
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 flow electrolysis – limiting impact of heat and gas evolution in the electrolyte gap on current density†
Christina
Martens
 *ab,
Bernhard
Schmid
*a,
Hermann
Tempel
a and
Rüdiger-A.
Eichel
ab
*ab,
Bernhard
Schmid
*a,
Hermann
Tempel
a and
Rüdiger-A.
Eichel
ab
aInstitute of Energy and Climate Research – Fundamental Electrochemistry (IEK-9), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: c.martens@fz-juelich.de; b.schmid@fz-juelich.de
bInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52074 Aachen, Germany
First published on 31st August 2023
Abstract
Research in CO2 electro-reduction with the aim of providing green chemical feedstock (e.g., CO) has been driven towards optimization of individual components such as CO2-reducing gas diffusion electrodes (GDEs) to achieve stable electrolysis processes. Moving forward, investigation into the performance of electrodes at a cell- and system-level is needed to identify key operational parameters that enhance electrode efficiency. In this study, we characterize self-regulated steady-states within an electrolytic cell. Additionally, we explore the circumstances under which the current density passing through the cell becomes self-limiting. GDE-relevant system parameters and their impact on the overall electrode durability during electrolysis at high current densities up to −1.2 A cm−2 were analyzed on an intermediate time scale. Integration of inline sensors to the electrolysis test setup enabled close monitoring of changes in the electrolyte temperature and electrolyte pH, as well as the detection of pressure changes around the cathode. In the presented study, the GDE did not appear to be the bottleneck to achieving high current density CO2-electrolysis. Instead, electrolyte heating and gas evolution within the electrolyte gap limited the maximum current densities that could be applied to a GDE flow cell. Our results suggest that electrode performance (selectivity, durability) can sometimes be underestimated when electrolysis cells and their periphery are not optimally suited for operation with GDEs yet, thus preventing performance windows from being reached.
1 Introduction
CO2 is a thermodynamically stable molecule1 that has been accumulating in the Earth's atmosphere over decades alongside human industrial development and the use of fossil carbon as an energy source.2 The resulting imbalance in the carbon cycle due to surplus CO2 emissions in addition to naturally occurring CO2 is directly linked to global warming and climate change.3,4 Therefore, to preserve life on this planet, a transition from the current model of a linear economy to a circular economy is imperative.5–7 One step towards mending the anthropogenically caused imbalance in global carbon cycles is to re-introduce CO2 to the market in the form of value-added green chemicals and energy carriers. A promising approach to facilitate this conversion is CO2 electrolysis. Electricity from renewable sources is supplied to an electrochemical reactor where it is used for the cathodic reduction of CO2 into a multitude of products, e.g., CO, ethene (C2H4), and ethanol (C2H5OH).8 This reaction is typically coupled with anode-sided water oxidation to oxygen.9 Producing universal educts like CO or syngas (CO, H2) using CO2 as a resource contributes to reaching carbon neutrality in many products of daily living, such as acetic acid household cleaners.10 Sourcing these educt streams from fossil sources, such as gas steam reforming or coal gasification, which are used to obtain CO and syngas for the chemical industry,10 could be avoided. Furthermore, this technology does not leave any critical waste or reaction byproducts behind. CO2 electrolysis can be considered a green process as its process media are aqueous solutions of environmentally benign salts and inorganic acids and the process is operated at ambient conditions.From the discovery of CO2 reduction and first studies on its kinetics in the 1960s,11 the development of CO2 electrolyzers progressed in response to a growing understanding of process and reaction requirements. CO2 reduction has since been studied using various reactor systems and with different focus. Whereas catalyst research requires a very controlled reaction environment that can be achieved, e.g., by using a rotating disc electrode (RDE)12 or a cell that encompasses static electrolytes like the H-cell,13 continuously run flow cell electrolyzers are required by the industry once CO2 electrolyzers are to be scaled up for commercial use.14,15 Currently, the state-of-the-art reactor for doing this are gas diffusion electrode (GDE) flow cells, which are studied in a variety of electrode, electrolyte and membrane configurations.16–18 Through the use of a GDE, this type of reactor comes with the advantage of supplying all educts (CO2, electrons and charge carriers) to the catalyst directly. The limiting slow mass transport of CO2 through aqueous electrolyte media19–22 is accelerated in GDEs, as diffusion pathways for CO2 are substantially shortened.16 When GDEs were first introduced, it was assumed that the reaction at the electrode takes place in a three-phase interface of the gaseous CO2, catalyst surface, and liquid electrolyte.23 Contrary to this assumption, recent studies indicate that CO2 reduction in such a system takes place within the two-phase interface of the catalyst and dissolved aqueous CO2.24 Due to accelerated educt transport, GDE flow cells surpass other cell designs not only in terms of selectivity, but also in terms of an extended current density range that can be applied to such reactors.15,25 As researchers explored the use of GDEs in this flexible cell concept, GDE flooding – the gradual (and sometimes quite spontaneous) intrusion of liquid electrolyte into the highly porous GDE structure and gas channel – emerged as the next big obstacle in realizing high current density CO2 electrolysis.26 Electrode flooding results in a series of unfavorable consequences, such as the blockage of passageways for gas exchange, a generally reduced electrode stability, and a loss in CO2-derived product selectivity favoring H2 evolution by water-splitting, among others.27,28 This general limitation often hindered research into continuous steady-state operation at high current densities, as experiments were often limited to minutes by GDE flooding.29 Systematic studies regarding different GDL types and binder polymers for GDE preparation are numerous9,16 and not the aim of this study. Currently, there are both flooding-resistant GDEs and the approach of intermittently washing off GDEs during electrolysis to extend their lifetime.30–32
By combining known electrode optimization trends,33–36 we prepared GDEs that were flooding-resistant within the necessary duration of experiments conducted for this study, making it possible for us to study system steady-states of CO2 electrolysis at high current densities in a GDE flow cell. In such reactors, local and bulk system conditions vary greatly, both in the gas stream and in the liquid catholyte. While the buffer chemistry of the catholyte does not allow concentration gradients parallel to the GDE plane, the current will still lead to the formation of concentration gradients in the catholyte gap along the axis towards the anode.37 By using CO2 in excess, concentration gradients in the gas flow field can be eliminated. Nevertheless, local CO2 depletion in the catalyst layer can be expected at high current densities, reducing selectivity for CO.38 However, this limitation is solely a function of the GDE properties and structure when CO2 excess is ensured over the entire GDE area. We expect high conversion rates in the cell to strongly impact local conditions. In order to obtain a stable and continuous operation, the conditions and gradients formed due to the current density must converge to a steady-state. This raises the question as to where and what the steady-states of such a system are. A more sophisticated analysis test-setup monitoring these additional, GDE-relevant system variables expected to impact GDE performance was assembled to answer this question.
This study explores the windows of steady-state electrolysis beyond GDE flooding, in which reversible operation dynamics can be achieved using a single electrode. Identification of operating regimes for optimal electrode performance, as well as the presentation of means to expand such stable windows of operation by tuning individual system parameters, complement previous studies on GDE design and material optimization.9,16,27,34,35 Within the experimental time frame, the entire electrolyzer system was allowed to reach steady-state conditions, while long-term degradation mechanisms were not yet in effect. Previously conducted theoretical studies of individual parameters, such as cell pressures, pH gradients around the GDE, or GDE-poisoning by CO2 reduction products, are available and pointing to the existence of limitations in GDE flow cells at high CO2 conversion rates.38,39 This work presents experimental data on the direct and indirect effects of high current densities in GDE flow cells. We further identify inherent system limitations and report a new method of identifying windows of steady-state operation for GDE flow cell systems.
2 Experimental
2.1 Electrode materials and preparation
The CO2-reducing electrodes used in this study are prepared from commercially available gas diffusion layer sheets (GDL) made of macroporous carbon paper and a dense microporous carbon black layer (MPL), treated with polytetrafluoroethylene (PTFE) (Sigracet-38 BC, SGL Carbon). Sheets were cut into rectangles, 4 cm × 5 cm in size. Catalyst-ionomer-ink was applied to the MPL with a hand-held airbrush (0.3 mm nozzle diameter, 0.75 bar N2 carrier gas). The catalyst layer consisted of Ag-nanoparticles (2 mg cm−2, <150 nm, Iolitec) bound to the electrode surface by an anion exchange resin (7.5 wt% of the catalyst, Aemion HNN-8, Ionomr). The precursor ink was prepared in an ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone-mixture (0.65:0.35 wt%) and ultrasonicated for 40 min before spraying. Prepared electrodes were stored in an argon atmosphere.
:acetone-mixture (0.65:0.35 wt%) and ultrasonicated for 40 min before spraying. Prepared electrodes were stored in an argon atmosphere.
