
Bridging the gap: viable reaction pathways from tetrahedrane to benzyne†
Taylor A.
Cole
,
Steven R.
Davis
 ,
Athena R.
Flint
and
Ryan C.
Fortenberry
*
,
Athena R.
Flint
and
Ryan C.
Fortenberry
*
Department of Chemistry and Biochemistry, University of Mississippi, University, Mississippi, 38677, USA. E-mail: r410@olemiss.edu
First published on 8th April 2024
Abstract
The addition of sp-carbon-containing molecules to polycyclic sp3 tetrahedrane (c-C4H4) results in the formation of both o-benzyne (c-C6H4) and benzene (c-C6H6). Since both c-C6H4 and c-C6H6 have been detected in the interstellar medium (ISM), providing additional pathways for their possible astrochemical formation mechanisms can lead to the discovery of other molecules, such as c-C4H4, benzvalyne, and vinylidene (:CCH2). Addition of diatomic carbon (C2), the ethynyl radical (C2H), vinylidene, and acetylene (HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) to c-C4H4 is undertaken in individual pathways through high-level quantum chemical computations at the CCSD(T)-F12b/cc-pVTZ-F12 level of theory. The resulting C2 addition pathway proceeds barrierlessly through benzvalyne as an intermediate and reaches a true minimum at c-C6H4, but no leaving groups are produced which is required to dissipate excess energy within an interstellar chemical scheme. Similarly, the C2H addition to c-C4H4 produces benzvalyne as well as its related isomers. This pathway allows for the loss of a hydrogen leaving group to dissipate the resulting energy. Lastly, the HCCH and :CCH2 addition pathways follow through both benzvalene and benzvalyne in order to reach c-C6H6 (benzene) and c-C6H4 (o-benzyne) as well as H2 as the required leaving group. Although there is a barrier to the HCCH addition, the :CCH2 addition presents the contrary with only submerged barriers. These proposed mechanisms provide alternative possibilities for the formation of complex organic molecules in space.
CH) to c-C4H4 is undertaken in individual pathways through high-level quantum chemical computations at the CCSD(T)-F12b/cc-pVTZ-F12 level of theory. The resulting C2 addition pathway proceeds barrierlessly through benzvalyne as an intermediate and reaches a true minimum at c-C6H4, but no leaving groups are produced which is required to dissipate excess energy within an interstellar chemical scheme. Similarly, the C2H addition to c-C4H4 produces benzvalyne as well as its related isomers. This pathway allows for the loss of a hydrogen leaving group to dissipate the resulting energy. Lastly, the HCCH and :CCH2 addition pathways follow through both benzvalene and benzvalyne in order to reach c-C6H6 (benzene) and c-C6H4 (o-benzyne) as well as H2 as the required leaving group. Although there is a barrier to the HCCH addition, the :CCH2 addition presents the contrary with only submerged barriers. These proposed mechanisms provide alternative possibilities for the formation of complex organic molecules in space.
1 Introduction
Hydrocarbons are known to exist in the interstellar medium (ISM)1,2 with polycyclic aromatic hydrocarbons (PAHs) potentially responsible for around a quarter of the carbon molecularly sequestered in the ISM.3 Recent research confirms that PAHs are the primary precursors to soot particles,4 showcasing that these molecules are broadly applicable and not just limited to astrochemistry. This narrative enriches our comprehension of PAHs and underscores their universal presence and significance, both in earthly and cosmic environments. Their aromatic nature and relation to recently detected molecules in the ISM1,2,5,6 promote their discussion in current astrochemical conversations.7,8 As the astrochemical significance of PAHs increases, understanding their involved precursors and reaction pathways are crucial for a full picture of the organic and prebiotic chemistry of the ISM.Many cyclic hydrocarbons, such as c-C6H6 (benzene)9,10 and c-C6H4 (o-benzyne),6 have been detected toward various astronomical regions, and their existence opens the possibility for new chemistry to be explored. Several C6H4 isomers lie along the potential energy surface between low-energy aromatic structures. One example is benzvalene, which was first synthesized via photochemical reactions of c-C6H6 in the laboratory nearly 60 years ago.11 Other studies delve into the potential mechanisms relating c-C6H6 to c-C6H4 and their additional isomers and derivatives.12,13 This research provides a possible foundation for how such reactions could take place in the gas phase with astrochemical applications clearly in view. Resulting reaction intermediates could serve as possible targets for the QUIJOTE1 or GOTHAM line surveys and the exploration of PAHs in molecular clouds, such as TMC-1.14–20 Beyond PAHs and their related species, other notable sources of carbon in the universe include C2 and its hydrogenated C2-containing affiliates, such as C2H (ethynyl radical), HCCH (acetylene), and :CCH2 (vinylidene).21–24 The diatomic carbon molecule (C2) has been detected in the near IR spectrum of Cyg OB2 No. 12,21 and HCCH has been previously detected in the IR spectrum of IRC +10° 216, and molecular clouds GL 2591, W3 1RS 5, and OMC-1 IRc2.23,24 :CCH2 is reported to have strong IR intensities for the ν1 CH symmetric stretching at 3025 cm−1, ν2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1635 cm−1, ν3 CH2 scissoring at 1165 cm−1, and ν6 CH2 rocking at 450 cm−1.25–27 Although the :CCH2 isomer has not been detected, its strong vibrational intensities provide potential for identification. These hydrocarbons could contribute materials to support complex chemistry and the creation of variable organic structures in the ISM since small molecules like C2 can become potential building blocks for the formation of c-C6H6 and other, more complex, PAHs.28 The growth of such chemistry through the interaction of these molecules may help to provide insights into astronomical spectral observations that have eluded explanation for decades.
C stretching at 1635 cm−1, ν3 CH2 scissoring at 1165 cm−1, and ν6 CH2 rocking at 450 cm−1.25–27 Although the :CCH2 isomer has not been detected, its strong vibrational intensities provide potential for identification. These hydrocarbons could contribute materials to support complex chemistry and the creation of variable organic structures in the ISM since small molecules like C2 can become potential building blocks for the formation of c-C6H6 and other, more complex, PAHs.28 The growth of such chemistry through the interaction of these molecules may help to provide insights into astronomical spectral observations that have eluded explanation for decades.
Most notably, the unidentified infrared bands (UIRs) are spectral features that have not been definitively linked to any certain molecule to date and are observed in the infrared region of the electromagnetic spectrum toward various astronomical objects.29–32 They were first reported from observations of the planetary nebulae NGC 7027, BD + 30° 3639, and NGC 6572.33 The current hypotheses suggest PAHs as contributing factors to these spectral lines with a possible inclusion of their aliphatic counterparts or mixed aliphatic-aromatics hydrocarbons.34–39 Beyond the infrared, ultraviolet wavelengths may also be applicable to PAHs. In 1965, a 2175 Å ultraviolet extinction bump was discovered in the ISM40 and further supported by the Orbiting Astronomical Observatory 2.41 Observations of this so-called “UV-Bump” have since shown this feature to be widespread and distinct in the Milky Way Galaxy.42 Similarly to the UIRs, this interstellar extinction bump has no known current rationale that explains its existence. However, some speculation as to its provenance includes carbonaceous molecules and PAHs.43–45
One such hypothesis for the origin of the UV-Bump is that of carbonaceous molecular clusters exhibiting a T-carbon molecular structure in which the vertices of a cubic diamond lattice are replaced with a tetrahedral arrangement of carbons. Such an sp3 carbon motif is theorized to be responsible for at least part of the UV-Bump.46 Considering the abundance of neutral hydrogen in the interstellar cosmos,47,48 T-carbons may exist in a hydrogenated form with 40 carbon and 16 hydrogen atoms, subsequently denoted as hydrogenated T-carbons (HTCs). The variable sizes of carbonaceous molecules and clusters in interstellar dust49 may enable HTCs to exist as a mixture of clusters. These HTCs produce a striking absorption peak at roughly the same wavelength as the extinction bump. Then, when combined with other hypothesized molecular clusters such as graphite, MgSiO3, and Fe2SiO4, a well-fitted mixed model is obtained with startling correlation to the UV-Bump.50 Whether HTCs contribute to this 2175 Å feature remains to be seen, but their role in the astronomical carbon budget has not been explored in as much detail as PAHs or their derivatives.
