
Tuning valence tautomerism in a family of dinuclear cobalt complexes incorporating a conjugated bridging bis(dioxolene) ligand with weak communication†
Tristan E.
Fischer
a,
Jett T.
Janetzki
 a,
F. Zahra
M. Zahir
a,
Robert W.
Gable
a,
Alyona A.
Starikova
b and
Colette
Boskovic
*a
a,
F. Zahra
M. Zahir
a,
Robert W.
Gable
a,
Alyona A.
Starikova
b and
Colette
Boskovic
*a
aSchool of Chemistry, University of Melbourne, Parkville, 3010 Victoria, Australia. E-mail: c.boskovic@unimelb.edu.au
bInstitute of Physical and Organic Chemistry, Southern Federal University, 344090, Rostov-on-Don, Russian Federation
First published on 8th January 2024
Abstract
Valence tautomerism (VT) involves the stimulated reversible intramolecular electron transfer between a redox-active metal and ligand. Dinuclear cobalt complexes bridged by bis(dioxolene) ligands can undergo thermally-induced VT with access to {CoIII-cat-cat-CoIII}, {CoIII-cat-SQ-CoII} and {CoII-SQ-SQ-CoII} states (cat2− = catecholate, SQ˙− = semiquinonate, CoIII refers to low spin CoIII, CoII refers to high spin CoII). The resulting potential for two-step VT interconversions offers increased functionality over mononuclear examples. In this study, the bis(dioxolene) ligand 3,3′,4,4′-tetrahydroxy-5,5′-dimethoxy-benzaldazine (thMH4) was paired with Mentpa (tpa = tris(2-pyridylmethyl)amine, n = 0–3 corresponds to methylation at 6-position of the pyridine rings) to afford [{Co(Mentpa)}2(thM)](PF6)2 (1a, n = 0; 2a, n = 2; 3a, n = 3). Structural, magnetic susceptibility and spectroscopic data show that 1a and 3a remain in the temperature invariant {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} forms in the solid state, respectively. In contrast, 2a exhibits incomplete thermally-induced VT between these two tautomeric forms via the mixed {CoIII-cat-SQ-CoII} tautomer. In solution, room temperature electronic absorption spectra are consistent with the assignments from the solid-state, with VT observed only for 2a. From electrochemistry, the proximity of the two 1e−-processes for the thMn− ligand indicates weak electronic communication between the two dioxolene units, supporting the potential for a two-step VT interconversion in thMn− containing complexes. Comparison of the redox potentials of the Co and thMn− processes suggests that only 2a has these processes in sufficient proximity to afford the thermally-induced VT observed experimentally. Density functional theory calculations are consistent with the prerequisite energy ordering for a two-step transition for 2a, and temperature invariant {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} states for 1a and 3a, respectively. This work presents the third example, and the first formally conjugated example, of a bridging bis(dioxolene) ligand that can afford two-step VT in a Co complex, suggesting new possibilities towards applications based on multistep switching.
Introduction
Valence tautomerism (VT) involves a reversible stimulated intramolecular electron transfer between a redox-active metal and redox-active ligand. The ability for valence tautomeric complexes to interconvert between distinct electronic states confers potential applications in displays, sensors or molecular electronics.1–3 Cobalt complexes that incorporate dioxolene (diox) ligands in the catecholate (cat2−) or semiquinonate (SQ˙−) state are the most widely studied examples of VT. In Co-dioxolene VT systems, a low spin Co(III) center coordinated to a catecholate ligand (LS-CoIII-cat) undergoes an intramolecular electron transfer and concerted spin transition, affording a high spin Co(II)-semiquinonate (HS-CoII-SQ) tautomer. Valence tautomerism in Co-dioxolene systems is entropically driven and can be induced by temperature, light, pressure, or magnetic fields and results in changes to the magnetic moment, color, structure, and polarization.1Complexes with multiple metal centers offer the possibility of multi-step switching between three or more electronic states, potentially enabling more complex logic processes in molecular spintronics and electronics and ternary data storage.4–9 Well reported examples of multiswitchability are provided by dinuclear Fe(II) complexes that undergo two-step spin crossover (SCO).10,11 Two-step SCO is also observed for mononuclear compounds in which symmetry breaking results in two or more inequivalent SCO centers at an intermediate phase.12–14 Two-step valence tautomerism has only been observed for two dinuclear cobalt complexes bridged by bis(dioxolene) ligands.15–18 The first reported example was [{Co(Me2tpa)}2(spiro)](ClO4)2 (spiroH4 = 3,3,3′,3′-tetramethyl-1,1′-spirobi(indan)-5,5′,6,6′-tetraol, Me2tpa = bis(6-methyl-2-pyridylmethyl)(2-pyridylmethyl)amine).15,16 This complex displays a two-step VT interconversion in both the solid- and solution-states, indicating a molecular phenomenon, involving the transition {LS-CoIII-cat-cat-LS-CoIII} ⇌ {HS-CoII-SQ-cat-LS-CoIII} ⇌ {HS-CoII-SQ-SQ-HS-CoII} (CoIII refers to low spin (LS) CoIII, CoII refers to high spin (HS) CoII).16 A detailed follow up study established a range of thermodynamic criteria for bis(dioxolene) cobalt complexes that governs the nature of the VT transition;17 the matching of the cobalt and dioxolene redox potentials and the degree of electronic communication within the bis(dioxolene) ligand. Unsuitable cobalt/dioxolene redox matching leads to temperature invariant charge distributions.16,17,19 Too strong communication leads to a partial or incomplete transition,17,20 whereas if this is too weak, a concerted transition is observed.20,21
A useful guiding principle for achieving two-step VT is to use a bis(dioxolene) ligand with sufficiently weak, but non-zero, intramolecular electronic communication, paired with an appropriately chosen ancillary ligand. A correlation of parameters obtained from electrochemical measurements: Δox-red (separation between the first one electron oxidation and first one electron reduction when one process is metal- and the other ligand-based) and Δdiox (separation between successive bis(dioxolene) ligand processes cat2−–cat2−/cat2−–SQ˙− and cat2−–SQ˙−/SQ˙−–SQ˙−) provides a phase-like diagram to map examples of cobalt bis(dioxolene) complexes and their associated VT behaviours (Fig. S1†).17 This affords a predictive tool for targeting two-step VT complexes, indicating that bis(dioxolene) ligands with Δdiox between 50 and 200 mV should be utilised. The compound [{Co(bpy)2}2(thM)]2+ (bpy = 2,2′-bipyridine, thMH4 = 3,3′,4,4′-tetrahydroxy-5,5′-dimethoxy-benzaldazine) was reported to undergo thermally-induced VT in MeCN.19 However, bpy over-stabilises the LS-Co(III) center such that only the onset of the transition was observed up to 353 K. The thM4− ligand appears to have sufficiently weak electronic communication and therefore a small Δdiox (∼120 mV) that has the potential to support two-step VT.17,19 We hypothesised that replacement of bpy with an ancillary ligand that increases the stabilisation of HS-Co(II) should afford a two-step VT complex. We therefore targeted dinuclear cobalt complexes bridged by thM4− with the Mentpa ancillary ligands (tpa = tris(2-pyridylmethyl)amine, n = 0–3 corresponds to methylation of the 6-position of the pyridine rings), which have been used to access numerous mononuclear and dinuclear Co-dioxolene VT compounds.16,17,22,23 The target complexes [{Co(tpa)}2(thM)]2+ (12+) [{Co(Me2tpa)}2(thM)]2+ (22+), and [{Co(Me3tpa)}2(thM)]2+ (32+) were isolated as the PF6− salts (1a, 2a, 3a, Chart 1) and analyzed using structural, magnetic, spectroscopic, electrochemical and computational methods.
 | ||
| Chart 1 Dinuclear cobalt complexes [{Co(tpa)}2(thM)]2+ (12+), [{Co(Me2tpa)}2(thM)]2+ (22+), and [{Co(Me3tpa)}2(thM)] (32+) isolated as the PF6− salts in 1a, 2a and 3a, respectively; R = H or Me for Mentpa; n = 0, 2 or 3. | ||
Results and discussion
Synthesis
The compounds [{Co(tpa)}2(thM)](PF6)2 (1a), [{Co(Me2tpa)}2(thM)](PF6)2 (2a), [{Co(Me3tpa)}2(thm)](PF6)2 (3a) (Chart 1) were synthesised under a N2 atmosphere by reacting two equivalents of cobalt(II) chloride and the Mentpa ancillary ligand, to preform {CoII(Mentpa)}2+, followed by addition of one equivalent of thMH4 deprotonated with four equivalents of triethylamine (Et3N). Addition of two equivalents of ferrocenium hexafluorophosphate (FcPF6) to the suspension resulted in visible color changes as oxidation occurred and resulted in formation of the family [{Co(Mentpa)}2thM](PF6)2 (n = 0, 2, 3). Layering an acetonitrile (MeCN) solution of 1a with toluene resulted in diffraction quality crystals, analyzed as 1a·2toluene·2MeCN, with the bulk sample obtained in a similar manner. Upon collection, the sample partially desolvates and H2O is adsorbed. Elemental analysis (EA) and thermogravimetric analysis (TGA, Fig. S2†) data confirm the formula 1a·0.5MeCN·H2O for the bulk sample. The powder X-ray diffraction (PXRD) pattern for 1a·0.5MeCN·H2O differs slightly from the simulated pattern for the 1a·2toluene·2MeCN crystal structure (Fig. S3†), consistent with solvent exchange. Single crystals of compound 2a were attained by layering a dichloromethane (CH2Cl2) solution with diethyl ether (Et2O), analyzing as 2a·1.8CH2Cl2. Single crystals can also be grown from layering CH2Cl2 with hexane; however, the crystals were too weakly diffracting to obtain a quality crystal structure. Attempts to recrystallise 2a for a bulk sample by layering CH2Cl2 with either Et2O or hexane led to a mixture of crystalline and amorphous solid. Instead, microcrystalline bulk sample was obtained by the slow addition of Et2O or hexane to a rapidly stirring concentrated CH2Cl2 solution to yield 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane, respectively, as confirmed by EA and TGA (Fig. S2†). Single crystals of 3a were grown by layering an acetone solution with hexane, analyzing as 3a·2acetone. Like 2a, a bulk sample was obtained by adding hexane to a stirred acetone solution, which affords a dark green crystalline solid, analyzed as 3a·0.5acetone using EA and TGA (Fig. S2†). Although consistent with diffractograms calculated from the single crystal, PXRD data indicate that the bulk samples of 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane and 3a·0.5acetone are poorly crystalline (Fig. S3†) due to rapid precipitation.Structure description
The solid-state structures of 1a·2toluene·2MeCN, 2a·1.8CH2Cl2, and 3a·2acetone were determined by single crystal X-ray diffraction at 100 K (Fig. 1 and Table S1†). Compound 1a·2toluene·2MeCN crystallises as dark green rectangular crystals in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with half a molecule of [{Co(tpa)}2(thM)]2+ and one PF6− in the asymmetric unit. The remaining cavities in the asymmetric unit are filled with solvent; one molecule of toluene and one molecule of MeCN. Compound 2a·1.8CH2Cl2 crystallises in the monoclinic P21/n space group as brown needles, with the asymmetric unit consisting of half a molecule of [{Co(Me2tpa)}2(thM)]2+, one PF6− and a disordered CH2Cl2 molecule. Compound 3a·2acetone also crystallises in the P21/n space group as dark green plates, with half a [{Co(Me3tpa)}2(thM)]2+, one PF6− and an acetone molecule in the asymmetric unit. In 1a·2toluene·2MeCN, 2a·1.8CH2Cl2, and 3a·2acetone, each six-coordinate cobalt center is coordinated by one Mentpa ligand and bridged by thMn− (Fig. 1). The two halves of the dinuclear complex are related by inversion. For compound 2a·1.8CH2Cl2, the two methyl groups of Me2tpa are approximately evenly disordered over three sites. Due to the inequivalence of the O1 and O2 oxygen atoms in thMn−, geometric isomerisation is possible; O1 is in closer proximity to the methoxy group on the ring (proximal), whilst O2 is further away (distal). In all three of 1a·2toluene·2MeCN, 2a·1.8CH2Cl2, and 3a·2acetone, as the distal O2 oxygen are trans to the tertiary-amine nitrogen (N1) of the Mentpa ligand at both cobalt centers, the isomer is distal–distal. This likely occurs to minimise the steric clash between the methoxy group and the pyridine ring containing N2.
