
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small lectin ligands as a basis for applications in glycoscience and glycomedicine†
Paul V.
Murphy
 *abc,
Ashis
Dhara
ab,
Liam S.
Fitzgerald
abc,
Eoin
Hever
a,
Saidulu
Konda
a and
Kishan
Mandal
a
*abc,
Ashis
Dhara
ab,
Liam S.
Fitzgerald
abc,
Eoin
Hever
a,
Saidulu
Konda
a and
Kishan
Mandal
a
aSchool of Biological and Chemical Sciences, Galway, H91TK33, Ireland. E-mail: paul.v.murphy@universityofgalway.ie
bSSPC, SFI Research Centre for Pharmaceuticals, Galway, H91TK33, Ireland
cCÚRAM, SFI Research Centre for Medical Devices, University of Galway, University Road, Galway, H91TK33, Ireland
First published on 20th August 2024
Abstract
Glycan recognition by lectins mediates important biological events. This Tutorial Review aims to introduce lectin–ligand interactions and show how these molecular recognition events inspire innovations such as: (i) glycomimetic ligands; (ii) multivalent ligand agonists/antagonists; (iii) ligands for precision delivery of therapies to cells, where therapies include vaccines, siRNA and LYTACs (iv) development of diagnostics. A small number of case studies are selected to demonstrate principles for development of new ligands for applications inspired by knowledge of natural glycan ligand structure and function.
 Paul V. Murphy | Paul Murphy received his BSc and PhD from University College Galway of the National University of Ireland. After appointment as a Chiroscience Postdoctoral Fellow at University of York, UK, he was appointed as a Lecturer and later as an Associate Professor in Organic Chemistry at University College Dublin and subsequently as an Established Professor of Chemistry at the University of Galway. His research interests are now focused on design and synthesis of glycomimetics and glycoclusters and includes synthesis method development for carbohydrate chemistry. |
 Ashis Dhara | Ashis Dhara received an MSc degree in Chemistry from Dr Shakuntala Misra National Rehabilitation University, India. He earned his PhD in 2024 from the University of Galway, Ireland, supervised by Professor Paul V. Murphy. Currently, he is a Postdoctoral Researcher in the Science Foundation Ireland Research Centre for Pharmaceuticals (SSPC) at University of Galway. His research focuses on design, synthesis, and evaluation of glycomimetics as lectin inhibitors. |
 Liam S. Fitzgerald | Liam Fitzgerald obtained his BSc from Maynooth University in 2018. Afterwards he pursued a PhD at the University of Galway supervised by Dr Miriam O’Duill and Prof. Paul Murphy and graduated in 2023. During this time, he worked on developing methodology for deuterating indoles as well as the development of multivalent sialic acid derivatives as inhibitors of influenza hemagglutinin. He will shortly begin work as a postdoctoral research fellow at the Krembil Research Institute with Prof. Donald Weaver. |
 Eoin Hever | Eoin Hever received BSc and MSc (Research) degree from National University of Ireland Galway. Eoin subsequently completed his PhD research at University of Galway under the supervision of Professor Paul Murphy in 2023. Eoin's research focused on the design and synthesis of novel constrained glycomimetics as inhibitors of galectins and galactosidases. During the PhD he had research visits to University of Copenhagen and University of Lund, the latter supported by an EMBO travel award. Eoin is now working as a Research Chemist in Merck Sharp & Dohme, Ireland. |
 Saidulu Konda | Saidulu Konda was born in Telangana, India (1985), and is currently Senior Chemist at Arran Chemical Company, Athlone (Ireland). He received his BSc from Osmania University in 2007 and MSc in Organic Chemistry from M.N.R. P.G. Collage (Affiliated to Osmania University) in 2010. He got his PhD in Stereoselective Synthesis of C27-C35 Eribulin Fragment and Related Macrocycles, at the University of Hyderabad (UGC Research Fellow) in 2015 under the supervison of Professor Prabhat Arya. After four years (2016–2020) industrial experience in GVK Biosciences, Bengaluru he was appointed as a postdoctoral researcher for three years (2020–2023) supported by SFI and the Irish Research Council at NUI Galway under the supervision of Professor Paul Murphy. His research interests are (i) the synthesis of natural product-inspired macrocycles, (ii) total synthesis of natural products, and (iii) carbohydrate research. |
 Kishan Mandal | Kishan Mandal was awarded his BSc in Chemistry from Burdwan University, India, in 2015, and his MSc in Organic Chemistry from DHSGSU, India, in 2017. He worked in the laboratory of Dr Chandan K. Jana at IIT Guwahati, India, as a Junior Research Fellow from February 2019 to June 2019, focusing on the total synthesis of bioactive natural products. Currently, he is pursuing his PhD under the supervision of Prof. Paul Murphy at the University of Galway, Ireland. His research is centred around the design and synthesis of new inhibitors of galectins and siglecs. |
Key learning points• Introduction to glycan and glycoconjugate structure.• Factors influencing ligand–lectin interactions. • Principles for glycomimetic research. • Principles for multivalent ligand design to increase avidity and selectivity. • Applications in chemical biology, biomedical science. |
1. Introduction
Glycans are mostly found on cell surfaces of organisms. These carbohydrates are conjugated to proteins (glycoproteins) and lipids (glycolipids). Glycoconjugate-protein recognition modulates biological events including cell adhesion, disease progression, immunological responses and infection.1 Lectins are one of two distinct classes of glycan binding proteins, the second being proteins that bind sulfated glycosaminoglycans. Lectins are classified into evolutionarily related families identified by carbohydrate-recognition domains (CRDs) based on primary and/or three-dimensional structural similarities. Lectins typically recognize terminal groups on glycan ligands, which fit into CRDs.2 Multivalency occurs, when multiple copies of ligands are found on natural scaffolds, as for glycans on surfaces. Lectins are often also multimeric, which results from either noncovalent or covalent association of two or more monomers with CRDs. Interaction between multimeric lectins and multimeric ligands gives more avid interaction.3 Ligand lectin recognition is central to the sugar code in biology (Fig. 1).4 | ||
| Fig. 1 Cartoon illustration (not to scale) of selected lectin ligand mediated events at a cell surface. | ||
This Tutorial Review focuses on three small lectin ligand types: natural glycan ligands; glycomimetics and glycocluster ligands. We provide knowledge of glycan structure (Section 2) and features of their interaction with lectins.5 Design of bioavailable glycomimetic ligands (Section 3) and small glycoclusters (Section 4) with biomedical or chemical biological applications are emerging from research. We have selected case studies to demonstrate how novel lectin ligand inhibitors are being developed, including some that are entering or have entered clinical trials. We discuss how research on lectin ligands is leading to innovations enabling precision delivery of vaccines and short interfering RNA (siRNA) to cell types and for chemical biology or sensing applications based on glycoclusters.
2. Natural glycans and interaction with lectins
2.1 Introduction to glycans
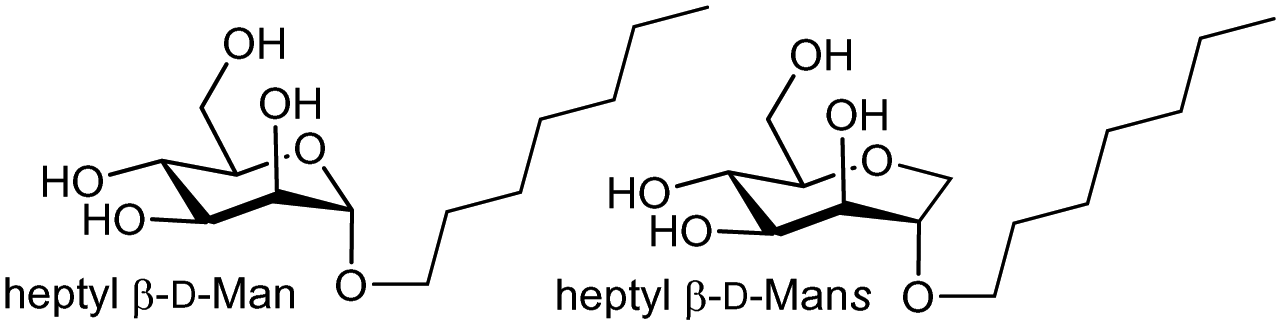
Mammalian cell surface glycans (Fig. 2),6 are comprised of monosaccharides in pyranose forms (Fig. S1, ESI†), including mannose (Man), galactose (Gal) or N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), fucose (Fuc) and N-acetylneuraminic acid or sialic acid (NeuAc). Herein it is assumed that abbreviations such as Gal, GalNAc refer to the more common pyranose form unless otherwise stated (e.g. Man = mannopyranose, Mans = mannoseptanose). The position of linkage between saccharides and anomeric configuration generated by glycosyltransferases influences the properties of glycans produced, including their interaction with lectins. | ||
| Fig. 2 Selected examples of lectin ligands. SNFG nomenclature was generated using glycoglyph.8 The N-glycan and O-glycans shown are bivalent glycoclusters where two LacNAc disaccharides (blue) are presented on saccharide scaffolds (black). | ||
2.2 Pyranose ring conformation is influenced by steric interactions and torsional strain
Pyranoses adopt chair conformations (Fig. S4, ESI†) to minimise strain with most substituents equatorial to minimise steric repulsive interactions; this, for example, gives the 4C1 conformation for Gal depicted in Fig. 2 and Fig. S4A (ESI†); its 1C4 conformer has both destabilising 1,3-diaxial interactions and Hassel-Ottar (1,3-syndiaxial or syn-pentane) interactions. The boat is not preferred as it displays torsional strain.2.3 The exo-anomeric effect influences conformation
The α- and β-D-glucopyranoses are the main populated forms for glucose in water and are in equilibrium and a preference is observed for β-D-Glc (equatorial OH at C-1). However, there is ∼3-fold increase for the OH group at C-1 in Glc to be axial compared to cyclohexanol; this increased preference for the axial anomer is the original definition of the endo-anomeric effect.7The endo-anomeric effect9 is stereoelectronic, requiring an electron withdrawing heteroatom (O-5) in the ring and an electron withdrawing atom (O-1) at C-1. One model proposed to explain the endo-anomeric effect is hyperconjugation (Fig. S2, ESI†), possible when the anomeric substituent is axial, but, not when equatorial. Another proposal to explain the effect is based on minimisation of electrostatic repulsion between oxygen atoms. Explaining anomeric preference is, however, more complex and influenced by multiple factors10 such as solvent, and the extent of steric repulsions, which are reduced by electron withdrawing substituents.11 The endo-anomeric effect gives rise to the exo-anomeric effect (Fig. S3, ESI†), which is considered when accurately calculating energies of ligand conformers that interact with lectins.
2.4 The gauche effect and conformation
The gauche effect12 is a stereoelectronic based increase in the preference for the gauche arrangement of F, and to a lesser degree, of O substituents on adjacent carbon atoms and is thus relevant to carbohydrates, particularly the ω torsion in 1,6 disaccharide and related linkages (Fig. S5, ESI†).2.5 Interglycosidic torsion preferences determine glycan conformation
The 3D structure of glycans is determined primarily by the conformational preferences of the bonds between saccharide residues (interglycosidic torsions, Φ, Ψ, ω), influenced by the exoanomeric effect, gauche effect and steric factors. The topic has been reviewed.13 Disaccharide conformation based on crystal structure data of glycans bound to proteins/antibodies, collected by Wormwald and co-workers provides experimentally derived information, which includes Φ–Ψ and Φ–Ψ–ω plots.14 Woods's glycam.org is a reliable source of low energy structures of glycans.15 We provide additional notes and Fig. S3–S8 (ESI†) on glycan conformation in the ESI† file as well as selected energy surface plots reproduced from literature sources in Fig. S9–S12 (ESI†).16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 gg and gt conformers, accounting for ∼95% of all ω torsion conformers.
:50 gg and gt conformers, accounting for ∼95% of all ω torsion conformers.