2.2 Electrode characterization
Cross-sectional and top-view images of GDE surfaces before and after electrolysis were acquired using a Thermo Scientific Quanta FEG 650 scanning electron microscope (SEM) with an accelerating voltage of 5–20 kV and a beam current of 1.19 × 10−7 pA and 170 μA. Representative cross-sections of the GDEs have been prepared by freezing and breaking GDE material in liquid nitrogen.2.3 Electrolyzer and periphery
Electrochemical testing was performed in a modified micro-flow cell (ElectroCell) consisting of three compartments: two liquid electrolyte compartments containing an acidic anolyte (H2SO4 of different concentrations, Honeywell Fluka) and a neutral buffered catholyte (KHCO3 of different concentrations, VWR Chemicals). A Nafion 117 membrane (Fuel Cell Store) was used to separate the electrolyte compartments. The dimensions of each electrolyte compartment were 30 × 34 × 3 mm3. The GDE separated the liquid catholyte compartment from the gas compartment. The gas compartment consisted of a titanium flow field with a meandering channel.18 This flow field also served as the electrical contact for the GDE. A zero-gap counter electrode consisting of an Ir-MMO coated titanium mesh (mesh size: 2 mm × 4 mm, Metakem) was used in the cell. The effective distance between the electrodes corresponded to the thickness of the catholyte compartment. The cell allowed for an active geometric electrode area of 10 cm2. Electric current was supplied via an Autolab PGSTAT302N potentiostat/galvanostat (Metrohm) coupled to a 20 A Booster (Metrohm). RE-1CP (Ag/AgCl/saturated KCl, ALS Japan) were used as reference electrodes and connected to the cell via a Haber-Luggin capillary through the catholyte supply line. 10-channel data loggers (Almemo-710, Ahlborn) were used to record the signals of sensors installed in the system, assessing the evolution of environmental system parameters. The test stand included two pH-electrodes with integrated Pt1000 (LL Unitrode Pt1000 WOC, Metrohm) to directly measure changes in temperature and pH in the catholyte entering (pHin) and exiting the electrolysis cell (pHout), two K-type thermocouples (Ahlborn) acquiring data on the temperature in each of the electrolyte reservoirs (bulk electrolyte temperatures, Tbulk), two differential pressure sensors (FDA602D, Ahlborn) assessing changes within the catholyte gap (entry vs. exit, Δpliq./liq.) and the gas channel (catholyte entry vs. gas channel, Δpliq./gas), two relative pressure sensors (FDAD3302R, Ahlborn) measuring the overpressure in the product gas stream (prel.,PG) as well as in the catholyte gas stream (prel.,CSG), and a galvanically separated electrometer (ZAD900AB3, Ahlborn). Catholyte and anolyte were recycled through the cell at a pump rate of 50 mL min−1 using peristaltic pumps for the catholyte and anolyte (Catholyte: Ismatec Ecoline ISM1089C peristaltic pump; Anolyte: Ismatec Reglo Digital piston pump). They were kept in external electrolyte reservoirs containing a volume of 200 mL equipped with cooling jackets. The reservoirs were connected to a water chiller (Corio CD-200F, Julabo). CO2 of 99.995% purity was used as a feed gas and supplied to the gas channel in single-pass mode before being analyzed by an online-gas chromatograph. The headspace of the catholyte reservoir was purged with an argon stream to analyze gases formed in the catholyte compartment. The volumetric flow rates of both gas streams exiting the experimental setup were determined using drum-type gas flow meters (TG05, Ritter). All reported relative and differential pressures are given as a total, including all components in the respective stream. Partial pressures were not discussed in this study. Pressure control was achieved using membrane back pressure regulators (LF-series, Equilibar) downstream the cell. Further information on the assembly of the electrochemical setup is provided in the ESI.†2.4 Product analysis
The product gases from the gas- and electrolyte-side of the GDE were simultaneously analyzed using a Trace 1310 gas chromatograph (Thermo Fischer) with two sample inlets. Each inlet was connected to two parallel gas analyzer channels. CO2 and CO were separated on a two-dimensional channel using a Hayesep-Q packed primary column and a 5 Å molecular sieves-packed secondary column with He carrier gas coupled to a thermal conductivity detector (TCD). H2 was separated on a 5 Å molecular sieves-packed column with argon carrier gas coupled to a TCD. Samples were injected from sample loops directly connected to the experiment via transfer lines.Two indicators were defined to evaluate cell performance: (1) the Faraday efficiency (FE) or the equivalent CO-specific current density (jCO), and (2) the maximum applicable current density, at which the cell stayed fully operational (jmax). The product-specific current density (ji) is directly derived from the more commonly presented FE for the electrochemical conversion of a species i. FEi and ji are calculated as follows:40
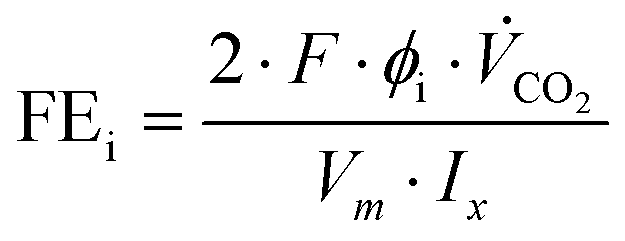 | (1) |
| ji = jx·FEi | (2) |
With the Faraday constant (F), the molar gas fractions in the product gas stream (ϕi), the product gas flow rate (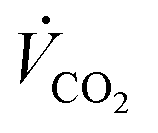 ) and the current that is applied to the cell (Ix) according to the list of settings given in Fig. 1. For eqn (2), the current density (jx) corresponds to the applied cell current (see Fig. 1) and the GDE's active geometrical size.
) and the current that is applied to the cell (Ix) according to the list of settings given in Fig. 1. For eqn (2), the current density (jx) corresponds to the applied cell current (see Fig. 1) and the GDE's active geometrical size.
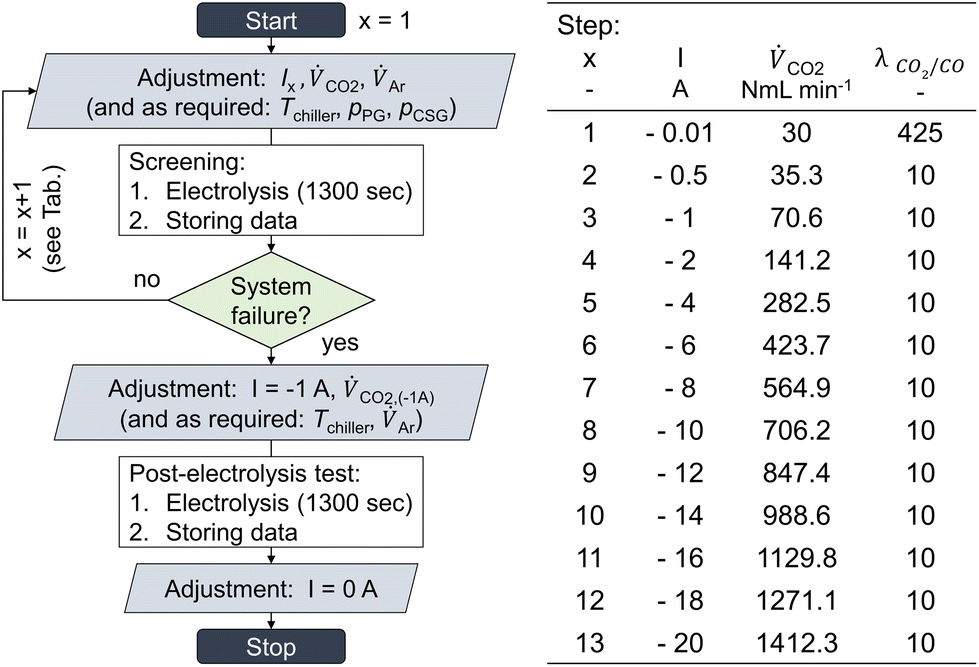 | ||
| Fig. 1 Electrochemical GDE testing parameters and procedure. Chrono-potentiometric screening continued until either GDE flooding occurred or cell voltage peaks exceeded the potentiostat's compliance voltage (±20 V). | ||
2.5 Electrochemical testing procedure
To better understand the limits of GDE flow cells to high-current density CO2 electrolysis, GDEs were tested via a chrono-potentiometric staircase screening method under varied environmental cell conditions. A screening run was carried out using a single GDE. After each staircase screening, a post-electrolysis test was performed at −0.1 A cm−2 to assess the GDE's remaining functionality. This protocol was validated by conducting additional GDE screenings, in which the order of applied current loads was shuffled.For this screening, jx was increased step-wise, until either the electrode seized to produce CO (e.g., due to flooding), or the potentiostat started to counter-regulate the applied current density to a lower value. The latter case was caused by cell voltages over-proportionally increasing in relation to the increase of jx, eventually exceeding the potentiostat's compliance voltage of ±20 V. Either of the two conditions was indicative of an unstable process operation and led to a termination of the experiment.
CO2 was supplied to the cell in 9-fold excess compared to its maximum achievable conversion rate to CO (λ = 10). It was increased proportionally to the applied current density (see ref. 41). Each current density step was applied for 1300 s in a continuous series (see table and scheme in Fig. 1). CO2 conversion in the GDE flow cell was allowed to reach equilibrium within 5 min, followed by catholyte gas and product gas stream analysis. Three gas samples were analyzed for each current density step, and the results were averaged. Error bars in graphs show the standard deviation between duplicate experiments. To assess whether a GDE was still functional after system limitations have been reached, a post-screening-electrolysis test was performed after the screening. For this, the current density was once again set to a low value (−0.1 A cm−2). The obtained data from the post-test was compared to the product selectivity obtained during the main screening, and the production of CO was an indicator of a not-yet-dysfunctional GDE.
