
Suppressing the energetic disorder of all-polymer solar cells enables over 18% efficiency†
Tao
Zhang
ab,
Ye
Xu
af,
Huifeng
Yao
 *a,
Jianqi
Zhang
c,
Pengqing
Bi
a,
Zhihao
Chen
a,
Jingwen
Wang
ab,
Yong
Cui
a,
Lijiao
Ma
a,
Kaihu
Xian
e,
Zi
Li
a,
Xiaotao
Hao
d,
Zhixiang
Wei
c and
Jianhui
Hou
*ab
*a,
Jianqi
Zhang
c,
Pengqing
Bi
a,
Zhihao
Chen
a,
Jingwen
Wang
ab,
Yong
Cui
a,
Lijiao
Ma
a,
Kaihu
Xian
e,
Zi
Li
a,
Xiaotao
Hao
d,
Zhixiang
Wei
c and
Jianhui
Hou
*ab
aState Key Laboratory of Polymer Physics and Chemistry, Beijing National Laboratory for Molecular Sciences, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, 100190 Beijing, P. R. China. E-mail: yaohf@iccas.ac.cn; hjhzlz@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, 100049 Beijing, P. R. China
cCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, 100190 Beijing, China
dSchool of Physics, State Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong 250100, P. R. China
eSchool of Materials Science and Engineering, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300350, P. R. China
fState Key Laboratory of Photovoltaic Science and Technology, Trina Solar, Changzhou, 213031, China
First published on 3rd February 2023
Abstract
All-polymer solar cells (all-PSCs) usually have a complex blend morphology due to their higher probability of chain entanglement. Under these conditions, the larger energetic disorder is one of the most important factors that hinder the increase in power conversion efficiency (PCE). Here, to take full advantage of two highly efficient polymer donors, PBDB-TF and PBQx-TF, we design and synthesize three ternary copolymers, PQB-x (x represents 1–3). The results suggest that the optical absorption spectrum and molecular energy levels can be linearly tuned by adjusting the content of the acceptor component. Interestingly, ternary copolymerization changes the molecular orientation from the coexistence of face-on and edge-on to face-on only and accordingly suppresses the energetic disorder (44, 47, and 53 meV for PQB-2, PBQx-TF, and PBDB-TF). In cell devices based on a polymer acceptor PY-IT, PQB-2 shows decreased energy loss and thus an improved open-circuit voltage of 0.942 V. Transient absorption measurements reveal that the structural component ascribed to PBQx-TF provides an efficient charge transfer channel with PY-IT. As a result, the PQB-2:PY-IT-based device yields a maximum PCE of 18.1%, which is an outstanding value among all-PSCs. This study demonstrates that suppressing the energetic disorder by molecular design may be a feasible method for further improving the PCEs of all-PSCs.
Broader contextOrganic solar cells (OSCs) are one of the most promising photovoltaic technologies to convert light energy into electricity. Over the past few years, the development of new materials has contributed to the rapid growth of power conversion efficiency (PCE). The all-polymer solar cells (all-PSCs) that consist of polymer donors and polymer acceptors have unique advantages of intrinsic flexibility, excellent mechanical properties, and morphology stability. However, for a long time, the PCEs of all-PSCs have been far behind the small-molecule-containing OSCs due to the lack of efficient materials and the huge challenge in morphology control. Here, by designing new terpolymers, we suppress the energetic disorder of all-polymer blends. The donor:acceptor combination shows decreased energy loss unexpectedly and thus improves the open-circuit voltage. As a result, the best device yields a maximum PCE of 18.1% (certified as 17.6%), which is one of the highest values among all-PSCs. |
Introduction
All-polymer solar cells (all-PSCs) have attracted considerable research attention for their intrinsic flexibility, excellent mechanical properties, and good morphology stability.1–4 However, the development of all-PSCs has long lagged far behind that of other types of organic solar cells based on small-molecule acceptors.5–7 One of the most important reasons is the lack of highly efficient polymer acceptors (PAs). In early studies, material design mainly focused on diimide building blocks based on perylene and naphehalene.8–10 However, the resulting PAs usually display severe aggregation and thus limited charge generation. For example, N2200 is a representative diimide PA, which has shown a power conversion efficiency (PCE) of approximately 11% in all-PSCs.11–13 Researchers have also developed PAs based on other electron-deficient building blocks, such as B ← N embedded (hetero)arene and dicyano-substituted benzothiadiazole,12–15 but the corresponding materials failed to show a significant improvement in PCEs. In recent years, polymerized small-molecule PAs have achieved great success as they inherit the excellent properties of highly efficient non-fullerene acceptors (NFAs).16–20 For instance, when this method was applied to Y-series NFAs,21,22 low-bandgap PAs could be designed. The corresponding all-PSCs showed high PCEs around 18% (Table S1, ESI†), substantially narrowing the efficiency gap with the top-performing organic solar cells based on small-molecule NFAs.23 At present, material design for all-PSCs is mainly focusing on PAs, while matched donors are considerably less synergistic. Only a few polymer donors, mainly PBDB-TF and its derivatives, can achieve excellent photovoltaic performance with typical PAs such as PY-IT.24–26 Even more recently, we have reported that the copolymer termed PQM-Cl reached an impressive PCE of approximately 18%, thus highlighting that polymer donors play a key role in improving the photovoltaic performance.23 Therefore, developing new polymer donors possessing matched properties, such as the absorption spectrum, molecular energy levels, and miscibility, with PAs is highly necessary.As organic semiconductor materials are commonly amorphous or poor crystalline, they have a relatively larger energetic disorder than inorganic semiconductors, leading to severe non-radiative recombination and thus high energy loss (Eloss).27–30 In small-molecule systems, suppressing the energetic disorder has been regarded as an effective method for improving the photovoltaic performance.31–33 For example, highly efficient NFAs like Y11 displayed a markedly lower Urbach energy (Eu) than the fullerene acceptor PCBM (25.6 vs 46.5 meV), which greatly accounts for their efficient charge generation at low Eloss.34 For polymer:polymer blends, the chain entanglement is getting more complex, and photovoltaic performance is more dependent on the blend morphology. Under these conditions, energetic disorder effects on charge transport and recombination prevail.11,35,36 For instance, P3HT disentanglement can enhance intra- and inter-chain interactions, thereby markedly improving hole mobility by ∼20 times.37 As such, enhancing the crystallinity of polymer donors without causing oversized aggregation may be a feasible way to suppress the energetic disorder of all-polymer blends. In our previous studies, we designed PBQx-TF(Cl) copolymers consisting of alternating dithieno[3,2-f:2′,3′-h]quinoxaline (DTQx) and halogen-substituted benzo[1,2-b:4,5-b′]dithiophene (BDT-TF or BDT-TCl), which provided highly encouraging PCEs under different light illumination conditions for their appropriate aggregation morphology and thus low energetic disorder.7,38 Relative to PBDB-TF, the highest occupied molecular orbital (HOMO) energy level is upshifted slightly, leading to a little decrease in the open-circuit voltage (Voc) in the corresponding devices. Thus, introducing further structural modifications to PBQx-TF(Cl) could improve PCEs and open up new applications in all-PSCs. Ternary polymerization has been successfully applied in polymer donor:NFA systems to tune the optical absorption, energy level, and intermolecular packing.39 Because of PBDB-TF and PBQx-TF, the resulting terpolymers will have gradually tuned optoelectronic characteristics and provide a good example to study the impacts of energy disorder on the device photovoltaic performance of all-PSCs.
