
Synergistic catalysis for promoting selective C–C/C–O cleavage in plastic waste: structure–activity relationship and rational design of heterogeneous catalysts for liquid hydrocarbon production†
Arjun K.
Manal‡
a,
Atal
Shivhare‡
a,
Sharad
Lande
b and
Rajendra
Srivastava
 *a
*a
aCatalysis Research Laboratory, Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar-140001, Punjab, India. E-mail: rajendra@iitrpr.ac.in
bResearch & Development, Reliance Industries Ltd, Thane Belapur Road, Ghansoli, Navi Mumbai-400701, India
First published on 1st October 2024
Abstract
Ever-increasing consumption of plastic products and poor waste management infrastructure have resulted in a massive accumulation of plastic waste in environments, causing adverse effects on climate and living organisms. Although contributing ∼10% towards the total plastic waste management infrastructure, the chemical recycling of plastic waste is considered a viable option to valorize plastic waste into platform chemicals and liquid fuels. Among the various chemical upcycling processes, catalytic hydroprocessing has attracted interest due to its potential to offer higher selectivity than other thermal-based approaches. Heterogeneous catalytic hydroprocessing reactions offer routes for converting plastic waste into essential industrially important molecules. However, the functional group similarities in the plastic polymers frequently constrain reaction selectivity. Therefore, a fundamental understanding of metal selection for targeted bond activation and plastic interaction on solid surfaces is essential for catalyst design and reaction engineering. In this review, we critically assess the structure–activity relationship of catalysts used in the hydroprocessing of plastic waste for the selective production of liquid hydrocarbons. We discuss the significance of C–C/C–O bond activation in plastic waste through active site modulation and surface modification to elucidate reaction networks and pathways for achieving selective bond activation and cleavage. Finally, we highlight current challenges and future opportunities in catalyst design to upcycle real-life plastic waste and produce selective liquid hydrocarbons.
 Arjun K. Manal | Arjun K. Manal completed his master's degree in Organic Chemistry from MES's ACS College Sonai, Maharashtra, India, in 2018. He qualified CSIR-NET in 2019 and the Graduate Aptitude Test in Engineering (GATE) in 2020. Following this, he became a postgraduate student, working towards a PhD degree under the guidance of Prof. Rajendra Srivastava, in the Department of Chemistry, Indian Institute of Technology Ropar, Punjab, India, in 2021. He has been awarded India's prestigious Prime Minister Research Fellowship for doctoral study. His research focuses on the development of heterogeneous catalytic processes for sustainable upgradation of lignocellulose biomass and plastic waste. |
 Atal Shivhare | Dr Atal Shivhare obtained his PhD in 2015 under the supervision of Prof. Robert Scott at the University of Saskatchewan. During his PhD, he worked on atomically precise Au and bimetallic clusters for catalysis. Then, he joined the research group of Prof. Adam F Lee and Prof. Karen Wilson at Aston University in 2015 as a postdoctoral fellow. Then, he moved to the group of Prof. Rajendra Srivastava, IIT Ropar, and worked for 4 years as a postdoctoral fellow. At present, he is working as a Development Editor, at ACS Publications, India. |
 Sharad Lande | Dr Sharad Lande did a PhD in Chemistry from ICT (UDCT) Mumbai and did post-doctorate research at the CCRI, University of Ottawa, Canada, for 2 years. He joined Reliance R&D in 2007 and is currently working as Assistant Vice-President at Corporate R&D, Mumbai. Dr Lande has more than 19 years of research experience in heterogeneous catalysis for hydrocarbon conversion, adsorptive purification, and CO2/green hydrogen purification. He has significantly contributed to commercialization of 9 in-house technologies, 7 on a pilot scale, and has 60 patents filed and several publications/awards. He is a Fellow of the Royal Society of Chemistry, London. |
 Rajendra Srivastava | Dr Rajendra Srivastava is a Professor at the Department of Chemistry, IIT Ropar. He acquired a PhD from CSIR-NCL Pune. He worked as a postdoctoral fellow at KAIST, South Korea, and as a JSPS fellow at Hokkaido University, Japan. He received the Best Thesis award, the NASI-SCOPUS Young Scientist, the Young Scientist award from the Catalysis Society of India, and the Research awards from IIT Ropar and Punjab University. His research interest includes the synthesis of nanostructured functional materials for fuel and chemical production. He has published more than 195 peer-reviewed research articles in reputed international journals and nine patents. |
1. Introduction
Plastics have been widely used since the 1950s and are ubiquitous in our daily lives. The wide range of plastic materials used in daily life find major applications across various sectors, including packaging, building and construction, transportation, textiles, consumer and institutional products, medical, and electrical/electronic industries (Fig. 1(a)–(c)).1–5 Among these, the packaging is the biggest (∼33% in 2018), whereas building & construction and textiles are the second and third largest plastic-consuming sectors.6 | ||
| Fig. 1 (a) Commonly employed plastics and (b) their annual production along with the amount of waste generated from the respective plastics in 2019. Data collected and reproduced with permission from ref. 7. Copyright 2020, Elsevier B.V. (c) The percentage consumption of plastics by various sectors in 2018. Data collected and reproduced with permission from ref. 6. Copyright 2021, Elsevier B.V. | ||
The favourable characteristics of plastics, such as low cost, light weight, formability, bio-inertness, and design versatility, make them an ideal feedstock for a diverse range of valuable products currently being manufactured.6,8,9 Further, with the fast-growing population and rapid urbanization, the consumption of plastic-based products has also increased significantly. Based on a report published by the International Energy Agency World Energy Outlook, the annual plastic consumption in the packaging sector alone will be 318 million tonnes by 2050, representing a four fold increase from the current level, and is also higher than the plastics consumed by the entire plastic industry today.10 For example, based on the Ellen McArthur Foundation, approximately 14% of the plastic waste is currently recycled, with 8% being “cascaded recycled” into materials of lower quality, and only 2% being closed-loop recycled, where the plastic polymer is converted into starting monomers, which can be used to produce virgin plastic. In contrast, 84%, a significant amount, is either disposed of via landfilling or mismanaged dumping or subjected to incineration.11–13 Further, the accumulated plastic waste causes several harmful effects.14–18 The incineration of plastic waste emits CO2, contributing to a significant proportion (∼2%, 37.5 giga-tons CO2 in 2018) of the total global CO2 emission.19,20 Considering this aspect, plastic waste recycling has been proposed as a viable solution to significantly limit the exposure and harmful impact of waste plastic on the environment. Currently, plastic waste recycling constitutes only a minor fraction (∼10%) of the plastic waste management sector.11 Mechanical plastic waste recycling is usually the preferred choice due to the lower energy requirements than chemical recycling.21 However, mechanical recycling is limited to repurposing plastic waste into lower-quality commodity products.
Chemical recycling can convert plastic waste into higher-value products such as liquid fuels, platform chemicals, and virgin plastics.13,22–24 Various chemical recycling approaches, including pyrolysis, gasification, hydrogenolysis, solvolysis, cracking, etc., have been developed to valorize plastic waste.23,25–27 Among these approaches, solvolysis and hydrolysis involve the depolymerization of plastic polymers into their respective monomers, which are subsequently processed to produce virgin plastics. Pyrolysis and gasification operate at higher temperatures compared to solvolysis and involve the conversion of plastic polymers into liquid oil, gases, and biochar. In contrast, catalytic hydrogenolysis and hydrocracking operate under milder conditions than pyrolysis and gasification, offering higher selectivity towards liquid hydrocarbons and valuable products.13,22–28 Several review articles in the literature describe these processes to valorize plastic waste using different technologies. For example, Zheng et al. and Wang et al. cover processes for upcycling plastic waste, such as pyrolysis, solvolysis (only briefly), microwave catalysis, photocatalysis, electrocatalysis, and biocatalysis.29,30 Chu et al. and Roy et al. review the depolymerization of the PET polymer using homogeneous and heterogeneous catalysts, and they also discuss the catalytic depolymerization of polyolefins, highlighting a limited number of examples involving hydrocracking and hydrogenolysis reactions.31,32 Mishra et al. summarize the literature on zeolite-based and metal oxide/hydroxide-based catalysts, discussing advancements in the catalytic pyrolysis of plastic waste.33 Su et al. focus on plastic waste degradation using plasma, Fenton, and electrochemical technologies, providing an up-to-date review of these emerging methods.34 Chen et al. offer a comprehensive summary of catalytic hydrogenolysis of polyolefins, photo-reforming of plastic waste to generate H2 and C2 chemicals, pyrolysis to produce liquid fuels and functional carbon materials, and solvolysis using homogeneous and heterogeneous catalysts.35 Tan et al. concentrate on heterogeneous catalysts for upcycling waste plastic through various techniques, summarizing several catalytic processes, including hydrogenolysis of polyolefins, cross-alkane metathesis of polyolefins, conversion of PET, PC, PS, and PPO into jet fuels and arenes, and the production of carbon materials and H2via thermocatalysis, microwave- and plasma-assisted catalysis, photocatalysis, and electrocatalysis.36 While these reviews comprehensively cover different upcycling processes using heterogeneous catalysts, they do not examine the structure–activity relationship of C–O/C–C bonds in polyaromatics or the terminal and internal C–C bonds in polyolefins. We seek to highlight the importance of underlying reaction mechanisms and surface chemistry.
Chemical upcycling via hydroprocessing offers a novel approach for converting plastic waste directly into liquid hydrocarbons. Heterogeneous catalysts are advantageous over homogeneous ones, reducing both energy consumption and environmental impact. The varied molecular structures of different plastics lead to diverse upcycling products. Aromatic plastics (e.g., PET, PC, PPO, PS) with C–C and C–O linkages can be transformed into arenes, phenolics, and cyclic hydrocarbons.25,37–47 In contrast, aliphatic plastics (e.g., HDPE, LDPE, PP) can be converted into liquid hydrocarbons such as fuels and lubricants. Selective hydrogenolysis of aromatic plastics to specific arenes is challenging, requiring catalysts that can selectively cleave C–O and/or C–C bonds while preserving aromatic rings. Catalysts suitable for jet fuel-range cyclic hydrocarbons involve complete hydrogenation of aromatic rings and C–O bond cleavage.48–72 For polyolefins, selective production of liquid hydrocarbons requires terminal C–C bond cleavage and methane formation, necessitating precise manipulation of catalytic properties to optimize interactions between active metal species and supports.
This review critically evaluates the structure–function relationship of catalysts for the selective cleavage of C–O and C–C bonds in common plastic waste polymers like PET, PC, and POs, converting them into value-added chemicals and fuels. It compiles heterogeneous catalytic hydroprocessing studies, focusing on active sites for specific transformations, and details catalysts that effectively cleave these bonds, discussing mechanistic pathways and active site requirements. The review covers various classes of catalysts, including monometallic and bimetallic systems, and examines the role of active sites, metal–support interactions, and the incorporation of secondary metals. It concludes with a discussion on catalyst design principles, emerging opportunities, and outlooks for enhancing bond cleavage selectivity and producing liquid hydrocarbons.
2. Hydroprocessing of polyaromatic polymers to produce liquid hydrocarbons
Hydrogenolysis/hydrodeoxygenation of aromatic plastic waste has gained significant attention in synthesizing value-added chemicals and fuels.25,37,38,73 Hydrogenolysis/hydrodeoxygenation will be referred to as hydroprocessing in this section. Hydroprocessing involves the cleavage of C–O/C–C linkages in plastic polymers to produce monomers and oligomers.25,37 The experimental conditions employed during hydroprocessing may also result in hydrolysis, de-carbonylation, hydrogenation, and hydrogenolysis reactions (Scheme 1). Further, the hydroprocessing of plastic is performed to recover monomers and value-added chemicals that can be used as either platform chemicals or liquid fuel additives.39–41 However, due to the accompanying side reactions during hydroprocessing, as mentioned above, improving the selectivity of the desired product has been challenging and is currently the major focus of heterogeneous catalyst design. Herein, we have summarized the literature examples involving the hydroprocessing of PET and PC over heterogeneous catalysts. This section is divided into two parts: the first part includes the hydroprocessing of polyaromatic polymers into oxygenates/aromatic compounds, and the second part consists of hydroprocessing of polyaromatic polymers into saturated cyclic hydrocarbons that can be employed as jet fuels. The main focus of the heterogeneous catalyst design has been to tune the metal–support interface, metal particle size, support acidity, and porosity to selectively produce aromatic compounds or saturated cyclic hydrocarbons. | ||
| Scheme 1 Schematic depiction of various catalytic routes operated during the hydrogenolysis/hydrodeoxygenation of (a) PET and (b) PC.25,42–47,74–81 | ||
2.1. C–C/C–O activation for selective arene production
Hydroprocessing of polyaromatics such as PET, PC, PPO, and PS to produce aromatic compounds involves breaking C–O and C–C bonds. This process requires careful optimization of the metal–support interface, metal particle size, and experimental parameters to selectively activate these bonds while minimizing the formation of side products, such as saturated cyclic hydrocarbons.Several metal–support combinations have been investigated for the selective production of aromatics from waste PET and PC (Table S1, ESI†). However, very few studies have been conducted on PPO and PS. Jing et al. found that the proper selection of metal–support combinations is crucial for regulating the metal particle size for effective C–O and C–C bond activation, thereby achieving high arene selectivity. PET hydrogenolysis was conducted over several bi-functional catalysts comprised of the combinations of metals (Ru, Pd, and Pt) and supports (Nb2O5, ZrO2, TiO2, and HZSM-5) (Fig. 2(a) and (b)).25 The PET hydrogenolysis, performed at 200 °C and 0.3 MPa H2 for 12 h, afforded exceptional monomer yield (95.2%) and high arene selectivity (87.1%) over the Ru/Nb2O5 catalyst. In contrast, Pd/Nb2O5 and Pt/Nb2O5 catalysts afforded a significantly lower monomer yield of ∼20% (Fig. 2(b)). The support effect showed that Ru/ZrO2, Ru/TiO2, and Ru/HZSM-5 were much less active, affording <80% of total monomer yield with significant percentages of saturated hydrocarbons (∼25% in each case). The DFT-calculated XANES spectra revealed that Ru species on ZrO2, TiO2, and HZSM-5 exhibit XANES spectra consistent with Ru coordination numbers of 9 or 12, aligning with EXAFS data. Notably, the XANES spectrum of Ru/Nb2O5 closely matches simulations for Ru with coordination numbers of 5 or 6, indicating that the primary Ru species on Nb2O5 have low coordination numbers, suggesting the presence of edge sites and sub-nanometer-sized Ru particles. Ru sub-nano particles with a low coordination number (C.N. = 5–6) inhibit benzene ring hydrogenation and result in distinct selectivity for arenes. Conversely, Ru/HZSM-5 predominantly produces ring-saturated products, indicating a preference for benzene ring hydrogenation over the desired C–C bond cleavage. This finding is supported by DRIFTS analysis of toluene adsorption. Ru species on Nb2O5 exhibit ultra-small particle sizes, which inhibits the hydrogenation of aromatic rings when compared to other supports. Ru combined with NbOx species facilitated C–O bond activation, and Brønsted acid sites promoted C–C bond cleavage, resulting in the desired reactivity for the depolymerization of aromatic plastics. Moreover, the use of hydrogen transfer agents as a reductant instead of the use of H2 gas can simplify the process and reduce costs. The group used the same catalyst to produce H2 that can participate in the hydrogenolysis of the PET polymer, hence eliminating the use of externally supplied H2 gas. Lu et al. discovered that the source of H2 can be ethylene glycol produced during PET hydrolysis over acid sites.37 They employed two catalysts, Ru/Nb2O5 and Ru/NiAl2O4, to selectively upgrade PET into BTX at 220 °C under an N2 atmosphere, employing ethylene glycol as a reaction medium and a H2 source. The catalytic data revealed that the major products produced were toluene and xylene via hydrogenolysis over Ru/Nb2O5. On the other hand, benzene, produced via decarboxylation reaction, was the major product over Ru/NiAl2O4 (Fig. 2(c)). Based on the data obtained from the in situ experiments, including DRIFTS and TPSR, the authors proposed that Ru(0) species, abundantly available in the Ru/NiAl2O4 catalyst, were responsible for the decarboxylation reaction. In contrast, the NbOx species in the Ru/Nb2O5 catalyst were involved in activating the C–O bond, resulting in toluene and xylene via hydrogenolysis as the major products.
