
Modulating the electronic structure of Ni(OH)2 by coupling with low-content Pt for boosting the urea oxidation reaction enables significantly promoted energy-saving hydrogen production†
Mengxiao
Zhong
a,
Meijiao
Xu
a,
Siyu
Ren
a,
Weimo
Li
a,
Ce
Wang
 a,
Mingbin
Gao
*b and
Xiaofeng
Lu
*a
a,
Mingbin
Gao
*b and
Xiaofeng
Lu
*a
aAlan G. MacDiarmid Institute, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: xflu@jlu.edu.cn
bNational Engineering Research Center of Lower-Carbon Catalysis Technology, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: mbgao@dicp.ac.cn
First published on 30th January 2024
Abstract
Replacing the high-potential oxygen evolution reaction (OER) by the low-potential nucleophile oxidation reaction (NOR) is an essential way to further promote the production rate of hydrogen during water electrolysis. Here, low-content Pt-anchored Ni(OH)2 on the surface of nickel-carbon nanofibers (Ni-CNFs) are prepared via a simple electrospinning–electrodeposition strategy to achieve the alkaline urea oxidation reaction (UOR) electrocatalysis, and they are optimized by controlling different concentrations of deposition solutions during the electrodeposition process. Owing to the Pt loading on the Ni(OH)2 surface, the dehydrogenation processes of both Ni(OH)2 to Ni(OH)O and urea molecules to N2 and CO2 are promoted, leading to a greatly enhanced UOR performance with low potentials of 1.363 and 1.422 V vs. RHE at current densities of 10 and 100 mA cm−2 for the optimized Pt–Ni(OH)2@Ni-CNFs-2 catalyst, which is significantly better than many reported representative Ni-based catalysts. Moreover, considering the superior hydrogen evolution reaction (HER) activity of the optimized Pt@Ni-CNFs-2 catalyst through a similar synthetic procedure, a home-made urea-assisted water splitting device is constructed with the Pt–Ni(OH)2@Ni-CNFs-2 and Pt@Ni-CNFs-2 catalysts serving as the anode and cathode, respectively, which exhibits a voltage of 1.40 V at 10 mA cm−2, surpassing many reported representative urea-assisted water splitting electrolyzers. The as-fabricated electrolyzer displays a significantly promoted H2 production rate of 10-fold that of the overall water splitting, demonstrating the prominent prospects of our catalysts in urea-assisted water splitting for energy-saving H2 generation.
Broader contextHydrogen has been considered an ideal energy carrier because of its favorable gravimetric energy density, zero contamination and exceptional recyclability. Therefore, the green and environmentally friendly strategy of electrocatalytic overall water splitting has attracted much attention in the field of hydrogen generation. However, the anodic oxygen evolution reaction (OER) of overall water splitting is critical for efficiently utilizing renewable energy resources due to the sluggish kinetics, which greatly hinders the efficiency of hydrogen production. Replacing the high-potential OER process by the low-potential nucleophile oxidation reaction (NOR) is a novel approach to achieve energy-saving hydrogen production. Urea has been proposed as a hydrogen source because of the high gravimetric hydrogen content of 6.71%. In addition, urea has the characteristics of low cost, non-toxic and stable chemical properties, which are widely derived from human/animal urine and industrial production. Thus, replacing OER by the urea oxidation reaction (UOR) during water splitting can not only reduce the energy consumption of hydrogen generation but also treat the urea-rich wastewater. In this work, we demonstrate a Pt-anchored Ni(OH)2 surface to facilitate the capture of urea and further enhance the hydrogen production rate, which also provides a low-cost and suitable approach for energy-saving hydrogen production. |
Introduction
With the rising demand for energy and increasing environmental concerns, there exists an urgent need for eco-friendly renewable energy sources to alleviate the consumption of fossil fuels.1–4 Hydrogen (H2) is frequently proposed as a prominent energy carrier because of its favorable gravimetric energy density, zero contamination and exceptional recyclability.5 However, several strategies for producing H2, such as coal gasification and steam methane reforming, suffer from the drawbacks of low H2 purity, high energy consumption and unsatisfactory efficiency.6 Thus promising approaches for generating H2 have been proposed, such as electrocatalytic water splitting,7 photoelectrochemical (PEC) water splitting,8,9 and so on. In line with the goal of sustainable development, electrocatalytic water splitting has garnered substantial attention for its highly efficient H2 production that is both economical and pollution-free.10 The process of overall water splitting involves the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER), which requires a high thermodynamic voltage of 1.23 V.11,12 Nevertheless, the actual operating voltage for water splitting is usually much higher due to the limitation of the sluggish kinetics of the OER.11 To minimize the overpotential during water splitting, the rational design of advanced electrocatalysts for boosting HER and OER activity is an essential approach for enhancing the efficiency of H2 generation.In addition to developing highly efficient HER and OER electrocatalysts, it is crucial to replace the anodic OER in overall water splitting by the electrooxidation reaction of small organic molecules for dramatically enhanced energy-saving H2 production.13 Generally, among various kinds of organic molecules, those with nucleophilic groups and active hydrogen are essential to act as substrates for electrooxidation.14 Thus, the electrooxidation of organic substrates containing hydroxyl, aldehyde and amino groups can be defined as the nucleophile oxidation reaction (NOR), including the urea oxidation reaction (UOR), ethanol oxidation reaction (EOR) and hydrazine oxidation reaction (HzOR), which can be coupled with the HER to reduce the energy consumption in H2 production.14 Moreover, the excessive production and use of nitrogen-containing compounds (including urea) can lead to environmental problems such as water eutrophication and disruption of the nitrogen cycle.15–17 Hence, the NOR process for the degradation of excessive available nitrogen has gained much attention as an environmentally friendly and gentle alternative for artificially balancing the nitrogen cycle in recent years.
Among these NOR processes, UOR offers great prospects for the replacement of the OER, as it can remarkably reduce the theoretical thermodynamic potential from 1.23 V to 0.37 V.18,19 Therefore, the anodic UOR can be coupled with the HER to construct a urea-assisted water splitting configuration in an alkaline environment:
| Anode: CO(NH2)2 + 6OH− → N2 + 5H2O + CO2 + 6e− |
| Cathode: 6H2O + 6e− → 3H2 + 6OH− |
| Overall reaction: CO(NH2)2 + H2O → N2 + 3H2 + CO2 |
Herein, further inspired by the formation of the Pt–H bond at the active sites to promote the HER performance, we report the fabrication of Pt-anchored Ni(OH)2 on the surface of nickel-carbon nanofibers (Pt–Ni(OH)2@Ni-CNFs) and Pt-decorated Ni-CNFs (Pt@Ni-CNFs) via a simple electrospinning and electrodeposition technique, which are used as highly efficient UOR and HER electrocatalysts, respectively. Owing to the interfacial interactions between the Pt nanoparticles and Ni(OH)2, the prepared Pt–Ni(OH)2@Ni-CNFs catalyst exhibits a prominent UOR performance with low potentials of 1.363 and 1.422 V vs. RHE at a current density of 10 and 100 mA cm−2 in a 1 M KOH/0.33 M urea system, which is better than many other reported representative UOR electrocatalysts. In situ characterizations and density functional theory (DFT) calculations reveal that Pt loading can significantly reduce the energy barrier during the UOR process, further promoting electrocatalytic performance. In addition, the Pt@Ni-CNFs catalyst presents excellent HER performance with an ultralow overpotential of 20.8 mV at a current density of 10 mA cm−2 in 1 M KOH, even surpassing that of the benchmark Pt/C catalyst. As a result of the superior UOR and HER performances, a urea-assisted water splitting electrolyzer is assembled, in which Pt–Ni(OH)2@Ni-CNFs and Pt@Ni-CNFs are served as anodic and cathodic electrodes, respectively. The electrolyzer delivers a low voltage of 1.40 V at 10 mA cm−2, and presents an enhanced 10-fold H2 generation rate compared with the overall water splitting device, which illustrates prominent prospects for energy-saving H2 production.