2.6 What glycan conformation is recognised by a lectin?
The conformation of a ligand bound to the lectin CRD may be similar to that determined in the gas phase, in solution or in crystal structures.20 Summarised in Table S1 (ESI†) are the glycosidic torsions observed in a subset of crystal structures, where lactose or its derivatives are bound to various galectin CRDs; the bound ligands all have the exo-syn Φ and syn-Ψ conformation, which is like unbound lactose conformers observed in gas/solution/solid state.There is evidence also for multiple modes of binding, where lectins recognise different ligand conformers, which arise if multiple conformers are populated. The interaction between a Manα1-3Man and rat mannose binding protein (rMBP) was studied by NMR spectroscopy and MD.21 The unbound Manα1-3Man showed population of two conformers, where the interglycosidic torsions were: (i) exo-syn Φ and syn-Ψ and (ii) exo-syn Φ and anti-Ψ and the two conformers were detected to bind to rMBP.
Induced fitting of lectin may occur on binding, and ligand conformer selection is known.22 One interesting example of induced fitting is in catch bonding. Catch bonds are biological interactions that are enhanced by mechanical force and occur when protein–ligand binding mode changes, due to conformational changes, leading to altered affinity. Catch bonding is associated with cell–cell adhesion under shear stress.
2.7 Non-covalent interactions in lectin ligand complexes
Hydrogen bonding of saccharide hydroxyl groups, including water mediated H-bonding between ligand and lectin is prevalent. For NeuAc containing glycans, there is frequently salt bridging or charged-charged or charged-neutral H-bonding to arginine, interactions which also occur for sulfated glycans. Hydrophobic interactions and/or CH–Pi interactions31 are frequently involved. Ligand hydroxyl groups can coordinate to metal ions (e.g. Ca2+) which induce charge transfer interactions from saccharide to metal ion.32 Fig. S3–S5 (ESI†) shows interactions in selected complexes between ligands and lectins. Fig. S3 (ESI†) shows the interaction of sulfatide with galectin-8, with recent crystallographic evidence showing that sulfatide sphingosine residue's hydroxyl group has indirect water mediated H-bonding interaction with the N-terminal domain;33 this shows contribution to lectin–ligand interaction of the sphingosine component of the glycolipid and contributes to understanding how sulfatide recognises lectins. We discuss below the full ligand for P-selectin, which is a glycosulfopeptide of the P-selectin glycoprotein ligand (PSGL). Thus, lectin–ligand recognition does not always involve only the glycan residue.2.8 Thermodynamics of lectin–ligand interaction
Low affinity lectin–monosaccharide interactions are prevalent, and a contributing factor is that the binding pocket (CRD) is often shallow, with a substantial area of bound ligand surrounded by solvent water, contributing to a relatively high dielectric constant, ε, for the CRD. Coulomb's law implies the force between electrostatic based interactions, which include H-bonding,34 is weaker in a pocket with higher ε and this increases Kd or reduces affinity. Higher affinity can arise where pockets are deeper and have lower ε as for FimH in its high affinity binding conformation as occurs in catch bonding. Ernst and co-workers indicate35 the strength of a H-bond could increase by ∼10 fold in a pocket with ε = 5–10 compared to one with ε = 20. Another feature is that Kd depends upon ligand binding on and off rates (Kd = koff/kon). It is easier for a ligand to dislodge from a shallow pocket than from an enclosed one and, thus, koff is reduced in the deeper pocket, reducing Kd.The binding free energy (−ΔG) increases (Kd reduces) as affinity increases, when comparing ligands and it has contributions from enthalpy (−ΔH) and entropy (+TΔS) according to the equation −ΔG = −ΔH + TΔS. Ligand binding is often enthalpy driven with the entropy change usually unfavourable, although there are exceptions such as for the sLeX-E-selectin interaction.36
Increasing −ΔH is associated with stronger interactions or extended site interactions, although it could be reduced if the binding mode must adopt an energetically less favourable conformation. Extended site interactions arise, for example, for a larger oligosaccharide binding to a lectin, when compared to a monosaccharide.37
It has been proposed that when performing research to optimise ligand affinities, required during drug discovery, to focus on measuring or computing free energies of protein–ligand interactions, rather than trying to solely focus on improving enthalpy or entropy alone, as enthalpy–entropy compensation can frustrate ligand design.38
2.8.1 Case study – dissection of entropy contributions to ΔS in FimH–ligand binding
The entropy contribution to the free energy of binding is a sum of changes in translational, rotational, conformational, and solvation entropies. A study by Peczuh, Ernst and co-workers provides an informative analysis of various entropy contributions in the binding of monosaccharide ligands to the high affinity state of FimH.39Conformational entropy, due to induced fitting, results from a ligand and/or lectin reorganising from a non-binding to binding conformation. The degree to which reorganisation takes place influences the magnitude of ΔS and is unfavourable entropically and reduces −ΔG. Two high affinity ligands for FimH, heptyl β-D-Man, and a septanoside, heptyl β-D-Mans, were studied, with ITC investigation indicating the difference in Kd (ΔΔG = 5.5 kJ mol−1 or ∼1.3 kcal mol−1) between the two ligands is mainly due to TΔS (see Table 1).
|
|
||||
|---|---|---|---|---|
| Ligand | K d (nM) | −ΔG (kJ mol−1) | −ΔH (kJ mol−1) | +TΔS (kJ mol−1) |
| heptyl β-D-Man | 29 (25.8–32.3) | +43.0 | +50.3 (+50.2 to +50.7) | −7.3 |
| heptyl β-D-Mans | 264 (245–284) | +37.5 | +49.4 (+48.9 to +49.8) | −11.8 |
Metadynamics simulations showed that the septanoside's ring is more flexible than of heptyl β-D-Man. The crystal structures of both ligands with FimH revealed identical H-bonding networks in their interaction, consistent with near identical enthalpy change measured for the two ligands. There is a conformation difference when orientation of heptyl groups is compared in the two co-crystal structures, but DFT calculations indicated that the binding energy contribution by the heptyl groups are equivalent in both crystal structures and that only one of the septanoside ring conformers is bound by the lectin. The TΔS due to losses in rotational and translational entropy on binding were considered equal in both cases (−10 kJ mol−1). The authors concluded that the entropy difference is associated with either solvation or conformational entropy. ITC measurements at different temperatures revealed almost identical ΔCp values for both ligands, which enabled establishment that the desolvation entropies (+TΔSsolv = +69.1 kJ mol−1 & +68.8 kJ mol−1) are nearly identical for the two ligands. The authors concluded that the difference in conformational entropy (+TΔSconf = +66.4 kJ mol−1) for heptyl β-D-Man vs. +70.6 kJ mol−1 for heptyl β-D-Mans accounts for most of the 10-fold difference in affinity (ΔΔSconf = 4.2 kJ mol−1, ∼1.0 kcal mol−1).
3. Glycomimetic ligands for galectins, selectins and siglecs
For drug discovery, substances that maintain a therapeutic concentration over an extended period, metabolic stability, and slow and prolonged renal excretion are required,40 however, these requirements are usually not met by natural glycan ligands, but may be met by synthetic mimics or glycomimetics, which requires acquiring fundamental knowledge of lectin–ligand interaction, such as identification of the key groups involved in interactions and 3D structure.41,42 High affinity ligands, ideally with Kd ∼ 50 nM or lower, is desired for drug discovery projects along with good selectivity profile for the lectin of interest. Oral bioavailability for glycomimetics is desired but is not essential. The development of glycomimetic drugs, substances that are clinically approved by regulatory bodies, has been relatively slow compared to other types of drugs. Tamiflu, although not a lectin inhibitor, is an exemplar synthetic glycomimetic,43 which gained regulatory approval as a drug. Tamiflu is a glycosidase (neuraminidase) inhibitor, for therapy or prevention of influenza infection. Tamiflu's development started from 2,3-dehydro-2-deoxy-NeuAc, which is a mimetic of the transition state of the neuraminidase catalysed reaction. One lesson from Tamiflu's development was that the systematic elimination of polar groups and their replacement with a more lipophilic group did not compromise affinity. Oral bioavailability was achieved by esterification of the acid group to give a lipophilic prodrug. A general strategy employed by medicinal chemists in lectin ligand development, is to identify core structural features in the native ligand essential for lectin recognition with subsequent removal of unnecessary polar groups, while appending groups capable of additional interactions or give desired pharmacological properties. Such approaches have, in recent years, yielded candidates that have entered clinical trials as E-selectin and galectin-3 inhibitors.3.1 Glycan ligands for galectins
Galectins44 are tissue lectins, of which 15 mammalian members are known, that play roles in cancer and inflammation and they generally bind β-galactopyranosides.45 There are three types of galectin: prototype (e.g. galectins-1,7), chimeric (galectin-3) and tandem repeat (e.g. galectin-4,8,9). Tandem repeat galectins have two unique CRDs, referred to as the N and C-terminal domain, separated by a covalently bound peptide linker. The CRDs have a conserved β-sandwich core sequence containing ∼130 amino acids which contains a groove long enough to hold a linear tetrasaccharide. Binding of the galactopyranoside within the galectin CRD's groove is the most conserved feature of ligand recognition and includes cooperative H-bonding of the 4-OH and 6-OH with His, Arg and Asp residues; there are CH–Pi interactions of the α-face of Gal with Trp. Lactose is considered a minimal ligand with affinity in the mM range,46 although binding has been detected for some galectins at mM or high mM concentrations for simpler galactopyranosides (see Table S2, ESI†). The various galectins bind lactose's exo-syn-Φ/syn-Ψ conformer. For some galectins, the affinity is increased for LacNAc derivatives compared to lactose with extended site interaction involving the acetamide explaining this. Gal-8,47 shows high affinity (2.7 μM, from SPR) for 3-O-sialylated lactose compared to LacNAc (type 1 or 2) or lactose (79–420 μM), with the increased affinity due to the interaction of the NeuAc carboxylate with sidechains of Arg59 and Gln47 in the N-terminal CRD (Gal-8N).48 Thus while galectins minimally recognise β-galactopyranosides, the affinity and selectivity varies between different galectins for different glycans, with synthesis being used to develop more potent glycomimetic ligands.3.2 Glycomimetics for galectins
Nilsson et al. generated 3′-amino-LacNAc, where the galactopyranoside 3-OH is replaced with an amino group and subsequent amide synthesis from the amino group led to the p-methoxytetrafluorobenzamide, which was ∼50-fold more potent than LacNAc; this showed the benefit from modifying galactopyranoside's 3-position.49 Nilsson and co-workers later synthesised 3-substituted derivatives of thiodigalactoside (TDG, Fig. 3). TDG was first synthesised more than 100 years ago50 and is stable to the action of galactosidases due to its S-glycoside and, thus, has bioavailability. TDG inhibits several galectins51 (Table S2, ESI†) with higher affinity than lactose/LacNAc; it has similar affinity for galectin-1 and galectin-3 and has proven to be useful for study of their biological function.52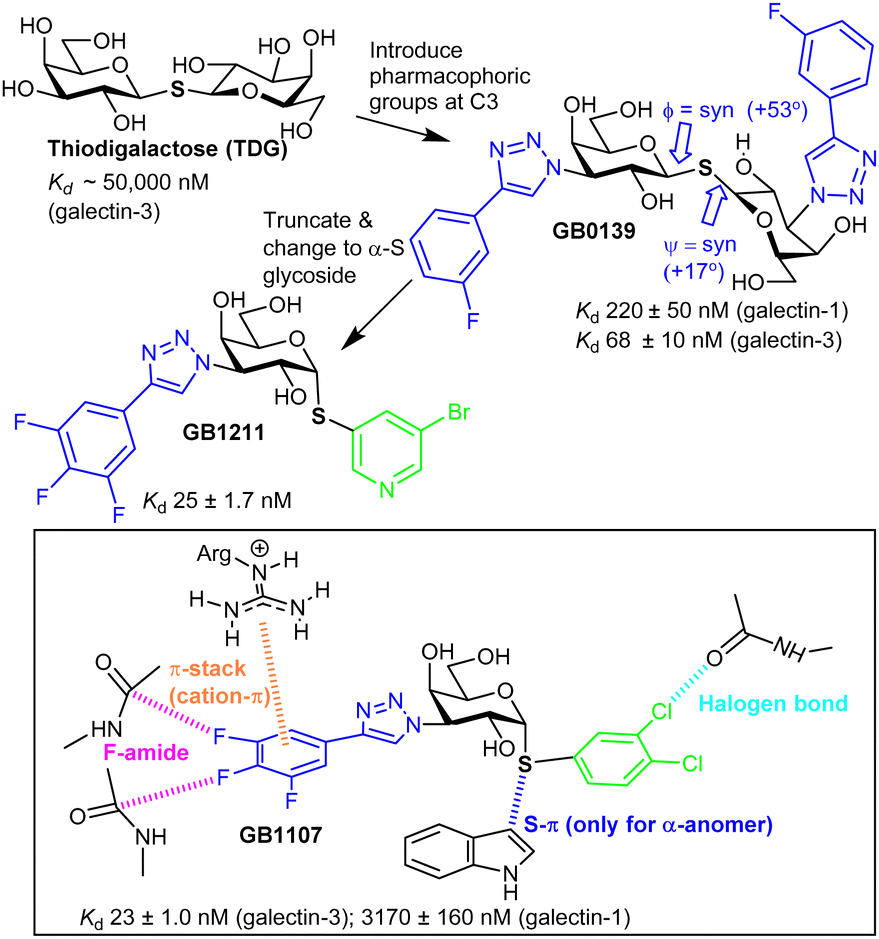 | ||
| Fig. 3 Evolution of galectin glycomimetic ligands. | ||
TDG has a Kd ∼ 50 μM for galectin-3, and subsequent replacement of its two 3-OH groups with aryltriazoles led to GB1039,53 with a much improved Kd, 68 ± 10 nM.54S-Glycosidic linkages may have more flexibility than an O-glycoside,55 yet, galectin-3 selects a conformer of GB1039 with an inter-residue torsion profile similar to lactose.56 GB0139 entered clinical trials as an inhaled, not orally available, inhibitor of idiopathic pulmonary fibrosis.57 However the GB1039's Phase 2b trial was not successful and Galecto, Inc. have since discontinued its development.58 Research inspired by GB1039 has, however, led to discovery of GB121159,60 and GB1107, which are both orally available. GB1211 is a nM range inhibitor, with selectivity for galectin-3 and its affinity is explained by interactions like those shown in Fig. 3 for GB1107.61 The cell permeability of GB1211 is linked to its lower polar surface area, when compared to GB1039 (114 vs. 201 Å2). GB1121 has a better safety profile than GB1107 and Galecto, Inc. are now pursuing clinical evaluation of GB1121 for oncology and liver fibrosis.
