
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies for arene dissociation from transition metal η6-arene complexes
Brett
Akana-Schneider†
 ,
Yahui
Guo†
,
Bryan
Parnitzke
and
Joseph
Derosa
*
,
Yahui
Guo†
,
Bryan
Parnitzke
and
Joseph
Derosa
*
Boston University, Department of Chemistry, Boston, MA 02215, USA. E-mail: derosaj@bu.edu
First published on 9th October 2024
Abstract
Transition metal η6-arene complexes have unique properties that facilitate a variety of arene substitution reactions, rendering π-activation a powerful approach for arene functionalization. For decades, these complexes have been studied in the context of coordination chemistry and synthetic methodology via stoichiometric reactivity; one central challenge in expanding the utility of arene functionalization via transition-metal-π-activation is the dissociation of the arene product that remains bound to the transition metal. In this perspective, we highlight representative strategies and methods for the removal and/or exchange of arenes from such complexes. Recent studies that implement these strategies toward catalytic processes are discussed, along with remaining challenges in this area.
Introduction
Organotransition metal complexes have served as powerful reagents and intermediates in a wide range of synthetic contexts. Complementary to conventional transition metal-catalyzed bond formations in which the metal is directly responsible for making and breaking carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds (i.e. cross-coupling),1–4 electronic activation of C–C π-bonds via coordination of transition metals is a powerful approach for introducing new functional groups (i.e. nucleometalation).5,6 Comparatively less developed than alkene and alkyne counterparts, transition metal π-complexes with arenes offer a powerful platform for modular functionalization of aromatic compounds. For decades, metal–arene π-complexes have been studied under the lens of coordination chemistry to probe the unique properties that η6-binding has on the bound arene.7 Depending on the identity of the metal center, oxidation state of the metal, and ligand environment, η6-bound arenes can be activated as substrates toward a variety of nucleophiles and/or electrophiles.8 This “π-activation” strategy has been demonstrated as a powerful method for accessing reactivity that traditional vicarious nucleophilic substitution (VNS), nucleophilic aromatic substitution (SNAr), and electrophilic aromatic substitution (SEAr) cannot. Using this reaction manifold, a wide range of C–C and C–N/O bonds can be installed in both SNAr- and C–H functionalization-type pathways. Despite the attractiveness of this approach, this reaction manifold is largely limited to conditions that employ stoichiometric preformed metal–arene complexes.When envisioning a catalytic cycle, arene exchange from the transition metal is critical. The first step in π-activation-mediated functionalization, addition of the desired coupling partner to the bound arene, generally leads to an η5-bound intermediate that rearomatizes to the product η6-bound arene (Scheme 1A). At this point, the bound arene must dissociate from the metal to yield the desired product. This key step presents a major challenge: selective dissociation of the product arene over starting material. This perspective highlights representative methods and strategies for arene dissociation from transition metal η6-arene complexes, with an outlook toward further adapting and innovating these protocols in catalytic contexts. Methods for arene decomplexation are generally organized based on: (1) photolytic or thermolytic cleavage in air, (2) use of chemical oxidants, and (3) ancillary ligand-assisted dissociation (Scheme 1B). Furthermore, we highlight recent efforts toward catalytic SNAr that utilize a combination of these strategies. While some functionalization reactions will be discussed in the context of subsequent arene dissociation, this perspective will not include exhaustive details on the types of compatible nucleophiles/electrophiles in this reactivity mode.9,10
 | ||
| Scheme 1 (A) General depiction of transition metal η6-arene complex undergoing nucleophilic substitution followed by rearomatization and dissociation of the arene product. (B) Overview of highlighted methods for arene dissociation discussed in this perspective. | ||
Thermolytic and photolytic arene dissociation
In 1970, pioneering work by Helling and coworkers demonstrated that bis(η6-mesitylene)Fe(II) complexes could undergo addition reactions with organolithium reagents at room temperature; in the absence of bound Fe(II), even temperatures as high as 165 °C led only to slow conversion.11 Subsequent heating of the corresponding (η6-mesitylene)(η5-arene)Fe(II) at 216 °C under air released the functionalized mesitylene product and mesitylene (Fig. 1). In this case, thermolysis under air served to both rearomatize the product and remove the bound Fe(II). Alternatively, the authors report that a solution of (η6-mesitylene)(η5-adduct)Fe(II) in pentane could be treated with chemical oxidants to affect the same sequence (vide infra). | ||
| Fig. 1 Helling and coworkers’ thermolysis under air to rearomatize and release Fe(II). | ||
Shortly after Helling's reports, a cyclopentadienyl (Cp) Fe(II) arene platform emerged as a more stable alternative to dicationic bis(arene)Fe(II) compounds. In 1982, Lee and coworkers showcased a double SNAr reaction using a CpFe(II)(η6-o-dichlorobenzene) complex with catechol to generate the corresponding η6-9,10-dioxaanthracene complex (Fig. 2A).12 Arene removal was achieved via pyrolytic sublimation at 200–250 °C at 1 torr, delivering 9,10-dioxaanthracene in 91% yield. Under these conditions, as shown by Nesmeyanov and coworkers,13 ferrocene and Fe(II) salts were generated by disproportionation. In 1997, Roberts and coworkers utilized CpFe(II)(η6-fluorobenzene) as a building block for SNAr with chiral amine substituents; they utilized flaked graphite in an 850 W microwave to yield N-phenyl-(s)-α-methylbenzylamine (Fig. 2B).14 Pearson and colleagues conducted a similar SNAr reaction using CpFe(II)(η6-p-dichlorobenzene) with phenolates and amines, and achieved decomplexation of the desired product after light irradiation for 4 h in acetonitrile (Fig. 2C).15
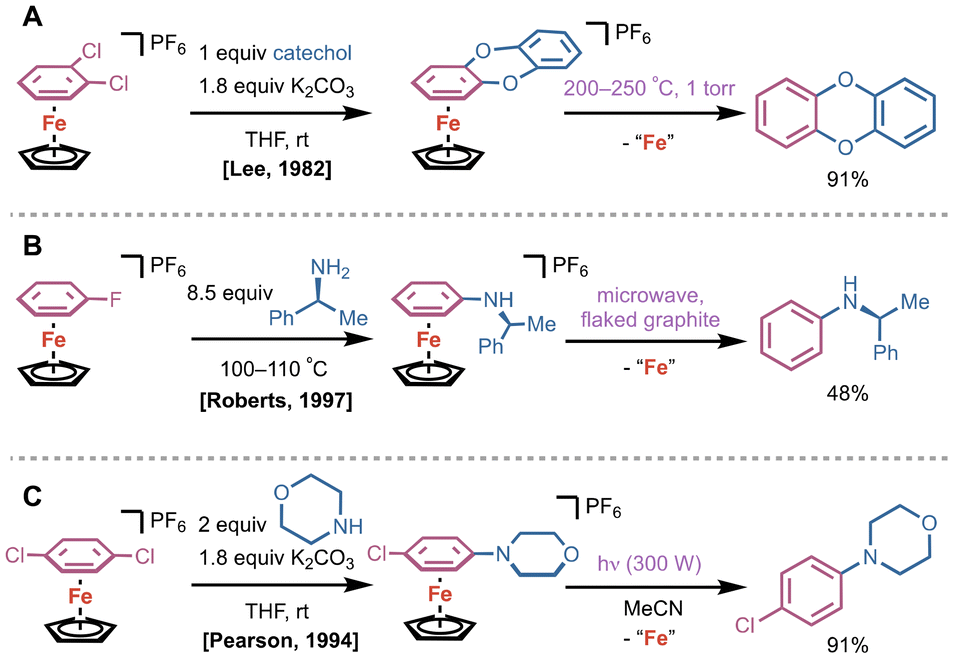 | ||
| Fig. 2 (A) Lee and coworkers’ synthesis of 9,10-dioxaanthracene using pyrolytic sublimation from the corresponding CpFe(II)(η6-arene). (B) Example of arene removal using flaked graphite in a microwave by Roberts. (C) Photolysis of a CpFe(II)(η6-arene) in MeCN solvent to yield desired N-aryl morpholine product. | ||
Using a related CpRu(II)(η6-arene) complex, Pearson later showcased the power of a π-activation strategy in an impressive multistep peptide coupling and macrocyclization en route to a ristocetin A model substrate.16 The synthesis relies on a CpRu(II) complex bound to a phenylalanine derivative which undergoes sequential peptide couplings and an intramolecular SNAr reaction with a phenol from the D ring of the structure. The combined yield of the SNAr reaction and demetallation using light was 46%, illustrating the robustness of this decomplexation protocol (Fig. 3A). In 2022, the same photolytic protocol was used with a related CpRu(II)(η6-arene) complex to dissociate SNAr products from 1,3-dione nucleophiles (Fig. 3B).
