
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotothermal catalytic C1 conversion on supported catalysts
Hui
Liu
a,
Liangliang
Han
 a,
Xiaoguang
Duan
b,
Hongqi
Sun
c,
Shaobin
Wang
b and
Jinqiang
Zhang
*b
a,
Xiaoguang
Duan
b,
Hongqi
Sun
c,
Shaobin
Wang
b and
Jinqiang
Zhang
*b
aCollege of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, China
bSchool of Chemical Engineering, The University of Adelaide, North Terrace, Adelaide, SA 5005, Australia. E-mail: jinqiang.zhang@adelaide.edu.au
cSchool of Molecular Sciences, The University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia
First published on 6th September 2023
Abstract
Photothermal catalysis has emerged as a robust approach for converting C1 feedstocks into valuable chemicals, providing a clean and sustainable alternative that reduces the carbon footprint and enables energy-efficient green synthesis. Supported metal materials are promising candidates for scaling up photothermal catalysis, as their photothermal properties can be precisely tailored to optimize energy conversion efficiency and product distribution for specific processes. In this review, we provide an in-depth examination of the application of supported catalysts in photothermal catalytic C1 chemistry, which begins with an introduction of the mechanism of photothermal catalysis and elucidation of the fundamental principles behind this process. An overview of the design strategies for supported catalysts follows, encompassing various techniques for tailoring their properties to meet specific catalytic requirements. Advancements made in utilizing supported catalysts for photothermal catalytic reactions in C1 chemistry are then highlighted. Finally, challenges in the fledgling field are summarized and valuable insights into the future development directions and focuses are provided. This comprehensive review is anticipated to offer a deeper understanding of the design, performance, and potential applications of supported catalysts in photothermal catalysis for C1 chemistry, transforming conventional chemical processes into ‘green’.
 Hui Liu | Dr Hui Liu received her PhD in chemical engineering and technology from China University of Petroleum (East China). She is now a lecturer at the College of Chemistry & Chemical Engineering, Yantai University, China. Her research interests primarily focus on the synthesis and application of nanomaterials for catalysis, encompassing electrocatalysis and photoelectrochemical catalysis for energy conversion and utilization. |
 Jinqiang Zhang | Dr Jinqiang Zhang received his PhD from Edith Cowan University, Australia. He is now a research fellow at the School of Chemical Engineering, The University of Adelaide, Australia. His research interests focus on photo-mediated catalysis, including photocatalysis, photoelectrochemical catalysis and photothermal catalysis, for energy preparation and fossil fuel upgrading. |
1. Introduction
The current chemical industry faces a critical challenge as it heavily relies on fossil fuels to produce commodity chemicals and energy sources. However, this dependence on non-renewable resources is not sustainable due to limited reserves and the release of harmful greenhouse gases into the atmosphere. To address these issues, the industry must find alternative ways to manufacture chemicals without solely relying on fossil fuels. C1 chemistry offers a revolutionary approach by converting simple C1 molecules such as CO, CO2, CH4, and CH3OH into advanced fuels and chemicals.1,2 This process can also produce high-quality hydrogen and premium specialty chemicals from synthesis gas or methanol. By harnessing the power of C1 chemistry, a sustainable supply model can be created, reducing dependence on traditional fossil fuels. Currently, the industry typically requires harsh reaction conditions, including high temperature and pressure, to achieve significant conversion of C1 molecules. For example, methane, the most stable alkane molecule,3,4 necessitates high temperatures (600–1100 °C) for effective activation and conversion.5 However, these conditions result in high energy consumption, environmental issues, and catalyst deactivation. Therefore, it is crucial to seek mild reaction conditions to reduce energy input, lower temperature requirements, and achieve robust C1 transformation at atmospheric pressure.In recent years, photothermal catalysis has emerged as a promising approach that utilizes solar energy to replace conventional heating means to trigger catalytic reactions.6,7 This innovative method offers several advantages over traditional thermal catalysis, notably the ability to drive reactions at moderate temperature and pressure using solar energy.8 This reduces the need for costly equipment and infrastructure while promoting sustainability by minimizing reliance on non-renewable resources and reducing harmful emissions. Despite significant advances in solar technology, there is still room for improvement in enhancing the efficiency of solar energy conversion. To address this challenge, researchers are actively exploring new materials and innovative designs to enhance optical properties and minimize heat losses during catalysis.
Supported metal catalysts with dispersed nanoparticles have garnered significant interest, as their photothermal properties and catalytic capabilities can be precisely tuned by tailoring the geometrical configuration of the metal and support, and their interactions.9 For example, reducing the size of metal nanoparticles can enhance solar-to-thermal conversion efficiency by increasing the number of accessible active sites. However, small metal species often suffer from poor stability and recyclability due to severe aggregation during catalysis, particularly under harsh conditions.10 Loading catalysts onto appropriate supports has emerged as an attractive strategy to reduce costs and maximize atom utilization. However, the understanding of the structure–activity relationship and reaction mechanisms in composite systems is still in its early stages.11 Supported catalysts are more intricate than their single-phase counterparts as they involve catalytically active phases and supports that work in tandem. Understanding their electronic/chemical properties, strong metal–support interactions (SMSIs), and working principles is essential to comprehend these intricate catalysts and their potential applications.
In this comprehensive review, we delve into the fascinating world of photothermal catalysis and explore its intricate mechanisms. Our aim is to provide a detailed understanding of the photochemical and photothermal effects that underpin this cutting-edge technology, as well as their synergistic coupling. Then, we explore the two important components (active and support components) in supported metal catalysts and conduct an in-depth exploration of their design strategies. Additionally, we comprehensively examine the recent advancements in supported metal catalysts for photothermal catalytic C1 chemistry. This includes the conversion of CO (such as Fischer–Tropsch synthesis and water-gas-shift reaction), CO2 (reverse water-gas-shift reaction, methanation, methanol and higher hydrocarbon production), and methane (oxidation, non-oxidative coupling, steam and dry reforming) (Fig. 1). Finally, we provide a concise summary of the current research challenges and prospects within this field. This review contributes fundamental knowledge to the emerging field and holds the potential to revolutionize conventional chemical processes in a green and sustainable manner.
 | ||
| Fig. 1 Summary of photothermal catalytic C1 conversion using supported metal catalysts in this work. | ||
2. Mechanism of photothermal catalytic C1 chemistry
Photothermal catalysis represents an emerging technology that harnesses the synergy between photo and thermal energy to enhance catalytic activity and selectivity, enabling alternative reaction pathways. In the realm of C1 chemistry, the conversion of C1 molecules necessitates the cleavage of C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–H bonds, demanding a significant driving force. Photothermal catalysis can be categorized into three modes, depending on the primary driving force: photo-assisted thermocatalysis, thermal-assisted photocatalysis, and photothermal synergistic catalysis.6,12 In order to facilitate the design and development of more resilient catalysts, we, in this section, present a comprehensive analysis of the mechanisms governing photothermal catalytic C1 chemistry, elucidating its intricate and multifaceted steps.
O/C–H bonds, demanding a significant driving force. Photothermal catalysis can be categorized into three modes, depending on the primary driving force: photo-assisted thermocatalysis, thermal-assisted photocatalysis, and photothermal synergistic catalysis.6,12 In order to facilitate the design and development of more resilient catalysts, we, in this section, present a comprehensive analysis of the mechanisms governing photothermal catalytic C1 chemistry, elucidating its intricate and multifaceted steps.
2.1 Photochemical effect
Photochemical processes involve the excitation of semiconductors, leading to the generation of charge carriers (photogenerated electrons and holes) that migrate to the semiconductor surface and participate in chemical reactions with adsorbed molecules.13 Since the groundbreaking work of Fujijima and Honda in 1972, significant advancements have been made in the field of photocatalysis.14 However, most photocatalytic processes still suffer from low efficiency, primarily due to rapid carrier recombination and limited solar spectrum absorption and utilization by conventional wide-band gap semiconductors. This limitation has driven the rise of photothermal catalysis as a technology to improve photocatalytic activity.15In addition, noble metal nanoparticles, such as Au, Ag, and Cu, can harness the wide solar spectrum (including visible and infrared light regions) through the strong localized surface plasmon resonance (LSPR) effect.16 These nanoparticles then undergo energy dissipation through three pathways: First, a significant portion of energy is absorbed within the metal nanoparticles, generating energetic hot charge carriers, primarily hot electrons, through non-radiative decay (Landau damping process).17 These energetic hot charge carriers further induce chemical reactions. Second, a small fraction of the non-radiative decay energy transfers to the metal lattice phonons, resulting in localized heating known as plasmonic-induced thermal effects (Section 2.2). The remaining minor amount of energy is re-emitted as photons through radiation decay, augmenting the optical near-field. It is worth noting that the first two pathways play crucial roles in photothermal catalysis.
2.2 Photothermal effect
The photothermal effect is a phenomenon frequently observed in photo-driven thermal catalysis. In this process, the catalyst utilizes incident light to generate thermal energy (heat) upon irradiation, thereby elevating the surface temperature to facilitate catalytic reactions. Photothermal catalysis stands out among other solar energy harnessing technologies due to its exceptional efficiency in energy conversion. The photothermal effect in the catalyst arises from various mechanisms, including non-radiative relaxation in semiconductors, plasmonic localized heating, and thermal vibration of molecules.18Fig. 2 demonstrates that different photothermal materials can convert light into heat through one or more pathways of photothermal conversion.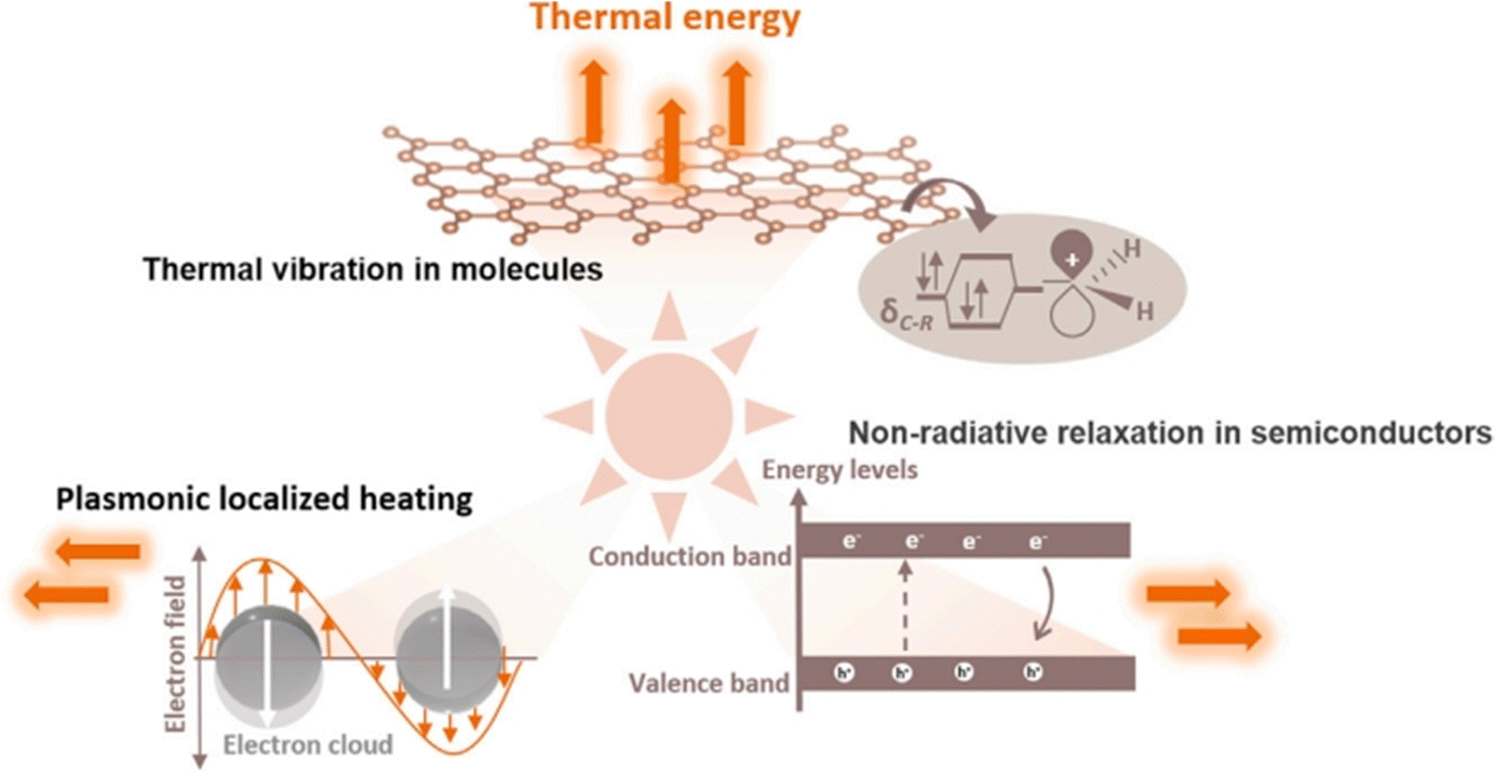 | ||
| Fig. 2 Different mechanisms of photo-to-thermal process in various materials.18 | ||
When light illuminates a semiconductor, photons with energy equal to or greater than the bandgap can excite the semiconductor, resulting in the generation of electron–hole pairs. These partial electrons and holes have the ability to migrate to the surface of a catalyst and participate in redox reactions. A significant portion of the electron–hole pairs releases energy through both radiative and non-radiative dissipation mechanisms. The non-radiative dissipation process induces phonons, which leads to heat energy generation through lattice vibration. This phenomenon, known as the photothermal effect in semiconductors, relies on the generation of charge carriers through photoexcitation. Similarly, the LSPR effect of plasmonic nanomaterials generates charge carriers, including high-energy hot electrons, that drive photochemical reactions. The non-radiative relaxation in plasmonic materials also contributes to local heating through electron–phonon scattering, thereby enhancing the activity of photothermal catalysis.
In addition to semiconductors and plasmas, photothermal effects can be observed in other materials with strong light absorption capacities, such as carbon-based materials (carbon nanotubes, graphene, amorphous carbon, etc.), metal–organic frameworks (MOFs), and mexenes.19–30 When these materials are excited by light, electrons undergo transitions from the highest occupied molecular orbital (HOMO) of the ground state to the lowest unoccupied molecular orbital (LUMO) of a higher energy state. Subsequently, through electron-vibrational relaxation processes, heat energy is generated. This photothermal conversion primarily occurs through thermal vibration in molecules.
2.3 Coupling of photochemical and photothermal effects
The photothermal catalytic process involves a complex interplay between the photochemical effect and photothermal effect, as these two phenomena are intricately connected and jointly determine the overall outcome. In the photochemical process, carriers excited by semiconductor photocatalysts or plasmonic materials act as driving forces for chemical reactions. The hot carriers generated through light irradiation actively participate in catalytic reactions, constituting the photochemical effect. On the other hand, the thermocatalytic process in photothermal catalysis relies on the non-radiative relaxation of semiconductors, plasmonic localized heating, and thermal vibration in molecules. These factors play pivotal roles in converting light into heat. The photothermal effect enables efficient dissipation of absorbed photon energy as heat under light irradiation, facilitating carrier transfer and enhancing catalytic activity. Furthermore, photothermal catalysis has the advantage of reducing or eliminating the need for additional heat input and energy consumption. The process concentrates most of the heat on the catalyst's surface, leading to high energy utilization rates. In certain cases, photothermal catalysis effectively prevents catalyst deactivation and enhances the selectivity of desired products. Therefore, the reaction mechanism of heterogeneous photothermal catalysis typically involves the coupling of both photochemical and photothermal effects, representing the interplay between these phenomena.313. Design of supported metallic photothermal catalysts
Supported catalysts are materials where the active component is uniformly dispersed and supported on a carefully chosen substrate. Typically, supported catalysts consist of two main elements: the active component and the support component. In certain cases, the support component can also exhibit catalytic activity. These two components work together synergistically, resulting in a combined effect that exceeds the sum of their individual contributions. An effective photothermal catalyst should possess specific capabilities, including (1) strong light absorption, (2) high efficiency in photothermal conversion and heat retention, and (3) excellent catalytic activity, product selectivity, and stability. Therefore, the design and modification of catalysts primarily focus on enhancing light absorption across the solar spectrum, promoting efficient heat generation and transfer, and improving catalytic kinetics. In the following section, we will discuss the design strategies for the active and support components in supported catalysts separately, considering their respective roles and functionalities.3.1 Design of active components
Metals possess exceptional properties which make them indispensable in various industries. Their efficient conductivity of electricity and heat makes them ideal for applications such as electrical wiring, power generation systems, and sustainable heating solutions. Additionally, metals exhibit efficient solar energy absorption and conversion capabilities, making them increasingly popular as a sustainable heat source for homes and buildings. By harnessing the power of the sun through metal-based technologies like solar panels or thermal collectors, we can reduce our reliance on non-renewable energy sources such as fossil fuels and significantly decrease our carbon footprint. Therefore, metals are versatile and valuable resources that play an essential role in the functioning of modern society. Their unique combination of conductivity and solar absorption capabilities makes them vital components in numerous critical applications today while offering promising solutions for future renewable energy needs.The LSPR effect plays a crucial role in determining the optical properties of metal nanomaterials. When nanometals are exposed to light with suitable energy, they induce the generation of energetic hot carriers and localized thermal energy, triggering chemical reactions. Previous research studies have demonstrated the remarkable responsiveness of plasmonic metal nanostructures to visible and near-infrared light.32,33 The LSPR effect involves three pathways (Section 2.1) which provide the necessary energy for catalytic reactions to occur on the surface of metal nanostructures. Enhancing the LSPR effect of plasmonic nanomaterials is an effective strategy for broadening the optical absorption spectrum, improving photothermal conversion rates, and enhancing catalytic performance.
