
A high-efficiency and stable organic solar cell with balanced crystallization kinetics†
Weichao
Zhang
ac,
Yaochang
Yue
a,
Rongsheng
Yang
a,
Yingyu
Zhang
c,
Wenna
Du
 c,
Guanghao
Lu
d,
Jianqi
Zhang
c,
Huiqiong
Zhou
*c,
Xuning
Zhang
*b and
Yuan
Zhang
*a
c,
Guanghao
Lu
d,
Jianqi
Zhang
c,
Huiqiong
Zhou
*c,
Xuning
Zhang
*b and
Yuan
Zhang
*a
aSchool of Chemistry, Beijing Advanced Innovation Center for Biomedical Engineering, Beihang University, Beijing 100191, China. E-mail: yuanzhang@buaa.edu.cn
bCollege of Physics Science & Technology, Hebei University, Baoding 071002, Heibei Province, China. E-mail: zhangxn@hbu.edu.cn
cCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: zhouhq@nanoctr.cn
dFrontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710054, Shaaxi Province, China
First published on 1st February 2024
Abstract
Obtaining controllable morphology in organic solar cells (OSCs) has long been sought to improve the photovoltaic efficiency and long-term stability for meaningful applications. Herein, we report a conceptual multiple acceptor OSC based on co-acceptor guests. Through monitoring the solution phase to solid-state film transition of the multi-acceptor based bulk heterojunctions (BHJs) by in situ optical spectroscopy, we show that the introduced co-acceptor guests featuring opposite crystallization trends are highly beneficial for attaining synergies in balancing the crystallization kinetics and modifying phase separation and charge carrier transport behaviors. Our combined investigation with GIWAXS, solid-state NMR, depth-sensitive optical spectroscopy and transient opto-electrical measurements further reveals the central functionality of the co-acceptor guests in leveraging and optimizing the crystallization dynamics and vertical phase separation while maintaining the favored short-range structural order. Based on a multi-acceptor model system, PM6:BTP-eC9:Y6-1O:PC71BM, we achieved a champion power conversion efficiency (PCE) of 19.35%, showing impressive photostability with the PCE decaying by <20% after ∼30 days of continuous irradiation. The demonstrated multi-component approach provides a valuable opportunity for fine optimization of the phase morphology and long-term device stability in OSCs toward realistic energy conversion applications.
Broader contextAlthough it is possible to improve the efficiency of organic solar cells (OSC) via the ternary (or quaternary) approach, our current understanding on how the third (or fourth) component modulates the phase morphology of OSCs and explicitly the structure–function relationship in the presence of molecular guests remains to be fully clarified. Herein, we employ combined experimental techniques, including in situ optical spectroscopy and solid-state nuclear magnetic resonance to reveal the essential role of introducing co-mediators comprising a non-fullerene and a fullerene electron acceptor in regulating the morphology and photovoltaic behaviors of OSCs. This not only enriches our insights into the factors governing morphology control but also allows us to achieve an impressive photovoltaic efficiency exceeding 19.3%. The demonstrated quaternary heterojunction exhibits an excellent morphology that translates in a more satisfactory operational stability of OSCs under irradiation, which helps push forward the OSC technology toward realistic energy-related applications. |
Solution-processed organic solar cells (OSCs), a next-generation photovoltaic technology for converting solar energy into electricity, have attracted enormous attention due to their structural simplicity, light weight, mechanical flexibility, low cost, and capability to be fabricated using large-area printing techniques. In the past decades, there has been tremendous progress in the design of new electron-donor and acceptor materials along with innovations in device engineering, pushing forward the power conversion efficiency (PCE) of OSCs to the 19% regime.1–6
In order to render OSCs suitable for meaningful applications, efficiencies above the necessitated threshold PCE values should be achieved. However, due to the narrow absorption characteristics of organic semiconductor materials (typically with an FWHM bandwidth of 200 nm), it is difficult to achieve a full utilization of the solar spectrum in donor–acceptor based binary devices, which limits the improvement of PCEs. To overcome this, ternary blend strategies have been employed. While this approach enables the enhancement of PCEs,3,7–10 complexities in optimizing the phase morphology of heterojunction (BHJ) films are often encountered. For example, the introduction of guest components may accelerate (or decelerate) the crystallization of host components in an unfavorable way,11,12 resulting in a scenario where the phase separation scale or crystallinity of blends is sub-optimal to fully realize the material potential.13–15 Moreover, the addition of single-component based guests (for ternary blends) tends to restrain the absorption complementarity of photoactive layers.16–18 To widen the absorption of OSCs and better control the nanomorphology and carrier transport,18–23 the approach based on quaternary blends has been proposed.24–28 For example, by introducing a co-donor and a co-acceptor (co-DA) into the host blends, the inter-percolated charge transport networks in the quaternary OSCs were improved, leading to a certified power conversion efficiency (PCE) of over 19%.5 Also, the co-DA method enabled achieving a branch-like morphology in the photoactive layer, which further reduced the voltage loss due to non-radiative charge recombination.29 On the basis of multiple polymeric donor components, a proposal was made for improving the hole transport and light-harvesting efficiency in OSCs.30 In this attempt, the multi-donor blends were thought to allow for the increase in the population of D–A interfaces while photo-generated holes were transported independently through the multiple donor phases.31–33 To rationally construct and optimize the quaternary target systems, various properties/parameters need to be considered, including the absorption profile, energetic level, inter-component compatibility, molecular interaction and crystallinity.33 To date, the commonly adopted approaches in this regard rely on trial-and-error, which to a large extent is due to the lack of effective pre-monitoring and control methods for the development of nanomorphology.
In this work, based on the widely used polymer donor PM6 and the non-fullerene acceptor (NFA) BTP-eC9 as the binary hosts, we present a proof-of-concept study for fine morphology regulation and prediction based on a multiple acceptor-containing quaternary system. We chose NFA Y6-1O34 and fullerene acceptor PC71BM as co-acceptor guests. Given the donor's proneness to crystallize rapidly and the similarity of melting rings between the NFAs of BTP-eC935 and Y6-1O, the introduction of the Y6-1O guest results in a competing crystallization process where the unfavorable large domain growth of PM6 donor is suppressed. Meanwhile, the PC71BM guest with a high phase crystallinity facilitates the NFA molecules to form dense π–π stacking, improving electron transport. The targeted morphological development in multi-acceptor-containing blends was monitored by in situ optical spectroscopy, revealing the bidirectionally compensatory role of the modified aggregating status of D–A molecules to achieve balanced crystallization kinetics during the film formation process. Furthermore, we employed solid-state nuclear magnetic resonance (NMR) spectroscopy to understand molecular order at the atomic level. The results evidence that in the presence of co-acceptor guests, the microscopic short-range structural order of the PM6 donor can be retained, which provides a foundation for improvements in crystallization. With the multi-acceptor concept, a PCE of 19.35% was attained and the champion quaternary device exhibited an impressive morphology stability, with the PCE decaying by less than 20% after nearly 30 days of continuous irradiation. The demonstrated approach provides an enabler for fine optimization of nanomorphology and maximization of light-harvesting in OSCs toward meaningful applications.
