
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClassic vs. C–H functionalization strategies in the synthesis of APIs: a sustainability comparison
Francesco
Ferlin
 ,
Giulia
Brufani
,
Gabriele
Rossini
and
Luigi
Vaccaro
*
,
Giulia
Brufani
,
Gabriele
Rossini
and
Luigi
Vaccaro
*
Laboratory of Green S.O.C., Dipartimento di Chimica Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://www.dcbb.unipg.it/greensoc
First published on 25th August 2023
Abstract
The landscape of organic synthesis has changed in the last two decades, especially in areas such as pharmaceutical synthesis and large-scale industrial synthesis. The progress and development of synthetic methodologies together with increasing attention to environmental and safety problems, have led many researchers to study procedures that could be compatible with the sustainable development of industrial production. Until a few years ago, the production of APIs was substantially dominated by reliable and solid chemistry but which nevertheless attracted some criticism from the point of view of the length of the synthetic procedures in terms of steps, and therefore in terms of waste production. In recent years, however, we have been witnessing the adoption, even by large industrial groups, of step-economical methodologies based on direct C–H functionalization reactions which, compared to reactions such as cross-coupling or aromatic nucleophilic substitutions, promise better sustainaibility as they do not require pre-functionalized substrates, and therefore offer the intriguing possibility of reducing the number of steps to attain the target. This review presents exemplary case studies analyzed holistically on the basis of waste production (E-factor) and on the basis of environmental and safety hazard scores (ES and SHS scores) with the aim of quantifying whether the adoption of direct C–H functionalization technology is actually always the most effective strategy compared to classic approaches. It will, therefore, ultimately give the reader a quantitative evaluation of how promising in terms of sustainability the current direction of chemical production actually is compared to previous methodologies.
1. Introduction
The definition of efficient routes of synthesis (ROS) for active pharmaceutical ingredients (APIs) is an extraordinary challenge. In different yet fascinating aspects and peculiarities, this is true both at the small-scale/discovery stage and at the larger/industrial commercial scale.To consolidate and intensify the most efficient and convenient ROS, numerous variables must be considered. Of course special attention is paid to optimization of the synthetic tools that often need to be tailored for the specific task, including a long-term rational and practical strategy.1–3 In this context, the cost of raw materials, the overall validity and duration of patents associated with the synthetic process, its toxicity and safety are crucial factors determining the investment capacity and its final success in an efficient, long-lasting production.4–6 Thanks to the great attention paid to safety and sustainability, there have been great changes in the synthetic chemistry mentality, and therefore in the industrialization of the production of fine chemicals or APIs.7,8
The roots of the API industry date back to the 19th century. The industrial revolution boosted experimentation as a method of investigation, and the production of goods was profoundly transformed.9
In those years, Merck in Germany and GlaxoSmithKline in the UK, arose among the very first industries that focused on the manufacture and sale of medicines. During the second half of the 19th century, realizing the antiseptic properties of some dyes,10 Swiss entrepreneurs began to sell them as pharmaceutical products, and Sandoz, Ciba-Geigy and Roche were then established.
Meanwhile in the United States, Pfizer—already dealing with general chemicals during the American Civil War—expanded its business to answer the demand for antiseptics and painkillers.
In this panorama, Colonel Eli Lilly, a chemist and probably the archetype of the 19th-century American industrialist, after a military career opened his own pharmaceutical company. Edward Robinson Squibb opened his own laboratory, thus laying the foundations for today's Bristol Myers Squibb. The same happened with Bayer, which at the end of the 20th century, with the successful commercialization of aspirin11 became a leading company.
The period between the two world wars saw the advent of two fundamental discoveries and their development: insulin12,13 and penicillin.14,15 The former was industrially produced and distributed by Eli Lilly and the latter by a joint international collaboration (including Pfizer, Squibb, Merck, and others) which saved thousands of lives during the 2nd World War.
These two pharma milestones with their molecular complexity and need for a huge-scale production brought about a new era in the development of APIs.
Chemical companies started to collaborate and, in many cases, outsourced research led by external experts in various fields, sometimes also with governments involved in this process.16–18
This was the historical and scientific–cultural context in which many chemical commodities were developed which are still on the market today.
To access the desired APIs, two main synthetic pillars have long dominated, and still dominate, API preparation: (i) nucleophilic aromatic substitution19–24 and (ii) cross-coupling reactions.25–27 The first represents a useful and reliable tool for the functionalization of aryl moieties and for the construction of C–heteroatom bonds, while the latter is widely used for the formation of C–C bonds, especially between aryl and/or heteroaryl units. Specifically, cross-coupling reactions and palladium chemistry have had an enormous impact on medicinal chemistry and drug discovery.28 Cross-coupling based methods are sound and generally feature fast reaction times. On the other hand, they require the use of pre-functionalized arenes and, especially with the growing attention to sustainability, this represents one of the major drawbacks of these procedures.
In this context, C–H functionalization technology has arisen as an efficient synthetic methodology for the construction of C–C bonds.29–33 Direct C–H functionalization does not necessarily require pre-functionalized/oxidized materials, promising great advantages in terms of step-economy over classic cross-coupling reactions. However, site-selectivity during the C–H activation event can be troublesome, and it is generally mastered with the help of a directing group34–36 (that may eventually need to be removed). Major drawbacks of C–H functionalization reactions are commonly the use of harsh reaction conditions (elevated temperature, additives) and/or the utilization of reagents of high price, high toxicity, and low availability, which intrinsically reduce the utility of such methodologies, especially for large-scale production.
Considering the environmental and societal challenges that we are facing nowadays, a particularly important topic is to precisely address and quantify the sustainability of novel procedures to boost an improvement in synthetic chemistry in the right direction.37–39
In recent years organic synthesis has made remarkable advances in terms of overall sustainability and the progress made with the definition of the direct C–H functionalization reaction is one example.40–49 Anyway, our impression is that the benefits of employing C–H functionalization technologies are certainly conceptual. At the moment, in real practical applications, there is still little quantitative data that clearly supports the advantages associated with the use of C–H functionalization over classic methodologies.
Our impression is that the intrinsic benefit of employing C–H functionalization technologies lies mainly in its conceptual value, as quantitative evidence of its practical applications is currently limited.
On the other hand, cross-coupling reactions, which have been continuously studied over the years,50–57 are indeed very robust and well-established synthetic tools. Moreover, chemists have learned how to handle changes in solvents, additives, and conditions in order to answer some sustainability issues and preserve the high yields from the process. Therefore, these reactions appear to be more ready than C–H functionalization reactions to define sustainable synthetic strategies.
The goal of this review is to measure and compare different classic and C–H functionalization synthetic strategies to access key relevant APIs, evaluating the improvements in terms of overall process efficiency and possibly drawing some general conclusions to understand what the key parameters are to improve access to an overall sustainable synthetic route (Fig. 1).
 | ||
| Fig. 1 Classic vs. C–H functionalization approaches. | ||
With this aim, we have considered 9 largely produced and popular APIs that have drawn the attention of both industry and academia in recent years. For all these APIs we have considered the more common classic production methodologies (generally patented) (giving preference to a cross-coupling based synthetic strategy if available) and a "novel" route based on C–H functionalization technology. We first quantified these synthetic strategies by calculating the E-factor,58,59 a direct measure of the waste generated by each synthesis. Thus, a second analysis was made by considering the Environmental Score60 (ES) and the Safety Hazard Score61 (SHS) for the input solvents used either as reaction media or for the work-up procedure, separately. More precisely, the Environmental Score is based on an evaluation of the Bioaccumulation Potential (BAP), the Bioconcentration Potential (BCP) and the Inhalation Toxicity Potential (INHTP), while for the Safety Hazard Score we considered the Flammability Potential (FP), the Corrosiveness Potential as a Gas (CGP), the Corrosiveness Potential as a Liquid/solid (CLP), the Occupational Exposure Limit Potential (OELP), and the Risk Phrase Potential (RPP). In this manner, each procedure has been assigned a final score given by the sum of each ES and SHS, which gives a numerical quantification (the higher, the worse) of its environmental and safety compatibility. A third level of analysis was also based on consideration of CHEM2162 scores associated with each solvent used (both as reaction media and in the work-up), to allow a more detailed final assessment.
2. Amiodarone (antiarrhythmic)
Amiodarone (Scheme 1) is an antiarrhythmic API which is currently used to treat several cardiac diseases. Amiodarone was first synthetized beginning of the 1960s to be used to treat angina-pectoris and later in the 1970s to treat arrythmia. Amiodarone is among the most prescribed APIs and has been listed in the World Health Organization's List of Essential Medicines.63 | ||
| Scheme 1 Amiodarone chemical structure. | ||
Given its popularity, amiodarone received attention from the scientific community with many researchers studying innovative methods for its production. Scheme 2 describes the initial synthetic pathways first patented in 1966.64 The route started from preformed 2,3-disubstituted benzofuran bearing a phenol functionality 1 that can be deprotonated by NaOMe. This intermediate undergoes O-alkylation via an SN2 reaction in the presence of 2-chloro-N,N′-diethylethan-1-amine (2) to give direct access to amiodarone hydrochloride in over 77% yield.
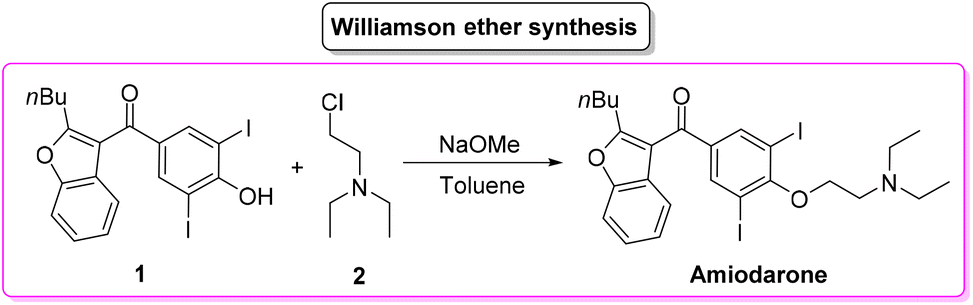 | ||
| Scheme 2 Patented synthesis of amiodarone via Williamson ether synthesis. | ||
Calculation of the E-factor associated with this synthetic protocol led to a very low value of 6.17, but it should be noted that the preparation of the starting benzofuran was not described and therefore a fair and overall E-factor cannot actually be calculated.
The influence of benzofuran synthesis on the E-factor values and on the efficiency of the overall process can be evaluated by considering the following examples.
In 2019, Gu and collaborators,65 described the total synthesis of amiodarone (Scheme 3) starting from 3,4-dihydro-2H-pyran-2-carbaldehyde (3) and 1,3-dicarbonyl derivative 4. The key-step is the synthesis of the benzofuran core via Lewis-acid catalysis involving a combination of AlCl3 and N-bromosuccinimide. The following steps are demethylation, iodination and O-alkylation to give amiodarone in a total yield of 46.2%. The first key-step (synthesis of benzofuran core) itself features an E-factor value of 149.15. The three subsequent steps have values of 220.1, 41.1, and 13.9, respectively. The overall E-factor is 337.9 for the four steps, with a total reaction time of 46 h.
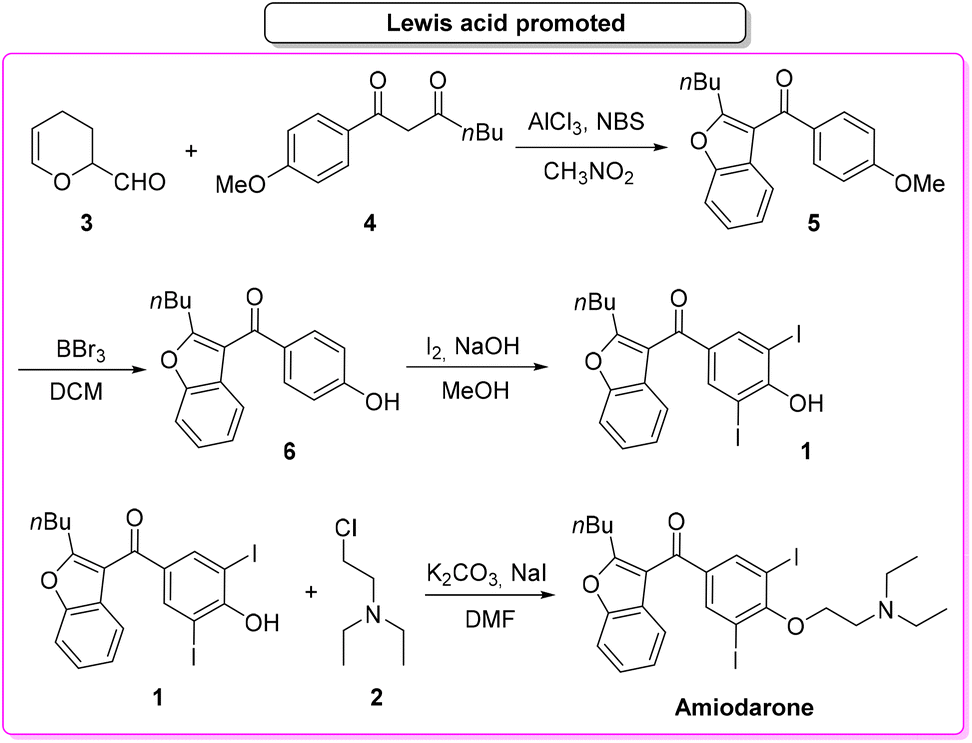 | ||
| Scheme 3 Lewis-acid-promoted synthesis of amiodarone. | ||
In the same year Maiti, Cho and co-workers66 described a synthesis of amiodarone in which the benzofuran scaffold was assembled via nickel-catalysed arylative cyclization reacting 2-alkynylphenols 7 and phenylboronic acid 8 (Scheme 4).
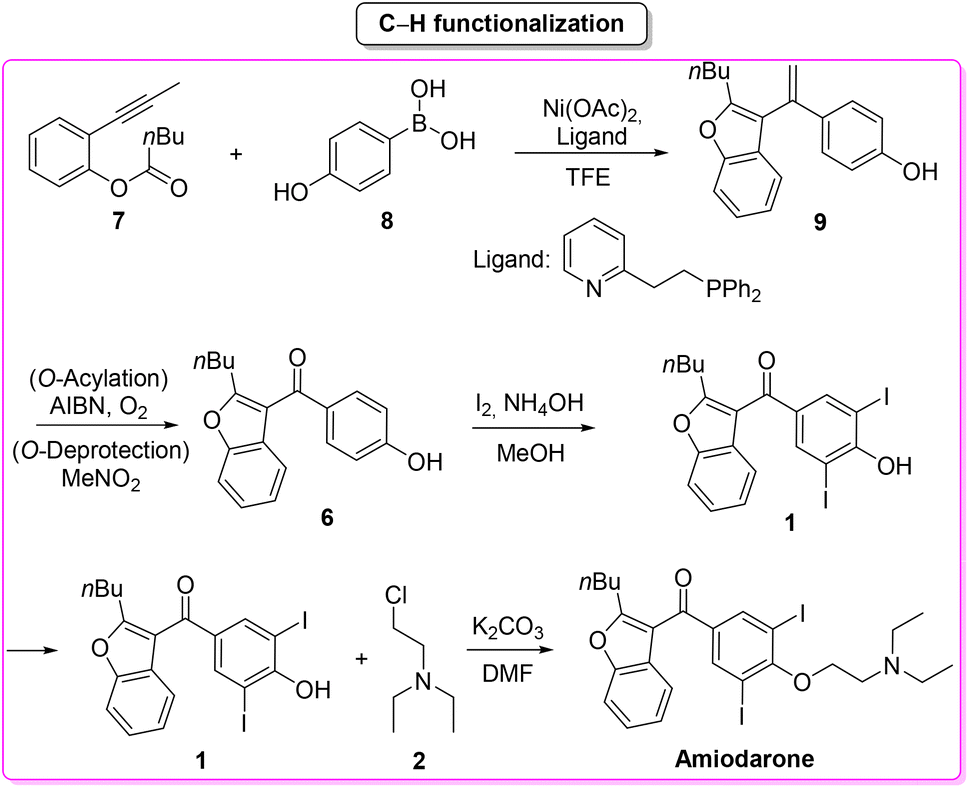 | ||
| Scheme 4 C–H functionalization toward the synthesis of amiodarone. | ||
The resulting vinylated methyl-benzofuran 9 was subsequently oxidized to give ketone intermediate 6 that after iodination and O-alkylation allowed access to amiodarone in 17% overall yield for the four steps. The E-factor associated with the formation of the benzofuran ring is 54.8, which is significantly lower than the previous Lewis-acid-catalysed example, as well as the E-factor for the three subsequent steps being 47.9, 48.0, and 19.0, respectively. The overall E-factor value for this arylative cyclization pathway is therefore 238.1.
Furthermore, in Table 1 we have compared the input SHS and ES values and reported the CHEM21 assessment for the solvent used as a reaction medium and in the work-up procedures.
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Williamson ether synthesis | |||||||
| DEC | 1.73 | 2.42 |

