
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative tungsten centers in polymeric carbon nitride for efficient overall photosynthesis of hydrogen peroxide†
Chengyang
Feng
 a,
Jun
Luo
b,
Cailing
Chen
a,
Shouwei
Zuo
a,
Yuanfu
Ren
a,
Zhi-Peng
Wu
a,
Miao
Hu
a,
Samy
Ould-Chikh
a,
Javier
Ruiz-Martínez
a,
Yu
Han
*a and
Huabin
Zhang
*a
a,
Jun
Luo
b,
Cailing
Chen
a,
Shouwei
Zuo
a,
Yuanfu
Ren
a,
Zhi-Peng
Wu
a,
Miao
Hu
a,
Samy
Ould-Chikh
a,
Javier
Ruiz-Martínez
a,
Yu
Han
*a and
Huabin
Zhang
*a
aKAUST Catalysis Center (KCC), Division of Physical Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia. E-mail: yu.han@kaust.edu.sa; huabin.zhang@kaust.edu.sa
bState Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, MOE Key Laboratory of New Processing Technology for Nonferrous Metals and Materials, School of Resources, Environment and Materials, Guangxi University, Nanning 530004, China
First published on 10th January 2024
Abstract
The artificial photosynthesis of hydrogen peroxide (H2O2) is of great interest, yet simultaneously boosting the oxygen reduction reaction (ORR) while maintaining an efficient water oxidation reaction (WOR) poses a significant challenge. Herein, we present an innovative strategy to construct precisely engineered photocatalysts with adjacent dual active sites, which effectively promote both the ORR and WOR. Specifically, isolated tungsten (W) atoms are strategically positioned next to the triazine rings of polymeric carbon nitride, creating synergistic reactive regions with complementary ORR and WOR activities. Through a combination of experimental investigations and theoretical simulations, we demonstrate that by tuning the isolated W species, the adjacent coordinating triazine units can activate a highly selective two-electron ORR pathway, facilitating the production of H2O2. Simultaneously, the WOR occurs at the dedicated W site, and the in situ generated O2 is rapidly consumed by the adjacent ORR active sites. This interplay between the two active sites is crucial for improving the overall reaction kinetics. The optimized catalyst exhibits exceptional performance, with a high activity of 556 μmol g−1 h−1 for overall H2O2 production with an apparent quantum yield of 8.53% and a remarkable solar-to-chemical conversion efficiency of 0.31%, outperforming most previously reported catalysts under similar conditions. The promising efficiency in H2O2 generation, accomplished through the intricate design of catalysts with cooperative dual sites, broadens the avenue for converting solar energy into valuable chemical products in a sustainable and environmentally friendly manner.
Broader contextHydrogen peroxide (H2O2) is sustainable, resource-efficient, and one of the cleanest chemicals today, and its importance is witnessed by the annual production of 4.5 million tons in 2022 with an annual growth rate of 5.52%. However, this widely used green chemical is currently produced through the energy-intensive and environmentally polluting anthraquinone process. Photocatalytic H2O2 synthesis driven by solar energy is considered to be an energy-saving and environmentally friendly sustainable process; however, most of the previously reported photocatalytic H2O2 synthesis systems only focus on the optimization of the O2 reduction reaction (ORR), resulting in the overall reaction efficiency being limited by the water oxidation reaction (WOR) process. In our contribution, we propose an alternative way to construct photocatalysts with adjacent ORR and WOR sites, which significantly improves the mass transfer efficiency and overall reaction rate. This study opens an attractive avenue for efficient photocatalytic H2O2 production. |
Introduction
Hydrogen peroxide (H2O2) is considered to have a significant energy density (3.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MJl−1 in 60 wt% H2O2), higher than compressed hydrogen gas, making it a promising alternative energy source for hydrocarbon fuels.1–4 However, traditional methods for H2O2 production, such as the anthraquinone method and electrochemical synthesis, face limitations due to their high energy consumption and high toxicity of by-products.5–8 As a result, there is a growing interest in developing green and sustainable technologies for H2O2 production. In recent decades, the photosynthesis of H2O2 has gained growing attention as a clean and carbon-neutral technology for renewable energy production.9–13 The overall process of H2O2 photosynthesis involves two half-reactions: the two-electron oxygen reduction reaction (ORR: O2 + 2H+ + 2e− → H2O2) and the water oxidation reaction (WOR: 2H2O + 4h+ → O2 + 4H+). To enhance the efficiency of H2O2 photosynthesis, it is crucial to design photocatalysts with specific reaction sites that can facilitate both the ORR and WOR pathways.14–18 However, most of the previous studies have primarily focused on optimizing the ORR pathway, while the WOR pathway has been largely ignored.19–21 Although the 2e− ORR has been achieved using various semiconductor photocatalysts with proton donor sacrificial agents, the overall efficiency of H2O2 production remains unsatisfactory.22 In general, the overall photosynthesis of H2O2 is limited by two main factors: the rate of the WOR process and the mass transfer efficiency of oxygen and protons from the WOR site to the ORR site. To overcome these limitations, researchers are exploring the construction of photocatalysts with adjacent ORR and WOR sites. This approach aims to synergistically enhance the overall reaction kinetics and address the bottleneck in H2O2 photosynthesis.
MJl−1 in 60 wt% H2O2), higher than compressed hydrogen gas, making it a promising alternative energy source for hydrocarbon fuels.1–4 However, traditional methods for H2O2 production, such as the anthraquinone method and electrochemical synthesis, face limitations due to their high energy consumption and high toxicity of by-products.5–8 As a result, there is a growing interest in developing green and sustainable technologies for H2O2 production. In recent decades, the photosynthesis of H2O2 has gained growing attention as a clean and carbon-neutral technology for renewable energy production.9–13 The overall process of H2O2 photosynthesis involves two half-reactions: the two-electron oxygen reduction reaction (ORR: O2 + 2H+ + 2e− → H2O2) and the water oxidation reaction (WOR: 2H2O + 4h+ → O2 + 4H+). To enhance the efficiency of H2O2 photosynthesis, it is crucial to design photocatalysts with specific reaction sites that can facilitate both the ORR and WOR pathways.14–18 However, most of the previous studies have primarily focused on optimizing the ORR pathway, while the WOR pathway has been largely ignored.19–21 Although the 2e− ORR has been achieved using various semiconductor photocatalysts with proton donor sacrificial agents, the overall efficiency of H2O2 production remains unsatisfactory.22 In general, the overall photosynthesis of H2O2 is limited by two main factors: the rate of the WOR process and the mass transfer efficiency of oxygen and protons from the WOR site to the ORR site. To overcome these limitations, researchers are exploring the construction of photocatalysts with adjacent ORR and WOR sites. This approach aims to synergistically enhance the overall reaction kinetics and address the bottleneck in H2O2 photosynthesis.
Among various photocatalysts, polymeric carbon nitride (PCN) has shown promise for H2O2 synthesis due to its adjustable two-electron oxygen reduction efficiency and abundant ORR active sites (Fig. S1, ESI†).23–25 However, most PCN-based photocatalysts face limitations in achieving overall H2O2 photosynthesis due to a lack of sufficient WOR sites.26,27 In addition, the severe carrier recombination in PCN materials negatively impacts their photocatalytic activity.28,29 To address these limitations, it is crucial to create WOR sites near the intrinsic ORR sites on PCN and simultaneously improve the separation efficiency of photogenerated carriers. One approach to achieve this is through the introduction of additional isolated active sites with excellent WOR functionality. Single-atom-catalysts (SACs) with precise and adjustable coordination structures have been shown to act as co-catalytic active sites and/or good charge separation centres to improve the catalytic efficiency in most photocatalytic systems.30 Research findings indicate that tungsten (W) stands as a promising candidate for the photocatalytic synthesis of H2O2.31,32 Therefore, it becomes feasible to overcome the limitation of overall H2O2 production in photocatalysis by precisely tuning the W anchoring sites to facilitate the construction of adjacent active regions for the WOR and ORR.
Here, we have developed a W single-atom photocatalyst by introducing well-defined W active sites onto PCN (CNW) for the overall photocatalytic synthesis of H2O2 in pure water under an O2 atmosphere. The presence of monodisperse W species coordinated next to the intrinsic ORR active site of PCN was confirmed through high-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) and X-ray absorption fine structure (XAFS) spectroscopy. Analysis of the band structure showed that the W site serves as a region rich in photogenerated holes, which is advantageous for boosting the charge separation and facilitating the WOR process. By incorporating pairs of adjacent ORR and WOR sites into the photocatalyst, we achieved significant synergistic effects that significantly enhanced the kinetics of H2O2 evolution. This dual-site synergy thus led to an outstanding time yield of 1889.3 μg L−1 h−1 for the overall photosynthesis of H2O2, as well as an apparent quantum efficiency (AQE) yield of 8.53% at 420 nm and a solar-to-chemical conversion (SCC) efficiency of 0.31%. Importantly, the strategy employed to realize overall artificial photosynthetic systems by constructing cooperative dual sites in one matrix holds promise for expanding its applications to other photocatalytic applications.
Results and discussion
The CNW photocatalysts are fabricated by a pre-coordination calcination method. This involves homogeneous blending and recrystallization, resulting in a composite structure where the W atom is coordinated and encapsulated by the melamine precursor (Fig. S2–S5, ESI†). Subsequently, isolated W species are restricted in the catalyst during the calcination process. According to the amount of WCl6 added (0.05, 0.1, 0.2, 0.3, 0.4 or 0.5 mmol), the CNW samples are denoted as CNWx (x = 005, 01, 02, 03, 04 or 05). The formation of isolated W sites is confirmed through HAADF–STEM analysis, where atomically dispersed bright spots (circled) corresponding to the W sites have been observed (Fig. 1a). Further characterizations using high-resolution transmission electron microscopy (HR-TEM) (Fig. 1b), energy-dispersive X-ray spectroscopy (Fig. S6, ESI†), and elemental mapping images (Fig. 1c) confirm the homogeneous distribution of carbon, nitrogen, and tungsten elements in the CNW catalyst. The incorporation of W into the PCN structure can regulate the polymerization process (Fig. S7 and S8, ESI†) and significantly increase the specific surface area and pore volume (Fig. S9, ESI†), which is beneficial for exposing more active sites. However, excessive loading of W can lead to agglomeration and hence inhibit the catalytic activity (Fig. S10, ESI†). The crystalline structure of CNW remains largely unchanged compared to pristine PCN, as evidenced by X-ray diffraction (XRD) patterns (Fig. 1d) and Fourier-transform infrared spectroscopy (FTIR, Fig. S11, ESI†). An increased amount of W doping causes a shift in the diffraction peak, representing the interlayer stacking structure to a smaller angle (Fig. 1e), and indicating that the doping of W single atoms expands the interlayer spacing of PCN. | ||
| Fig. 1 (a) High-magnification HAADF-STEM image of CNW03. (b) HR-TEM image of CNW03. (c) Elemental mapping images of CNW03. (d) XRD patterns and (e) the local contrast XRD patterns around the (002) peak of the PCN and CNW samples. High-resolution XPS spectra of (f) N 1s and (g) C 1s of PCN and CNW03. (h) High-resolution W 4f XPS spectrum of CNW03. | ||
The interaction between the isolated W atoms and the PCN framework is investigated by X-ray photoelectron spectroscopy (XPS) measurements. The introduction of isolated W species results in a noticeable shift in the main characteristic peaks of N and C towards lower binding energies in the XPS spectrum of the CNW03 sample (Fig. 1f and g), which indicates the electron transfer between the isolated W atoms and its surrounding coordination environment. This rearrangement of charge distribution plays a crucial role in promoting carrier separation, which is essential for efficient photocatalytic performance. The XPS peaks representing W 4f are detected (Fig. 1h), further confirming the successful loading of W onto the CNW photocatalyst. However, no Cl signals can be detected, indicating the successful removal of chlorine elements after the calcination process (Fig. S12, ESI†).
To gain further insights into the local atomic structure and coordination sphere of the isolated W atoms in CNW, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS) have been conducted (Fig. S13, ESI†).33 The oxidation state of the W atoms is determined by the XANES technique (Fig. 2a and b), showing a valence state of about +4.2 for the W element in CNW03, which has the potential to serve as a WOR active site. The k2-weighted Fourier transform EXAFS spectrum obtained from the W LIII-edge of CNW03 in K space shows only one peak at around 1.73Å, with no W–W bond at 2.62Å observed, suggesting that the W atoms in CNW03 are atomically dispersed (Fig. 2c). The Fourier-transform EXAFS curves and fitting results indicate that W atoms have a coordination number of 4.1 for the W–N path in the first shell (Fig. 2d, e and Table S1, ESI†). In order to obtain a more accurate coordination structure, we performed a peak-splitting analysis on the signals in the R and K spaces. As shown in Fig. 2f, two similar W–N coordination signals can be obtained in the first shell, which correspond to the edge N sites of adjacent triazine units in the model. Moreover, only the W–C structure is obtained in the signal of the second shell, which is highly consistent with the DFT fitting result (Fig. S14, ESI†). Furthermore, wavelet transform EXAFS (WT-EXAFS) spectra show similar contour plots for CNW03, WO2 and WO3, with only one intensity maximum (Fig. 2g). These results further confirm the formation of atomically dispersed W atoms with W–N coordination in CNW03.
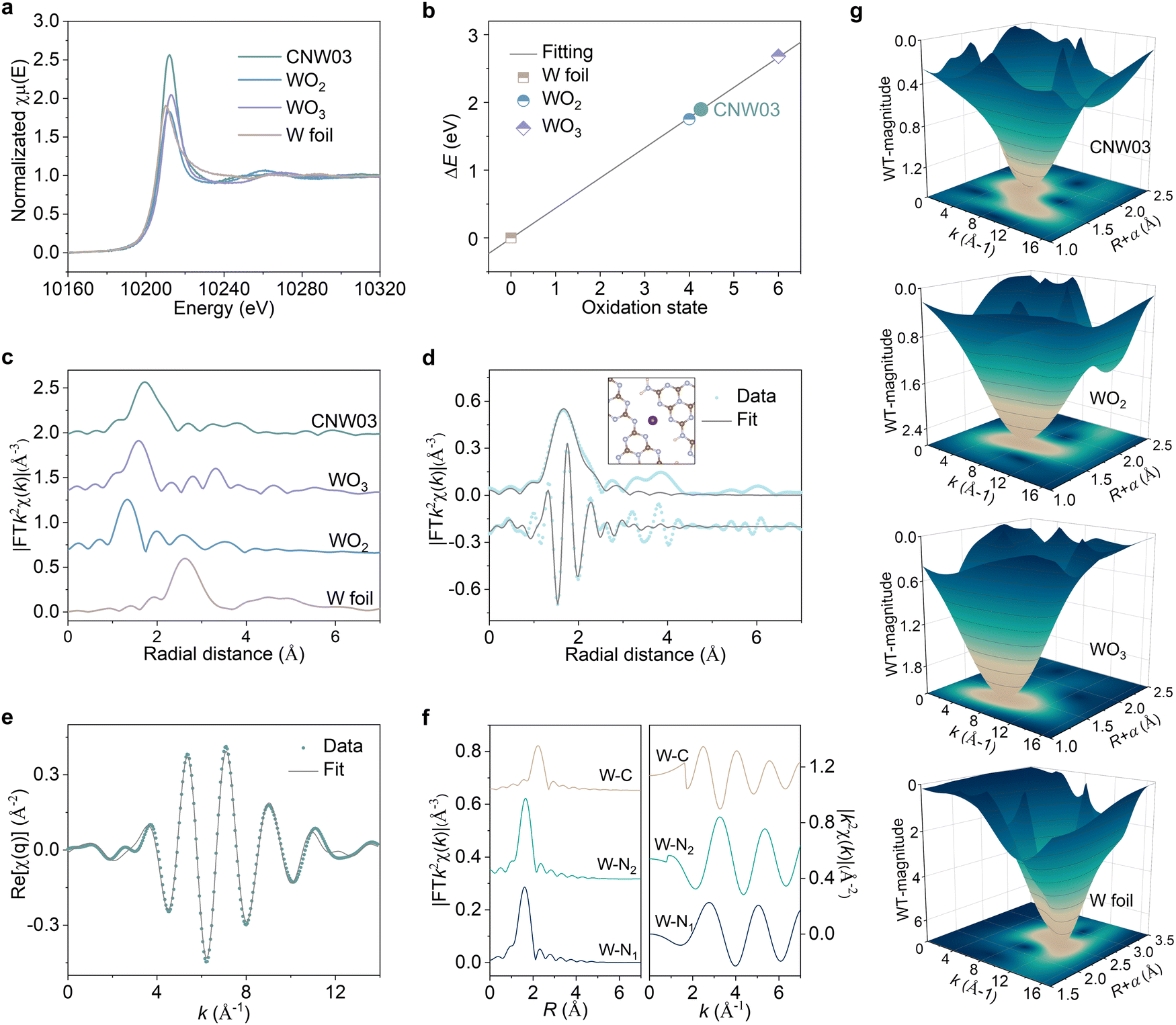 | ||
| Fig. 2 (a) W L-edge XANES experimental spectra of CNW03, WO2, WO3 and W foil. (b) The corresponding first derivative of the absorbing edge. (c) W L-edge Fourier-transformed EXAFS spectra of CNW03, WO2, WO3 and W foil. Fitting of the EXAFS data of the CNW03 based on the model obtained from DFT simulation in (d) R space and (e) K space. (f) W L-edge EXAFS analysis of CNW03 in R (left) and K (right) species: curves from bottom to top are first shell (W–N1, W–N2) and second shell (W–C) signals split from the total signal. (g) WT for the k3-weighted EXAFS signal of CNW03, WO2, WO3 and W foil. | ||
The performance of photocatalytic H2O2 production is evaluated through titration in pure water under visible light irradiation (λ > 420 nm) with continuous bubbling of O2 (Fig. S15 and S16, ESI†).34–36Fig. 3a shows that all W-loaded CNW photocatalysts display higher H2O2 production activities compared to the pristine PCN photocatalyst (319.0 μg L−1 h−1). The maximized H2O2 production rate of 1889.3 μg L−1 h−1 is achieved on CNW03, which is approximately 5.9 times higher than that of pristine PCN. The W loading contained in CNW03 is confirmed to be 1.18 wt% (Fig. S17, ESI†). The accumulation of H2O2 is determined by the competitive formation (Kf) and decomposition (Kd) kinetics of H2O2. By assuming zero-order kinetics for H2O2 formation and a first-order for H2O2 decomposition, the value of Kf and Kd constants can be obtained (Fig. S18, ESI†). As shown in Fig. 3b, CNW03 exhibited 4.4 times higher H2O2 generation rate and 1/3 the H2O2 decomposition rate compared to CN, indicating that CNW03 not only possesses enhanced catalytic activity but also exhibits higher stability towards the formation of H2O2.
 | ||
| Fig. 3 (a) Photocatalytic H2O2 production on pristine PCN and CNW samples. (b) The corresponding fitted formation rate constants (Kf) and decomposition rate constants (Kd). (c) Photocatalytic H2O2 production in an oxygen-free environment with an electron acceptor as the reaction trigger under a 1 bar Ar atmosphere. (d) Photocatalytic O2 evolution and H2O2 production over the CNW03 sample with an electron acceptor under a 0.6 bar Ar atmosphere. (e) AQE of CNW03 as a function of wavelength. (f) SCC efficiency of CNW03 in pure water. (g) Activity comparison of CNW03 with reported state-of-the-art photocatalysts. (h) Cycling stability of the CNW03 catalyst. (i) Long timescale H2O2 production over the CNW03 catalyst in flow mode and (j) the schematic diagram of the corresponding continuous-flow apparatus. | ||
To quantitatively investigate the synergy between the WOR and ORR, photocatalytic H2O2 production experiments in an O2-free environment have been conducted with an electron acceptor as the reaction trigger. As shown in Fig. 3c, PCN exhibits almost no H2O2 production activity, while H2O2 can be detected in the CNW03 system. However, in a low-pressure reactor setting, the system yielded O2 instead of H2O2 during an identical experiment (Fig. 3d). This observation underscores that the water oxidation reaction (WOR) process facilitated by CNW03 results in the generation of O2. Following this, the produced O2 is promptly sequestered by the neighboring oxygen reduction reaction (ORR) site under normal pressure, ultimately undergoing conversion into H2O2. The conducted photocatalytic oxygen evolution experiment establishes that W functions as a potential active site for the WOR process (Fig. S19, ESI†). Furthermore, the evaluation of the oxygen reduction half-reaction on CNW03 was carried out in a 10% isopropanol (IPA) aqueous solution. As depicted in Fig. S20 (ESI†), the oxygen reduction reaction (ORR) activity of CNW03 surpasses that of PCN, signifying that the incorporation of the W site exerts a positive influence on the active area for the ORR on the triazine unit. Moreover, the hydrogen evolution test eliminates the impact of potential competing reactions on the ORR activity (Fig. S21, ESI†). Comparing the promotion levels of the overall reaction, reduction half-reaction, and oxidation half-reaction, it can be confirmed that the significant enhancement of W species in the WOR reaction and the optimization of the active area for the ORR are key factors in improving the overall reaction efficiency.
After optimizing the reaction conditions, the apparent quantum yield (AQY) of CNW03 for H2O2 photosynthesis has been measured under monochromatic light irradiation (Fig. S22, ESI†). The AQYs of CNW03 at 420, 450, 485, and 535 nm are determined to be 8.53%, 5.73%, 2.45%, and 0.63%, respectively (Fig. 3e). These values are higher than those reported for most photocatalysts under similar reaction conditions (Table S2, ESI†). More importantly, the SCC efficiency of CNW03 reaches an impressive value of 0.31% (Fig. 3f), surpassing the SCC of plant natural photosynthesis (0.1%).37 In comparison with other state-of-the-art photocatalysts, the AQY and SCC of CNW03 outperform the majority (Fig. 3g), highlighting the superiority of the strategy employed to construct adjacent dual active sites. In addition to its high activity, CNW03 demonstrates excellent stability, showing no significant loss of catalytic activity after five consecutive photocatalytic runs (Fig. 3h). Based on the obtained high activity and stability of CNW03, we further developed a continuous-flow reaction process, where a flow of O2-saturated water (10 mL h−1) is passed through a packed layer of CNW03 catalyst under irradiation (Fig. 3j and Fig. S23, ESI†). It can be observed that the CNW03 catalyst can maintain a stable and efficient H2O2 production efficiency for the duration of the 60-hour experiment (Fig. 3i), demonstrating the great promise of CNW03 in practical applications. The characteristics of CNW03 after continuous reaction for four cycles remain almost the same as the fresh samples (Fig. S24, ESI†), confirming the catalyst's excellent stability.
The electronic band structure of the as-prepared catalysts was first investigated to analyze the photoexcitation behavior. Fig. 4a displays the UV-vis diffuse reflectance spectra (DRS) and calculated band gaps for pristine PCN and CNW samples, revealing the enhanced optical absorption of the CNW samples in the 250–450 nm range due to improved π–π* transitions.38,39 Additionally, a new absorbance band appears in the 450–600nm region, which can be attributed to the excitation of unpaired electrons from W4+ sites (n–π* transition).38,40 The band gaps of the samples, determined from the transformed Kubelka–Munk function, progressively narrow from 2.77 to 2.59 eV, with CNW03 exhibiting a band gap of 2.66 eV. To explain the narrower band gap, valence band (VB) XPS spectra were collected (Fig. S25, ESI†).41 The VB maxima of PCN and CNW03 were determined to be 1.51 and 1.37 eV, respectively. The conduction band (CB) potentials of PCN and CNW03 were calculated to be −1.26 and −1.29V (vs. NHE), respectively, indicating the thermodynamic feasibility of O2 reduction to H2O2 by CB electrons (Fig. 4b). The band structure of the as-prepared samples was further revealed by Mott–Schottky plots. All samples exhibit positive slopes, suggesting their n-type semiconductor characteristics (Fig. S26, ESI†). Moreover, CNW03 shows a flat-band potential of −0.82V, which is a 0.04eV downshift compared to that of PCN (−0.86V). This trend is consistent with the calculated CB potential above (Fig. 4b). The upshifted CB level in CNW03 facilitates the generation of photoexcited electrons with stronger reduction ability, providing the initial driving force for the overall solar energy conversion process.
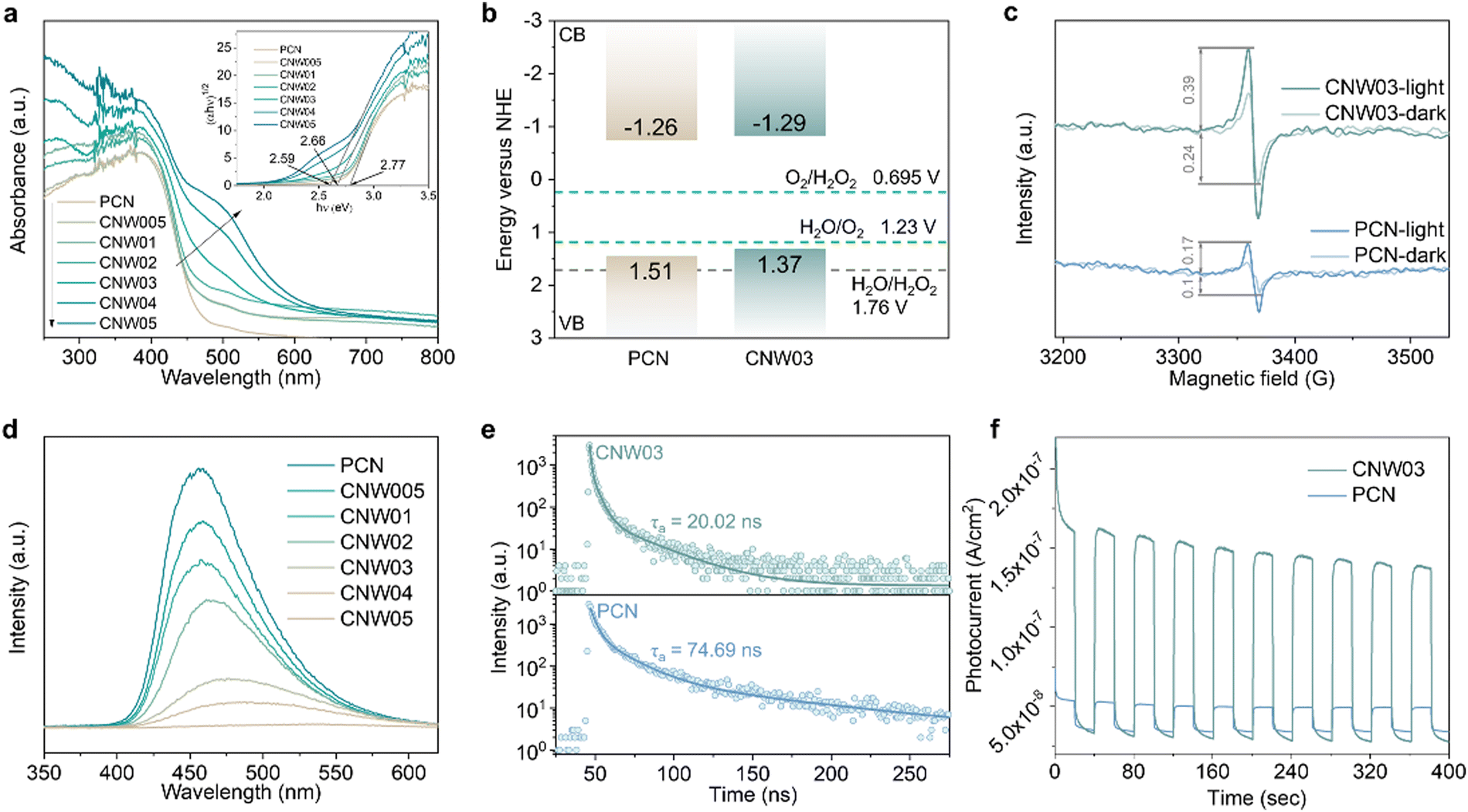 | ||
| Fig. 4 (a) Ultraviolet-visible spectra of PCN and CNW samples (inset: corresponding Tauc plots). (b) Energy band diagrams of PCN and CNW03. (c) EPR signals of PCN and CNW03 under dark and light conditions. (d) Photoluminescence spectra of PCN and CNW samples. (e) Time-resolved photoluminescence spectra of PCN and CNW03. (f) Photocurrent responses of PCN and CNW03. | ||
Furthermore, the influence of isolated W species on the generation, separation, and transfer of photoexcited charge carriers has been investigated using electron paramagnetic resonance (EPR), photoluminescence analysis (PL), and photoelectrochemical techniques. Under visible light irradiation, CNW03 displays a stronger EPR signal amplification compared to pristine PCN (Fig. 4c), indicating its superior ability to generate photogenerated electrons.42 PL analysis of pristine PCN under visible-light irradiation (λ = 400 nm) reveals an intense fluorescence signal (Fig. 4d), which gradually decreases in intensity and exhibits a redshift with increasing W loading content. This substantiates that the presence of isolated W species is highly beneficial for the separation of photoexcited charge carriers in CNW. The dynamic change in charge transfer induced by isolated W species is further elucidated by time-resolved PL spectra (Fig. 4e and Table S3, ESI†). Fitting the data with a tri-exponential equation, it is observed that CNW03 exhibits a much shorter average lifetime of charge carriers (20.02 ns) compared to pristine PCN (74.69 ns), implying more efficient charge transfer.43,44 These findings are further corroborated by photoelectrochemical tests. CNW03 exhibits a smaller arc radius in the electrochemical impedance spectroscopy plot (Fig. S27, ESI†) and a significantly enhanced photocurrent response (Fig. 4f), providing further confirmation of the boosting effect of the isolated W species on the excitation, separation, and transfer of photoexcited charge carriers.45,46
To gain insight into the high activity and selectivity of H2O2 production over the CNW photocatalyst, the reaction mechanism has been investigated. Control experiments were conducted to understand the essential factors contributing to H2O2 production. As displayed in Fig. 5a, replacing the feed gas (O2) with air or argon significantly decreases the H2O2 yield, and under vacuum conditions, the H2O2 production is nearly undetectable. These results indicate that dissolved oxygen is a necessary feedstock for H2O2 production. Additionally, the addition of the superoxide radical quencher (1,4-benzoquinone, BQ) effectively inhibits the progress of the reaction, confirming the involvement of a two-step two-electron oxygen reduction reaction with *OOH as an intermediate (O2 + e− + H+ → *OOH; *OOH + e− + H+ → H2O2). The presence of *OOH is further confirmed by in situ EPR measurements. CNW03 exhibits an enhanced DMPO–*OOH signal compared to pristine PCN (Fig. 5b), indicating that the molecular oxygen activation is greatly promoted upon loading isolated W species. To further understand the oxygen reduction mechanism, a rotating ring-disk electrode (RRDE) test has been performed to investigate the number of electrons (n) transferred and the H2O2 selectivity in the oxygen reduction process (Fig. S28 and S29, ESI†). Fig. 5c shows the disk current for oxygen reduction and the ring current for H2O2 oxidation in an O2-saturated 0.1 M KOH solution under visible light irradiation. The estimated value of n CNW03 is closer to 2 compared to PCN, suggesting that the CNW03 catalyst has a higher selectivity of the two-electron oxygen reduction pathway (Fig. 5d). The H2O2 selectivity of PCN and CNW03 is calculated to be approximately 65% and over 90%, respectively, further confirming that the two-electron oxygen reduction dominates on CNW03.
 | ||
| Fig. 5 (a) Photocatalytic H2O2 production over CNW03 with different reaction gases or sacrificial agents. (b) In situ EPR signals of DMPO–*OOH over PCN and CNW03 under visible light irradiation (λ > 420nm, irradiation time: 0 min, 5 min and 10 min). (c) RRDE voltammograms of PCN and CNW03 in O2-saturated solution. (d) Percentage of peroxide and the electron transfer numbers (inset) of PCN and CNW03 at various potentials. (e) Raman spectra for PCN and CNW03 recorded after 2 h of visible light irradiation in an O2-saturated propanol/water mixture. (i) PCN before irradiation, (ii) PCN after irradiation, (iii) CNW03 before irradiation, and (iv) CNW03 after irradiation. In situ DRIFT spectra of O2 photocatalytic reduction over (f) CNW03 and (g) PCN. (h) GCMS spectra of O2 transformed from H2O2 formed by H216O + 18O2 (upper) or H218O (lower). | ||
To provide further evidence for the formation of the intermediate species, Raman spectroscopy measurements have been conducted (Fig. S30a, ESI†). Both PCN and CNW03 exhibit two bands at 712 and 989 cm−1 before light irradiation (Fig. 5e), corresponding to the breathing modes of the triazine ring.47 After light irradiation, a new band appears at 896 cm−1, which is assigned to the C–O vibration and O–O stretching modes of the endoperoxide formed on the triazine ring.26 Notably, CNW03 exhibits an additional band at 851 cm−1, which can be attributed to the O–O stretching mode of *OOH species and is not observed in PCN. This observation suggests that the presence of isolated W species enhances the conversion of endoperoxide to *OOH, thereby promoting the formation of H2O2.24 To further confirm the O2 conversion process, in situ diffuse-reflectance infrared Fourier transform spectroscopy (DRIFT) has been employed to investigate the adsorption behaviour of *OOH and H2O2 on CNW03 and PCN (Fig. S30b, ESI†). As displayed in Fig. 5f, a prominent band at approximately 1230 cm−1, attributed to the O–O stretching mode of surface-adsorbed *OOH (OOHad), and a weaker band at around 1390 cm−1, corresponding to the OOH bending mode of surface-adsorbed *H2O2 (HOOHad) emerge and increase with the reaction time for CNW03.48,49 In comparison, pristine PCN exhibits a weaker OOHad band, and the HOOHad band is barely detectable (Fig. 5g), which suggests that H2O2 may be unstable in CN and prone to dissociation, aligning with our earlier fitting results (Fig. 3b). These findings substantiate that isolated W species can significantly promote the generation of the *OOH intermediate on the carbon nitride framework, thereby enhancing the yield of two-electron oxygen reduction to H2O2.
Subsequently, isotope-controlled experiments have been performed to elucidate the reaction pathway (Fig. 5h). In one set of experiments, a reaction mixture containing H216O solution saturated with 18O2 is used as the reactant (Fig. S31, ESI†). The gaseous product obtained after the reaction exhibited both an 18O2 (m/z = 36) peak and a 16O2 (m/z = 32) peak, providing evidence that the generated H2O2 originates from the dissolved O2. Although a 16O2 signal is also detected, the presence of atmospheric 16O2 cannot be completely ruled out as a potential source of error. To overcome this limitation, an ingenious condition-controlled experiment is designed using H218O as the reactant with NaIO3 as the electron acceptor (Fig. S32, ESI†).50,51 In this experimental setup, H2O2 can only be produced through the reduction of 18O2 generated from water oxidation. Notably, a distinct 18O2 signal is detected in this system, indicating that the O2 generated from water oxidation in the CNW03 system has been rapidly consumed by the two-electron oxygen reduction process to produce H2O2.
Density functional theory (DFT) calculations have been performed to gain further insight into the mechanism and the high selectivity of the two-electron route in the photocatalytic production of H2O2 on the CNW catalysts. The fully relaxed adsorption model shows two different O2 adsorption states on PCN and CNW, respectively. On pristine PCN, O2 is found to preferentially adsorb at the 1,4-sites, which is consistent with previous studies.35 However, upon coordination of the W atom with the triazine unit, the adsorption mode shifts to the 1,2-sites (Fig. S33 and 34, ESI†). Analysis of the charge density map reveals that O2 adsorbed on pristine PCN exhibits weak interaction with the 1,4-sites, resulting in inefficient activation (Fig. 6a). In contrast, O2 adsorbed on CNW displays a strong interaction with the 1,4 sites of the triazine unit (Fig. 6b), which facilitates a lower reaction energy barrier. This change in adsorption mode implies that the presence of W species regulates the electronic structure of the adjacent triazine unit, leading to a decrease in adsorption energy (Fig. 6c) and an enhancement of charge transfer (Fig. 6d). The isolated W species not only promote localized charge separation but also facilitate charge transfer from the triazine ring to O2, effectively promoting O2 activation. Furthermore, the energy profile of the oxygen reduction process is depicted based on the calculated Gibbs free energies of the proposed reaction steps (Fig. 6e). The results demonstrate that the process of O2 extracting the H atom to form *OOH on the CNW surface has a much lower energy of –4.909 eV compared to –3.637 eV on the pristine PCN surface. This indicates that the presence of W species facilitates the formation of the primary product *OOH. The conversion of *OOH to H2O2 is a crucial step that determines the selectivity of the reaction. In Fig. 6e, it can be observed that the transition state of *OOH to H2O2 has a significantly lower energy barrier on the CNW surface (0.371 eV) compared to pristine PCN (1.209 eV). This indicates that the coordination of the triazine unit with the isolated W species effectively promotes the conversion of *OOH to H2O2 through the two-electron pathway and inhibits the formation of H2O. This feature greatly improves the overall selectivity of the photocatalytic process. Furthermore, upon separation of photogenerated electron–hole pairs at the isolated W sites, the photogenerated holes accumulate on the W species and participate in the oxidizing of water (Fig. S35, ESI†). This process is crucial for lowering the overall reaction barrier and enhancing the photocatalytic production of H2O2. Moreover, CNW03 displays a higher absolute value of zeta potential compared to PCN (Fig. S36, ESI†), indicating a stronger ability for H2O dissociation. This characteristic is beneficial for the oxidation of H2O. Thus, the calculations of the Gibbs free energy change for the dissociation of H2O molecules and the release of protons on PCN and CNW are pursued (Fig. 6f). These computations affirm that the presence of the W site effectively diminishes the energy barrier associated with the water oxidation process, which is expected to expedite the dissociation of H2O and facilitate the provision of protons, thereby promoting the direct reduction of O2 to H2O2. Based on the above results, a mechanism for the photocatalytic synthesis of H2O2 over the adjacent triazine units and isolated W species via the two-electron ORR and four-electron WOR pathways are proposed. Firstly, under light irradiation, photogenerated electrons migrate from the W sites to the adjacent triazine units, promoting charge separation. The photogenerated electrons and holes accumulate on the triazine unit and W species, respectively, creating adjacent active regions for the ORR and the WOR reactions. Then, H2O undergoes in situ dissociation on the isolated W sites, producing O2 and H+ species. Simultaneously, O2 adsorbed on the triazine unit reacts with H+ species to produce H2O2 through the two-electron ORR pathway (Fig. 6g). It is important to note that due to the proximity of the active sites, the in situ formed O2via WOR can rapidly migrate to the triazine unit, replenishing the consumed O2 and boosting the overall reaction kinetics (Fig. S37, ESI†).
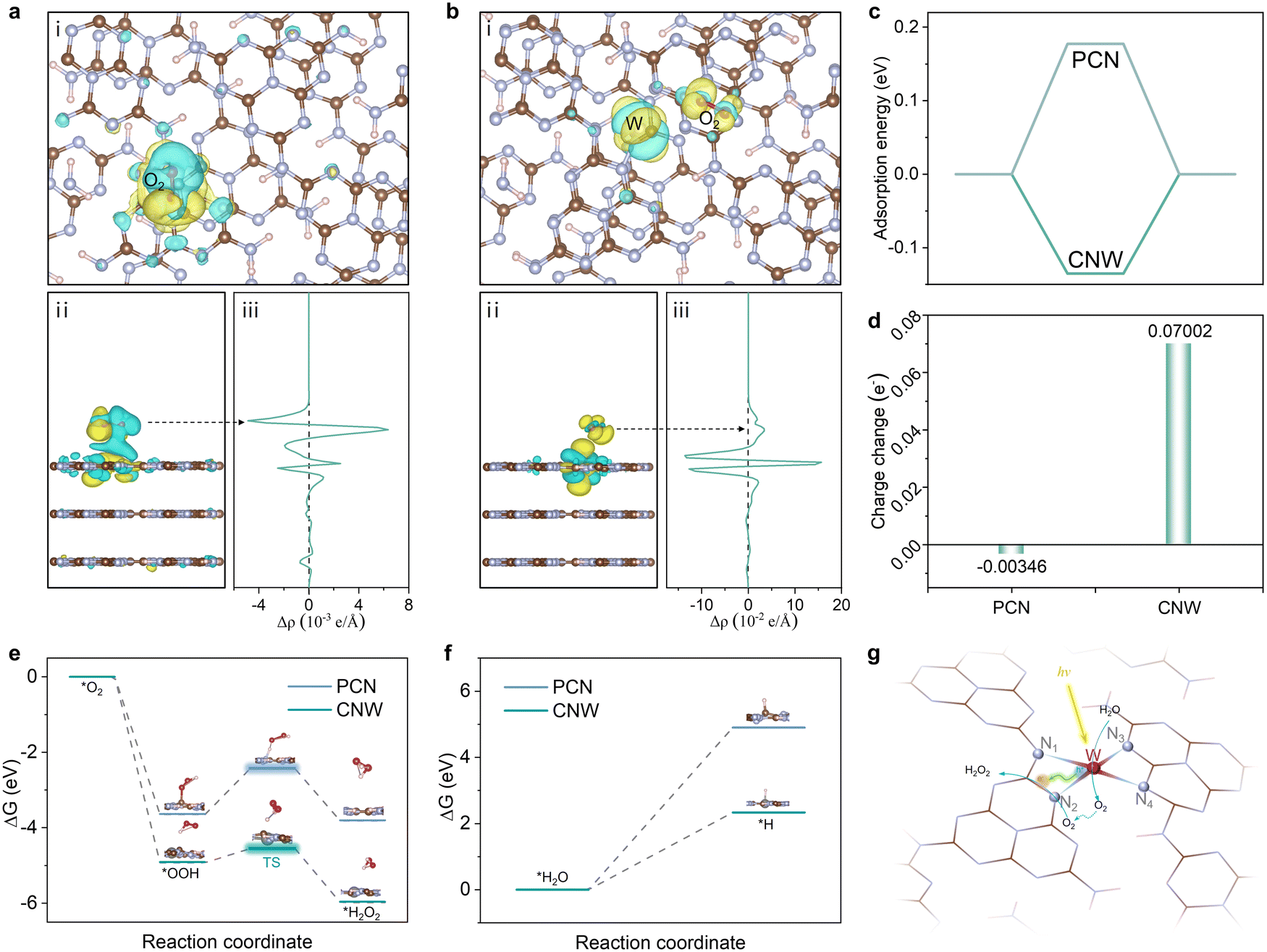 | ||
| Fig. 6 Charge density distribution of O2 molecules absorbed on (a) PCN and (b) CNW. (i) Top view, (ii) side view, and (iii) average charge density along the z axis. (c) Calculated adsorption energies of O2 molecules on PCN and CNW. (d) Calculated charge changes of O2 molecules absorbed on PCN and CNW. (e) Energy profiles of O2 reduction to H2O2 on PCN and CNW. (f) Energy profiles of H2O dehydrogenation to *H on PCN and CNW. (g) Proposed mechanism of photocatalytic H2O2 production over adjacent dual active sites on the CNW photocatalyst. | ||
Conclusions
In summary, this work introduces an innovative approach by anchoring W-N4 atomic sites on carbon nitride photocatalysts. This unique configuration exhibits remarkable properties in terms of charge excitation and migration, enabling highly efficient photosynthesis of H2O2. The combination of experimental characterizations and theoretical simulations collectively indicates that the collaborative effect between the isolated W sites and the support plays a crucial role in promoting both oxidation and reduction half-reactions, resulting in enhanced overall reaction kinetics. Under light irradiation, the W sites effectively generate and harness photogenerated electrons, optimizing the charge density and distribution of adjacent triazine units. This optimization facilitates the reduction of O2 on the triazine units, while the accumulated photogenerated holes on the W sites expedite the water oxidation kinetics, ultimately enhancing the overall H2O2 production. This work not only provides a rational catalyst design strategy, but also offers a profound understanding of the underlying mechanisms. Consequently, our findings pave the way for designing advanced SACs for various photocatalytic reactions in energy conversion and sustainable production.Author contributions
C. F. Y. H. and H. Z. constructed and planned the whole project. C. F. and Z. W. carried out the synthesis of the samples and photocatalytic experiments. J. L. conducted the DFT calculations. S. Z. and S. O.-C. carried out the XANES and EXAFS characterizations under the guidance of J. R.-M. C. C. and Y. R. performed electron microscope imaging. M. H. performed the EPR tests. C. F. wrote the manuscript. H. Z. reviewed and edited the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work received financial support from King Abdullah University of Science and Technology (KAUST).References
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- K. P. Bryliakov, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley and A. Y. Borisevich, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- N. M. Wilson and D. W. Flaherty, J. Am. Chem. Soc., 2016, 138, 574–586 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Colic, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park and K.-S. Lee, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao and M. Shao, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed.
- A. Gopakumar, P. Ren, J. Chen, B. V. Manzolli Rodrigues, H. Vincent Ching, A. Jaworski, S. V. Doorslaer, A. Rokicińska, P. Kuśtrowski and G. Barcaro, J. Am. Chem. Soc., 2022, 144, 2603–2613 CrossRef CAS PubMed.
- Y. Zhao, P. Zhang, Z. Yang, L. Li, J. Gao, S. Chen, T. Xie, C. Diao, S. Xi and B. Xiao, Nat. Commun., 2021, 12, 3701 CrossRef CAS PubMed.
- T. Weller, J. Timm, L. Deilmann, T. S. Doerr, C. Greve, A. S. Cherevan, P. A. Beaucage, U. B. Wiesner, E. M. Herzig and D. Eder, Small Struct., 2023, 2200184 CrossRef CAS.
- K. He, E. Campbell, Z. Huang, R. Shen, Q. Li, S. Zhang, Y. L. Zhong, P. Zhang and X. Li, Small Struct., 2022, 3, 2200104 CrossRef CAS.
- J. Luo, Y. Liu, C. Fan, L. Tang, S. Yang, M. Liu, M. Wang, C. Feng, X. Ouyang and L. Wang, ACS Catal., 2021, 11, 11440–11450 CrossRef CAS.
- J. Luo, C. Feng, C. Fan, L. Tang, Y. Liu, Z. Gong, T. Wu, X. Zhen, H. Feng and M. Yan, J. Catal., 2022, 413, 1132–1145 CrossRef CAS.
- C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich and E. Stavitski, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6376–6382 CrossRef CAS PubMed.
- C. Feng, Z. P. Wu, K. W. Huang, J. Ye and H. Zhang, Adv. Mater., 2022, 34, 2200180 CrossRef CAS PubMed.
- W. Wang, Q. Song, Q. Luo, L. Li, X. Huo, S. Chen, J. Li, Y. Li, S. Shi and Y. Yuan, Nat. Commun., 2023, 14, 2493 CrossRef CAS PubMed.
- W. Fan, B. Zhang, X. Wang, W. Ma, D. Li, Z. Wang, M. Dupuis, J. Shi, S. Liao and C. Li, Energy Environ. Sci., 2020, 13, 238–245 RSC.
- P. Zhang, Y. Tong, Y. Liu, J. J. M. Vequizo, H. Sun, C. Yang, A. Yamakata, F. Fan, W. Lin and X. Wang, Angew. Chem., Int. Ed., 2020, 132, 16343–16351 CrossRef.
- C. Feng, L. Tang, Y. Deng, J. Wang, J. Luo, Y. Liu, X. Ouyang, H. Yang, J. Yu and J. Wang, Adv. Funct. Mater., 2020, 30, 2001922 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058–3066 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu, X. Ouyang, H. Yang, J. Yu and J. Wang, Appl. Catal., B, 2021, 281, 119539 CrossRef CAS.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 7021–7029 CrossRef CAS.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang and X. Zhou, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- L. Chen, C. Chen, Z. Yang, S. Li, C. Chu and B. Chen, Adv. Funct. Mater., 2021, 31, 2105731 CrossRef CAS.
- Z.-H. Xue, D. Luan, H. Zhang and X. W. D. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- C. Zou, W. Ahmad, L. Yan, C. Qian, M. Ling, L. Zheng and S. Zhou, Cell Rep. Phys. Sci., 2023, 4, 101288 CrossRef CAS.
- E. P. Alsaç, K. Zhou, W. Rong, S. Salamon, J. Landers, H. Wende and R. D. Smith, Energy Environ. Sci., 2022, 15, 2638–2652 RSC.
- D. Ryoo, X. Xu, Y. Li, J. A. Tang, J. Zhang, P. C. Van Zijl and G. Liu, Anal. Chem., 2017, 89, 7758–7764 CrossRef CAS PubMed.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 129, 13630–13634 CrossRef.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- Y. Duan, Y. Wang, L. Gan, J. Meng, Y. Feng, K. Wang, K. Zhou, C. Wang, X. Han and X. Zhou, Adv. Energy Mater., 2021, 11, 2004001 CrossRef CAS.
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang and J. Wang, Nat. Commun., 2021, 12, 4412 CrossRef CAS PubMed.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- J. Chen, C. L. Dong, D. Zhao, Y. C. Huang, X. Wang, L. Samad, L. Dang, M. Shearer, S. Shen and L. Guo, Adv. Mater., 2017, 29, 1606198 CrossRef PubMed.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- S. Nayak, I. J. McPherson and K. A. Vincent, Angew. Chem., Int. Ed., 2018, 130, 13037–13040 CrossRef.
- R. Li, H. Han, F. Zhang, D. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369–1376 RSC.
- S. Ye, R. Chen, Y. Xu, F. Fan, P. Du, F. Zhang, X. Zong, T. Chen, Y. Qi and P. Chen, J. Catal., 2016, 338, 168–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information accompanying this paper is available online. See DOI: https://doi.org/10.1039/d3ee03032f |
| This journal is © The Royal Society of Chemistry 2024 |