The main HTC molecular building block, c-C4H4 (tetrahedrane), has been subjected to intense research for years due to its high angle strain and kinetic stability. Unlike other platonic species, including cubane and dodecahedrane, tetrahedrane has been elusive in the synthesis process, fostering further research.51–53 The uptick in attention towards increasingly complex hydrocarbons in interstellar environments has brought c-C4H4 some research attention due to its structural prominence and theorized implementation in larger, more complex molecules in the ISM.54–56 This c-C4H4 aliphatic monomer has shown to be a minimum on a potential energy surface57 and is likely stable in cold, low pressure environments, i.e. the ISM. More recently, its rovibrational data obtained from quartic force field calculations reveal two intense vibrational frequencies close in proximity to UIRs, providing possibility for detection in both the ISM and the laboratory.58–60 Because of tetrahedrane's correlation with HTCs, UIRs, and the UV-Bump, further astrochemically-minded research into this organic molecule is warranted. Since c-C4H4 is so strained, reactions with simple, C2-containing molecules could lead to the known interstellar molecule c-C6H4, and may have implication in the larger chemistry of PAHs.
A likely intermediate along the reaction pathway of c-C4H4 and C2 is benzvalyne, another theoretical hydrocarbon. Previous work has shown this structure to be a minimum with a significant dipole moment of 2.6 D and formative correspondence to already-detected interstellar molecules like c-C6H4 along the reaction pathway.12,13 Benzvalyne is shown to be a required intermediate between c-C4H4, c-C6H6, and c-C6H4 in a potential energy surface (PES) that links these species. This bicyclic isomer of c-C6H4 can offer an internal perspective for the pathways of sophisticated hydrocarbons in interstellar environments. Any correlation between these detected and undetected organic molecules can provide additional advantageous results for future detection signal acquisitions in the ISM and understanding of their overall chemistry. Hence, this work will explore reactions of c-C4H4 with C2, C2H, HCCH, and :CCH2. While such reactions are believed to play an important role in astrochemistry, their role in combustion chemistry cannot be neglected as these molecules and their derivatives are known to inflict harmful consequences towards the climate, environment, and overall human health.4,61–65
2 Computational methods
The small size of the systems under study within this work allows for the use of highly-accurate quantum chemical methods to be employed. The current “gold-standard” method within computational chemistry is CCSD(T),66–71 often paired with the cc-pVXZ basis sets of Dunning and coworkers.72–74 The CCSD(T)/cc-pVXZ level of theory can be improved through the addition of an explicit-correlation (F12) correction for a negligible increase in computational cost.75–79 The CCSD(T)-F12b/cc-pVXZ-F12 level of theory, through the inclusion of short-range interelectronic interactions, is shown to increase calculation accuracy significantly; addition of the F12 correction to the CCSD(T)/cc-pVXZ level of theory is shown to produce results of CCSD(T)/cc-pV(X + 2)Z accuracy.80 Throughout this work, the primary level of theory will be CCSD(T)-F12b/cc-pVTZ-F12, hereafter abbreviated as F12-TZ.All reactant, product, and intermediate structures are optimized, alongside harmonic frequency calculations, at the F12-TZ level of theory using the MOLPRO 2022.3 quantum chemical software.81–83 Transition state structures are optimized, followed by the requisite frequency calculations, at the B3LYP84–88/aug-cc-pVTZ72,73,89 level of theory using the Gaussian1690 software package. In order to correctly place these structures along the reaction pathway, a single-point energy calculation is done at the F12-TZ level of theory in MOLPRO at the B3LYP transition state optimized geometry, a method shown to work well for explaining observed, experimental reactions.91,92 Intrinsic reaction coordinate (IRC) calculations93 are performed at the B3LYP/aug-cc-pVTZ level of theory in Gaussian so that the computed transition states correspond to the desired motion along the reaction coordinate.
In some cases, the energy profile of the pathway may not be able to be described through a transition state, or a transition state structure may be difficult to locate. In such situations, a relaxed potential energy surface (PES) scan can be created in order to evaluate the potential surface more rigorously. Relaxed potential energy scans follow the variable corresponding to a given reaction coordinate, such as an interatomic distance or angle, by constraining the geometric variable to a series of values between two points of interest. At each value of this variable, the remainder of the structure is allowed to optimize around it. The electronic energies of each partially-constrained geometry along the coordinate can be plotted and used to visualize a two-dimensional slice of the potential energy surface. Points within each relaxed PES scan are optimized at the B3LYP/aug-cc-pVTZ level of theory, followed by single-point energy calculations at the F12-TZ level of theory, in MOLPRO.
In the addition of C2H to c-C4H4, a PES is generated to model the change in energy as the terminal carbon of C2H approaches a carbon vertex in c-C4H4. Points are created over a 1.0–4.0 Å interatomic range in increments of 0.1 Å. On the C6H6 potential surface, two PESs are constructed. One PES is created to visualize the change in energy as the angle between the migratory hydrogen of TS1a (see below) and the two terminal carbon atoms increases. Points for this scan are generated over a 30°–150° range in increments of 0.3°. The final PES scans over the distance between a carbon vertex in c-C4H4 and a dummy atom placed within the carbon–carbon bond of HCCH. The X–C reaction coordinate variable is varied over a 1.0–4.3 Å range in increments of 0.1 Å.