In photothermal catalysis, metals in photothermal catalysts primarily include precious metals (Au,34 Ag,35 Ru,36 Rh,37,38 Pd,39,40 Ir, and Pt41) and non-precious metals (Fe,42 Co,43 Ni,44 Cu,45 Al,46,47 and Nb28). By precisely controlling their sizes, shapes, and compositions, their photothermal catalytic performance can be tailored. In a recent study, Yang et al. achieved continuous size regulation of monodisperse Pd nanoparticles ranging from 2.8 to 8.1 nm (Fig. 3a). Through precise size control and manipulation of the surface electronic structure, the 6.3 nm Pd nanoparticles exhibited the highest catalytic activity in the photothermal catalytic hydrogenation of CO2 to CO.39 These findings illustrate that the catalytic activity of size-regulated nanoparticles is primarily influenced by the quantum size effect, metal–support interaction, and photothermal effect.
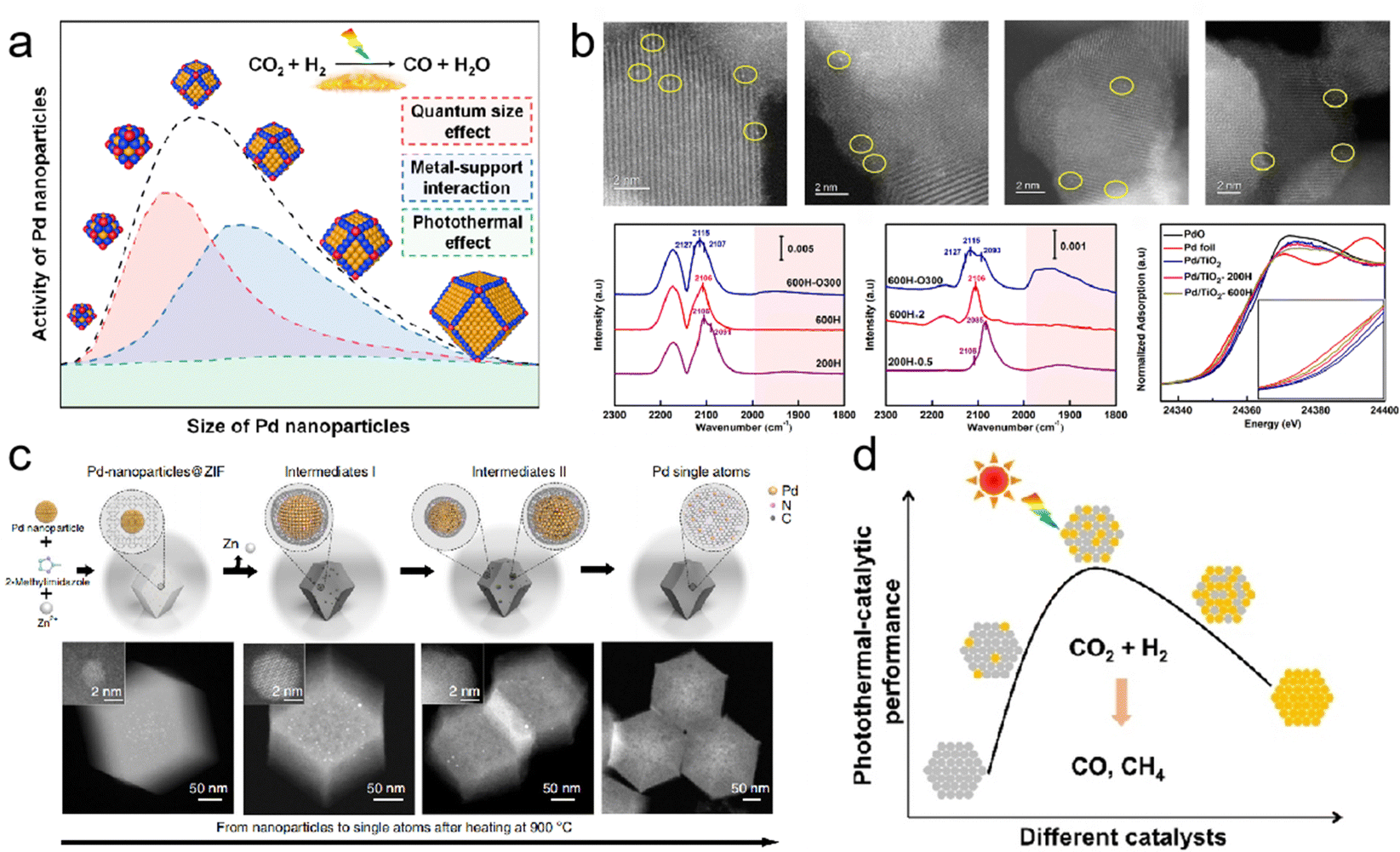 | ||
| Fig. 3 (a) Monodispersed Pd nanoparticles with a narrow size distribution supported on TiO2.39 (b) Structural characterization of Pd/TiO2 serial catalysts.40 (c) Scheme of the transformation of nanoparticles to single atoms and structural characterization of Pd single atoms.50 (d) The influence of the composition of bimetallic nanocrystals on photothermal catalytic performance.51 | ||
Single-atom catalysts (SACs) have emerged as promising candidates in catalysis due to their exceptional atomic utilization and unique electronic structures. Researchers have shown a growing interest in exploring the potential applications of SACs. In a study conducted by Guo et al., they constructed Pd1/TiO2 SACs through the selective encapsulation of co-existing Pd nanoclusters/nanoparticles based on different conditions of SMSI occurrence (Fig. 3b).40 By utilizing detailed characterization techniques and density functional theory (DFT) calculations, the team gained valuable insights into the behavior of these SACs. The results were intriguing, as photo-induced electrons transferred from TiO2 to adjacent Pd atoms, facilitating acetylene activation and highlighting the potential of these SACs in various catalytic reactions.
However, the excessive surface free energy of single-atom metals makes them susceptible to agglomeration at high temperatures, resulting in a significant reduction in the exposed metal surface area, a phenomenon known as sintering. This process adversely affects catalytic reactivity and, in some cases, can render the catalyst inactive. Metal sintering occurs through two main pathways: migration between individual metal atomic nanoparticles or the coalescence of adjacent metal nanoparticles. It is worth noting that these pathways are not mutually exclusive and often occur simultaneously, particularly at elevated temperatures. To address this issue, researchers have explored strategies such as the introduction of SMSI or ligands to thermodynamically stabilize metal atoms. These approaches effectively reduce surface energy and limit the migration tendency of individual atoms. However, the use of certain ligands may have toxic effects, leading to deactivation of the catalyst. Alternatively, preventing collisions and coalescence kinetically can be achieved by confining metal nanoparticles within microporous solids such as zeolites and metal–organic frameworks.48,49 A groundbreaking study by Wei et al. achieved a significant breakthrough in the field of nanotechnology.50 The researchers successfully reversed the sintering process of nanoparticles by utilizing nitrogen-doped carbon (CN) as an anchoring substrate to capture migrating metal atoms at high temperatures (Fig. 3c). They employed ZIF-8 as the framework and transformed noble metal nanoparticles into highly active single atoms, including Pd, Pt, and Au. The resulting thermally stable particles exhibited exceptional catalytic activity. During the process of recording films with in situ environmental transmission electron microscopy (ETEM), an intriguing observation was made. Despite exposure to high temperatures, sintering occurred, causing smaller particles to fuse and form larger ones. However, as the Pd nanoparticle collided with the substrate, it gradually shrank and transformed into a single atom. The resulting thermally stable Pd single atoms exhibited superior catalytic activity and selectivity for acetylene semi-hydrogenation compared to Pd nanoparticles. This study presents a user-friendly top–down approach for obtaining single-atom catalysts from nanoparticles and even reactivating sintered industrial precious metal nanocatalysts by dispersing them into individual atomic sites. These findings provide valuable insights for the development of high-performance, heat-stable catalysts.
In addition to the aforementioned strategies, the preparation of dispersed supported bimetallic nanocrystalline catalysts represents another feasible solution to enhance the performance of single-component metal catalysts. Chen et al. reported on the photothermal hydrogenation of CO2 using Al2O3-supported CoFe alloy catalysts.46 Their research demonstrated the effectiveness of these catalysts in the conversion process. Similarly, Zhu et al. developed a monodispersed Pd-Ni nanoalloys/SiO2 catalyst, where the inclusion of Ni improved the optical absorption and photothermal conversion properties, while the presence of Pd enhanced the selectivity of the catalyst in converting CO2 to CO (Fig. 3d).51 These findings highlight the potential of dispersed supported bimetallic nanocrystalline catalysts in enhancing the performance and efficiency of catalytic processes.
However, the synthesis of these metal materials presents several challenges that need to be considered. Factors such as the wide range of metal materials available, the need for controllable morphology, environmental impact, high cost, and complex preparation processes should not be overlooked. Additionally, metal nanoparticles can undergo structural distortion when exposed to acids, bases, or salts, and they are susceptible to agglomeration under harsh conditions or contamination by trace impurities. Therefore, the careful selection of a suitable support material becomes crucial in ensuring the stability and functionality of the catalyst.
3.2 Design of support components
The support components of a catalyst play a crucial role in determining its overall performance, serving three primary functions that contribute to its efficiency. First, the support component provides an effective surface area and pore structure, which enhances catalytic activity by increasing the available surface area for reactions to occur. It also helps prevent the sintering or aggregation of the active component, which can lead to catalyst deactivation over time. Additionally, the support component increases the mechanical strength of the catalyst, making it more durable and resistant to wear and tear. Second, support components can provide additional active centers, particularly in bifunctional catalysts where multiple reactions occur simultaneously. These additional active centers contribute to improved reaction rates and selectivity. They can promote specific reaction pathways or provide sites for intermediate species adsorption, thereby enhancing the overall catalytic performance. Finally, the interaction between the support and active component is crucial in determining the catalytic activity and selectivity. The nature of this interaction depends on factors such as chemical composition, morphology, and size distribution, all of which influence the enhancement or inhibition of catalytic performance. Optimal support materials should exhibit strong interaction with the active component, facilitating charge transfer, promoting adsorption–desorption processes, and optimizing reactant accessibility.Currently, inorganic semiconductors serve as the primary foundation for active metal components, providing a robust support system for photothermal catalysts. However, there are several other types of support materials commonly used in addition to semiconductors. These alternative supports include materials such as carbon, MXenes, and metal–organic frameworks (MOFs), each offering unique properties and advantages for specific photothermal applications.
To address these challenges, specific design strategies have been employed in the investigation of inorganic semiconductors. One prevalent method involves doping and/or introducing defects or vacancies to broaden the light absorption range and accelerate the separation of photogenerated carriers.52,53 For example, treating molybdenum trioxide (m-WO3) with pure hydrogen generates oxygen-deficient WO3 (m-WO3−x) catalysts, exhibiting broader absorption from UV to near-infrared (NIR) light regions and narrower band gaps.54 This transformation is attributed to the formation of oxygen vacancy-induced defect states in m-WO3−x, which not only enhances catalytic activity but also expands the potential for photothermal conversion. Similarly, the introduction of defects into Bi2O3−x induces the LSPR effect in the near-infrared wavelength range, enhancing CO2 molecule adsorption and enabling efficient photocatalysis of CO2-to-CO even under low-intensity near-IR light irradiation.55 Careful engineering of these imperfections allows customization of electronic and optical properties to meet specific requirements.
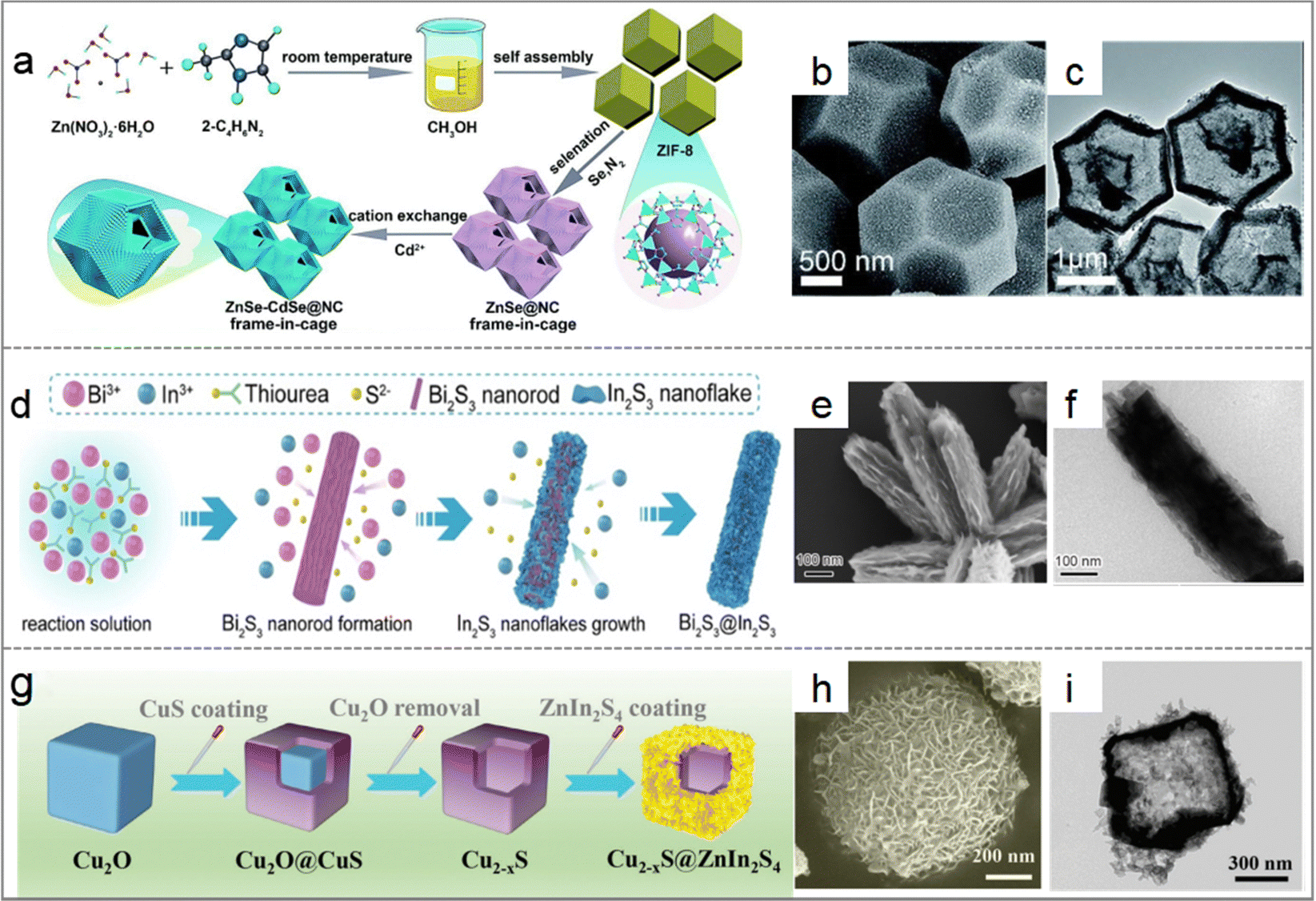
Another effective strategy for enhancing photothermal catalytic performance is coupling two types of inorganic semiconductors with different photocatalytic and thermal catalytic properties. Constructing heterojunctions with atomic arrangement and lattice mismatches can lead to remarkable light-to-hot carriers and light-to-thermal efficiency. S-scheme heterojunctions, in particular, have shown excellent photo-mediated conversion abilities (Fig. 4).56–60 For example, Wang et al. synthesized a core–shell Cu2−xS@ZnIn2S4 S-scheme heterojunction through a simple hydrothermal reaction, which broadened the absorption range of photocatalytic response, increased infrared light absorption, exhibited excellent photothermal effects, and promoted the photocatalytic reaction.61,62 However, developing novel, facile, and clean approaches to produce engineered heterojunction materials with specific composites, sizes, and shapes remains a challenging task.