In this study, we design and synthesize three random terpolymers (PQB-x, x = 1, 2, and 3) using BDT-TF as the donor unit and benzo[1,2-c:4,5-c′] dithiophene-4,8-dione (BDD) and DTQx as the acceptor unit. Increasing the BDD content of PQB-x gradually downshifts the molecular energy level and redshifts the absorption spectrum. Unexpectedly, the terpolymer PQB-2 yields the highest voltage in the devices although its HOMO level is located in the middle between the binary polymers (PBDB-TF and PBQx-TF). The morphology characterization suggests that PQB-2 shows a preferential face-on orientation in the film, while the binary polymers have higher percentages of the edge-on orientation. Relative to the PBQx-TF:PY-IT and PBDB-TF:PY-IT blends (42 and 45 meV), the PQB-2:PY-IT blend displays a suppressed hole disorder of 36 meV. The transient absorption measurements suggest that the DTQx unit in PQB-2 provides a fast and efficient hole transfer channel with PY-IT. These features lead to a considerably decreased non-radiative Eloss of 0.179 eV. As a result, the PQB-2:PY-IT-based device yields a maximum PCE of 18.1% (certified as 17.6%), which is one of the top values among all-PSCs. We anticipate that more highly efficient donors or acceptors can be developed by suppressing the energetic disorder and the PCEs of all-PSCs can be further improved.
Results and discussion
The chemical structures of PBDB-TF, PBQx-TF, and PQB-x are shown in Fig. 1a. In the terpolymers, the BDD:DTQx ratio is tuned to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 (PQB-1), 5:5 (PQB-2), and 8:2 (PQB-3). We performed the molecular simulation using density functional theory (DFT) at the B3LYP/6-31G (d, p) level to study the geometries and frontier orbitals of the simplified dimers. Fig. 1b displays the optimized molecular geometry of PQB-2, where we can find that the dihedral angle (11.9°) between BDT and BDD is much narrower than that between BDT and DTQx (20.9°). This feature may affect the intermolecular packing in the films. In Fig. 1c, we plotted the total density of states for the PQB-2 dimer and separated the individual contributions of the BDT-BDD and BDT-DTQx fragments. The two fragments have a similar impact on the HOMO, while the BDT-BDD moiety has a more pronounced effect on the lowest unoccupied molecular orbital (LUMO) than the BDT-DTQx unit. For the dimers, the energy levels and wave function distributions of the HOMO and the LUMO are illustrated in Fig. 1d. When compared to PBQx-TF, the terpolymer PQB-2 has down-shifted HOMO/LUMO levels with a decreased bandgap. All polymers were prepared via the Stille coupling reaction (Scheme S1, ESI†), and the detailed procedures are provided in the ESI.† The molecular weights were measured by gel permeation chromatography. For PBQx-TF, PQB-1, PQB-2, PQB-3, and PBDB-TF, the number-average molecular weights are 47, 41, 50, 47, and 56 KDa, and the polydispersity indices are 2.7, 2.8, 2.5, 3.7, and 3.5.
:8 (PQB-1), 5:5 (PQB-2), and 8:2 (PQB-3). We performed the molecular simulation using density functional theory (DFT) at the B3LYP/6-31G (d, p) level to study the geometries and frontier orbitals of the simplified dimers. Fig. 1b displays the optimized molecular geometry of PQB-2, where we can find that the dihedral angle (11.9°) between BDT and BDD is much narrower than that between BDT and DTQx (20.9°). This feature may affect the intermolecular packing in the films. In Fig. 1c, we plotted the total density of states for the PQB-2 dimer and separated the individual contributions of the BDT-BDD and BDT-DTQx fragments. The two fragments have a similar impact on the HOMO, while the BDT-BDD moiety has a more pronounced effect on the lowest unoccupied molecular orbital (LUMO) than the BDT-DTQx unit. For the dimers, the energy levels and wave function distributions of the HOMO and the LUMO are illustrated in Fig. 1d. When compared to PBQx-TF, the terpolymer PQB-2 has down-shifted HOMO/LUMO levels with a decreased bandgap. All polymers were prepared via the Stille coupling reaction (Scheme S1, ESI†), and the detailed procedures are provided in the ESI.† The molecular weights were measured by gel permeation chromatography. For PBQx-TF, PQB-1, PQB-2, PQB-3, and PBDB-TF, the number-average molecular weights are 47, 41, 50, 47, and 56 KDa, and the polydispersity indices are 2.7, 2.8, 2.5, 3.7, and 3.5.