 | ||
| Fig. 3 (A) Photolytic dissociation of a CpRu(II)(η6-arene) with a macrocyclic peptide after sequential peptide coupling and SNAr. (B) Recent example of photolytic dissociation by Walton to generate substituted arenes via 1,3-dione addition. | ||
Photolytic dissociation methods have also been employed for arene dissociation from Cr-based complexes, particularly with respect to (CO)3Cr(η6-arene) systems. These typically involve initial photodissociation of a CO ligand, which promotes subsequent arene dissociation. In 1980, Trahanovsky demonstrated ortho-lithiation reactions of (CO)3Cr(η6-anisole) with n-BuLi at −40 °C followed by addition of iodomethane to yield (CO)3Cr(η6-2,6-dimethylanisole); the reaction was also shown to proceed with η6-bound fluorobenzene.17 Exposure to UV light resulted in quantitative decomplexation of 2,6-dimethylanisole (Fig. 4A). Various developments using this ortho-lithiation protocol emerged shortly after; in 1983, Uemura and coworkers used CO2 as an electrophile with (CO)3Cr(η6-benzyl alcohol) to synthesize a variety of phthalides and tetralones, albeit as minor products compared to methyl benzoates (Fig. 4B).18,19 Photoirradiation under air yielded the desired cyclization products in 65–71% yield depending on the substrate. Similar indanone-derived spirocyclic products could be generated via a stereocontrolled formal [3 + 2] annulation from ortho-trimethylsilyl benzaldehyde (CO)3Cr(η6-arene) complexes; in 2002, Moser and colleagues demonstrated that these spirocyclic products could be generated by sequential aldol condensation, Brook rearrangement and cyclization, followed by photoirradiation in Et2O (Fig. 4C).20 In an interesting demethoxylation reaction using LiAlH4 reported by Hacksell in 1991, the resultant (CO)3Cr(η6-arene) is dissociated using a combination of photoirradiation and ligand-induced displacement with gaseous ammonia.21
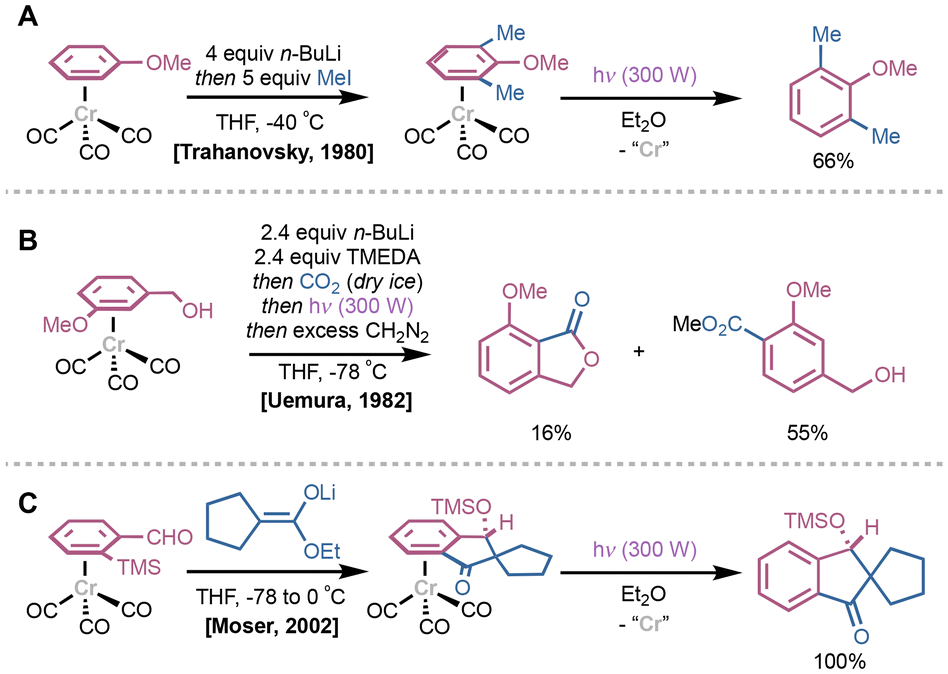 | ||
| Fig. 4 (A) Example of an ortho-lithiation/methylation sequence using (CO)3Cr(η6-anisole) and decomplexation under photoirradiation. (B) Tetralone and benzoate synthesis using CO2 as an electrophile after ortho-lithiation of a (CO)3Cr(η6-arene) followed by photolysis to release product. (C) Formal [3 + 2] to access substituted indanones following photodissociation from product (CO)3Cr(η6-arene) complex. | ||
Reagent-driven arene dissociation
Along with photo- and thermolytic methods, subsequent addition of a chemical reagent is an alternative strategy for arene dissociation. These are most commonly in the form of stoichiometric oxidants or Lewis bases that drive dissociative ligand exchange. In 1979, Semmelhack and coworkers conducted a study on substituent effects of (CO)3Cr(η6-arene) complexes on lithium carbanion addition; the intermediate Meisenheimer-like (CO)3Cr(η5-bound) species could be simultaneously rearomatized and released from the Cr center by treatment with excess I2 (Fig. 5A).22 Notably, one of the first examples of ortho-lithiation of (CO)3Cr(η6-arene) complexes and subsequent treatment of electrophiles reported by Semmelhack utilized the same strategy.23 In 1991, Kündig and coworkers reported a similar protocol for decomplexation using I2 after nucleophilic addition of dithianyllithium to a (CO)3Cr(η6-naphthalene) complex.24 Interestingly, this report disclosed that difunctionalized products could be accessed in a stepwise fashion starting from (CO)3Cr(η6-OMe-naphthalene) and treatment with dithianyllithium followed by iodomethane under CO atmosphere. To prevent rearomatization but release product, PPh3 was added to drive decomplexation via ligand exchange. In addition to I2, N-bromo succinimide (NBS) is commonly used as a chemical oxidant to rearomatize η5-bound species and/or dissociate the desired arene for a variety of transition metal complexes. Trahanovsky demonstrated treatment with NBS at −40 °C as an alternative to photoirradiation on a series of (CO)3Cr(η6-arene) complexes.17 In 1981, Rose and coworkers generated either CpFe(II)(η6-arene) or the dissociated arene from a CpFe(II)(η5-adduct) by controlling the stoichiometry of NBS; one equivalent of NBS carried out rearomatization of the bound arene, while excess NBS directly released product (Fig. 5B).25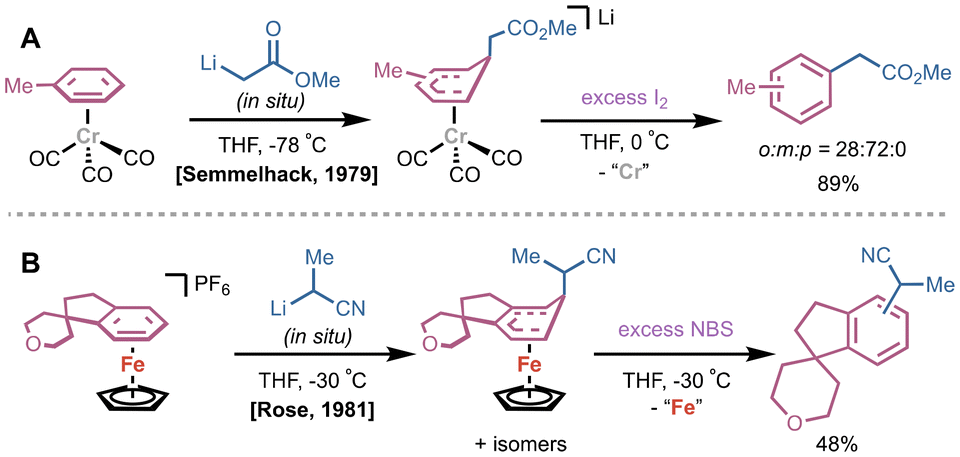 | ||
