
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-assisted electrochemical CO2 reduction at a boron-doped diamond cathode†
Goki
Iwai
 ,
Andrea
Fiorani
*,
Jinglun
Du
and
Yasuaki
Einaga
*
,
Andrea
Fiorani
*,
Jinglun
Du
and
Yasuaki
Einaga
*
Department of Chemistry, Keio University, Yokohama 223-8522, Japan. E-mail: andrea.fiorani@keio.jp; einaga@chem.keio.ac.jp
First published on 7th April 2023
Abstract
We report on the coupling of photoelectrochemical water oxidation and electrochemical CO2 reduction to formic acid for a photoelectrochemical system capable of light and electrochemical energy conversion to chemical energy. Titanium oxide nanotubes (TiO2 NT) and boron-doped diamond (BDD) electrodes were used as a photoanode and a cathode, respectively. From CO2 electrochemical reduction at BDD, formic acid was obtained with a faradaic efficiency of about 86% and the system provided an overall energy conversion efficiency of 5.5%. From previous CO2 reduction at a dark electrolyzer based on a BDD cathode, the cell voltage was reduced from 2.7 V to 1.4 V with 52% saving of the electrical energy input. This resulted in an increase from 50% to nearly 80% of electrical-to-chemical energy conversion efficiency.
Introduction
The utilization of carbon-based materials (e.g., lignite, carbon monoxide, etc.) for the production of hydrocarbons has long been established, namely in 1913 with the Bergius process,1 and in 1925 with the Fischer–Tropsch process.2The evolution of these processes is nowadays pursued by the utilization of CO2 as an alternative carbon feedstock,3,4 therefore transformation into high-added value chemicals can be a favourable pathway for this C1-synthon utilization in chemical/organic synthesis.5–9
If the synthesis is conducted using electrochemical methods, the products of CO2 reduction (e.g., CO, formic acid, methane, C2+ hydrocarbons, and oxygenates) might find application in the production of fine chemicals,10,11 and in addition as carriers or storage materials for renewable electrical energy. Furthermore, CO2 electrochemical reduction can be performed under ambient conditions, even at the industrial-scale.3,12,13 In this case, to be competitive, this reaction must retain most of the energy in the final chemical product i.e., high energy conversion efficiency, or low activation energy for CO2,14 and electrochemical (EC) and photoelectrochemical (PEC) approaches have been investigated for CO2 reduction.15 This may help produce energy with a lower environmental impact,16–18 as in the case of hydrogen production from water electrolysis.19–22
In a (photo)electrochemical system, energy efficiency is affected by several energy losses, mainly ascribed to the electrochemical cell (e.g., ohmic drop, electrolyte, mass transport, and separators),23,24 which can be reduced by a careful engineering design, and the overpotential that is intrinsic for the reaction and electrocatalyst/electrode materials used. In particular, when a reduction reaction is coupled with the oxygen evolution reaction (OER) from water oxidation, this 4-electron reaction may account for the largest loss of energy in the electrochemical system due to overpotential.25
This problem has been tackled by the use of several metal oxides as highly active electrocatalysts for the OER.26,27
In this context, we demonstrated previously that in a CO2 electrolyser the right choice of the anode material improved the electric-to-chemical energy conversion efficiency up to 50%.28
To further decrease the reliance on electrical energy, we present here a photoelectrochemical system comprising titanium oxide nanotubes (TiO2 NTs) and boron-doped diamond (BDD) electrodes as a photoanode and a cathode, respectively. A BDD electrode has been selected for its high faradaic efficiency for formic acid (FA) production from CO2 reduction, and in fact it can prevent the side reaction of proton reduction which competes with CO2 reduction by its wide potential window and high overpotential for hydrogen evolution,28–30 and long term stability.31,32 TiO2 NTs were selected as a photoanode because they can drive water oxidation at negative potentials under light illumination compared to conventional metal oxide electrodes,33 and in addition, nanotubes can expose a larger surface area than flat electrodes.34,35
The objective was to develop a PEC system with the ability of photo-assisted water oxidation at the TiO2 NT electrode coupled with electrochemical CO2 reduction to formic acid, and evaluate its energy efficiency as the main figure of merit. Finally, the energy efficiency of the present PEC system was compared with that of a full EC system that we developed previously28,29 to evaluate the benefits of using photo-assisted electrochemical water oxidation and the viability of BDD as a cathode for CO2 electrochemical reduction.