Ren et al. developed a hierarchical graphene foam as a solar absorber (Fig. 5a), exhibiting high absorption across the solar spectrum (295–2500 nm) with minimal transmittance (≈ 0%) and an exceptional photo-thermal conversion efficiency of up to 93.4%.64 This innovative design maximizes the utilization of incoming sunlight for energy conversion. Similarly, Ito et al. designed a versatile porous graphene material with excellent heat localization and steam generation capabilities, achieving an impressive energy conversion rate of 80% through solar radiation (Fig. 5b).65 These advancements highlight the potential of carbon-based materials in photothermal catalysis. However, one challenge associated with graphene sheets is the shading effect due to their light-blocking nature. This issue can be mitigated by controlling the dosage of graphene sheets and utilizing small-pore semiconductors or in-plane pore materials for structural adjustments, allowing for improved light exposure and efficient catalytic performance.
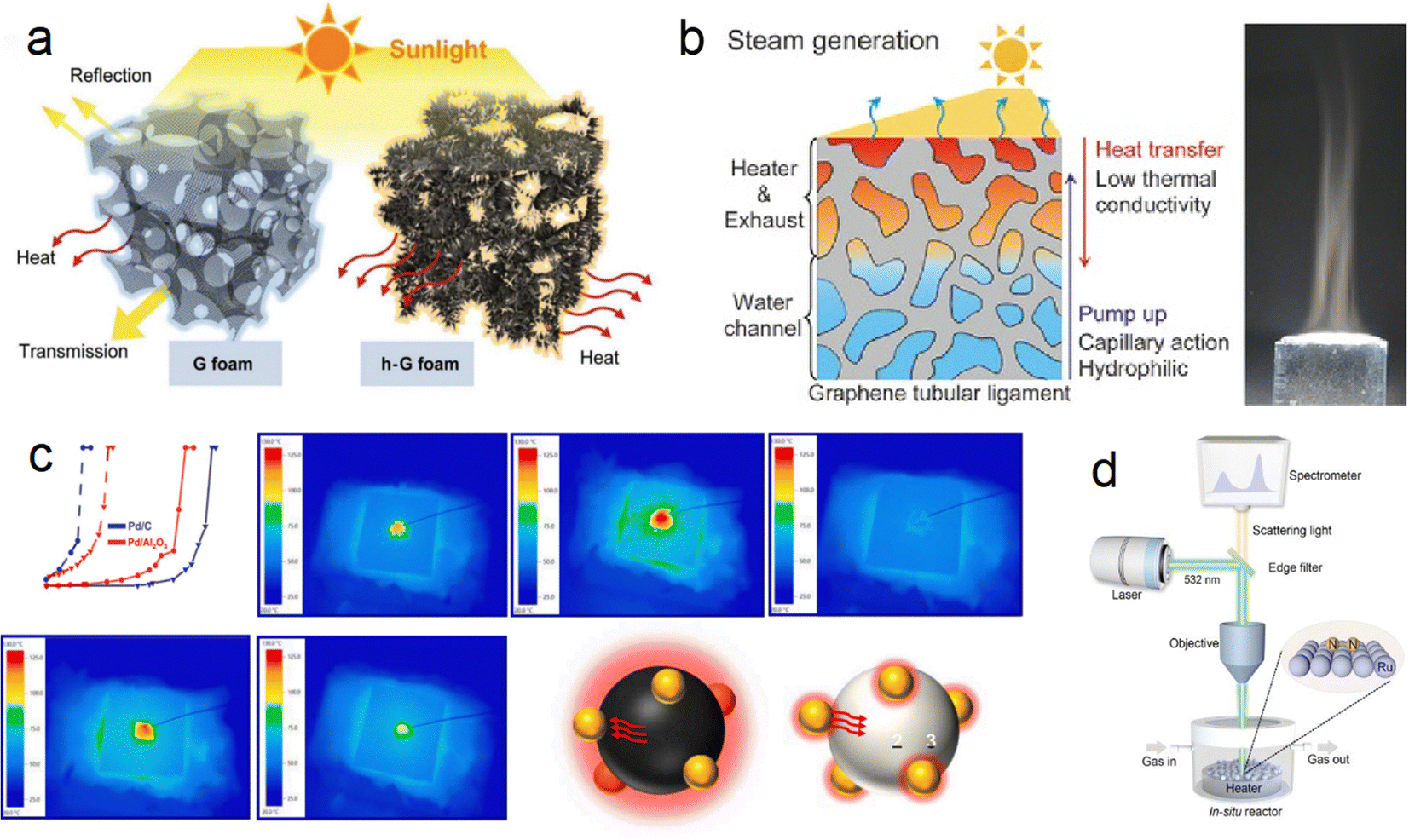 | ||
| Fig. 5 (a) Difference in solar-thermal conversion processes between ordinary graphene foam and hierarchical graphene foam.64 (b) Multifunctional nanoporous graphene converting solar illumination into high-energy steam.65 (c) Heat transfer path in CO conversion proved by IR monitoring images (carbon support → Pd → Al2O3).66 (d) Schematic illustration of the customized in situ Raman system.67 | ||
Although significant advancements have been made in the field of graphene-based photothermal catalysis, it is important to acknowledge that these materials still lag behind noble metal/oxide catalysts due to their weaker interactions with reactants. However, carbon emerges as a promising candidate for exploring photothermal catalysis when utilized as both a catalyst support and a photothermal material with full-spectrum light absorption capabilities. Liu et al. synthesized a Pd/C catalyst using commercial carbon (XC-72R) as a catalyst support through a reduction and impregnation method and applied it in the photothermal catalytic oxidation of CO reaction.66 The Pd/C catalyst demonstrated excellent light absorption ability, exhibiting significantly better catalytic performance under light conditions compared to dark conditions. Under a light intensity of 900 mW cm2, the temperature for achieving 100% CO conversion (TF100, furnace temperature) for the 1% Pd/C catalyst decreased from 146 to 30 °C, realizing complete CO conversion at room temperature (Fig. 5c). This exceptional catalytic performance is attributed to the ultra-high local temperature generated by the excellent photothermal conversion ability of carbon. Furthermore, infrared light was identified as the primary contributor to the improved catalytic performance. This work opens new possibilities for harnessing solar energy using carbon-based materials.
In a different study, Bian et al. measured the actual reaction temperature of photothermal synthesis of ammonia on carbon-supported Ru catalysts to quantify the contribution of thermal electrons, employing Le Chatelier's principle (Fig. 5d).67 By excluding the local heating effect, they found that the activation energy of photothermal catalysis is significantly lower than that of thermal catalysis (54.9 kJ mol−1 compared to 126.0 kJ mol−1). This reduction in activation energy is attributed to the phenomenon of hot electron injection, which lowers the energy barrier for N2 dissociation and intermediate product hydrogenation. Additionally, the injection of hot electrons suppressed the methanation reaction of carbon carriers, resulting in excellent operational stability of the catalyst over 1000 hours.
For example, Wu et al. utilized a chemical stripping method to obtain two-dimensional Mo2TiC2 nanosheets, which were then functionalized with Ru clusters (Ru/Mo2TiC2) (Fig. 6a).68 These MXene-supported clusters exhibited remarkable photothermal conversion performance, with the surface temperature of Ru/Mo2TiC2 rapidly reaching 360 °C within 30 seconds under sunlight. After 5 minutes of illumination, the CO production rate reached 3.9 mol gRu−1 h−1, accompanied by a CO selectivity of 90% (Fig. 6b and c). This achievement positions Ru/Mo2TiC2 as one of the most effective photothermal catalysts reported for the reverse water-gas shift (RWGS) reaction. Another advantage of MXene materials lies in their tunable optical, electronic, thermal, and surface properties, which can be adjusted by modifying their compositions, structures, and terminations. This inherent versatility expands the range of available MXene-based materials. Notably, recent work by Wu et al. demonstrated that two representative MXene materials, Nb2C and Ti3C2, enhance the photothermal effect, thereby boosting the photothermal catalytic activity of Ni nanoparticles (Fig. 6e and f).28
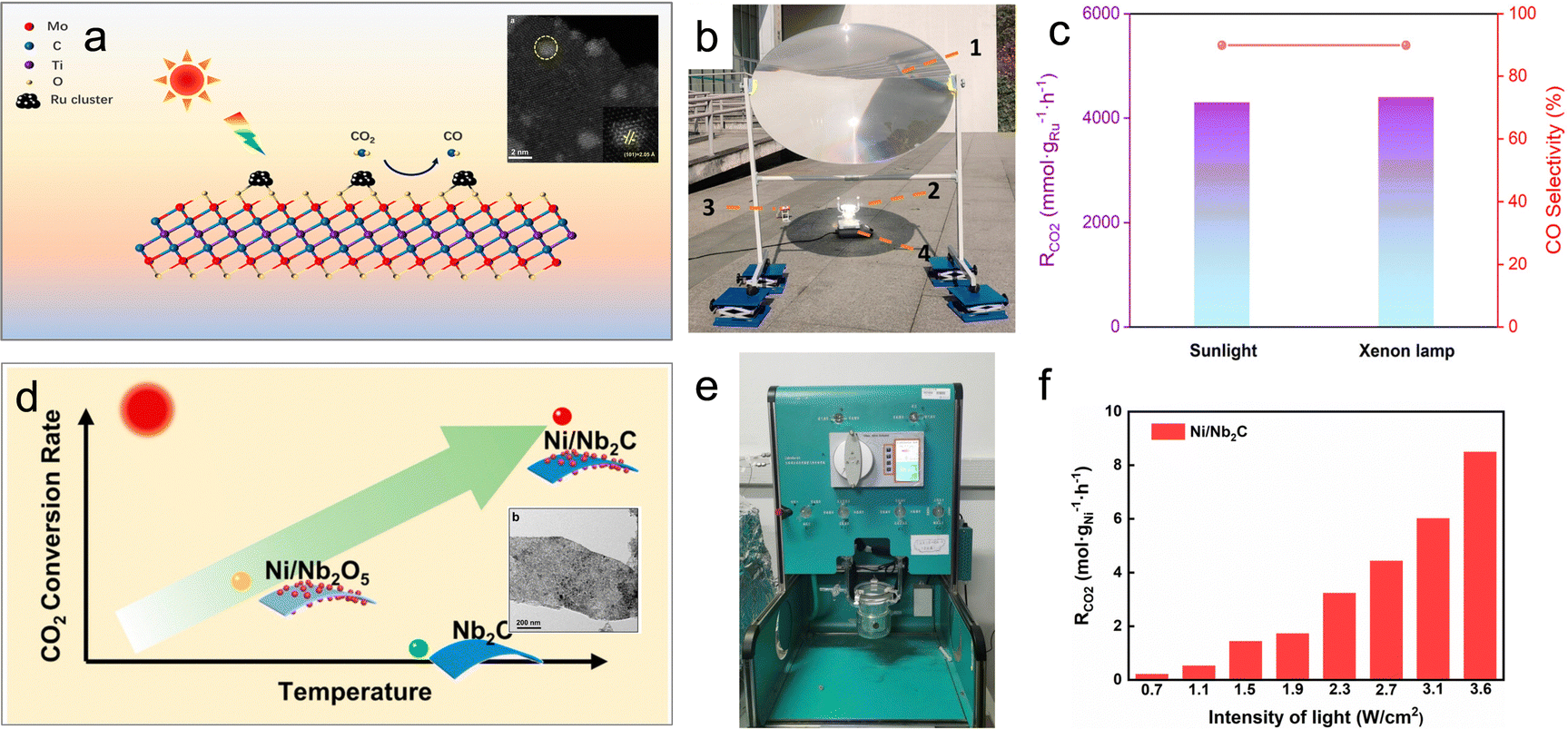 | ||
| Fig. 6 (a) Schematic illustration and TEM image of the Ru/Mo2TiC2 photothermal catalyst. (b) Setup of the outdoor photothermal catalysis. (c) CO2 conversion and product selectivity of Ru/Mo2TiC2 in photothermal catalytic RWGS.68 (d) Schematic illustration and TEM image of the Ni/Ni2C photothermal catalyst. (e) Photograph of the batch reactor. (f) Photothermal catalytic activity of Ni/Nb2C in photothermal catalytic RWGS.28 | ||
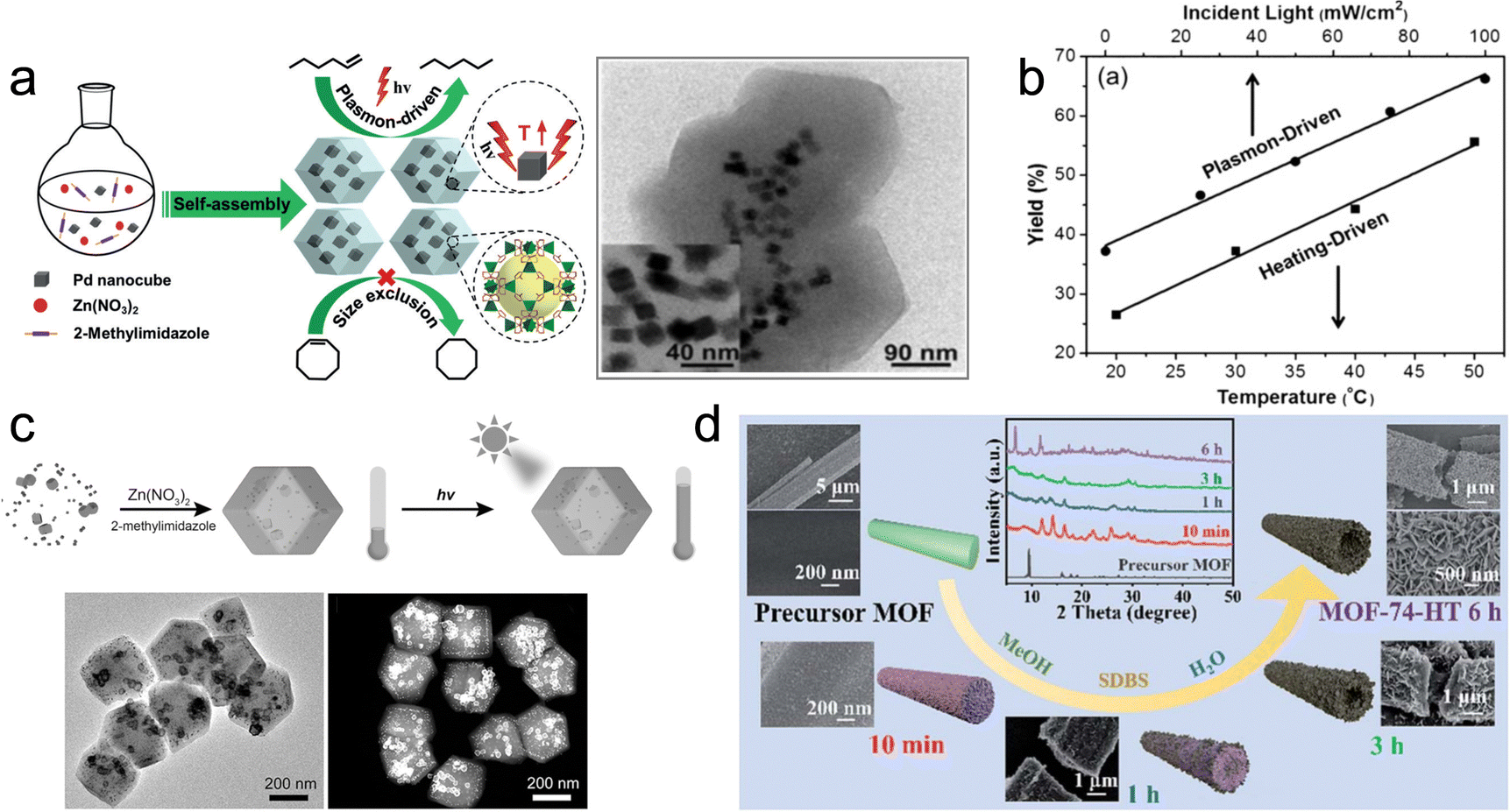 | ||
| Fig. 7 (a) Self-assembly of Pd NCs@ZIF-8 and plasmon-driven selective catalysis of hydrogenation of olefins. (b) The hydrogenation performance of Pd NCs@ZIF-8.69 (c) Schematic illustration of the formation of Au&Pt@ZIF and temperature variation under light irradiation.70 (d) Phase transformation from the MOF precursor to MOF-74-HT.71 | ||
Overall, although semiconductor-based supports are widely used in photothermal catalyst applications, ongoing research aims to explore new materials and approaches to further advance this field.36,72,73 In these approaches, heat energy is harnessed to drive catalytic reactions instead of being dissipated to the surrounding environment through convection and conduction. Through structural control, interface engineering, multifunctional assembly and nanoscale effects for meticulous selection and optimization of both the active component and support structure, researchers can develop highly efficient systems capable of catalyzing complex chemical transformations with exceptional precision and control. This continuous exploration and refinement of materials and strategies hold great promise for pushing the boundaries of photothermal catalysis and unlocking new possibilities in diverse areas of chemistry and beyond.