| Fig. 5 (A) Early report by Semmelhack utilizing I2 to rearomatize and dissociate arene. (B) Example by Rose of NBS oxidation of a CpFe(II)(η5-adduct) where excess NBS releases rearomatized product. | ||
In cases where a halogen-based oxidant may limit functional group compatibility on the arene fragment, alternative oxidants have been employed. In Helling's 1970 report on the functionalization of [bis(η6-mesitylene)]Fe(II) complexes with organolithium nucleophiles (vide supra), it was shown that quenching with dilute aqueous KMnO4 or ceric ammonium nitrate (CAN) yielded desired mesityl derivatives in up to 95% yield.11 In 1989, Miles and coworkers reacted lithium enolate nucleophiles with (CO)3Mn(I)(η6-arene) complexes at 0 °C for 1 hour, yielding the (CO)3Mn(I)(η5-bound) addition intermediate. Importantly, this was extended to lithium enolates bearing chiral auxiliaries to generate enantiopure α-arylation products. Upon treatment with 2 equivalents of 2,3-dichloro-5,6-dicyanobenzquinone (DDQ), rearomatization and Mn removal occurred to deliver the α-arylation product (Fig. 6A).26 Woodgate reported a DDQ rearomatization/dissociation sequence on Fe(II)- and Ru(II)(η6-arene) complexes after SNAr with silyl-group-containing nucleophiles.27 Recently in 2017, Miles and coworkers highlighted a unique site-selectivity of direct lithio-benzofuran addition to a (CO)3Mn(I)(η6-2,6-dimethoxytoluene) complex en route to the stemofuran family of natural products (Fig. 6B).28 Subsequent treatment with CAN or DDQ delivered stemofuran precursors as a single isomer in 93% yield.
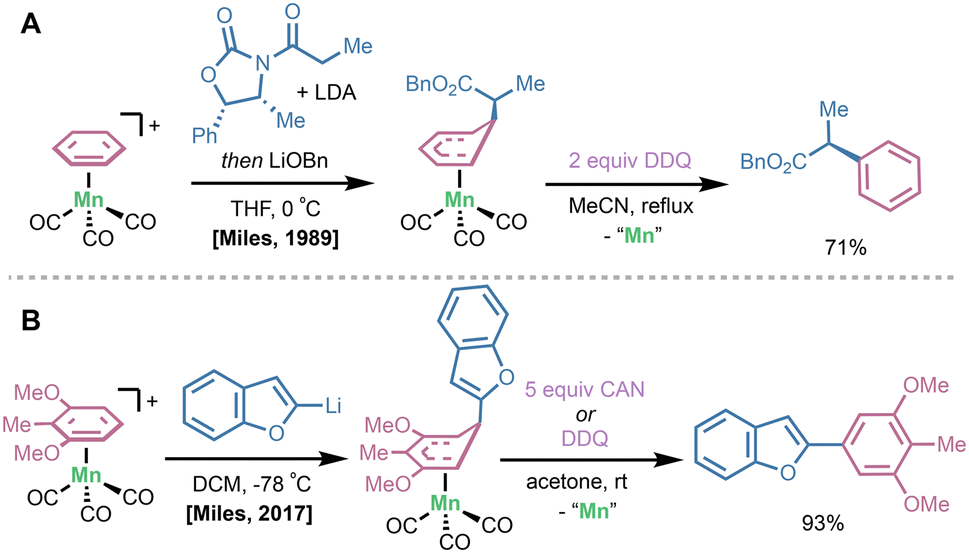 | ||
| Fig. 6 (A) Decomplexation via DDQ oxidation to yield enantiopure α-arylation products derived from chiral enolate addition to (CO)3Mn(I)(η6-arene) complexes. (B) Stemofuran precursor synthesis by Miles and coworkers demonstrating CAN oxidation to rearomatize and dissociate product arene. | ||
Ligand-based arene dissociation
Given that many reagent-based arene dissociation strategies render the metal center unreactive toward further π-complexation, one attractive strategy to enable catalytic π-activation of arenes is the design of ligands that accelerate arene dissociation and exchange. The resulting ligand systems can be generally categorized in one of two ways: (1) hemilabile ligands that stabilize the transition from the η6-to η4-bound arene; or (2) η5-ligands that enable direct dissociation or accelerate association of other donors.In 2005 Semmelhack and coworkers reported their efforts in the design of tris(pyrrolyl)phosphine ligands bearing additional chelating groups on one pyrrole substituent.29 Based on the acceleration of arene exchange by coordinating solvents or additives (Fig. 7A), they hypothesized that incorporation of a coordinating side chain would greatly accelerate arene exchange while minimizing the possibility of deactivating the metal center via coordinative saturation.30,31 They found that ligands bearing groups with coordinating lone pairs and low-lying π* orbitals—such as 2-pyridyl or an ester—greatly accelerated arene exchange in LCr(CO)2(η6-fluorobenzene) complexes compared to the parent tris(pyrrolyl)phosphine (Fig. 7B). While the unfunctionalized tris(pyrrolyl)phosphine provided slow exchange at 150 °C, the fastest chelating ligands enabled measurable rates at room temperature, and unmeasurably fast exchange at 70 °C.
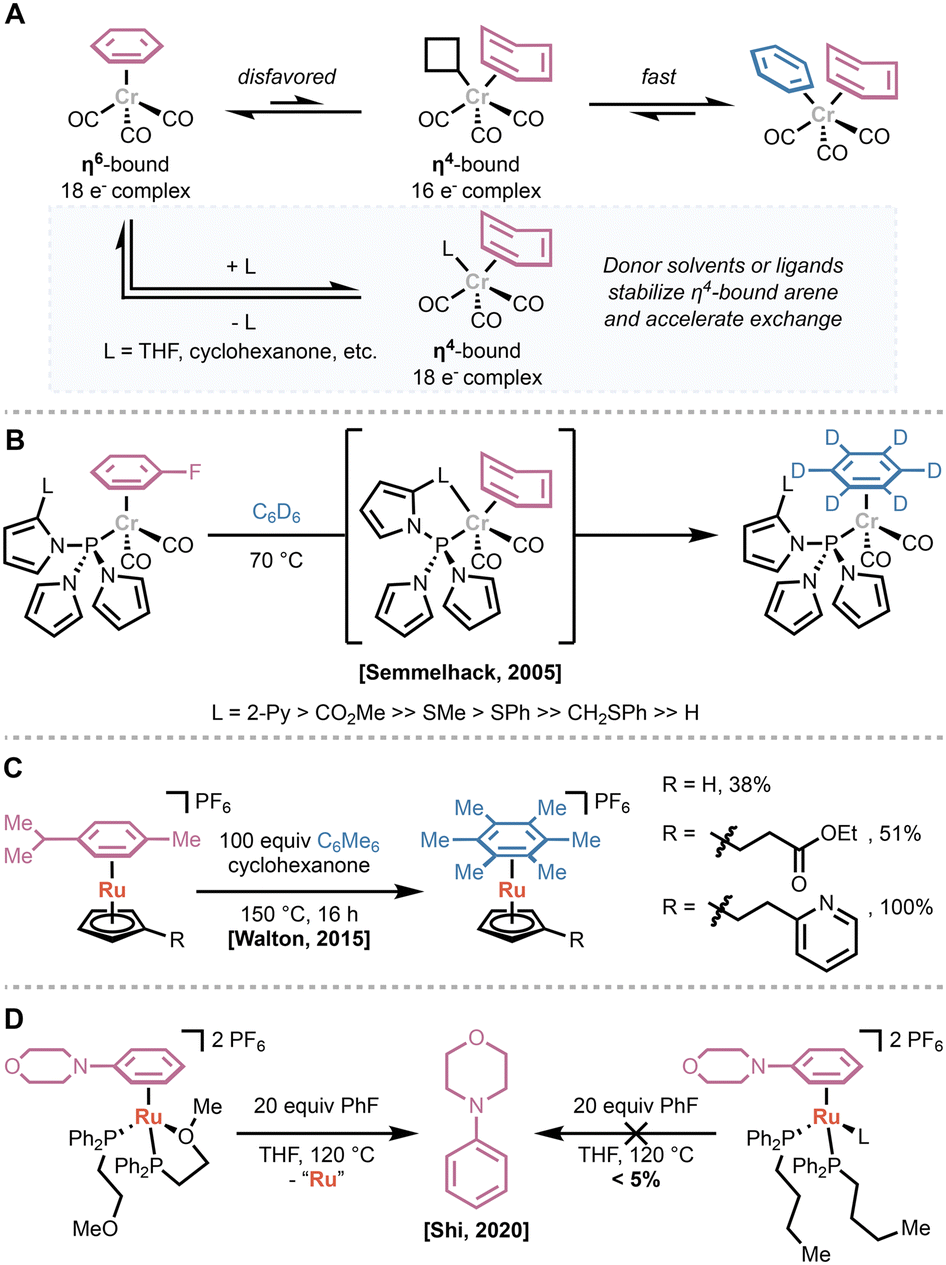 | ||
| Fig. 7 (A) Rate limiting dissociation of the η6-arene ligand to form an η4-bound species can be accelerated by addition of donor solvents or ligands. (B) Hemilabile tris(pyrrolyl)phosphine ligands accelerate arene exchange from 18-electron chromium complexes. (C) Incorporation of chelating groups to Cp ligands accelerates arene exchange in ruthenium(II) complexes. (D) Demonstration of Shi's hemilabile phosphine ligands that enable exchange of arylamines from ruthenium(II) complexes. | ||
In 2015, Walton and Williams applied a similar approach to the development of Cp-style ligands to accelerate the ruthenium-catalyzed nucleophilic aromatic substitution of chloroarenes (vide supra).32 They found that tethering donor ligands—with an appropriately long carbon spacer, such as carbonyl derivatives or a 2-pyridyl group—increased the rate of arene exchange by up to 18-fold compared to the unfunctionalized CpRu(II)(η6-arene) catalyst (Fig. 7C). Despite this acceleration, they did not see a concomitant increase in the rate or yield of catalytic reactions. Finally, Shi and coworkers developed hemilabile phosphine ligands to promote arene exchange in LRu(II)I(η6-arene) complexes (Fig. 7D).33 They found that incorporating a pendant methoxy group on the phosphine enables dissociation, even when the leaving arene is significantly more tightly binding than the incoming arene.