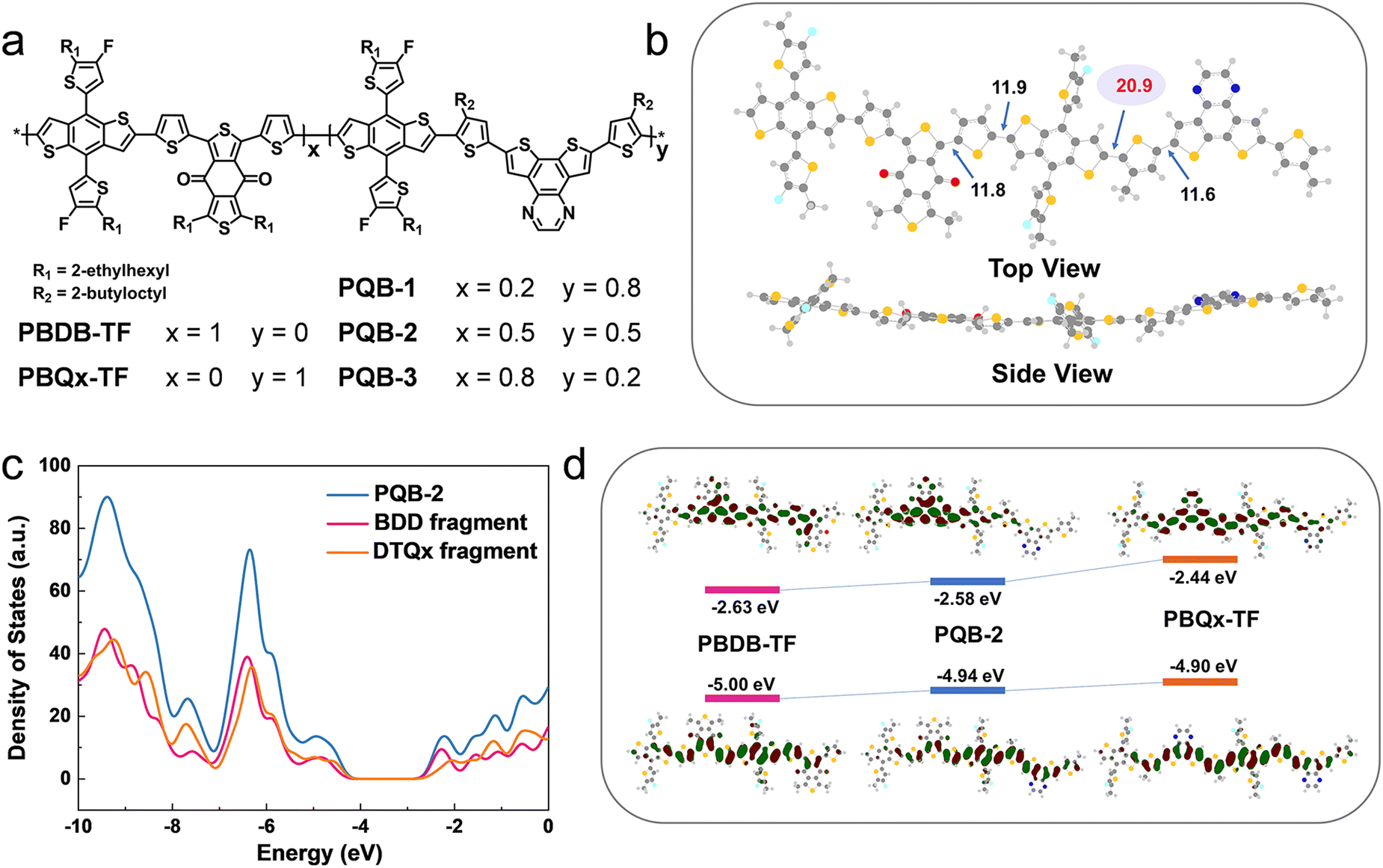 | ||
| Fig. 1 (a) Molecular structures of the polymers. (b) Simulated chemical geometry of the molecular model for the copolymer with a BDD: DTQx ratio of 1:1. (c) Simulated density of states of BDD, DTQx fragment, and PQB-2. (d) Molecular energy levels and wavefunction distributions of the frontier orbits for the polymer models. | ||
The pre-aggregation effect in solution has proved to be one of the important features for polymer donors to make sure of a suitable morphology when blending with acceptors.40,41 As displayed in Fig. S1 (ESI†), we measured the temperature-dependent absorption spectra of the polymers in toluene. At increased temperatures, all diluted solutions show blue-shifted absorption peaks with decreased intensity. From solutions (at room temperature) to thin films, the absorption peaks display small redshifts (Fig. 2a and Fig. S2, ESI†). The absorption intensity of the PQB-2 film in the 600–700 nm range lies between that of the two binary polymers. By contrast, the PQB-2 film has a larger absorption half-peak width. As shown in Fig. 2b and Fig. S3 (ESI†), we evaluated the energy levels of the polymers by performing cyclic voltammetry measurements. The polymers with a more BDD content show gradually downshifted ionized potentials, which is consistent with the calculation results. In cell devices, the terpolymers are expected to have Vocs higher than PBQx-TF, but the values should be lower than PBDB-TF based on experience.
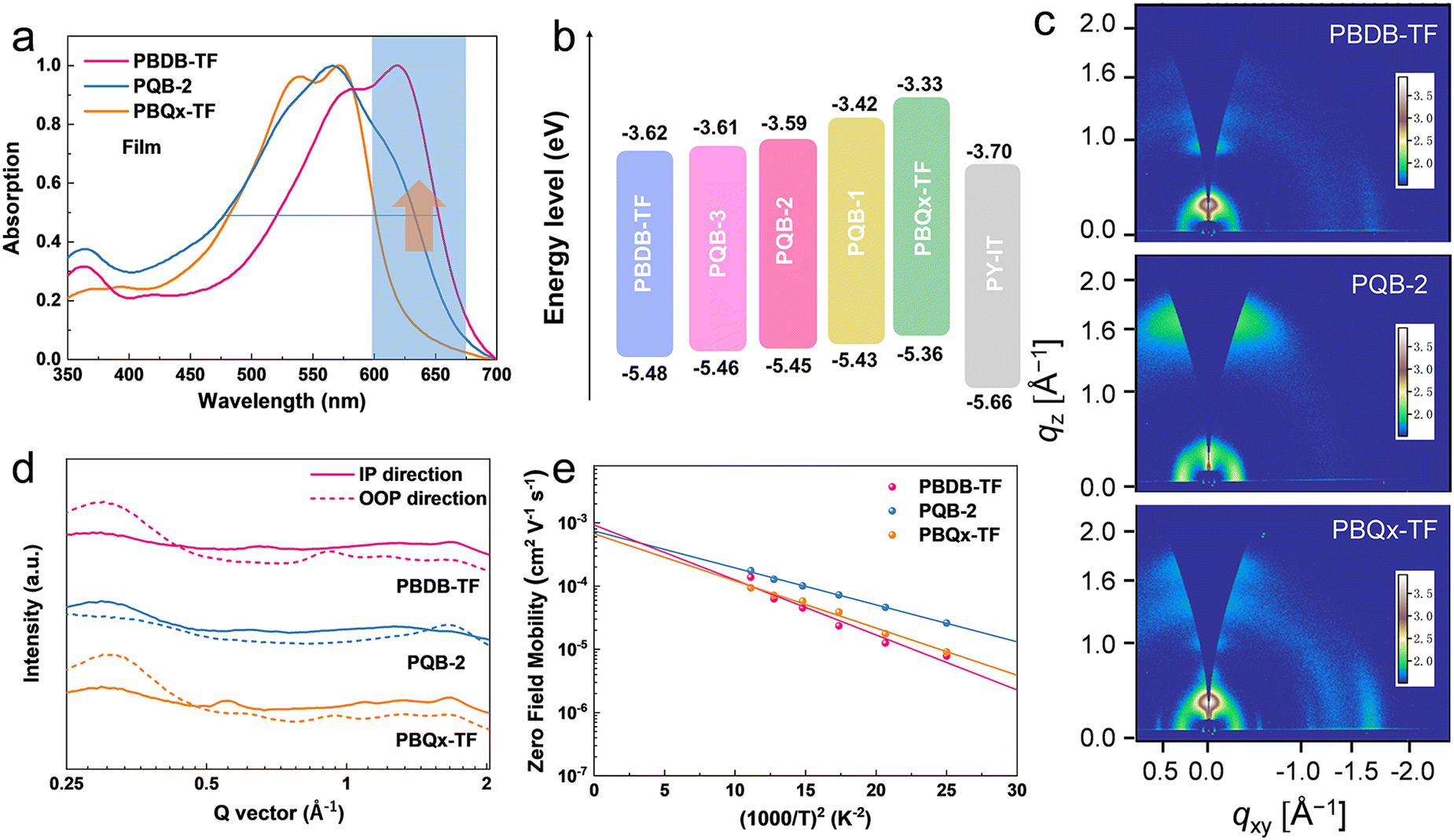 | ||
| Fig. 2 (a) Absorption spectra of PBDB-TF, PQB-2, and PBQx-TF films. (b) Energy level alignment of the donors and acceptors. (c) 2D-GIWAXS patterns of PBDB-TF, PQB-2 and PBQx-TF films. (d) The GIWAXS profiles of PBDB-TF, PQB-2, and PBQx-TF films in IP and OOP directions. (e) Hole mobilities of the PBDB-TF, PQB-2, and PBQx-TF films as a function of 1/T2 using SCLC derived data. | ||
To assess the impact of the ternary structure on the crystalline properties, we performed grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements on the three terpolymers and the two binary polymers, PBQx-TF and PBDB-TF. From the two-dimensional (2D) patterns displayed in Fig. 2c and Fig. S4a, b (ESI†), the (010) diffraction signals appear in both in-plane (IP) and out-of-plane (OOP) directions for the binary polymers, suggesting that they have coexistence of face-on and edge-on orientations relative to the substrate.42,43 The one-dimensional (1D) line-cut profiles are plotted in Fig. 2d and 4c, d. As strong (010) and (100) diffractions appear in the IP and OOP directions, respectively, the binary polymers have higher percentages of edge-on orientations. Interestingly, all the terpolymers display a preferential face-on orientation in the films. The PQB-2 film exhibits the strongest diffraction peak in the OOP direction compared to PQB-1 and PQB-3. This result implies the coexistence of BDD and DTQx units in the backbone greatly changes the molecular packing and may facilitate carrier transport in the vertical direction. The five polymers show very close (010) diffraction locations (1.67 Å−1), corresponding to a π–π distance of 3.76 Å. The (010) coherence length (CL) of PBDB-TF is calculated to be 17.5 Å, which is smaller than that of PBQx-TF (22.4 Å). This result indicates that the crystallinity of PBDB-TF is weaker than that of PBQx-TF in toluene solution. We then studied the energetic disorders of the polymer donors by measuring the variation of carrier mobility as a function of temperature. Using the Gaussian disorder model (GDM) (eqn (1)), the hole disorder of the polymers can be assessed.
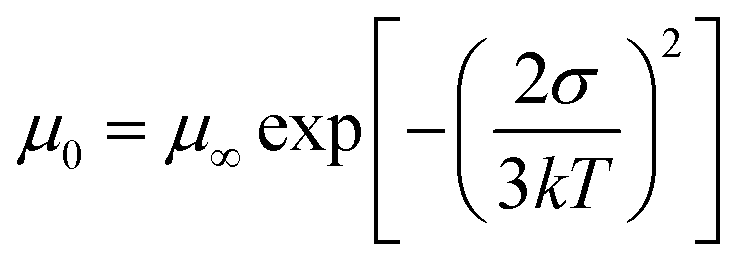 | (1) |
