer, I. Kamps, X. Tian, A. Hepp, T. Pape, R. Fröhlich and N. W. Mitzel, Angew. Chem., Int. Ed., 2007, 46, 4176–4179, DOI:10.1002/anie.200700113
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281–8283, DOI:10.1002/anie.200702116
- T. Kottke and D. Stalke, Angew. Chem., Int. Ed. Engl., 1993, 32, 580–582, DOI:10.1002/anie.199305801
- O. Tai, R. Hopson and P. G. Williard, Org. Lett., 2017, 19, 3966–3969, DOI:10.1021/acs.orglett.7b01644
- J. F. McGarrity and C. A. Ogle, J. Am. Chem. Soc., 1985, 107, 1805–1810, DOI:10.1021/ja00293a001
- A. C. Jones, A. W. Sanders, M. J. Bevan and H. J. Reich, J. Am. Chem. Soc., 2007, 129, 3492–3493, DOI:10.1021/ja0689334
- H. J. Reich, J. Org. Chem., 2012, 77, 5471–5491, DOI:10.1021/jo3005155
- C. D. Broaddus, J. Org. Chem., 1970, 35, 10–15, DOI:10.1021/jo00826a003
- F. Rebiere, O. Samuel and H. B. Kagan, Tetrahedron Lett., 1990, 31, 3121–3124, DOI:10.1016/S0040-4039(00)94710-5
- R. Sanders and U. T. Mueller-Westerhoff, J. Organomet. Chem., 1996, 512, 219–224, DOI:10.1016/0022-328X(95)05914-B
- M. D. Rausch, G. A. Moser and C. F. Meade, J. Organomet. Chem., 1973, 51, 1–11, DOI:10.1016/S0022-328X(00)93496-X
- I. R. Butler, W. R. Cullen, J. Ni and S. J. Rettig, Organometallics, 1985, 4, 2196–2201, DOI:10.1021/om00131a023
- D. Waldmüller, B. J. Kotsatos, M. A. Nichols and P. G. Williard, J. Am. Chem. Soc., 1997, 119, 5479–5480, DOI:10.1021/ja970557n
- J. L. C. Rutherford, D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 2002, 124, 264–271, DOI:10.1021/ja002979u
- W. Bauer and P. v. R. Schleyer, J. Am. Chem. Soc., 1989, 111, 7191–7198, DOI:10.1021/ja00200a044
- D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 1998, 120, 5810–5811, DOI:10.1021/ja971512p
- M. A. Nichols and P. G. Williard, J. Am. Chem. Soc., 1993, 115, 1568–1572, DOI:10.1021/ja00057a050
- N. D. R. Barnett, R. E. Mulvey, W. Clegg and P. A. O'Neil, J. Am. Chem. Soc., 1993, 115, 1573–1574, DOI:10.1021/ja00057a051
- C. Strohmann, K. Strohfeldt and D. Schildbach, J. Am. Chem. Soc., 2003, 125, 13672–13673, DOI:10.1021/ja0374372
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2008, 130, 11719–11725, DOI:10.1021/ja8017187
- M. A. Nichols, R. M. Sobinsky, A. D. Hunter and M. Zeller, J. Chem. Crystallogr., 2007, 37, 433–438, DOI:10.1007/s10870-007-9188-7
- G. Fraenkel, M. Henrichs, M. Hewitt and B. M. Su, J. Am. Chem. Soc., 1984, 106, 255–256, DOI:10.1021/ja00313a052
- W. Bauer, W. R. Winchester and P. v. R. Schleyer, Organometallics, 1987, 6, 2371–2379, DOI:10.1021/om00154a017
- R. D. Thomas, M. T. Clarke, R. M. Jensen and T. C. Young, Organometallics, 1986, 5, 1851–1857, DOI:10.1021/om00140a016
- C. Strohmann, T. Seibel and K. Strohfeldt, Angew. Chem., Int. Ed., 2003, 42, 4531–4533, DOI:10.1002/anie.200351308
- M. Hülsmann, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2014, 46–50, DOI:10.1002/ejic.201301267
-
V. H. Gessner, Ideas in Chemistry and Molecular Sciences, 2010, ch. 5, pp. 95–113. DOI:10.1002/9783527630554
- D. Bojer, I. Kamps, X. Tian, A. Hepp, T. Pape, R. Fröhlich and N. W. Mitzel, Angew. Chem., Int. Ed., 2007, 46, 4176–4179, DOI:10.1002/anie.200700113
- C. Strohmann and V. H. Gessner, Chem. – Asian J., 2008, 3, 1929–1934, DOI:10.1002/asia.200800213
- C. B. Bi Ekogha, O. Ruel and S. A. Julia, Tetrahedron Lett., 1983, 24, 4825–4828, DOI:10.1016/S0040-4039(00)94017-6
- P. Beak and A. I. Meyers, Acc. Chem. Res., 1986, 19, 356–363, DOI:10.1021/ar00131a005
- M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206–2225, DOI:10.1002/anie.200300590
- G. W. Klumpp, Recl. Trav. Chim. Pays-Bas, 1986, 105, 1–21, DOI:10.1002/recl.19861050102
- J. Clayden and S. A. Yasin, New J. Chem., 2002, 26, 191–192, 10.1039/B109604D
- M. P. Crockett, J. Piña, A. R. Gogoi, R. F. Lalisse, A. V. Nguyen, O. Gutierrez and A. A. Thomas, J. Am. Chem. Soc., 2023, 145, 10743–10755, DOI:10.1021/jacs.2c13047
- M. Hage, C. A. Ogle, T. L. Rathman and J. L. Hubbard, Main Group Met. Chem., 1998, 21, 777–782, DOI:10.1515/MGMC.1998.21.12.777
- M. A. Beno, H. Hope, M. M. Olmstead and P. P. Power, Organometallics, 1985, 4, 2117–2121, DOI:10.1021/om00131a009
- L. Brieger, C. Unkelbach and C. Strohmann, Chem. – Eur. J., 2021, 27, 17780–17784, DOI:10.1002/chem.202103430
- C. Unkelbach, D. F. O'Shea and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 553–556, DOI:10.1002/anie.201306884
- M. Westerhausen and W. Schwarz, Z. Naturforsch., B: J. Chem. Sci., 1998, 53, 625–627, DOI:10.1515/znb-1998-5-620
- W. Zarges, M. Marsch, K. Harms and G. Boche, Chem. Ber., 1989, 122, 2303–2309, DOI:10.1002/cber.19891221217
- C. Schade, P. v. R. Schleyer, H. Dietrich and W. Mahdi, J. Am. Chem. Soc., 1986, 108, 2484–2485, DOI:10.1021/ja00269a078
- T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 6666–6669, DOI:10.1002/anie.201102068
- D. Hoffmann, W. Bauer, F. Hampel, N. J. R. van Eikema Hommes, P. v. R. Schleyer, P. Otto, U. Pieper, D. Stalke, D. S. Wright and R. Snaith, J. Am. Chem. Soc., 1994, 116, 528–536, DOI:10.1021/ja00081a013
- D. R. Baker, W. Clegg, L. Horsburgh and R. E. Mulvey, Organometallics, 1994, 13, 4170–4172, DOI:10.1021/om00023a017
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364–3369, DOI:10.1002/chem.201003493
- D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Inorg. Chem., 2013, 52, 12023–12032, DOI:10.1021/ic401777x
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167, DOI:10.1038/s41570-020-00249-y
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19, DOI:10.1021/acscentsci.6b00277
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094, 10.1039/C7SC05371A
- N. Davison, J. A. Quirk, F. Tuna, D. Collison, C. L. McMullin, H. Michaels, G. H. Morritt, P. G. Waddell, J. A. Gould, M. Freitag, J. A. Dawson and E. Lu, Chem, 2023, 9, 576–591, DOI:10.1016/j.chempr.2022.11.006
- Y. Gao, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2023, 62, e202217723, DOI:10.1002/anie.202217723
| This journal is © The Royal Society of Chemistry 2023 |