Experimental
Materials
Potassium chloride (KCl), potassium hydroxide (KOH), sulphuric acid (H2SO4), acetone (C3H6O), and isopropanol (C3H8O) were purchased from Wako Pure Chemical Industries Ltd. Ammonium dihydrogen phosphate [(NH4)H2PO4] and ammonium fluoride–hydrofluoric acid (NH4F·HF) were purchased from Sigma-Aldrich. Formic acid for HPLC calibration was obtained from FUJIFILM Wako Pure Chemical Corporation, while hydrogen and carbon monoxide for GC calibration were obtained from GL Sciences. Carbon dioxide (purity over 99.990 vol%) was purchased from Resonac Gas Products Corporation, and nitrogen gas (purity over 99.99995 vol%) was purchased from Taiyo Nippon Sanso Corporation. All reagents were used as received. Titanium (thickness 0.2 mm) and platinum (thickness 1 mm, Φ 50 mm) plates were purchased from Nilaco Corp. (Japan). Ultrapure water was from a Simply-Lab water system (DIRECT-Q 3 UV, Millipore) with a resistivity of 18.2 MΩ·cm at 25 °C. Experiments were performed at room temperature (23–25 °C) and atmospheric pressure.BDD fabrication and characterization: cathode
The polycrystalline BDD film was deposited on a Si (100) wafer substrate with a microwave plasma-assisted chemical vapor deposition (MPCVD) system (AX6500, Cornes Technologies Ltd.). The concentration of boron in the BDD film was set to be 0.1% according to the ratio between the carbon source (methane) and the boron source (trimethylborane).28 The surface morphology of the BDD film was imaged with a scanning electron microscope (JCM-6000, JEOL) (Fig. S1, ESI†). The Raman spectrum was recorded using an Acton SP2500 (Princeton Instruments) with a 532 nm laser to confirm the diamond nature, boron inclusion and exclude the presence of sp2 carbon (Fig. S1, ESI†).28,29TiO2 NT fabrication and characterization: photoanode
Titanium oxide nanotubes (TiO2 NTs) were prepared by titanium foil oxidation in a fluorine electrolyte, a procedure adapted from previous reports.35,36 A titanium plate was sonicated in acetone, isopropanol and water 10 minutes each, and dried under a nitrogen stream. The electrolyte solution consisted of 1 M (NH4)H2PO4 and 0.5 wt% NH4F·HF in ultrapure water. Using a DC power supply, a constant voltage of 30 V was applied between the Ti plate and Pt plate for 3 hours, followed by rinsing with ultrapure water. The TiO2 NT plate was annealed at 450 °C for 1 hour (Program electric Furnace SMF-1, AS ONE Co.). The crystalline phase was investigated using an X-ray diffractometer (D8 ADVANCE, Bruker) with Cu-Kα radiation, and the surface was imaged with a field emission scanning electron microscope (FE-SEM-H2 SU70, Hitachi High-Tech Co.). The band gap was measured from the reflectance spectra obtained using a UV-3600 Plus UV-Vis-NIR spectrophotometer (Shimadzu). Details of the optimisation procedure to obtain TiO2 NTs and characterization are available in the supporting information (Fig. S2, ESI†).Electrochemistry
Electrochemical measurements were performed with a potentiostat/galvanostat system PGSTAT204 (Metrohm Autolab). Ag/AgCl, KCl sat’d was the reference electrode in all electrochemical measurements, and all potentials are referred to this electrode, except otherwise stated. Before each experiment, the BDD electrode was sonicated in isopropanol and ultrapure water for 10 min each. We proved that electrochemical pretreatment ensured surface cleanliness and the optimal electrochemical response of the BDD,28,29 therefore several cyclic voltammetry (CV) scans (10 cycles from −3.5 V to 3.5 V, and 20 cycles from 0 V to 3.5 V in 0.1 M H2SO4 with a scan rate of 0.5 V s−1) were performed before CO2 reduction. Electrochemical measurements were conducted in a two-chamber polytetrafluoroethylene (PTFE) flow cell, both equipped with a reference electrode and separated by a Nafion NRE-212 (Sigma-Aldrich). BDD (0.1% B/C) and TiO2 NTs were set as working and counter electrodes, with geometrical areas of 9.62 cm−2 and 12.56 cm−2, respectively (Fig. S3, ESI†). This photoelectrochemical cell can be operated in a three-electrode configuration (a BDD working electrode, an Ag/AgCl, KCl sat’d reference electrode, and a TiO2 NT counter electrode), and in a two-electrode configuration (a BDD cathode working electrode, and a TiO2 NT photoanode counter electrode). The catholyte and anolyte were 0.5 M KCl (50 mL) and 0.5 M KOH (90 mL), respectively. Oxygen was removed from the catholyte by N2 purging for 30 min and then CO2 was bubbled for 60 min at 200 mL min−1, resulting in a CO2-sauturated solution. During electrolysis, CO2 flow was set to be 30 mL min−1. Each experiment has been repeated three times to confirm the results.Product analysis