To investigate the photovoltaic application of the terpolymers in all-PSCs, we fabricated single-junction cells with a traditional device configuration of ITO/PEDOT:PSS/active layer/PDINN/Ag (with ITO for indium tin oxide, PEDOT:PSS for poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) and PDINN for N,N′-Bis{3-[3-(Dimethylamino)propylamino]propyl}perylene-3,4,9,10-tetracarboxylic diimide), where PY-IT was the PA. The changes in photovoltaic parameters, including VOC, short-circuit current density (JSC), fill factor (FF), and PCE, are shown in Fig. S6a and Table S2 (ESI†). The PBQx-TF-based device provides a lowest VOC of 0.917 V and the terpolymer-based devices show increased VOCs. Note that PQB-2 and PQB-3 even achieve larger VOCs than PBDB-TF although they possess higher HOMO levels, implying decreased Elosss in the devices. The FFs of the devices based on PBQx-TF and terpolymers are as high as ∼80%, which are much higher than the PBDB-TF-containing device. The J–V curves of the optimized devices based on PBDB-TF, PQB-2, and PBQx-TF are plotted in Fig. 3a, and the detailed parameters are listed in Table 1. Under simulated AM 1.5G (100 mW cm−2) illumination, the PBQx-TF:PY-IT-based device achieved a PCE of 17.2%, which is comparable with our previous work. Encouragingly, the PQB-2:PY-IT-based device provides a maximum PCE of 18.1% with a VOC of 0.942, a JSC of 24.2 mA cm−2, and an FF of 0.795. The best device reached a certified PCE of 17.6% in the National Institute of Metrology (NIM), China, which is one of the top values among all-PSCs (Fig. S7, ESI†). The small differences in VOC values may result from the device stability and different test conditions. We also fabricated PQB-2:PY-IT-based devices with increasing film thickness from 100 to 350 nm. As collected in Table S3 (ESI†), we can find that the device based on PQB-2:PY-IT still maintains a good PCE of 15.5% at a film thickness of 350 nm, which may be a good feature for adapting industrial printing techniques. We carried out the stability measurements of devices under light irradiation (simulated one-sun intensity using an LED array) and thermal annealing (50 °C) conditions. From Fig. S8 (ESI†), it can be found that the device stability is not very good, especially under thermal conditions. To find out the reason for the efficiency decay, we recorded the absorption spectra of the blend film. As proved in Fig. S9 (ESI†), we can find that the absorption intensity in the 400–700 nm is gradually decreased, suggesting that there is a disaggregation process for the polymer donor. More studies will be conducted to solve this stability problem in the future. To study the reasons behind the unexpected change in the VOC of terpolymers, we measured the highly sensitive EQE (s-EQE) and electroluminescence external quantum efficiency (EQEEL) of the devices.45 From the intersections of fitted spectra shown in Fig. S10 (ESI†), the bandgaps (Egs) are determined as 1.45 eV for PBDB-TF and PQB-2-based polymers and 1.46 eV for PBQx-TF-based devices, which are close to charge transfer energy (1.44 eV) and imply a negligible Eloss caused by charge generation. As shown in Fig. 3b, the PQB-2-based device gives an EQEEL of 9.7 × 10−4 and corresponds to a non-radiative Eloss of 0.179 eV, which is a very small value among the highly efficient organic solar cells. By contrast, the binary polymer-based devices exhibit lower EQEEL values (6.6 × 10−4 for the PBDB-TF-based device and 3.9 × 10−4 for the PBQx-TF-based device), and the non-radiative Eloss values were calculated to be 0.189 eV for the PBDB-TF-based device and 0.203 eV for the PBQx-TF-based device. The decreased Eloss may be connected to the suppressed energetic disorder and is the key factor that accounts for the higher VOC of the PQB-2-based device. The EQE spectra of the devices are depicted in Fig. 3c and Fig. S6b (ESI†). All the devices show efficient photon–electron response in 300–900 nm with maximum EQE values over 80%. In the short-wavelength region (300–680 nm), both the donor and the acceptor contribute to EQE profiles. Differently, the EQE response in the long-wavelength region (680–900 nm) is only attributed to the light absorption of PY-IT. For the PBQx-TF-based device, we can find that there is a plump in the EQE curve from 600 to 750 nm. By contrast, the EQE values are much higher for the PQB-2-based device in the same range, which should be ascribed to the enhanced optical absorption. Besides, the PQB-2:PY-IT-based PSC shows a higher EQE value in the long-wavelength region, which implies that the hole transport from PY-IY to PQB-2 is more efficient. The integrated current densities are 23.2, 23.3, 23.5, 23.7, and 23.5 mA cm−2 for PBDB-TF-, PBQx-TF-, PQB-1, PQB-2 and PQB-3-based devices, which agree well with the values obtained from the J–V tests.
