
X-ray crystallographic analysis of the antiferromagnetic low-temperature phase of galvinoxyl: investigating magnetic duality in organic radicals†
Rie
Suizu
 ab,
Yoshiaki
Shuku
a,
Vincent
Robert
c,
Pablo
Roseiro
c,
Nadia
Ben Amor
d,
Zain
Khawar
e,
Neil
Robertson
e and
Kunio
Awaga
*a
ab,
Yoshiaki
Shuku
a,
Vincent
Robert
c,
Pablo
Roseiro
c,
Nadia
Ben Amor
d,
Zain
Khawar
e,
Neil
Robertson
e and
Kunio
Awaga
*a
aDepartment of Chemistry & Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: awaga.kunio.h8@f.mail.nagoya-u.ac.jp
bJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cUniversité de Strasbourg, CNRS, Laboratoire de Chimie Quantique UMR 7177, 67000 Strasbourg, France
dLaboratoire de Physique et Chimie Quantiques, UMR 5626 Université Paul Sabatier, 118 route de Narbonne, 31062 Toulouse, France
eSchool of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK
First published on 13th December 2023
Abstract
Galvinoxyl, as one of the most extensively studied organic stable free radicals, exhibits a notable phase transition from a high-temperature (HT) phase with a ferromagnetic (FM) intermolecular interaction to a low-temperature (LT) phase with an antiferromagnetic (AFM) coupling at 85 K. Despite significant research efforts, the crystal structure of the AFM LT phase has remained elusive. This study successfully elucidates the crystal structure of the LT phase, which belongs to the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The crystal structure of the LT phase is found to consist of a distorted dimer, wherein the distortion arises from the formation of short intermolecular distances between anti-node carbons in the singly-occupied molecular orbital (SOMO). Starting from the structure of the LT phase, wave function calculations show that the AFM coupling 2J/kB varies significantly from −1069 K to −54 K due to a parallel shift of the molecular planes within the dimer.
space group. The crystal structure of the LT phase is found to consist of a distorted dimer, wherein the distortion arises from the formation of short intermolecular distances between anti-node carbons in the singly-occupied molecular orbital (SOMO). Starting from the structure of the LT phase, wave function calculations show that the AFM coupling 2J/kB varies significantly from −1069 K to −54 K due to a parallel shift of the molecular planes within the dimer.
Introduction
Stable organic radicals have garnered significant attention in a range of research fields, encompassing biology, chemistry, and physics, due to their exceptional reactivity and physical properties arising from their open-shell electronic structures. Among the diverse array of organic radicals,1 galvinoxyl (4-[[3,5-bis(1,1-dimethylethyl)-4-oxo-2,5-cyclohexadien-1-ylidene]methyl]-2,6-bis(1,1-dimethylethyl)phenoxy, see Scheme 1), a phenoxyl radical synthesized in 1957,2,3 stands out as an extraordinarily unique case. While most stable radicals, such as nitroxyl radicals, possess a localized unpaired electron on a specific functional group, galvinoxyl exhibits a captivating characteristic: the unpaired electron is extensively delocalized along an odd alternating π conjugation, and the nodes of the singly-occupied molecular orbital (SOMO) alternately appear on the molecular skeleton (Scheme 1). Galvinoxyl has a rich research history, initially serving as an excellent sample for magnetic resonance research in solution. Electron paramagnetic resonance (EPR) spectroscopy revealed a substantial spin polarization on the molecular skeleton of galvinoxyl,4,5 and the first successful electron nuclear double resonance (ENDOR) measurements were conducted on this molecule.6 Subsequently, galvinoxyl has found utility in various applications, including as a target molecule for radical scavengers in food chemistry,7,8 an additive material in organic solar cells,9,10 and a redox-active material in rechargeable batteries.11 Notably, galvinoxyl's most remarkable features lie in its magnetic intermolecular interactions and pronounced phase transition. At room temperature, galvinoxyl exhibits ferromagnetic (FM) intermolecular interaction,12–14 distinguishing it from the majority of organic radicals that display antiferromagnetic (AFM) interactions in their solids. Additionally, galvinoxyl undergoes a first-order structural phase transition below 85 K,12–14 resulting in a change of the intermolecular magnetic coupling to AFM. Although switchable FM/AFM coupling has been observed in intermolecular systems,15,16 it is quite unique of galvinoxyl that FM and AFM intermolecular couplings switch across the phase transition. The crystal structure of the high-temperature (HT) phase was determined in 1969.17 However, the crystal structure of the low-temperature (LT) phase still eludes resolution. Pioneering efforts in single-crystal X-ray analysis faltered due to crystal disintegration during the phase transition.18 In contrast, EPR measurements indicated the formation of an AFM dimer (S = 0) in conjunction with a thermally accessible S = 1 state in the LT phase.19 This means that galvinoxyl exhibits the structural phase transition from an FM 1D chain to an AFM dimer. Given its historical significance, it is crucial to uncover the hidden aspects of galvinoxyl's dual nature, particularly its molecular and crystal structures in the LT phase. Understanding these aspects is essential for comprehending the distinct properties and reactivity exhibited by galvinoxyl.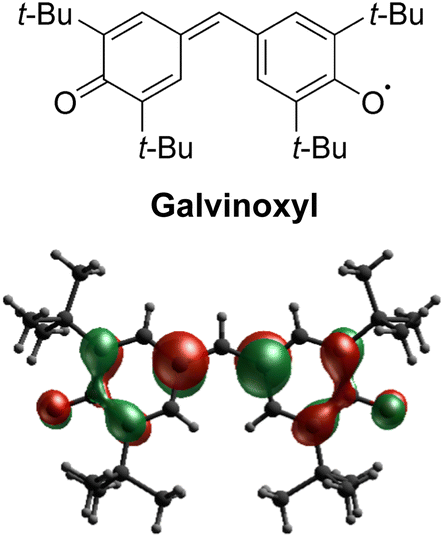 | ||
| Scheme 1 Molecular structure of galvinoxyl and its SOMO calculated from a doublet CASSCF calculation. | ||
In the present investigation, our aim was to determine the crystal structure of the LT phase of galvinoxyl. Through the diligent resolution of various technical challenges, we ultimately achieved success in the structural analysis. The results obtained settled a long-standing unresolved issue of more than 30 years, unveiling a radical dimer structure that is formed by intermolecular, interatomic contacts among the anti-node carbons.
Results and discussion
Crystal structure of HT phase
The galvinoxyl radical was prepared employing the procedure described in the literature,2,3 and its needle-shaped crystals were obtained via recrystallization from solutions in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol/diethyl ether mixed solvent at −20 °C. Fig. S1† illustrates the temperature dependence of the paramagnetic susceptibility χp, which is in agreement with previously reported data.12–14 Specifically, in the HT phase, χp conforms to the Curie–Weiss law with a Curie constant of 0.374 emu K mol−1 and a Weiss constant of 12.2 K. In contrast, in the LT phase, χp is substantially diminished.
:1 methanol/diethyl ether mixed solvent at −20 °C. Fig. S1† illustrates the temperature dependence of the paramagnetic susceptibility χp, which is in agreement with previously reported data.12–14 Specifically, in the HT phase, χp conforms to the Curie–Weiss law with a Curie constant of 0.374 emu K mol−1 and a Weiss constant of 12.2 K. In contrast, in the LT phase, χp is substantially diminished.
We conducted a re-examination of the crystal structure in the HT phase at 123 K for the purpose of comparison with the LT phase. The results were consistent with the reported ones.17,18 The lattice parameters for the HT phase are listed in Table S1.† This phase belongs to the monoclinic I2/a space group.‡Fig. 1a depicts the molecular structure in this phase, where a C2 axis is present, making half of this unit crystallographically independent. The selected bond distances are listed in Table S2.† The π molecular plane displays notable planarity, while the phenoxyl ring manifests bond alternation akin to that observed in benzoquinone. Notably, within the three methyl groups of the t-butyl moiety, two form intramolecular hydrogen bonds with the oxygen atom, while the C–C bond in the remaining methyl group lies coplanar with the phenoxyl ring. This characteristic is also evident in the molecular structures of mono-, di-, and tri-substituted t-butylbenzenes (refer to Fig. S3†), suggesting a probable steric effect arising from the hydrogen atoms in the methyl group and the hydrogen atoms on the phenoxyl ring. In Fig. 2a, the presence of a one-dimensional (1D) π-stacking chain in the HT phase is apparent, featuring intermolecular and interatomic distance of 3.903 Å for C6 and C6 (red dotted line). Fig. 3a depicts the intermolecular arrangement within the 1D chain of the HT phase. The discussion regarding the magneto-structural correlation will be presented subsequently.
 | ||
| Fig. 1 Comparison between the molecular structures of galvinoxyl in the HT (a) and LT (b) phases. While half of the molecule is crystallographically independent in the HT phase, two molecules are crystallographically independent in the LT phase. | ||
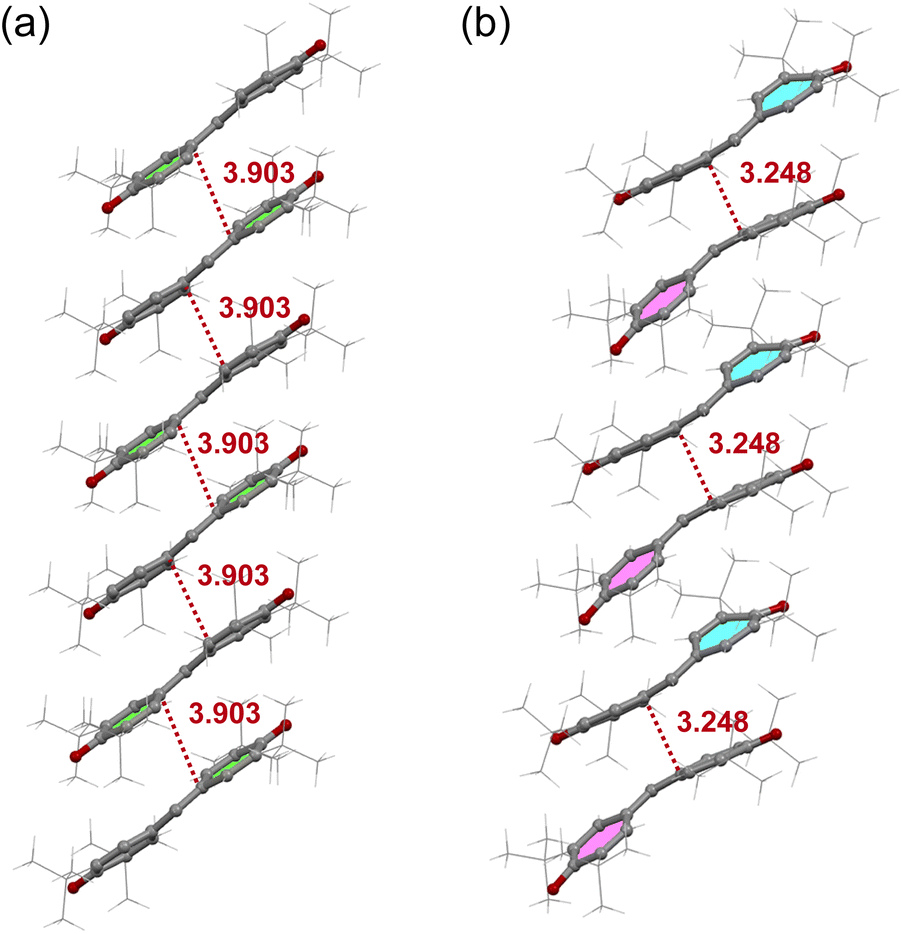 | ||
| Fig. 2 1D stacking of galvinoxyl in the HT (a) and LT (b) phases. | ||
 | ||
| Fig. 3 Nearest neighbor intermolecular arrangements in the HT (a) and LT (b) phases. | ||
Space-filling views of the intermolecular arrangement in the HT phase are provided in Fig. 4. It is likely that the intermolecular arrangement in the HT phase is determined to avoid steric hindrance between the bulky t-butyl groups. This suggests that the intermolecular arrangement in the HT phase is not caused by the FM interaction in this phase, but rather is obtained accidentally due to the steric hindrance between the t-butyl groups. This is supported by the fact that galvinoxyl can form isostructural mixed crystals with its corresponding closed-shell compound, hydrogalvinoxyl.18
 | ||
| Fig. 4 Space-filling views for the intermolecular arrangements in the HT phase. | ||
Crystal structure of LT phase
We conducted X-ray crystallographic analysis of the LT phase of galvinoxyl at 50 K using an X-ray diffractometer equipped with a He-gas open-flow cryostat. Initially, we opted for large single crystals for the experiments. However, during the cooling and/or at the phase transition, microcrystals detached from the parent crystal and remained on the crystal surface, compromising the diffraction data. To address this issue, we selected a flake-shaped crystal, which resulted in the detachment of split microcrystals from the parent crystal. Consequently, we obtained diffraction data exclusively from this flake-shaped crystal. Twinning of the crystals due to the phase transition also posed a challenge. Nonetheless, we successfully tackled this obstacle by employing structural analysis that considered twinning. Through these approaches, we achieved the resolution and refinement of the crystal structure of the LT phase.The crystal parameters for the LT phase are presented in Table S1.† This phase belongs to the triclinic P space group, featuring two crystallographically independent molecules. The structures of these molecules, designated as A and B, are illustrated in Fig. 1b. Table S2† provides the selected bond distances. In comparison to the molecular structure observed in the HT phase, the π skeletons of molecules A and B in the LT phase exhibit more pronounced bending. However, there are no significant differences in the corresponding bond distances among the molecules A and B in the LT phase compared to those in the HT phase. It is worth noting, however, that one t-butyl group in molecule B deviates from the coplanar rule. Fig. 2b depicts the distinct alternation observed in the stacking chain of the LT phase.
Fig. 3 illustrates a comparison of the nearest neighbor intermolecular arrangements observed in the HT and LT phases. In the HT one, the intermolecular distance between C6 and the midpoint between C4 and C5 (3.809 Å) of the neighboring molecule is shorter than that between C6 position (3.903 Å) (see Fig. 3a). Since galvinoxyl is an odd-alternant π conjugated compound, the SOMO manifests nodes at C1, C4, and C5. Therefore, the short distance between C6 and C4 or C5 indicates an overlap between the node and anti-node carbons (Type-A in Fig. S6†).20 This and absence of short distance between anti-node carbon atoms suggests a nearly orthogonal relationship between the SOMOs and a FM coupling, resulting from the potential exchange interaction. In contrast, the nearest-neighbor intermolecular arrangement in the LT phase reveals a short interatomic distance of 3.248 Å between C6A and C6B (Fig. 3b), implying close contact between anti-node carbons, which is expected to induce an AFM coupling (Type-B in Fig. S6†).
Magneto-structural correlation
While the existence of FM coupling was confirmed by ab initio calculations,21 we performed CAS[2,2]SCF + DDCI3 wavefunction calculations to estimate the energy differences between the S = 0 and S = 1 states ΔST, utilizing simplified nearest-neighbor dimer geometries in the two phases (see SI). This value is correlated to the exchange coupling constant J, as ΔST = 2J. For the HT phase, the calculation concluded an S = 1 ground state with 2J/kB = 36 K. This value is in reasonably good agreement with the positive Weiss constant, and is slightly larger than the value obtained by the EPR measurement (17 ± 8 K).19 On the other hand, the calculations for the LT structure concluded on an S = 0 ground state with 2J/kB = −1069 K, which is nearly the double of the EPR data (−580 K).19 To comprehend this disparity, we computed ΔST for hypothetical dimer structures, wherein one molecule of the dimer in the LT phase was incrementally shifted in parallel (see the inset in Table S4†). The outcomes are presented in Table S4,† where the structure at δ = 0 Å represents the LT phase. Notably, a minor shift of Δδ ≈ 0.6 Å leads to a substantial 2J/kB alteration by a factor of twenty. Taking into account this sensitivity to the structure, experimental errors, and temperature effects,21,22 the current calculations provide a reasonable approximation of the experimental results. Fig. 5 illustrates the isoelectronic density surfaces of the highest occupied molecular orbital (HOMO), representing the bonding orbital between the SOMOs. | ||
| Fig. 5 HOMO of the galvinoxyl dimer in the LT phase. | ||
Conclusions
In conclusion, we have successfully resolved a long-standing problem related to the extensively studied galvinoxyl radical, which had remained unsolved. While it is known that this molecule exhibits FM and AFM interactions in the HT and LT phases, respectively, we were able to elucidate the crystal structure of the AFM LT phase. In contrast to the high symmetry of the HT phase, which belongs to the I2/a space group, the symmetry of the LT phase dropped to the P space group. Moreover, the FM 1D chains in the HT phase underwent noticeable dimerization in the LT phase, leading to a distorted geometry in the LT phase dimer. This distortion was attributed to the formation of short distances between the anti-node carbons in the SOMO. The wavefunction calculations for the LT phase dimer indicated a strong and structural-sensitive AFM coupling 2J/kB that varies by more than one order of magnitude by a parallel shift of Δδ ≈ 0.6 Å. These findings shed light on the unique properties and interactions of the galvinoxyl radical in different phases, providing valuable insights for further research in this field.
Author contributions
R. S. carried out single crystal structure analysis. Y. S. and Z. K. performed the synthesis and magnetic measurements. V. R., P. R., and N. B. A. carried out theoretical model calculations. All authors contributed to writing and reviewing the manuscript. N. R. and K. A. directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was conducted in Institute for Molecular Science, supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Proposal Number JPMXP1222MS1089. The authors acknowledge Japan Society for the Promotion of Science (JSPS) KAKENHI Grants 19K15520 (to Y. S.), 20H02707 (to R. S.) and 20H05621 (K. A.), and Japan Science and Technology Agency (JST) PRESTO Grant JPMJPR21A9 (to R. S.) for funding this work.References
- Z. X. Chen, Y. Li and F. Huang, Chem., 2021, 7, 288–332 CAS.
- G. M. Coppinger, J. Am. Chem. Soc., 1957, 79, 501–502 CrossRef CAS.
- M. S. Kharasch and B. S. Joshi, J. Org. Chem., 1957, 22, 1435–1438 CrossRef CAS.
- C. Besev, A. Lund, T. Vänngård and R. Håkansson, Acta Chem. Scand., 1963, 17, 2281–2284 CrossRef CAS.
- G. R. Luckhurst, Mol. Phys., 1966, 11, 205–211 CrossRef CAS.
- J. S. Hyde and A. H. Maki, J. Chem. Phys., 1964, 40, 3117–3118 CrossRef CAS.
- T. Esatbeyoglu, A. E. Wagner, R. Motafakkerazad, Y. Nakajima, S. Matsugo and G. Rimbach, Food Chem. Toxicol., 2014, 73, 119–126 CrossRef CAS PubMed.
- J. Dose, S. Matsugo, H. Yokokawa, Y. Koshida, S. Okazaki, U. Seidel, M. Eggersdorfer, G. Rimbach and T. Esatbeyoglu, Int. J. Mol. Sci., 2016, 17, 103 CrossRef PubMed.
- Y. Zhang, T. P. Basel, B. R. Gautam, X. M. Yang, D. J. Mascaro, F. Liu and Z. V. Vardeny, Nat.Commun., 2012, 3, 1043 CrossRef PubMed.
- T. Basel, U. Huynh, T. Zheng, T. Xu, L. Yu and Z. V. Vardeny, Adv. Funct. Mater., 2015, 25, 1895–1902 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- K. Awaga, T. Sugano and M. Kinoshita, J. Chem. Phys., 1986, 85, 2211–2218 CrossRef CAS.
- K. Mukai, H. Nishiguchi and Y. Deguchi, J. Phys. Soc. Jpn., 1967, 23, 125 CrossRef CAS.
- K. Mukai, Bull. Chem. Soc. Jpn., 1969, 42(1), 40–46 CrossRef CAS.
- A. Izuoka, S. Murata, T. Sugawara and H. Iwamura, J. Am. Chem. Soc., 1987, 109, 2631–2639 CrossRef CAS.
- A. Rajca, K. Lu, S. Rajca and C. R. Ross II, Chem. Commun., 1999, 1249–1250 RSC.
- D. E. Williams, Mol. Phys., 1969, 16, 145–151 CrossRef CAS.
- K. M. Chi, J. C. Calabrese, J. S. Miller and S. I. Khan, Mol. Cryst. Liq. Cryst., 1989, 176, 185–198 CrossRef CAS.
- K. Awaga, T. Sugano and M. Kinoshita, Chem. Phys. Lett., 1986, 128, 587–590 CrossRef CAS.
- K. Awaga, T. Sugano and M. Kinoshita, Chem. Phys. Lett., 1987, 141, 540–544 CrossRef CAS.
- S. J. Luo and K. L. Yao, J. Magn. Magn. Mater., 2003, 257, 11–14 CrossRef CAS.
- A. A. Leitch, X. Yu, S. M. Winter, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2009, 131, 7112–7125 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the magnetic data, crystal structure analysis, and MO calculations. CCDC 2296534 and 2296535. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03601d |
| ‡ Note that in the previous reports, the structure had been designated as monoclinic C2/c (ref. 17 and 18). While the space group remains consistent (#15), IUCr defines the standard orientation of monoclinic crystal system to take β closer to 90 degrees. (https://journals.iucr.org/services/cif/checking/PLAT128.html), and a recent recommendation advises adopting the axis orientation of I2/a. |
| This journal is © The Royal Society of Chemistry 2024 |