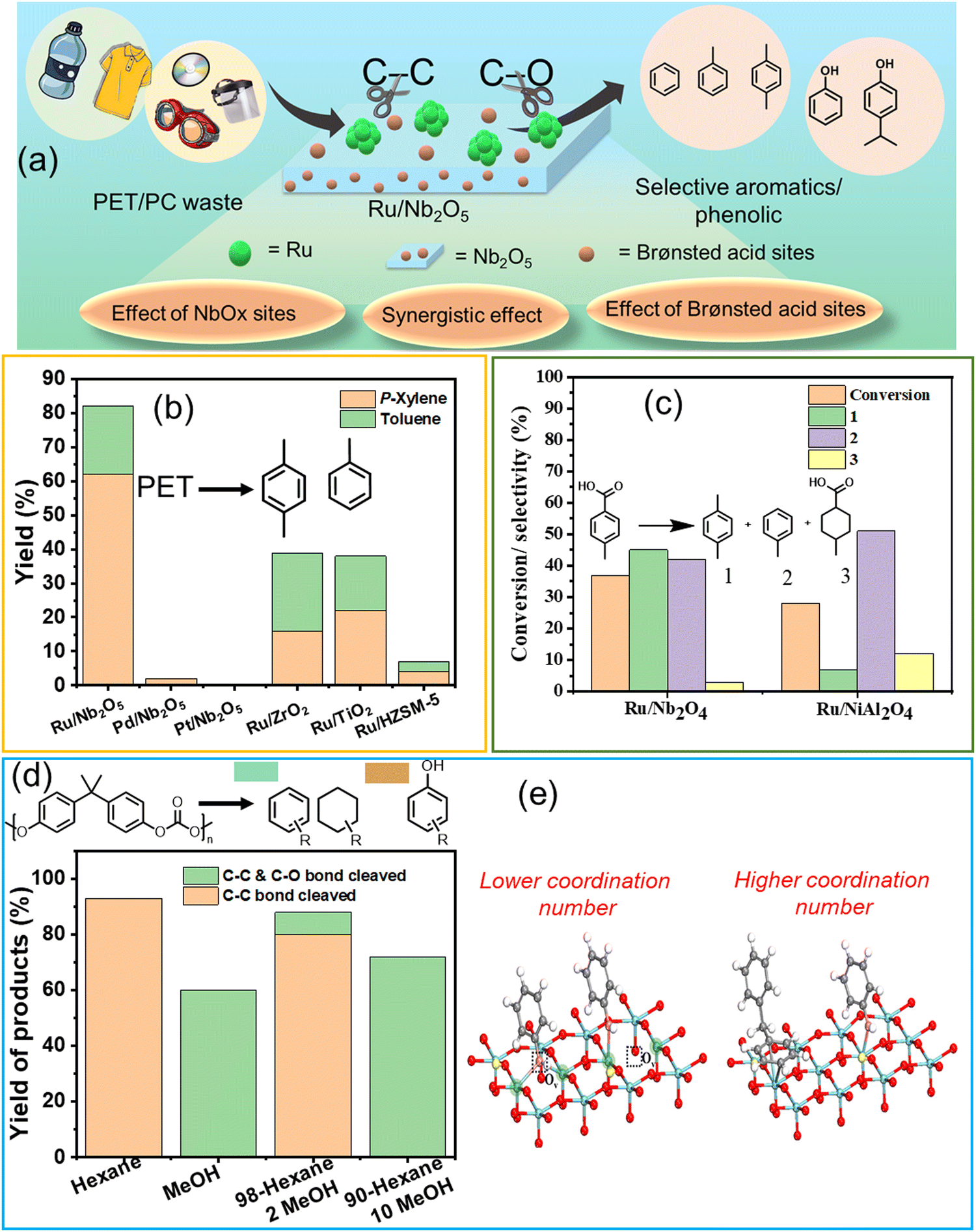 | ||
| Fig. 2 (a) Schematic representation of cleavage of the C–O/C–C bond in polyaromatics using Ru/Nb2O5. (b) PET conversion and product distribution over different catalysts. Data collected and reproduced with permission from ref. 25. Copyright 2021, John Wiley and Sons. (c) Hydrogenolysis and decarboxylation reaction results. Data collected and reproduced with permission from ref. 37. Copyright 2021, John Wiley and Sons. (d) Conversion of PC in various solvent media. (e) Adsorption models of the C–O bond and benzene ring on different Nb2O5 surfaces. Data collected and reproduced with permission from ref. 42. Copyright 2022 American Chemical Society. | ||
Selective poisoning of the C–O bond to favor the cleavage of the C–C bond in polycarbonate (PC) has been demonstrated.42 The hydrogenolysis of PC was investigated at 270 °C and 0.7 MPa H2 in n-hexane, affording ∼95% yield of arene monomers via the cleavage of the C–C and C–O bonds in PC, whereas upon changing the reaction medium to methanol, aromatic oxygenates possessing hydroxyl functional groups were produced in >70% yield (Fig. 2(d)). DRIFT, XANES, and control experiments revealed that methanol selectively poisoned the NbOx active sites responsible for C–O activation, hence preventing the cleavage of C–O linkages, improving aromatic oxygenate yield, and simultaneously affording lower arene yields (Fig. 2(e)). These investigations unveil that NbOx species comprising the support exhibit significant capability in activating C–O bonds in plastics, thereby playing a crucial role in C–O bond cleavage. Nevertheless, selective cleavage of the C–C bond can be attained through the inhibition of the C–O bond, achieved by introducing a small quantity of alcohol solvent. Manal A. K. et al. recently reported a transition metal-free method for selectively cleaving C–C bonds to produce phenol from polycarbonate plastics.82 Commercial zeolites, including HZSM-5, HBeta, and HY with varying Si/Al ratios, were employed in the transformation process. Among the zeolites tested (HZSM-5, HBeta, and HY), HY with a Si/Al ratio of 15 showed superior performance, yielding ∼75% phenol and ∼15% acetone. The efficiency of HY (Si/Al = 15) was attributed to its topology and access to Brønsted acid sites. A correlation coefficient of 0.82 between Brønsted acid sites and BPA conversion highlighted the strong positive correlation with catalytic efficiency. Surface area and pore volume, with coefficients of 0.72 and 0.64, respectively, played a secondary role by enhancing mass transfer. The mechanistic pathway, supported by experimental and DFT analysis, revealed that the chemical environment at the Cα position was key to deconstructing the Csp2–Csp3 bond under mild conditions.
A study conducted by Zhang et al. demonstrates the role of the geometric structure of Ru metal in selective aromatic production. The impact of the geometric structure of Ru metal was explored in PET hydrogenolysis, utilizing a TiO2 support.43 Zhang et al. used an inert TiO2 support to synthesize Ru/TiO2 catalysts and investigate the effect of Ru metal during PET hydrogenolysis. Ru/TiO2 catalysts with varying Ru loadings (0.1–10 wt%) were prepared via wet-impregnation. Electron microscopy and EXAFS analyses showed that decreasing Ru loading from 10 wt% to 0.1 wt% reduced the average Ru coordination number due to smaller Ru particle sizes. The truncated hexagonal bipyramid model revealed a decrease in the ratio of terrace to edge/corner atoms as the particle size decreased (Fig. 3(a)). In PET hydrogenolysis at 230 °C and 0.3 MPa H2, the catalytic activity varied with Ru concentration, and gave a volcano-type curve for BTX yield (Fig. 3(b)). The change in Ru concentration resulted in different particle sizes. 3.1 nm Ru particles (10 wt% Ru/TiO2) produced ∼35% arenes (benzene, toluene), ∼10% methyl cyclohexane, and ∼20% gaseous products. 1.1 nm Ru particles (0.5 wt% Ru/TiO2) resulted in an increased arene yield of ∼75% with lower gaseous products (∼10%) and negligible methyl cyclohexane. However, 0.8 nm Ru particles (0.1 wt% Ru/TiO2) resulted in ∼45% arenes and ∼25% gaseous products, indicating a volcano relationship, with 1.1 nm Ru particles affording the highest yield of arenes (Fig. 3(b)). DFT calculations showed that methyl p-toluate adsorbed flat on Ru(0001) terrace sites, forming ring-saturated products, and upright on Ru(1015) edge sites, forming arenes. This explained higher arene productivity with 1.1 nm Ru particles. Smaller Ru particles led to more gaseous products due to over-hydrogenolysis, while larger particles favored ring-opening reactions.
 | ||
| Fig. 3 (a) The fraction of various types of surface atoms as a function of Ru particle size and (b) catalytic data showing the selectivity towards various products as a function of Ru particle size. Data collected and reproduced with permission from ref. 43. Copyright 2023, John Wiley and Sons. Catalytic activity of Cu/SiO2 catalysts (c), and XAES and XPS spectra of Cu/SiO2 (d) and CuNa/SiO2 (e) catalysts. Data collected and reproduced with permission from ref. 38. Copyright 2023. | ||
Alternative catalysts derived from non-noble metals were also investigated to enhance economic viability and industrial feasibility. Hongkailers et al. synthesized a TiO2 supported 5 wt% Co catalyst via wet-impregnation and employed it in the hydrogenolysis of PET to arenes.40 Arenes were produced as the major products with ∼75% yield at 320 °C and 3 MPa H2 pressure. PET first undergoes depolymerization via C–O bond cleavage over Co/TiO2, followed by deoxygenation to produce arenes. The activity was achieved due to the Co NPs at the external surface of the TiO2 support, enabling the direct interaction with large PET molecules and the co-presence of the Co3O4 phase, providing acid sites. Gao et al. discovered that the concentration of metal species significantly influences non-noble catalyzed hydrogenolysis reactions. Specifically, active copper species exhibited higher hydrogenolysis activity, demonstrating exceptional efficacy in hydrogen production from methanol. Several Cu/SiO2 catalysts were synthesized using different techniques, including Cu/SiO2-IM (impregnation), Cu/SiO2-HT (hydrothermal), Cu/SiO2-DPU (deposition precipitation with urea), and Cu/SiO2-DPU (deposition precipitation with ammonia), and employed for PET hydrogenolysis in methanol at 210 °C for 6 h.38 All catalysts prepared by various techniques, except Cu/SiO2-HT, selectively produced 100% DMT. Notably, Cu/SiO2-HT, synthesized via the hydrothermal method, yielded 73% para-xylene (PX) (Fig. 3(c)). The authors attributed this outcome to the higher proportion of Cu+ and Cu0 species in the Cu/SiO2-HT catalyst, which played a crucial role in H2 production from methanol. To enhance catalytic performance, an alkali metal salt was added to maintain the balance of Cu+ and Cu0 species. Specifically, the CuNa/SiO2 catalyst, prepared by introducing NaCl during hydrothermal synthesis, achieved a 100% PX yield. The excellent performance of CuNa/SiO2 in PX production was attributed to the promotional effect of Na in facilitating the production of copper silicate with a higher Cu+/Cu0 (1.87) ratio for H2 production from methanol (Fig. 3(d) and (e)).
The synergy between two metals in supported bimetallic catalysts has been demonstrated to favour higher PET conversion and improved product selectivity. For example, Wu et al. synthesized a nitrogen-doped-carbon supported CoMo bimetallic catalyst (CoMo@NC) via the pyrolysis of Mo@ZIF-CoZn at 900 °C.74 The resulting CoMo@NC catalyst was employed for PET hydrogenolysis and it afforded an impressive 91% terephthalic acid (PTA) yield at 260 °C under atmospheric H2 in 10 h. In contrast, the monometallic catalysts Co@NC and Mo@NC, derived from the pyrolysis of ZIF-CoZn and Mo@ZIF-Zn, respectively, demonstrated significantly lower performance, yielding only 30% and 18% PTA, respectively. The superior catalytic activity of the bimetallic CoMo@NC catalyst can be attributed to the synergistic interaction between Co and Mo. In this bimetallic system, the Mo-sites play a crucial role in activating PET and facilitating the β-scission of the bridging glycol C–O bond, while the Co-sites are responsible for activating hydrogen and catalyzing the hydrogenolysis step. This coordinated interaction between the two metals significantly enhanced the catalytic efficiency for PET depolymerization.
2.2. C–C/C–O activation for selective cyclic hydrocarbon production
Cyclic hydrocarbons are generally employed as jet fuels due to their high volumetric heat value, helping aircraft to fly and acquire more speed with minimum fuel storage infrastructure.76–81 PC and PET polymers can be converted into cyclic hydrocarbons over bi-functional catalysts (Scheme 2). The reactions proceed through multiple steps such as depolymerization, hydrogenation, and deoxygenation. The overall reaction pathway involved is termed the hydrodeoxygenation pathway. It involves cooperative catalytic interactions between multiple distinct active sites, particularly a synergy between metal sites with pronounced ring hydrogenation capabilities (such as Pt, Pd, Ru, Rh, Ni, Co, etc.) and acidic supports (zeolites, metal oxides) exhibiting robust C–O/C–C bond activation properties.54–57 The breaking of the C–O bond and the consequent formation of fully deoxygenated products are facilitated by the effective collaboration between the metal and the support and the support itself, underscoring the pivotal role of the support in this conversion. The optimal achievement of this ambitious objective relies on the judicious selection of active supports capable of activating C–O bonds while ensuring effective adsorption of the plastic substrate. | ||
| Scheme 2 Catalytic hydrodeoxygenation of PC (a) and PET (b) to cyclic hydrocarbons over heterogeneous catalysts.44–47,83 | ||
Tang et al. investigated the production of jet-fuel range polycyclic hydrocarbons from PC over supported metal catalysts.44 PC first underwent depolymerization via methanolysis at 180 °C, producing bisphenol A. Subsequently, bisphenol A underwent hydrogenation followed by hydrodeoxygenation over supported metal catalysts and produced jet-fuel range cyclic hydrocarbons. Carbon-supported transition metal catalysts, including Pt/C, Pd/C, and Ru/C, were found to be active for hydrogenating bisphenol A (Scheme 2a). Among these catalysts, Pt/C afforded the highest yield (∼90%) due to higher hydrogenation activity, with the predominant formation of cyclic alcohols. To perform the C–O cleavage of cyclic alcohol, Pt/C was combined with Brønsted acid H-β zeolite, and the integrated catalytic system afforded ∼80% yield of cyclic hydrocarbons (Scheme 2a). H-β zeolite provides strong Brønsted acid sites and a large surface area to facilitate mass transfer, resulting in higher C–O bond cleavage efficiency than HZSM-5 and HMOR. To avoid the multi-step process, Wang et al. investigated the catalytic performance of supported Rh catalysts in the hydrodeoxygenation of direct PC pellets.45 The catalytic data revealed that Rh/C afforded nearly complete PC conversion at 200 °C and 3.5 MPa H2 in 12 h. However, the main products formed were C15 oxygenates (26.9% yield), including bisphenol A and 4-benzylcyclohexane-1-ol, and a small quantity of propane-2,2-diyldicyclohexane (3.8% yield). Moreover, a physical mixture of Rh/C and H-USY zeolite (Rh/C + H-USY) afforded ∼95% yield of propane-2,2-cyclohexane (jet-fuel range cyclic hydrocarbon). The study shows that Rh/C and H-USY were active in catalyzing the hydrolysis of PC pellets in water to produce C15 oxygenates. Subsequently, the hydrogenation of these oxygenates over Rh sites, dehydration over acid sites, and finally, hydrogenation over Rh sites resulted in the production of propane-2,2-diyldicyclohexane. Later, the same group employed RANEY® metal catalysts, RANEY®@Cu, RANEY®@Fe, RANEY®@Co, and RANEY®@Ni, in PC hydrodeoxygenation at 190 °C using iso-propanol as the H-donor.46 Among these catalysts, RANEY®@Ni exhibited the maximum activity with nearly complete PC conversion and a total ∼95% yield of several products, including C6–C15 oxygenates as the major products (∼65% yield), C6–C15 aromatics (∼20% yield), and C6–C15 cycloalkanes (∼10% yield) (Scheme 2a). To increase the yield of C6–C15 cycloalkanes, RANEY®@Ni was combined with various solid acids, USY, H-ZSM-5, and Al2O3, and the combination of RANEY®@Ni and USY afforded the maximum C6–C15 cycloalkane yield (∼20%).