4. Photothermal catalytic C1 conversion using supported metal catalysts
When it comes to harnessing the power of solar energy, photothermal catalytic C1 chemistry has emerged as a highly promising alternative to traditional thermocatalysis. This innovative approach capitalizes on abundant and clean energy sources, and it has made remarkable progress in recent years. Over the past decade, significant advancements have been achieved in the field of photothermal catalytic C1 conversion. From improving photothermal catalytic CO conversion to groundbreaking developments in CO2 hydrogenation, and conversions of methane and methanol, researchers are continuously pushing the boundaries of what can be accomplished with this transformative technology. The performance of supported catalysts for photothermal catalytic reactions in C1 chemistry has been summarized in Table 1. By harnessing the unique properties of light and heat, photothermal catalysis offers a more efficient and sustainable method for converting raw materials into valuable products. This exciting field holds immense potential for revolutionizing the energy landscape and enabling a more sustainable and greener future.| Catalysts | Reactant | Conversion/% | Products | Selectivity or mass specific activity | Ref. |
|---|---|---|---|---|---|
| Fe5C2/NC600 | CO | 22.3 | C2–4 |
55.3% | 75 |
| Ni/MnO | CO | 14.9 | Light olefins | 33% | 76 |
| Fe-500/ZnO–Al2O3 | CO | 20.9 | Light olefins | 42.4% | 77 |
| Co–Co3O4/ZnO–Al2O3 | CO | 15.4 | Light olefins | 36% | 78 |
| TiO2-supported Ni2P/Ni | CO | 20.7 | C2+ | 70.7% | 79 |
| Ru1Co-SAA | CO | 58.6 | C5+ | 75.8% | 80 |
| Pt/α-MoC | CO | 22 | H2 | 83 | |
| CuOx/Al2O3 | CO | >95 | H2 | 86 | |
| CeO2/Cu1.5Mn1.5O4 | CO | 96.6 | H2 | 87 | |
| Ni12P5/SiO2 | CO2 | 960 ± 12 (mmol gcat−1 h−1) | H2 | Near 100% | 100 |
| K+–Co–C | CO2 | 758 (mmol gcat−1 h−1) | CO | 99.8% | 105 |
| Ru–Al2O3 | CO2 | 95.75 | CH4 | 99.22% | 38 |
| Rh–Al2O3 | |||||
| Ni–Al2O3 | |||||
| Fe3O4 | CO2 | 11.3 (mmol gcat−1 h−1) | CO | 100% | 42 |
| Fe3C | CH4 | 97% | |||
| Pd-loaded WN-WO3 | CO2 | 40.6 ± 0.7 (mmol gcat−1 h−1) | CH4 | 100% | 107 |
| Ru–Al2O3−x-L | CO2 | 87.68 | CH4 | Near 100% | 108 |
| SA Ni/Y2O3 | CO2 | 80 | CH4 | 7.5 L m−2 h−1 | 110 |
| Au–Ru/TiO2 | CO2 | CH4 | 99% | 116 | |
| Fe/FeOx supported on MgO–Al2O3 | CO2 | 50.1 | C2+ | 52.9% | 117 |
| AuCu–ZnO | CH4 | CH3OH, CH3OOH and HCHO | 100% | 120 | |
| Pd-def-In2O3 | CH4 | CH3OH and CH3OOH | 82.5% | 121 | |
| Au/In2O3 | CH4 | CH3OH | 97.62% | 122 | |
| HCHO | 89.42% | ||||
| Pt@BT-O | CH4 | 8.2 | propane | 65% | 123 |
| Au/ZnO | CH4 | C2H6 | 124 | ||
| TiO2-supported Rh NPs | CH4 | CO2, H2 | 125 | ||
| TiO2-supported Pt | CH4 | H2 | 185 (mmol g−1 h−1) | 126 | |
| Single-layer Ni cluster stabilized by Al2O3 | CH4 | CO | 9614.26 (mmol gNi−1 min−1) | 128 | |
| H2 | 8572.96 (mmol gNi−1 min−1) | ||||
| Ni/Ga2O3 | CH4 | CO2, H2 | 129 | ||
| Cu–Ru/MgO–Al2O3 | CH4 | H2 | 34 (mol molRu−1 s−1) | 130 | |
| Pt/CeO2 | CH4 | CO | 22.7 (mmol gcat−1 h−1) | 131 | |
| Rh/CexWO3 | CH4 | H2 | 88.5 (mmol gRh−1 h−1) | 3 | |
| CO | 152.3 (mmol gRh−1 h−1) |
4.1 CO conversion
) from syngas, a process known as Fischer–Tropsch to olefins (FTO). Typically, harsh conditions (200–400 °C and 2–5 MPa) are required to lower the energy barrier for CO molecule activation and the C–C coupling process in the FTO process. However, solar-driven FTS has emerged as a promising alternative to conventional thermocatalytic FTS, as it enables operation under milder reaction conditions with lower energy requirements.
Recent studies have shown that Fe-based supported catalysts exhibit high selectivity for C2–4 products and demonstrate excellent reaction activity in the FTO process. For instance, Ou's group synthesized a Fe5C2-loaded N-doped carbon microsphere catalyst (Fe5C2/NC600) using a wet chemistry method.75 This catalyst exhibited broad sunlight absorption ability, exceptional C2–4 selectivity of 55.3% (CO2-free) at 22.3% CO conversion, and superior catalytic activity, which was 3.5 times higher than that of pristine Fe5C2 in the photothermal catalytic FTO process. Characterization techniques such as XPS, XAFS, CO-TPD, and CO pulse reaction revealed that pyrrolic N in the N-doped carbon support acted as an electron donor, transferring electrons to the surface of the active Fe5C2 phase and modifying its electronic structure. This electron transfer process created electron-rich active sites, enhancing the CO adsorption and dissociation ability of the Fe5C2-based catalyst, thereby increasing the CO conversion rate (Fig. 8a). The author further investigated a series of Fe5C2-based catalysts with different supports (hydrophobic silica, hydrophilic silica, α-Al2O3, γ-Al2O3, and α,γ-Al2O3) via a one-step wet-chemical method to explore the influence of metal–support interaction on the photothermal catalytic FTO process. It was found that the weak interaction between the active phase and α-Al2O3 supports favored the formation of Fe5C2, contributing to high selectivity for light olefins.
 | ||
| Fig. 8 (a) Scheme and CO adsorption ability of Fe5C2/NC600.75 (b) Characterization data and product selectivity of the Ni/MnO catalyst.76 (c) The CO hydrogenation selectivities of the Fe-x photocatalysts derived from ZnFeAl–LDH nanosheets at different temperatures.77 (d) Comparison of FTS performance of Ru1Co-SAA with other catalysts.80 | ||
Research on Ni-based catalysts for the photothermal FTO process has also been conducted. One study focused on developing Ni-containing nanoparticles supported on MnO catalysts (Ni/MnO) derived from the reduction of NiMn mixed metal oxide.76 The optimized Ni/MnO catalyst with a Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ratio of 2:1 demonstrated excellent performance in FTO, achieving a high selectivity of light olefins (33%) at 14.9% CO conversion under UV-visible light irradiation (Fig. 8b). Characterization techniques such as XAFS and XPS analysis of the Ni-500 catalyst revealed the presence of MnO-supported Ni nanoparticles, which induced modifications in the electronic structure and valence of the Ni nanoparticles, resulting in the formation of Niδ+ sites. This slight change in nickel valence hindered the strong hydrogenation ability typically associated with Ni0, leading to C–C coupling reactions that produced alkenes.
:Mn ratio of 2:1 demonstrated excellent performance in FTO, achieving a high selectivity of light olefins (33%) at 14.9% CO conversion under UV-visible light irradiation (Fig. 8b). Characterization techniques such as XAFS and XPS analysis of the Ni-500 catalyst revealed the presence of MnO-supported Ni nanoparticles, which induced modifications in the electronic structure and valence of the Ni nanoparticles, resulting in the formation of Niδ+ sites. This slight change in nickel valence hindered the strong hydrogenation ability typically associated with Ni0, leading to C–C coupling reactions that produced alkenes.
In one-step FTO processes, preventing the high selectivity for the by-product of CO2 resulting from the WGS reaction remains a challenge. To address this issue, Zhang's group developed a series of Fe-based heterostructured photocatalysts by H2 reduction of ZnFeAl–LDH precursor nanosheets at different temperatures (300–650 °C).77 The Fe-500 catalyst (reduced at 500 °C), comprising Fe and FeOx supported on ZnO and Al2O3, exhibited high selectivity (42%) for light olefins and low selectivity (11.4%) for CO2 at a conversion of 20.9% in the photothermal FTO reaction. DFT calculations indicated that the abundant interface between Fe and FeOx in the Fe-500 heterostructured catalysts, along with moderate intermediate binding energies, contributed to the high selectivity for light olefins and suppressed CO2 evolution (Fig. 8c). The author further investigated Co-based heterostructured catalysts derived from ZnCoAl–LDH precursor nanosheets.78 Surprisingly, the Co–Co3O4/ZnO–Al2O3 catalyst exhibited high selectivity (36%) for light olefins at a CO conversion of 15.4%. The heterogeneous structure of oxide-modified metallic Co nanoparticles in this catalyst weakened the hydrogenation capacity of olefins, resulting in high selectivity for low carbon olefins.
In addition to light olefins, higher hydrocarbons (C2+) are valuable products in the FTS process. Zhang et al. reported a partial phosphidation strategy for preparing TiO2-supported Ni2P/Ni heterostructure catalysts for photothermal FTS reactions.79 Precise control over the degree of phosphating in Ni/Ni2P catalysts was achieved by adjusting the input amount of NaH2PO2 during synthesis. The optimized Ni/Ni2P-based catalyst for CO hydrogenation exhibited remarkable C2+ selectivity of 70.7% at a CO conversion rate of 20.7% under Xe lamp irradiation and demonstrated excellent stability in flow reaction system testing. DFT calculations indicated that metallic Ni facilitated CO activation, while Ni2P/Ni favored C–C coupling. The coexistence of both metals provided an efficient pathway for CO hydrogenation to C2+ products, inhibiting over-hydrogenation to methane.
Metal alloys such as Fe, Co, Ni, and Ru can also achieve high CO conversion rates and selectivity for specific products. Zhang's group recently developed an Al2O3-supported RuCo single atom (Ru1Co-SAA) catalyst derived from LDH topological transformation for converting syngas into C5+ liquid fuels via the photothermal catalytic FTS process.80 Characterization of the Ru1Co-SAA structure confirmed the successful preparation of isolated Ru atoms on the surface of metallic Co nanoparticles. The Ru1Co-SAA catalyst exhibited higher CO conversion activity (58.6%) and C5+ product selectivity (75.8%) compared to Al2O3-supported Co nanoparticles or RuCo nanoalloys (Fig. 8d). Furthermore, the FTS performance of Ru1Co-SAA at low pressure was comparable to that of traditional FTS catalysts under higher pressure conditions. Chemisorption experiments and DFT calculations revealed that the Ru-Co coordination environment in Ru1Co-SAA promoted CO dissociation and C–C coupling of 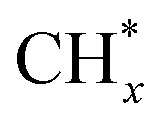 intermediates while inhibiting H2 activation and over-hydrogenation of
intermediates while inhibiting H2 activation and over-hydrogenation of  intermediates. This promoted the effective growth of carbon chains to liquid products. This work is expected to inspire the design of more bimetallic catalysts for photothermal catalytic systems to convert small C1 molecules into high-value chemicals.
intermediates. This promoted the effective growth of carbon chains to liquid products. This work is expected to inspire the design of more bimetallic catalysts for photothermal catalytic systems to convert small C1 molecules into high-value chemicals.
