
Enhanced electrochemical nitrate reduction to ammonia with nanostructured Mo2C on carbon nanotube-reduced graphene oxide hybrid support†
So Eun
Jang
 a,
Jae Young
Kim
*b and
Duck Hyun
Youn
*a
a,
Jae Young
Kim
*b and
Duck Hyun
Youn
*a
aDepartment of Chemical Engineering, Department of Integrative Engineering for Hydrogen Safety, Kangwon National University, Cheucheon 24341, South Korea. E-mail: youndh@kangwon.ac.kr
bKorea Research Institute of Chemical Technology, Daejeon 34114, South Korea. E-mail: jaeykim@krict.re.kr
First published on 24th October 2024
Abstract
The electrochemical nitrate reduction reaction (NO3−RR) is emerging as a promising method for ammonia production under ambient conditions while simultaneously addressing nitrate pollution. Due to the complexity of NO3−RR, which involves multi-electron/proton transfer and competes with the hydrogen evolution reaction (HER), the development of efficient electrocatalysts with high activity and stability is crucial. In this study, we report the use of Mo2C nanoparticles homogeneously dispersed on a carbon nanotube-reduced graphene oxide hybrid support (Mo2C/CNT-RGO) as an effective electrocatalyst for NO3−RR. The three-dimensional CNT-RGO hybrid provides a large surface area for electrolyte contact, enhanced electrical conductivity, and prevents the aggregation of Mo2C nanoparticles. Consequently, the Mo2C/CNT-RGO electrocatalyst demonstrated high NO3−RR performance, achieving a maximum NH3 production rate of 5.23 mg h−1 cm−2 with a faradaic efficiency of 95.9%. Mo2C/CNT-RGO also exhibited excellent long-term stability during consecutive cycling tests. This work presents a promising strategy for developing high-performance and durable NO3−RR electrocatalysts.
Introduction
Ammonia is an indispensable chemical in various industries, including fertilizers, explosives, pharmaceuticals, and plastics.1,2 Additionally, it is considered a promising carbon-free energy carrier, offering a high energy density of 4.32 kW h L−1.3–6 However, industrial-scale ammonia synthesis remains reliant on the energy-intensive Haber–Bosch process, which requires high temperatures (400–600 °C) and high pressures (200–350 atm). This process is heavily dependent on fossil fuels,7–10 contributing to 1–2% of the world's energy consumption and approximately 1% of global annual greenhouse gas emissions.2,11 Therefore, developing an eco-friendly and sustainable approach for NH3 production is of great importance.12–14Recently, the electrochemical nitrogen reduction reaction (NRR) has attracted significant interest as it utilizes water as a sustainable proton source under mild conditions.15–17 However, NRR faces significant challenges, such as extremely low faradaic efficiency and NH3 yield, attributable to the poor solubility of N2 in water, highly stable N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (941 kJ mol−1), and competing hydrogen evolution reaction (HER).18–22 As an alternative to NRR, the research on electrochemical nitrate reduction reaction (NO3−RR) is growing extensively.23–25 This growth is driven by substantially higher NH3 production rates compared to NRR, owing to the high solubility of NO3− in aqueous solutions and the lower bond energy of N
N bond (941 kJ mol−1), and competing hydrogen evolution reaction (HER).18–22 As an alternative to NRR, the research on electrochemical nitrate reduction reaction (NO3−RR) is growing extensively.23–25 This growth is driven by substantially higher NH3 production rates compared to NRR, owing to the high solubility of NO3− in aqueous solutions and the lower bond energy of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (204 kJ mol−1).26–28 Moreover, NO3− is abundantly present as a contaminant in surface and groundwater due to the excessive use of nitrogen-containing fertilizers and chemicals, posing significant health risks, such as blue baby syndrome, non-Hodgkin's lymphoma, and other cancers.26,29 Therefore, NO3−RR offers a “two birds-one stone” solution by converting nitrate pollutants into value-added ammonia while simultaneously purifying water. However, the NO3−RR is a complex process involving an eight-electron and nine-proton transfer with sluggish kinetics, and it includes several intermediates (e.g., NO2−, N2, and N2H4).30–35 Thus, the development of efficient electrocatalysts with high activity and stability for NO3−RR is imperative.
O (204 kJ mol−1).26–28 Moreover, NO3− is abundantly present as a contaminant in surface and groundwater due to the excessive use of nitrogen-containing fertilizers and chemicals, posing significant health risks, such as blue baby syndrome, non-Hodgkin's lymphoma, and other cancers.26,29 Therefore, NO3−RR offers a “two birds-one stone” solution by converting nitrate pollutants into value-added ammonia while simultaneously purifying water. However, the NO3−RR is a complex process involving an eight-electron and nine-proton transfer with sluggish kinetics, and it includes several intermediates (e.g., NO2−, N2, and N2H4).30–35 Thus, the development of efficient electrocatalysts with high activity and stability for NO3−RR is imperative.
Various noble metal-based catalysts, including Pt, Rh, and Pd, have been employed as NO3−RR catalysts, demonstrating high faradaic efficiency and stability.36–38 However, their high costs and limited availability hinder the large-scale application of NO3−RR systems. Transition metal carbides (TMCs) have emerged as promising alternatives to Pt-group metals in various electrocatalytic processes due to their electronic structures, which resemble those of noble metals.39–41 Among TMCs, molybdenum carbides (Mo2C) have been employed as catalysts for both NRR and NO3−RR owing to their high adsorption capacity and catalytic hydrogenation ability for electron-rich nitrogenous molecules.42,43 Nevertheless, the aggregation and large particle size of Mo2C, typically resulting from high synthesis temperatures, reduce the exposure of active sites, and consequently impair catalytic activity.44 Therefore, there remains significant scope for improving the activity and stability of Mo2C, particularly for NO3−RR applications.
To enhance the electrocatalytic activity of Mo2C, one effective strategy is to compound it with carbonaceous materials as catalyst supports. Carbonaceous materials, such as carbon nanotubes (CNTs) and graphene, offer a large surface area and high electrical conductivity, which can enhance the performance of loaded Mo2C.45–49 However, when used individually, graphene tends to stack due to van der Waals forces and strong π–π interactions between nanosheets.50,51 Similarly, CNTs have a tendency to bundle together, which reduces their surface area and electrochemical performance.52 The combination of CNTs and graphene (forming a CNT-RGO hybrid) addresses the stacking and bundling challenges associated with their individual use by creating a three-dimensional structure. This hybrid CNT-RGO framework not only provides a larger contact area with the electrolyte but also facilitates improved electron transfer, thereby enhancing the overall electrocatalytic activity.
In this study, nanostructured Mo2C supported on a CNT-RGO hybrid (Mo2C/CNT-RGO) was prepared using a modified urea-glass route for NO3−RR.53,54 The Mo2C/CNT-RGO composite comprised uniformly dispersed 7 nm Mo2C nanoparticles on a three-dimensional CNT-RGO hybrid support. The resulting Mo2C/CNT-RGO demonstrated high NO3−RR activity, achieving a maximum NH3 production rate of 5.23 mg h−1 cm−2 and a faradaic efficiency (FE) of 95.9%, outperforming the Mo2C/CNT and Mo2C/RGO catalysts. Additionally, it exhibited excellent long-term stability over ten consecutive cycling tests. The enhanced NO3−RR performance of Mo2C/CNT-RGO can be attributed to the synergistic interaction between the active Mo2C nanoparticles and the CNT-RGO hybrid support, which provided a high surface area and enhanced electrical conductivity.
Experimental
Materials
Molybdenum(V) chloride (MoCl5, 99.6%) was obtained from Alfa Aesar. GO was prepared using Hummers’ method,55 and CNT (CM-95) was sourced from Hanwha Nanotech. Dimethyl sulfoxide-d6 (DMSO-d6, 99.9 atom% D), ammonium chloride (NH4Cl, ≥99.5%), ammonium-15N chloride (15NH4Cl, ≥98 atom% 15N), and potassium sodium tartrate tetrahydrate (KNaC4H4O6, 99%) were purchased from Sigma-Aldrich. High-purity N2 (99.999%) and Ar (99.999%) gases were purchased from Hyundai Energy. Urea (99.0%), ethyl alcohol (C2H5OH, 99.9%, anhydrous), Nessler's reagent, and hydrochloric acid (HCl, 35.0–37.0%) were procured from Samchun Chemicals. Nickel foam (NF, 99.5%) was obtained from Goodfellow.Synthesis of Mo2C/CNT-RGO
Mo2C/CNT-RGO composites were fabricated via a modified urea-glass route. GO and CNT were first dispersed in 15 mL of ethanol at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio. Separately, 1.0 g MoCl5 was dissolved in 2.53 mL of ethanol and added to the CNT-GO solution under vigorous stirring. After 30 min of stirring, 1.75 g of urea (molar ratio of urea/Mo = 8) was introduced into the mixture, which was subsequently stirred for an additional hour. The resulting solution was dried at 100 °C in an oven to remove excess ethanol and then calcined at 750 °C (with a ramp rate of 3 °C min−1) for 3 h under N2 flow. For comparison, Mo2C/CNT and Mo2C/RGO were synthesized using only CNT and RGO, respectively, following the same procedure. The nominal weight content of Mo2C in each sample was fixed at ca. 80 wt%.
:1 weight ratio. Separately, 1.0 g MoCl5 was dissolved in 2.53 mL of ethanol and added to the CNT-GO solution under vigorous stirring. After 30 min of stirring, 1.75 g of urea (molar ratio of urea/Mo = 8) was introduced into the mixture, which was subsequently stirred for an additional hour. The resulting solution was dried at 100 °C in an oven to remove excess ethanol and then calcined at 750 °C (with a ramp rate of 3 °C min−1) for 3 h under N2 flow. For comparison, Mo2C/CNT and Mo2C/RGO were synthesized using only CNT and RGO, respectively, following the same procedure. The nominal weight content of Mo2C in each sample was fixed at ca. 80 wt%.
Characterization
The surface morphologies of the prepared catalysts were examined using field-emission transmission electron microscopy (FE-TEM, JEOL, JEM-2100F). The crystalline structures were characterized by X-ray diffraction (XRD, Rigaku, MiniFlex 600) with Cu-Kα (1.54 Å) radiation. X-ray photoelectron spectroscopy (XPS, Thermo-Scientific, K-alpha) was employed to analyze the chemical states of the samples. The produced ammonia was detected using a UV-Vis spectrophotometer (SHIMADZU, UV 2600i), while the gaseous products from the electrochemical experiments were analyzed by gas chromatography (GC, Agilent, 8890 GC) equipped with thermal conductivity detectors (TCD) using nitrogen as the carrier gas. Proton nuclear magnetic resonance (1H NMR, Bruker, Advance Neo 600 MHz) was conducted to identify the nitrogen source in the ammonia. Electrical conductivity measurements were performed using a four-point probe (AIT, CMT-100S).Electrochemical measurements
Electrochemical measurements were conducted using a potentiostat (AMETEK, VersaSTAT 3) in a three-electrode cell system. To prepare the working electrode, 30 mg of the synthesized catalyst was dispersed in a mixture of ethanol and deionized (DI) water (4 mL:2 mL), followed by ultrasonication for 20 min to ensure uniform dispersion. The resulting catalyst ink was then loaded onto a 1 × 1 cm2 nickel foam substrate, achieving a mass loading of 3 mg cm−2. The Pt wire served as the counter electrode, while an Ag/AgCl (3 M KCl) electrode was used as the reference electrode. All potentials were converted to the reversible hydrogen electrode (RHE) using the equation: ERHE = EAg/AgCl + 0.059 pH + 0.197. Prior to measurements, the electrolyte (0.1 M KOH with 0.1 M NaNO3) was purged with Ar gas for 30 min to remove dissolved O2. Cyclic voltammetry (CV) was conducted until steady-state polarization curves were achieved, using a scan rate of 50 mV s−1. Linear sweep voltammetry (LSV) measurements were performed at a scan rate of 5 mV s−1, and chronoamperometry tests were conducted for 1 h at various applied potentials. Electrochemical impedance spectroscopy (EIS) was performed over a frequency range from 100 kHz to 0.1 Hz with a 5 mV amplitude, and the resulting EIS data were fitted using Z-view software. The electrochemical double-layer capacitance (EDLC) was evaluated using CV at scan rates of 20–60 mV s−1 in the non-faradaic potential region (0.1–0.2 V vs. RHE).
Results and discussion
Fig. 1 presents a schematic illustration of the synthesis process for Mo2C/CNT-RGO. Molybdenum precursor (MoCl5) in ethanol solution was mixed with the CNT-GO solution under vigorous stirring. After adding urea, a Mo–urea complex was formed on the CNT-GO support. This complex was then annealed at 750 °C for 3 h under N2 flow to produce the Mo2C/CNT-RGO composite. During annealing, Mo2C crystallization occurred, along with the simultaneous reduction of GO to RGO, thus eliminating the need for toxic gases (e.g., methane) for carbide formation or an additional reduction step for GO. This facile method resulted in the uniform dispersion of Mo2C nanoparticles on the CNT-RGO support. For comparison, Mo2C/CNT and Mo2C/RGO were synthesized using only CNT and RGO, respectively, following the same procedure.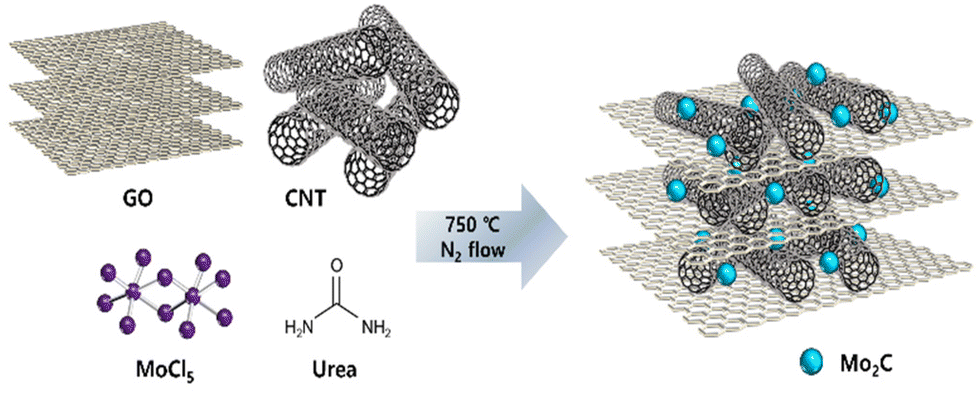 | ||
| Fig. 1 Schematic illustration of the synthetic method for Mo2C/CNT-RGO. | ||
Fig. 2a shows the TEM image of the Mo2C/CNT-RGO catalyst, where CNTs are randomly distributed across the RGO layers, and spherical Mo2C particles are uniformly dispersed on the CNT-RGO hybrid support without aggregation. The average particle size of Mo2C is approximately 7 nm. The high-resolution TEM (HRTEM) image in Fig. 2b reveals lattice fringes of 0.224 and 0.240 nm, corresponding to the (101) and (002) facets of the Mo2C phase, respectively. Fig. 2c and e present the TEM images of Mo2C/CNT and Mo2C/RGO, showing that ∼8 nm Mo2C nanoparticles are exclusively anchored onto the CNTs and RGO, respectively. Similar to Fig. 2b, lattice spacings of 0.224 and 0.240 nm were detected, confirming the presence of the Mo2C phase (Fig. 2d and f).
 | ||
| Fig. 2 TEM and HRTEM images of (a and b) Mo2C/CNT-RGO; (c and d) Mo2C/CNT; (e and f) Mo2C/RGO. | ||
The XRD patterns of the prepared catalysts are presented in Fig. 3a. All catalysts exhibit identical XRD patterns, aligning with the reference XRD patterns of hexagonal Mo2C (JCPDS No. 00-035-0787). The observed peaks at 34.4°, 37.9°, 39.4°, 52.2°, 61.6°, 69.6°, 74.6°, and 75.5° correspond to the (100), (002), (101), (102), (110), (103), (112), and (201) planes of Mo2C, respectively. No impurity peaks, such as Mo metal or MoOx, were detected, confirming the successful synthesis of pure Mo2C on the CNT-RGO, CNT, and RGO supports. The absence of peaks around 10° further indicates the conversion of GO to RGO during thermal annealing.56
 | ||
| Fig. 3 (a) XRD patterns of the synthesized catalysts. XPS spectra of Mo2C/CNT-RGO for (b) C 1s, (c) Mo 3d, and (d) N 1s. | ||
The chemical states of Mo2C/CNT-RGO were investigated via XPS, as shown in Fig. 3b–d. The survey spectrum of Mo2C/CNT-RGO confirms the presence of C, Mo, N, and O elements (Fig. S1a†). In the C 1s spectrum (Fig. 3b), the deconvoluted peaks at 283.2, 284.6, 285.6, and 286.5 eV correspond to the Mo2C, C–C, C–N, and C–O bands, respectively.57,58 Fig. S1b† shows the C 1s spectrum for GO, where broad peaks observed at 280–290 eV indicate the presence of oxygen-containing functional groups such as hydroxyl, carboxyl, and epoxy groups. The significantly reduced peak intensities in the C 1s spectrum of Mo2C/CNT-RGO suggest the conversion of GO to RGO during synthesis. The Mo 3d spectrum in Fig. 3c is deconvoluted into Mo2+ (228.3/231.5 eV), Mo4+ (229.2/233.6 eV), and Mo6+ (232.5/235.6 eV).59 The Mo4+ and Mo6+ species are attributed to MoO2 and MoO3, respectively, due to unavoidable surface oxidation,42,43,60 while the Mo2+ species stem from the Mo2C phase.61–63 The N 1s spectrum (Fig. 3d) exhibits three main peaks at 396.1, 398.3, and 400.6 eV, corresponding to N–Mo, pyridinic N, and graphitic N, respectively. The peak observed at 394.4 eV is assigned to Mo 3p due to binding energy overlap.57 Fig. S2 and S3† display the XPS results of Mo2C/CNT and Mo2C/RGO, with their C 1s, Mo 3d, and N 1s spectra showing similarities to Mo2C/CNT-RGO, indicating comparable chemical states among these catalysts.
The electrical conductivities of the catalysts were measured using the four-point probe method, with results summarized in Table 1. Among the catalysts, Mo2C/CNT-RGO exhibited the highest electrical conductivity of 3.04 × 103 S m−1, followed by Mo2C/CNT (1.98 × 103 S m−1), and Mo2C/RGO (0.64 × 103 S m−1).
| Sample | Resistivity [ohm cm] | Electrical conductivity × 103 [S m−1] |
|---|---|---|
| Mo2C/CNT-RGO | 3.28 × 10−2 | 3.04 (±0.14) |
| Mo2C/CNT | 5.05 × 10−2 | 1.98 (±0.57) |
| Mo2C/RGO | 1.58 × 10−1 | 0.64 (±0.07) |
The high electrical conductivity, which is crucial for enhanced NO3−RR activity, was effectively achieved by employing the CNT-RGO hybrid support for Mo2C.
Electrochemical NO3−RR measurements were conducted in a 0.1 M KOH solution with 0.1 M NaNO3 under ambient conditions. LSV curves were used to investigate the electrocatalytic responses of the catalysts both in the presence and absence of NO3− (Fig. S4a†). In 0.1 M KOH (dashed line), the LSV curves displayed a delayed onset potential. However, the addition of 0.1 M NaNO3 (solid line) resulted in significant changes, including an increase in current density and a reduction in onset potential, indicating a strong NO3− reduction response. As shown in Fig. 4a, the LSV curves for the prepared catalysts in the presence of 0.1 M NaNO3 demonstrated that Mo2C/CNT-RGO exhibited a higher current density across the entire potential range, indicating its superior electrocatalytic activity for nitrate reduction.
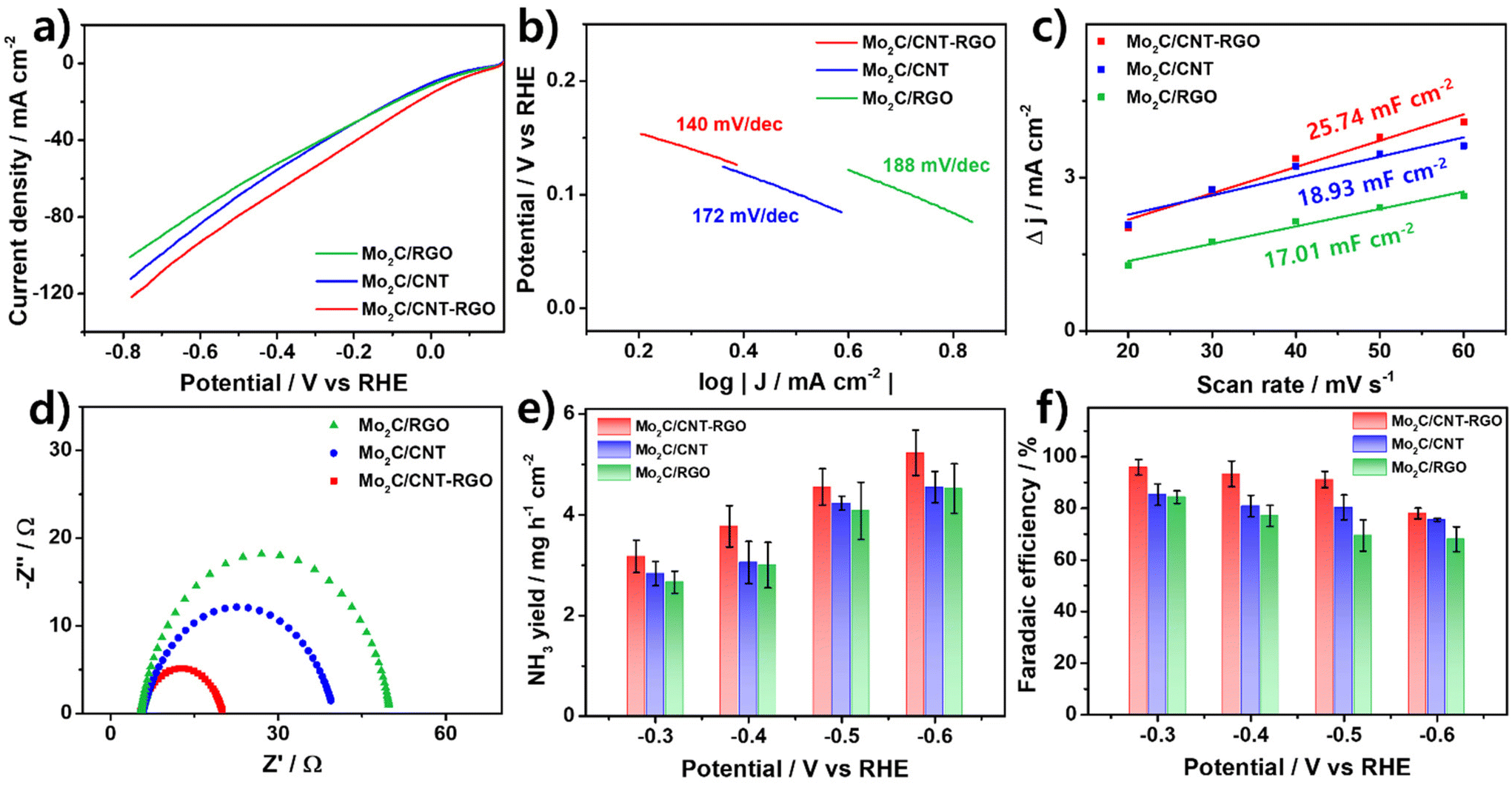 | ||
| Fig. 4 Electrochemical nitrate reduction performance of the prepared catalysts: (a) LSV curves in 0.1 M KOH with 0.1 M NaNO3, (b) Tafel plots, (c) measured capacitive currents as a function of scan rate, (d) Nyquist plots, (e) NH3 yields, and (f) FENH3 at each applied potential for 1 h. | ||
Fig. 4b presents the Tafel plots of the prepared catalysts fitted to the Tafel equation (η = b log|J| + a, where b is the Tafel slope and J is the current density). A lower Tafel slope typically signifies better reaction kinetics. The Tafel slope of Mo2C/CNT-RGO was 140 mV dec−1, which is smaller than that of Mo2C/CNT (172 mV dec−1) and Mo2C/RGO (188 mV dec−1), suggesting that Mo2C/CNT-RGO possesses higher electrocatalytic activity.
The cyclic voltammogram (CV) curves of the catalysts at various scan rates in the non-faradaic region are shown in Fig. S5,† and the corresponding electrochemical double-layer capacitance (Cdl) values are provided in Fig. 4c. Mo2C/CNT-RGO exhibited a larger Cdl of 25.74 mF cm−2 compared to Mo2C/CNT (18.93 mF cm−2) and Mo2C/RGO (17.01 mF cm−2). The Cdl is generally proportional to the contact area between the catalyst and electrolyte. Employing the CNT-RGO hybrid support leads to a larger contact area due to its 3D network structure, contributing to the enhanced NO3−RR activity of Mo2C/CNT-RGO.64,65 This is further supported by the adsorption capacity (qe, the adsorbed NO3− amount per unit catalyst amount) measurements, which confirm the effectiveness of the CNT-RGO hybrid.66 In Fig. S6,† the Mo2C/CNT-RGO recorded a much higher qe for NO3− (67.5 g gcat−1) compared to Mo2C/CNT (42. 9 g gcat−1) and Mo2C/RGO (32.8 g gcat−1), indicating that CNT-RGO provides an enhanced surface area for reactant adsorption.
Fig. 4d shows the Nyquist plots of the catalysts obtained from electrochemical impedance spectroscopy (EIS). The charge transfer resistance (Rct) at the catalyst-electrolyte interface is represented by the semicircle in the Nyquist plot and is inversely proportional to the electrocatalytic activity. The measured Rct values were 16.2, 33.5, and 47.5 Ω for Mo2C/CNT-RGO, Mo2C/CNT, and Mo2C/RGO, respectively, suggesting faster electron transfer and thus enhanced NO3−RR activity of Mo2C/CNT-RGO.
Chronoamperometry (CA) tests were conducted over a potential range from −0.3 V to −0.6 V for 1 h (Fig. S4b†). The concentrations of NO2−, NO3−, and NH3 products were quantified using the colorimetric method, with their standard calibration curves shown in Fig. S7.† As illustrated in Fig. 4e and f, the Mo2C/CNT-RGO demonstrated a higher NH3 yield and FENH3 across the entire potential range compared to Mo2C/CNT and Mo2C/RGO. The NH3 yield for Mo2C/CNT-RGO increased with more negative potentials, reaching a maximum yield of 5.23 mg h−1 cm−2 at −0.6 V (Fig. 4e). In comparison, the maximum NH3 yields for Mo2C/CNT and Mo2C/RGO were 4.55 and 4.53 mg h−1 cm−2, respectively. Additionally, Mo2C/CNT-RGO achieved a maximum FENH3 of 95.92% at −0.3 V and maintained FENH3 values above 90% up to −0.5 V (Fig. 4f), demonstrating superior performance at relatively less negative potentials compared to other reported Mo-based catalysts (Table S1†). A decreasing trend in FENH3 was observed at −0.6 V, primarily due to the increasing dominance of HER at more negative potentials. Although Mo2C/CNT and Mo2C/RGO also demonstrated notable performance with FENH3 values of 85.25% and 84.39% at −0.3 V, respectively, their overall activity was significantly lower than that of Mo2C/CNT-RGO. The three-dimensional CNT-RGO hybrid support offers a larger contact area with the electrolyte (as evidenced by Fig. 4c and Fig. S6†) and exhibits higher electrical conductivity (Table 1). Consequently, when combined with the active Mo2C, this hybrid support structure contributes to the superior NO3−RR performance of Mo2C/CNT-RGO compared to Mo2C/CNT and Mo2C/RGO.
Long-term stability is crucial in determining the performance of catalysts. To evaluate the stability of Mo2C/CNT-RGO, ten consecutive electrolytic cycles were conducted at −0.3 V. As shown in Fig. 5a, both the NH3 yield and FENH3 remained stable throughout the cycling tests, indicating the catalyst's good stability. The morphology of Mo2C/CNT-RGO after the cycling test was examined using TEM. As shown in Fig. S8a,† the Mo2C particles remain well-dispersed on the CNT-RGO hybrid support without significant aggregation. Additionally, the lattice spacings of 0.224 nm and 0.240 nm observed in Fig. S8b† correspond to the (101) and (002) planes of Mo2C, respectively. The similar morphology of Mo2C/CNT-RGO after the cycling test suggests its structural stability.
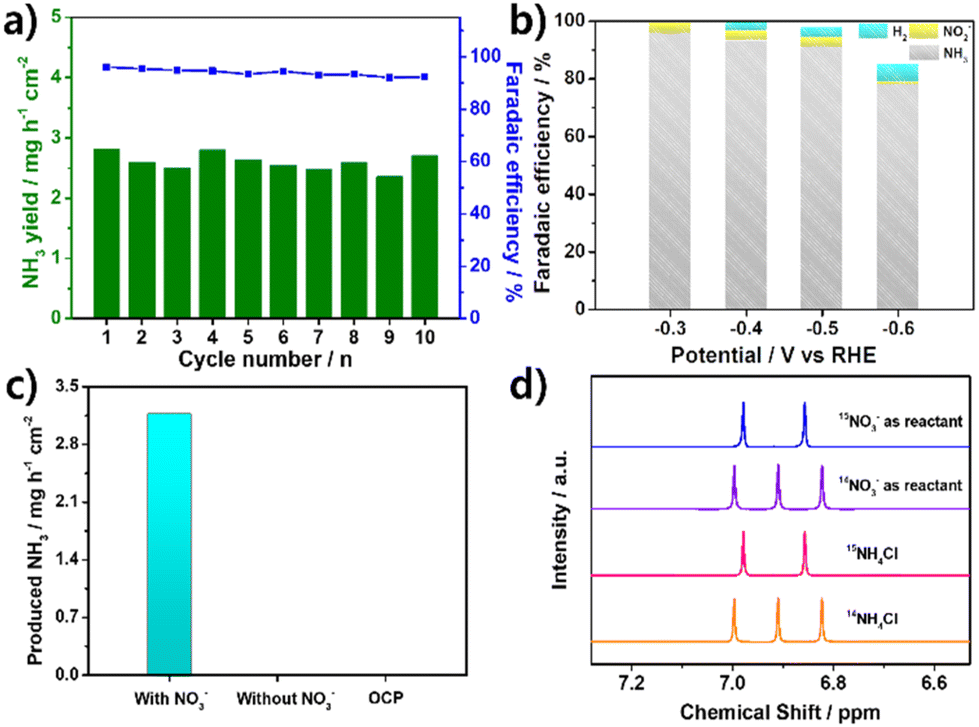 | ||
| Fig. 5 (a) Consecutive cycling test of Mo2C/CNT-RGO at −0.3 V, (b) FEs of different products after NO3−RR electrolysis, (c) amounts of NH3 produced at −0.3 V for 1 h under different conditions, and (d) 1H NMR spectra of standard solutions of 14NH4Cl and 15NH4Cl, along with electrolyte after NO3−RR using 15NO3− and 14NO3− as nitrogen sources. | ||
Possible by-products such as NO2− and H2 were also measured (Fig. 5b). NO2− was detected only in the potential range of −0.3 to −0.5 V and was nearly absent at −0.6 V. As the potential becomes more negative, the FE of H2 gradually increased due to the competing HER. Nevertheless, Mo2C/CNT-RGO maintained high selectivity for NH3 generation across all potentials.
To identify the nitrogen source in the produced NH3, several control experiments were performed. Conducting CA tests at −0.3 V for 1 h in the absence of NO3− and under open-circuit potential (OCP) conditions resulted in negligible ammonia formation (Fig. 5c), confirming that the produced NH3 originated from the NO3−RR. Furthermore, a 15N isotopic labeling experiment was conducted using 1H-NMR. In Fig. 5d, standard solutions of 14NH4Cl and 15NH4Cl exhibited characteristic triplet peaks of 14NH4+ and doublet peaks of 15NH4+, respectively. When 14NO3− (from Na14NO3) was used as the reactant, triplet peaks corresponding to 14NH4+ were observed, while doublet peaks corresponding to 15NH4+ appeared when 15NO3− (from Na15NO3) was used as the reactant. These results strongly confirm that the NH4+ detected in the electrolyte originated from NO3−RR and not from other contaminants.
Conclusions
In summary, nanostructured Mo2C homogeneously dispersed on a CNT-RGO hybrid support was employed as a catalyst for NO3−RR. The three-dimensional CNT-RGO hybrid offered a high surface area for electrolyte contact, enhanced electrical conductivity, and effectively dispersed the Mo2C nanoparticles, preventing aggregation. Due to these advantages, the Mo2C/CNT-RGO catalyst exhibited superior performance compared to Mo2C/CNT and Mo2C/RGO, achieving a FENH3 of 95.9% and a maximum ammonia yield of 5.23 mg h−1 cm−2. Additionally, the Mo2C/CNT-RGO demonstrated good stability over ten consecutive cycling tests. Control experiments and the 15N isotopic labeling experiment confirmed that the generated ammonia originated from NO3−, further validating the reliability of the results. This work presents a promising strategy for designing high-performance, stable NO3−RR electrocatalysts.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20224000000080). This work was also supported by the research fund of Hanyang University and KRICT (HY-202400000003499).References
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- A. E. Yüzbaşıoğlu, C. Avşar and A. O. Gezerman, Curr. Res. Green Sustainable Chem., 2022, 5, 100307 CrossRef.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS.
- C. H. Christensen, T. Johannessen, R. Z. Sørensen and J. K. Nørskov, Catal. Today, 2006, 111, 140–144 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 RSC.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- K. Chen, G. Wang, Y. Guo, D. Ma and K. Chu, Nano Res., 2023, 16, 8737–8742 CrossRef CAS.
- D. Wu, K. Chen, P. Lv, Z. Ma, K. Chu and D. Ma, Nano Lett., 2024, 24, 8502–8509 CrossRef CAS PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. Y. Park, S. E. Jang, C. W. Kim, Y. J. Jang and D. H. Youn, RSC Adv., 2023, 13, 34410–34415 RSC.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 6073–6076 CrossRef CAS PubMed.
- J. Deng, J. A. Iñiguez and C. Liu, Joule, 2018, 2, 846–856 CrossRef CAS.
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- M. Arif, G. Yasin, L. Luo, W. Ye, M. A. Mushtaq, X. Fang, X. Xiang, S. Ji and D. Yan, Appl. Catal., B, 2020, 265, 118559 CrossRef CAS.
- G. Zhang, G. Wang, Y. Wan, X. Liu and K. Chu, ACS Nano, 2023, 17, 21328–21336 CrossRef PubMed.
- M. Yang, T. Wei, J. He, Q. Liu, L. Feng, H. Li, J. Luo and X. Liu, Nano Res., 2024, 17, 1209–1216 CrossRef CAS.
- Y. Qi, X. Hou, Z. He, F. He, T. Wei, G. Meng, H. Hu, Q. Liu, G. Hu and X. Liu, Chem. Commun., 2024, 60, 8728–8731 RSC.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- A. Kumar, J. Lee, M. G. Kim, B. Debnath, X. Liu, Y. Hwang, Y. Wang, X. Shao, A. R. Jadhav, Y. Liu, H. Tüysüz and H. Lee, ACS Nano, 2022, 16, 15297–15309 CrossRef CAS PubMed.
- S. Xu, D. C. Ashley, H.-Y. Kwon, G. R. Ware, C.-H. Chen, Y. Losovyj, X. Gao, E. Jakubikova and J. M. Smith, Chem. Sci., 2018, 9, 4950–4958 RSC.
- D. P. Butcher and A. A. Gewirth, Nano Energy, 2016, 29, 457–465 CrossRef CAS.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- W. Siriwatcharapiboon, Y. Kwon, J. Yang, R. L. Chantry, Z. Li, S. L. Horswell and M. T. M. Koper, ChemElectroChem, 2014, 1, 172–179 CrossRef.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- K. Chen, D. Ma, Y. Zhang, F. Wang, X. Yang, X. Wang, H. Zhang, X. Liu, R. Bao and K. Chu, Adv. Mater., 2024, 36, 2402160 CrossRef CAS PubMed.
- W. Du, Z. Sun, K. Chen, Y. Wei, R. Bao and K. Chu, Adv. Energy Mater., 2024, 2401765 CrossRef.
- G. A. Cerrón-Calle, A. S. Fajardo, C. M. Sánchez-Sánchez and S. Garcia-Segura, Appl. Catal., B, 2022, 302, 120844 CrossRef.
- H. Liu, J. Timoshenko, L. Bai, Q. Li, M. Rüscher, C. Sun, B. R. Cuenya and J. Luo, ACS Catal., 2023, 13, 1513–1521 CrossRef CAS.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- T. Qin, Z. Wang, Y. Wang, F. Besenbacher, M. Otyepka and M. Dong, Nano-Micro Lett., 2021, 13, 183 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- J. B. Claridge, A. P. E. York, A. J. Brungs, C. Marquez-Alvarez, J. Sloan, S. C. Tsang and M. L. H. Green, J. Catal., 1998, 180, 85–100 CrossRef CAS.
- B. Fan, H. Wang, H. Zhang, Y. Song, X. Zheng, C. Li, Y. Tan, X. Han, Y. Deng and W. Hu, Adv. Funct. Mater., 2022, 32, 2110783 CrossRef CAS.
- H. Cheng, L.-X. Ding, G.-F. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- Y. Hu, D.-G. Guan, B. Yu, W. Hou, B. Zheng, W. Zhang and Y. Chen, Electrochim. Acta, 2018, 263, 192–200 CrossRef CAS.
- K. Ojha, S. Saha, H. Kolev, B. Kumar and A. K. Ganguli, Electrochim. Acta, 2016, 193, 268–274 CrossRef CAS.
- L. F. Pan, Y. H. Li, S. Yang, P. F. Liu, M. Q. Yu and H. G. Yang, Chem. Commun., 2014, 50, 13135–13137 RSC.
- L. Huo, B. Liu, G. Zhang and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 18107–18118 CrossRef CAS PubMed.
- B. Šljukić, M. Vujković, L. Amaral, D. M. F. Santos, R. P. Rocha, C. A. C. Sequeira and J. L. Figueiredo, J. Mater. Chem. A, 2015, 3, 15505–15512 RSC.
- K. Zhang, Y. Zhao, D. Fu and Y. Chen, J. Mater. Chem. A, 2015, 3, 5783–5788 RSC.
- B. Wang, Y. Qin, W. Tan, Y. Tao and Y. Kong, Electrochim. Acta, 2017, 241, 1–9 CrossRef CAS.
- X. Du, Z. Qin and Z. Li, Nanomaterials, 2021, 11, 1420 CrossRef CAS.
- B. Ding, D. Guo, Y. Wang, X. Wu and Z. Fan, J. Power Sources, 2018, 398, 113–119 CrossRef CAS.
- D. H. Youn, S. Han, J. Y. Kim, J. Y. Kim, H. Park, S. H. Choi and J. S. Lee, ACS Nano, 2014, 8, 5164–5173 CrossRef CAS PubMed.
- G. H. Lee, M. H. Lee, Y. Kim, H.-K. Lim and D. H. Youn, J. Alloys Compd., 2019, 805, 113–119 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- C. Lu, D. Tranca, J. Zhang, F. R. Hernández, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933–3942 CrossRef CAS PubMed.
- A. Griboval-Constant, J.-M. Giraudon, G. Leclercq and L. Leclercq, Appl. Catal., A, 2004, 260, 35–45 CrossRef CAS.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399–3405 RSC.
- X. Zhang, J. Wang, T. Guo, T. Liu, Z. Wu, L. Cavallo, Z. Cao and D. Wang, Appl. Catal., B, 2019, 247, 78–85 CrossRef CAS.
- Y. Wang, Z. Shi, Q. Mo, B. Gao, B. Liu, L. Wang, Y. Zhang, Q. Gao and Y. Tang, ChemElectroChem, 2017, 4, 2169–2177 CrossRef CAS.
- J. Zhang, L. Zhao, A. Liu, X. Li, H. Wu and C. Lu, Electrochim. Acta, 2015, 182, 652–658 CrossRef CAS.
- L. Huang, Y. Hou, Z. Yu, Z. Peng, L. Wang, J. Huang, B. Zhang, L. Qian, L. Wu and Z. Li, Int. J. Hydrogen Energy, 2017, 42, 9458–9466 CrossRef CAS.
- Y. Liu, J. Wei, Z. Yang, L. Zheng, J. Zhao, Z. Song, Y. Zhou, J. Cheng, J. Meng, Z. Geng and J. Zeng, Nat. Commun., 2024, 15, 3619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02817a |
| This journal is © The Royal Society of Chemistry 2024 |