Results
Materials properties and device performance
The chemical structures of the utilized PM6, BTP-eC9, Y6-1O and PC71BM are shown in Fig. 1(a). Given the similar chemical structures between BTP-eC9 and Y6-1O, these two NFAs were expected to form alloy-like composites within the blends.36–38 As an affirmation, we performed contact angle (CA) measurements on pure donor and acceptor films (see results in Fig. 1(b)). The surface tension (γ) values for pure films of PM6, BTP-eC9, Y6-1O and PC71BM were determined to be 35.32, 40.99, 40.76 and 46.49 mJ m−2, respectively (Fig. 1(c)). BTP-eC9 and Y6-1O exhibit very similar surface tension values, suggesting that these two NFA components likely form alloy acceptors. We further estimated the miscibility between the donor and acceptors by calculating the Flory Huggins interaction parameter χD–A (= (γ1/2D − γ1/2A)2, see data in Table S1, ESI†). The χD–A values between PM6 and BTP-eC9, Y6-1O and PC71BM are 0.21, 0.19 and 0.77, respectively; while they decrease to 0.15 (PM6 and BTP-eC9:Y6-1O) and 0.18 (PM6 and BTP-eC9:Y6-1O:PC71BM) (see results in Fig. S1, ESI†). Obviously, PM6 shows better molecular compatibility with BTP-eC9 or Y6-1O than with PC71BM. Therefore, appropriate intermolecular miscibility between NFAs can be retained and meanwhile excessive aggregations of donor/acceptor materials are suppressed through PC71BM.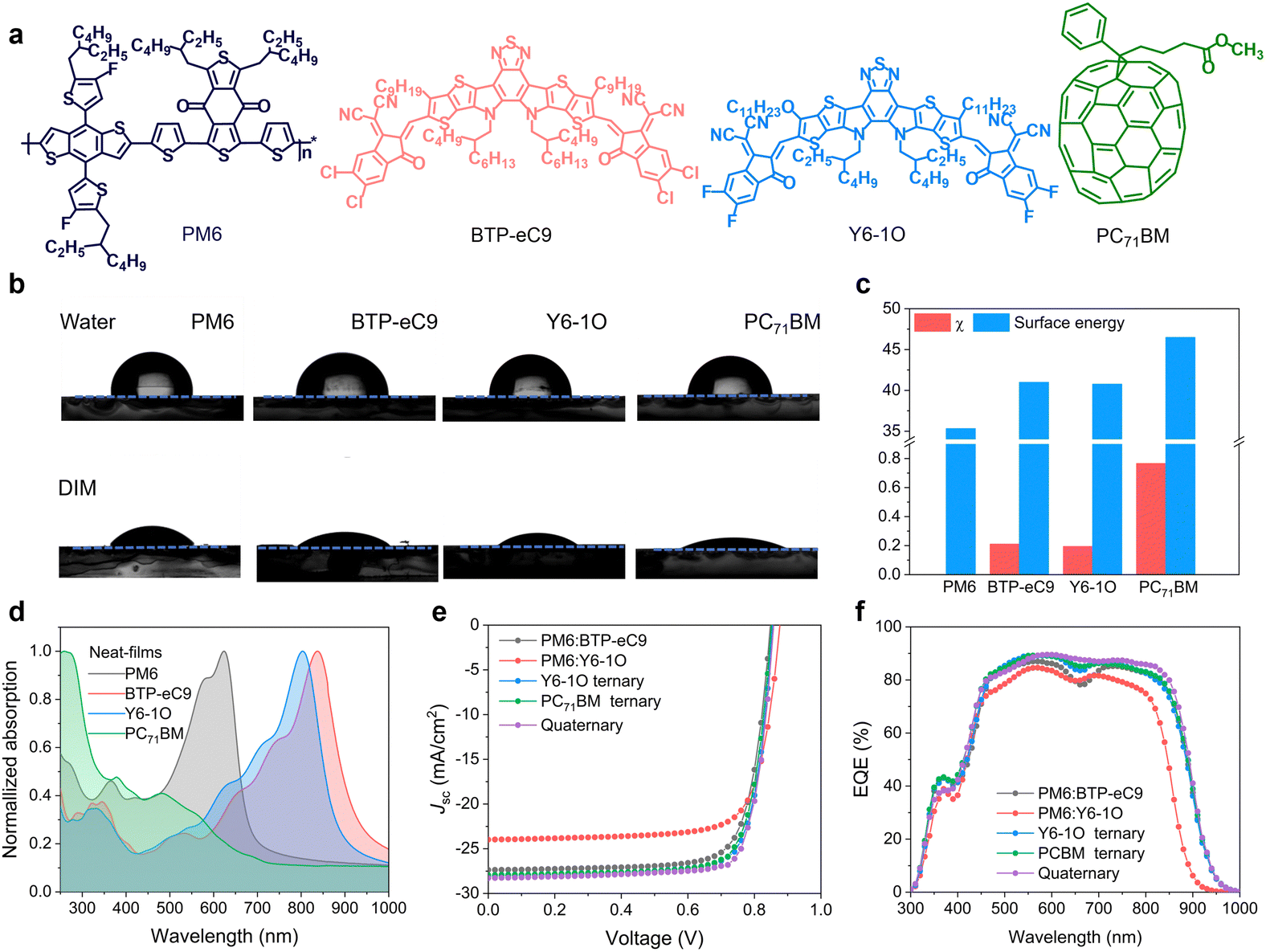 | ||
| Fig. 1 (a) Chemical structures of utilized materials in this study. (b) Snapshots of water and DIM droplets on the top surfaces of different pure films. (c) Surface energy of neat films and χ values of relevant components. (d) UV-vis absorption spectra of PM6, BTP-eC9, Y6-1O and PC71BM based neat films. (e) Current density versus voltage characteristics of binary, ternary, and quaternary solar cells under AM 1.5 g illumination. (f) EQE curves of the corresponding devices. | ||
Fig. 1(d) and Fig. S2 (ESI†) show normalized absorption spectra of neat and blend films. As seen from Fig. 1(d), the concerned donor and acceptor materials exhibit complementary light absorption, covering a wide spectral range from 350 to 950 nm. As shown in Fig. S3a, ESI,† due to the large spectral overlap between the absorption of BTP-eC9 and the photoluminescence (PL) of Y6-1O, along with the fact that the emission of Y6-1O has higher energy than the absorption of BTP-eC9, there is a tendency for energy transfer between these two NFAs, which was considered beneficial for the photon-harvesting of acceptor components.13 As a support, we measured PL spectra of neat films of BTP-eC9, Y6-1O and BTP-eC9:Y6-1O blend films (Fig. S3b, ESI†). The PL spectra of neat BTP-eC9 and Y6-1O are positioned between 780 nm and 950 nm with emission maxima at 862 nm (BTP-eC9) and 872 nm (Y6-1O). The emission peak of the BTP-eC9:Y6-1O blend film is at 870 nm, seemingly pointing to the energy transfer from Y6-1O to BTP-eC9.42,43 We further note that there is a cascade energetic level arrangement across these photo-absorbing materials (Fig. S4, ESI†). This feature provides merits for electron transfers between acceptors and charge transport through the device.44
To explore the impact of the introduced acceptor co-guests on photovoltaic performance, OSC devices were fabricated with a conventional device architecture of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS)/active layer/poly[[2,7-bis(2-ethylhexyl)-1,2,3,6,7,8-hexahydro-1,3,6,8-tetraoxobenzo[lmn]phenanthroline-4,9-diyl]-2,5-thiophenediyl[9,9-bis [3-(dimethylamino)propyl]-9H-fluorene-2,7-diyl]-2, 5-thiophenediyl] (PNDIT-F3N)/Ag. Fig. 1(E) shows current density versus voltage (J–V) characteristics of OSC devices under AM 1.5 G (100 mW cm−2) illumination. The extracted photovoltaic parameters can be found in Table 1. The PM6:BTP-eC9 binary device delivered a PCE of 17.86% (Jsc = 27.36 mA cm−2, Voc = 0.848 V, and FF = 76.85%), which is consistent with the results reported previously.37 For comparison, the PM6:Y6-1O binary device yielded a PCE of 15.90% (Voc = 0.877 V, Jsc = 23.98 mA cm−2, and a FF of 75.56%). We also fabricated ternary solar cells by adding Y6-1O or PC71BM as the third component. The maximum efficiencies are 18.81% with Y6-1O (Voc = 0.855 V, Jsc = 28.06 mA cm−2, and a FF of 78.15%) and 18.66% with PC71BM (Voc = 0.851 V, Jsc = 27.94 mA cm−2, and a FF of 78.32%). Of note, upon adding the two guest acceptors simultaneously, the PCE (19.35%) increased considerably (Voc = 0.858 V, Jsc = 28.27 mA cm−2, and a FF of 79.65%), owing to the concurrent enhancements of cell parameters (see Table 1). We observed a 10 meV increase in the Voc of the four-component device compared to the binary device (PM6:BTP-eC9). Further evidence supporting the voltage gain was obtained by analyzing the external quantum efficiency (EQE) of electroluminescence (EQEEL, see Fig. S5, ESI†) of solar cells operated like a light-emitting diode. The results of EQEEL confirm that non-radiative voltage losses are reduced in the four-component device, contributing to the higher Voc. In Fig. 1(f), we show the EQE spectra of the corresponding binary, ternary and quaternary devices. The Jsc values integrated from the EQE are consistent with the values obtained from illuminated J–V curves. Apart from the retention of of high EQEs in the donor absorption regime, the quaternary device displays a pronounced EQE enhancement in the acceptor absorption regime (reaching a peak value approaching 90%), showing a more flattened spectral response. This is in contrast to the pronouncedly reduced EQEs around the absorption valley (around 700 nm) in the binary control cell. By comparing the absolute absorption of respective BHJ films (Fig. S2, ESI†), the EQE gains in the quaternary device cannot be solely attributed to the (slight) changes in the absorbance of photoactive layers but other attributes, which are likely related to the improved transport and/or mitigated charge recombination (will be further discussed below).