space group with half a molecule of [{Co(tpa)}2(thM)]2+ and one PF6− in the asymmetric unit. The remaining cavities in the asymmetric unit are filled with solvent; one molecule of toluene and one molecule of MeCN. Compound 2a·1.8CH2Cl2 crystallises in the monoclinic P21/n space group as brown needles, with the asymmetric unit consisting of half a molecule of [{Co(Me2tpa)}2(thM)]2+, one PF6− and a disordered CH2Cl2 molecule. Compound 3a·2acetone also crystallises in the P21/n space group as dark green plates, with half a [{Co(Me3tpa)}2(thM)]2+, one PF6− and an acetone molecule in the asymmetric unit. In 1a·2toluene·2MeCN, 2a·1.8CH2Cl2, and 3a·2acetone, each six-coordinate cobalt center is coordinated by one Mentpa ligand and bridged by thMn− (Fig. 1). The two halves of the dinuclear complex are related by inversion. For compound 2a·1.8CH2Cl2, the two methyl groups of Me2tpa are approximately evenly disordered over three sites. Due to the inequivalence of the O1 and O2 oxygen atoms in thMn−, geometric isomerisation is possible; O1 is in closer proximity to the methoxy group on the ring (proximal), whilst O2 is further away (distal). In all three of 1a·2toluene·2MeCN, 2a·1.8CH2Cl2, and 3a·2acetone, as the distal O2 oxygen are trans to the tertiary-amine nitrogen (N1) of the Mentpa ligand at both cobalt centers, the isomer is distal–distal. This likely occurs to minimise the steric clash between the methoxy group and the pyridine ring containing N2.
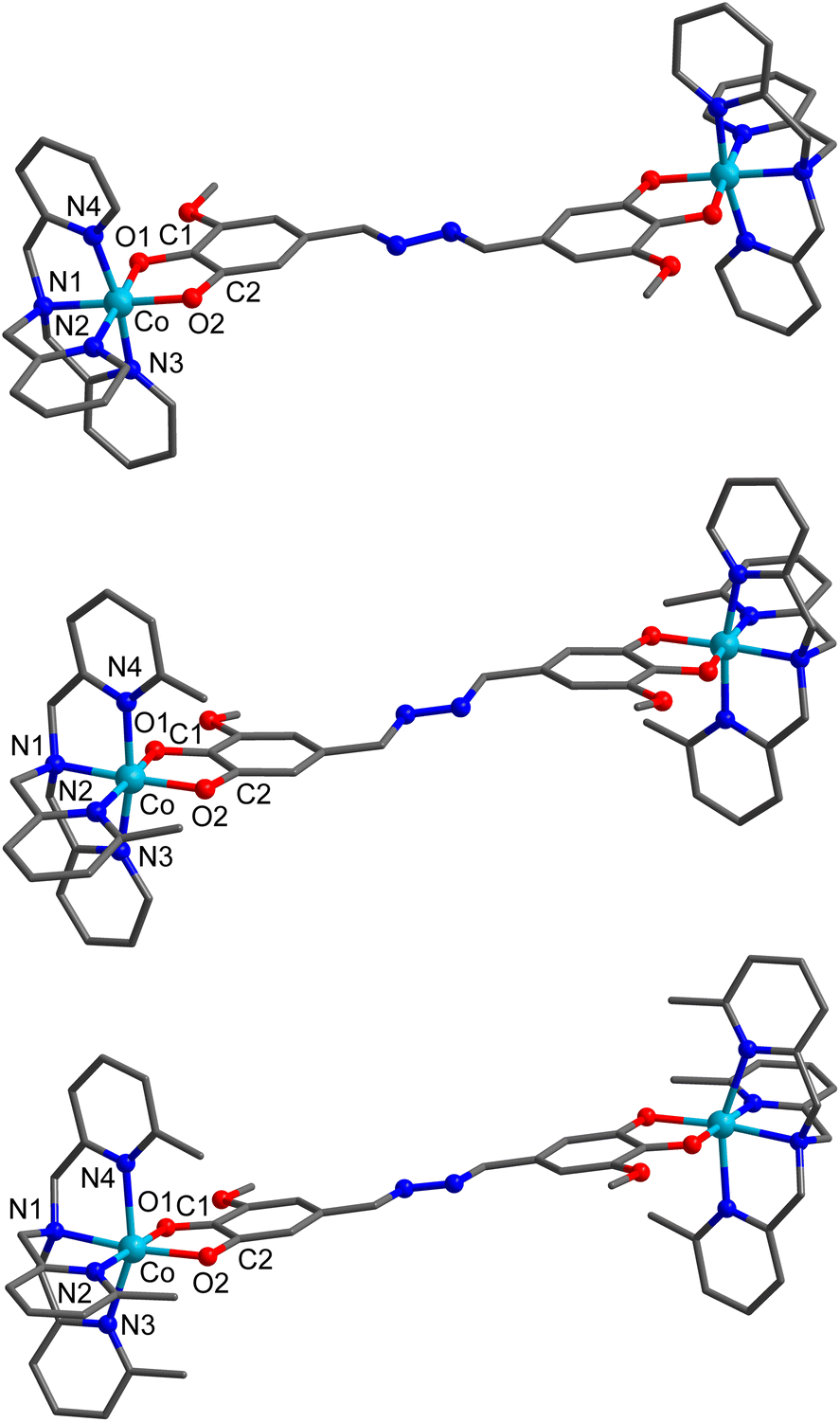 | ||
| Fig. 1 Complex cations in 1a·2toluene·2MeCN (top), 2a·1.8CH2Cl2 (middle), and 3a·2acetone (bottom) collected at 100 K. Hydrogen atoms have been omitted for clarity. Color code: C (dark grey), N (blue), O (red), Co (light blue). | ||
The oxidation state of the cobalt centers can be determined from the Co–N and Co–O bond lengths and octahedral distortion parameters (octahedral SHAPE, Σ, Θ).24,25 The SHAPE index calculated by SHAPE 2.1 represents the distortion of the coordination sphere from an ideal polyhedron,24,26 and the Σ and Θ parameters calculated using OctaDist represent the angles of distortion in an octahedron.25 In general, Co–O/N bond lengths and octahedral distortions increase following the trend LS-CoIII < LS-CoII < HS-CoII.27,28 Typical Co–O, Co–Namine, and Co–Npyridine bond lengths for CoIII/CoII with Mentpa ligands are 1.85–1.91/1.99–2.11, 1.91–1.97/2.09–2.13 and 1.87–2.03/2.14–2.29 Å respectively.16,17,22,23 The typical octahedral SHAPE index for CoII and CoIII with Mentpa and a dioxolene ligand are 1.4–1.8 and 0.1–0.5, respectively.17,23 The oxidation state of the dioxolene moiety can be determined via the C–C and C–O bond lengths,29 such that the least-squares fit aids in the determination of an apparent metrical oxidation state (MOS);30 catecholate ligands have a MOS of around −2 with longer C–O and shorter C1–C2 bond lengths, whilst semiquinonate ligands have a MOS around −1 with shorter and longer C–O and C1–C2 bond lengths, respectively.
At 100 K, 1a·2toluene·2MeCN and 2a·1.8CH2Cl2 have short Co–O/N bond distances and low octahedral distortion (Table 1), consistent with LS-Co(III). These data, together with the calculated MOS values (Table 1), suggest both 1a·2toluene·2MeCN and 2a·1.8CH2Cl2 adopt the {CoIII-cat-cat-CoIII} charge distribution at 100 K. For compound 3a·2acetone, elongated Co–O/N bond lengths and increased octahedral distortion (Table 1) now indicate of HS-Co(II), with MOS reflective of SQ˙−. Therefore at 100 K, 3a·2acetone exists as {CoII-SQ-SQ-CoII}. Overall, structural analysis at 100 K suggests that 1a·2toluene·2MeCN and 2a·1.8CH2Cl2 exist in the {CoIII-cat-cat-CoIII} state, whereas 3a·2acetone adopts a {CoII-SQ-SQ-CoII} charge distribution.
| 1a·2toluene·2MeCN | 2a·1.8CH2Cl2 | 3a·2acetone | |
|---|---|---|---|
| a SHAPE index for octahedral geometry in SHAPE 2.1.24,26 A value of 0 represents a perfect octahedron. b Σ = sum of the deviation of the 12 N/O–Co–N/O angles from 90°. Θ = sum of the deviation of 24 unique torsional angles between the N/O atoms on opposite triangular faces of the octahedron from 60°, providing the degree of trigonal distortion from an octahedron to trigonal prism. These were calculated using OctaDist – a program for determining the structural distortion of the octahedral complexes. For a perfect octahedron, Σ and Θ are zero.25 | |||
| Interatomic distances/Å | |||
| Co–O1 | 1.877(2) | 1.894(3) | 2.171(2) |
| Co–O2 | 1.880(2) | 1.874(3) | 1.988(2) |
| Co–N1 | 1.955(2) | 1.948(4) | 2.129(2) |
| Co–N2 | 1.929(3) | 1.983(4) | 2.163(2) |
| Co–N3 | 1.926(3) | 1.987(4) | 2.227(2) |
| Co–N4 | 1.932(3) | 1.977(4) | 2.243(2) |
| C1–O1 | 1.342(4) | 1.343(5) | 1.252(3) |
| C2–O2 | 1.357(4) | 1.346(6) | 1.293(3) |
| Co⋯Co | 15.233(7) | 15.807(4) | 16.207(5) |
| Distortion parameters | |||
| SHAPE (Oh)a | 0.167 | 0.306 | 1.545 |
Σ/°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
31.7 | 45.6 | 93.6 |
|
Θ/°b |
84.9 | 110.0 | 250.7 |
| MOS | −1.8(1) | −1.9(1) | −0.88(7) |
Infrared spectroscopy
Infrared (IR) spectra of 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane and 3a·0.5acetone were acquired in the solid state (ATR; 4000–400 cm−1; Fig. S4 and S5†), with tabulated data and assignments in Table S2.† The IR spectra confirm that 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane are isostructural and differ only by lattice solvation (Fig. S5†). Previous density functional theory (DFT) and transient IR studied determined the key bands associated with catecholate and semiquinonate oxidation states.31,32 Compound 1a·0.5MeCN·H2O displays characteristics catecholate bands between 1200–1400 cm−1 (1248, 1296, 1335, 1428 cm−1) and a lack of semiquinonate bands, suggesting thM4− is in the cat2–cat2− oxidation state at room temperature. Compounds 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane also show absorption bands consistent with a dominant cat2–cat2− oxidation state at room temperature (1249, 1296, 1340, 1428 cm−1). However, there is a band at 1455 cm−1 that arises from a semiquinonate ligand. The IR spectrum of compound 3a·0.5acetone displays the characteristic semiquinonate peak at 1455 cm−1, as well as no obvious catecholate peaks, suggesting a SQ˙−–SQ˙− oxidation state of thM2− at room temperature. Following 12+ > 22+ > 32+, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations location around 1580–1600 cm−1 moves to lower wavenumbers with decreased intensity and separation as the thMn− oxidation state shifts from cat2−–cat2− to SQ˙−–SQ˙−.
C vibrations location around 1580–1600 cm−1 moves to lower wavenumbers with decreased intensity and separation as the thMn− oxidation state shifts from cat2−–cat2− to SQ˙−–SQ˙−.
Magnetic measurements
Variable temperature magnetic susceptibility data for 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane and 3a·0.5acetone are plotted as χMT vs. T (Fig. 2 and Fig. S6†), where χM is the molar magnetic susceptibility. Variable-field magnetisation measurements are presented in Fig. S7.† Compounds 1a·0.5MeCN·H2O, 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane were measured from 300–1.8 K, followed by warming to 400 K, followed by a subsequent cooling to 1.8 K and warming to 400 K (Fig. S6†). Compound 3a·0.5acetone was measured from 300 to 1.8 K, followed by warming to 300 K and cooling to 1.8 K (Fig. S6†).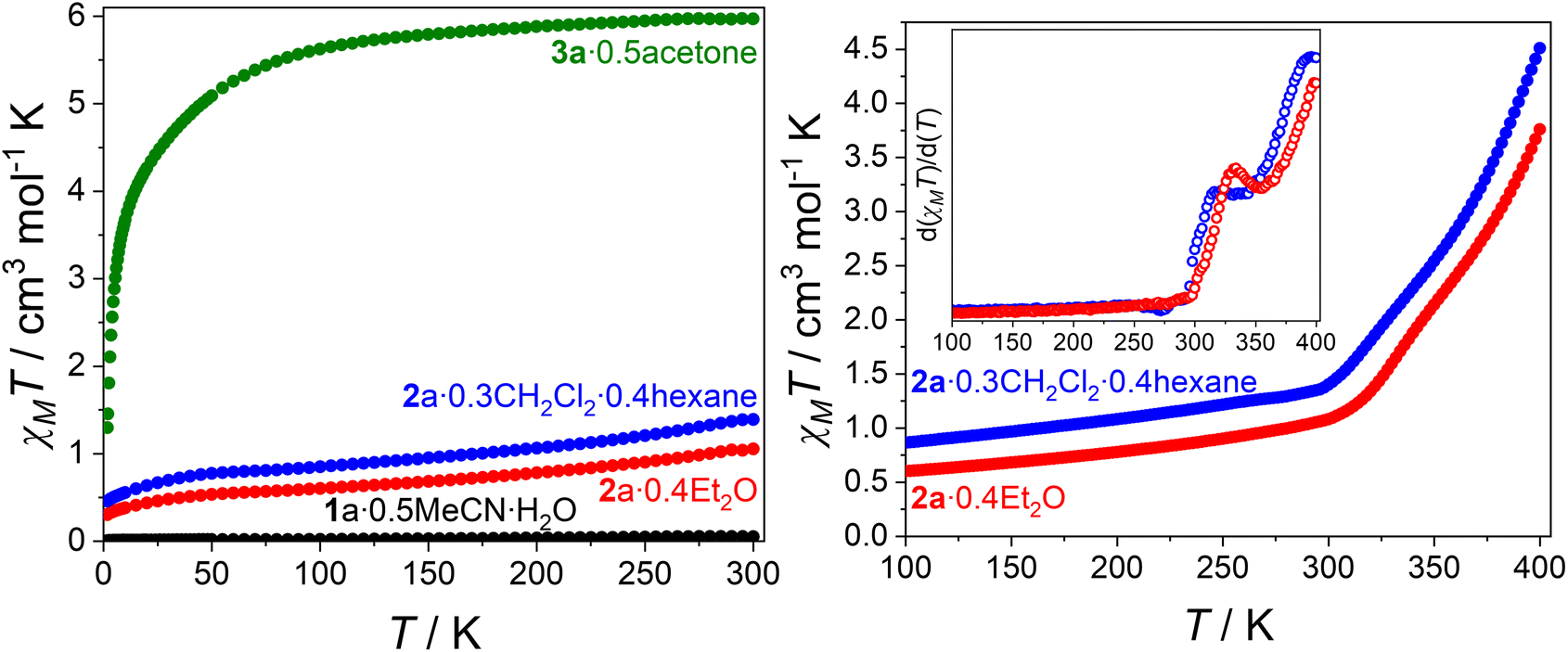 | ||
| Fig. 2 (left) Plots of χMT vs. T for the first cooling cycle between 300 and 1.8 K of 1a·0.5MeCN·H2O (black), 2a·0.4Et2O (red), 2a·0.3CH2Cl2·0.4hexane (blue) and 3a·0.5acetone (green) in 5 K steps. (right) Plots of χMT vs. T for the first heating cycle between 1.8 and 400 K for 2a·0.4Et2O (red), 2a·0.3CH2Cl2·0.4hexane (blue) (shown between 100 and 400 K) in 2 K steps. Inset: first derivative plot d(χMT)/d(T) for 2a·0.4Et2O (red), 2a·0.3CH2Cl2·0.4hexane (blue), highlighting the two-step VT behaviour and the plateau temperature. | ||
The χMT value for compound 1a·0.5MeCN·H2O (Fig. 2) remains a constant 0.0–0.05 cm3 mol−1 K between 300 and 1.8 K, indicating a temperature invariant {CoIII-cat-cat-CoIII} state, consistent with structural data. Heating to 400 K causes a small increase to 0.2 cm3 mol−1 K (Fig. S6†), which is attributed to structural changes upon solvent loss. Compound 3a·0.5acetone exhibits a constant χMT value of 5.6–6.0 cm3 mol−1 K between 100 and 300 K (Fig. 2), similar to other {CoII-SQ-SQ-CoII} complexes (6.1–7.4 cm3 mol−1 K),16,17,21 indicating a {CoII-SQ-SQ-CoII} state with two non-interacting SQ˙− ligands and two HS-CoII centers. Below 100 K, a rapid decrease in χMT to 1.3 cm3 mol−1 K at 1.8 K is due to depopulation of the HS-CoII spin–orbit coupled states as well as possible intramolecular antiferromagnetic coupling. The subsequent heating and cooling cycles overlay within error.
The small χMT values for 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane at 1.8 K of 0.3 and 0.5 cm3 mol−1 K (Fig. 2), respectively, indicate a predominantly diamagnetic {CoIII-cat-cat-CoIII} charge distribution. This is consistent with the 100 K structural data for 2a·1.8CH2Cl2. Increasing the temperature to 300 K results in a gradual increase to 1.1 and 1.4 cm3 mol−1 K for 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane, respectively. This increase might be at least partially due to temperature independent paramagnetism (TIP), commonly observed for Co(III).17,33 Above 300 K, both 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane exhibit a rapid increase in χMT to 3.8 and 4.5 cm3 mol−1 K at 400 K, respectively, resulting from a thermally induced VT transition. Based on the χMT value of 3a·0.5acetone at 300 K, 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane reach ∼60% and ∼75% HS-CoII-SQ, respectively, at 400 K. Interestingly, both compounds exhibit a plateau (355 K for 2a·0.4Et2O, 344 K for 2a·0.3CH2Cl2·0.4hexane) at χMT values of 2.3 and 2.4 cm3 mol−1 K for 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane, respectively (Fig. 2). This represents ∼40% HS-CoII-SQ character. The transitions observed for 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane are comparable to that of [{Co(Me2tpa)}2(spiro)](ClO4)2,16 albeit with the transition shifted ∼50 K higher. Further cooling–heating cycles resulted in a much more gradual VT transition, with a larger trapped fraction of HS-CoII-SQ, due to thermal trapping and loss of lattice solvent upon heating.
