
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-centered radical based dynamic covalent chemistry for stimuli-responsive chromic materials
Tingting
Xu
,
Jun
Zhu
,
Yi
Han
and
Chunyan
Chi
 *
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore, Singapore. E-mail: chmcc@nus.edu.sg
First published on 28th April 2023
Abstract
As an essential smart material, stimuli-responsive chromic materials have received increasing research attention because of their visualization effects induced by external stimuli, including temperature, light, and mechanical stress. According to the types of stimuli, they can be categorized as thermochromic, photochromic, and mechanochromic materials, respectively. In recent studies, organic radical-based dynamic covalent chemistry (DCC) has demonstrated great potential to serve as a prototype for the preparation of stimuli-responsive chromic materials because the process is facilitated by simple radical–radical coupling reactions without the addition of catalysts or the generation of by-products and is usually accompanied by a distinct color change. This review aims to highlight carbon-centered radicals that can associate and dissociate reversibly with external stimuli and explore their potential as building blocks for the preparation of stimuli-responsive chromic materials.
 Tingting Xu | Tingting Xu was born in Anhui, China, in 1994. She received her bachelor's degree from Anhui Normal University in 2015 and her master's degree from University of Science and Technology of China in 2018. She then obtained her PhD degree from National University of Singapore in 2022 under the supervision of Prof. Chunyan Chi and joined Prof. Chi's research group as a research fellow in 2022. Her current research focuses on the design and synthesis of organic monoradical and diradicaloid based materials. |
 Jun Zhu | Jun Zhu was born in Wuhan, China, in 1992. He received his bachelor's degree from the National University of Singapore in 2018. He is currently completing his PhD under the supervision of Prof. Wu Jishan at the same university. His research is focused on the exploration and development of new synthetic strategies for the preparation of challenging molecular architectures. |
 Yi Han | Yi Han was born in Shanghai, China, in 1994. He received his bachelor's degree in Polymer Materials and Engineering from Fudan University in 2016. He then performed doctoral research on the organic synthesis of topologically unique polycyclic π-conjugated systems with Prof. Chi Chunyan at National University of Singapore and obtained his PhD in chemistry in 2021. He is now a postdoc research fellow in the same group, focusing on the research of synthetic carbon nanotubes and fullerene analogues. |
 Chunyan Chi | Chunyan Chi conducted her PhD research at the Max-Planck Institute for Polymer Research under the supervision of Professor Gerhard Wegner from 2001–2004. She then carried out postdoctoral work with Professor Guillermo Bazan at the University of California at Santa Barbara. She is now a tenured associate professor in the Department of Chemistry, National University of Singapore. Her research interests include the synthesis of extended π-electron systems with novel structures, aromaticity, organic diradicaloids, dyes, liquid crystals and functional materials for organic electronics. |
10th Anniversary StatementThis year marks the 10th anniversary of Journal of Materials Chemistry C. It has been a great honor for me to serve as an international advisory board member since 2017. During my service, I have been impressed by many high-quality works in the research field of organic functional materials from diversified research groups all over the world. In this duty, it has been my honour to witness the growth and development of both this flagship journal of materials science and our research field. With the growing requirement for “smart” materials, there has been increasing research interest in dynamic covalent chemistry compared to classical covalent chemistry and supramolecular chemistry. I would want to take this opportunity to present our latest review “Carbon-centered Radical Based Dynamic Covalent Chemistry for Stimuli-responsive Chromic Materials”. We hope this review will draw more attention to this rising research topic. |
1. Introduction
Chromic materials have gained increasing research attention in the past few decades because color change phenomena can be easily perceived with the naked eye, giving rise to a wide range of applications, including smart windows,1–3 optical switches,4–6 sensors,7,8etc.9–11 Stimuli-responsive chromic materials are defined as dyes that exhibit a distinct color change when exposed to an external stimulus. These color change phenomena are classified and named according to the stimulus which causes the change, like photochromism, thermochromism, mechanochromism and electrochromism.12–16 These chromic materials are useful and valuable in daily life and industry, especially for materials in which the color change is reversible and controllable.The synthesis of organic molecules is generally dominated by kinetically controlled reactions, which irreversibly form strong covalent bonds. In the reactions, the reactants are prone to generating a product with a more stable transition state instead of others. Once the product is formed, it can never revert to the reactants under the same conditions because of the irreversible nature of the reaction. However, the covalent bonds can be formed and cleaved reversibly in dynamic covalent chemistry (DCC)17 because the dynamic process allows the exchange of molecular components at equilibrium to achieve thermodynamic minima of the system. It has been widely employed by chemists from different research fields for the preparation of complex molecular assemblies, including molecular knots,18 macrocycles,19–21 polymers22,23 and covalent organic frameworks.24–27 Meanwhile, the thermodynamic equilibrium can be disturbed dramatically by external conditions, like temperature, light, electricity, mechanical stress, etc., which makes it suitable for self-healing systems, sensors, and actuators.28–30