Quantification of the CO2 reduction products was performed at the end of each experiment. Formic acid was quantified by high-performance liquid chromatography (HPLC, CDD-10A, Shimadzu Corp.). The gaseous products (CO and H2) were collected in an aluminium gas bag (GL Sciences) and quantified by gas chromatography (GC-2014, Shimadzu Corp.). Calibration lines, LOD and Min–Max concentrations detected are available in Fig. S4 (ESI†).Light source
Light irradiation was carried out using a SP11 Spot-cure (USHIO Inc., Japan). The lamp emission spectrum is available in Fig. S5 (ESI†). Light intensity was measured using a power meter (Solar power meter TM-207, MK Scientific, Inc.).Energy conversion efficiency
To take into account the electric and light power injected into this photoelectrochemical system which provides a net conversion of sunlight in the form of a reduced electric bias, the energy throughput conversion efficiency for photo-assisted electrolysis (ηPAE) can be calculated using eqn (1):37,38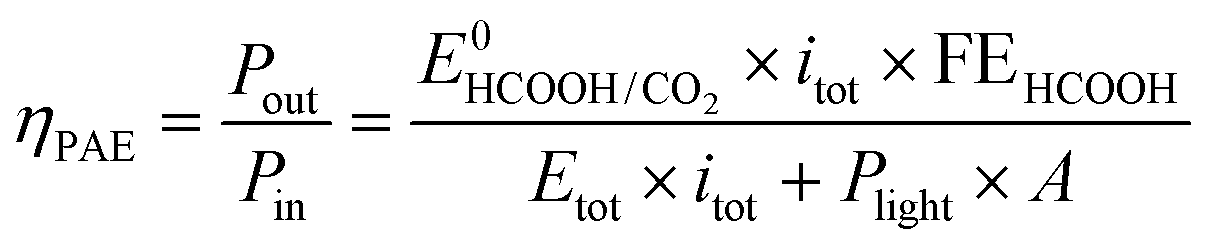 | (1) |
The electrical-to-chemical energy (ECE) conversion efficiency (ηECE) can be calculated using eqn (2).29
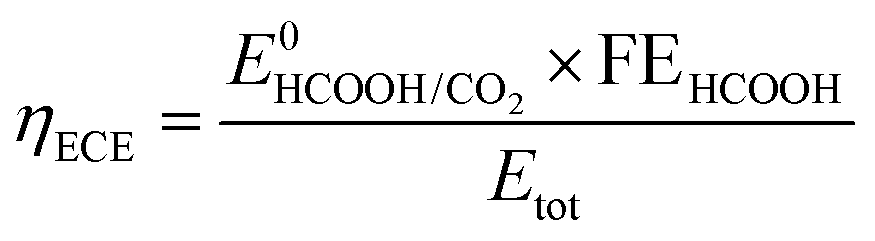 | (2) |
Results and discussion
The photoelectrochemical system capable of reducing CO2 to formic acid developed here consists of a BDD cathode and a TiO2 NT photoanode. We demonstrated the viability of electrochemical reduction of CO2 on BDD achieving an optimised and stable process.28,29 To further decrease the dependence on electrical energy for the whole electrochemical process of water oxidation (anode) and CO2 reduction (cathode), we substituted the metal oxide anode with a TiO2 NT photoanode. The TiO2 NT electrode was fabricated by titanium oxidation and dissolution in a fluorine electrolyte, with optimization of parameters (Fig. S2, ESI†), such as electrolytic solution and oxidation time. The structure resulted in nanotubes of 200 nm in diameter (Fig. 1A) and the crystalline phase has been confirmed as anatase using XRD (Fig. 1B, and SI 2.1, ESI†). The average band gap, measured using the diffuse reflectance spectra according to the Kubelka–Munk theory,39 was 3.4 ± 0.1 eV (Fig. 1C and Fig. S2, ESI†), which is in agreement with the typical band gap energy of TiO2 (anatase).40 We should mention that band gaps down to 2 eV can be effectively obtained for TiO2, and this has implication on the energy adsorption (e.g., from the solar radiation). This requires further processing of TiO2 by doping with non-metals as carbon at high temperature,34,35,41 or by metal doping as in SrTiO3:Al.42 However, this is beyond the scope of our present investigation. Finally, under our experimental conditions, the addition of HF compared to NH4F only gave better results in terms of water oxidation current (Fig. 1D). | ||
| Fig. 1 Characterisation of TiO2 NTs. (A) SEM micrograph, scale bar is 1 μm. (B) XRD spectrum. (C) Kubelka–Munk plot. (D) Photoelectrochemical response measured by cyclic voltammetry at 100 mV s−1 in 0.5 M KOH with light applied of 6.5 mW cm−2, for TiO2 NTs fabricated with addition of NH4F (blue) or NH4F·HF (red and black). Reference electrode: Ag/AgCl, KCl sat’d. | ||
The flat band potential (EFB) of TiO2 NTs was evaluated by open circuit potential (OCP) measurements upon increasing the light intensity (Fig. S6, ESI†) because the OCP shifts towards the EFB theoretically reaching it at sufficiently high illumination intensities.43–45 The EFB set at 0.1 V (vs. normal hydrogen electrode, NHE) at pH 0 with a Nernstian dependence as a function of pH (54 mV pH−1, Fig. 2A) indicates that H+ and OH− are potential determining ions (PDIs) adsorbed on the metal oxide surface within the Helmholtz layer.46,47 The EFB value is also in agreement with the potential for the onset of the photocurrent (Fig. 1D and 2B).45,48 From this characterization, we assessed the fine quality of the TiO2 NTs.