Thus 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane appear to be undergoing a gradual two-step VT transition: {CoIII-cat-cat-CoIII} ⇌ {CoIII-cat-SQ-CoII} ⇌ {CoII-SQ-SQ-CoII}, which remains incomplete up to 400 K. New samples of 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane measured between 1.8–400 K in 5 K steps also displayed plateaus at 345 K (Fig. S8†). However, as demonstrated by the change in magnetic profile upon desolvation (Fig. S6†), these observed plateaus could be due to structural changes upon loss of lattice solvent rather than a two-step VT transition. To examine if the plateaus remained without lattice solvent, 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane were desolvated in vacuo for 48 hours, with TGA confirming majority removal of solvent (Fig. S9†). For desolvated 2a·0.4Et2O (Fig. S10†), the plateau is less pronounced, appearing as an inflection point centered at ∼355 K. For desolvated 2a·0.3CH2Cl2·0.4hexane (Fig. S11†), the plateau is even flatter. Desolvation leading to a more gradual two-step VT process has been observed before,15–18 and highlights the importance of lattice solvation in influencing the VT profile. Further loss of crystallinity upon solvent removal might remove cooperative effects that make the transitions sharper. The retention of a two-step profile upon desolvation, albeit less pronounced, is consistent with 2a undergoing a two-step VT transition. The nature of the very small plateau for 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane necessitates caution in describing the exact nature of the VT transition; a two-step VT transition might be occurring, but with a combination of the three possible spin-states at the inflection point. What is clear is that compound 2a undergoes VT at both cobalt centers.
In summary, compound 1a·0.5MeCN·H2O adopts the temperature invariant {CoIII-cat-cat-CoIII} state between 1.8 and 400 K, whilst 3a·0.5acetone exists as temperature invariant {CoII-SQ-SQ-CoII}. Compounds 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane both undergo an incomplete and gradual thermally-induced VT transition from {CoIII-cat-cat-CoIII} to {CoII-SQ-SQ-CoII} that appears to involve a discrete {CoIII-cat-SQ-CoII} species via a two-step process.
Electronic absorption spectroscopy
Electronic absorption spectra were recorded for solutions of 1a, 2a, and 3a in MeCN and 1,2-dichloroethane (DCE) (Fig. 3 and Fig. S12–S14†), with absorption bands and tabulated assignments in Table S3.† The spectrum for free protonated thMH4 was also recorded in MeCN and DCE (Fig. S15†). The stability of compounds 1a, 2a, and 3a in MeCN in air was assessed by collecting ultraviolet-visible-near-infrared (UV-Vis-NIR) spectra over a period of hours; all compounds displayed small visible changes visible by eye after 2–3 hours. | ||
| Fig. 3 UV-Vis-NIR absorption spectra for MeCN (top) and DCE (bottom) solutions of 1a (black), 2a (red), and 3a (blue) at 298 K. | ||
In MeCN (Fig. 3), the absorption spectrum of 12+ display absorption features characteristic of a LS-CoIII-cat species,17,23 the most notable being a ligand-to-metal-charge-transfer (LMCT) at 684 nm. The spectrum is dominated by an intense thM4− ligand centered catecholate feature at 418 nm, that is observed in the free thMH4 spectrum (Fig. S15†). Therefore, 12+ can be assigned as the {CoIII-cat-cat-CoIII} state in MeCN at room temperature, in agreement with the solid-state magnetic data. Compound 32+ in MeCN no longer displays the intense thM4− ligand centered catecholate peak at 418 nm (Fig. 3), ruling out a catecholate state. Instead, a ligand-centered semiquinonate band at 580 nm and a metal-to-ligand-charge-transfer (MLCT) process at 406 nm are observed, indicating that 32+ adopts a {CoII-SQ-SQ-CoII} charge distribution in MeCN at room temperature, again consistent with the solid-state magnetic behaviour. In DCE, both 12+ and 32+ display essentially the same UV-Vis-NIR spectra as MeCN (Fig. 3), indicating no solvent dependent charge distribution.
In MeCN (Fig. 3), 22+ displays the same intense thM4− ligand centered peak at 418 nm as 12+, indicating a catecholate oxidation state is present. However, 22+ also displays absorption bands at 522 and 570 nm that can be assigned as ligand-centered semiquinonate absorptions.17,23 The broad shoulder between 620 and 800 nm is assigned as a MLCT processes. The absorptions for 22+ indicate a mixture of LS-CoIII-cat and HS-CoII-SQ, arising from an ongoing valence tautomeric transition. No NIR peak is observed at energies below 1100 nm, possibly discounting a mixed valent state. In DCE (Fig. 3), compound 22+ displays a drastically different spectrum than observed in MeCN, akin to the spectrum displayed by 32+. The intense thM4− ligand-centered peak at 418 nm is now absent, and an intense semiquinonate band at 585 nm appears, consistent with the presence of a semiquinonate oxidation state. This suggests that 22+ adopts a {CoII-SQ-SQ-CoII} charge distribution in DCE at room temperature, indicating solvent-dependent charge distribution. Stabilisation of the the HS-CoII-SQ state by chlorinated solvents has been reported previously for mononuclear VT analogues.23,28,34
We measured variable temperature UV-Vis spectra of 22+ in butyronitrile (BuCN) (Fig. 4), chosen for its high boiling point, to determine if 22+ undergoes a two-step VT transition in solution. When prepared under N2, 22+ is stable in BuCN for over 3 hours. Unfortunately, heating above 323 K results in visible thermal degradation, preventing measurements of the full VT interconversion that could provide information about the two-step transition in solution. At room temperature, 22+ exhibits a spectrum between that in MeCN and DCE (Fig. S14†), suggesting a mixture of {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII}. Heating from 268 K to 323 K, the peak at ∼410 nm associated with {CoIII-cat-cat-CoIII} decreases in intensity whilst the peak at ∼570 nm associated with {CoII-SQ-SQ-CoII} increases in intensity (Fig. 4). Isosbestic points at 465, 350 and 304 nm indicating multiple absorbing species. The 298 K spectrum was recovered after the full measurement, demonstrating reversible interconversions and thermal stability of the complex (Fig. S16†). Compound 22+ therefore undergoes a VT interconversion in solution, with a transition temperature above 298 K, incomplete up to 323 K. With the data collected only to 323 K, we cannot determine if 22+ undergoes two-step VT in solution. The VT observed for 22+ occurs at a higher temperature than that measured for [{Co(Me2tpa)}2(spiro)]2+ (250 K),15,16 qualitatively mirroring the higher transition temperature of 2a compared to [{Co(Me2tpa)}2(spiro)](ClO4)2 in the solid state.
 | ||
| Fig. 4 Variable-temperature absorption spectra of 22+ in BuCN between 268 (blue) and 323 K (red) in 5 K increments. | ||
In solution, compounds 12+ and 32+ exists as the {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} charge distribution at room temperature, respectively, mirroring the behaviour observed in the solid state. For compound 22+, the spectrum in MeCN indicates majority {CoIII-cat-cat-CoIII}, whilst in DCE majority {CoII-SQ-SQ-CoII}. In BuCN, 22+ undergoes a VT interconversion centered above room temperature.
Electrochemistry
The redox and electronic communication properties of 1a, 2a, and 3a were investigated by electrochemistry. Cyclic (CV) and rotating disk electrode (RDE) voltammograms were recorded for MeCN solutions of 1a, 2a, and 3a (Fig. 5 and Table 2). A DCE solution of 2a (Fig. S17 and Table S4†) was measured, as previous studies show solvent can affect the electrochemical profile.35 Midpoint potentials (Em) for reversible processes, peak potentials (EP) for irreversible processes, peak-to-peak separations (ΔEp), half-wave potentials (E1/2) and limiting currents (iL) are listed in Table 2 and Table S4.†| Cyclic voltammetry Em or Ep/V (ΔEp/mV) | Rotating disk electrode voltammetry E1/2/V (iL/μA) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I/I′ | II/II′ | III | IV | V | I/I′ | II/II′ | III | IV | V | |
| a 1.0 mM in MeCN, 0.25 M Bu4NPF6, scan rate 100 mV s−1. Potentials reported vs. Fc/Fc+ couple. b E p rather than Em. | ||||||||||
| 12+ | 0.070 (75) | 0.168 (80) | — | 0.975 (145) | −1.268b | 0.08 (27) | 0.22 (55) | — | 1.02 (90) | −1.23 (10) |
| 22+ | 0.008b | 0.155b | — | 1.093b | −0.573 (130) | 0.01 (21) | 0.18 (43) | — | 1.10 (97) | −0.53 (27) |
| 32+ | −0.524 (80) | −0.663 (60) | 0.245b | — | — | −0.49(25) | −0.63(14) | 0.23(40) | ||
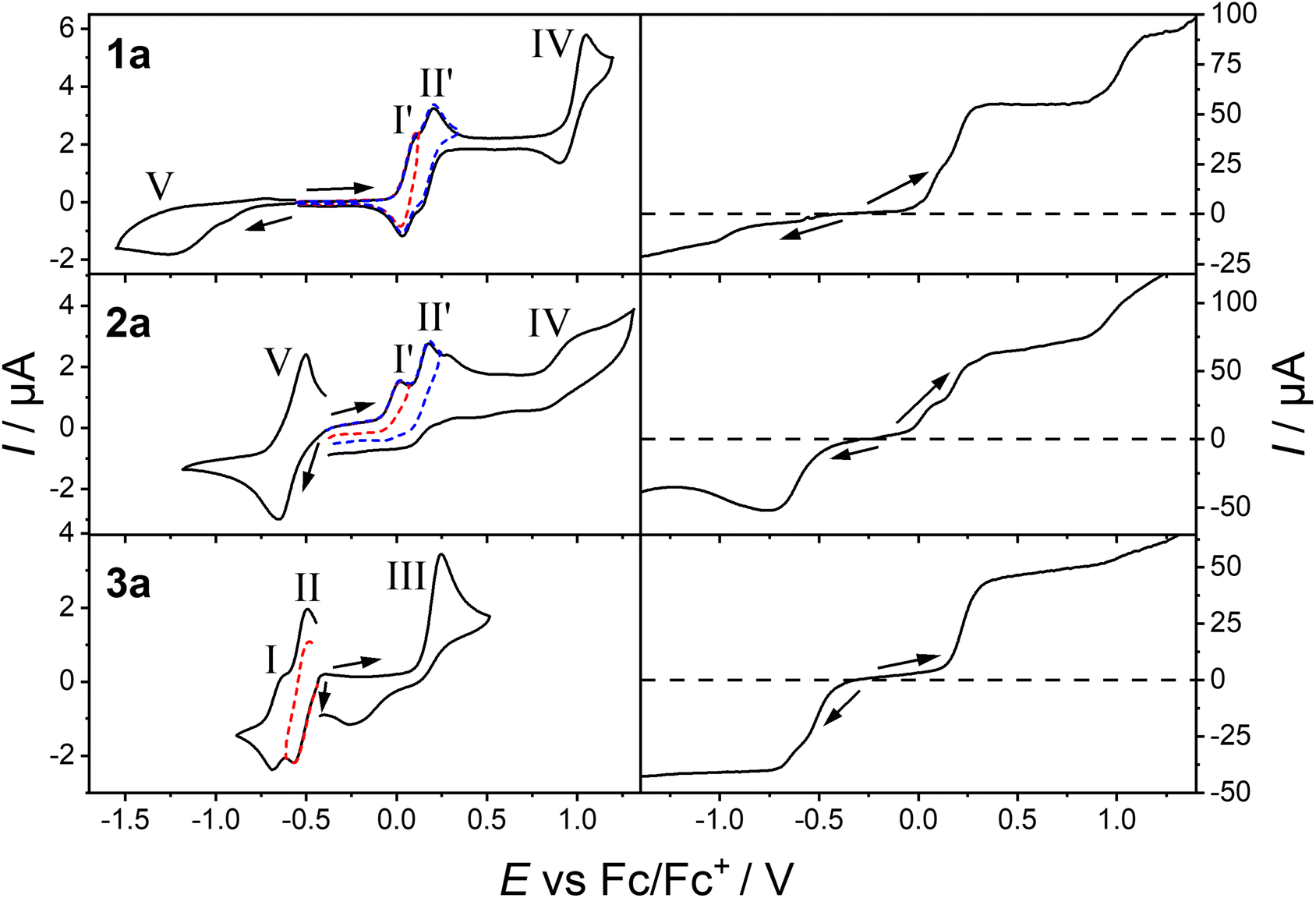 | ||
| Fig. 5 Cyclic voltammograms of MeCN solutions of compounds 1a, 2a and 3a (1.0 mM with 0.25 M Bu4NPF6) at a scan rate of 100 mV s−1 (left). Corresponding rotating disc electrode voltammogram at a scan rate of 50 mV s−1 and a rotation rate of 500 rpm. Arrows indicate the starting potential and direction of the scan. | ||
Complexes 12+ and 22+ display four redox processes and 32+ displays three redox processes in the measured potential range (Fig. 5). For 12+ and 22+, there are two closely spaced one-electron oxidations (I′ and II′), a two-electron oxidation (IV), and a two-electron reduction (V). For 32+ (Fig. 5), two closely spaced one-electron reductions (I and II), and a two-electron oxidation (III) are observed instead. A second oxidation process (IV) for 32+ occurs that overlaps with the MeCN solvent window. The position of zero current in the RDE voltammogram identifies I′, II′, III and IV as oxidations, whilst I, II and V are reductions. For 32+, the onset of a second oxidation (IV) is observed in the RDE, whereas in the CV it coincides with the solvent window. These processes are qualitatively similar to those observed for analogous cobalt complexes [{Co(Mentpa)}2(spiro)]2+ and [{Co(Mentpa)}2(Br4spiro)]2+ (n = 0, 2, 3; Br4spiroH4 = 3,3,3′,3′-tetramethyl-1,1′-spirobi(indan)-4,4′,7,7′-tetrabromo-5,5′,6,6′-tetraol) with different bis(dioxolene) ligands.16,17 For complex 12+, processes I′, II′ and IV are diffusion controlled and chemically reversible with peak-to-peak separations (ΔEp) of 75, 80 and 145 mV, respectively (ferrocene/ferrocenium has measured ΔEp of 75 mV) indicating process I′ and II′ are one-electron and process IV is a two-electron process. Process V is clearly irreversible with no anodic current in the reverse sweep. In compound 22+, process I′ and II′ are now irreversible, process IV quasi-reversible, and process V reversible (ΔEp = 140 mV). In DCE, process I′ and II′ for 22+ are now quasi-reversible, with IV remaining irreversible, with such an effect observed in cobalt complexes.17,36 Between MeCN and DCE, 22+ displays similar redox properties. In compound 32+, processes I and II are quasi-reversible one-electron reductions (ΔEp = 60 and 95, respectively), and III is an irreversible two-electron oxidation.
As established by UV-Vis-NIR measurements in MeCN, compounds 12+ and 22+ exist in the {CoIII-cat-cat-CoIII} state. Process I′ and II′ are therefore sequential thM4− oxidation processes following cat2−–cat2−/cat2−–SQ˙− and cat2−–SQ˙−/SQ˙−–SQ˙−, respectively (Scheme 1). Process V is a Co(III)/Co(II) reduction process. Process IV is a concerted SQ˙−–SQ˙−/Q–Q (Q = quinone) oxidation. The potentials for processes I′ and II′ in 12+ and 22+ are the same within error (Table 2). However, process V in 22+ is shifted approximately +1 V compared to 12+, as the increased steric bulk of Me2tpa stabilises the Co(II) state compared to tpa. The potentials of processes I′, II′, IV and V for 22+ in MeCN and DCE remain similar (Table S4†). Compared to the analogous I′ and II′ processes (Table S5†)measured for [{Co(tpa)}2spiro]2+ (−0.15 and 0.01 V) and [{Co(tpa)}2Br4spiro]2+ (0.17 and 0.30 V), the redox potentials for the thM4− processes in 12+ lie between those for spiro4− and Br4spiro4−.16,17 This is consistent with the VT transition temperature for 2a in the solid state being higher than for [{Co(Me2tpa)}2(spiro)](ClO4)2 but lower than for [{Co(Me2tpa)}2(Br4spiro)](PF6)2.
 | ||
| Scheme 1 Assigned redox processes for complexes 12+, 22+ and 32+. | ||
Compound 32+ adopts a {CoII-SQ-SQ-CoII} state in MeCN at room temperature. Therefore, process I and II are sequential reductions of the bis(semiquinonate) following SQ˙−–SQ˙−/cat2−–SQ˙− and cat2−–SQ˙−/cat2−–cat2−, opposite to what we see for 12+ and 22+ (Scheme 1). Process III, which might be assumed to be a Co(II)/Co(III) oxidation process, was previously established for related bis(dioxolene) bridged cobalt complexes as a ligand-based oxidation of the bis(semiquinonate) to the bis(quinone) to give {CoII-Q-Q-CoII}4+,17 which undergoes rapid interconversion to {CoIII-SQ-SQ-CoIII}4+ (Scheme 1). Given the similarity of the electrochemical behavior of 32+ with related cobalt complexes, process III is also assigned as an oxidation of the bis(semiquinonate) to bis(quinone), followed by conversion to {CoIII-SQ-SQ-CoIII}4+. This assignment is only tentative, with confirmation requiring a zinc analogue and/or spectroelectrochemical measurements.
The parameter Δox-red is defined as the separation between the midpoint potentials (when reversible) of the first one-electron oxidation and first one-electron reduction where one is metal-based and the other ligand-based. For irreversible processes, Δox-red can only be estimated. Previous work on mononuclear compounds established the approximate rule that Δox-red < 740 mV is required for VT to occur in a thermally accessible range.23 Complexes 12+ and 22+ have a ligand-based oxidation and metal-based reduction processes and the Δox-red value can be determined (due to the irreversibility of some processes, the values are only estimates). The values are 1340 and 580 mV for 12+ and 22+, respectively (Table S6†). For complex 32+, the first oxidation and reduction are both ligand based. Therefore, the Δox-red value of 770 mV for 32+ is a rough estimate using process II and III, given that {CoII-Q-Q-CoII}4+ undergoes rapid conversion to {CoIII-SQ-SQ-CoIII}4+. Therefore, only 22+ has a Δox-red < 740 mV and is therefore the only complex expected to show VT, with 12+ and 32+ remaining as temperature invariant {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII}, respectively.
The strength of the intramolecular electronic communication within a bis(dioxolene) ligand can be correlated with the Δdiox value, the separation between processes I and II or I′ and II′. Previous work for {Co(Mentpa)} moieties bridged by various bis(dioxolene) ligands gave Δdiox ranging 80–440 mV,17 with lower values indicating reduced communication. Values below 140 mV indicated mixed-valent (MV) class II, whilst values between 140 and 175 mV are borderline between MV II and II–III.17 The Δdiox value exhibited by the two-step VT compound [{Co(Me2tpa)}2(spiro)]2+ was 120 mV. For 12+, 22+ and 32+ the Δdiox values are approximately 100, 150 and 140 mV in MeCN (Table S6†), respectively. These values are consistent with the value for 120 mV displayed by [{Co(bpy)2}2(thM)]2+, as well as the values for the [{Co(Mentpa)}2(spiro)]2+ and [{Co(Mentpa)}2(Br4spiro)]2+ (n = 0, 1, 3) families, suggesting weak electronic communication between MV class II and II–III.16,17,19
From a previous proposal,17 two-step VT occurs in bis(dioxolene) cobalt complexes when the d-orbital energies of cobalt in {CoII(L)} and {CoIII(L)} (L = ancillary ligand) are similar to the energies of the frontier orbitals of the bis(dioxolene) in all three of the cat2−–cat2−, cat2−–SQ˙− and SQ˙−–SQ˙− states. This is achieved via tuning using Mentpa (Δox-red). The likelihood of matching the metal and ligand orbital energy is also enhanced when the energy separation between the dioxolene orbitals is minimised (smaller Δdiox); therefore, the smaller the electronic coupling in the bis(dioxolene), whilst remaining non-zero, the higher the chance of a two-step transition. This results in the previously proposed phase diagram in Fig. 6 plotting Δox-red and Δdiox. Compounds 12+ and 32+ are in the temperature independent {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} regions, respectively, ruling VT out entirely. However, 22+ occurs in the region that borders both two-step and incomplete VT, which suggests that 22+ should show thermally-induced VT. The nature of this VT process is therefore predicted to be a two-step transition at a higher temperature compared to [{Co(Me2tpa)}2(spiro)]2+, which exhibits a plateau at 300 K.15,16 This matches the magnetic data measured for 2a, which presents a clear VT interconversion above 300 K with an apparent plateau that suggests a two-step process. This result further highlights the predictive power of the correlation diagram of Fig. 6, and the importance of matching both the Co and dioxolene frontier orbital energies and minimising the electronic communication across the bis(dioxolene).
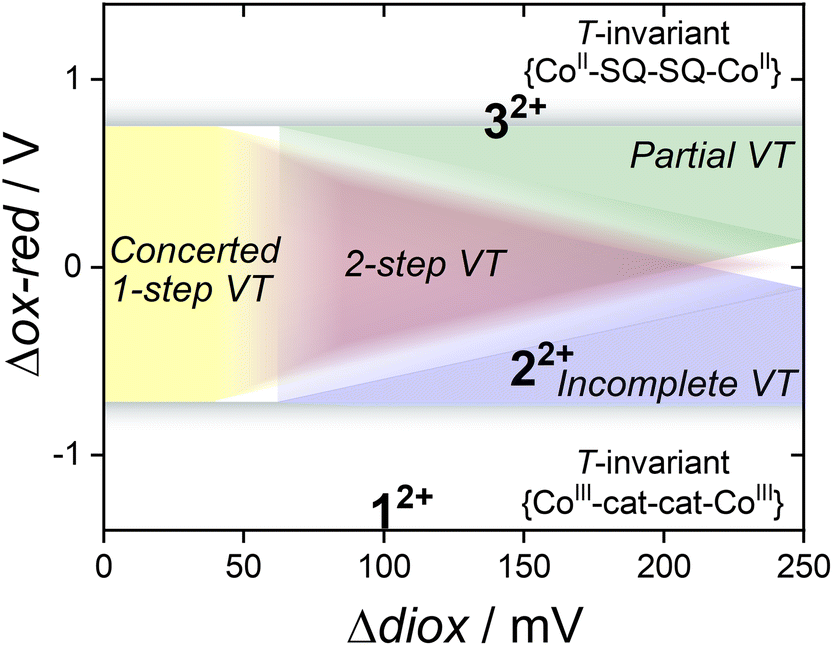 | ||
| Fig. 6 The correlation between the Δdiox and Δox-red redox parameters, types of VT transitions displayed by binuclear cobalt bis(dioxolene) complexes, and the placement of complexes 12+, 22+ and 32+via the electrochemical data measured in MeCN (1 mM analyte concentration, 0.25 M Bu4NPF)6. Figure adapted with permission from G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704.17 Copyright 2020 American Chemical Society. | ||
Density functional theory calculations
Density functional theory (DFT) calculations have played an important role in studying VT exhibited by dinuclear cobalt complexes bridged by bis(dioxolene) ligands.17,18,37 Calculations were performed on complexes 1a, 2a and 3a in the gas phase to examine the relative energies of the different tautomeric states (Fig. 7 and Tables S7, S8, Fig. S18–S20†). Calculations were performed with inclusion of the PF6− anions, which has been shown to aid in the correct calculation of the spin state energetics.17,33,38,39 For a dinuclear VT complex, a two-step VT transition can only occur if the three tautomeric states are thermally-accessible and have the energy ordering: {CoIII-cat-cat-CoIII} < {CoIII-cat-SQ-CoII} < {CoII-SQ-SQ-CoII}. In addition, the likelihood of a two-step transition occurring via {CoIII-cat-SQ-CoII} is described by the thermochemical parameter ρ, calculated by ρ = W/ΔH, where ΔH is the enthalpy difference between the {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} states and W is the energetic stabilisation of the {CoIII-cat-SQ-CoII} state relative to the average energy of the {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} states (ΔH/2).10,17 When ρ < 0, a two-step transition is favoured, i.e. energy of {CoIII-cat-SQ-CoII} lies below halfway energy between {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII}. The more negative ρ, the more likely a two-step transition will be observed. A ρ > 0 indicates a two-electron one-step VT transition. | ||
| Fig. 7 Schematic showing the relative energies of different electronic states of 1a, 2a and 3a calculated by the DFT UTPSSh/6-311++G(d,p) method with zero point energy. | ||
Calculations of compound 1a (Fig. 7) indicate the structure on the singlet potential energy surface (PES) corresponding to the {CoIII-cat-cat-CoIII} tautomer is the ground state, in agreement with the experimental results. The calculated Co–O/N and dioxolene bond lengths are in excellent agreement with the 100 K X-ray structural data for 1a·2toluene·2MeCN (Fig. S21†). The {CoIII-cat-SQ-CoII} (quintet PES) and {CoII-SQ-SQ-CoII} (nonet PES) tautomers are destabilised by 7.4 and 19.2 kcal mol−1, respectively, and are predicted to be thermally inaccessible. The ρ value of −0.11 suggests a two-step VT transition could occur, but will not be observed in an experimentally accessible temperature range. The DFT calculations are consistent with the temperature invariant {CoIII-cat-cat-CoIII} state observed for 1a.
For compound 2a, the ground state was calculated to be {CoIII-cat-cat-CoIII}, followed by {CoIII-cat-SQ-CoII} (0.3 kcal mol−1) and {CoII-SQ-SQ-CoII} (4.1 kcal mol−1) at thermally-accessible energies (Fig. 7). The DFT optimised geometry of the {CoIII-cat-cat-CoIII} matches well with the experimentally determined 2a·1.8CH2Cl2 structure (Fig. S21†). The ρ value of −0.43 is consistent with the ability to observe two separate interconversion steps. The calculations therefore imply that 2a should show a two-step VT transition, suggesting that the observed interconversion between {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} occurs via {CoIII-cat-SQ-CoII} in a two-step fashion.
Calculations of compound 3a suggest a {CoII-SQ-SQ-CoII} ground state, consistent with experimental data (Fig. 7), with the calculated bond lengths consistent with the structure of 3a·2acetone at 100 K (Fig. S21†). The {CoIII-cat-SQ-CoII} and {CoIII-cat-cat-CoIII} tautomers are destabilised by 0.5 and 6.7 kcal mol−1, respectively. Despite the small energy difference between the {CoII-SQ-SQ-CoII} and {CoIII-cat-SQ-CoII} states, no transition is expected to occur as thermally-induced VT is an entropy-driven process. The calculations are therefore consistent with the temperature invariant {CoII-SQ-SQ-CoII} state for 3a.
The isotropic exchange coupling parameters J (−2JŜ1·Ŝ2 formalism) for 1a, 2a and 3a were calculated using the broken symmetry (BS) approximation.40 For the {CoIII-cat-SQ-CoII} and {CoII-SQ-SQ-CoII} states, calculations suggest strong ferromagnetic interactions between the HS-CoII center and the coordinated semiquinonate radical (Table S9†). These calculations do not consider spin–orbit coupling contributions, which is known to affect the exchange parameters,41 preventing an exact value of the exchange to be conclusively determined. However, these calculations are consistent with previous calculations,17,18,37,38 with experimental support of ferromagnetic exchange in the {CoII-SQ-SQ-CoII} state.37 For the {CoII-SQ-SQ-CoII} tautomer, strong antiferromagnetic coupling is calculated between the two semiquinonate centers of thM2− (Table S9†). The calculated exchange interactions between the two HS-CoII centers, and between the HS-CoII and non-coordinated SQ˙−, are negligibly small and are within the error of the DFT method.
Considering the large distance between the dioxolene moieties in thMn− (∼11.9 Å distance between the O1–C1–C2–O2 centroids) compared to spiron− (∼7.2 Å)16 and Br4spiron− (∼7.4 Å),17,42 which have weak communication, even smaller electronic communication might be expected between the two dioxolene moieties. Analysis of the magnetic orbital of the {CoII-SQ-SQ-CoII} state for 1a (Fig. 8) shows the formation of a π-conjugated system facilitated by the planar structure of thM2−. This formally conjugated nature of thMn− allows for the observed weak, non-zero, communication between the two dioxolene moieties despite the extended distance. The conjugation results in spin delocalisation (Fig. S22†) that produces the calculated strong SQ–SQ antiferromagnetic coupling. The formal conjugation via a longer pathway in thMn− provides an alternative route to weak communication to the spiroconjugation via a shorter path that exists for spiron− and Br4spiron−.
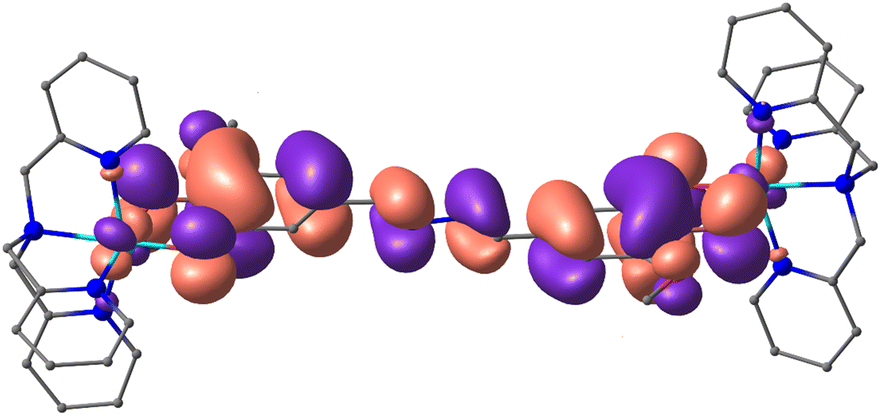 | ||
| Fig. 8 The shape of magnetic orbital (contour value = 0.015 e Å−3) of the {CoII-SQ-SQ-CoII} state of 1a as calculated by the DFT UTPSSh/6-311++G(d,p) method. Hydrogen atoms and PF6− anions are omitted. | ||
Concluding remarks
Informed by previous work that identified that bis(dioxolene) ligands with weak, but non-zero, electronic communication increases the likelihood of two-step VT in dinuclear cobalt complexes, we report three new dinuclear cobalt complexes incorporating a thMn− bridging ligand. This ligand was chosen for its apparent weak electronic communication, postulated to support two-step VT. The complexes, [{Co(Mentpa)2(thM)}](PF6)2, incorporate varying degrees of methylation of the Mentpa ligand with n = 0 (1a), n = 2 (2a), and n = 3 (3a). Structural analysis at 100 K shows 1a and 2a exist in the {CoIII-cat-cat-CoIII} state, while 3a adopts a {CoII-SQ-SQ-CoII} charge distribution. In MeCN at room temperature, the same charge distribution suggested by the structural data is observed for 12+, 22+ and 32+. In DCE, however, 22+ instead adopts a {CoII-SQ-SQ-CoII} state, with variable temperature UV-Vis in BuCN showing that 22+ undergoes VT in the solution state.Magnetochemical analysis shows that 1a·0.5MeCN·H2O and 3a·0.5acetone adopt temperature invariant {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} states, respectively, consistent with the structural and spectroscopic analysis. For the two isolated samples of 2a, 2a·0.4Et2O and 2a·0.3CH2Cl2·0.4hexane, a thermally-induced VT transition above room temperature is apparent, which appears to proceed via a plateau between 345 and 355 K. The nature of the plateau is affected by the lattice solvation. The χMT values, and observation of the plateau, suggest an incomplete two-step VT conversion from {CoIII-cat-cat-CoIII} to {CoII-SQ-SQ-CoII}, via {CoIII-cat-SQ-CoII}.
Electrochemical analysis of 12+ and 22+ in MeCN shows closely spaced sequential cat2−–cat2−/cat2−–SQ˙− and cat2−–SQ˙−/SQ˙−–SQ˙− oxidation processes and a Co(III)/Co(II) reduction, whereas 32+ instead displays sequential SQ˙−–SQ˙−/cat2−–SQ˙− and cat2−–SQ˙−/cat2−–cat2− reduction processes and a Co(II)/Co(III) oxidation. Importantly, the Δox-red values of 1340, 580 and 770 mV for 12+, 22+ and 32+, respectively, indicate that the energy match between the cobalt and dioxolene frontier orbitals that would support VT is only present for 22+. The thMn− was found to have weak electronic communication, parameterised by the Δdiox value of 150 mV for 22+, that should support two-step VT. Combining the Δox-red and Δdiox parameters for 22+ allows the prediction of two-step VT above room temperature, matching the observed magnetic behaviour. It is important to note that the use of solution-based electrochemical prediction to predict solid state VT behaviour does not account for lattice effects that also influence the switching behaviour. This is evidenced by 2a displaying different plateaus depending on the lattice solvation. Therefore, the correlation diagram (Fig. 6 ) can only serve as a guiding tool for predicting VT.
Density functional theory calculations suggest temperature invariant {CoIII-cat-cat-CoIII} and {CoII-SQ-SQ-CoII} states for 1a and 3a, respectively, mirroring experimental observations. For 2a, DFT calculation affords the correct energy ordering of the three tautomeric states for two-step VT behavior. Orbital analysis demonstrates that despite the large distance between the dioxolene moieties in thMn−, non-zero communication occurs due to formal conjugation. This results in a bis(dioxolene) ligand that minimises communication, while keeping it non-zero, to a degree that supports two-step VT.
Here, we have shown that two-step VT in dinuclear cobalt complexes can be achieved via judicious combination of a bis(dioxolene) and ancillary ligands. The choice of the thMn− ligand, with its weak electronic communication, coupled with the Me2tpa ancillary ligand ensures that the {CoII(Me2tpa)}/{CoIII(Me2tpa)} redox couple matches with both cat2−/cat2−–SQ˙− and cat2−–SQ˙−/SQ˙−–SQ˙− redox couples, as required for two-step VT. The direct control bis(dioxolene) bridging ligands impart over the electronic communication between the two VT centers allows a rational strategy to achieve desired VT switching behaviour, with thMn− predicted as a ligand that should give two-step VT prior to experimental investigation. With the wide libraries of other available bis(dioxolene) and ancillary ligands, multiple cobalt complexes with two-step VT that are more abrupt and with larger plateaus can foreseeably be achieved.
Experimental section
Synthesis
All manipulations, unless otherwise indicated, were performed under anaerobic conditions on a Schlenk line using standard Schlenk techniques. All chemicals purchased were of reagent grade or higher and used as received. Solvents were dried over molecular sieves (3 Å) for a minimum of 3 days and stored under N2 on sieves until used. Solvents were degassed prior to use by sparging with N2 for 45 minutes. Molecular sieves were activated by heating at 300 °C for 48 hours and then cooled under vacuum. Ligands Me2tpa, Me3tpa, and thMH4 were prepared as previously reported.17,19,22000 L mol−1 cm−1).
000 L mol−1 cm−1).
000), 400 (10000 L mol−1 cm−1).
X-ray data collection and structure solution
Single crystal X-ray diffraction data were collected for 1a·2toluene·2MeCN on a Rigaku XtaLAB Synergy-S Dual Microfocus X-ray diffractometer using Cu-Kα (λ = 1.5418 Å) radiation at 100 K. Data for 2a·1.8CH2Cl2 were collected at 100 K on the MX243 beamline at the Australian Synchrotron fitted with a silicon double crystal monochromator and Dectris Eiger 16 M detector, the wavelength being tuned to approximate Mo-Kα radiation (λ = 0.71073 Å), while for 3a·2acetone data were collected at 100 K on the MX144 beamline at the Australian Synchrotron, fitted with a silicon double crystal monochromator and Dectris Eiger2 9M detector, the wavelength being tuned to approximate Mo-Kα radiation (λ = 0.71092 Å). Crystals were transferred directly from the mother liquor into cryoprotective oil and then mounted on the diffractometer at 100 K to prevent solvent loss and loss of crystallinity. The data for 1a·2toluene·2MeCN were processed using CrysAlisPro,45 while data reduction for 2a·1.8CH2Cl2 and 3a·2acetone were performed using XDS,46 using strong multi-scan absorption correction in SADABS.47 Structures were solved with SHELXT48 and refined using a full matrix least squared procedure based on F2 using SHELXL within Olex2.49,50 All non-hydrogen atoms were refined using anisotropic displacement factors. Hydrogen atoms were placed at geometrical positions and refined using the riding model. For 2a·1.8CH2Cl2, the two methyl groups on the pyridine rings were almost equally distributed over the three different positions. Each of the occupancy factors were allowed to refine with the sum of the three factors being fixed at 2. The PF6− anion was disordered. Refinement was carried out with the anion distributed over three orientations with the geometries and displacement parameters restrained to be similar. The occupancy factors were allowed to refine with the sum of the occupancy factors being constrained to be 1. There were a number of peaks attributed to a disordered molecule of CH2Cl2. Attempts to model this disorder were unsuccessful and so refinement was continued with the contribution of the solvent being modelled using the OLEX2 solvent mask routine.51 The electron density was consistent with 0.9 molecules of CH2Cl2 per asymmetric unit, or 1.8 molecules per formula unit.
Powder X-ray diffraction (PXRD) data were measured on a Rigaku synergy dual wavelength rotating anode X-ray diffractometer system using Cu-Kα (λ = 1.5418 Å) at 100 K. Powder samples were prepared by crushing the sample gently and loading them into a borosilicate glass capillary for measurement. Data were collected at 2θ = 50° with an exposure time of 60 seconds per frame and processed using CrysAlisPro.45
Solid-state magnetic measurements
The dc susceptibility and magnetisation measurements for samples 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane and 3a·0.5acetone were performed on a quantum design MPMS 3 SQUID magnetometer measured between 1.8 and 300 or 400 K with an applied field of 1000 Oe. Samples 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane were loaded into a plastic cap. Sample 3a·0.5acetone was loaded into a gel cap and restrained in eicosane. The data was corrected for the diamagnetic contribution of the plastic cap for 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane and for the gel cap and eicosane for 3a·0.5acetone, and of the sample using Pascal's constants.52 Samples 1a·0.5MeCN·H2O, 2a·0.4Et2O, 2a·0.3CH2Cl2·0.4hexane were measured from 300 to 1.8 K, before measuring to 400 K, followed by measurement to 1.8 K, followed by measurement to 400 K. Sample 3a·0.5acetone was measured from 300 to 1.8 K, before measuring back to 300 K. Magnetisation (M) vs. field (H) measurements were performed between 0 and 7 T at 1.8, 4 and 7 K.Electronic and infrared spectroscopy, elemental analysis and thermogravimetric analysis
Attenuated total reflectance infrared spectra were measured on a Bruker Alpha spectrometer and normalised as absorbance spectra. Solution state UV-Visible-NIR spectra were measured on a PerkinElmer UV-Vis-NIR spectrometer Lambda 1050. Variable temperature UV-Vis spectra were measured on an Agilent Cary UV–visible Multicell Peltier Spectrometer, with a flow of N2 gas to remove condensation. Samples used for variable temperature UV-Vis measurements were prepared in an M-Braun N2-atmosphere glove box and placed in a gastight cuvette. Elemental analyses (C, H, N) were performed at the Macquarie Analytical and Fabrication Facility, Macquarie University. Thermogravimetric analyses were performed on a PerkinElmer TGA 8000 thermogravimetry analyzer using a ramp rate of 5 °C per minute from 30 °C up to a maximum temperature of 400 °C under an N2 atmosphere.Electrochemistry
Electrochemical measurements were performed in MeCN at room temperature using a standard three-electrode configuration connected to an eDAQ computer-controlled potentiostat. Measurements were performed under a constant flow of N2. For cyclic voltammetry measurements, the three-electrode system consisted of a 1.0 mm diameter glassy carbon electrode (Cypress Systems), a platinum/titanium auxiliary electrode (eDAQ), and an Ag/AgCl reference electrode (eDAQ). For steady state voltammetry measurements, the working electrode was replaced with a 3.0 mm diameter glassy carbon rotating disk electrode with external controller (Metrohm). Analyte solutions of 1.0 mM were prepared using anaerobic techniques in 5 ml MeCN containing 0.25 M Bu4NPF6 as the supporting electrolyte. All potentials have been references versus the ferrocene/ferrocenium redox couple, measured immediately afterwards. The cyclic voltammetry (CV) midpoint potentials (Em) are calculated by taking the average of the anodic and cathodic peak potentials (Ep). The half-wave potentials (E1/2) are calculated as the potential at half the limiting current (iL). Reversible processes are identified by peak-to-peak separations (ΔEp) close to the value measured under the same conditions for ferrocene at a scan rate of 100 mV s−1 (75 mV in MeCN).Density functional theory (DFT) calculations
The DFT calculations were performed using the Gaussian 16 program package53 with the UTPSSh54,55 functional and the standard 6-311++G(d,p) basis set including diffuse and polarisation functions at all atoms, with inclusion of zero point energy (ZPE). This methodology was shown to provide a good reproduction of the energetic characteristics and magnetic properties of magnetically bistable transition metal compounds, in particular cobalt complexes with redox-active ligands.17,18,27,33,38,56–58 Since the calculated (Mulliken, Lowdin, NBO, Hirshfeld) charges do not provide reliable conclusions about the oxidation state of the central ion in complexes with redox-active ligands,59 the isomers of complexes 1a, 2a and 3a were assigned to the corresponding electronic states on the basis of spin density distribution. Geometry optimisations were performed with the crystal structure of 1a, 2a and 3a as the starting geometry. It has been earlier demonstrated that calculations of cationic structures of such type complexes led to the significant underestimation of energies of the low-spin electromers.17,33,38,39 Therefore, the calculations have been performed with inclusion into the computational scheme of outer-sphere counteranions, the positions of which were taken from X-ray data. The stationary points on the potential energy surfaces (PESs) were localised by full geometry optimisation with subsequent confirmation of the stabilities of the DFT wave function and calculation of force constant matrices. The estimation of exchange coupling parameters J (in cm−1; Ĥ = −2JS1·S2) was carried out by calculation of all possible charge distributions in the framework of the “broken symmetry” (BS) formalism40 with the use of generalised spin-projection method developed by Yamaguchi.60 For the search of the BS states, the Gaussian 16 procedure (guess = fragment = N) has been employed. This technique implies a possibility to assume spin states and signs along with charges for separate parts of a molecule on the stage of generation of the initial guess. Structural visualisations were prepared using the ChemCraft61 software with the calculated atomic coordinates as input parameters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Australian Research Council for financial support (DP220100398) and an equipment grant (LE210100009). T. E. F. thanks the University of Melbourne for a Dr Jim Desmarchelier Scholarship. J. T. J. acknowledges support from the Australian Government for a Research Training Stipend. A. A. S. thanks the Ministry of Science and Higher Education of the Russian Federation (State assignment in the field of scientific activity, project no. FENW-2023-0017). This work was performed, in part, at the Trace Analysis for Chemical, Earth and Environmental Sciences (TrACEES) Platform at the University of Melbourne. This research was undertaken, in part, using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, Victoria, Australia and made use of the Australian Cancer Research Foundation (ACRF) detector (for MX2). We are grateful to Dr Gemma Gransbury for her invaluable feedback and discussions, and for providing Fig. S1†/Fig. 6.References
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- M. M. Paquette, D. Plaul, A. Kurimoto, B. O. Patrick, N. L. Frank and B. O. Patrick, J. Am. Chem. Soc., 2018, 140, 14990–15000 CrossRef CAS PubMed.
- A. Dei, D. Gatteschi, C. Sangregorio and L. Sorace, Acc. Chem. Res., 2004, 37, 827–835 CrossRef CAS PubMed.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- C. Simão, M. Mas-Torrent, J. Casado-Montenegro, F. Otón, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2011, 133, 13256–13259 CrossRef PubMed.
- S. Fortier, J. J. Le Roy, C. H. Chen, V. Vieru, M. Murugesu, L. F. Chibotaru, D. J. Mindiola and K. G. Caulton, J. Am. Chem. Soc., 2013, 135, 14670–14678 CrossRef CAS PubMed.
- J. Andréasson, U. Pischel, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 11641–11648 Search PubMed.
- X. Feng, C. Mathonière, I. R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef CAS PubMed.
- B. Doistau, L. Benda, J. L. Cantin, L. M. Chamoreau, E. Ruiz, V. Marvaud, B. Hasenknopf and G. Vives, J. Am. Chem. Soc., 2017, 139, 9213–9220 CrossRef CAS PubMed.
- J. A. Real, H. Bolvin, A. Bousseksou, A. Dworkin, O. Kahn, F. Varret and J. Zarembowitch, J. Am. Chem. Soc., 1992, 114, 4650–4658 Search PubMed.
- J. Cirera and E. Ruiz, J. Mater. Chem. C, 2015, 3, 7954–7961 RSC.
- D. Chernyshov, M. Hostettler, K. W. Törnroos and H. B. Bürgi, Angew. Chem., Int. Ed., 2003, 42, 3825–3830 CrossRef CAS PubMed.
- C. Zheng, S. Jia, Y. Dong, J. Xu, H. Sui, F. Wang and D. Li, Inorg. Chem., 2019, 58, 14316–14324 CrossRef CAS PubMed.
- E. Dobbelaar, V. B. Jakobsen, E. Trzop, M. Lee, S. Chikara, X. Ding, H. Müller-Bunz, K. Esien, S. Felton, M. A. Carpenter, E. Collet, G. G. Morgan and V. S. Zapf, Angew. Chem., 2022, 134, 1–9 CrossRef.
- K. G. Alley, G. Poneti, J. B. Aitken, R. K. Hocking, B. Moubaraki, K. S. Murray, B. F. Abrahams, H. H. Harris, L. Sorace and C. Boskovic, Inorg. Chem., 2012, 51, 3944–3946 CrossRef CAS PubMed.
- K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 Search PubMed.
- G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS PubMed.
- J. P. Wang, W. T. Liu, M. Yu, X. Y. Ji, J. L. Liu, M. Z. Chi, A. A. Starikova and J. Tao, Inorg. Chem., 2022, 61, 4428–4441 CrossRef CAS PubMed.
- Y. Suenaga and C. G. Pierpont, Inorg. Chem., 2005, 44, 6183–6191 CrossRef CAS PubMed.
- Y. Suenaga, T. Mibu, T. Okubo, M. Maekawa, T. Kuroda-Sowa and K. Sugimoto, Dalton Trans., 2021, 50, 9833–9841 RSC.
- G. Poneti, M. Mannini, B. Cortigiani, L. Poggini, L. Sorace, E. Otero, P. Sainctavit, R. Sessoli and A. Dei, Inorg. Chem., 2013, 52, 11798–11805 CrossRef CAS PubMed.
- A. Beni, A. Dei, S. Laschi, M. Rizzitano and L. Sorace, Chem. – Eur. J., 2008, 14, 1804–1813 CrossRef CAS PubMed.
- G. K. Gransbury, M. E. Boulon, S. Petrie, R. W. Gable, R. J. Mulder, L. Sorace, R. Stranger and C. Boskovic, Inorg. Chem., 2019, 58, 4230–4243 CrossRef CAS PubMed.
- S. Llunell, M. Casanova, D. Cirera, J. Alemany and P. Alvarez, SHAPE 2.1, 2013 Search PubMed.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
- S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996–1009 Search PubMed.
- M. A. Hay, J. T. Janetzki, V. J. Kumar, R. W. Gable, R. Clérac, A. A. Starikova, P. J. Low and C. Boskovic, Inorg. Chem., 2022, 61, 17609–17622 CrossRef CAS PubMed.
- T. Tezgerevska, E. Rousset, R. W. Gable, G. N. L. Jameson, E. C. Sañudo, A. Starikova and C. Boskovic, Dalton Trans., 2019, 48, 11674–11689 RSC.
- O. Carugo, C. B. Castellani, K. Djinovic and M. Rizzi, Dalton Trans., 1992, 837–841 RSC.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251–1260 CrossRef CAS PubMed.
- P. Tourón Touceda, S. Mosquera Vázquez, M. Lima, A. Lapini, P. Foggi, A. Dei and R. Righini, Phys. Chem. Chem. Phys., 2012, 14, 1038–1047 RSC.
- F. Rupp, K. Chevalier, M. Graf, M. Schmitz, H. Kelm, A. Grün, M. Zimmer, M. Gerhards, C. van Wüllen, H.-J. Krüger and R. Diller, Chem. – Eur. J., 2017, 23, 2119–2132 CrossRef CAS PubMed.
- T. Tezgerevska, E. Rousset, R. W. Gable, G. N. L. Jameson, E. C. Sañudo, A. Starikova and C. Boskovic, Dalton Trans., 2019, 48, 11674–11689 RSC.
- V. L. Nadurata and C. Boskovic, Inorg. Chem. Front., 2021, 8, 1840–1864 RSC.
- A. Hildebrandt, D. Miesel, Q. Yuan, J. Freytag, J. Mahrholdt and H. Lang, Dalton Trans., 2019, 48, 13162–13168 RSC.
- V. L. Nadurata, M. A. Hay, J. T. Janetzki, G. K. Gransbury and C. Boskovic, Dalton Trans., 2021, 50, 16631–16646 RSC.
- Y. Zhao, J. Wang, X. Chen, M. Yu, A. A. Starikova and J. Tao, Inorg. Chem. Front., 2023, 10, 7251–7264 RSC.
- A. A. Starikova, M. G. Chegerev, A. G. Starikov and V. I. Minkin, Comput. Theor. Chem., 2018, 1124, 15–22 CrossRef CAS.
- A. A. Starikova, M. G. Chegerev and A. G. Starikov, Chem. Phys. Lett., 2021, 762, 138128 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- G. K. Gransbury, M.-E. Boulon, R. A. Mole, R. W. Gable, B. Moubaraki, K. S. Murray, L. Sorace, A. Soncini and C. Boskovic, Chem. Sci., 2019, 10, 8855–8871 RSC.
- J. T. Janetzki, M. G. Chegerev, G. K. Gransbury, R. W. Gable, J. K. Clegg, R. J. Mulder, G. N. L. Jameson, A. A. Starikova and C. Boskovic, Inorg. Chem., 2023, 62, 15719–15735 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 Search PubMed.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- J. P. Glusker, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1993, 49, 1–1 CrossRef.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Adv., 1990, 46, 194–201 CrossRef.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. Cirera, M. Via-Nadal and E. Ruiz, Inorg. Chem., 2018, 57, 14097–14105 CrossRef CAS PubMed.
- M. G. Chegerev, D. V. Korchagin, G. V. Shilov, N. N. Efimov, A. G. Starikov, A. V. Piskunov, A. V. Chernyshev, A. N. Bulgakov, V. I. Minkin, A. V. Palii and S. M. Aldoshin, Dalton Trans., 2022, 51, 16876–16889 RSC.
- A. Bannwarth, S. O. Schmidt, G. Peters, F. D. Sönnichsen, W. Thimm, R. Herges and F. Tuczek, Eur. J. Inorg. Chem., 2012, 2776–2783 CrossRef CAS.
- V. I. Minkin, A. G. Starikov and A. A. Starikova, Pure Appl. Chem., 2018, 90, 811–824 CrossRef CAS.
- M. Shoji, K. Koizumi, Y. Kitagawa, T. Kawakami, S. Yamanaka, M. Okumura and K. Yamaguchi, Chem. Phys. Lett., 2006, 432, 343–347 CrossRef CAS.
- G. A. Andrienko, Chemcraft - Graphical Software for Visualization of Quantum Chemistry Computations Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric analysis, PXRD, crystallographic parameters, IR, additional magnetic data, additional electronic spectroscopy, additional electrochemistry, additional DFT data, crystallographic files, DFT files. CCDC 2309300–2309302. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04162j |
| This journal is © The Royal Society of Chemistry 2024 |