 | ||
| Fig. 3 (a) J–V curves in AM 1.5G, (b) EQEEL curves, (c) EQE curves and (d) Photo-CELIV curves of the PBDB-TF:PY-IT, PQB-2:PY-IT and PBQx-TF:PY-IT-based all PSCs. (e) Hole mobilities of the PBDB-TF:PY-IT, PQB-2:PY-IT and PBQx-TF:PY-IT-based films as a function of 1/T2 using SCLC derived data. (f) Defect density of states of the blended films and the corresponding Gaussian fitting results. | ||
| Active layer | V OC (V) | J SC (mA cm−2) | J cal (mA cm−2) | FF | PCEa (%) |
|---|---|---|---|---|---|
| a Average values with standard deviation are obtained from 10 independent cells. | |||||
| PBQx-TF:PY-IT | 0.917 (0.917 ± 0.003) | 23.6 (23.5 ± 0.3) | 23.3 | 0.796 (0.786 ± 0.012) | 17.2 (16.9 ± 0.2) |
| PQB-2:PY-IT | 0.942 (0.941 ± 0.002) | 24.2 (24.2 ± 0.3) | 23.7 | 0.795 (0.780 ± 0.011) | 18.1 (17.8 ± 0.2) |
| PBDB-TF:PY-IT | 0.938 (0.936 ± 0.003) | 23.5 (23.4 ± 0.3) | 23.2 | 0.729 (0.725 ± 0.010) | 16.1 (15.9 ± 0.2) |
We then investigated the fast carrier mobilities of the devices by performing photon-induced charge carrier extraction in linearly increasing voltage (photo-CELIV) measurements. From the curves shown in Fig. 3d, the calculated carrier mobility of the PQB-2:PY-IT device is 2.3 × 10−4 cm2 V−1 s−1, which is higher than that of devices based on binary polymer donors (∼1.0 × 10−4 cm2 V−1 s−1). To further evaluate the energetic disorders of all-PSCs, we measured the temperature dependence of hole mobilities and plotted the results in Fig. S11 (ESI†) and Fig. 3e. The energetic disorder σ of the PQB-2:PY-IT device is estimated to be 36 meV. By contrast, the PBQx-TF- and PBDB-TF-based devices show higher σ values of 42 and 45 meV, respectively. Moreover, Mott–Schottky analysis was used to probe the density of states (DOS). The DOS distributions were further fitted using eqn (2).
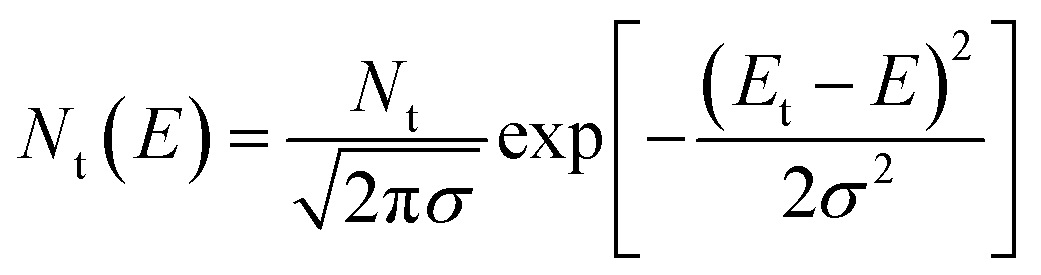 | (2) |
To investigate the dynamics of exciton and charge carriers, we conducted transient absorption (TA) spectra measurements. Neat polymer donors are pumped at 400 nm, and the 2D TA images are illustrated in Fig. S13 (ESI†). Both PBDB-TF and PBQx-TF films possess two peaks in ground-state bleaching (GSB) signals. The GSB signal of PBDB-TF is located in the range of 550–660 nm while PBQx-TF has a blue-shifted GSB signal in the range of 500–600 nm. For the PQB-2 film, a significantly broadened GSB signal covering 500–670 nm can be observed, which is almost the coherent overlap of its composing precursors. The TA spectrum of the PY-IT film is also investigated (Fig. S13d, ESI†), showing a wide-distributed GSB signal between 650 and 810 nm. To acquire the details of charge-transfer dynamics among the donor:acceptor blends, an 800 nm pump was selected to solely excite PY-IT, triggering the hole transfer process (Fig. 4a–c and Fig. S14, ESI†). It can be observed that both PBDB-TF and PBQx-TF can accept the hole generated from PY-IT since the GSB signals of PBDB-TF and PBQx-TF exhibit delayed rising in the corresponding blends. For the PQB-2:PY-IT film, the donor GSB signal is majorly distributed in the short wavelength region of 460–530 nm, while the GSB signal between 550 and 600 nm can hardly be distinguished. This indicates that the BDT-DTQx fragments in the conjugated backbone provide favorable hole-transporting sites.48,49