The development of a mono-catalytic system for the selective hydrodeoxygenation of PC holds significant importance in enhancing economic viability and industrial feasibility. Recently, Manal et al. reported the synthesis of a bifunctional Ru–Ni supported H-beta catalyst for the hydrodeoxygenation (HDO) of bisphenol A (BPA) and PC to high-density propane-2,2-diyldicyclohexane under mild conditions.47 The study suggested that the alloy formed between Ru and Ni was responsible for the enhanced catalytic activity. Catalytic data indicated that the selectivity for propane-2,2-diyldicyclohexane was dependent on the Ru/Ni weight ratio. A volcano plot demonstrated that a Ni/Ru weight ratio of 0.5 was optimal, achieving high selectivity for propane-2,2-diyldicyclohexane. Higher Ni/Ru ratios (>0.5) resulted in a larger domain of Ni species, which significantly influenced selectivity and promoted the formation of lower-range hydrocarbons via C–C bond cleavage. Ru sites on the catalyst facilitated H2 dissociation, which was transferred to the aromatic ring at the Ru–Ni alloy/support interface, resulting in BPA hydrogenation to form ring-hydrogenated cyclic alcohol. Additionally, Brønsted acid sites associated with the Ru–Ni alloy phases played a crucial role in cleaving Csp3–OH, leading to the formation of propane-2,2-diyldicyclohexane (Scheme 3).
 | ||
| Scheme 3 Reaction mechanism for the HDO of PC/BPA over the 1Ru0.5Ni/H-Beta catalyst. Data collected and reproduced with permission from ref. 47. Copyright 2023, Elsevier B.V. | ||
Tang et al. investigated upcycling of PET into cyclic hydrocarbons over supported transition metal catalysts.83 Under catalyst-free conditions, PET was depolymerized via methanolysis to dimethyl terephthalate (DMT) at 200 °C in 3.5 h. Subsequently, the hydrogenation of DMT was investigated over carbon-supported metal catalysts, Pt/C, Ru/C, and Pd/C, at 100 °C and 5 MPa H2 for 7 h. The maximum DMT conversion (73.1% conversion) into dimethyl cyclohexane-1,4-dicarboxylate (DMCD, 71.1%) was obtained over the Pt/C catalyst (Scheme 2b). Subsequently, the hydrodeoxygenation of DMCD was investigated over Ru/SiO2, Cu/SiO2, and Ru–Cu/SiO2 catalysts at 400 °C and 4 MPa H2. Among these catalysts, Ru–Cu/SiO2 afforded the highest yield of C7–C8 cycloalkanes, which was attributed to the smaller size Ru–Cu alloy particles showing higher hydrodeoxygenation efficiency than Cu and lower methanation activity than Ru.
The insights gained from the mechanistic studies of polyaromatic hydrogenolysis/hydrodeoxygenation shed light on how the reactant structures, bond linkages, nanoparticle sizes, support effects, and the role of secondary metal catalytic functions influence the selective cleavage of C–O and C–C bonds to liquid hydrocarbons. Among various transition metals, Ru has been identified as the most effective for arene production, with smaller Ru particles (∼1.0 nm) exhibiting higher activity than larger ones. Ru species with low coordination numbers (C.N. = 5–6) prevent benzene ring hydrogenation, thereby enhancing selectivity for arenes. Supported catalysts with specific states of metal species demonstrate elevated activity in generating arenes under mild conditions, using methanol as the hydrogen donor. Such higher activity is attributed to an optimized Cu+/Cu0 ratio, which enhances both hydrogenolysis and arene yield. However, maintaining an optimal Cu+/Cu0 ratio is crucial due to the stability concerns associated with Cu+ and Cu0 states. The choice of support material is also critical for selectively activating C–C and C–O bonds and producing arenes. Oxide supports with oxophilic metals possessing Lewis-Brønsted acid sites significantly activate C–C and C–O bonds. However, a small amount of alcohol can occupy Brønsted acid sites, inhibiting C–O bond cleavage and selectively facilitating C–C bond cleavage. Producing jet-fuel-range cyclic hydrocarbons over heterogeneous catalysts requires multifunctional catalytic systems. In these systems, metal sites are responsible for hydrogenation reactions, while the acid sites of the support facilitate deoxygenation reactions. The effectiveness with hydrogenation metals (e.g., Pt, Pd, Rh, Ru, Ni, Co, etc.) is supported by robust Brønsted acid materials predominantly employed for this purpose. The Brønsted acid sites in zeolites play a dual role: they facilitate the depolymerization of PET or PC into its monomers and activate the C–O bond of cyclic alcohols to cyclic hydrocarbons. Additionally, zeolites, with their high surface area and well-defined pore structure, provide easy access to active sites on the external surface and within the pores. This enhances mass transfer, contributing to their remarkable catalytic efficiency.
3. Hydroprocessing of polyolefins to produce liquid hydrocarbons
Polypropylene (PP) and polyethylene (PE) are the two most common polyolefins (POs), with a market share of 76 Mt and 109 Mt, respectively, in 2021, and POs represent more than 50% of the globally produced plastic.62 PP finds application in the production of packaging materials, hinged caps, thermoplastic pipes, bank notes, automotive components, etc.10,84–88 PE is mainly used in toys, bottles, houseware appliances, reusable bags, cling wraps, food packages, etc.89 Currently, the industrial processes for the recycling of PO waste include pyrolysis, plastic waste purification, aerobic oxidation, etc.13 Pyrolysis and aerobic oxidation processes non-selectively convert PO waste into shorter-chain alkanes and oxygenates. On the other hand, the hydroconversion processes are more selective toward producing liquid fuels, lubricants/wax, and platform gases (Fig. 4).90–92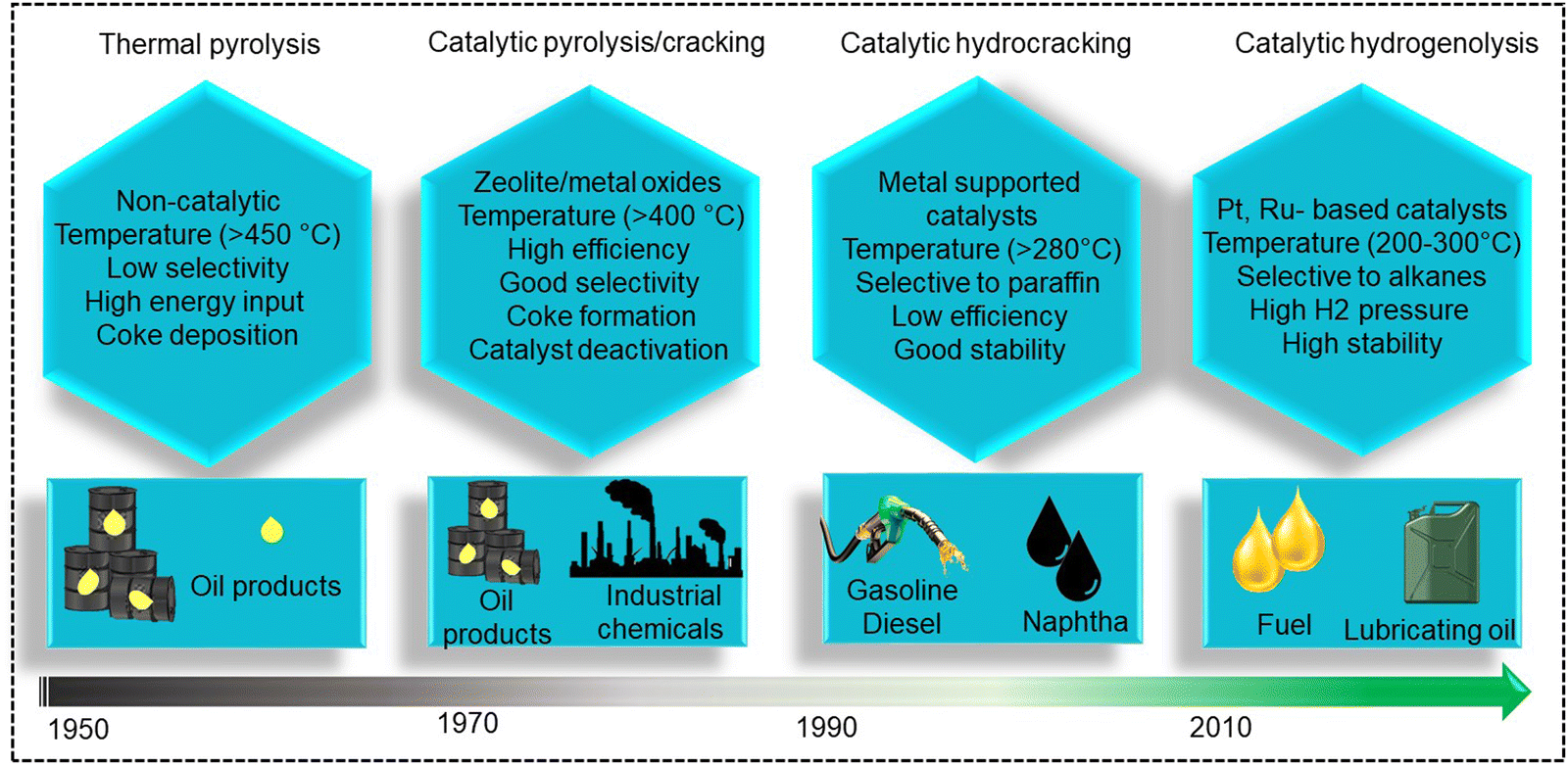 | ||
| Fig. 4 Evolution of industrial processes over time for the processing of polyolefins. | ||
Hydroprocessing entails the catalytic conversion of polyolefins into shorter hydrocarbon chains through hydrogenolysis and hydrocracking mechanisms. Hydrogenolysis, facilitated by metal sites, involves breaking the H–H bond in H2 and the C–C bond in the substrates, leading to the formation of two new terminal C–H bonds (Scheme 4a). Conversely, hydrocracking necessitates bifunctional catalysts comprising metal particles and acid supports (Scheme 4b). Metal sites catalyze the dehydrogenation step, generating an olefin intermediate. This intermediate diffuses onto Brønsted acid sites, where it undergoes isomerization and C–C cracking via a carbenium ion intermediate. Literature examples of these processes are summarized in Table S2 (ESI†), with a primary emphasis on selectively producing hydrocarbons within the liquid fuel range. Nevertheless, scant reports address polyolefin hydrogenolysis, where selectively producing valuable gases and lubricant/wax is the primary goal. To establish a benchmark for hydrogenolysis and hydrocracking, various examples were examined for product distribution at different temperatures and durations. Comparative analysis revealed that hydrogenolysis typically occurs at relatively low temperatures (200–350 °C) than hydrocracking. Lower hydrogenolysis temperatures corresponded to higher yields of liquid fuel products compared to hydrocracking, albeit at the expense of longer reaction times. Additionally, hydrocracking at elevated temperatures (350–600 °C), while reducing reaction times, accelerates catalyst deactivation due to coking, limiting practical applications and increasing gaseous product yields while decreasing liquid fuel yields (Fig. 5(a)–(d)). Comparing parameters such as temperature, duration, liquid yield, and gas yield for hydrocracking and hydrogenolysis of polyolefins reveals a distinct disparity between the two processes, highlighting the efficacy of selective product distribution.
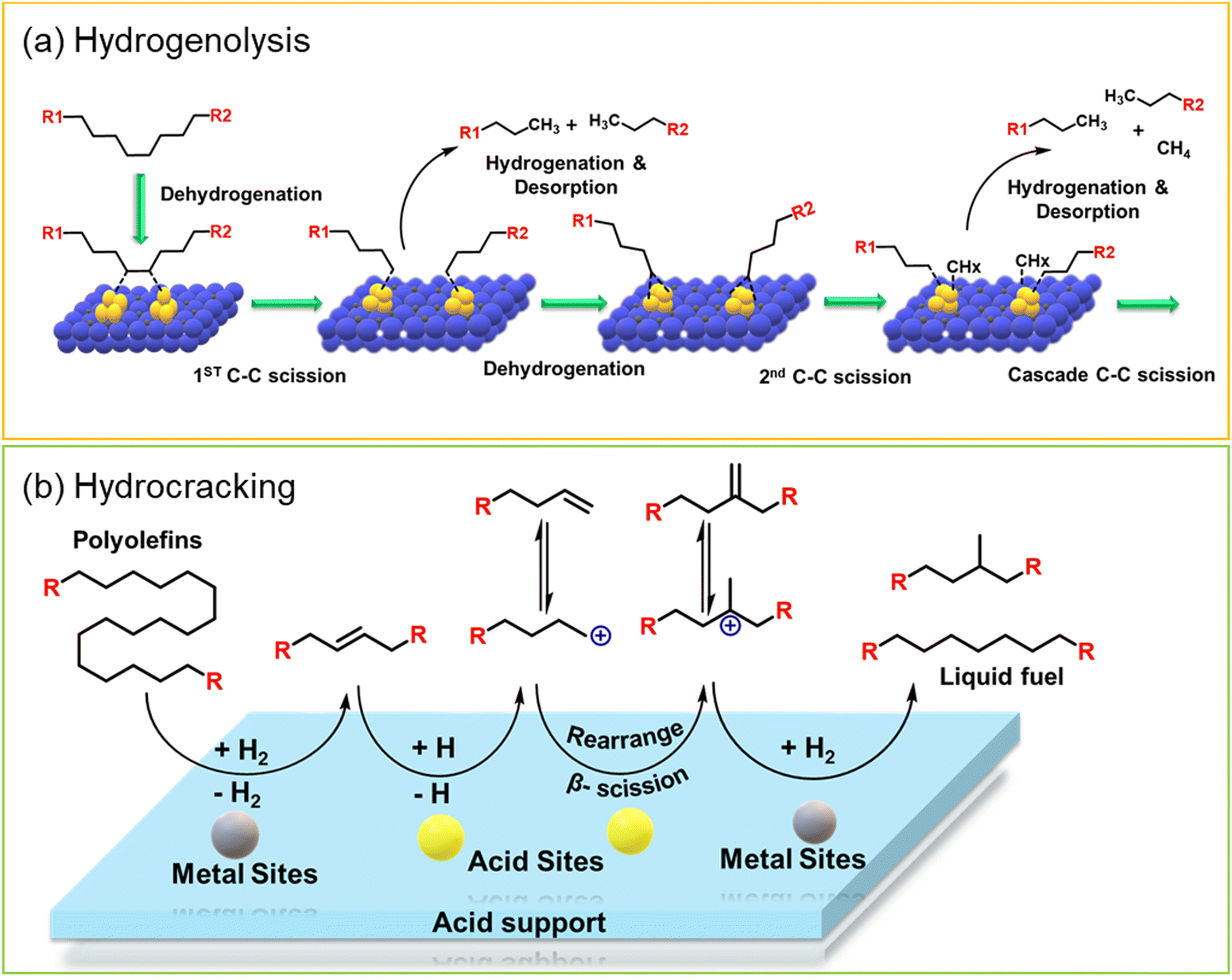 | ||
| Scheme 4 (a) Reaction mechanism of the catalytic hydrogenolysis of polyolefins over metal catalysts, and (b) the reaction mechanism of the catalytic hydrocracking of polyolefins over metal-acid bi-functional catalysts. | ||
 | ||
| Fig. 5 (a) Relationship between reaction temperature and time for PO recycling via hydrogenolysis and hydrocracking at complete conversion; (b) comparison of gaseous product yield as a function of reaction temperature; (c) comparison of liquid product yield as a function of reaction temperature; and (d) evaluation of the catalytic efficiency of diverse catalysts in PO hydrocracking and hydrogenolysis processes. The rate of formation of liquid products was calculated as follows: rate of formation of liquid = mass (PE or PP) × liquid yield/(mass (catalyst) × t). | ||
3.1. C–C activation and hydrogenolysis of polyolefins to C12–C22 hydrocarbons
Hydrogenolysis of polyolefins is catalyzed by the metal sites. Various catalysts comprised of transition metal nanoparticles and oxide supports have been investigated to find the appropriate metal–support combination, suitable metal particle size, and the appropriate experimental conditions to improve the desired product selectivity (Table S2, entries 1–14, ESI†). For example, Tamura et al. scrutinized the structure–activity relationship of supported Ru catalysts in the hydrogenolysis of LDPE at 240 °C and 6 MPa H2.93 Among several supports employed, CeO2, ZrO2, TiO2, γ-Al2O3, SiO2, and Nb2O5, the highest yield of liquid fuel (C5–C21) was obtained over Ru/ZrO2, prepared via the impregnation of Ru on the ZrO2 support, which was pre-calcined at 800 °C. The catalytic data revealed a volcano relationship between the Ru particle size (varying between 1 and 9 nm) and the liquid fuel yield. The highest liquid fuel productivity was obtained over the 7 wt% Ru/ZrO2 catalyst, which was attributed to the appropriate Ru metal size (∼2.5 nm), showing higher hydrogenolysis activity. The correlation between Ru particle size and catalytic performance (activity and selectivity) was investigated using these Ru catalysts. Good correlations were observed, with one notable exception being the Ru/γ-Al2O3 catalyst, which exhibited a higher production of gas (C1–C4) compared to other supported Ru catalysts.48 Furthermore, Chen et al. demonstrated that the Ru particle size, influenced by the Ru metal loading, directly impacted catalytic efficiency, thereby affecting the product selectivity. The investigation was conducted at 260 °C and 30 bar H2 over Ru/CeO2 catalysts with varying Ru loadings between 0.05 and 2.0 wt%.49 A unique structure-dependent catalytic behavior was observed. For instance, the supported Ru particles with a higher loading range (0.5 to 2.0 wt%) were less active towards PP hydrogenolysis into liquid fuel and more selective towards CH4 formation. Further, the CH4 selectivity increased with Ru loading from 0.5 to 2.0 wt%. In contrast, the supported Ru particles with a lower loading range (<0.5 wt%) were more efficient for PP hydrogenolysis into liquid products and less selective for CH4 formation, where the selectivity toward liquid products increased to ∼70%, and CH4 selectivity decreased to ∼20% over the 0.05 wt% Ru/CeO2 catalyst. Similar trends in product selectivity were observed for LDPE. The authors suggested that the higher Ru loading resulting in large particle size and low metal dispersion attributed to lower intrinsic efficiency and terminal C–C cleavage leads to the formation of CH4 with high selectivity. However, the lower Ru loading, resulting in high metal dispersion and small NPs, leads to a higher coverage of adsorbed hydrogen (*H) and high regioselectivity towards selective internal C–C cleavage, contributing to high liquid hydrocarbon selectivity. EXAFS, XANES, and STEM investigations revealed that the abundance of sub-nanometre cationic Ru species with 2D raft-like structure was responsible for higher catalytic activity in the hydrogenolysis of PP and LDPE into liquid hydrocarbons in the lower loading range. Nakaji et al. investigated the performances of several supported transition metal catalysts, including M/CeO2 (M = Ru, Ir, Rh, Pt, Pd, and Cu), in the hydrogenolysis of LDPE (Mn: ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7000, Mw: ∼4000 Da) at 240 °C and 6 MPa H2 for 5 h.50 Among these catalysts, only Ru/CeO2 was active for hydrocracking, affording 76% LDPE conversion and higher yields of liquid fuel (54%) and wax (15%). The higher activity of Ru/CeO2 among other M/CeO2 catalysts was not due to the difference in particle size or oxidation states, but it was due to the Ru metal itself. Further, the authors also investigated the effect of various supports, including carbon, TiO2, MgO, and ZrO2, and based on the catalytic data, and the Ru/ZrO2 catalyst showed activity (83% LDPE conversion, 61% liquid fuel yield, and 12% wax yield) comparable to that of the Ru/CeO2 catalyst. The basic ZrO2 and CeO2 supports were able to stabilize smaller particles (∼1.5 nm) compared to other supports, which were more selective towards the internal C–C bond scission, resulting in lower gaseous production.
7000, Mw: ∼4000 Da) at 240 °C and 6 MPa H2 for 5 h.50 Among these catalysts, only Ru/CeO2 was active for hydrocracking, affording 76% LDPE conversion and higher yields of liquid fuel (54%) and wax (15%). The higher activity of Ru/CeO2 among other M/CeO2 catalysts was not due to the difference in particle size or oxidation states, but it was due to the Ru metal itself. Further, the authors also investigated the effect of various supports, including carbon, TiO2, MgO, and ZrO2, and based on the catalytic data, and the Ru/ZrO2 catalyst showed activity (83% LDPE conversion, 61% liquid fuel yield, and 12% wax yield) comparable to that of the Ru/CeO2 catalyst. The basic ZrO2 and CeO2 supports were able to stabilize smaller particles (∼1.5 nm) compared to other supports, which were more selective towards the internal C–C bond scission, resulting in lower gaseous production.
Du et al. synthesized a series of bi-functional Rh-based catalysts (Rh/Nb2O5, Rh/ZrO2, Rh/TiO2, and Rh/Al2O3) via wet-impregnation methodology and investigated their catalytic performances in the hydrogenolysis of LLDPE at 300 °C and 3 MPa H2 for 6 h.94 Among these catalysts, Rh/Nb2O5 was the most active, affording complete LLDPE conversion and approximately 80% yield of the liquid products. The liquid products comprising high-value iso-alkanes (68.4%) were obtained. The higher number of Brønsted acid sites (39 μmol g−1) and the strong Brønsted acidity (21 μmol g−1) were responsible for the impressive performance of the Rh/Nb2O5 catalyst.
The activation of C–H and C–C bonds using regulated size Ru supported on CeO2 was investigated by H. Ji et al.51 The study revealed a distinctive trend in the activity of CeO2-supported Ru catalysts, ranging from single atoms to nanoclusters and nanoparticles, in the hydrogenolysis of low-density polyethylene (LDPE). The synthesis of Ru/CeO2 catalysts involved hydrothermal and impregnation methods, with variations in stirring time and reducing agent being critical for controlling the size of Ru species. Among these catalysts, CeO2-supported Ru nanoclusters (NC) demonstrated the highest conversion efficiency and superior selectivity towards liquid alkanes (56.8%) (Fig. 6(a)). The reduction in Ru size promoted metal–support interactions (MSI) while diminishing the hydrogen spillover effect during the reaction. MSI, influenced by the surface states of Ru, is particularly benefitted from more electronegative Ru centers for the activation of C–H and C–C bonds (Fig. 6(b)). Consequently, the competition between MSI and hydrogen spillover underscores the optimal catalytic performance of moderately sized Ru nanoclusters supported on CeO2. S. Lu et al. explored the selective hydrogenolysis of internal and terminal C–C bonds in polyolefins (POs) using Ru/CeO2 catalysts.52 They prepared two distinct CeO2 phases, denoted as CeO2 (R) and CeO2 (O), and impregnated Ru onto them via a simple wet impregnation method. Their investigation elucidates the role of metal–support interactions in tuning the chemical states of Ru within the catalyst. Positively charged Ru species (Ru/CeO2-R) exhibit a preference for the hydrogenolysis of internal C–C bonds, thus suppressing methane production (Fig. 6(a) and (d)). This selectivity arises from the propensity of Ruδ+ species to selectively bond with internal carbons possessing higher electron density, facilitated by the electron-donating effect of adjacent alkyl groups rather than terminal carbons. The plot of CH4 selectivity vs the ratio of Ruδ+/(Ruδ+ + Ru0) revealed that a lower Ruδ+ content corresponds to higher CH4 selectivity (Fig. 6(e)). Comparing these Ru/CeO2 catalysts in terms of Ru particle size and liquid yield indicates that the higher liquid yield can be attributed to the smaller Ru particle size (Fig. 6(f)). Similarly, Sun J. A. et al. investigated the impact of particle size on PO hydrogenolysis by examining Ru/C catalysts with varying particle sizes. They found that increasing the Ru cluster size from 0.8 nm to 1.2 nm led to a significant increase in liquid product yield, from 39% to 78%.53 However, further increasing the particle size from 1.2 nm to 2.3 nm resulted in a dramatic decrease in liquid product yield to 8%. The larger crystalline Ru nanoparticles (∼2.3 nm, 2% Ru/C–He) exhibit lower activity for C–C bond scission but still catalyze stereoisomerization reactions. Intermediate-sized Ru particles maintain consistent activity and produce a broader range of products, resulting in a high yield of liquid hydrocarbons. X-ray absorption spectroscopy (XAS), XPS, and TPR revealed that smaller Ru particles contain more disordered Ruδ+ species, which correlates with their higher hydrogenolysis activity.
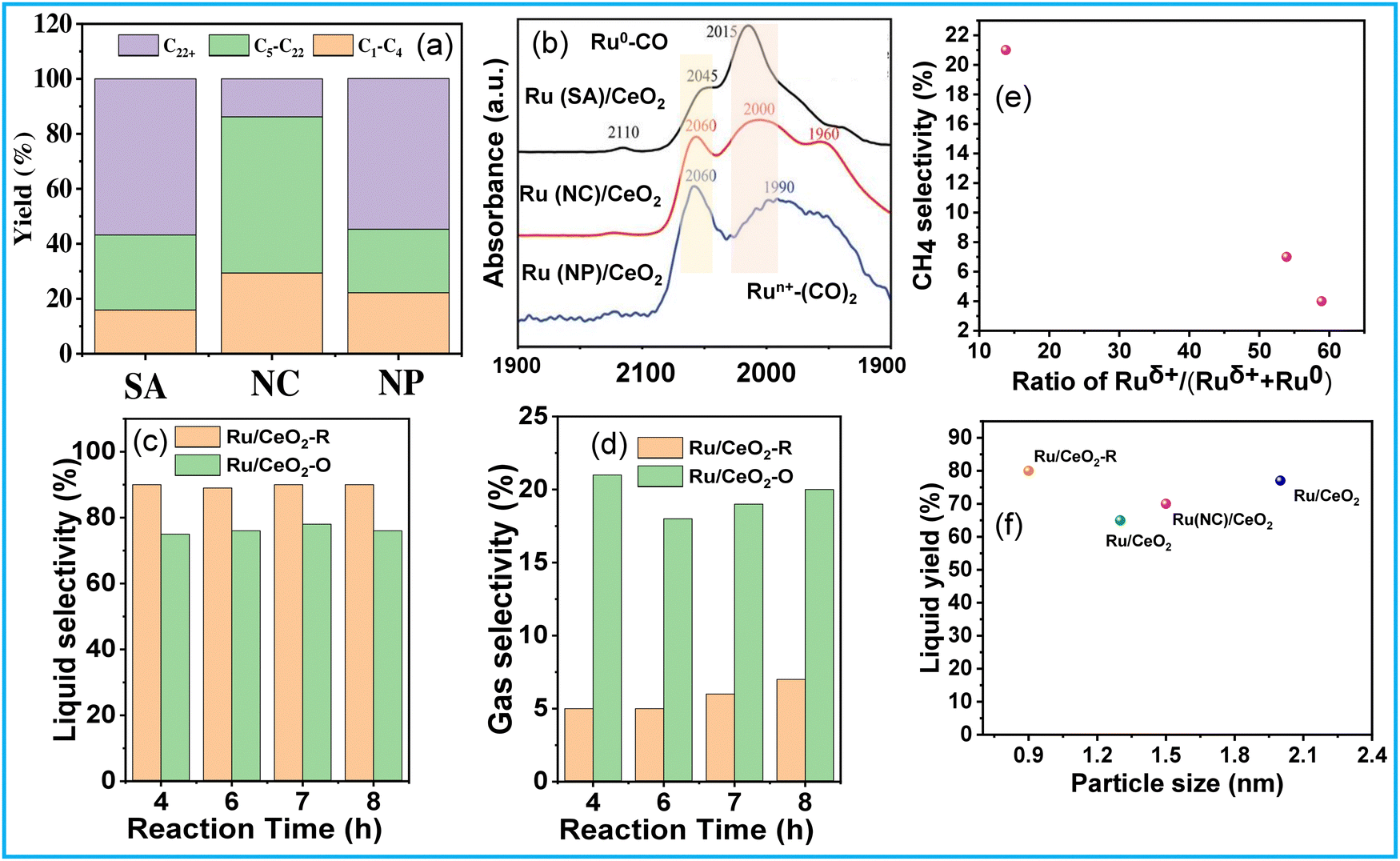 | ||
| Fig. 6 (a) Hydrogenolysis of LDPE over different catalysts and (b) CO DRIFTS spectra of Ru/CeO2 catalysts; data collected and reproduced with permission from ref. 51. Copyright 2023, John Wiley and Sons. (c) and (d) Catalytic performance and product selectivity over Ru/CeO2-R and Ru/CeO2-O; and (e) CH4 selectivity vs the ratio of Ruδ+/(Ruδ+ + Ru0). Data collected and reproduced with permission from ref. 52. Copyright 2022, John Wiley and Sons. Comparing particle size and liquid product yield of Ru/CeO2 (f). | ||
The addition of secondary metals could provide additional hydrogen through reverse spillover, thereby facilitating the rate-limiting hydrogenation/desorption processes on the active metal surface or at the interface and also altering the polymer–catalyst interactions, leading to enhanced catalytic activity and selectivity, and potentially changing the overall reaction mechanism. In this regard, Vlachos and co-workers incorporated WOx in Ru/ZrO2 and investigated the difference in the catalytic activity of Ru–Zr and tungstate zirconia-supported Ru (Ru–WZr) catalysts towards LDPE hydrogenolysis at 250 °C and 50 bar H2 for 2 h.54 Interestingly, the CH4 yield was ∼16% over the Ru–Zr catalyst and decreased to ∼7.5% and ∼4.9% over Ru–15WZr and Ru–25WZr catalysts, respectively. When moving from Ru–Zr to Ru–25WZr catalyst, the gasoline selectivity increased from 25% to 28%, jet fuel selectivity increased from 33% to 42%, diesel fuel selectivity increased from 47% to 66%, and the selectivity towards wax/lubricant based-oil increased from 22% to 35%. Such interesting trends demonstrate that the incorporation of WOx in the Ru–Zr catalyst decreased the formation of gaseous CH4 and increased the formation of value-added liquid hydrocarbons. The hydrogenolysis of LDPE over a supported Ru surface (Ru–Zr) is shown in Fig. 7(a) and (b). First, dehydrogenation of the polyethylene chain occurs on the Ru surface, followed by Ru-catalyzed C–C bond scission to produce short-chain alkyl intermediates. These intermediates can either undergo hydrogenation/desorption to produce shorter chain alkanes, or they can undergo further dehydrogenation followed by C–C bond scission to produce even shorter chain alkyl intermediates. The continuation of dehydrogenation and C–C bond scission processes on the Ru surface eventually produces CH4. The hydrogenation/desorption of alkyl intermediates was slower than the dehydrogenation followed by C–C bond scission steps under low H2 pressure, resulting in a relatively high CH4 yield. This hypothesis was also supported by the hydrogenolysis experiment performed at higher H2 pressure (130 bar), demonstrating a significant reduction in CH4 formation. Interestingly, (WOx)n clusters provide additional hydrogen through reverse spillover, facilitating the rate-limiting hydrogenation/desorption processes on the Ru/ZrO2 interface and also altering the polymer–catalyst interactions, leading to enhanced catalytic activity and affording lower CH4 yield even at low H2 pressure. H2 first dissociates on the Ru surface and subsequently spills over to the (WOx)n clusters and is stored as surface hydroxyls (Fig. 7(b)). These hydroxyl groups act as hydrogen storage sites and provide active hydrogen to the Ru sites via the reverse spillover effect, facilitating the hydrogenation of alkyl intermediate species. Hence, WOx clusters increase the hydrogen-storage capacity and increase the rate of hydrogenation/desorption of alkyl intermediates under lower H2 pressure, significantly reducing CH4 formation.
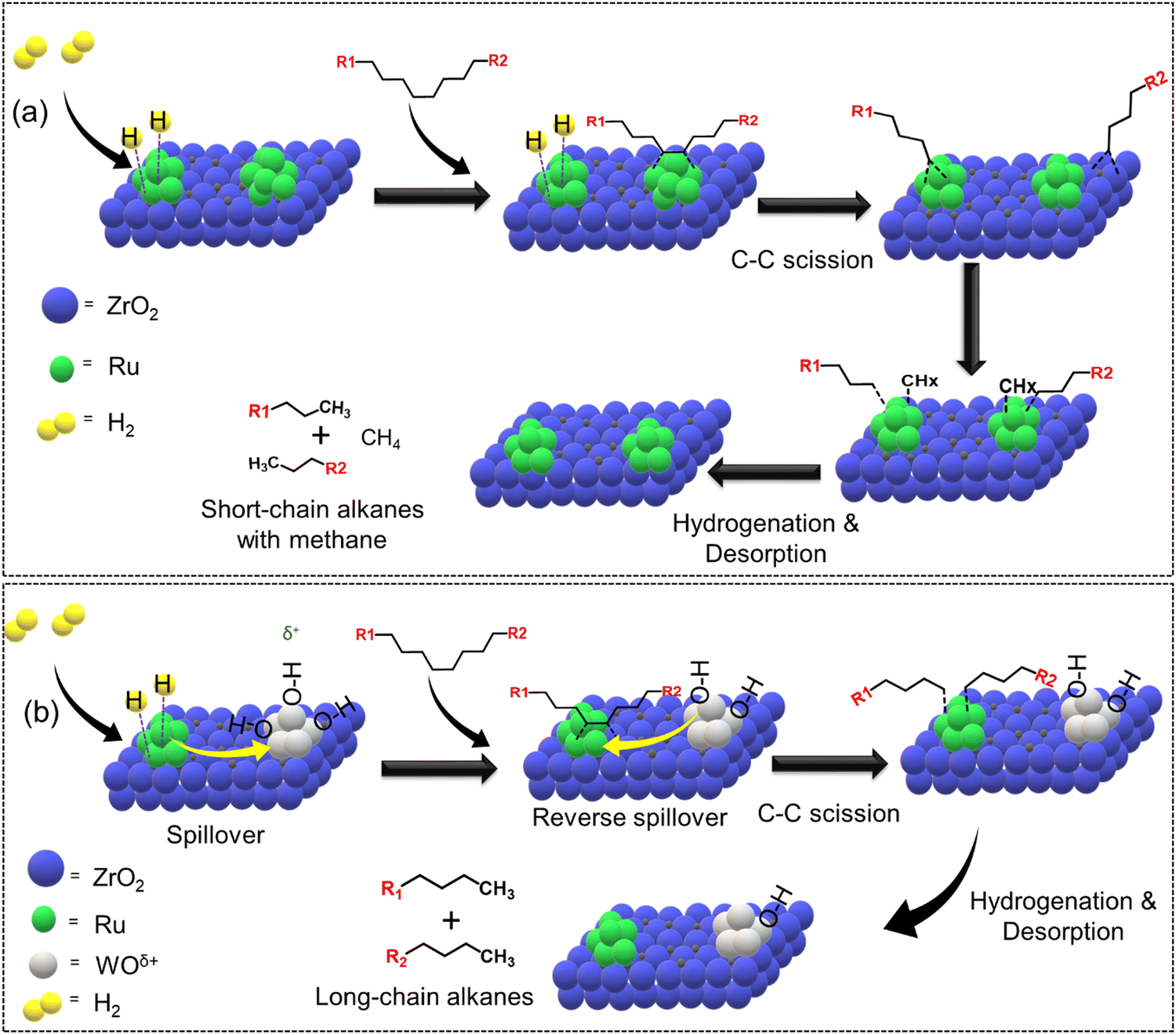 | ||
| Fig. 7 Selective hydrogenolysis of LDPE over Ru–Zr and Ru supported on a tungstated zirconia (Ru–WZr) catalyst. (a) Proposed LDPE hydrogenolysis mechanism over Ru–Zr and (b) Ru–W–Zr catalysts. Data collected and reproduced with permission from ref. 54. Copyright 2021 American Chemical Society. | ||
In addition to the metal oxide support, a neutral carbon support was also employed in this transformation. Rorrer et al. investigated the hydrogenolysis of PP of different molecular weights and a mixture of PP and HDPE over a Ru/C catalyst.55 Under the optimized experimental conditions of 225 °C and 20 bar H2, 700 mg of PP pallets (Mw = ∼12 kDa) was converted into iso-alkanes of carbon chain length ranging from C8 to C42 with C24 as the major product (504 mg), along with various gases as minor products (148 mg). The authors also optimized the experimental conditions (including temperature, pressure, and time) for the hydrogenolysis of isotactic PP (Mw = 340000 Da), and observed the maximum liquid hydrocarbon (iso-alkane) yields under the optimized experimental conditions of 225 °C, 50 bar H2 and 24 h. Finally, the hydrogenolysis of mixed plastic waste (PP + HDPE) under optimized experimental conditions was also investigated, and linear and branched alkanes were observed in the product stream.
Jia et al. investigated HDPE hydrogenolysis to find suitable conditions to produce liquid hydrocarbon fuels and lubricants over carbon-supported transition metal catalysts.56 Several carbon-supported metal catalysts (M/C) were employed, including 5 wt% M/C (M = Cu, Fe, Ni, Pt, Pd, Rh, and Ru) at 220 °C and 3 MPa H2 for 1 h, and among these catalysts, only 5 wt% Ru/C was active, affording 60.8 wt% C8–C16 hydrocarbons (jet fuel range liquid hydrocarbons) and 14.1 wt% C17–C22 hydrocarbons (diesel fuels). The most interesting results were obtained while studying the effect of solvent. Almost negligible HDPE conversions were observed in H2O, n-pentane, and decalin, whereas n-hexane and methylcyclohexane were highly effective. In n-hexane, ∼60 wt% C8–C16 jet-fuel range liquid hydrocarbons and ∼10 wt% C17–C22 diesel fuel range hydrocarbons were obtained, whereas in methylcyclohexane, ∼38 wt% C23–C28 lubricant range hydrocarbons were observed as the major products, along with ∼15 wt% C17–C22 and ∼20 wt% C8–C16 range hydrocarbons. The solvation ability of HDPE in various solvents was key in determining the product distributions. For instance, HDPE has negligible solubility in H2O, thereby resulting in negligible product formation. Similarly, HDPE has lower solubility in supercritical pentane at 220 °C (the critical temperature of pentane is 196.45 °C, which is lower than the reaction temperature); therefore, negligible products were formed in pentane.57 HDPE adopts a fully extended conformation in decalin due to its high affinity towards decalin, which prevents HDPE from reaching the catalyst surface, resulting in negligible conversion. In contrast to these solvents, higher reactivity in n-hexane and methylcyclohexane suggests that the lower affinity of HDPE with these solvents forces the HDPE molecule to turn into a coil, which then sieves through the solvents to reach the catalyst surface and undergo cracking.
Other than Ru-based catalysts, Pt-based catalysts were also used in this transformation. Celik et al. synthesized Pt/SrTiO3 catalysts with varying Pt particle sizes (1.2, 2.3, and 2.9 nm) via the atomic layer deposition (ALD) process, wherein by simply increasing the ALD cycles they were able to increase the particle size. They subsequently investigated the catalytic performances of these materials in the solvent-free hydrogenolysis of PE by employing 170 psi H2 pressure at 300 °C for 96 h.58 The catalytic data revealed complete PE conversion into gaseous hydrocarbons (C1–C8) over 1.2 nm Pt particles. On the other hand, high-quality liquid products with narrow size distribution were produced over 2.3 nm and 2.9 nm Pt particles with 42% and 95% yields, respectively. The formation of high-quality liquid products with narrow size distribution over bigger particles suggested that bigger Pt particles were active for the internal C–C bond cleavage in the PET molecules. Higher gaseous product formation over smaller particles suggested that smaller Pt particles were active for the terminal C–C bond cleavage. The authors also compared the activity of 2.9 nm SrTiO3 supported Pt particles with that of the commercially available Pt/Al2O3 catalyst and found that the Pt/Al2O3 catalyst afforded a broad distribution of liquid products along with a significant proportion of gaseous products compared to the Pt/SrTiO3 catalyst. Additionally, Pt particle size increased from 1.2 nm to 1.6 nm during PE hydrogenolysis in the case of the Pt/Al2O3 catalyst. On the other hand, only a minor increment in Pt particle size, from 2.0 nm to 2.1 nm, was observed in the case of the Pt/SrTiO3 catalyst. The authors proposed that the epitaxial growth of Pt particles over SrTiO3 due to the close lattice match between cubic SrTiO3 and FCC Pt resulted in the strong metal–support interaction, hence controlling the sintering of Pt particles during PE hydrogenolysis. Moreover, the authors also proposed that the higher selectivity towards liquid products could be due to the unique geometrical features of Pt particles that were obtained via the epitaxial growth. Later, the same group demonstrated that the hydrogenolysis activity of the Pt/SrTiO3 catalyst at 300 °C and 170 psi H2 pressure significantly varies depending on the microstructure of the polymer chain, which is defined as the length and density of branching in the polymer.59,60 These interesting findings suggested that comprehensive structural characterization of the polymers present in anthropogenic plastic waste is important for subsequent optimization of catalyst structure and valorization processes.
The size-dependent property of encapsulated Pt NPs was investigated for wax formation. Wu et al. synthesized mesoporous silica shell-surrounded Pt NPs supported on a solid silica sphere (mSiO2@Pt-X/SiO2; X is the mean NP diameter), and employed them in PE hydrogenolysis at 300 °C and 0.89 MPa H2 pressure. The authors’ main objective was to investigate the combined effects of the mesoporous shell, confinement of the Pt particles inside the mesoporous shell wall, and the effect of Pt NP size on PE conversion and product distribution (Fig. 8).61 PE conversion and the yield of extracted wax were inversely proportional to the size of encapsulated Pt particles, such that after 12 h, mSiO2/Pt-1.7/SiO2 afforded 62% PE hydrogenolysis and 50.5% extractable wax, whereas mSiO2/Pt-5.0/SiO2 afforded 26% PE hydrogenolysis and 17.5% extractable wax. In contrast, the carbon number distribution of the extracted wax was independent of the size of Pt NPs; the mean size of the carbon chain was C23 in the extracted wax in all cases. The hydrogenolysis performance of the non-encapsulated Pt particles (Pt/SiO2) was also scrutinized and was found to be much lower than that of mSiO2/Pt-X/SiO2. The higher hydrogenolysis activity of mSiO2/Pt/SiO2 compared to the non-encapsulated Pt/SiO2 catalyst was attributed to the protective features of the mesoporous SiO2 shell preventing the sintering of Pt particles. The conformation of the PE chain governed the average carbon chain length in the extracted wax adsorbed inside the mesopores.
 | ||
| Fig. 8 TEM images of the mSiO2/Pt/SiO2 catalyst; product distribution as a function of Pt particle size; and the mechanism of the hydrogenolysis of the PE molecule. Data collected and reproduced with permission from ref. 61. Copyright 2022 American Chemical Society. | ||
Similarly, Li et al. synthesized Pt-encapsulated and Pt-supported SiO2 catalysts.62 The first material was prepared by supporting Pt NPs on the external surface of a silica support (Pt/Si-1), and the second material was prepared by encapsulating Pt NPs inside the mesoporous silica shell (Pt@Si-1). These materials were combined with beta zeolite via mechanical grinding, and the resulting composites were employed as catalysts in LDPE hydrocracking at 250 °C and 3 MPa H2. Interestingly, the catalytic data revealed higher liquid hydrocarbon yield (∼90%) with narrow alkane distribution (C5–C9) in the case of the Pt@Si-1 and zeolite beta composite catalyst. In contrast, the Pt/Si-1-based composite, where Pt NPs were present on the external silica surface, afforded only ∼70% yield of liquid hydrocarbons and the formation of gaseous and solid products. The authors proposed that when Pt particles were present on the external silica surface (Pt/Si-1), all olefin intermediates produced via hydrocracking underwent hydrogenation to produce liquid, solid, and gaseous products. However, when Pt particles were encapsulated inside the silica shell, then only the selected olefin intermediates that could fit inside the silica mesopores were able to reach the Pt particle surface, hence resulting in improved selectivity towards specific liquid hydrocarbon products (C5–C9).
The gaseous product formation over the supported Co catalysts has been investigated. For example, Zichittella et al. investigated cheaper Co-based catalysts, including Co/SIRAL (SIRAL = amorphous silica-alumina), Co/SiO2, Co/CeO2, Co/ZrO2, Co/TiO2, and Co/ZSM-5, for the hydrogenolysis of PE (Mw = 4000 Da) at 250 °C and 4 MPa H2 for 20 h.63 Among these catalysts, Co3O4 and Co/ZSM-5 afforded the highest weight percentage of gaseous products (∼85–90%), whereas other supported Co catalysts were active towards the formation of solid products (∼80–90%). Further, Co3O4 afforded CH4 (∼90 wt%) and Co/ZSM-5 afforded C3H8 (∼85 wt%) as the major gases. The analytical results revealed that the higher dispersion of oxidic Co2+ on ZSM-5 and the higher acidic strength of the ZSM-5 support were responsible for the scission of the internal C–C bond in the PE polymer, forming high-value C3H8 gas. Vance et al. investigated the effect of Ni loading (1–100 wt%) on SiO2 and found that 15 wt% Ni/SiO2 afforded the highest liquid yield (∼50%) during LDPE hydrogenolysis at 300 °C and 3 MPa H2.95 The LDPE hydrogenolysis occurred via internal scission due to the higher probability of interior C–C bonds on the catalyst surface.
3.2. C–C activation and hydrocracking of polyolefins to C5–C12 hydrocarbons
Hydrocracking requires bi-functional catalysts, where metal sites perform dehydrogenation to form an olefin intermediate. The olefin intermediate is then protonated by the Brønsted acid sites to produce a carbenium ion, which undergoes cracking and isomerization followed by hydrogenation over metal sites to produce shorter chain hydrocarbons. Although hydrocracking requires relatively high temperatures than hydrogenolysis, the obtained liquid product has shorter hydrocarbon chains in the gasoline range and a narrow distribution of carbon chain length (Table S2, entries 15–22, ESI†).The effect of transition metals and the textural properties of acidic supports in bi-functional catalysts have been investigated. Kots et al. investigated the catalytic behavior of several TiO2-supported metal nanoparticles, including Pd, Rh, Ir, Ni, Pt, and Ru, in the hydrocracking of isotactic polypropylene (i-pp) at 250 °C and 3 MPa H2 for 16 h.64 Among these catalysts, only Ru/TiO2 afforded the highest liquid hydrocarbon yield (65.6%). Linxiao et al. reported an efficient polyolefin hydrocracking over Ru and Ni supported on a sol–gel anatase TiO2 (TiO2-A-SG).65 The findings revealed that the limited Brønsted acid sites on TiO2-A-SG significantly enhance hydrocracking activity, thereby improving selectivity towards high-value C4–C20 hydrocarbons and increasing isomerization efficiency in comparison to Ru-based monofunctional hydrogenolysis catalysts. Hydrocracking on Ru/TiO2-A-SG is more effective under lower H2 pressures, reduced Ru loadings, and with substrates that have a higher degree of branching. The abundance of tertiary carbons (3C) in these substrates promotes rapid β-scissions and suppresses hydrogenolysis. Furthermore, the authors observed that Ni supported on TiO2-A-SG, a non-noble metal catalyst, exhibits superior activity and selectivity for polypropylene (PP) hydrocracking compared to conventional Brønsted-acid supports like zeolites.
Utami et al. synthesized several ZrO2-based catalysts, including sulphated-ZrO2 (SZ) and platinum-supported sulfated-zirconia (Pt/SZ).66 These catalysts were employed for the hydrocracking of low-density polyethylene waste (LDPE) at 250 °C under a H2 flow rate of 20 mL min−1. The catalytic data revealed that Pt/SZ catalysts were highly active for the hydrocracking of LDPE into gasoline-range hydrocarbons (C5–C12 hydrocarbons including olefins, linear paraffins, isoparaffins, and naphthenes). Further, the yield of gasoline-range hydrocarbons increased with an increase in Pt loading from 1 wt% (Pt1/SZ) to 3 wt% (Pt3/SZ), with the Pt3/SZ catalyst affording 67.5% C5–C12 hydrocarbon yield. The higher hydrocracking efficiency of Pt/SZ catalysts was attributed to the higher acidity compared to the bare ZrO2 and SZ supports. Further, the acidity increased with Pt loading. Hence Pt3/SZ afforded the highest hydrocracking efficiency and C5–C12 hydrocarbon yield. H2 dissociation on the Pt surface produced H-atoms, which spilled over to the SZ support and reacted with Lewis acid sites to produce H+ species acting as acid sites.
Liu et al. synthesized multi-functional catalysts by combining Pt supported on tungstate-zirconia (Pt/WO3/ZrO2) and zeolites possessing different Si/Al ratios and micropore sizes and investigated the effect of the multi-functional characteristics of these catalysts on product distribution during LDPE hydrocracking at 250 °C under H2 flow (100 mL min−1 equimolar flow of H2 and He) for 2 h.67 Pt/WO3/ZrO2 afforded ∼20% yield of lighter hydrocarbons (C1–C12), ∼9% yield of intermediate hydrocarbons (C13–C14), and ∼10% yield of heavier hydrocarbons (>C17) along with a huge amount of unconverted solids (∼40% yield). HY zeolite, on the other hand, was much less active, affording less than 10% yield of lighter hydrocarbons, mostly between C1 and C12, along with a significant amount of unreacted solids (greater than 90%). Interestingly, the yield of unreacted solids reduced to ∼2% and the yield of gasoline-range liquid hydrocarbons (C5–C12) rose to ∼80% along with a small percentage of lighter hydrocarbons (C1–C4) and diesel-range hydrocarbons (C13–C16) with ∼7% and ∼3% yields, respectively, over the composite catalyst (Fig. 9(a)). The plastic polymer first undergoes hydrocracking into intermediate range hydrocarbons (C∼13) over the bi-functional Pt/WO3/ZrO2 catalyst; subsequently, these intermediate hydrocarbons further undergo hydrocracking inside the micropores of HY zeolites, producing gasoline-range hydrocarbons (Fig. 9(c)). HZSM-5, with a smaller pore size than HY, was selective for C1–C4 range lighter hydrocarbons (70% selectivity), suggesting that the product distribution during the hydrocracking of C∼13 hydrocarbons can be regulated by tuning the pore size of zeolites (Fig. 9(b)).
 | ||
| Fig. 9 Hydrocracking of LDPE using Pt/WOx/ZrO2 and solid acid catalysts. (a) The product distribution of different zeolites mixed with Pt/WO3/ZrO2 in LDPE hydrocracking; (b) effect of solid acid catalyst pore diameter on C1–C4 hydrocarbon yield; (c) illustration of key intermediates diffusing over the Pt/WO3/ZrO2 + HY(30) catalyst. Data collected and reproduced with permission from ref. 67. Copyright 2021, AAAs. | ||
To systematically develop bifunctional metal/zeolite catalysts for the hydrocracking of hydrocarbons, it is essential to balance the metal and acid sites. Jumah et al. investigated the effect of support acidity on liquid hydrocarbon production.68 For example, they synthesized Pt-impregnated zeolite beta (Si/Albulk 12.5 and 175) and zeolite USY (Si/Albulk 6 and 15) with different Si/Al ratios in the hydrocracking of squalene and LDPE (Mw ∼ 150000 g mol−1). The hydrocracking of squalene over 1 wt% Pt-beta catalyst with a lower Si/Albulk ratio (12.5) at 275 °C and 2 MPa H2 afforded a greater yield of lighter hydrocarbons (C3 to C6). The catalyst with a higher Si/Albulk ratio (300) afforded greater yields of heavier hydrocarbons (C7–C11). In contrast, an opposite trend was observed over 1 wt% Pt-USY catalyst. In this case, the catalyst with a higher Si/Albulk ratio of 15 afforded a higher yield of lighter and intermediate hydrocarbons (C5–C9). On the other hand, the catalyst with a lower Si/Albulk ratio of 6 afforded a higher yield of heavier hydrocarbons (C11–C20). The authors observed that the variation in the textural and acidic properties of zeolite with different Si/Al ratios across various zeolite frameworks leads to differences in product selectivity. The hydrocracking of LDPE at 315 °C and 2 MPa H2 was also investigated, and 1% Pt-beta was more active than 1% Pt-USY in producing gasoline range hydrocarbons.
Rorrer et al. synthesized supported Ru catalysts with 5 wt% metal loading, and investigated the effect of support acidity on product selectivity during the hydrogenolysis of PE (avg. Mw 4000 Da) at 200 °C under 3 MPa H2 for 16 h.69 Based on the catalytic data, Ru/HBEA and Ru/FAU possessing a higher number of acid sites afforded liquid alkanes (C5–C33) with 51% and 67% mass yields, respectively, compared to the catalysts with lower acidity, Ru/SiO2, Ru/SIRAL30, and Ru/MWW. The authors proposed that catalysts with a higher number of acid sites promote the internal C–C bond cleavage, resulting in the formation of liquid hydrocarbons. Lee et al. investigated the activity of silica-alumina catalysts for hydrocracking n-hexadecane (as a model substrate) and polyethylene. Initially, the hydrocracking performances of SiO2–Al2O3, HY, and ZSM-5 supports were investigated at three different temperatures, 275 °C, 325 °C, and 375 °C, at 4.5 MPa H2 for 2 h.70 The n-hexadecane conversion ranges between ∼2 and 4% over SiO2–Al2O3 at different temperatures. Over HY, the n-hexadecane conversion increased from 4.5% to 26.7% on increasing the temperature from 275 °C to 375 °C. ZSM-5 was demonstrated to be the most active catalyst, with conversion increasing from 13.7% to 98.0% on increasing the temperature from 275 °C to 325 °C. Interestingly, the most active ZSM-5 afforded a higher yield of C1–C4 hydrocarbons (77.3%) than C5–C16 hydrocarbons (20.8% yield). ZSM-5 was further chosen as a support to prepare Co-, Ni-, and Ru-supported ZSM-5 bi-functional catalysts. Among these, Ru/ZSM-5 afforded the maximum 99.9% conversion and 99.2% yield for C1–C4 range hydrocarbons. Ding et al. synthesized Ni sulfide loaded on a hybrid support, which contains a mixture of H-ZSM-5 and silica-alumina (the hybrid support abbreviated as HSiAl).71 The resulting catalyst, Ni/HSiAl, was employed for the hydrocracking of HDPE and real plastic waste at 375 °C and 1000 psig H2. The catalyst afforded complete HDPE conversion and ∼40% liquid hydrocarbon yield in 1 h. Similarly, the catalyst also afforded the complete conversion of post-consumer plastic waste and gave ∼50% liquid hydrocarbon yield in 1 h. The excellent performance of the Ni/HSiAl catalyst was attributed to the higher resistance to sulfur, nitrogen, and impurities present in post-consumer plastic waste. The catalyst was easily recycled by calcination followed by sulfidation, and the recycled catalyst exhibited a similar activity as the original catalyst.
Regulating the morphology of zeolites, including the use of nanocrystals and plate-like structures, in addition to modifying their pore structure, is an effective approach for reducing diffusion limitations and improving catalyst efficiency in polyolefin cracking. Munir et al. synthesized the composites of zeolite beta and mesoporous silica and investigated the effect of the co-presence of micropores and mesopores in composite catalysts on their hydrocracking performance (Fig. 10(a)).72 Six catalysts, including BC300 (commercially available zeolite beta), BC11.1, BC5.7, and BC0.7 (prepared by desilication), and BC48 and BC27 (prepared by without alkali pre-treatment), were employed for the hydrocracking of a model plastic mixture containing 20 wt% PS (MW = 192000 g mol−1), 30 wt% PP (MW = 250000 g mol−1), 10 wt% LDPE, and 40 wt% HDPE at 360 °C and 2 MPa H2. BC48 afforded a higher yield of liquid hydrocarbons than the BC300 catalyst, which was attributed to the presence of micropores and mesopores in the BC48 catalyst with microporous and mesoporous surface areas of 229.1 m2 g−1 and 198.1 m2 g−1 respectively. Micropores provided acidic sites for hydrocracking, and mesopores facilitated the easier diffusion of the liquid hydrocarbon chain away from the active sites. Zhang et al. demonstrated the self-assembled nanocrystalline ZSM-5 for LDPE hydrocracking. Nano-ZSM-5 provided accessible strong acid sites due to its high external surface area and framework defect (Fig. 10(b)).96 By distinguishing acid strength from acid amount, nano-ZSM-5 exhibited much higher catalytic activity in PP cracking than conventional ZSM-5, even at lower reaction temperatures. Similarly, Duan et al. demonstrated the efficiency of ZSM-5 nanosheets (s-ZSM-5) to achieve light hydrocarbon (C1–C7) yield of up to 74.6% and 83.9% of C3–C6 olefins with minimal coke formation (<1 wt% weight loss) at 280 °C.97 In contrast, nano-ZSM-5 (n-ZSM-5) showed approximately 27% weight loss due to coke formation. This work indicates the significant potential of ZSM-5 zeolite with controlled b-axis direction growth for polyethylene (PE) upcycling (Fig. 10(c)). The shorter diffusion path of s-ZSM-5 compared to nano-ZSM-5 efficiently suppressed intermediate accumulation on the zeolite surface, minimizing coke formation and providing a viable approach for handling PE plastic waste. Furthermore, the involvement of hydrogen in the hydrocracking process inhibits the condensation and formation of polycyclic molecules within the micropores and facilitates rapid diffusion.
 | ||
| Fig. 10 (a) Catalytic performance of plastic mixture conversion over different catalysts at 360 °C, and textual properties of the catalysts. Data collected and reproduced with permission from ref. 72. Copyright 2020. (b) NH3-TPD profiles (a = CZ5, b = N-Z5, C = NS-Z5) and textual properties of the catalysts. Data collected and reproduced with permission from ref. 96. Copyright 2013 Elsevier B.V. (c) Depolymerization of PE over various zeolite catalysts; picture of PE and SEM image of s-ZSM-5 displaying its panel morphology, along with a schematic illustration depicting the cascade cracking steps occurring on the external surface and within the micropores of n-ZSM-5 and s-ZSM-5 zeolite catalysts. Data collected and reproduced with permission from ref. 97. Copyright 2022 American Chemical Society. | ||
From the above discussion, it is evident that hydrogenolysis and hydrocracking of POs are conducted using supported monometallic and bifunctional catalysts, with the goal of selectively producing liquid fuel-range hydrocarbons. Comparative analyses of reaction temperature and duration across various studies have revealed that hydrogenolysis occurs at relatively mild temperatures, resulting in higher liquid product yields than hydrocracking. Noble metals such as Ru, Ni, Pt, and Rh are commonly employed to produce liquid hydrocarbons under these mild conditions. Hydrogenolysis of POs results in a broad distribution of liquid fuel-range hydrocarbons with relatively high carbon numbers than hydrocracking. Studies have focused on the effects of metal particle size and metal–support interactions on liquid hydrocarbon yields. Supported Ru- and Pt-based catalysts with optimal nanoparticle sizes have shown higher liquid hydrocarbon productivity during polyolefin hydrogenolysis. The catalytic selectivity is influenced by the active metal's chemical state, which is modulated by metal–support interactions. Positively charged metal species, generated by strong metal–support interactions, facilitate internal C–C bond cleavage, reducing methane production. The metal–support interaction and size can be fine-tuned by using different supports, supports with various morphologies, or by adjusting the support calcination temperature, all of which significantly influence product selectivity. Similar to hydrogenolysis, the main focus of PO hydrocracking is also the selective production of liquid fuel-range hydrocarbons. This process investigates bifunctional catalysts comprising various transition metal nanoparticles and acidic supports with different textural properties. The choice of metal and acidic support, along with the support's surface area and porosity, greatly affects the productivity of short-chain hydrocarbons during hydrocracking.
4. Summary and future outlook
The chemical upcycling of waste plastic into valuable carbon feedstocks is crucial for advancing a sustainable and circular carbon economy. Catalytic hydrogenolysis and hydrocracking are key techniques for producing selective gases, liquid hydrocarbons, and high-value aromatic chemicals. A major challenge is to identify catalysts that selectively cleave C–O and/or C–C bonds while minimizing unwanted byproducts. This review highlights research efforts to develop efficient catalysts and catalytic systems for converting aromatic plastics and polyolefins into value-added chemicals and liquid fuels. Aromatic plastics, with C–O and C–C bonded aromatic units, require selective bond cleavage and preservation of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functionalities to produce arenes. For jet fuel-range cycloalkanes, complete hydrogenation of the CC functionality and cleavage of the C–O bond are necessary, often forming undesired products. Polyolefins contain internal and terminal C–C bonds, with internal C–C bond cleavage yielding liquid fuels and lubricants, and terminal C–C bond cleavage predominantly producing low-value methane. Thus, promoting internal C–C bond cleavage is crucial for selectively enhancing fuel and lubricant production, highlighting the importance of understanding the factors influencing C–C bond cleavage positions. The review compiles the literature on hydroprocessing of common plastic polymers, such as PET, PC, and POs, thoroughly investigating the structure–function relationships of catalysts used to produce selective liquid hydrocarbons. It emphasizes the key characteristics of catalysts that enhance the selective activation of C–C and C–O bonds, improving catalytic efficiency. The review also showcases state-of-the-art chemical recycling processes and aims to provide new insights for developing novel heterogeneous catalysts for future advancements in chemical recycling. To advance a sustainable future for plastics, a stronger focus on catalytic design and multidisciplinary collaboration among science, engineering, and materials is required. To support the sustainable processing of real-life plastic waste and enhance economic viability and industrial feasibility, we propose designing new catalysts and catalytic pathways based on the current knowledge of C–O and C–C bond cleavage in plastic waste.
C functionalities to produce arenes. For jet fuel-range cycloalkanes, complete hydrogenation of the CC functionality and cleavage of the C–O bond are necessary, often forming undesired products. Polyolefins contain internal and terminal C–C bonds, with internal C–C bond cleavage yielding liquid fuels and lubricants, and terminal C–C bond cleavage predominantly producing low-value methane. Thus, promoting internal C–C bond cleavage is crucial for selectively enhancing fuel and lubricant production, highlighting the importance of understanding the factors influencing C–C bond cleavage positions. The review compiles the literature on hydroprocessing of common plastic polymers, such as PET, PC, and POs, thoroughly investigating the structure–function relationships of catalysts used to produce selective liquid hydrocarbons. It emphasizes the key characteristics of catalysts that enhance the selective activation of C–C and C–O bonds, improving catalytic efficiency. The review also showcases state-of-the-art chemical recycling processes and aims to provide new insights for developing novel heterogeneous catalysts for future advancements in chemical recycling. To advance a sustainable future for plastics, a stronger focus on catalytic design and multidisciplinary collaboration among science, engineering, and materials is required. To support the sustainable processing of real-life plastic waste and enhance economic viability and industrial feasibility, we propose designing new catalysts and catalytic pathways based on the current knowledge of C–O and C–C bond cleavage in plastic waste.
4.1. Current understanding of C–O and C–C activation and production of liquid hydrocarbons from plastic waste
Polyolefins consist of two types of C–C bonds: internal Csecondary–Csecondary and terminal Csecondary–Cprimary bonds.51,52 Selectively cleaving these bonds is challenging due to the inertness of C–C and C–H bonds, and the mechanisms of their activation is somewhat unclear. Aromatic plastics such as PC, PET, PPO, and PS, which have C–O and C–C bond linkages, can produce various products through selective bond cleavage.25,37–42Hydrogenolysis of plastic waste cleaves C–O and C–C bonds, generating reactive intermediates often formed by breaking C–C/C–O bonds in polyolefins and polyaromatics. On late transition metal surfaces and nanoparticles, these reactions involve the loss of several H atoms, facilitating charge transfer between the metal surface and reactive intermediates, which lowers the intrinsic ΔH‡ for breaking strong bonds. This process also produces hydrogen gas, reducing ΔS‡ through entropic contributions.98,99 For hydrogenolysis of aromatic plastics and C–C/C–O bond cleavage, Ru- or Co-supported catalysts have been found to be more active and selective for arene formation than Pt and Pd. Supports with Lewis acid/Brønsted acid sites enhance C–O/C–C bond activation and cleavage. For example, a Ru/Nb2O5 catalyst achieved a high total yield of arenes (approximately 80%) during PET hydrogenolysis. Sub-nanometer Ru particles prevented aromatic ring hydrogenation, while Nb2O5 Lewis acid/Brønsted acid sites facilitated C–O/C–C bond activation.25 Positively charged Ru species favored hydrogenolysis over decarboxylation. The geometric and electronic effects of Ru, combined with support effects, influence catalytic activity and selectivity for C–C and C–O bond cleavage. In addition, the concentration of Ru NPs on the edge or terrace sites of the support significantly affects the energy barrier for ring hydrogenation and ring opening, thereby altering catalytic efficiency and product selectivity.43 The addition of a poisoning solvent and the use of metal or acid-functionalized catalysts can enhance selectivity towards C–C bond cleavage over C–O bond cleavage. This is achieved by modifying the stability and coordination preferences of reactive surface species and introducing a secondary solvent (e.g. containing oxygen functionality) that alters the reaction mechanism. For example, a small amount of alcohol incorporated in hexane can occupy Nb2O5 Brønsted acid sites, inhibiting C–O bond cleavage. Moreover, producing jet fuel range hydrocarbons from polyaromatics requires catalytic sites for aromatic ring hydrogenation and C–C/C–O bond cleavage. Multifunctional catalysts, including bimetallic, metal–oxophilic metal oxide, and composite or physically mixed catalysts, show promise for the selective production of jet fuel range cyclic hydrocarbons. Metals with strong hydrogenation capabilities, such as Pt, Pd, Rh, Ru, and Ni, combined with strong Brønsted acid supports like zeolite, provide distinct binding sites (e.g., metal sites associated with acid sites). These configurations modify the adsorption of reactive intermediates, activate sterically hindered C–O bonds, and alter the reaction pathways (e.g., hydrogenation, dehydration, and hydrodeoxygenation) to produce cyclic hydrocarbons. Furthermore, active metals combined with oxophilic metal oxides exhibit unique properties that stabilize transition states and reactive intermediates, enhancing the selective production of cyclic hydrocarbons. For example, oxophilic metal oxides like ReOx, modified with noble metals such as Ru, show proximity between Ru and ReOx species. This promotes a synergistic effect that is highly effective for the cleavage of C–C and C–O bonds and hydrodeoxygenation of polyaromatics.100
The cleavage of C–C bonds in polyolefins and the production of liquid hydrocarbons are achieved using bifunctional catalysts composed of transition metals, acidic surfaces, and nanoparticles. Hydrogenolysis of internal Csec–Csec bonds minimizes methane production and enhances selectivity for liquid hydrocarbons. Despite significant interest in catalytic hydrogenolysis of polyolefins to liquid alkanes, our understanding remains limited due to factors influencing the cleavage positions of internal Csec–Csec and terminal Csec–Cpri bonds. Catalytic selectivity is influenced by the particle size and chemical state of the active metal, which are modulated by support and metal–support interactions. Small, disordered nanoclusters are effective in selective C–C bond scission, whereas larger metal nanoparticles promote stereoisomerization. However, the ultra-small nanoclusters were observed to be less active for Csec–Csec bond session (Fig. 11). Spectral characterization shows that as Ru particle size increases, the intensity of metal–support interaction (MSI) decreases while the hydrogen spillover effect increases. The balance between these two effects enables moderately sized Ru (1.0–1.5 nm) catalysts to achieve optimal catalytic performance. Strong metal–support interactions generate positively charged metal species that facilitate internal Csec–Csec bond cleavage, thereby reducing methane production.52 Metal oxides with identical composition, but different sizes and shapes exhibit variations in oxygen vacancies and electronic properties, leading to distinct metal–support interactions and subsequent differences in catalytic activity. Incorporating a second metal or acid functionality alters activation pathways of reactive surface species and introduces additional catalytic functions, enhancing selectivity towards liquid hydrocarbons. For example, augmenting transition metal catalysts with an oxophilic metal or an acid function increases the hydrogen storage capacity of surface hydroxyls through hydrogen spillover from the active metal phase.54 This increased hydrogen adsorption capacity is essential for hydrogenolysis of long-chain alkyl intermediates, promoting their desorption before over-cracking into methane. Metal nanoparticle size and dispersion, influenced by metal loading, also affect product selectivity. Lower metal loading results in smaller nanoparticles and higher metal dispersion, increasing liquid hydrocarbon yield (Fig. 11(a)–(c)). In the hydrogenolysis of polyolefins on supported metal catalysts, metal nanoclusters demonstrate the highest conversion efficiency and superior selectivity toward liquid alkanes. Reduced metal size enhances metal–support interactions (MSIs) while reducing hydrogen spillover during the reaction. MSI benefits more electronegative metal centers for the activation of C–H and C–C bonds. Moreover, the metal sites are critical for achieving selective liquid hydrocarbon production (Fig. 11(d)). The literature reviewed in this study indicates that Ru- and Pt-based catalysts are most frequently studied and consistently demonstrate high catalytic performance. Additionally, although less explored, Rh-based catalysts have also been found to be effective for liquid hydrocarbon production. Multifunctional metal/zeolite catalysts promote catalytic C–C bond cleavage in polyolefins. In these catalysts, hydrogenation/dehydrogenation components, such as noble metals (Pt, Rh, or Ru), and Lewis-Brønsted acid zeolites, which balance metal and acid sites, are all crucial for selective liquid hydrocarbon production.66,68,69 Acid density plays a significant role in determining mechanistic pathways and product distribution. High Brønsted acid site density promotes an ideal C–C bond cleavage mechanism, favoring non-terminal C–C bond cleavage, preventing methane formation, and accelerating selectivity towards liquid products.69
 | ||
| Fig. 11 Effect of catalyst calcination temperature on particle size and liquid product yield (a); effect of particle size (b) and (c), and effect of metal (d) on liquid product yield. | ||
Consequently, future research should focus on developing multifunctional, selective, and cost-effective catalysts. It includes evaluating and studying the selective cleavage of C–C and C–O bonds, particularly in polyaromatics and polyolefin plastic waste.
4.2. Rational design and optimization of catalysts for future research
The activation of C–C and C–O bonds over heterogeneous catalysts presents substantial opportunities for the selective production of liquid aromatics and hydrocarbons. Hence, advancing plastic chemical recycling technologies relies on developing efficient catalysts characterized by high activity, selectivity, and stability. Single-atom catalysts (SACs), which consist of isolated single atoms or metal nanoclusters of active metal species, have gained prominence over the past decade due to their high metal dispersion and electronic metal–support interaction (EMSI).101–104 These characteristics confer superior activity and high selectivity, along with 100% active site utilization. Compared to nanoparticle-supported catalysts, SACs with low coordination numbers significantly enhance metal utilization, thereby improving both activity and selectivity, particularly in the production of arenes.105–107 The selection and ratio of metal catalysts are also critical for the selective depolymerization of plastic polymers. Economic transition metals such as Co, Cu, Ni, W, Re, or Fe can be combined or alloyed with precious metals like Pt, Pd, or Ru to potentially enhance activity and selectivity compared to their monometallic counterparts.108 Moreover, the catalyst support plays a vital role in influencing catalytic activity through steric and electronic effects.109 Thus, the design of catalyst systems that integrate optimal metal combinations and supports is essential for efficient plastic recycling processes.Moreover, ensuring the stability of heterogeneous catalysts is a challenge in upcycling waste plastics into valuable chemicals and fuels. Recent advances in designing stable catalysts are predominantly derived from investigations of high-temperature gas-phase reactions employing oxide materials, including zeolites, Al2O3, SiO2, etc. These materials are found to have excellent thermal stability and are optimized for durability in petrochemical processes. Oxide materials combined with oxophilic metals (e.g., Nb, Ti, Zr, W, Mo) can facilitate the activation of plastic materials through strong adsorption. For instance, the selective cleavage of C–O and C–C bonds in aromatic plastics (e.g., PET, PC, PS, PPO) is critical for producing aromatics while preserving aromatic functionalities.25 Niobium-based porous solids, which contain NbOx and Brønsted acid sites, have shown the ability to catalyze the cleavage of C–O and C–C bonds under an H2 atmosphere, achieving an arene selectivity of up to 70%.25,43 Valuable insights for designing new catalysts emerge from these promising systems: (1) enhancement of C–O and C–C bond cleavage can be achieved by constructing cooperative catalytic sites, such as the combination of early transition metals with strong Brønsted acid sites, (2) preventing the hydrogenation of benzene rings requires weakening their adsorption on catalyst supports and (3) employing bimetallic nanoparticles to optimize active sites significantly improves hydrogen dissociation. However, Nb is costly and has low abundance. Nonetheless, beyond Nb2O5, other oxides such as TiO2, ZrO2, WOx, and MoOx can also hold potential for achieving this goal due to their ability to activate the C–C/C–O bond and high oxophilicity.
Zeolites, characterized by their crystalline nanoporous structure, possess significant specific surface areas, well-defined porous channels, and adjustable acid sites.95,110,111 Pd, Pt, Ru, Ni, and Co supported on high surface area zeolites with abundant acid sites (e.g., ZSM-5, H-Beta, H-Y) can facilitate hydrogenolysis, deoxygenation, isomerization, and cracking of hydrocarbon chains in single-step processes.112 Zeolites exhibit strong Brønsted acid sites, facilitate the activation of C–OH bonds in cyclic alcohols, and enhance selectivity towards cycloalkanes.55,113 Adjusting parameters such as acidity and porosity in zeolites can significantly enhance catalytic activity and product selectivity under relatively mild conditions. Introducing hierarchical porosity into zeolites, either by incorporating mono-quaternary or multi-quaternary structure-directing agents (SDAs) or using growth inhibitors like silanes, can induce mesoporosity within the zeolite framework. This modification increases the material's surface area, improves mass transfer capabilities, and facilitates the transformation of bulky molecules. Acid treatments, such as dealumination, and base treatment, such as desilication, modify the zeolite structure and its physicochemical properties, reducing extra framework aluminum species and the formation of larger pores, collectively enhancing the zeolite's stability and product selectivity.114–116
Hydroprocessing of polyolefins (POs) via hydrogenolysis and hydrocracking produces liquid fuels, gases, and lubricants/waxes, differing in experimental conditions and active sites for C–C bond cleavage. Hydrogenolysis occurs at lower temperatures with metallic active sites, cleaving H–H bonds in H2 and C–C bonds in POs to form C–H terminal bonds.51,52 Hydrocracking requires bifunctional catalysts: metal sites dehydrogenate POs to olefins, which are then protonated on Brønsted acid sites to form carbocations.68,69 These carbocations undergo C–C bond cracking and hydrogenation on metal sites (e.g., Ru, Pt, Rh, Ni, Co), yielding shorter hydrocarbons. Hydrogenolysis generally produces hydrocarbons with a broad chain length distribution (C12–C22), typical of diesel fuel. Conversely, hydrocracking typically yields hydrocarbons with narrower chain lengths (C5–C12), typical of gasoline. Reducing methane (CH4) formation and improving selective C–C bond cleavage efficiency remain as challenges. Studies show that the chemical states of metals and metal–support interactions (MSI) correlate with selective internal and terminal C–C bond cleavage. Positively charged metal species (Mδ+) with strong MSI preferentially activate internal C–C bonds, reducing methane production. Ruthenium (Ru) is particularly effective for C–C bond activation in POs and is more cost-effective than platinum (Pt), presenting industrial opportunities.52–72,96–99,101,102 The amphoteric nature of oxide supports (e.g., Al2O3, CeO2, MgO, TiO2, ZrO2, ZnO, Fe3O4) can modulate the chemical state of metal sites through strong MSI, enhancing internal C–C bond cleavage and inhibiting methane production.
4.3. Experimental condition and analytical techniques for catalyst's activity
The experimental setup depends on the process type: hydrogenolysis and hydrocracking typically occur in batch reactors at high temperatures and H2 pressures. Key factors influencing catalytic activity include the choice of catalyst calcination temperature and catalyst reduction temperature. Catalyst calcination temperature affects metal particle size and dispersion, while catalyst reduction temperature impacts the M0/Mδ+ ratio (Fig. 11(a)).93 Positively charged metal species (Mδ+) coupled with M0 favor internal C–C bond cleavage, reducing methane production. Optimal reaction conditions, such as temperature, time, H2 pressure, and solvent, are essential for selectivity.52 For example, polyaromatic hydrogenolysis is effective between 220 and 280 °C, while lower H2 pressures (0.2–0.5 MPa) yield selective aromatics and higher pressures (2–5 MPa) produce hydrocarbons.40–43 Polyolefin (PO) C–C cleavage during hydrogenolysis occurs at 200–350 °C. Lower temperatures yield liquid fuels, whereas longer reaction times result in lower alkanes and gases. Plastic polymer properties like molecular weight, crystallinity, and impurity levels affect catalytic efficiency, complicating catalyst comparisons based on conversion and selectivity. Several research groups have employed turnover frequency (TOF) values by normalizing the amount of plastic polymer converted to the number of catalyst's active sites per unit of time. However, this value again does not consider the molecular weight variations in polymers obtained from different sources, even if we ignore factors such as crystallinity and impurity level. For example, the weight percentage conversion of the same polymer with different molecular weights will be different over the same catalyst. Vlachos and coworkers proposed solving this problem by calculating the number of C–C and C–O bond cleavage in different polymers by employing a suitable analytical probe and normalizing it to the number of catalyst's active sites.91 In the case of aromatic polymers, including PET, PC, PS, etc., the final product is usually the monomeric building blocks. Therefore, the exact determination of the number of C–C and C–O bonds cleaved can easily facilitate the comparison between the catalyst's activities. However, it is not so simple in the case of PO's hydrogenolysis and hydrocracking, where the formation of solid, liquid, and gaseous products depends upon the point of C–C cleavage. For instance, the terminal C–C bond cleavage in POs results in gases and waxes, whereas the internal C–C bond cleavage forms liquid products.The analysis must include the chromatographic techniques to determine the number average and weight average molecular weights of the polymers, the analytical probes to determine the number of C–C bonds present in the substrate, and the products necessary to calculate the number of broken C–C bonds according to eqn (1), and suitable characterization techniques to quantify the amount of solid (C18–C33), liquid (C5–C17), and gaseous products (C1–C5) formed, especially in the case of POs.
 | (1) |
4.4. Environmental impact and techno-economic assessment of plastic upcycling
While various thermo-chemical upcycling processes exist, comprehensive assessments of sustainability factors and techno-economic assessment (TEA) guidelines for selecting the most appropriate technology are still lacking. Incorporating sustainability metrics such as global warming potential (GWP), acidification potential, ecotoxicity, and E-factor can facilitate the comparison between different plastic upcycling processes and guide the selection of the most suitable options for industrial implementation.117–119 Therefore, comprehensive evaluations of the environmental impacts of these processes, based on sustainability metrics, are essential. Furthermore, TEA is essential for evaluating the economic feasibility of these technologies, comparing their viability, and guiding the selection of the most suitable options for industrial implementation.120–122 Therefore, evaluating the following parameters is crucial for selecting the optimal recycling process for plastic waste.• Equipment cost scaling:
| I = Ib(Q|Qb)m | (2) |
• Total installation cost (TIC):
| Total installation cost (TIC) = installation factor × total equipment cost |
• Total direct cost (TDC):
| TDC = TIC + warehouse cost + site development cost + other cost |
Comparing the sustainability and techno-economic analysis of hydroprocessing and other plastic waste recycling processes will provide guidelines on selecting the most appropriate plastic waste recycling processes and render large-scale plastic waste recycling economical with the least environmental impact.
Finally, significant research has been dedicated to developing sustainable catalysts and catalytic processes for recycling model plastic polymers. Although heterogeneous catalysis has led to substantial advances in plastic upcycling, several challenging issues remain. The specific functions of various plastics arise from their unique chemical structures, resulting in different tolerances to depolymerization environments, thereby making the depolymerization of plastic waste technically challenging. Consequently, more versatile catalysts are needed to be designed to upgrade plastic mixtures. This is further complicated by the additives (such as food residues, dyes, and plasticizers) and the complexity of mixed-waste streams. In addition to the limited activity of catalysts in mixed plastic waste, another challenge is the ineffective interaction between the solid-state catalyst and solid plastic under ambient conditions. Addressing this may involve developing new catalysts capable of selective C–C/C–O bond activation or using appropriate solvents. The design of advanced catalytic routes and catalysts for plastic waste upcycling could also apply to biomass valorization. Biomass, composed of cellulose, hemicellulose, and lignin interconnected by C–C and C–O bonds, presents challenges similar to plastic waste. The study discussed here offers an innovative approach to reducing and removing oxygen functional groups in biomass-derived molecules by cleaving C–O and C–C bonds and production of value-added chemicals and fuels. Thus, catalytic systems, methodologies, and strategies that have proven effective for plastic transformation could be adapted for biomass conversion. Additionally, using a mixture of plastic waste and oxygen-rich biomass as co-feedstock in catalytic systems may provide further opportunities for plastic upcycling, addressing significant socio-environmental issues of the 21st century.
Data availability
This is a review article. It is our assessment of the published articles by various researchers in this domain. The figures and schemes presented in this review are prepared by the first author of this review. The required copyright permission has also been taken.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AKM acknowledges the Ministry of Education, Government of India, for the PMRF fellowship (PMRF-ID, 2902500), and RS expresses his gratitude for the Research and Innovation Award received from IIT Ropar.References
- H. A. Maddah, J. Polym. Sci., Polym. Phys. Ed., 2016, 6, 1–11 CAS
.
-
A. Emblem, Packaging Technology, Elsevier, 2012, pp. 287–309 Search PubMed
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 1–5 CrossRef
- N. C. Paxton, M. C. Allenby, P. M. Lewis and M. A. Woodruff, Eur. Polym. J., 2019, 118, 412–428 CrossRef CAS
- C. Soutis, Mater. Sci. Eng., A, 2005, 412, 171–176 CrossRef
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, Sustainable Mater. Technol., 2020, 25, e00188 CrossRef CAS
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. Harmelen, P. Wild, G. P. Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed
- F. N. Hill, F. E. Bailey and J. T. Fitzpatrick, Ind. Eng. Chem., 1958, 50, 5–7 CrossRef CAS
- I. Agenda, 2016, https://es.weforum.org/reports/the-new-plastics-economy-rethinking-the-future-of-plastics.
- Foundation, E. M. Plastics and the Circular Economy. https://archive.ellenmacarthurfoundation.org/explore/plastics-and-the-circular-economy.
- C. J. Rhodes, Sci. Prog., 2018, 101, 207–260 CrossRef PubMed
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC
- A. Okunola A, O. Kehinde I, A. Oluwaseun and A. Olufiropo E, J. Toxicol. Risk Assess., 2019, 5, 1–13 Search PubMed
- L. Adane and D. Muleta, J. Toxicol. Environ. Health Sci., 2011, 3, 234–248 Search PubMed
- S. M. Nxumalo, S. D. Mabaso, S. F. Mamba and S. S. Singwane, Soc. Sci. Humanit. Open, 2020, 2, 100066 Search PubMed
- J. Fuller, D. White, H. Yi, J. Colley, Z. Vickery and S. Liu, Chemosphere, 2020, 260, 127589 CrossRef CAS
- Y. Chae and Y.-J. An, Environ. Pollut., 2018, 240, 387–395 CrossRef CAS PubMed
- J. P. Lange, ACS Sustainable Chem. Eng., 2021, 9, 15722–15738 CrossRef CAS
- A. W. Larsen and T. Astrup, J. Waste Manage, 2011, 31, 1597–1605 CrossRef CAS PubMed
- J. Huang, A. Veksha, W. P. Chan, A. Giannis and G. Lisak, Renewable Sustainable Energy Rev., 2022, 154, 111866 CrossRef CAS
- M. Solis and S. Silveira, J. Waste Manage., 2020, 105, 128–138 CrossRef CAS PubMed
- K. Ragaert, L. Delva and K. Van Geem, J. Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed
- X. Wei, J. Yu, J. Du and L. Sun, Energy Fuels, 2021, 35, 5026–5038 CrossRef CAS
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed
- A. K. Manal, G. Saini and R. Srivastava, Green Chem., 2024, 26, 3814–3831 RSC
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, Energy Convers. Manage., 2016, 115, 308–326 CrossRef CAS
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC
- C. Wang, H. Han, Y. Wu and D. Astruc, Coord. Chem. Rev., 2022, 458, 214422 CrossRef CAS
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS
- P. S. Roy, G. Garnier, F. Allais and K. Saito, ChemSusChem, 2021, 14, 4007–4027 CrossRef CAS
- R. Mishra, A. Kumar, E. Singh and S. Kumar, ACS Sustainable Chem. Eng., 2023, 11, 2033–2049 CrossRef CAS
- J. Su, T. Li, W. Xie, C. Wang, L. Yin, T. Yan and K. Wang, ACS Sustainable Chem. Eng., 2023, 11, 8176–8192 CrossRef CAS
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522 CrossRef CAS
- S. Lu, Y. Jing, B. Feng, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2021, 14, 4242–4250 Search PubMed
- Z. Gao, B. Ma, S. Chen, J. Tian and C. Zhao, Nat. Commun., 2022, 13, 3343 Search PubMed
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC
- S. Hongkailers, Y. Jing, Y. Wang, N. Hinchiranan and N. Yan, ChemSusChem, 2021, 14, 4330–4339 CrossRef CAS
- K. L. Sánchez-Rivera and G. W. Huber, ACS Cent. Sci., 2021, 7, 17–19 CrossRef PubMed
- Y. Jing, M. Shakouri, X. Liu, Y. Hu, Y. Guo and Y. Wang, ACS Catal., 2022, 12, 10690–10699 CrossRef CAS
- M. Ye, Y. Li, Z. Yang, C. Yao, W. Sun, X. Zhang, W. Chen, G. Qian, X. Duan, Y. Cao, L. Li, X. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202301024 Search PubMed
- H. Tang, Y. Hu, G. Li, A. Wang, G. Xu, C. Yu, X. Wang, T. Zhang and N. Li, Green Chem., 2019, 21, 3789–3795 RSC
- L. Wang, G. Li, Y. Cong, A. Wang, X. Wang, T. Zhang and N. Li, Green Chem., 2021, 23, 3693–3699 RSC
- L. Wang, F. Han, G. Li, M. Zheng, A. Wang, X. Wang, T. Zhang, Y. Cong and N. Li, Green Chem., 2021, 23, 912–919 RSC
- A. K. Manal, G. V. Shanbhag and R. Srivastava, Appl. Catal., B, 2023, 338, 123021 CrossRef CAS
- B. C. Vance, P. A. Kots, C. Wang, J. E. Granite and D. G. Vlachos, Appl. Catal., B, 2023, 322, 122138 CrossRef CAS
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS
- H. Ji, X. Wang, X. Wei, Y. Peng, S. Zhang, S. Song and H. Zhang, Small, 2023, 19, 2300903 CrossRef CAS
- S. Lu, Y. Jing, S. Jia, M. Shakouri, Y. Hu, X. Liu, Y. Guo and Y. Wang, ChemCatChem, 2023, 15, e202201375 CrossRef CAS
- J. A. Sun, P. A. Kots, Z. R. Hinton, N. S. Marinkovic, L. Ma, S. N. Ehrlich, W. Zheng, T. H. Epps, L. T. J. Korley and D. G. Vlachos, ACS Catal., 2024, 5, 3228–3240 CrossRef
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Chem. Catal., 2021, 1, 437–455 CrossRef CAS
- B. Coq, R. Dutartre, F. Figueras and T. Tazi, J. Catal., 1990, 122, 438–447 CrossRef CAS
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed
- R. A. Hackler, J. V. Lamb, I. L. Peczak, R. M. Kennedy, U. Kanbur, A. M. LaPointe, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, Macromolecules, 2022, 55, 6801–6810 CrossRef CAS
- I. L. Peczak, R. M. Kennedy, R. A. Hackler, R. Wang, Y. Shin, M. Delferro and K. R. Poeppelmeier, ACS Appl. Mater. Interfaces, 2021, 13, 58691–58700 CrossRef CAS
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS
- L. Li, H. Luo, Z. Shao, H. Zhou, J. Lu, J. Chen, C. Huang, S. Zhang, X. Liu, L. Xia, J. Li, H. Wang and Y. Sun, J. Am. Chem. Soc., 2023, 145, 1847–1854 CrossRef CAS
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, JACS Au, 2022, 2, 2259–2268 CrossRef CAS PubMed
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CrossRef CAS
- M. Utami, K. Wijaya and W. Trisunaryanti, Mater. Chem. Phys., 2018, 213, 548–555 CrossRef CAS
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2023, 863, 160934 Search PubMed
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CrossRef CAS
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone, G. T. Beckham and Y. Román-Leshkov, ACS Catal., 2022, 12, 13969–13979 CrossRef CAS
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13, 4850 CrossRef CAS
- W. Ding, J. Liang and L. L. Anderson, Energy Fuels, 1997, 11, 1219–1224 CrossRef CAS
- D. Munir, H. Amer, R. Aslam, M. Bououdina and M. R. Usman, Mater. Renewable Sustainable Energy, 2020, 9, 9 CrossRef
- P. Wu, G. Lu and C. Cai, Green Chem., 2021, 23, 8666–8672 RSC
- P. Wu, G. Lu and C. Cai, Green Chem., 2021, 23, 8666–8672 RSC
- B. Feng, Y. Jing, Y. Guo, X. Liu and Y. Wang, Green Chem., 2021, 23, 9640–9645 RSC
- Y. Chen, C. Shi, T. Jia, Q. Cai, L. Pan, J. Xie, L. Wang, X. Zhang and J.-J. Zou, Fuel, 2022, 308, 122077 CrossRef CAS
- Y. Liu, C. Ma, C. Shi, L. Pan, J. Xie, S. Gong, Y.-C. Zhang, G. Nie, X. Zhang and J.-J. Zou, Fuel Process. Technol., 2020, 201, 106339 CrossRef CAS
- H. S. Chung, C. S. H. Chen, R. A. Kremer, J. R. Boulton and G. W. Burdette, Energy Fuels, 1999, 13, 641–649 CrossRef CAS
- K. T. Serijan and P. H. Wise, J. Am. Chem. Soc., 1951, 73, 4766e9 Search PubMed
- B. G. Harvey, W. W. Merriman and T. A. Koontz, Energy Fuels, 2015, 29, 2431–2436 CrossRef CAS
- A. Rana, L. Cid Gomes, J. S. Rodrigues, D. M. M. Yacout, H. Arrou-Vignod, J. Sjölander, N. Proos Vedin, O. El Bakouri, K. Stensjö, P. Lindblad, L. Andersson, C. Sundberg, M. Berglund, P. Lindberg and H. Ottosson, Green Chem., 2022, 24, 9602–9619 RSC
- A. K. Manal, D. R. Kanchan, A. Banerjee, J. Zhao and R. Srivastava, ChemSusChem, 2024, e202401146 CrossRef
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709–2719 RSC
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer and S. L. Scott, J. Am. Chem. Soc., 2022, 144, 18526–18531 CrossRef CAS PubMed
- G. Viscusi, V. Bugatti, V. Vittoria and G. Gorrasi, Future Foods, 2021, 4, 100063 CrossRef CAS
- in Handbook of Polypropylene and Polypropylene Composites, Revised and Expanded, ed. Karian, CRC Press, 2003 Search PubMed
- M. Jambrich and P. Hodul, in Polypropylene: An AZ reference, ed. J. Karger-Kocsis, Springer, Dordrecht, The Netherlands, 1999, pp. 806–812 Search PubMed.
- S. Moritomi, T. Watanabe and S. Kanzaki, Sumitomo Kagaku, 2010, 1, 1–16 Search PubMed
- S. L. Aggarwal and O. J. Sweeting, Chem. Rev., 1957, 57, 665–742 CrossRef CAS
- P. A. Kots, B. C. Vance and D. G. Vlachos, React. Chem. Eng., 2022, 7, 41–54 RSC
- J. Wei, J. Liu, W. Zeng, Z. Dong, J. Song, S. Liu and G. Liu, Catal. Sci. Technol., 2023, 13, 1258–1280 RSC
- D. Balducci, S. Guidetti, F. Scavello, N. Vecchini and P. Mariani, US Pat., 20220041937, 2022 Search PubMed.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS
- B. Du, X. Chen, Y. Ling, T. Niu, W. Guan, J. Meng, H. Hu, C. Tsang and C. Liang, ChemSusChem, 2023, 16, e202202035 CrossRef CAS
- N. E. R. Zimmermann and M. Haranczyk, Cryst. Growth Des., 2016, 16, 3043–3048 CrossRef CAS
- H. Zhang, Y. Ma, K. Song, Y. Zhang and Y. Tang, J. Catal., 2013, 302, 115–125 CrossRef CAS
- J. Duan, W. Chen, C. Wang, L. Wang, Z. Liu, X. Yi, W. Fang, H. Wang, H. Wei, S. Xu, Y. Yang, Q. Yang, Z. Bao, Z. Zhang, Q. Ren, H. Zhou, X. Qin, A. Zheng and F. S. Xiao, J. Am. Chem. Soc., 2022, 144(31), 14269–14277 CrossRef CAS PubMed
- A. Almithn and D. Hibbitts, J. Phys. Chem. C, 2019, 123, 5421–5432 CrossRef CAS
- A. Almithn and D. Hibbitts, ACS Catal., 2018, 8, 6375–6387 CrossRef CAS
- J. Wei, M. Zhu, B. Liu, N. Wang, J. Liu, K. Tomishige, S. Liu and G. Liu, Angew. Chem., Int. Ed., 2023, 62, e202310505 CrossRef CAS
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- W. Wang, S. Li, Q. Qiang, K. Wu, X. Pan, W. Su, J. Cai, Z. Shen, Y. Yang, C. Li and T. Zhang, Angew. Chem., Int. Ed., 2024, e202404683 CAS
- G. S. Parkinson, Catal. Lett., 2019, 149, 1137–1146 CrossRef CAS PubMed
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS
- T. Li, N. Ji, Z. Jia, X. Diao, Z. Wang, Q. Liu, C. Song and X. Lu, ChemCatChem, 2020, 12, 5288–5302 CrossRef CAS
- F. Solymosi, Catal. Rev., 1968, 1, 233–255 CrossRef
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, J. Messinger Robert, F. Chmelka Bradley and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS
- A. K. Manal, J. H. Advani, N. Kanna and P. Kumar, Chem. – Asian J., 2023, 18, e202300294 CrossRef CAS
- P. Rani, R. Srivastava and B. Satpati, Cryst. Growth Des., 2016, 16, 3323–3333 CrossRef CAS
- Q. Sun, N. Wang and J. Yu, Adv. Mater., 2021, 33, 2104442 CrossRef CAS PubMed
- E. Soghrati, T. K. C. Ong, C. K. Poh, S. Kawi and A. Borgna, Appl. Catal., B, 2018, 235, 130–142 CrossRef CAS
- A. Zukal, V. Patzelova and U. Lohse, Zeolites, 1986, 6, 133–136 CrossRef CAS
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS
- B. Hernández, P. Kots, E. Selvam, D. G. Vlachos and M. G. Ierapetritou, ACS Sustainable Chem. Eng., 2023, 11, 7170–7181 CrossRef
- L. Pires Costa, D. M. Vaz de Miranda and J. C. Pinto, ACS Sustainable Chem. Eng., 2022, 10, 3799–3807 CrossRef CAS
- P. T. Anastas and R. L. Lankey, ‘Life Cycle Assessment and green chemistry: the yin and yang of industrial ecology’, Green Chem., 2000, 2, 289 RSC
- G. Yadav, A. Singh, A. Dutta, T. Uekert, J. S. DesVeaux, S. R. Nicholson, E. C. Tan, C. Mukarakate, J. A. Schaidle, C. J. Wrasman and A. C. Carpenter, Energy Environ. Sci., 2023, 16, 3638–3653 RSC
- V. Cappello, P. Sun, G. Zang, S. Kumar, R. Hackler, H. E. Delgado, A. Elgowainy, M. Delferro and T. Krause, Green Chem., 2022, 24, 6306–6318 RSC
- U. Salahuddin, J. Sun, C. Zhu, M. Wu, B. Zhao and P. X. Gao, Adv. Sustainable Syst., 2023, 7, 2200471 CrossRef
- W. Wu, H. Xu, B. Shi and P. C. Kuo, CEP:PI., 2023, 184, 109297 CAS
- P. Huang, A. Ahamed, R. Sun, G. X. De Hoe, J. Pitcher, A. Mushing, F. Lourenço and M. P. Shaver, ACS Sustainable Chem. Eng., 2023, 11, 15328–15337 CrossRef CAS PubMed
- J. R. Cruce, A. Beattie, P. Chen, D. Quiroz, M. Somers, S. Compton, K. DeRose, B. Beckstrom and J. C. Quinn, Algal Res., 2021, 54, 102169 CrossRef
- P. Fasahati, R. Dickson, C. M. Saffron, H. C. Woo and J. J. Liu, Renewable Sustainable Energy Rev., 2022, 157, 112011 CrossRef CAS
- J. Ma, P. A. Tominac, H. A. Aguirre-Villegas, O. O. Olafasakin, M. M. Wright, C. H. Benson, G. W. Huber and V. M. Zavala, Green Chem., 2023, 25, 1032–1044 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03261f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |