
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the reductive CO2 fixation with amines and hydrosilanes using readily available Cu(II) NHC–phenolate catalyst precursors†
Giammarco
Meloni
ab,
Luca
Morgan
a,
David
Cappelletti
a,
Matteo
Bevilacqua
a,
Claudia
Graiff
 c,
Piermaria
Pinter
d,
Andrea
Biffis
ab,
Cristina
Tubaro
*ab and
Marco
Baron
*ab
c,
Piermaria
Pinter
d,
Andrea
Biffis
ab,
Cristina
Tubaro
*ab and
Marco
Baron
*ab
aDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: cristina.tubaro@unipd.it; marco.baron@unipd.it
bConsorzio Interuniversitario per le Reattività Chimiche e la Catalisi, Unità di Ricerca di Padova, via Marzolo 1, 35131 Padova, Italy
cDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università degli Studi di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy
dNovaled GmbH, Elisabeth-Boer-Straße 9, 01099 Dresden, Germany
First published on 23rd October 2024
Abstract
N-Methylation of amines is of great interest in the synthesis of pharmaceuticals and valuable compounds, and the possibility to perform this reaction with an inexpensive and non-toxic substrate like CO2 and its derivatives is quite appealing. Herein, the synthesis of four novel homoleptic Cu(II) complexes with hybrid NHC–phenolate (NHC = N-Heterocyclic Carbene) ligands is reported, and their use in the catalytic N-methylation of amines with CO2 in the presence of hydrosilanes is explored. Both bidentate or tetradentate ligands can be used in the preparation of the complexes provided that the structural requirement that the two NHC and the two phenolate donors in the metal coordination sphere are mutually in trans is fulfilled. A new reaction protocol to perform the N-methylation of secondary aromatic amines and dibenzylamine in high yield under mild reaction conditions is developed, using the ionic liquid [BMMIM][NTf2] (1-butyl-2,3-dimethylimidazolium bis(trifluoromethylsulfonyl)imide) as solvent and the catalyst precursor [Cu(L2)2]. Reactivity studies indicate that the reaction follows two different pathways with different hydrosilanes, and that the starting Cu(II) complexes are reduced under the catalytic conditions.
Introduction
Copper has always attracted considerable interest in coordination chemistry, especially in its monovalent (+1) and bivalent (+2) oxidation states. A plethora of different applications with copper-containing compounds, ranging from catalysis, to medicinal and agricultural chemistry is documented in the literature.1–5 Even though most of the reported studies involve nitrogen- and phosphorus-based ligands, copper complexes with N-Heterocyclic Carbene ligands (NHCs) are increasingly getting more studied, following the first report by Arduengo in 1993.6 The NHCs, thanks to their soft character, prefer to coordinate to the softer Cu(I) rather than to the borderline–hard Cu(II) metal centre. Fundamental contributions in the field of homogeneous catalysis with Cu(I) NHC complexes were reported by the groups of Hoveyda,7–11 Nolan,12–15 and Cazin.16–18 A huge variety of organic reactions is catalysed by Cu(I) NHC complexes,19 and exceptional results in terms of stability, activity, regio- and enantioselectivity are achieved, if compared to the similar phosphine-based Cu(I) complexes.18,20,21 Thanks to the copper natural abundance and low toxicity,22–24 Cu(I) NHCs are more environmentally sustainable catalysts compared to those commonly adopted in organometallic chemistry (e.g. platinum group metals). Furthermore, thanks to the d10 ground state configuration, Cu(I) NHC complexes exhibit high-quality photophysical properties: the fast nonradiative decay towards low-lying metal-centred states is absent and they can be applied for their fluorescence or phosphorescence properties.25–31 Instead, the chemistry of Cu(II) NHCs has been less explored and a lower number of complexes has been reported to date.32–42 The first Cu(II) NHC complex was reported by Meyer's group in 2003, with an hybrid tripodal NHC-imine ligand (NHC–N).32 Thanks to the hybrid nature of the ligand the resulting complex is air stable in the solid state, a marked difference compared to monodentate NHC Cu(II) complexes, which in general are moisture and water sensitive.35 A similar behaviour is observed with several hybrid multidentate NHC–N and NHC–O ligands. Among them, although the examples of Cu(II) complexes with NHC–phenolates (NHC–O) are limited (Scheme 1),43–45 they have been recently reported to be air and moisture stable.43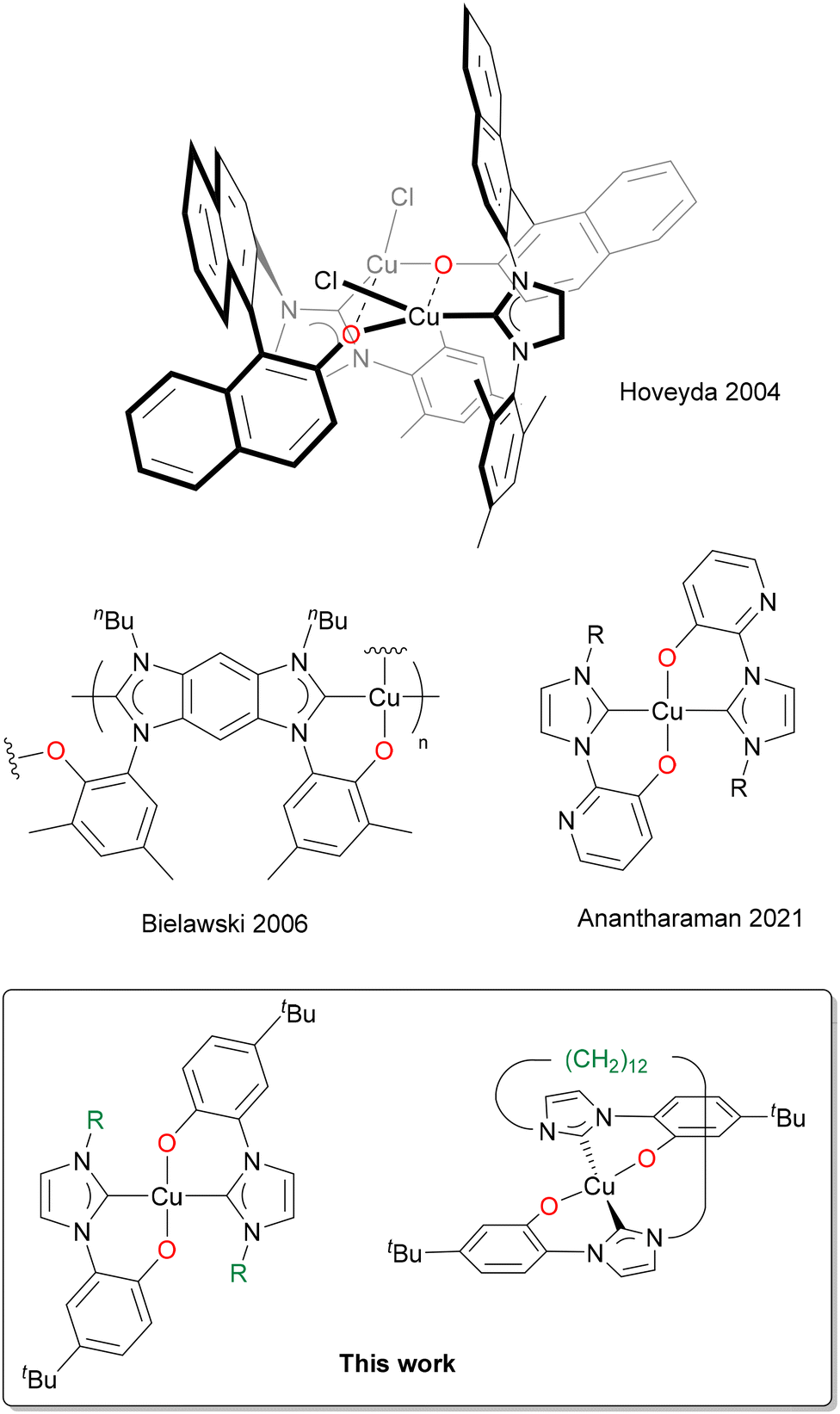 | ||
| Scheme 1 Cu(II) NHC–phenolate complexes reported in the literature (top), and in this study (bottom).43–45 | ||
One interesting catalytic reaction is the reductive N-formylation and N-methylation of amines with CO2 or formic acid in the presence of hydrosilanes. In this frame it is worth mentioning that in industrial processes the Eschweiler–Clarke reaction, using formaldehyde as the C1 source, prevails among the N-methylation of amine methodologies,46–49 and it is still an active research area,50–53 considering that N-methylation of amines is of great interest in the synthesis of pharmaceuticals and valuable compounds.54,55 Anyway, the possibility to perform this reaction with an inexpensive and non-toxic substrate like CO2 and its derivatives is quite appealing.56–63
The reductive N-formylation and N-methylation of amines with CO2 or formic acid in the presence of hydrosilanes was first reported by Cantat in 2013 catalysed by Zn(II) salts in combination with neutral donor ligands, such phosphines and NHCs.56 Organocatalysts, such as free NHCs, are also active in this transformation.64
Cu(I) NHC complexes were also reported to be active in this reaction but, to the best of our knowledge, Cu(II) NHC complexes have never been studied yet. Up to now, in fact only Cu(II) metal salts in combination with phosphine ligands have been reported as catalysts for the cited reaction.65–68 In this work we report the synthesis of three novel bidentate NHC–phenolate and one tetradentate bis(NHC)–bis(phenolate) ligand precursors, and their corresponding Cu(II) complexes (Scheme 1). The catalytic activity of the complexes in the reductive N-formylation/N-methylation of amines with CO2 and hydrosilanes is also explored, by comparing the activity with mono- and bis(NHC) Cu(I) complexes and through a reactivity study aimed at shedding light on the possible active species involved in the catalytic process. We can anticipate that the catalytic performance of the Cu(II) complexes is superior in terms of N-methylated product yield compared to what we observed in previous studies with Mn(I) complexes, even working under milder conditions.59
Experimental
Materials and methods
Unless specified otherwise, all manipulations were performed under argon atmosphere using standard Schlenk techniques, and all the utilized glassware was oven-dried at 110 °C prior to use. Methanol, dichloromethane, and n-hexane used for synthesis were argon-degassed and stored under inert gas and molecular sieves. All the solvents used for catalytic tests were stored over molecular sieves and degassed by three consecutive freeze–pump–thaw cycles before their use. All the chemicals were purchased from commercial supplier and used without further purification. 1-(5-tert-butyl-2-methoxyphenyl)-imidazole was prepared following reported literature procedures.69,70 NMR spectra were recorded at 298 K on a Bruker Avance 300 MHz, operating at 300.1, 75.5 and 121.5 MHz, respectively for 1H, 13C and 31P NMR nuclei. Chemical shifts are reported in parts per million and calibrated respect to the solvent residue. 1H NMR signals are labelled as s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sept = septet, m = multiplet and br = broad. ESI mass spectra were recorded on a Finnigan Thermo LCQ-Duo ESI mass spectrometer operating in positive ion mode; sample solutions were prepared by dissolving the compounds in acetonitrile and were directly infused into the ESI source by a syringe pump at 8 μL min−1 flow rate. Elemental analyses were performed by the microanalytical laboratory of Chemical Sciences Department (University of Padova) with a ThermoScientific FLASH 2000 elemental analyzer. FT-IR spectra were recorded at 25 °C, with a Bruker Tensor 27 spectrometer, preparing KBr pellets for the samples. The IR spectra were recorded in the mid-infrared using a 4000–400 cm−1 wavenumber range and a spectral resolution of 2 cm−1. Absorption peaks are labelled based on their intensity, vs = very strong, s = strong, m = medium, w = weak, vw = very weak, b = broad. UV-vis absorption spectra were recorded in dichloromethane at 25 °C using a Varian Cary 100 Bio spectrophotometer, with 1 cm optical path quartz cells, from 900 to 190 nm after baseline correction.Synthesis of the proligands
Ha1Br – alkyl halide: benzyl bromide, Yield: 95% (white solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 9.80 (s, 1H, NCHN), 8.08 (s, 1H, NCH), 8.00 (s, 1H, NCH), 7.64–7.27 (m, 8H, Ar), 5.54 (s, 2H, CH2), 3.85 (s, 3H, OCH3), 1.30 (s, 9H, CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 149.9 (Ar), 143.9 (Ar), 137.3 (NCHN), 134.6 (Ar), 129.0 (Ar), 128.9 (Ar), 128.5 (Ar), 128.3 (Ar), 124.3 (NCH), 123.2 (Ar), 122.8 (Ar), 122.0 (NCH), 112.8 (Ar), 56.4 (CH2), 52.2 (OCH3), 34.2 (C), 31.1 (CH3) ppm.
Ha2Br – alkyl halide: 1-bromohexane, Yield: 91% (hygroscopic white solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 9.56 (s, 1H, NCHN), 8.07 (t, J = 1.7 Hz, 1H, NCH), 8.00 (t, J = 1.7 Hz, 1H, NCH), 7.63–7.57 (m, 2H, Ar), 7.28 (d, J = 9.5 Hz, 1H, Ar), 4.25 (t, J = 7.2 Hz, 2H, NCH2), 3.84 (s, 3H, OCH3), 1.87 (quint, J = 7.2 Hz, 2H, CH2), 1.30 (m, 15H, CH3 + CH2), 0.87 (t, J = 6.5 Hz, 3H, hex-CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 150.0 (Ar), 143.9 (Ar), 137.1 (NCHN), 128.3 (Ar), 124.0 (NCH), 123.3 (Ar), 122.9 (Ar), 122.1 (NCH), 112.9 (Ar), 56.4 (OCH3), 49.2 (NCH2), 34.2 (C), 31.1 (CH3), 30.6 (CH2), 29.2 (CH2), 25.2 (CH2), 22.0 (CH2), 13.9 (hex-CH3) ppm.
Ha3I – alkyl halide: methyl iodide, Yield: 93% (hygroscopic light–yellow solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 9.49 (s, 1H, NCHN), 8.04 (s, 1H, NCH), 7.90 (s, 1H, NCH), 7.61–7.58 (m, 2H, Ar), 7.28 (d, J = 9.5 Hz, 1H, Ar), 3.95 (s, 3H, NCH3), 3.85 (s, 3H, OCH3), 1.30 (s, 9H, CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 149.9 (Ar), 143.9 (Ar), 137.6 (NCHN), 128.2 (Ar), 123.7 (NCH), 123.3 (NCH), 123.2 (Ar), 122.8 (Ar), 112.8 (Ar). 56.4 (OCH3), 36.0 (NCH3), 34.1 (C), 31.0 (CH3) ppm.
H2a4Br2 – Yield: 94% (brownish solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 9.62 (t, J = 1.5 Hz, 2H, NCHN), 8.07 (t, J = 1.5 Hz, 2H, NCH), 8.00 (t, J = 1.5 Hz, 2H, NCH), 7.60–7.58 (m, 4H, Ar), 7.28 (d, J = 9.5 Hz, 2H, Ar), 4.27 (t, J = 7.4 Hz, 4H, NCH2), 3.84 (s, 6H, OCH3), 1.87 (br, 4H, CH2), 1.30–1.26 (m, 34H, CH3 + CH2) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 150.0 (Ar), 144.0 (Ar), 137.1 (NCHN), 128.3 (Ar), 123.9 (NCH), 123.2 (Ar), 122.9 (Ar), 122.2 (NCH), 112.9 (Ar), 56.4 (OCH3), 49.2 (NCH2), 34.2 (C), 31.1 (CH3), 29.3 (CH2), 29.0 (CH2), 28.9 (CH2), 28.4 (CH2), 25.6 (CH2) ppm.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HBr:acetic acid mixture and heated at 125 °C for 16 h. The solvent was then removed at reduced pressure, and the product recrystallized from acetone/diethyl ether, washed with diethyl ether (3 × 15 mL) and dried under reduced pressure.
:1 HBr:acetic acid mixture and heated at 125 °C for 16 h. The solvent was then removed at reduced pressure, and the product recrystallized from acetone/diethyl ether, washed with diethyl ether (3 × 15 mL) and dried under reduced pressure.
H2L1Br – Yield: 80% (light brown solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 10.62 (s, 1H, OH), 9.86 (s, 1H, NCHN), 8.09 (s, 1H, NCH), 8.03 (s, 1H, NCH), 7.56–7.10 (m, 7H, Ar), 7.11 (d, J = 8.5 Hz, 1H, Ar), 5.58 (s, 2H, NCH2), 1.28 (s, 9H, CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 148.1 (Ar), 142.5 (Ar), 137.0 (NCHN), 134.8 (Ar), 129.0 (Ar), 128.8 (Ar), 128.5 (Ar), 128.0 (Ar), 124.0 (NCH), 122.5 (Ar), 122.0 (Ar), 121.6 (NCH), 116.7 (Ar), 52.0 (NCH2), 34.0 (C), 31.1 (CH3) ppm. FT-IR ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) : 3062 (vs), 2960 (s), 1622 (m), 1549 (s), 1513 (s), 1456 (m), 1363 (m), 1274 (s), 1139 (m), 1072 (m), 823 (s), 710 (vs), 650 (m), 472 (w) cm−1.
: 3062 (vs), 2960 (s), 1622 (m), 1549 (s), 1513 (s), 1456 (m), 1363 (m), 1274 (s), 1139 (m), 1072 (m), 823 (s), 710 (vs), 650 (m), 472 (w) cm−1.
H2L2Br – Yield: 85% (hygroscopic white solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 10.60 (s, 1H, OH), 9.67 (s, 1H, NCHN), 8.07 (s, 1H, NCH), 8.00 (s, 1H, NCH), 7.50–7.41 (m, 2H, Ar), 7.12 (d, J = 8.50 Hz, 1H, Ar), 4.30 (t, J = 7.2 Hz, 2H, NCH2), 1.87 (br, 2H, CH2), 1.27 (br, 15H, CH3 + CH2), 0.85 (s, 3H, hex-CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 148.2 (Ar), 142.5 (Ar), 136.8 (NCHN), 128.0 (Ar), 123.6 (NCH), 122.6 (Ar), 122.1 (NCH), 121.7 (Ar), 116.7 (Ar), 49.1 (NCH2), 34.0 (C), 31.1 (CH3), 30.6 (CH2), 29.3 (CH2), 25.2 (CH2), 21.9 (CH2), 13.9 (hex-CH3) ppm.
H2L3Br – Yield: 92% (hygroscopic light yellow solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 10.60 (s, 1H, OH), 9.48 (s, 1H, NCHN), 8.04 (t, J = 1.8 Hz, 1H, NCH), 7.88 (t, J = 1.8 Hz, 1H, NCH), 7.47–7.42 (m, 2H, Ar), 7.07 (d, J = 8.5 Hz, 1H, Ar), 3.95 (s, 3H, NCH3), 1.28 (s, 9H, CH3) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 148.2 (Ar), 142.5 (Ar), 137.4 (NCHN), 128.1 (Ar), 123.5 (NCH), 123.3 (NCH), 122.6 (Ar), 121.7 (Ar), 116.7 (Ar), 36.0 (NCH3), 34.0 (C), 31.1 (CH3) ppm.
H4L4Br2 – Yield: 83% (brownish solid). 1H NMR (300.1 MHz, DMSO-d6): δ = 10.59 (s, 2H, OH), 9.61 (s, 2H, NCHN), 8.07 (s, 2H, NCH), 8.01 (s, 1H, NCH), 7.49–7.42 (m, 4H, Ar), 7.09 (d, J = 8.50 Hz, 2H, Ar), 4.27 (t, J = 7.4 Hz, 4H, NCH2), 1.87 (br, 4H, CH2), 1.30 (br, 34H, CH3 + CH2) ppm. 13C NMR (75.5 MHz, DMSO-d6): δ = 148.2 (Ar), 142.5 (Ar), 136.8 (NCHN), 128.1 (Ar), 123.7 (NCH), 122.6 (NCH), 122.1 (Ar), 121.7 (Ar), 116.7 (Ar), 49.1 (NCH2), 34.0 (C), 31.1 (CH3), 29.3 (CH2), 30.0 (CH2), 28.9 (CH2), 28.4 (CH2), 25.6 (CH2) ppm.
Ha1PF6 – Starting from Ha1Br, Yield: 90% (white solid). 1H NMR (300.1 MHz, CD3CN): δ = 8.92 (s, 1H, NCHN), 7.62 (s, 1H, NCH), 7.60 (m, 1H, Ar), 7.52 (s, 1H, NCH), 7.49–7.47 (m, 6H, Ar), 5.43 (s, 2H, CH2), 3.87 (s, 3H, OCH3), 1.33 (s, 9H, CH3) ppm. 31P NMR (121 MHz, CD3CN): δ = −144.62 (sept, J = 710 Hz, PF6) ppm. 19F NMR (189 MHz, CD3CN): δ = −70.57 (d, J = 710 Hz, PF6) ppm.
H2L1PF6 – Starting from H2L1Br, Yield: 85% (white solid). 1H NMR (300.1 MHz, CD3CN): δ = 9.06 (s, 1H, NCHN), 7.55–7.46 (m, 10H, Ar + NCH), 7.35 (d, J = 8.4 Hz, 1H, Ar), 5.47 (s, 2H, NCH2), 1.34 (s, 9H, CH3) ppm. 31P NMR (121 MHz, CD3CN): δ = −144.6 (sept, J = 710 Hz, PF6) ppm. 19F NMR (189 MHz, CD3CN): δ = −70.6 (d, J = 710 Hz, PF6) ppm.
Synthesis of the copper complexes
[Cu(L1)2] – Yield: 80% (dark brownish-purple powder). Elemental analysis calcd (%) for C40H42CuN4O2·H2O: C 69.39, H 6.41, N 8.09. Found: C 69.77, H 6.75, N 7.92. ESI(+)-MS (m/z): 674.17 [[Cu(L1)2] + H]+. FT-IR : 3089 (vw), 3062 (vw), 3025 (vw), 2960 (s), 2863 (w), 1610 (m), 1499 (vs), 1455 (m), 1411 (m), 1492 (w), 1356 (m), 1306 (vs), 1262 (s), 1147 (m), 1109 (w), 866 (w), 834 (s), 776 (w), 737 (s), 721 (m), 709 (m), 667 (w), 619 (vw), 595 (vw), 557 (vw), 447 (vw), 411 (vw) cm−1. UV-vis (CH2Cl2): λ (ε/M−1 cm−1) = 230 (44100), 318 (14400), 455 (970) nm. Crystals suitable for single crystal X-ray diffraction (SC-XRD) analysis were obtained by slow evaporation of a concentrated solution of the complex in acetone.
[Cu(L2)2] – Yield: 60% (purple powder). Elemental analysis calcd (%) for C38H54CuN4O2·1.5H2O: C 66.20, H 8.33, N 8.13. Found: C 66.19, H 8.42, N 8.05. ESI(+)-MS (m/z): 662.21 [[Cu(L2)2] + H]+. FT–IR : 3159 (vw), 3124 (vw), 3087 (vw), 2958 (vs), 2928 (vs), 2959 (s), 1609 (m), 1500 (vs), 1464 (m), 1392 (m), 1362 (m), 1349 (m), 1303 (vs), 1262 (s), 1148 (m), 1105 (w), 1171 (vw), 871 (w), 828 (s), 737 (s), 671 (w), 620 (vw), 595 (vw), 445 (vw), 410 (vw) cm−1. UV-vis (CH2Cl2): λ (ε/M−1 cm−1) = 230 (37100), 319 (12500), 460 (830) nm.
[Cu(L3)2] – Yield: 55% (dark brown powder). ESI(+)-MS (m/z): 522.12 [[Cu(L3)2] + H]+. FT–IR : 3174 (vw), 3145 (vw), 3063 (vw), 2958 (s), 2903 (m), 2864 (m), 1675 (w), 1609 (m), 1501 (vs), 1460 (s), 1402 (m), 1353 (m), 1315 (vs), 1264 (s), 1149 (m), 1118 (w), 1083 (w), 1020 (vw), 873 (w), 835 (s), 819 (s), 751 (s), 723 (s), 666 (m), 617 (vw), 592 (vw), 524 (vw), 445 (vw), 403 (w) cm−1. UV-vis (CH2Cl2): λ (ε/M−1 cm−1) = 230 (35700), 320 (9400), 454 (580) nm.
[Cu(L4)] – In this synthesis 0.26 mmol of H4L4Br2 were used (1:1 molar ratio between Cu and ligand precursor). Yield: 58% (dark purple powder). Elemental analysis calcd (%) for C38H52CuN4O2·H2O: C 67.28, H 8.02, N 8.26. Found: C 67.23, H 7.73, N 8.06. ESI(+)-MS (m/z): 660.26 [[Cu(L4)] + H]+. FT–IR : 3171 (vw), 3135 (vw), 3069 (vw), 2952 (vs), 2925 (vs), 2854 (s), 1688 (w), 1609 (m), 1500 (vs), 1462 (m), 1414 (m), 1392 (m), 1362 (m), 1350 (m), 1315 (s), 1262 (s), 1202 (w), 1147 (m), 1107 (w), 1070 (w), 1027 (vw), 874 (w), 862 (w), 834 (s), 832 (s), 737 (m), 720 (m), 669 (m), 619 (vw), 528 (vw), 404 (w) cm−1. UV-vis (CH2Cl2): λ (ε/M−1 cm−1) = 230 (32500), 318 (10400), 465 (980) nm.
Computational details
All calculations were performed with ORCA v5.0.3.71,72 Molecular geometries were optimized without any symmetry constrain in the gas phase using the PBE0 functional.73 Scalar relativistic effects were modeled using the Zeroth Order Regular Approximation (ZORA)74 with the ZORA-Def2-SVP75 basis set and the ZORA-Def2-TZVP75 for copper with the RI76 approximations with the related auxiliary basis sets (SARC/J).77,78 The D379 dispersion correction with Becke-Johnson80 damping was used. All optimized structures were verified as true minima by the absence of negative eigenvalues in the harmonic vibrational frequency analysis.Single point calculations were performed at the same level of theory with a ZORA-Def2-TZVPP basis set for all atoms. The absorption spectra were modeled by time-dependent DFT calculations (TD-DFT) with the ZORA-def2-TZVPP basis set and PBE0 functional. For the TD-DFT calculations 20 roots were computed and solvation effects were included with the SMD solvation model with (DCM)81 as solvent. The Tamm-Dancoff82 approximation was used to speed up the calculations. Intrinsic bond orbitals (IBOs)83 were calculated, and were visualized using Chemcraft84 and IBOview.85
General procedure for catalytic tests
The catalytic tests were carried out in a 10 mL Schlenk tube or, when pressure higher than 1 bar was used, in a 35 mL Fischer-Porter reactor. To the magnetic stirring bar equipped tube, after three vacuum–argon cycles, the catalyst, the solvent, the amine (substrate), and the silane were added in the order. Then, CO2 was fluxed to saturate the environment and the reactor was closed, maintaining a CO2 balloon connected to the reactor. For the high-pressure experiments, the reactor was simply sealed and maintained at the target pressure. Subsequently, the reactor was dipped into a thermostatic bath fixed to the target temperature. The magnetic stirring and the reactor immersion level were maintained identical in all performed tests. The starting time of the tests was set at five minutes after the reactor immersion. After the reaction time, the reactor was cooled to room temperature, depressurized, and 2,5-dimethylfuran was added with a micro syringe as internal standard (1 equiv. respect to the substrate). The reaction mixture was then thoroughly mixed and the 1H NMR spectrum of a sample in CDCl3 was recorded. Conversions and yields were calculated by 1H NMR spectroscopy. In some cases, the addition of a small quantity of potassium carbonate to the NMR tube helped to obtain a better spectrum. Every test was performed three times and the reported conversion and yield are the resulting average value. The yield obtained in the replication of the same test varied in the range ±1%.X-Ray crystallography
The crystallographic data for compounds [Cu(L1)2] and [CuBr(a1)] were obtained by mounting a single crystal of each sample on a loop fiber and transferring it to a Bruker D8 Venture Photon II single crystal diffractometer equipped with an area detector, working with monochromatic MoKα radiation in the case of [Cu(L1)2] and CuKα radiation in the case of [CuBr(a1)]. The APEX 3 program package was used to obtain the unit-cell geometrical parameters for both structures. The raw frame data were processed using SAINT and SADABS to obtain the data file of the reflections. The structures were solved using SHELXT (Intrinsic Phasing method in the APEX 3 program).86 The refinement of the structures (based on F2 by full-matrix least-squares techniques) was carried out using the SHELXTL-2018/3 program.87 The hydrogen atoms were introduced in the refinement in defined geometry and refined “riding” on the corresponding carbon atoms. Deposition numbers 2371863 and 2371864 contain the supplementary crystallographic data for compound [Cu(L1)2] and [CuBr(a1)] respectively.Results and discussion
Synthesis and characterization
The hybrid bidentate H2L1–3Br and tetradentate H4L4Br2 ligand precursors were prepared by a two-step synthesis (Scheme 2), adaptation of a previously reported procedure,69,88 consisting in the quaternarization of 1-(5-tert-butyl-2-methoxyphenyl)-imidazole with the selected alkyl halide, followed by the demethylation of the methoxy moieties using HBr. The imidazolium salts Ha1–2Br, Ha3I and H2a4Br2 obtained after the first reaction step, can be used as NHC precursors as well (vide infra). From the imidazolium salts Ha1Br and H2L1Br, the respective hexafluorophosphate salts Ha1PF6 and H2L1PF6 were obtained in high yield, performing the anion exchange in water using KPF6. All the proligands were isolated with an overall yield >80%.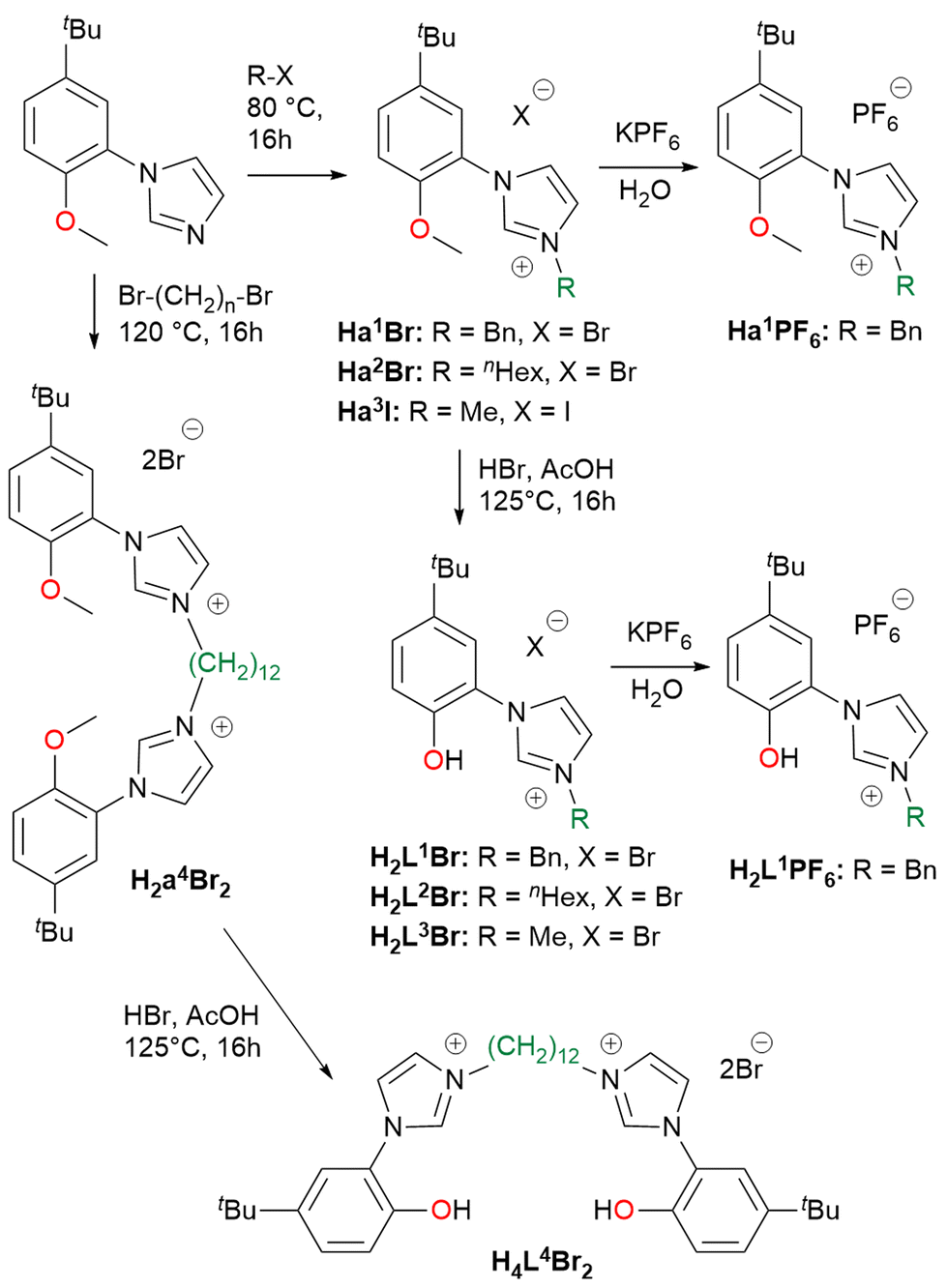 | ||
| Scheme 2 Synthesis of the ligand precursors used in this study. | ||
The ligand precursors H2L1–3Br and H4L4Br2 were reacted with Cu(OAc)2·H2O and K2CO3 in methanol to afford the corresponding Cu(II) complexes of general formula [Cu(L)2] or [Cu(L)] depending on the use of bidentate (L1–3) or tetradentate (L4) ligands (Scheme 3). By using tetradentate ligand precursors with shorter linkers, methylene or dimethylene bridging groups,69,70 it was not possible to isolate the corresponding Cu(II) complexes. The complexes [Cu(L1–3)2] are soluble in common organic solvents, while [Cu(L4)] is soluble only in dichloromethane, chloroform, and sparingly soluble in acetone. Complexes [Cu(L1–3)2] and [Cu(L4)] were characterized by means of ESI-MS spectrometry, FT-IR and UV-Vis spectroscopy and in the case of [Cu(L1)2] single crystal X-ray diffraction. In the ESI-MS spectra, the presence of the peaks attributed to the species [Cu(L1–3)2 + H+]+ and [Cu(L4) + H+]+ is diagnostic of the formation of the Cu(II) complexes.
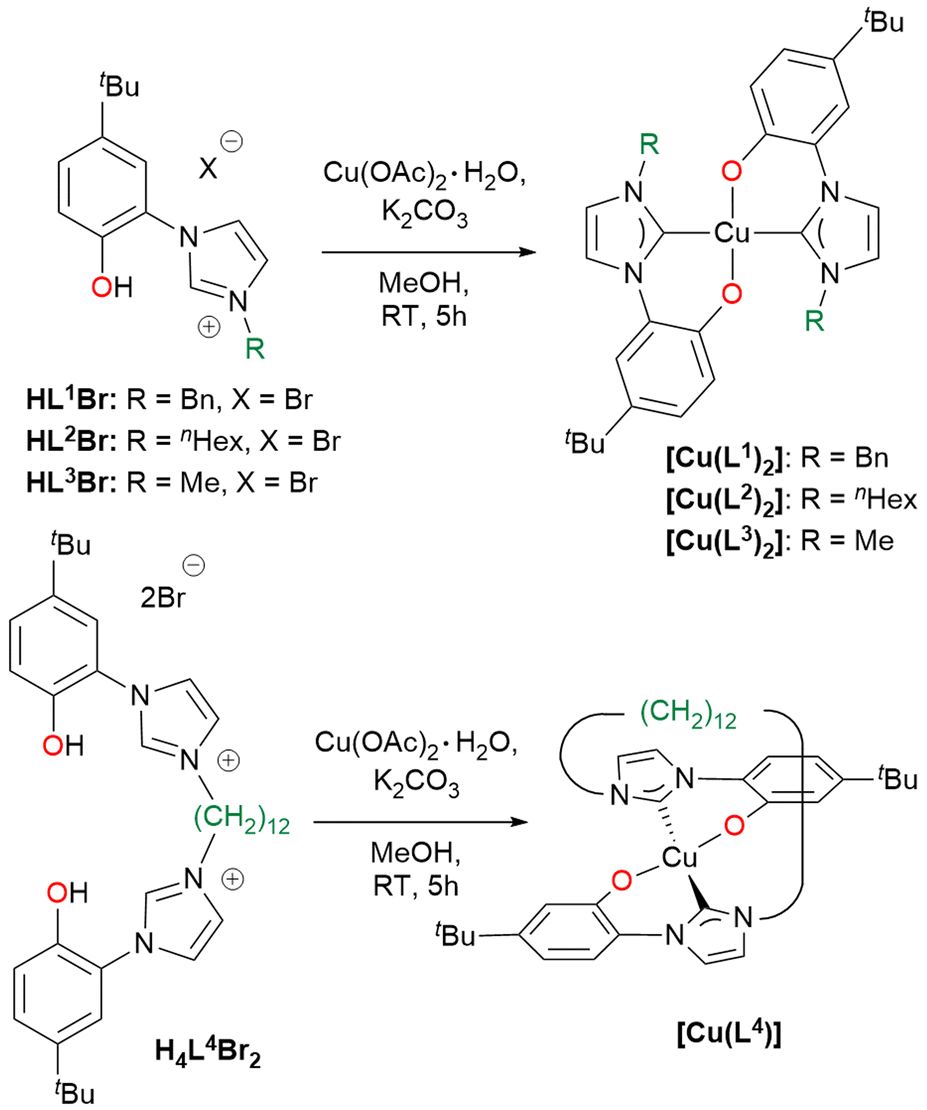 | ||
| Scheme 3 Synthesis of the Cu(II) complexes prepared in this work. | ||
In the FT-IR spectra of the complexes (Fig. S35–38†), the lack of the PhO–H stretching signal at ca. 3060 cm−1, clearly visible for proligand H2L1Br (Fig. S34†), agrees with the deprotonation of the phenol moieties upon metal coordination. The UV-visible absorption spectra of the Cu(II) complexes were registered in dichloromethane solution, showing similar features for the four compounds with three well defined absorption maxima at ca. 230, 320 and 460 nm (Fig. S39–42†). Finally in the case of complex [Cu(L1)2], the molecular structure was confirmed by X-ray diffraction on a single crystal obtained by slow evaporation of an acetone solution of the complex (Fig. 1). The complex crystallizes in the monoclinic P21 space group and presents a slightly distorted square planar geometry, as indicated by the value close to 0 (0.05) of the τ4 geometry index.89 In the coordination sphere of the Cu(II) centre the two NHC and the two phenolate donors are mutually in trans. The two NHC rings are almost coplanar, with a dihedral angle of 5.75(13)° between them, and are slightly tilted with respect to the mean metal coordination plane with angles of 26.82(9) and 23.03(9)° respectively. The Cu–O and Cu–C bond lengths are comparable to similar copper(II) NHC–phenolate complexes.43 Two intramolecular C–H⋯O hydrogen bonds are present in the structure. The donor-H⋯acceptor distances (D) are 2.939(4) and 3.015 (4) for C24–H⋯O1 and C4–H⋯O2 respectively. The C–H⋯O angles (θ) are 124.03(17)° and 137.83(19)° for C24–H⋯O1 and C4–H⋯O2 respectively. Thus, D and θ values are within the ranges of those reported in the literature for this type of interaction.90
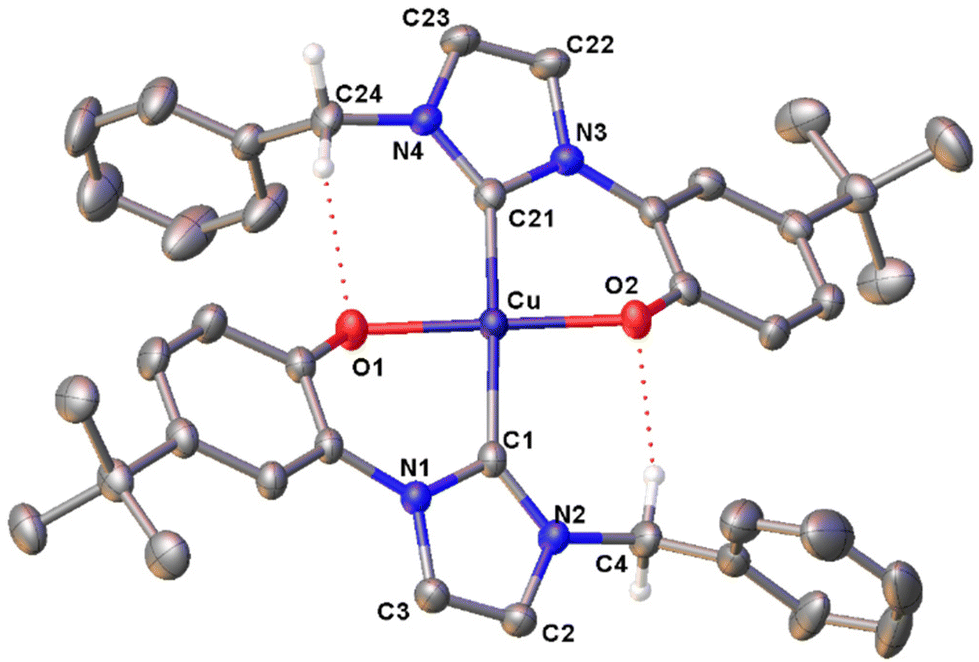 | ||
| Fig. 1 ORTEP drawing of complex [Cu(L1)2]. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°): Cu–C1 2.005(3), Cu–C21 2.007(3), Cu–O1 1.946(2), Cu–O2 1.958(2), C1–Cu–C21 174.1(1), O1–Cu–O2 179.2(1), C1–Cu–O1 86.7(1), C21–Cu–O2 88.3(1), C1–Cu–O2 179.2(1), C21–Cu–O1 91.6(1). | ||
The number of homoleptic Cu(II) complexes with hybrid NHC–phenolate ligands reported in the literature up to now is very limited.43–45 All of them exhibit a square planar coordination geometry, with different degrees of angular distortion and a trans arrangement. The formation of cis isomer has not been documented yet in these systems. Instead, with group 10 metal centres both the cis and trans isomers can be obtained selectively, mainly depending on the steric bulkiness of the NHC wingtip substituent. With methyl wingtip the cis isomer is obtained, whereas with bulkier Dipp (2,6-diisopropyl-phenyl) or Mes (mesityl) wingtips the trans isomer is isolated.91 Our results align with the literature results for Cu(II), as indicated by the molecular structure of [Cu(L1)2] and by the fact that in the case of the tetradentate ligands, only the use of a long and flexible bridging group, allowing the formation of the trans isomer, brings to the isolation of a stable Cu(II) complex. Differently, in a previous work we successfully coordinated tetradentate bis(NHC)–bis(phenolate) ligands with the shorter CH2 and CH2CH2 linkers to Ni(II), obtaining the complexes cis-[NiL].70
To gain more insight on the experimental selective formation of the trans isomer for our Cu(II) complexes, the electronic and structural properties of the complex with the benzyl substituent were investigated by means of computational methods. In order to save computational resources, the tBu group on the phenolate ring was substituted with a methyl one, and the truncated complex is reported as [Cu(L1)2]Me in the text. DFT calculations in the case of trans-[Cu(L1)2]Me well reproduced the structural parameters observed in the solid-state structure of [Cu(L1)2] (Fig. 2, left). The calculated τ4 geometry index is 0.00, so perfectly square planar, close to the 0.05 value measured by XRD. Next, the geometry of the cis isomer was optimized at the same level of theory, obtaining for cis-[Cu(L1)2]Me a highly distorted structure as indicated by the τ4 geometry index of 0.51 (Fig. 2, right). The deviation from a square planar coordination is imposed by the arrangement of the ligands forced by the steric hindrance of the benzyl wingtip substituents. A consequence of the forced cis geometry is the loss of the hydrogen bonding interaction evident in the X-ray structure of the trans isomer. In gas phase, the trans isomer is predicted thermodynamically more stable than the cis one (ΔGcis–trans = +6 kJ mol−1), but the energy difference is too small to justify the selective trans isomer formation, thus kinetic factors cannot be excluded in determining the output of the syntheses.
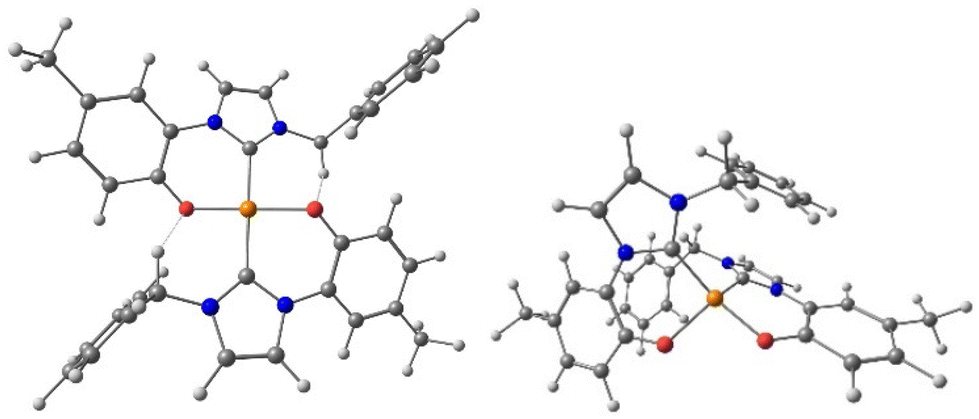 | ||
| Fig. 2 Optimized structures at the ZORA PBE0 ZORA-Def2-TZVPP D3BJ level of theory level for the trans (left) and cis (right) isomers of [Cu(L1)2]Me. We highlight the formation of H-bonds for the trans isomer. Colour codes: Cu, orange; O, red; N, light blue; C, grey; H, white. | ||
Next, we investigated the electronic structure of the [Cu(L1)2]Me complex, and the spin-density is located mainly on the metal centre with small contribution from the ligands (see Fig. 3).
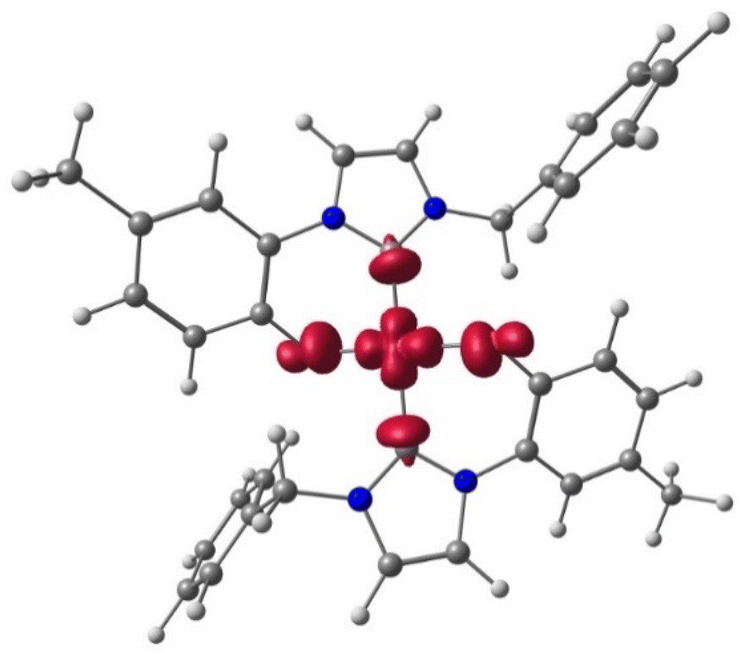 | ||
| Fig. 3 Spin density plot of trans-[Cu(L1)2]Me ZORA PBE0 ZORA-Def2-TZVPP D3BJ level of theory (iso-surface 0.005). | ||
Intrinsic bonding orbital (IBO) analysis shows that in complex trans-[Cu(L1)2]Me the ligand forms two sigma bonds with the metal centre (see Fig. 4). In addition, TD-DFT calculations on the cis and trans isomers of [Cu(L1)2]Me predict a weak absorption band for the cis isomer at λabs. = 537 nm (fosc = 0.016) which is not observed in the experimental UV-Vis spectrum (see Fig. S67†). Differently, the TD-DFT spectrum of the trans isomer well reproduces the experimental absorption spectrum with a predicted λabs. = 405 nm (fosc = 0.033) in good agreement with the experimental value of 455 nm. Therefore, we tentatively suggest that also in solution the trans isomer is favoured.
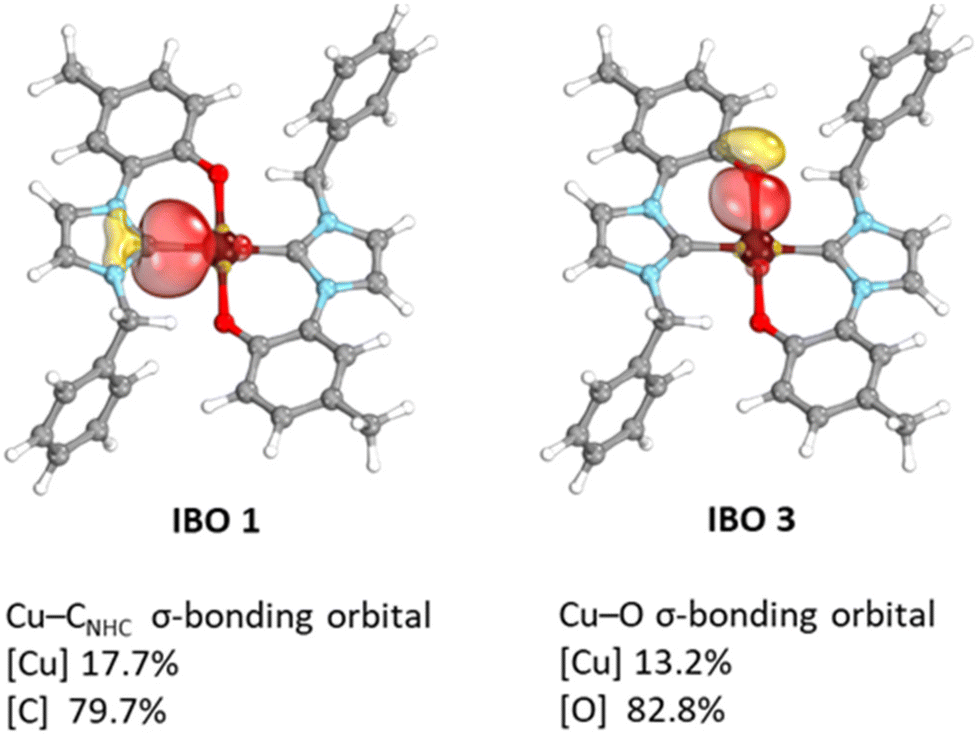 | ||
| Fig. 4 IBO analysis of trans-[Cu(L1)2]Me complex. The metal–ligand σ-bonding orbitals are shown and the partial-charge distribution of a given IBO at ZORA PBE0 ZORA-Def2-TZVPP D3BJ level of theory is given below. Structural depictions were made using IboView. Considering the centrosymmetric structure of the complex only one Cu–C and one Cu–O σ-bonding orbitals are depicted. | ||
Successively, with the aim of comparing the catalytic activity of Cu(II) and Cu(I) complexes (vide infra), we tried to coordinate the hybrid ligands L1–4 to Cu(I), but none of the complexation attempts led to the desired product. Differently, by using potassium carbonate as base, [CuBr(SMe2)] as metal precursor, and performing the reaction in acetone the Cu(I) complex [CuBr(a1)] was obtained starting from the ligand precursor Ha1Br (Scheme 4).12 Furthermore, starting from Ha1PF6 the synthesis of the bis(NHC) complex [Cu(a1)2]PF6 was achieved in acetonitrile with [Cu(CH3CN)4](PF6) as metal source (Scheme 4). The Cu(I) complexes were isolated as crystalline light-yellow solids in moderate yields. Purity of the complexes was confirmed by elemental analysis, and from their 1H NMR spectra. In the case of complex [Cu(a1)2]PF6 the carbene carbon chemical shift was identified at 179.0 ppm via1H–13C HMBC NMR experiment, a value in agreement with other reported bis-NHC Cu(I) cationic complexes.92
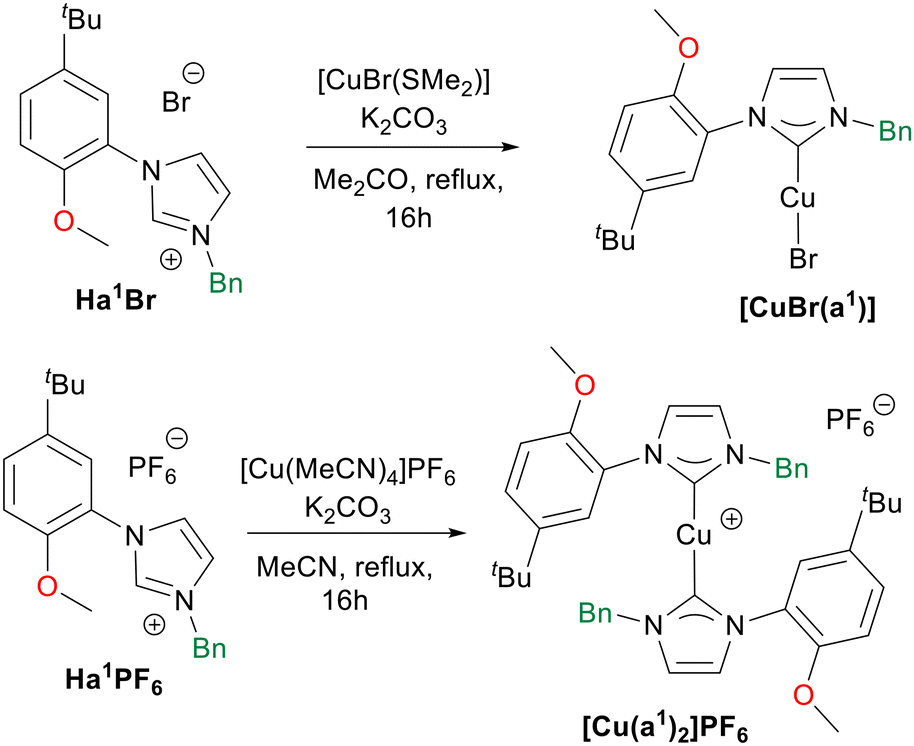 | ||
| Scheme 4 Synthesis of the Cu(I) complexes prepared in this work. | ||
The molecular structure of the complex [CuBr(a1)] was confirmed by single crystal X-ray diffraction (Fig. 5), from crystals obtained from layering of diethylether on a dichloromethane solution of the complex at −18 °C. As expected, the copper(I) centre presents a slightly distorted linear geometry (C1–Cu–Br 178.3(2)°). The Cu–C and Cu–Br bond distances of 1.890(6) and 2.2273(9) Å respectively are perfectly in agreement with similar [CuBr(NHC)] complexes reported in the literature.93 No interaction is observed between the oxygen atom of the methoxy group and the copper centre, with the substituted phenyl wingtip oriented in the way that minimizes steric repulsion around the metal centre.
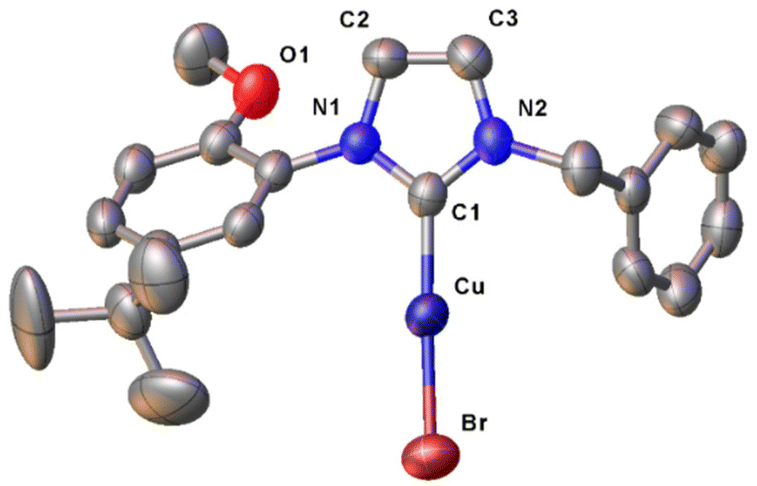 | ||
| Fig. 5 ORTEP drawing of complex [CuBr(a1)]. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°): Cu–C1 1.890(6), Cu–Br 2.2273(9), C1–Cu–Br 178.3(2). | ||
Catalytic studies
The obtained Cu(II) complexes were tested as catalyst precursors in the reductive N-formylation and N-methylation of amines with CO2, a reaction that we have recently studied with Mn(I) complexes as catalysts.59 A preliminary screening of the reaction conditions was carried out using N-ethylaniline (1a) as substrate, [Cu(L1)2], 3 equivalents of PhSiH3 as reducing agent, at 40 °C and varying the reaction solvent and CO2 pressure (Scheme 5). | ||
| Scheme 5 Benchmark reaction for the N-methylation and N-formylation of amines with CO2. Reaction conditions: N-ethylaniline 0.40 mmol, PhSiH3 (3 equiv.), [Cu(L1)2] 1 mol%, 40 °C, 5 h, in 1 mL of solvent. | ||
The results of the preliminary catalytic experiments are reported in Table S2.† Under the adopted reaction conditions, the proligand H4L1Br2 and Cu(OAc)2·H2O were not able to promote any product formation. Instead, using [Cu(L1)2] at a 1 mol% loading with respect to the aniline 1a a complete conversion can be achieved in 5 h at 40 °C and a 3 bar CO2 pressure, using DMF or the ionic liquid 1-butyl-2,3-dimethylimidazolium bis(trifluoromethylsulfonyl)imide ([BMMIM][NTf2]) as solvent. A product mixture of 2a and 3a was obtained, with 3a as the main product in all the performed tests. A blank experiment carried out using [BMMIM][NTf2] showed that the ionic liquid is not able to promote the products formation in the absence of the copper complex (Table 1, entry 1). Successively, to further increase the methylated (2a) product yield, the temperature was increased to 60 °C, the CO2 pressure was lowered to 1 bar, and the reaction time was extended to 16 h, performing the reaction in [BMMIM][NTf2] (Table 1). Under these conditions the selectivity was reversed with 2a being always the main product. This is not surprising since it is known that a decrease in the CO2 pressure and an increase in the temperature shift the selectivity towards the methylated species.59 Then the activity of the different Cu(II) complexes was evaluated (Table 1, entries 2–5). A marked difference was observed among the different complexes, with complexes bearing ligands with bulkier wingtip substituents on the NHC showing a better catalytic performance. The catalysts screening shows that [Cu(L2)2] is the most active and selective towards the N-methyl product, leading to full conversion of the aniline 1a and an 81% yield of 2a (Table 1, entry 3). The performance of [Cu(L1)2] is slightly lower compared to that of [Cu(L2)2], whereas [Cu(L3)2] and [Cu(L4)] were found to be significantly less active. The differences in activity among the Cu(II) complexes become more evident looking at Fig. 6a, in which the reagent conversion is reported against the reaction time. After 7 hours, [Cu(L2)2] achieved an 80% conversion, [Cu(L1)2] 43%, and complexes [Cu(L3)2] and [Cu(L4)] slightly less than 20%. In Fig. 6b, the yields of 2a and 3a and the residual PhSiH3 over time using [Cu(L2)2] can be observed. After 24 hours, no residual PhSiH3 remains in the reaction mixture. The Cu(I) complexes [CuBr(a1)] and [Cu(a1)2]PF6 were also evaluated for this reaction (Table 1, entries 6 and 7), but their performance was way lower compared to the Cu(II) complexes. The most active complex [Cu(L2)2] was also tested in the presence of other hydrosilanes, namely Ph2SiH2, Ph3SiH, Me2PhSiH and PMHS (polymethylhydrosiloxane). Only with Ph2SiH2 a poor conversion was observed (13%), interestingly with complete selectivity towards 2a (Table 1, entry 8).
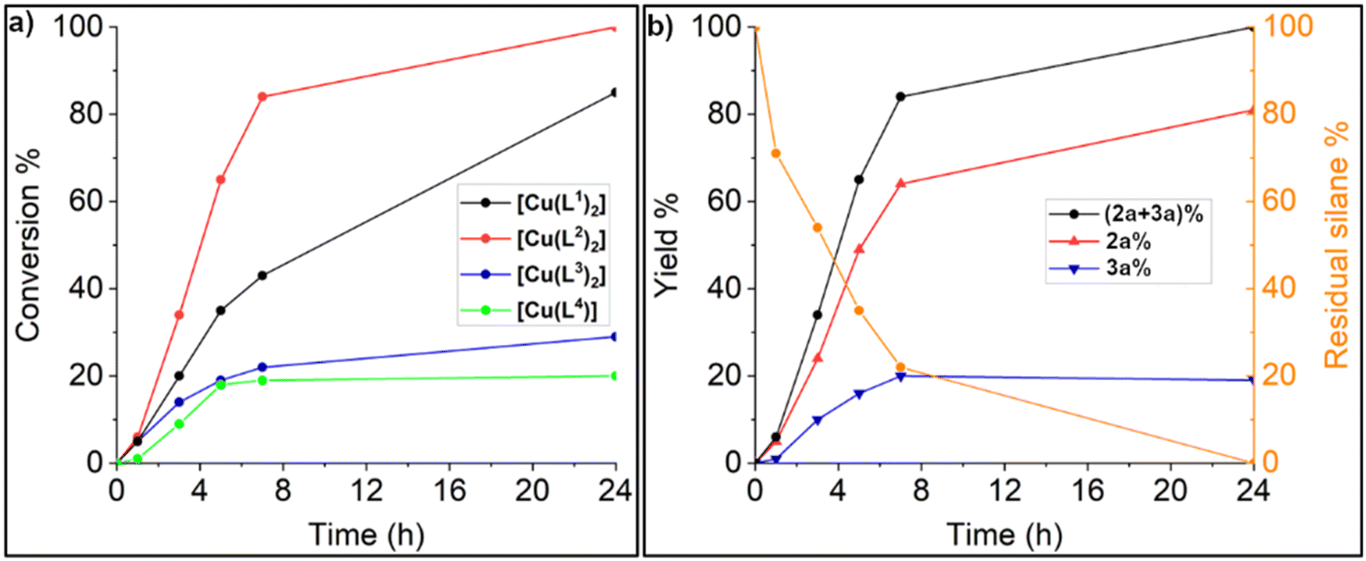 | ||
| Fig. 6 (a) Total conversion of N-ethylaniline versus time using the Cu(II) complexes; (b) total conversion of N-ethylaniline, yield of 2a and 3a and residual silane versus time using [Cu(L2)2] as catalyst, complete data reported in Table S3.† Reaction conditions for all the experiments: N-ethylaniline 0.40 mmol, PhSiH3 (3 equiv.), catalyst loading 1 mol%, p(CO2) = 1 bar (balloon), 60 °C, in 1 mL of [BMMIM][NTf2]. | ||
| Entry | Catalyst | Silane (equiv.) | Conv.a/% | 2a /% | 3a /% |
|---|---|---|---|---|---|
| Reaction conditions: N-ethylaniline 0.40 mmol, hydrosilane, catalyst loading 1 mol%, p(CO2) = 1 bar (balloon), 60 °C, 24 h, in 1 mL of [BMMIM][NTf2].a Yield determined by 1H NMR using 2,5-dimethylfuran as an internal standard.b T = 80 °C.c Catalyst loading 2 mol%, t = 16 h. | |||||
| 1 | — | PhSiH3 (3) | 0 | 0 | 0 |
| 2 | [Cu(L1)2] | PhSiH3 (3) | 84 | 62 | 22 |
| 3 | [Cu(L2)2] | PhSiH3 (3) | 100 | 81 | 19 |
| 4 | [Cu(L3)2] | PhSiH3 (3) | 29 | 15 | 14 |
| 5 | [Cu(L4)] | PhSiH3 (3) | 20 | 15 | 5 |
| 6 | [CuBr(a1)] | PhSiH3 (3) | 5 | 2 | 3 |
| 7 | [Cu(a1)2]PF6 | PhSiH3 (3) | 2 | 0 | 2 |
| 8 | [Cu(L2)2] | Ph2SiH2 (3) | 13 | 13 | 0 |
| 9b | [Cu(L2)2] | PhSiH3 (3) | 68 | 59 | 9 |
| 10b | [Cu(L2)2] | Ph2SiH2 (3) | 39 | 39 | 0 |
| 11 | [Cu(L2)2] | Ph2SiH2 (9) | 75 | 75 | Traces |
| 12 | [Cu(L2)2] | PhSiH3 (6) | 100 | 85 | 15 |
| 13c | [Cu(L2)2] | PhSiH3 (3) | 100 | 93 | 7 |
By increasing the temperature to 80 °C a different behaviour was observed with PhSiH3 and Ph2SiH2. In the case of PhSiH3 lower conversion and yields were obtained (Table 1, entry 9), probably as consequence of a faster silane consumption in side-reactions. It has been observed that at 80 °C, PhSiH3 already reached 90% consumption after the first three hours (Table S3 and Fig. S46b†). Differently, in the case of Ph2SiH2, a higher temperature led to higher conversion and yield (39%), maintaining the full 2a selectivity (Table 1, entry 10). Better performances can be obtained increasing the silane equivalents. With 9 Ph2SiH2 equivalents the 2a yield increased to 75% (Table 1, entry 11) maintaining an almost complete selectivity. With 6 PhSiH3 equivalents the 2a yield increased to 85% (Table 1, entry 12). A better improvement was achieved maintaining 3 equivalents of silane and increasing the catalyst loading to 2 mol%, obtaining a 2a with 93% yield (Table 1, entry 13). Under the latter conditions a preliminary substrate scope was carried out (Scheme 6).
 | ||
| Scheme 6 N-Methylation of secondary amines using catalyst [Cu(L2)2], yield in parenthesis. 2a: N-ethyl-N-methylaniline, 2b: N,N-dimethylaniline, 2c: N,N-dimethyl-p-methoxyaniline, 2d: N-methyl-N,N-dibenzylamine. | ||
Under the adopted conditions a full conversion of the selected secondary amines was always reached, and the respective N-formyl and N-methyl products were obtained in different ratio. The methylation of the N-methylaniline (1b) was achieved with a 92% yield. Instead, the reaction carried out using N-methyl-p-methoxyaniline (1c) or dibenzylamine (1d) led to a lower methylated product yield of 80% and 75% respectively.
Mechanistic insights
To obtain information about the mechanism of the reaction, specific reactivity tests were performed. For this reaction, the mechanism is still under debate and might vary for different catalysts and hydrosilanes. Both Beller and Cantat proposed that the reaction proceeds through an initial formation of silylformate from CO2 and hydrosilane, then formation of N-formyl product by reaction with the amine and finally the N-methyl product derives from a further reduction (Scheme 7, route a).56,57 The N-formyl product can be formed also via reduction of a carbamate salt or via formation of a silyl carbamate.94,95 Alternatively, as reported by Zhang and Wang, the silylformate can react with PhSiH3 to form a silyl methoxide intermediate that is attacked by the nucleophilic amine to yield the desired N-methylamine (Scheme 7, route b).94,96,97 To verify if the N-formyl to N-methyl reduction mechanism (route a) is involved in our case, a test was carried out using N-methyl-formanilide 3a as the substrate under catalytic conditions, showing no reaction (Scheme 8, top). Successively, two NMR scale reactions were carried out under slightly different conditions and using both PhSiH3 and Ph2SiH2 (Scheme 8, bottom). With PhSiH3 almost no reaction was observed, whereas with Ph2SiH2 complete conversion of 3a into 2a was achieved. | ||
| Scheme 7 Supposed reaction routes involved in the N-formylation and N-methylation of amines with CO2 and hydrosilanes according to the literature.56,57,94–97 | ||
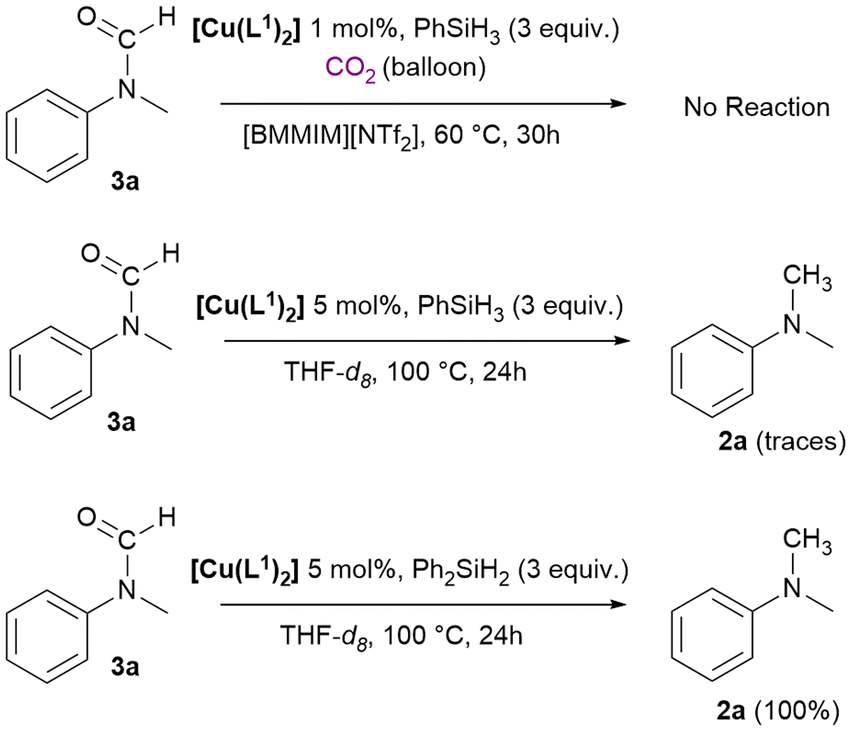 | ||
| Scheme 8 Reduction attempt of N-methylformanilide 3a using the catalyst [Cu(L1)2] with hydrosilanes. | ||
These results suggest that the reaction follows two different mechanisms with the different hydrosilanes, and this is consistent with what was observed in the catalytic study. In fact, using Ph2SiH2 we selectively obtained product 2a (Table 1, entries 8, 10 and 11); if formation of 3a is involved in the mechanism, it might be rapidly converted into 2a when using Ph2SiH2, whereas its conversion does not take place with PhSiH3 and for this reason a mixture of 2a and 3a is observed in the catalytic tests with this silane. We then tried to get information on the nature of the active catalytic species. In this frame, free NHCs are reported to be able of catalysing the N-methylation of amines with CO2.64 To assess whether the reaction mechanism involves the formation of free carbenes, experiments with the commercially available 1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene (IMes) were performed and reported in Table 2. The use of IMes (2 mol%) led to complete conversion of 1a with 92% yield in 2a (Table 2, entry 2) under the used reaction conditions.
| Entry | Catalyst (mol%) | Conv.a/% | 2a /% | 3a /% |
|---|---|---|---|---|
| Reaction conditions: N-ethylaniline 0.40 mmol, PhSiH3 (3 equiv.), p(CO2) = 1 bar (balloon), 60 °C, 16 h, in 1 mL of [BMMIM][NTf2].a Yield determined by 1H NMR using 2,5-dimethylfuran as an internal standard.b IMes = 1,3-Bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene.c Addition of S8 0.05 mol%. | ||||
| 1 | — | 0 | 0 | 0 |
| 2 | IMesb (2) | 100 | 92 | 8 |
| 3c | IMesb (2) | 12 | 3 | 9 |
| 4 | [Cu(L2)2] (1) | 100 | 81 | 19 |
| 5c | [Cu(L2)2] (1) | 100 | 81 | 19 |
An analogous test carried out with the addition of elemental sulphur, resulted in a much lower yield (only 3% of 2a). Sulphur, in fact, can trap the free carbenes, by forming the catalytically inactive thioureas.98 The same tests were carried out using complex [Cu(L2)2] (1 mol%), leading to the same conversions and product yields, with or without the addition of sulphur (Table 2, entries 4 and 5). This result suggests that free carbenes are not the active catalytic species in our system.
Successively, stoichiometric experiments were performed to test the stability of the Cu(II) complexes under reductive conditions. In fact, reductive organic transformations catalysed by Cu–NHC complexes in the presence of hydrosilanes typically involve the formation of Cu(I)–NHC hydride species [Cu(H)(NHC)] as key intermediates.99–101 Dimeric Cu(I) NHC hydride complexes were obtained also treating Cu(II) species in the presence of free NHCs and hydrosilanes.102 Complex [Cu(L2)2] was treated with PhSiH3 in NMR scale experiments in CD3CN. The 1H NMR spectrum of the complex alone did not show signals, as expected for a paramagnetic d9 complex. After the addition of PhSiH3 (4 equiv.) bubbling and precipitation of a dark solid were observed, while the mixture colour shifted from brown to light yellow. Few minutes after the addition, the 1H NMR spectrum showed a complex mixture of signals that evolved to a simpler and stable spectrum after 1 h at 60 °C (Fig. S54†). Two sets of signals were detected, that by comparison with the literature were attributed to two different L ligands.70Via1H–13C HMBC experiments it was possible to detect the carbene carbon signals of these two ligands at 173.1 and 173.4 ppm (Fig. S59†). The presence of dihydrogen in solution was also observed.103 Furthermore, a new singlet at 4.61 ppm was found, attributed to a Si–H group by 1H–29Si HMBC experiments (Fig. S54†). In the corresponding cross-peak in the1H–29Si HMBC experiments, the 29Si signal was found at −197.7 ppm. A similar 29Si chemical shift was recently reported by Bellemin-Laponnaz, Mauro et al., and attributed to a hexacoordinated Si(IV) NHC–O species (Fig. S60†).104 Furthermore, in the 1H–1H NOESY we observed a cross-peak between the N–CH2– protons of the NHC wingtip substituent and the new singlet at 4.61 ppm attributed to the Si–H group (Fig. S57†). All the above considerations are consistent with the formation of a species of formula [PhSiH(L2)2]. The same behaviour was also observed in the reaction between the complex [Cu(L1)2] and PhSiH3, for which the formation of the analogous silicon adduct is probably involved, whereas with [Cu(L1)2] that is much less active in catalysis, the silicon adduct is not observed in the NMR experiments.
Summarizing, the performed experiments seem to indicate that the Cu(II) complexes are reduced in the presence of PhSiH3. However, formation of Cu(I) hydride species typically observed in Cu(I) NHC catalysis seems not to be followed with the hybrid NHC–phenolate ligand system, whereas the formation of a silicon adduct seems to be more likely. Further studies are ongoing to clarify the details of the reaction mechanism.
Conclusions
Four new Cu(II) complexes with hybrid NHC–phenolate ligands were obtained by a convenient synthetic procedure using readily available Cu(OAc)2·H2O as metal precursor under mild reaction conditions. Either bidentate (L1–3) or tetradentate (L4) NHC–phenolate ligands can be used in the complexes preparation of general formula [Cu(L1–3)2] and [Cu(L4)] provided that the two NHC and the two phenolate donors are mutually in trans in the Cu(II) coordination sphere. This restricts the use of tetradentate ligands only to those presenting a long and fluxional linker between the two NHC donors, such as the dodecamethylene used in L4.Complexes [Cu(L1–3)2] and [CuL4] were used in the catalytic reductive N-formylation and N-methylation of amines with CO2 and hydrosilanes, with complex [Cu(L2)2] delivering the best catalytic performance among the series of complexes synthesised in this study. An efficient reaction protocol was developed using the ionic liquid [BMMIM][NTf2] as solvent, 60 °C, 1 bar CO2 and 3 equivalents of PhSiH3. Under these conditions, it was possible to perform the N-methylation of secondary aromatic amines and dibenzylamine in high yield. Reactivity tests indicated that under the adopted conditions the N-methylation of amines follows two different reaction routes with different hydrosilanes. With PhSiH3, N-methylation occurs probably via a silyl methoxide path (route b, Scheme 7), whereas with Ph2SiH2 the stepwise N-formylation to N-methylation is also possible (route a, Scheme 7). The study of the behaviour of the Cu(II) complexes under reductive conditions in the presence of controlled amounts of PhSiH3, revealed that the starting complexes are reduced to diamagnetic species that through NMR studies were assigned to be Si(IV) adducts. Overall, the use of hybrid NHC–phenolate ligands in the reductive catalytic N-methylation on amines with CO2 and hydrosilanes bring to a different reactivity compared to standard NHCs, opening new possibilities to develop innovative catalytic systems. Further studies are ongoing to fully elucidate the reaction mechanism to fully exploit the potential of these systems in catalysis.
Author contributions
Giammarco Meloni: conceptualization, investigation, data curation, writing – original draft. Luca Morgan: investigation. David Cappelletti: investigation. Matteo Bevilacqua: data curation, writing – review & editing. Claudia Graiff: investigation, writing – review & editing. Piermaria Pinter: investigation, writing – original draft. Andrea Biffis: writing – review & editing. Cristina Tubaro: conceptualization, supervision, writing – review & editing. Marco Baron: conceptualization, supervision, writing – original draft, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.† Deposition numbers 2371863 and 2371864† contain the supplementary crystallographic data for compound [Cu(L1)2] and [CuBr(a1)] respectively. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We dedicate this work to our mentor prof. Marino Basato on the occasion of his 80th birthday. This work was supported by the European Union - Next Generation EU - Bando PRIN 2022 – M4.C2.1.1, project 20224PJT7C - MEGs, and by the Department of Chemical Sciences of the University of Padova [P-DiSC#01BIRD2019-UNIPD]. Prof. Paolo Sgarbossa (University of Padova) is kindly acknowledged for the ESI-MS measurements.References
- A. Das, Y. Ren, C. Hessin and M. D.-E. Murr, Beilstein J. Org. Chem., 2020, 16, 858–870 CrossRef CAS PubMed.
- L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- Y. Wang, T. Tang, Y. Yuan, N. Li, X. Wang and J. Guan, ChemMedChem, 2024, 19, e202400060 CrossRef CAS PubMed.
- Y. Han, N. Xie and W. Zhou, Adv. Ther., 2024, 7, 2300305 CrossRef CAS.
- J. R. Lamichhane, E. Osdaghi, F. Behlau, J. Köhl, J. B. Jones and J.-N. Aubertot, Agron. Sustainable Dev., 2018, 38, 28 CrossRef.
- A. J. I. Arduengo, H. V. R. Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405–3409 CrossRef CAS.
- A. H. Hoveyda, Y. Zhou, Y. Shi, M. K. Brown, H. Wu and S. Torker, Angew. Chem., Int. Ed., 2020, 59, 21304–21359 CrossRef CAS PubMed.
- K. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823–7826 CrossRef CAS PubMed.
- J. A. Dabrowski, F. Haeffner and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 7694–7699 CrossRef CAS PubMed.
- F. Gao, J. L. Carr and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 2149–2161 CrossRef CAS PubMed.
- N. W. Mszar, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 3362–3365 CrossRef CAS PubMed.
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC.
- S. Díez-González, E. D. Stevens and S. P. Nolan, Chem. Commun., 2008, 4747–4749 RSC.
- S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2006, 12, 7558–7564 CrossRef PubMed.
- H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157–1160 CrossRef CAS.
- M. R. L. Furst and C. S. J. Cazin, Chem. Commun., 2010, 46, 6924–6925 RSC.
- F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS.
- F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293–294, 48–79 CrossRef CAS.
- J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912–926 RSC.
- O. Santoro, F. Lazreg, D. B. Cordes, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2016, 45, 4970–4973 RSC.
- H. D. Velázquez, Y. R. García, M. Vandichel, A. Madder and F. Verpoort, Org. Biomol. Chem., 2014, 12, 9350–9356 RSC.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- K. M. P. Wheelhouse, R. L. Webster and G. L. Beutner, Organometallics, 2023, 42, 1677–1679 CrossRef CAS.
- M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619–5621 CrossRef CAS.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti and A. Steffen, Chem. Commun., 2016, 52, 2932–2935 RSC.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J.-F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J.-L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678–14691 CrossRef CAS PubMed.
- M. Gernert, U. Müller, M. Haehnel, J. Pflaum and A. Steffen, Chem. – Eur. J., 2017, 23, 2206–2216 CrossRef CAS PubMed.
- J. Föller, C. Ganter, A. Steffen and C. M. Marian, Inorg. Chem., 2019, 58, 5446–5456 CrossRef PubMed.
- C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 4456–4482 RSC.
- X. Hu, I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2003, 125, 12237–12245 CrossRef CAS PubMed.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431–2433 RSC.
- Y. Liu, S. G. Resch, I. Klawitter, G. E. Cutsail III, S. Demeshko, S. Dechert, F. E. Kühn, S. DeBeer and F. Meyer, Angew. Chem., Int. Ed., 2020, 59, 5696–5705 CrossRef CAS PubMed.
- N. Ségaud, J. McMaster, G. van Koten and M. Albrecht, Inorg. Chem., 2019, 58, 16047–16058 CrossRef PubMed.
- B. Liu, B. Liu, Y. Zhou and W. Chen, Organometallics, 2010, 29, 1457–1464 CrossRef CAS.
- B. R. M. Lake and C. E. Willans, Organometallics, 2014, 33, 2027–2038 CrossRef CAS.
- M. Sharma, B. Adhikari, R. F. Awoyemi, A. M. Perkins, A. K. Duckworth, B. Donnadieu, D. O. Wipf, S. L. Stokes and J. P. Emerson, Chemistry, 2022, 4, 560–575 CrossRef CAS PubMed.
- J. Cheng, L. Wang, P. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed.
- E. Jürgens, O. Back, J. J. Mayer, K. Heinze and D. Kunz, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 1011–1018 CrossRef.
- D. J. O'Hearn and R. D. Singer, Organometallics, 2017, 36, 3175–3177 CrossRef.
- M. Sharma, A. M. Perkins, R. F. Awoyemi, A. N. Schmittou, S. Raju, B. S. Pierce, B. Donnadieu, D. O. Wipf, S. L. Stokes and J. P. Emerson, Dalton Trans., 2024, 53, 3180–3190 RSC.
- I. Ahmad Bhat, I. Avinash, S. Kumar Sachan, S. Singh and G. Anantharaman, Eur. J. Inorg. Chem., 2021, 2021, 4560–4565 CrossRef CAS.
- A. O. Larsen, W. Leu, C. N. Oberhuber, J. E. Campbell and A. H. Hoveyda, J. Am. Chem. Soc., 2004, 126, 11130–11131 CrossRef CAS PubMed.
- A. J. Boydston and C. W. Bielawski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2006, 47, 177–178 CAS.
- W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 38, 880–882 CrossRef.
- H. T. Clarke, H. B. Gillespie and S. Z. Weisshaus, J. Am. Chem. Soc., 1933, 55, 4571–4587 CrossRef CAS.
- E. Farkas and C. J. Sunman, J. Org. Chem., 1985, 50, 1110–1112 CrossRef CAS.
- J. R. Harding, J. R. Jones, S.-Y. Lu and R. Wood, Tetrahedron Lett., 2002, 43, 9487–9488 CrossRef CAS.
- V. Goyal, G. Naik, A. Narani, K. Natte and R. V. Jagadeesh, Tetrahedron, 2021, 98, 132414 CrossRef CAS.
- Z. Guo, T. Pang, L. Yan, X. Wei, J. Chao and C. Xi, Green Chem., 2021, 23, 7534–7538 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- D. Wang, W. Lang, W. Wang, Q. Zou, C. Yang, F. Liu and T. Zhao, ACS Omega, 2023, 8, 30640–30645 CrossRef CAS PubMed.
- W. Tarpey, H. Hauptmann, B. M. Tolbert and H. Rapoport, J. Am. Chem. Soc., 1950, 72, 5126–5127 CrossRef CAS.
- W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 38, 880–882 CrossRef.
- O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed.
- C. Masaro, G. Meloni, M. Baron, C. Graiff, C. Tubaro and B. Royo, Chem. – Eur. J., 2023, 29, e202302273 CrossRef CAS PubMed.
- T. Oku, Y. Arita, H. Tsuneki and T. Ikariya, J. Am. Chem. Soc., 2004, 126, 7368–7377 CrossRef CAS PubMed.
- I. Sorribes, K. Junge and M. Beller, Chem. – Eur. J., 2014, 20, 7878–7883 CrossRef CAS PubMed.
- S. Savourey, G. Lefèvre, J.-C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC.
- Y. Li, I. Sorribes, C. Vicent, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 16759–16763 CrossRef CAS PubMed.
- S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS PubMed.
- K. Motokura, N. Takahashi, A. Miyaji, Y. Sakamoto, S. Yamaguchi and T. Baba, Tetrahedron, 2014, 70, 6951–6956 CrossRef CAS.
- Z. Song, J. Liu, S. Xing, X. Shao, J. Li, J. Peng and Y. Bai, Org. Biomol. Chem., 2023, 21, 832–837 RSC.
- S. Zhang, Q. Mei, H. Liu, H. Liu, Z. Zhang and B. Han, RSC Adv., 2016, 6, 32370–32373 RSC.
- C. Qiao, X.-F. Liu, X. Liu and L.-N. He, Org. Lett., 2017, 19, 1490–1493 CrossRef CAS PubMed.
- G. Meloni, L. Beghetto, M. Baron, A. Biffis, P. Sgarbossa, M. Mba, P. Centomo, L. Orian, C. Graiff and C. Tubaro, Mol. Catal., 2023, 538, 113006 CrossRef CAS.
- G. Meloni, M. Bevilacqua, C. Graiff, A. Biffis, M. Baron and C. Tubaro, Inorg. Chim. Acta, 2024, 569, 122096 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- J. D. Rolfes, F. Neese and D. A. Pantazis, J. Comput. Chem., 2020, 41, 1842–1849 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 760–768 CrossRef CAS PubMed.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- Chemcraft – graphical software for visualization of quantum chemistry computations. Version 1.8, build 682. https://www.chemcraftprog.com.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- N. Komiya, A. Yoshida and T. Naota, Inorg. Chem. Commun., 2013, 27, 122–126 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS PubMed.
- I. A. Bhat, I. Avinash and G. Anantharaman, Organometallics, 2019, 38, 1699–1708 CrossRef CAS.
- A. Banerjee, S. Sarkar, J. A. Shah, N. C. Frederiks, E. A. Bazan-Bergamino, C. J. Johnson and M.-Y. Ngai, Angew. Chem., Int. Ed., 2022, 61, e202113841 CrossRef CAS PubMed.
- M. Delgado-Rebollo, C. García-Morales, C. Maya, A. Prieto, A. M. Echavarren and P. J. Pérez, J. Organomet. Chem., 2019, 898, 120856 CrossRef CAS.
- H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545–6548 RSC.
- Z.-Z. Yang, B. Yu, H. Zhang, Y. Zhao, G. Ji and Z. Liu, RSC Adv., 2015, 5, 19613–19619 RSC.
- S. N. Riduan, J. Y. Ying and Y. Zhang, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS PubMed.
- H. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222–3224 RSC.
- O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. J. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC.
- S. Díez-González, E. D. Stevens, N. M. Scott, J. L. Petersen and S. P. Nolan, Chem. – Eur. J., 2008, 14, 158–168 CrossRef PubMed.
- M. Trose, F. Lazreg, T. Chang, F. Nahra, D. B. Cordes, A. M. Z. Slawin and C. S. J. Cazin, ACS Catal., 2017, 7, 238–242 CrossRef CAS.
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS.
- S. Semwal, A. Kumar and J. Choudhury, Catal. Sci. Technol., 2018, 8, 6137–6142 RSC.
- T. Thierry, V. Giuso, F. Polo, P. Mercandelli, Y.-T. Chen, C.-H. Chang, M. Mauro and S. Bellemin-Laponnaz, Dalton Trans., 2024, 53, 6445–6450 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FT-IR, UV-Vis and ESI-MS spectra, SC-XRD and computational details, additional catalytic data, stoichiometric reactions NMR spectra. CCDC 2371863 and 2371864. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02936d |
| This journal is © The Royal Society of Chemistry 2024 |