3 Results and discussion
3.1 Electrochemical performance of flooding-resistant GDEs under baseline conditions
The GDE flow cell was operated over a range of current densities according to Fig. 2a. With increasing current density, FECO reached a maximum of 83.6% at −0.2 A cm−2 and consistently decreased with higher current densities. The cell was operable up to −0.8 A cm−2 at FECO of 44.3%. Higher current densities led to over-proportional increases of the cell voltage that terminated the measurements. Except at the lowest current density (−0.001 A cm−2), the total FE added up to 100% with deviations that fall into the combined error margin of the equipment used for gas analysis and quantification. The data indicates that electrons provided to the electrolyzer, which did not contribute to CO generation, were utilized to generate H2, the only by-product detected in the process.‡ In addition to the FECO, Fig. 2b depicts the corresponding jCO, as an equivalent performance indicator expressing reaction selectivity. The graph shows saturation behavior at −0.6 A cm−2, after which jCO decreases as H2 formation at the GDE increases. This observation is consistent with previously reported, and expected, CO2 and CO mass transport limitations towards high current density electrolysis.38 Besides showing information about product-selectivity (jCO) and the current density range (jmax), this graph also contains information about the operating regimes of the cell at different currents. We differentiate two process regimes that can be reached by the system, as well as an inaccessible third region. The two working regimes are (I) steady-state process conditions, and (II) process states with accelerated degradation.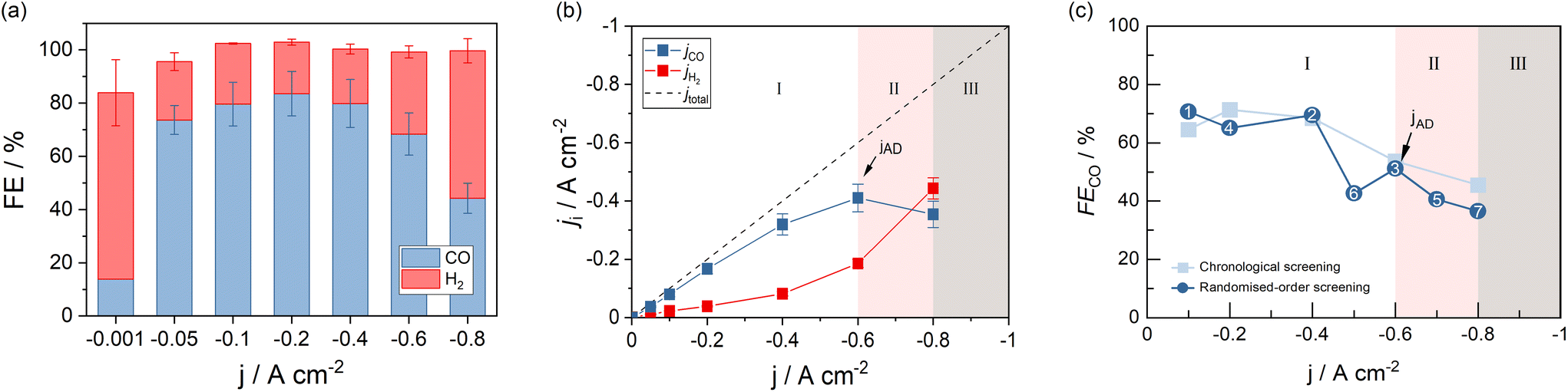 | ||
| Fig. 2 GDE performance measured in terms of (a) the CO-Faraday efficiency, (b) the product-specific current density, and jAD (indication arrow). Randomized-order screenings confirmed the representativity of the data (c). | ||
Operating regime I (white area in Fig. 2b) may be characterized as the current density range, in which current switching to higher or lower jx does not significantly alter the process's performance at either current density. Thus, reaction selectivity is a GDE property in regime I, independent of its previous operation. It is exclusively a function of the applied current density. Operating regime II (pink area in Fig. 2b), on the other hand, is characterized by gradual GDE degradation or sudden GDE failure (flooding). Continuous and irreversible degradation in this regime is verified when electrolysis is subsequently performed in regime I and the previously achieved CO selectivity no longer reached by the same electrode. Nevertheless, it is possible to operate the flow cell in a stable and meaningful manner in regime II, if accelerated degradation is acceptable. When a further increase of jx leads to erratic data and uncontrollable parameters, the system has reached one or more limitations, under which the electrolyzer can no longer be operated (regime III).
The three states of operation can be identified from the slope of jCO. A positive slope of jCO with increasing jx is indicative of the stable, steady-state regime I. Once the slope of jCO turns negative, regime II is reached by the system. If there is no option to record meaningful data at higher current densities, the inaccessible region III has been reached (see Fig. 2b). We define the current density, at which the system changes from regime I to regime II, as the first point of accelerated degradation (jAD) of the GDE hereon. This point is identifiable as the first data point at which the slope of jCO changes from positive to negative. In Fig. 2b, the slope of jCO turns negative for current densities above −0.6 A cm−2. In the present GDE flow cell, −0.6 A cm−2 was therefore identified as jAD (arrow indication in Fig. 2b). At −0.8 A cm−2, the production of CO starts decreasing although jx is increased and, for the first time, less CO is being produced despite a higher electron and CO2 supply to the GDE (pink area in Fig. 2b). This behavior is not explicable by electrochemical reaction kinetics. jAD and the current density operating regimes is only visible in the jCO-plot. It is not distinct in the FECO-plot.
To rule out the possibility of the tested electrodes degrading in the low-current density regime, we performed randomized order screenings where the order of the previously chronologically applied current density steps was shuffled. Fig. 2c shows the obtained FECO for both a chronological (light blue) and a randomized-order screening (dark blue). The numbers in the data points obtained for the randomized-order screening represent the order in which the current density steps have been applied to the cell in this case. The order differs from the chronological staircase screening but was arranged in that order for comparison (see Fig. S1 of the ESI†).§ Operating points 1 and 2 were both measured in regime I, without entering regime II, and show comparable CO selectivity to the step-wise increased chronological screening results at the respective current densities. Operating point 3 coincides with jAD in the chronological screening: the GDE still reduces CO2 at similar rates as observed for electrodes in the staircase screenings. Subsequently, lowering jx and moving the operating state back to regime I shows a minimal decrease in selectivity by −6.3% FECO compared to the previously achieved rate (71.3% FECO for the chronological screening). Deviations from the chronological screening up to and including point 4 are within the error margins of the instruments used for product analysis in our system. The following point 5 (40.6% FECO) fell far within the range of operating regime II. This caused the electrode to experience accelerated degradation. Subsequently, shifting the state of cell operation into regime I (point 6) once again increased the FECO. However, the observable GDE performance stayed well below the efficiency observed for electrodes that were not yet operated in regime II (42.6% FECO in the randomized screening vs. interpolated 61.0% FECO in the chronological screening). A performance decrease in terms of FECO was evident for all successive data points recorded within the lower current density regime.
Continued electrolysis in regime II resulted in additional degradation of the GDE (point 7) and further decrease of FECO (36.6% FECO at point 7). The results indicate that GDE performance is not affected by the applied current load below a current density of −0.6 A cm−2, from which point onwards it lost its peak performance irreversibly. Therefore, the data generated by the chronological GDE screening method is representative within the current density regime I, up to jAD, the first point of accelerated GDE degradation. In the following, the newly introduced parameters jAD and jmax will be used to assess the impact of process parameters on the GDE performance in CO2 reduction.
Microscopic analysis of the GDEs before and after the electrochemical screening procedures revealed no morphological changes in either the catalyst layer or in the gas diffusion layer structure (see Fig. 3b vs. c). This further indicates that the electrodes could not have lost all functionality when the system ceased to operate in a controlled manner.
 | ||
| Fig. 3 SEM micrographs of Ag-coated electrodes (a and b) before and (c) after electrolysis. (a) 45° top view of the catalyst layer on the electrode surface (BSE-detector). (b) Catalyst layer cross-section of the GDE before electrolysis (BSE-detector). (c) Catalyst layer cross-section of the GDE after electrolysis (BSE-detector). | ||
From these observations, we conclude that the GDE is not necessarily the single process-limiting component for high current density CO2 electrolysis. Thus, the system is constrained by additional factors stemming from the design of the electrolyzer and the evolution of its reaction environment. To elucidate this, we changed the GDE environment in terms of the electrolyte concentration (1 M, 2 M, 3 M), the electrolyte temperature (with or without cooling), by augmenting system pressures (no pressure regulation, liquid-sided overpressure at the GDE, fully pressurized system), and by comparing two settings for the CO2 supply (gas supply as a constant CO2-to-electron ratio with 9-fold excess of CO2 and a constant volumetric CO2 flow rate over the entire range of jx). We were specifically looking for mechanisms that led to the observed instable and uncontrolled process state that terminated experiments at high current densities in the flow cell. The following sections present how changes in the electrode environment affect the process and performance of GDEs in their ability to reduce CO2.
3.2 Current density dependent steady-states of the process
To monitor the evolution of system parameters with increasing jx, such as the catholyte temperature and system pressures, a number of inline sensors have been built into the GDE flow cell according to Fig. 5. An example of a complete dataset obtained by the chrono-potentiometric screening procedure is depicted in Fig. 4. Individual parameters are discussed in detail in the following sections.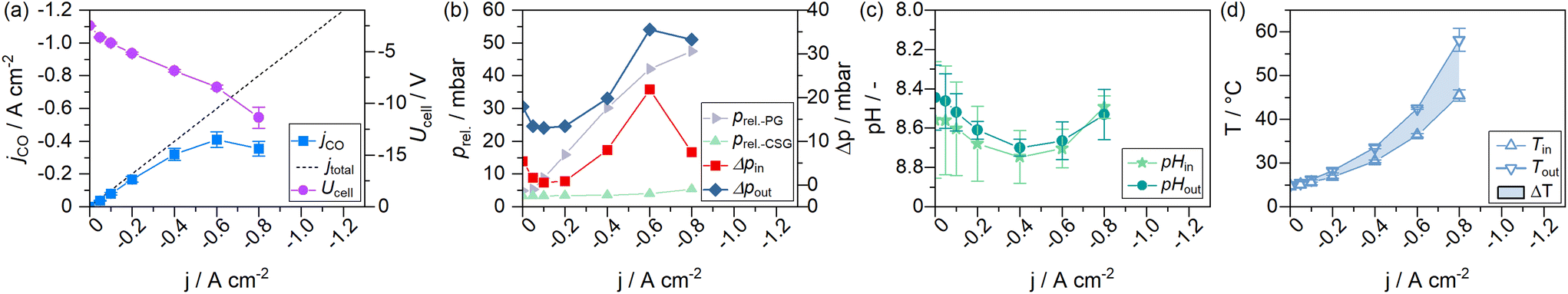 | ||
| Fig. 4 Current-dependent steady-states of a GDE flow cell operated with 1 M electrolytes. (a) Voltage and CO-specific current density, (b) relative system pressures (PG – product gas, CSG – catholyte sweep gas), (c) and the catholyte pH entering and exiting the GDE flow cell, and (d) the catholyte temperatures entering and exiting the flow cell. | ||
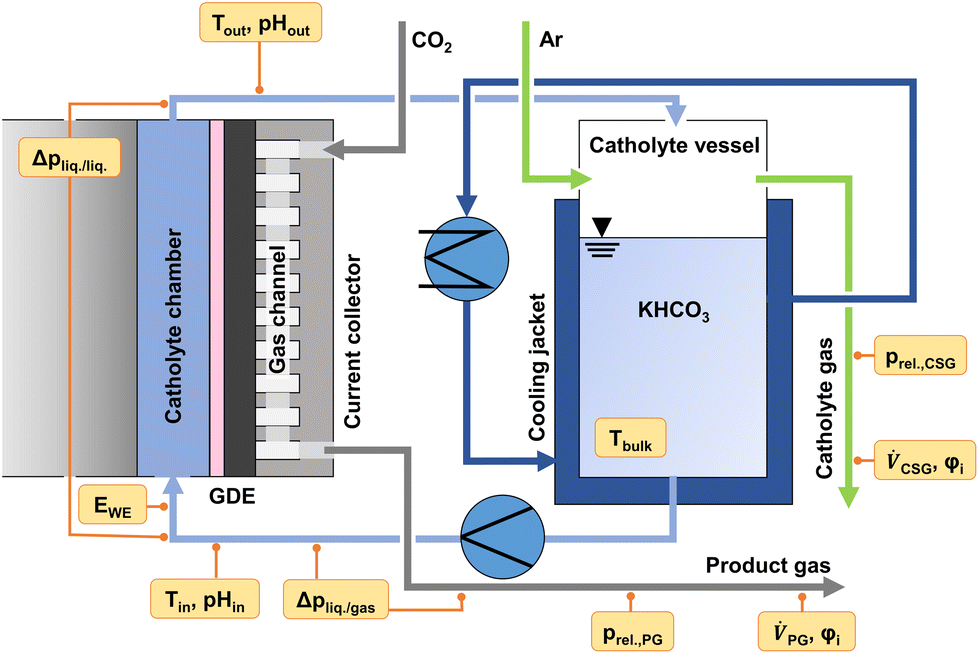 | ||
| Fig. 5 GDE cell environment and inline sensors built into the GDE flow cell setup (WE: working electrode, PG: product gas, CSG: catholyte sweep gas). | ||
It is evident that almost all measured parameters are dependent on jx. While for static cells, the assumption of near-constant process conditions around the electrodes may still be a realistic approximation, the parameters in GDE flow cells change profoundly with jx (to the point of sign inversion in some parameters, as presented in the following sections). A significant reason for this is the ratio of electrolyte to the applied current in the catholyte gap.
The overall electrolyte volume in an H-Cell and a flow cell setup may be quite similar, while the GDE flow cell may operate at 100–1000 times higher current densities. This is exacerbated in flow cells with thin electrolyte gaps, resulting in a very small electrolyte volume present in the cell itself, which amplifies induced changes to the electrode environment by accelerated reaction rates. The catholyte bulk pH could be identified as the only near-constant parameter in our cell setup (see Fig. S4 of the ESI†). It was measured in the electrolyte stream entering and exiting the GDE flow cell. It is, therefore, only representative of bulk electrolyte properties. Only minor variations in the catholyte pH were expected in the presence of a bicarbonate buffer system (1 M KHCO3). Fig. 4c shows the catholyte pH to remain at a constant value of 8.6 ± 0.15 over the entire screening range, with a deviation of ΔpH 0.1 within the catholyte cell compartment.
3.3 Impact of electrolyte concentration on temperature evolution and selectivity
The ionic resistance in the cell is largely given by the electrolyte concentration. With a higher electrolyte concentration, less energy is lost to heating, resulting in a higher energy efficiency.42 From experiments with single-pass electrolytes it has been reported that electrolytes with higher concentrations result in higher CO selectivity.43 To elucidate how the catholyte concentration affects CO selectivity and the steady-states of cell operation in a system with cycled electrolytes, we conducted the staircase screening experiments with 1 M, 2 M, and 3 M electrolytes, respectively.Fig. 6 shows a similar trend for jCO up to a current density of −0.6 A cm−2 for all concentrations. The CO selectivity for experiments conducted with 2 M and 3 M electrolytes was identical within the error bars. Yet, when using 1 M catholytes, the selectivity was consistently higher. Above −0.6 A cm−2 the cell reached jAD when 1 M catholytes were used, whereas 2 M catholytes enabled a shift of jAD to −1 A cm−2, and −0.8 A cm−2 for 3 M catholytes. Following the same trend, jmax was increased from −0.8 A cm−2 at 1 M, to −1.2 A cm−2 at 2 M, and −1.0 A cm−2 at 3 M electrolytes.
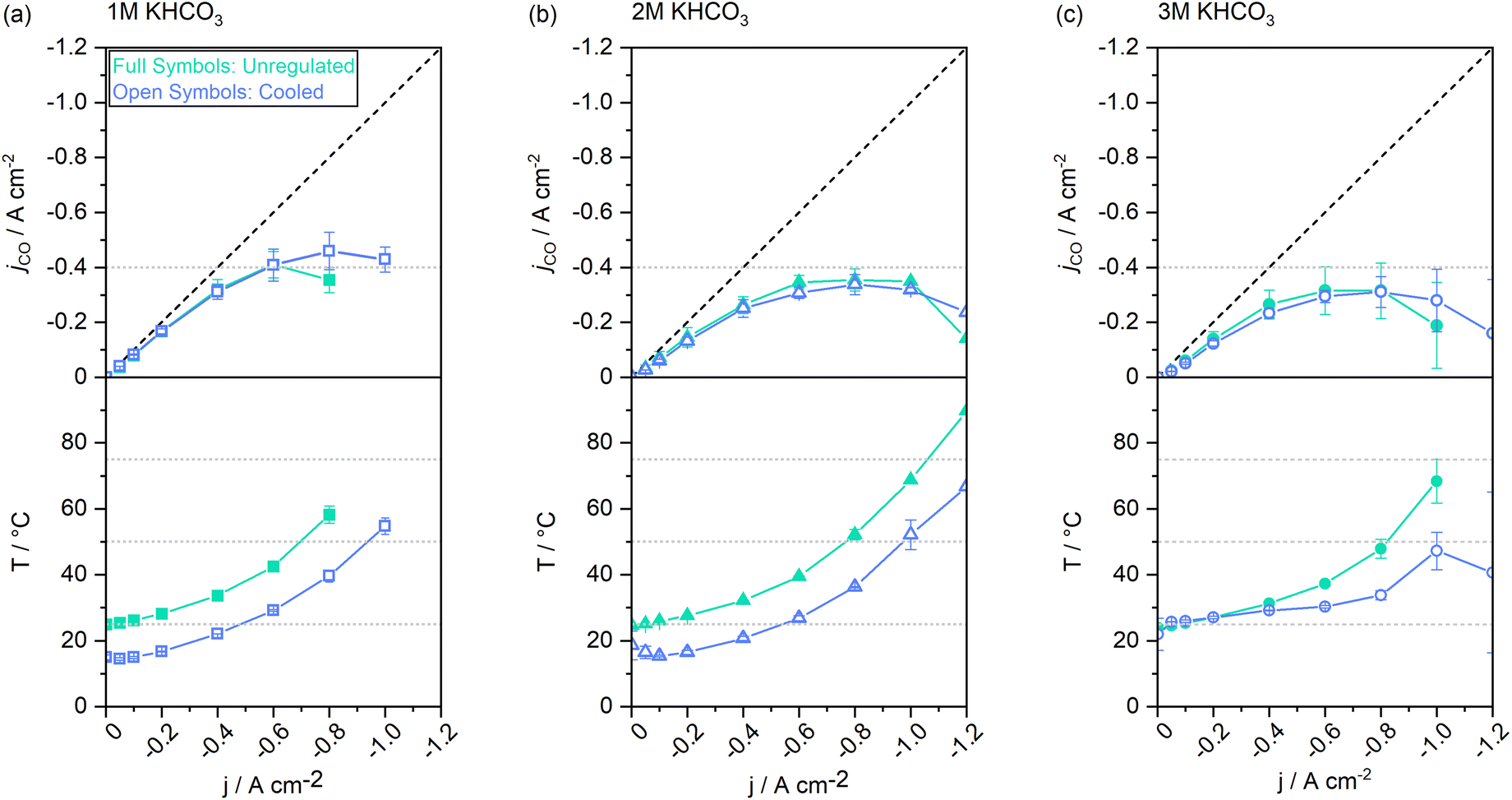 | ||
| Fig. 6 CO-specific current densities and temperature evolution throughout the chrono-potentiometric screenings at varied electrolyte concentrations with vs without electrolyte cooling. (a) 1 M electrolytes, (b) 2 M electrolytes, and (c) 3 M electrolytes. | ||
In contrast to previous observations with non-cycled electrolytes,43 the selectivity moderately declined for the higher electrolyte concentrations. Additional effects, such as the decreasing solubility of CO2 at increased salt concentrations,44–46 may have limited the benefits of increasing electrolyte concentrations beyond an optimum.¶ It has also previously been demonstrated that at high current densities, higher cation and HCO3− concentrations may negatively impact the kinetics of the electro-catalytic conversion at the catalyst itself.47 Because electrolytes are cycled in this study, heat accumulation in the electrolyte needs to be addressed.
In the lower half of Fig. 6 the measured catholyte temperatures at the cell outlet are shown. Upon reaching jmax, the cell operated with 1 M electrolytes reached temperatures up to 58 °C. At the same current density, the 2 M experiment reached a temperature of 52 °C. The 3 M experiment remained the lowest with 48 °C. Thus, the use of 2 M electrolytes reduced the steady-state catholyte temperature at −0.8 A cm−2 by 6.0 K, 3 M electrolytes further reduced the temperature by 4.3 K. At their respective jmax, the 2 M experiment reached a maximum of 90 °C, and 68 °C were reached in the catholyte gap of the 3 M experiment. As the electrolyte volume in the catholyte gap that absorbs all heat generated from the cell, is only 3 mL (1.5% of the total catholyte volume), higher jx also result in a larger differential between catholyte cell entry and cell exit temperatures. A maximum temperature difference of up to 20 K at −0.8 A cm−2 was measured in the cell's catholyte gap for 1 M experiments (see Fig. 4d).
Furthermore, higher electrolyte concentrations led, as expected, to a reduction in cell voltage in regime I (see Fig. 7). When the catholyte temperature increased, the Ohmic resistance in the cell was lowered, which benefitted energy efficiency in the electrolyzer (see Fig. 7, regime I). In regime II, an abrupt increase in the Ohmic resistance of the cell could be observed. We attribute this to an increased gas content in the liquid electrolyte due to bubble formation and the resulting progressive insulation in the cell. jAD coincides with the current density beyond which the cell voltage sees this extraordinary voltage increase (arrow indications in Fig. 7). The presence of gas bubbles likely reduced the electrode–electrolyte contact, causing local current density overloads in the catalyst layer. These hotspots presumably contributed to the accelerated GDE degradation, irreversibly damaging the catalyst layer.
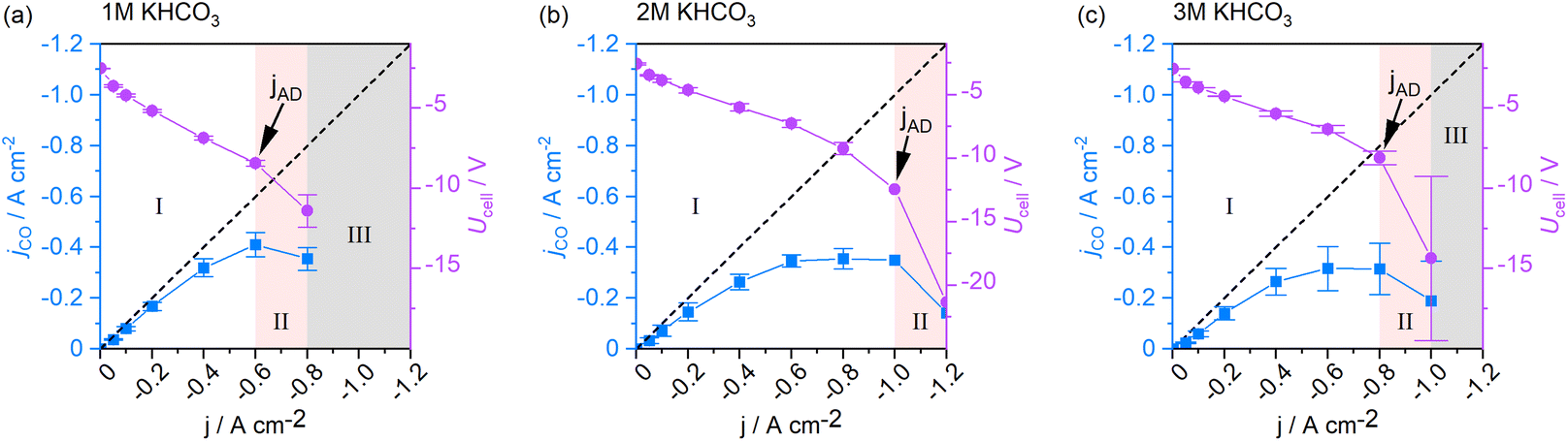 | ||
| Fig. 7 Cell voltage and CO-specific current density throughout GDE screenings using (a) 1 M, (b) 2 M, and (c) 3 M electrolytes. Regime I: steady-state cell operation, regime II: cell operation with accelerated GDE degradation, regime III: inaccessible region. | ||
We conclude that by using electrolytes with an increased salt concentration, trade-offs in CO conversion efficiency can be made in favor of electrolysis over an extended current range. Maximum possible electrolyte concentrations, however, do not benefit either of the performance indicators, as overlapping effects such as a reduced CO2 solubility44,48 come into play. We found the intermediate electrolyte concentration of 2 M KHCO3 to perform optimally in our system.
3.4 Benefits of electrolyte cooling on process stability and selectivity
In attempts to reduce the cell temperature, thus keeping CO2 solubility in the catholyte high, the experiments described in the previous section were repeated while cooling jackets actively cooled both electrolyte vessels. As a result, the overall electrolyte temperature could be kept below 70 °C for all jx, whereas previous experiments reached temperatures of >90 °C (see Fig. 6). Similar to the reduced heating effect through the increase in electrolyte concentration, active cooling of the electrolytes led to an extension of the current density range applicable to our GDE flow cell.On average, the catholyte temperature in the actively cooled cell got reduced by 8 to 12 K compared to the cell without electrolyte cooling jackets. jCO in the cell operated with 1 M electrolytes improved by an additional −0.11 A cm−2 at −0.8 A cm−2. Electrolysis could be performed at −1.0 A cm−2, whereas the non-cooled system could not sustain this current through the cell without voltage overloads. For systems operated with 2 M and 3 M electrolytes, the active cooling system did not lead to significant changes in product selectivity, as the CO-specific current density at any given data point was comparable.||
Our data suggest that active electrolyte cooling has no discernible impact on product selectivity. Thus, the lowered temperature and the resulting increase in CO2 solubility had no significant benefits on the selectivity of the catalyst. However, the current density range that can be applied to a GDE flow cell could be substantially extended.
3.5 Effects of gas pressure control on the system
The differential pressure of liquid catholyte and CO2 feed gas within the GDE is decisive to the electrode's catalyst efficiency. If the gaseous phase does not reach the catalyst layer, CO2 supply to the catalyst particles is limited, which leads to larger amounts of water being reduced to H2. If the catalyst particles are not sufficiently wetted by the catholyte phase on the other hand, the available electro-chemically active surface area is reduced.Depending on the differential pressure within the GDE, two operation modes can be distinguished, in which the CO2 gas stream may be supplied to the catalyst layer: flow-by and flow-through.16 Flow-by conditions exist when the gas–liquid interface is formed within the catalyst layer and there is no gas flow into the catholyte gap. CO2 molecules diffuse into the catalyst layer towards the catalytically active sites, and the gaseous products diffuse back into the gas channel. This condition is favorable. The second operation mode, flow-through, occurs when the gas-sided overpressure towards the electrolyte exceeds the GDE's bubble point pressure (ΔpGDE = pliquid − pgas < pbubble point < 0).49 Flow-through of CO2 into the catholyte gap is unfavorable for many reasons. First of all, the presence of gas bubbles adds to the overall cell resistance and may cause current fluctuations. Secondly, the GDE catalyst layer may take damage from the additional stress caused by the bubbles and current hotspots. This may result in early accelerated degradation, effectively reducing jAD. A third reason is the acidification of the aqueous electrolyte by increased amounts of CO2 dissolving (electrolyte degradation).16 We, therefore, aim to operate our GDE flow cell at flow-by conditions only.
Fig. 8a depicts the evolution of system pressures during an experiment without any pressure regulation. With increasing current density and correspondingly increasing CO2 feed flows in the system (const. CO2 excess with λ = 10) the gas-sided pressure at the GDE increased almost linearly. The sweep gas that was used to flush out gaseous products from the catholyte cycle, was kept at a constant feed rate. Correspondingly, pressure in the sweep gas did not change between the current steps. For the GDE, liquid-sided overpressure could be observed during the screening. This overpressure increased with increasing current density until jAD was reached. From −0.6 A cm−2 to −0.8 A cm−2, we observed a collapsing liquid–gas differential pressure at the cell inlet (Δpin in Fig. 8a). At the same time, a slight drop in the slope of the feed gas pressure (prel.-PG) was observed. Additionally, a slight increase in the relative catholyte sweep gas pressure (prel.-CSG) was observed. Because no pressure regulation was conducted, the pressure is expected to be proportional to the gas flow. Hence, a drop in the slope of the feed gas pressure means that the gas stream pressure has not increased by the expected amount and that the total flow in the sweep gas stream must have been increased. We resume that part of the CO2 feed gas stream must have passed through the GDE into the catholyte gap, indicating CO2 flow-through conditions.
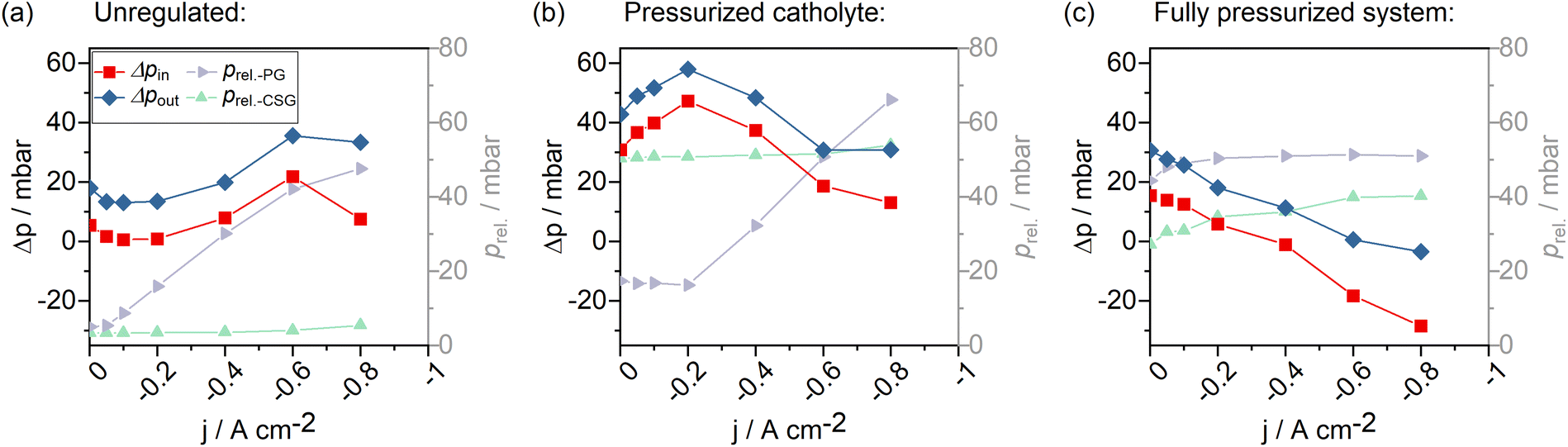 | ||
| Fig. 8 Pressure evolution in the cell with different pressure control settings (1 M electrolytes). (a) System without pressure control, (b) back pressure regulation of catholyte sweep gas stream, (c) back pressure regulation of both catholyte sweep gas stream and product gas stream. Δpin: Differential liquid-to-gas pressure at the GDE – cell entry, Δpin: differential liquid-to-gas pressure at the GDE – cell exit, prel.-PG: relative pressure in the product gas stream, prel.-CSG: relative pressure in the catholyte sweep gas stream. | ||
Applying an overpressure of 40 mbar** to the catholyte sweep gas pressure resulted in an increase in pressure within the liquid catholyte gap (overall increase of Δpin to liquid-sided overpressures in Fig. 8b). We found that it is possible to regulate the pressure of the liquid catholyte by pressurizing the headspace gas in the electrolyte reservoir. Pressurizing both the catholyte sweep gas and the product gas to 40 mbar and 60 mbar, respectively, caused the differential gas–liquid pressure within the GDE to gradually change from a liquid-sided overpressure to a gas-sided overpressure, alongside reduced CO-production and an increased flow-through of the CO2 feed gas into the catholyte compartment (Fig. 8c). This further increased the gas content in the catholyte gap.
All in all, we observed the pressure gradient within the electrode to depend strongly on the applied current density, when the electrolyzer is operated at a constant CO2 excess. A liquid-sided overpressure ensured CO2 flow-by conditions. We further observed that, by regulating the catholyte headspace gas pressure, it is possible to directly tune the GDE-liquid sided catholyte pressure.
3.6 Comparison of CO2 feed settings
The experiments to this point have been carried out with a constant 9-fold over supply of CO2 to avoid effects from CO2 concentration gradients in the gas channel. This was done because a 9-fold excess of an educt in a general second-order order reaction (reaction rate dependency on the availability of CO2 and electrons) may be regarded as a pseudo-first-order reaction (only dependent on the applied current density jx).50 CO2 concentration gradients within the gas channel could be neglected. To assess the necessity of a constant CO2 supply ratio and its impact on the formation of steady-states, we performed electrolysis with a CO2 feed of 282.5 mLn min−1 that was kept constant over the entire electrochemical screening range. This setting corresponds to the previously used λ = 10 at −0.4 A cm−2. With a constant CO2 flow rate, λ changes throughout the measurement, e.g., to λ = 20 at −0.2 A cm−2 and λ = 5 at −0.8 A cm−2.Fig. 9a shows that the values for jCO were much lower for cells supplied with a constant CO2 feed stream, than previously observed for cells supplied with a constant CO2 excess of λ = 10 (see Fig. 4a), at otherwise identical conditions. At −0.2 A cm−2 a jCO of −0.11 A cm−2 could be observed (56.9% FECO), whereas a nearly 40% higher efficiency was observed in a system with 9-fold CO2 excess (−0.16 A cm−2jCO, 83.6% FECO). At a current density of −0.8 A cm−2, a selectivity jCO of −0.24 A cm−2 could be observed at λ = 5 and −0.39 A cm−2 for λ = 10. The catholyte temperature evolved the same way for both experiments (see Fig. 4d and 9c). Similarly, the CO2 excess had no discernible impact on the pH in the catholyte gap (see Fig. 9c).
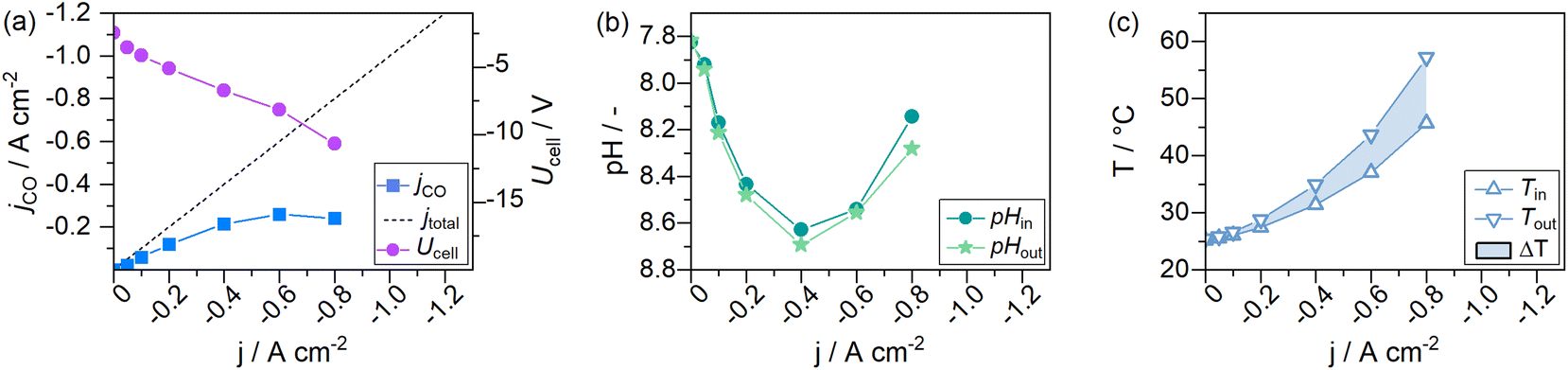 | ||
Fig. 9 Current-dependent steady-states of a GDE flow cell operated at a constant CO2 feed rate (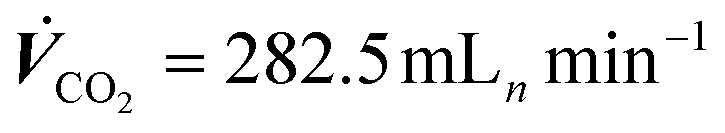 ). (a) Cell voltage and CO-specific current density, (b) catholyte pH entering and exiting the GDE flow cell, and (c) catholyte temperatures entering and exiting the flow cell. ). (a) Cell voltage and CO-specific current density, (b) catholyte pH entering and exiting the GDE flow cell, and (c) catholyte temperatures entering and exiting the flow cell. | ||
From this, we conclude that a constant CO2 feed ratio is beneficial when operating CO2 electrolyzers at different current loads.
3.7 Prospect of passive pressure regulation in large-scale electrolyzers
Fig. 8a shows a near-linear pressure build-up with increasing current density when the experiment was conducted with constant λ. For experiments with constant gas flow, the product-gas pressure was nearly constant (see Fig. S2 of the ESI†). Hence, the observed pressure evolution originated from the flow-resistance downstream the cell and the product gas flow and viscosity. Operating the cell with constant λ has not only provided a constant CO2 excess but also provided a gas-sided, self-regulated pressure adjustment according to the applied current. Attempts to keep the gas-sided pressure at a constant value or keeping the CO2 feed flow constant did not prove beneficial to GDE performance. Hence, adding a flow-restriction downstream the cell in combination with a constant CO2 excess may be used to improve the dynamic range of an electrolyzer without having to resort to expensive, digitally controlled pressure regulators.3.8 Interdependence of process parameters
In a highly complex process like CO2 electrolysis, it is rather unlikely that the process parameters are independent. We looked into the effects of changing from 1 M to 3 M catholytes on the other process parameters to evaluate these dependencies.Comparing the two scenarios, an increase in electrolyte concentration did not significantly change the observed relative system pressures in the catholyte sweep gas stream and product gas stream. However, a notable difference could be observed in the pressure difference within the liquid catholyte gap (Δpin at the cell entry vs. Δpout at the cell exit, Fig. 10). As gas solubility in aqueous solutions decreases with an increasing salt concentration,45,46 the increased pressure gradient within the catholyte gap using a 3 M catholyte has likely been caused by more significant amounts of gas leaving the liquid solution in a gaseous form (additional bubble formation) compared to the experiment using 1 M electrolytes.
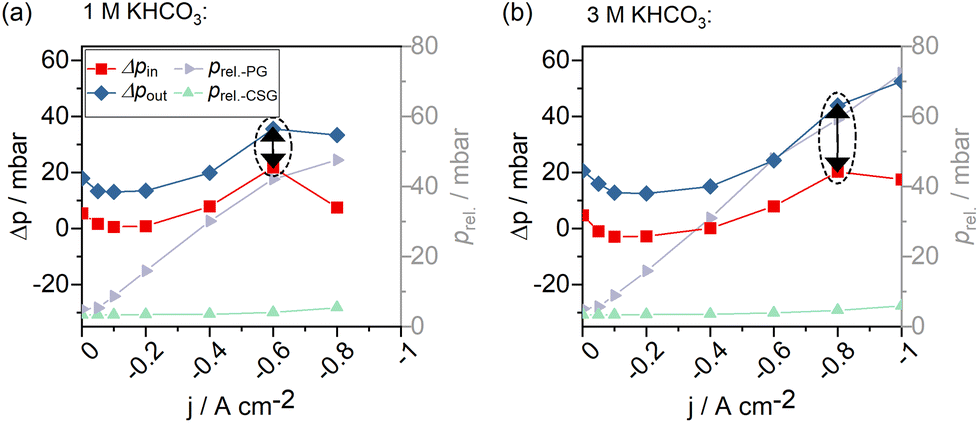 | ||
| Fig. 10 Pressure evolution in a GDE flow cell operated with (a) 1 M electrolytes and (b) 3 M electrolytes. Δpin: Differential liquid-to-gas pressure at the GDE – cell entry, Δpin: differential liquid-to-gas pressure at the GDE – cell exit, prel.-PG: relative pressure in the product gas stream, prel.-CSG: relative pressure in the catholyte sweep gas stream. | ||
At the end of section 3 we stated that there can be multiple reasons for a reduced CO selectivity when operating the electrolyzer with highly concentrated 3 M catholyte (see also Fig. 7c). When increasing the catholyte concentration, effects from the catalyst, temperature, and gas bubble formation due to the salting-out effect cannot be clearly distinguished yet. We have further observed that the pressure gradient in the catholyte gap is directly impacted by the catholyte concentration as well (Fig. 10). The catholyte concentration may, therefore, also have an indirect effect on the GDE selectivity via the pressure in the catholyte gap.
Changing one parameter (electrolyte concentration) caused both changes in the differential pressure within the GDE and a deterioration in reaction selectivity. We note that any change in the cell system (e.g., a change in the electrolyte concentration) could have the potential to impact all other GDE-, cell- and system-level parameters. This dependency must be recognized, and the GDE performance must be isolated. Without resolved parameter effects, cause-and-effect questions cannot be answered.
3.9 Salt formation and flooding
Despite recent advances, flooding and salt formation within the GDE are still commonly reported in the literature.26,51 We did not observe any salt depositions on the gas-side of the GDEs or notable amounts of permeate. In experiments in which the catholyte was pressurized, small amounts of salt crystals were found in the gas flow channel. This may be due to minor amounts of electrolyte permeating through cracks in the GDL.52,53 The permeate did not stick and dry on the gas-side of the GDL. We attribute this to the high hydrophobicity of the PTFE-treated carbon paper.4 Conclusions
In this work, we investigated the steady-states of GDE-relevant process parameters in the high current density regime and explore the limitations of CO2 electrolysis.It became apparent that process limitations are linked to the catholyte medium and arise from processes taking place in the catholyte gap. Increased reaction rates at high current densities generate significant amounts of heat around the electrodes that is transferred into the electrolytes. In the catholyte, this leads to bubble formation through degassing and water vapor formation. In addition, high CO2 flow rates at increased current densities may result in a gas-sided overpressure within the GDE that leads to undesirable CO2 flow-through conditions. Insufficient contact between the GDE and the catholyte causes over-proportional increases in the cell voltage. As soon as the gas-to-liquid ratio in the catholyte gap becomes too high, currents can no longer be passed through the cell in a controlled way.
By increasing electrolyte conductivity and by electrolyte cooling, it has been possible to extend the operating current range of a lab-scale flow cell from −8 A to −12 A on a geometrical active GDE area of 10 cm2. Results are summarized in Table 1.
| Electrolyte molarity and temperature: | j AD | j max | FECO at −0.4 A cm−2 |
|---|---|---|---|
| 1 M | −0.6 A cm−2 (68.3% FECO) | −0.8 A cm−2 (44.2% FECO) | 79.9% FECO |
| 2 M | −1.0 A cm−2 (34.8% FECO) | −1.2 A cm−2 (11.8% FECO) | 65.8% FECO |
| 3 M | −0.8 A cm−2 (39.4% FECO) | −1.0 A cm−2 (18.9% FECO) | 66.1% FECO |
| Cooled 1 M | −0.8 A cm−2 (57.5% FECO) | −1.0 A cm−2 (42.9% FECO) | 77.7% FECO |
| Cooled 2 M | −0.8 A cm−2 (42.3% FECO) | −1.2 A cm−2 (19.7% FECO) | 62.7% FECO |
| Cooled 3 M | −0.8 A cm−2 (38.8% FECO) | −1.2 A cm−2 (22.7% FECO) | 58.3% FECO |
| System pressures: | j AD | j max | FECO at −0.4 A cm−2 |
|---|---|---|---|
| Unregulated | −0.6 A cm−2 (68.3% FECO) | −0.8 A cm−2 (44.2% FECO) | 79.9% FECO |
| Electrolyte-sided overpressure | −0.8 A cm−2 (55.8% FECO) | −0.8 A cm−2 (55.8% FECO) | 80.8% FECO |
| Fully pressurized | −0.6 A cm−2 (46.3% FECO) | −0.8 A cm−2 (27.5% FECO) | 62.7% FECO |
| CO2 feed setting: | j AD | j max | FECO at −0.4 A cm−2 |
|---|---|---|---|
| Const. λ | −0.6 A cm−2 (68.3% FECO) | −0.8 A cm−2 (44.2% FECO) | 79.9% FECO |
| Const. CO2 flow rate | −0.6 A cm−2 (40.5% FECO) | −0.8 A cm−2 (29.5% FECO) | 52.8% FECO |
We resume that the GDE is not the singular critical component moving toward high current density CO2 electrolysis. To realize long-term experiments, one must address not only GDE improvements, but also the entire cell and system design. We consider the design of the catholyte gap the necessary next step in the development of CO2 flow electrolyzers intended to operate at elevated current densities. This study highlights the importance of monitoring not only electrode performance but also GDE-relevant process environment variables. Electrode benchmarking in a single cell with a single set of experimental parameters does not necessarily reflect a GDE's true performance unless cell limitations can be excluded and the reactor's steady-state operation windows are known. This study complements studies optimizing the GDE design and materials by presenting and analyzing performance enhancing aspects stemming from the cell conditions surrounding the GDE. Thus, we take a further step towards producing green CO as well as syngas (CO, H2), whereby downstream and end products are also de-fossilized.