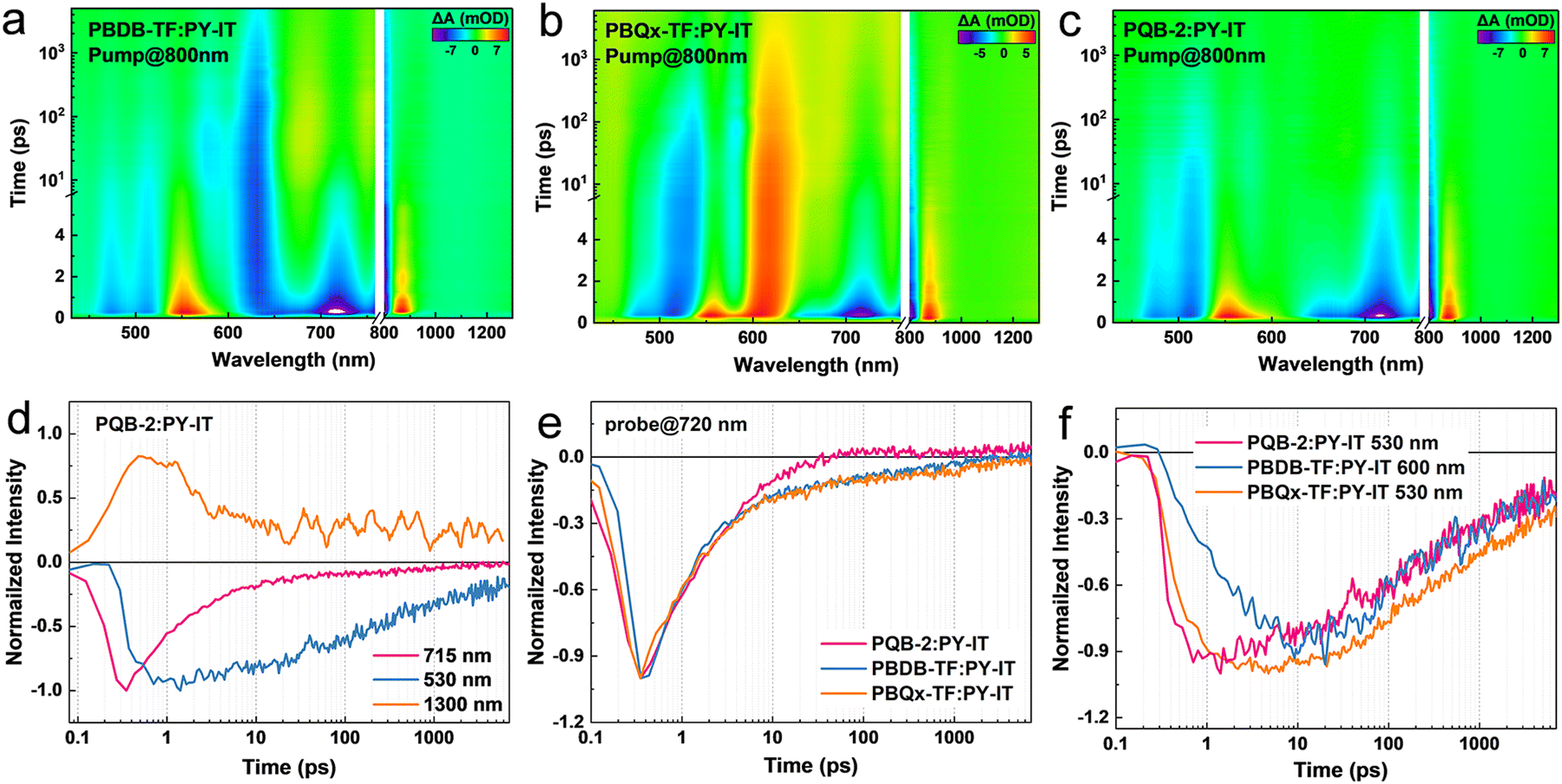 | ||
| Fig. 4 (a–c) 2D TA profiles of PBDB-TF:PY-IT, PBQx-TF:PY-IT, and PQB-2:PY-IT films. (d) TA dynamics of the PQB-2:PY-IT film. (e) GSB quenching of PY-IT and (f) GSB rising dynamics of polymer donors in the blend films. | ||
We extracted TA dynamics to further assess the hole-transfer kinetics. Fig. 4d shows the dynamics traced in the PQB-2:PY-IT film. After the excitation, PY-IT exhibits a rapid rise in the GSB peak at 715 nm and is accompanied by a slow-accumulated PQB-2 GSB signal at 530 nm. A photo-induced absorption (PIA) signal can be observed at 1300 nm, which could be ascribed to the intermediated charge-transfer state during the hole transfer.48 In Fig. 4e and f, the GSB quenching of PY-IT and GSB rising of the donors are compared. The PQB-2-based system exhibits the shortest PY-IT exciton lifetime while the fast-rising of donor GSB dynamics saturates at ∼1 ps. By contrast, the hole-transfer rate in the PBQx-TF-based blend is very close while that in the PBDB-TF-based system exhibits a severely delayed dynamic. These results indicate that the BDT-DTQx fragment is beneficial for hole transfer, leading to a fast and efficient transfer channel in the ternary copolymerization polymers.
We characterized the microstructure of the films by atomic force microscopy (AFM) and GIWAXS. Fig. 5a–c and Fig. S15 (ESI†) show the AFM height images of the blend films, where we can find the terpolymer:PY-IT films show slightly lower root-mean-squares (Rqs) (1.13 nm for PQB-1:PY-IT, 1.65 nm for PQB-2:PY-IT, and 1.33 nm for PQB-3:PY-IT). By contrast, the Rq values of PBQx-TF:PY-IT and PBDB-TF:PY-IT films are 2.23 and 1.80 nm, respectively. The AFM phase patterns are provided in Fig. S16 (ESI†). All three blend films exhibit a fibrillar morphology, which should be associated with the pre-aggregation effect of the polymer donors. Relative to the PBQx-TF:PY-IT and PBDB-TF:PY-IT films, the terpolymer:PY-IT films show densely distributed fibrils with a decreased width. Fig. 5d–f and Fig. S17a, b (ESI†) display the 2D GIWAXS patterns of the blend films. The corresponding line-cut profiles are plotted in Fig. 5g and Fig. S17c, d (ESI†). The neat PY-IT film was also measured for comparison. As shown in Fig. S18 (ESI†), PY-IT adopts a preferred face-on orientation. The (010) diffraction peak is located at 1.30 Å−1, and the π–π distance is estimated to be 4.85 Å. All blend films show strong (010) diffraction peaks in the OOP directions, indicating a preferential face-on orientation. The π–π stacking distances are estimated to be ∼3.8 Å for all blend films. The calculated CL values are 19.9, 17.5, 15.2, 16.9, and 15.6 Å for PBQx-TF:PY-IT, PQB-1:PY-IT, PQB-2:PY-IT, PQB-3:PY-IT and PBDB-TF:PY-IT blend films, respectively. These results indicate that PQB-2:PY-IT has an appropriate fibril domain.