3.3 Selectin glycomimetic ligands
P-, E- and L-selectin are glycoprotein receptors that are involved in adherence and subsequent rolling of leukocytes on endothelium during inflammation or immune response before their secure attachment and recruitment into tissues and sLeX is a minimal glycan ligand for the selectins. There have been sustained efforts over 30 years to develop pharmacologically effective sLeX mimetics.The binding of sLeX to E-selectin is dependent on bidentate coordination of L-fucopyranoside to Ca2+ and interaction of NeuAc's carboxylate with Tyr and Arg. sLeX is preorganised into its bioactive conformation with high and low affinity binding modes identified (Section 2.5.2 above).62 Interaction of sLex to E-selectin is enthalpically unfavourable and entropically driven (−ΔH = −5.4 ± 0.7 kJ mol−1, +TΔS = +23 ± 1 kJ mol−1).
Regarding P-selectin, a high affinity natural ligand has been identified, PSGL-1. PSGL-1 is a homodimeric transmembrane protein that presents sLeX and sulfated tyrosine residues. The soluble recombinant form of PSGL-1 has a Kd value for P-selectin of ∼800 nM, which has much higher affinity than sLeX alone (Kd = 7.8 mM). The affinity of PSGL-1 compared well to that of a truncated sulfated glycopeptide derived from the PSGL1, presenting the sLeX and three sulphated tyrosine residues. Crystal structure of the glycosulfopeptide ligand bound to P-selectin showed interactions of the sulfate groups with Arg85 and His114, residues not replicated for E-selectin, explaining the increased affinity of PSGL-1 for P-selectin.63
Knowledge regarding ligand–selectin interactions have informed glycomimetic development. Binder, Ernst, and co-workers reported mimetics BEM1 and BEM2 (Fig. 4), where the NeuAc residue is replaced by the S-cyclohexyl lactate group and the GlcNAc residue is replaced by a substituted cyclohexane. The main purpose of the S-cyclohexyl lactate is to ensure optimal conformational preorganisation of the carboxylate group, relative to that of the L-Fuc for binding; the use of the R-cyclohexyl lactate produces a conformation that is not preorganised correctly. Preorganisation of ligand pharmacophoric groups correlates with affinity for E-selectin.64 BEM2 showed a Kd (ITC) of 19 ± 2 μM for E-selectin compared to 878 ± 93 μM (ITC) for sLeX, whereas BEM1 was ∼3-fold less potent than BEM2. When compared with sLeX, the affinity gain for BEM2 is almost entirely due a more favourable enthalpic contribution, while showing a similar entropic contribution observed for sLeX. The major difference between BEM1 and BEM2 is in the entropy contribution, which we speculate may be linked to more flexibility in the cyclohexane in BEM1, reduced by the additional methyl substituent in BEM2.
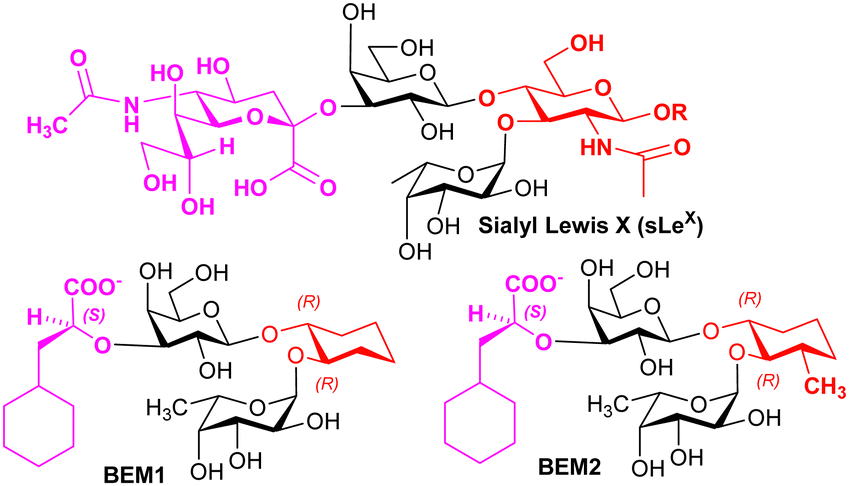 | ||
| Fig. 4 Structures of sLeX and mimetics. | ||
Based on their observations, Ernst, Binder and co-workers proposed that glycomimetic design should involve replacing glycan components with predominantly structural (scaffold) roles with hydrophobic groups that can mimic the same structural role, to improve the enthalpic contributions to binding while reducing solvation penalties;65 this assumes that the glycan component being replaced has purely a scaffolding role with no important interaction with the lectin. This very important finding informs strategy to contribute to optimising free energy of binding of glycomimetics. Co-crystallisation of BEM2 with E-selectn (PDB = 4C16) enabled confirmation of the structural role for the cyclohexane.66 Furthermore, ligands for E-selectin with low nM range affinity have been synthesised, incorporating groups that introduce binding at a second site.67
3.4 Rivipansel and Uproleselan
Rivipansel,68 and Uproleselan are sLex mimetics containing the essential minimal pharmacophoric groups for selectin recognition (Fig. 5), and both contain features found in the BEM2 glycomimetic. In Rivipansel, sulfate groups were incorporated with the aim to generate an improved L- and P-selectin ligand. Thus, Rivipansel is as a pan-selectin inhibitor and it showed increase in affinity for all three selectins compared to sLeX. Nevertheless, evidence indicates Rivipansel's effectiveness is linked to selective inhibition of E-selectin; in an ELISA involving immobilised selectins, was >78-fold better (∼4.3 μM) for E- than P- (423 μM) and L-selectin (337 μM). Also, Rivipansel inhibited E-selectin mediated rolling in a flow chamber cell-based assay more effectively than L- and P-selectin. Neutrophil adhesion to vascular endothelium is mediated by E-selectin and is a driver of acute vaso-occlusive crisis (VOC) in sickle cell disease. P-selectin is also upregulated on endothelial cells and both it and E-selectin bind adhesion molecules on red blood cells surfaces. Rivipansel was phase-III trialled in human patients with sickle cell disease (SCD) who were hospitalized for VOC and required treatment with intravenous opioids to reduce severe pain from having this condition. In a full analysis the median time to readiness for discharge (primary end point) was not different comparing treatment with Rivipansel and placebo and thus regulatory approval was not granted. However, a later statistical analysis (post hoc) showed early Rivipansel treatment within 26.4 hours of VOC pain onset had reduced median time to readiness for discharge by 41.5 hours, and reduced median time to discontinuation of IV opioids by 50.5 hours, compared with placebo (all P < 0.05), indicating that timing of Rivipansel administration after pain onset may be critical to achieving accelerated resolution of acute VOC in sickle cell disease patients.69 More data was presented from the clinical study to support the proposal that Rivipansel targets E-selectin in patients, and it seems clear that the sulfate groups of Rivipansel do not interact optimally with P-selectin, compared to PSGL-1.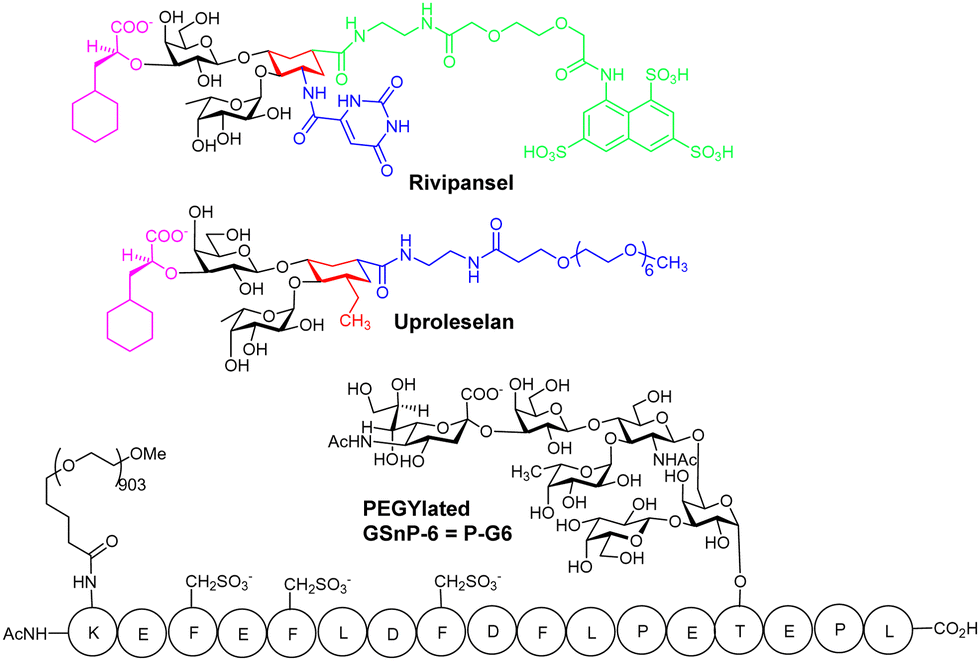 | ||
| Fig. 5 Structures of Rivipansel, Uproleselan and P-G6. Amino acids are shown in single letter code form. Tyr-OSO3− in PSGL-1 are replaced with CH2SO3− groups to get G-SnP-6. PEGylation of GSnP-6 at the side chain of the N-terminal Lys gives P-G6. | ||
Uproleselan is another sLeX mimetic, modified with a polyethylene glycol (PEG) fragment. Although Uproleselan is not orally available, its pharmacokinetics and pharmacodynamics are much improved by the PEG residue. A phase 1/2 clinical study has recently completed where Uproleselan was added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia (AML). The preliminary clinical study with Uproleselan reported promising initial outcomes such as a reduction in mucositis, a debilitating side effect of chemotherapy,70 but, GlycoMimetics, Inc. recently reported that the primary endpoint was not attained.