Other studies have focused on identifying η5-, Cp-style ligands that promote arene dissociation and exchange. Loginov and coworkers used arene exchange as a model for catalyst initiation in rhodium-catalyzed reductive amination using carbon monoxide as a reductant.34 They found that more donating, permethylated Cp* and indenyl ligands provided faster exchange than unfunctionalized variants (Fig. 8A). Given the large atomic radius of rhodium, they concluded that the increase in the rate of exchange is due to electronic factors rather than steric effects or indenyl slippage. Computational studies suggested that the more donating ligands stabilize the [LRh(III)]2+ fragment, decreasing the strength of the Rh–(η6-arene) bond.
 | ||
| Fig. 8 (A) Electron-rich η5-ligands provide improved rates of arene exchange. (B) Ritter's η5-phenoxo ligands provide improved rates of arene exchange due to ligand–arene interactions and anion-promoted η6–η2 transition. | ||
Recently, the Ritter group described the systematic study and design of η5-phenoxo ligands to promote arene exchange in LRu(II)(η6-arene) complexes.35 They found that η5-phenoxo ligands provide a greater than 20-fold increase in the rate of arene exchange when compared to the isolobal CpRu(II)I(η6-arene) complex (Fig. 8B). Incorporating an electron-donating substituent in the 4-position of the ligand led to an increase in the rate of arene exchange due to a productive interaction between the donor substituent and the counterion. Adding halide ligands to the 2- and 6-positions of the phenoxo also led to an increase in the exchange rate due to a stabilizing hydrogen bond interaction between the halide and the dissociating arene proton in the transition state between η6- and η2-arene binding. Additionally, they found that more coordinating sulfonate anions increased the rate of arene exchange by interacting with both the leaving arene and the metal center (Fig. 8B).
Other studies have highlighted the ability of specific ligand classes to promote rapid arene exchange or have compared the relative rates of arene dissociation between ligands in a specific class. The lower prevalence of many of these ligands compared to phosphines or cyclopentadienyl ligands and their unproven application to catalytic η6-activation make them less applicable to this perspective.10,36,37
Catalytic examples of arene dissociation
Despite the expansive study of stoichiometric activation and decomplexation of η6-bound arenes, catalytic applications remain limited. Incompatibility inherent to solvolysis or chemical methods render the metal center inactive for additional turnover. As a result, many examples of catalytic π-activation of arenes leverage a combination of: (1) accelerated rates of arene exchange on metal centers with large atomic radii; (2) electron-rich Cp*-style ligands; and (3) inherent thermodynamic favorability due to a decrease in the donicity of the arene following functionalization. One area that experienced exciting developments from a π-activation standpoint is benzylic functionalization; however, we elected to instead focus on a rapidly emerging area that builds on catalyst development strategies.38–43Beyond α-functionalization of arenes, the other category of reactions in which π-activation has seen significant interest is in promoting SNAr reactions.44 The strong electron-withdrawing properties of η6-bound metal cations enable substitutions that are typically thermodynamically inaccessible or require alternative strategies involving C–X oxidative addition or arene oxidation. While attractive, catalytic η6-activation is complicated by possible deactivation of the metal center by the nucleophile and a strong thermodynamic preference for binding the product arene over the starting material due to the increase in donicity following substitution of the nucleofuge.45 The earliest example of catalytic η6-activation was reported by Houghton, Voyle, and Price in 1984.46 They described the rhodium-catalyzed cyclization of 3-(2-fluorophenyl)propanols to generate chromanes (Fig. 9A). While previous work had found that simple tricarbonyl-metal complexes promoted the desired SNAr reaction under basic conditions, the necessity of strong associating bases prevented catalytic turnover.47 They found that ethyl-tetramethylcyclopentadienyl rhodium(III) salts were capable of promoting cyclization even under neutral conditions, enabling catalytic turnover. Notably, the identity of the counterion impacted the activity of the catalyst significantly, with the PF6 salts often providing a 1.2–1.8 times increase in turnover compared to the BF4 equivalent. Altogether, this early report demonstrated that careful selection of the metal, supporting ligand, and solvent can enable catalytic π-activation strategies, even in the presence of nucleophilic functionalities.
 | ||
| Fig. 9 (A) Houghton's synthesis of chromanes via η6-activation using Rh(III) catalysts. (B) Shibata's two catalyst systems enable amination of unactivated fluoroarenes using either a silane/base mixture or molecular sieves. (C) The active catalyst in Shibata's DPPPent system is the L2Ru(II)(H)(η6-arene), which is stable and catalytically active, while the cyclometallated species formed in the absence of a silane is not. (D) An example of how π-activation using Shi's hemilabile ruthenium(II) catalyst system provides SNAr orthogonal to traditional thermochemical SNAr reactions. | ||
Despite the early success of Houghton and Price, no advances in catalytic π-activation-promoted SNAr were published for 25 years. In 2009, Shibata and coworkers published the first ruthenium-catalyzed amination of unactivated fluoroarenes (Fig. 9B).48 This system overcame many obstacles: the large thermodynamic preference for binding of the product over the starting material; possible deactivation of the ruthenium(II) center via saturation with the nucleophile; and deactivation due to the production of HF. They found that DPPPent—a wide-bite-angle bisphosphine ligand—in combination with stoichiometric triethylamine and triethylsilane provided the highest yield. To overcome the thermodynamic preference for binding of the product N-arylmorpholine over the starting fluoroarenes, they employed an excess of the fluoroarene starting material. Shibata later published an alternative catalytic system that utilized two equivalents of the monodentate tris(4-fluorophenyl)phosphine rather than DPPPent, and 4 Å molecular sieves to adsorb the formed HF.49 Notably, both wide-bite-angle bisphosphines and bis-monodentate phosphinyl ligand systems have been shown to accelerate arene dissociation in related rhodium(I) complexes.50 In-depth mechanistic analysis by Schley and Mueller demonstrated that the silane additive functioned not as an HF or F− adsorbent, but to convert the ruthenium catalyst into the η6-product-bound ruthenium(II) hydride (Fig. 9C).45 This ruthenium(II) hydride was identified as the resting state of the catalyst, and was stable and soluble under the reaction conditions, unlike the κ3 C–H-activated ruthenocycle. Kinetic and equilibrium measurements established that the rate-limiting step of the reaction was arene exchange, and that the product arene had a binding affinity approximately 2000 times greater than the fluoroarene. Finally, they established that triethylamine was unnecessary, only functioning to generate the triflate anion, which is critical to enable arene exchange. Overall, the work by Shibata revitalized interest in catalytic π-activation and provided a basis for further advancements.
In 2020, Shi and coworkers published the development and application of hemilabile phosphine ligands that accelerate arene exchange in L2Ru(II)(η6-arene) complexes (vide supra).33 The use of these ligands enabled amination of un- or deactivated fluoroarenes with excess amine rather than fluoroarene. As a result, Shi's system could be used to maximize the yield of amination products with respect to highly functionalized arenes. Notably, neither bisphosphines nor non-hemilabile phosphine ligands provided significant yield of the desired product (<2% yield). Additionally, the preference for complexation of electron-rich arenes enabled reactivity orthogonal to traditional SNAr reactions (Fig. 9D).
