
Probing local charge transfer processes of Pt–Au heterodimers in plasmon-enhanced electrochemistry by CO stripping techniques†
Chenxi
Guo
ab,
Shiyu
Xia
ab,
Yu
Tian
a,
Fenghua
Li
 a,
Guobao
Xu
ab,
Fengxia
Wu
*a and
Wenxin
Niu
*ab
a,
Guobao
Xu
ab,
Fengxia
Wu
*a and
Wenxin
Niu
*ab
aChangchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: niuwx@ciac.ac.cn; wufengxia@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, China
First published on 30th January 2024
Abstract
CO-stripping experiments are employed as a highly structure-sensitive and in situ strategy to explore the mechanisms of plasmon-enhanced electrooxidation reactions. By using Pt–Au heterodimers as a model catalyst, the plasmon-induced current and potential changes on Pt and Au sites can be identified and explained.
Local surface plasmon resonance (LSPR), a unique optical property of plasmonic nanomaterials (e.g., gold, silver, and copper), is a collective free electron oscillation confined in nanomaterials when they are excited by light of the corresponding resonance wavelength.1–5 Electrocatalysis enhanced by the LSPR effect has received extensive attention owing to its potential applications in efficient solar energy utilization.6–9 However, the time-scale mismatch between the ultrafast dynamics of resonance energy relaxation (femtoseconds to nanoseconds) and electrocatalytic processes (milliseconds to minutes) hampers the effective utilisation of the resonance energy in catalytic reactions.10–12 One key strategy to enhance the performance of plasmon-enhanced catalysis is the construction of heterostructures, wherein materials with distinct energy level differences are selectively chosen to enable the migration of hot electrons to adjacent components.13,14 This phenomenon prolongs the lifetime of hot carriers and enhances the efficiency of the catalytic processes, making it a highly desirable approach for solar energy conversion.9,15
In spite of the promising application of metallic heterostructures in plasmon-enhanced catalysis, the precise construction of plasmonic catalysts with well-defined heterointerfaces remains challenging. Furthermore, directly probing the local charge transfer processes of heterostructures at the nanoscale is of vital importance for the elucidation of their catalytic mechanisms. Previously, several analytical methods using transient absorption (TA),16in situ electron paramagnetic resonance (EPR),17 photoluminescence (PL),18 surface-enhanced Raman spectroscopy (SERS),19 and Kelvin probe force microscopy (KPFM)20 techniques have been utilized to characterize the plasmonic processes. However, most of these analytical methods still face challenges in probing the plasmon-enhanced electrocatalytic reactions in situ or at the nanoscale, and their implementation is complicated and time-consuming. A convenient in situ measurement that can reflect the local plasmon-enhanced electrocatalytic processes needs to be exploited.
In this study, we propose a strategy using carbon monoxide stripping (CO-stripping) experiments,21 a highly structure-sensitive reaction, as a means to probe the mechanisms of plasmon-enhanced electrooxidation reactions by using heterogeneous Pt–Au dimer nanostructures as a model catalyst. Pt is one of the most commonly used catalysts for fuel cells due to its remarkable electrocatalytic activity.22–24 To increase the resistance to CO poisoning of Pt,25 the method of plasmon-enhanced electrooxidation is employed,26,27 wherein Au is introduced because of its strong plasmonic properties and excellent stability.28 This proposed CO-stripping strategy is devoted to enabling the in situ differentiation of local reactions that occur on Pt and Au sites and thus can be developed as a general strategy to probe the fundamental mechanisms in metallic heterogeneous plasmonic electrocatalysts.
A schematic diagram of the synthesis of the Pt–Au heterodimer catalyst is shown in Fig. 1(a). Pt–Au heterodimers with well-defined interfaces were synthesized through a seed-mediated growth method with platinum (Pt) nanocubes as seeds.29 The morphology of the Pt nanocrystals is a concave cube with {hk0} high-index facets with an average size of ∼29.9 nm (Fig. S1, ESI†), which have been proven to be active sites for CO-stripping reaction.30,31Fig. 1(b)–(d) presents the transmission electron microscopy (TEM) images of the Pt–Au heterodimers. These heterodimers have a high purity and are composed of two distinct components. Scanning electron microscopy (SEM) studies show that the Pt component is a concave nanocube and the Au component exhibits a rhombic dodecahedral (RD) morphology with slight truncation (Fig. S2, ESI†). The formation of the RD shape enclosed with {100} facets is consistent with previous results of Au nanocrystals grown in N,N-dimethylformamide solvent.32 The geometric model is shown in Fig. 1(c) for a direct exhibition. Au, a widely investigated plasmonic metal, can also provide a stable LSPR effect for the following research.33 High-resolution TEM (HRTEM) images (Fig. 1(e)–(g)) show the lattices of Pt, Au, and their interfaces. The lattice matching shown in Fig. 1(h) plays an important role in forming the heterostructures of the Pt–Au heterodimers with excellent photocatalytic performance.34 The heterogeneous structure and composition of the Pt–Au heterodimers can also be confirmed by dark field scanning transmission electron microscopy (STEM) images (Fig. 1(h)). The dark field STEM of a single Pt–Au heterodimer (Fig. 1(i)) and corresponding energy dispersive X-ray spectroscopy mapping (EDS-mapping) (Fig. 1(j)) show a clear heterogeneous structure. Modulation of the Pt/Au ratio in the heterodimers is also investigated (Fig. S3, ESI†). Our results show that both a low Au content (weak plasmonic properties) and a high Au content (blocked Pt surfaces) are not suitable for the plasmon-enhanced electrochemistry.
 | ||
| Fig. 1 (a) Schematic diagram of the synthesis of Pt–Au heterodimers. (b)–(d) TEM images and a geometric model (inset) of the Pt–Au heterodimers at different magnifications. Corresponding HRTEM images of (e) Au, (f) Pt, and (g) the Pt–Au interface marked with green squares in panel (d). (h) Dark field STEM image of Pt–Au heterodimers. (i) Dark field STEM image of a single Pt–Au heterodimer and (j) corresponding EDS-mapping of the single Pt–Au heterodimer. | ||
Compared with Pt concave nanocubes, the Pt–Au heterodimers exhibit a strong plasmonic peak located at 560 nm, similar to that of Au nanocubes, whose extinction peak is tuned by size regulation (Fig. 2(a)). Dimer structures can minimize the effect of plasmonic damping by non-plasmonic metals such as Pt and Pd; therefore, the Pt–Au heterodimers serve as an ideal candidate for plasmon-enhanced electrocatalysis.35,36 The X-ray diffraction (XRD) patterns (Fig. 2(b)) show that the diffraction peaks of the Pt–Au heterodimers consist of cubic Pt (PDF# 04-0784) and Au (PDF# 87-0646), respectively. The Pt diffraction peaks of the Pt–Au heterodimers match well with the diffraction peaks of the Pt concave nanocubes without any shift, indicating the formation of heterostructures rather than an alloy. The X-ray photoelectron spectroscopy (XPS) results of the Au 4f and Pt 4f XPS spectra of the Pt concave nanocubes and Pt–Au heterodimers are displayed in Fig. 2(c) and (d). The Au 4f spectrum of the Pt–Au heterodimers can be divided into two different peaks that belong to 4f7/2 (83.95 eV) and 4f5/2 (87.65 eV) species. The Pt 4f spectrum of the Pt–Au heterodimers can be deconvoluted into four peaks that belong to Pt0 4f7/2 (70.75 eV), Pt2+ 4f7/2 (71.55 eV), Pt0 4f5/2 (74.10 eV), and Pt2+ 4f7/2 (74.90 eV). The Pt2+ should come from the Pt oxidized by O2 in the air. After heterostructure formation, the Pt 4f peaks integrally shift to higher binding energy compared with that of the Au nanocubes, while the Au 4f peaks shift to lower binding energy. These results indicate electron transfer from Pt to Au exists in the Pt–Au heterodimers.