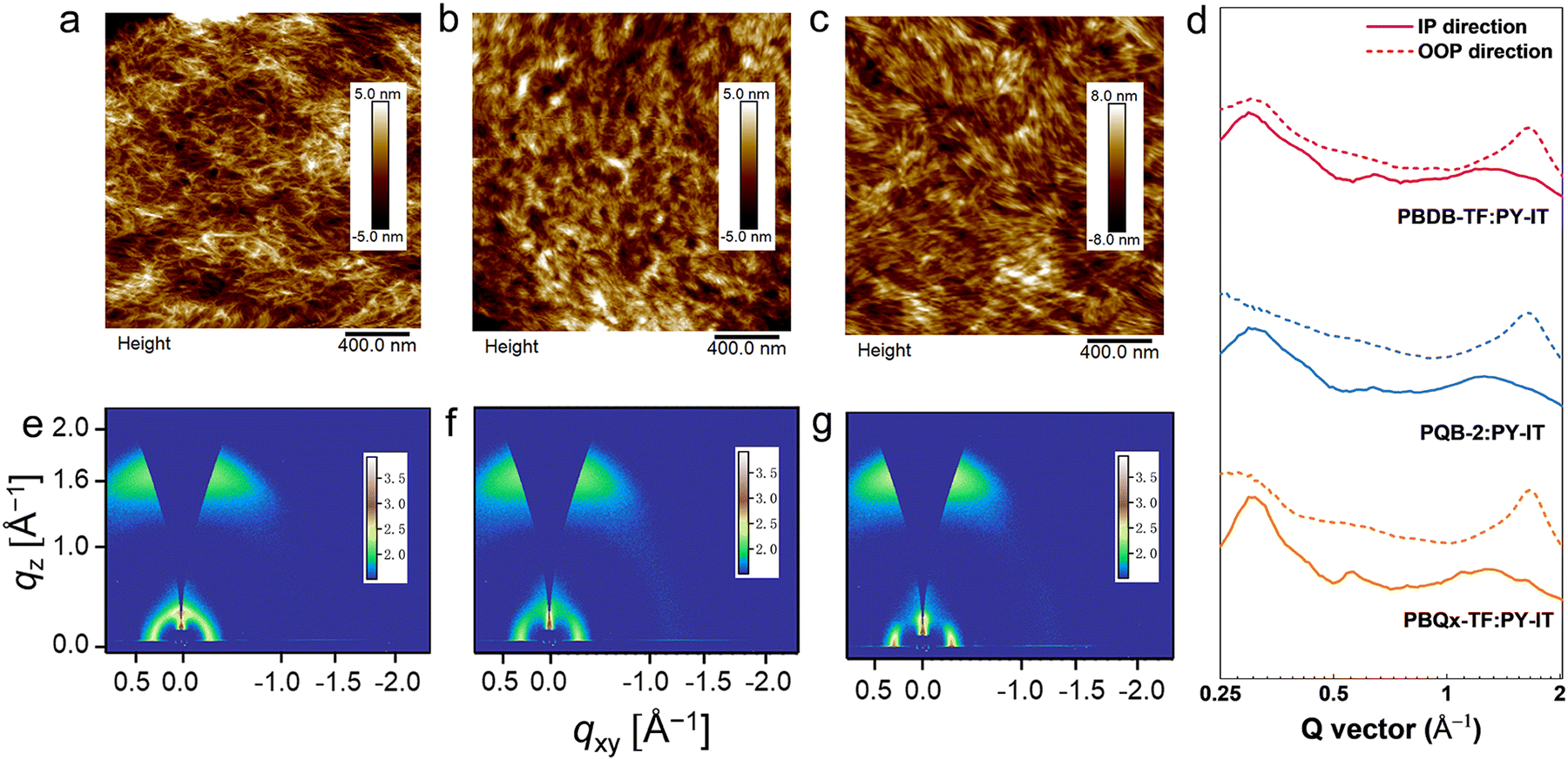 | ||
| Fig. 5 AFM height images of the (a) PBDB-TF:PY-IT, (b) PQB-2:PY-IT, and (c) PBQx-TF: PY-IT-based films. (d) The GIWAXS profiles of PBDB-TF:PY-IT, PQB-2:PY-IT, and PBQx-TF:PY-IT-based films in IP and OOP directions. 2D-GIWAXS patterns of (e) PBDB-TF:PY-IT, (f) PQB-2:PY-IT, and (g) PBQx-TF: PY-IT-based films. | ||
Conclusions
In conclusion, we successfully demonstrated an outstanding efficiency of 18.1% in all-PSCs. By combining the advantages of BDD and DTQx blocks, we can not only effectively modulate the absorption spectra and molecular energy levels but also optimize the aggregation properties of the binary polymers PBQx-TF and PBDB-TF. Ternary copolymerization changed the molecular orientation from the coexistence of face-on and edge-on to face-on only, which is beneficial for decreasing the energetic disorder. When blending with a polymer acceptor of PY-IT, PQB-2 shows an improved fibrillar morphology with an appropriate domain size. The TA results indicate that PQB-2 possesses more efficient charge generation and transport processes than binary polymer-based systems, thus contributing to decreased non-radiative Eloss. As a result, the PQB-2-based device gives the highest VOC although its HOMO level locates in the middle of binary polymers, which is rarely seen in the molecular design of organic photovoltaic materials. This work suggests that ternary copolymerization is not only a molecular design strategy for adjusting the optical and electronic properties but also a feasible way to suppress the energetic disorder of all-PSCs, which plays a critical role in boosting the photovoltaic performance. We think that further molecular design should consider energetic disorder for highly efficient all-PSCs.Author contributions
T. Z., H. Y. and J. H. conceived the idea. T. Z., L. M., and Y. C. carried out the device fabrication and characterization. J. Z. and W. Z. performed the GIWAXS measurements. Y. X., J. W., and Z. L. synthesized the polymers. P. B., Z. C. and X. H. performed the TA characterization. K. X. analyzed the GIWAXS data. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22122905, 21835006, 22075301, and 52073068). J. H. was supported by the Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-201903) and the “Dual Carbon” Science and Technology Innovation of Jiangsu Province (Industrial Prospect and Key Technology Research program) (BE2022021).Notes and references
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- S. Ma, H. Zhang, K. Feng and X. Guo, Chem. – Eur. J., 2022, 28, e202200222 CAS.
- Q. Wu, W. Wang, Y. Wu, Z. Chen, J. Guo, R. Sun, J. Guo, Y. Yang and J. Min, Adv. Funct. Mater., 2021, 31, 2010411 CrossRef CAS.
- P. Bi, S. Zhang, Z. Chen, Y. Xu, Y. Cui, T. Zhang, J. Ren, J. Qin, L. Hong, X. Hao and J. Hou, Joule, 2021, 5, 2408–2419 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. Tang and E. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- S. Dai, S. Huang, H. Yu, Q. Ling and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 682–689 CrossRef CAS.
- L. Zhu, W. Zhong, C. Qiu, B. Lyu, Z. Zhou, M. Zhang, J. Song, J. Xu, J. Wang, J. Ali, W. Feng, Z. Shi, X. Gu, L. Ying, Y. Zhang and F. Liu, Adv. Mater., 2019, 31, 1902899 CrossRef CAS PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- P. Bi and X. Hao, Sol. RRL, 2019, 3, 1800263 CrossRef.
- R. Zhao, B. Lin, J. Feng, C. Dou, Z. Ding, W. Ma, J. Liu and L. Wang, Macromolecules, 2019, 52, 7081–7088 CrossRef CAS.
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- P. Bi, S. Zhang, J. Ren, Z. Chen, Z. Zheng, Y. Cui, J. Wang, S. Wang, T. Zhang, J. Li, Y. Xu, J. Qin, C. An, W. Ma, X. Hao and J. Hou, Adv. Mater., 2022, 34, 2108090 CrossRef CAS PubMed.
- Q. Fan, W. Su, S. Chen, T. Liu, W. Zhuang, R. Ma, X. Wen, Z. Yin, Z. Luo, X. Guo, L. Hou, K. Moth-Poulsen, Y. Li, Z. Zhang, C. Yang, D. Yu, H. Yan, M. Zhang and E. Wang, Angew. Chem., Int. Ed., 2020, 59, 19835–19840 CrossRef CAS PubMed.
- Z.-G. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422–4433 CrossRef CAS PubMed.
- J. Yuan, T. Huang, P. Cheng, Y. Zou, H. Zhang, J. L. Yang, S.-Y. Chang, Z. Zhang, W. Huang, R. Wang, D. Meng, F. Gao and Y. Yang, Nat. Commun., 2019, 10, 570 CrossRef CAS PubMed.
- Y. Yang, ACS Nano, 2021, 15, 18679–18682 CrossRef CAS PubMed.
- J. Wang, Y. Cui, Y. Xu, K. Xian, P. Bi, Z. Chen, K. Zhou, L. Ma, T. Zhang, Y. Yang, Y. Zu, H. Yao, X. Hao, L. Ye and J. Hou, Adv. Mater., 2022, 34, 2205009 CrossRef CAS PubMed.
- T. Liu, T. Yang, R. Ma, L. Zhan, Z. Luo, G. Zhang, Y. Li, K. Gao, Y. Xiao, J. Yu, X. Zou, H. Sun, M. Zhang, T. A. Dela Peña, Z. Xing, H. Liu, X. Li, G. Li, J. Huang, C. Duan, K. S. Wong, X. Lu, X. Guo, F. Gao, H. Chen, F. Huang, Y. Li, Y. Li, Y. Cao, B. Tang and H. Yan, Joule, 2021, 5, 914–930 CrossRef CAS.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS PubMed.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- C. Zhang, S. Mahadevan, J. Yuan, J. K. W. Ho, Y. Gao, W. Liu, H. Zhong, H. Yan, Y. Zou, S.-W. Tsang and S. K. So, ACS Energy Lett., 2022, 7, 1971–1979 CrossRef CAS.
- P. Bi, J. Ren, S. Zhang, J. Wang, Z. Chen, M. Gao, Y. Cui, T. Zhang, J. Qin, Z. Zheng, L. Ye, X. Hao and J. Hou, Nano Energy, 2022, 100, 107463 CrossRef CAS.
- J. Yuan, C. Zhang, B. Qiu, W. Liu, S. K. So, M. Mainville, M. Leclerc, S. Shoaee, D. Neher and Y. Zou, Energy Environ. Sci., 2022, 15, 2806–2818 RSC.
- X. Zhang, L. Qin, Y. Li, J. Yu, H. Chen, X. Gu, Y. Wei, X. Lu, F. Gao and H. Huang, Adv. Funct. Mater., 2022, 32, 2112433 CrossRef CAS.
- J. Yuan, H. Zhang, R. Zhang, Y. Wang, J. Hou, M. Leclerc, X. Zhan, F. Huang, F. Gao, Y. Zou and Y. Li, Chem, 2020, 6, 2147–2161 CAS.
- L. Zhu, M. Zhang, W. Zhong, S. Leng, G. Zhou, Y. Zou, X. Su, H. Ding, P. Gu, F. Liu and Y. Zhang, Energy Environ. Sci., 2021, 14, 4341–4357 RSC.
- F. Zhao, C. Wang and X. Zhan, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- H. Hu, K. Zhao, N. Fernandes, P. Boufflet, J. H. Bannock, L. Yu, J. C. de Mello, N. Stingelin, M. Heeney, E. P. Giannelis and A. Amassian, J. Mater. Chem. C, 2015, 3, 7394–7404 RSC.
- Y. Xu, Y. Cui, H. Yao, T. Zhang, J. Zhang, L. Ma, J. Wang, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2101090 CrossRef CAS PubMed.
- D. Dang, D. Yu and E. Wang, Adv. Mater., 2019, 31, 1807019 CrossRef PubMed.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. A. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- P. Bi, J. Ren, S. Zhang, J. Wang and J. Hou, Polymer, 2020, 209, 122900 CrossRef CAS.
- Y. Li, Q. Li, Y. Cai, H. Jin, J. Zhang, Z. Tang, C. Zhang, Z. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 3854–3861 RSC.
- T. Zhang, C. An, Y. Cui, J. Zhang, P. Bi, C. Yang, S. Zhang and J. Hou, Adv. Mater., 2022, 34, 2105803 CrossRef CAS PubMed.
- M. Sajjad, A. Ruseckas and I. Samuel, Matter, 2020, 3, 341–354 CrossRef.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- J. Xu, S. B. Jo, X. Chen, G. Zhou, M. Zhang, X. Shi, F. Lin, L. Zhu, T. Hao, K. Gao, Y. Zou, X. Su, W. Feng, A. K. Y. Jen, Y. Zhang and F. Liu, Adv. Mater., 2022, 34, 2108317 CrossRef CAS PubMed.
- F.-Z. Cui, Z.-H. Chen, J.-W. Qiao, T. Wang, G.-H. Lu, H. Yin and X.-T. Hao, Adv. Funct. Mater., 2022, 32, 2200478 CrossRef CAS.
- G. Zhou, M. Zhang, Z. Chen, J. Zhang, L. Zhan, S. Li, L. Zhu, Z. Wang, X. Zhu, H. Chen, L. Wang, F. Liu and H. Zhu, ACS Energy Lett., 2021, 6, 2971–2981 CrossRef CAS.
- P. Bi, T. Xiao, X. Yang, M. Niu, Z. Wen, K. Zhang, W. Qin, S. K. So, G. Lu, X. Hao and H. Liu, Nano Energy, 2018, 46, 81–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03535a |
| This journal is © The Royal Society of Chemistry 2023 |