Other studies focused on conversion of chloroarenes rather than fluoroarenes, utilizing Cp-style ligands to promote the desired substitution. In 2015, Walton and Williams described the use of CpRu(II)(η6-arene) catalysts to afford amination of 4-chlorotoluene.32 They found that the efficiency of the reaction depended greatly on the solvent employed. While reactions in cyclohexanone provided 16% conversion after 18 h, using 1-octanol improved conversion to 25%. Increasing the reaction time to 14 days led to 90% conversion of 4-chlorotoluene. In the same year, Grushin and coworkers published the fluorination of haloarenes with catalytic Cp*Ru(η6-naphthalene)BF4.51 They found that reactions provided the highest yield when run neat at 180 °C. While turnover numbers were relatively low (3–9 TON in many cases), these results demonstrate that π-activation can be used with diverse arene electrophiles.
Extension of existing ruthenium(II) catalyst systems to engage more diverse nucleophiles was hampered by the low overall electrophilicity imparted by the monocationic ruthenium(II) center in CpRu(II)(η6-arene) complexes.52 In 2021, Shi and coworkers demonstrated that neither ruthenium(II) catalysts utilizing their hemilabile phosphine ligands nor Cp* were capable of activating fluorobenzene such that substitution with water occurred under neutral conditions.53 While using a hydroxide nucleophile did provide the substituted product, the resulting η6-bound phenol was rapidly deprotonated to form the non-exchangeable η5-phenoxo complex, preventing possible catalytic turnover (Fig. 10A). They found that a strategy analogous to that published by Houghton—the use of highly withdrawing rhodium(III) catalysts—allowed for the substitution of bound fluoroarenes under neutral conditions. This strategy allowed for the catalytic hydroxylation or alkoxylation of a variety of fluoroarenes with only three equivalents of water or an alcohol coupling partner (Fig. 10B). Later work by Shi leveraged the strong activating effects of rhodium(III) centers to allow for hexafluoroisopropoxylation of less electrophilic chloro- or bromoarenes using the typically non-nucleophilic hexafluoroisopropanol (HFIP) as a coupling partner.54 Since binding of the nucleofuge would deactivate the rhodium(III) catalyst, they found that adding stoichiometric basic silver salts, such as Ag2CO3 or Ag2O, was necessary to provide the desired product.
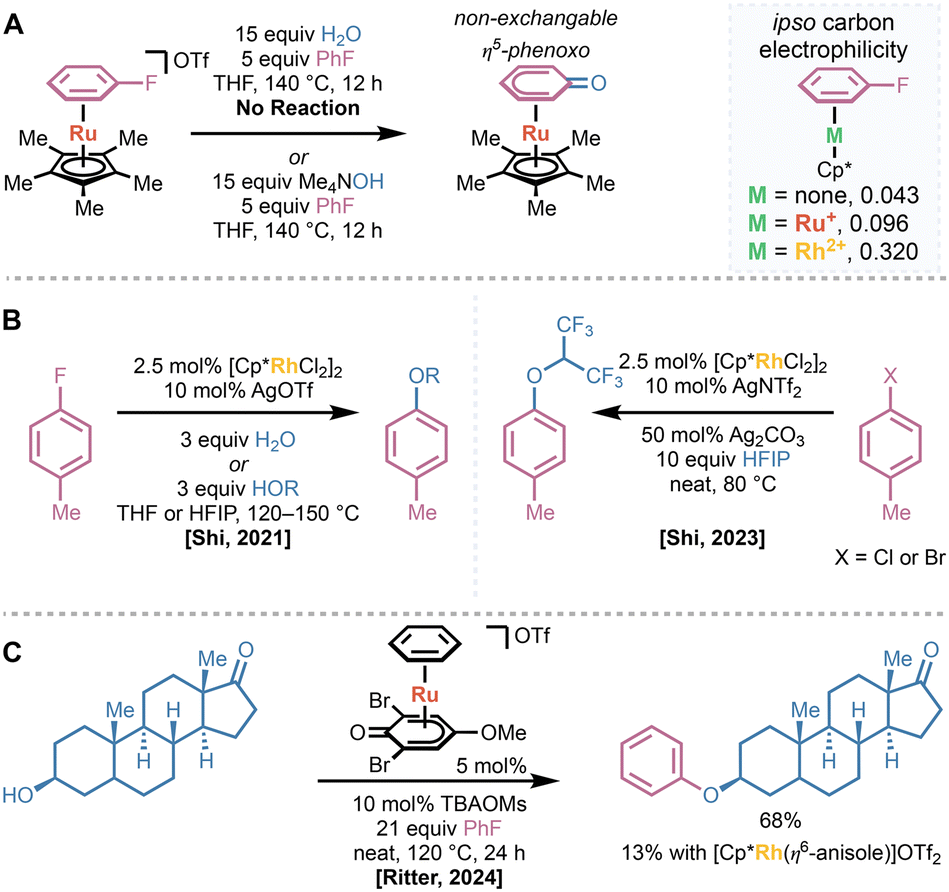 | ||
| Fig. 10 (A) Many ruthenium(II) catalyst systems are insufficiently activating to promote hydroxylation of fluoroarenes under neutral conditions. Electrophilicity calculations demonstrate that using a more oxidized rhodium(III) center is significantly more activating. (B) Examples of hydroxylation and alkoxylation of fluoroarenes using rhodium(III) catalysts. (C) Ritter's phenoxo ligands are sufficiently withdrawing such that ruthenium(II) catalysts can promote alkoxylation of fluoroarenes, even with secondary alcohols. | ||
In 2024, Ritter demonstrated that utilizing more withdrawing ruthenium(II) catalysts can also provide alkoxylation of fluoroarenes, obviating the need for expensive rhodium-based catalysts.35 They found that η5-phenoxo ligands were sufficiently activating to enable substitution of bound fluoroarenes even with secondary alcohols under neutral conditions. When used in concert with catalytic amounts of tetrabutylammonium mesylate (TBAOMs)—which accelerated arene exchange compared to triflate—their optimized catalyst provided arylation of epandrosterone, in significantly higher yield than [Cp*Rh(III)(η6-anisole)]OTf2 (Fig. 10C). Further, they demonstrated that the same catalyst can promote thermal decarboxylation of phenylacetic acid derivatives at significantly lower temperatures than traditional methods.
Recently, the Shi group has shown that substitution of 2-aminopyridines with amines can occur in the presence of 1,2,4-tri-tert-butylcyclopentadienyl ruthenium(II) catalysts.55 While the identity of the ligand is necessary to modulate the efficiency of the substitution, it is unclear if the electronic and steric properties of the Cp(t-Bu)3 ligand promote arene exchange, or if a combination of κ-N binding of the starting material or nucleophile accelerate arene dissociation. Binding studies suggest that, while κ-N coordination of the starting 2-aminopyridine can occur, transition to the η6-coordination mode occurs rapidly under the reaction conditions.