 | ||
| Fig. 2 (a) UV-vis extinction spectra of Pt concave nanocubes, Au nanocubes and Pt–Au heterodimers. (b) XRD patterns recorded from Pt concave nanocubes and Pt–Au heterodimers. (c) Au 4f and (d) Pt 4f XPS spectra of Pt concave nanocubes, Au nanocubes, and Pt–Au heterodimers. | ||
The Pt–Au heterodimers are used as a model catalyst to probe the local charge transfer processes in plasmon-enhanced electrochemistry using CO-stripping experiments, with Pt concave nanocubes (Fig. S1, ESI†) and Au nanocubes (Fig. S6, ESI†) as contrast samples. The CO-stripping reaction has been proposed as a well-documented example of structure-sensitive reactions in electrocatalysis, which involves an oxidation process between adsorbed CO (COads) and active oxygen species, such as hydroxyl species (OHads) from water oxidation. This means that CO-stripping is highly sensitive to both COads and active oxygen species on the surface of the catalysts (COads + OHads = CO2 + Hads).21 The oxidation peaks on Pt and Au appear at different potentials, allowing for the distinguishing of the reactions occurring on different components of the heterostructures.
The CO-stripping experiments were conducted in a single cell with 1.0 M potassium hydroxide (KOH) electrolyte under ambient conditions. A catalyst-supported glassy carbon electrode, a saturated calomel electrode (SCE), and a Pt net electrode were used as the working electrode, reference electrode, and counter electrode, respectively. All the potentials were reported on a reversible hydrogen electrode (RHE) scale by the Nernst equation (ERHE = ESCE + 0.0591 × pH + 0.241).37 The cut-on 515 nm optical filter was chosen to control the light wavelength according to the extinction spectra of the heterodimers. All of these catalysts were activated at the electrolyte before CO-stripping testing. The electrodes were first adsorbed with CO in the CO-saturated electrolyte with CO bubbling at a potential of 0.1 V vs. RHE for sufficient adsorption.21,38 Then the CO-stripping with or without light irradiation was conducted after removing CO from the electrolyte by high-purity nitrogen through the cyclic voltammetry (CV) technique at a scan rate of 50 mV s−1. The CO-stripping curves on the Pt concave nanocubes, Au nanocubes, and Pt–Au heterodimers are shown in Fig. 3(a)–(c), respectively. The peak at ∼0.73 V vs. RHE belongs to CO-oxidation on Pt (blue boxes), while the peak at ∼1.1 V vs. RHE belongs to CO-oxidation on Au (orange boxes).21,39 After visible light irritation, the current of the Pt concave nanocubes slightly deceases (−8.2%) (Fig. 3(a)), while the oxidation current on the Au nanocubes slightly increases (18.2%) (Fig. 3(b)). In the case of Pt–Au heterodimers (Fig. 3(c)), the CO-oxidation current on Au increased more (29%) than on the Au nanocubes but the potential remained the same. Meanwhile, the current of the CO-oxidation peak on Pt decreases (−26.6%), while the potential shifts negatively (−3%) upon light irradiation. These changes of Pt concave nanocubes, Au nanocubes, and Pt–Au heterodimers are presented in column charts (Fig. 3(d)–(f), respectively).
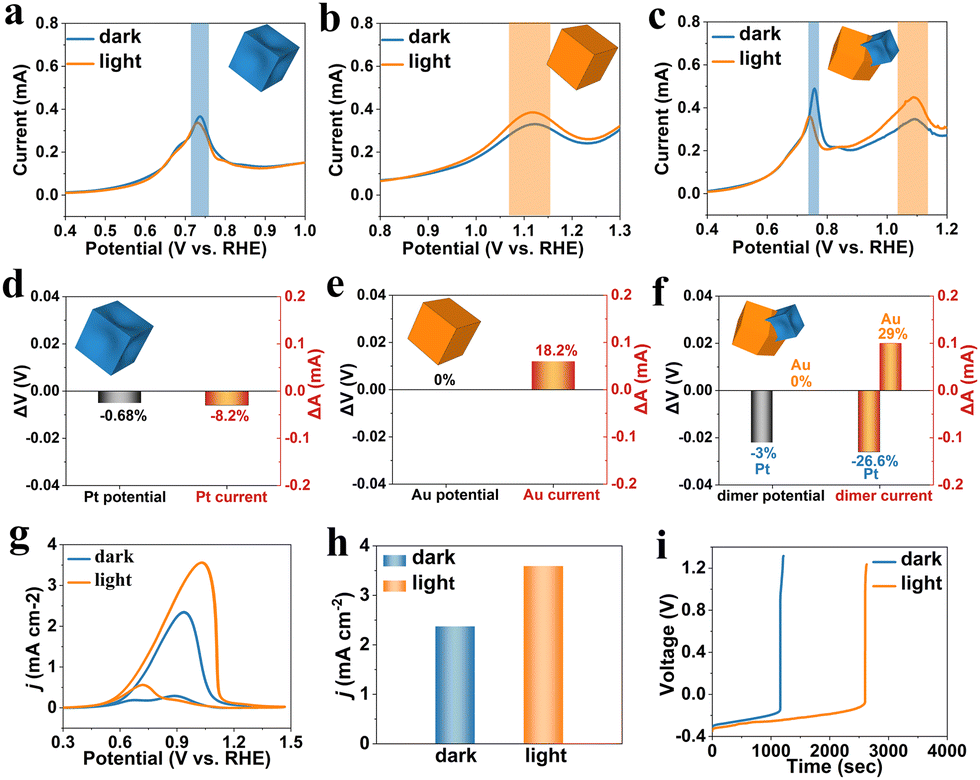 | ||
| Fig. 3 Electrocatalytic CO-oxidation curves of (a) Pt concave nanocubes, (b) Au nanocubes, and (c) Pt–Au heterodimers with (orange lines) and without (blue lines) light. Potential and current changes of (d) Pt concave nanocubes, (e) Au nanocubes, and (f) Pt–Au heterodimers. (g) Electrocatalyst MOR curves of Pt–Au heterodimers and (h) corresponding peak current column chart with (orange) and without (blue) light. (i) Chronoamperometry curves of Pt–Au heterodimers with (orange) and without (blue) light. | ||
These detailed in situ local changes provide a basis for understanding the plasmonic effects in electrooxidation reactions. To further illustrate the charge transfer processes obtained from plasmon-enhanced CO-stripping experiments, the Pt–Au heterodimers are also employed in the plasmon-enhanced methanol oxidation reaction (MOR), where intermediate COads may cause poisoning. The MOR was conducted under the same conditions, but the electrolyte was changed to 1.0 M KOH + 1.0 M methanol. Fig. 3(g) shows that the current density (3.57 mA cm−2) on Pt with light irritation is obviously larger than that (2.35 mA cm−2) without light. The column chart (Fig. 3(h)) gives a clearer sight of the oxidation peak current density, and the ratio of current density between light and dark conditions is 152%. Furthermore, the anti-poisoning ability was also assessed by chrono-potentiometry curves at a current of 1 mA for the MOR. The potential jump to a higher value (∼1.2 V) can be understood as poisoning of the catalysts.40,41 As Fig. 3(i) shows, the time before poisoning of the Pt–Au heterodimers upon light irradiation (2600 s) was 2.26 times longer than that under dark conditions (1150 s). These results indicate that the plasmonic effect can increase the anti-poisoning ability of the Pt–Au heterodimers. The Pt concave nanocubes (Fig. S7a, ESI†) and Au nanocubes (Fig. S7b, ESI†) were also studied for comparison.
For plasmon-enhanced catalytic reactions, three typical effects are commonly considered for the enhancement, namely the hot charge carriers, local photothermal heating13,42 and strong electromagnetic fields.4,43 In the case of plasmon-enhanced electrochemistry based on heterostructure catalysts, hot charge carriers (e–h pairs) are the major contributors.44,45 For Au nanocubes, the current increases slightly upon plasmonic excitation because the generated hot carriers tend to have a short lifetime and decay in a short time.20,46 In contrast, the current on the Au component of the Pt–Au heterodimers increases greater than the Au nanocubes, due to the fact that heterostructures can enable the migration of hot electrons to adjacent components and extend their lifetimes.15,47 As to the CO-oxidation peak of the Pt component on the Pt–Au heterodimers, the decrease of current originates from the work function difference-induced electron transfer.48,49 Hot electrons tend to transfer to Pt because Pt has a higher work function than Au, which hampers the capability of withdrawing electrons from CO and thus inhibits the efficiency of CO-oxidation.50,51 The negative shift of oxidation potential on the Pt component can be attributed to the adsorption of CO on the active sites.52 As the hot electrons are injected, the electron occupancy of the d-band of Pt increases (d-band centre downshifts relatively),53,54 reducing the adsorption of CO molecules onto the Pt atoms.55,56 The weaker adsorption will result in the negative shift of the CO-oxidation potential.52,57 The mechanisms can be concluded as a schematic diagram (Fig. 4), and these mechanistic insights illustrate that plasmonic excitation can enhance the CO-oxidation efficiency.
 | ||
| Fig. 4 Schematic illustration of the plasmon-enhanced catalytic processes of Pt–Au heterodimers probed by CO-stripping tests. | ||
In summary, we propose a strategy using a structure-sensitive CO-stripping reaction as a means to probe the mechanisms of plasmon-enhanced electrooxidation reactions, using Pt–Au heterodimers with well-defined interfaces as a model catalyst. Taking advantage of this strategy, the current and potential changes of the CO-oxidation peaks on different components of the heterodimers can be precisely monitored and thus provide insights into the hot carrier separation and migration upon light excitation. Specifically, the oxidation current of the Au component is enhanced, while the oxidation current of the Pt component decreases but shifts to a lower potential. These in situ local changes can be understood in terms of efficient separation of hot charge carriers and subsequent hot electron injection. This study deepens our understanding of plasmonic enhancement of heterogeneous electrocatalysis and provides a convenient strategy to study plasmonic electrocatalytic processes.
This work is supported by the National Natural Science Foundation of China (no. 22374144, 22072144, 22102171, and 22204160) and Natural Science Foundation of Jilin Province (no. YDZJ202201ZYTS341).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- S. Kim, J. Kim, J. Park and J. Nam, Adv. Mater., 2018, 30, 1704528 CrossRef PubMed.
- F. Wu, F. Li, Y. Tian, X. Lv, X. Luan, G. Xu and W. Niu, Nano Lett., 2023, 23, 8233–8240 CrossRef CAS PubMed.
- P. Han, X. Mao, Y. Jin, S. Sarina, J. Jia, E. R. Waclawik, A. Du, S. E. Bottle, J. C. Zhao and H. Y. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202215201 CrossRef CAS PubMed.
- L. Liu, H. Zhang, S. Xing, Y. Zhang, L. Shangguan, C. Wei, F. Peng and X. Liu, Adv. Sci., 2023, 10, 2207342 CrossRef CAS PubMed.
- K. Song, H. Lee, M. Lee and J. Y. Park, ACS Energy Lett., 2021, 6, 1333–1339 CrossRef CAS.
- Y. Kim, E. B. Creel, E. R. Corson, B. D. McCloskey, J. J. Urban and R. Kostecki, Adv. Energy Mater., 2018, 8, 1800363 CrossRef.
- J. Wang, J. Heo, C. Chen, A. J. Wilson and P. K. Jain, Angew. Chem., Int. Ed., 2020, 59, 18430–18434 CrossRef CAS.
- X. Sang, S. Xia, L. Cheng, F. Wu, Y. Tian, C. Guo, G. Xu, Y. Yuan and W. Niu, Small, 2023, 19, 2305369 Search PubMed.
- V. Jain, R. K. Kashyap and P. P. Pillai, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- R. Wan, S. Liu, Y. Wang, Y. Yang, Y. Tian, P. K. Jain and X. Kang, Nano Lett., 2022, 22, 7819–7825 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- F. Wu, S. Xia, J. Wei, W. Gao, F. Li and W. Niu, Metallic Heterostructures for Plasmon-Enhanced Electrocatalysis, ChemPhysChem, 2023, 24, e202200881 CrossRef CAS PubMed.
- V. G. Rao, U. Aslam and S. Linic, J. Am. Chem. Soc., 2019, 141, 643–647 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed.
- J. Guo, Y. Zhang, L. Shi, Y. Zhu, M. F. Mideksa, K. Hou, W. Zhao, D. Wang, M. Zhao, X. Zhang, J. Lv, J. Zhang, X. Wang and Z. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- J. Huang, W. Guo, S. He, J. R. Mulcahy, A. Montoya, J. Goodsell, N. Wijerathne, A. Angerhofer and W. D. Wei, ACS Nano, 2023, 17, 7813–7820 CrossRef CAS PubMed.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- S. Wang, Y. Gao, S. Miao, T. Liu, L. Mu, R. Li, F. Fan and C. Li, J. Am. Chem. Soc., 2017, 139, 11771–11778 CrossRef CAS PubMed.
- A. Cuesta and M. Escudero, Phys. Chem. Chem. Phys., 2008, 10, 3628 RSC.
- Y. Yu, S. J. Lee, J. Theerthagiri, Y. Lee and M. Y. Choi, Appl. Catal., B, 2022, 316, 121603 CrossRef CAS.
- Y. Lee, Y. Yu, H. Tanaya Das, J. Theerthagiri, S. Jun Lee, A. Min, G. Kim, H. C. Choi and M. Y. Choi, Fuel, 2023, 332, 126164 CrossRef CAS.
- S. Shankar Naik, J. Theerthagiri, F. S. Nogueira, S. J. Lee, A. Min, G. Kim, G. Maia, L. M. C. Pinto and M. Y. Choi, ACS Catal., 2023, 13, 1477–1491 CrossRef CAS.
- H. J. Kim, C. J. Moon, S. Lee, J. Theerthagiri, J. W. Hong, M. Y. Choi and Y. W. Lee, J. Mater. Sci. Technol., 2023, 165, 153–160 CrossRef.
- M. P. S. Rodrigues, A. H. B. Dourado, K. Krischer and S. I. C. Torresi, Electrochim. Acta, 2022, 420, 140439 CrossRef CAS.
- C. Tang, G. Chen, Y. Liu, J. Wang, X. He, C. Xie, Z. He and J. Huang, J. Mater. Chem. A, 2023, 11, 16671–16682 RSC.
- S. J. Lee, H. Lee, T. Begildayeva, Y. Yu, J. Theerthagiri, Y. Kim, Y. W. Lee, S. W. Han and M. Y. Choi, Biosens. Bioelectron., 2022, 197, 113766 CrossRef CAS PubMed.
- Z. Zhang, J. Hui, Z. Liu, X. Zhang, J. Zhuang and X. Wang, Langmuir, 2012, 28, 14845–14848 CrossRef CAS PubMed.
- M. T. M. Koper, Nanoscale, 2011, 3, 2054–2073 RSC.
- P. Urchaga, S. Baranton, C. Coutanceau and G. Jerkiewicz, Langmuir, 2012, 28, 3658–3663 CrossRef CAS PubMed.
- W. Niu, W. Zhang, S. Firdoz and X. Lu, J. Am. Chem. Soc., 2014, 136, 3010–3012 CrossRef CAS PubMed.
- Y. Kim, J. G. Smith and P. K. Jain, Nat. Chem., 2018, 10, 763–769 CrossRef CAS PubMed.
- Z. Yuan, Y. Cao, Y. Meng, G. Pan, Y. Zheng, Z. Ni and S. Xia, J. Hazard. Mater., 2023, 458, 131895 CrossRef CAS PubMed.
- L. Cheng, F. Wu, Y. Tian, X. Lv, F. Li, G. Xu, H. Hsu, Y. Zhang and W. Niu, Nano Res., 2023, 16, 8961–8969 CrossRef CAS.
- Y. Zhao, F. Wu, J. Wei, H. Sun, Y. Yuan, H. Bao, F. Li, Z. Zhang, S. Han and W. Niu, Chem. – Eur. J., 2022, 28, e202200494 CrossRef CAS PubMed.
- M. Hao, B. D. Assresahegn, A. Abdellah, L. Miner, A. Al Hejami, N. Zaker, J. Gaudet, L. Roué, G. A. Botton, D. Beauchemin, D. C. Higgins, S. Thorpe, D. A. Harrington and D. Guay, ACS Catal., 2023, 13, 1726–1739 CrossRef CAS.
- P. Urchaga, S. Baranton, C. Coutanceau and G. Jerkiewicz, Langmuir, 2012, 28, 3658–3663 CrossRef CAS PubMed.
- B. B. Blizanac, M. Arenz, P. N. Ross and N. M. Marković, J. Am. Chem. Soc., 2004, 126, 10130–10141 CrossRef CAS PubMed.
- S. K. Meher and G. R. Rao, ACS Catal., 2012, 2, 2795–2809 CrossRef CAS.
- G. Chen, M. Sun, J. Li, M. Zhu, Z. Lou and B. Li, Nanoscale, 2019, 11, 18874–18880 RSC.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed.
- C. H. Choi, K. Chung, T. H. Nguyen and D. H. Kim, ACS Energy Lett., 2018, 3, 1415–1433 CrossRef CAS.
- B. Y. Zheng, H. Zhao, A. Manjavacas, M. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2015, 6, 7797 CrossRef PubMed.
- C. Zhan, M. Moskovits and Z. Tian, Matter, 2020, 3, 42–56 CrossRef.
- L. Huang, J. Zou, J. Y. Ye, Z. Y. Zhou, Z. Lin, X. Kang, P. K. Jain and S. Chen, Angew. Chem., Int. Ed., 2019, 58, 8794–8798 CrossRef CAS PubMed.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard III and H. A. Atwater, Nat. Commun., 2014, 5, 5788 CrossRef CAS PubMed.
- Z. Liu, W. Gao, L. Liu, S. Luo, C. Zhang, T. Yue, J. Sun, M. Zhu and J. Wang, J. Hazard. Mater., 2023, 442, 130036 CrossRef CAS PubMed.
- W. Nie, X. Wang, Z. Wang, Y. Gao, R. Chen, Y. Liu, C. Li and F. Fan, J. Phys. Chem. Lett., 2021, 12, 10829–10836 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, H. Xue, J. Gao, Y. Peng, W. Song and L. Ge, Chin. J. Catal., 2021, 42, 1677–1688 CrossRef CAS.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard III and H. A. Atwater, Nat. Commun., 2014, 5, 5788 CrossRef CAS PubMed.
- J. Yuan, G. Yang, H. Wang, Y. Cao, H. Wang, F. Peng and H. Yu, Chem. Eng. Sci., 2023, 276, 118818 CrossRef CAS.
- Z. Zhang, X. Tian, B. Zhang, L. Huang, F. Zhu, X. Qu, L. Liu, S. Liu, Y. Jiang and S. Sun, Nano Energy, 2017, 34, 224–232 CrossRef CAS.
- R. Wan, M. Luo, J. Wen, S. Liu, X. Kang and Y. Tian, J. Energy Chem., 2022, 69, 44–53 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- S. Chen, N. Liu, J. Zhong, R. Yang, B. Yan, L. Gan, P. Yu, X. Gui, H. Yang, D. Yu, Z. Zeng and G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202209693 CrossRef CAS PubMed.
- W. F. Lin, T. Iwasita and W. Vielstich, J. Phys. Chem. B, 1999, 103, 3250–3257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section, and additional figures (Fig. S1–S7). See DOI: https://doi.org/10.1039/d3cp05624d |
| This journal is © the Owner Societies 2024 |