Results and discussion
Fabrication and characterization of catalysts
As illustrated in Fig. 1, the Ni precursor-polyacrylonitrile (PAN) nanofibers are firstly prepared via an electrospinning technology with diameters in the range of 490–550 nm (Fig. S1, ESI†). Then, after the high temperature carbonization process, the obtained Ni-CNFs membranes significantly shrink to 470–520 nm, which results from the pyrolysis of the polymer (Fig. S2a and b, ESI†). Thanks to the soft and flexible nature of the Ni-CNF membranes, they contribute as self-supporting working electrodes for electrodeposition reactions. Based on the standard electrode potential, the reduction of NO3− in the Ni(NO3)2 solution is given priority over H2O and Ni2+ on the cathode, which is depicted as follows:| NO3− + 7H2O + 8e− → NH4+ + 10OH− |
| PtCl62− + 4e− → Pt + 6Cl− |
 | ||
| Fig. 1 Schematic diagram of the fabrication of Pt–Ni(OH)2@Ni-CNFs. | ||
We have prepared a range of deposition solutions with varying concentrations for the electrodeposition process, where the deposition solutions for Ni(OH)2@Ni-CNFs and Pt–Ni(OH)2@Ni-CNFs samples include 20 mL of aqueous solution with 15 mM Ni(NO3)2 and varied concentrations of H2PtCl6 (0, 1.25, 2.5 and 3.75 mM). Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) images reveal that the presence of the Ni precursor in the solution results in the hierarchical nanosheets coating on the fiber surface of Ni-CNFs (Fig. 2a, b and Fig. S2c–h, ESI†). Notably, a thicker surface nanosheet-like coating is observed when the Pt content in the deposition solution is low, which may be attributed to the deposition sequence of Ni and Pt species on the cathode. In addition, it is evident that many Ni nanoparticles are evenly embedded throughout the entire nanofibers, contributing to the promotion of the electron transfer properties. As shown in Fig. 2c, the diffraction peaks of Ni-CNFs sample in the X-ray diffraction (XRD) pattern at 44.6, 52.1 and 76.5° can be indexed to the (111), (200) and (220) planes of metallic Ni (JCPDS No. 04-0850), which are still maintained after the electrodeposition process. With increasing Pt concentration in the deposition solution, a broad peak appears at 39.8°, corresponding to the (111) plane of metallic Pt (JCPDS No. 04-0802).40–42 Although there is no obvious diffraction peak associated with Pt in the XRD pattern of Pt–Ni(OH)2@Ni-CNFs-2, the lattice fringe of the surface nanoparticle with a spacing of 0.224 nm in the high-resolution (HRTEM) image ascribes to the (111) plane of metallic Pt (Fig. 2d). Moreover, the distinct flexural lattice fringe of C (002) with a d-spacing of 0.360 nm can be observed, indicating the high graphitization of the carbon substrate, which facilitates electron transport.18 Fig. S3 (ESI†) shows the energy dispersive X-ray (EDX) pattern that further confirms the existence of the Pt element. And EDX mappings also present the uniform distribution of Pt, Ni, O and C elements (Fig. 2e and Fig. S4, ESI†). To identify the compositions of surface nanosheets, we employ a Pt-free Ni(OH)2@Ni-CNFs sample that is prepared in a more concentrated solution as reference, and its XRD pattern shows the diffraction peaks pointing to α-Ni(OH)2 (JCPDS No. 38-0715) (Fig. S5, ESI†). Furthermore, Raman spectra show that the peak located at around 510 cm−1 in the Pt-free and Pt–Ni(OH)2@Ni-CNFs samples corresponds to the Ni(II)–O bond of Ni(OH)2 (Fig. 2f), which confirms the formation of Ni(OH)2.43 Nevertheless, another peak attributed to the Pt–O bond appears at 560 cm−1 in the Pt-containing samples, suggesting that the Pt species are attached to Ni(OH)2.44,45 To further analyze the chemical composition of the nanofibers, X-ray photoelectron spectroscopy (XPS) is conducted to depict the surface valence states of the prepared samples. The XPS survey spectrum of Pt–Ni(OH)2@Ni-CNFs-2 shows the coexistence of Pt, Ni, C and O elements (Fig. S6a, ESI†). In detail, as shown in Fig. 2g, the Pt–Ni(OH)2@Ni-CNFs-2 sample exhibits prominent characteristic peaks at binding energies of 71.2 and 74.6 eV, corresponding to Pt 4f7/2 and 4f5/2 of Pt0, respectively.40,46,47 Furthermore, the peaks at 72.1 and 75.9 eV are assigned to the high-valence state of Ptδ+, confirming the successful introduction of the Pt species.40,46,47 Notably, the binding energy peaks of Pt 4f and Ni 3p partially overlap, whereas the peak with a binding energy of 68.5 eV corresponds to Ni 3p.40 In the Ni 2p spectrum of the Pt–Ni(OH)2@Ni-CNFs-2 sample, the characteristic peaks of Ni 2p3/2 and 2p1/2 corresponding to Ni2+ in Ni(OH)2 appear at 856.0 and 873.4 eV (Fig. 2h).48 Compared with the Ni(OH)2@Ni-CNFs sample without Pt loading, there is an obvious positive shift of the Ni 2p peaks, implying that the introduction of Pt changes the electron density around the Ni atom, and thereby improves the electrocatalytic oxidative activity.36,49 In addition, the doublets at 857.4 and 874.7 eV belong to the Ni 2p3/2 and 2p1/2 of Ni3+. The characteristic peak in the narrow-scan O 1s spectrum of Pt–Ni(OH)2@Ni-CNFs-2 shifts in the direction of high binding energy compared to the Pt-free sample, which is mainly due to the formation of the Pt–O bond.40,50,51 On the other hand, the noticeable peaks of Pt–Ni(OH)2@Ni-CNFs at 284.8, 285.8 and 289.1 eV in the C 1s spectrum belong to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O and CO bonds, respectively, which is originated from the carbon network after PAN carbonization (Fig. S6b, ESI†).52,53
C, C–O and CO bonds, respectively, which is originated from the carbon network after PAN carbonization (Fig. S6b, ESI†).52,53
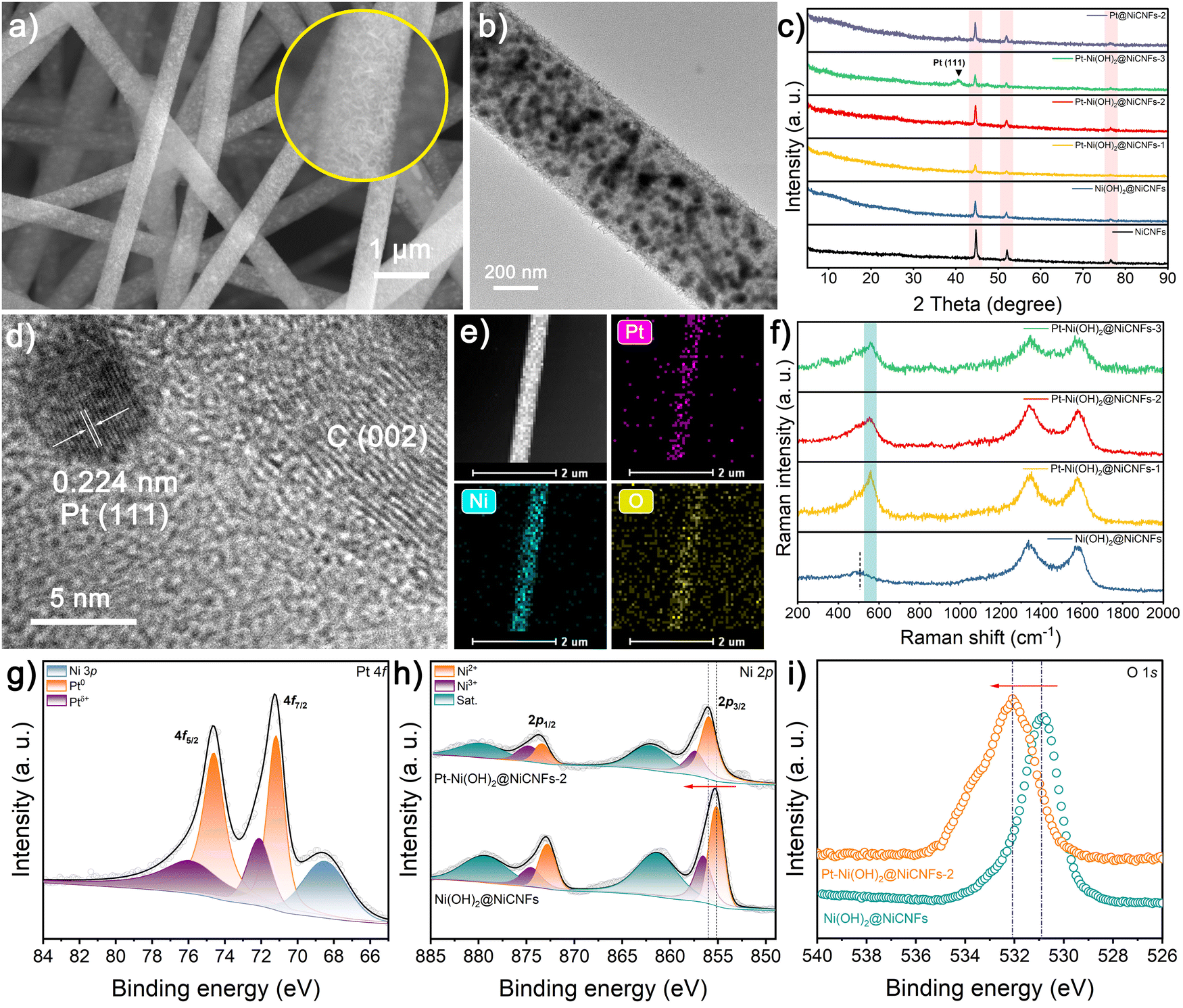 | ||
| Fig. 2 (a) SEM and (b) TEM images of Pt–Ni(OH)2@Ni-CNFs-2. (c) XRD patterns of different samples. (d) HRTEM image, (e) HAADF-STEM micrograph and EDX mappings of Pt, Ni and O elements of Pt–Ni(OH)2@Ni-CNFs-2. (f) Raman spectra of different samples. (g) Narrow-scan XPS spectrum of Pt 4f of Pt–Ni(OH)2@Ni-CNFs-2. Narrow-scan (h) Ni 2p and (i) O 1s XPS spectra of Pt–Ni(OH)2@Ni-CNFs-2 and Ni(OH)2@Ni-CNFs. | ||
For comparison, Pt@Ni-CNFs samples are also fabricated through a similar electrodeposition process in Ni precursor-free solution. No significant changes are observed in the surface morphology and the diameter of the final nanofibers compared with the neat Ni-CNFs, because the singlet Pt primarily exists in the form of small nanoparticles, which have minimal impact on the changes in surface morphology and fiber diameter (Fig. S7a, b and S8, ESI†). The XRD pattern shown in Fig. S9 (ESI†) demonstrates that in addition to the intense diffraction peaks of metallic Ni, only the sample with high Pt loading can detect the characteristic peak of metallic Pt(111). However, a lattice spacing of 0.224 nm is still observed in the Pt@Ni-CNFs-2 sample, which is indexed to the (111) plane of metallic Pt (Fig. S7c, ESI†). And the appearance of the Pt signal in the EDX pattern further confirms the successful formation of Pt nanoparticles on Ni-CNFs (Fig. S7d, ESI†). Additionally, the HAADF-STEM micrograph and the corresponding EDX mapping illustrate the uniform distribution of Pt, Ni and C elements (Fig. S7e, ESI†). The XPS survey spectra of Pt@Ni-CNFs-2 and Ni-CNFs show the presence of Ni and C elements in the nanofibers, and a clear characteristic peak of Pt 4f in Pt@Ni-CNFs-2, demonstrating the successful loading of Pt in the Pt@Ni-CNFs-2 sample (Fig. S10a, ESI†). The binding energy peaks of Pt 4f observed in Fig. S10b (ESI†) can be deconvoluted into Pt 4f7/2 (71.5 eV) and Pt 4f5/2 (74.9 eV) for metallic Pt, and Pt 4f7/2 (72.5 eV) and Pt 4f5/2 (76.0 eV) for Ptδ+, where the binding energy peak located at 68.1 eV corresponds to Ni 3p. In the Ni 2p narrow-scan spectrum of Pt@Ni-CNFs-2, two typical peaks at 853.2 and 870.6 eV correspond to Ni 2p3/2 and 2p1/2 of metallic Ni, respectively (Fig. S10c, ESI†). Moreover, the noticeable peaks located at 856.2 and 874.1 eV are attributed to Ni 2p3/2 and 2p1/2 of Ni2+ derived from the surface oxidation, and they are accompanied by a pair of satellite peaks. It is noteworthy that the characteristic peaks of metallic Ni in Pt@Ni-CNFs-2 illustrate a negative shift compared with the Pt-free Ni-CNF sample, demonstrating that Pt loading modulates the electron density of Ni sites. Finally, the narrow-scan C 1s spectrum in Fig. S10d (ESI†) can also be fitted to C–C/CC (284.8 eV), C–O (285.6 eV) and CO (288.5 eV). The mass loading of Pt and Ni in catalysts is further determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) measurement, in which the Pt–Ni(OH)2@Ni-CNFs-2 and Pt@Ni-CNFs-2 contain only 6.1 and 6.5 wt% metallic Pt, respectively (Table S1, ESI†).
UOR performance in a three-electrode configuration
The UOR performance of the as-prepared varied catalysts is assessed by a simple three-electrode system. As shown in Fig. 3a, the linear sweep voltammetry (LSV) curve of Pt–Ni(OH)2@Ni-CNFs-2 toward the UOR exhibits a potential advantage of 274 mV over the OER at a current density of 50 mA cm−2, indicating that the UOR catalysis is more favorable and energy-efficient than the OER based on the fabricated Pt–Ni(OH)2@Ni-CNFs-2 catalyst. In addition, the oxidation peak at a potential of 1.36 V vs. RHE in the LSV curve of the OER corresponds to the Ni2+/Ni3+ redox couple in the anodic potential range for most nickel-based catalysts, which approximates the onset potential of the UOR. This suggests that UOR catalysis may also be related to the electrochemical oxidation of the nickel active site. The cyclic voltammetry (CV) curves for the UOR at different scan rates are obtained with reduction peaks corresponding to the reduction process of Ni2+/Ni3+ redox pairs, and the reduction peak current density is plotted versus the square root of the scan rate, which shows a good linear relationship as a function, indicating that the redox of Ni2+/Ni3+ is a diffusion-controlled process (Fig. S11, ESI†).54 Moreover, the much lower Tafel slope of the UOR (13.7 mV dec−1) compared with the OER (84.8 mV dec−1) further confirms the faster kinetics of the UOR process for the fabricated Pt–Ni(OH)2@Ni-CNFs-2 catalyst (Fig. S12, ESI†). In the following, the impact of the electrolyte concentration on the UOR activity has been investigated. As shown in Fig. S13a (ESI†), the polarization curves of Pt–Ni(OH)2@Ni-CNFs-2 in 1 M KOH electrolyte containing different concentrations of urea reveal that the current density increases gradually in the low concentration range and reaches the maximum value at 0.33 M urea concentration. In the electrolyte with a high concentration of urea, the surface of Pt–Ni(OH)2@Ni-CNFs-2 is heavily covered by urea molecules and catalytic intermediates, which hampers the contact of OH− species with the active sites, thereby reducing the urea oxidation rate.55,56 In addition, excess urea undergoes hydrolysis to generate CO2, which in turn mitigates the current density.55 As a result, a concentration of 0.33 M urea is used in the UOR process to attain the maximum current density, which is also equivalent to the concentration of urea in human urine. On the other hand, the UOR performance of the catalyst is also explored by the LSV method in the KOH system with varied concentrations (Fig. S13b, ESI†). The onset potential shifts towards the negative range with increasing KOH concentration, indicating that higher KOH concentrations can stimulate the formation of active species.57 Similarly, the anodic current density increases with KOH concentration, which is mainly due to the increased electrolyte conductivity, thereby facilitating current transport. However, a substantial reduction in current density is observed at high potentials when the KOH concentration reaches 6 M, which is likely attributed to the complete coverage of OH− on the surface of the electrocatalyst, hindering the contact between urea molecules and the electrode to yield decreased activity.55,56 Thus, the subsequent UOR experiments are performed employing the 1 M KOH/0.33 M urea system.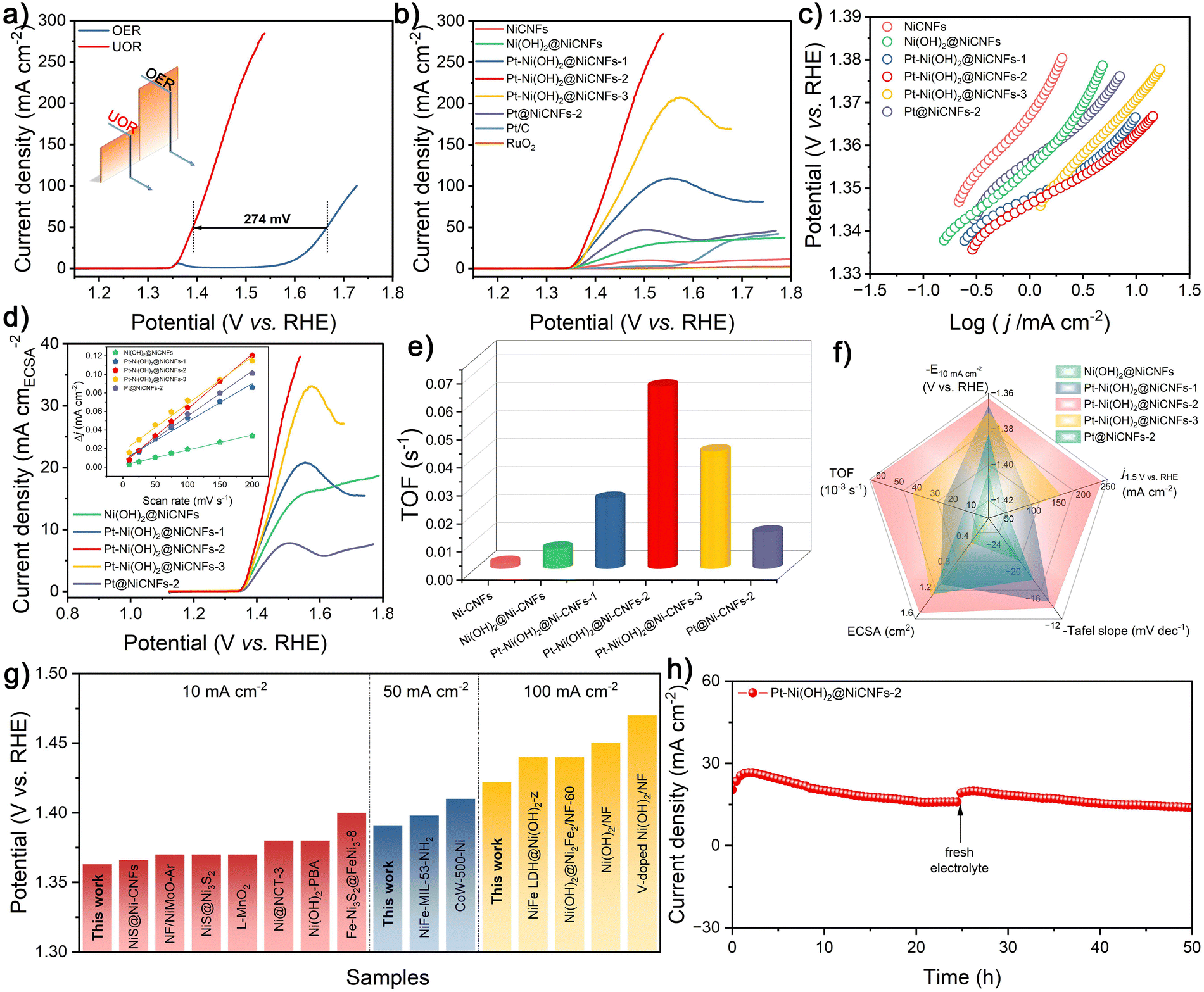 | ||
| Fig. 3 (a) Polarization curves of Pt–Ni(OH)2@Ni-CNFs-2 for the UOR and OER in 1 M KOH/0.33 M urea and 1 M KOH, respectively. (b) Polarization curves and (c) corresponding Tafel plots of different catalysts. (d) ECSA-normalized polarization curves and (d, inset) capacitive current density of different catalysts with linear fitting with different scan rates of different catalysts. (e) Comparison of TOF values of different control catalysts. (f) Comprehensive performance comparison of different catalysts. (g) Comparison of some representative catalysts for UOR activity. (h) i–t curve of Pt–Ni(OH)2@Ni-CNFs-2 for the UOR at a potential of 0.47 V vs. Hg/HgO. | ||
For further comparison, the UOR activities of other control samples are explored. As shown in Fig. 3b, the Ni(OH)2@Ni-CNFs catalyst requires 1.404 V vs. RHE to attain a current density of 10 mA cm−2. However, it is observed that the current densities of Pt–Ni(OH)2@Ni-CNFs catalysts are dramatically enhanced upon the addition of Pt solution to the electrodeposition system; among them, the Pt–Ni(OH)2@Ni-CNFs-2 catalyst only requires 1.363 V vs. RHE to drive a current density of 10 mA cm−2, which surpasses those of Pt–Ni(OH)2@Ni-CNFs-1 (1.367 V vs. RHE) and Pt–Ni(OH)2@Ni-CNFs-3 (1.371 V vs. RHE). Moreover, the polarization curve of the sample with individual electrodeposited Pt nanoparticles (Pt@Ni-CNFs-2) presents a potential of 1.383 V vs. RHE at a current density of 10 mA cm−2. It can be seen that the UOR performances of both Ni(OH)2@Ni-CNFs and Pt@Ni-CNFs-2 are lower than those of the Pt–Ni(OH)2@Ni-CNF samples, suggesting that the excellent electrocatalytic UOR performance of our prepared catalyst is due to the mutual synergistic effect between surface Pt nanoparticles and Ni(OH)2.
Additionally, the Pt–Ni(OH)2@Ni-CNFs-2 catalyst presents a current density of 238.4 mA cm−2 at 1.5 V vs. RHE, which displays a significant enhancement compared with the Pt-free Ni(OH)2@Ni-CNFs catalyst (27.5 mA cm−2). The Tafel slope of Pt–Ni(OH)2@Ni-CNFs-2 for the UOR process is calculated as 13.7 mV dec−1, which is lower than those of Pt–Ni(OH)2@Ni-CNFs-1 (14.6 mV dec−1), Pt–Ni(OH)2@Ni-CNFs-3 (26.5 mV dec−1), Ni(OH)2@Ni-CNFs (20.7 mV dec−1), Pt–Ni-CNFs-2 (17.9 mV dec−1) and Ni-CNFs (26.2 mV dec−1), suggesting the enhanced reaction kinetics of Pt–Ni(OH)2@Ni-CNFs-2 for the UOR (Fig. 3c). The kinetics of this catalyst for the UOR can be further investigated via EIS measurements. According to the Nyquist plot illustrated in Fig. S14 (ESI†), the Pt–NiS@Ni-CNFs-2.5 catalyst displays the smallest semicircle among all the prepared catalysts, signifying the lowest charge transfer resistance (Rct).
The lower Rct value represents a faster charge transfer capability of the catalysts for improving the electrocatalytic performance. Remarkably, all Pt-loaded catalysts have substantially reduced charge transfer resistance. Based on the previous XPS characterization, it can be inferred that Pt loading modulates electron density at the nickel sites, which promotes electron transfer and enhances the electrocatalytic activity. Furthermore, the electrochemical surface area (ECSA) can be evaluated through the double layer capacitance (Cdl) to study the intrinsic activity of catalysts. As depicted in the Fig. S15 (ESI†) and the inset in Fig. 3d, the calculated Cdl of Pt–Ni(OH)2@Ni-CNFs-2 is 0.3 mF cm−2, which is higher than that of Pt–Ni(OH)2@Ni-CNFs-1 (0.21 mF cm−2), Pt–Ni(OH)2@Ni-CNFs-3 (0.25 mF cm−2), Ni(OH)2@ Ni-CNFs (0.08 mF cm−2) and Pt@Ni-CNFs-2 (0.24 mF cm−2). In general, the ECSA and roughness factor (RF) can be estimated by Cdl, where the higher value of Pt–Ni(OH)2@Ni-CNFs-2 reveals its more exposed active sites (Table S2, ESI†).58,59 Additionally, the ECSA-normalized polarization curves indicate that the Pt–Ni(OH)2@Ni-CNFs-2 still exceeds other prepared catalysts, indicating its superior intrinsic activity due to the synergistic interaction between Pt and Ni(OH)2 components (Fig. 3d).58 Based on the molar ratio of metal components in the catalysts given in Table S1 (ESI†), the turnover frequency (TOF) values of each catalyst at various potentials are calculated (Fig. 3e and Fig. S16, ESI†). The Pt–Ni(OH)2@Ni-CNFs-2 possesses the highest TOF of 0.065 s−1 at a potential of 1.5 V vs. RHE, further demonstrating the faster kinetics and higher intrinsic activity of our prepared catalyst.60 The overall performance comparison of the catalysts in Fig. 3f demonstrates that the prepared Pt–Ni(OH)2@Ni-CNFs-2 has the best electrocatalytic performance for the UOR. Moreover, the prepared Pt–Ni(OH)2@Ni-CNFs-2 catalyst exhibits excellent activity at different current densities, surpassing many reported representative Ni-based catalysts (Fig. 3g and Table S3, ESI†).15,22,48,61–72
Stability is a critical aspect in assessing the catalyst performance. The long-term i–t curve in Fig. 3h exhibits that the Pt–Ni(OH)2@Ni-CNFs-2 catalyst maintains its electrocatalytic activity in 1 M KOH/0.33 M urea system over 50 h at a continuous current density of 20 mA cm−2, demonstrating its exceptional long-term stability. It is noteworthy that after replacing with the fresh electrolyte, the current density recovers, proving that the current decrease might partially result from the consumption of urea and the formation of carbonate byproduct from the adsorption of CO2 in the electrolytic system (Fig. 3h and Fig. S17, ESI†). A series of examinations of Pt–Ni(OH)2@Ni-CNFs-2 after the UOR process has been carried out to further understand the origin of the stability (Fig. S18, ESI†). SEM image shows that Pt–Ni(OH)2@Ni-CNFs-2 still retains its fibrous morphology and the nanosheet structure, suggesting its structural robustness. The XRD patterns present no significant changes before and after the UOR process. Moreover, XPS analysis confirms the unchanged chemical compositions of the catalyst after the UOR process. The characteristic peaks in the narrow-scan spectra of Pt 4f and Ni 2p remain consistent with those prior to UOR. In addition, the O 1s spectrum confirms the composition stability of Pt–Ni(OH)2@Ni-CNFs-2 with the characteristic peaks corresponding to the metal–O bond, the O–H bond, and the H2O molecule on the surface of the catalyst, respectively. Notably, the binding energy peak located at 535.7 eV stems from the ether-oxygen bond in Nafion. Furthermore, the ICP results present the low losses of Ni (1.8%) and Pt (6.1%) contents for the Pt–Ni(OH)2@Ni-CNFs-2 after the i–t test, resulting in its excellent long-term UOR performance.
Catalytic mechanism for the high UOR performance
It is essential to analyze the phase transition of Pt–Ni(OH)2@Ni-CNFs-2 during the UOR process to figure out its reaction mechanism. In situ Raman spectra are recorded to monitor the change of the chemical structure of the catalyst and its surface species (Fig. 4a). When the applied potential exceeds 0.45 V vs. Hg/HgO (1.372 V vs. RHE) in 1 M KOH for the OER, two distinct Raman peaks appear at 478 and 554 cm−1, which belongs to the Ni3+–O bending and stretching vibrations of Ni2+δOxHy species.43 However, these characteristic peaks disappear during the UOR process in the 1 M KOH/0.33 M urea system (Fig. 4b). According to the reported literature, Ni(OH)2 is usually converted to Ni(OH)O intermediate through the dehydrogenation progress (Ni(OH)2 + OH− → Ni(OH)O + H2O + e−) under the oxidation potential in an alkaline electrolyte, initially arising at approximately 1.35–1.37 V vs. RHE.15 Subsequently, the Ni(OH)O intermediates undergo different reactions depending on the electrolyte. In the 1 M KOH electrolyte for the OER process, Ni(OH)O accumulates to form Raman sensitive Ni2+δOxHy species (NiOOH and NiOx).14 Conversely, in an electrolyte containing a nucleophile, the generated Ni(OH)O intermediates will be filled with hydrogen from the nucleophile through the spontaneous reduction, thus, leading to their conversion back to Ni(OH)2, which is difficult to be identified by Raman spectra.15,40 Therefore, it can be inferred from the absence of the bending and stretching vibrations of Ni3+–O that the accumulation of Ni(OH)O intermediates to Ni2+δOxHy species is inhibited during the UOR process. | ||
| Fig. 4 (a) Schematic diagram of the in situ Raman device. (b) In situ Raman spectra of Pt–Ni(OH)2@Ni-CNFs-2 for the OER and UOR processes. Bode plots of Pt–Ni(OH)2@Ni-CNFs-2 at different applied potentials for the (c) OER (1 M KOH) and (d) UOR (1 M KOH/0.15 M urea), respectively. (e) The energy barrier for Ni-OH dehydrogenation at the surface of Ni(OH)2 and Pt–Ni(OH)2 catalysts. Here, H, O, Ni and Pt atoms are indicated by white, red, cyan and dark blue spheres, respectively. (f) Calculated Gibbs free energy diagrams for UOR steps on the surface of Ni(OH)2 and Pt–Ni(OH)2 catalysts. | ||
To further probe the interface behavior during OER and UOR processes, the electrochemical impedance spectroscopy (EIS) measurement is adopted under diverse potentials. As illustrated in the Bode plots in Fig. 4c, there are two peaks emerging at different frequency ranges when the applied potential exceeds 1.34 V vs. RHE in 1 M KOH. Given the mechanism of alkaline OER on nickel-based catalysts, the peak reflected in the high frequency range is related to the electrocatalytic oxidation of the catalyst (formation of Ni2+δOxHy species), which is associated with the first semicircle in the Nyquist plots (Fig. S19a and b, ESI†). On the other hand, the peak observed in the low frequency range, corresponding to the second semicircle, is indexed to the OER. Beyond the applied potential of 1.46 V vs. RHE, the phase angle in the low frequency range drops gradually, representing the progressive faster electron transfer during the OER process with increasing applied potential. In comparison, Fig. 4d shows the Bode plots from the EIS measurement of the UOR process. Unlike the OER process, the Bode plots of the UOR manifest only one peak in the entire applied frequency range, which corresponds to one semicircle in Nyquist plots (Fig. S19c and d, ESI†), implying the charge transfer process during UOR. Consistent with the in situ Raman results, the intervention of urea hinders the accumulation of Ni(OH)O to generate Ni2+δOxHy. Furthermore, the phase angle exhibits a sharp drop with an applied potential exceeding 1.34 V vs. RHE, suggesting a faster charge transfer for the UOR process. Notably, the phase angle starts to rise when the applied potential increases above 1.46 V vs. RHE due to the passive reaction on the surface of the catalyst.20,34
As shown in Fig. 3b, the polarization curves indicate that the bare Pt/C cannot catalyze urea oxidation. Therefore, it is possible that the incorporation of Pt can facilitate the dehydrogenation of Ni(OH)2 to Ni(OH)O, thereby promoting the UOR. To gain a better understanding of the acceleration of Pt loading for dehydrogenation kinetics, density functional theory (DFT) calculations are performed. First, the energy barrier of the dehydrogenation process (ΔE) of Ni–OH is revealed in Fig. 4e and Fig. S20 (ESI†). Pt–Ni(OH)2 is found to exhibit a ΔE value of 1.25 eV under Pt–O–Ni–OH coordination conditions, which is lower than the bare Ni(OH)2 surface (2.53 eV), suggesting that Pt loading favors the dehydrogenation of the Ni(OH)2 surface to form the vacancies. Pt loading can significantly reduce the dehydrogenation energy barrier in the oxidation progress of Ni(OH)2, ultimately promoting the thermodynamic behavior of the UOR. Furthermore, to validate the adsorption behavior of urea, the adsorption energies (ΔEads) of –NH2 and –CO groups in urea molecules on the catalyst surface are investigated (Fig. S21, ESI†). Our findings depict that the –NH2 group has a stronger adsorption ability than the –CO group on both dehydrogenated Ni(OH)2 and Pt–Ni(OH)2 catalysts. Therefore, the Gibbs free energy diagrams for UOR steps on Ni(OH)2 and Pt–Ni(OH)2 catalysts are calculated based on the adsorption behavior. As illustrated in Fig. 4f and Fig. S22 (ESI†), the dehydrogenation of *CONNH2 intermediates is the rate-determining step (RDS) during the entire UOR process. Evidently, the calculated Gibbs free energy change (ΔG) of the RDS on Pt–Ni(OH)2 is 1.64 eV, which is lower than that on Ni(OH)2 (2.90 eV). In addition, the results indicate that Pt loading promotes the adsorption of the *CO intermediate while not conducive to the adsorption of N2, enabling the prompt desorption of N2, and thus facilitating the formation of *COOH from *CO coupling with OH−. These findings suggest that the incorporation of Pt species can manipulate the electronic structure and significantly decrease the energy barrier, ultimately resulting in excellent UOR activity.
HER performance in a three-electrode configuration
Bifunctional catalysts have attracted much attention for the development of efficient H2 production; the UOR serves as an excellent anodic OER replacement reaction for overall water splitting. Hence, the HER performances of a series of the prepared catalysts are investigated. It is found that the Pt@Ni-CNFs-2 catalyst presents the best HER activity, surpassing Pt–Ni(OH)2@Ni-CNFs-2 and even the benchmark Pt/C catalyst (Fig. 5a and b). In detail, Pt@Ni-CNFs-2 shows an overpotential of 20.8 and 107.8 mV at current densities of 10 and 200 mA cm−2, which are lower than those of Pt@Ni-CNFs-1 (26.5 and 142.6 mV), Pt@Ni-CNFs-3 (27.3 and 202.3 mV), Pt–Ni(OH)2@Ni-CNFs-2 (25.5 and 210.9 mV), Ni-CNFs (213.7 and 424.5 mV) and commercial Pt/C (20.9 and 139.3 mV), suggesting that Pt loading on Ni-CNFs promotes HER activity. Meanwhile, the excellent HER activity of Pt@Ni-CNFs-2 is also better than many reported representative catalysts (Table S4, ESI†). The mass activity (MA) of Pt@Ni-CNFs-2 is calculated and further compared with that of commercial Pt/C based on the Pt content of the sample given in Table S1 (ESI†). As shown in Fig. 5c, the MA of Pt@Ni-CNFs-2 at 100 mV is 2706.6 A g−1, which is 4.3-fold that of commercial Pt/C (629.6 A g−1), indicating the realization of low Pt loading of our prepared Pt@Ni-CNFs-2 while still achieving a high activity. This result implies that the excellent HER catalytic activity of Pt@Ni-CNFs-2 may originate not only from the active sites provided by the Pt nanoparticles, but also from the synergistic interaction between Pt nanoparticles and Ni nanoparticles within CNFs (Fig. S7c, ESI†), which has been further revealed by DFT calculations. In Fig. 5d and Fig. S23 (ESI†), the relatively high water dissociation energy barriers of 1.90 and 1.44 eV on Pt(111) and Ni(111) catalysts can be observed, respectively, demonstrating that Pt(111) and Ni(111) catalysts are inefficient for the water dissociation step under alkaline conditions. However, the activation barriers for water dissociation are remarkably reduced to 1.22 eV on the Pt NPs/Ni(111) catalyst, indicating the synergistic interaction between the Pt NPs and the Ni surface within CNFs is effective for the cleavage of H-OH bonds. After the formation of *H intermediates in the previous water dissociation step, *H intermediates are desorbed from the metal surface, thus yielding H2. The calculated hydrogen adsorption free energy value of *H intermediates (ΔG*H) of Pt NPs/Ni(111) (−0.19 eV) is higher than that of the Pt(111) surface (−0.34 eV) (Fig. 5e). This suggests that the desorption from *H intermediates to H2 on Pt NPs/Ni(111) can be faster than that on Pt(111) surface (when ΔG*H < 0, the desorption from *H intermediates to H2 is the rate-determined step), which can be beneficial for the re-exposure of active sites. This combined with a low energy barrier of water dissociation and fast desorption of *H intermediates on Pt NPs/Ni(111) facilitates highly efficient HER performance under alkaline conditions. The Tafel slope is further evaluated as an important parameter to understand the kinetics of the catalytic reaction (Fig. S24, ESI†). It is found that Pt@Ni-CNFs-2 has the lowest Tafel slope of 20.1 mV dec−1, lower than those of Pt@Ni-CNFs-1 (24.3 mV dec−1), Pt@Ni-CNFs-3 (20.6 mV dec−1), Ni-CNFs (150.4 mV dec−1), Pt–Ni(OH)2@Ni-CNFs-2 (47.4 mV dec−1), and commercial Pt/C (21.3 mV dec−1), demonstrating excellent kinetic merits and superiority over many reported representative HER catalysts (Fig. 5f and Table S4, ESI†). In the HER process, the mechanism of the reaction and its RDS for a catalyst can be approximately estimated based on its Tafel slope. Herein, the Tafel slope of 20.1 mV dec−1 for the Pt@Ni-CNFs-2 catalyst suggests its Volmer–Tafel mechanism, with the RDS being the Tafel step (2H* + 2e− → H2). | ||
| Fig. 5 HER measurements in 1 M KOH. (a) Polarization curves of different catalysts. (b) Overpotentials of different catalysts at 10 and 200 mA cm−2. (c) Comparison of mass activity of Pt@Ni-CNFs-2 and commercial Pt/C at an overpotential of 100 mV. (d) Calculated free energy diagrams for water dissociation on the surface of Pt(111), Ni(111) and Pt NPs/Ni(111) for the alkaline HER process. (e) Calculated free energy diagrams for hydrogen desorption steps on the surface of Pt(111), Ni(111) and Pt NPs/Ni(111). (f) Comparison of some representative catalysts for HER properties. (g) TOF plots of different catalysts. (h) Polarization curves before and after 2000 CV cycles of Pt@Ni-CNFs-2. (i) i–t curves of Pt@Ni-CNFs-2 and commercial Pt/C at a potential of −0.97 V vs. Hg/HgO. | ||
The kinetic information of the catalysts for the HER process is further explored by the EIS measurement. As illustrated in Fig. S25 (ESI†), after fitting by equivalent circuit, the Rct of Pt@Ni-CNFs-2 is calculated as 6.07 Ω, which is lower than that of other control samples including Pt@Ni-CNFs-1 (6.67 Ω), Pt@Ni-CNFs-3 (8.78 Ω), Ni-CNFs (494.7 Ω) and Pt–Ni(OH)2@Ni-CNFs-2 (12.41 Ω). The smaller Rct value further confirms that Pt@Ni-CNFs-2 possesses faster charge transfer capability and outstanding kinetic advantages, which is beneficial for the synergistic effect between surface Pt nanoparticles and the embedded Ni nanoparticles within CNFs. Moreover, according to the ICP results in Table S1 (ESI†), the TOF plots of the prepared catalysts are obtained by assuming that both Pt and Ni species are participating in the HER catalytic process (Fig. 5g). At an overpotential of 150 mV, the TOF value of the Pt@Ni-CNFs-2 catalyst is 0.311 s−1, which exceeds that of Pt@Ni-CNFs-1 (0.199 s−1), Pt@Ni-CNFs-3 (0.117 s−1), Ni-CNFs (0.003 s−1) and Pt–Ni(OH)2@Ni-CNFs-2 (0.095 s−1), indicating the excellent reaction kinetics of the Pt@Ni-CNFs-2 catalyst and its high intrinsic activity. Fig. 5h and 5i illustrate the stability of the Pt@Ni-CNFs-2 catalyst toward the HER in an alkaline system. First, the polarization curves of Pt@Ni-CNFs-2 prior to and post 2000 CV cycles almost overlap, indicating that our prepared catalyst has excellent cycling stability. Second, in the continuous i–t test with a constant applied potential, Pt@Ni-CNFs-2 undergoes an HER progress for 50 h with a slight decline in current density, while the commercial Pt/C decays fast within 20 h, further confirming favorable long-term stability of the Pt@Ni-CNFs-2 catalyst. It brings a promising application prospect in the practical water electrolysis for H2 production. To gain insight into the structural stability of the Pt@Ni-CNFs-2 catalyst during the HER process, a series of characterizations after i–t testing are performed. As illustrated in Fig. S26a (ESI†), the SEM image of the Pt@Ni-CNFs-2 catalyst post HER shows morphological and structural robustness. In addition, its XRD pattern reveals no obvious phase changes, further realizing the stability of the crystal structure of the Pt@Ni-CNFs-2 catalyst (Fig. S26b, ESI†). The surface chemical valence states of the catalyst post HER process are further investigated through the XPS spectra (Fig. S26c and d, ESI†). Compared with the Ni 2p and Pt 4f spectra of the Pt@Ni-CNFs-2 catalyst before the HER process, neither of them shows significant differences. The ICP results further reveal that Pt loss for the Pt@Ni-CNFs-2 sample (4.6%) is much lower than that of the commercial Pt/C catalyst (22.2%), thus contributing to the better chemical stability of Pt@Ni-CNFs-2 during the HER process.
Urea-assisted overall water splitting in a two-electrode configuration
In light of the remarkable UOR activity of Pt–Ni(OH)2@Ni-CNFs-2 and the highly efficient HER performance of Pt@Ni-CNFs-2, a urea-assisted water splitting electrolyzer is assembled in the 1 M KOH/0.33 M urea system with the two catalysts as the anode and cathode, respectively (Fig. 6a). For comparison, an overall water splitting electrolzyer of Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs-2 is also prepared in 1 M KOH. As illustrated in Fig. 6b and c, the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs-2 electrolyzer for overall water splitting requires voltages of 1.635 and 1.841 V to reach the current densities of 10 and 50 mA cm−2. While for the urea-assisted water splitting with the anodic reaction replaced by the UOR, the same current density can be attained with a voltage of merely 1.400 and 1.544 V. It reveals that replacing the anodic OER process with UOR can achieve the desired current density at a significantly lower voltage, which greatly reduces the energy consumption in the H2 production process. Consequently, based on the LSV curves of the urea-assisted water splitting and overall water splitting, the power consumptions at different current densities are calculated and presented in Fig. S27 (ESI†). The significantly decreased power consumption from 4.27 to 3.58 kW h m−3 H2 at 80 mA cm−2 can be clearly observed due to the replacement of the OER by the UOR, and it can be concluded that urea-assisted water splitting needs considerably less electrical energy than overall water splitting to achieve the energy-saving hydrogen production.73,74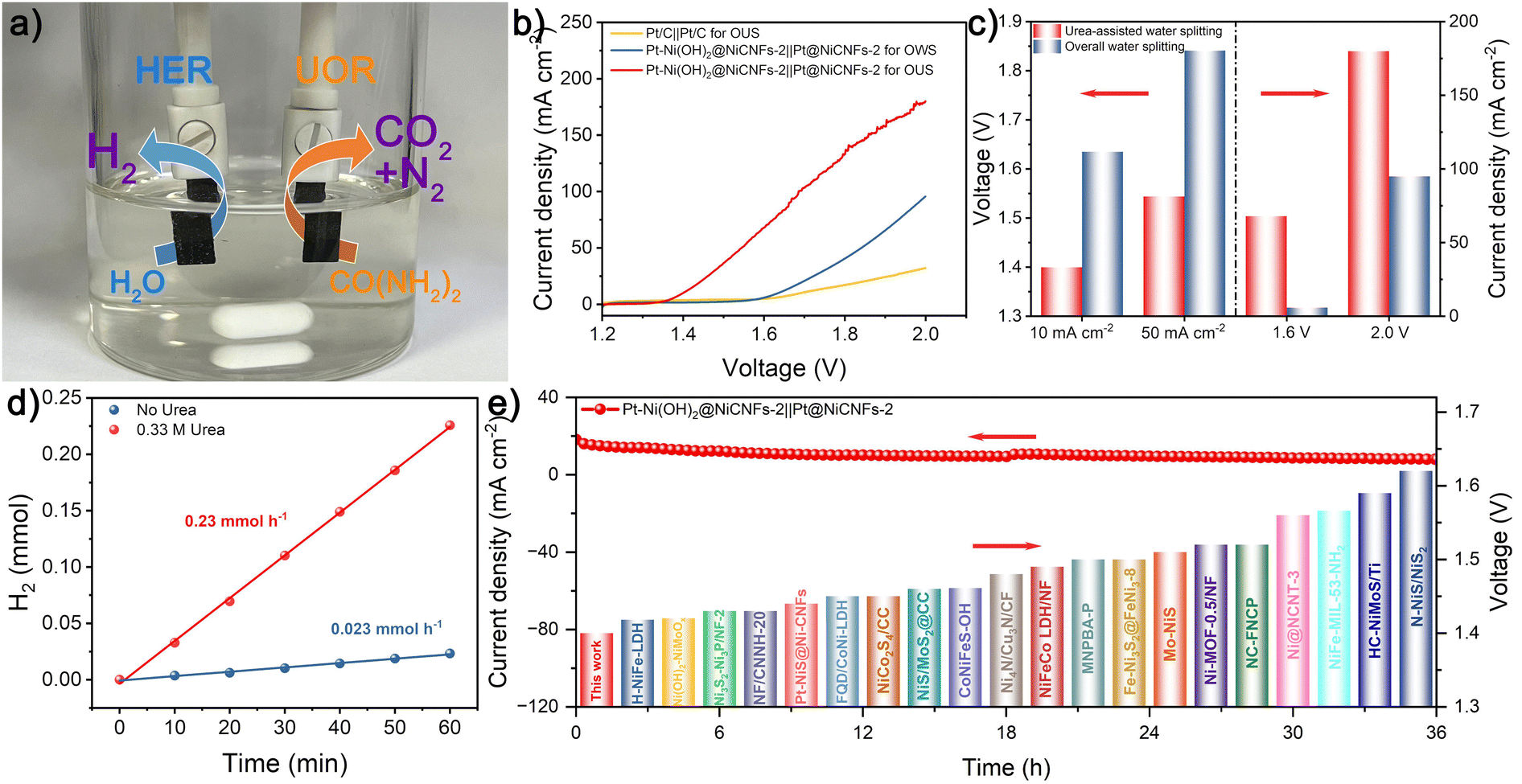 | ||
| Fig. 6 (a) Digital photograph of the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs electrolyzer. (b) Polarization curves of different electrolyzers for overall water splitting and urea-assisted water splitting without iR-compensation. (c) Activity comparison and (d) cathodic H2 evolution of the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs electrolyzer for overall water splitting and urea-assisted water splitting. (e) I–T curve of the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNF electrolyzer at a voltage of 1.44 V, and the comparison of the urea-assisted water splitting activity for Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs with other reported similar electrolyzers. | ||
Therefore, to highlight the advantages of the urea-assisted electrolyzer in practical H2 production, we have collected the cathodic H2 from the constructed electrolyzer. The H2 production rate of 0.23 mmol h−1 is attained at a voltage of 1.60 V for urea-assisted water splitting, which is 10-fold that of overall water splitting without the addition of urea (Fig. 6d). In addition, the urea-assisted water splitting electrolyzer assembled with commercial Pt/C as both the cathode and anode requires a voltage of 1.690 V to attain a current density of 10 mA cm−2, which is much higher than the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs-2 electrolyzer (1.400 V). Furthermore, the cell voltage based on the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs-2 electrolyzer for urea-assisted water splitting also outperforms many reported representative electrolyzers at a current density of 10 mA cm−2 (Fig. 6e and Table S5, ESI†). Fig. 6e also presents the stability test of the Pt–Ni(OH)2@Ni-CNFs-2‖Pt@Ni-CNFs-2 electrolyzer at a constant voltage, during which only a slight decline is observed after 36 h, demonstrating its excellent long-term stability.
Conclusions
In summary, we have successfully synthesized highly efficient electrocatalysts for the UOR and HER via a simple electrospinning-electrodeposition strategy. The prepared Pt–Ni(OH)2@Ni-CNFs catalyst only requires a potential of 1.363 and 1.422 V vs. RHE to achieve a current density of 10 and 100 mA cm−2. In situ characterization reveals an indirect mechanism on the Pt–Ni(OH)2@Ni-CNF catalyst toward the UOR, that is, Ni(OH)2 is firstly electrooxidized to Ni(OH)O followed by the subsequent spontaneous dehydrogenation of the urea molecule at the vacancy site of Ni(OH)O, and then Ni(OH)O is simultaneously converted back to Ni(OH)2. DFT calculations confirm that Pt loading facilitates the electrooxidation of Ni(OH)2 and significantly decreases the energy barrier of the RDS, boosting the UOR activity efficiently. On the other hand, the obtained Pt@Ni-CNF catalyst exhibits an extremely higher HER activity than the commercial Pt/C catalyst in an alkaline medium. Therefore, the two-electrode urea-assisted water splitting electrolyzer with Pt–Ni(OH)2@Ni-CNFs and Pt@Ni-CNFs serving as the anode and cathode, respectively, renders a much lower working voltage in H2 production compared with the overall water splitting electrolyzer. The H2 production rate of urea-assisted water splitting is 10-fold that of water splitting, demonstrating the enormous prospective of replacing the OER with UOR in the field of energy-saving and sustainable H2 production.Author contributions
Xiaofeng Lu and Ce Wang proposed the idea and supervised the program. Mengxiao Zhong performed the experiments and analyzed the data. Meijiao Xu, Siyu Ren and Weimo Li participated in the data analysis and discussions. Mingbin Gao performed the DFT calculations. Mengxiao Zhong wrote the manuscript and Xiaofeng Lu revised it.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52273056) and the Science and Technology Development of Jilin Province, China (20220402008GH). We thank Dr Junyu Yang in Dalian Institute of Chemical Physics, Chinese Academy of Sciences for his help with the DFT calculations on the HER property of Pt@Ni-CNFs.References
- J. Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- C. McGlade and P. Ekins, Nature, 2015, 517, 187–190 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS PubMed.
- M. Zhong, W. Li, J. Chen, S. Ren, R. Qi, C. Wang and X. Lu, Sep. Purif. Technol., 2023, 310, 123164 CrossRef CAS.
- T. Xiong, Z. Zhu, Y. He, M. S. Balogun and Y. Huang, Small Methods, 2023, 7, 2201472 CrossRef CAS PubMed.
- Y. Wang, D. Chen, J. Zhang, M. S. Balogun, P. Wang, Y. Tong and Y. Huang, Adv. Funct. Mater., 2022, 32, 2112738 CrossRef CAS.
- X. Lu, K.-h Ye, S. Zhang, J. Zhang, J. Yang, Y. Huang and H. Ji, Chem. Eng. J., 2022, 428, 131027 CrossRef CAS.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- W. Li, M. Li, C. Wang and X. Lu, Sci. China: Mater., 2023, 66, 1024–1032 CAS.
- A. Kumar, X. Liu, J. Lee, B. Debnath, A. R. Jadhav, X. Shao, V. Q. Bui, Y. Hwang, Y. Liu, M. G. Kim and H. Lee, Energy Environ. Sci., 2021, 14, 6494–6505 RSC.
- H. Xu, Y. Liao, Z. Gao, Y. Qing, Y. Wu and L. Xia, J. Mater. Chem. A, 2021, 9, 3418–3426 RSC.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- W. Chen, L. Xu, X. Zhu, Y. C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C. L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 CrossRef CAS PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- X. Xu, H. Ullah, M. Humayun, L. Li, X. Zhang, M. Bououdina, D. P. Debecker, K. Huo, D. Wang and C. Wang, Adv. Funct. Mater., 2023, 33, 2303986 CrossRef CAS.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- F. Guo, K. Ye, M. Du, X. Huang, K. Cheng, G. Wang and D. Cao, Electrochim. Acta, 2016, 210, 474–482 CrossRef CAS.
- P. Li, W. Li, Y. Huang, Q. Huang, F. Li and S. Tian, Small, 2023, 19, 2305585 CrossRef CAS PubMed.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2023, 33, 2209698 CrossRef CAS.
- K. Zhang, S. Wang, X. Li, H. Li and Y. Ni, Small, 2023, 19, 2300959 CrossRef CAS PubMed.
- P. Li, W. Li, Y. Huang, Q. Huang and S. Tian, Small, 2023, 19, 2300725 CrossRef CAS PubMed.
- M. Ranjani, N. Senthilkumar, G. Gnana kumar and A. Manthiram, J. Mater. Chem. A, 2018, 6, 23019–23027 RSC.
- N. Senthilkumar, G. Gnana kumar and A. Manthiram, Adv. Energy Mater., 2017, 8, 1702207 CrossRef.
- S. Huang, Q. Zhang, P. Xin, J. Zhang, Q. Chen, J. Fu, Z. Jin, Q. Wang and Z. Hu, Small, 2022, 18, 2106841 CrossRef CAS PubMed.
- M. Zhong, W. Li, C. Wang and X. Lu, Appl. Surf. Sci., 2022, 575, 151708 CrossRef CAS.
- N. Chen, Y.-X. Du, G. Zhang, W.-T. Lu and F.-F. Cao, Nano Energy, 2021, 81, 105605 CrossRef CAS.
- R.-Q. Li, Q. Liu, Y. Zhou, M. Lu, J. Hou, K. Qu, Y. Zhu and O. Fontaine, J. Mater. Chem. A, 2021, 9, 4159–4166 RSC.
- Y. Dong, Y. Wu, X. Wang, H. Wang, J. Ren, P. Wang, L. Pan, G. Wang and R. Wang, Nanoscale, 2023, 15, 1813–1823 RSC.
- Y. Ding, Y. Li, Y. Xue, B. Miao, S. Li, Y. Jiang, X. Liu and Y. Chen, Nanoscale, 2019, 11, 1058–1064 RSC.
- B. Zhu, Z. Liang and R. Zou, Small, 2020, 16, 1906133 CrossRef CAS PubMed.
- C. Liu, X. R. Shi, K. Yue, P. Wang, K. Zhan, X. Wang, B. Y. Xia and Y. Yan, Adv. Mater., 2023, 35, 2211177 CrossRef CAS PubMed.
- Y. Sun, H. Shin, F. Wang, B. Tian, C. W. Chiang, S. Liu, X. Li, Y. Wang, L. Tang, W. A. I. Goddard and M. Ding, J. Am. Chem. Soc., 2022, 144, 15185–15192 CrossRef CAS PubMed.
- J. Zhang, J. Dang, X. Zhu, J. Ma, M. Ouyang and F. Yang, Appl. Catal., B, 2023, 325, 122296 CrossRef CAS.
- Z. Yin, R. He, Y. Zhang, L. Feng, X. Wu, T. Wågberg and G. Hu, J. Energy Chem., 2022, 69, 585–592 CrossRef CAS.
- Y. J. Lee and S. K. Park, Small, 2022, 18, 2200586 CrossRef CAS PubMed.
- A. Pei, R. Xie, Y. Zhang, Y. Feng, W. Wang, S. Zhang, Z. Huang, L. Zhu, G. Chai, Z. Yang, Q. Gao, H. Ye, C. Shang, B. H. Chen and Z. Guo, Energy Environ. Sci., 2023, 16, 1035–1048 RSC.
- B. Zhou, Y. Li, Y. Zou, W. Chen, W. Zhou, M. Song, Y. Wu, Y. Lu, J. Liu, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 22908–22914 CrossRef CAS PubMed.
- Z. Liu, J. Li, S. Xue, S. Zhou, K. Qu, Y. Li and W. Cai, J. Energy Chem., 2020, 47, 317–323 CrossRef.
- H. Zhang, F. Wan, X. Li, X. Chen, S. Xiong and B. Xi, Adv. Funct. Mater., 2023, 33, 2306340 CrossRef CAS.
- C. Hu, Y. Hu, C. Fan, L. Yang, Y. Zhang, H. Li and W. Xie, Angew. Chem., Int. Ed., 2021, 60, 19774–19778 CrossRef CAS PubMed.
- D. Y. Wei, M. F. Yue, S. N. Qin, S. Zhang, Y. F. Wu, G. Y. Xu, H. Zhang, Z. Q. Tian and J. F. Li, J. Am. Chem. Soc., 2021, 143, 15635–15643 CrossRef CAS PubMed.
- S. Lin, H. Ze, X. G. Zhang, Y. J. Zhang, J. Song, H. Zhang, H. L. Zhong, Z. L. Yang, C. Yang, J. F. Li and Z. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202203511 CrossRef CAS PubMed.
- A. Mosallanezhad, C. Wei, P. Ahmadian Koudakan, Y. Fang, S. Niu, Z. Bian, B. Liu, T. Huang, H. Pan and G. Wang, Appl. Catal., B, 2022, 315, 121534 CrossRef CAS.
- S. Vijayapradeep, N. Logeshwaran, S. Ramakrishnan, A. Rhan Kim, P. Sampath, D. Hwan Kim and D. Jin Yoo, Chem. Eng. J., 2023, 473, 145348 CrossRef CAS.
- W. Zhang, Q. Jia, H. Liang, L. Cui, D. Wei and J. Liu, Chem. Eng. J., 2020, 396, 125315 CrossRef CAS.
- T. Wang, L. Miao, S. Zheng, H. Qin, X. Cao, L. Yang and L. Jiao, ACS Catal., 2023, 13, 4091–4100 CrossRef CAS.
- K. L. Zhou, C. Wang, Z. Wang, C. B. Han, Q. Zhang, X. Ke, J. Liu and H. Wang, Energy Environ. Sci., 2020, 13, 3082–3092 RSC.
- Y. Tian, M. Wen, A. Huang, Q. Wu, Z. Wang, Q. Zhu, T. Zhou and Y. Fu, Small, 2023, 19, 2207569 CrossRef CAS PubMed.
- B. Tang, X. Yang, Z. Kang and L. Feng, Appl. Catal., B, 2020, 278, 119281 CrossRef CAS.
- M. Li, H. Wang, W. Zhu, W. Li, C. Wang and X. Lu, Adv. Sci., 2020, 7, 1901833 CrossRef CAS PubMed.
- Z. Xu, Q. Chen, Q. Chen, P. Wang, J. Wang, C. Guo, X. Qiu, X. Han and J. Hao, J. Mater. Chem. A, 2022, 10, 24137–24146 RSC.
- L. Sha, K. Ye, J. Yin, K. Zhu, K. Cheng, J. Yan, G. Wang and D. Cao, Chem. Eng. J., 2020, 381, 122603 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2012, 81, 292–300 CrossRef CAS.
- A. V. Munde, B. B. Mulik, P. P. Chavan and B. R. Sathe, Electrochim. Acta, 2020, 349, 136386 CrossRef CAS.
- D. Voiry, M. Chhowalla, Y. Gogotsi, N. A. Kotov, Y. Li, R. M. Penner, R. E. Schaak and P. S. Weiss, ACS Nano, 2018, 12, 9635–9638 CrossRef CAS PubMed.
- C. Liu, H. Ma, M. Yuan, Z. Yu, J. Li, K. Shi, Z. Liang, Y. Yang, T. Zhu, G. Sun, H. Li and S. Ma, Electrochim. Acta, 2018, 286, 195–204 CrossRef CAS.
- X. Fu, R. Shi, S. Jiao, M. Li and Q. Li, J. Energy Chem., 2022, 70, 129–153 CrossRef CAS.
- S. W. Tatarchuk, J. J. Medvedev, F. Li, Y. Tobolovskaya and A. Klinkova, Angew. Chem., Int. Ed., 2022, 61, e202209839 CrossRef CAS PubMed.
- R. Li, H. Xu, P. Yang, D. Wang, Y. Li, L. Xiao, X. Lu, B. Wang, J. Zhang and M. An, Nano-Micro Lett., 2021, 13, 120 CrossRef CAS PubMed.
- Z. Gao, Y. Wang, L. Xu, Q. Tao, X. Wang, Z. Zhou, Y. Luo, J. Yu and Y. Huang, Chem. Eng. J., 2022, 433, 133515 CrossRef CAS.
- Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Energy Environ. Sci., 2018, 11, 1890–1897 RSC.
- Q. Zhang, F. M. D. Kazim, S. Ma, K. Qu, M. Li, Y. Wang, H. Hu, W. Cai and Z. Yang, Appl. Catal., B, 2021, 280, 119436 CrossRef CAS.
- M. Zhong, J. Yang, M. Xu, S. Ren, X. Chen, C. Wang, M. Gao and X. Lu, Small, 2023, 2304782, DOI:10.1002/smll.202304782.
- L. Sha, T. Liu, K. Ye, K. Zhu, J. Yan, J. Yin, G. Wang and D. Cao, J. Mater. Chem. A, 2020, 8, 18055–18063 RSC.
- S. Chen, J. Duan, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 3804–3808 CrossRef CAS PubMed.
- H. Xu, K. Ye, K. Zhu, Y. Gao, J. Yin, J. Yan, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2020, 8, 16037–16045 CrossRef CAS.
- Y. Ye, Y. Gan, R. Cai, X. Dai, X. Yin, F. Nie, Z. Ren, B. Wu, Y. Cao and X. Zhang, J. Alloys Compd., 2022, 921, 166145 CrossRef CAS.
- L. Zhang, T. Wang, H. Wu, H. Wang and F. Wang, J. Alloys Compd., 2022, 918, 165564 CrossRef CAS.
- I. M. A. Mohamed, P. Kanagaraj, A. S. Yasin, W. Iqbal and C. Liu, J. Alloys Compd., 2020, 816, 152513 CrossRef CAS.
- Y. Yang, J. Kim, H. Jo, A. Seong, M. Lee, H.-K. Min, M.-G. Seo, Y. Choi and G. Kim, J. Mater. Chem. A, 2021, 9, 11571–11579 RSC.
- C. Sanchez, F. J. Espinos, A. Barjola, J. Escorihuela and V. Compan, Polymers, 2022, 14, 4500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03398h |
| This journal is © The Royal Society of Chemistry 2024 |