While deprotonation of phenols to form η5-phenoxo complexes is degradative in direct SNAr reactions, it provides an alternative pathway for functionalization. In 2022, the Shi group showed that, when bound to a rhodium(III) center, η5-phenoxo compounds can undergo dehydrative condensation with amines to give an η5-iminium, which tautomerizes to form an η6-bound aniline (Fig. 11A).56 The resulting catalytic reaction functions as a pseudo-SNAr of phenols with amines (Fig. 11B). Notably, while exchange of an aniline for a phenol is electronically unfavorable, deprotonation to from the η5-phenoxo provides a thermodynamic driving force for the desired exchange. Later work from Shi and coworkers expanded this type of reaction to employ ruthenium(II) catalysts.57 Concurrent with the Ritter group, they discovered that the use of η5-phenoxo ligands both accelerated arene exchange and improved the electrophilicity of the bound arene (Fig. 11C). They demonstrated the change in the electrophilicity of the bound phenol ligand by reacting both Ru(II)(p-cresol)2 and CpRu(I)(p-cresol) with iodomethane under basic conditions. They found that the Cp* complex exclusively provided the anisole derivative, indicating that the phenol was nucleophilic. Contrastingly, the bis(p-cresol) complex yielded the neutral Ru(II)(η5-p-Me-phenoxo) compound. Methylation was only possible upon heating the complex to 80 °C with excess iodomethane. Interestingly, the use of η6-phenol/η5-phenoxo ligands allowed Shi and coworkers to provide amination products in an effectively ligandless system, where the starting phenol derivative (used in slight excess) functioned as both the ligand and the substrate. This method simplified the reaction setup and prevented any possible conversion of an exogenous phenoxo ligand to the substituted aniline.
 | ||
| Fig. 11 (A) Outline of the strategy that promotes arene exchange and dehydrative amination of phenols using (Me4RCp)Rh(III)(η6-phenol) complexes. (B) Examples of Shi's dehydrative amination of phenols using both rhodium and ruthenium catalysts. (C) Comparison of the relative nucleophilicity and electrophilicity of LRu(II)(η6-p-cresol) complexes. aR = 3,5-bis(trifluoromethyl)phenyl. bX = 1. cX = 2. | ||
Together, the advances in nucleophilic substitution reactions catalyzed by η6-binding of arenes to ruthenium(II) and rhodium(III) centers highlight many of the approaches used to promote arene exchange. The design of new hemilabile or phenoxo ligands, careful selection of solvent, and changes in thermodynamic preference due to favorable tautomerization all provide interesting approaches that enable their specific reaction. Moving forward, it seems likely that further advances will continue to improve the scope, practicality, and applicability of these strategies to existing and new functionalizations.
Conclusions
Arene removal and exchange from transition metal η6-arene complexes serves as a critical enabling feature of modular functionalization via transition metal π-complexes. The examples mentioned here highlight the strategies by which dissociation of the functionalized arene from a transition metal η6-arene complex is promoted.Early work done by Helling and Trahanovsky showcased rearomatization and dissociation of the functionalized arene product via thermolysis or photolysis. Alternatively, chemical reagents—most commonly chemical oxidants such as iodine or NBS—may also be used to rearomatize and dissociate the arene product. While these early strategies helped develop the understanding of the conditions necessary to promote arene dissociation and allowed for the use of stoichiometric π-activation in synthetic campaigns, milder strategies that were compatible with diverse metal centers and reagents were necessary to enable applications of π-activation in catalytic contexts.
Guided by ligand electronics and chelating effects, recent advances in the design of catalysts that accelerate arene displacement have revitalized interest in catalytic SNAr enabled by π-activation of arenes. Early work by Houghton demonstrated that leveraging the synergistic effects of metal identity, ligand electronics, counterion effects, and solvent identity could overcome the unfavorable thermodynamics of product inhibition in π-arene SNAr. Following pioneering work by Shibata in 2009, the understanding of each of these effects has grown due to fundamental and catalytic studies by many groups. Most recently, Shi and Ritter demonstrated that new scaffolds such as η5-phenoxo ligands can accelerate arene exchange via new factors, such as favorable tautomerization and ligand–arene interactions. Together, the field is poised to largely overcome the issues of product inhibition, slow exchange rates, and insufficient activation that have plagued the development of new π-activation methods.
Contributions over the past decade in SNAr promoted by catalytic π-activation have yielded insight into underexplored strategies of catalytic arene displacement. While these advances have laid the groundwork for future applications of catalytic π-activation, additions to and careful navigation of the existing schema will be necessary to afford efficient and novel catalytic functionalization of arenes via transition metal η6-arene complexes. However, further advances are still required to extend catalytic insights summarized herein toward the development of first-row transition metals as competent π-activation catalysts.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Boston University (BU) for financial support.References
- C. C. C. Johansson Seechurn, M. O. Kitching, T. H. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef PubMed.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef PubMed.
- L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef PubMed.
- M. A. Bennett, Coord. Chem. Rev., 1997, 225–254 CrossRef.
- M. Rosillo, G. Dominguez and J. Pérez-Castells, Chem. Soc. Rev., 2007, 37, 1589–1604 RSC.
- L. J. Williams, Y. Bhonoah, L. A. Wilkinson and J. W. Walton, Chem. – Eur. J., 2020, 27, 3650–3660 CrossRef PubMed.
- N. V. Shvydkiy and D. S. Perekalin, Coord. Chem. Rev., 2020, 411, 213238 CrossRef.
- J. F. Helling and D. M. Braitsch, J. Am. Chem. Soc., 1970, 92, 7207–7209 CrossRef.
- R. G. Sutherland, A. Piórko, U. S. Gill and C. C. Lee, J. Heterocycl. Chem., 1982, 19, 801–803 CrossRef.
- A. N. Nesmeyanov, N. A. Vol'kenau and I. N. Bolesova, Dokl. Akad. Nauk SSSR, 1967, 175, 606–611 Search PubMed.
- Q. Dabirmanesh, S. I. S. Fernando and R. M. G. Roberts, J. Chem. Soc., Perkin Trans. 1, 1995, 743–747 RSC.
- A. J. Pearson and A. M. Gelormini, J. Org. Chem., 1994, 59, 4561–4570 CrossRef.
- A. J. Pearson and M. V. Chelliah, J. Org. Chem., 1998, 63, 3087–3098 CrossRef.
- R. J. Card and W. S. Trahanovsky, J. Org. Chem., 1980, 45, 2560–2566 CrossRef.
- M. Uemura, N. Nishikawa, K. Take, M. Ohnishi, K. Hirotsu, T. Higuchi and Y. Hayashi, J. Org. Chem., 1983, 48, 2349–2356 CrossRef.
- M. Uemura, K. Take, K. Isobe, M. T. Minami and Y. Hayashi, J. Org. Chem., 1985, 41, 5771–5778 Search PubMed.
- W. H. Moser, J. Zhang, C. S. Lecher, T. L. Frazier and M. Pink, Org. Lett., 2002, 4, 1981–1984 CrossRef PubMed.
- M. Persson, U. Hacksell and I. Csöregh, J. Chem. Soc., Perkin Trans. 1, 1991,(6), 1453–1459 RSC.
- M. F. Semmelhack, G. R. Clark, R. Farina and M. Saeman, J. Am. Chem. Soc., 1979, 101, 217–218 CrossRef.
- M. F. Semmelhack, J. Bisaha and M. Czarny, J. Am. Chem. Soc., 1979, 101, 768–770 CrossRef.
- E. P. Kündig, M. Inage and G. Bernardinelli, Organometallics, 1991, 10, 2921–2930 CrossRef.
- J. C. Boutonnet and E. Rose, J. Organomet. Chem., 1981, 221, 157–163 CrossRef.
- W. H. Miles, P. M. Smiley and H. R. Brinkman, J. Chem. Soc., Chem. Commun., 1989, 24, 1897–1899 RSC.
- R. C. Cambie, S. A. Coulson, L. G. Mackay, S. J. Janssen, P. S. Rutledge and P. D. Woodgate, J. Organomet. Chem., 1991, 409, 385–409 CrossRef.
- W. H. Miles, C. M. Madison, C. Y. Kim, D. J. Sweitzer, S. D. Valent and D. M. Thamattoor, J. Organomet. Chem., 2017, 851, 218–224 CrossRef.
- M. F. Semmelhack, A. Chlenov and D. M. Ho, J. Am. Chem. Soc., 2005, 127, 7759–7773 CrossRef.
- P. E. Kündig, M. Kondratenko and P. Romanens, Angew. Chem., Int. Ed., 1998, 37, 3146–3148 CrossRef.
- T. G. Traylor, K. Stewart and M. Goldberg, J. Am. Chem. Soc., 1984, 106, 4445–4454 CrossRef.
- J. W. Walton and J. M. J. Williams, Chem. Commun., 2015, 51, 2786–2789 RSC.
- Q.-K. Kang, Y. Lin, Y. Li and H. Shi, J. Am. Chem. Soc., 2020, 142, 3706–3711 CrossRef PubMed.
- V. B. Kharitonov, M. Makarova, M. A. Arsenov, Y. V. Nelyubina, O. Chusova, A. S. Peregudov, S. S. Zlotskii, D. Chusov and D. A. Loginov, Organometallics, 2018, 37, 2553–2562 CrossRef.
- T. Schulte, Z. Wang, C.-C. Li, A. Hamad, F. Waldbach, J. Pampel, R. Petzold, M. Leutzsch, F. Bahns and T. Ritter, J. Am. Chem. Soc., 2024, 146, 15825–15832 CrossRef PubMed.
- N. V. Shvydkiy, E. A. Trifonova, A. M. Shved, Y. V. Nelyubina, D. Chusov, D. S. Perekalin and A. R. Kudinov, Organometallics, 2016, 35, 3025–3031 CrossRef.
- H. Tobita, K. Hasegawa, J. J. G. Minglana, L.-S. Luh, M. Okazaki and H. Ogino, Organometallics, 1999, 18, 2058–2060 CrossRef.
- S. Takemoto, E. Shibata, M. Nakajima, Y. Yumoto, M. Shimamoto and H. Matsuzaka, J. Am. Chem. Soc., 2016, 138, 14836–14839 CrossRef PubMed.
- Q. Kang, Y. Li, K. Chen, H. Zhu, W. Wu, Y. Lin and H. Shi, Angew. Chem., Int. Ed., 2022, 61, e202117381 CrossRef PubMed.
- Y. Li, W. Wu, H. Zhu, Q. Kang, L. Xu and H. Shi, Angew. Chem., Int. Ed., 2022, 61, e202203820 CrossRef PubMed.
- Y. Li and H. Shi, Chin. Chem. Lett., 2024, 35, 108650 CrossRef.
- W.-Q. Wu, Y. Lin, Y. Li and H. Shi, J. Am. Chem. Soc., 2023, 145, 9464–9470 CrossRef PubMed.
- Y. Lin and H. Shi, J. Am. Chem. Soc., 2023, 145, 22753–22761 CrossRef PubMed.
- K. Chen and H. Shi, Acc. Chem. Res., 2024, 57, 2194–2206 CrossRef CAS.
- B. R. J. Mueller and N. D. Schley, Dalton Trans., 2020, 49, 10114–10119 RSC.
- R. P. Houghton, M. Voyle and R. Price, J. Chem. Soc., Perkin Trans. 1, 1984, 925, 10.1039/p19840000925.
- R. P. Houghton, M. Voyle and R. Price, J. Organomet. Chem., 1983, 259, 183–188 CrossRef CAS.
- M. Otsuka, K. Endo and T. Shibata, Chem. Commun., 2010, 46, 336–338 RSC.
- M. Otsuka, H. Yokoyama, K. Endo and T. Shibata, Synlett, 2010, 2601–2606 Search PubMed.
- S. D. Pike, I. Pernik, R. Theron, J. S. McIndoe and A. S. Weller, J. Organomet. Chem., 2015, 784, 75–83 CrossRef.
- A. I. Konovalov, E. O. Gorbacheva, F. M. Miloserdov and V. V. Grushin, Chem. Commun., 2015, 51, 13527–13530 RSC.
- Other ligands such as phosphines have been shown to dignificantly decrease the activating properties of η6-arene complexes. For example, see: M. F. Semmelhack, G. Hilt and J. H. Colley, Tetrahedron Lett., 1998, 39, 7683–7686 CrossRef.
- Q. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Angew. Chem., Int. Ed., 2021, 60, 20391–20399 CrossRef PubMed.
- J. Su, K. Chen, Q. Kang and H. Shi, Angew. Chem., Int. Ed., 2023, 62, e202302908 CrossRef PubMed.
- J. Chen, Y. Lin, W.-Q. Wu, W.-Q. Hu, J. Xu and H. Shi, J. Am. Chem. Soc., 2024, 146, 22906–22912 CrossRef PubMed.
- K. Chen, Q.-K. Kang, Y. Li, W.-Q. Wu, H. Zhu and H. Shi, J. Am. Chem. Soc., 2022, 144, 1144–1151 CrossRef PubMed.
- K. Chen, Y. Ma, Y. Lin, J.-Y. Li and H. Shi, J. Am. Chem. Soc., 2024, 146, 15833–15842 CrossRef PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |