
Chemistry of organometallic nucleic acid components: personal perspectives and prospects for the future
Mateusz
Klarek
 and
Konrad
Kowalski
*
and
Konrad
Kowalski
*
University of Łódź, Faculty of Chemistry, Department of Organic Chemistry, Tamka 12, 91-403 Łódź, Poland. E-mail: konrad.kowalski@chemia.uni.lodz.pl
First published on 1st November 2024
Abstract
Organometallic modifications of biologically important compounds such as drugs, secondary natural products, peptides, and nucleic acids, to name just a few, represent a well-established strategy for the development of new anticancer and antimicrobial agents. Supported by these reasons, over 12 years ago, we initiated a research program into organometallic modifications of nucleic acid components. This account summarizes key results regarding the synthetic chemistry and biological activities of the obtained compounds. As synthetic chemists, our main goal over the last 12 years has been to develop new strategies that allow for the exploration of the chemical space of organometallic nucleic acid components. Accordingly, we have developed a Michael addition reaction-based methodology that enabled the synthesis of an entirely new class of glycol nucleic acid (GNA) constituents. Concerning GNA chemistry, we also reported the synthesis of the first-ever ferrocenyl GNA-RNA “mixed” dinucleoside phosphate analog. Recently, we developed a Cu(I)-catalyzed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction-based approach for the synthesis of novel 1,2,3-triazole-linked (“click”) nucleosides. The high value of this approach is because it allows for the introduction of functional (e.g., luminescent and redox-active) groups that protrude from the main oligomer sequence. With respect to biological activity studies, we identified several promising anticancer and antimicrobial compounds. Furthermore, we found that simple ferrocenyl-nucleobase conjugates have potential as modulators of Aβ21–40 amyloid aggregation. The final section of this article serves as a guide for future studies, as it presents some challenging goals yet to be achieved within the rapidly growing field of nucleic acid chemistry.
 Mateusz Klarek | Mateusz Klarek was born in 1994 in Poznań, Poland. He studied chemistry at the Faculty of Chemistry, Adam Mickiewicz University in Poznań, where he received his PhD degree in 2023. During his PhD studies, he carried out a short research program at the Institut Charles Gerhardt in Montpellier, France. During his PhD studies, he worked as a researcher at the Institute of Bioorganic Chemistry, Polish Academy of Sciences in Poznań, where he gained experience in the modification of solid supports for the synthesis of oligonucleotides. In March 2024, he joined the group of Prof. Konrad Kowalski at the University of Łódź. His research interests include the chemistry of luminescent compounds for bioimaging and therapeutic applications. |
 Konrad Kowalski | Konrad Kowalski was born in 1974 in Łódź, Poland. He received his PhD under the supervision of Prof. Janusz Zakrzewski in 2003 and DSc (habilitation) in 2011 and was promoted to a Full Professor of Chemistry in 2020. He was a recipient of the Marie Skłodowska-Curie Fellowship (École Nationale Supérieure de Chimie de Paris, France, under the supervision of Prof. Gérard Jaouen). He worked as a postdoctoral fellow at Imperial College London, UK, under the supervision of Prof. Nicholas J. Long. Afterwards, supported by the Alexander von Humboldt Foundation scholarship, he joined the group of Prof. Rainer. F. Winter (University of Regensburg, Germany). He is the co-author of more than 80 scientific publications, including 8 review articles and a book chapter. During his work at ENSCP, Kowalski took part in the discovery of ferrocifen type anticancer agents. His main current scientific interests include synthetic chemistry and biological activity studies of XNA components. He is also interested in the development of new luminescent complexes for bioimaging and therapeutic applications. |
Introduction
The last few decades have witnessed tremendous progress in the chemistry of nucleic acids, which has allowed for the broadening of their applications in biology and medicine. This is well illustrated by the diverse synthetic nucleoside analogues that have been marketed as antiviral and anticancer drugs,1–5 the discovery of ribozymes,6 riboswitches,7 alarmones,8 epigenetic modifications,9 CRISPR-Cas910 and so forth.11 On a separate note, there are studies on the chemical origins of nucleic acids themselves12–15 and those focused on the chemistry and biology of xeno-nucleic acids (XNAs).16–29 Both of these areas are fundamentally relevant to questions about the origin of life on our planet and others in the universe. Last but not least, nucleic acids have been used as digital information storage carriers30,31 and as building blocks for nanoscale molecular architectures.32,33Metals have gained an important role in therapeutic applications, mainly due to the cisplatin family of anticancer drugs.34–36 Their clinical success has stimulated further multidirectional studies in the chemistry and biology of metal complexes and organometallic compounds (those with a direct metal–carbon bond), which, after years of effort, resulted in the establishment of a new subfield referred to as bioorganometallic chemistry.37–58
Due to their fundamental biochemical importance, nucleic acid components such as nucleobases, nucleosides and nucleotides were early on recognised as attractive scaffolds for organometallic functionalisation.59–62 Accordingly, Fig. 1 shows two examples of (η5-C5H5)Fe(CO)2 conjugates, 1 and 2, reported by Bergstrom in the 1990s.61
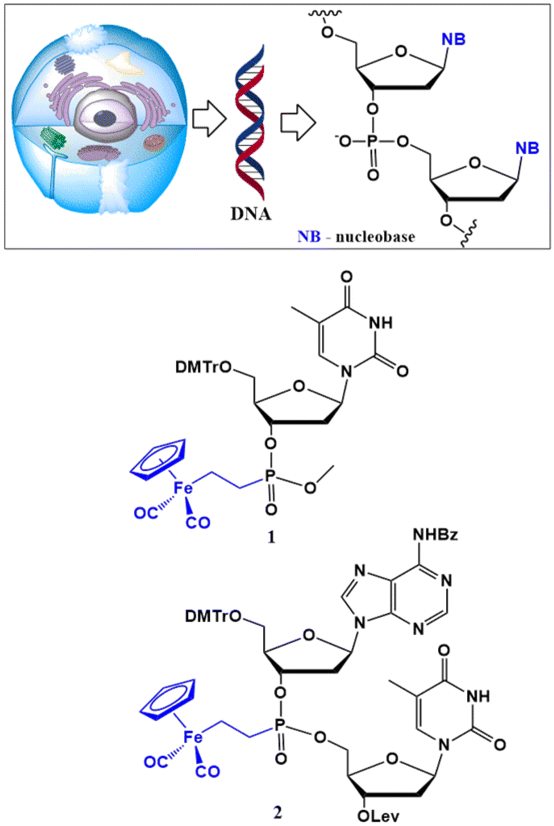 | ||
| Fig. 1 Simplified structure of DNA and structures of DNA components modified with a (η5-C5H5)Fe(CO)2 entity at the phosphorus (1) and at the nonlinking position of the phosphodiester bridge (2).61 | ||
Since then, the area of organometallic derivatives of nucleic acid constituents has steadily advanced,63–69 and it is now comparable in significance to the more mature field of coordination (Werner-type) metal complexes of nucleic acids.70–75 Depending on the modification site, organometallic derivatives of nucleosides can be categorised into three general classes of compounds: A, B and C (Fig. 2).66
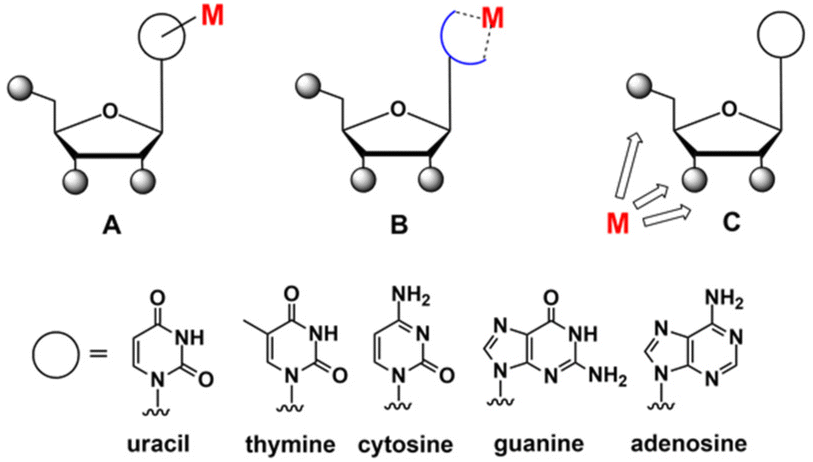 | ||
| Fig. 2 Structures of organometallic derivatives of nucleosides belonging to classes A, B and C. | ||
The most abundant are compounds of class A, in which the metal-containing group (“M”) is attached to the canonical nucleobase moiety. Class B comprises compounds where the base itself has been chemically modified to act as a chelate ligand.66,76 In compounds of class C, the “M” entity is attached either directly or via specific linkers to a relevant position in the ribose moiety77,78 (see compounds 1 and 2 in Fig. 1 for examples). This categorisation is, of course, tentative, but it helps to introduce order into the structurally diversified realm of organometallic nucleic acid components.
It has been shown that the insertion of metal atoms into nucleic acids often results in significant changes in their molecular structural organisation and chemical properties. Arguably the most striking examples supporting this statement are oligonucleotides with metal-mediated base pairs.70,71,73,75,79–81 Furthermore, direct metallisation of click-modified DNA with a Tollens reagent (an aqueous Ag(NH3)2OH solution) provides an opportunity to construct conductive DNA nanodevices and circuits.82
Likewise, organometallic modifications of nucleic acids and their components confer new activities and functions.63,64,66,69 This field has been dominated by the redox-active ferrocenyl ((η5-C5H5)Fe(η5-C5H4); Fc) derivatives of nucleobases, nucleosides, nucleotides and oligonucleotides. These conjugates have been studied mainly as electrochemically detectable probes and sensors.83–92 Furthermore, the Fc entity can act as a reactive oxygen species (ROS) generator, thereby exerting cytotoxic activity in cancer cells.93 In other cases, the anticancer activity of Fc-containing nucleoside analogues has more elaborate origins, as shown by the excellent example of nucleoside (S,Rp)-3 (Fig. 3) reported by Tucker, Hodges and co-workers.94 Detailed studies on 3 in a panel of pancreatic ductal adenocarcinoma cells (PDACs) revealed that its mechanism of action involves inhibition of DNA replication, S-phase cell cycle arrest and stalling of DNA replication forks. All of these biochemical traits correspond to the transcriptional activation or repression of specific cell-cycle regulating genes. Another excellent contribution from Tucker's group to the field of ferrocenyl nucleic acid constituents pertains to the multistep synthesis of the ferrocene nucleic acid oligomer 4 (Fig. 3), which mimics DNA.95 The gap between its cyclopentadienyl decks in 4 (3.3 Å) is very similar to the distance between adjacent nucleobase pairs in B-DNA (3.4 Å). Oligomer 4 contains eight phosphodiester-connected 1,2,1′,2′-tetrasubstituted ferrocene units, each with precisely defined stereochemistry. The ferrocene-derived units in 4 replace the deoxyriboses of natural DNA and confer electrochemical activity to the oligonucleotide.
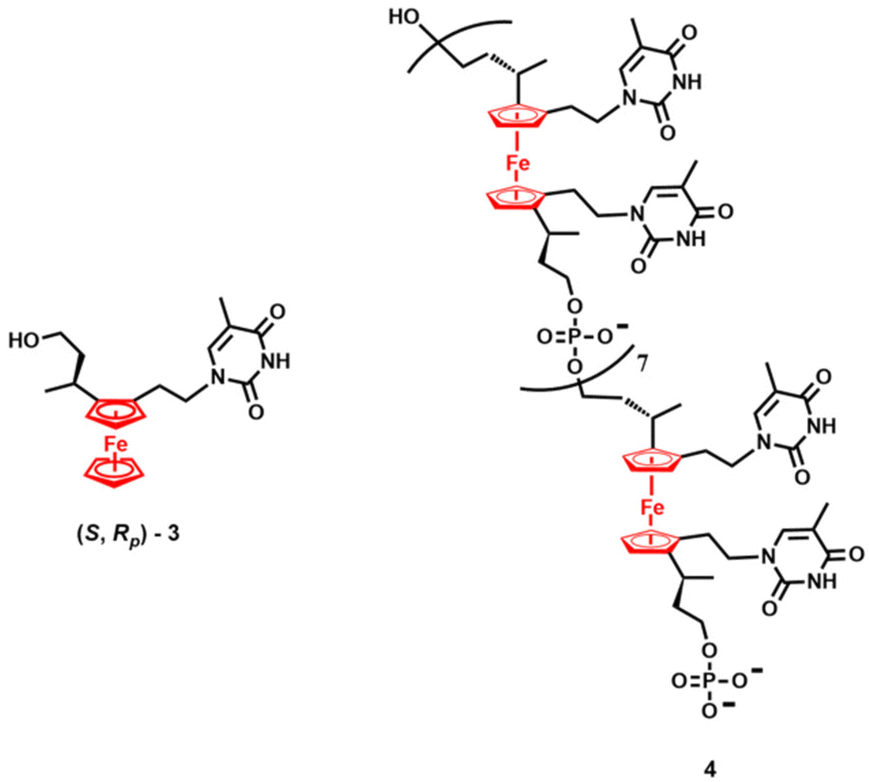 | ||
| Fig. 3 Structures of (S,Rp)-3 and oligomer 4.94,95 | ||
The field of Fc-nucleobase conjugates was reviewed in 2016,67 but since then, new publications have appeared. For example, Vrček and co-workers96,97 reported in-depth mechanistic studies pertaining to transacylation reactions in Fc-purines 5 and 6 (Fig. 4), while the Tucker group98–100 comprehensively studied the anticancer activity of various metallocenyl congeners of compound 3.
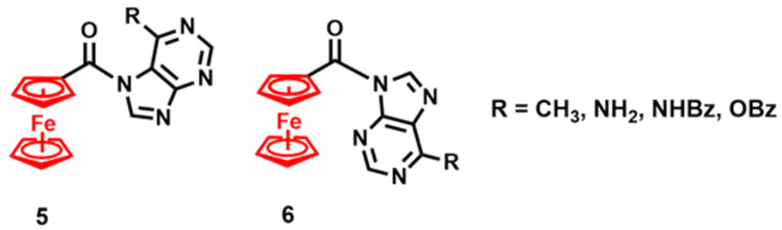 | ||
| Fig. 4 Structures of Fc-purines 5 and 6.96,97 | ||
It must be emphasised that metallocenes are not the only organometallic derivatives used for the derivatisation of nucleic acids and their components. Advances in the chemistry of non-metallocenyl organometallic nucleic acid constituents and oligomers have been reviewed in two articles published in the last 4 years, with the most recent appearing in 2024.68,69 Thus, this field will not be discussed further here.
The purpose of this account is to summarise the work on organometallic nucleic acid components carried out in our laboratory over the past 12 years. It is also important to present some ideas that await experimental verification in the future.
Early studies
The beginning of interest in organometallic nucleic acid chemistry in our lab dates back to 2003, when the corresponding author (then a PhD student) conducted photochemical reactions of (η5-C5H5)Fe(CO)2I with protected uridine and thymidine nucleosides.101 The reactions were carried out in benzene containing a small amount of diisopropylamine and led to the formation of complexes 7–9 (Fig. 5). These, belonging to class A pyrimidine derivatives, were modestly stable against light and temperature. The iron-N3-uracil coordination mode prevents 7–9 from forming Watson–Crick base–base pairs. Despite these drawbacks, metal carbonyl complexes 7–9 have the advantage of displaying sharp and intense absorption bands in the IR (1900–2150 cm−1) spectral region, making them potentially attractive as probes for CMIA applications.102 | ||
| Fig. 5 Structures of 7–9.101 | ||
Furthermore, detritylation of 8 afforded the corresponding unprotected thymidine nucleoside in quantitative yield.101 The biological activity of complexes 7–9 has not been studied since then.
Organometallic-nucleobase adducts and related compounds
In 2012, a paper on the synthesis of ferrocenyl thymine nucleoside 12 (Scheme 1) marked the revival of interest in the chemistry of organometallic nucleic acid components in our laboratory.103 The key to the synthesis was a Michael addition reaction between acryloyl ferrocene 10 (Michael acceptor) and thymine (Michael donor), as shown in Scheme 1. The reaction yielded adduct 11 with a 64% yield. Although stable in the solid state, compound 11 underwent a retro-Michael reaction in solution. This problem was easily addressed by reducing the carbonyl function with NaBH4 (Scheme 1).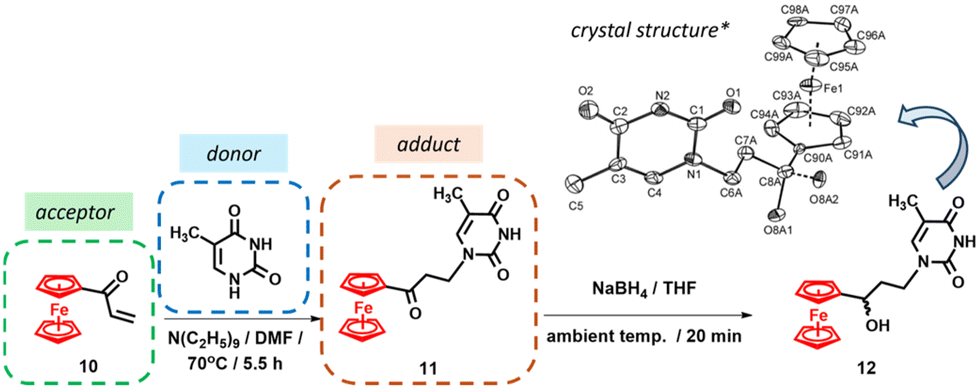 | ||
| Scheme 1 Synthesis of nucleoside 12.103 *Crystal structure adapted with permission from ref. 103. Copyright 2012 Elsevier. | ||
Finally, the structure of nucleoside 12 was unambiguously confirmed by X-ray crystal structure analysis103 (see the inset in Scheme 1).
The synthetic approach shown in Scheme 1 was soon improved by developing a procedure in which organometallic Michael acceptors are generated in situ from the corresponding 3-chloropropionyl metallocene (metallocene = ferrocene or ruthenocene) precursors 13 or 14 (Scheme 2).104 This procedure enabled the synthesis of ferrocenyl and ruthenocenyl adducts 11 and 16–18, which were then transformed into the corresponding nucleosides 19–21 and subsequently dehydrated by treatment with a catalytic amount of H2SO4 to afford olefins 22–24 (Scheme 2).104 Likewise, the dehydration of the previously reported nucleoside 12 afforded olefine 25.
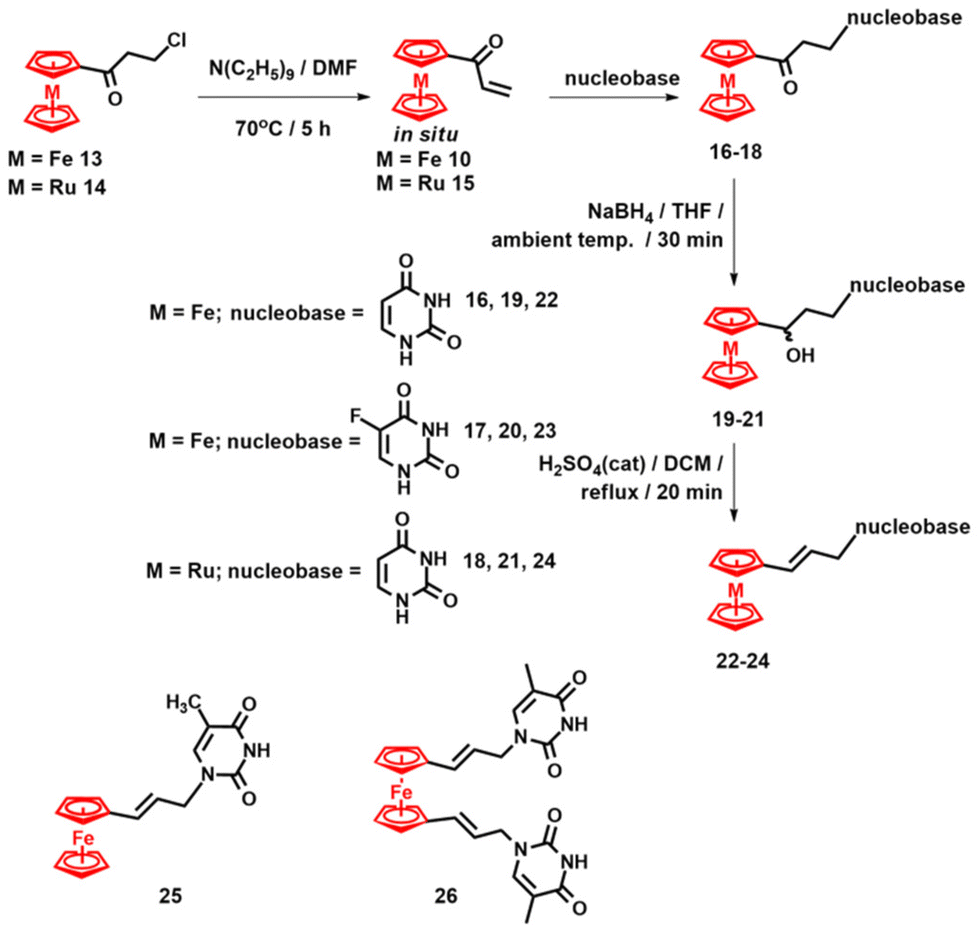 | ||
| Scheme 2 Synthesis of compounds 22–24 and structures of 25 and 26.104–106 | ||
Compound 25 was active against breast cancer MCF-7 cells with an IC50 value of 23.8 ± 0.1 μM (Table 1), whereas the 5-fluorouracil adduct 17 showed activity against methicillin-resistant and vancomycin-resistant strains of Staphylococcus aureus and Staphylococcus epidermidis (Table 2).104 Compound 25 and its disubstituted congener 26 also showed moderate activity as inhibitors of S/ACE2 interactions106 and acted as modulators of Aβ21–40 amyloid aggregation.107 In particular, conjugate 26 is so effective as an Aβ21–40 aggregation enhancer that micrometric oligomers of this peptide were observed.107 The key to these properties is the combination of lipophilicity conferred by the ferrocenyl moiety and the hydrogen-bonding ability of the thymine fragment.
| Comp. | MCF-7 | MCF-7/DOX | A549 | MDA-MB-231 | HT-29 | H-1975 | Ref. |
|---|---|---|---|---|---|---|---|
| 25 (cisPt) | 23.8 ± 0.1 (2.0) | nd | nd | nd | >100 (7.0) | nd | 104 |
| 35 (DOX) | 119.45 ± 2.97 (0.439 ± 0.026) | nd | 7.24 ± 0.34 (0.300 ± 0.018) | >150 (1.073 ± 0.062) | nd | nd | 109 |
| 41 (DOX) | 57.78 ± 2.89 (0.439 ± 0.026) | 39.92 ± 1.41 (48.50 ± 2.78) | 20.35 ± 1.09 (0.300 ± 0.018) | 35.73 ± 1.44 (1.073 ± 0.062) | nd | nd | 109 |
| 62 (cisPt) | nd | nd | nd | 16.1 ± 8.0 (7.7 ± 0.4) | 4.3 ± 0.7 (7.5 ± 0.4) | nd | 113 |
| 66 (cisPt) | 5.1 ± 1.4 (8.3 ± 2.7) | nd | nd | 8.3 ± 0.6 (18.6 ± 5.3) | 23.2 ± 4.0 (7.5 ± 1.8) | nd | 115 |
| 81 (cisPt) | nd | nd | 57 ± 18 (108 ± 12) | nd | nd | 5 ± 2 (4 ± 0.1) | 142 |
| 97 (cisPt) | nd | nd | 10.55 ± 5.11 (14.70 ± 6.186) | nd | nd | nd | 145 |
| Comp. | S. aureus ATCC 29213 | S. epidermidis ATCC 12228 | T. brucei | Ref. |
|---|---|---|---|---|
| 17 | 32 | 16 | nd | 104 |
| 27 | nd | nd | 100 (3.60 ± 0.66) | 108 |
| 28 | nd | nd | 10 (4.41 ± 1.02) | 108 |
| 29 | 64 | 8 | 10 (3.02 ± 0.14) | 109 |
| 30 | nd | nd | 10 (3.55 ± 0.13) | 108 |
| 31 | nd | nd | 10 (3.36 ± 0.39) | 108 |
| 40 | 32 | 16 | 10 (1.67 ± 0.37) | 108 |
| Penicillin | 2 | 2 | nd | 108 |
| Suramin | nd | nd | 0.1–1 (0.039 ± 0.003) | 108 |
The procedure presented in Scheme 2 is operationally simple and robust. It has become a synthetic workhorse in our laboratory, as it avoids the time-consuming isolation and handling of often unstable acryloyl intermediates (such as 10 in Scheme 1). This method also provided access to a series of half-sandwich cymantrenyl and cyrhetrenyl adducts 27–32 (Fig. 6), which were further transformed into nucleosides 33–41 (Fig. 6).108,109
 | ||
| Fig. 6 Structures of compounds 27–41.108,109 | ||
Adducts 27–31 showed activity against the bloodstream forms of Trypanosoma brucei, a protozoan parasite (Table 2), whereas nucleosides 33, 34, 36, 37 and 41 were less active.108 With respect to antimicrobial activity, the adenine nucleoside 40 and the 5-fluorouracil adduct 29 exhibited high activity against MRSA bacterial strains, including clinical isolates (Table 2).109 Also noteworthy is the activity of 35 against human lung carcinoma A549 cells (IC50 = 7.24 ± 0.34 μM) and its triphenylphosphine congener 41 against oestrogen-responsive and doxorubicin-resistant breast adenocarcinoma MCF-7/DOX cells (IC50 = 39.92 ± 1.41 μM) (Table 1).109
On a separate note, the “in situ” procedure presented in Scheme 2 enabled the synthesis of organic nucleobase adducts 42–44, which were further transformed into luminescent probes 45–48 (Fig. 7).110–112
 | ||
| Fig. 7 Structures of compounds 42–48.110–113 | ||
The luminescence of these compounds was utilised for their detection inside cancer HeLa cells using confocal microscopy.110–113
As shown by the above examples, the “in situ” Michael addition approach tolerated structurally diverse Michael acceptors. We also examined its usefulness with 5-iodouracil as the Michael donor, due to the potential for further functionalisation of the 5-iodo position of the uracil base.113,114 Accordingly, the reaction of precursors 13, 14 and 49 with 5-iodouracil afforded ferrocenyl, ruthenocenyl and binuclear adducts 50, 51 and 52 with yields of 77%, 51% and 84%, respectively (Scheme 3).
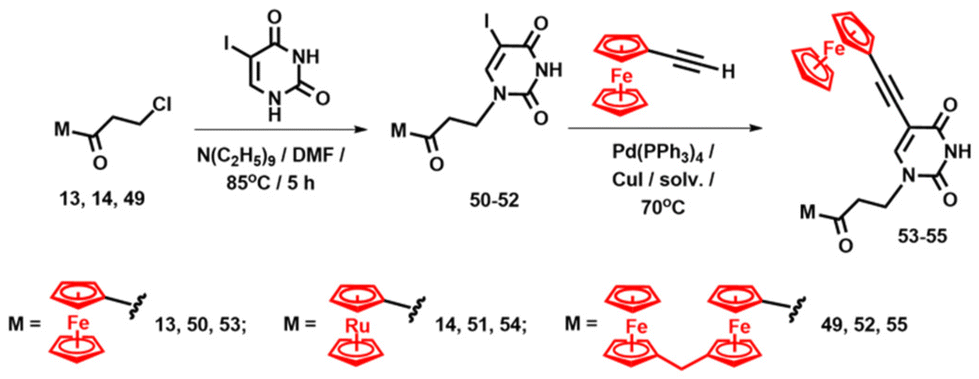 | ||
| Scheme 3 Synthesis of compounds 53–55.113 | ||
In the next step, the Sonogashira cross-coupling reaction of 50–52 with ethynylferrocene yielded dinuclear and trinuclear uracil conjugates 53–55 (Scheme 3).113 We further capitalised on the 5-iodouracil adducts 50, 51 and their [2.2]paracyclophane congener 56 by obtaining three closely related pyrenyl compounds 57–59 (Fig. 8).114
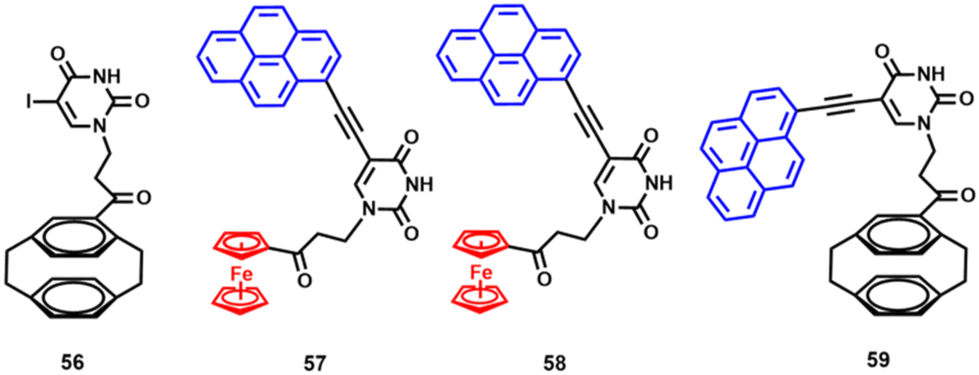 | ||
| Fig. 8 Structures of compounds 56–59.114 | ||
Adducts 57 and 58 are not luminescent, but their [2.2]paracyclophane analogue 59 shows characteristic “pyrenyl” emission with λem/max at 428 nm in ethanol solution. Compound 59 represents an interesting non-organometallic congener of 57 and 58. It has been shown to bind to the complementary DNA template (dA)_20 but not to (dT)_20.114 This binding is accompanied by broad green emission (λem/max = 505 nm) due to pyrene excimer formation. Furthermore, it has been shown that 59 is internalised by HeLa cells and localises mainly in their membranes and lipophilic cytoplasmic compartments. Due to the similarity between 57, 58 and 59, the latter was proposed as a metal-free luminescent probe for the indirect bioimaging of the former in cells.114
Chemical transformations of adduct 11 were investigated in more detail. During these studies, it was found that the treatment of 11 with PEMB in acetic acid at ambient temperature afforded compound 60, with a saturated linker connecting the thyminyl and ferrocenyl end groups (Scheme 4).113 These mild reduction conditions were equally effective in the synthesis of derivatives 61 and 62 (Scheme 4).113
 | ||
| Scheme 4 Synthesis of compound 60–62.113 | ||
Furthermore, it was found that the carbonyl function in 11 can be easily transformed into a thioketone group. The thionation reaction of 11 was carried out using either Lawesson's reagent or diphosphorus pentasulfide (P2S5), as shown in Scheme 5.115 Lawesson's reagent afforded disulphur product 63 with a 77% yield, along with trace amounts of thioketone 64. In contrast, when P2S5 was used, only compound 64 was obtained, with a 68% yield. In the following step, the hetero-Diels–Alder reaction of 63 and 64 with 2,3-dimethyl-1,3-butadiene resulted in the formation of thiopyranes 65 and 66 (Scheme 5).115
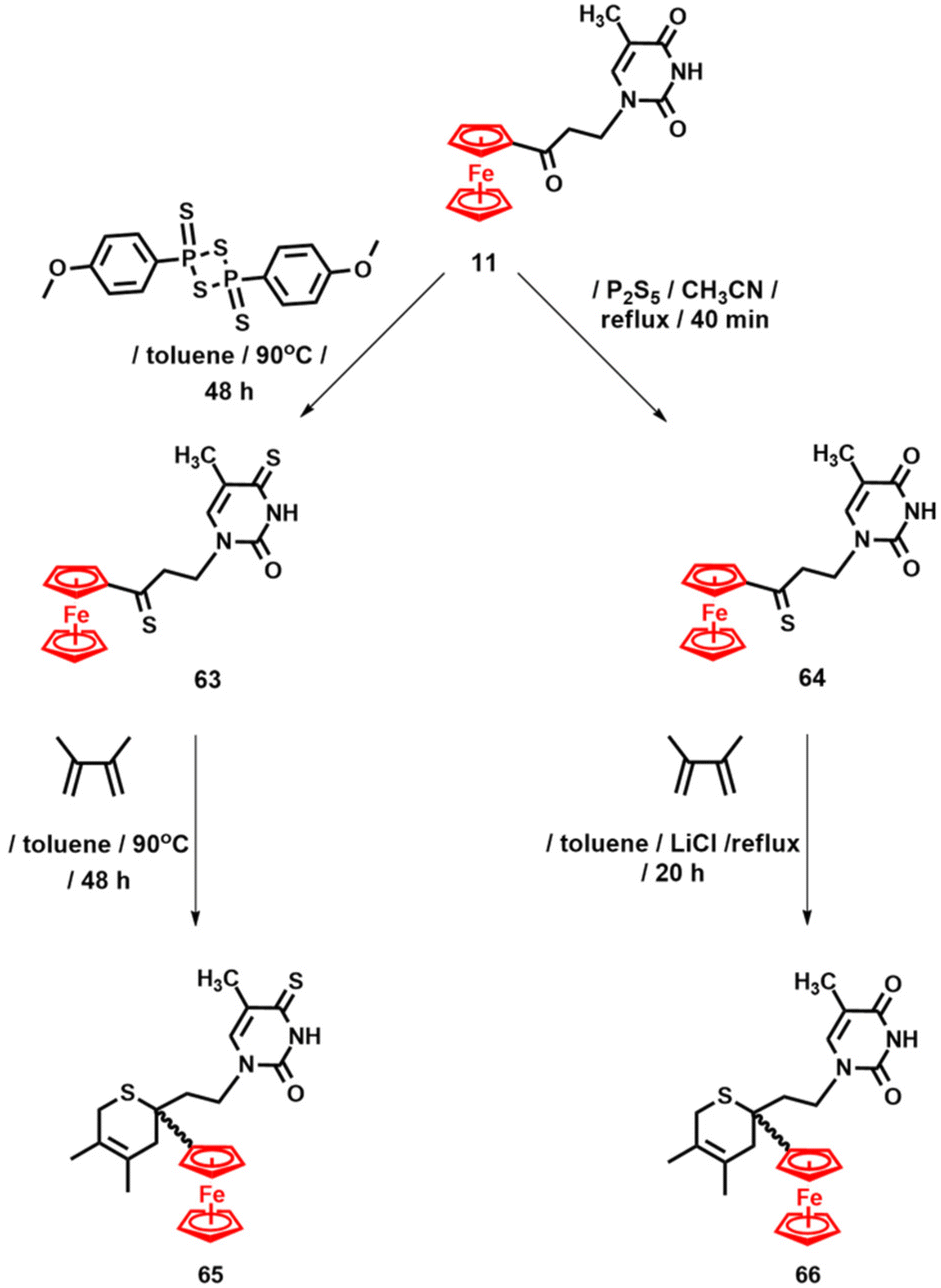 | ||
| Scheme 5 Synthesis of compounds 63–66.115 | ||
Compounds 63 and 64 represent rare examples of sulphur-containing ferrocenyl–nucleobase conjugates. Their thermal and chemical stability is also noteworthy, contrasting with the limited stability of organic thioketones. The molecular structure of thiopyrane 65 was unambiguously confirmed by X-ray diffraction analysis.115 Anticancer activity studies of compounds 66 and 65 against a panel of human cancer cell lines (HT-29, MCF-7, MDA-MB-231, HL-60 and MonoMac6) indicated that the number of sulphur atoms affects the activity. The mono-sulphur compound 66 was significantly more active than its disulphur congener 65 against the entire panel of cancer cells tested. More importantly, compound 66 rivals the cisPt reference drug in terms of activity. For example, the IC50 value of 66 against mammary cancer cells MCF-7 and MDA-MB-231 was 5.1 ± 1.4 and 8.3 ± 0.6 μM, respectively, compared to 8.3 ± 2.7 and 18.6 ± 5.3 μM for cisPt (Table 1). Similarly, the IC50 value of 66 against leukaemia cells HL-60 and MonoMac6 was 4.5 ± 0.3 and 17.4 ± 4.2 μM, respectively, which is within the range of cisPt (3.6 ± 0.2 and 36.5 ± 10.2 μM for HL-60 and MonoMac6, respectively).115 Based on these results, further anticancer activity studies of sulphur-containing ferrocenyl nucleoside analogues are definitely warranted.
Ferrocenylated glycol nucleic acid (GNA) nucleosides
The next advancement in our research involved modifications of glycol nucleic acid (GNA) nucleosides. GNA is the simplest known representative of XNA.116–127 It is composed of a three-carbon 1,2-propanediol (propylene glycol) backbone connected to a nucleobase (Fig. 9 shows uridine GNA nucleoside 67 as an example).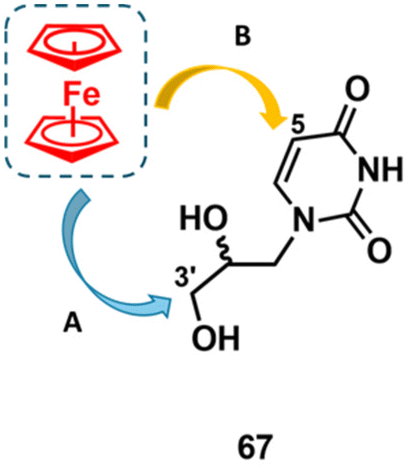 | ||
| Fig. 9 Structure of uracil GNA nucleoside 67 and strategies A and B applied for its modification. | ||
The GNA nucleoside backbone has a single carbon atom stereocentre, which implies the existence of either (S)- or (R)-enantiomeric forms. Similar to DNA, single strands of GNA form antiparallel homoduplexes with their thermodynamic and thermal stability exceeding that of analogous DNA and RNA duplexes.127 GNA is a promising molecule for bio-medical applications. This is illustrated by the finding that (S)-GNA modifications of siRNAs enhance the safety of RNAi therapeutics while maintaining potency.128 Equally intriguing is the finding that GNA can serve as a template for enzymatic DNA synthesis.129 This reaction does not require stable duplex formation between the product and the template. Moreover, GNA has been applied in the assembly of molecular objects with complex architectures, such as mirror-image 4-helix junctions.130 Further details on GNA chemistry and applications can be found in recent review articles.124–126
By the time we initiated our own research programme into ferrocenyl GNA chemistry, we had not encountered any previous reports in this field. Introducing the ferrocenyl entity into the GNA scaffold can potentially confer useful redox and cytotoxic activities, which could be beneficial for sensing, materials, anticancer and antimicrobial applications. Encouraged by these promising possibilities, we aimed to combine the redox-active ferrocenyl moiety with uridine GNA as a model nucleoside. To this end, strategies A and B were designed (Fig. 9). In strategy A, the ferrocenyl entity was introduced into the pseudo C3′ position of the propylene glycol backbone, while in strategy B, it was attached to C5 of the uracil base. The practical implementation of strategy A was announced in 2018, when the preparation of nucleosides (S,R)-68 and (R,S)-68, as well as their methylated congeners (S,R)-69 and (R,S)-69, was reported (Scheme 6).131 The key to the synthesis was a Sharpless catalytic asymmetric dihydroxylation of alkene 25.
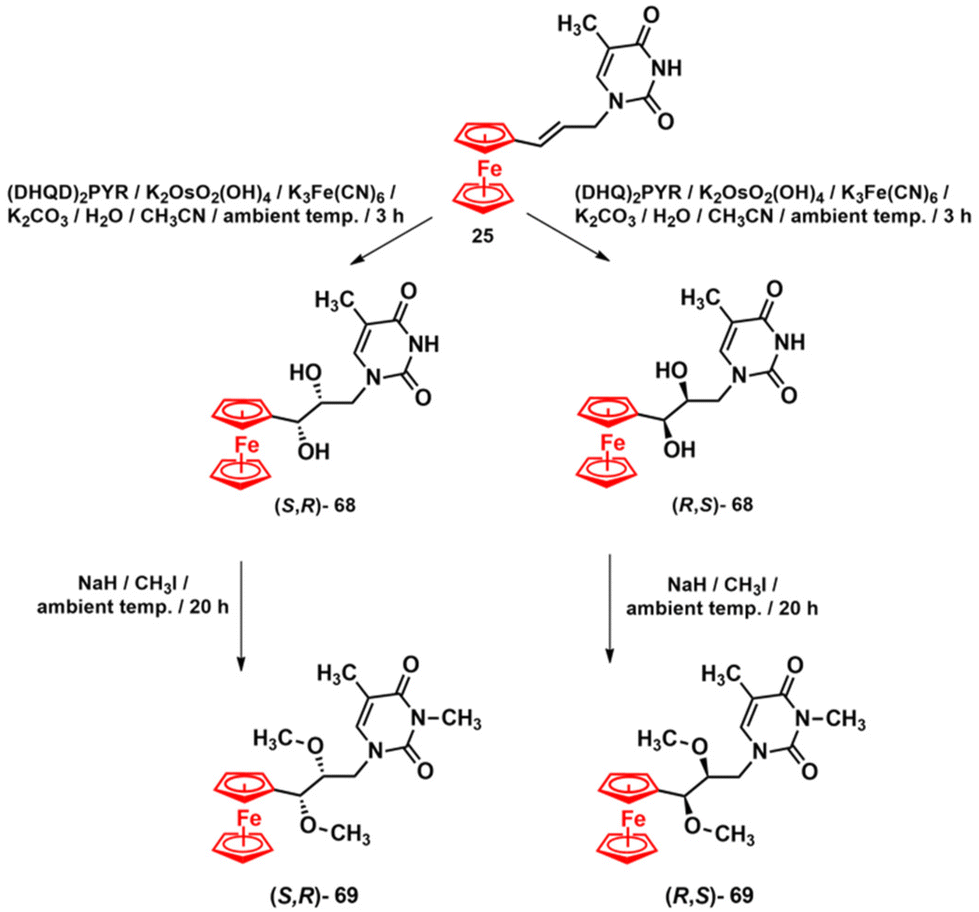 | ||
| Scheme 6 Synthesis of compounds 68 and 69.131 | ||
The absolute configurations of nucleosides 68 were assigned after their transformation into crystalline methyl derivatives 69 by X-ray diffraction analysis. In a subsequent, unsuccessful attempt, we tried to transform nucleosides 68 into the corresponding nucleotides. The failure of these attempts could be attributed to the interplay between the steric hindrance provided by the ferrocenyl moiety and the propensity for forming a stable α-ferrocenium carbenium-type ion. Both factors may have contributed to the experimentally observed decomposition of nucleosides 68 during phosphorylation reactions. To address this issue, we synthesised nucleoside 70 with an elongated hydroxyl side chain in the pseudo C3′ position using a ytterbium-mediated etherification reaction (Scheme 7).132 This reaction involves the attack of the ethylene glycol nucleophile on a stable α-ferrocenium carbenium-type ion derived from nucleosides 68. The obtained diastereomers (R,R)-70 and (S,R)-70 were separated by HPLC and then used to synthesise the corresponding nucleoside ethyl esters via a reaction with diethyl chlorophosphite. Only the reaction with (R,R)-70 yielded the desired nucleotide product (R,R)-71 in analytically pure form (Scheme 7).132
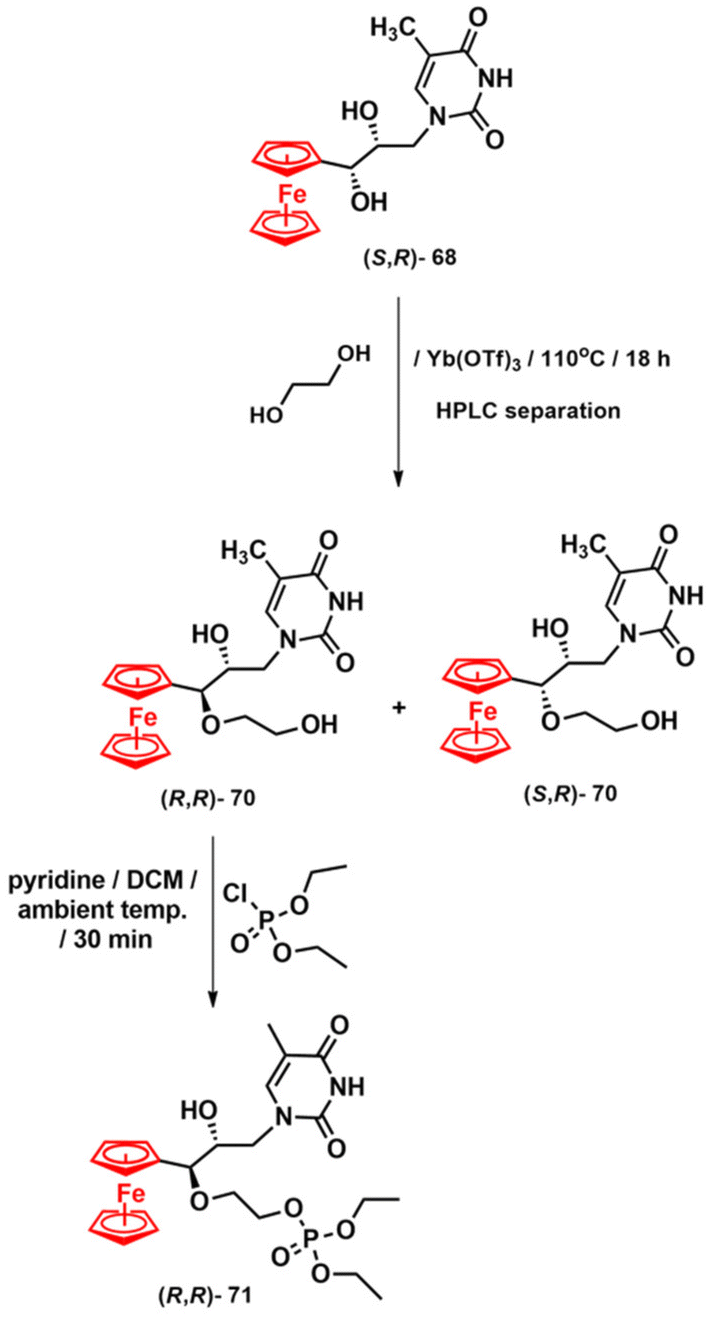 | ||
| Scheme 7 Synthesis of compounds 70 and 71.132 | ||
Keeping in mind the potential applications of redox-active GNA oligonucleotides as analytical tools or molecular wires, the electrochemical properties of 68, 69, 70 and 71 were studied by cyclic voltammetry (CV). All compounds underwent a single Nernstian one-electron ferrocenyl-centered redox process at E0/+1/2 = −25 mV (68, DMSO solution),131 75 mV (69, DMSO solution),131 14/15 mV ((R,R)-70/(S,R)-70, DCM solution)132 and 29 mV (71, DCM solution)132versus the FcH/FcH+ couple. These electrochemical properties confirm their potential relevance to the aforementioned applications.
Anticancer activity studies of 68, 70 and 71 were also carried out using human endometrial Ishikawa and cervical HeLa cancer cells, as well as non-tumorigenic L929 cells.132 The compounds showed negligible activity against cancer cells and no activity against L929 cells (IC50 > 100 μM). The most active compound was (S,R)-70, with IC50 values of 66.0 ± 1.0 μM against HeLa cells and 62.3 ± 3.1 μM against Ishikawa cells. At first glance, this result may seem undesirable. However, it is in fact promising with respect to the future use of Fc-GNA oligonucleotides in complex biological systems, such as vertebrate animal models. The results concerning strategy B were published in 2020.133 In this account, we reported the linear synthesis of Fc-GNA nucleoside 74, followed by its transformation into dinucleoside phosphate 77. The key step in the synthesis of nucleoside 74 was a Sonogashira cross-coupling reaction of protected nucleoside 72 (obtained in two steps from 5-iodo-3-benzoyluracil and (S)-2,3-O-isopropylidene glycerol) with ethynylferrocene (Scheme 8). This reaction afforded synthon 73, which, after the cleavage of the N3-benzoyl group, yielded ferrocenyl GNA nucleoside 74 in 70% yield. In a subsequent two-step process, 74 was first protected with a 4,4′-dimethoxytrityl group (compound 75) and then transformed into phosphoramidite 76. In the final step, compound 76 was coupled with commercially available N6-benzoyl-2′,3′-O-isopropylideneadenosine to afford the desired dinucleoside phosphate 77 in 47% yield (Scheme 8).133
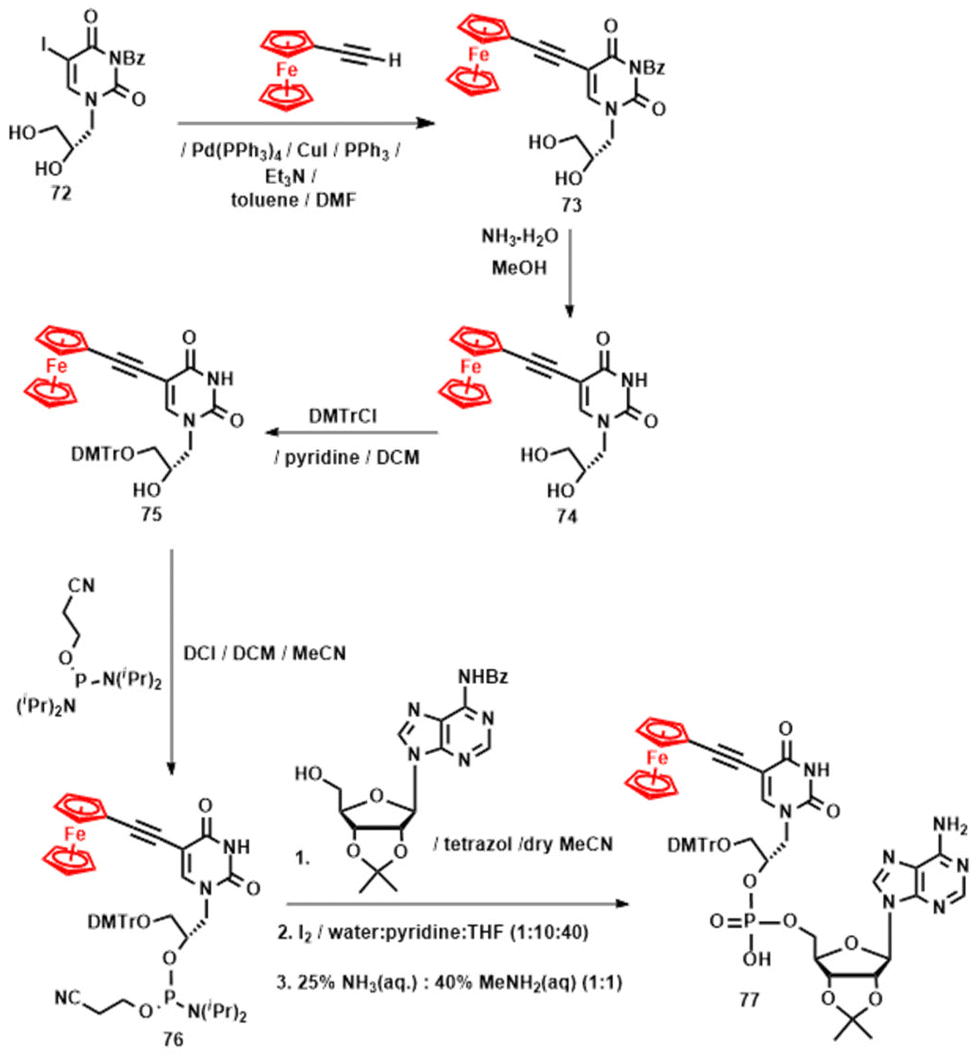 | ||
| Scheme 8 Synthesis of compound 77.133 | ||
To the best of our knowledge, compound 77 represents the first-ever hybrid between canonical RNA and organometallic GNA nucleosides reported to date. Ferrocenyl GNA components 74 and 77 underwent a single Nernstian one-electron ferrocenyl-centred redox process, making them similar to the GNA compounds 68, 69, 70 and 71 discussed above. Ferrocenyl GNA derivatives 74 and 77 were not active against cancer HeLa or non-tumorigenic L929 cells.133 The lack of toxicity of dinucleoside phosphate 77 could be relevant for the future development of Fc-capped RNA oligonucleotides. It can be expected that such hypothetical molecules will exhibit desirable resistance against exonuclease enzymes present in cells. In this context, it is worth noting that strategy A was also successfully applied for the preparation of luminescent enantiomeric pyrenyl GNA nucleosides 78, whereas strategy B allowed the synthesis of luminescent phenanthrenyl GNA constituents 79 and 80 (Fig. 10).111,134
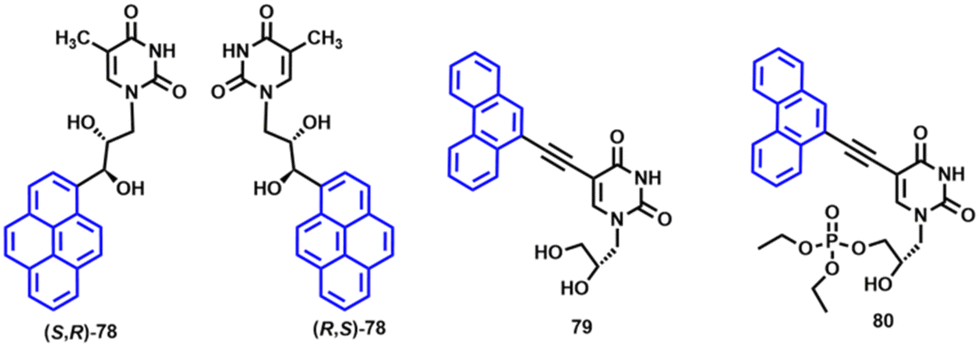 | ||
| Fig. 10 Structure of luminescent GNA components 78, 79 and 80.111,134 | ||
Organometallic “click” modifications
Recently, we have turned our attention towards 1H-1,2,3-triazolyl (hereafter abbreviated as “click”)-modified organometallic nucleic acid components. This choice was justified by the rapidly growing importance of the Cu(I)-catalysed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction (CuAAC) in nucleic acid chemistry.135,136 Our interest was directed in two ways. First, we aimed to obtain a 1H-1,2,3-triazolyl-bridged biferrocene scaffold connected to a model canonical nucleoside vector for anticancer activity studies. The second goal was to replace the canonical phosphodiester linker with a charge-neutral organometallic-1H-1,2,3-triazolyl-derived linker. The first idea was motivated by our earlier observation of high anticancer activity in heterobimetallic complexes and our intention to explore this field further with new, more biologically relevant compounds.137,138 The second idea originated from excellent reports by Brown and co-workers, who demonstrated the compatibility of 1H-1,2,3-triazolyl linkers with processes related to nucleic acid biology, such as transcription,139 gene assembly,140 and replication,141 among others.136 Accordingly, to obtain the binuclear thymidine “click” conjugate 81 and the mononuclear complex 82, a CuAAC reaction of AZT with ethynylferrocene was carried out under the conditions shown in Scheme 9.142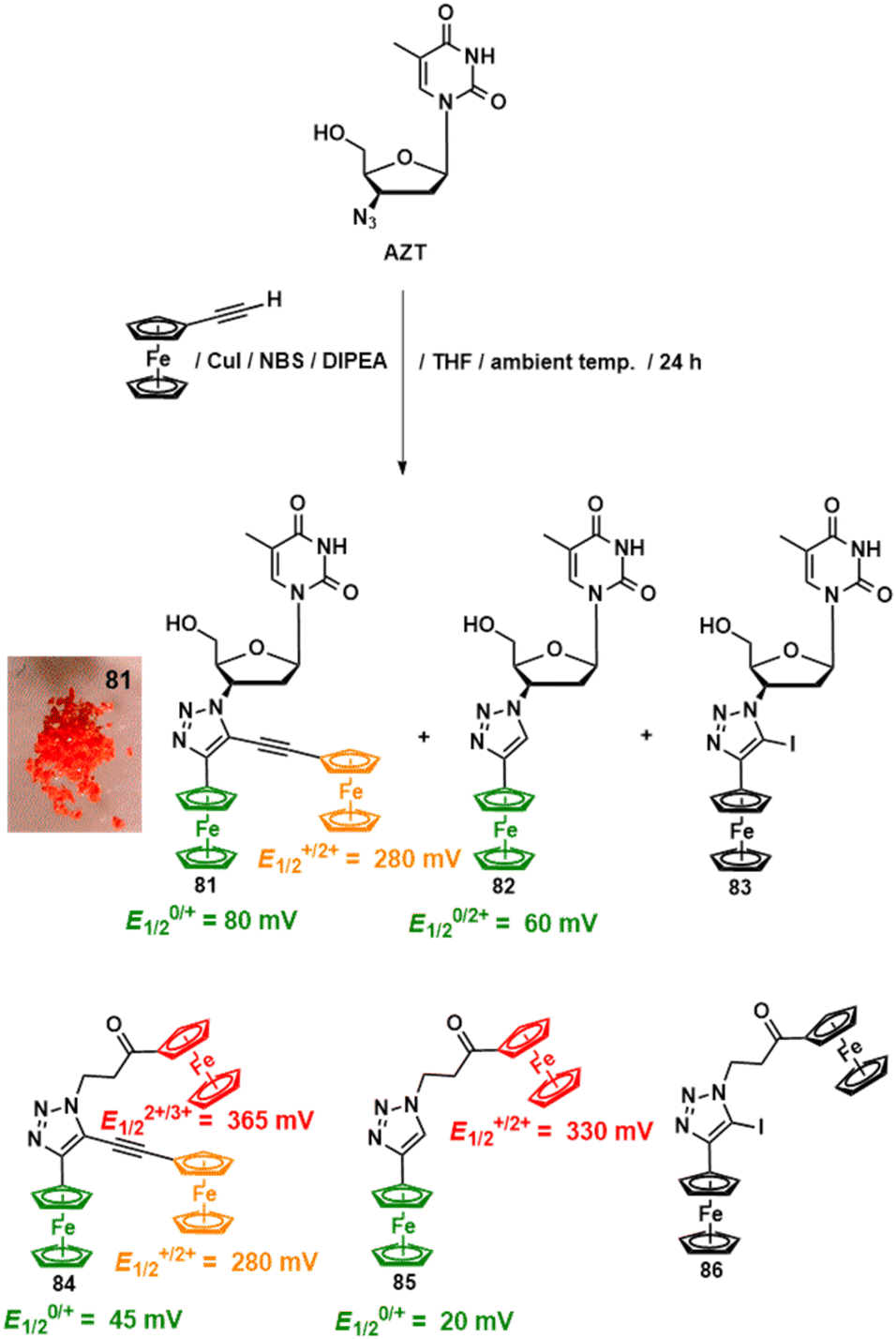 | ||
| Scheme 9 Synthesis and electrochemical properties of compounds 81–86.142 Electrochemical measurements were performed at 25 °C in anhydrous CH2Cl2.142 Potentials are referenced to the FcH/FcH+ redox couple. Inset photo shows crystals of 81, which can be obtained in gram amounts. | ||
Apart from compounds 81 and 82, which were obtained in 39% and 6% yields, the reaction also afforded an iodo-derivative, 83 (in 9% yield). Using the same reaction but with 3-azidopropionylferrocene instead of AZT, compounds 84, 85, and 86 were synthesized in 22%, 15%, and 24% yields, respectively (Scheme 9).142 At first sight, iodo complex 83 can be considered as an intermediate for binuclear compound 81. Unexpectedly, our attempts to transform it into 81via the Sonogashira cross-coupling reaction with ethynylferrocene failed. Biferrocenyl and triferrocenyl compounds 81 and 84 undergo two and three consecutive Nernstian one-electron ferrocenyl-centered processes with formal potentials given in Scheme 9.
(Spectro)electrochemical studies using an optically transparent thin-layer electrode (OTTLE) cell showed that the 1H-1,2,3-triazolyl group facilitates electronic communication between the two ferrocenyl entities in the radical cations 81˙+ and 84˙+.142 Based on OTTLE measurements, 81˙+ and 84˙+ were categorised as class II mixed-valence species, according to the Robin and Day classification.143 Considering the potential positive correlation between ROS generation and anticancer activity, electron paramagnetic resonance (EPR) spectroscopic studies of 81, 82, and 84 were carried out. They showed that the binuclear compound 81 and its trinuclear congener 84 indeed act as ROS generators, with higher ROS generation capacity than the mononuclear complex 82. Anticancer activity studies of 81, 82, 84 and 85 were performed against human lung cancer A549 and H1975 cells, as well as non-tumorigenic human bronchial epithelial BEAS-2B cells. The two most active compounds were 81 and 84. However, the trinuclear compound 84 showed activity only against H1975 cells (IC50 = 84 ± 5 μM), while the thymidine conjugate 81, with an IC50 of 57 ± 18 and 5 ± 2 μM against A549 and H1975 cells, respectively, exhibited activity comparable to cisPt (IC50 = 108 ± 12 and 4 ± 0.1 μM against A549 and H1975 cells, respectively, Table 1). Mononuclear compound 82 and binuclear compound 85 exhibited IC50 values of over 100 μM in all the cells tested. A remarkable feature of compound 81 was its lack of activity against BEAS-2B cells, contrasting with the toxicity exhibited by cisPt (IC50 = 3 ± 0.1 μM), tamoxifen (IC50 = 9 ± 0.2 μM), and 5-fluorouracil (IC50 = 6 ± 0.1 μM). The ROS generation ability of 81 and mononuclear complex 82 in A549 and H1975 cells revealed the superior activity of the former compound. It generated approximately 1.6 and 2.5 times more ROS than 82 in H1975 and A549 cells, respectively. The treatment of the cells with N-acetyl cysteine (NAC), a known free-radical scavenger, had a protective effect against 81, substantiating the ROS-dependent mechanism of its action. This finding is also corroborated by the EPR studies. It can then be hypothesised that the electronic communication in 81˙+ potentiates its anticancer activity. On the other hand, cation 84˙+, which also exhibits electronic coupling, demonstrated less anticancer activity. This leads to the conclusion that the thymidinyl entity present in 81, but not in 84, plays a decisive role in its antiproliferative activity. Detailed anticancer activity studies are underway to gain further insight into the mechanism of action of compound 81.
In relation to the development of “click” internucleotide linkers to replace natural phosphodiesters, we designed a general synthetic methodology that was first successfully utilised in the synthesis of luminescent [Re2(μ-Cl)2(CO)6(μ-pyridazine)] dinucleosides 90–92 (Scheme 10).144 In the same work, [Re2(μ-Cl)2(CO)6(μ-pyridazine)] conjugates 93 and 94 were also reported (Scheme 10).144
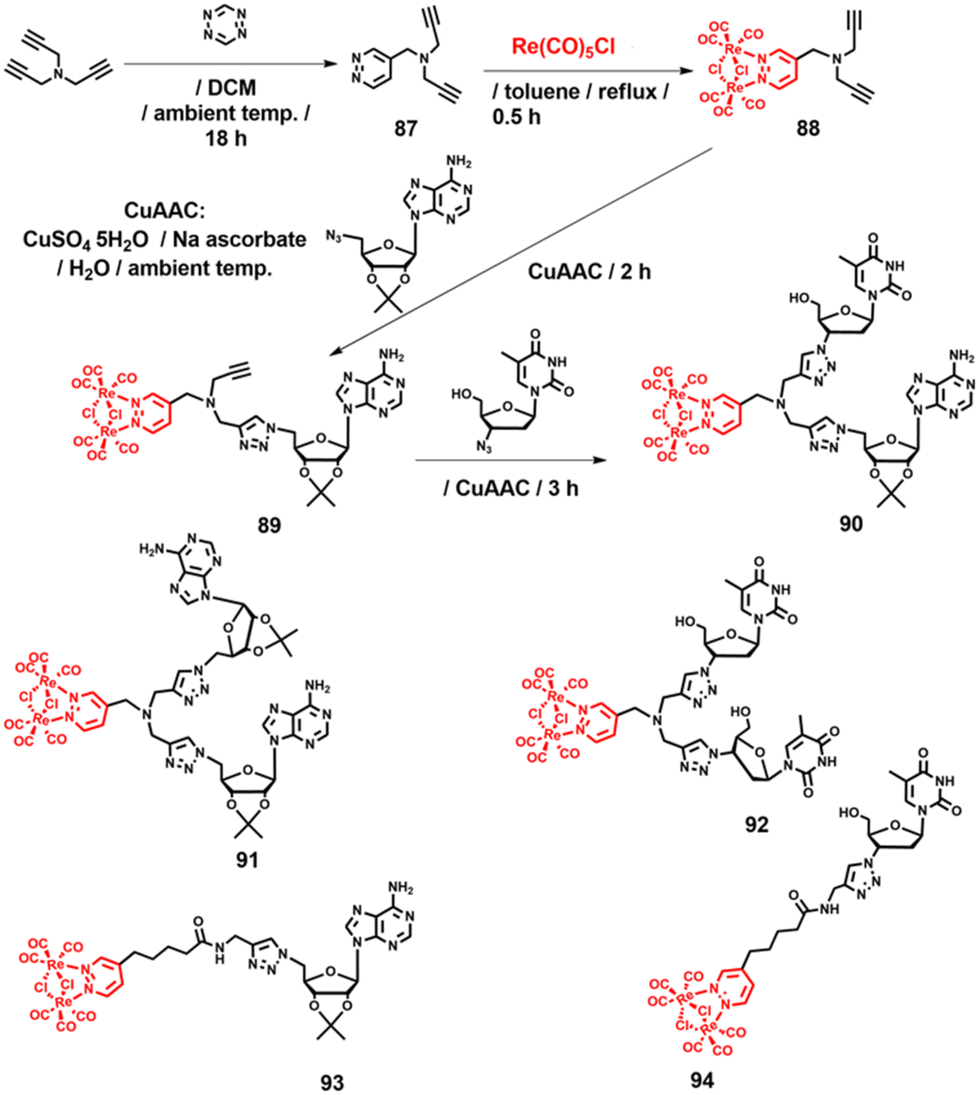 | ||
| Scheme 10 Synthesis of compounds 89–94.144 | ||
Conceptually, our approach was based on using tripropargylamine as a simple “clickable” linker synthon. In the synthesis of compounds 90–92, one of its three propargyl arms underwent an inverse-electron-demand Diels-Alder (IEDDA) [4 + 2]-cycloaddition reaction with 1,2,4,5-tetrazine to afford the pyridazine 87 ligand, which was then coordinated with [Re2(μ-Cl)2(CO)6] to produce intermediate 88. In the subsequent two steps, CuAAC reactions were performed. First, intermediate 88 reacted with 5′-azido-2′,3′-O-isopropylidene adenosine to yield the unstable compound 89. Subsequently, the CuAAC reaction of 89 with AZT provided the desired dinucleoside 90 in 40% yield. A similar approach, carried out twice with either 5′-azido-2′,3′-O-isopropylidene adenosine or AZT, was used to obtain dinucleosides 91 and 92, respectively.144 Compounds 90–92 and 94 were not anticancer active against HeLa and Ishikawa cells (IC50 > 100 μM), while 93 showed moderate activity (IC50ca. 42 μM). The lack of toxicity was advantageous, as it allowed us to examine the intracellular uptake and distribution of phosphorescent compounds 90–94 in HeLa cells using confocal microscopy. Fig. 11 shows the localisation of compound 92 within the interior of HeLa cells. It was localised in the plasmalemma, nuclear envelope (hollow arrows in Fig. 11), and filopodia (loop in Fig. 11), and around the nucleus. Additionally, cells responded to 92 by encapsulating it inside lysosomes or vesicular-like structures (inverted arrows in Fig. 11). Furthermore, dinucleoside 92 was localised in mitochondria (full-tip arrows in Fig. 11) and in the nucleus (asterisk in Fig. 11). The nuclear localisation of conjugates 93 and 94 was also confirmed.
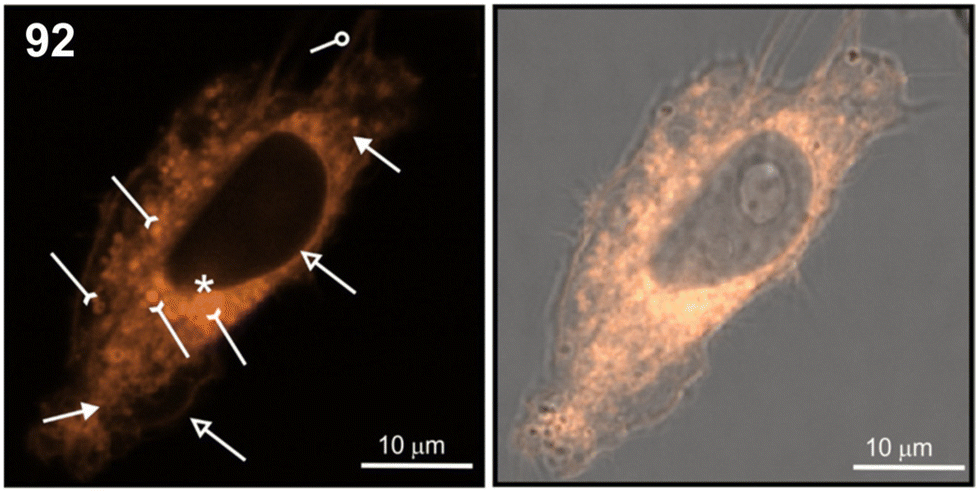 | ||
| Fig. 11 Cellular distribution of dinucleoside 92 in a viable HeLa cell. The images were taken after a 30 min treatment. The left photo shows the luminescence image, and the right photo displays the merged images of luminescence and transmitted light. Lysosomes/vesicles are denoted by inverted arrows; the plasmalemma and nuclear envelope are denoted by hollow arrows; filopodia are denoted by a loop; mitochondria are denoted by full-tip arrows. The compound in the nucleus is denoted by an asterisk. Reproduced from ref. 144 with permission from the Royal Society of Chemistry. | ||
The uptake of dinucleosides 90–94 was also observed inside E. coli and S. aureus bacterial cells, further confirming the practical value of these luminescent nucleic acid analogues. In recent studies, the "click" approach presented in Scheme 10 was re-designed and applied to synthesise redox-active Fc-dinucleosides 97 and 98, as well as trinucleoside 100 with a T–T–A nucleobase sequence (Fig. 12).145
 | ||
| Fig. 12 Structure of compounds 97, 98 and 100.145 | ||
The synthesis of 97 and 98 relied entirely on CuAAC reactions of synthons such as tripropargylamine, 3-azidopropanoylferrocene, AZT, 5′-azidoadenosine or its 2′,3′-O-isopropylidene protected derivative and proceeded through intermediates 95 and 96 according to Scheme 11.145 This approach is cheap and operationally simple, meets the atom-economy criteria and most importantly is universal as it allows for the use of versatile intermediates in place of building block 95.
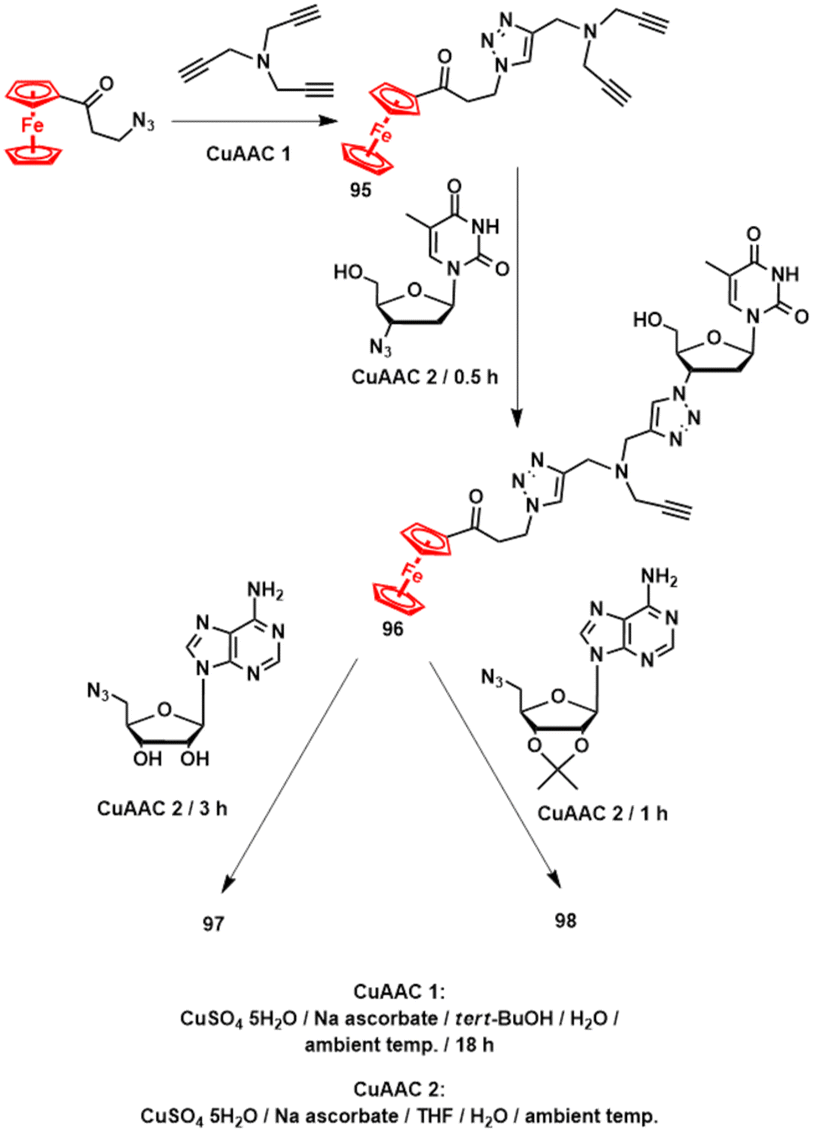 | ||
| Scheme 11 Synthesis of compounds 97 and 98.145 | ||
The synthesis of artificial trinucleotide 100 was achieved via esterification of 98 with azidoacetic acid, followed by the CuAAC reaction of intermediate 99 with 96 (Scheme 12).145
 | ||
| Scheme 12 Synthesis of compound 100.145 | ||
Antiproliferative activity studies of compounds 97, 98 and 100 against A549 cancer cells and non-cancerous human embryonic kidney HEK293T cells show that the deprotected dinucleoside 97 is more active (IC50 = 10.55 ± 5.11 μM) than cisPt (IC50 = 14.70 ± 6.186 μM) in A549 cells and less toxic than cisPt (13.82 ± 1.47 vs. 1.21 ± 0.54 μM) in HEK293T cells. Although the mechanism of 97 anticancer action has not been studied, it seems that the presence of two hydroxyl groups in C2′ and C3′ plays a key role. This hypothesis is supported by the fact that the isopropylidene-protected nucleosides 98 and 100 were less active than 97. The mechanism of antiproliferative activity of protected vs. unprotected “click” nucleosides, such as 90–92, 97, 98 and 100, definitely requires further examination.
Conclusions and prospects for the future
Over more than 12 years of studying organometallic nucleic acid components, we have achieved two major synthetic goals. The first goal pertains to the development and optimisation of a Michael addition reaction-based methodology, which has enabled the synthesis of versatile organometallic and organic GNA components. The second goal concerns the development of “click” internucleotide linkers that replace natural phosphodiesters in canonical oligonucleotides. This approach allows for the introduction of virtually any tripropargylamine-derived linker into the oligomer sequence. We have demonstrated this by synthesising luminescent rhenium compounds 90–92 and redox-active ferrocenyl derivatives 97, 98 and 100.We believe that the concept of “click oligo” linkers, exemplified by 90–92, 97, 98 and 100, will attract attention from the nucleic acid chemistry and biology communities. Overall, our synthetic research programme has resulted in compounds with steadily increasing molecular complexity. Complexity increase pertains not only to the overall size of compounds but also to their stereochemical architecture (compare 12 with 71).
Regarding biological studies, it has been shown that the vast majority of our organometallic nucleic acid components are not promising as anticancer drugs. However, there are some exceptions, such as the compounds listed in Table 1, particularly 41, 66, 81 and 97 (Fig. 13), which are promising candidates for future studies.
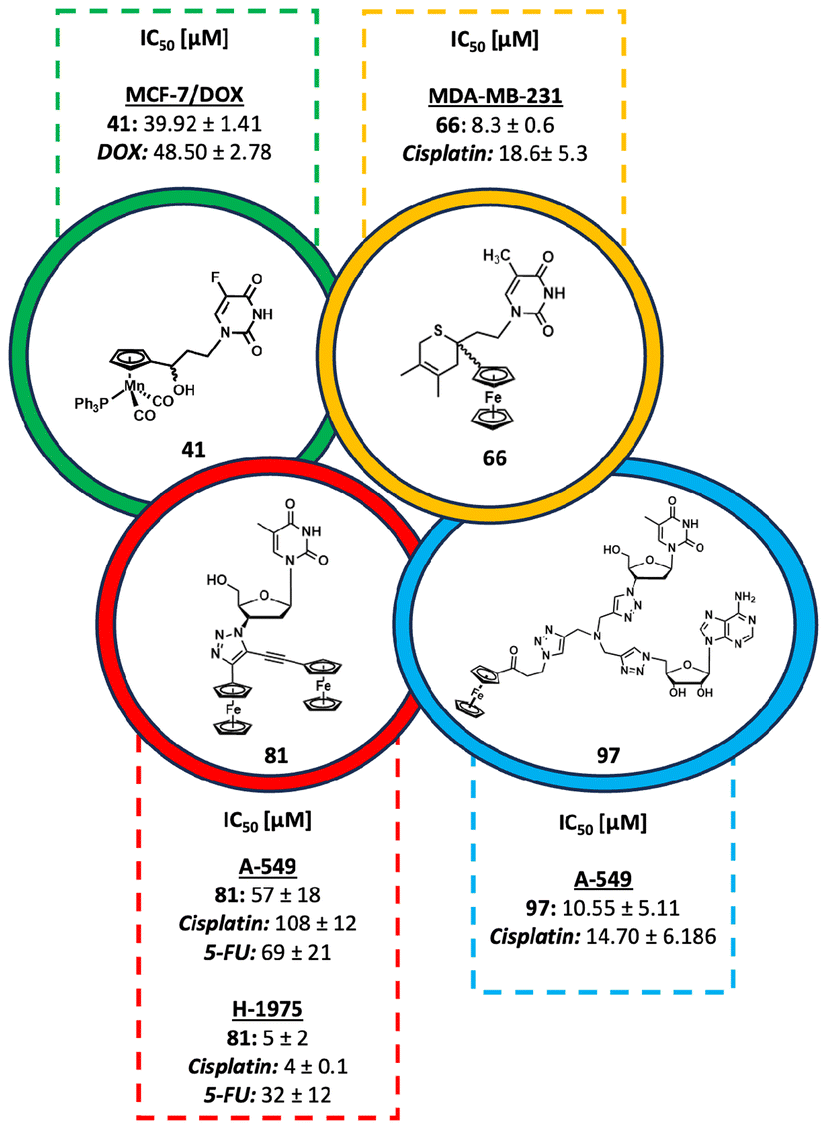 | ||
| Fig. 13 Structures of the most promising anticancer active compounds 41, 66, 81 and 97. | ||
In relation to neglected tropical diseases, the activity of half-sandwich pyrimidine conjugates 27–31 against the bloodstream forms of the T. brucei parasite is also worth noting. Additionally, the observation that compounds 25 and 26 show some activity as inhibitors of S/ACE2 interactions and as modulators of Aβ21–40 aggregation is intriguing and warrants further study. They may be relevant for the development of organometallic coronavirus entry inhibitors and anti-Alzheimer's disease agents. Future studies are likely to focus on the synthesis of functional click-type oligonucleotides with longer nucleobase sequences. Achieving this goal using conventional linear synthesis in solution will be challenging; therefore, the ligation of pre-synthesised shorter fragments or synthesis on solid supports would be more desirable. Click-type oligonucleotides obtained by either method can be studied as redox-active polymers, luminescent markers, and immuno- or radiotracers (Scheme 13). Also, they can be potentially used in hybridisation experiments or employed as self-organising structures for nanotechnology applications. Scheme 13 shows the hybridisation of a hypothetical oligonucleotide 101 with a complementary “click” oligonucleotide strand (the helical-like structure is speculative).
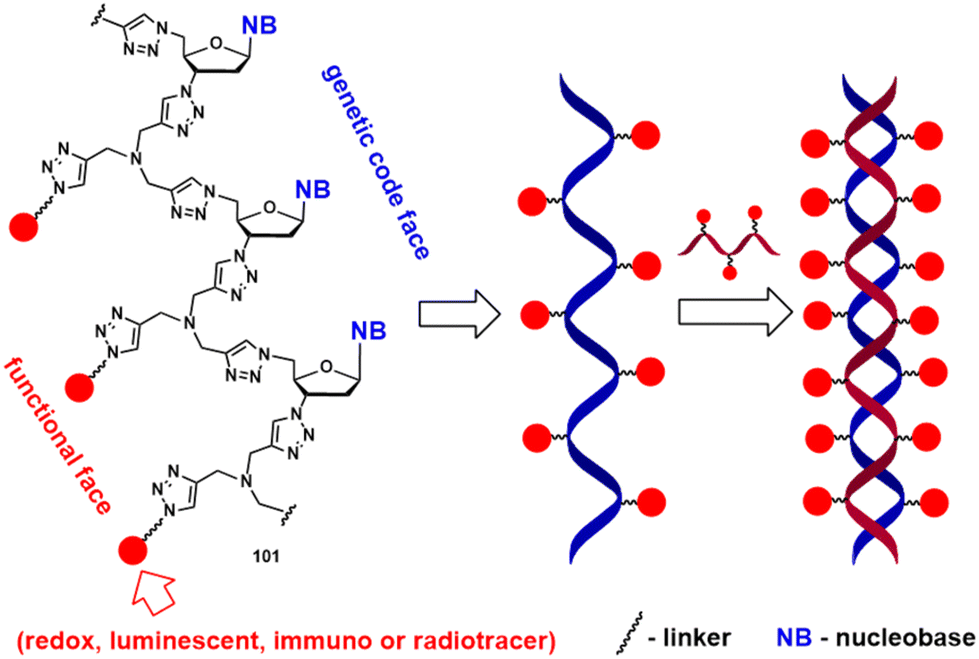 | ||
| Scheme 13 Hybridisation of click-type oligonucleotide 101 with a complementary oligonucleotide sequence. | ||
PEX reactions represent another challenge for future studies on organometallic nucleic acid components. Scheme 14 shows the general conditions for a PEX reaction, using Fc-gNTP 102 as an example of an organometallic triphosphate. Practical implementation of this goal requires access to properly engineered polymerases and therefore it can be only possible to achieve in a few laboratories in the world.
 | ||
| Scheme 14 General scheme for the PEX reaction with Fc-GNA nucleoside triphosphate 102. | ||
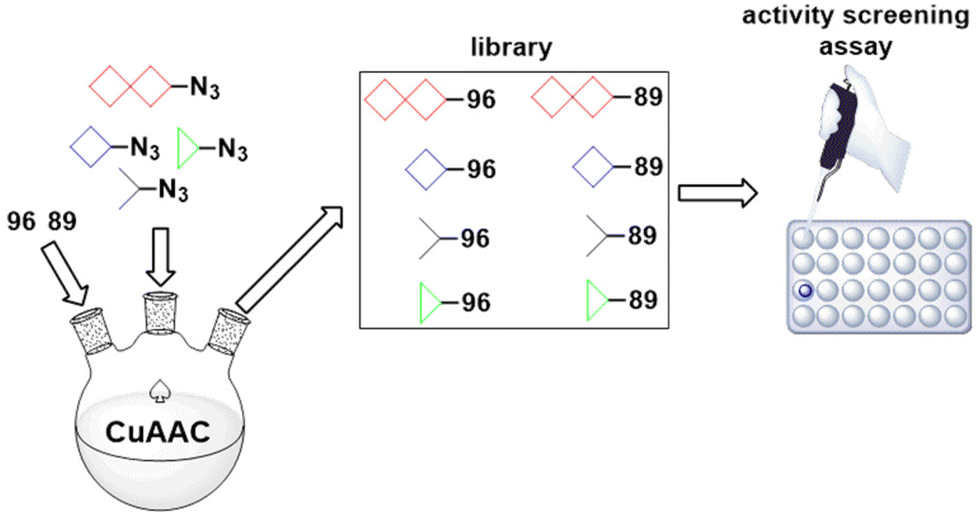
Yet another attractive area for future research involves organometallic “click” modifications of the 5′ mRNA cap structure.146–148 Due to its role as an inhibitor of mRNA enzymatic degradation, a regulator of mRNA nuclear export and a promoter of translation,149–151 the cap represents an interesting target for bioorganometallic chemistry. Accordingly, Fig. 14 shows four hypothetical derivatives of the m7GpppN (N stands for the first transcribed nucleotide) cap modified with click-ferrocenyl entities. As shown in Fig. 14 compounds 103–106 can potentially act as translation inhibitors (e.g. via eIF4E binding)147 or mRNA stability enhancers, both of which are desirable in the development of new anticancer agents or vaccines. Last but not least, it should be emphasized that acetylenic intermediates such as 89 (Scheme 10) and 96 (Scheme 11) hold application potential for combinatorial chemistry with azides as reaction partners. Libraries of compounds obtained in this way can then be biologically screened in many directions (Scheme 15).
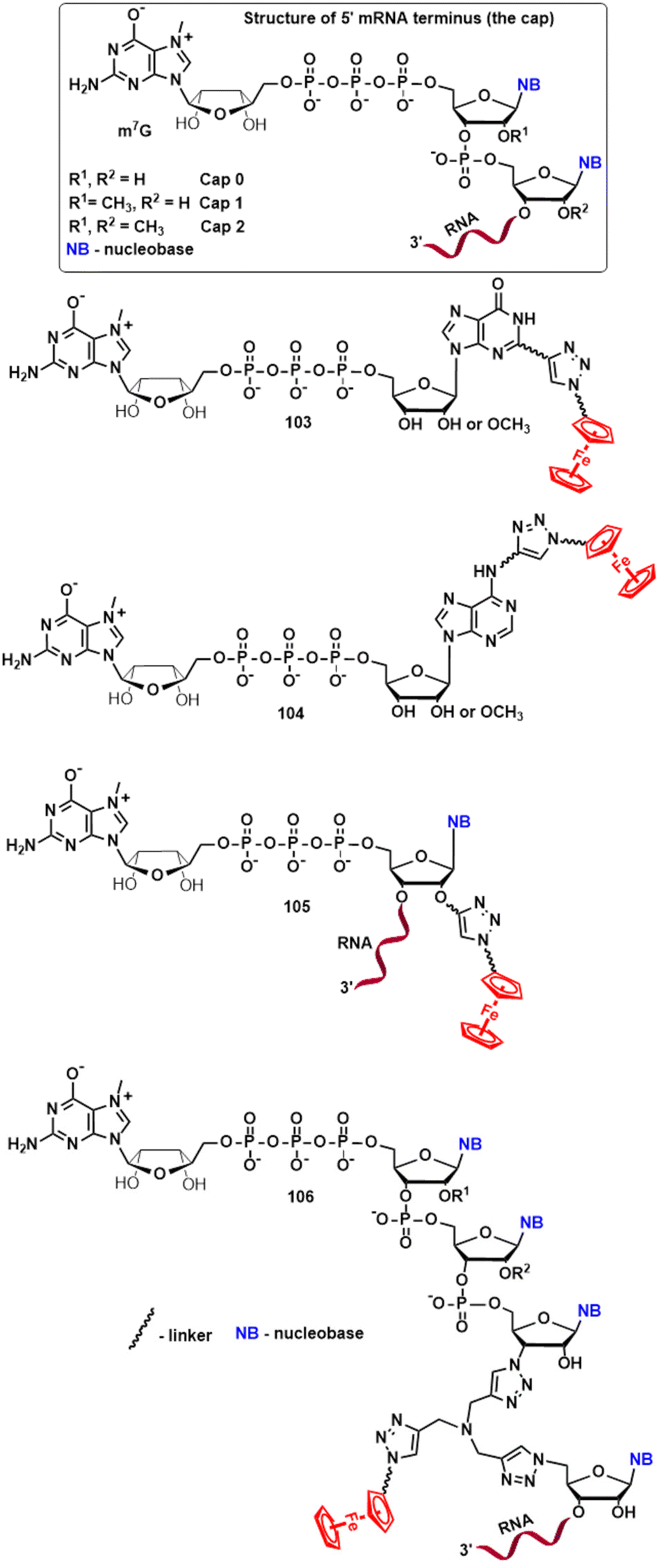 | ||
| Fig. 14 Structures of the 5′ mRNA cap and hypothetical click-ferrocenyl cap conjugates 103–106 with potential for biological applications. | ||
 | ||
| Scheme 15 Combinatorial chemistry with 89 and 96 compounds and azides. | ||
Apart from synthetic chemistry, future studies will also need to focus on deciphering the biological mechanisms of action of organometallic nucleic acid components using modern proteomics and metallomics techniques. To summarize, over the last 12 years we have developed synthetic approaches that have enabled the synthesis of increasingly complex organometallic nucleic acid constituents. Some of these have shown promising biological activities, while others are emerging as building blocks for more elaborate molecular structures. Although progress has been made, further exploration of chemical space is still needed in the quest for new compounds with better activities.
Author contributions
M.K. wrote the manuscript and K.K. designed the concept of the manuscript and wrote the manuscript. All authors have read and approved the final version of the manuscript.Abbreviations
| A549 | Human non-small cell lung carcinoma cells |
| Ac | Acetyl |
| ACE2 | Angiotensin-converting enzyme 2 |
| AZT | 3′-Azido-3′-deoxythymidine |
| Aβ21–40 | The C-terminal region of amyloid-β |
| BEAS-2B | Non-tumorigenic human bronchial epithelial cells |
| Bz | Benzoyl |
| cisPt | Cisplatin |
| CMIA | Carbonyl metallo immunoassay |
| Cp | Cyclopentadienyl (C5H5) ring |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| CuAAC | Cu(I)-Catalysed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction |
| CV | Cyclic voltammetry |
| dA | Deoxyadenosine |
| DCM | Dichloromethane |
| DHQ | Dihydroquinine |
| DHQD | Dihydroquinidine |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DMTr | Dimethoxytrityl |
| DNA | Deoxyribonucleic acid |
| DOX | Doxorubicin |
| dT | Deoxythymidine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| eIF4E | Eukaryotic initiation factor 4E |
| EPR | Electron paramagnetic resonance |
| ESI-MS | Electrospray Ionisation Mass Spectrometry |
| Fc | Ferrocenyl |
| Fc-gTNPs | Ferrocenyl-glycol nucleoside triphosphates |
| FcH | Ferrocene |
| 5-FU | 5-Fluorouracil |
| GI50 | Growth inhibition 50%—the concentration of a compound that reduces total cell growth by 50% |
| GNA | Glycol nucleic acid |
| H1975 | Erlotinib-resistant lung cancer cells |
| HEK293T | Human embryonic kidney cells |
| HeLa | Cervical carcinoma cells |
| HL-60 | Human promyelocytic leukaemia cells |
| HOBt | 1-Hydroxybenzotriazole |
| HPLC | High-performance liquid chromatography |
| HT-29 | Human colon carcinoma cells |
| IC50 | Half maximal inhibitory concentration |
| IEDDA | Inverse-electron-demand Diels–Alder reaction |
| iPr | Isopropyl |
| Ishikawa | Human endometrial adenocarcinoma cells |
| L929 | Non-tumorigenic murine fibroblast cells |
| Lev | Levulinoyl |
| m7G | An N7 methylated guanine |
| MCF-7 | Oestrogen receptor-responsive human breast adenocarcinoma cells |
| MCF-7/DOX | Oestrogen receptor-responsive human breast adenocarcinoma cells resistant to doxorubicin |
| MDA-MB-231 | Oestrogen-negative human breast adenocarcinoma cells |
| MIC | Minimal inhibitory concentration |
| MonoMac6 | Human monocytic leukaemia cells |
| mRNA | Messenger RNA |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAC | N-Acetyl-L-cysteine |
| NBS | N-Bromosuccinimide |
| OTTLE | Optically transparent thin-layer electrode cell |
| PDAC | Pancreatic ductal adenocarcinoma cells |
| PEMB | 5-Ethyl-2-methylpyridine borane |
| PEX | Primer extension reaction |
| PYR | Pyrimidine |
| RNA | Ribonucleic acid |
| RNAi | RNA interference |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| S | Spike protein of SARS-CoV-2 virus |
| THF | Tetrahydrofuran |
| X-Ray | X-Radiation |
| XNA | Xeno-nucleic acid |
Data availability
This is a Perspective article and thus no new experimental data were generated here.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the University of Łódź and the National Science Center in Cracow, Poland is gratefully acknowledged. We would like to thank all the co-workers who over the last 12 years have contributed to our program in the chemistry of organometallic nucleic acid components.References
- J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs, Chem. Rev., 2016, 116, 14379–14455, DOI:10.1021/acs.chemrev.6b00209
.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazzi, Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs, Chem. Rev., 2014, 114, 9154–9218, DOI:10.1021/cr5002035
- E. De Clercq, Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections, Clin. Microbiol. Rev., 2003, 16, 569–596, DOI:10.1128/cmr.16.4.569-596.2003
- W. Yin, C. Mao, X. Luan, D.-D. Shen, H. Su, X. Wang, F. Zhou, W. Zhao, M. Gao, S. Chang, Y.-C. Xie, G. Tian, H.-W. Jiang, S.-C. Tao, J. Shen, Y. Jiang, H. Jiang, Y. Xu, S. Zhang, Y. Zhang and H. E. Xu, Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir, Science, 2020, 368, 1499–1504, DOI:10.1126/science.abc1560
- K. W. Knouse, J. N. de Gruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. Mcdonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Unlocking P(V): Reagents for chiral phosphorothioate synthesis, Science, 2018, 361, 1234–1238, DOI:10.1126/science.aau3369
- K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling and T. R. Cech, Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena, Cell, 1982, 31, 147–157, DOI:10.1016/0092-8674(82)90414-7
- T. E. Edwards, D. J. Klein and A. R. Ferre-D'Amare, Riboswitches: small-molecule recognition by gene regulatory RNAs, Curr. Opin. Struct. Biol., 2007, 17, 273–279, DOI:10.1016/j.sbi.2007.05.004
- S. Diez, J. Ryu, K. Caban, R. L. Gonzalez Jr and J. Dworkin, The alarmones (p)ppGpp directly regulate translation initiation during entry into quiescence, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15565–15572, DOI:10.1073/pnas.1920013117
- T. Carell, C. Brandmayr, A. Hienzsch, M. Müller, D. Pearson, V. Reiter, I. Thoma, P. Thumbs and M. Wagner, Structure and Function of Noncanonical Nucleobases, Angew. Chem., Int. Ed., 2012, 51, 7110–7131, DOI:10.1002/anie.201201193
- J. R. Hamilton, E. Chen, B. S. Perez, C. R. Sandoval Espinoza, M. H. Kang, M. Trinidad, W. Ngo and J. A. Doudna, In vivo human T cell engineering with enveloped delivery vehicles, Nat. Biotechnol., 2024, 1–9, DOI:10.1038/s41587-023-02085-z
- W. Brad Wan and P. P. Seth, The Medicinal Chemistry of Therapeutic Oligonucleotides, J. Med. Chem., 2016, 59, 9645–9667, DOI:10.1021/acs.jmedchem.6b00551
- J. W. Szostak, The Narrow Road to the Deep Past: In Search of the Chemistry of the Origin of Life, Angew. Chem., Int. Ed., 2017, 56, 11037–11043, DOI:10.1002/anie.201704048
- H. Mutschler, A. Wochner and P. Holliger, Freeze–thaw cycles as drivers of complex ribozyme assembly, Nat. Chem., 2015, 7, 502–508, DOI:10.1038/nchem.2251
- S. Stairs, A. Nikmal, D.-K. Bučar, S.-L. Zheng, J. W. Szostak and M. W. Powner, Divergent prebiotic synthesis of pyrimidine and 8-oxo-purine ribonucleotides, Nat. Commun., 2017, 8, 15270, DOI:10.1038/ncomms15270
- S. Becker, C. Schneider, H. Okamura, A. Crisp, T. Amatov, M. Dejmek and T. Carell, Wet-dry cycles enable the parallel origin of canonical and non-canonical nucleosides by continuous synthesis, Nat. Commun., 2018, 9, 163, DOI:10.1038/s41467-017-02639-1
- P. Herdewijn and P. Marlière, Toward Safe Genetically Modified Organisms through the Chemical Diversification of Nucleic Acids, Chem. Biodivers., 2009, 6, 791–808, DOI:10.1002/cbdv.200900083
- J. C. Chaput and P. Herdewijn, What Is XNA?, Angew. Chem., Int. Ed., 2019, 58, 11570–11572, DOI:10.1002/anie.201905999
- J. C. Chaput, P. Herdewijn and M. Hollenstein, Orthogonal Genetic Systems, ChemBioChem, 2020, 21, 1408–1411, DOI:10.1002/cbic.201900725
- N. Budisa, V. Kubyshkin and M. Schmidt, Xenobiology: A Journey towards Parallel Life Forms, ChemBioChem, 2020, 21, 2228–2231, DOI:10.1002/cbic.202000141
- M. Schmidt, Xenobiology: A new form of life as the ultimate biosafety tool, BioEssays, 2010, 32, 322–331, DOI:10.1002/bies.200900147
- V. B. Pinheiro and P. Holliger, The XNA world: progress towards replication and evolution of synthetic genetic polymers, Curr. Opin. Chem. Biol., 2012, 16, 245–252, DOI:10.1016/j.cbpa.2012.05.198
- J. C. Chaput, H. Yu and S. Zhang, The Emerging World of Synthetic Genetics, Chem. Biol., 2012, 19, 1360–1371, DOI:10.1016/j.chembiol.2012.10.011
- J. C. Chaput, Redesigning the Genetic Polymers of Life, Acc. Chem. Res., 2021, 54, 1056–1065, DOI:10.1021/acs.accounts.0c00886
- G. Houlihan, S. Arangundy-Franklin, B. T. Porebski, N. Subramanian, A. I. Taylor and P. Holliger, Discovery and evolution of RNA and XNA reverse transcriptase function and fidelity, Nat. Chem., 2020, 12, 683–690, DOI:10.1038/s41557-020-0502-8
- G. Houlihan, S. Arangundy-Franklin and P. Holliger, Exploring the Chemistry of Genetic Information Storage and Propagation through Polymerase Engineering, Acc. Chem. Res., 2017, 50, 1079–1087, DOI:10.1021/acs.accounts.7b00056
- E. Meggers and L. Zhang, Synthesis and Properties of the Simplified Nucleic Acid Glycol Nucleic Acid, Acc. Chem. Res., 2010, 43, 1092–1102, DOI:10.1021/ar900292q
- P. Kumar and M. H. Caruthers, DNA Analogues Modified at the Nonlinking Positions of Phosphorus, Acc. Chem. Res., 2020, 53, 2152–2166, DOI:10.1021/acs.accounts.0c00078
- M. Flamme, S. Hanlon, I. Marzuoli, K. Püntener, F. Sladojevich and M. Hollenstein, Evaluation of 3′-phosphate as a transient protecting group for controlled enzymatic synthesis of DNA and XNA oligonucleotides, Nat. Commun. Chem., 2022, 5, 68, DOI:10.1038/s42004-022-00685-5
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Recent progress in non-native nucleic acid modifications, Chem. Soc. Rev., 2021, 50, 5126–5164, 10.1039/D0CS01430C
- M. Yu, X. Tang, Z. Li, W. Wang, S. Wang, M. Li, Q. Yu, S. Xie, X. Zuo and C. Chen, High-throughput DNA synthesis for data storage, Chem. Soc. Rev., 2024, 53, 4463–4489, 10.1039/D3CS00469D
- K. Yang, C. M. McCloskey and J. C. Chaput, Reading and Writing Digital Information in TNA, ACS Synth. Biol., 2020, 9, 2936–2942, DOI:10.1021/acssynbio.0c00361
- Y. Wang, Y. Xiong, K. Shi, C. Y. Effah, L. Song, L. He and J. Liu, DNA nanostructures for exploring
cell–cell communication, Chem. Soc. Rev., 2024, 53, 4020–4044, 10.1039/D3CS00944K
- F. Li, J. Li, B. Dong, F. Wang, C. Fan and X. Zuo, DNA nanotechnology-empowered nanoscopic imaging of biomolecules, Chem. Soc. Rev., 2021, 50, 5650–5667, 10.1039/D0CS01281E
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7, 573–584, DOI:10.1038/nrc2167
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs, Chem. Rev., 2016, 116, 3436–3486, DOI:10.1021/acs.chemrev.5b00597
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Metal-based anticancer agents as immunogenic cell death inducers: the past, present, and future, Chem. Soc. Rev., 2022, 51, 1212–1233, 10.1039/D1CS00417D
-
Bioorganometallics: Biomolecules, Labeling, Medicine, ed. G. Jaouen, Wiley-VCH, Weinheim, 2006 Search PubMed
- B. Albada and N. Metzler-Nolte, Organometallic–Peptide Bioconjugates: Synthetic Strategies and Medicinal Applications, Chem. Rev., 2016, 116, 11797–11839, DOI:10.1021/acs.chemrev.6b00166
- G. Gasser, I. Ott and N. Metzler-Nolte, Organometallic Anticancer Compounds, J. Med. Chem., 2011, 54, 3–25, DOI:10.1021/jm100020w
- M. Patra and G. Gasser, The medicinal chemistry of ferrocene and its derivatives, Nat. Rev. Chem., 2017, 1, 0066, DOI:10.1038/s41570-017-0066
- G. Jaouen, A. Vessières and S. Top, Ferrocifen type anti cancer drugs, Chem. Soc. Rev., 2015, 44, 8802–8817, 10.1039/C5CS00486A
- K. D. Mjos and C. Orvig, Metallodrugs in Medicinal Inorganic Chemistry, Chem. Rev., 2014, 114, 4540–4563, DOI:10.1021/cr400460s
- K. Kowalski, Recent developments in the chemistry of ferrocenyl secondary natural product conjugates, Coord. Chem. Rev., 2018, 366, 91–108, DOI:10.1016/j.ccr.2018.04.008
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, The development of RAPTA compounds for the treatment of tumors, Coord. Chem. Rev., 2016, 306, 86–114, DOI:10.1016/j.ccr.2015.06.014
- A. Merlino, Interactions between proteins and Ru compounds of medicinal interest: A structural perspective, Coord. Chem. Rev., 2016, 326, 111–134, DOI:10.1016/j.ccr.2016.08.001
- L. E. Jennings and N. J. Long, ‘Two is better than one’—probes for dual-modality molecular imaging, Chem. Commun., 2009, 3511–3524, 10.1039/B821903F
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Organometallic and coordination rhenium compounds and their potential in cancer therapy, Coord. Chem. Rev., 2019, 393, 79–117, DOI:10.1016/j.ccr.2019.04.014
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics?, J. Med. Chem., 2018, 61, 5805–5821, DOI:10.1021/acs.jmedchem.7b01689
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Challenges and Opportunities in the Development of Organometallic Anticancer Drugs, Organometallics, 2012, 31, 5677–5685, DOI:10.1021/om300373t
- P. Chellan and P. J. Sadler, Enhancing the Activity of Drugs by Conjugation to Organometallic Fragments, Chem. – Eur. J., 2020, 26, 8676–8688, DOI:10.1002/chem.201904699
- A. Vessières, Y. Wang, M. J. McGlinchey and G. Jaouen, Multifaceted chemical behaviour of metallocene (M = Fe, Os) quinone methides. Their contribution to biology, Coord. Chem. Rev., 2021, 430, 213658, DOI:10.1016/j.ccr.2020.213658
- B. Sharma and V. Kumar, Has Ferrocene Really Delivered Its Role in Accentuating the Bioactivity of Organic Scaffolds?, J. Med.
Chem., 2021, 64, 16865–16921, DOI:10.1021/acs.jmedchem.1c00390
- K. K-W. Lo, Molecular Design of Bioorthogonal Probes and Imaging Reagents Derived from Photofunctional Transition Metal Complexes, Acc. Chem. Res., 2020, 53, 32–44, DOI:10.1021/acs.accounts.9b00416
- U. Schatzschneider and N. Metzler-Nolte, New Principles in Medicinal Organometallic Chemistry, Angew. Chem., Int. Ed., 2006, 45, 1504–1507, DOI:10.1002/anie.200504604
- C. E. Carraher Jr., Condensation Metallocene Polymers, J. Inorg. Polym. Mater., 2005, 15, 121–145, DOI:10.1007/s10904-004-2382-6
- D. R. van Staveren and N. Metzler-Nolte, Bioorganometallic Chemistry of Ferrocene, Chem. Rev., 2004, 104, 5931–5986, DOI:10.1021/cr0101510
- D. Astruc, Why is Ferrocene so Exceptional?, Eur. J. Inorg. Chem., 2017, 2017, 6–29, DOI:10.1002/ejic.201600983
- P. Štěpnička, Forever young: the first seventy years of ferrocene, Dalton Trans., 2022, 51, 8085–8102, 10.1039/D2DT00903J
- D. Bergstrom, P. Beal and R. Lind, Synthesis of (Dicarbonyl)(η5-cyclopentadienyl)Manganese Complex Stabilized Nucleoside Phosphite Esters, Nucleosides Nucleotides, 1989, 8, 1061–1063, DOI:10.1080/07328318908054281
- D. Bergstrom and T. Schmaltz, Synthesis of (Dicarbonyl)(η5-cyclopentadienyl)iron-Derived Nucleoside Phosphonate Esters, Nucleosides Nucleotides, 1989, 8, 1057–1059, DOI:10.1080/07328318908054280
- D. Bergstrom and T. Schmaltz, Organoiron-mediated alkylation of phosphite esters: synthesis of (dicarbonyl)(η5-cyclopentadienyl)iron-derived nucleoside phosphonate esters, J. Org. Chem., 1992, 57, 873–876, DOI:10.1021/jo00029a017
- J. M. D. R. Toma and D. E. Bergstrom, Transition Metal Labeling of Oligodeoxyribonucleotides: Synthesis and Characterization of (Pentacarbonyl)tungsten(0) Nucleoside Phosphites, J. Org. Chem., 1994, 59, 2418–2422, DOI:10.1021/jo00088a024
- J.-L. H. A. Duprey and J. H. R. Tucker, Metal–Carbon Bonds in Biopolymer Conjugates: Bioorganometallic Nucleic Acid Chemistry, Chem. Lett., 2014, 43, 157–163, DOI:10.1246/cl.131019
- J. M. Lynam, Nucleobase-containing transition metal complexes as building blocks for biological markers and supramolecular structures, Dalton Trans., 2008, 4067–4078, 10.1039/B802347F
- F. Boisten, I. Maisuls, T. Schäfer, C. A. Strassert and J. Müller, Site-specific covalent metalation of DNA oligonucleotides with phosphorescent platinum(II) complexes, Chem. Sci., 2023, 14, 2399–2404, 10.1039/D2SC05916A
- K. Kowalski, Organometallic nucleosides—Synthesis, transformations, and applications, Coord. Chem. Rev., 2021, 432, 213705, DOI:10.1016/j.ccr.2020.213705
- K. Kowalski, Ferrocenyl-nucleobase complexes: Synthesis, chemistry and applications, Coord. Chem. Rev., 2016, 317, 132–156, DOI:10.1016/j.ccr.2016.02.008
- D. Ukale and T. Lönnberg, Organomercury Nucleic Acids: Past, Present and Future, ChemBioChem, 2021, 22, 1733–1739, DOI:10.1002/cbic.202000821
- T. K. Kotammagari, L. Y. Saleh and T. Lönnberg, Organometallic modification confers oligonucleotides new functionalities, Chem. Commun., 2024, 60, 3118–3128, 10.1039/D4CC00305E
- J. Müller, Nucleic acid duplexes with metal-mediated base pairs and their structures, Coord. Chem. Rev., 2019, 393, 37–47, DOI:10.1016/j.ccr.2019.05.007
- B. Jash and J. Müller, Metal-Mediated Base Pairs: From Characterization to Application, Chem. – Eur. J., 2017, 23, 17166–17178, DOI:10.1002/chem.201703518
- A. Rousina-Webb, C. Lachance-Brais, F. J. Rizzuto, M. S. Askari and H. F. Sleiman, Transition-Metal-Functionalized DNA Double-Crossover Tiles: Enhanced Stability and Chirality Transfer to Metal Centers, Angew. Chem., Int. Ed., 2020, 59, 4091–4098, DOI:10.1002/anie.201913956
- S. Ghosh and E. Defrancq, Metal-Complex/DNA Conjugates: A Versatile Building Block for DNA Nanoarrays, Chem. – Eur. J., 2010, 16, 12780–12787, DOI:10.1002/chem.201001590
- H. Yang, K. L. Metera and H. F. Sleiman, DNA modified with metal complexes: Applications in the construction of higher order metal–DNA nanostructures, Coord. Chem. Rev., 2010, 254, 2403–2415, DOI:10.1016/j.ccr.2010.02.026
- B. Lippert and P. J. S. Miguel, The Renaissance of Metal–Pyrimidine Nucleobase Coordination Chemistry, Acc. Chem. Res., 2016, 49, 1537–1545, DOI:10.1021/acs.accounts.6b00253
- A. Collado, M. Gómez-Gallego and M. A. Sierra, Nucleobases Having M–C Bonds: An Emerging Bio-Organometallic Field, Eur. J. Org. Chem., 2018, 1617–1623, DOI:10.1002/ejoc.201800135
- D. Schwalbe, A. Majdalani, J. Velcicky, E. Heßler, T. Wieder, A. Prokop and H.-G. Schmalz, Iron-Containing Nucleoside Analogues with Pronounced Apoptosis-Inducing Activity, Angew. Chem., Int. Ed., 2004, 43, 1731–1734, DOI:10.1002/anie.200353132
- C. Hirschhäuser, J. Velcicky, D. Schwalbe, E. Hessler, A. Majdalani, J.-M. Neudörfl, A. Prokop, T. Wieder and H.-G. Schmalz, Nucleoside Analogues with a 1,3-Diene-Fe(CO)3 Substructure: Stereoselective Synthesis, Configurational Assignment, and Apoptosis-Inducing Activity, Chem. – Eur. J., 2013, 19, 13017–13029, DOI:10.1002/chem.201301672
- Y. Takezawa and M. Shionoya, Metal-Mediated DNA Base Pairing: Alternatives to Hydrogen-Bonded Watson–Crick Base Pairs, Acc. Chem. Res., 2012, 45, 2066–2076, DOI:10.1021/ar200313h
- M. Flamme, P. Röthlisberger, F. Levi-Acobas, M. Chawla, R. Oliva, L. Cavallo, G. Gasser, P. Marlière, P. Herdewijn and M. Hollenstein, Enzymatic Formation of an Artificial Base Pair Using a Modified Purine Nucleoside Triphosphate, ACS Chem. Biol., 2020, 15, 2872–2884, DOI:10.1021/acschembio.0c00396
- J. Kondo, Y. Tada, T. Dairaku, Y. Hattori, H. Saneyoshi, A. Ono and Y. Tanaka, A metallo-DNA nanowire with uninterrupted one-dimensional silver array, Nat. Chem., 2017, 9, 956–960, DOI:10.1038/nchem.2808
- G. A. Burley, J. Gierlich, M. R. Mofid, H. Nir, S. Tal, Y. Eichen and T. Carell, Directed DNA Metallization, J. Am. Chem. Soc., 2006, 128, 1398–1399, DOI:10.1021/ja055517v
- H. Song, X. Li, Y. Long, G. Schatte and H.-B. Kraatz, Ferrocene-modified pyrimidine nucleosides: synthesis, structure and electrochemistry, Dalton Trans., 2006, 39, 4696–4701, 10.1039/B608077D
- A. Simonova, I. Magriñá, V. Sýkorová, R. Pohl, M. Ortiz, L. Havran, M. Fojta, C. K. O'Sullivan and M. Hocek, Tuning of Oxidation Potential of Ferrocene for Ratiometric Redox Labeling and Coding of Nucleotides and DNA, Chem. – Eur. J., 2020, 26, 1286–1291, DOI:10.1002/chem.201904700
- P. Brázdilová, M. Vrábel, R. Pohl, H. Pivoňková, L. Havran, M. Hocek and M. Fojta, Ferrocenylethynyl Derivatives of Nucleoside Triphosphates: Synthesis, Incorporation, Electrochemistry, and Bioanalytical Applications, Chem. – Eur. J., 2007, 13, 9527–9533, DOI:10.1002/chem.200701249
- C. Figazzolo, Y. Ma, J. H. R. Tucker and M. Hollenstein, Ferrocene as a potential electrochemical reporting surrogate of abasic sites in DNA, Org. Biomol. Chem., 2022, 20, 8125–8135, 10.1039/D2OB01540D
- M. Hocek, P. Štěpnička, J. Ludvík, I. Císařová, I. Votruba, D. Řeha and P. Hobza, Ferrocene-Modified Purines as Potential Electrochemical Markers: Synthesis, Crystal Structures, Electrochemistry and Cytostatic Activity of (Ferrocenylethynyl)- and (Ferrocenylethyl)purines, Chem. – Eur. J., 2004, 10, 2058–2066, DOI:10.1002/chem.200305621
- G. Chatelain, A. Meyer, F. Morvan, J.-J. Vasseur and C. Chaix, Electrochemical detection of nucleic acids using pentaferrocenyl phosphoramidate α-oligonucleotides, New J. Chem., 2011, 35, 893–901, 10.1039/C0NJ00902D
- A. R. Pike, L. C. Ryder, B. R. Horrocks, W. Clegg, B. A. Connolly and A. Houlton, Ferrocenyl-Modified DNA: Synthesis, Characterization and Integration with Semiconductor Electrodes, Chem. – Eur. J., 2005, 11, 344–353, DOI:10.1002/chem.200400632
- W. A. Wlassoff and G. C. King, Ferrocene conjugates of dUTP for enzymatic redox labelling of DNA, Nucleic Acids Res., 2022, 30, e58, DOI:10.1093/nar/gnf058
- F. Patolsky, Y. Weizmann and I. Willner, Redox-Active Nucleic-Acid Replica for the Amplified Bioelectrocatalytic Detection of Viral DNA, J. Am. Chem. Soc., 2002, 124, 770–772, DOI:10.1021/ja0119752
- C. J. Yu, Y. Wan, H. Yowanto, J. Li, C. Tao, M. D. James, C. L. Tan, G. F. Blackburn and T. J. Meade, Electronic Detection of Single-Base Mismatches in DNA with Ferrocene-Modified Probes, J. Am. Chem. Soc., 2001, 123, 11155–11161, DOI:10.1021/ja010045f
- S. Djaković, L. Glavaš-Obrovac, J. Lapić, S. Maračić, J. Kirchofer, M. Knežević, M. Jukić and S. Raić-Malić, Synthesis and biological evaluations of mono- and bis-ferrocene uracil derivatives, Appl. Organomet. Chem., 2021, 35, e6052, DOI:10.1002/aoc.6052
- M. Rana, A. Perotti, L. M. Bisset, J. D. Smith, E. Lamden, Z. Khan, M. K. Ismail, K. Ellis, K. A. Armstrong, S. L. Hodder, C. Bertoli, L. Meneguello, R. A. M. de Bruin, J. R. Morris, I. Romero-Canelon, J. H. R. Tucker and N. J. Hodges, A ferrocene-containing nucleoside analogue targets DNA replication in pancreatic cancer cells, Metallomics, 2022, 14, mfac041, DOI:10.1093/mtomcs/mfac041
- H. V. Nguyen, Z. Zhao, A. Sallustrau, S. L. Horswell, L. Male, A. Mulas and J. H. R. Tucker, A ferrocene nucleic acid oligomer as an organometallic structural mimic of DNA, Chem. Commun., 2012, 48, 12165–12167, 10.1039/C2CC36428J
- M. Toma, L. Božičević, J. Lapić, S. Djaković, D. Šakić, T. Tandarić, R. Vianello and V. Vrček, Transacylation in Ferrocenoyl-Purines. NMR and Computational Study of the Isomerization Mechanism, J. Org. Chem., 2019, 84, 12471–12480, DOI:10.1021/acs.joc.9b01944
- M. Toma, G. Zubčić, J. Lapić, S. Djaković, D. Šakić and V. Vrček, Ferrocenoyl-adenines: substituent effects on regioselective acylation, Beilstein J. Org. Chem., 2022, 18, 1270–1277, DOI:10.3762/bjoc.18.133
- M. K. Ismail, K. A. Armstrong, S. L. Hodder, S. L. Horswell, L. Male, H. V. Nguyen, E. A. Wilkinson, N. J. Hodges and J. H. R. Tucker, Organometallic nucleoside analogues: effect of the metallocene metal atom on cancer cell line toxicity, Dalton Trans., 2020, 49, 1181–1190, 10.1039/C9DT04174E
- M. K. Ismail, Z. Khan, M. Rana, S. L. Horswell, L. Male, H. V. Nguyen, A. Perotti, I. Romero-Canelón, E. A. Wilkinson, N. J. Hodges and J. H. R. Tucker, Effect of Regiochemistry and Methylation on the Anticancer Activity of a Ferrocene-Containing Organometallic Nucleoside Analogue, ChemBioChem, 2020, 21, 2487–2494, DOI:10.1002/cbic.202000124
- J. L. Kedge, H. V. Nguyen, Z. Khan, L. Male, M. K. Ismail, H. V. Roberts, N. J. Hodges, S. L. Horswell, Y. Mehellou and J. H. R. Tucker, Organometallic Nucleoside Analogues: Effect of Hydroxyalkyl Linker Length on Cancer Cell Line Toxicity, Eur. J. Inorg. Chem., 2017, 2, 466–476, DOI:10.1002/ejic.201600853
- K. Kowalski and J. Zakrzewski, (η5-C5H5)Fe(CO)2-complexes of uridine and thymidine, J. Organomet. Chem., 2003, 668, 91–94, DOI:10.1016/S0022-328X(02)02144-7
- A. Varenne, A. Vessières, P. Brossier and G. Jaouen, Application of the non- radioisotopic carbonyl metalloimmunoassay (CMIA) to diphenylhydantoin, Res. Commun. Chem. Pathol. Pharmacol., 1994, 84, 81–92, DOI:10.1016/abio.1996.0450
- K. Kowalski, A. Koceva-Chyła, A. Pieniążek, J. Bernasińska, J. Skiba, A. J. Rybarczyk-Pirek and Z. Jóźwiak, The synthesis, structure, electrochemistry and in vitro anticancer activity studies of ferrocenyl-thymine conjugates, J. Organomet. Chem., 2012, 700, 58–68, DOI:10.1016/j.jorganchem.2011.11.014
- K. Kowalski, J. Skiba, L. Oehninger, I. Ott, J. Solecka, A. Rajnisz and B. Therrien, Metallocene-Modified Uracils: Synthesis, Structure, and Biological Activity, Organometallics, 2013, 32, 5766–5773, DOI:10.1021/om400294s
- I. Anisimov, S. Saloman, A. Hildebrandt, H. Lang, D. Trzybiński, K. Woźniak, D. Šakić, V. Vrček and K. Kowalski, 1,1′-Bis(thymine)ferrocene Nucleoside: Synthesis and Study of Its Stereoselective Formation, ChemPlusChem, 2017, 82, 859–866, DOI:10.1002/cplu.201700215
- M. Gil-Moles, S. Türck, U. Basu, A. Pettenuzzo, S. Bhattacharya, A. Rajan, X. Ma, R. Büssing, J. Wölker, H. Burmeister, H. Hoffmeister, P. Schneeberg, A. Prause, P. Lippmann, J. Kusi-Nimarko, S. Hassell-Hart, A. McGown, D. Guest, Y. Lin, A. Notaro, R. Vinck, J. Karges, K. Cariou, K. Peng, X. Qin, X. Wang, J. Skiba, Ł. Szczupak, K. Kowalski, U. Schatzschneider, C. Hemmert, H. Gornitzka, E. R. Milaeva, A. A. Nazarov, G. Gasser, J. Spencer, L. Ronconi, U. Kortz, J. Cinatl, D. Bojkova and I. Ott, Metallodrug Profiling against SARS-CoV-2 Target Proteins Identifies Highly Potent Inhibitors of the S/ACE2 interaction and the Papain-like Protease PLpro, Chem. – Eur. J., 2021, 27, 17928–17940, DOI:10.1002/chem.202103258
- S. La Manna, C. Di Natale, V. Panzetta, P. A. Netti, A. Merlino, K. Kowalski and D. Marasco, Enhancers of amyloid aggregation: novel ferrocene-based compounds selective toward amyloid models, Inorg. Chem. Front., 2024, 11, 6577–6587, 10.1039/D4QI01854K
- K. Kowalski, Ł. Szczupak, S. Saloman, D. Steverding, A. Jabłoński, V. Vrček, A. Hildebrandt, H. Lang and A. Rybarczyk-Pirek, Cymantrene, Cyrhetrene and Ferrocene Nucleobase Conjugates: Synthesis, Structure, Computational Study, Electrochemistry and Antitrypanosomal Activity, ChemPlusChem, 2017, 82, 303–314, DOI:10.1002/cplu.201600462
- A. Jabłoński, K. Matczak, A. Koceva-Chyła, K. Durka, D. Steverding, K. Jakubiec-Krześniak, J. Solecka, D. Trzybiński, K. Woźniak, V. Andreu, G. Mendoza, M. Arruebo, K. Kochel, B. Krawczyk, D. Szczukocki and K. Kowalski, Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies, Molecules, 2017, 22, 2220, DOI:10.3390/molecules22122220
- A. Jabłoński, Y. Fritz, H.-A. Wagenknecht, R. Czerwieniec, T. Bernaś, D. Trzybiński, K. Woźniak and K. Kowalski, Pyrene–nucleobase conjugates: synthesis, oligonucleotide binding and confocal bioimaging studies, Beilstein J. Org. Chem., 2017, 13, 2521–2534, DOI:10.3762/bjoc.13.249
- J. Skiba, A. Kowalczyk, M. A. Fik, M. Gapińska, D. Trzybiński, K. Woźniak, V. Vrček, R. Czerwieniec and K. Kowalski, Luminescent pyrenyl-GNA nucleosides: synthesis, photophysics and confocal microscopy studies in cancer HeLa cells, Photochem. Photobiol. Sci., 2019, 18, 2449–2460, 10.1039/c9pp00271e
- A. Jabłoński, A. Kowalczyk, M. A. Fik, D. Trzybiński, K. Woźniak, K. Vinogradova, S. Glińska, V. Vrček, R. Czerwieniec and K. Kowalski, Anthracene-thymine luminophores: Synthesis, photophysical properties, and imaging in living HeLa cells, Dyes Pigm., 2019, 170, 107554, DOI:10.1016/j.dyepig.2019.107554
- J. Skiba, K. Kowalski, A. Prochnicka, I. Ott, J. Solecka, A. Rajnisz and B. Therrien, Metallocene-uracil conjugates: Synthesis and biological evaluation of novel mono-, di- and tri-nuclear systems, J. Organomet. Chem., 2015, 782, 52–61, DOI:10.1016/j.jorganchem.2014.11.017
- J. Skiba, C. Schmidt, P. Lippmann, P. Ensslen, H.-A. Wagenknecht, R. Czerwieniec, F. Brandl, I. Ott, T. Bernaś, B. Krawczyk, D. Szczukocki and K. Kowalski, Substitution of Metallocenes with [2.2]Paracyclophane to Enable Confocal Microscopy Imaging in Living Cells, Eur. J. Inorg. Chem., 2017, 297–305, DOI:10.1002/ejic.201600281
- J. Skiba, R. Karpowicz, I. Szabó, B. Therrien and K. Kowalski, Synthesis and anticancer activity studies of ferrocenyl-thymine-3,6-dihydro-2H-thiopyranes – A new class of metallocene-nucleobase derivatives, J. Organomet. Chem., 2015, 794, 216–222, DOI:10.1016/j.jorganchem.2015.07.012
- T. Seita, K. Yamauchi, M. Kinoshita and M. Imoto, The Synthesis of Nucleoside and Nucleotide Analogs, Bull. Chem. Soc. Jpn., 1972, 45, 926–928, DOI:10.1246/bcsj.45.926
- A. Holý, Aliphatic analogues of nucleosides, nucleotides, and oligonucleotides, Collect. Czech. Chem. Commun., 1975, 40, 187–214, DOI:10.1135/cccc19750187
- T. Seita, M. Kinoshita and M. Imoto, Synthesis of Some Substituted Nucleoside Analogs, Bull. Chem. Soc. Jpn., 1973, 46, 1572–1573, DOI:10.1246/bcsj.46.1572
- A. Holý and G. S. Ivanova, Aliphatic analogues of nucleotides: synthesis and affinity towards nucleases, Nucleic Acid Res., 1974, 1, 19–34, DOI:10.1093/nar/1.1.19
- M. K. Schlegel, X. Xie, L. Zhang and E. Meggers, Insight into the High Duplex Stability of the Simplified Nucleic Acid GNA, Angew. Chem., Int. Ed., 2009, 48, 960–963, DOI:10.1002/ange.200803472
- M. K. Schlegel, L.-O. Essen and E. Meggers, Duplex Structure of a Minimal Nucleic Acid, J. Am. Chem. Soc., 2008, 130, 8158–8159, DOI:10.1021/ja802788g
- M. K. Schlegel, L.-O. Essen and E. Meggers, Atomic resolution duplex structure of the simplified nucleic acidGNA, Chem. Commun., 2010, 46, 1094–1096, 10.1039/B916851F
- M. K. Schlegel, L. Zhang, N. Pagano and E. Meggers, Metal-mediated base pairing within the simplified nucleic acid GNA, Org. Biomol. Chem., 2009, 7, 476–482, 10.1039/B816142A
- K. Murayama and H. Asanuma, Design and Hybridization Properties of Acyclic Xeno Nucleic Acid Oligomers, ChemBioChem, 2021, 22, 2507–2515, DOI:10.1002/cbic.202100184
- E. Meggers and L. Zhang, Synthesis and Properties of the Simplified Nucleic Acid Glycol Nucleic Acid, Acc. Chem. Res., 2010, 43, 1092–1102, DOI:10.1021/ar900292q
- K. Kowalski, Synthesis and chemical transformations of glycol nucleic acid (GNA) nucleosides, Bioorg. Chem., 2023, 141, 106921, DOI:10.1016/j.bioorg.2023.106921
- M. K. Schlegel, A. E. Peritz, K. Kittigowittana, L. Zhang and E. Meggers, Duplex Formation of the Simplified Nucleic Acid GNA, ChemBioChem, 2007, 8, 927–932, DOI:10.1002/cbic.200600435
- M. Egli, M. K. Schlegel and M. Manoharan, Acyclic (S)-glycol nucleic acid (S-GNA) modification of siRNAs improves the safety of RNAi therapeutics while maintaining potency, RNA, 2023, 29, 402–414, DOI:10.1261/rna.079526.122
- C.-H. Tsai, J. Chen and J. W. Szostak, Enzymatic synthesis of DNA on glycerol nucleic acid templates without stable duplex formation between product and template, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14598–14603, DOI:10.1073/pnas.0704211104
- R. S. Zhang, E. O. McCullum and J. C. Chaput, Synthesis of Two Mirror Image 4-Helix Junctions Derived from Glycerol Nucleic Acid, J. Am. Chem. Soc., 2008, 130, 5846–5847, DOI:10.1021/ja800079j
- J. Skiba, Q. Yuan, A. Hildebrandt, H. Lang, D. Trzybiński, K. Woźniak, R. K. Balogh, B. Gyurcsik, V. Vrček and K. Kowalski, Ferrocenyl GNA Nucleosides: A Bridge between Organic and Organometallic Xeno-nucleic Acids, ChemPlusChem, 2018, 83, 77–86, DOI:10.1002/cplu.201700551
- J. Skiba, A. Kowalczyk, D. Trzybiński, K. Woźniak, V. Vrček, M. Gapińska and K. Kowalski, Stereo-Defined Ferrocenyl Glycol Nucleic Acid (Fc-GNA) Constituents: Synthesis, Electrochemistry, Mechanism of Formation, and Anticancer Activity Studies, Eur. J. Inorg. Chem., 2021, 2171–2181, DOI:10.1002/ejic.202100193
- M. Piotrowicz, A. Kowalczyk, D. Trzybiński, K. Woźniak and K. Kowalski, Redox-Active Glycol Nucleic Acid (GNA) Components: Synthesis and Properties of the Ferrocenyl-GNA Nucleoside, Phosphoramidite, and Semicanonical Dinucleoside Phosphate, Organometallics, 2020, 39, 813–823, DOI:10.1021/acs.organomet.9b00851
- A. Kowalczyk, M. Piotrowicz, M. Gapińska, D. Trzybiński, K. Woźniak, T. M. Golding, T. Stringer, G. S. Smith, R. Czerwieniec and K. Kowalski, Chemistry of glycol nucleic acid (GNA): Synthesis, photophysical characterization and insight into the biological activity of phenanthrenyl GNA constituents, Bioorg. Chem., 2022, 125, 105847, DOI:10.1016/j.bioorg.2022.105847
- F. Amblard, J. H. Cho and R. F. Schinazi, Cu(I)-Catalyzed Huisgen Azide−Alkyne 1,3-Dipolar Cycloaddition Reaction in Nucleoside, Nucleotide, and Oligonucleotide Chemistry, Chem. Rev., 2009, 109, 4207–4220, DOI:10.1021/cr9001462
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, A Hitchhiker's Guide to Click-Chemistry with Nucleic Acids, Chem. Rev., 2021, 121, 7122–7154, DOI:10.1021/acs.chemrev.0c00928
- K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H.-J. Krüger, B. Sarkar and W. Kaim, Charge Delocalization in a Heterobimetallic Ferrocene−(Vinyl)Ru(CO)Cl(PiPr3)2 System, Organometallics, 2009, 28, 4196–4209, DOI:10.1021/om9002945
- I. Ott, K. Kowalski, R. Gust, J. Maurer, P. Mücke and R. Winter, Comparative biological evaluation of two ethylene linked mixed binuclear ferrocene/ruthenium organometallic species, Bioorg. Med. Chem. Lett., 2010, 20, 866–869, DOI:10.1016/j.bmcl.2009.12.080
- C. N. Birts, A. P. Sanzone, A. H. El-Sagheer, J. P. Blaydes, T. Brown and A. Tavassoli, Transcription of Click-Linked DNA in Human Cells, Angew. Chem., Int. Ed., 2014, 53, 2362–2365, DOI:10.1002/anie.201308691
- M. Kukwikila, N. Gale, A. H. El-Sagheer, T. Brown and A. Tavassoli, Assembly of a biocompatible triazole-linked gene by one-pot click-DNA ligation, Nat. Chem., 2017, 9, 1089–1098, DOI:10.1038/nchem.2850
- S. Epple, A. Modi, Y. R. Baker, E. Węgrzyn, D. Traoré, P. Wanat, A. E. S. Tyburn, A. Shivalingam, L. Taemaitree, A. H. El-Sagheer and T. Brown, A New 1,5-Disubstituted Triazole DNA Backbone Mimic with Enhanced Polymerase Compatibility, J. Am. Chem. Soc., 2021, 143, 16292–16301, DOI:10.1021/jacs.1c08057
- P. Biegański, E. Kovalski, N. Israel, E. Dmitrieva, D. Trzybiński, K. Woźniak, V. Vrček, M. Godel, C. Riganti, J. Kopecka, H. Lang and K. Kowalski, Electronic Coupling in 1,2,3-Triazole Bridged Ferrocenes and Its Impact on Reactive Oxygen Species Generation and Deleterious Activity in Cancer Cells, Inorg. Chem., 2022, 61, 9650–9666, DOI:10.1021/acs.inorgchem.2c01110
- M. B. Robin and P. Day, Mixed Valence Chemistry-A Survey and Classification, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422, DOI:10.1016/S0065-2792(08)60179-X
- J. Skiba, A. Kowalczyk, A. Gorski, N. Dutkiewicz, M. Gapińska, J. Stróżek, K. Woźniak, D. Trzybiński and K. Kowalski, Replacement of the phosphodiester backbone between canonical nucleosides with a dirhenium carbonyl “click” linker—a new class of luminescent organometallic dinucleoside phosphate mimics, Dalton Trans., 2023, 52, 1551–1567, 10.1039/D2DT03995H
- J. Skiba, M. Hirschfeld, H. Lang, D. Trzybiński, K. Woźniak, M. Gazecka, P. Zmora and K. Kowalski, Click—ferrocenyl nucleotides—synthesis, electrochemistry, and antiproliferative activity studies, J. Organomet. Chem., 2024, 123242, DOI:10.1016/j.jorganchem.2024.123242
- Y. Furuichi, Discovery of m(7)G-cap in eukaryotic mRNAs, Proc. Jpn. Acad., Ser. B, 2015, 91, 394–409, DOI:10.2183/pjab.91.394
- K. Piecyk, P. Pietrow, T. Arnold, R. Worch, N. L. Korneeva and M. Jankowska-Anyszka, Effect of HIV-1 TAT peptide fusion on 5′ mRNA cap analogs cell membrane permeability and translation inhibition, Bioconjugate Chem., 2020, 31(4), 1156–1166, DOI:10.1021/acs.bioconjchem.0c00080
- J. Jemielity, J. Kowalska, A. M. Rydzyk and E. Darzynkiewicz, Synthetic mRNA cap analogs with a modified triphosphate bridge-synthesis, applications and prospects, New J. Chem., 2010, 34, 829–844, 10.1039/c0nj00041h
- A. C. Gingras, B. Raught and N. Sonenberg, eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation, Annu. Rev. Biochem., 1999, 68, 913–963, DOI:10.1146/annurev.biochem.68.1.913
- J. D. Lewis and E. Izaurralde, The role of the cap structure in RNA processing and nuclear export, Eur. J. Biochem., 1997, 247, 461–469, DOI:10.1111/j.1432-1033.1997.00461.x
- R. E. Rhoads, The cap structure of eukaryotic messenger RNA and its interaction with cap-binding protein, Prog. Mol. Subcell. Biol., 1985, 9, 104–155, DOI:10.1007/978-3-642-70203-7_3
| This journal is © The Royal Society of Chemistry 2024 |