| Active layer | V oc (V) | J sc (mA cm−2) | J cal (mA cm−2) | FF (%) | PCEb (%) |
|---|---|---|---|---|---|
| a Integrated current densities from EQE spectra. b Average PCEs based on 10 devices. | |||||
| PM6:BTP-eC9 | 0.848 (0.845 ± 0.002) | 27.36 (27.08 ± 0.12) | 26.49 | 76.85 (76.49 ± 0.27) | 17.86 (17.52 ± 0.15) |
| PM6:Y6-1O | 0.877 (0.876 ± 0.003) | 23.98 (23.45 ± 0.23) | 23.52 | 75.56 (75.13 ± 0.15) | 15.90 (15.60 ± 0.30) |
| PM6:BTP-eC9:Y6-1O | 0.855 (0.852 ± 0.002) | 28.06 (27.76 ± 0.17) | 26.97 | 78.15 (77.65 ± 0.41) | 18.81 (18.35 ± 0.19) |
| PM6:BTP-eC9:PC71BM | 0.851 (0.850 ± 0.002) | 27.94 (27.42 ± 0.38) | 26.89 | 78.32 (78.96 ± 0.21) | 18.66 (18.52 ± 0.16) |
| PM6:BTP-eC9:Y6-1O:PC71BM | 0.858 (0.854 ± 0.003) | 28.27 (28.04 ± 0.26) | 27.25 | 79.65 (78.96 ± 0.38) | 19.35 (19.13 ± 0.12) |
In situ monitoring of film-formation and morphology development
To fully understand and control the morphology, monitoring the film formation process of BHJ films is of critical importance. Here, we utilized in situ UV-vis and PL spectroscopic measurements to gain insights into the effects of the introduced co-guest components on morphological evolution.12,39,40Fig. 2(a) shows the temporal evolution of UV-vis absorption contour maps of different blends (see raw data of in situ spectra in Fig. S6, ESI†), where the colors represent the intensity of reflectance signals at a specific time and wavelength. According to literature reports,12 the crystallization process can be divided into three temporal stages, namely, the solvent evaporation stage, nucleation and crystal growth stage, and eventually the dried film stage. Given that the absorption of PM6 and BTP-eC9 is distinguishable, we separately plotted the absorption peak positions of the two hosts without and with adding the guest components as a function of time (see Fig. 2(b) and (c)). By doing so, useful information on the evolution of aggregating status or the crystallization rate was obtained. As seen, the absorption peaks of PM6 and BTP-eC9 in all blend films present different degrees of red shifts (from 603 to 613 nm for PM6 and from 739 nm to 802 nm for BTP-eC9). This tendency implies the presence of orderly packing in these molecules.49 We further note that the three stages in the event of film-formation vary among different material systems and the role of the two acceptor guests in mediating the film-formation is discernable. By including only the Y6-1O guest, the crystallization in the donor and acceptor hosts tends to accelerate, as revealed by the shortened drying time (0. 84 s for the donor and 0.96 s for acceptor). Upon adding the PC71BM guest only, the crystallization time of the donor decreases slightly while the crystallization time of the acceptor host remains unchanged. This stark contrast strongly suggests that Y6-1O has a greater impact on expediting the crystallization of blends while PC71BM tends to decelerate the crystallization, at least for the donor. These behaviors are in line with the observation that the film morphology of the blends containing only Y6-1O or PC71BM exhibits an inversely changing trend compared to that of the reference binary blend, which will be discussed below.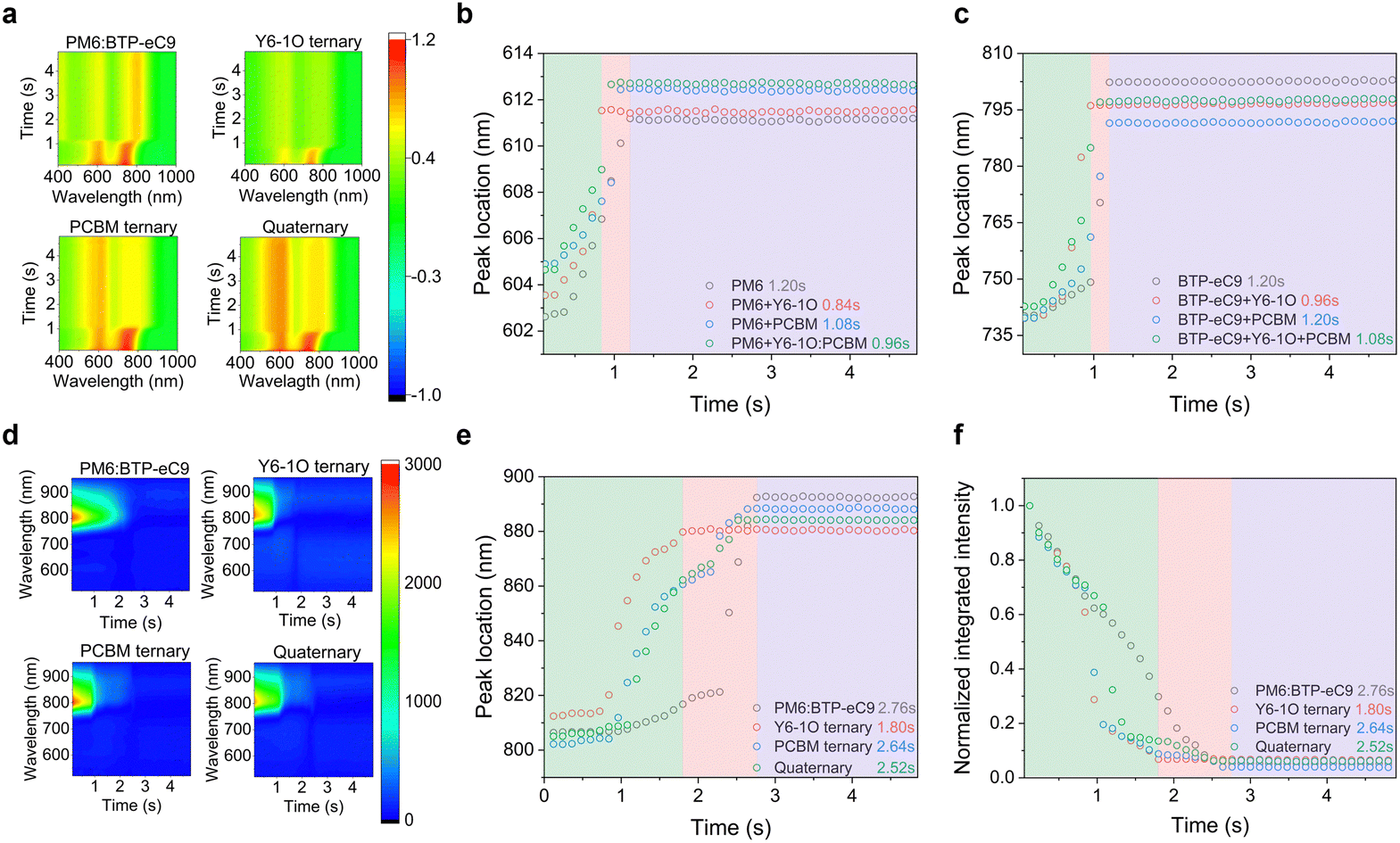 | ||
| Fig. 2 (a) Time-dependent contour maps of UV-vis absorption spectra measured on different blends. Plots of spectral evolution probed at the (b) host donor and (c) acceptor absorption peaks based on the results in (a). (d) Time-dependent contour maps of PL spectra based on various BHJ films. (e), (f) Time evolution of peak location and normalized PL intensity for optimal BHJ films. | ||
After elaborating the key functionalities of the introduced acceptor guests, it will be of interest to examine how the crystallization kinetics is affected with the simultaneous incorporation of Y6-1O and PC71BM as co-guests. As evidenced in Fig. 2(b) and (c), for the quaternary blends the crystallization time of the host donor (0.96 s) and acceptor (1.08 s) is modified within a window between that of the two ternary blends. Based on this, we consider that the introduced co-guest acceptors promote the balance between the crystallization rate of the donor and acceptor molecules. Generally speaking, a more balanced aggregation kinetics can lead to more uniform growth of donor–acceptor phase domains, which can contribute to the improvement of light-harvesting efficiency in OSCs (given the comparable diffusion distance of excitons generated in the D and A components), as well as the charge transport balance.41,42
We further evaluated the quenching kinetics through in situ PL characterization. Fig. 2(d) shows the contour maps for PL evolution (see in situ spectra in Fig. S7, ESI†). According to the characterization results in Figure S7, the extracted temporal evolutions of the integrated PL intensity and peak position are displayed in Fig. 2(e) and (f) respectively, which are correlated with the spatial distribution of fluorophores and quenchers in the blends. The evolution trend of the PL peak position resembles that in the in situ absorption spectra, characteristic of three stages. For PM6:BTP-eC9, the PL peak location during the first stage (0–0.84 s) remains unchanged with time, while the PL intensity decreases slowly with the evaporation of solvent. For PM6:BTP-eC9:Y6-1O and PM6:BTP-eC9:PC71BM ternary blends, the PL peak retention time slightly differs during the first stage (0.72 s vs. 0.84 s) while both are shorter than the corresponding time for the binary blend film. For the quaternary system, the retention time of the PL peak position is further shortened (0–0.60 s). It points to two facts: (i) the combined interactions involving Y6-1O and PC71BM tend to accelerate the growth rate of the film and (ii) the fluorescence quenching is more efficient when multi-acceptors are present (see Fig. S8, ESI†). In the second stage, we note that the rate of change of the PL intensity is significantly larger than that of the PL peak position. Such rapid drops in the signal can be ascribed to the interaction between the fluorophore and the quencher.43 For the PM6:BTP-eC9 binary blend, the PL quenching (0.84–2.76 s) presents a simple process that decreases over time. In contrast, the quenching of the PM6:BTP-eC9:Y6-1O blend exhibits a two-part feature (0.72–1.80 s), including a rapid quenching process (0.72–1.08 s) followed by a slow quenching process (1.08–1.80 s). The 1st step can result from a further shortening of the distance between the donor and acceptor molecules as the solvent volatilizes.44 The 2nd process corresponds to the slowly occurring aggregation in BTP-eC9:Y6-1O after the rapid aggregation is completed. The peculiar quenching behaviors are also observed in the PM6:BTP-eC9:PC71BM ternary blends. Referring to the quaternary system, the changing rate of PL intensity is pronouncedly higher. This phenomenon is explained by the increase in the probability of contact between the polymer donor and muti-acceptors when Y6-1O and PC71BM are simultaneously introduced. The results also imply another aspect related to energy transfer, which is likely promoted in the presence of the co-acceptor guests. The last stage of PL evolution is similar to the evolution of absorption, representing the solvent removal toward the stabilization of phase morphology in the solid state (Fig. 2(b) and (c)). The in situ PL measurements affirm the modified interdiffusion process between the donor and acceptor molecules during film evolution, leading to favorable crystallization and morphology.
Depth-dependent phase distribution and exciton generation rate
To further investigate the vertical phase distribution in these blends, we performed FLAS (film-depth-dependent light absorption spectroscopy) characterization.45 The FLAS results are shown in Fig. S9 (ESI†) where the characteristic absorption peaks of the donor and acceptor materials display different intensities along the depth direction of the film. Based on a comparative analysis of the absorption of neat D and A components (Fig. 1(d)) alongside the results of FLAS, we provide the quantitative assessments on the vertical distribution of each component in these blend films (Fig. 3(a)–(d)). We observe a unique feature in the quaternary system that the donor and acceptor components are distributed more homogeneously along the film growth direction, e.g., in the 20–80 nm depth region. This particularity can contribute to the reduction of bimolecular charge recombination, thus helpful for the improvement of the photocurrent and device fill factor.46 On top of this, more pronounced enrichment of the acceptor host in the quaternary blend film is indicated (Fig. 3(d)), providing benefits for the suppression on interfacial charge accumulation and recombination.25 Based on the results of FLAS, we further simulated the distribution in exciton generation rate density (Gd, see 2D maps of Gd in Fig. S8E–H, ESI†). The integrated Gd values along the depth direction of the active layer are comparatively plotted in Fig. 3(e). The results show that the exciton generation rate in the quaternary blends is, on average, higher inside the bottom to mid-film area, showing a Gd maximum value of 1.532 × 1030 m−2 s−1 (versus Gd maxima of 1.470 × 1030 m−2 s−1 for PM6:BTP-eC9, 1.525 × 1030 m−2 s−1 for PM6:BTP-eC9:Y6-1O and 1.510 × 1030 m−2 s−1 for PM6:BTP-eC9:PC71BM). In Fig. 3(f), we show the total exciton generation rate density obtained by integrating the 3D spatial distribution of Gd. Consistently, the quaternary blends are associated with the highest Gd value (compared to that of the other blends), meaning the highest efficiency in exciton generation throughout the entire photoactive layer.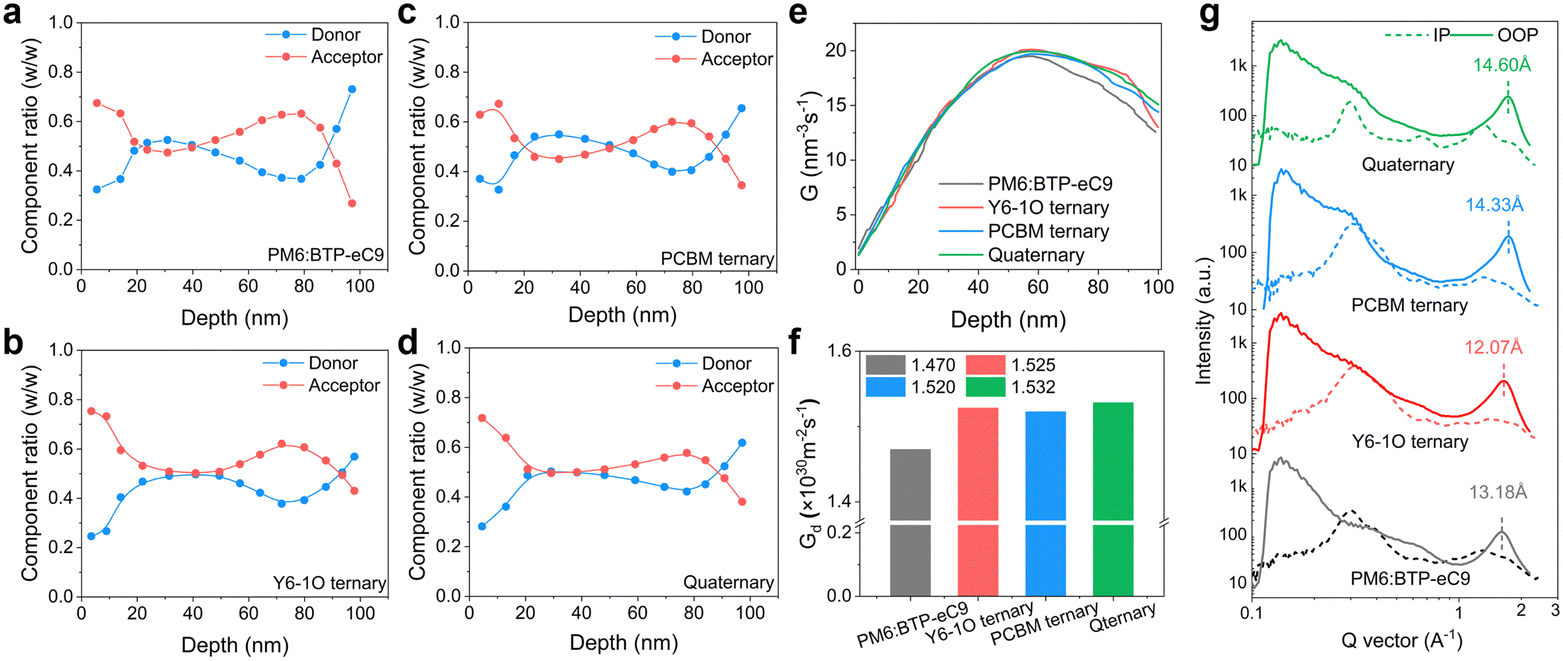 | ||
| Fig. 3 (a)–(d) Composition ratio of donor and acceptor components along the vertical direction of the active layer. (e) Integrated generation rate in the vertical direction of blend films. (f) Calculated exciton generation rate through the entire active layer of solar cells. (g) GIWAXS intensity profiles of different BHJ films along the in-plane (dashed lines) and out-of-plane (solid lines) directions. | ||
Combined analyses on phase morphology
Next, we examined the properties of molecular packing and crystallinity by grazing incidence wide-angle X-ray diffraction (GIWAXS, see 2D GIWAXS maps in Fig. S10, ESI†). The line-cut profile curves of various blend films are shown in Fig. 3(g) (see GIWAXS results of neat PM6 films in Fig. S11, ESI†). The related morphology parameters determined from the GIWAXS measurements are summarized in Table S2 (ESI†). The (010) peaks corresponding to the binary, ternary and quaternary blend films are positioned at 23.15, 24.75, 24.95 and 24.9 Å, corresponding to the π–π distances of 1.63, 1.74, 1.75, and 1.75 Å−1, respectively. Regarding the quaternary blends, we note an increase in the crystal coherence length (CCL = 14.60 nm versus CCLs of 13.18, 12.07, and 14.33 Å for PM6:BTP-eC9, PM6:BTP-eC9:Y6-1O, and PM6:BTP-eC9:PC71BM, respectively) along the out-of-plane direction. The feature is helpful for the enhancement of charge carrier transport in the OSC. The modified intermolecular packing in the quaternary blends corresponds to a reduction in the Urbach energy (Fig. S12, ESI†) or energetic disorder in tail states.47,48To gain a better understanding of the photophysical and morphological properties in these blends, transient absorption (TA) spectroscopy was performed to examine the hole transfer (HT) kinetics and exciton diffusion characteristics. The pronounced ground state bleaching (GSB) bands (500–670 nm) originate from the PM6 donor (see Fig. S13(e) and (f), ESI†). Based on the 2D maps of TA spectra (Fig. S11(a)–(d), ESI†), we focus on the decay kinetics of GSB bands (probe at 635 nm). As seen in Fig. 4(a) inset, the decays feature a fast rising followed by a slow decay part characterized by the time constants τ1 and τ2, respectively. The fast component τ1 is commonly assigned to the dissociation of acceptor-generated excitons formed at the D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A interface. The second component τ2 is a representation of the diffusion-limited dissociation process involving excitons generated in the bulk film.49 The concerned binary, ternary and quaternary films exhibit a similar time constant τ1 (see Fig. 4(a) and Table S3 in ESI†), indicative of the temporally comparable photoexcitation in these blends. The decay time constant τ2 is associated with the domain size and phase separation.50 Of interest, by adding a single acceptor guest of Y6-1O or PC71BM to the binary hosts, the decay time τ2 sharply reduces to 239 ps or adversely increases to 311 ps. These trends outline a scenario where the diffusion-limited dissociation occurs more rapidly with the addition of only Y6-1O, hinting a shrinkage of phase separation. Oppositely, the diffusion toward interfacial dissociation may take a longer time with the addition of only PC71BM, implicative of an enlargement of phase-separation or grain sizes. In comparison to the ternary blends, we observe an intermediate τ2 value (250 ps) in the quaternary blends (Fig. 4(a)). Based on these results, we consider that the co-introduction of Y6-1O and PC71BM guest components synergistically modifies the phase sizes, such that over-phase separation or over-intermixing could be avoided. These merits can be attributed to the improved balance in the crystallization kinetics in the multicomponent-based blends. The surface morphology captured by atomic force microscopy confirms the modified phase separation (see results of AFM in Fig. 4(b)–(e)). The binary blend film PM6:BTP-eC9 has a surface roughness (Rq) of 1.33 nm (a Rq of 2.06 nm was determined on pure PM6 films, see Fig. S14, ESI†). The addition of Y6-1O or PC71BM guests results in a different changing trend in the surface topography and roughness (Rq = 1.21 nm with Y6-1O versus Rq = 1.64 nm with PC71BM). Compared to the binary or ternary blends, the quaternary blend film presents an intermediate change in surface roughness (Rq = 1.45 nm), associated with a finely tuned structure with more ordered features. This can be attributed to the improved crystallization dynamics or the balance in crystallization rates.
:A interface. The second component τ2 is a representation of the diffusion-limited dissociation process involving excitons generated in the bulk film.49 The concerned binary, ternary and quaternary films exhibit a similar time constant τ1 (see Fig. 4(a) and Table S3 in ESI†), indicative of the temporally comparable photoexcitation in these blends. The decay time constant τ2 is associated with the domain size and phase separation.50 Of interest, by adding a single acceptor guest of Y6-1O or PC71BM to the binary hosts, the decay time τ2 sharply reduces to 239 ps or adversely increases to 311 ps. These trends outline a scenario where the diffusion-limited dissociation occurs more rapidly with the addition of only Y6-1O, hinting a shrinkage of phase separation. Oppositely, the diffusion toward interfacial dissociation may take a longer time with the addition of only PC71BM, implicative of an enlargement of phase-separation or grain sizes. In comparison to the ternary blends, we observe an intermediate τ2 value (250 ps) in the quaternary blends (Fig. 4(a)). Based on these results, we consider that the co-introduction of Y6-1O and PC71BM guest components synergistically modifies the phase sizes, such that over-phase separation or over-intermixing could be avoided. These merits can be attributed to the improved balance in the crystallization kinetics in the multicomponent-based blends. The surface morphology captured by atomic force microscopy confirms the modified phase separation (see results of AFM in Fig. 4(b)–(e)). The binary blend film PM6:BTP-eC9 has a surface roughness (Rq) of 1.33 nm (a Rq of 2.06 nm was determined on pure PM6 films, see Fig. S14, ESI†). The addition of Y6-1O or PC71BM guests results in a different changing trend in the surface topography and roughness (Rq = 1.21 nm with Y6-1O versus Rq = 1.64 nm with PC71BM). Compared to the binary or ternary blends, the quaternary blend film presents an intermediate change in surface roughness (Rq = 1.45 nm), associated with a finely tuned structure with more ordered features. This can be attributed to the improved crystallization dynamics or the balance in crystallization rates.
 | ||
| Fig. 4 (a) Comparison of fitting results to the transient absorption (TA) decay kinetics of PM6:BTP-eC9, Y6-1O ternary, PCBM ternary, and quaternary blends. Inset: TA kinetic curves probe at 635 nm. (b)–(e) AFM phase images of M6:BTP-eC9, ternary and quaternary films. | ||
Physical and microscopic impacts of the introduced co-guests
To correlate the improved nanomorphology with the modulation of opto-electrical behaviors in solar cells, we performed irradiation intensity (Plight) dependent photocurrent and photovoltage measurements in a steady state. The Jphversus effective voltage (Veff) curves are shown in Fig. S15 (ESI†), based on which the Jph values under the conditions of short-circuit (Jph,sc), maximal power point (MPP), and saturated points were determined (see results in Table S4, ESI†). It is noted that the ratios of Jph,MPP/Jph,sat and Jph,sc/Jph,sat increase with Y6-1O as the third or fourth component. The results are indicative of the enhanced charge collection in the quaternary device, especially when the internal electrical field is low.51–53 In Fig. S16a (ESI†), we show the Jscversus Plight characteristics (in a double-logarithm plot). The slopes determined on the quaternary device approach unity (0.998, versus slopes of 0.992, 0.994, and 0.995 in the PM6:BTP-eC9, PM6:BTP-eC9:Y6-1O and PM6:BTP-eC9:PC71BM cells, respectively). The slopes of Vocversus Plight characteristics (Fig. S16b, ESI†) were determined to be 1.24, 1.28, 1.19, 1.18 and 1.13kT/q (k is the Boltzmann constant, T is the absolute temperature, q is the elemental charge). These tendencies indicate a reduction in bimolecular charge recombination in the quaternary blends, providing an interpretation for the enhancements in device FF and Jsc. We further employed transient optoelectrical analysis to explore charge carrier behaviors under irradiation. The decay traces of transient photovoltage (TPV, open-circuit condition) and transient photocurrent (TPC, short-circuit condition) based on different blends are shown in Fig. S17 (ESI†). The TPV decay time (τTPV) was determined to be 25.22 μs for PM6:BTP-eC9, 26.77 μs for PM6:BTP-eC9:Y6-1O, 28.42 μs for PM6:BTP-eC9:PC71BM, and 36.66 μs for PM6:BTP-eC9:Y6-1O:PC71BM. The elongated τTPV of the quaternary device confirms the retardation of charge recombination.28 As seen from the TPC decay traces (Fig. S17b, ESI†), the shortest charge extraction time was found in the quaternary device (24.48 ns) and this trend aligns well with the promoted charge extraction and gains in the device FF. Since charge recombination and extraction properties are closely related to the charge carrier mobility (μ), we further examined the electron (μe) and hole (μh) mobilities using the well-established space charge-limited current (SCLC) method (see results in Fig. S18 and Table S5 in ESI†). Upon adding the Y6-1O and PC71BM acceptor co-guests, both μe and μh are enhanced, and meanwhile, the transport balance between the two carriers increases, as revealed by the mobility ratio that approaches unity (μe/μh = 1.03). We consider that the modified carrier transport plays a role in the mitigation of charge recombination at D:A interfaces, which provides a solid explanation for the enhancement of Jsc and FF in the multi-acceptor based solar cell.54In order to explore the effect of the introduced guest components on the structural order of blends, we performed solid-state 19F magic angle spinning nuclear magnetic microscopy (19F MAS-NMR) measurements (see results in Fig. 5(a)). In the concerned materials, both PM6 and Y6-1O are fluorinated and the signal appearing at −130.9 ppm originates from the 19F sites on the fluorinated thiophene of PM6.42 For the 19F sites on the fluorinated and thiol-substituted thiophene of Y6-1O, the 19F signal appears at −124.5 ppm and −122 ppm, respectively. As can be seen from the peak deconvolution (Fig. 5(b)), the resolved two 19F signals are attributed to the 19F sites on different end groups of Y6-1O, rather than the 19F sites at the ortho-positions within the same one end group of Y6-1O (the chemical environment around the F terminal group at both ends of Y6-1O is different).33 Since BTP-eC9 has no F element, the position of the 19F signal remains unchanged (131.2 ppm) upon mixing PM6 with BTP-eC9, while the bandwidth changes slightly (full-width-at-the half-maximum or FWHM reduced from 5.9 ppm to 5.8 ppm). It indicates that the short-ranged structural order of PM6 phase is enhanced with the interaction between PM6 and BTP-eC9. With the introduction of the Y6-1O guest, a new acceptor phase is generated atop the original PM6 and BTP-eC9 phases. At the same time, an additional local environment is created for the 19F sites on the PM6 molecule. We further note that in the presence of Y6-1O, the 19F signal of PM6 remains at 130. 9 ppm and the bandwidth become wider (5.8–6.0 ppm, see Fig. 5(b)). The tendency is understood as follows: due to the similar chemical structures of Y6-1O and BTP-eC9, the guest phase can penetrate well into the PM6:BTP-eC9 phase interface, so a part of PM6 does not interact with the BTP-eC9 host but with Y6-1O. The situation differs with the addition of PC71BM to PM6:BTP-eC9:Y6-1O: the bandwidth adversely decreases (6.0–5.8 ppm) while the 19F signal of the donor remains at 130.9 ppm. The reduction of the bandwidth again suggests the preservation of the short-range structural order of PM6 when PC71BM and Y6-1O guests are simultaneously present. We would like to further mention that tuning on the local environments of the 19F sites on PM6 via adding the guests is associated with rearrangements in the aggregating structure of PM6, which is reflected by the red shifting in the absorption spectra (the donor absorption peak at 624 nm shifts to 625 nm in the PM6:BTP-eC9 binary blend and further to 626 nm with the addition of Y6-1O and Y6-1O:PC71BM co-guests, see Fig. S19, ESI†).
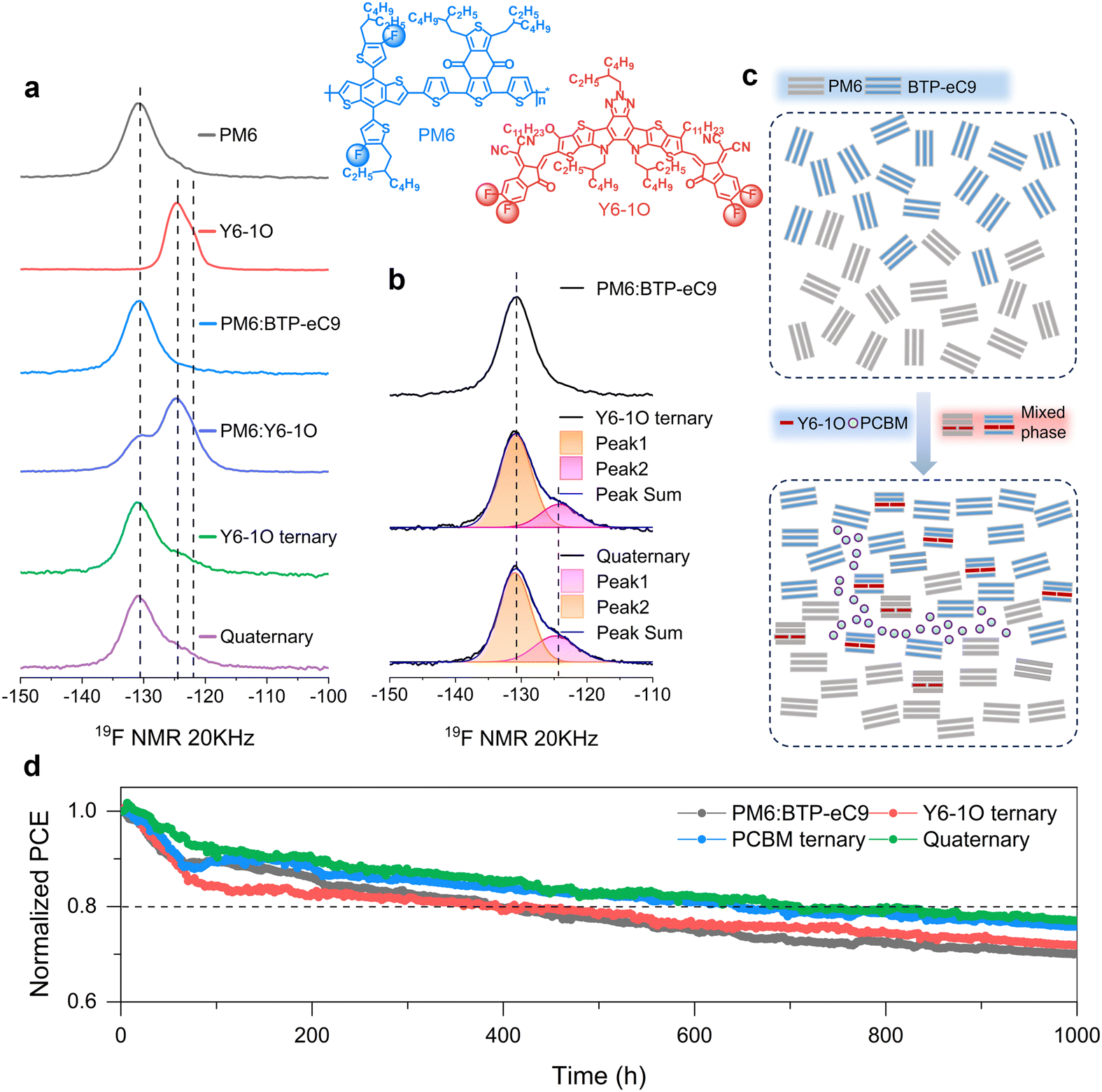 | ||
| Fig. 5 (a) Solid-state 19F MAS NMR spectra of neat PM6, Y6-1O and various blends (b) used in this study. Also shown by the lines are the two-peaks fitting curves based on the solid-state 19F MAS NMR spectra. (c) Illustrated diagrams for the aggregated structures of the PM6, BTP-eC9, Y6-1O, and PC71BM phases in the binary, ternary and quaternary blends. Formation of the individual Y6-1O and PC71BM phases introduce additional interactions to the hosts, creating new local environments for the 19F sites on PM6, which accounts for the changes observed in the solid-state 19F MAS NMR spectra. (d) Stability test on different solar cells showing normalized PCEs as a function of light-soaking time under MPP tracking. | ||
Combining these analyses, we attempt to provide the diagrams illustrating the aggregating structures of the control PM6:BTP-eC9 binary blends and PM6:BTP-eC9:Y6-1O:PC71BM quaternary blends (Fig. 5(c)). As indicated by the results of solid-state NMR, the formation of individual Y6-1O and PC71BM phases can introduce additional interactions to the hosts, creating new local environments for the 19F sites on PM6. Importantly, the short-range structural order of the PM6 donor is favorably retained in the presence of co-guest acceptors, which provides a microscopic explanation for the achievement of the finely tuned phase-separated morphology and superior charge transport in the bulk heterojunction film.
Morphology variation and device stability under irradiation
Finally, we demonstrate the merit of the proposed multi-acceptor concept for the enhancement of morphology and device stability. In this regard, we measured the efficiency decay of solar cells under continuous irradiation. Fig. 5(d) displays the tracking characteristics of the maximum power point (MPP) based on the binary, ternary, and quaternary devices under continuous AM 1.5 G illumination (100 mW cm−2). As clearly seen, the degradation of binary and ternary devices is more pronounced than that of the quaternary devices. For the control binary and Y6-1O ternary devices, the initial PCE losses exceed 20% after around 400 hours of light soaking. In contrast, the multi-acceptor-based device retains 80% of the initial PCE after more than 708 hours of aging, indicating enhanced photostability. At the same time, we also performed thermal stability tests (60 °C) on these devices. As seen from the results shown in Fig. S20 (ESI†), the quaternary device retained 85% of the initial PCE after 240 hours of thermal annealing, while the efficiency of the binary control cell dropped to ∼75% of the initial value. These observations seem to be counterintuitive, given that complex systems are prone to experience heavier morphological changes upon external stimuli.51To gain a more in-depth understanding on the improved stability, we aged the blend films under AM 1.5 G (100 mW cm−2) illumination and performed UV-vis spectroscopy on the aged films. As shown in Fig. S21 (ESI†), the absorption peak arising from the acceptor host (BTP-eC9) displays redshifts after light soaking and the degree of redshifts varies among different systems and is aging-time dependent. Moreover, upon aging, we observe a slight broadening in the acceptor absorption peak, which could be related to the increase in disorder.55 The key difference lies in that, compared to the binary or ternary blends, the quaternary blend film presents the least spectral variation after aging, namely, the absorption curves are largely overlapped in a wide spectra range (350–1000 nm). These behaviors strongly indicate that excessive aggregation could be suppressed in the quaternary blends.
To affirm this, we carried out thin film morphology aging characterization on the same set of thin film samples. As shown in the AFM images (Fig. S22, ESI†), the surface roughness of the aged (336 hours) quaternary blend film slightly increases (Rq = 1.56 nm versus Rq = 1.45 nm before aging), showing a surface morphology resembling that of the freshly prepared film. As a comparison, the control binary and ternary blend films experience substantial increases in the surface roughness after aging, especially for the sample containing only PC71BM as an acceptor guest. The contrast emphasizes the necessity of the introduced co-acceptor guests for suppressing the light-driven aggregation in the blends. Additional insights into the changes of disorder are gained from AFM characterization. For example, the fibril structure is maintained in the aged quaternary blends whereas in the other blends, structureless features tend to become more visible. We consider that the preservation of the favored molecular order together with the minimization of self-aggregating provides a fundamental merit for the enhanced morphology and long-term photostability in the multi-acceptor solar cell.
Conclusions
We have proposed a multiple acceptor-based strategy for OSCs via introducing non-fullerene Y6-1O and PC71BM as co-guest acceptors to achieve enhanced photovoltaic device performance. The combinatorial investigation with in situ optical spectroscopy, transient opto-electrical characterization and solid-state NMR reveals the central functionality of the co-guests in balancing the crystallization kinetics of D:A components and suppressing excessive aggregations. As a result, a finely optimized nanomorphology of photoactive layers has been attained, while maintaining the favorable short-range molecular order. These modifications have translated into a boost in PCE to 19.35% in the OSC, showing enhanced photon harvesting efficiencies within a wide spectral range. Notably, the champion multi-acceptor device exhibits impressive photostability, with the PCE decaying by <20% after ∼30 days of continuous 1 sun irradiation. The attractiveness can be attributed to the enhanced morphological robustness of the quaternary blends in which the light-driven aggregation of molecules is mitigated. This work not only contributes to attaining an in-depth understanding of morphology control, but also provides a new avenue to fully exploit the material potential toward realistic applications of OSCs.Author contributions
Huiqiong Zhou, Xuning Zhang and Yuan Zhang proposed this research and guided the project. Weichao Zhang and Xuning Zhang designed and performed the experiments and drafted the original manuscript. Yaochang Yue and Rongshen Yang carried out device physical characterization. Yingyu Zhang and Wenna Du performed AFM and TA measurements. Guanghao Lu and Jianqi Zhang conducted FLAS and GIWAXS measurements and analyzed the data. Yuan Zhang revised the manuscript. All authors contributed to the general discussion.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Ministry of Science and Technology of China (No. 2022YFB4200400), National Natural Science Foundation of China (NSFC) (No. 22279003). H. Zhou thanks National Key Research and Development Program of China (2022YFB3805203), the National Natural Science Foundation of China (No. 52273245), and the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB36000000). X. Zhang thanks the support from The Central Guidance on Local Science and Technology Development Fund Project of Hebei Province (No. 226Z4306G) and Natural Science Foundation of Hebei Province (F2023201005).References
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin and J. Ren, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed
.
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, 2109516 CrossRef CAS PubMed
- R. Sun, Y. Wu, X. Yang, Y. Gao, Z. Chen, K. Li, J. Qiao, T. Wang, J. Guo and C. Liu, Adv. Mater., 2022, 34, 2110147 CrossRef CAS PubMed
- J. Wang, Z. Zheng, Y. Zu, Y. Wang, X. Liu, S. Zhang, M. Zhang and J. Hou, Adv. Mater., 2021, 33, 2102787 CrossRef CAS PubMed
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song and X. Xue, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed
- J. Fu, P. W. K. Fong, H. Liu, C.-S. Huang, X. Lu, S. Lu, M. Abdelsamie, T. Kodalle, C. M. Sutter-Fella, Y. Yang and G. Li, Nat. Commun., 2023, 14, 1760 CrossRef CAS PubMed
- Z. Yao, X. Cao, X. Bi, T. He, Y. Li, X. Jia, H. Liang, Y. Guo, G. Long and B. Kan, Angew. Chem., Int. Ed., 2023, e202312630 CAS
- C. Chen, L. Wang, Y. Sun, Y. Fu, C. Guo, B. Zhou, Z. Gan, D. Liu, W. Li and T. Wang, Adv. Funct. Mater., 2023, 2305765 CrossRef CAS
- Y. Wei, Y. Cai, X. Gu, G. Yao, Z. Fu, Y. Zhu, J. Yang, J. Dai, J. Zhang and X. Zhang, Adv. Mater., 2023, 2304225 Search PubMed
- H. Liu, Z. Chen, R. Peng, Y. Qiu, J. Shi, J. Zhu, Y. Meng, Z. Tang, J. Zhang and F. Chen, Chem. Eng. J., 2023, 474, 145807 CrossRef CAS
- Y. Zhang, J. Deng, S. You, X. Huang, J. Liu, Y. Cheng, B. Huang, X. Chen, Z. Sun and C. Yang, Adv. Funct. Mater., 2023, 2308151 Search PubMed
- R. Ma, C. Yan, P. W.-K. Fong, J. Yu, H. Liu, J. Yin, J. Huang, X. Lu, H. Yan and G. Li, Energy Environ. Sci., 2022, 15, 2479–2488 RSC
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC
- Q. An, J. Wang, W. Gao, X. Ma, Z. Hu, J. Gao, C. Xu, M. Hao, X. Zhang, C. Yang and F. Zhang, Sci. Bull., 2020, 65, 538–545 CrossRef CAS PubMed
- Y. Xie, F. Yang, Y. Li, M. A. Uddin, P. Bi, B. Fan, Y. Cai, X. Hao, H. Y. Woo, W. Li, F. Liu and Y. Sun, Adv. Mater., 2018, 30, 1803045 CrossRef PubMed
- A. Lan, J. Zhu, Z. Zhang, Y. Lv, H. Lu, N. Zhao, H. Do, Z.-K. Chen and F. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 39657–39668 CrossRef CAS PubMed
- Y. Chen, R. Ma, T. Liu, Y. Xiao, H. K. Kim, J. Zhang, C. Ma, H. Sun, F. Bai and X. Guo, Adv. Energy Mater., 2021, 11, 2003777 CrossRef CAS
- X. Yuan, Y. Zhao, D. Xie, L. Pan, X. Liu, C. Duan, F. Huang and Y. Cao, Joule, 2022, 6, 647–661 CrossRef CAS
- X. Song, P. Sun, D. Sun, Y. Xu, Y. Liu and W. Zhu, Nano Energy, 2022, 91, 106678 CrossRef CAS
- J. Ge, L. Hong, W. Song, L. Xie, J. Zhang, Z. Chen, K. Yu, R. Peng, X. Zhang and Z. Ge, Adv. Energy Mater., 2021, 11, 2100800 CrossRef CAS
- S. Li, Z. Jia, Q. Ma, Y. Wu, Q. Meng, J. Zhang, B. Qiu, J. Qiao and Y. Li, Sol. RRL, 2022, 6, 2200496 CrossRef CAS
- Y. Wang, J. Xue, H. Zhong, C. R. Everett, X. Jiang, M. A. Reus, A. Chumakov, S. V. Roth, M. A. Adedeji and N. Jili, Adv. Energy Mater., 2023, 13, 2203496 CrossRef CAS
- X. Xu, L. Yu, H. Meng, L. Dai, H. Yan, R. Li and Q. Peng, Adv. Funct. Mater., 2022, 32, 2108797 CrossRef CAS
- Z. Bi, Q. Zhu, X. Xu, H. B. Naveed, X. Sui, J. Xin, L. Zhang, T. Li, K. Zhou and X. Liu, Adv. Funct. Mater., 2019, 29, 1806804 CrossRef
- L. Arunagiri, Z. Peng, X. Zou, H. Yu, G. Zhang, Z. Wang, J. Y. L. Lai, J. Zhang, Y. Zheng and C. Cui, Joule, 2020, 4, 1790–1805 CrossRef CAS
- Z. Bi, H. B. Naveed, X. Sui, Q. Zhu, X. Xu, L. Gou, Y. Liu, K. Zhou, L. Zhang and F. Zhang, Nano Energy, 2019, 66, 104176 CrossRef CAS
- K. Zhang, Z. Jiang, J. Qiao, P. Lu, C. Qin, H. Yin, X. Du, W. Qin and X. Hao, Energy Environ. Sci., 2023, 16, 3350–3362 RSC
- X. Xu, W. Jing, H. Meng, Y. Guo, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2208997 CrossRef CAS PubMed
- H. Kang, B. Zheng, Y. Li, S. Li, H. Zhou, L. Huo and Y. Zhang, Sol. RRL, 2022, 6, 2101040 CrossRef
- J. Gao, N. Yu, Z. Chen, Y. Wei, C. Li, T. Liu, X. Gu, J. Zhang, Z. Wei, Z. Tang, X. Hao, F. Zhang, X. Zhang and H. Huang, Adv. Sci., 2022, 9, 2203606 CrossRef CAS PubMed
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS
- G. Zhou, M. Zhang, Z. Chen, J. Zhang, L. Zhan, S. Li, L. Zhu, Z. Wang, X. Zhu and H. Chen, ACS Energy Lett., 2021, 6, 2971–2981 CrossRef CAS
- W. Zhang, J. Huang, J. Xu, M. Han, D. Su, N. Wu, C. Zhang, A. Xu and C. Zhan, Adv. Energy Mater., 2020, 10, 2001436 CrossRef CAS
- Y. Chen, F. Bai, Z. Peng, L. Zhu, J. Zhang, X. Zou, Y. Qin, H. K. Kim, J. Yuan and L. K. Ma, Adv. Energy Mater., 2021, 11, 2003141 CrossRef CAS
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma and C. An, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed
- H. R. Bai, Q. An, M. Jiang, H. S. Ryu, J. Yang, X. J. Zhou, H. F. Zhi, C. Yang, X. Li and H. Y. Woo, Adv. Funct. Mater., 2022, 32, 2200807 CrossRef CAS
- R. Yu, S. Zhang, H. Yao, B. Guo, S. Li, H. Zhang, M. Zhang and J. Hou, Adv. Mater., 2017, 29, 1700437 CrossRef PubMed
- C. Zhang, M. Zhang, Q. Zhou, S. Chen, S. Kim, J. Yao, Z. Zhang, Y. Bai, Q. Chen and B. Chang, Adv. Funct. Mater., 2023, 2214392 CrossRef CAS
- J. Xue, H. Zhao, B. Lin, Y. Wang, Q. Zhu, G. Lu, B. Wu, Z. Bi, X. Zhou and C. Zhao, Adv. Mater., 2022, 34, 2202659 CrossRef CAS PubMed
- J. Zhang, L. Zhang, X. Wang, Z. Xie, L. Hu, H. Mao, G. Xu, L. Tan and Y. Chen, Adv. Energy Mater., 2022, 12, 2200165 CrossRef CAS
- Y. Cai, Q. Li, G. Lu, H. S. Ryu, Y. Li, H. Jin, Z. Chen, Z. Tang, G. Lu and X. Hao, Nat. Commun., 2022, 13, 2369 CrossRef CAS PubMed
- D. Luo, Z. Jiang, W. L. Tan, L. Zhang, L. Li, C. Shan, C. R. McNeill, P. Sonar, B. Xu and A. K. K. Kyaw, Adv. Energy Mater., 2023, 13, 2203402 CrossRef CAS
- Y. Wang, X. Wang, B. Lin, Z. Bi, X. Zhou, H. B. Naveed, K. Zhou, H. Yan, Z. Tang and W. Ma, Adv. Energy Mater., 2020, 10, 2000826 CrossRef CAS
- Q. Zhu, J. Xue, G. Lu, B. Lin, H. B. Naveed, Z. Bi, G. Lu and W. Ma, Nano Energy, 2022, 97, 107194 CrossRef CAS
- C. Zhao, J. Yi, L. Wang, G. Lu, H. Huang, H. K. Kim, H. Yu, C. Xie, P. You and G. Lu, Nano Energy, 2022, 104, 107872 CrossRef CAS
- S. Li, T. Jiang, H. Zhang, Y. Li, Q. Cheng, H. Kang, Y.-N. Jing, L. Xiao, X. Zhang and G. Lu, Sol. RRL, 2023, 7, 2201011 CrossRef CAS
- X. Si, Y. Huang, W. Shi, R. Wang, K. Ma, Y. Zhang, S. Wu, Z. Yao, C. Li and X. Wan, Adv. Funct. Mater., 2023, 2306471 CrossRef CAS
- H. Kang, Y. Jing, Y. Zhang, Y. Li, H. Zhang, H. Zhou and Y. Zhang, Sol. RRL, 2023, 7, 2201084 CrossRef CAS
- L. Zhan, S. Yin, Y. Li, S. Li, T. Chen, R. Sun, J. Min, G. Zhou, H. Zhu and Y. Chen, Adv. Mater., 2022, 34, 2206269 CrossRef CAS PubMed
- C. He, Y. Pan, Y. Ouyang, Q. Shen, Y. Gao, K. Yan, J. Fang, Y. Chen, C.-Q. Ma and J. Min, Energy Environ. Sci., 2022, 15, 2537–2544 RSC
- X. Li, L. Zhou, X. Lu, L. Cao, X. Du, H. Lin, C. Zheng and S. Tao, Mater. Chem. Front., 2021, 5, 3850–3858 RSC
- Z. Bi, H. B. Naveed, X. Sui, Q. Zhu, X. Xu, L. Gou, Y. Liu, K. Zhou, L. Zhang, F. Zhang, X. Liu and W. Ma, Nano Energy, 2019, 66, 104176 CrossRef CAS
- Z. Bi, Q. Zhu, X. Xu, H. B. Naveed, X. Sui, J. Xin, L. Zhang, T. Li, K. Zhou, X. Liu, X. Zhan and W. Ma, Adv. Funct. Mater., 2019, 29, 1806804 CrossRef
- P. Bi, S. Zhang, T. Xiao, M. Cui, Z. Chen, J. Ren, C. Qin, G. Lu, X. Hao and J. Hou, Sci. China: Chem., 2021, 64, 599–607 CrossRef CAS
- K. An, W. Zhong, F. Peng, W. Deng, Y. Shang, H. Quan, H. Qiu, C. Wang, F. Liu, H. Wu, N. Li, F. Huang and L. Ying, Nat. Commun., 2023, 14, 2688 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04169g |
| This journal is © The Royal Society of Chemistry 2024 |