|
Acetone | 0.35 | 2.33 |

|
| MeOH | 0.14 | 3.77 |

|
||||
| Total | 1.88 | 6.19 | 0.35 | 2.33 | |||
| E-Factor: 6.2 (only for SN 2 reaction) | |||||||
| Lewis-acid-promoted synthesis | |||||||
| DEC | 1.73 | 2.42 |

|
EtOAc | 0.23 | 2.23 |

|
| MeOH | 0.14 | 3.77 |

|
DCM | 0.80 | 6.28 |

|
| DCM | 0.80 | 6.28 |

|
||||
| DMF | 0.02 | 12.60 |

|
||||
| Nitromethane | 1.96 | 7.81 |

|
||||
| Total | 2.70 | 25.08 | 1.03 | 8.51 | |||
| E-Factor: 337.9 | |||||||
| C–H functionalization pathway | |||||||
| MeOH | 0.14 | 3.77 |

|
Et2O | 0.59 | 3.27 |

|
| DMF | 0.02 | 12.60 |

|
EtOAc | 0.23 | 2.23 |

|
| TrifluoroEtOH | 3.66 | 48.07 |

|
||||
| Total | 3.82 | 64.45 | 0.82 | 5.50 | |||
| E-Factor: 238.1 | |||||||
Concerning the key-step, which is the formation of the benzofuran scaffold, the route to amiodarone through Lewis-acid catalysis features both lower SHS and ES scores compared to the procedure based on the C–H activation strategy. A major criticism of the latter is the use of TFE as solvent (Scheme 5). It is also worth noting that once the benzofuran core has been formed, better performance in terms of safety/benign indexes and E-factor is achieved with the 1966 patented procedure.
 | ||
| Scheme 5 Summarised assessment for amiodarone synthesis. | ||
Drawing a conclusion on the synthesis of amiodarone, from the waste point of view, the C–H functionalization strategy is a better option than the classic one based on Lewis-acid catalysis. The opposite result is obtained when the environmental and safety features are evaluated, making the classic route superior to the innovative C–H functionalization strategy. Amiodarone is an interesting example proving that the comparisons proposed in this review do not have an obvious outcome.
3. Anacetrapib (cholesterol modulator)
Anacetrapib (Scheme 6) is an API used as a cholesterylester transfer protein (CETP) inhibitor to prevent heart attacks while modulating cholesterol levels.67 Anacetrapib was first synthesized by Merck researchers in 2011 within a structure–activity study that led to the discovery of its potent CETP inhibition. The route of synthesis (ROS) to anacetrapib68 was based on the borylation of substituted aryl bromide 10 followed by a Suzuki cross-coupling. The resultant biaryl product 13 was subsequently chlorinated and then N-alkylated using an oxazolidine partner that completes the anacetrapib molecular structure (Scheme 7). The total synthesis described in the initial patent consists of seven steps overall that include the preparation of materials. Within this review and for the sake of a comparison with other procedures, only the borylation and consecutive cross-coupling steps have been considered. Borylation involves the lithiation of arylbromide 10 which is left to react with triisopropyl borate. This process is associated with an E-factor value of 12.8. The following palladium-catalysed Suzuki cross-coupling proceeded in the presence of a phosphine ligand and with the use of a mixture of acetonitrile/3 M potassium carbonate reaction medium and it features an E-factor value of 11.6. The total reaction time for the two steps is 1 h and 40 min with yields of 83% and 95%, respectively. The overall E-factor is 17.1. Given the fact that the two steps were based on different amounts of starting materials, it is not appropriate to precisely assess the overall E-factor for the two combined steps.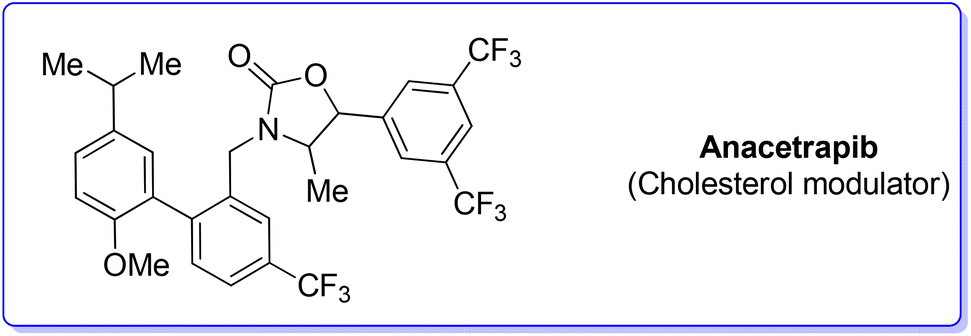 | ||
| Scheme 6 Anacetrapib chemical structure. | ||
 | ||
| Scheme 7 Merck's synthetic approach to anacetrapib via cross-coupling. | ||
In the year of anacetrapib's discovery, the Merck laboratories also developed a different route based on a ruthenium-catalysed direct C–H arylation technology69 (Scheme 8). By exploiting oxazolidine ring 15 as a directing group to steer the ruthenium catalyst towards the desired C–H bond, the two aryl moieties were connected to replace the cross-coupling strategy. Biaryl oxazoline 16 was obtained in 96% yield in 8 h of reaction time with an E-factor value of 20.5. Importantly, both procedures were concluded with a water precipitation work-up with obvious advantages in terms of the overall sustainability the synthesis.
 | ||
| Scheme 8 Merck's synthetic approach to anacetrapib via C–H functionalization. | ||
In Table 2 we report the data to compare the two approaches in terms of safety hazard and benign index of the solvents input. From this analysis it is evident that even if the C–H functionalization technology utilizes harmful N-methyl pyrrolidone (NMP) as a reaction medium, the reduction of the synthetic step toward the final target leads to overall lower ES/SHS scores (Scheme 9). This is a quantified example of the possible advantages of C–H functionalization over cross-coupling synthetic technologies in terms of atom efficiency and sustainability. It is also noteworthy that efforts to develop greener solvents adequate for performing sustainable C–H functionalization strategies, will make this approach greener and more applicable.70–72
 | ||
| Scheme 9 Summarised assessment for anacetrapib synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Cross-coupling approach | |||||||
| Toluene | 2.99 | 4.84 |

|
Water |

|
||
| Acetonitrile | 1.81 | 2.32 |

|
||||
| Total | 4.81 | 7.16 | |||||
| E-Factor: 12.8, 11.6 | |||||||
| Overall E-factor: 17.1 | |||||||
| C–H functionalization approach | |||||||
| NMP | 0.01 | 5.35 |

|
Water |

|
||
| Total | 0.01 | 5.35 | — | — | |||
| E-Factor: 20.5 | |||||||
4. Ivachtin (caspase-3-inhibitor)
During an organism life's cycle there are key events such as programmed cell death (e.g. apoptosis) that are mediated by cysteine-aspartic proteases (caspases), present in different forms. Caspase-3 is essential for normal brain development and is important or essential in other apoptotic scenarios in a remarkable tissue-, cell-type- or death-stimulus-specific manner. Thus, inhibitors of caspase-3 were described as promising cardioprotectants, neuroprotectants and hepatoprotectants.73,74Among the many caspase-3-inhibitors, in 2005, Ivachtchenko and coworkers, identified a potent small molecule featuring a morpholine moiety, Ivachtin (Scheme 10) and developed its synthesis.75 It starts from sulfonyl amide 17 which was refluxed in an AcOH/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture to afford isatin derivative 18, followed by preparation of dicarboxylic acids 19via a Pfitzinger reaction with a keto ester. The diacid was then converted into furandione 20 upon reaction with acetic anhydride, which was then transformed into the final product after reaction with 2-aminoethanol and protection with acetic anhydride (Scheme 11).
:1 v/v) mixture to afford isatin derivative 18, followed by preparation of dicarboxylic acids 19via a Pfitzinger reaction with a keto ester. The diacid was then converted into furandione 20 upon reaction with acetic anhydride, which was then transformed into the final product after reaction with 2-aminoethanol and protection with acetic anhydride (Scheme 11).
 | ||
| Scheme 10 Ivachtin chemical structure. | ||
 | ||
| Scheme 11 Ivachtin common strategy. | ||
To allow for comparison with other methodologies, the step related to the installation of a morpholine moiety is excluded. The four steps described above are associated with the following E-factor values: 43.0, 18.0, 15.0, 3.2, respectively, with an overall E-factor of 269.1. The yields for each step are 89%, 60%, 88% and 60%, respectively, with a total yield of 28% over four steps and a total reaction time of 32.5 h.
In 2020, Kang and collaborators described a concise synthesis of the key-intermediate of the caspase-3-inhibitor described above. This strategy76 (Scheme 12) relies on the generation of an iron-catalysed α-aminoalkyl radical and its condensation with electron-deficient alkenes. The pyrrolo[3,4-c]quinolone-1,3-dione (23) formed was first reduced in the presence of hydrogen and a palladium catalyst to remove the N-protecting group and subsequently re-oxidized to fused-quinoline 25. The latter was reacted with aminoethanol to give caspase-3-inhibitor key-intermediate 26. The E-factor values of these four steps are 101.1 (yield: 27%), 93.7 (yield: 55%), 419.3 (yield: 80%), 864.7 (yield: 75%), respectively, with a total reaction time of 33 h and an overall E-factor of 3675.8.
 | ||
| Scheme 12 Ivachtin route via C–H functionalization. | ||
By comparing the two approaches, it is evident that the C–H functionalization procedure furnishes a direct method for the construction of pyrrolo-fused-quinoline 26, but it also produces larger amounts of waste than that coming from the classical procedure. By considering the ES/SHS scores (Table 3), the C–H functionalization synthetic pathways show only slightly lower performance related to the reaction media. On the other hand, it is worth noting that the standard procedure suffers from the utilization of harmful diethyl ether and hexane as work-up solvents while the C–H functionalization includes the use of more benign and safer ethyl acetate (Scheme 13).
 | ||
| Scheme 13 Summarised assessment for Ivachtin synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Pfitzinger reaction pathway | |||||||
| Water |

|
Et2O | 0.59 | 3.27 |

|
||
| AcOH | 0.05 | 12.04 |

|
Hexane | 22.85 | 5.39 |

|
| Total | 0.05 | 12.04 | 23.44 | 8.66 | |||
| E-Factor: 43.0, 18.0, 15.0, 3.2 | |||||||
| Overall E-factor: 269.1 | |||||||
| C–H functionalization pathway | |||||||
| Toluene | 2.99 | 4.84 |

|
EtOAC | 0.23 | 2.23 |

|
| Acetonitrile | 1.81 | 2.32 |

|
||||
| EtOH | 0.15 | 1.53 |

|
||||
| Total | 4.96 | 8.70 | 0.23 | 2.23 | |||
| E-Factor: 101.1, 93.7, 419.3,864.7 | |||||||
| Overall E-factor: 3675.8 | |||||||
5. Omarigliptin (diabetes)
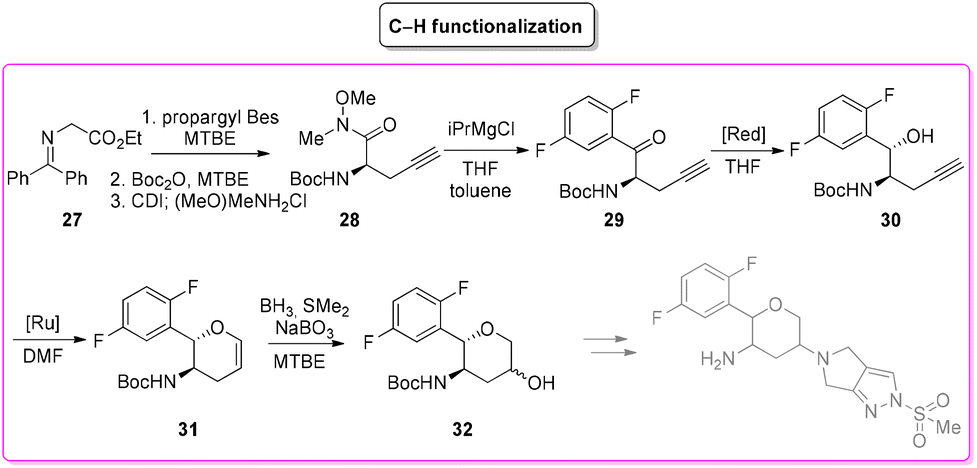
Among the many diseases that are showing increasing incidence worldwide, type-2 diabetes mellitus is certainly one of the most relevant, being itself or through related complications, one of the leading causes of death globally.77 In 2006, the FDA approved the first-in-class DPP-4 inhibitor, sitagliptin, as a daily treatment for patients with type-2 diabetes.78 Since then, considering the clinical success of DPP-4 inhibitors, there has been an ongoing interest in the development of other drug candidates with longer half-lives that could decrease the dosing frequency. In this arena, Merck Laboratories developed omarigliptin (also known as MK-3102, Scheme 14) which has already received authorization for marketing as a once-weekly remediation for type-2 diabetes. Over the years Merck Laboratories have released two main processes for the synthesis of omarigliptin, in a very rare case, the first79 (in chronological order) is based on an asymmetric reduction followed by a C–H functionalization reaction to build the key pyranol core 32, while the second starts with an initial asymmetric Henry reaction and proceeds with more classical acrolein-based C–C bond formation chemistry toward the final target. Considering the fact that omarigliptin is a chiral API, this example could also give further insight into the sustainability assessment of two distinct asymmetric approaches. The C–H functionalization strategy (Scheme 15) starts with alkylation of glycine benzophenone imine 27 with propargyl besylate and subsequent Boc-protection followed by Grignard installation of difluoro aryl moiety 29 and asymmetric reduction. The as-formed aminoalcohol 30 undergoes ruthenium-catalysed C–H cyclization to form pyranol ring 31 with subsequent hydroboration to form key intermediate 32. This four-step procedure features the following four calculated E-factor values: 49.8 (yield: 75%), 61.1 (yield: 89%), 171.5 (yield: 80%) and 54.2 (yield: 89%), with an overall E-factor of 402.9. | ||
| Scheme 14 Omarigliptin chemical structure. | ||
 | ||
| Scheme 15 Omarigliptin synthesis via C–H functionalization. | ||
Two years later, the same group improved the above-described methodology with an alternative approach80 (Scheme 16) to produce a pyranol scaffold which begins with an asymmetric Henry reaction (E-factor: 23.2; yield: 92%) between benzaldehyde and nitromethane. The nitro aldol product 33 undergoes cascade nitro-Michael/lactonization/dehydration with acrolein to afford dihydropyran 36 (E-factor: 74.2; yield: 79%), which is separated from the enantiomer (E-factor: 16.6; yield: 83%) and then hydroboration/oxidation (E-factor: 29.6; yield: 93.5%) and nitro reduction/Boc-protection (E-factor: 88.5; yield: 84%) afforded the target pyranol 32 with an overall E-factor over the four steps of 1484.8.
 | ||
| Scheme 16 Omarigliptin synthesis via nitro-Michael/lactonization/dehydration. | ||
In terms of waste production, the two procedures are similar, since the high E-factor of the C–H functionalization step in the initial methodology is compensated by the larger number of steps of the latter, while in terms of overall E-factor, the C–H functionalization procedure turns out to be better. A similar situation can be extrapolated by analysing the ES score (Table 4) for the reaction media which are very similar as well as the ES and SHS scores for the solvents used during work-up. Anyway, the situation clearly changes after considering the SHS score of the reaction media, which turns in favour of the new procedure (Scheme 17).
 | ||
| Scheme 17 Summarised assessment for omarigliptin synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| C–H functionalization | |||||||
| DMF | 0.01 | 12.60 |

|
MTBE | 0.60 | 5.18 |

|
| MTBE | 0.60 | 5.18 |

|
Heptane | 114.85 | 1.97 |

|
| THF | 2.38 | 3.31 |

|
Toluene | 2.997 | 4.84 |

|
| iPrOH | 0.12 | 2.47 |

|
||||
| Total | 3.00 | 21.10 | 118.57 | 14.45 | |||
| E-Factor: 49.8, 61.1, 171.5, 54.2 | |||||||
| Overall E-factor: 402.9 | |||||||
| Lactonization/dehydration pathway | |||||||
| EtOH | 0.15 | 1.53 |

|
MTBE | 0.60 | 5.18 |

|
| MTBE | 0.60 | 5.18 |

|
Heptane | 114.85 | 1.97 |

|
| THF | 2.38 | 3.31 |

|
iPrOH | 0.12 | 2.47 |

|
| iPrOH | 0.12 | 2.44 |

|
THf | 2.38 | 3.31 |

|
| EtOH | 0.15 | 1.53 |

|
||||
| Total | 3.26 | 12.48 | 118.11 | 14.45 | |||
| E-Factor: 74.2, 16.6, 29.6, 88.5 | |||||||
| Overall E-factor: 1484.8 | |||||||
6. Reboxetine (antidepressant)
Reboxetine (Scheme 18) is a popular API used to treat depression and attention deficit hyperactivity disorder (ADHD). It is a norepinephrine reuptake inhibitor (NRI), unique to this class of drugs.81 Reboxetine was first discovered by Farmitalia-Carlo Erba which in 2003 was acquired by Pfizer. The first synthesis of reboxetine82 was based on a three-step procedure (formally seven steps but many of them without isolation) in which cinnamyl alcohol 38 was first epoxidized to (R,R)-phenylglycidol (39) and subsequently reacted without isolation with 2-ethoxyphenol. Further steps are the key asymmetric epoxidation followed by acylation in Schotten–Baumann conditions. Finally, S,S-reboxetine succinate was directly obtained via crystallization of the free base. The yields for the three steps were 58%, 60%, and 54.6%, respectively. The corresponding E-factor values are 57.6, 182.3, and 290.0 with an overall E-factor of 1212.4 and the total reaction time to obtain the final S,S-reboxetine is 18 h (Scheme 19). | ||
| Scheme 18 Reboxetine chemical structure. | ||
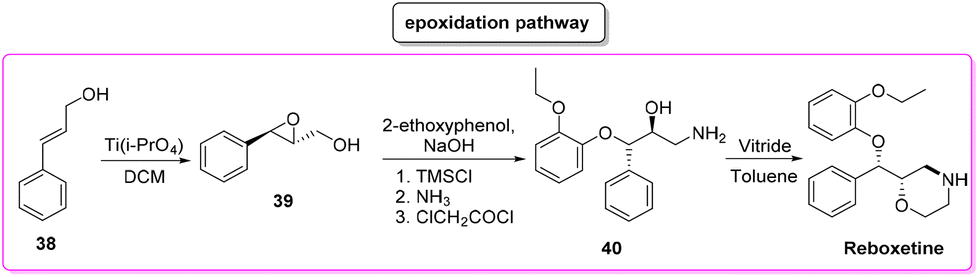 | ||
| Scheme 19 Reboxetine synthesis using the Pfizer process. | ||
A direct comparison of this methodology can be made with the process reported by Christina White and coworkers (Scheme 20), who discovered a smart way to access enantiopure reboxetine via a C(sp3)–N fragment coupling reaction between terminal olefins and N-triflyl protected aliphatic and aromatic amines using a palladium-catalyzed intermolecular allylic C–H amination reaction.83
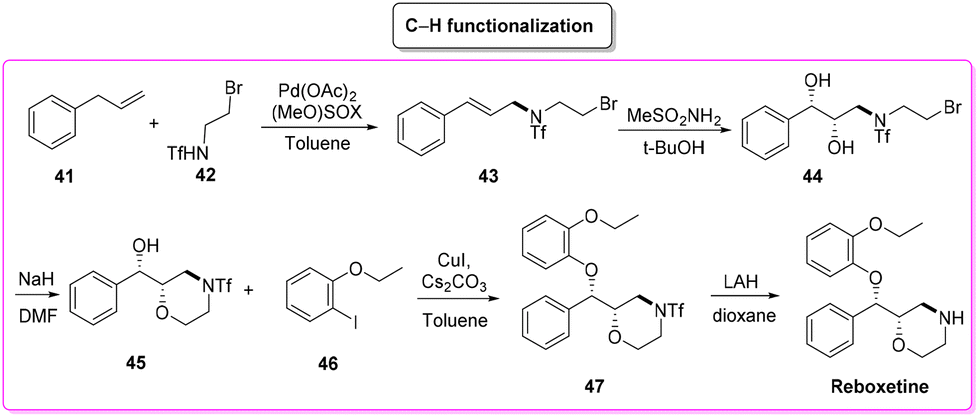 | ||
| Scheme 20 Reboxetine synthesis via C–H functionalization. | ||
The synthetic strategy relies on allylic C–H amination between allylbenzene 41 and N-triflyl 2-bromoethylamine (42), furnishing E-allylic amine 43. At this stage, Sharpless asymmetric dihydroxylation furnishes chiral amino diol 44 that upon exposure to sodium hydride cyclizes to the key morpholine moiety 45. Copper-catalysed etherification followed by lithium aluminium hydride triflate removal furnishes (S,S)-reboxetine in 96% ee. This five-step sequence allows for a 48% overall yield, which compares favourably with the seven-step industrial route (41% overall yield). Considering the E-factor, the C–H amination route features the following values: 35.8, 353.5 (for the 2nd and 3rd steps without intermediate isolation), 18.2, and 151.2 with a total reaction time of 122 h and an overall E-factor of 1772.8.
In terms of safety and environmental compatibility (Table 5), a comparison of these two processes is truly significant. The ES scores achieved with the C–H functionalization path are a little lower than the Pfizer procedure when considering both the reaction media and the solvents used for work-up. While, with regard to the SHS scores the standard procedure has much lower values in terms of reaction media but higher numbers for the utilization of work-up solvents. In these examples, it is possible to note that, even if the C–H functionalization strategy allows for an easier work-up methodology, is concise and gives smart access to reboxetine, the epoxidation pathway results in globally better sustainability (Scheme 21). Nonetheless, this is another example of how a global sustainability assessment can also take into account the contributions of diverse asymmetric approaches utilizing the asymmetric step at different stages of the total synthesis of the API.
 | ||
| Scheme 21 Summarised assessment for reboxetine synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Epoxidation pathway | |||||||
| DCM | 0.80 | 6.28 |

|
MeOH | 0.14 | 3.77 |

|
| Toluene | 2.99 | 4.84 |

|
iPrOH | 0.12 | 2.47 |

|
| 3.80 | 11.12 | EtOAc | 0.23 | 2.23 |

|
||
| MTBE | 0.60 | 5.18 |

|
||||
| Total | 3.01 | 21.10 | 1.09 | 13.63 | |||
| E-Factor: 57.6, 182.3, 290.0 | |||||||
| Overall E-factor: 1212.4 | |||||||
| C–H functionalization | |||||||
| DMF | 0.02 | 12.60 |

|
EtOAc | 0.23 | 2.23 |

|
| Toluene | 2.99 | 4.84 |

|
Acetone | 0.35 | 2.33 |

|
| t-BuOH | 0.48 | 3.54 |

|
Et2O | 0.59 | 3.27 |

|
| Dioxane | 0.25 | 30.32 |

|
||||
| Total | 3.75 | 51.31 | 1.17 | 7.84 | |||
| E-Factor: 35.8, 353.5, 18.2, 151.2 | |||||||
| Overall E-factor: 1772.8 | |||||||
7. Dantrolene (malignant hyperthermia)
Dantrolene (Scheme 22) appeared in the scientific literature in 1967, among many hydantoin-based muscle relaxants.84 After extensive studies, the mode of action of dantrolene on skeletal muscle was elucidated in depth, showing an ability to depress excitation–contraction coupling, a complex process in which muscles transduce a chemical signal at the neuromuscular junction into a muscle contraction. Therefore, dantrolene (under the brand name Dantrium®), while initially discovered as an efficient muscle myorelaxant, has now became the only clinically available agent for the treatment of malignant hyperthermia (MH).85 The original synthesis was studied by Davis and Snyder86 and it was patented by Norwich Pharma Co. This synthetic approach relies on a modified Meerwein arylation in which p-nitro aniline undergoes diazotization and subsequent copper-catalysed arylation with furfural followed by reaction with hydantoin.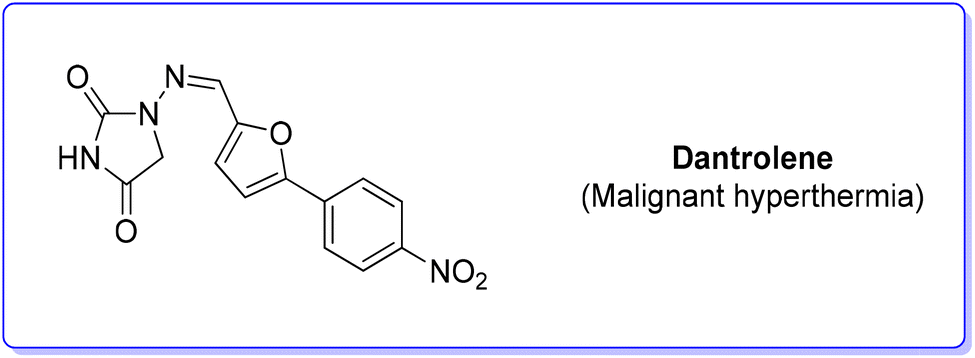 | ||
| Scheme 22 Dantrolene chemical structure. | ||
A new synthesis87 of dantrolene was proposed in 2020 by Forgione and coworkers with the smart utilization of underutilized C6-platform 5-hydroxymethyl furfural through a decarboxylative cross-coupling reaction. Unfortunately, due to lack of information regarding the details of the synthetic strategy, this approach cannot be discussed in the present review.
Another approach based on cross-coupling,88 relies on the use of a Stille reaction between p-iodo-anisole 48 and 5-(tri-n-butylstannyl)-2-furaldehyde (49) to furnish arylated furfural 50 which reacts with amino-hydantoine 52 in the presence of chloridric acid to obtain dantrolene in 71% overall yield and in 19 h of total reaction time (Scheme 23). The E-factors calculated for the two steps are 146.8 (Stille coupling, 75% yield) and 608.9 (hydantoine condensation, 94% yield), respectively, with an overall E-factor of 1012.0.
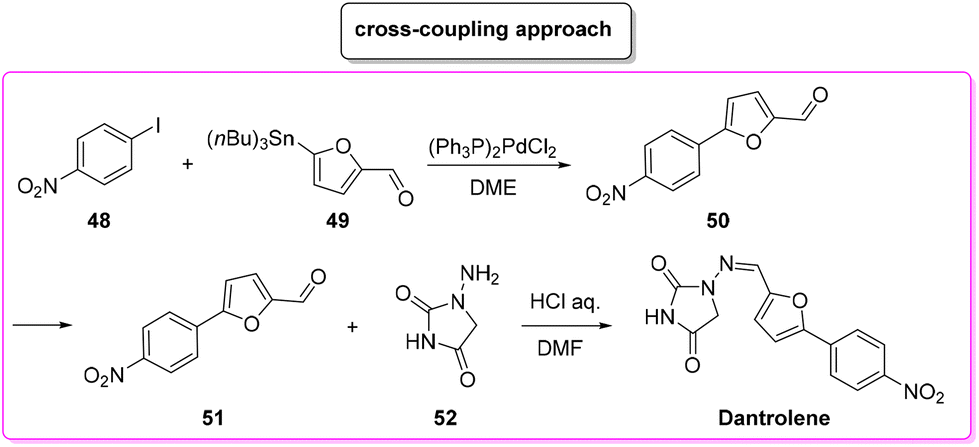 | ||
| Scheme 23 Dantrolene synthesis via cross-coupling. | ||
In 2018, Colacino, Porcheddu and coworkers proposed an innovative synthesis89 (Scheme 24) of dantrolene, adopting a mechanochemical methodology to achieve the condensation of a preformed 4-arylated furfural 51 with hydantoine 52, which resulted in a procedure with a very low E-factor (0.3). Importantly, by means of a mechanochemical apparatus the authors avoided the use of any solvent as a reaction medium. Application of this procedure has been tested to a limited scale of 6.6 mmol.
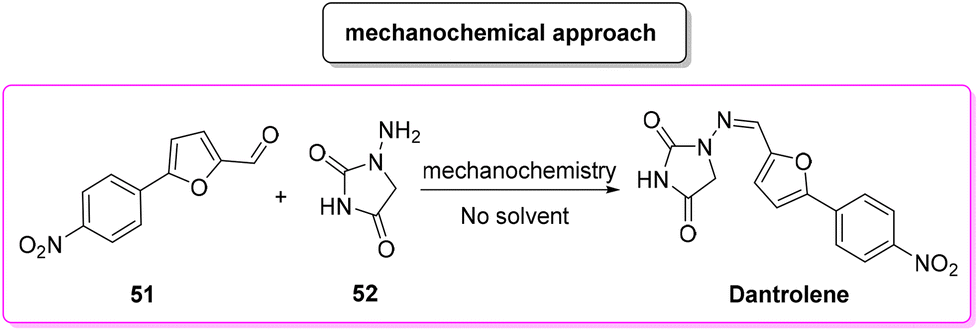 | ||
| Scheme 24 Mechanochemical approach to dantrolene synthesis. | ||
In the same year, Ackermann and coworkers developed the synthesis of dantrolene via a C–H functionalization pathway by using a flow-photochemical apparatus (Scheme 25).90 The synthetic sequence reflects the original Meerwein arylation in which the diazonium salts of aryl moiety 53 undergo manganese photocatalyzed C–H functionalization with furfural 54 (65% yield). Subsequent condensation of the obtained furfural derivatives furnishes dantrolene in 94% yield. The E-factor values associated with the two steps are 1102.0 and 66.9, respectively, with an overall E-factor of 1393.0.
 | ||
| Scheme 25 Dantrolene synthesis via C–H functionalization. | ||
Analysis of the ES/SHS scores (Table 6) revealed that, ruling out the mechanochemical process which exploited the greenest situation with regard to the hydantoine condensation step, the ES scores of the cross-coupling procedure and the C–H functionalization are very similar with a slight advantage to the former. Anyway, the SHS score strongly suggests safer conditions for the C–H functionalization synthetic pathways (Scheme 26).
 | ||
| Scheme 26 Summarised assessment for dantrolene synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Cross-coupling approach | |||||||
| DMF | 0.02 | 12.60 |

|
EtOAc | 0.23 | 2.23 |

|
| DME | 1.28 | 205.48 |

|
||||
| Total | 1.30 | 218.09 | 0.23 | 2.23 | |||
| E-Factor: 146.8, 608.9 | |||||||
| Overall E-factor: 1012.0 | |||||||
| Mechanochemical approach | |||||||
| No solvent | –— | — | No solvent | — | — | ||
| Total | — | — | — | — | |||
| E-Factor: 0.3 | |||||||
| C–H functionalization approach | |||||||
| Acetonitrile | 1.81 | 2.32 |

|
EtOAc | 0.23 | 2.23 |

|
| DMSO | 0.001 | 1.66 |

|
||||
| Total | 1.81 | 3.99 | 0.23 | 2.23 | |||
| E-Factor: 1102.0, 66.9 | |||||||
| Overall E-factor: 1393.0 | |||||||
8. Tafamidis (transthyretin amyloidosis)
Tafamidis (Scheme 27) is a first-in-class FDA-approved medication to treat the disease progression of transthyretin amyloidosis, caused by the deposition of transthyretin fibrils in the myocardium. Tafamidis binds to transthyretin and stabilizes its quaternary structure, preventing amyloidogenesis.91 | ||
| Scheme 27 Tafamidis chemical structure. | ||
Tafamidis was first developed at the Scripps Research Institute by the group of J. W. Kelly through a structure–property relation drug design and its structure was first published in 2003. In the same year J. W. Kelly and S. Lindquist (Massachusetts Institute of Technology) cofounded FoldRX to receive market approval for tafamidis. In 2010, FoldRX was acquired by Pfizer which is now marketing tafamidis in two formulations under the brand names Vyndaqel and Vyndamax. The patented synthesis92 of tafamidis consists of a three-step procedure starting from 3,5-dichlorobenzoic acid which is transformed into its chloride and then reacted with 4-amino-3-hydroxybenzoic acid to form the respective amide that undergoes cyclization to tafamidis. In 2011, Itami and coworkers developed a C–H functionalization strategy93 toward the synthesis of tafamidis which consists of three steps (Scheme 28). In the first step the formation of the acid chloride of benzoxazole-6-benzoic acid (55) and its subsequent reaction with morpholine 56 take place, leading to protected benzoxazole core 57 (E-factor: 50.8; yield: 56%) which undergoes C–H arylation with 3,5-dichloro iodobenzene (58) (E-factor: 14.7; yield: 74%) and subsequent deprotection (E-factor: 40.5; yield: 94%) to finally access tafamidis with an overall E-factor of 2802.2.
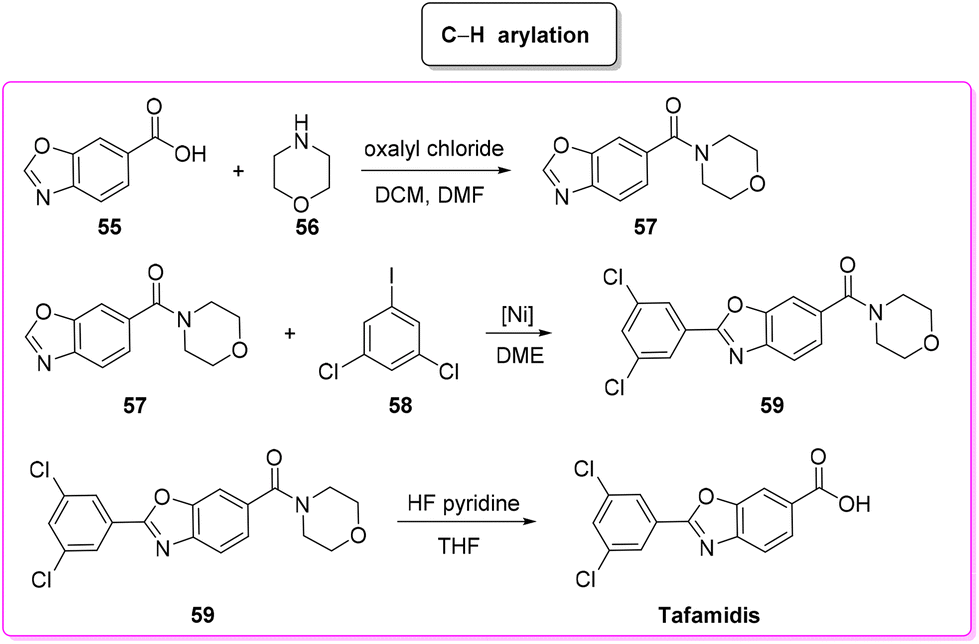 | ||
| Scheme 28 Tafamidis synthesis via C–H functionalization. | ||
In 2019, Vaccaro and coworkers developed a heterogeneous manganese-catalyzed C–H cyclization strategy94 in a flow strategy (Scheme 29) which led to the synthesis of tafamidis in a concise (1 h) manner starting from 3,5-dichlorobenzyl alcohol (60) and 4-amino-3-hydroxybenzoic acid (61) through the formation of the imine and its subsequent cyclization in environmentally friendly CPME as medium. This methodology has an overall calculated E-factor value of 4.4.
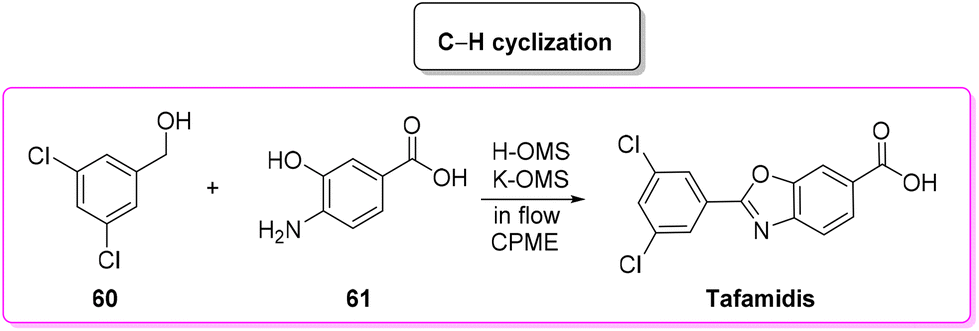 | ||
| Scheme 29 Continuous flow C–H functionalization approach to tafamidis. | ||
By considering the above reported E-factor values, it is possible to note that the protocol by Itami features low E-factor values, while the optimized flow strategy, also considering that the solvent is continuously recycled, is more efficient from a sustainability point of view. The same consideration can be made regarding the ES/SHS analysis (Table 7) in which, for the protocol by Itami there are high values due to the utilization of harmful and toxic petrol-based solvents (Scheme 30).
 | ||
| Scheme 30 Summarised assessment for tafamidis synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| C–H arylation pathway | |||||||
| DCM | 0.80 | 6.28 |

|
EtOAc | 0.23 | 2.23 |

|
| DMF | 0.02 | 12.60 |

|
||||
| THF | 2.38 | 3.31 |

|
||||
| DME | 1.28 | 205.48 |

|
||||
| Total | 4.49 | 227.68 | 0.23 | 2.23 | |||
| E-Factor: 50.8, 14.7, 40.5 | |||||||
| Overall E-factor: 2802.2 | |||||||
| C–H cyclization pathway | |||||||
| CPME | 2.39 | 28.78 |

|
EtOH | 0.15 | 1.54 |

|
| Total | 2.39 | 28.78 | 0.15 | 1.54 | |||
| E-Factor: 4.4 | |||||||
9. Olmesartan (antihypertensive)
Olmesartan (Scheme 31) is an angiotensin II receptor blocker, with main uses in the treatment of hypertension, diabetic nephropathy, and heart failure. It is among the most prescribed medicines to treat blood pressure related diseases. Olmesartan, as well as the other members of the sartan family, selectively blocks the functionalization of the angiotensin receptors causing vasodilation, reduction of secretion of vasopressin and aldosterone, and this combination of events reduces blood pressure.95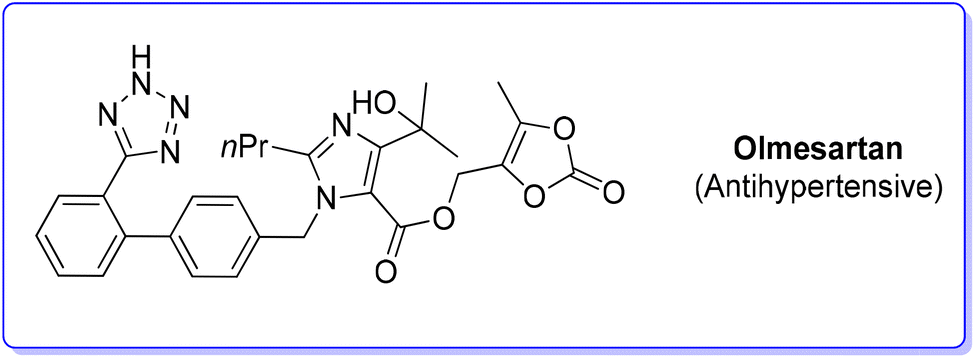 | ||
| Scheme 31 Olmesartan chemical structure. | ||
Given its popularity, the synthesis of olmesartan has continuously attracted the interest of many researchers. For the purposes of this review, we selected two exemplificative strategies, the first of which is based on nucleophilic substitution and the second of which uses the tetrazole ring as a directing group for a C–H functionalization process. In more detail, the classic approach, published in 2009,96 involves eight synthetic steps (Scheme 32) and it starts with the synthesis of the imidazole core, followed by nucleophilic substitution to form a C–N bond with the biphenyl-tetrazole unit and then the process is completed with final decoration of the side chain using dioxolone-ester. The E-factor values for the eight steps and their relative yields are: 5.6 (yield: 98%), 26.3 (yield: 95%), 14.4 (yield: 96%), 7.1 (yield: 99%), 5.7 (yield: 99%), 20.1 (yield: 99%), 25.6 (yield: 80%), 206.9 (yield: 80%), with an overall E-factor of 839.3.
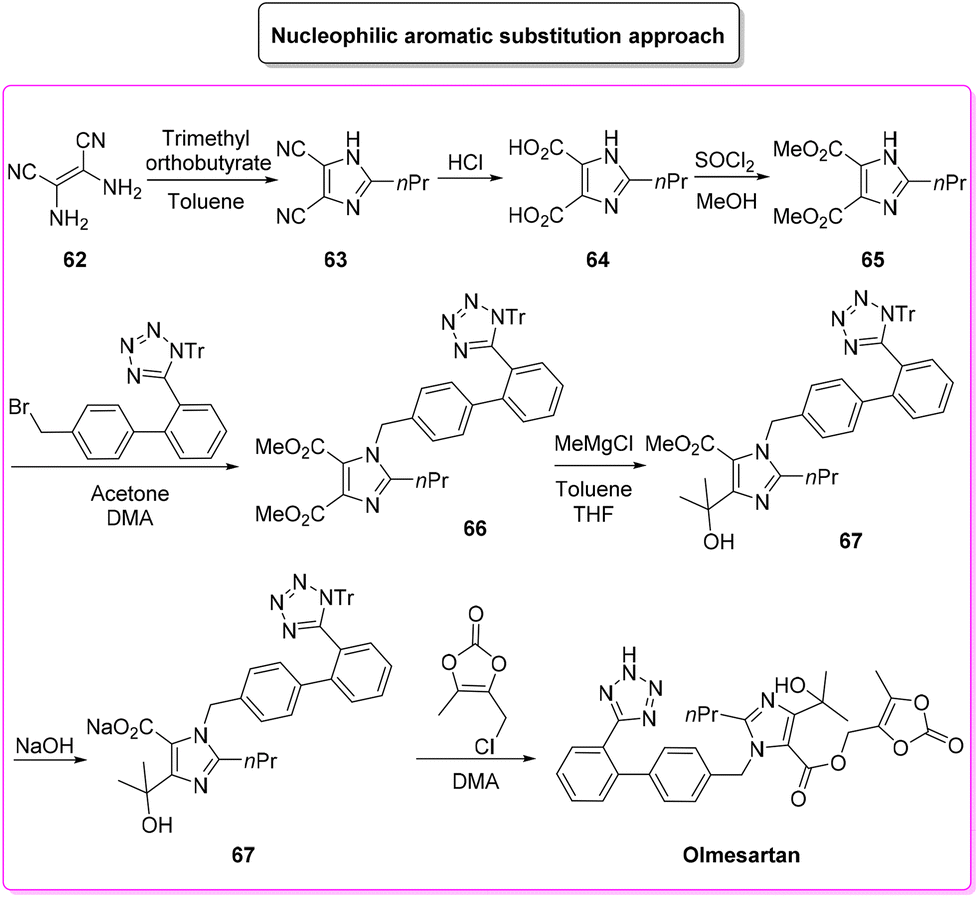 | ||
| Scheme 32 General approach to olmesartan. | ||
The whole procedure, ruling out step 3 in which esterification takes place, shows low values of the E-factor indicating good management of the waste produced. Indeed, most of the steps are performed with stochiometric amounts of reagents and/or slightly concentrated conditions (>1 M).
The second procedure97 (Scheme 33) considered in this evaluation was published in 2015 by Seki and coworkers and involves six consecutive steps. The synthesis begins with a C–H arylation reaction to form a biphenyl-tetrazole unit (E-factor: 53.7; yield: 55%) a product which undergoes deprotection of the benzyl group and chlorination (E-factor: 112.4; yield: 68%). Subsequent steps involve C–N coupling (E-factor: 90.4; yield: 50%), hydrolysis (E-factor: 244.9; yield: 87%), esterification (E-factor: 33.8; yield: 74%), and deprotection (E-factor: 230.8; yield: 98%) leading to olmesartan with an overall E-factor of 5821.6.
 | ||
| Scheme 33 Olmesartan synthesis via C–H functionalization. | ||
This latter described synthesis features higher E-factor values due to utilization of more solvent compared to the previous synthesis. It is also noteworthy that the C–H functionalization step (the 1st step) is associated with an acceptable E-factor value as low as 53.7 which assumes even more importance considering that only 55% yield was achieved in this step. By considering the ES/SHS analysis (Table 8) instead, the situation is different. The classic procedure, based on eight synthetic steps (vs. six for the C–H functionalization strategy) showed higher values for ES and SHS associated with both reaction media and work-up solvents (Scheme 34).
 | ||
| Scheme 34 Summarised assessment for olmesartan synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Nucleophilic substitution approach | |||||||
| MeOH | 0.14 | 3.77 |

|
Cyclohexane | 10.25 | 2.43 |

|
| Toluene | 2.99 | 4.84 |

|
AcOH | 0.05 | 12.04 |

|
| Acetone | 0.35 | 2.33 |

|
EtOAc | 0.22 | 2.23 |

|
| AcOH | 0.05 | 12.04 |

|
Toluene | 2.99 | 4.84 |

|
| DMA | 0.01 | 13.49 |

|
DCM | 0.80 | 6.28 |

|
| Total | 3.56 | 36.48 | 14.33 | 27.83 | |||
| E-Factor: 5.6, 26.3, 14.4, 7.1, 5.7, 20.1, 25.6, 206.9 | |||||||
| Overall E-factor: 839.3 | |||||||
| C–H functionalization pathway | |||||||
| Methanol | 0.14 | 3.77 |

|
Toluene | 2.99 | 4.84 |

|
| DCM | 0.80 | 6.28 |

|
DCM | 0.80 | 6.28 |

|
| NMP | 0.01 | 5.35 |

|
MTBE | 0.60 | 5.18 |

|
| Acetone | 0.35 | 2.33 |

|
||||
| Total | 1.32 | 17.74 | 4.40 | 16.31 | |||
| E-Factor: 53.7, 112.4, 90.4, 244.9, 33.8, 230.8 | |||||||
| Overall E-factor: 5821.6 | |||||||
10. Lapatinib (anticancer)
Lapatinib (Scheme 35) is a dual tyrosine kinase inhibitor, developed in 2004 by GlaxoSmithKline for the treatment of patients with advanced or metastatic breast cancer.98 | ||
| Scheme 35 Lapatinib chemical structure. | ||
In 2007, lapatinib was approved in combination with capecitabine and later, in 2013, the approval was extended to a chemotherapy-free combination with trastuzumab. Herein we cover a patented route of synthesis,99 a cross-coupling based synthetic pathway100 and a C–H functionalization strategy101 toward the synthesis of lapatinib.
The patented procedure99 (Scheme 36) relies on the initial iodination of 2-cyano-aniline (E-factor: 26.5) followed by formamidine formation (E-factor: 2.4) which undergoes subsequent acid-promoted aromatic nucleophilic substitution (E-factor: 11.7) and a Suzuki–Miyaura reaction with borylated furfural (E-factor: 73.8). The route of synthesis leads to 28 g of pure lapatinb (base) in a total reaction time of 45 h with an overall E-factor of 124.4. Afterward, there are two steps (E-factor: 17.4 and 91.4, respectively) in which sulfonyl-ethyl-imine is formed and reduced, but these have not been considered in this review to allow a closer comparison with the other synthetic strategies.
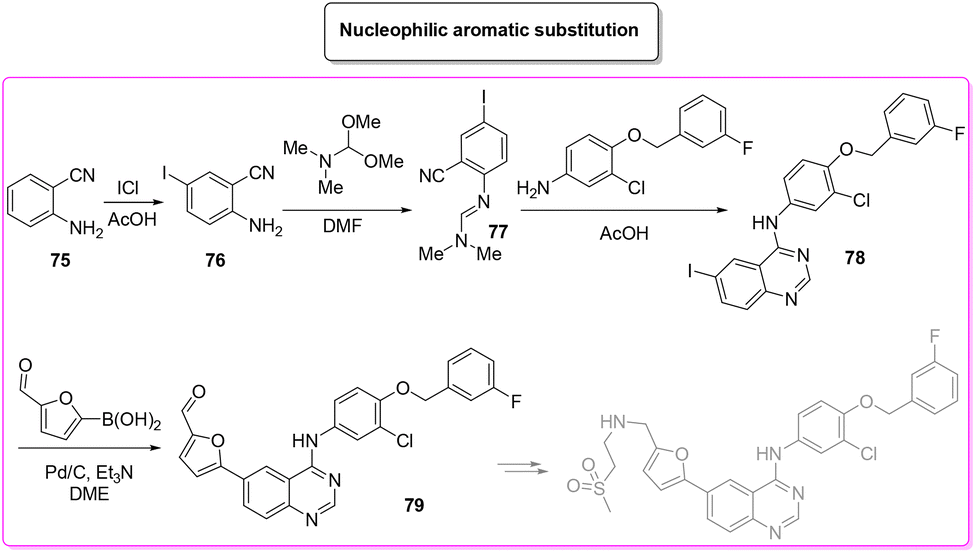 | ||
| Scheme 36 Lapatinib patented procedure. | ||
The second protocol was published in 2020 by Lipshutz and coworkers and is still based on classic cross-coupling chemistry.100 Lipshutz revisited the patent by changing solvents, bases and additive and bypassing the isolation of some intermediates in order to establish a concise four-step one-pot protocol with an E-factor of 67.4 and an almost quantitative yield of lapatinib base (Scheme 37). It is worth mentioning that the authors also continued the synthesis towards the final lapatinib ditosylate monohydrate. In addition, in order to allow a better comparison, it must be considered that Lipshutz and collaborators also attempted the synthesis by separating each reaction in a four-step consecutive procedure and for each step the E-factor values are: 8.4, 44.8, 4.0, 18.3.
 | ||
| Scheme 37 Lapatinib synthesis via cross-coupling. | ||
A very recent approach was reported by Hii and coworkers in 2022.101 This elegant example presents a revisited route for lapatinib to define its sustainable manufacture in South Africa. As the reported synthesis was developed to work closely to solubility limits in order to favour precipitation of the obtained products, no utilization of chromatographic purification was needed through the different steps. It is worth noting that the synthesis considers the manufacture of quinazoline 84 from anthranilic acid 83 (84% yield) with a very low E-factor of 14.7. This is a very important feature in view of a reduction of the starting material supply chain. From the obtained quinazoline 84 after chlorination (E-factor: 86.7; yield: 87%), the nucleophilic aromatic substitution was performed elegantly with a very simple filtration work-up leading to 94% yield of 78 with a E-factor value as low as 9.3. The subsequent palladium-catalysed Suzuki–Miyaura cross-coupling affords 79 in 91% yield with an E-factor of 86.0. The process needs 51 h to achieve key-intermediate 79 with an overall E-factor of 121.3. The synthesis follows the final steps required to obtain lapatinib which for the sake of comparison with other syntheses reported herein have been neglected. It is important to highlight that attempts to increase the environmental features was done by telescoping the many reaction steps. These advantages mainly occur in the final step and therefore are outside this evaluation (Scheme 38).
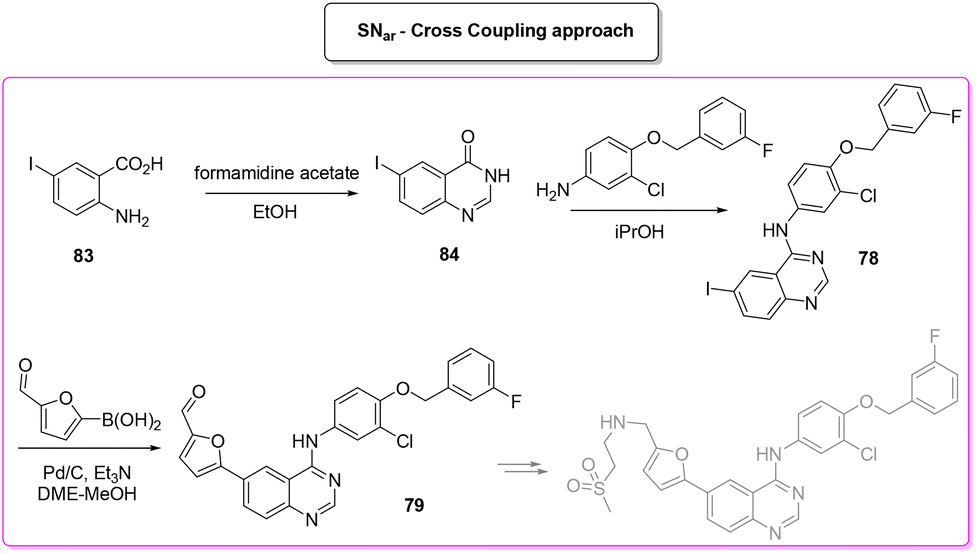 | ||
| Scheme 38 Lapatinib novel approach for South Africa manufacture. | ||
The last synthetic pathway is based on a C–H functionalization strategy102 (Scheme 39) and takes into account a three-step two-pot methodology. The first step (two steps in the same reactor) is the chlorination of the quinazoline ring and its subsequent nucleophilic aromatic substitution with a preformed benzyl-phenyl ether moiety (E-factor: 23.2; yield: 93%). The product undergoes a C–H functionalization reaction with furfural (which is also used as solvent) leading to lapatinib (base) in 63% yield with an E-factor of 35.8, and with an overall E-factor over the two steps of 984.8.
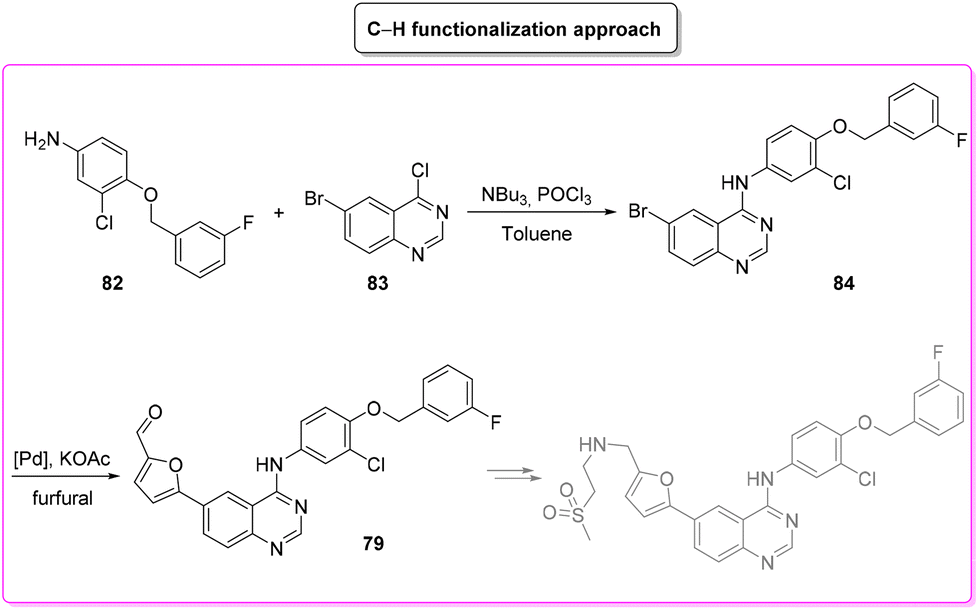 | ||
| Scheme 39 Lapatinib synthesis via C–H functionalization. | ||
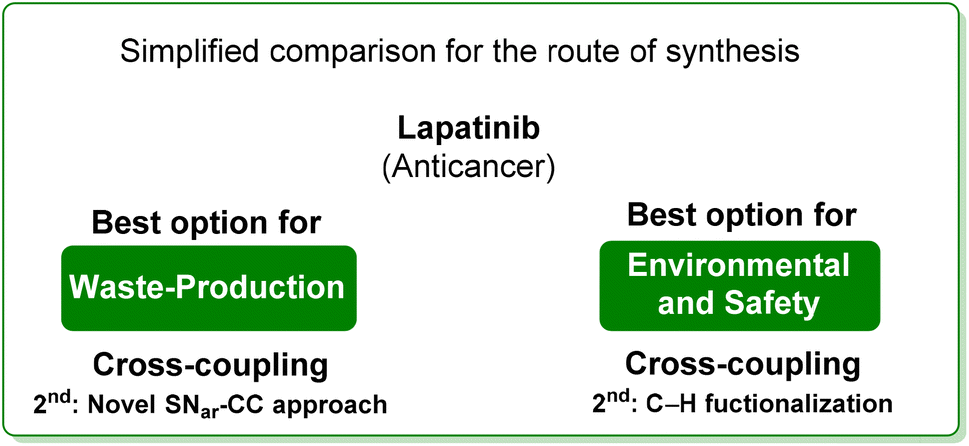
From the two analyses of the E-factor and the ES/SHS scores (Table 9), the synthesis by Lipshutz based on cross-coupling chemistry is truly sustainable, even if the surfactant solvent system used does not yet provide the information to be ranked in CHEM21 or be evaluated by ES/SHS analysis. The whole synthesis features low E-factor values and only safe and benign solvents (EtOAc, EtOH) were used as reaction media and/or in the work-up. The C–H functionalization synthetic pathway can be ranked as second best, only in terms of environmental and safety data, with good ES/SHS scores for the work-up solvent. Anyway, regarding the reaction media ES/SHS score, it must be considered that the use of toluene in the first step and furfural in the C–H functionalization process led to high values and therefore to the worst ES score among the strategies considered. The novel SNar/CC approach can be ranked as second best in terms of waste production, possessing relatively low overall E-factor value and also good CHEM21 solvent utilization (making an exception for the use of DME and Et2O). The patented synthesis of lapatinib represents the worst situation in terms of overall ES/SHS score (making an exception for the reaction media ES score as detailed above) while the E-factor values are not roughly comparable to those of the other procedures considered (Scheme 40).
 | ||
| Scheme 40 Summarised assessment for lapatinib synthesis. | ||
| Reaction media | Work-up solvents | ||||||
|---|---|---|---|---|---|---|---|
| ES | SHS | ES | SHS | ||||
| Nucleophilic aromatic substitution | |||||||
| AcOH | 0.05 | 12.04 |

|
EtOAc | 0.22 | 2.23 |

|
| DMF | 0.02 | 12.60 |

|
Toluene | 2.99 | 4.84 |

|
| DME | 1.28 | 205.48 |

|
Hexane | 22.85 | 5.39 |

|
| MeOH | 0.14 | 3.77 |

|
MeOH | 0.14 | 3.77 |

|
| iPrOH | 0.12 | 2.44 |

|
||||
| Total | 1.620 | 236.35 | 26.22 | 16.23 | |||
| E-Factor: 26.5, 2.4, 11.7, 73.8 | |||||||
| Overall E-factor: 124.4 | |||||||
| Cross-coupling approach | |||||||
| TPGS | — | — |

|
EtOH | 0.15 | 1.53 |

|
| EtOAc | 0.22 | 2.23 |

|
||||
| EtOH | 0.15 | 1.53 |

|
||||
| Total | 0.38 | 3.76 | 0.15 | 1.53 | |||
| E-Factor: 67.4 | |||||||
| Novel SN ar –CC approach | |||||||
| EtOH | 0.15 | 1.53 |

|
EtOH | 0.15 | 1.53 |

|
| Toluene | 2.99 | 4.84 |

|
Et2O | 0.59 | 3.27 |

|
| iPrOH | 0.12 | 2.44 |

|
iPrOH | 0.12 | 2.44 |

|
| DME | 1.28 | 205.48 |

|
MeOH | 0.14 | 3.77 |

|
| MeOH | 0.14 | 3.77 |

|
||||
| Total | 4.68 | 218.06 | 1.00 | 11.01 | |||
| E-Factor: 14.8, 86.7, 9.3, 86.0 | |||||||
| Overall E-factor: 121.3 | |||||||
| C–H functionalization approach | |||||||
| Toluene | 2.99 | 4.84 |

|
EtOH | 0.153 | 1.53 |

|
| Furfural | 3.31 | 58.95 |

|
Acetone | 0.359 | 2.33 |

|
| Total | 6.31 | 63.80 | 0.511 | 3.87 | |||
| E-Factor: 23.2, 36.7 | |||||||
| Overall E-factor: 984.8 | |||||||
11. Conclusions
In conclusion, after evaluating all the processes within their respective routes of synthesis, we can appreciate that in terms of waste production, C–H functionalization generally provides higher E-factor values (except for amiodarone and anacetrapib). This is the result of a generalised tendency to adopt, in the case of C–H functionalization, more dilute conditions and/or the use of a larger amount of solvents in the work-up procedures. While, unexpectedly, considering the environmental and safety analysis, most of the C–H functionalization synthetic pathways resulted in a more positive route of synthesis. The reasons for this result can be found in the fact that the C–H activation-based synthetic routes are generally associated with a smaller number of synthetic steps and therefore the number of factors that count in the total sum toward target achievement is inherently reduced. Nonetheless, it must also be considered that the C–H functionalization reactions were, for the most part, chronologically "novel" syntheses, which therefore have already been influenced by some behavioural and conceptual changes in organic synthesis, which are taking root in both the academy and industry. In addition, it is worth noting that the adoption of green technologies and strategies is continuously helping the transition towards more sustainable C–H functionalization methodologies103–111 and, in these terms, the constant efforts in calculation and measurement of the greenness in its various declinations could be of importance for next-generation synthetic chemistry. Importantly, the development of techno-economic studies on the subject of the present review could certainly be of general interest and assist in an even more precise way the development of new chemical methodologies.Abbreviations
| DEC | Diethyl carbonate |
| MeOH | Methanol |
| DCM | Dichloromethane |
| DMF | N,N-Dimethylformamide |
| TrifluoroEtOH or TFE | 2,2,2-Trifluoroethanol |
| EtOH | Ethanol |
| iPrOH | Isopropanol |
| t-BuOH | tert-Butanol |
| EtOAc | Ethyl acetate |
| Et2O | Diethyl ether |
| MTBE | Methyl tert-butylether |
| NMP | N-Methyl-2-pirrolidone |
| AcOH | Acetic acid |
| THF | Tetrahydrofuran |
| DMSO | Dimethyl sulfoxide |
| CPME | Cyclopenthyl methylether |
| NBS | N-Bromosuccinimide |
| AIBN | Azobisisobutyronitrile |
| TMEDA | N,N,N′,N′-Tetramethylethylenediamine |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| LAH | Lithium aluminium hydride |
| TPGS-750-M | DL-α-Tocopherol methoxypolyethylene glycol succinate solution |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality. The University of Perugia is acknowledged for financial support to the university project “Fondo Ricerca di Ateneo, edizione 2021”. E. C. thanks the EU NextGenerationEU program in the context of PNRR for financial support.References
- K. Lovato, P. S. Fier and K. M. Maloney, Nat. Rev. Chem., 2021, 5, 546–563 CrossRef CAS PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- C. J. Testa, C. Hu, K. Shvedova, W. Wu, R. Sayin, F. Casati, B. S. Halkude, P. Hermant, D. E. Shen, A. Ramnath, Q. Su, S. C. Born, B. Takizawa, S. Chattopadhyay, T. F. O'Connor, X. Yang, S. Ramanujam and S. Mascia, Org. Process Res. Dev., 2020, 24, 2874–2889 CrossRef CAS.
- P. J. Kitson, G. Marie, J.-P. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS PubMed.
- J. C. McWilliams, A. D. Allian, S. M. Opalka, S. A. May, M. Journet and T. M. Braden, Org. Process Res. Dev., 2018, 22, 1143–1166 CrossRef CAS.
- N. S. Arden, A. C. Fischer, K. Tyner, L. Yu, S. L. Lee and M. Kopcha, Int. J. Pharm., 2021, 602, 120554 CrossRef CAS PubMed.
- S. Stegeman, Eur. J. Pharm. Sci., 2016, 90, 8–13 CrossRef PubMed.
- J. L. McGuire, H. Hasskarl, R. Krestzschmar, K.-J. Hahn and M. Zahn, Pharmaceuticals, General Survey, in Ullmann's Encyclopedia of Industrial chemistry, Wiley, Goettingen, 2000 Search PubMed.
- C. Browning, Nature, 1922, 109, 750–751 CrossRef CAS.
- P. Elwood, Aspirin yesterday, aspirin today, aspirin tomorrow: a history of prophylactic aspirin, 2017 Search PubMed.
- C. C. Quianzon and I. Cheikh, JCHIMP, 2012, 2, 18701 Search PubMed.
- C. R. Kahn, Annu. Rev. Med., 1985, 36, 429–451 CrossRef CAS PubMed.
- A. Fleming, Br. Med. J., 1941, 2, 386 CrossRef.
- N. Kardos and A. L. Demain, Appl. Microbiol. Biotechnol., 2011, 92, 677–687 CrossRef CAS PubMed.
- For General worldwide reading of interest: https://www.cbo.gov/publication/57126. And reference cited herein.
- For updated reading regarding Europe: https://www.efpia.eu/media/554521/efpia_pharmafigures_2020_web.pdf; https://specialty-chemicals.eu/apic-active-pharmaceutical-ingredients-committee/.
- For updated reading regarding US: https://www.fda.gov/news-events/congressional-testimony/safeguarding-pharmaceutical-supply-chains-global-economy-10302019. And reference cited herein.
- E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Nat. Chem., 2018, 10, 917–923 CrossRef CAS PubMed.
- P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 10139–10147 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- J. Lu, I. Paci and D. C. Leitch, Chem. Sci., 2022, 13, 12681–12695 RSC.
- E. Buncel, J. M. Dust and F. Terrier, Chem. Rev., 1995, 95, 2262–2280 CrossRef.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberthm, Green Chem., 2018, 20, 5082–5103 RSC.
- J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maesa and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- D. P. Debecker, K. Kuok Hii, A. Moores, L. M. Rossi, B. Sels, D. T. Allen and B. Subramaniam, ACS Sustainable Chem. Eng., 2021, 9, 4936–4940 CrossRef CAS.
- L. J. Diorazio, P. Richardson, H. F. Sneddon, A. Moores, C. Briddell and I. Martinez, ACS Sustainable Chem. Eng., 2021, 9, 16862–16864 CrossRef CAS.
- V. Hessel, M. Escribà-Gelonch, J. Bricout, N. N. Tran, A. Anastasopoulou, F. Ferlin, F. Valentini, D. Lanari and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 9508–9540 CrossRef CAS.
- A. Mondal and M. van Gemmeren, Angew. Chem., Int. Ed., 2022, 61, e20221082 Search PubMed.
- R. Laskar, T. Pal, T. Bhattacharya, S. Maiti, M. Akita and D. Maiti, Green Chem., 2022, 24, 2296–2320 RSC.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- S. Santoro, F. Ferlin, L. Ackermann and L. Vaccaro, Chem. Soc. Rev., 2019, 48, 2767–2782 RSC.
- F. Valentini, G. Brufani, L. Latterini and L. Vaccaro, ACS Symp. Ser., 2020, 1359, 513–543 CrossRef CAS.
- N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS.
- D. Sciosci, F. Valentini, F. Ferlin, S. Chen, Y. Gu, O. Piermatti and L. Vaccaro, Green Chem., 2020, 22, 6560–6566 RSC.
- J. Osorio-Tejada, F. Ferlin, L. Vaccaro and V. Hessel, Green Chem., 2022, 24, 325–337 RSC.
- F. Ferlin, I. Anastasiou, L. Carpisassi and L. Vaccaro, Green Chem., 2021, 23, 6576–6582 RSC.
- N. Kaplaneris, M. Vilches-Herrera, J. Wu and L. Ackermann, ACS Sustainable Chem. Eng., 2022, 10, 6871–6888 CrossRef CAS.
- H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen and C.-J. Li, ACS Catal., 2016, 7, 510–519 CrossRef.
- S. Vásquez-Céspedes, R. C. Betori, M. A. Cismesia, J. K. Kirsch and Q. Yang, Org. Process Res. Dev., 2021, 25, 740–753 CrossRef.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- F. Valentini, L. Carpisassi, A. Comes, C. Aprile and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10(37), 12386–12393 CrossRef CAS.
- F. Valentini, F. Ferlin, S. Lilli, A. Marrocchi, L. Ping, Y. Gu and L. Vaccaro, Green Chem., 2021, 23, 5887–5895 RSC.
- F. Valentini, F. Ferlin, E. Tomarelli, H. Mahmoudi, M. Bagherzadeh, M. Calamante and L. Vaccaro, ChemSusChem, 2021, 14, 3359–3366 CrossRef CAS PubMed.
- H. Pang, Y. Hu, J. Yu, F. Gallou and B. H. Lipshutz, J. Am. Chem. Soc., 2021, 143, 3373–3382 CrossRef CAS PubMed.
- A. B. Wood, S. Plummer, R. I. Robinson, M. Smith, J. Chang, F. Gallou and B. H. Lipshutz, Green Chem., 2021, 23, 7724–7730 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- J. Andraos, Org. Process Res. Dev., 2012, 16, 1482–1506 CrossRef CAS.
- J. Andraos, Org. Process Res. Dev., 2013, 17, 175–192 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. Soar, G. D. Perkins, I. Maconochie, B. W. Bottiger, C. D. Deakin, C. Sandroni, T. M. Olasveengen, J. Wyllie, R. Greif, A. Lockey, F. Semeraro, P. Van de Voorde, C. Lott, L. Bossaert, K. G. Monsieurs and J. P. Nolan, Resuscitation, 2019, 134, 99–103 CrossRef PubMed.
- R. Tondeur and F. Binon, Patent Number: US3248401, 1966.
- W. Huang, J. Xu, C. Liu, Z. Chen and Y. Gu, J. Org. Chem., 2019, 84, 2941–2950 CrossRef CAS PubMed.
- N. Iqbal, N. Iqbal, D. Maiti and E. J. Cho, Angew. Chem., 2019, 131, 15955–15959 CrossRef.
- P. J. Barter and K. A. Rye, Arterioscler. Thromb. Vasc. Biol., 2016, 36, 439–441 CrossRef CAS PubMed.
- C. K. Chung, G. R. Humphrey, P. E. Maligres and T. J. Wright, Merck Sharpe & Dohme Corp, Patent Number: US9145348, 2015.
- S. G. Ouellet, A. Roy, C. Molinaro, R. Angelaud, J.-F. Marcoux, P. D. O'Shea and I. W. Davies, J. Org. Chem., 2011, 76, 1436–1439 CrossRef CAS PubMed.
- P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023–8040 CrossRef CAS.
- S. Santoro, A. Marrocchi, D. Lanari, L. Ackermann and L. Vaccaro, Chem. – Eur. J., 2018, 24, 13383–13390 CrossRef CAS PubMed.
- F. Valentini, G. Brufani, B. Di Erasmo and L. Vaccaro, Curr. Opin. Green Sustainable Chem., 2022, 36, 100634 CrossRef CAS.
- J. Chapman, W. Magee, H. Stukenbrok, G. Beckius, A. Milici and W. Tracey, Eur. J. Pharmacol., 2002, 456, 59–68 CrossRef CAS PubMed.
- C. W. Scott, C. Sobotka-Briner, D. E. Wilkins, R. T. Jacobs, J. J. Folmer, W. J. Frazee, R. V. Bhat, S. V. Ghanekar and D. Aharony, J. Pharmacol. Exp. Ther., 2003, 304, 433–440 CrossRef CAS PubMed.
- D. V. Kravchenko, Y. A. Kuzovkova, V. M. Kysil, S. E. Tkachenko, S. Maliarchouk, I. M. Okun, K. V. Balakin and A. V. Ivachtchenko, J. Med. Chem., 2005, 48, 3680–3683 CrossRef CAS PubMed.
- J. Y. Hwang, A. Y. Ji, S. H. Lee and E. J. Kang, Org. Lett., 2020, 22, 16–21 CrossRef CAS PubMed.
- https://www.who.int/news-room/fact-sheets/detail/diabetes .
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021995s019lbl.pdf .
- F. Peng, Y. Chen, C. Chen, P. G. Dormer, A. Kassim, M. McLaughlin, R. A. Reamer, E. C. Sherer, Z. J. Song, L. Tan, M. T. Tudge, B. Wan and J. Y. L. Chung, J. Org. Chem., 2017, 82, 9023–9029 CrossRef CAS PubMed.
- J. Y. L. Chung, J. P. Scott, C. Andersson, B. Bishop, N. Bremeyer, Y. Cao, Q. Chen, R. Dunn, A. Kassim, D. Lieberman, A. J. Moment, F. Sheen and M. Zacuto, Org. Process Res. Dev., 2015, 19, 1760–1768 CrossRef CAS.
- M. Hajos, J. C. Fleishaker, J. K. Filipiak-Reisner, M. T. Brown and E. H. F. Wong, CNS Drug Rev., 2004, 10, 23 CrossRef CAS PubMed.
- K. E. Henegar and M. Cebula, Org. Process Res. Dev., 2007, 11, 354–358 CrossRef CAS.
- R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS PubMed.
- T. Krause, M. U. Gerbershagen, M. Fiege, R. Weisshorn and F. Wappler, Anaesthesia, 2004, 59, 364–373 CrossRef CAS PubMed.
- F. Amiri, M. Rashidi and M. Savaei, Hum. Pathol., 2023, 31, 300693 Search PubMed.
- C. S. Davis and H. R. Snyder, Patent Number: US3415821A, 1968.
- F. Chacón-Huete, J. D. Lasso, P. Szavay, J. Covone and P. Forgione, J. Org. Chem., 2021, 86, 515–524 CrossRef PubMed.
- T. Hosoya, H. Aoyama, T. Ikemoto, Y. Kihara, T. Hiramatsu, M. Endo and M. Suzuki, Bioorg. Med. Chem., 2003, 11, 663–673 CrossRef CAS PubMed.
- E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, F. Delogu, R. Guerra and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC.
- Y. F. Liang, R. Steinbock, L. Yang and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10625–10629 CrossRef CAS PubMed.
- J. Park, U. Egolum, S. Parker, E. Andrews, D. Ombengi and H. Ling, Ann. Pharmacother., 2020, 54, 470–477 CrossRef PubMed.
- K. Barseghyan, D. Hambardzumyan, G. Gevorgyan, V. Karapetyan, K. Nerkararyan and T. Maier, Patent Number: WO2019175263A1, 2018.
- T. Yamamoto, K. Muto, M. Komiyama, J. Canivet, J. Yamaguchi and K. Itami, Chem. – Eur. J., 2011, 17, 10113–10122 CrossRef CAS PubMed.
- F. Ferlin, M. K. Van der Hulst, S. Santoro, D. Lanari and L. Vaccaro, Green Chem., 2019, 21, 5298–5305 RSC.
- J. Agata, N. Ura, H. Yoshida, Y. Shinshi, H. Sasaki, M. Hyakkoku, S. Taniguchi and K. Shimamoto, Hypertens. Res., 2006, 29, 865–874 CrossRef CAS PubMed.
- K. S. Babu, M. S. Reddy, A. R. Tagore, G. S. Reddy, S. Sebastian, M. S. Varma, G. Venkateswarlu, A. Bhatta-charya, P. P. Reddy and R. V. Anand, Synth. Commun., 2008, 39, 291–298 CrossRef.
- M. Seki, Synthesis, 2015, 47, 2985–2990 CrossRef CAS.
- B. Paul, J. A. Trovato and J. Thompson, Am. J. Health-Syst. Pharm., 2008, 65, 1703–1710 CrossRef CAS PubMed.
- J. Prasad, A. K. Satya and V. Chowdary, Patent Number: WO2011/039759A1, 2011.
- J. Yu, K. S. Iyer and B. H. Lipshutz, Green Chem., 2022, 24, 3640–3643 RSC.
- R. T. Stark, D. R. Pye, W. Chen, O. J. Newton, B. J. Deadman, P. W. Miller, J.-L. Panayides, D. L. Riley, K. Hellgardt and K. K. Hii, React. Chem. Eng., 2022, 7, 2420–2426 RSC.
- G. Erickson, J. S. Guo, M. McClure, M. Mitchell, M. C. Salaun and A. Whitehead, Tetrahedron Lett., 2014, 55, 6007–6010 CrossRef CAS.
- F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- D. Cantillo, Chem. Commun., 2022, 58, 619–628 RSC.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC.
- G. Brufani, F. Valentini, F. Sabatelli, B. Di Erasmo, A. Afanasenko, C.-J. Li and L. Vaccaro, Green Chem., 2022, 24, 9094–9100 RSC.
- B. Martin-Matute, M. A. R. Meier, T. X. Metro, S. G. Koenig, H. F. Sneddon, P. Sudarsanam and P. Watts, ACS Sustainable Chem. Eng., 2021, 9, 13395–13398 CrossRef CAS.
- F. Ferlin, I. Anastasiou, N. Salameh, T. Miyakoshi, O. Baudoin and L. Vaccaro, ChemSusChem, 2022, 15, e2021027 CrossRef PubMed.
- S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC.
- C. Espro, E. Paone, F. Mauriello, R. Gotti, E. Uliassi, M. L. Bolognesi, D. Rodríguez-Padrón and R. Luque, Chem. Soc. Rev., 2021, 50, 11191–11207 RSC.
| This journal is © The Royal Society of Chemistry 2023 |