The PESs discussed above can reveal internal conversions between structures with differing orbital configurations. To visualize how the reaction proceeds through these internal conversions, correlation diagrams are created by plotting the frontier orbitals of structures before and after the internal conversion on the potential surface. Molecular orbital calculations on these structures are done at the RHF/aug-cc-pVTZ level of theory (for closed-shell references) or the ROHF/aug-cc-pVTZ level of theory (for open-shell references) with Gaussian16. Molecular orbital visualizations are created in Gabedit.94,95
3 Results and discussion
3.1 C2 addition to c-C4H4
The addition of C2 to c-C4H4 results in the reaction pathway given in Fig. 1. This reaction runs barrierlessly to an intermediate at −111.56 kcal mol−1 below the starting materials. This intermediate passes through a transition state with a small, 4.7 kcal mol−1 increase and settles at benzvalyne, −118.20 kcal mol−1 relative to the initial reactants.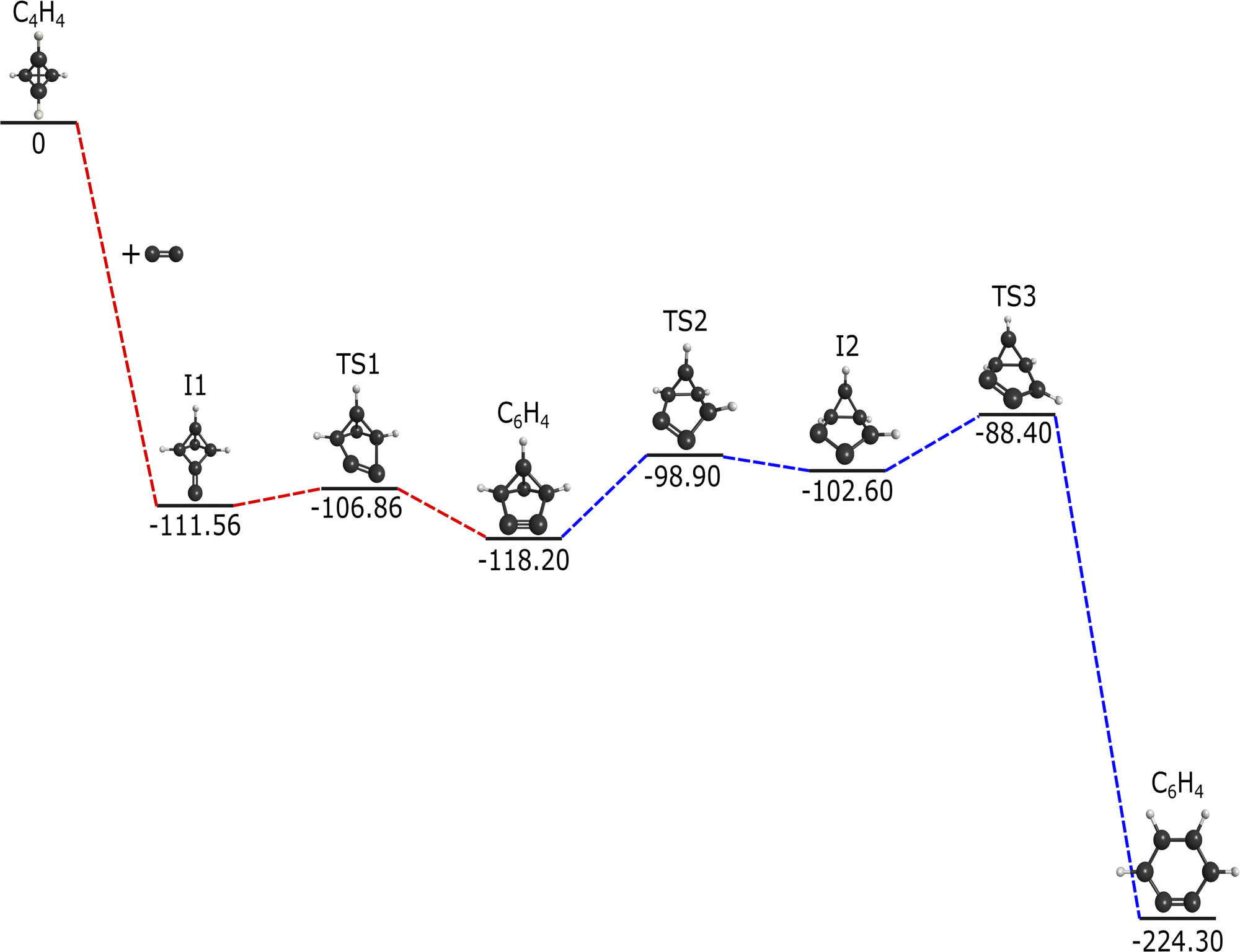 | ||
| Fig. 1 Reaction pathway for the formation of c-C6H4via C2 addition to c-C4H4 in kcal mol−1. Red dashed lines represent this research, and blue dashed lines from Poland et al.12 | ||
The imaginary vibrational mode of TS1 has a frequency of 342.5i cm−1 and shifts the two foremost carbons back and forth, providing a connection between I1 and benzvalyne. The −118.2 kcal mol−1 decrease from the reactants implies that additional energy is available for further chemistry after this initial creation of benzvalyne. Also, benzvalyne has been shown to be a possible intermediate towards the creation of c-C6H4.12 Due to these two factors, this previously-researched pathway included in blue in Fig. 1 in order to formulate the connection from benzvalyne to c-C6H4.12 This additional route is recomputed to match the current level of theory and encompasses a multi-transition state barrier from benzvalyne. Benzvalyne, I1, and TS1 are all more than 100 kcal mol−1 lower than the reactants, providing the thermodynamics necessary for chemistry to take place in low-temperature and low-pressure environments. Additionally, the relatively low energy barriers from intermediates to TSs imply that the kinetics should also be favorable.
Alone, this reaction would not be feasible in the ISM or most low-pressure gas phase environments, unfortunately. When combined with the reactive steps stemming from c-C4H4 and C2, an overall favorable reaction mechanism is formed. Notably, c-C6H4 is −224.3 kcal mol−1 lower than the combination of c-C4H4 and C2. However, since no leaving group is present that would result in isolation of c-C6H4, this formation pathway is less feasible.96,97 While radiative stabilization of c-C6H4 is possible in theory, such processes have yet to be observed experimentally for neutral–neutral reactions.98 As procession through an available bimolecular exit channel, if it exists, is often much faster than the corresponding radiative stabilization process for small species,99c-C6H4 is more likely to re-dissociate into reactants or generate nontargeted products on this pathway.
Using the acquired information about the C2 addition reaction pathway, there is potential for c-C4H4 to be a feasible reactant of the already-detected c-C6H4 if some form of a leaving group presents itself through additional reactive connections. However, the most natural place to expect a leaving group would come from the incident molecule and not the c-C4H4, making a hydrogen atom from the closely-related C2H triatomic molecule100 the next step for collisions with the present HTC standin utilized here, c-C4H4.
3.2 C2H addition to c-C4H4
The supplemental hydrogen from the addition of C2H, a molecule first detected in the Orion Nebula with the National Radio Astronomy Observatory,22 creates a unique reaction pathway in comparison to that of the addition of C2. This reaction, present in Fig. 2, proceeds barrierlessly towards the first submerged-well intermediate, I1, at −84.68 kcal mol−1 relative to the reactants akin to the formation of I1 in Fig. 2. Similar organic molecules appear throughout these reaction pathways even if I1 in the C2H addition is higher than I1 in the C2 reaction. Both I1 structures in Fig. 1 and 2 are qualitatively equivalent in structure, but the additional hydrogen in the C2H I1 creates an odd number of electrons, resulting in free radical behavior and thus increased reactivity. Intermediate I2 in Fig. 2 is also structurally similar to benzvalyne showcasing consistent behavior as would be expected for systems that only differ by a single hydrogen atom.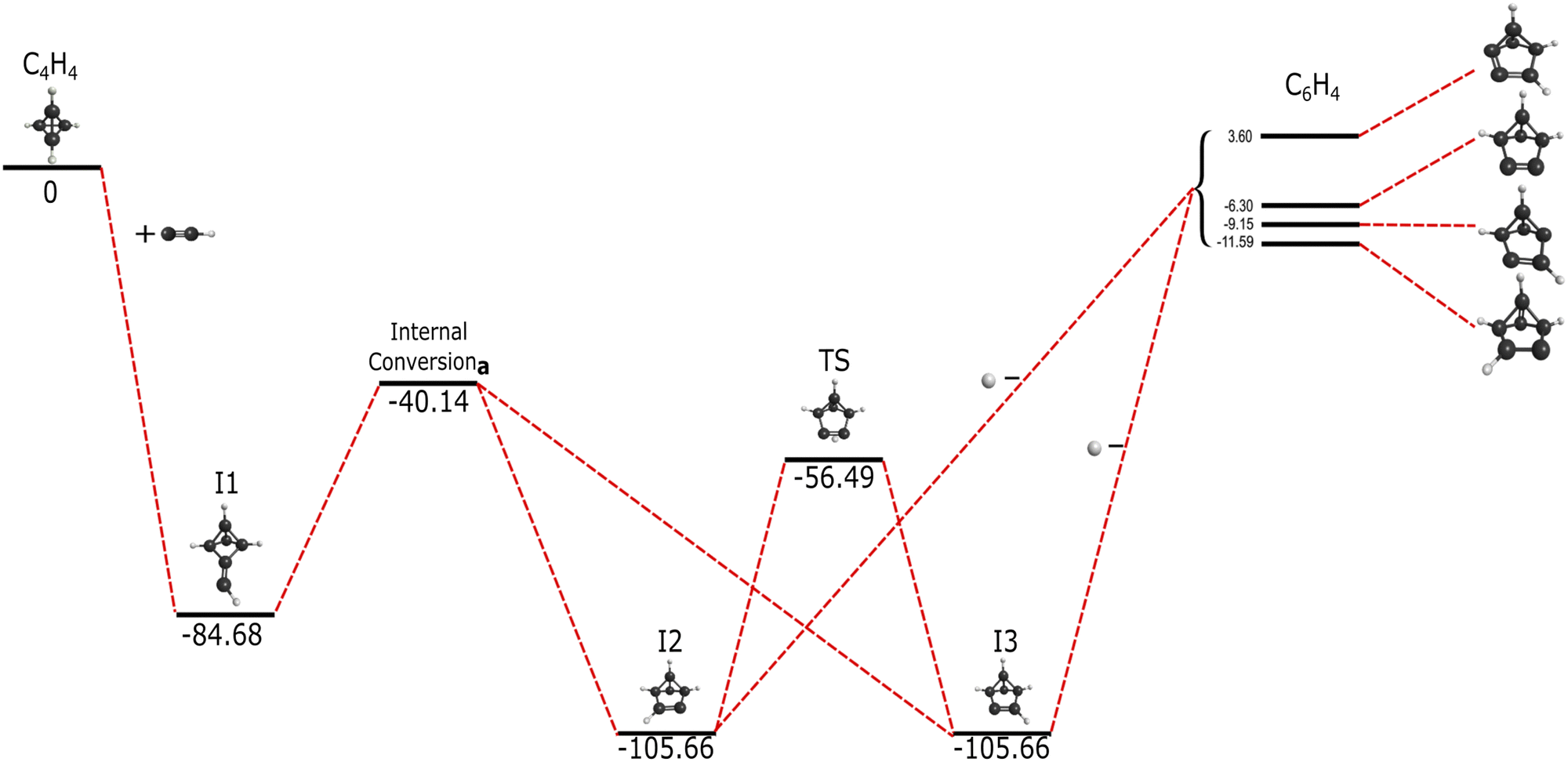 | ||
| Fig. 2 Reaction pathway for the formation of benzvalyne via C2H addition to c-C4H4 in kcal mol−1. Internal conversiona represents the intersection seen in Fig. 3. | ||
In contrast to the C6H4 potential surface described previously where a transition state separates I1 and benzvalyne, this C6H5 potential surface (where the ethynyl radical and tetrahedrane react) allows I1 and I2 to interconvert through an internal conversion of molecular structures as shown in the scan depicted in Fig. 3. This varied distance between the terminal carbon on the C2H and a carbon on tetrahedrane produces two potential energy wells as described in Section 2 which cross when the distance reaches approximately 2.2 Å. This internal conversion can promote the radiationless interchange between structures of the same orbital symmetry.101–104 While I1 and I2 appear to be structurally different, their orbital spaces are actually closely related. When observing the geometries closest to the exact point of conversion, the two highest-energy occupied molecular orbitals (HOMOs) and the lowest-energy unoccupied molecular orbitals (LUMOs) retain their original symmetry and ordering through this internal conversion as given in Fig. 4. While I1 represents a minimum, I2 is a lower energy isomer. The orbital topologies, ordering, and occupation remain consistent between I1 and I2 as shown in Fig. 4 on both sides of the internal conversion, but the repositioning of the carbon atoms in I2 (to the right of the internal conversion in Fig. 3) optimizes the orbitals slightly differently and produces the lower-energy state. As such, the molecule can shift surfaces readily when the interacting carbon atoms are placed at roughly 2.2 Å from one another. This, of course, could be true for any tetrahedrane carbon as all are equivalent by symmetry.
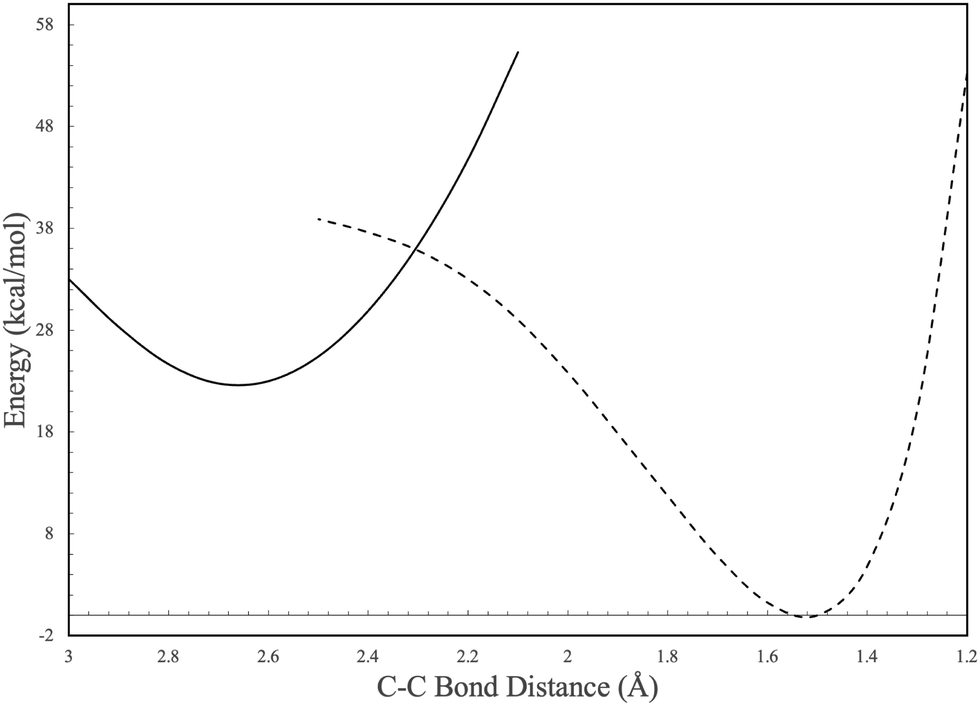 | ||
| Fig. 3 Potential energy scan of the C–C bond between C2H and c-C4H4. Solid and dashed lines represent individual molecular configurations correlating to I1 and I2 in Fig. 2. | ||
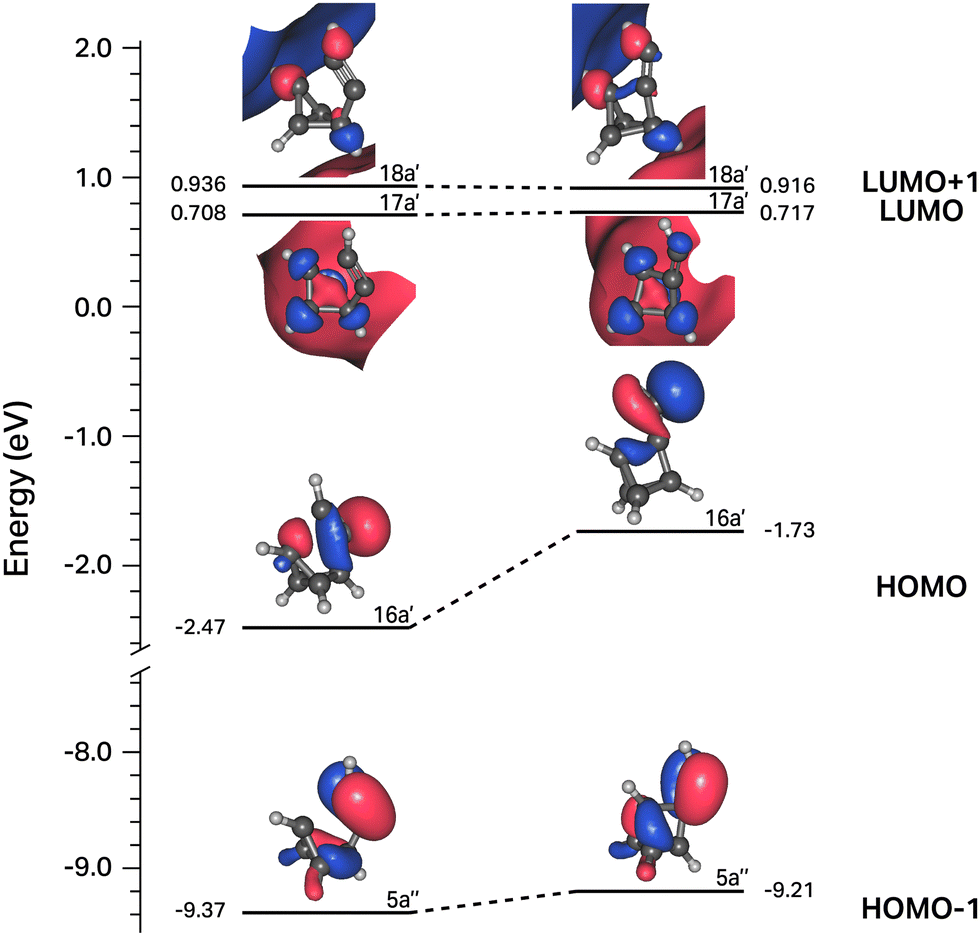 | ||
| Fig. 4 Correlation diagram for the four frontier orbitals of structures to the right and left of the internal conversion shown in Fig. 3. | ||
Moving on to the relationship between I2 and I3 in Fig. 2, the connectivity and achirality of both I2 and I3 show that they are inherently the same molecule with the same energies and capabilities. As I2 at −105.66 kcal mol−1 isomerizes towards I3 through a transition state, either conformation can be created from I1, and both can expel a hydrogen atom as a leaving group.
The departure of the H atom from either I2 or I3 creates four different, bicyclic isomers of c-C6H6. Benzvalyne comes third in stability at −6.3 kcal mol−1 below the reactants, where two other C6H4 isomers lie lower at −9.15 kcal mol−1 and −11.59 kcal mol−1 as given in the upper-right of Fig. 1. In addition to these three, there is an additional isomer of C6H4 that lies above the reactants at 3.6 kcal mol−1 which is unlikely be accessed in the gas phase as it will be higher in energy than the reactants and would require more than 1600 K in ambient temperature. The transition state energies for the interconversion between these isomers creates a further thermodynamic barrier as they are dozens of kcal mol−1 above the reactants’ energy. Hence, once these isomers are made in the gas phase, they cannot interconvert between one another if they are formed in this pathway. The likely Boltzmann behavior for the distribution of these isomer populations implies that benzvalyne will still be able to contribute to the formation of o-benzyne, but it will not be the dominant player. Other reactions would be able to contribute moreso to their populations.
3.3 HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CH and :CCH2 addition to c-C4H4
CH and :CCH2 addition to c-C4H4
The last pathway follows the inclusion of the isomers, :CCH2 and HCCH, as separate reactants with c-C4H4, creating two interrelataed pathways. The reaction proceeds towards the creation of c-C6H6 and c-C6H4 shown in Fig. 5 largely with submerged barriers. Acetylene's previous detection in the IR within the ISM and vinylidene's potential existence implies that such a pathway may influence the interstellar production of benzene and o-benzyne and, hence, that of PAHs in the gas phase.
 | ||
| Fig. 5 Reaction pathway for the formation of c-C6H6 and c-C6H4via HCCH and :CCH2 addition to c-C4H4 in kcal mol−1. TS1b represents the transition state analyzed in Fig. 6. Internal conversionc represents the intersection seen in Fig. 7. Red dashed lines represent this research, blue dashed lines are based on Poland et al.,12 and green dashed lines are inspired from Kislov et al.14 | ||
The addition of :CCH2 to c-C4H4 leads to I2, which is a recognizable motif from the aforementioned intermediates of the previous reaction pathways. The inclusion of two hydrogens from the reactant separates this intermediate from the previous structures of similar connectivities. The creation of I2 from :CCH2 and c-C4H4 allows for the formation of the cyclic C6H6 isomers and promotes stabilization from a diatomic hydrogen leaving group. While I2 can also be formed from acetylene combined with tetrahedrane, this reaction may be a non-starter as another step must take place before I2 is created.
HCCH reacting with c-C4H4 leads to I1 at −68.37 kcal mol−1, which then requires an increase of 100.98 kcal mol−1 in order to reach the first transition state, lying at 32.61 kcal mol−1 above the reactants. This transition state, denoted as “TS1b”, is represented as the crossing point between the two potential energy curves seen in Fig. 6 with I1 and I2 as the minima. This scan follows the change in angle of the hydride shift from the bottom to the top-most carbon seen in I1 and I2. The point of crossing occurs at a bond angle of roughly 70° and serves as a maximum between the two intermediates, which interconvert via TS1b. At this geometry the hydrogen migrates across two carbon atoms in a concerted motion according the reaction coordinate and the imaginary frequency normal mode coordinate. However, the height of TS1b not only precludes the formation of I2 from acetylene and tetrahedrane, but it also keeps I1 from being created as any backwards pathways from I2. Hence, once I2 is made from :CCH2 and c-C4H4, its most likely pathway is forward, away from either sets of reactants.
 | ||
| Fig. 6 Potential energy scan of H–C–C bond angle in TS1 of Fig. 5. Solid and dashed lines represent individual electronic states correlating to I2 and I1 in Fig. 5. | ||
Consistent with the C2H pathway, another internal conversion immediately after I2, denoted as “internal conversionc” in Fig. 5, rests at −10.33 kcal mol−1 relative to acetylene or −54.18 kcal mol−1 relative vinylidene as the starting materials, and internal conversionc occurs between I2 and the c-C6H6 (benzvalene) isomer. In scanning across the bond distance defined from the mid point of the acetylene and a tetrahedrane carbon vertex, two evident energetic wells (I2 and benzvalene) are formed and have their surfaces cross at an approximate bond length of 3.0 Å (Fig. 7). Subsequently, benzvalene sits at a minimum −88.62 kcal mol−1 below the reactants. Further analysis of internal conversionc showcases similar results as Fig. 4. At the geometries adjacent to the conversion maximum, the HOMO, HOMO−1, LUMO, and LUMO+1 retain their orbital symmetry and occupation as given in Fig. 8. The HOMO and the HOMO−1 switch positions between the two isomers as the carbon atoms from the two reactants interact with one another, but the orbital topologies are largely unchanged. This internal conversion is similar to that for the C6H5 PES described in the previous section but with the occupied orbitals shifting positions here. This produces the two minima shown in Fig. 7 which correspond to I2 and benzvalene, respectively from left-to-right, and are given in their stations in Fig. 5.
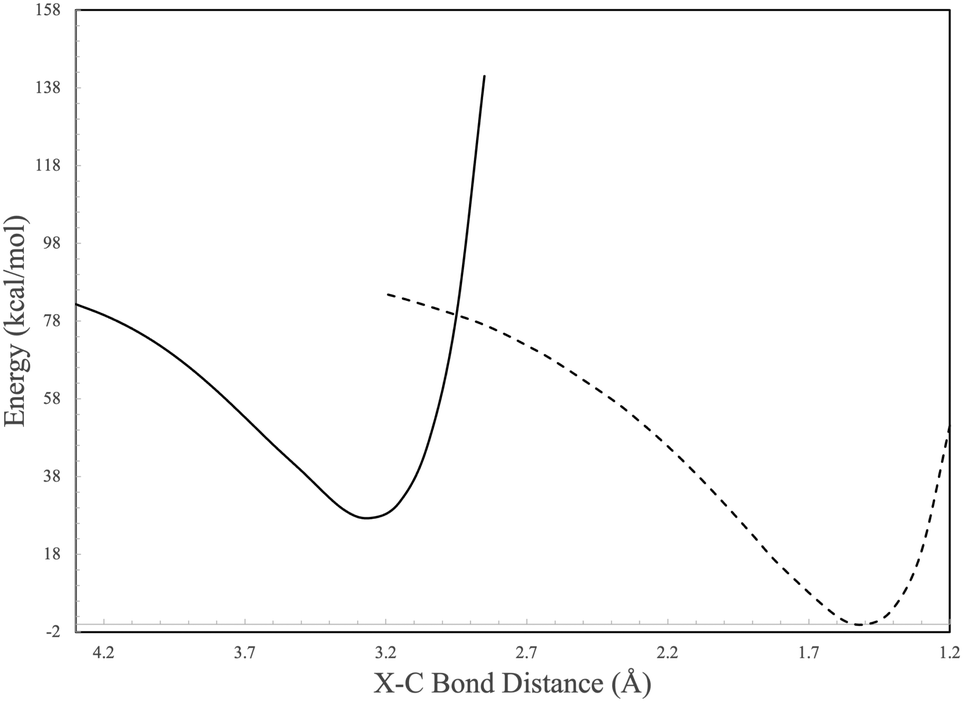 | ||
| Fig. 7 Potential energy scan of X–C bond of HCCH and c-C4H4. Solid and dashed lines represent individual electronic states correlating to I2 and C6H6 in Fig. 5. | ||
 | ||
| Fig. 8 Correlation diagram for the four frontier orbitals of structures to the right and left of the internal conversion shown in Fig. 7. | ||
Once benzvalene forms, it can then progress through three different isomerization pathways. The lower energy two have benzene as an intermediate, but all can lead to o-benzyne. The most energetically favorable pathway progresses through TS8 and then goes down significantly to benzene at −160.9 kcal mol−1. TS6 is higher in energy and leads to I5 before a slight uphill to TS7 and onto benzene. TS6, TS7, and TS8 are all qualitatively similar, but the structures are slightly different with the five-member ring showing different puckering characteristics. However, the most notable difference are the normal coordinates for the imaginary frequencies. Regardless, the formation of benzene is energetically favorable and proceeds through these pinched six-membered rings. Formation of o-benzyne from benzene has been previously shown by Kislov et al.,14 and this is verified here by more modern methods. I6 with two hydrogens on a single carbon is a minimum even with TS9 being very nearly degenerate. All materials lie below all starting materials, even TS10 before ushering off H2 to generate o-benzyne.
The upper pathway from benzvalene to o-benzyne in Fig. 5 proceeds through TS2 and climbs up to TS3. This structure has an energy of −4.55 kcal mol−1 relative to vinylidene and tetrahedrane as the starting materials. Even if the acetylene reaction could produce I2, this pathway would not be energetically accessible. However, :CCH2 as a reactant appears to be necessary to drive this reaction forward in the gas phase. TS3 loses H2 at this step, and creates benzvalyne. The same process to produce o-benzyne as given in the C6H4 pathway is enacted, but TS5 here is 1.77 kcal mol−1 above even the :CCH2 + c-C4H4 starting materials. However, ambient temperatures of 890 K would overcome this barrier, and even room temperature could get notable percentages (∼5%) of I4 to overcome the energy of TS5. Hence, even this upper pathway is not unreasonable but would require warm environments especially for astrochemical considerations.
4 Conclusions
In utilizing tetrahedrane as a stand-in for HTCs, this work shows that the reaction of vinylidene with HTCs can readily produce benzene, but benzene will likely only be an intermediate from this reaction since there is no leaving group in the reaction pathway. However, o-benzyne, a known interstellar molecule, could be formed in various pathways. Addition of the ethynyl radical to HTCs could also produce o-benzyne, but acetylene or C2 reacting with HTCs likely will not. The former is due to energetics and the latter due to the lack of a leaving group to dissipate the excess energy kinetically. However, this work is showing that strained species can lead to other strained species before more stable molecules are produced. Such reactions certainly have applications to astrochemistry, and the possible interstellar observation of benzvalene or benzvalyne would certainly allow for more complicated organic chemistry as shown in this work. In any case, reactions of HTCs, as modeled by tetrahedrane, with common, dicarbon molecules produce common, cyclic aromatic hydrocarbons as final products but also generate less well examined intermediates before doing so.Author contributions
Taylor A. Cole: methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. Steven R. Davis: conceptualization, methodology, validation, formal analysis, project administration, resources, software, supervision, writing – review & editing. Athena R. Flint: methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization. Ryan C. Fortenberry: conceptualization, methodology, validation, formal analysis, project administration, resources, software, supervision, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank funding from NASA Grants 22-A22ISFM-0009 & NNH22ZHA004C and from the University of Mississippi's College of Liberal Arts and Department of Chemistry. The computing resources utilized in this work were from the Mississippi Center for Supercomputing Research supported, in part, by NSF Grant OIA-1757220.References
- B. A. McGuire, A. M. Burkhardt, S. Kalenskii, C. N. Shingledecker, A. J. Remijan, E. Herbst and M. C. McCarthy, Science, 2018, 359, 202–205 CrossRef CAS PubMed.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265 CrossRef CAS PubMed.
- C. Joblin and A. G. G. M. Tielens, PAHs and the Universe, EDP Sciences, 2011 Search PubMed.
- C. Shao, Q. Wang, W. Zhang, A. Bennett, Y. Li, J. Guo, H. G. Im, W. L. Roberts, A. Violi and S. M. Sarathy, Commun. Chem., 2023, 6, 223 CrossRef CAS PubMed.
- A. M. Burkhardt, K. L. K. Lee, P. B. Changala, C. N. Shingledecker, I. R. Cooke, R. A. Loomis, H. Wei, S. B. Charnley, E. Herbst, M. C. McCarthy and B. A. McGuire, Astrophys. J., Lett., 2021, 913, L18 CrossRef CAS.
- J. Cernicharo, M. Agúndez, R. I. Kaiser, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 652, L9 CrossRef CAS.
- R. F. Knacke, Nature, 1977, 269, 132–134 CrossRef CAS.
- M. Gatchell, J. Ameixa, M. Ji, M. H. Stockett, A. Simonsson, S. Denifl, H. Cederquist, H. T. Schmidt and H. Zettergren, Nat. Commun., 2021, 12, 6646 CrossRef CAS PubMed.
- J. Cernicharo, A. M. Heras, A. G. G. M. Tielens, J. R. Pardo, F. Herpin, M. Guélin and L. B. F. M. Waters, Infrared Space Observatory's Discovery of C4H2, C6H2, and Benzene in CRL 618, 2001.
- E. F. van Dishoeck, S. Grant, B. Tabone, M. van Gelder, L. Francis, L. Tychoniec, G. Bettoni, A. M. Arabhavi, D. Gasman, P. Nazari, M. Vlasblom, P. Kavanagh, V. Christiaens, P. Klaassen, H. Beuther, T. Henning and I. Kamp, Faraday Discuss., 2023, 245, 52–79 RSC.
- K. E. Wilzbach, J. S. Ritscher and L. Kaplan, J. Am. Chem. Soc., 1967, 89, 1031–1032 CrossRef CAS.
- K. N. Poland, W. Yang, R. C. Fortenberry and S. R. Davis, Phys. Chem. Chem. Phys., 2022, 24, 14573–14578 RSC.
- K. N. Poland, B. R. Westbrook, D. H. Magers, R. C. Fortenberry and S. R. Davis, J. Chem. Phys., 2022, 156, 24302 CrossRef CAS PubMed.
- V. V. Kislov, T. L. Nguyen, A. M. Mebel, S. H. Lin and S. C. Smith, J. Chem. Phys., 2004, 120, 7008–7017 CrossRef CAS PubMed.
- H. Wang, A. Laskin, N. W. Moriarty and M. Frenklach, On Unimolecular Decomposition of Phenyl Radical, 2000 Search PubMed.
- A. Matsugi and A. Miyoshi, Phys. Chem. Chem. Phys., 2012, 14, 9722–9728 RSC.
- A. Comandini and K. Brezinsky, J. Phys. Chem. A, 2012, 116, 1183–1190 CrossRef CAS PubMed.
- C. W. Bauschlicher and A. Ricca, Chem. Phys. Lett., 2013, 566, 1–3 CrossRef CAS.
- L. V. Moskaleva, L. K. Madden and M. C. Lin, Unimolecular isomerization/decomposition of ortho-benzyne: ab initio MO/statistical theory study, 1999.
- G. Ghigo, A. Maranzana and G. Tonachini, Phys. Chem. Chem. Phys., 2014, 16, 23944–23951 RSC.
- S. P. Souza and B. L. Lutz, Detection of C2 in the interstellar spectrum of Cygnus OB2 Number 12 (IV Cygni Number 12), 1977.
- K. D. Tucker, M. L. Kutner and P. Thaddeus, The Ethynyl Radical C2H-A New Interstellar Molecule, 1974 Search PubMed.
- S. T. Ridgway, D. N. B. Hall, S. G. Kleinmann, D. A. Weinberger and R. S. Wojslaw, Nature, 1976, 264, 345–346 CrossRef CAS.
- J. H. Lacy, N. J. Evans, J. M. Achtermann, D. E. Bruce, J. F. Arens and J. S. Carr, Astrophys. J., Lett., 1989, 342, L43 CrossRef CAS.
- J. M. Smith, M. Nikow, M. J. Wilhelm and H. L. Dai, J. Phys. Chem. A, 2023, 127, 8782–8793 CrossRef CAS PubMed.
- J. Chang, L. Guo, R. Wang, J. Mou, H. Ren, J. Ma and H. Guo, J. Phys. Chem. A, 2019, 123, 4232–4240 CrossRef CAS PubMed.
- R. L. Hayes, E. Fattal, N. Govind and E. A. Carter, J. Am. Chem. Soc., 2001, 123, 641–657 CrossRef CAS PubMed.
- R. I. Kaiser and N. Hansen, An Aromatic Universe-A Physical Chemistry Perspective, 2021.
- F. C. Gillett, W. J. Forrest and K. M. Merrill, Astrophys. J., 1973, 183, 87 CrossRef CAS.
- A. G. G. M. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- E. Peeters, L. J. Allamandola, D. M. Hudgins, S. Hony and A. G. G. M. Tielens, Astrophysics of Dust, ASP Conference Series, Astronomical Society of the Pacific, San Francisco, CA, 2004, vol. 309, pp. 141–162 Search PubMed.
- E. Peeters, C. Mackie, A. Candian and A. G. G. M. Tielens, Acc. Chem. Res., 2021, 54, 1921–1933 CrossRef CAS PubMed.
- E. Peeters, S. Hony, C. Van Kerckhoven, A. G. G. M. Tielens, L. J. Allamandola, D. M. Hudgins and C. W. Bauschlicher, Astron. Astrophys., 2002, 390, 1089–1113 CrossRef CAS.
- E. Peeters, Proceedings of the International Astronomical Union, 2011, vol. 7, pp. 149-161.
- G. C. Sloan, M. Jura, W. W. Duley, K. E. Kraemer, J. Bernard-Salas, W. J. Forrest, B. Sargent, A. Li, D. J. Barry, C. J. Bohac, D. M. Watson and J. R. Houck, The Unusual Hydrocarbon Emission From the Early Carbon Star HD 100764: The Connection Between Aromatics and Aliphatics, 2007.
- L. D. Keller, G. C. Sloan, W. J. Forrest, S. Ayala, P. D'alessio, S. Shah, N. Calvet, J. Najita, A. Li, L. Hartmann, B. Sargent, D. M. Watson and C. H. Chen, PAH Emission From Herbig Ae/Be Stars, 2008.
- M. S. Murga, S. A. Khoperskov and D. S. Wiebe, Astron. Rep., 2016, 60, 233–251 CrossRef CAS.
- B. Acke, J. Bouwman, A. Juhász, T. Henning, M. E. V. D. Ancker, G. Meeus, A. G. Tielens and L. B. Waters, Astrophys. J., 2010, 718, 558–574 CrossRef CAS.
- S. Kwok and Y. Zhang, Nature, 2011, 479, 80–83 CrossRef PubMed.
- T. P. Stecher, Astrophys. J., 1965, 142, 1683 CrossRef.
- R. C. Bless and B. D. Savage, Astrophys. J., 1972, 171, 293 CrossRef CAS.
- T. Zafar, K. E. Heintz, J. P. U. Fynbo, D. Malesani, J. Bolmer, C. Ledoux, M. Arabsalmani, L. Kaper, S. Campana, R. L. C. Starling, J. Selsing, D. A. Kann, A. de Ugarte Postigo, T. Schweyer, L. Christensen, P. Møller, J. Japelj, D. Perley, N. R. Tanvir, P. DAvanzo, D. H. Hartmann, J. Hjorth, S. Covino, B. Sbarufatti, P. Jakobsson, L. Izzo, R. Salvaterra, V. DElia and D. Xu, Astrophys. J., 2018, 860, L21 CrossRef.
- B. Draine, Interstellar Dust, 1989, p. 313.
- B. Donn, Astrophys. J., Lett., 1968, 152, L129 CrossRef CAS.
- G. P. Vclovykin, Interstellar Dust and Aromatic Carbon, 1969 Search PubMed.
- X.-L. Sheng, Q.-B. Yan, F. Ye, Q.-R. Zheng and G. Su, Phys. Rev. Lett., 2011, 106, 155703 CrossRef PubMed.
- P. M. W. Kalberla, W. B. Burton, D. Hartmann, E. M. Arnal, E. Bajaja, R. Morras and W. G. L. Pöppel, The Leiden/Argentine/Bonn (LAB) Survey of Galactic HI: Final data release of the combined LDS and IAR surveys with improved stray-radiation corrections, 2005 Search PubMed.
- P. M. Kalberla and J. Kerp, Annu. Rev. Astron. Astrophys., 2009, 47, 27–61 CrossRef CAS.
- V. J. Herrero, M. Jiménez-Redondo, R. J. Peláez, B. Maté and I. Tanarro, Structure and evolution of interstellar carbonaceous dust. Insights from the laboratory, 2022.
- X. Y. Ma, Y. Y. Zhu, Q. B. Yan, J. Y. You and G. Su, Mon. Not. R. Astron. Soc., 2020, 497, 2190–2200 CrossRef CAS.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 3157–3158 CrossRef CAS.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 962–964 CrossRef CAS.
- L. A. Paquette, R. J. Ternansky, D. W. Balogh and G. Kentgen1, Total Synthesis of Dodecahedrane, 1983 Search PubMed.
- J. M. Schulman and T. J. Venanzi, J. Am. Chem. Soc., 1974, 96, 4739–4746 CrossRef CAS.
- Y. Ozaki, S. Saito, K.-I. Kondo, N. S. Zefirov, A. S. Koz'min and A. V. Abramenkov, The Problem of Tetrahedrane The Problem of Tone Reproduction in Offset Lithography V G W Harrison-The Problem of Hydrostatic Pressure Generation using a Piston-Cylinder Device The Problem of Tetrahedrane, 1978 Search PubMed.
- A. Nemirowski, H. P. Reisenauer and P. R. Schreiner, Chem. – Eur. J., 2006, 12, 7411–7420 CrossRef CAS PubMed.
- A. M. Mebel, V. V. KisIov and R. I. Kaiser, J. Chem. Phys., 2006, 125, 133113 CrossRef CAS PubMed.
- M. B. Gardner, B. R. Westbrook, R. C. Fortenberry and T. J. Lee, Spectrochim. Acta, Part A, 2021, 248, 119184 CrossRef CAS PubMed.
- R. C. Fortenberry and T. J. Lee, in Chapter Six - Computational vibrational spectroscopy for the detection of molecules in space, ed. D. A. Dixon, Elsevier, 2019, vol. 15, pp. 173–202 Search PubMed.
- B. R. Westbrook, G. M. Beasley and R. C. Fortenberry, Phys. Chem. Chem. Phys., 2022, 24, 14348–14353 RSC.
- F. Karagulian, C. A. Belis, C. F. C. Dora, A. M. Prüss-Ustün, S. Bonjour, H. Adair-Rohani and M. Amann, Atmos. Environ., 2015, 120, 475–483 CrossRef CAS.
- P. Pant and R. M. Harrison, Atmos. Environ., 2013, 77, 78–97 CrossRef CAS.
- U. Lohmann, F. Friebel, Z. A. Kanji, F. Mahrt, A. A. Mensah and D. Neubauer, Nat. Geosci., 2020, 13, 674–680 CrossRef CAS.
- M. Antiñolo, M. D. Willis, S. Zhou and J. P. D. Abbatt, Nat. Commun., 2015, 6, 6812 CrossRef PubMed.
- H. Wang, Proceedings of the Combustion Institute, 2011, 33, 41–67.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, A Fifth-Order Perturbation Comparison of Electron Correlation Theories, 1989 Search PubMed.
- C. Hampel, K. A. Peterson and H.-J. Werner, Chem. Phys. Lett., 1992, 190, 1–12 CrossRef CAS.
- P. J. Knowles, C. Hampel and H.-J. Werner, J. Chem. Phys., 1993, 99, 5219–5227 CrossRef CAS.
- M. J. O. Deegan and P. J. Knowles, Perturbative corrections to account for triple excitations in closed and open shell coupled cluster theories, 1994 Search PubMed.
- T. D. Crawford and H. F. Schaefer III, Rev. Comput. Chem., 2000, 14, 33–136 CAS.
- I. Shavitt and R. J. Bartlett, Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory, Cambridge University Press, Cambridge, 2009 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 1995, 102, 2032–2041 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. E. Yousaf and K. A. Peterson, J. Chem. Phys., 2008, 129, 184108 CrossRef PubMed.
- J. G. Hill and K. A. Peterson, Phys. Chem. Chem. Phys., 2010, 12, 10460–10468 RSC.
- W. Györffy and H.-J. Werner, J. Chem. Phys., 2018, 148, 114104 CrossRef PubMed.
- H. J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- H.-J. Werner and P. J. Knowles, et al., MOLPRO, version, a package of ab initio programs, 2023 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- W. Yang, R. G. Parr and C. Lee, Phys. Rev. A, 1986, 34, 4586–4590 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. M. Turner, A. S. Koutsogiannis, N. F. Kleimeier, A. Bergantini, C. Zhu, R. C. Fortenberry and R. I. Kaiser, Astrophys. J., 2020, 896, 88 CrossRef CAS.
- A. M. Turner, S. Chandra, R. C. Fortenberry and R. I. Kaiser, ChemPhysChem, 2021, 22, 985–994 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A.-R. Allouche, J. Comput. Chem., 2010, 32, 174–182 CrossRef PubMed.
- A. Allouche, Gabedit, 2017 Search PubMed.
- C. Puzzarini, Front. Astron. Space Sci., 2022, 8, 811342 CrossRef.
- L. Tinacci, S. Ferrada-Chamorro, C. Ceccarelli, S. Pantaleone, D. Ascenzi, A. Maranzana, N. Balucani and P. Ugliengo, Astrophys. J., Suppl. Ser., 2023, 266, 38 CrossRef.
- E. Herbst, Front. Astron. Space Sci., 2021, 8, 776942 CrossRef.
- E. Hébrard, M. Dobrijevic, J. C. Loison, A. Bergeat, K. M. Hickson and F. Caralp, Astron. Astrophys., 2013, 552, A132 CrossRef.
- R. C. Fortenberry, Astrophys. J., 2021, 921, 132 CrossRef CAS.
- A. Devaquet, AVOIDED CROSSINGS IN PHOTOCHEMISTRY Search PubMed.
- A. Devaquet, A. Sevin and B. Bigot, J. Am. Chem. Soc., 1978, 100, 2009–2011 CrossRef CAS.
- S. Shaik, J. Mol. Liq., 1994, 61, 49–79 CrossRef CAS.
- L. Salem, C. Leforestier, G. Segal and R. Wetmore, J. Am. Chem. Soc., 1975, 97, 479–487 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Containing cartesian coordinates for all geometric structures. See DOI: https://doi.org/10.1039/d3cp06199j |
| This journal is © the Owner Societies 2024 |