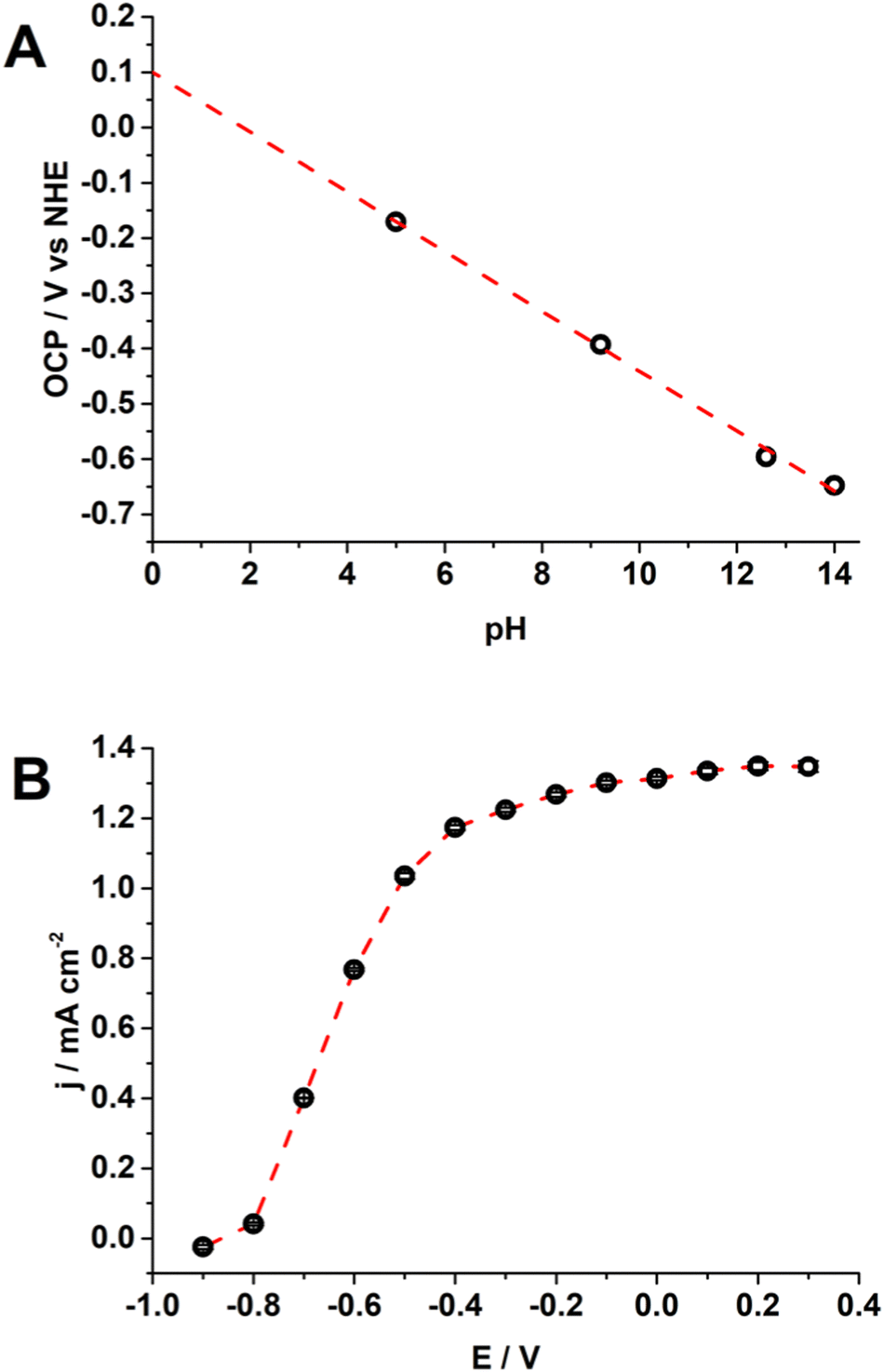 | ||
| Fig. 2 (A) OCP under illumination in 0.1 M phosphate buffer, while pH 14 is 0.5 M KOH; reference electrode: NHE = Ag/AgCl, KCl sat’d + 0.197 V; standard error: 0.01 V. (B) Limiting current from chronoamperometry at selected potentials in KOH 0.5 M (light power: 25.2 mW cm−2); reference electrode: Ag/AgCl, KCl (sat’d); the error bar shows the standard deviation (n = 3). | ||
The electrochemical cell assembly for CO2 reduction is depicted in Fig. 3 and Fig. S3 (ESI†). We used two configurations: (1) the potential at the BDD cathode was fixed compared to the Ag/AgCl, KCl (sat’d) reference electrode (i.e., the potential at the TiO2 NT electrode was floating, that is a three-electrode configuration), namely Ecathode; (2) the total voltage between the BDD cathode and the TiO2 NT photoanode was fixed (i.e., both TiO2 NT and BDD potentials were floating, that is a two-electrode configuration), namely Etot.
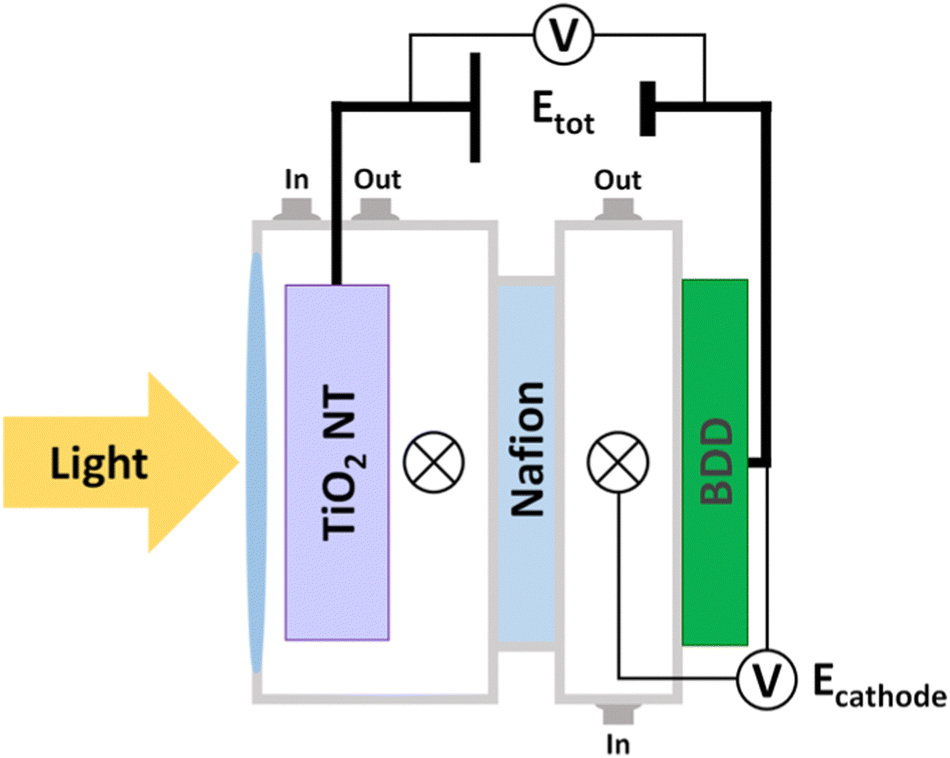 | ||
| Fig. 3 Schematic diagram of the photoelectrochemical cell. The symbol ⊗ represents the reference electrode: Ag/AgCl, KCl (sat’d). Ecathode means that the potential at the BDD cathode is fixed with respect to the reference electrode. Etot means that the potential is fixed between the BDD and TiO2 NT electrodes. The reference electrode in the TiO2 NT electrode compartment is used to monitor the potential at TiO2 NTs. Black lines are cable connections. In and Out are connecting tubes for electrolyte circulation. A quartz window permits the light to reach the TiO2 NT photoanode. | ||
Configuration 1, that is a three-electrode configuration, of Ecathode at −2.15 V vs. Ag/AgCl, KCl (sat’d) is meant to demonstrate that the faradaic efficiency for formic acid production can reach an adequate level (nearly 90%), as the potential at BDD (Ecathode) is in the optimal range, as we observed previously,28,29 therefore the effectiveness of the photoelectrochemical cell can be assessed and the stability of the TiO2 NT photoanode can be tested. ηPAE was measured at different light powers (from 6.5 to 35 mW cm−2) and was compared with theoretical efficiency computed for 90% faradaic efficiency, combined with Etot and current as in the range of values measured experimentally, from 1.4 V to 3 V and from 0.3 mA cm−2 to 0.7 mA cm−2, respectively (Fig. S7 and S8, ESI†).
The potential at TiO2 NTs is stabilised to around −0.55 V to −0.75 V vs. Ag/AgCl, KCl sat’d (Fig. S7B, ESI†), enough to provide the current measured at the BDD cathode (Fig. S7D and Fig. 2B, ESI†), and is therefore limited only by the kinetics of CO2 reduction at BDD. The faradaic efficiency for formic acid production could reach an adequate value of 90% (range 79–94%, Table S1, ESI†) which confirmed the appropriate operation of the electrochemical cell in the cathodic reaction. A particular case is the irradiation with 6.5 mW cm−2 which is not sufficient to sustain water oxidation at a stable potential that gradually shifted positive, from −0.6 V to 2.6 V vs. Ag/AgCl, KCl sat’d (Fig. S7, ESI†).
When the photoelectrochemical cell is operated in configuration 2, that is a two-electrode configuration, we performed the CO2 reduction at different Etot, from 1 V to 2.4 V, and a light input of 25.2 mW cm−2 was selected from the highest efficiency obtained from configuration 1 (Fig. S8, ESI†). Moreover, this light power could maintain a stable potential at TiO2 NTs during the CO2 reduction (Fig. S7, ESI†).
The voltage range applied is 0.3 to 1.7 V lower than that of a conventional electrochemical cell without photo-assisted electrolysis (i.e., Etot = 2.7 V at the BDD cathode and Ir oxide anode).28 Concerning this fundamental aspect, we have to recall that the cell does not convert directly light energy into chemical energy (i.e., the photocurrent is zero without an external bias, Fig. S9, ESI†), however the overall effect is to lower Etot and decrease the electrical power input.
In the range of Etot investigated the current increased up to 1.7 mA cm−2, and the faradaic efficiency for formic acid (the main product, together with a small amount of carbon monoxide and hydrogen, Table S2 and Fig. S10, S11 and S12) reached an average and stable value of 86% from 1.4 V onward (Fig. 4A).
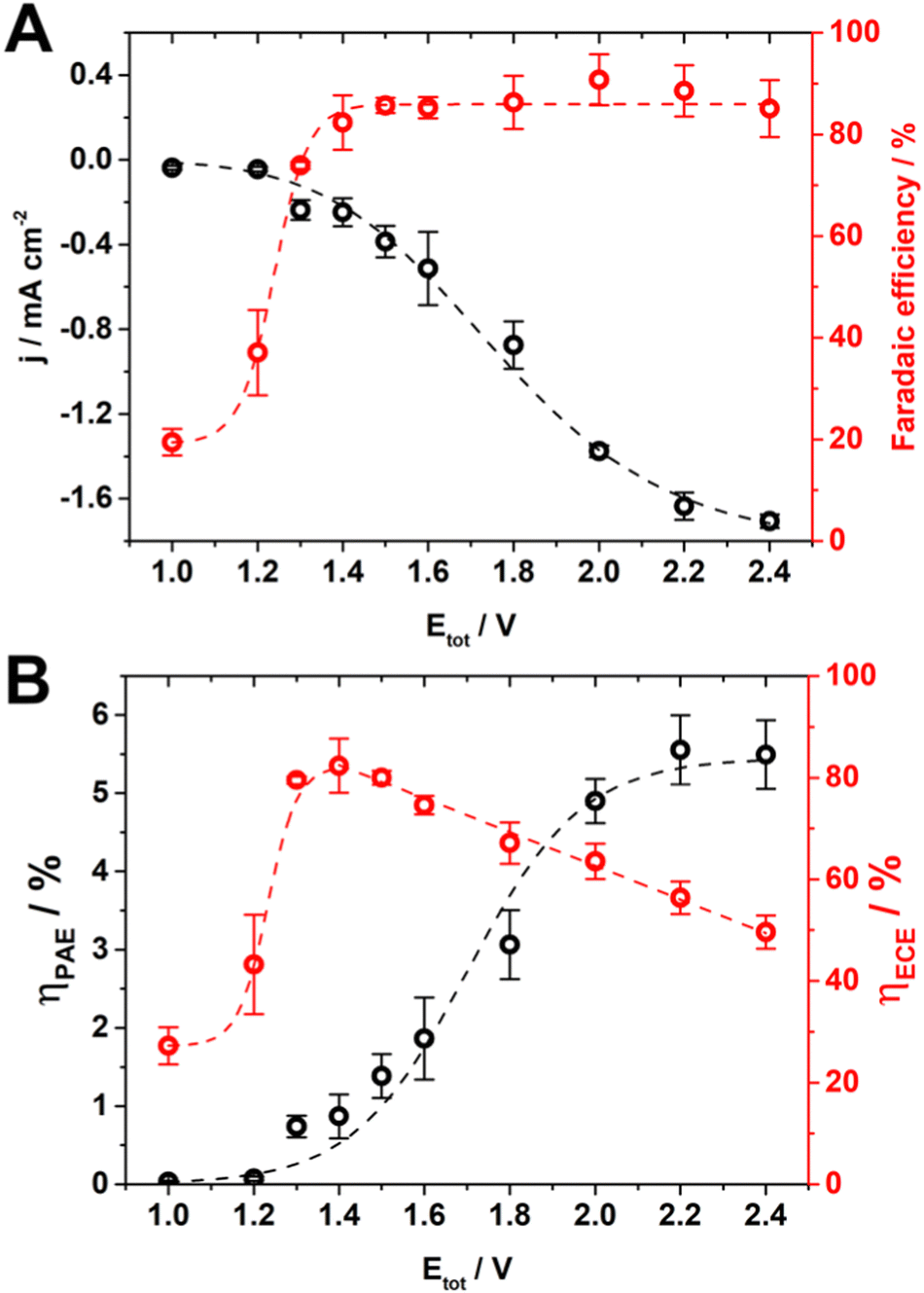 | ||
| Fig. 4 (A) Average current measured at the BDD cathode (black) and faradaic efficiency for the production of formic acid (red). (B) Energy throughput conversion efficiency for photo-assisted electrolysis (ηPAE, black), and electric-to-chemical energy conversion efficiency (ηECE, red) as a function of Etot in configuration 2. Light intensity: 25.2 mW cm−2. Lines are only a guide for the eye. The error bar shows the standard deviation (n = 3). | ||
At Etot of 1.4 V, the potential measured at BDD was −2.04 V which is near the limit for CO2 reduction to formic acid. In fact, for a lower potential at BDD (i.e., < 2.0 V vs. Ag/AgCl, KCl sat’d, Fig. S10, ESI†), the kinetics for the formation of a CO2 radical anion becomes slow, and this is the rate determining step on an inert electrode such as BDD where the CO2 radical anion is loosely absorbed30,32 with a consequent increase of proton reduction to hydrogen. On the other hand, Etot higher than 1.4 V increased only the reduction rate of CO2 (i.e., the current increased along with the potential at BDD), while did not enhance the faradaic efficiency further.