3.5 Glycosulfopeptide mimetics for P-selectin
Chaikof and co-workers have rationally designed a more optimal PSGL-1 mimetic, GSnP-6, as a synthetic high-affinity specific P-selectin ligand for human therapy (Kd = 22 nM).71 The hydrolytically unstable tyrosine OSO3− groups of PSGL-1 are replaced with chemically stable CH2SO3− groups in GSnP-6; these modifications yield ∼3 fold improved affinity for P-selectin, as compared to that for PSGL-1. Introduction of OSO3− groups by chemoenzymatic synthesis is also problematic due to stability issues with 3′-phosphoadenosine 5′-phosphosulfate, the required source of sulfate for all eukaryotic sulfations, providing another advantage of using the CH2SO3− modification in the peptide.MD simulations were used to predict the stability of GSnP-6 bound to a P-selectin model and the structure of the calculated complex was comparable, only under conditions where His114 was fully protonated, to that of the crystal structure of P-selectin bound to PSGL-1, retaining all key non-covalent interactions. GSnP-6 also bound to E- and L-selectin (low μM range) albeit with lower affinity than for P-selectin.
PEGylation of GSnP-6 gave P-G6 (Fig. 5), which led to substantial increase in terminal half-life in plasma from several minutes for non-PEGylated structures to 15.65 ± 3.55 hours for P-G6, a very important development, giving a candidate with plasma half-life in the range of orally used anticoagulants. In preclinical study P-G6 inhibited P-selectin binding to murine and human leukocytes in a dose-dependent manner and reduced platelet–leukocyte aggregation in vitro and in vivo. P-G6 inhibited venous thrombosis in a preclinical model without impairing haemostasis.72 Chaikof and co-workers described recently the gram scale synthesis GSnP-6 with a view to scale up synthesis for preclinical evaluation; they also show GSnP-6 displays dose-dependent inhibition of venous thrombosis in vivo and inhibits vaso-occlusive like events on a microvasculature-on-a-chip model system.73
3.6 Glycomimetics for siglecs
Sialic acid binding immunoglobulin-like lectins (siglecs) are immune cell lectins, found on the surfaces of most immune cells and are cell biomarkers.74 Restricted expression patterns of siglecs have enabled therapies, such as an FDA antibody–drug conjugate, to be targeted to defined cell populations.75 Liposomes decorated with siglec-1 ligands have been used to target antigen to macrophages.76 Siglec-2 (CD-22), restricted to B-cells, is an endocytic receptor that is internalized and recycled to the cell surface.77 Thus, siglecs have clear potential for selective delivery of drugs or other cargo to immune cells.78 Siglecs are also targets for therapeutic intervention79 as they inhibit immune cell activation and are associated with inflammatory diseases. Multivalency enhances ligand avidity for siglecs,80 but, is associated with agonism of function, not antagonism. Features typical in binding of siglecs to sialic acid derivatives are evident in the crystal structure of mouse sialoadhesin (siglec 1) bonded to NeuAcα2-3Galβ1-4Glc (3′SL, 3′-sialyllactose), which discloses that most contacts are with the NeuAc residue (Fig. S3, ESI†).81 NeuAc's carboxylate is coplanar with the guanidino group of Arg-97, a residue conserved across siglecs, and engaged in charged-charged H-bonding interactions.The interaction of α-NeuAc is critical to recognition of mouse siglec-1 as the corresponding NeuAcβ-OMe failed to show evidence of binding by NMR study,82 which is explained by the carboxyl group in NeuAcβ-OMe losing the interaction with the conserved Arg. Other interactions that occur are that the acetamide methyl group has contact with the indole ring of Trp, and the terminal carbon of the glycerol side chain (C9) is close to the indole of another Trp residue. The 9-OH establishes important H-bonding with the amide NH and carbonyl of a Leu residue of the siglec, while the amide nitrogen of the N-acetyl group forms a hydrogen bond with the main chain carbonyl of an Arg; these H-bond interactions are with residues in a β-strand peptide conserved among siglecs and likely sustained in glycomimetics (Fig. S3, ESI†).
Small molecule monomeric inhibitors of siglec-2, siglec-783,84 have been designed and synthesised. Identification of monomeric glycomimetic inhibitors of siglecs is considered highly important including comparison of their biological effects with multivalent ligands or antibodies. The compounds shown in Fig. 6 show significant enhancements in affinity for siglec-2/7 compared to NeuAc, the latter being conserved in siglec ligands.
 | ||
| Fig. 6 Siglec-2 and siglec-7 glycomimetic ligands developed by Brossman and co-workers. | ||
Siglec-8, is an eosinophil- and mast-cell associated cell-surface receptor and is a target for the treatment of asthma. Siglec-8 has a role in accumulation and delayed apoptosis of activated eosinophils in the airways and a cause of persistent inflammation and tissue damage. Glycan microarray analyses85 have revealed that the 6′-sulfo-sLeX was selective for siglec-8 and structural information was generated by NMR86 and more recently by X-ray crystallography (3.3 Å resolution).87 Ernst and co-workers have successfully developed a higher affinity glycomimetic shown in Fig. 7,88 which has a sulfonamide substituent in the 9-position and the polar Gal is replaced with a reduced polar carbocyclic scaffold to present the required sulfate. Compared to the 6′-sulfo-sLeX, the affinity was improved ∼20-fold. An induced fit binding mode where a hydrophobic pocket is created in siglec-8 to accommodate the naphthyl group is supported by the crystal structure. That monomeric inhibitors antagonise siglec-8 function, in contrast to multivalent ligands was recently demonstrated,89 giving evidence of the importance to develop potent monomeric ligands as antagonists of siglecs.
 | ||
| Fig. 7 Evolution of a glycomimetic inhibitor for siglec-8. | ||
4. Multivalent ligand–lectin interactions
4.1 Mechanisms for avidity gain
Strong interactions are needed to promote adhesion between cells/particles or induce and sustain signal transduction and one mechanism by which this is achieved is via multivalency. Multivalent ligands are collections of small lectin ligands displayed on a scaffold and they tend to show enhancement in affinity compared to monomeric ligand, on a per mole of ligand basis,90 which is termed the “glycoside cluster effect.”91 Investigations by Lee et al. using the asialoglycoprotein receptor (ASGPR) showed the distance between ligands (Gal or GalNAc) had to be enough (∼20 Å) to enable simultaneous interaction with the trivalent ASGPR CRDs so that chelate type binding could arise (Fig. 8(B)). If spacing between the ligands and the CRDs are complimentary then large affinity gains are observed due to simultaneous binding of ligands.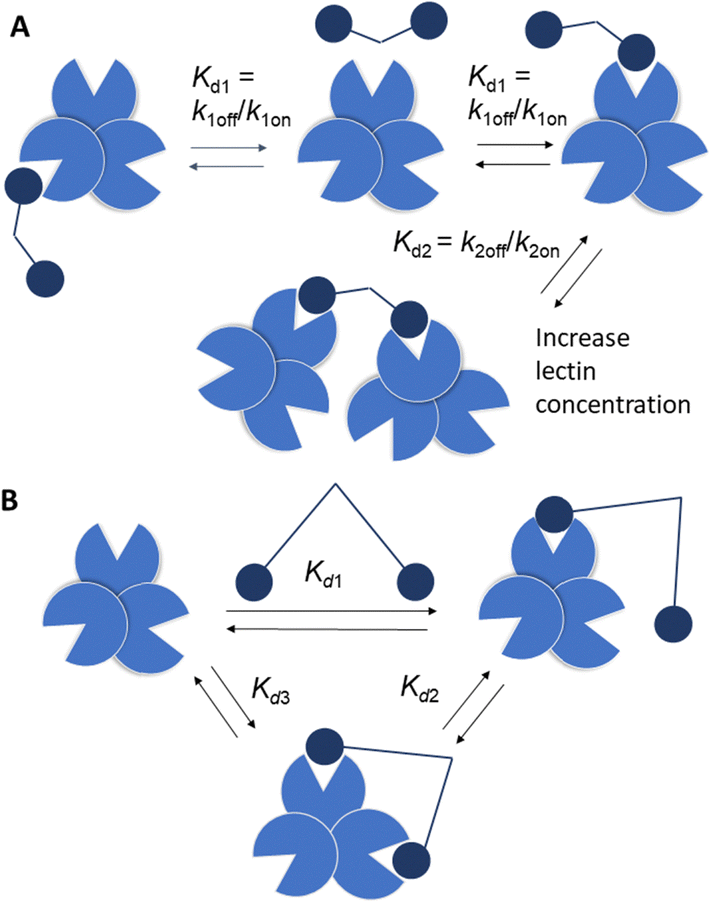 | ||
| Fig. 8 (A) Bind and jump mechanism (statistical rebinding). Kd1 decreases for a bivalent ligand compared to a monomer due to increase of kon relative to koff. Kd2 > Kd1 as number of available ligand binding sites is reduced and there may be increased steric interactions, reducing kon. (B) Simultaneous binding to two CRDs is reflected by Kd3 with Kd3 < Kd1. | ||
There are various dissociation constants (Kd) for multivalent interaction. The Kd3 (Fig. 8(B)) for the simultaneous binding interaction is the avidity.92 The Kd3 is lower than Kd1 when the glycoside cluster effect (chelate binding) occurs. The Kd1 is not a true measure of a single ligand–single lectin interaction as Kd1 is also an avidity constant as the ligand and lectin are multivalent. The Kd1 avidity arises due to statistical rebinding or the bind and jump mechanism (Fig. 8) when simultaneous binding of two ligands to two CRDs is not possible and is due to an increase in the on rate (kon) relative to off rate (koff) due to increased local ligand/lectin concentration because of multivalency.93 The true single ligand single lectin interaction Kd, i.e. the affinity, would require the ligand and lectin to be monomeric.
A third mechanism involves crosslinking promoted by multivalent ligands and multivalent lectins that leads to aggregation, driving formation of a precipitate, which can complicate the picture given in Fig. 11.94 Cross-linking can take place in the presence of a soluble lectin or at a cell surface and provides the basis for classical hemagglutination assays. At cell surfaces, cross-linking (Fig. 9) leads to receptor clustering, which leads to stronger and more prolonged signal transduction. Structure of crosslinked aggregates has biological relevance and was found, to influence galectin-1 mediated apoptosis.95 The crosslinking with galectins, for example, can explain their role in bringing ligands to cell surfaces, such as occurs for tandem repeat type galectins (e.g. Gal4/8) and their interaction with sulfatide at cell surfaces (see Fig. 1).
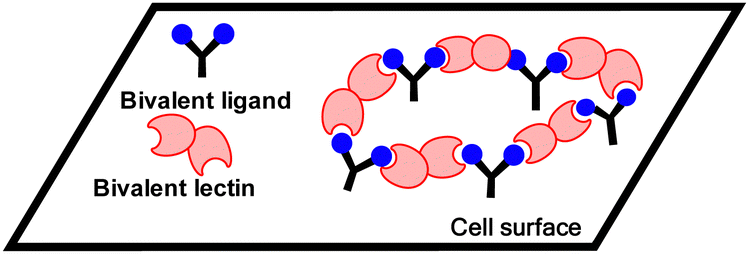 | ||
| Fig. 9 Crosslinking by bivalent ligands and bivalent lectins at a cell surface. | ||
4.2 Statistical rebinding glycocluster for modular vaccine design
The macrophage galactose C-type lectin (MGL) is a receptor on antigen presenting cells (APCs) that shows preferential binding for GalNAc. MGL is a trimeric C-type lectin receptor with distances estimated at ∼82 Å between its CRDs, ∼4-fold longer, than between the CRDs in its related trimer, ASGPR.96 The tumour associated Tn antigen (GalNAcα1-O-Ser/Thr) is a ligand for MGL. The folding of MGL CRD and binding of GalNAc is Ca2+ ion dependant and involves chelation to the ion via its 3- and 4-OH groups. This coordination likely involves charge transfer to the Ca2+ ion enhancing the ligand CH polarization and strength of CH–π interaction, which has an electrostatic component.97 The GalNAc–MGL binding98 also includes CH–Pi interaction of 4-, 5- and 6-CH groups with the indole of Trp 271 (Fig. S5, ESI†). The N-acetyl group engages in CH–Pi interaction with the indole of Tyr-236 as well as carbonyl H-bonding with the amino group of Lys-264 and His-286's imidazole, explaining the ∼70-fold increase in affinity of GalNAc relative to Gal for hMGL.99 The Kd measured for Gal by ITC was 210 μM.100 The binding of GalNAc as determined by ITC is enthalpy driven (Kd = 17.7 μM, −ΔH = +47.2 kJ mol−1, TΔS = −20.11 kJ mol−1).101 As it has higher affinity than Gal, GalNAc was chosen as the monomeric ligand for multivalent ligand design to target MGL. Inhibition by a tetrameric tetraphenylethene (TPE) based glycocluster, which displays GalNAc, of binding of MGL to a surface neoglycoprotein presenting GalNAc by ELISA showed a remarkable ∼125000-fold increase when compared to free GalNAc.102 The bind and jump mechanism appears to contribute to enhancement of avidity for MGL as the distances between GalNAc residues in the TPE glycocluster are not sufficient to accommodate simultaneous binding. The TPE glycocluster did not inhibit a lectin specific for GlcNAc, and showed only low cross reactivity for galectins; it was shown to block MGL adherence to tissue sections.103 Recently, a tripartite vaccine candidate (Fig. 10), which incorporated a trimeric version of the TPE glycocluster, conjugated to a tumour glycopeptide antigen and T-helper epitope showed greater uptake into DCs due to its avidity for MGL; the novel vaccine prototype also showed enhanced humoral response, demonstrating the potential for highly avid glycoclusters targeting MGL on immune cells for vaccine research. The tumour antigen segment of the prototype, by itself, has good avidity for MGL as it already presents GalNAc residues on the glycopeptide. However, when the trimeric TPE glycocluster was conjugated there was ∼10-fold further avidity improvement.104
 | ||
| Fig. 10 Modular vaccine design using TPE glycocluster to engage MGL at APC surface. | ||
4.3 Simultaneous binding of glycoclusters to block influenza
The influenza virus hemagglutinin (HA) is a homotrimeric surface lectin that interacts with NeuAc containing ligands. NeuAc shows a network of electrostatic interactions, including H-bonding and indole–CH3 contact (Fig. S3, ESI†).105 Research on developing small molecule ligands for HA based on NeuAc, has, thus far, not yielded very potent inhibitors. A serendipitously identified HA ligand, N-cyclohexyltaurine, was found to mimic interactions of broadly neutralizing antibodies and that of NeuAc, which might offer potential to identify low molecular weight ligands with no carbohydrate character.106Recognition by HA of host cell glycans has been associated with human type influenza specificity for the NeuAcα-2,6-Gal compared to NeuAcα2,3-Gal, the latter being associated with avian influenza.107 Higher avidity is observed in the recognition of biantennary N-glycans, containing NeuAcα2,6GalNAc of the human airway by HA compared to those based on NeuAcα2,3GalNAc. The Kd for NeuAcα2,3GalNAc shows only ∼1.5-fold lower potency than the 2,6-disaccharide (3.1 ± 0.4 mM vs. 2.0 ± 0.2 mM) for binding to the HA of the H3N2 viral strain A/HongKong/1/1968.108 The NeuAcα2,6Gal canonical specificity is explained by extensive presentation of this disaccharide on glycans of epithelial cells of the human airway, where multivalency leads to increase selectivity for NeuAcα-2,6Gal.109 Recent work has provided insight into the origin of an evolved specificity increase for a subtype of biantennary N-glycans.110 Specifically, two NeuAcα2,6Gal residues are projected in an optimal geometry for simultaneous binding to two CRDs in the HA trimer; the HA ligand binding sites are spaced about 45 Å apart in the HA trimer and the chelate binding accounts for >100-fold enhancement of avidity as measured by ELISA. This avidity and specificity gain has been maintained by influenza virus HA variants for biantennary N-glycans containing multiple LacNAc repeat units found in airway tissue glycomes. The high avidity associated with chelate type binding to bivalent N-glycans is due to NeuAcα2,6Gal being located at the end of chains having 3/4 LacNAc spacer groups. The spacing between NeuAc residues for simultaneous binding is not accessible either for N-glycans presenting NeuAcα2,3Gal with 3/4 LacNAc spacer groups or N-glycans presenting NeuAcα2,6Gal with only 1 or 2 spacer LacNAcs because the latter cannot simultaneously bind to two CRDs (Fig. 11). Influenza virus has caused several pandemics111 and the development of infection blockers is of interest. Such insights from how native glycans interact are, thus, enabling design and synthesis of glycocluster ligands designed with the appropriate spacing to enable simultaneous binding to at least two of HA's CRDs.112 Pieters and co-workers prepared the dimeric NeuAc presenting glycocluster in Fig. 12 that showed an IC50 of 0.7 μM, representing a 428-fold enhancement over the reference ligand. Thus, precisely designed multivalent ligands may address need to develop influenza infection blockers.
 | ||
| Fig. 11 Affinity/avidity for selected glycans for influenza virus HA. Weak binding is observed for single ligands and for bivalent ligands where the spacing between NeuAc residues does not facilitate simultaneous ligand binding. Strong binding is observed for biantennary extended N-glycans with 3/4 LN spacer groups like 6SLN4-N, but not 3SLN4-N. | ||
 | ||
| Fig. 12 HA ligands for structure activity relationships, including chelating ligands. | ||
4.4 Avidity boosting by combining statistical rebinding with simultaneous binding for langerin and DCSIGN
Recent attempts have been made to combine statistical rebinding with simultaneous binding to boost avidity of multivalent ligands. Seitz and co-workers produced DNA–peptide nucleic acid (PNA) complexes as scaffolds to precisely display two glycoclusters, to probe the impact of distance on binding to langerin, a trimeric cell surface lectin with three CRDs located at ∼42 Å distances apart.113 The DNA–PNA complexes form by Watson–Crick hybridization of 39 nucleotide long DNA template strands in the presence of three complimentary 13 nt peptide nucleic acid (PNA) strands. A selection of the construct is shown in Fig. 13, based on the langerin ligand GlcNHTs. The distances between the trimeric glycoclusters varied from ∼16 Å to 104 Å. | ||
| Fig. 13 DNA–PNA hybrids for targeting langerin. Strongest binding is observed for hexavalent compound where two trimeric glycoclusters are separated by distances of 42 Å, matching the distance of 42 Å between CRDs in langerin. | ||
The trimeric glycocluster presenting GlcNHTs shows avidity increase compared to GlcNHTs of ∼7-fold. The presentation of the GlcNHTs ligand as a monomer on the PNA–DNA hybrid showed an increase of ∼3-fold over GlcNHTs, while presentation of one trimeric glycocluster on PNA–DNA showed ∼10-fold avidity enhancement over the monomer on the PNA–DNA hybrid. Spacing of two trimeric glycoclusters at ∼23 Å on PNA–DNA gave a further enhancement of ∼13-fold; the distances between the trimeric glycocluster is substantially less than 42 Å and not sufficient to facilitate simultaneous binding to two langerin CRDs and the avidity gain is thus explained by statistical rebinding only (bind and jump). A hybrid with ∼42 Å distance between the glycoclusters, matching that between langerin CRDs, showed the highest avidity, which is explained to be due to the combination of statistical rebinding combined with chelate binding. Subsequent cell binding studies indicated the more potent hybrids could also bind to Langerhan cell surfaces, which express langerin. Ligands with high avidity for langerin have potential for targeting vaccines to Langerhans cells for improved therapeutic effect.
Bernardi, Fieschi and co-workers had earlier aimed to take advantage of both chelation and statistical rebinding in the modular design of ligands for DC-SIGN, which have two trivalent glycoclusters presented on a rigid core of defined length.114 DC-SIGN is a homotetrameric lectin with four CRDs at the C-terminus of each monomer in a square-like arrangement; the distance between the CRDs in DC-SIGN is ∼39 Å (width) and ∼52 Å (length) and ∼60 Å (across the diagonal), assuming the CRDs are at the corners of a rectangle.115 They synthesised the divalent binder construct, polyman26, with nanomolar range affinity (Fig. 14). The evaluation of polyman26 was recently updated using a surface that preserves the tetrameric form of DC-SIGN, accessibility to its CRDs and their topology and thus appropriate to dissect the contribution of statistical rebinding and chelate binding from ligands. Polyman26 had an apparent Kd = 11.5 ± 2.3 nM and was 570-fold more potent per mole of ligand than the reference compound (Kd = 39300 ± 3900 nM).116 MD simulations predicted that simultaneous binding of two headgroups from polyman26 was feasible to two DC-SIGN CRDs, contributing most to the observed avidity increase, boosted in turn by statistical rebinding. The chelate binding was facilitated by flexibility in the DC-SIGN structure allowing movement of the CRDs and induced fitting (Fig. 15).
 | ||
| Fig. 14 DC-SIGN ligands: monomer reference ligand and polyman26. | ||
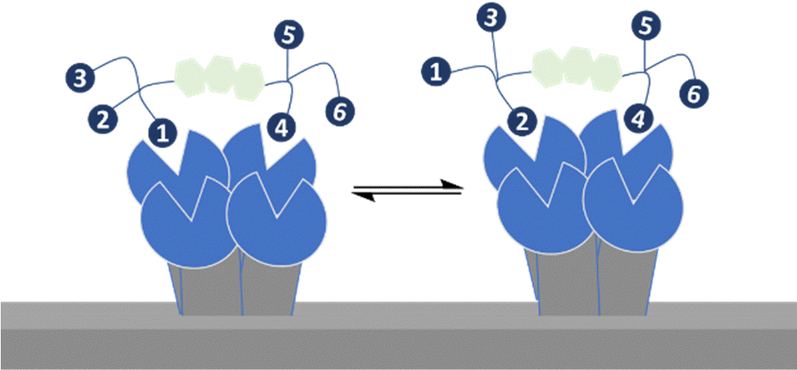 | ||
| Fig. 15 Cooperation of statistical rebinding and simultaneous binding. | ||
4.5 Promotion or prevention of receptor cross-linking in multivalent ligand design
Reymond and co-workers designed galactopyranoside based glycoclusters that were both chelating ligands for the LecA lectin of the Pseudomonas aeruginosa (PA) pathogen leading to high avidity gain, but which could also show the ability to promote aggregation. The combining of simultaneous binding of ligands to two LecA sites and crosslinking led to more effective biofilm inhibitors.117The formation of precipitates via crosslinking is associated with plaque formations and there are instances where it is not desired from a biomedical viewpoint. Wittman and co-workers synthesised compounds with high avidity for WGA, based on ligands that show simultaneous binding to four GlcNAc binding sites in a WGA dimer. The ligands had Kd values in the low nM range. One inline tetravalent ligand (Fig. 16) showed one to two million-fold avidity increase per GlcNAc residue. Various evidence, including EPR using spin labelled inline ligands, supported simultaneous binding of the four GlcNAc to the four WGA binding sites. There was no evidence for crosslinking of the inline tetravalent ligand, i.e. no precipitation was observed. Crosslinking was observed for ligands with shorter distances between the GlcNAcs when simultaneous binding is not possible.118
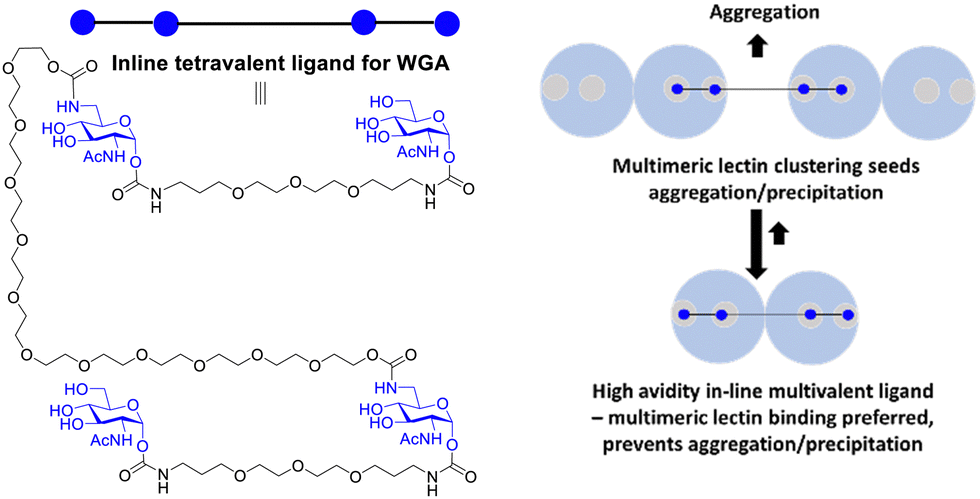 | ||
| Fig. 16 Multivalent lectin ligands with inline topology (iLecs) bind simultaneously to four GlcNAc binding sites in WGA leading to high avidity with no precipitation. | ||
4.6 Glycoclusters for LYTACs, cell targeting of antisense nucleotides and for diagnostics
Targeted protein degradation has emerged as a strategy for therapeutic development and as a tool for chemical biology, with proteolysis targeting chimeras (PROTACs) gaining most attention thus far.119 Bertozzi's group developed lysosome targeting chimeras (LYTACs) by conjugating a polyvalent ligand, based on Man-6-phosphate, to a small molecule or antibody that binds a protein to be degraded; the polyvalent ligand targets the cation-independent mannose-6-phosphate receptor (CI-M6PR),120 triggering internalization of the desired target through CI-M6PR mediated endocytosis, leading to transport to the lysosome and protein degradation. Lectin ASGPR is another lysosomal targeting receptor, with ASGPR responsible for clearing glycoproteins via endocytosis and lysosomal degradation, particularly in liver cells. Hence, Zhou et al. used high avidity ASGPR glycocluster ligands in LYTACs.121 In another development, ASGPR targeting glycoclusters increase the delivery of antisense nucleotides to hepatocytes enabling development of therapies for what had been considered undruggable protein targets.122,123 Givosiran is a small interfering ribonucleic acid (siRNA) that is conjugated to a trivalent GalNAc glycocluster to enhance the delivery and effectiveness of siRNA.124 Givosiran, obtained regulatory approval from the FDA for treatment of acute hepatic porphyria in 2019.125 Other siRNA–GalNAc glycocluster conjugates are in advanced clinical studies or approved126 (lumisiran, inclisiran, eplontersen). Another application developed for GalNAc glycocluster ligands is in synthesis of positron emission tomography (PET) diagnostic probes to measure ASGPR expression, reduced in nonalcoholic steatohepatitis, where early diagnosis may enable prevention of life-threatening diseases associated with this condition.127 In one other recent development, a luminescent glycocluster molecular sensor system, which showed reliable enhancement of lanthanide ion centred emission in the presence of unlabelled lectins have been developed. This work by Byrne and co-workers included sensing of the bacterial lectin LecA, which is lectin of the priority ESCAPE pathogen PA.1285. Conclusions and perspectives
Lectin–ligand interactions mediate important biological processes and research is leading to new clinically studied inhibitors, prototype vaccines, targeting agents for delivery, diagnostics and tools for chemical biology based on small monomeric ligand or glycocluster development.Glycans have well-defined 3D structural preferences and the conformation of the free ligand is often very similar to that bound by the lectin. The availability of structural information is enabling improved ligand design. There are relatively few X-ray crystal structures of oligosaccharides in the Cambridge Crystallographic Datacentre, although information is often accessible from co-crystal structural coordinates deposited at the Protein Data Bank. Moreover, computational tools are being developed and made openly available to non-specialists for generating low-energy conformers for glycans which can be used in new ligand design.129 The recent development of cryo-electron microscopy has the potential to give atomic-level detail of the interaction of glycoproteins/glycoclusters with lectins and reveal new strategies for ligand design,130 especially where there is very limited experimentally derived information, such as for structures of multivalent ligands bound to multimeric lectins, with reliance in many cases on computing simulations, including AI based alphafold131 to give potential geometries.132
Native glycan ligands do not have ideal properties as pharmaceuticals. However, glycans are inspiring the development of drug-like glycomimetics. The use of synthetic chemistry for producing such substances, is essential. Factors determining lectin–ligand binding affinities are complex, determined by type and strength of interaction, binding pocket shape and solvent effects. Glycomimetic design includes substituting part of natural ligand with other functional groups or scaffolds to (i) enhance affinity or selectivity/specificity for a target lectin; (ii) increase stability in vitro and in vivo such as with use of S-glycosides; (iii) increase other physicochemical and pharmacokinetic properties such as by reducing polar surface area of ligand to enhance oral availability or (iv) appending PEG groups to increase half-time in plasma. Binding free energies have been improved by strategies which include conformational preorganisation of binding groups. Ernst's group have shown that binding free energies can be more favourable after replacing part of the glycan which has a purely structural role by a more hydrophobic scaffold with reduced polarity. As typified for galectin-3 ligand development led by Nilsson, Leffler and co-workers the generation of new interactions contribute to affinity enhancement. Areas where monomeric ligand inhibitors continue to be investigated include for galectins, for siglecs as well as for lectins involved in anti-microbial resistance such as FimH,133 LecA134 and LecB.135
Conjugating multiple small lectin ligands to scaffolds to give multivalent ligands is a well-developed strategy to improve avidity and selectivity. There is now proven clinical success in use of multivalent ligands as a component of new siRNA medicines for delivery purposes which leads to enhanced uptake and efficacy,136 which is a milestone for the field. There is potential for expansion of this approach to other lectins and cell types. Multivalent ligands act as delivery agents and agonists for siglecs, in contrast to monomeric ligands, which act as antagonists, showing the need to optimise both monomeric and multivalent ligands, which will depend on the desired application.
Avidities of multivalent ligands are influenced by geometrical factors such as spacing between individual ligands and whether they can match the distances between CRDs. Higher avidity is observed for glycoclusters where chelate binding is enabled by matching spacing between ligands and CRDs. It has been shown that further benefits arise from combining statistical rebinding with chelate binding as seen more recently for DCSIGN and langerin. Developing chelate binding ligands is likely more challenging for a lectin like MGL where distances between CRDs are ∼80 Å, in contrast with distances ∼20–25 Å for ASGPR but developing approaches to targeting such CRDs with chelate binding ligands over large distances could be important. Despite the distance requirement for MGL potent glycocluster ligands have emerged based on a tetraphenylethene (TPE) scaffold, which can deliver glycopeptide tumour vaccines via MGL targeting and the resulting improved humoral response has been shown in vivo. How TPE ligands work effectively is not clear; reversible formation of small aggregates of TPE glycocluster in water that engage in chelate binding seems possible and could be contributing to improved avidity. The investigation of aggregating scaffolds could be a strategy to further explore for multivalent ligand design either with TPE or related aggregating scaffolds. It would also be interesting to evaluate aggregating glycoclusters for other lectins. Another multivalent ligand mechanism which is relevant for soluble lectins is cross-linking. Research from Wittman's group shows how precipitation, via crosslinking can be avoided by careful design of very potent inline multivalent ligands based on simultaneous binding with optimised geometries. On the other hand increasing the cross-linking efficiency was shown to be useful for developing biofilm inhibitors, which is important in antimicrobial research.
There are many opportunities as development of ligands for lectins is still at a relatively early stage, and potent ligands with desirable properties are still lacking for many lectin targets. It is expected that development of new ligands for application in glycoscience and glycomedicine will be a very active research area for the foreseeable future.
Author contributions
PVM sourced and analysed data, planned, wrote, and revised the manuscript. AD, SK, LF, EH, and KM sourced data and contributed to drafting and/or editing.Data availability statement
The data supporting this tutorial review have been included with the ESI† or are found in the cited references.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Science Foundation Ireland (16/IA/4419, PM, EH, KM), SSPC, The SFI Centre for Research on Pharmaceuticals (12/RC/2275_P2), CÚRAM, The SFI Centre for Research in Medical Devices and the European Regional Development Fund (13/RC/2073, 17/RC-PhD/3480, LF) and the Irish Research Council for a Government of Ireland Postdoctoral Research Fellowship (GOIPD/2020/155, SK).Notes and references
- A. Varki and S. Kornfeld Historical, Background and Overview, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 4th edn, 2022. ch. 1 Search PubMed.
- M. E. Taylor, K. Drickamer, A. Imberty, Y. van Kooyk, R. L. Schnaar, M. E. Etzler and A. Varki, Discovery and Classification of Glycan-Binding Proteins, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar, P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022, ch. 28 Search PubMed.
- (a) N. Jayaraman, Chem. Soc. Rev., 2009, 38, 3463–3483 RSC; (b) R. Roy, P. V. Murphy and H. J. Gabius, Molecules, 2016, 21, 629 CrossRef.
- H. J. Gabius, M. Cudic, T. Diercks, H. Kaltner, J. Kopitz, K. H. Mayo, P. V. Murphy, S. Oscarson, R. Roy, A. Schedlbauer, S. Toegel and A. Romero, ChemBioChem, 2022, 23, e2021003 CrossRef.
- V. S. R. Rao, P. K. Qasba, P. V. Balaji and R. Chandrasekaran, Conformation of Carbohydrates, Harwood Academic Publishers, 1998, pp. 309 Search PubMed.
- V. S. R. Rao, P. K. Qasba, P. V. Balaji and R. Chandrasekaran, Conformation of Carbohydrates, Harwood Academic Publishers, 1998, pp. 49–190 Search PubMed.
- A. Y. Mehta and R. D. Cummings, Bioinformatics, 2020, 36, 3613–3614 CrossRef CAS.
- (a) J. T. Edward, J. Chem. Ind., 1955, 1102–1104 CAS; (b) R. U. Lemieux, Rearrangements and Isomerizations in Carbohydrate Chemistry, in Molecular Rearrangements, ed. P. De Mayo, Interscience, New York, 1964, pp. 709–769 Search PubMed.
- I. V. Alabugin, L. Kuhn, N. V. Krivoshchapov, P. Mehaffy and M. G. Medvedev, Chem. Soc. Rev., 2021, 50, 10212–10252 RSC.
- K. B. Wiberg, W. F. Bailey, K. M. Lambert and Z. Stempel, J. Org. Chem., 2018, 83, 5242–5255 CrossRef CAS PubMed.
- L. Kerins, S. Byrne, A. Gabba and P. V. Murphy, J. Org. Chem., 2018, 83, 7714–7729 CrossRef CAS.
- For ethane-1,2-diol there is an increased preference for the gauche arrangement of the electron withdrawing oxygen atoms, sometimes attributed to intramolecular H-bonding. For 1,2-dimethoxyethane, where there is no H-bonding, there is still an increase in preference for the gauche arrangement. The gauche effect is stronger for fluorinated derivatives. See (a) S. Wolf, Acc. Chem. Res., 1970, 5, 102–111 CrossRef; (b) M. Andersson and G. Karlstrom, J. Phys. Chem., 1985, 89, 4957–4962 CrossRef CAS.
- Y. Yu and M. Delbianco, Chem. – Eur. J., 2020, 26, 9814 CrossRef CAS PubMed.
- A. J. Petrescu, S. M. Petrescu, R. A. Dwek and M. R. Wormald, Glycobiology, 1999, 9, 343–352 CrossRef CAS PubMed.
- Woods Group. (2005-XXXX) GLYCAM Web. Complex Carbohydrate Research Center, University of Georgia, Athens, GA. (https://legacy.glycam.org).
- (a) S. Kumar, M. Frank and R. Schwartz-Albiez, PLoS One, 2013, 8, e59761 CrossRef CAS; (b) V. Palivec, C. Johannessen, J. Kaminsky and H. Martinez-Seara, PLoS Comput. Biol., 2022, 18, e1009678 CrossRef CAS PubMed; (c) P. Vidal, J. Jiménez-Barbero and J. F. Espinosa, Carbohydr. Res., 2016, 433, 36–40 CrossRef CAS; (d) S. Sabesan, K. Bock and J. C. Paulson, Carbohydr. Res., 1991, 218, 27–54 CrossRef CAS.
- M. Martín-Pastor, A. Canales, F. Corzana, J. L. Asensio and J. Jiménez-Barbero, J. Am. Chem. Soc., 2005, 127, 3589–3595 CrossRef PubMed.
- (a) K. Hirotsu and A. Shimada, Bull. Chem. Soc. Jpn., 1974, 47, 1872–1879 CrossRef CAS; (b) Coordinates are available Cambridge Crystallographic Data Centre. https://www.ccdc.cam.ac.uk/CSDentry=BLACTO.
- U. Olsson, E. Sawen, R. Stenutz and G. Widmalm, Chem. – Eur. J., 2009, 15, 8886–8894 CrossRef CAS.
- C. W. von der Lieth, H. C. Siebert, T. Kožár, M. Burchert, M. Frank, M. Gilleron, H. Kaltner, G. Kayser, E. Tajkhorshid, N. V. Bovin, J. F. G. Vliegenthart and H. J. Gabius, Acta Anat., 1998, 161, 91–109 CrossRef CAS PubMed.
- (a) E. W. Sayers and J. H. Prestegard, Biophys. J., 2000, 79, 3313–3329 CrossRef CAS PubMed; (b) E. W. Sayers and J. H. Prestegard, Biophys. J., 2002, 82, 2683–2699 CrossRef CAS.
- T. Chandran, A. Sharma and M. Vijayan, Adv. Protein Chem. Struct. Biol., 2013, 92, 135–178 CAS.
- M. M. Sauer, R. P. Jakob, J. Eras, S. Baday, D. Eriş, G. Navarra, S. Bernèche, B. Ernst, T. Maier and R. Glockshuber, Nat. Commun., 2016, 7, 10738 CrossRef CAS PubMed.
- E. V. Sokurenko, V. Vogel and W. E. Thomas, Cell Host Microbe, 2008, 4, 314–323 CrossRef CAS.
- C. S. Hung, J. Bouckaert, D. Hung, J. Pinkner, C. Widberg, A. DeFusco, C. G. Auguste, R. Strouse, S. Langermann, G. Waksman and S. J. Hultgren, Mol. Microbiol., 2002, 44, 903–915 CrossRef CAS PubMed.
- A. Wellens, M. Lahmann, M. Touaibia, J. Vaucher, S. Oscarson, R. Roy, H. Remaut and J. Bouckaert, Biochemistry, 2012, 51, 4790–4799 CrossRef CAS PubMed.
- M. M. Sauer, R. P. Jakob, T. Luber, F. Canonica, G. Navarra, B. Ernst, C. Unverzagt, T. Maier and R. Glockshuber, J. Am. Chem. Soc., 2019, 141, 936–944 CrossRef CAS.
- L. A. Lasky, Annu. Rev. Biochem., 1995, 64, 113–139 CrossRef CAS PubMed.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS.
- R. C. Preston, R. P. Jakob, F. P. Binder, C. P. Sager, B. Ernst and T. Maier, J. Mol. Cell Biol., 2016, 8, 62–72 CrossRef CAS PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- E. P. Mitchell, C. Sabin, L. Šnajdrová, M. Pokorná, S. Perret, C. Gautier, C. Hofr, N. Gilboa-Garber, J. Koča, M. Wimmerová and A. Imberty, Proteins, 2005, 58, 735–746 CrossRef CAS.
- P. V. Murphy, A. Romero, Q. Xiao, A. K. Ludwig, S. Jogula, N. V. Shilova, T. Singh, A. Gabba, B. Javed, D. Zhang, F. J. Medrano, H. Kaltner, J. Kopitz, N. V. Bovin, A. M. Wu, M. L. Klein, V. Percec and H. J. Gabius, iScience, 2021, 24, 101919 CrossRef CAS.
- C. A. Fitch, D. A. Karp, K. K. Lee, W. E. Stites, E. E. Lattman and E. B. García-Moreno, Biophys. J., 2002, 82, 3289–3304 CrossRef CAS.
- C. P. Sager, D. Eriş, M. Smieško, R. Hevey and B. Ernst, Beilstein J. Org. Chem., 2017, 13, 2584–2595 CrossRef CAS.
- E. J. Toone, Curr. Opin. Struct. Biol., 1994, 4, 719–728 CrossRef CAS.
- D. Gupta, T. K. Dam, S. Oscarson and C. F. Brewer, J. Biol. Chem., 1997, 272, 6388–6392 CrossRef CAS.
- J. D. Chodera and D. L. Mobley, Annu. Rev. Biophys., 2013, 42, 121–142 CrossRef CAS PubMed.
- C. P. Sager, B. Fiege, P. Zihlmann, R. Vannam, S. Rabbani, R. P. Jakob, R. C. Preston, A. Zalewski, T. Maier, M. W. Peczuh and B. Ernst, Chem. Sci., 2018, 9, 646–654 RSC.
- L. Pang, J. Bezençon, S. Kleeb, S. Rabbani, A. Sigl, M. Smiesko, C. P. Sager, D. Eris, O. Schwardt and B. Ernst, Carbohdr. Chem., 2016, 42, 248–273 Search PubMed.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS PubMed.
- U. Schmitz and S. Swaminathan, Antiviral Ther., 2022, 27, 13596535211067598 Search PubMed.
- R. D. Cummings, F. T. Liu, G. A. Rabinovich, S. R. Stowell and G. R. Vasta, Galectins, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings and J. D. Eskoet al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022, ch. 36 Search PubMed.
- K. V. Mariño, A. J. Cagnoni, D. O. Croci and G. A. Rabinovich, Nat. Rev. Drug Discovery, 2023, 22, 295–316 CrossRef.
- R. P. M. Dings, M. C. Miller, R. J. Griffin and K. H. Mayo, Int. J. Mol. Sci., 2018, 19, 905 CrossRef PubMed.
- H. Ideo, A. Seko, I. Ishizuka and K. Yamashita, Glycobiology, 2003, 13, 713–723 CrossRef CAS PubMed.
- H. Yoshida, S. Yamashita, M. Teraoka, A. Itoh, S. I. Nakakita, N. Nishi and S. Kamitori, FEBS J., 2012, 279, 3937–3951 CrossRef CAS.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, J. Am. Chem. Soc., 2005, 127, 1737–1743 CrossRef PubMed.
- W. Schneider, Ber. Dtsch. Chem. Ges., 1919, 52B, 2135–2149 CrossRef CAS.
- H. Leffler and S. H. Barondes, J. Biol. Chem., 1986, 261, 10119–10126 CrossRef CAS.
- S. R. Stowell, Y. Qian, S. Karmakar, N. S. Koyama, M. Dias-Baruffi, H. Leffler, R. P. McEver and R. D. Cummings, J. Immunol., 2008, 180, 3091–3102 CrossRef CAS.
- A. MacKinnon, W. S. Chen, H. Leffler, N. Panjwani, H. Schambye, T. Sethi and U. J. Nilsson, Top. Med. Chem., 2014, 12, 95–121 CAS.
- T. J. Hsieh, H. Y. Lin, Z. Tu, T. C. Lin, S. C. Wu, Y. Y. Tseng, F. T. Liu, S. T. D. Hsu and C. H. Lin, Sci. Rep., 2016, 6, 29457 CrossRef CAS PubMed.
- E. Montero, A. García-Herrero, J. L. Asensio, K. Hirai, S. Ogawa, F. Santoyo-González, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Org. Chem., 2000, 1945–1952 CrossRef CAS.
- A. Kumar, M. Paul, M. Panda, S. Jayaram, N. Kalidindi, H. Sale, M. Vetrichelvan, A. Gupta, A. Mathur, B. Beno, A. Regueiro-Ren, D. Cheng, M. Ramarao and K. Ghosh, Glycobiology, 2021, 31, 1390–1400 CrossRef CAS PubMed.
- N. Hirani, A. C. MacKinnon, L. Nicol, P. Ford, H. Schambye, A. Pedersen, U. J. Nilsson, H. Leffler, T. Sethi, S. Tantawi, L. Gravelle, R. J. Slack, R. Mills, U. Karmakar, D. Humphries, F. Zetterberg, L. Keeling, L. Paul, P. L. Molyneaux, F. Li, W. Funston, I. A. Forrest, A. J. Simpson, M. A. Gibbons and T. M. Maher, Eur. Respir. J., 2021, 57, 2002559 CrossRef CAS PubMed.
- News release Aug. 15 2023. Downloaded 12/01/2024. https://ir.galecto.com/news-releases/news-release-details/galecto-announces-topline-results-phase-2b-galactic-1-trial.
- F. R. Zetterberg, A. MacKinnon, T. Brimert, L. Gravelle, R. E. Johnsson, B. Kahl-Knutson, H. Leffler, U. J. Nilsson, A. Pedersen, K. Peterson, J. A. Roper, H. Schambye, R. J. Slack and S. Tantawi, J. Med. Chem., 2022, 65, 12626–12638 CrossRef CAS PubMed.
- T. Brimert, R. Johnsson, H. Leffler, U. Nilsson and F. Zetterberg, WO2016120403, 2016.
- F. R. Zetterberg, K. Peterson, R. E. Johnsson, T. Brimert, M. Hakansson, D. T. Logan, H. Leffler and U. J. Nilsson, ChemMedChem, 2018, 13, 133–137 CrossRef CAS.
- F. P. C. Binder, K. Lemme, R. C. Prestin and B. Ernst, Angew. Chem., Int. Ed., 2012, 51, 7327–7331 CrossRef CAS.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS PubMed.
- H. C. Kolb and B. Ernst, Chem. – Eur. J., 1997, 3, 1571 CrossRef CAS.
- A. J. Ruben, Y. Kiso and E. Freire, Chem. Biol. Drug Design, 2006, 67, 2–4 CrossRef CAS PubMed.
- R. C. Preston, R. P. Jakob, F. P. C. Binder, C. P. Sager, B. Ernst and T. Maier, J. Mol. Cell Biol., 2016, 8, 62–72 CrossRef CAS.
- J. Egger, C. Weckerle, B. Cutting, O. Schwardt, S. Rabbani, K. Lemme and B. Ernst, J. Am. Chem. Soc., 2013, 135, 9820–9828 CrossRef CAS PubMed.
- (a) J. Chang, J. T. Patton, A. Sarkar, B. Ernst, J. L. Magnani and P. S. Frenette, Blood, 2010, 116, 1779–1786 CrossRef PubMed; (b) J. L. Magnani, J. T. Patton, A. K. Sarkar, S. A. Svarovsky and B. Ernst, WO2007028050A1, 2007.
- C. D. Dampier, M. J. Telen, T. Wun, R. Clark Brown, P. Desai, F. El Rassi, B. Fuh, J. Kanter, Y. Pastore, J. Rothman, J. G. Taylor, D. Readett, K. M. Sivamurthy, B. Tammara, L. J. Tseng, J. Nelson Lozier, H. Thackray, J. L. Magnani and K. L. Hassell, Blood, 2023, 141, 168–179 CrossRef CAS PubMed.
- D. J. DeAngelo, B. A. Jonas, J. L. Liesveld, D. L. Bixby, A. S. Advani, P. Marlton, J. L. Magnani, H. M. Thackray, E. J. Feldman, M. E. O’Dwyer and P. S. Becker, Blood, 2022, 139, 1135–1146 CrossRef CAS.
- V. Krishnamurthy, M. Sardar, Y. Ying, X. Song, C. Haller, E. Dai, X. Wang, D. Hanjaya-Putra, L. Sun, V. Morikis, S. I. Simon, R. J. Woods, R. D. Cummings and E. L. Chaikof, Nat. Commun., 2015, 6, 6387 CrossRef CAS PubMed.
- D. J. Wong, D. D. Park, S. S. Park, C. A. Haller, J. Chen, E. Dai, L. Liu, A. R. Mandhapati, P. Eradi, B. Dhakal, W. J. Wever, M. Hanes, L. Sun, R. D. Cummings and E. L. Chaikof, Blood, 2021, 138, 1182–1193 CrossRef CAS PubMed.
- B. Dhakal, A. Mandhapati, P. Eradi, S. Park, K. Fibben, K. Li, A. DeYong, S. Escopy, G. Karki, D. D. Park, C. A. Haller, E. Dai, L. Sun, W. A. Lam and E. L. Chaikof, J. Am. Chem. Soc., 2024, 146, 17414–17427 CrossRef CAS PubMed.
- M. S. Macauley, P. R. Crocker and J. C. Paulson, Nat. Rev. Immunol., 2014, 14, 653 CrossRef CAS PubMed.
- C. Selby, L. R. Yacko and A. E. Glode, J. Adv. Pract. Oncol., 2019, 10, 68–82 Search PubMed.
- W. C. Chen, N. Kawasaki, C. M. Nycholat, S. Han, J. Pilotte, P. R. Crocker and J. C. Paulson, PLoS One, 2012, 7, e39039 CrossRef CAS.
- M. K. O’Reilly, H. Tian and J. C. Paulson, J. Immunol., 2011, 186, 1554–1563 CrossRef.
- (a) P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS; (b) H. Läubli, K. Kawanishi, C. G. Vazhappilly, R. Matar, M. Merheb and S. S. Siddiqui, FEBS J., 2021, 288, 6206–6225 CrossRef.
- B. A. H. Smith and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2021, 20, 217–243 CrossRef CAS PubMed.
- M. K. O’Reilly and J. C. Paulson, Methods Enzymol., 2010, 478, 343–363 Search PubMed.
- A. P. May, R. C. Robinson, M. Vinson, P. R. Crocker and E. Y. Jones, Mol. Cell, 1998, 1, 719–728 CrossRef CAS.
- P. R. Crocker, M. Vinson and S. Kelm, Biochem. J., 1999, 341, 355–361 CrossRef CAS.
- (a) H. Prescher, M. Frank, S. Gütgemann, E. Kuhfeldt, A. Schweizer, L. Nitschke, C. Watz and R. Brossmer, J. Med. Chem., 2017, 60, 941–956 CrossRef CAS; (b) M. Frank, E. Kuhfeldt, J. Cramer, C. Watzl and H. Prescher, J. Med. Chem., 2023, 66, 14315–14334 CrossRef CAS.
- H. Prescher, A. Schweizer, E. Kuhfeldt, L. Nitschke and R. Brossmer, ACS Chem. Biol., 2014, 9, 1444–1450 CrossRef CAS PubMed.
- (a) H. Tateno, P. R. Crocker and J. C. Paulson, Glycobiology, 2005, 15, 1125–1135 CrossRef CAS PubMed; (b) B. S. Bochner, R. A. Alvarez, P. Mehta, N. V. Bovin, O. Blixt, J. R. White and R. L. Schnaar, J. Biol. Chem., 2005, 280, 4307–4312 CrossRef CAS.
- J. M. Pröpster, F. Yang, S. Rabbani, B. Ernst, F. H. T. Allain and M. Schubert, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4170–E4179 CrossRef PubMed.
- M. P. Lenza, U. Atxabal, C. Nycholat, I. Oyenarte, A. Franconetti, J. I. Quintana, S. Delgado, R. Núñez-Franco, C. T. G. Marroquín, H. Coelho, L. Unione, G. Jiménez-Oses, F. Marcelo, M. Schubert, J. C. Paulson, J. Jiménez-Barbero and J. Ereño-Orbea, JACS Au, 2023, 3, 204–215 CrossRef CAS.
- (a) C. M. Nycholat, S. Duan, E. Knuplez, C. Worth, M. Elich, A. Yao, J. O’Sullivan, R. McBride, Y. Wei, S. M. Fernandes, Z. Zhu, R. L. Schnaar, B. S. Bochner and J. C. Paulson, J. Am. Chem. Soc., 2019, 141, 14032–14037 CrossRef CAS PubMed; (b) B. S. Kroezen, G. Conti, B. Girardi, J. Cramer, X. Jiang, S. Rabbani, J. Müller, M. Kokot, E. Luisoni, D. Ricklin, O. Schwardt and B. Ernst, ChemMedChem, 2020, 15, 1706–1719 CrossRef CAS PubMed.
- G. Conti, A. Bärenwaldt, S. Rabbani, T. Mühlethaler, M. Sarcevic, X. Jiang, O. Schwardt, D. Ricklin, R. J. Pieters, H. Läubli and B. Ernst, Angew. Chem., Int. Ed., 2023, 62, e202314280 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 8, 321–327 CrossRef.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- (a) T. K. Dam, R. Roy, D. Pagé and C. F. Brewer, Biochemistry, 2002, 41, 1351–1358 CrossRef CAS; (b) T. K. Dam, T. A. Gerken and C. F. Brewer, Biochemistry, 2009, 48, 3822–3827 CrossRef CAS.
- L. R. Olsen, A. Dessen, D. Gupta, S. Sabesan and J. Sacchettini, Biochemistry, 1997, 36, 15073–15080 CrossRef CAS PubMed.
- K. E. Pace, C. Lee, P. L. Stewart and L. G. Baum, J. Immunol., 1999, 163, 3801–3811 CrossRef CAS.
- M. Abbas, M. Maalej, F. Nieto-Fabregat, M. Thépaut, J. P. Kleman, I. Ayala, A. Molinaro, J. P. Simorre, R. Marchetti, F. Fieschi and C. Laguri, PNAS Nexus, 2023, 2, 1–10 CrossRef CAS PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- A. Gabba, A. Bogucka, J. G. Luz, A. Diniz, H. Coelho, F. Corzana, F. J. Cañada, F. Marcelo, P. V. Murphy and G. Birrane, Biochemistry, 2021, 60, 1327–1336 CrossRef CAS.
- F. Marcelo, F. Garcia-Martin, T. Matsushita, J. Sardinha, H. Coelho, A. Oude-Vrielink, C. Koller, S. Andre, E. J. Cabrita, H. J. Gabius, S. I. Nishimura, J. Jimenez-Barbero and J. Canada, Chem. – Eur. J., 2014, 20, 16147–16155 CrossRef CAS PubMed.
- M. Sakakura, S. Oo-Puthinan, C. Moriyama, T. Kimura, J. Moriya, T. Irimura and I. Shimada, J. Biol. Chem., 2008, 283, 33665–33673 CrossRef CAS PubMed.
- D. M. Beckwith, F. G. FitzGerald, M. C. Rodriguez Benavente, E. R. Mercer, A. K. Ludwig, M. Michalak, H. Kaltner, J. Kopitz, H. J. Gabius and M. Cudic, Biochemistry, 2021, 60, 547–558 CrossRef CAS.
- S. André, S. O’Sullivan, C. Koller, P. V. Murphy and H. J. Gabius, Org. Biomol. Chem., 2015, 13, 4190 RSC.
- H. Kaltner, J. C. Manning, G. G. Caballero, C. Di Salvo, A. Gabba, L. L. Romero-Hernández, C. Knospe, D. Wu, H. C. Daly, D. F. O’Shea, H. J. Gabius and P. V. Murphy, RSC Adv., 2018, 8, 28716–28735 RSC.
- A. Gabba, R. Attariya, S. Behren, C. Pett, J. C. van der Horst, H. Yurugi, J. Yu, M. Urschbach, J. Sabin, G. Birrane, E. Schmitt, S. J. van Vliet, P. Besenius, U. Westerlind and P. V. Murphy, J. Am. Chem. Soc., 2023, 145, 13027–13037 CrossRef CAS.
- N. K. Sauter, J. E. Hanson, G. D. Glick, J. H. Brown, R. L. Crowther, S. J. Park, J. J. Skehel and D. C. Wiley, Biochemistry, 1992, 31, 9609–9621 CrossRef CAS PubMed.
- R. U. Kadam and I. A. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4240–4245 CrossRef CAS.
- A. J. Thompson and J. C. Paulson, J. Biol. Chem., 2021, 296, 100017 CrossRef CAS PubMed.
- Y. Ji, Y. J. B. White, J. A. Hadden, O. C. Grant and R. J. Woods, Curr. Opin. Struct. Biol., 2017, 44, 219–231 CrossRef CAS PubMed.
- (a) X. Xiong, P. Coombs, S. Martin, J. Liu, H. Xiao, J. W. McCauley, K. Locher, P. A. Walker, P. J. Collins, Y. Kawaoka, J. J. Skehel and S. J. Gamblin, Nature, 2013, 497, 392–396 CrossRef CAS; (b) N. K. Sauter, M. D. Bednarski, B. A. Wurzburg, J. E. Hanson, G. M. Whitesides, J. J. Skehel and D. C. Wiley, Biochemistry, 1989, 28, 8388–8396 CrossRef CAS.
- W. Peng, R. P. de Vries, O. C. Grant, A. J. Thompson, R. McBride, B. Tsogtbaatar, P. S. Lee, N. Razi, I. A. Wilson, R. J. Woods and J. C. Paulson, Cell Host Microbe, 2017, 21, 23–34 CrossRef CAS PubMed.
- K. J. Taubenberger and J. C. Kash, Cell Host Microbe, 2010, 7, 440–451 CrossRef PubMed.
- W. Lu, W. Du, V. J. Somovilla, G. Yu, D. Haksar, E. de Vries, G. J. Boons, R. P. de Vries, C. A. M. de Haan and R. J. Pieters, J. Med. Chem., 2019, 62, 6398–6404 CrossRef CAS.
- G. Bachem, E. C. Wamhoff, K. Silberreis, D. Kim, H. Baukmann, F. Fuchsberger, J. Dernedde, C. Rademacher and O. Seitz, Angew. Chem., Int. Ed., 2020, 59, 21016–21022 CrossRef CAS PubMed.
- S. Ordanini, N. Varga, V. Porkolab, M. Thepaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816–3819 RSC.
- G. Tabarani, M. Thepaut, D. Stroebel, C. Ebel, C. Vives, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS.
-
(a) V. Porkolab, C. Pifferi, I. Sutkeviciute, S. Ordanini, M. Taouai, M. Thépaut, C. Vivès, M. Benazza, A. Bernardi, O. Renaudet and F. Fieschi, Org. Biomol. Chem., 2020, 18, 4763–4772 RSC;
(b) V. Porkolab, M. Lep
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ík, S. Ordanini, A. St John, A. Le Roy, M. Thépaut, E. Paci, C. Ebel, A. Bernardi and F. Fieschi, ACS Cent. Sci., 2023, 9, 709–718 CrossRef CAS PubMed.
ík, S. Ordanini, A. St John, A. Le Roy, M. Thépaut, E. Paci, C. Ebel, A. Bernardi and F. Fieschi, ACS Cent. Sci., 2023, 9, 709–718 CrossRef CAS PubMed. - R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. E. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015, 10, 2455–2462 CrossRef CAS PubMed.
- P. Rohse, S. Weickert, M. Drescher and V. Wittmann, Chem. Sci., 2020, 11, 5227–5237 RSC.
- L. M. Luh, U. Scheib, K. Juenemann, L. Wortmann, M. Brands and P. M. Cromm, Angew. Chem., Int. Ed., 2020, 59, 15448–15466 CrossRef CAS.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, ACS Cent. Sci., 2021, 7, 499–506 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- V. Kumar and W. B. Turnbull, Chem. Soc. Rev., 2023, 52, 1273–1287 RSC.
- E. A. L. Biessen and T. J. C. van Berkel, Arterioscler., Thromb., Vasc. Biol., 2021, 41, 2855–2865 CrossRef CAS PubMed.
- D. M. Bissell, K. E. Anderson and H. L. Bonkovsky, N. Engl. J. Med., 2017, 377, 862–872 CrossRef CAS.
- N. H. Gosselin, V. J. A. Schuck, O. Barriere, K. Kulmatycki, A. Margolskee, P. Smith and Y. He, Clin. Pharmacol. Ther., 2023, 113, 328–338 CrossRef CAS PubMed.
- A. Mishra, T. R. Castañeda, E. Bader, B. Elshorst, S. Cummings, P. Scherer, D. S. Bangari, C. Loewe, H. Schreuder, C. Pöverlein, M. Helms, S. Jones, G. Zech, T. Licher, M. Wagner, M. Schudok, M. deHoop, A. T. Plowright, J. Atzrodt, A. Kannt, I. Laitinen and V. Derdau, Adv. Sci., 2020, 7, 2002997 CrossRef CAS PubMed.
- K. Wojtczak, E. Zahorska, I. J. Murphy, F. Koppel, G. Cooke, A. Titz and J. P. Byrne, Chem. Commun., 2023, 59, 8384–8387 RSC.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS.
- J. H. Lee, G. Ozorowski and A. B. Ward, Science, 2016, 351, 1043–1048 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- A. Gabba, P. V. Murphy, L. L. Kiessling and G. Birrane, Biochemistry, 2024, 63, 191–193 CrossRef.
- L. Mousavifar and R. Roy, Drug Discovery Today, 2021, 26, 2124–2137 CrossRef CAS.
- E. Zahorska, F. Rosato, K. Stober, S. Kuhaudomlarp, J. Meiers, D. Hauck, D. Reith, E. Gillon, K. Rox, A. Imberty, W. Römer and A. Titz, Angew. Chem., Int. Ed., 2023, 6, e202215535 Search PubMed.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E.-C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140(7), 2537–2545 CrossRef CAS PubMed.
- M. Egli and M. Manoharan, Nucleic Acids Res., 2023, 51, 2529–2573 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables. See DOI: https://doi.org/10.1039/d4cs00642a |
| This journal is © The Royal Society of Chemistry 2024 |