Unlike traditional supramolecular chemistry involving only weak non-covalent bonds, dynamic covalent chemistry deals with covalent bonds with higher bonding energy and lower kinetics of bond formation and dissociation. In the practical application of dynamic covalent chemistry, the lifetime of the dynamic covalent bonds, which is an essential factor for dynamic behavior, must be long enough for the detection and isolation of the product. When the lifetime is too short, the associated product does not exist, whereas too long a lifetime yields a product without the desired dynamic behavior. Practically, the lifetime should be in the range from 1 μs to 1 min31 to ensure reasonably fast kinetics of the reversible bonds for error-correction and error-proof during the reaction period. In some cases, the presence of a catalyst that promotes reversible bond formation and dissociation is crucial to maintain the fast kinetics of the reversible bonds to reach thermodynamic equilibrium within a reasonable period. In contrast, radical-based dynamic covalent chemistry does not involve a catalyst. The reversibility is induced by radical–radical coupling reactions to form dynamic covalent bonds that can break and form under certain conditions. In addition, the reactions do not generate any by-product so that the products can be simply isolated and purified. These specific features have made organic radicals promising motifs for dynamic covalent chemistry.32–40 The definition of organic radicals reveals that organic radicals are open-shelled molecules whose highest molecular orbital is occupied by an unpaired electron, which enables them to have special spectroscopic properties and largely expands the scope of their applications, especially as stimuli-responsive chromic materials (Fig. 1). Recently, stable organic radicals and their applications as functional materials have been actively explored. While there are many reviews focused on stable radicals and their applications,41–45 few reviews have discussed radical-based dynamic covalent chemistry on their potentials as stimuli-responsive chromic materials.46 Among all organic radical species, carbon-centered radicals are more attractive, because the trivalent feature of carbon-centered radicals guarantees structural diversities and intricate molecular designs.41 Therefore, this review aims to highlight the most reported carbon-centered radical-based dynamic covalent chemistry. With a focus on their structures and properties, the applications as stimuli-responsive chromic materials are also explored and discussed.
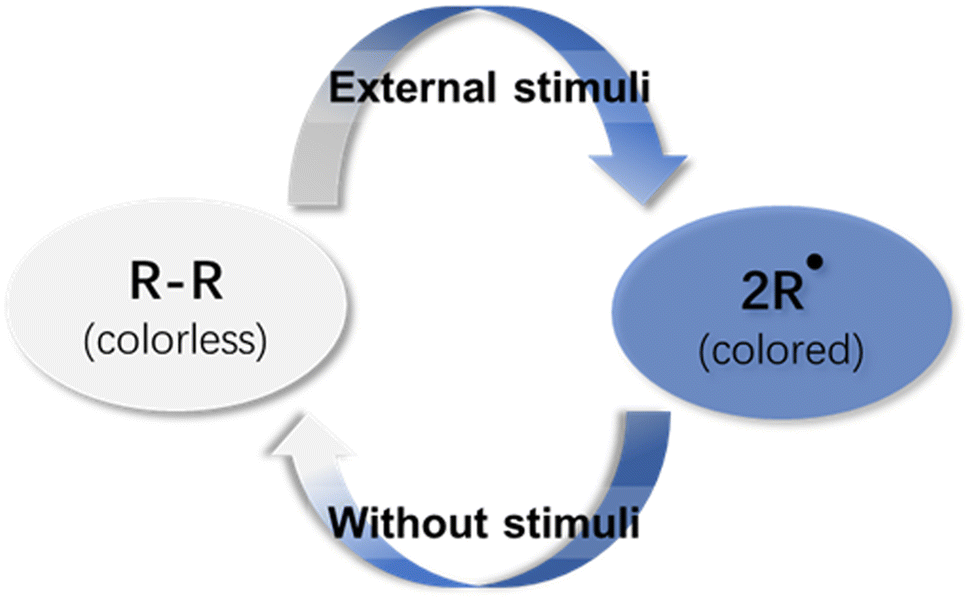 | ||
| Fig. 1 Schematic representations of radical-based dynamic covalent chemistry. | ||
2. Carbon-centered radical-based dynamic covalent chemistry
Ever since the successful isolation of the first stable organic radical, triphenylmethyl radical (1, Fig. 2(a)), by Gomberg in 1900,47 radical chemistry has become a popular research field. Much research effort has been dedicated to the development of stable organic radicals in recent decades due to their unique optoelectronic and magnetic properties that are crucial in many research fields.48–50 It has been reported that the triphenylmethyl radical (1) can dimerize spontaneously at its para-position to form its dimer 12 (Fig. 2(a)). After some pioneering studies on the monomer–dimer equilibrium with various para-substitutions (CF3, tBu, OMe, OPh, CN, COPh, COMe, Ph, SMe and NO2) of 1,51,52 the dynamic properties of radicals in the monomer–dimer equilibrium have been continuously explored. It was found that all substituents caused facilitated dissociation of the dimer, hence stabilizing the radical form. Seki's group reviewed many stable radicals based on dynamic covalent chemistry.46 The reversible formation of dynamic covalent bonds is mainly triggered by the dimerization and dissociation of organic radicals. Due to the fact that the intrinsic instability of organic radicals could largely restrict their practical applications, appropriate stabilization strategies to make the radicals less sensitive to water and oxygen are necessary. Kinetic stabilization and thermodynamic stabilization are two common approaches. Kinetic stabilization introduces bulky substituents on the active sites to avoid adverse side reactions while thermodynamic stabilization involves the facilitation of spin delocalization. A series of stable carbon-centered radicals have been reported based on these two approaches.41,43,44 However, some radicals are too stable to undergo dimerization to form covalent bonds in solution or solid state, for instance, (pentachlorophenyl)methyl radical53 and trioxoriangulene.54,55 Therefore, carbon-based organic radicals that possess good resistance to undesired side reactions, yet can still reversibly undergo association and dissociation, are fundamental. Representative stable carbon-centered radicals for dynamic covalent chemistry are discussed in this section, including fluorenyl radicals, benzofuranone radicals, dicyanomethyl radicals and others (Fig. 2(b) and Table 1).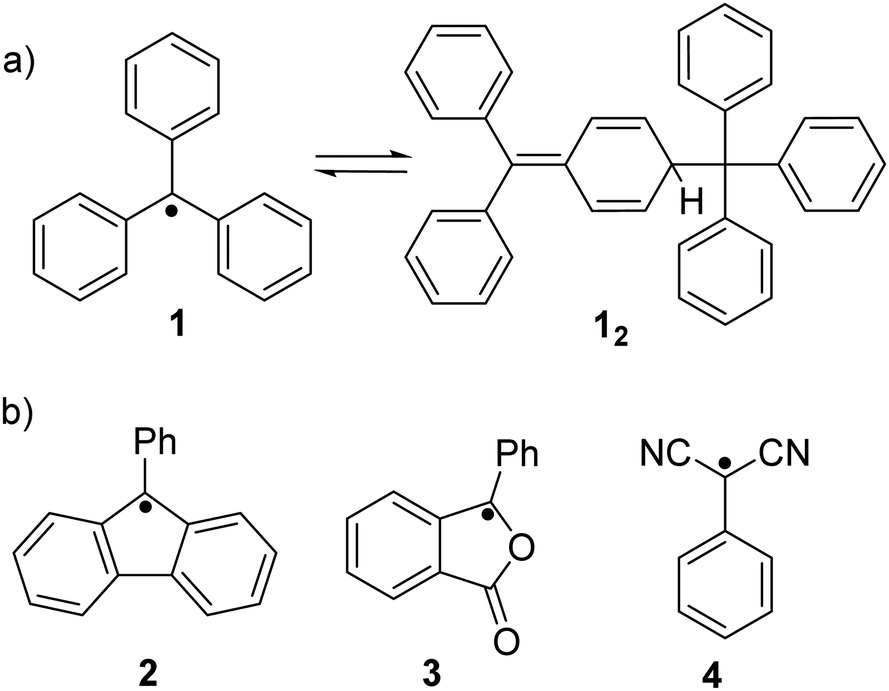 | ||
| Fig. 2 (a) Equilibrium between triphenylmethyl radical (1) and its dimer (12), and (b) chemical structures of most reported carbon-centered radicals that are unreactive to oxygen. | ||
| Radical | BDE (kcal mol−1) | Reacts with oxygen? | λ max of dimer (nm) |
|---|---|---|---|
| 1 | Not reported | Yes | 315 |
| 2 | 15.2 | No | 320 |
| 3 | 23.6 | No | 346 |
| 4 | Not reported | No | Not reported |
2.1 Fluorenyl radicals
The bond dissociation energy of 9,9′-diphenyl-9,9′-bi-9H-fluorene to 9-phenylfluorenyl radicals was theoretically calculated to be 15.2 kcal mol−1, which makes fluorenyl radicals suitable for dynamic covalent chemistry.56,57 The bulky substituents could offer kinetic stabilization to lower the bond dissociation energy and stabilize the radical form.58,59 In 1969, G. Wittig tried to synthesize quinoidal compound 5 (Fig. 3) by dehydrogenation of 1,4-bis(9-bromofluorenyl)benzene. Unexpectedly, tetramer macrocycle 54 was obtained as a colourless solid. It was striking to observe that the color of 54 can change from colourless to deep blue with external stimuli, including temperature and mechanical stress, which indicates the existence of diradical 5 with a deep blue color. Dissolving colorless 54 in organic solvents resulted in a deep blue solution, suggesting an equilibrium between 5 and 54 in solution.60 Ipaktschi et al., in 1999, investigated the self-assembly process of the substituted derivatives of Wittig's hydrocarbon 5. Reduction of the di-hydro precursor with acidic tin(II) chloride solution resulted in macrocycles with a high yield of 82%. The high yield could be attributed to dynamic covalent features, which favor the formation of more thermodynamically stable macrocyclic products. The substituted derivatives of Wittig's hydrocarbons can also produce a colored solution after dissolving in organic solvents. The structures of the tetramer macrocycles were unambiguously confirmed and characterized using X-ray crystallography.61,62 In 2016, Beaudoin et al. synthesized two giant carbocycles 6a6 and 6b4, which are formed from fluorenyl radical monomers using a reversible C–C bond. The precursor 6a/6b are spirobifluorene-substituted derivatives of Wittig's hydrocarbon 5 (Fig. 4(a)).63 Based on crystallographic analysis of the structures, it can be observed that the bifluorenyl units in the hexamer 6a6 adapt a gauche conformation which could have promoted the cyclization for macrocycle formation (Fig. 4(b)). Bond length analysis revealed that the C–C bond lengths between the monomers in 6a6 and 6b4 were significantly longer (>1.61 Å) than common C(sp3)–C(sp3) bonds, suggesting an evident feature of dynamic bonds. The selective formation of the cyclic hexamer can be attributed to intramolecular C–H⋯π networks among the peripheral fluorene units as donors and the spirobifluorenyl units as acceptors in 6a6. While the tert-butyl groups at the peripheral fluorene units in 6b disturb intramolecular C–H⋯π interactions, a more discrete tetramer macrocycle is formed instead. The results imply that the presence of weak interactions is critical for the self-assembled structures of DCC radicals.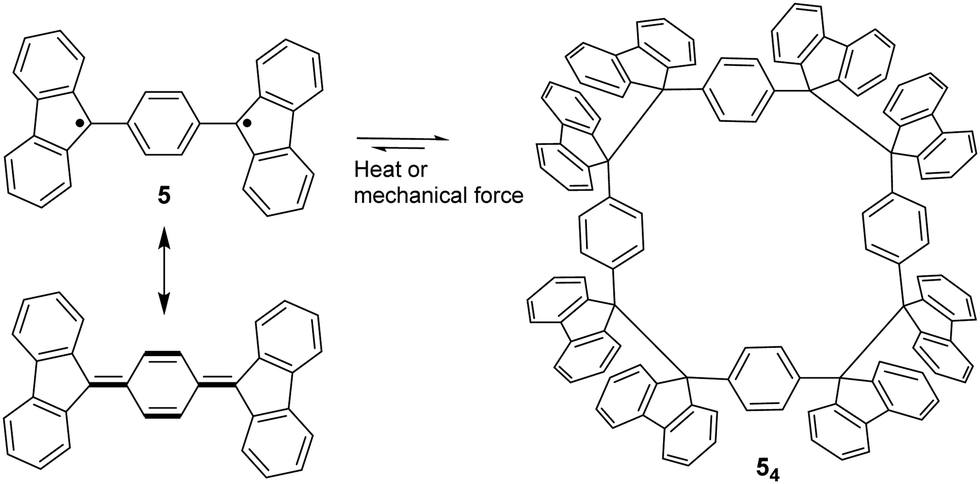 | ||
| Fig. 3 Resonance structure of Wittig's hydrocarbon 5, and the equilibrium between diradical 5 and its macrocyclic tetramer 54. | ||
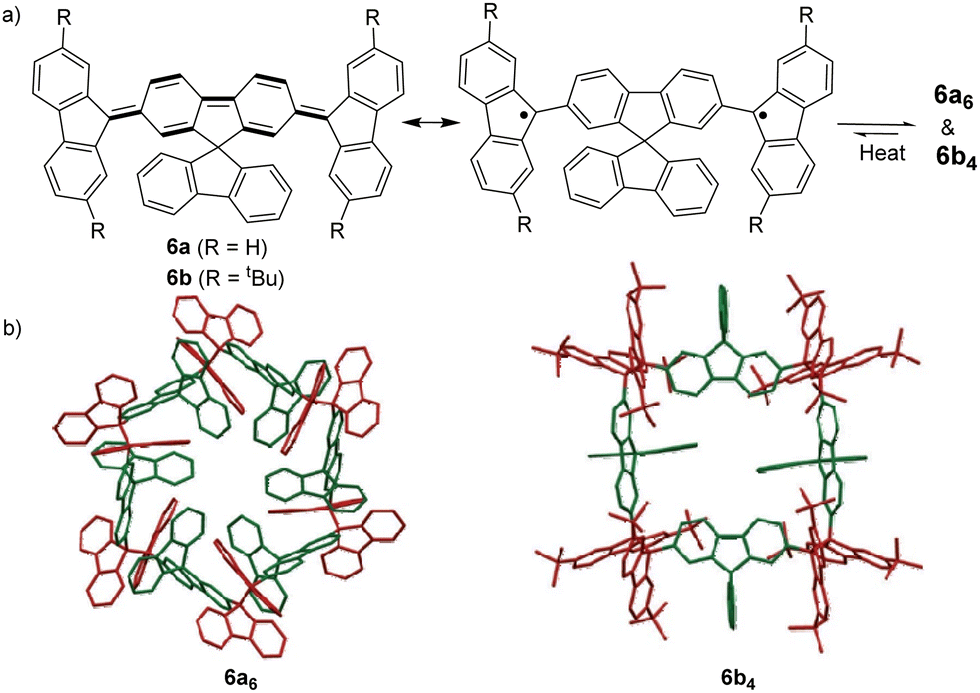 | ||
| Fig. 4 (a) Resonance structures of 6a/6b; (b) X-ray crystallographic structure of macrocyclic hexamer 6a6 and macrocyclic tetramer 6b4. Reprinted with permission from ref. 63. Copyright 2016 Wiley-VCH. | ||
Other than a phenyl substituent at the 9-position of fluorane, it can also be substituted with a cyano group to achieve stablization of the fluorenyl radical for dynamic covalent chemistry since the cyano group is a strong electro-withdrawing group that can induce spin delocalization. Besides, the cyano substituent can also offer kinetic stabilization to some extent. The reported 9-cyanofluorenyl radicals have demonstrated good stability, especially in their inertness toward oxygen.64 Meanwhile, the dynamic process between difluorenylsuccinonitrile (DFSN, 72), the dimer of cyanofluorene radicals, and its monomer (7) was investigated with application of external mechanical stress induced by grinding (Fig. 5). DFSN has been widely employed as thermally stable mechanophores when combined with polymers (such as 82), which can be potentially used as stress or damage detecting sensors.65–67
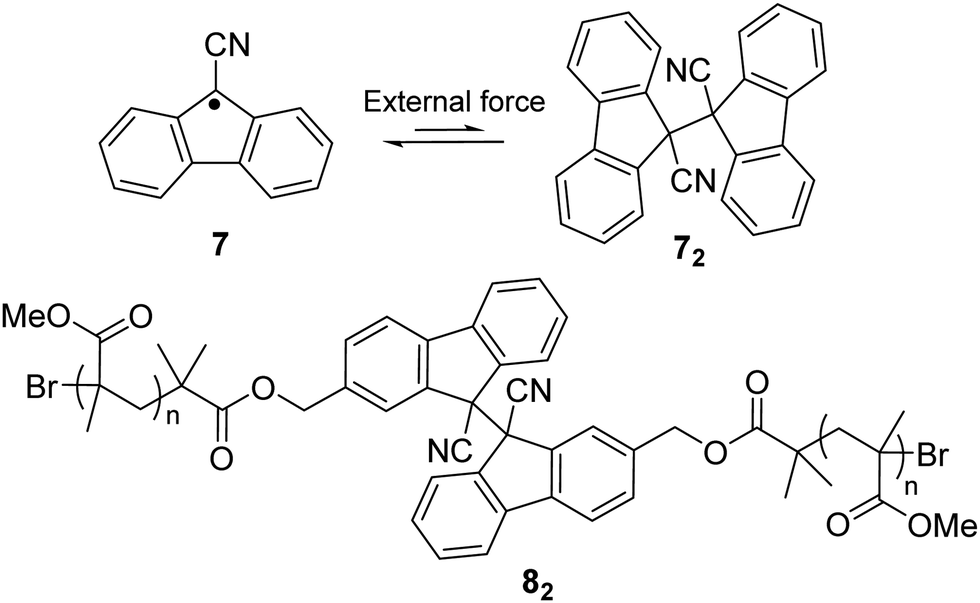 | ||
| Fig. 5 The equilibrium between cyanofluorene radical (7) and its dimer difluorenylsuccinonitrile (72) and the example of a polymer bearing cyanofluorene radical (82). | ||
2.2 Benzofuranone radicals
As a typical antioxidant, benzofuranone radicals exhibit excellent resistivity toward oxygen due to the electro-withdrawing nature of the lactone ring which offers a thermodynamic stabilization effect on the radical.68,69 As reported by Scaiano's group in 2004, the reversible dimerization and dissociation of benzofuranone radicals was observed without any degradation in air, which suggested the attenuated reactivity of the radicals toward oxygen.56 An experiment on the temperature dependent dissociation of the dimers was performed and the bond dissociation energy (BDE) of the central C(sp3)–C(sp3) bonds was estimated to be 23.6 kcal mol−1 (Fig. 6(a)). The crystallographic analysis results revealed the length of the C–C bonds between the two monomers was 1.596 Å, which is rather long for typical σ C(sp3)–C(sp3) bonds and suggests the bonds can be easily broken with external stimuli. Thermochromism of the dimers of 3 (32) and its derivatives was reported previously in 1925.70 A toluene or chloroform solution of 32 showed a pale-blue color at room temperature, and it turned colorless after cooling in an ice-water bath.71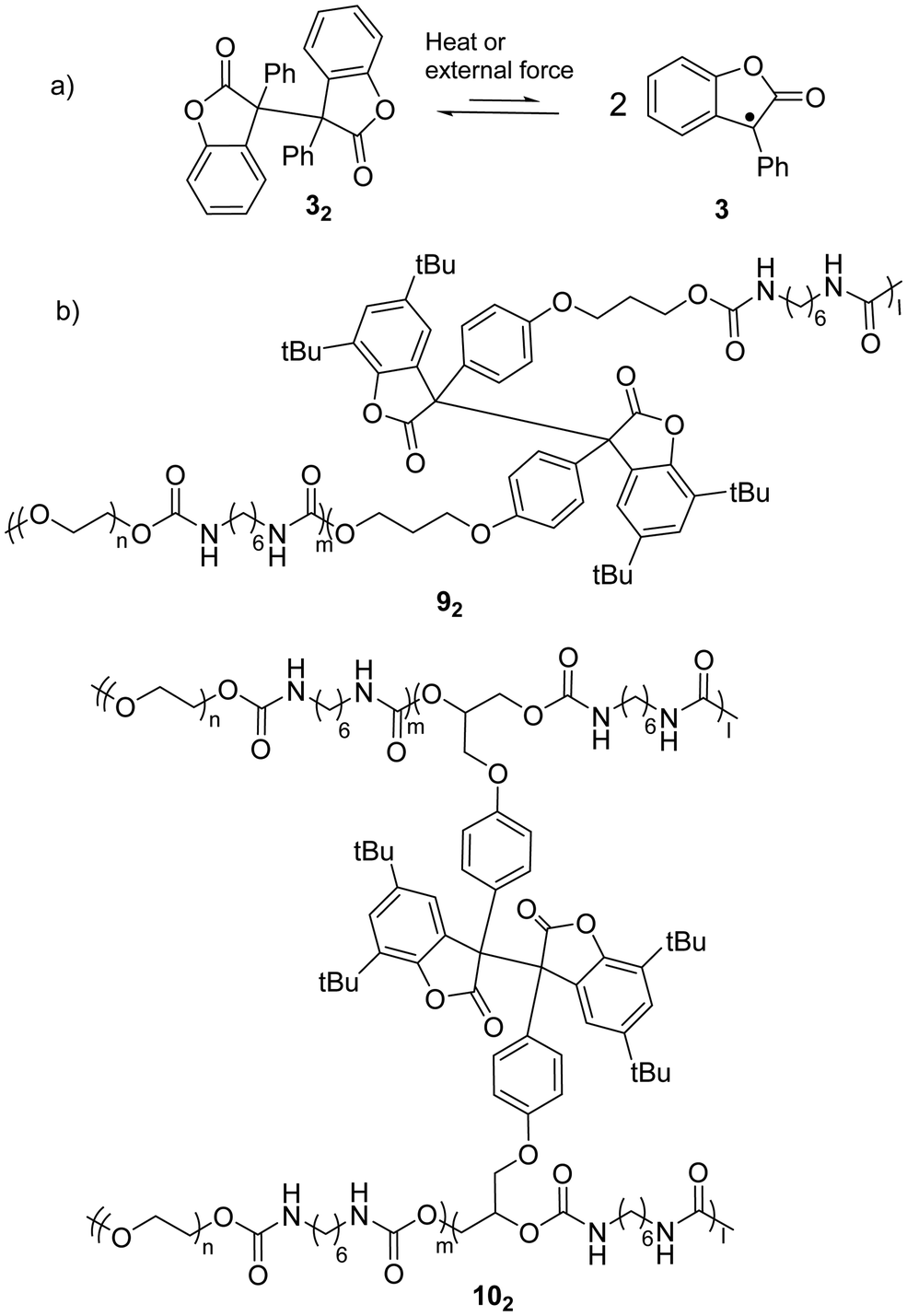 | ||
| Fig. 6 (a) The equilibrium between benzofuranone radical (3) and its dimer (32); (b) examples of polymers bearing the benzofuranone radical (92 and 102). | ||
When benzofuranone radicals are grafted into polymer chains, the materials display responsive self-healing properties.72 In 2012, Otsuka's group reported a polymeric gel whose polymer chains were crosslinked by diaryldibenzofuranone (dimer of arylbenzofuranone radicals). The gel was able to autonomously self-repair under mild conditions (in air and room temperature) without any external stimuli.73 Furthermore, a few polymeric stimuli-responsive chromic materials consisting of benzofuranone radical dimers as the chromophores have been successfully prepared including co-polymer 92 that is linearly linked with a benzofuranone radical dimer and polymer 102 crosslinked by the dimers (Fig. 6(b)).74 Details of the chromic process are elucidated in Section 3.
2.3 Dicyanomethyl radical
The dicyanomethyl radical is a commonly used carbon-centered radical in dynamic covalent chemistry. Recently, dicyanomethyl-substituted aromatic compounds with self-healing properties have been extensively studied and reported due to their effective reversible dimerization and dissociation process. The strong electro-withdrawing effect of two cyano groups largely improves the spin delocalization and provides thermodynamic stabilization of the correlated radical. In 1966, Hartzler et al. synthesized 1,2-diphenyl-1,1,2,2-tetracyanoethane (42) which could be considered the dimer of phenyl-dicyanomethyl radicals (4). As reported, there is an equilibrium between 42 and 4 in solution32 (Fig. 7(a)). Due to the high reactivity at the para-position of 4, the radical species can undergo dimerization at the para-position to form 42′ as an isomer of 42. Nevertheless, this dimerization can be prohibited by the introduction of a substituent at the reactive para-position75–77 (11, Fig. 7(b)). Thereafter, dicyanomethyl radicals have been widely employed in radical-based dynamic covalent chemistry because of their symmetric and sterically unhindered structures, which can avoid the formation of diastereomers during dimerization. However, dimers are the dominant form at equilibrium under room temperature, suggesting the radicals prone to dimerize and the association rate of dicyanomethyl radicals is much faster than the dissociation rate at room temperature. As emphasized, a sufficiently fast equilibration is necessary for dynamic covalent chemistry for practical applications.17 On this account, it is crucial to consider stabilization of dicyanomethyl radicals in their structural design. A straightforward strategy is to promote spin-delocalization to stabilize the radicals by introduction of an electron-donor into the radical system as the unpaired electron can spin-delocalize onto the donor group. Kobashi et al. successfully synthesized two N-substituted dicyanomethylphenyl radicals (12 and 13, Fig. 7(c)) as a triphenylamine and a carbazole derivative.78 The N-substituents serve as electron-donors on the para-position of the dicyanomethyl radicals, which can avoid isomeric dimerization and induce some degree of stabilization. The crystallographic analysis revealed that the C–C bond length between monomers was significantly longer than typical C(sp3)–C(sp3) bonds (1.616 Å for 122 and 1.626 Å for 132, respectively). During the bond dimerization and dissociation, the color of the solution was changed notably at elevated temperatures, evidently suggesting its thermochromic properties. The existence of radical species was confirmed by variable-temperature UV-vis absorption measurements, which shows increased absorption intensities when the temperature is increased. Besides, it was demonstrated that a corelation could be established between the bond dissociation energy (BDE) and the theoretically calculated spin density on the central carbon of the dicyanomethyl radical. In general, the central carbon with a lower spin density results in a smaller BDE. In 2017, Winter's group synthesized a series of phenyl-dicyanomethyl radicals with various substituents (14a–k, Fig. 7(d)), which were in equilibrium with their σ-dimers in solution. Meanwhile, thermodynamic parameters for the self-dimerization were evaluated to determine the effect of substituents on the phenyl-dicyanomethyl radicals.79 Their results revealed that the Hammett parameter was linearly correlated to the binding constants (Ka). Electro-donating substituents weaken the σ-dimers, whereas electron-withdrawing substituents strengthen the σ-dimers. Additionally, the solvent effect on the stability and delocalization of dicyanomethyl radicals was also explored by the same group,80 which revealed that captodative stabilization of the radicals was more pronounced in polar solvents than nonpolar solvents or in the gas phase. Interestingly, the dimerization behavior of the dicyanomethyl radicals can also be modulated through slight structural modifications. Two types of naphthodithiophenes consisting of two cyanoalkyloxycarbonyl-methyl radicals (15) and two dicyanomethyl radicals (16) have been reported by Takimiya and Casado et al., which can be regarded as the resonance hybrids between closed-shell quinoids and open-shell diradicals (Fig. 8). The two molecules behaved differently in the self-assembly process, in which 15 formed the cyclic dimer or higher oligomers bound via σ-bonds (σ-152), instead, a π-dimer (π-162) was formed for 16. In addition, σ-(15 + 16) and π-(15 + 16), as two hetero-dimers, were also observed in the mixture solution of 15 and 16.81 In 2017, Sakamaki and Seki et al. demonstrated the dimerization of dicyanomethyl radicals with a julolidine skeleton to form a π-dimer instead of a σ-dimer.82 It was found out that the electrostatic interactions played a key role in the π-dimer formation from neutral radicals with an intrinsic dipole moment. This finding provides new insights for the structural design of radicals for dynamic covalent chemistry. Later, Winter and co-workers reported systematic investigations of the structural and molecular effects on the dimerization behavior.83,84 It was claimed that the dimerization behavior was dictated by radical spin densities, radical polarizability, and London dispersion stabilization of the dimers.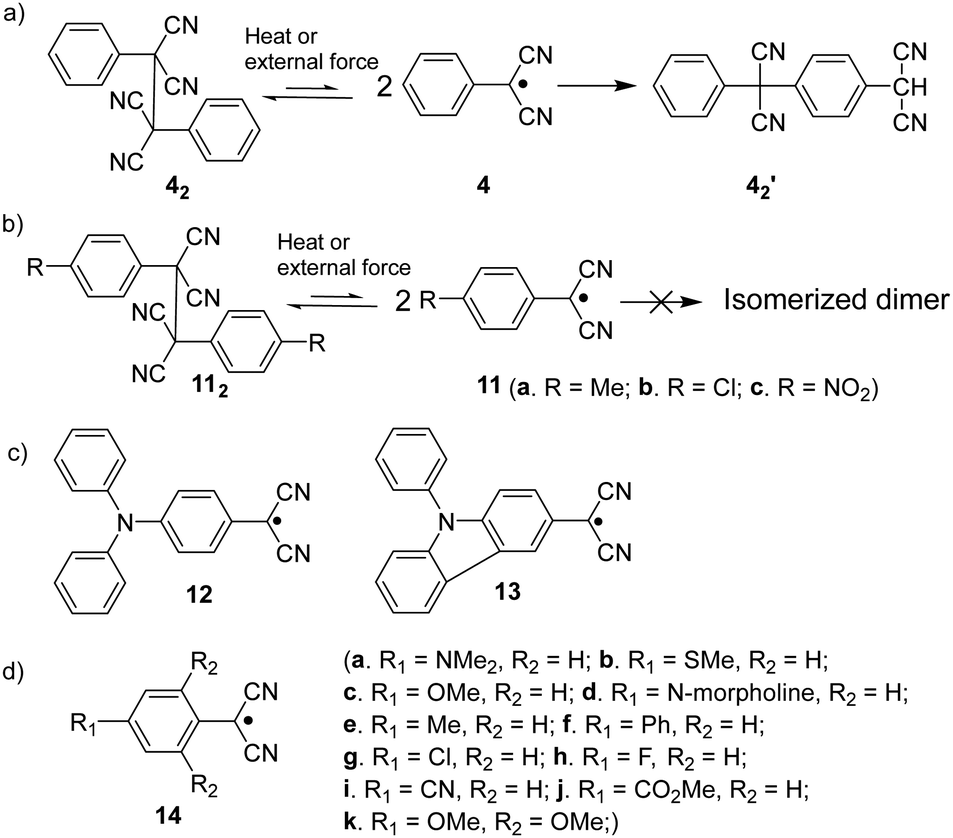 | ||
| Fig. 7 (a) The equilibrium between phenyl-dicyanomethyl radical (4) and 1,1,2,2-tetracyanoethane (42), and its isomerization to 42′; (b) the equilibrium between substituted phenyl-dicyanomethyl radicals (9) and their dimers (112); (c) structures of N-substituted dicyanomethylphenyl radicals (12 and 13); (d) structures of phenyl-dicyanomethyl radical with different substituents (14a–k). | ||
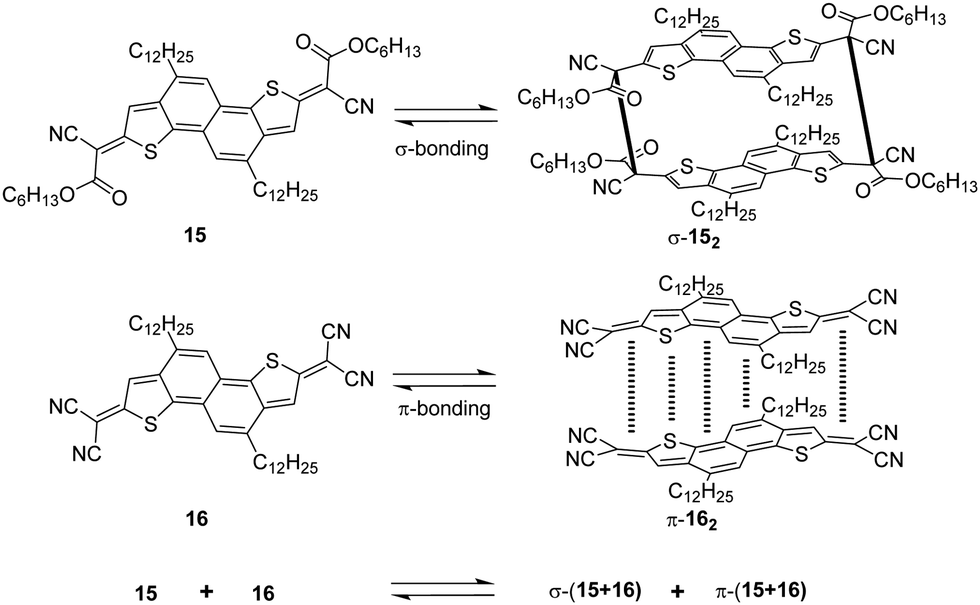 | ||
| Fig. 8 Structures of quinoidal naphthodithiophenes and the formation reactions of σ-152, π-162, σ-(15 + 16), and π-(15 + 16). | ||
It has been observed that cyclic oligomers can be formed when a monomer is designed to possess two dicyanomethyl radical centers. Seki et al. reported the synthesis of macrocyclic oligomers, 172, 183 and 184 (Fig. 9(a)), using triphenylamine (TPA) and carbazole derivatives with two dicyanomethyl as the monomers.78 These oligomers in the solid state are responsive to external stimuli, including temperature and mechanical stress, with a notable change of color from colorless or pale yellow to green or blue. The color change was reversible after the addition of a few drops of solvent onto the solids, which makes them potential candidates for thermochromic and mechanochromic materials.
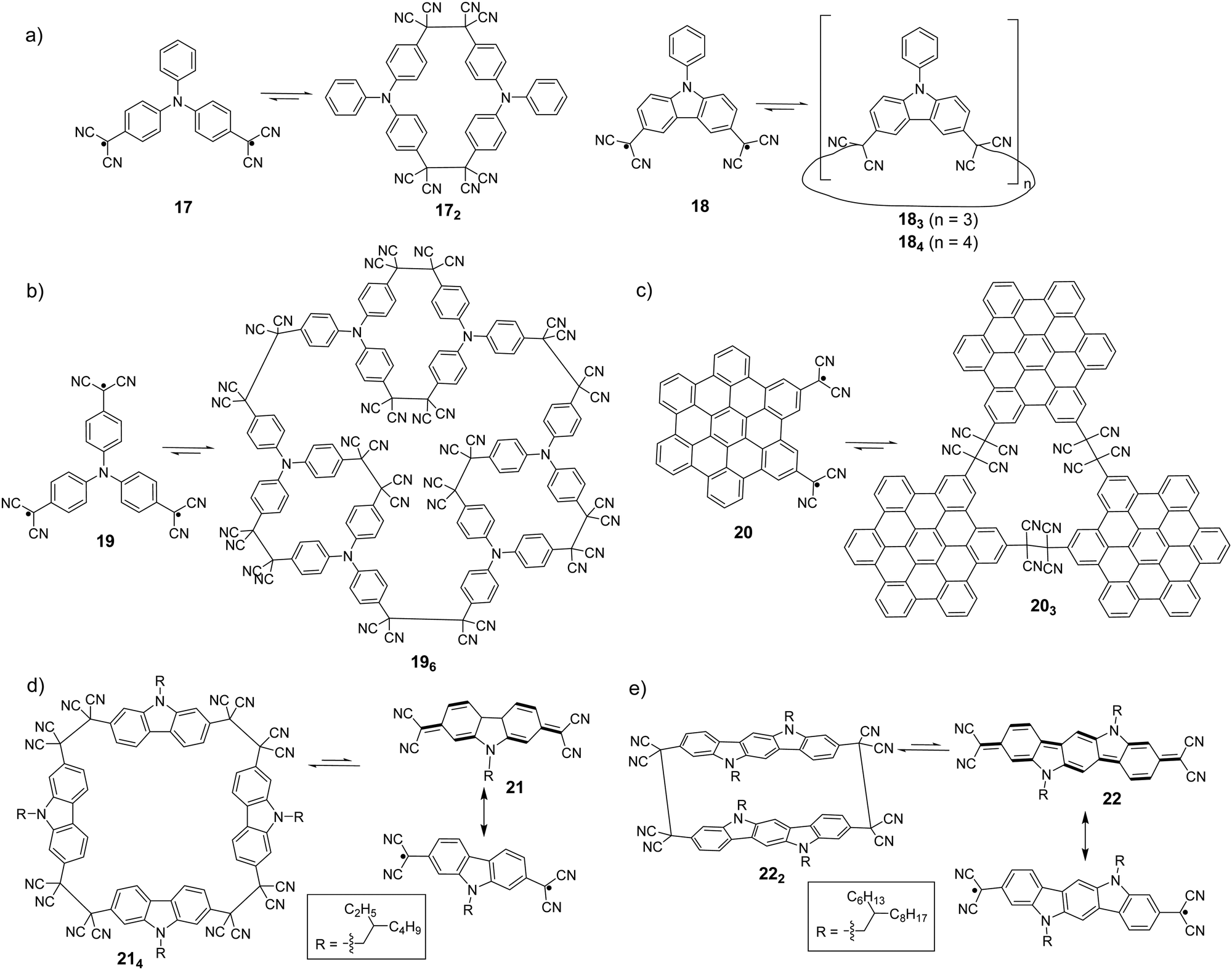 | ||
| Fig. 9 Equilibrium between dicyanomethyl diradicals/poly-radicals and their oligomers (all the conditions for oligomers to their corresponding monomers are a high temperature or external force). | ||
In addition, complex three dimensional macrocyclic structures based on aryl-dicyanomethyl radicals were reported in 2018 by Chi's group.85 The radical monomer was based on a triphenylamine substituted structure with three dicyanomethyl radical centers as the reactive sites, which subsequently underwent oligomerization to form a macrocycles-in-a-macrocycle superstructure with helical chirality. The crystallographic analysis indicated that the structure involved six monomers linked by nine elongated C(sp3)–C(sp3) bonds, depicting dynamic covalent characters (Fig. 9(b)). Firstly, the TPA-substituted dicyanomethyl radicals (19) are prone to form small cyclic dimers by a C(sp3)–C(sp3) bond with gauche-conformation, then three dimeric segments continue to link with each other to form a three-dimensional hexamer cage (196). The elongated C–C bonds that link the monomers were reversible and can undergo dissociation at elevated temperatures with a remarkable change of color. Hiroto and Shinokubo et al. synthesized a hexa-peri-hexabenzocoronene (HBC)-substituted dicyanomethyl radical with enhanced stability because of significant spin delocalisation over the large π-conjugated HBC unit.86 Firstly, a hydro-precursor consisting of one dicyanometyl radical center was oxidized with PbO2, and the resulting radical underwent association to afford a tetracyanoethylene-linked dimer. The structure of the dimer was ambiguously confirmed by X-ray crystallography. Subsequently, a HBC precursor with two dicyanomethyl radical centers at the 2,5-position (20) was employed, and it was found out that the radicals self-assemble into a cyclic trimer (203) at room temperature. The structure of 203 was confirmed by high-resolution mass spectroscopy, NMR and preliminary X-ray crystallography. The mechanochromic behavior of 203 was also investigated, in which the color of the powder sample of 203 changed from yellow to brown after grinding. The UV-vis-NIR absorption spectra in the solid state demonstrated that the dark color corresponded to the dissociated radical species 20. After the addition of one drop of dichloromethane onto the brown powder of 203, the color recovered back to the original yellow and the characteristic NIR absorption bands corresponding to radical 20 disappeared. In 2017, H. Li et al. reported the equilibrium between 2,7-dicyanomethylenecarbazole-based biradicaloid (21) and its corresponding cyclic tetramer 214 (Fig. 9(c)).87 X-ray crystallographic analysis revealed that four monomers were linked by elongated C(sp3)–C(sp3) bonds (ca. 1.631 Å), which were resulted from couplings of the unpaired electrons in biradicaloid 21, to form a cyclic tetramer. Based on the biradicaloid 18 and 21, the effect of different substitutional positions of the dicyanomethyl radical on the self-assembly of carbazole-based diradicals was investigated by Delgado et al.88 It was found out that the meta-substituted cyclophanes are more difficult to dissociate both in solution and solid state as compared to the para-substituted carbazole homologues, indicated by the formation of more stable macrocycles with shorter C–C bonds. Recently, an indolo[3,2-b]carbazole compound containing terminal dicyanomethylene groups in the 3,9-positions (22) was synthesized and characterized by Li and Delgado et al. as a continuing work to investigate how the expansion of the conjugated core can affect the intermolecular σ-bonding reactivity of carbazole-based diradicals.89 After comparing with shorter chain carbazole-based analogue 21, it was revealed that the larger conjugated core results in an increased diradical character as well as a smaller open-shell singlet–triplet gap. The biradicaloid 22 was isolated as a stable σ-dimer (222) in the solid state with two co-facial indolocarbazole units featuring attractive π–π interactions.
As another conjugated core extension of dicyanomethyl radicals for dynamic covalent chemistry, Osuka and co-workers prepared dicyanomethyl radicals containing porphyrins (Fig. 10). Firstly, a simpler subporphyrin molecule 23 with one dicyanomethyl radical center at the meso-position was synthesized to validate the feasibility for reversible σ- or π-dimerization in solutions or the solid state.90 However, 23 was not prone to undergo association because of its outstanding stability induced by the efficient spin delocalization over the subporphyrin skeleton. Next, another two types of porphyrins bearing a dicyanomethyl radical at the meso-position (24) and β-position (25) of the porphyrin ring were prepared.91 Similar to radical 23, the meso-substituted porphyrin 24 did not exhibit any dimerization behavior and remained as monomeric radicals both in solution and the solid state for the same reason. In contrast, the β-substituted porphyrin 25 was in equilibrium with its σ-dimers (252) in solution. The dynamic behavior was confirmed and characterized by variable temperature 1H-NMR, UV-vis and ESR measurements. It was observed that the monomeric radical species were predominant at high temperature while σ-dimerization was favorable at low temperature. The structures of monoradical 24 and the σ-dimer 252 were unequivocally confirmed by X-ray crystallography. The difference in the dynamic properties between 24 and 25 can be explained by the spin density distribution. The spin on 24 can be fully delocalized over the meso-substituted porphyrin skeleton, leading to excellent stabilization of the monoradicals. In contrast, the spin on 25 is relatively more localized on the dicyanomethylene moiety because it resides at the β-position of the porphyrin skeleton, which can only provide limited stabilization. Additionally, porphyrins with two dicyanomethyl radical centers were also reported.92,93 In 2020, Osuka et al. prepared 2,18-bis(dicyanomethyl)-substituted NiII porphyrin radical 26 and ZnII porphyrin radical 27, which can undergo dimerization to afford cyclophane type chlorin dimers 262 and 272, respectively.94 The X-ray crystallographic analysis revealed that dimer 262 adapted a syn-conformation with two distorted NiII chlorins while 272 displayed an anti-conformation with relatively planar ZnII chlorins. The equilibrium between monomers 26/27 and respective dimers 262/272 based on reversible radical–radical dissociation and a recombination process was observed at high temperature for 26/262 and at room temperature for 27/272. The existence of radical species during the dynamic process was detected by the broadening of 1H-NMR signals, changes in the UV-vis-NIR absorption spectra, and the active monoradical signals in the ESR spectra.
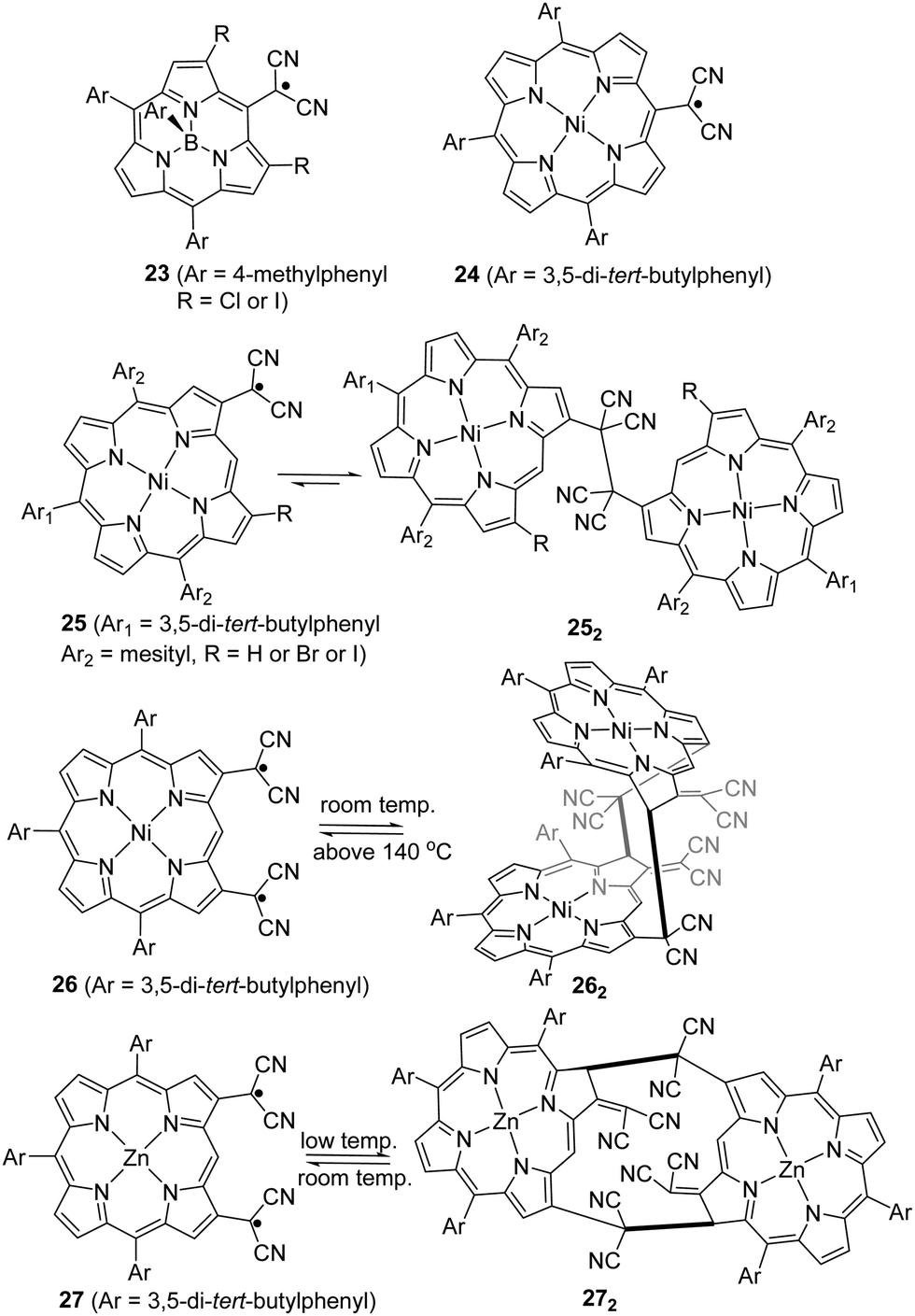 | ||
| Fig. 10 Mono or bi-dicyanomethyl radical-substituted (sub)porphyrins. | ||
2.4 Others
Apart from the fluorenyl radicals, benzofuranone radicals and dicyanomethyl radicals, there are some other carbon-centerd radicals showing dynamic covalent chemistry characters.46 For example, similar to benzofuranone radicals, the introduction of some heteroatoms can provide additional stabilization to the radicals because of promoted spin delocalization (Fig. 11), including 2,3,4-trisubstituted chromenyl radicals (28a–e),95,96 2-arly-3-benzothiophenonyl radicals (29a–d)39,97–99 and arylindolinone radicals (30a–f),100etc. These radicals were reported to undergo dimerization spontaneously to form the corresponding dimers, and the as-formed dimers can also dissociate into individual radicals by external stimuli.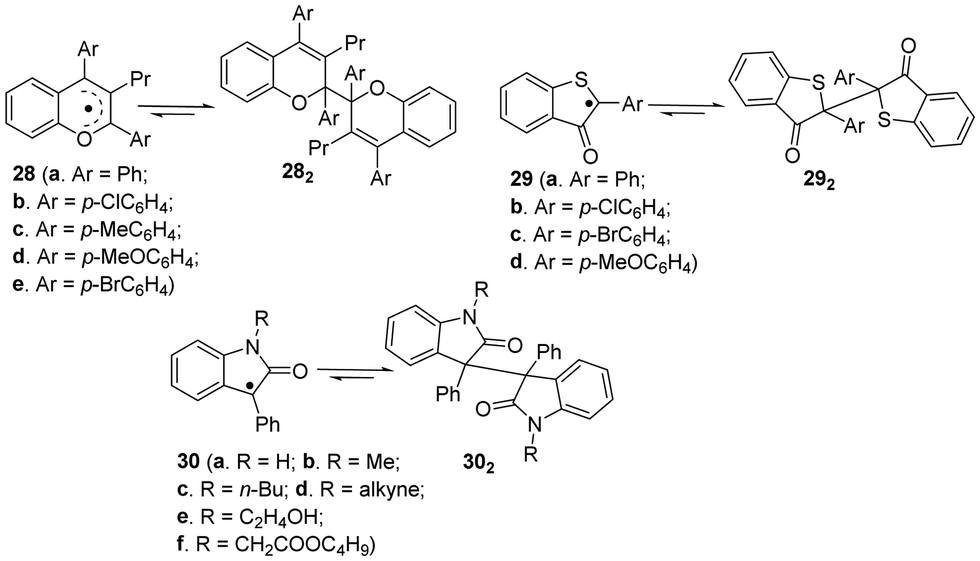 | ||
| Fig. 11 Carbon-centered radicals with heteroatoms in reversible equilibrium with the dimers (all the conditions for dimers to their corresponding monomers are external force). | ||
3. Carbon-centered radical as stimuli-responsive chromophores
Dimeric compounds are usually colorless or light colored, while the radical species exhibit a dark color due to their special spectroscopic properties. Taking advantage of the radical-based dynamic covalent nature, carbon-centered radicals have been used as promising stimuli-responsive chromophores to work as stimuli-responsive chromic materials.101–104 This kind of radical-based chromophore contains dynamic C–C “bridges” connecting two identical moieties and can undergo homolytic bond cleavage when an external stimulus is applied, leading to the formation of radical monomers together with a remarkable color change.Diarylbibenzofuranone (DABBF), the dimer of an arylbenzofuranone radical, has been widely used as a new mechanically sensitive probe in the preparation of stimuli-responsive polymer materials72,74,105–107 because the fast equilibrium between DABBF and its corresponding radicals occurs at room temperature and there is no byproduct formed during the process. In addition, the benzofuranone radicals show blue color as an obvious sensory signal.69–71 In 2015, Otsuka et al. reported the preparation of a linear polymer and a cross-linked polymer with DABBF linkages as the mechanophores. To estimate the magnitude of force required to cause the dynamic bond dissociation in the polymer chains, the BDEs for the central C–C bonds between the two monomers was calculated through ESR measurements under different conditions. As shown in Fig. 12(a), upon cooling, the color was changed from colorless to dark blue, suggesting there was a freezing-induced force that caused cleavage of the dynamic bonds between the radical monomers, and the materials can revert to colorless after thawing. The results indicated that DABBF derivatives could serve as reversible color-changing mechanophores. Furthermore, the system can be employed for the estimation of force induced by freezing of the polymer. Therefore, benzofranone radicals are promising materials for applications as a mechanical stress indicator and sensor.74
 | ||
| Fig. 12 (a) Photographs of the gel swollen with 1,4-dioxane before and after freezing with liquid nitrogen in the air. Reprinted with permission from ref. 74. Copyright 2015 Wiley-VCH. (b) Images of the thermochromic behavior of dicyanomethyl diradical 21 and its tetramer 214. Reprinted with permission from ref. 87. Copyright 2017 Wiley-VCH; (c) Images of the thermochromic behavior of dicyanomethyl diradical 22 and its dimer 222. Reprinted with permission from ref. 89. Copyright 2021 Wiley-VCH. | ||
As mentioned in Section 2, the mechanochromic and thermochromic behaviors of dicyanomethyl radical based dynamic covalent chemistry have also been frequently reported. For example, the elongated bonds in 214 can undergo dissociation when an external stimulus is applied, including light, temperature and mechanical stress.87 As shown in Fig. 12(b), when the purple solution of monomer 21 in chloroform was slowly cooled from 293 K to 210 K, the color faded gradually, with decreased absorption at 450 and 850 nm, along with increased UV absorption at 250–400 nm, typical to that of tetramer 214. The purple color recovered quickly when the solution was warmed up to room temperature, thus revealing the reversible thermochromic properties of these materials. Similar phenomenon was also observed in dimer 222.89 The weak C–C bonds between monomers in 222 are susceptible to thermal and mechanochemical cleavage both in the solid state and solution, which is accompanied by a strong chromism from light yellow to blue-green (Fig. 12(c)). TDDFT calculation results revealed that the blue-green color originated from the specific absorption of the cleaved diradical species 21. Sakamaki and Seki et al. prepared three novel dicyanomethyl radicals with a triphenylamine skeleton, which show thermochromism in the near-infrared (NIR) region based on reversible dimerization–dissociation reactions.108 A. H. Winter et al. synthesized a series of 14 tethered aryl dicyanomethyl diradicals and investigated their thermodynamic properties.109 Significant thermochromic behavior was observed, thus they determined that these diradicals are promising building blocks for stimuli-responsive polymers and plastics, as well as dynamic covalent assemblies, possessing remarkable stability. Very recently, L. Zhao, G. Frenking and X. Wang et al. reported a strategy not only to isolate a dynamically stable radical with tunable physical properties, but to efficiently regulate the radical dissociation with reversibility and photo controllability. The dissociation of the dimer and the formation of the radical adduct become photocontrollable through introducing a photo BCF generator.110 All these results demonstrated that dicyanomethyl radicals could potentially work as promising chromophores for stimuli-responsive materials.
The cyano substituted diphenylmethyl radicals (31, Fig. 13(a)) have been reported to demonstrate reversible equilibrium with its dimer (312). The bond dissociation energy of the C–C bond between monomers in 312 is theoretically calculated to be 26.2 kcal mol−1, which is quite low and suggests the bonds can be easily broken.56,111 In addition, radical 31 shows a characteristic pink color and emits yellow light under UV irradiation, making radical 31 and its derivatives promising materials for mechanochromism and mechanoluminescence.112 Interestingly, freezing-induced mechanoluminescence phenomenon was demonstrated for the first time by Otsuka's group in 2018, which enables the visualization of mechanical stress and chain cleavage induced by freezing indicated by changes in color and light emission.113 A cross-linked polyurethane consisting of tetraarylsuccinonitrile (TASN) moieties at the cross-linking points was successful prepared through polyaddition reaction. As shown in Fig. 13(b), when the as-prepared TASN gel swollen with 1,4-dioxane was frozen using liquid nitrogen, the color of the gel changed from colorless to passion pink with an emission of brilliant yellow light under UV irradiation (λex = 365 nm). The mechanochromism and mechanoluminescence can be attributed to the generation of radicals from dissociated TASN, which is induced by freezing of the swelling solvent. The freezing-induced mechanochromism and mechanoluminescence mechanisms were studied using ESR spectroscopy, which revealed that the affinity and freezing point of the swelling solvents are the critical factors affecting the mechanochromism and mechanoluminescence properties. The ultimate advantage of radical-based mechanochromic polymers involves not only qualitative visualization of the effect, but also its quantitative evaluation by ESR spectroscopy both in solution and solid state. The reports on freezing-induced mechanochromism and mechanoluminescence pave a new way for the design and preparation of radical-based polymers and materials. In 2022, Li's group chemically introduced a multi-stimuli-responsive tetraarylsuccinonitrile (TASN) chromophore into a liquid crystal elastomer (LCE) network through a facile thiol-ene photoaddition method.114 As shown in Fig. 13(c), the obtained TASN–LCE soft actuators not only exhibit reversible shape-morphing and reversible color-changing behavior in response to heat and mechanical compression, but also show excellent self-healing, reprogramming and recycling characteristics.
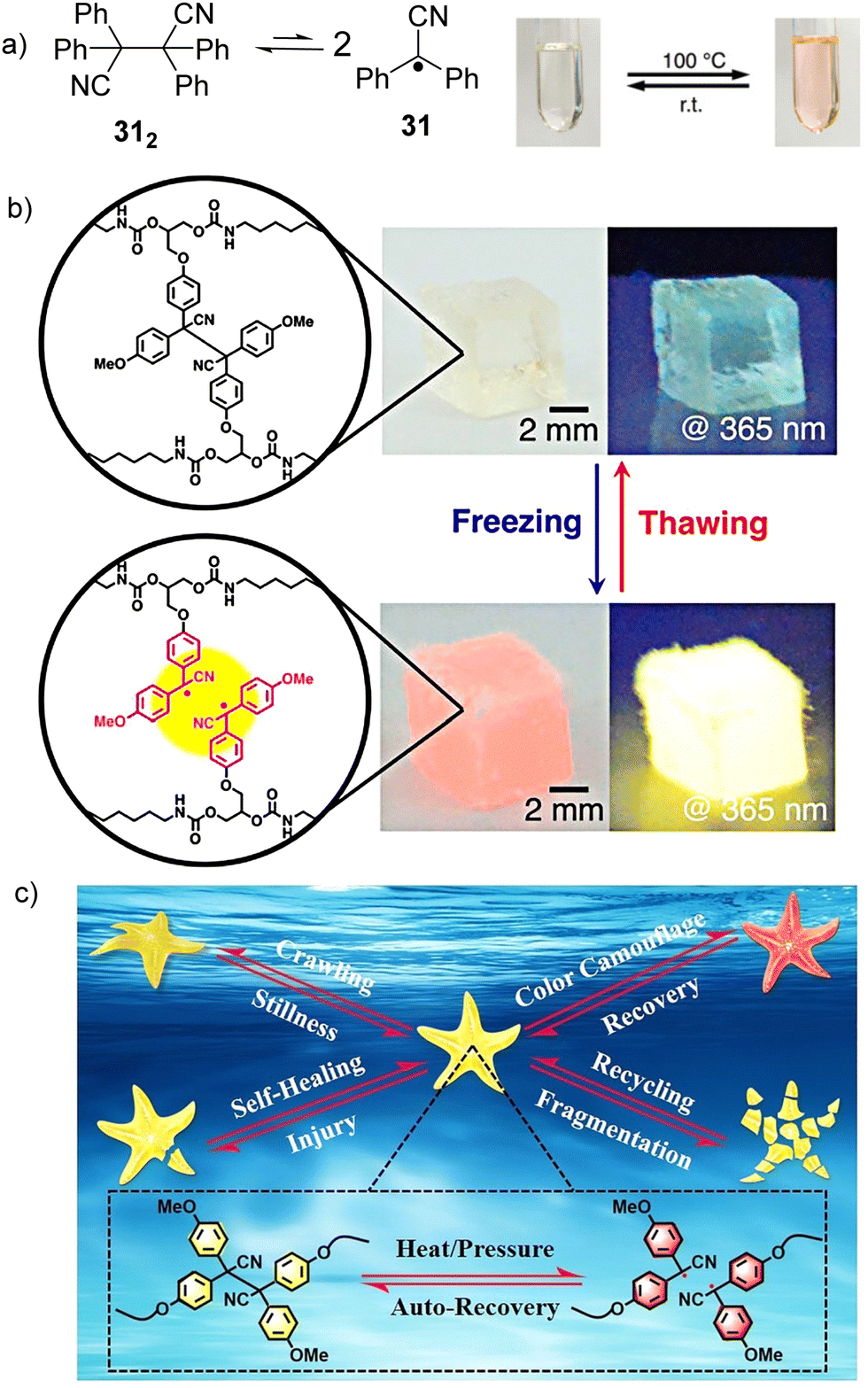 | ||
| Fig. 13 (a) Equilibrium between radical 31 and its dimer 312 and the images of the thermochromic behavior of radical 31 derivatives. Reprinted with permission from ref. 112. Copyright 2017 Royal Society of Chemistry. (b) freezing-introduced mechanochromism and mechanoluminescence of TASN gel swollen with 1,4-dioxane under ambient conditions and under UV irradiation. Reprinted with permission from ref. 113. Copyright 2018 American Chemical Society; (c) schematic illustration of the integration of shape morphing, color change, self-healing and recycling functions in a starfish-mimic soft actuator containing a tetraarylsuccinonitrile chromophore. Reprinted with permission from ref. 114. Copyright 2022 Wiley-VCH. | ||
Additionally, it has also been reported that a non-symmetric radical-based mechanophore can be prepared through molecular crossing between two different types of radical mechanophores.115 UV-vis absorption and ESR results have demonstrated that the newly formed daughter mechanophore exhibits properties intermediate to those of its parents. In this way, new radical-based mechanophores with tuneable properties, such as thermal stability, colors and mechanoresponsiveness, could be achieved through structural modifications on their parent radical-based mechanophores.116,117
4. Conclusions and outlook
Carbon-centered organic radicals as a promising motif for dynamic covalent chemistry have attracted increasing research attention during recent decades not only due to the simplicity of their synthesis, but also their remarkable color change in the dynamic process. Their applications as stimuli-responsive chromophores have been extensively explored. In this review, we scrutinized and summarized the recent progress of stable carbon-centered radicals showing reversible association/dissociation with a distinct change of color. The unique responsive chromic properties induced by external stimuli (light, mechanical stress, and temperature) have been receiving more recognition in radical-based dynamic covalent chemistry to serve as novel materials for applications in various fields, including but not limited to wide-range dyes, sensing, and spintronics. However, the types of carbon-centerd radicals based on dynamic covalent chemistry are still limited, and some of them are not stable enough within a wide range of practical conditions. Therefore, stable radicals with various new structures that are easy for further modification and functionalization are expected to largely expand their applications as stimuli-responsive materials. We hope this review outlines the current exploration progress of radical-based dynamic covalent chemistry and provides insights into the design and stabilization of new radicals for diverse structures and properties to broaden their applications as radical-based stimuli-responsive chromic materials.Author contributions
C. C. conceived the topic of this review and supervised the work. T. X. designed the structure of this review and wrote the manuscript. J. Z., Y. H. and C. C. contributed to the editing and reviewing of this work. All the authors participated in the discussion and revision of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. acknowledges financial support from Singapore MOE Tier 1 grant (A-8000992-00-00) and Tier 2 grant (MOE-MOET2EP10120-0006). T. X. thanks the scholarship support from the China Scholarship Council (CSC).Notes and references
- P. Kim, Y. Hu, J. Alvarenga, M. Kolle, Z. Suo and J. Aizenberg, Adv. Opt. Mater., 2013, 1, 381–388 CrossRef.
- S. Zeng, D. Zhang, W. Huang, Z. Wang, S. G. Freire, X. Yu, A. T. Smith, E. Y. Huang, H. Nguon and L. Sun, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- E. Lee, M. Zhang, Y. Cho, Y. Cui, J. Van der Spiegel, N. Engheta and S. Yang, Adv. Mater., 2014, 26, 4127–4133 CrossRef CAS PubMed.
- S. G. Lee, D. Y. Lee, H. S. Lim, D. H. Lee, S. Lee and K. Cho, Adv. Mater., 2010, 22, 5013–5017 CrossRef CAS PubMed.
- D. Ge, E. Lee, L. Yang, Y. Cho, M. Li, D. S. Gianola and S. Yang, Adv. Mater., 2015, 27, 2489–2495 CrossRef CAS PubMed.
- K. Bange and T. Gambke, Adv. Mater., 1990, 2, 10–16 CrossRef CAS.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918–2922 CrossRef CAS PubMed.
- J. Yoon, S. K. Chae and J.-M. Kim, J. Am. Chem. Soc., 2007, 129, 3038–3039 CrossRef CAS PubMed.
- C. M. Rodd and R. Agarwal, Nano Lett., 2011, 11, 3460–3467 CrossRef CAS PubMed.
- S. Zeng, R. Li, S. G. Freire, V. M. M. Garbellotto, E. Y. Huang, A. T. Smith, C. Hu, W. R. T. Tait, Z. Bian, G. Zheng, D. Zhang and L. Sun, Adv. Mater., 2017, 29, 1700828 CrossRef PubMed.
- P. Andersson, R. Forchheimer, P. Tehrani and M. Berggren, Adv. Funct. Mater., 2007, 17, 3074–3082 CrossRef CAS.
- P. Bamfield, Chromic phenomena: The Technological Applications of Colour Chemistry, ed. P. Bamfield and M. G. Hutchings, Royal Society of Chemistry, 2010, pp. 9–125 Search PubMed.
- M. Vik and A. P. Periyasamy, Chromic material: fundamentals, measurements, and applications, ed. M. Viková, CRC Press, 2018 Search PubMed.
- R. M. Christie, Chromic materials for technical textile applications, in Advances in the Dyeing and Finishing of Technical Textiles, 2013, pp. 3–36 Search PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- S. Lin, K. G. Gutierrez-Cuevas, X. Zhang, J. Guo and Q. Li, Adv. Funct. Mater., 2021, 31, 2007957 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantos and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed.
- J. W. Li, J. M. A. Carnall, M. C. A. Stuart and S. Otto, Angew. Chem., Int. Ed., 2011, 50, 8384–8386 CrossRef CAS PubMed.
- C. S. Hartley and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 11682–11683 CrossRef CAS PubMed.
- J. Zhu, S. Wu, X. Hou and J. Wu, Angew. Chem., Int. Ed., 2021, 60, 25323–25327 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 1785–1791 CrossRef CAS.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- X. Hou, K. Geng, J. Li, S. Wu and J. Wu, ACS Mater. Lett., 2022, 4, 1154–1159 CrossRef CAS.
- S. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- T. Zhang, G. Zhang and L. Chen, Acc. Chem. Res., 2022, 55, 795–808 CrossRef CAS PubMed.
- S. Huang, Y. Shen, H. K. Bisoyi, Y. Tao, Z. Liu, M. Wang, H. Yang and Q. Li, J. Am. Chem. Soc., 2021, 143, 12534–12551 Search PubMed.
- L. Chen, H. K. Bisoyi, Y. Huang, S. Huang, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2021, 60, 16394–16398 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- H. D. Hartzler, J. Org. Chem., 1966, 31, 2654–2658 CrossRef CAS.
- J. M. Wittman, R. Hayoun, W. Kaminsky, M. K. Coggins and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 12956–12959 CrossRef CAS PubMed.
- W. C. Danen and D. D. Newlirk, J. Am. Chem. Soc., 1976, 98, 516–520 CrossRef CAS.
- A. Nilsen and R. Braslau, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 697–717 CrossRef CAS.
- C. Harnack, W. Krull, M. Lehnig, W. P. Neumann and A. K. Zarkadis, J. Chem. Soc., Perkin Trans. 2, 1994, 1247–1252 RSC.
- F. M. Beringer, S. A. Galton and S. J. Huang, Tetrahedron, 1963, 19, 809–816 CrossRef CAS.
- B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1976, 98, 520–523 CrossRef CAS.
- Y. Mori, N. Yamada, M. Kanazawa, Y. Horikoshi, Y. Watanabe and K. Maeda, Bull. Chem. Soc. Jpn., 1996, 69, 2355–2359 CrossRef CAS.
- I. V. Khudyakov, A. I. Yasmenko and V. A. Kuzmin, Int. J. Chem. Kinet., 1979, 11, 621–633 CrossRef CAS.
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS PubMed.
- T. Kubo, Molecules, 2019, 24, 665 CrossRef PubMed.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368–7403 RSC.
- D. Sakamaki, S. Ghosh and S. Seki, Mater. Chem. Front., 2019, 3, 2270–2282 RSC.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC.
- H. Lankamp, W. Th Nauta and C. MacLean, Tetrahedron Lett., 1968, 9, 249–254 CrossRef.
- W. P. Neumann, A. Penenory, U. Stewen and M. Lehnig, J. Am. Chem. Soc., 1989, 111, 5845–5851 CrossRef CAS.
- O. Armet, J. Veciana, C. Riera, J. CastaÇer, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608 CrossRef CAS.
- G. Allinson, R. J. Bushby and J. L. Paillaud, J. Am. Chem. Soc., 1993, 115, 2062–2064 CrossRef CAS.
- G. Allinson, R. J. Bushby, J. L. Paillaud and M. Thornton-Pett, J. Chem. Soc., Perkin Trans. 1, 1995, 385–390 RSC.
- M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579–2582 CrossRef CAS PubMed.
- K. Rakus, S. P. Verevkin, J. Schätzer, H.-D. Beckhaus and C. Rüchardt, Ber. Dtsch. Chem. Ges., 1994, 127, 1095–1103 CrossRef CAS.
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. Lopez Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed.
- Y. Tian, K. Uchida, H. Kurata, Y. Hirao, T. Nishiuchi and T. Kubo, J. Am. Chem. Soc., 2014, 136, 12784–12793 CrossRef CAS PubMed.
- G. Wittig, E. Dreher, W. Reuther, H. Weidinger and R. Steinmetz, Justus Liebigs Ann. Chem., 1969, 726, 188–200 CrossRef.
- J. Ipaktschi, R. Hosseinzadeh, P. Schlaf, E. Dreiseidler and R. Goddard, Helv. Chim. Acta, 1998, 81, 1821–1834 CrossRef CAS.
- J. Ipaktschi, R. Hosseinzadeh and P. Schlaf, Angew. Chem., Int. Ed., 1999, 38, 1658–1660 CrossRef CAS PubMed.
- D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Angew. Chem., Int. Ed., 2016, 55, 894–898 CrossRef CAS PubMed.
- E. Font-Sanchis, C. Aliaga, K.-S. Focsaneanu and J. C. Scaiano, Chem. Commun., 2002, 1576–1577 RSC.
- H. Sakai, T. Sumi, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1359–1363 CrossRef CAS PubMed.
- H. Sakai, D. Aoki, K. Seshimo, K. Mayumi, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, ACS Macro Lett., 2020, 9, 1108–1113 CrossRef CAS PubMed.
- K. Seshimo, H. Sakai, T. Watabe, D. Aoki, H. Sugita, K. Mikami, Y. Mao, A. Ishigami, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Angew. Chem., Int. Ed., 2021, 60, 8406–8409 CrossRef PubMed.
- J. C. Scaiano, A. Martin, G. P. Yap and K. U. Ingold, Org. Lett., 2000, 2, 899–901 CrossRef CAS PubMed.
- E. V. Bejan, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2001, 3, 4059–4062 CrossRef CAS PubMed.
- A. Löwenbein and W. Folberth, Ber. Dtsch. Chem. Ges., 1925, 58, 601–609 CrossRef.
- T.-C. Liu and E. R. Wasserman, J. Am. Chem. Soc., 1953, 75, 2056–2058 CrossRef.
- K. Imato and H. Otsuka, Polymer, 2018, 137, 395–413 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- H. Suzuki, H. Koide and T. Ogawa, Bull. Chem. Soc. Jpn., 1988, 61, 501–504 CrossRef CAS.
- H. A. P. De Jongh, C. R. H. I. De Jonge and W. J. Mijs, J. Org. Chem., 1971, 36, 3160–3168 CrossRef CAS.
- H. A. P. De Jongh, C. R. H. I. De Jonge, H. J. M. Sinnige, W. J. De Klein, W. G. B. Huysmans and W. J. Mijs, J. Org. Chem., 1972, 37, 1960–1966 CrossRef CAS.
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634–8638 CrossRef CAS PubMed.
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS PubMed.
- J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed.
- J. L. Zafra, L. Qiu, N. Yanai, T. Mori, M. Nakano, M. PeÇa Alvarez, J. T. López Navarrete, C. J. Gómez-García, M. Kertesz, K. Takimiya and J. Casado, Angew. Chem., Int. Ed., 2016, 55, 14563–14568 CrossRef CAS PubMed.
- K. Okino, S. Hira, Y. Inoue, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2017, 56, 16597–16601 CrossRef CAS PubMed.
- J. P. Peterson and A. H. Winter, Org. Lett., 2020, 22, 6072–6076 CrossRef CAS PubMed.
- J. P. Peterson, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 5304–5313 CrossRef CAS PubMed.
- L. Yuan, Y. Han, T. Tao, H. Phan and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 9023–9027 CrossRef CAS PubMed.
- K. Oda, S. Hiroto and H. Shinokubo, J. Mater. Chem. C, 2017, 5, 5310–5315 RSC.
- D. Wang, C. C. Ferrón, J. Li, S. Gámez-Valenzuela, R. P. Ortiz, J. T. López Navarrete, V. H. Jolín, X. Yang, M. P. Ávarez, V. G. Baonza, F. Hartl, M. C. R. Delgado and H. Li, Chem. – Eur. J., 2017, 23, 13776–13783 CrossRef CAS PubMed.
- I. Badía-Domínguez, A. Pérez-Guardiola, J. C. Sancho-García, J. T. L. Navarrete, V. H. Jolín, H. Li, D. Sakamaki, S. Seki and M. C. R. Delgado, ACS Omega, 2019, 4, 4761–4769 CrossRef.
- I. Badía-Domínguez, M. P. Ávarez, D. Wang, A. Pérez-Guardiola, Y. Vida, S. R. González, J. T. López-Navarrete, V. H. Jolín, J. C. Sancho-García, V. García-Baonza, R. Nash, F. Hartl, H. Li and M. C. R. Delgado, Chem. – Eur. J., 2021, 27, 5509–5520 CrossRef PubMed.
- B. Adinarayana, D. Shimizu and A. Osuka, Chem. – Eur. J., 2019, 25, 1706–1710 CrossRef CAS.
- B. Adinarayana, D. Shimizu, K. Furukawa and A. Osuka, Chem. Sci., 2019, 10, 6007–6012 RSC.
- I. M. Blake, H. L. Anderson, D. Beljonne, J.-L. Brédas and W. Clegg, J. Am. Chem. Soc., 1998, 120, 10764–10765 CrossRef CAS.
- I. M. Blake, L. H. Rees, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 112, 1888–1891 CrossRef.
- B. Adinarayana, K. Kato, D. Shimizu, T. Tanaka, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2020, 132, 4350–4353 CrossRef.
- A. Löwenbein and B. Rosenbaum, Justus Liebigs Ann. Chem., 1926, 448, 223–248 CrossRef.
- J. Ohkanda, Y. Mori, K. Maeda and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1992, 59–63 RSC.
- R. W. Baldock, P. Hudson and A. R. Katritzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1422–1427 RSC.
- A. Margulis, I. V. Khudyakov, V. A. Kuzmin, A. I. Prokof’ev, A. I. Yasmenko and G. Smets, Int. J. Chem. Kinet., 1985, 17, 735–747 CrossRef.
- L. Kalb and J. Bayer, Ber. Dtsch. Chem. Ges., 1913, 46, 3879–3885 CrossRef.
- K. Kawasaki, D. Aoki and H. Otsuka, Macromol. Rapid Commun., 2020, 41, 1900460 CrossRef CAS PubMed.
- W. Qiu, J. M. P. Scofield, P. A. Gurr and G. G. Qiao, Macromol. Rapid Commun., 2022, 43, 2100866 CrossRef CAS PubMed.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- F. Verstraeten, R. Göstl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- D. Aoki and H. Otsuka, Mechanically Responsive Materials for Soft Robotics, Wiley-VCH, 2020, pp. 307–325 Search PubMed.
- T. Kosuge, K. Imato, R. Goseki and H. Otsuka, Macromolecules, 2016, 49, 5903–5911 CrossRef CAS.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS PubMed.
- K. Imato, T. Kanehara, S. Nojima, T. Ohishi, Y. Higaki, A. Takahara and H. Otsuka, Chem. Commun., 2016, 52, 10482–10485 RSC.
- K. Okino, D. Sakamaki and S. Seki, ACS Mater. Lett., 2019, 1, 25–29 CrossRef CAS.
- R. Zhang, J. P. Peterson, L. J. Fischer, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 14308–14313 CrossRef CAS PubMed.
- M. Liu, X. Yang, Q. Sun, T. Wang, R. Pei, X. Yang, Y. Zhao, L. Zhao, G. Frenking and X. Wang, Angew. Chem., Int. Ed., 2023, e202300068 Search PubMed.
- M. Frenette, P. D. MacLean, L. R. C. Barclay and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 16432–16433 CrossRef CAS PubMed.
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC.
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1087–1091 CrossRef CAS PubMed.
- Z. Liu, H. K. Bisoyi, Y. Huang, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202115755 CAS.
- K. Yanada, S. Kato, D. Aoki, K. Mikami, H. Sugita and H. Otsuka, Chem. Commun., 2021, 57, 2899–2902 RSC.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem. – Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed.
- K. Yanada, D. Aoki and H. Otsuka, Soft Matter, 2022, 18, 3218–3225 RSC.
| This journal is © The Royal Society of Chemistry 2023 |