It is possible to translate this combined effect of current and faradaic efficiency directly on the energy throughput conversion efficiency (Fig. 4B). ηPAE is limited by both faradaic efficiency and current below 1.4 V, while it is mainly limited by the current above 1.4 V. Therefore, the flattening of ηPAE at 5.5% was ascribed to the limited current available in this system and set by the TiO2 NT electrode (Fig. 2 and Fig. S10, ESI†).
While ηPAE is the optimal descriptor for a PEC system, combining both light and electrical energies in the conversion to chemical energy, we would present all figures of merit for the system under investigation.
The electrical-to-chemical conversion efficiency accounts only for the electrical energy input, however it can be reported also for PEC systems (Table S3, ESI†).49–51
This permits a direct comparison with our previous electrochemical CO2 reduction at BDD, furthermore evaluating explicitly the effect of the TiO2 NTs on water oxidation.
η ECE reaches a stable 80% in the range 1.3–1.5 V (Fig. 4B) compared to 50% reached previously without photo-assisted electrolysis at 2.7 V,28 which saved 52% of electrical energy. This energy saving is evident from the potential at TiO2 NTs for water oxidation in the range of −0.75 to −0.67 V (Fig. S10, ESI†) compared to 0.5 V observed experimentally28 (theoretical 0.21 V, see ESI† Section 15).
Conclusions
We developed an electrolyser to convert carbon dioxide to formic acid with faradaic efficiency reaching 86%. The system comprised a photoanode made of titanium oxide (anatase) nanotubes for photoelectrochemical water oxidation and a boron-doped diamond cathode for CO2 electrochemical reduction to formic acid. This photoelectrochemical cell could convert light power, in the form of a reduced electric bias, to chemical with an efficiency of 5.5%, reaching 80% of electrical-to-chemical conversion efficiency. A further improvement in this photoelectrochemical system will be the coupling of a photovoltaic module with an electrolyser consisting of an oxide anode and a BDD cathode to evaluate the solar to energy conversion efficiency and the viability of BDD as a profitable cathode for CO2 electrochemical reduction. This approach is currently under investigation in our laboratory.Author contributions
Goki Iwai: methodology, investigation, formal analysis, visualization, and writing – original draft. Andrea Fiorani: conceptualization, supervision, methodology, investigation, formal analysis, validation, and writing – original draft. Jinglun Du: methodology and validation. Yasuaki Einaga: conceptualization, supervision, and funding acquisition. All authors: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the New Energy and Industrial Technology Development Organization (NEDO) P16002 (to Y.E.).References
-
Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons Ltd., Hoboken, USA, 2009, 397–340 Search PubMed
.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed
- C. Chen, M. Garedew and S. W. Sheehan, ACS Energy Lett., 2022, 7, 988–992 CrossRef CAS
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
- C. M. Rayner, Org. Process Res. Dev., 2007, 11, 121–132 CrossRef CAS
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef PubMed
- L. Lombardi, A. Cerveri, L. Ceccon, R. Pedrazzani, M. Monari, G. Bertuzzi and M. Bandini, Chem. Commun., 2022, 58, 4071–4074 RSC
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC
- J. Seidler, A. Roth, L. Vieira and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2023, 11, 390–398 CrossRef CAS
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef PubMed
- Y. Cheng, P. Hou, X. Wang and P. Kang, Acc. Chem. Res., 2022, 55, 231–240 CrossRef CAS PubMed
- A. Löwe, M. Schmidt, F. Bienen, D. Kopljar, N. Wagner and E. Klemm, ACS Sustainable Chem. Eng., 2021, 9, 4213–4223 CrossRef
- S. Matsuda, M. Tanaka and M. Umeda, Anal. Methods, 2022, 14, 3280–3288 RSC
- J. He and C. Janaky, ACS Energy Lett., 2020, 5, 1996–2014 CrossRef CAS PubMed
- C. A. Martinez-Huitle, M. A. Rodrigo, I. Sires and O. Scialdone, Appl. Catal., B, 2023, 328, 122430 CrossRef CAS
- S. O. Ganiyu and C. A. Martinez-Huitle, Curr. Opin. Electrochem., 2020, 22, 211–220 CrossRef CAS
- S. O. Ganiyu, C. A. Martinez-Huitle and M. A. Rodrigo, Appl. Catal., B, 2020, 270, 118857 CrossRef CAS
- J. C. Ehlers, A. A. Feidenhans’l, K. T. Therkildsen and G. O. Larrazábal, ACS Energy Lett., 2023, 8, 1502–1509 CrossRef CAS
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
- J. C. Cardozo, D. R. da Silva, C. A. Martínez-Huitle, M. A. Quiroz and E. V. Dos Santos, Materials, 2022, 15, 7445 CrossRef PubMed
- J. E. L. Santos, D. R. da Silva, C. A. Martínez-Huitle, E. V. dos Santosa and M. A. Quiroz, RSC Adv., 2020, 10, 37947–37955 RSC
- Y. Kim, E. W. Lees and C. P. Berlinguette, ACS Energy
Lett., 2022, 7, 2382–2387 CrossRef CAS
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS
- M. D. Merrill and Ralph C. Dougherty, J. Phys. Chem. C, 2008, 112, 3655–3666 CrossRef CAS
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- J. Du, A. Fiorani, T. Inagaki, A. Otake, M. Murata, M. Hatanaka and Y. Einaga, JACS Au, 2022, 2, 1375–1382 CrossRef CAS PubMed
- J. Du, A. Fiorani and Y. Einaga, Sustainable Energy Fuels, 2021, 5, 2590–2594 RSC
- Y. Einaga, Electrochemistry, 2022, 90, 101002 CrossRef CAS
- N. Ikemiya, K. Natsui, K. Nakata and Y. Einaga, ACS Sustainable Chem. Eng., 2018, 6, 8108–8112 CrossRef CAS
- K. Natsui, H. Iwakawa, N. Ikemiya, K. Nakata and Y. Einaga, Angew. Chem., Int. Ed., 2018, 57, 2639–2643 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler Jr., Science, 2002, 297, 2243–2245 CrossRef CAS PubMed
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS PubMed
- A. Ghicov, H. Tsuchiya, J. M. Macak and P. Schmuki, Electrochem. Commun., 2005, 7, 505–509 CrossRef CAS
- B. Parkinson, Acc. Chem. Res., 1984, 17, 431–437 CrossRef CAS
- R. H. Coridan, A. C. Nielander, S. A. Francis, M. T. McDowell, V. Dix, S. M. Chatman and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2886–2901 RSC
- L. Salmon, S. Iran Rocha, F. Elisabete, V. Mikhail, C. Joaquim and T. Carlos José, Solid State Commun., 2022, 341, 114573 CrossRef
- T. D. Burleigh, Corrosion, 1989, 45, 464–472 CrossRef CAS
- E. Barborini, A. M. Conti, I. Kholmanov, P. Piseri, A. Podestà, P. Milani, C. Cepek, O. Sakho, R. Macovez and M. Sancrotti, Adv. Mater., 2005, 17, 1842–1846 CrossRef CAS
- Z. Zhao, R. V. Goncalves, S. K. Barman, E. J. Willard, E. Byle, R. Perry, Z. Wu, M. N. Huda, A. J. Moulé and F. E. Osterloh, Energy Environ. Sci., 2019, 12, 1385–1395 RSC
- Y. V. Pleskov, V. M. Mazin, Y. E. Evstefeeva, V. P. Varnin, I. G. Teremetskaya and V. A. Laptev, Electrochem. Solid-State Lett., 2000, 3, 141–143 CrossRef CAS
- R. Beranek, Adv. Phys. Chem., 2011, 786759 Search PubMed
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 RSC
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS
- M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS
- J. M. Bolts and M. S. Wrighton, J. Phys. Chem., 1976, 80, 2641–2645 CrossRef CAS
- Gurudayal, J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS
- S. Y. Chae, S. Y. Lee, S. G. Han, H. Kim, J. Ko, S. Park, O.-S. Joo, D. Kim, Y. Kang, U. Lee, Y. J. Hwang and B. K. Min, Sustainable Energy Fuels, 2020, 4, 199–212 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental setup, material characterisation, and electrochemical data. See DOI: https://doi.org/10.1039/d3ya00054k |
| This journal is © The Royal Society of Chemistry 2023 |