Author contributions
Conceptualization, C. M. and B. S.; data curation, C. M.; formal analysis, C. M. and B. S.; funding acquisition, B. S., H. T., R.-A. E.; investigation, C. M.; methodology, C. M. and B. S.; project administration, B. S., H. T., R.-A. E.; resources, R.-A. E.; supervision, B. S., H. T., R.-A. E.; visualization, C. M.; writing – original draft preparation, C. M. and B. S.; writing – review and editing, C. M., B. S., H. T., R.-A. E. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors kindly acknowledge funding by the Federal Ministry of Education and Research (BMBF), Project: iNEW2.0 “Inkubator Nachhaltige Elektrochemie” Funding code: 03SF0627A. The authors thank M. Quentmeier for designing the titanium flow frame,18 M. Turiaux for supplying gaskets to assemble the GDE flow cell, and A. Luft for his dedicated maintenance of the lab equipment.References
- Z. Jiang, T. Xiao, V. Kuznetsov and P. Edwards, Philos. Trans. R. Soc., A, 2010, 368, 3343–3364 CrossRef CAS PubMed.
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- IPCC, Summary for Policymakers, Cambridge University Press, 2022, pp. 1–24 Search PubMed.
- European Commission, Towards a circular economy: A zero waste programme for Europe, Communication, 2014, 52014DC0398 Search PubMed.
- D. C. Deselnicu, G. Militaru, V. Deselnicu, G. Zăinescu and L. Albu, Proceedings of the 7th International Conference on Advanced Materials and Systems, Bucharest, Romania, 2018, pp. 18–20.
- P. Ghisellini, C. Cialani and S. Ulgiati, J. Cleaner Prod., 2016, 114, 11–32 CrossRef.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustain. Chem., 2019, 16, 47–56 CrossRef.
- R. Pierantozzi, Carbon Monoxide, John Wiley & Sons, Ltd, 2000 Search PubMed.
- W. Paik, T. Andersen and H. Eyring, Electrochim. Acta, 1969, 14, 1217–1232 CrossRef CAS.
- G. Denuault, M. Sosna and K.-J. Williams, Handbook of Electrochemistry, Elsevier, Amsterdam, 2007, pp. 431–469 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- B. Endródi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- S. Hernandez-Aldave and E. Andreoli, Catalysts, 2020, 10, 713 CrossRef CAS.
- Y. Yang and F. Li, Curr. Opin. Green Sustain. Chem., 2021, 27, 100419 CrossRef CAS.
- M. Quentmeier, B. Schmid, H. Tempel, H. Kungl and R.-A. Eichel, ACS Sustainable Chem. Eng., 2023, 11, 679–688 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- J. Durst, A. Rudnev, A. Dutta, Y. Fu, J. Herranz, V. Kaliginedi, A. Kuzume, A. A. Permyakova, Y. Paratcha, P. Broekmann and T. J. Schmidt, Chimia, 2015, 69, 769–776 CrossRef CAS PubMed.
- J. J. Carroll, J. D. Slupsky and A. E. Mather, J. Phys. Chem. Ref. Data, 1991, 20, 1201–1209 CrossRef CAS.
- B. Jähne, G. Heinz and W. Dietrich, J. Geophys. Res.: Oceans, 1987, 92, 10767–10776 CrossRef.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 607 CrossRef CAS.
- N. T. Nesbitt, T. Burdyny, H. Simonson, D. Salvatore, D. Bohra, R. Kas and W. A. Smith, ACS Catal., 2020, 10, 14093–14106 CrossRef CAS.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Y. Wu, S. Garg, M. Li, M. N. Idros, Z. Li, R. Lin, J. Chen, G. Wang and T. E. Rufford, J. Power Sources, 2022, 522, 230998 CrossRef CAS.
- A. Lamibrac, J. Roth, M. Toulec, F. Marone, M. Stampanoni and F. N. Büchi, J. Electrochem. Soc., 2015, 163, F202–F209 CrossRef.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- B. Endródi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef PubMed.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- G. A. El-Nagar, F. Haun, S. Gupta, S. Stojkovikj and M. T. Mayer, Nat. Commun., 2023, 14, 2062 CrossRef CAS PubMed.
- J. Wicks, M. L. Jue, V. A. Beck, J. S. Oakdale, N. A. Dudukovic, A. L. Clemens, S. Liang, M. E. Ellis, G. Lee, S. E. Baker, E. B. Duoss and E. H. Sargent, Adv. Mater., 2021, 33, 2003855 CrossRef CAS PubMed.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- Q. Wang, H. Dong, H. Yu and H. Yu, J. Power Sources, 2015, 279, 1–5 CrossRef CAS.
- M. Schalenbach, B. Hecker, B. Schmid, Y. E. Durmus, H. Tempel, H. Kungl and R.-A. Eichel, Electrochem. Sci. Adv., 2023, 3, e2100189 CrossRef CAS.
- M. Löffelholz, J. Osiewacz, A. Lüken, K. Perrey, A. Bulan and T. Turek, Chem. Eng. J., 2022, 435, 134920 CrossRef.
- J. W. Blake, V. Konderla, L. M. Baumgartner, D. A. Vermaas, J. T. Padding and J. W. Haverkort, ACS Sustainable Chem. Eng., 2023, 11, 2840–2852 CrossRef CAS PubMed.
- T. F. Fuller and J. N. Harb, Electrochemical Engineering, John Wiley & Sons, 1st edn, 2018, pp. 8–9 Search PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- J.-B. Vennekötter, T. Scheuermann, R. Sengpiel and M. Wessling, J. CO2 Util., 2019, 32, 202–213 CrossRef.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- R. E. Zeebe and D. Wolf-Gladrow, in CO2 in Seawater: Equilibrium, Kinetics, Isotopes, Elsevier Science B.V., 1st edn, 2001, vol. 65, ch. 1, pp. 9–11 Search PubMed.
- M. König, J. Vaes, E. Klemm and D. Pant, iScience, 2019, 19, 135–160 CrossRef PubMed.
- M. R. Singh, E. L. Clark and A. T. Bell, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC.
- G. Marcandalli, A. Goyal and M. T. M. Koper, ACS Catal., 2021, 11, 4936–4945 CrossRef CAS PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- R. I. Peinador, O. Abba and J. I. Calvo, Membranes, 2022, 12, 212 CrossRef CAS PubMed.
- S. W. Benson, The Foundations of Chemical Kinetics, R.E. Krieger, 1982, pp. 81–82 Search PubMed.
- T. Zhang, Z. Li, X. Lyu, J. Raj, G. Zhang, H. Kim, X. Wang, S. Chae, L. Lemen, V. N. Shanov and J. Wu, J. Electrochem. Soc., 2022, 169, 104506 CrossRef CAS.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, Small Methods, 2022, 408, 1–8 CAS.
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, ACS Appl. Energy Mater., 2022, 5, 15125–15135 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02140h |
| ‡ Ion exclusion chromatography was used to detect charged products such as formate in the catholyte after the screening procedures. Analysis showed that the amount of liquid products remained well below 1%, which is within the error margins of the instrument and calibration data. It is therefore not reported in this study and regarded as noise. |
| § FEH2 is not displayed in Fig. 2c, but completes the balance up to 100% total FE. |
| ¶ While the CO2 is supplied to the GDE via the gas stream, recent studies report that the reduction reaction takes place in a liquid film on the catalyst.24 Hence, CO2 solubility matters. |
| || Information on the evolution of corresponding cell voltages can be found in Fig. S3 of the ESI.† |
| ** A pressure difference of +20 mbar in the product gas compartment compared to the catholyte headspace sweep gas has yielded the highest jCO at −0.2 A cm−2 in the reference experiment using 1 M electrolytes in a non-pressurized flow cell and without active electrolyte cooling. This gradient was artificially generated by pressurizing the catholyte headspace sweep gas to 40 mbar and the product gas to 60 mbar, with room to tune the pressure up or down as needed. |
| This journal is © The Royal Society of Chemistry 2023 |