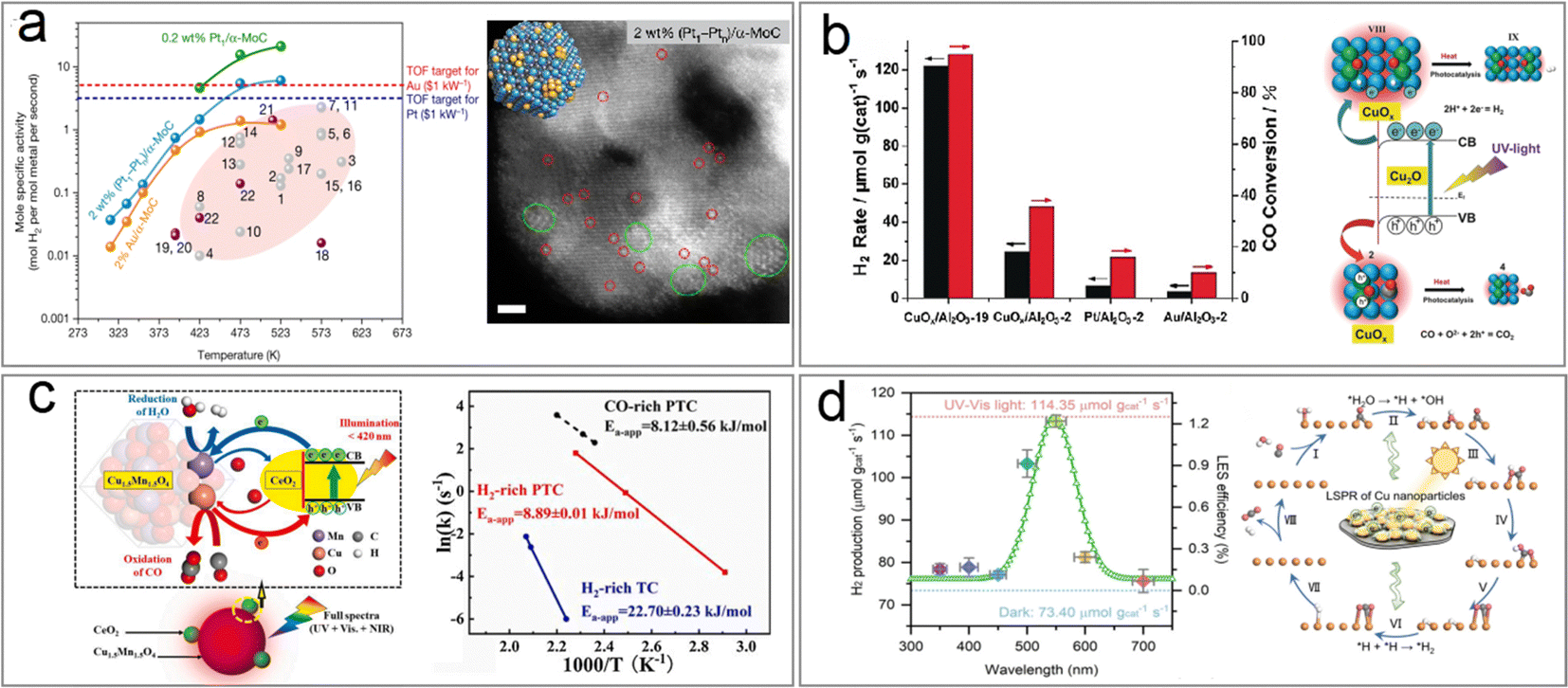
A recent study conducted by Zhang et al. presented a remarkable discovery regarding the activity of noble-metal Au clusters and Pt1–Ptn clusters on α-MoC in low-temperature WGS reactions, which achieved unprecedented rates of H2 production (Fig. 9a).83 However, the utilization of noble metals for large-scale H2 production is impractical due to their high costs and the substantial amount of heat energy required from electricity or fossil fuel combustion. Consequently, it becomes crucial to prioritize the design and development of non-noble metal catalysts capable of activating water and efficiently driving WGS reactions using clean energy at lower temperatures. Previous research has demonstrated that semiconductor photocatalysts can drive the WGS reaction at room temperature when exposed to light irradiation. Nevertheless, the hydrogen production rates achieved through photocatalysis fall significantly below industry standards.84–88
 | ||
| Fig. 9 (a) WGS performance and morphology of the Pt/α-MoC catalyst.83 (b) WGS activity and mechanism of the CuOx/Al2O3 catalyst.86 (c) WGS reaction mechanism and the apparent activation energy of the CeO2/Cu1.5Mn1.5O4 catalyst.87 (d) WGS reaction activity and mechanism of the LD-Cu catalyst.96 | ||
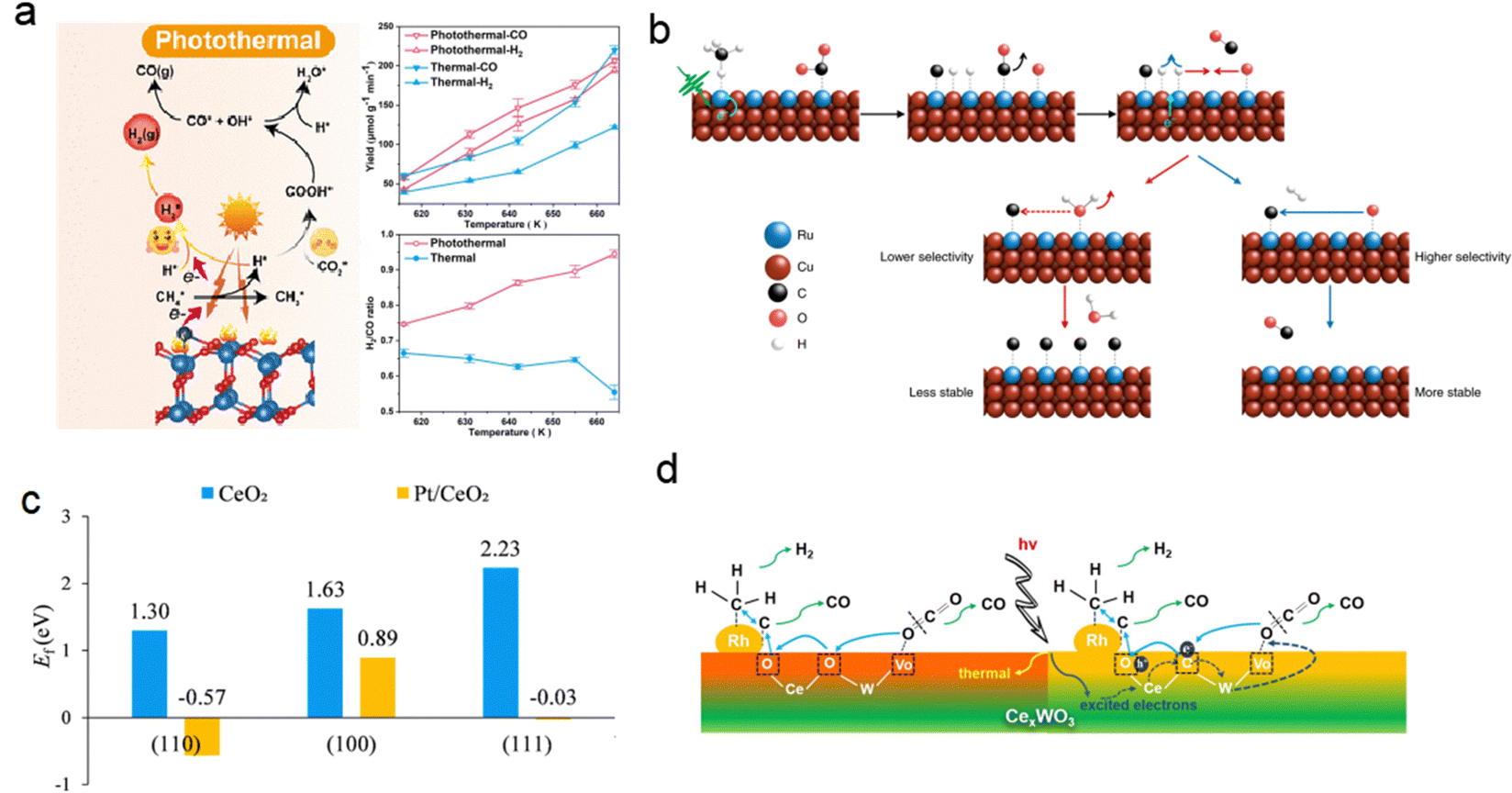
In contrast, Cu-based photothermal catalysts for WGS have shown the ability to produce hydrogen at rates comparable to those of traditional high-temperature thermal catalysts when operated under light irradiation at 250–350 °C.85,86,89,90 The group led by Ye reported that CuOx/Al2O3 exhibits excellent catalytic activity (122 μmol gcat−1 s−1 H2 evolution and >95% CO conversion) in the solar-driven WGS process under light irradiation, surpassing noble-metal-based catalysts (Au/Al2O3 and Pt/Al2O3) in efficiency (Fig. 9b).86 This solar-driven WGS process incurred no electric or thermal power costs while achieving 1.1% light-to-energy storage. However, the presence of an oxygen vacancy at the interface between Cu and Cu2O in this catalyst limited reaction kinetics due to its singularity. To address this, CeO2 was introduced as a promoter to facilitate oxygen transport from the H2O reduction active site to that of CO, resulting in a low-CO-residual WGS reaction under H2-rich conditions at a reaction temperature as low as 225 °C.87 Photocarrier-enhanced photothermal catalysis over the CeO2-loaded Cu1.5Mn1.5O4 catalyst injects photocarriers into active sites and significantly reduces the apparent activation energy (decreased by 61%) (Fig. 9c).
However, it should be noted that photothermal catalysts for the WGS process exhibited inferior performance at temperatures below 250 °C when compared to thermal noble metal-based catalysts.91–95 By leveraging the LSPR effects in metal nanoparticle-based catalysts, such as Au, Ag and Cu, it becomes possible to significantly lower the operating temperature of the WGS reaction. The LSPR excitation generated hot electrons from copper nanoparticles, which were then transferred to adsorbates like water molecules, leading to enhanced activation of these adsorbates. This approach showed promise in overcoming the high energy barrier for water dissociation, resulting in improved WGS reaction activity under mild conditions. Zhao et al. synthesized a novel LDH-derived Cu-based catalyst (LD-Cu) for photo-driven WGS reactions, consisting of Cu nanoparticles dispersed densely on an amorphous alumina support (Fig. 9d).96 Under UV/vis irradiation, LD-Cu exhibited excellent H2 production activity at low temperatures (160–240 °C), significantly enhancing WGS reaction activity compared to tests conducted under dark conditions. Further experiments with monochromatic light sources revealed that the heightened H2 production activity observed under UV/vis irradiation can be attributed to the excitation of LSPR in Cu nanoparticles, resulting in the generation of hot electrons. In situ diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) and DFT calculations further revealed that the light-induced LSPR effect promoted water activation and hydrogen generation. This study showcased the efficient driving of the WGS reaction at very low temperatures using plasmonic Cu nanoparticle-based catalysts, offering new possibilities for designing low-cost photo-driven catalysts for WGS reactions and other catalytic reactions requiring water activation.
4.2 CO2 conversion
CO2 capture and hydrogenation offer a promising approach to mitigate the greenhouse effect while simultaneously producing valuable chemicals. In addition to the product of CO, there is also interest in targeting methane, methanol, and higher carbon products (C2+). However, the key challenge lies in developing suitable catalysts that can activate stable CO2 molecules with high CO bond energy while controlling the selectivity of the desired products.97
 | ||
| Fig. 10 (a) CO production rate, (b) selectivity, and (c) long-term stability of Ni12P5/SiO2 catalyst in the photothermal RWGS reaction.100 (d) Structure and mechanism of plasmon-induced carrier-assisted RWGS on Al@Cu2O.47 | ||
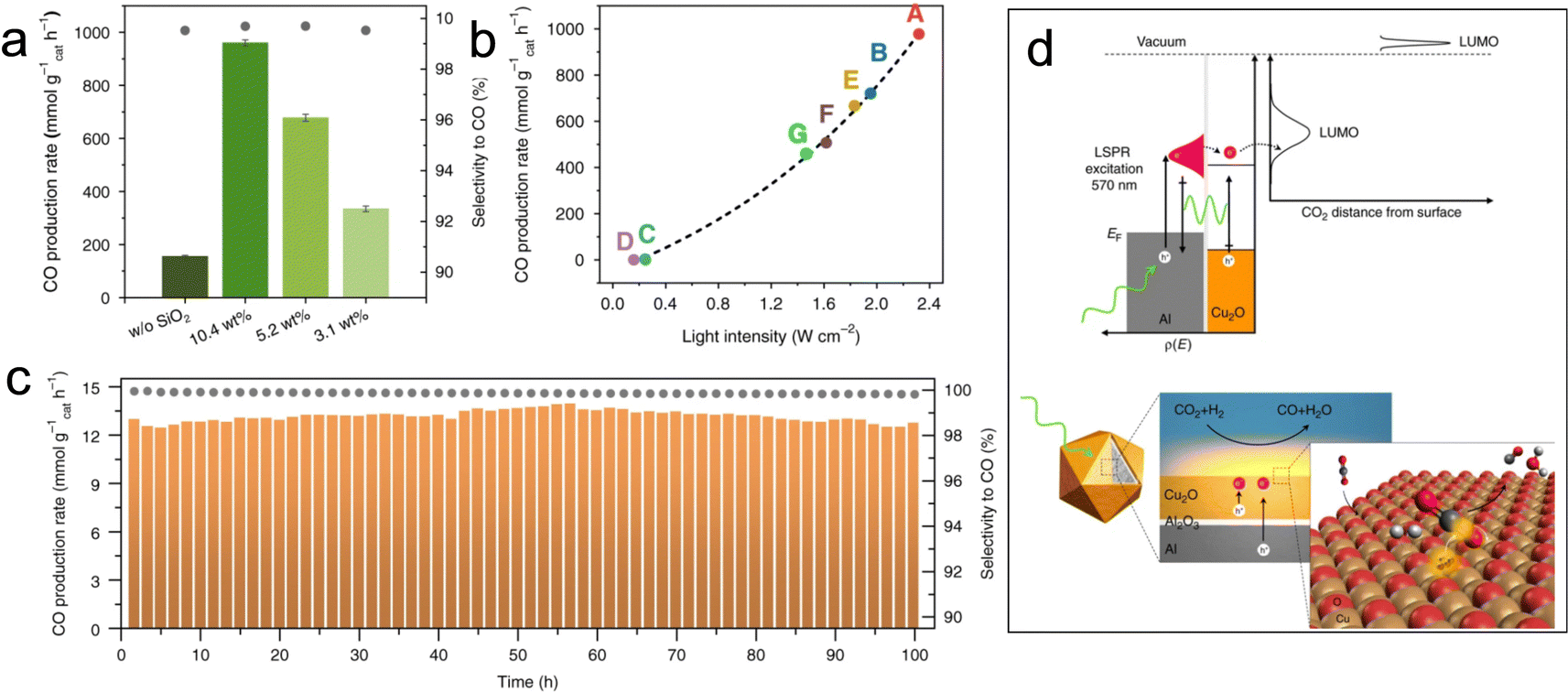
The photothermal catalysts dispersed on high surface-area supports, such as Si,101 Cu2O,47 CeO2,102 and Al2O3,103 also showed sufficient exposure of active sites and better photothermal conversion efficiency. For example, Robatjazi et al. presented novel plasmon-mediated heterogeneous catalysts utilizing earth-abundant Al embedded in Cu2O, which exhibit enhanced efficiency and selectivity for RWGS reaction under light illumination compared to conventional thermal conditions.47 The plasmon resonance of Al generates more energetic hot-carriers and enhances the optical absorption in Cu2O for selective conversion of CO2 to CO under visible light irradiation (Fig. 10d). This work also demonstrates the potential application of plasma photothermal catalysts in energy conversion.
The utilization of low-density carbon materials as supports can enhance the active mass loading and significantly improve the overall CO productivity.104 Very recently, Wang et al. presented a potassium-modified carbon-supported cobalt (K+–Co–C) catalyst that emulates the structure of a lotus pod.105 As a result of the designed lotus-pod structure, which incorporated an efficient photothermal C substrate with hierarchical pores, an intimate Co/C interface featuring covalent bonding, and exposed Co catalytic sites exhibiting optimized CO binding strength, the K+–Co–C catalyst exhibited exceptional performances in the photothermal RWGS reaction, achieving a record-breaking rate of 758 mmol gcat−1 h−1, accompanied by an exceptional selectivity for CO at 99.8%, surpassing typical photochemical CO2 reduction reactions by three orders of magnitude.
 | ||
| Fig. 11 (a) Preparation strategy for photothermal catalysts with varying degree of hydrogenation/carbonization.42 (b) Schematic illustration of the charge-carrier migration mechanism according to the type-II and Z-scheme heterojunction for WN–WO3.106 (c) (d) Schematic illustration of the flow reactor system and outdoor flow reactor system for photothermal CO2 methanation.108 (e) The proposed reaction pathways and B(OH)x surface frustrated Lewis pair reaction pathway for CO2 methanation.109 (f) and (g) Schematic of the nanoscale greenhouse effect in Ni@p-SiO2-30 and Tlocal of different catalysts under different illuminations.103 (h) and (i) Schematic of the new photothermal catalytic CO2 methanation system and the CO2 conversion rates of the catalysts.110 | ||
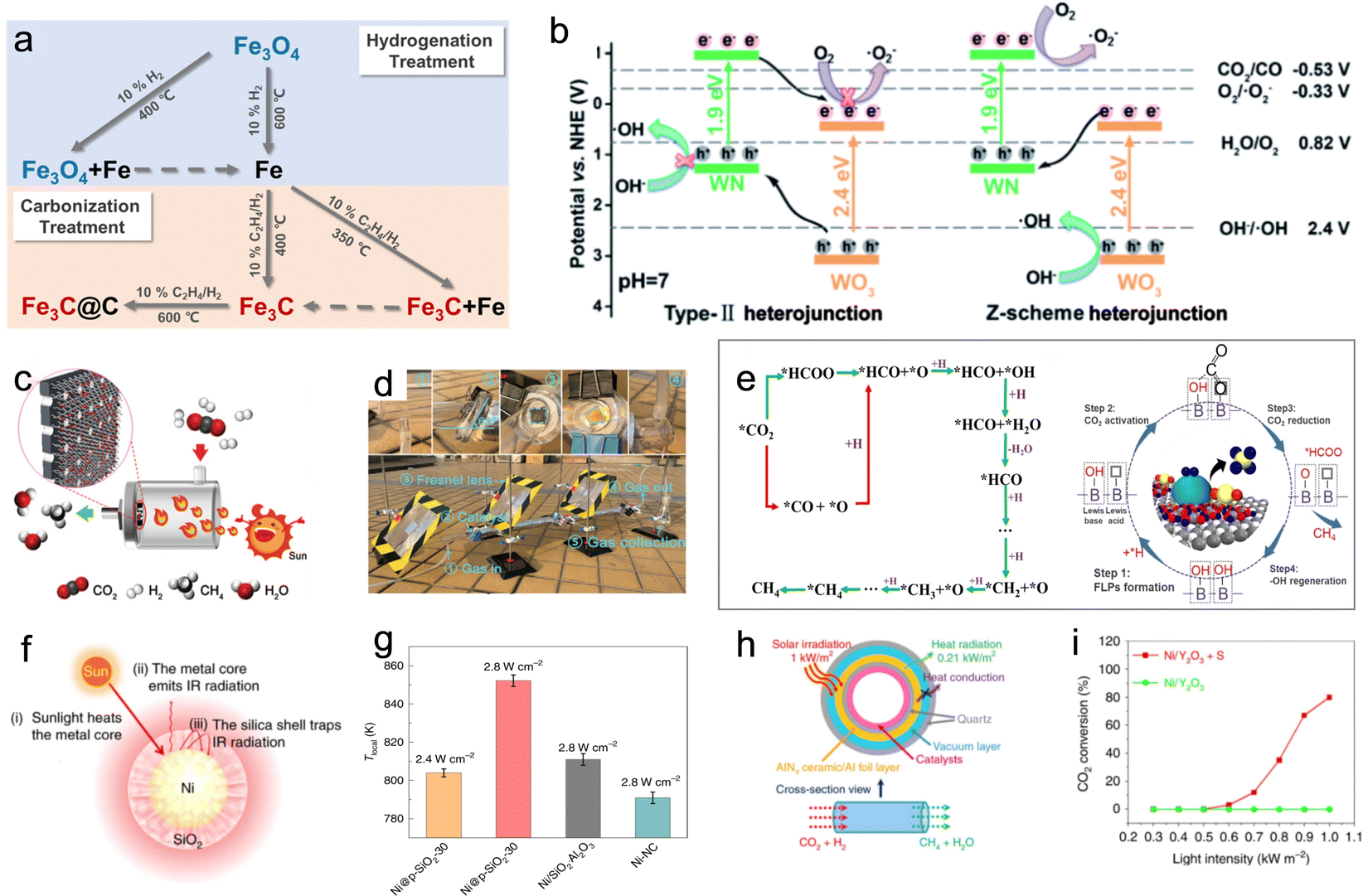
A recent development in the field involved a Pd-loaded WN-WO3 heterostructure, which acted as a direct Z-scheme catalyst, broadening the absorption spectrum of solar energy for the photothermal-assisted conversion of CO2 and H2O into CH4.106 The catalytic activity reached 40.6 ± 0.7 mmol h−1 g−1 for CH4 due to the unique carrier transfer mode of the Z-scheme catalyst. Under sunlight illumination, the WN-WO3 heterostructure can efficiently separate and migrate photoinduced carriers, resulting in more energetic hot electrons involved in the reaction (Fig. 11b). The local photothermal effect facilitated the migration of gas molecules and carriers, thereby enhancing CO2 conversion efficiency. Recent studies have also explored Ni nanoparticles supported on barium titanate perovskite for photothermal catalytic CO2 conversion, achieving high CH4 yields with nearly 100% selectivity. The dominant reactivity was attributed to a nonthermal hot-electron-driven pathway, aided by the local photothermal effect.107
To further advance photothermal catalytic CO2 hydrogenation, the effect of SMSI has also been investigated. For instance, Ru–Al2O3−x-L, which is rich in defects, was grown in situ on aluminum foil using an ultraviolet pulsed laser, and then ultra-small Ru nanoparticles were loaded in a controllable manner to prepare Ru–Al2O3−x-L, which achieved effective photothermal catalytic CO2 methanation.108 The willow fluff structured Al2O3−x-L can effectively capture light and ensure a high light adsorption capability of Ru–Al2O3−x-L. Defects in Al2O3−x-L serve as anchoring sites for Ru. The SMSI between the ultra-small Ru nanoparticles and Al2O3−x-L is established. An innovative flow reactor based on the one-piece Ru–Al2O3−x-L microchannel catalyst enhanced the CH4 yield to 14.04 mol gRu−1 h−1 due to local pressure on the edge of the microchannels (Fig. 11c). In addition, the feasibility of photothermal catalytic CO2 methanation (with a CH4 yield of 18.00 mmol min−1) was demonstrated through an outdoor setup (Fig. 11d). Zhu et al. reported a hexagonal boron nitride (h-BN) supported Ni particle (Ni/BN) as a photothermal catalyst for CO2 methanation, showing excellent CO2 conversion (87.68%), reaction rate (2.03 mol gNi−1 h−1) and CH4 production selectivity (near 100%) under ambient conditions.109 During the photothermal-assisted oxidation operation, the photothermal effect of Ni particles generated higher temperatures at the BN interface than in other regions. This caused slight oxidation of the h-BN surface defect, leading to the formation of B(OH)x species (Fig. 11e). The B atom in B(OH)x underwent a transformation in its bonding, creating a normal covalent bond after losing its π-conjugated structure. The electron-deficient B3+ site, due to its nature, accepted electrons from O in CO2, facilitated by the linear structure of the CO2 molecule. This created an electron-deficient B3+ center (Lewis acid site) and OH (Lewis base site), forming a frustrated Lewis acid–base pair (HOB⋯B). This pair provided the appropriate energy for CO2 adsorption and dissociation, serving as an activation site for photothermal catalytic methanation reactions. Furthermore, researchers have explored other exceptional photothermal supports, such as 2D MXenes. Wu et al. reported on Ni/Nb2C catalysts supported by MXenes, which exhibited outstanding photothermal effects and achieved a record-breaking CO2 conversion rate of 8.50 mol gNi−1 h−1 under 36 sun illumination, surpassing the reference of Ni/Nb2O5.28
In the realm of photothermal catalysis, the release of thermal radiation into the surrounding environment represents wasted heat energy. To address this issue, Cai et al. developed a catalyst composed of nickel nanocrystals encapsulated in nanoporous silica with 30 nm-thick shells, known as Ni@p-SiO2.103 This catalyst demonstrated superior photothermal catalytic efficiency for CO2 hydrogenation (Fig. 11f). When exposed to light illumination at 2.8 W cm−2, the Ni@p-SiO2 catalyst reached a maximum temperature of 334 °C, as measured using a contacting thermocouple. In comparison, the reference catalysts (Ni/SiO2·Al2O3 and Ni-NC) only achieved temperatures of 314 and 300 °C, respectively. These results highlight the enhanced photothermal catalytic efficiency conferred by the core–shell structure. As the thermocouple had limitations in precisely reflecting the local temperature (Tlocal) of the catalysts, the authors estimated Tlocal based on the gas composition under reaction equilibrium conditions. The findings revealed that Ni@p-SiO2 exhibited the highest Tlocal of 852 K, further confirming its potent photothermal effect (Fig. 11g). This supraphotothermal phenomenon originated from the greenhouse-like engineering of the SiO2 shell, which acted as both heat insulation and infrared shielding, minimizing heat loss from the illuminated Ni inner region to the external surroundings. Due to its supra-photothermal and spatial confinement effects, the Ni@p-SiO2 catalyst demonstrated improved photothermal catalytic performance and stability compared to Ni/SiO2·Al2O3 and Ni-NC, enabling its activity in both CO2 methanation and RWGS reactions.
In a different approach, Li et al. employed selective light absorbers to construct a photothermal system capable of generating high temperatures (up to 288 °C) under low solar exposure (1 kW m−2).110 This system exhibited three times higher efficiency than conventional photothermal catalytic systems (Fig. 11h). Moreover, ultra-thin amorphous Y2O3 nanosheets decorated with single nickel atoms (SA Ni/Y2O3) were synthesized and displayed superior activity in CO2 methanation. With the aid of a selective light absorber, SA Ni/Y2O3 achieved an CO2 conversion efficiency of 80% and a CH4 production rate of 7.5 L m−2 h−1 under solar irradiation ranging from 0.52 to 0.7 kW m−2 (Fig. 11i). These results demonstrate the potential of SA Ni/Y2O3 as a platform for the direct conversion of dispersed solar energy into valuable chemicals.
 | ||
| Fig. 12 (a) Illustration of selectivity in CO2 hydrogenation over different CoFe-x catalysts.46 (b) Coupling of thermocatalysis and photocatalysis in CO2 hydrogenation over Au–Ru/TiO2.116 (c) Potential-energy profile of possible pathways for CO2 conversion at active sites.117 (d) C–C coupling mechanism of Co0 and Co0–Coδ+ over Co–CoOx/MgAl2O4 by theoretical calculations.118 | ||
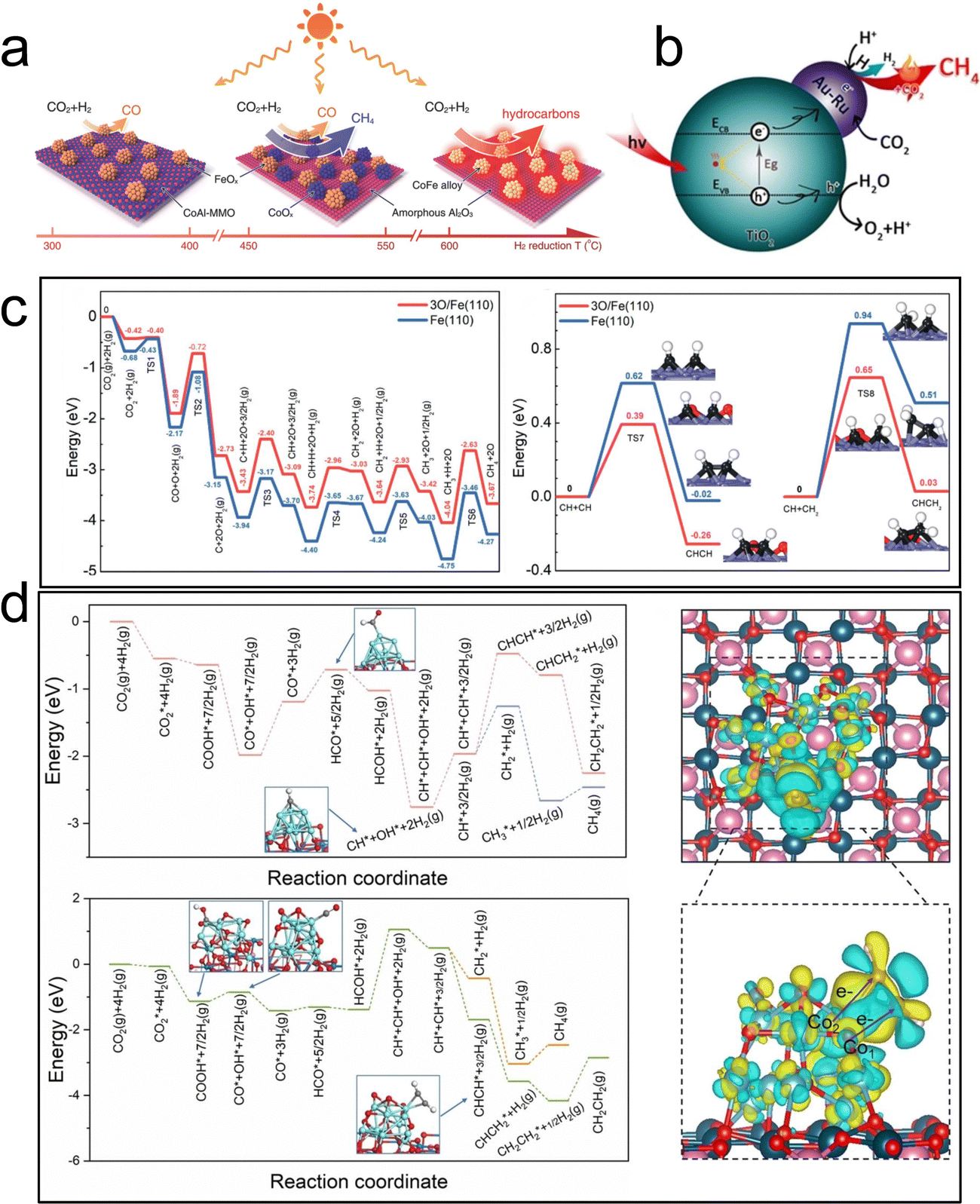
Recently, a Fe/FeOx heterogeneous structure supported on MgO–Al2O3 mixed metal oxide, derived from hydrogen reduction of the MgFeAl–LDH precursor, has also been employed for photothermal catalytic CO2 hydrogenation.117 The catalyst exhibited a surprising C2+ selectivity of 52.9% at a CO2 conversion rate of 50.1% under UV-visible light irradiation. The coexistence of Fe and FeOx was found to be favorable for promoting the C–C coupling reaction, thereby enhancing the selectivity of C2+ products (Fig. 12c). Notably, there was no discernible difference in performance between photothermal catalysis and thermal catalysis processes for CO2 hydrogenation to C2+.
In another work, Ning et al. prepared a Co0–Coδ+ structure catalyst through in situ photothermal activation. Co–CoOx/MgAl2O4 achieved a C2–4 yield of 1303 μmol g−1 h−1 under light irradiation.118 The selectivity of C2–4 total organic carbon reached 62.5%, and the ratio of olefin to alkane reached approximately 11. Experimental and theoretical results confirmed that the two active sites at the interface effectively adsorbed and activated CO2 to form C1 intermediates through the Co0 site. The introduction of the Coδ+ electron-deficient state effectively reduced the reaction energy of the key CHCH* intermediate, thus promoting the formation of low carbon olefin C2+ hydrocarbons (Fig. 12d). This study offers a novel approach to designing photothermal catalysts for the conversion of CO2 to C2+ hydrocarbons through photoreduction.
4.3 Methane conversion
Methane, a colorless and odorless gas, serves as a vital building block in the chemical industry. It is primarily known for being the main component of natural gas and methane clathrates, which are found deep beneath the ocean floor. This versatile hydrocarbon feedstock plays a crucial role in the synthesis of fuels and other essential chemicals. The commercial production of H2 heavily relies on catalytic steam reforming technology, along with partial oxidation and coal gasification methods. Therefore, it is evident that the conversion of CH4 holds immense practical significance for the chemical industry.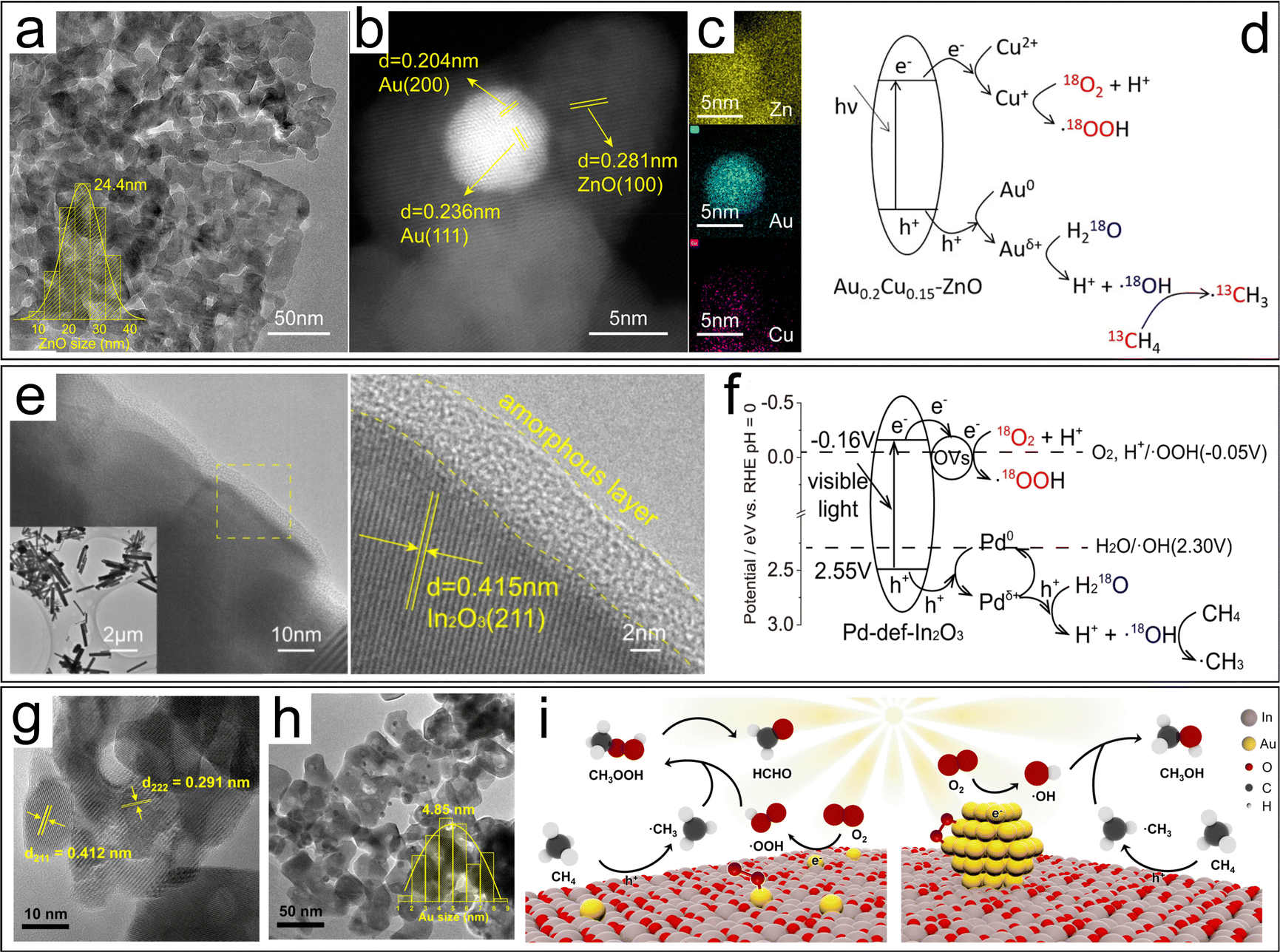 | ||
| Fig. 13 (a) TEM image of ZnO. (b) HAADF-STEM and (c) EDS-mapping images of Au0.2Cu0.15–ZnO. (d) Methane conversion mechanism of AuCu–ZnO.120 (e) HRTEM images and (f) CH4 conversion mechanism of Pd-def-In2O3.121 TEM images of (g) Au1/In2O3 and (h) AuNPs/In2O3. (i) Proposed mechanism of photocatalytic conversion of CH4 to HCHO or CH3OH on Au1/In2O3 and AuNPs/In2O3, respectively.122 | ||
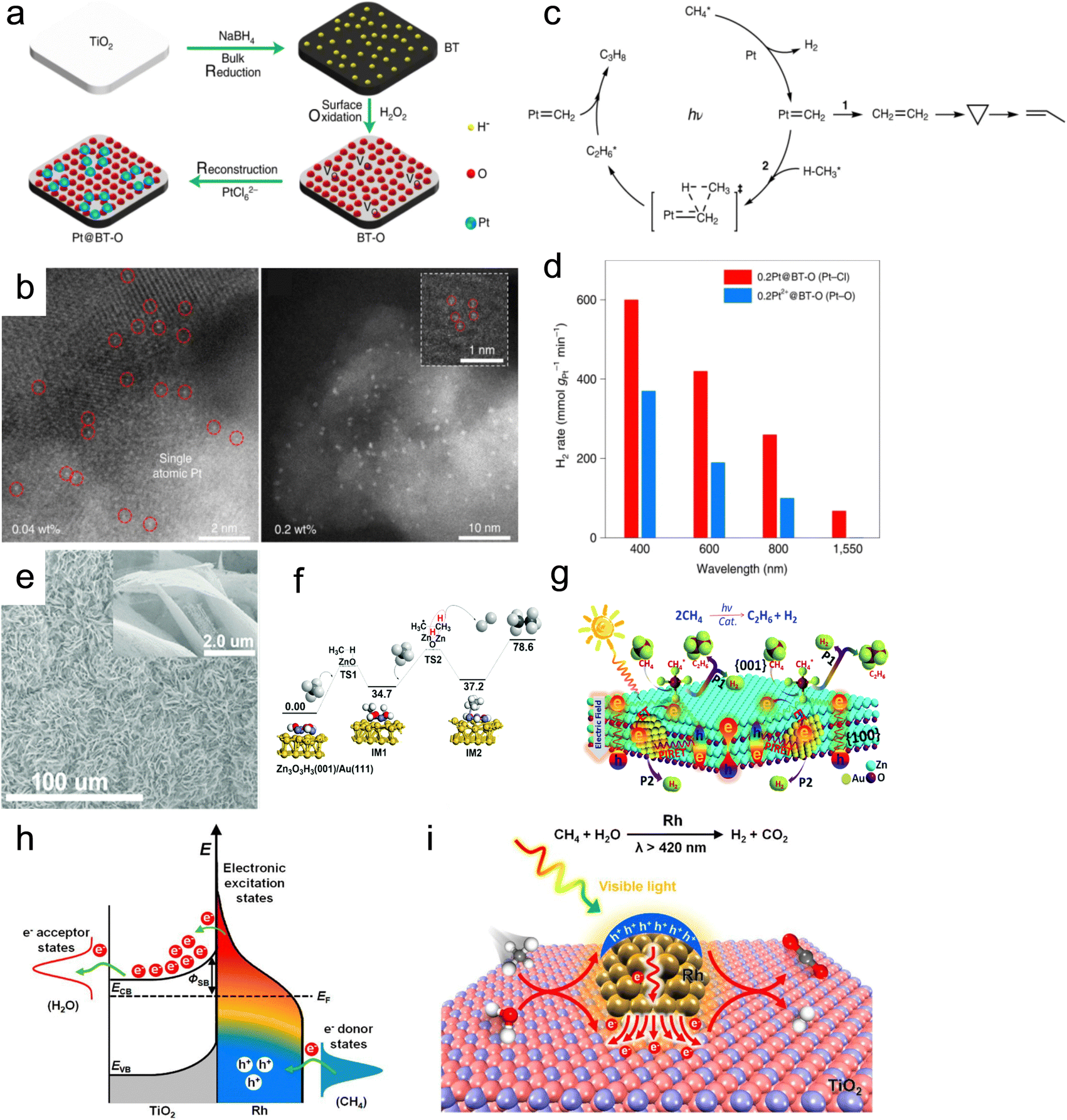 | ||
| Fig. 14 (a) Synthesis and (b) characterization of the Pt@BT-O catalyst. (c) Proposed mechanism and (d) catalytic activity of catalysts for cyclohexane dehydrogenation.123 (e) SEM image of the Au/ZnO photothermal catalyst. (f) Potential energy diagram for the methane coupling reaction on Zn3O3H3(001)/Au(111) clusters. (g) Schematic description for the dehydrogenative coupling of methane into ethane over Au/ZnO.124 (h) Schematic of energy transfer from photoexcited hot carriers to adsorbate states. (i) Mechanism description for the SRM reaction on Rh/TiO2 under visible light illumination.125 | ||
Furthermore, the DRM process was hindered by a H2/CO ratio of less than 1.0, primarily due to the simultaneous occurrence of the RWGS reaction. This reaction is not conducive to the formation of C5–20 liquid fuels via FTS. Therefore, the regulation of the reaction paths to promote H2 formation and suppress the RWGS reaction is a tremendous challenge. Zhou's group synthesized the Ni/Ga2O3 catalyst using a facile impregnation method.129 The direction of electron transfer from the Ga2O3 support to Ni can be reversed under light irradiation, resulting in the formation of Ni0 active sites. This generated abundant hot electrons through electronic interband transition of Ni, which enhanced H2 formation and desorption. As a result, the H2/CO ratio increased from 0.55 to 0.94 (Fig. 15a). Through in situ DRIFTS technology, it was discovered that light can promote the cracking of methane and accelerate the further cracking of methyl groups. Additionally, compared to thermal catalysis, photothermal catalysis resulted in a more significant reduction of methyl groups. Therefore, the introduction of light not only promoted methane pyrolysis but also facilitated continuous methyl group cracking. This study confirmed the non-thermal effects of light irradiation and the crucial role that light played in inducing directional electron migration.
 | ||
| Fig. 15 (a) Schematic and DRM performance of the Ni/Ga2O3 catalyst.129 (b) Schematic of enhanced selectivity and stability in Cu–Ru surface alloys.130 (c) Oxygen vacancy formation energy of the Pt/CeO2 catalyst.131 (d) Schematic diagram for the light-driven DRM reaction over the Rh/CexWO3 catalyst.3 | ||
In recent times, the integration of plasmonic ‘antenna’ metal nanoparticles with catalytically active ‘reactor’ materials has emerged as a remarkable strategy to enhance the efficiency, selectivity, and specificity of photocatalytic reactions. This innovative configuration harnesses the local optical field generated by the antenna to effectively convert the catalyst into a highly efficient photocatalyst. Zhou et al. synthesized a series of photothermal catalysts with different Ru loadings on the MgO–Al2O3 composite support by a coprecipitate method, and the impact of Ru concentration on the photothermal catalytic performance of Cu–Ru surface alloys was investigated, revealing that isolated Ru atoms on the Cu surfaces exhibited excellent DRM activity (Fig. 15b).130 Through temperature programmed oxidation and thermal catalysis of isotopic reactants, it was determined that methane was the sole source of coke production. The authors speculated that CH4 and CO2 molecules first dissociated on the catalyst surface to form Hads, Cads, Oads and COads, respectively. The desorption of H2 was accelerated by the hot carrier through the electron transition mechanism, which inhibited the reaction between Hads and Oads, leading to high photocatalytic selectivity. This inhibition can effectively promote the removal of carbon and improve the stability of the catalyst. In addition, Hads was more likely to react with Oads in the thermal catalytic process, and the lower decarbonization efficiency of Oads led to lower selectivity and stability of the thermal catalytic reaction. The excellent photothermal catalytic performances with long term stability (50 h) and high selectivity (>99%) can be attributed to the light-excited hot carriers and single-atom active sites.
The construction of interfaces between single atoms and oxide catalysts with a synergistic effect has been proven to be effective in improving atomic utilization, reducing reaction energy consumption, and enhancing catalytic activity. For instance, the DRM reaction was catalyzed by a Pt monatomic catalyst supported by CeO2 (Pt/CeO2) with different morphologies at low temperature (300–450 °C).131 Experimental and theoretical results demonstrated that rod-like CeO2-R with abundant defects provided better anchoring for Pt monatoms, while Pt doping induced surface reconfiguration of CeO2, significantly reducing the oxygen vacancy formation energy of CeO2-R. The Pt–O lattice structure at the interface facilitated CH4 oxygen-assisted dissociation, leading to CO formation through CHxO intermediates. CO2 was adsorbed and dissociated at the Pt–CeO2 interface, generating CO and O lattice species to complete the catalytic cycle (Fig. 15c). Similar photoinduced redox cycles can occur in other heteronuclear bimetallic photocatalysts with oxygen bridging bonds. Yang et al. reported a highly active and coke-resistant DRM photocatalyst consisting of 3 nm Rh nanoparticles uniformly dispersed on CexWO3 nanosheets.3 This catalyst utilized full-spectrum sunlight without the need for an external heat source and achieved a light-to-chemical energy efficiency of 4.65% under mild conditions. Metal-to-metal charge transfer induced by light facilitated the redox cycle, improving the catalyst's reaction activity and stability. Rapid oxygen migration assisted in the removal of carbon deposits and reduced the reaction start-up temperature. The authors attributed the catalyst's high performance, including H2 and CO evolution rates of 88.5 and 152.3 mmol gRh−1 h−1, respectively, to the strong CO2 absorption on the CexWO3 surface, the high mobility of oxygen and the electron transfer from Ce to W species (Fig. 15d). This construction of a heteronuclear bimetallic photocatalyst with oxo-bridge bonds provides valuable insights for the design of low-temperature DRM catalysts.
5. Summary and outlook
In summary, photothermal catalytic C1 chemistry offers a cleaner pathway for fuel synthesis using solar energy, which is particularly crucial in the face of climate change and the need for energy structure transformation. Supported catalysts with excellent optical properties and high specific surface area offer an ideal platform for photothermal catalysis. In this paper, we review the mechanism of photothermal catalysis, the design strategy for supported photothermal catalysts, and recent research progress in photothermal catalysis for C1 chemistry, including CO conversion (Fischer–Tropsch synthesis and water-gas-shift reaction), CO2 conversion (reverse water-gas-shift reaction, methanation, methanol and higher hydrocarbon production), and methane conversion (oxidation, non-oxidative coupling, steam reforming, and dry reforming). Although significant progress has been made in the development of supported photothermal catalysts with excellent optical properties and high specific surface area, the field of photothermal catalysis is still in its early stages. Several challenges and prospects for future work can be identified:(1) In order to further improve the performance of photothermal materials, it is still challenging to design materials with extremely high light absorption over a wide wavelength range, photothermal conversion ability, catalytic ability, and material stability. Advancements in this area are crucial to enable the cost-effective mass production of photothermal materials for everyday life and industrial applications.
(2) Distinguishing the differences between photocatalysis and thermal catalysis pathways in the context of photothermal processes is essential. Current temperature measurement methods, such as thermocouples or infrared cameras, provide macroscopic measurements but do not accurately reflect the local temperature of nanoparticles or fully capture the photothermal catalytic mechanism. Exploring new thermometry technologies, such as scanning thermal microscopy (SThM) and non-contact measurement techniques based on Raman spectroscopy/in situ infrared spectroscopy, is necessary to achieve high sensitivity, accuracy, spectral/temporal resolution, reproducibility, stability, biocompatibility, cost-effectiveness, and ease of operation.
(3) By harnessing the power of light and heat, we can transform harmful gases like CO2 and CH4 into valuable polycarbon alkanes/olefins and other high-value chemicals. But this technology has even greater potential than just reducing emissions – it can also help us tackle industrial waste gases that would otherwise go unused. With further research and development, we can unlock a whole new world of possibilities for creating sustainable, profitable products from what was once considered waste.
Author contributions
Hui Liu: writing – original draft and investigation. Liangliang Han: investigation and writing – review and editing. Xiaoguang Duan: investigation. Hongqi Sun: supervision. Shaobin Wang: writing – review and editing and supervision. Jinqiang Zhang: writing – review and editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Young Scholars Research Fund of Yantai University (HY22B73) and the National Natural Science Foundation of China (51873227).References
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6, 2497–2514 CAS
.
- J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2019, 9, 3026–3053 CrossRef CAS
- Y. Yang, Z. Chai, X. Qin, Z. Zhang, A. Muhetaer, C. Wang, H. Huang, C. Yang, D. Ma, Q. Li and D. Xu, Angew. Chem., Int. Ed., 2022, 61, e202200567 CrossRef CAS PubMed
- Q. Li, H. Wang, M. Zhang, G. Li, J. Chen and H. Jia, Angew. Chem., Int. Ed., 2023, 62, e202300129 CrossRef CAS PubMed
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC
- S. Du, X. Bian, Y. Zhao, R. Shi and T. Zhang, Chem. Res. Chin. Univ., 2022, 38, 723–734 CrossRef CAS
- H. W. Lin, S. N. Luo, H. B. Zhang and J. H. Ye, Joule, 2022, 6, 294–314 CrossRef CAS
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC
- M. Fan, L. Cui, X. He and X. Zou, Small Methods, 2022, 6, e2200855 CrossRef PubMed
- Y. Yang, S. Zhao, L. Cui, F. Bi, Y. Zhang, N. Liu, Y. Wang, F. Liu, C. He and X. Zhang, Green Energy Environ., 2023, 8, 654–672 CrossRef CAS
- Y. Y. Li, Y. S. Zhang, K. Qian and W. X. Huang, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- S. Q. Luo, X. H. Ren, H. W. Lin, H. Song and J. H. Ye, Chem. Sci., 2021, 12, 5701–5719 RSC
- X. M. Zhang, Y. L. Chen, R. S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS PubMed
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52–83 CrossRef CAS
- C. Choi, F. Zhao, J. L. Hart, Y. Gao, F. Menges, C. L. Rooney, N. J. Harmon, B. Shang, Z. Xu, S. Suo, Q. Sam, J. J. Cha, T. Lian and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302152 CrossRef CAS PubMed
- B. Han, Y. L. Zhang, Q. D. Chen and H. B. Sun, Adv. Funct. Mater., 2018, 28, 1802235 CrossRef
- D. Meng, S. Yang, L. Guo, G. Li, J. Ge, Y. Huang, C. W. Bielawski and J. Geng, Chem. Commun., 2014, 50, 14345–14348 RSC
- G. K. Lim, Z. L. Chen, J. Clark, R. G. Goh, W. H. Ng, H. W. Tan, R. H. Friend, P. K. Ho and L. L. Chua, Nat. Photonics, 2011, 5, 554–560 CrossRef CAS
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed
- J. Deng, J. Li, P. Chen, X. Fang, X. Sun, Y. Jiang, W. Weng, B. Wang and H. Peng, J. Am. Chem. Soc., 2016, 138, 225–230 CrossRef CAS PubMed
- X. Huang, Q. Qian, X. Zhang, W. Du, H. Xu and Y. Wang, Part. Part. Syst. Charact., 2013, 30, 235–240 CrossRef CAS
- C. Maggi, F. Saglimbeni, M. Dipalo, F. De Angelis and R. Di Leonardo, Nat. Commun., 2015, 6, 7855 CrossRef CAS PubMed
- C. X. Guo, X. J. Ma and B. Wang, Acta Chim. Sin., 2021, 79, 967–985 CrossRef CAS
- Z. Wu, C. Li, Z. Li, K. Feng, M. Cai, D. Zhang, S. Wang, M. Chu, C. Zhang, J. Shen, Z. Huang, Y. Xiao, G. A. Ozin, X. Zhang and L. He, ACS Nano, 2021, 15, 5696–5705 CrossRef CAS PubMed
- W. Feng, X. Han, R. Wang, X. Gao, P. Hu, W. Yue, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1805919 CrossRef PubMed
- K. Hantanasirisakul and Y. Gogotsi, Adv. Mater., 2018, 30, 1804779 CrossRef PubMed
- M. M. Gao, T. X. Zhang and G. W. Ho, Nano Res., 2022, 15, 9985–10005 CrossRef CAS
- F. Wang, C. Li, H. Chen, R. Jiang, L. D. Sun, Q. Li, J. Wang, J. C. Yu and C. H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed
- J. T. Li, S. K. Cushing, F. K. Meng, T. R. Senty, A. D. Bristow and N. Q. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed
- H. An, M. Li, R. Liu, Z. Gao and Z. Yin, Chem. Eng. J., 2020, 382, 122953 CrossRef CAS
- P. G. O'Brien, A. Sandhel, T. E. Wood, A. A. Jelle, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400001 CrossRef PubMed
- H. Liu, X. Meng, T. D. Dao, H. Zhang, P. Li, K. Chang, T. Wang, M. Li, T. Nagao and J. Ye, Angew. Chem., 2015, 127, 11707–11711 CrossRef
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed
- Z. Yang, T. Zhao, Y. Tang, Y. Jiang, H. Kitagawa, X. Wen and F. Wang, J. Catal., 2023, 424, 22–28 CrossRef CAS
- Y. Guo, Y. Huang, B. Zeng, B. Han, M. Akri, M. Shi, Y. Zhao, Q. Li, Y. Su, L. Li, Q. Jiang, Y. T. Cui, L. Li, R. Li, B. Qiao and T. Zhang, Nat. Commun., 2022, 13, 2648 CrossRef CAS PubMed
- Y. Zhao, Y. Wei, X. Wu, H. Zheng, Z. Zhao, J. Liu and J. Li, Appl. Catal., B, 2018, 226, 360–372 CrossRef CAS
- C. Song, X. Liu, M. Xu, D. Masi, Y. Wang, Y. Deng, M. Zhang, X. Qin, K. Feng, J. Yan, J. Leng, Z. Wang, Y. Xu, B. Yan, S. Jin, D. Xu, Z. Yin, D. Xiao and D. Ma, ACS Catal., 2020, 10, 10364–10374 CrossRef CAS
- T. Fujita, K. Higuchi, Y. Yamamoto, T. Tokunaga, S. Arai and H. Abe, Metals, 2017, 7, 406 CrossRef
- X. Liu, H. Shi, X. Meng, C. Sun, K. Zhang, L. Gao, Y. Ma, Z. Mu, Y. Ling and B. Cheng, Sol. RRL, 2021, 5, 2100185 CrossRef CAS
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim and S.-P. Cho, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS PubMed
- Z. Li, R. Yu, J. Huang, Y. Shi, D. Zhang, X. Zhong, D. Wang, Y. Wu and Y. Li, Nat. Commun., 2015, 6, 8248 CrossRef CAS PubMed
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed
- Z. Zhu, X. Hu, X. An, M. Xiao, L. Zhang, C. Li and L. He, Chem. – Asian J., 2022, 17, e202200993 CrossRef CAS PubMed
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef PubMed
- M. N. Ha, G. Lu, Z. Liu, L. Wang and Z. Zhao, J. Mater. Chem. A, 2016, 4, 13155–13165 RSC
- L. Wang, Y. Wang, Y. Cheng, Z. Liu, Q. Guo, M. N. Ha and Z. Zhao, J. Mater. Chem. A, 2016, 4, 5314–5322 RSC
- Y. Li, M. Wen, Y. Wang, G. Tian, C. Wang and J. Zhao, Angew. Chem., Int. Ed., 2021, 60, 910–916 CrossRef CAS PubMed
- Y. Yang, S. Zhao, F. Bi, J. Chen, Y. Wang, L. Cui, J. Xu and X. Zhang, Appl. Catal., B, 2022, 315, 121550 CrossRef CAS
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He and Q. Li, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed
- Z. Shi, L. Lan, Y. Li, Y. Yang, Q. Zhang, J. Wu, G. Zhang and X. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 16503–16514 CrossRef CAS
- W. Han, Y. Chen, Y. Jiao, S. Liang, W. Li and G. Tian, J. Mater. Chem. A, 2022, 10, 17642–17651 RSC
- K. Yan, D. Wu, T. Wang, C. Chen, S. Liu, Y. Hu, C. Gao, H. Chen and B. Li, ACS Catal., 2023, 13, 2302–2312 CrossRef CAS
- Y. Wang, M. Liu, C. Wu, J. Gao, M. Li, Z. Xing, Z. Li and W. Zhou, Small, 2022, 18, e2202544 CrossRef PubMed
- X. M. Gao, H. B. He, W. Zhu, C. M. Yang, K. X. Xu, B. B. Feng, Y. A. Hu and F. Fu, Small, 2023, 19, 2206225 CrossRef CAS PubMed
- J. U. Kim, S. Lee, S. J. Kang and T. I. Kim, Nanoscale, 2018, 10, 21555–21574 RSC
- H. Ren, M. Tang, B. Guan, K. Wang, J. Yang, F. Wang, M. Wang, J. Shan, Z. Chen, D. Wei, H. Peng and Z. Liu, Adv. Mater., 2017, 29, 1702590 CrossRef PubMed
- Y. Ito, Y. Tanabe, J. Han, T. Fujita, K. Tanigaki and M. Chen, Adv. Mater., 2015, 27, 4302–4307 CrossRef CAS PubMed
- Z. Liu, L. Niu, X. Zong, L. An, D. Qu, X. Wang and Z. Sun, Appl. Catal., B, 2022, 313, 121439 CrossRef CAS
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202304452 CrossRef
- Z. Wu, J. Shen, C. Li, C. Zhang, K. Feng, Z. Wang, X. Wang, D. M. Meira, M. Cai, D. Zhang, S. Wang, M. Chu, J. Chen, Y. Xi, L. Zhang, T. K. Sham, A. Genest, G. Rupprechter, X. Zhang and L. He, ACS Nano, 2022, 17, 1550–1559 CrossRef PubMed
- Q. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed
- W. Zhang, L. Wang, K. Wang, M. U. Khan, M. Wang, H. Li and J. Zeng, Small, 2017, 13, 1602583 CrossRef PubMed
- H. Chen, L. Shao, C. Wang and Y. Fu, J. Mater. Chem. A, 2022, 10, 17434–17439 RSC
- X. Feng, D. Liu, B. Yan, M. Shao, Z. Hao, G. Yuan, H. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2021, 133, 18700–18704 CrossRef
- Q. Chen, Z. Pei, Y. Xu, Z. Li, Y. Yang, Y. Wei and Y. Ji, Chem. Sci., 2018, 9, 623–628 RSC
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS
- R. Li, Y. Li, Z. Li, W. Wei, Q. Hao, Y. Shi, S. Ouyang, H. Yuan and T. Zhang, ACS Catal., 2022, 12, 5316–5326 CrossRef CAS
- Y. Wang, Y. Zhao, J. Liu, Z. Li, G. I. N. Waterhouse, R. Shi, X. Wen and T. Zhang, Adv. Energy Mater., 2019, 10, 1902860 CrossRef
- Y. Zhao, Z. Li, M. Li, J. Liu, X. Liu, G. I. N. Waterhouse, Y. Wang, J. Zhao, W. Gao, Z. Zhang, R. Long, Q. Zhang, L. Gu, X. Liu, X. Wen, D. Ma, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1803127 CrossRef PubMed
- Z. Li, J. Liu, Y. Zhao, G. I. N. Waterhouse, G. Chen, R. Shi, X. Zhang, X. Liu, Y. Wei, X. D. Wen, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1800527 CrossRef PubMed
- Z. Li, X. Zhang, J. Liu, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. Zhang, Adv. Mater., 2021, 33, e2103248 CrossRef PubMed
- J. Zhao, J. Liu, Z. Li, K. Wang, R. Shi, P. Wang, Q. Wang, G. I. N. Waterhouse, X. Wen and T. Zhang, Nat. Commun., 2023, 14, 1909 CrossRef CAS PubMed
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Perez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed
- J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Perez, Angew. Chem., Int. Ed., 2007, 46, 1329–1332 CrossRef CAS PubMed
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y. W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CrossRef CAS PubMed
- L. Millard and M. Bowker, J. Photochem. Photobiol., A, 2002, 148, 91–95 CrossRef CAS
- Z. Zhang, S. S. Wang, R. Song, T. Cao, L. Luo, X. Chen, Y. Gao, J. Lu, W. X. Li and W. Huang, Nat. Commun., 2017, 8, 488 CrossRef PubMed
- L. Zhao, Y. Qi, L. Song, S. Ning, S. Ouyang, H. Xu and J. Ye, Angew. Chem., Int. Ed., 2019, 131, 7790–7794 CrossRef
- Y. Tong, L. Song, S. Ning, S. Ouyang and J. Ye, Appl. Catal., B, 2021, 298, 120551 CrossRef CAS
- S. M. Fang, B. H. Chen and J. White, J. Phys. Chem., 1982, 86, 3126–3130 CrossRef CAS
- F. Liu, L. Song, S. Ouyang and H. Xu, Catal. Sci. Technol., 2019, 9, 2125–2131 RSC
- C. Shi, D. Yuan, L. Ma, Y. Li, Y. Lu, L. Gao, X. San, S. Wang and G. Fu, J. Mater. Chem. A, 2020, 8, 19467–19472 RSC
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu and B. Chen, Science, 2017, 357, 389–393 CrossRef CAS PubMed
- N. Liu, M. Xu, Y. Yang, S. Zhang, J. Zhang, W. Wang, L. Zheng, S. Hong and M. Wei, ACS Catal., 2019, 9, 2707–2717 CrossRef CAS
- L. Sun, J. Xu, X. Liu, B. Qiao, L. Li, Y. Ren, Q. Wan, J. Lin, S. Lin, X. Wang, H. Guo and T. Zhang, ACS Catal., 2021, 11, 5942–5950 CrossRef CAS
- N. Yi, R. Si, H. Saltsburg and M. Flytzani-Stephanopoulos, Energy Environ. Sci., 2010, 3, 831–837 RSC
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed
- J. Zhao, Y. Bai, Z. Li, J. Liu, W. Wang, P. Wang, B. Yang, R. Shi, G. I. Waterhouse and X. D. Wen, Angew. Chem., Int. Ed., 2023, 62, e202219299 CrossRef CAS PubMed
- J. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Nano Energy, 2022, 102, 107650 CrossRef CAS
- J. Yu, A. Muhetaer, X. Gao, Z. Zhang, Y. Yang, Q. Li, L. Chen, H. Liu and D. Xu, Angew. Chem., Int. Ed., 2023, 62, e202303135 CrossRef CAS PubMed
- M. Gonzalez-Castano, B. Dorneanu and H. Arellano-Garcia, React. Chem. Eng., 2021, 6, 954–976 RSC
- Y. F. Xu, P. N. Duchesne, L. Wang, A. Tavasoli, A. A. Jelle, M. Xia, J. F. Liao, D. B. Kuang and G. A. Ozin, Nat. Commun., 2020, 11, 5149 CrossRef CAS PubMed
- X. Shen, C. Li, Z. Wu, R. Tang, J. Shen, M. Chu, A.-B. Xu, B. Zhang, L. He and X. Zhang, Nanoscale, 2022, 14, 11568–11574 RSC
- B. Deng, H. Song, K. Peng, Q. Li and J. Ye, Appl. Catal., B, 2021, 298, 120519 CrossRef CAS
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng, A.-B. Xu, S. Tang, A. Tavasoli, M. Peng, W. Liu, A. S. Helmy, L. He, G. A. Ozin and X. Zhang, Nat. Energy, 2021, 6, 807–814 CrossRef CAS
- S. Ning, H. Xu, Y. Qi, L. Song, Q. Zhang, S. Ouyang and J. Ye, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS
- H. Wang, S. Fu, B. Shang, S. Jeon, Y. Zhong, N. J. Harmon, C. Choi, E. A. Stach and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305251 CrossRef CAS PubMed
- D. Liu, Y. Xu, M. Sun, Y. Huang, Y. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 1077–1083 RSC
- D. Mateo, N. Morlanes, P. Maity, G. Shterk, O. F. Mohammed and J. Gascon, Adv. Funct. Mater., 2020, 31, 2008244 CrossRef
- X. Liu, C. Xing, F. Yang, Z. Liu, Y. Wang, T. Dong, L. Zhao, H. Liu and W. Zhou, Adv. Energy Mater., 2022, 12, 2201009 CrossRef CAS
- X. Zhu, H. Zong, C. J. V. Perez, H. Miao, W. Sun, Z. Yuan, S. Wang, G. Zeng, H. Xu, Z. Jiang and G. A. Ozin, Angew. Chem., Int. Ed., 2023, 62, e202218694 CrossRef CAS PubMed
- Y. Li, J. Hao, H. Song, F. Zhang, X. Bai, X. Meng, H. Zhang, S. Wang, Y. Hu and J. Ye, Nat. Commun., 2019, 10, 2359 CrossRef PubMed
- A. Iglesias-Juez and J. M. Coronado, Chem, 2018, 4, 1490–1491 CAS
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed
- B. Xie, R. J. Wong, T. H. Tan, M. Higham, E. K. Gibson, D. Decarolis, J. Callison, K. F. Aguey-Zinsou, M. Bowker, C. R. A. Catlow, J. Scott and R. Amal, Nat. Commun., 2020, 11, 1615 CrossRef CAS PubMed
- B. Xie, P. Kumar, T. H. Tan, A. A. Esmailpour, K.-F. Aguey-Zinsou, J. Scott and R. Amal, ACS Catal., 2021, 11, 5818–5828 CrossRef CAS
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed
- L. Zhang, G. Kong, Y. Meng, J. Tian, L. Zhang, S. Wan, J. Lin and Y. Wang, ChemSusChem, 2017, 10, 4709–4714 CrossRef CAS PubMed
- Z. Li, J. Liu, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. Zhang, Adv. Energy Mater., 2021, 11, 2002783 CrossRef CAS
- S. Ning, H. Ou, Y. Li, C. Lv, S. Wang, D. Wang and J. Ye, Angew. Chem., Int. Ed., 2023, 135, e202302253 CrossRef
- A. Caballero and P. J. Perez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC
- L. Luo, Z. Gong, Y. Xu, J. Ma, H. Liu, J. Xing and J. Tang, J. Am. Chem. Soc., 2022, 144, 740–750 CrossRef CAS PubMed
- L. Luo, L. Fu, H. Liu, Y. Xu, J. Xing, C. R. Chang, D. Y. Yang and J. Tang, Nat. Commun., 2022, 13, 2930 CrossRef CAS PubMed
- Y. Jiang, S. Li, S. Wang, Y. Zhang, C. Long, J. Xie, X. Fan, W. Zhao, P. Xu, Y. Fan, C. Cui and Z. Tang, J. Am. Chem. Soc., 2023, 145, 2698–2707 CrossRef CAS PubMed
- L. Zhang, L. Liu, Z. Pan, R. Zhang, Z. Gao, G. Wang, K. Huang, X. Mu, F. Bai, Y. Wang, W. Zhang, Z. Cui and L. Li, Nat. Energy, 2022, 7, 1042–1051 CrossRef CAS
- L. Meng, Z. Chen, Z. Ma, S. He, Y. Hou, H.-H. Li, R. Yuan, X.-H. Huang, X. Wang, X. Wang and J. Long, Energy Environ. Sci., 2018, 11, 294–298 RSC
- H. Song, X. Meng, Z.-J. Wang, Z. Wang, H. Chen, Y. Weng, F. Ichihara, M. Oshikiri, T. Kako and J. Ye, ACS Catal., 2018, 8, 7556–7565 CrossRef CAS
- B. Han, W. Wei, M. Li, K. Sun and Y. H. Hu, Chem. Commun., 2019, 55, 7816–7819 RSC
- J. Zhao, X. Guo, R. Shi, G. I. N. Waterhouse, X. Zhang, Q. Dai and T. Zhang, Adv. Funct. Mater., 2022, 32, 2204056 CrossRef CAS
- Q. Hu, Y. Li, J. Wu, Y. Hu, H. Cao and Y. Yang, Adv. Energy Mater., 2023, 13, 2300071 CrossRef CAS
- Z. Rao, Y. Cao, Z. Huang, Z. Yin, W. Wan, M. Ma, Y. Wu, J. Wang, G. Yang, Y. Cui, Z. Gong and Y. Zhou, ACS Catal., 2021, 11, 4730–4738 CrossRef CAS
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS
- D. Shen, Z. Li, J. Shan, G. Yu, X. Wang, Y. Zhang, C. Liu, S. Lyu, J. Li and L. Li, Appl. Catal., B, 2022, 318, 121809 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |