
Porphyrin-based metal–organic frameworks for photo(electro)catalytic CO2 reduction
Guixiang
Ding†
a,
Chunxue
Li†
 b,
Lihui
Chen
a and
Guangfu
Liao
*a
b,
Lihui
Chen
a and
Guangfu
Liao
*a
aNational Forestry and Grassland Administration Key Laboratory of Plant Fiber Functional Materials, College of Materials Engineering, Fujian Agriculture and Forestry University, Fuzhou 350002, China. E-mail: liaogf@mail2.sysu.edu.cn
bCollege of Ecological Environment and Urban Construction, Fujian University of Technology, Fuzhou 350118, China
First published on 17th June 2024
Abstract
Amidst the significant challenges posed by global climate change and the need for sustainable resource recycling, there is a pressing demand for the development of new materials that offer high cost-effectiveness, exceptional conversion efficiency, and robust stability to facilitate the conversion of CO2 into high-value products. Metal–organic frameworks (MOFs), which incorporate versatile porphyrin fragments, have emerged as promising candidates in the realm of photo(electro)catalytic CO2 reduction. Given their straightforward synthesis, tunable structure, chemical stability, and abundance of active sites, MOFs enriched with specific functional porphyrins can be precisely incorporated through in situ self-assembly or post-synthesis techniques, thereby broadening the scope of design possibilities for porphyrin-based MOFs. This comprehensive review delves into the rational design principles and recent advancements in the utilization of porphyrin-based MOFs for photo(electro)catalytic CO2 reduction. It elucidates the synthesis methods and CO2 interaction mechanisms, and delves into the latest research endeavors encompassing catalyst structure optimization, activity enhancement, selectivity control, and other pivotal breakthroughs. Furthermore, it conducts a detailed analysis of the current technical challenges, future trends, and the promising prospects of porphyrin-based MOFs in driving the industrialization of this technology. By offering a thorough theoretical framework and practical insights, this review serves as a valuable resource for enhancing the understanding and application of porphyrin-based MOFs in high-value conversion of CO2, thereby paving the way for improved catalytic performance and the successful implementation of this technology.
 Guixiang Ding | Guixiang Ding received his MS degree from the College of Chemical Engineering of Liaoning Normal University in 2022. His current research focuses on the development of emerging materials for photocatalytic CO2 reduction. |
 Chunxue Li | Chunxue Li obtained her BSc (2016) and MS (2019) degree from Jilin Normal University. She then obtained her PhD degree from Jiangsu University. Presently, she is a lecturer in Fujian University of Technology. Her current research focuses on the synthesis and application of semiconductor nanomaterials for photocatalytic hydrogen evolution. She has received some awards including Graduate Student National Scholarship, Xue Liang Scholarship, and Excellent Graduation Thesis of Jilin Normal University. |
 Guangfu Liao | Guangfu Liao received his PhD degree in Material Physics & Chemistry from Sun Yat-sen University in 2020. Then he joined the laboratory of Prof. Yi-Chun Lu at The Chinese University of Hong Kong working as a research associate. Subsequently, he was a researcher at China University of Geosciences. Now, he is a professor in Fujian Agriculture and Forestry University. His research interests involve photo and electro catalysis, polymer synthesis and applications, polymer membranes, nanoporous and nanostructured materials, biomaterials, gas storage and energy conversion, etc. The broader impacts of his research include polymeric photocatalysts, polymer design and synthesis, nanostructure engineering, and biomaterials. So far, he has published more than 80 high profile SCI papers such as Matter, Progress in Materials Science, Energy & Environmental Science, Physics Reports, Applied Catalysis B: Environmental, Nano Energy, ACS Catalysis, Chemical Science, etc. The awards he received up to now include Young Elite Scientists Sponsorship Program by CAST, 2022 Journal of Materials Chemistry A Emerging Investigators, 2023 Chemical Communications Emerging Investigators, etc. In addition, he also serves as Youth Editorial Board for Advanced Fiber Materials, Exploration, etc. |
Broader contextThe technology of photo(electro)catalytic reduction of CO2 aims to mimic the natural photosynthesis mechanism by using light or electrical energy to convert excess CO2 in the atmosphere into valuable chemicals like C1, C2, and other hydrocarbons. This process helps in efficiently utilizing carbon resources and reducing greenhouse gas emissions. Porphyrin-based metal–organic frameworks (MOFs) have attracted much attention in the field of CO2 photo(electro)reduction due to their many advantages such as superior CO2 adsorption and tunable photoelectrochemical properties, strong light absorption and response abilities, large specific surface area, and abundant and evenly distributed active sites. This review presents a panorama of the latest developments of the emerging porphyrin-based MOF photo(electro)catalysts for CO2 reduction. It starts with the synthesis and design of porphyrin-based MOFs. After that, the basic principle of CO2 reduction is illustrated. Later, the recent advancements in the field of porphyrin-based MOFs for application in photo(electro)catalytic CO2 reduction are exemplified. In the end, this review also offers some new views into the major challenges, opportunities, and heuristic perspectives for future research in this emerging field. |
1. Introduction
The greenhouse effect caused by excessive CO2 emissions has emerged as a significant threat to human survival.1–5 The conversion of CO2 into energy is an effective strategy that addresses two issues simultaneously: protecting the environment and conserving non-renewable energy sources.6–8 Among different approaches to CO2 energy conversion, photo(electro)catalytic CO2 reduction has gained considerable attention.9–11 This method offers favorable reaction conditions and enables the production of fuels and chemicals, thereby mitigating the environmental impact of excessive CO2 emissions.12 However, the symmetric linear CO2 molecular structure means the process of activation and transformation requires more input energy and lower overpotential to overcome the sluggish reaction kinetics.13,14 For photocatalytic CO2 reduction, appropriate conduction band potential and effective chemisorption are desirable in photocatalysts.15 However, low carrier separation efficiency and poor electron conductivity limit the photocatalytic activity.16,17 Numerous reports have suggested various approaches to enhance the photocatalytic activity, which can be broadly categorized as structural engineering (doping elements,18–20 defect engineering,21–23etc.) and co-catalytic design (heterojunction,24–27 loading noble metals,28,29 single atom catalysis,30,31etc.). On the other hand, for electrochemical CO2 reduction reaction (CO2RR), electrocatalysts are expected to have lower overpotential and higher efficiency in CO2RR.32 However, the hydrogen evolution reaction (HER) competes with CO2RR in aqueous systems, as HER occurs in a similar potential range.33–35 Furthermore, the selectivity of CO2RR is reduced by the multi-electron and multi-proton reactions, which makes it satisfactory. Both structure engineering and surface decoration are key approaches to enhance the selectivity and catalytic activity of electrocatalysts. It is important to note that selecting the right photo(electro)catalytic materials is also crucial for optimizing selectivity and maintaining high performance in catalytic CO2 reduction.Recently, research on metal–organic frameworks (MOFs) for energy conversion of CO2 has become a hot discussion and a lot of scientific achievements have been published in some top journals.36,37 As we know, MOFs are a class of porous crystal materials consisting of inorganic metal ions or clusters and organic linkers.38–40 The notion of MOFs was first introduced by Yaghi et al. in 1995.41 With thorough research on the properties and synthesis strategies of MOFs, more outstanding merits have been discovered for their application in the field of energy conversion, gas storage, separation, and sensors.42,43 These merits can be attributed to their large specific surface area, tunable open channels, and abundant active sites.44 In addition, the flexible composition of metal nodes and the organic linkers in MOF materials allows for excellent functional expansion and regulation.45–48 Thus, MOFs have long been considered as a promising platform for research and application in the area of energy conversion of CO2.49
Porphyrins are widespread in nature and play an irreplaceable role in the metabolism of living organisms, such as in photosynthesis as chlorophyll, in the transportation of oxygen in the blood in the form of hemoglobin, in the production of vitamin B12 in red blood cells, etc.50–53 Porphyrins are a class of aromatic organics with macrocyclic conjugated planar structures, and the center is a porphyrin core of 18-π conjugate electrons, and substituents are observed on the bridging hypomethyl (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2–) groups.54 Meanwhile, nitrogen atoms existing on porphine can coordinate with most metal ions, and ultimately, porphyrins transform into metalloporphyrins.55–57 Porphyrin materials serve as the active centers in diverse bioenzymatic catalysts and environmentally friendly biomimetic catalysts, and show outstanding performance and superior selectivity in catalytic reaction.41,58–60 Noteworthily, metalloporphyrins present stronger catalytic and photoelectric activity than porphyrins, which is mainly attributed to the fact that the addition of the coordinating metal ions changes the configuration of electrons in porphyrins.56,61,62 With increasing research on porphyrins and porphyrin derivatives for photo(electro)catalytic application, more drawbacks related to their use, such as small specific surface area, non-recyclability of homogeneous systems, and low catalytic efficiency, are exposed during the catalytic process.63–65 In this case, the strategy of integration of porphyrins and porphyrin derivatives into MOFs has received high attention in the field of photo(electro)catalytic CO2 reduction. In addition, porphyrin MOFs stand out with excellent light absorption and a precise active center, facilitating tailored design and exhibiting great potential in advanced photo(electro)catalytic applications compared to conventional MOFs.
CH2–) groups.54 Meanwhile, nitrogen atoms existing on porphine can coordinate with most metal ions, and ultimately, porphyrins transform into metalloporphyrins.55–57 Porphyrin materials serve as the active centers in diverse bioenzymatic catalysts and environmentally friendly biomimetic catalysts, and show outstanding performance and superior selectivity in catalytic reaction.41,58–60 Noteworthily, metalloporphyrins present stronger catalytic and photoelectric activity than porphyrins, which is mainly attributed to the fact that the addition of the coordinating metal ions changes the configuration of electrons in porphyrins.56,61,62 With increasing research on porphyrins and porphyrin derivatives for photo(electro)catalytic application, more drawbacks related to their use, such as small specific surface area, non-recyclability of homogeneous systems, and low catalytic efficiency, are exposed during the catalytic process.63–65 In this case, the strategy of integration of porphyrins and porphyrin derivatives into MOFs has received high attention in the field of photo(electro)catalytic CO2 reduction. In addition, porphyrin MOFs stand out with excellent light absorption and a precise active center, facilitating tailored design and exhibiting great potential in advanced photo(electro)catalytic applications compared to conventional MOFs.
Over the past few years, the emerging porphyrin-based MOFs have been widely reported for photo(electro)catalytic CO2 reduction. It is an opportune moment to offer a review on porphyrin-based MOFs for photo(electro)catalytic CO2 reduction. Up to now, although there have been many inspiring reviews on porphyrin-based MOFs for various applications,66–72 a comprehensive review focusing on porphyrin-based MOFs for photo(electro)catalytic CO2 reduction is inexistent. In this review, the synthetic approaches of porphyrin-based MOFs and the basic principle of CO2 reduction are firstly described. Subsequently, the recent advancements in the utilization of porphyrin-based MOFs in photo(electro)catalytic CO2 reduction are outlined. Lastly, the challenges and prospects of this class of emerging materials for application in photo(electro)catalytic CO2 reduction are discussed.
2. Synthesis and design of porphyrin-based MOFs
A diverse set of MOFs, such as zeolite imidazolate frameworks (ZIFs),73–75 isoreticular metal–organic frameworks (IRMOFs),76 materials of institute Lavoisier frameworks (MILs),55 and pocket-channel frameworks (PCNs),77 have been successfully designed and synthesized. According to the past experiences, the development trend of the synthesis and design of porphyrin-based MOFs generally follows the MOFs’ guideline. Toward the target of pragmatization, the design and synthesis of porphyrin-based MOFs meet the characteristics of excellent photo(electro)chemical property and chemical stability. Meanwhile, combined with the porous and large specific surface area of the framework structure of MOFs, this enables the development of porphyrin-based MOFs in CO2 capture with great potential. As research on the application of porphyrin-based MOFs in CO2 catalysis advances, numerous design concepts and structural engineering approaches for porphyrin-based MOFs have been proposed. These concepts have been thoroughly validated, demonstrating their excellent catalytic activities. The synthesis, construction, and development of porphyrin-based MOFs have been described in recent years through two design directions, namely porphyrinic MOFs and porphyrin@MOFs.2.1. Porphyrinic MOFs
Porphyrins are heterocyclic macromolecular compounds that can act as functional organic linkers and coordinate with metal ions or secondary building units (SBUs) to form porphyrinic MOFs; the secondary building units (SBUs) refer to the fundamental repeating units that constitute the crystalline structure of porphyrinic MOFs. These units typically arise from the coordination between metal ions (such as zinc, copper, iron, etc.) and organic ligands (commonly carboxylic acids, imidazoles, bipyridines, etc.), forming simpler polyhedral structures. Acting as building blocks, SBUs assemble into larger and more intricate three-dimensional networks through various connecting modes, including vertex-sharing or edge-sharing, to create the complex architectures of porphyrinic MOFs; common secondary building units include octahedral structures formed by four metal ions and six organic ligands, denoted as [M4(O/R)6], where M represents metal ions, and O or R signifies oxygen atoms or functional groups within the organic ligands or metal nodes; and SBUs have significant influences on the structural and physicochemical properties of porphyrinic MOFs.44,60 Some common porphyrin linkers are shown in Fig. 1b. In addition, porphyrinic MOFs can also be further modified by incorporating other functionalized components, such as quantum dots and photoelectronic nanoparticles. This modification transforms them into an exceptionally advanced photoelectrocatalytic platform. Besides composition, the structure of porphyrinic MOFs is crucial in the field of energy-conversion catalysis.78–80 In this section, we introduce the synthesis strategy of porphyrinic MOFs from topological engineering, metal–organic cage engineering and pillar-layer engineering. Meanwhile, the structural stability of porphyrinic MOFs with different synthetic ideas is elaborated. | ||
| Fig. 1 (a) Representative molecular structures of the porphyrin core, porphyrins, and metalloporphyins. (b) Porphyrin-based organic linkers for the synthesis of porphyrin-based MOFs. | ||
As shown in Fig. 1, porphyrin consists of a central heterocyclic ring and external functional groups; the structure and length of these functional groups can be easily modified. Thus, porphyrin functions as a linker usually with a large size of about 2.5 nm, which facilitates the generation of large pores inside the organic framework, ultimately forming mesoporous porphyrinic MOF materials. Here, Zhou86 and his group developed a series of porphyrinic MOFs consisting of tetratopic carboxylate porphyrin ligand H4TCPP (tetrakis (4-carboxyphenyl)-porphyrin) with D4h symmetry and the 12-connected Zr6 cluster with Oh symmetry; topological connection of those two nodes to each other could ideally give rise to a highly connected ftw-a network (Fig. 2a). Herein, the desired organic linkers were elongated by arranging the vicinal phenyl ring and the carboxylate group. Ultimately, a series of mesoporous porphyrinic MOFs with ftw-a topology, namely PCN-228, PCN-229, and PCN-230 (PCN is the abbreviation for porous coordination network), were synthesized by the assembly of organic linkers and the 12-connected Zr6 cluster. The mesopore size of porphyrinic MOFs (PCN-228, PCN-229, and PCN-230) was 2.5, 2.8, and 3.8 nm, respectively (Fig. 2b), and PCN-229 presented the highest specific surface area according to the Brunauer–Emmett–Teller (BET) test (Fig. 2c). Noteworthily, the longer the linker, the lower the stability; however, PCN-230 showed unexpected stability over the pH range of 0–12 in aqueous solution, which demonstrated PCN-230's excellent pH tolerance and chemical stability (Fig. 2d). The adjustment of linker length in porphyrin-based MOFs is expected to broaden their application potential, particularly in gas adsorption and catalytic site distribution. This modification will help overcome stability issues, enhancing the versatility and feasibility of their use.
 | ||
| Fig. 2 (a) Assembly of Oh and D4h nodes into an ftw-a network. (b) Structures and crystals of PCN-230. (c) and (d) N2 adsorption isotherms of PCN-230 after different treatments. Reproduced from ref. 86. Copyright 2022 ACS. | ||
In addition, the interaction between metal nodes and porphyrinic linkers plays a crucial role in the chemical stability of porphyrinic MOFs. By adjusting the hardness of metal ions and porphyrin ligands, the chemical stability of porphyrinic MOFs can be controlled in accordance with the soft and hard acid–base theory (HSAB).87 Especially, porphyrinic MOFs consist of coordination centers with high-valent metal ions and linkers with carboxylate terminal groups exhibit a significant degree of variability when exposed to water. Zhou and co-workers88 guided by a top-down topological analysis rationally designed and synthesized a MOF, namely PCN-601, based on the pyrazolate-based (PZ-based) porphyrinic ligand. The porphyrinic MOF with ftw-a topology is composed of the Oh symmetric 12-connected [Ni8(OH)4(H2O)2Pz12] (which is denoted as [Ni8]) nodes and the D4h symmetric 4-connected 5,10,15,20-tetra(1H-pyrazol-4-yl)porphyrin (H4TPP) ligands (Fig. 3a). Then, a series of tests were carried out to identify the chemical stability of PCN-601. According to X-ray diffraction (XRD) and the BET specific surface area test, the crystallinity and porosity of PCN-601 can be greatly maintained between room temperature and 100 °C in saturated NaOH solution (20 mol L−1) (Fig. 3b and c). The experimental results suggest that designing porphyrin-based MOFs based on HSAB theory is crucial for the creation of acid and alkali resistant, as well as thermally stable, materials. This highlights the potential use of porphyrin-based MOFs in demanding catalytic environments in the future.
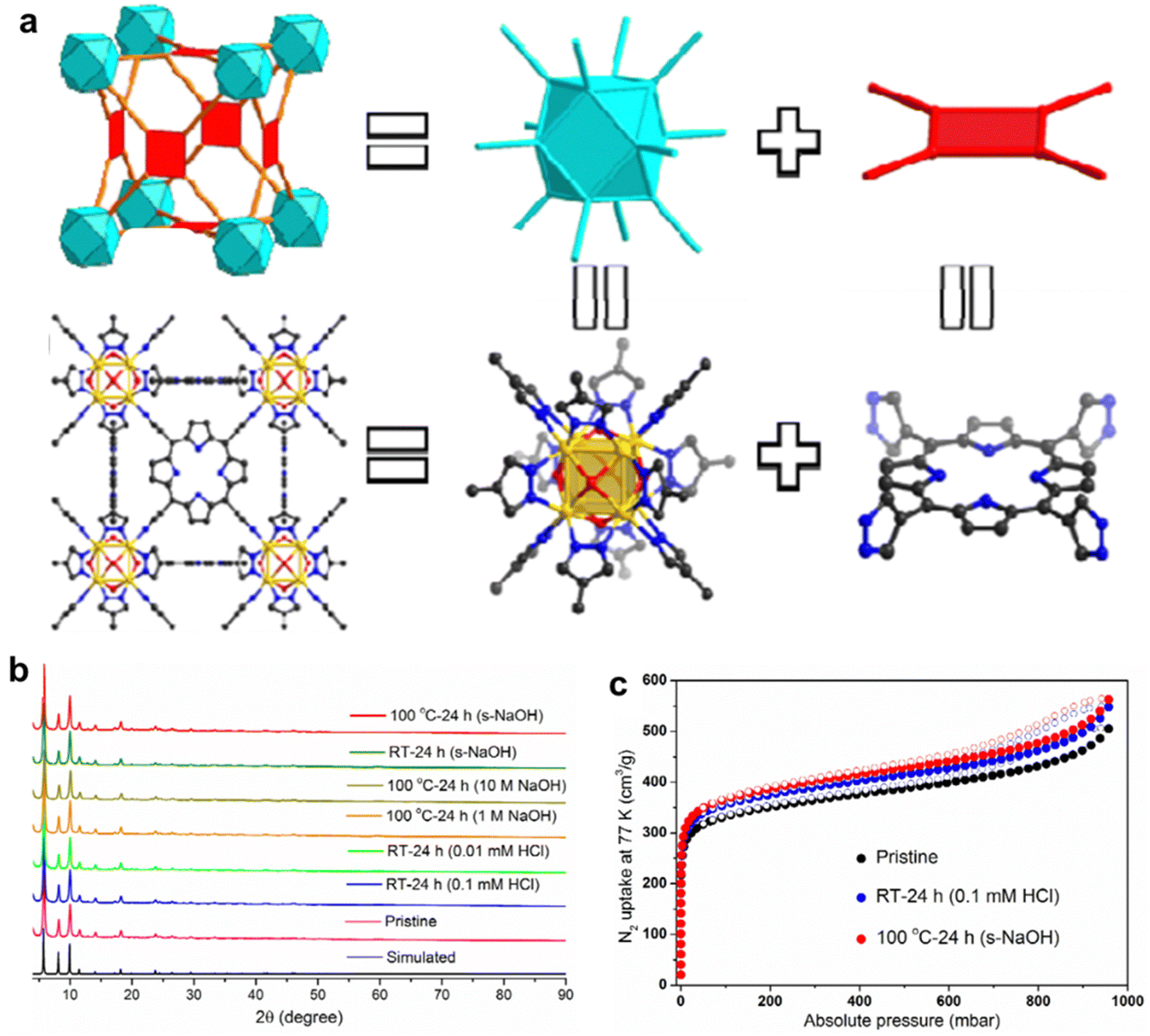 | ||
| Fig. 3 (a) Structural analysis of PCN-601. (b) PXRD patterns for simulated, pristine PCN-601, and PCN-601 samples treated under different conditions. (c) N2 adsorption/desorption isotherms at 77 K of pristine PCN-601 and acid and base treated PCN-601 samples. Reproduced from ref. 88. Copyright 2022 ACS. | ||
Due to the distinctive structure of the porphyrin nucleus, the nitrogen atoms within the porphyrin cores of metalloporphyrin MOFs display a high reactivity towards coordination reactions with metal ions. By adjusting the metal ions within the ligands, changes in the photoelectronic properties of porphyrin MOFs can be achieved. Therefore, controlling the coordination of metal ions proves to be a valuable method for improving the catalytic efficiency of metalloporphyrin MOFs. Gu and his team89 reported a series of 2D MOF Zn2[TCPP(M)] (named ZnTCPP(M), M = Zn, Mn, Fe, and H2) films used in photodetectors and revealed the photodetection response of metalloporphyrin MOFs (Fig. 4a and b). Among ZnTCPP(H2), ZnTCPP(Fe), ZnTCPP(Mn) and ZnTCPP(Zn), ZnTCPP(Zn) exhibited notable enhancement in photoresponse and light harvesting, which suggests its great potential for research and application in photoelectrocatalysis (Fig. 4c and d). Furthermore, the topological guidance of the metalloporphyrin MOFs increases the number of ligand metal ions, promoting a uniform distribution of metal active sites. In addition, integrating porphyrins containing different metal centers into porphyrin-based MOFs is expected to be a prominent research focus in the future. The metal-dependent photoelectrochemical properties are anticipated to play a key role in CO2 catalytic reduction reactions.
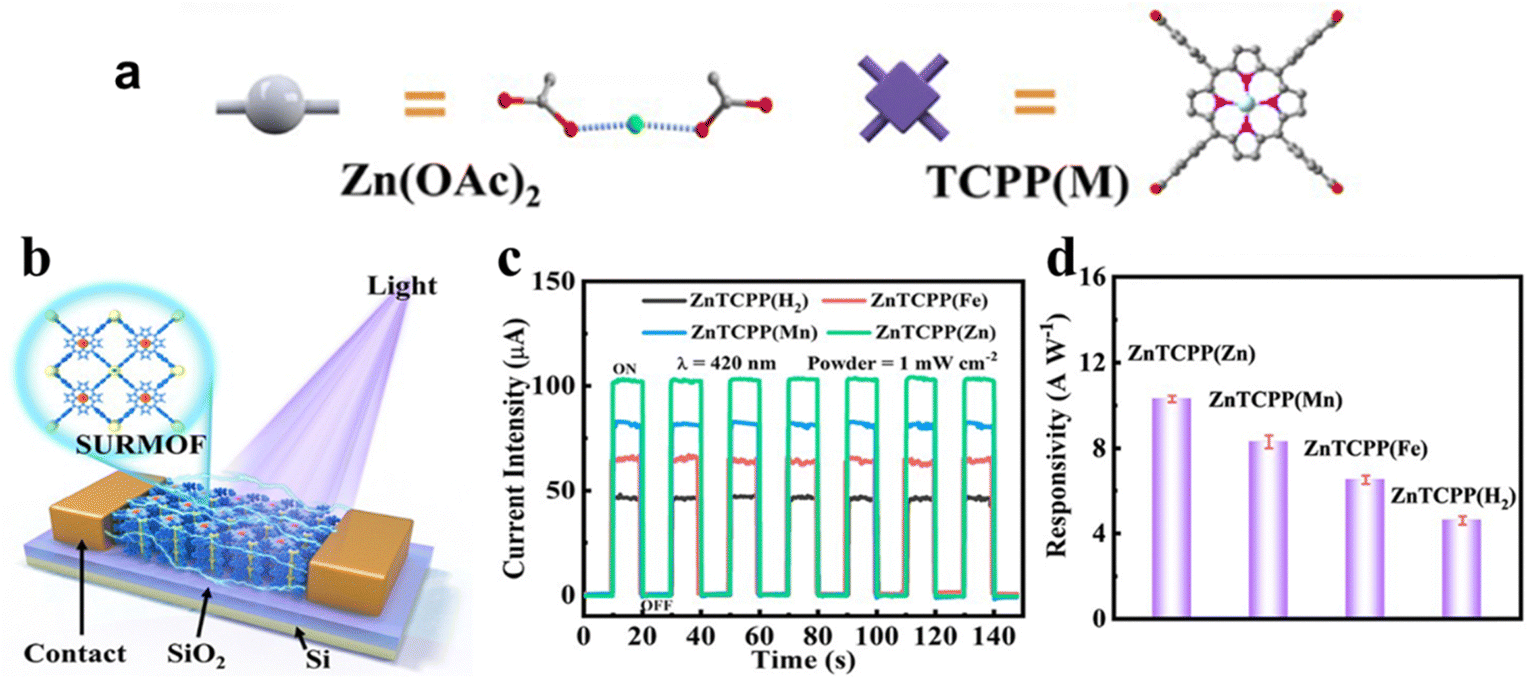 | ||
| Fig. 4 (a) Preparation process of ZnTCPP(M) films. (b) Schematic diagram of ZnTCPP(M)-based photodetectors. (c) Photocurrent curve and (d) responsivity of the ZnTCPP(M)-based photodetector. Reproduced from ref. 89. Copyright 2022 ACS. | ||
 | ||
| Fig. 5 (a) The large cubic cage constructed from six porphyrin ligands and eight Zr6O4(OH)4 clusters. (b) The small octahedral cage constructed from four porphyrin ligands and two Zr6O4(OH)4 clusters. (c) Packing of the two kinds of cages. (d) Experimental N2 adsorption isotherms for FJI-H6, FJI-H6(Cu), FJI-H7 and FJI-H7(Cu). (e) Cycloaddition reactions of CO2 with epoxide catalyzed by FJI-H6(Cu), FJI-H7 and FJI-H7(Cu). Reproduced from ref. 92. Copyright 2022 RSC. | ||
 | ||
| Fig. 6 Design of mixed-linker Zr-MOFs: (a) and (b) PCN-134 and PCN-224 formed by the 6-connected Zr6 cluster and TCPP. (c) The XRD patterns and (d) the N2 isotherms after immersion in aqueous solutions with different pH values at room temperature for 24 h. (e) Effects of TCPP defects of the PCN-134 system on N2 uptake measured at 77 K. Reproduced from ref. 98. Copyright 2022 ACS. | ||
2.2. Porphyrin@MOFs
To create porphyrin@MOFs, we usually trap free-base porphyrins or metallo-porphyrins inside the pores or attach them to the surface of MOFs, and employ in situ formation or post-synthesis methods to integrate porphyrins into MOFs.47 Because porphyrin molecules self-degrade and aggregate, porphyrin or metalloporphyrin-based catalysts are typically utilized in homogeneous catalytic systems, which significantly reduces their catalytic efficiency.55,99 The catalytic activity and stability of porphyrin can be enhanced by incorporating porphyrins into MOFs to create heterogeneous porphyrin@MOFs.PCN-134 is a porphyrin-based MOF consisting of Zr-BTB and porphyrinic linkers with a typical pillar-layer structure as described in the introduction of Section 2.1.3. Zhou et al. successfully synthesized PCN-134-2D (functionalized MOF 2D nanosheet) and PCN-134-3D (functionalized MOF 3D nanosheet) through one-pot reaction and step-wise synthesis, and the processes are illustrated in Fig. 7a. Subsequent tests on the acid and alkali resistance of PNC-134-2D, modified with varied porphyrin groups, demonstrated that PNC-134-2D synthesized using the step-wise method remained stable in the pH range of 1–10. PNC-134-2D produced through TCPP synthesis was found to be even more stable, likely due to its greater number of coordination groups. Abbreviations will be explained when first used. Yi and his group synthesized the stable metal organic framework PCN-224 through post-modification and embedded Mn3+ in it to form PCN-224(Mn). Moreover, compared with PCN-224 with a specific surface area of 2630 m2 g−1, PCN-224(Mn) almost retains the same specific surface area as PNC-24 while introducing a metal active center. The metalloporphyrin-based MOFs obtained through the post-synthesis method maintained structural stability during acid and alkali tests, with no detachment of metal ions observed.
 | ||
| Fig. 7 (a) Schematic representation showing the one-pot synthesis of PCN-134-3D and the stepwise synthesis of PCN-134-2D nanosheets with accessible catalytic sites. (b) PXRD of Zr-BTB and PCN-134-2D treated by aqueous solutions with different pH values. (c) Stability test of Zr-BTB modified by TCPP, DCPP, and TPP treated by aqueous solutions with different pH values. Reproduced from ref. 100. Copyright 2022 Wiley. | ||
Guided by topological engineering, metal–organic cage engineering, pillar-layer engineering and post-synthesis strategies, substantial numbers of porphyrin-based MOFs have been synthesized and modified by purpose, presenting tremendous potential for research and application. High-value conversion of CO2 necessitates a uniform distribution of active sites, a large specific surface area, and stability of the catalyst. Metal–organic cage engineering can offer a significantly large specific surface area and uniformly distributed active sites for porphyrin-based MOFs. However, the metal nodes of the metal–organic cage are generally classified as soft acids, which restricts the intensity of interactions between the ligand and center metals, thereby compromising stability. This issue has been subject to numerous criticisms. However, a notable advantage of topological engineering is its wider range of metals available for metal nodes, which can be used to build stable-structured MOFs based on porphyrins. The issue of some functional groups being unable to be introduced directly during in situ synthesis is resolved by introducing them to fine-tune the properties in the porphyrin-based MOFs or to functionalize the porphyrin-based MOFs on a larger scale. However, often the introduction of functional groups leads to a reduction in the specific surface area of porphyrin-based MOFs.
3. Basic principle of CO2 reduction
The high symmetry and linear structure of CO2 molecules result in evenly distributed electronic charges and spatial arrangement, leading to a low-energy state. Consequently, activating CO2 necessitates the input of substantial energy. The photo(electro)catalytic CO2 reduction using porphyrin-based MOFs is classified as a prototypical heterogeneous catalytic reaction, wherein CO2 molecules undergo activation and subsequent evolution of intermediates at the porphyrin MOF surface.101 Consequently, an in-depth investigation into the catalytic mechanisms and structure–activity relationships of porphyrin MOFs is of paramount importance.102 Below is a detailed explanation of the CO2 activation, the mechanisms involved and the structure–activity relationship in these systems.3.1. Surface interface catalysis mechanism
The processes of photocatalytic CO2 reduction are generally divided into light absorption, electron injection, CO2 activation and evolution of intermediates. Porphyrin rings possess 26-π electrons, forming a highly conjugated electronic system. Conjugated systems enable π electrons to delocalize along the chain, creating a uniform electron cloud and molecular orbitals π and π* with distinct energy levels. When the energy of photons matches the gap between these levels, absorption occurs. The continuous band-like energy structure of conjugated π electrons broadens their light absorption range, especially in the visible spectrum, allowing these compounds to capture diverse wavelengths.103 Thus, porphyrin functions as a vital constituent of porphyrin-based MOFs, endowing porphyrin-based MOF materials with exceptional absorption properties in the visible spectrum, enabling them to efficiently capture photons and undergo transitions to excited states. The electrons excited by photo energy are injected into the conduction band of the porphyrin-based MOFs, leaving positive holes (h+) in the porphyrin. The essence of this process lies in the transfer of photogenerated electrons to active sites on the porphyrin-based MOF, where they can engage in reduction reactions. Then, the injected electrons reduce adsorbed CO2, creating reactive intermediates (COOH*, CO*, OCHO*) that are amenable to further reduction steps. Subsequently, the intermediates are converted to target products; the sufficient reaction kinetics and the availability of photogenerated electrons are crucial factors for this process.104Porphyrin-based MOFs functioning as electrocatalysts in the electrochemical CO2 reduction undergo a process akin to the general mechanism of CO2 electroreduction, albeit with variations attributed to the specific properties of porphyrin moieties and metal centers.105 The process initiates with the adsorption and activation of CO2.106 Then, driven by an applied electric field or photoexcitation, electrons migrate from the electrode to the metal center and further to the CO2 molecule.2 Finally, a series of intermediates and target products are formed. Additionally, in the case of carefully designed porphyrin-based MOFs, their inherent structural resilience and the capacity of their metal centers to undergo reversible redox transformations are pivotal to a phenomenon known as catalyst regeneration, which is essential for upholding catalytic activity after multiple operational cycles.107 And porphyrin-based MOFs excel in photocatalytic applications primarily due to their conjugated structures and tunable pore architectures, which collectively enhance both conductivity and mass transfer efficiency.
It is widely acknowledged that CO2 adsorption is significantly influenced by factors such as specific surface area, pore dimensions, and functional groups attached to porphyrins.109 The synthesis of porphyrin-based MOFs predominantly involves strategies like topological engineering, layer-pillar schemes, and cage construction methodologies, thereby bestowing unique advantages in designing porphyrin MOFs with high surface areas and appropriately sized pores.110–112 Furthermore, the tunability of porphyrin linkers implies enhanced regulatory capacity over CO2 adsorption in porphyrin-based MOFs, offering improved modulation of their catalytic performance. Zhou and his team prepared PCN-601 composed of reactive Ni-oxo cluster nodes and light-harvesting metalloporphyrin ligands connected via the pyrazolyl group, as a catalyst for gas-phase overall CO2 photoreduction with H2O vapor at room temperature.113 Comparison through BET analysis and CO2 capture experiments has revealed that porphyrin-based MOFs generally possess substantial specific surface areas. While surface areas have a notable impact on CO2 adsorption, a critical factor in the kinetics of heterogeneous gas–solid reactions, particularly the affinities between reactants and the catalyst, was meticulously assessed in the case of PCN-601. Consequently, the substantial specific surface area provides an abundance of adsorption sites for CO2, and efficient adsorption is further augmented by the generation of photogenerated electrons under light irradiation. Additionally, this study confirmed that in PCN-601, these photogenerated electrons utilize Ni as a conduit, facilitating accelerated electron transfer and thereby enhancing the catalytic efficiency (Fig. 8a).
 | ||
| Fig. 8 (a) The BET surface area and the CO2 uptake of PCN-601 and the structure of PCN-601 and PCN-222. Reproduced from ref. 113. Copyright 2022 ACS. (b) The electron-transfer mechanisms of SC-Bi-PMOFs. Reproduced from ref. 114. Copyright 2022 Elsevier. (c) The mechanisms of ET-PT and PCET and the pattern of log(jCO) vs. log(HCO3−). Reproduced from ref. 115. Copyright 2022 Wiley. | ||
The activation of CO2 is a pivotal step in its conversion into value-added chemicals and fuels. This process typically involves breaking the stable double bond between the carbon and oxygen atoms in CO2, rendering it reactive enough to participate in subsequent reduction reactions. Strategies to provide suitable reaction sites and lower the energy barrier for CO2 activation in porphyrin-based MOFs primarily involve several methods: constructing metal ions coordinated within the porphyrin ring, designing frameworks with synergistic multiple reactive sites, and engineering metal nodes to enhance conductivity. Xu demonstrated that CO2 coordination can drive low temperature rapid synthesis of porphyrin-based bismuth-MOFs (Bi-PMOFs) by utilizing synergistic physical and chemical properties of supercritical CO2.114 And SC-Bi-PMOFs combined many advantages of highly accessible active sites, fast electron/mass transfer capability, and a unique coordination environment around active sites, which endowed them with superior photocatalytic CO2 reduction activity (Fig. 8b). In both photocatalytic and electrocatalytic CO2 reduction, the provision of electrons with sufficient kinetic energy to the active sites is crucial for disrupting the inherent symmetry of CO2 molecules, thereby facilitating the activation of CO2. This step is pivotal in converting CO2 into value-added chemicals or fuels.
| One step: *COOH + e− → CO + OH− |
| Two steps: *COOH + e− → *CO + OH− → CO + H2O |
| *CO + e− + H+ → *CHOH → CH3OH |
| Multi-step: *CO + 2e− + 2H+ → *CHOHCH2 → C2H4 |
| *CO + 2e− + 2H+ → *CHOHCH2OH → CH3CH2OH |
Generally, redox-active sites of porphyrin-based MOFs are typically situated at the metal centers within their structure, where these centers possess a unique electronic configuration enabling participation in redox reactions and facilitation of electron transfer. Zhang and his team prepared porphyrin-based MOFs (Cu-SAs@Ir-PCN-222-PA) with dual active sites of Ir-porphyrin and Cu-SAs through the pre-coordination confinement strategy. Catalytic results disclosed that Cu-SAs@Ir-PCN-222-PA could drive the reduction of CO2 to C2H4 with a high faradaic efficiency (FE) (70.9%) with a current density of 20.4 mA cm−2. And the DFT calculation was carried out to deduce the electrocatalytic CO2 reduction mechanism (2CO2 + 8e− + 8H+ → C2H4 + 2H2O). The reduction of CO2 molecules at the catalyst surface involves a sequential acquisition of electrons and protons, namely, a stepwise electron transfer proton transfer (ET-PT).
Distint from the traditional redox mechanism, in proton-coupled electron transfer (PCET), the transfer of electrons is tightly coupled with that of protons, occurring virtually simultaneously, which contrasts with traditional redox reactions where electron and proton transfers may occur as separate steps.114 Moreover, the thermodynamic and kinetic properties of PCET reactions can vastly differ from those of isolated electron or proton transfers. This synergistic transfer can impact the free energy change, activation energy, and reaction rate, sometimes rendering reactions feasible that would otherwise be difficult to proceed. Sarazen and co-workers demonstrated that the well-dispersed iron-porphyrin-based MOF (PCN-222(Fe)) on carbon-based electrodes exhibited optimal turnover frequencies for CO2 electroreduction to CO at 1 wt% catalyst loading, beyond which the intrinsic catalyst activity declined due to CO2 mass transport limitations.115 The pattern of log(jCO) vs. log(HCO3−) indicates that the CO2 electroreduction rate is not dependent on the transfer of protons. This finding excludes both PT in the stepwise ET-PT pathway and PCET as the rate-determining step of the reaction, suggesting that ET is the rate-determining step (Fig. 8c).
3.2. Structure–activity relationship
Porphyrin-based MOFs have emerged as prominent catalysts in the realm of advanced materials, primarily due to their integration of unique structural elements: a porphyrin core offering tunable electronic properties, strategically selected metal nodes that facilitate electron transfer, meticulously designed linkers enabling porosity tuning, and a highly ordered porous structure providing enhanced surface area. This sophisticated design not only fosters exceptional photo(electro)catalytic functionality for efficient CO2 conversion into valuable chemicals and fuels but also underscores their stability and recyclability, which are pivotal for sustainable carbon capture and utilization technologies. The synergy of these features positions porphyrin-based MOFs as prime candidates in the quest for optimized photocatalytic and electrocatalytic systems, thereby driving advancements in green chemistry and climate change mitigation strategies.The porphyrin core serves as a common reactive site in porphyrin-based MOFs for photo(electro)catalytic CO2 reduction, primarily due to its facile coordination capabilities and ability to easily accept and transfer electrons. When the macrocyclic porphyrin structure coordinates with metal ions, it effectively captures light energy and facilitates electron excitation and transfer, which is vital for activating CO2 molecules. With metal ions situated at the center of the porphyrin ring, they can modulate charge distribution, influencing the electronic structure of the catalytic site and, consequently, altering the adsorption and activation abilities towards reactants such as CO2. Furthermore, tuning the metal species within the porphyrin core allows for further optimization of catalytic performance, as different metal centers impart distinct electronic characteristics and activities, advantageous in breaking the symmetry of CO2 and promoting its conversion into valuable chemicals and fuels during photocatalysis or electrocatalysis.29,117 Su and his team contrastively investigated the catalytic performance of two-dimensional (2D) cobaltporphyrin-based organic frameworks linked with phenyl, CoO-cluster, ZnO-cluster, and ZrO-cluster, as CO2 reduction photocatalysts (abbreviated as CoP, Co-PMOF, ZnPMOF, and Zr-PMOF, respectively) using DFT computations. The calculated results suggest that the CoP, Co-PMOF, Zn-PMOF, and Zr-PMOF monolayers are all semiconductors with band gaps of 1.63, 1.21, 1.72, and 1.68 eV, respectively.118 A narrower bandgap implies superior light-harvesting capability; hence modifications to the porphyrin core are conducive to enhancing the electronic environment around the porphyrin, thereby elevating the catalytic performance of porphyrin-based MOFs. The selection of metal nodes in porphyrin-based MOFs not only directly influences the electronic structure of the MOFs but also interacts synergistically with the porphyrin units, collectively enhancing the catalytic performance of porphyrin-based MOFs. Sheng fabricated a Cu-ZnTCPP MOF, in which the first Cu refers to the carboxylate-coordinated Cu2(COO)4 node in a paddle-wheel structure.119 The research suggested that the enhanced CO2 reduction rate in the presence of O2 benefited from the promoted activity on the higher coordination Cu node (HO-Cu node) relative to that on the lower coordination Cu node (LC-Cu node) with a significantly lower activation barrier (0.55 eV vs. 1.42 eV), as well as the role of –OH in capturing and activating the gas-phase CO2. The judicious introduction of functional groups markedly enhances the photocatalytic performance of porphyrin-based MOFs by widening the light absorption range, refining charge transport efficiency, creating and tuning active sites, and fostering synergies. Additionally, it bolsters their electrocatalytic capabilities through improvements in conductivity and modulation of local charge distribution, thereby comprehensively elevating photo(electro)catalytic efficiencies. Chen synthesized a series of functional group-containing (–F, –NH2 and –NO3−) MOF-808 catalysts and investigated their adsorption behavior toward CO2. F-MOF-808 exhibited super catalytic performance for CO2, which underscores the significance of rationally tuning the electronic structure and defective metal centers. Furthermore, the introduction of fluorine significantly enhanced the selectivity towards CO production for F-MOF-808, reaching up to 97.8%.120
Porphyrin-based MOFs have demonstrated remarkable potential in the field of photo(electro)catalysis for CO2 reduction, with the exploration of their high-performance characteristics primarily centered around three core aspects: CO2 adsorption and activation, evolution of intermediates, and the relationship between structure and activity. Firstly, the adsorption and activation of CO2 mark the inception of the entire catalytic process.121–123 The unique porous architecture of porphyrin-based MOFs, coupled with functionalized ligands, enhances both the physical adsorption and chemical activation of CO2 molecules. Particularly, tuning the functional groups on the porphyrin ring can optimize the binding mode of CO2 at active sites, reducing its activation energy barrier and facilitating the initial step of CO2 conversion. Secondly, the evolution of intermediates is pivotal to gaining an in-depth understanding of the catalytic mechanism. Under photo or electrostimulation, the formation, stabilization, and transformation pathways of intermediates generated post-CO2 activation, such as *COOH and *CHO, directly influence the selectivity and yield of the final products. Furthermore, the proposition of PCET mechanisms offers profound insights into the intricate interplay of electron and proton migration in photo(electro)catalytic CO2 reduction, facilitating a deeper understanding of the reaction pathways involved. Lastly, the investigation of structure–activity relationships elucidates the microcosmic principles underlying catalyst design. Variations in metal nodes, linking strategies, and degrees of functionalization not only impact the electronic structure and photo(electro)responsive properties of the materials but also directly relate to the distribution and efficiency of catalytically active sites. Systematically adjusting these structural parameters uncovers new avenues for enhancing catalytic performance. In summary, by focusing on CO2 adsorption and activation, tracing the progression of intermediates, and delving into the intrinsic connections between structure and activity, research on porphyrin-based MOFs for photocatalytic and electrocatalytic CO2 reduction is progressively advancing our comprehension of efficient CO2 conversion mechanisms. This endeavor provides a scientific foundation and innovative perspectives for the development of sustainable carbon cycle utilization technologies.
4. Porphyrin-based MOFs for photocatalytic CO2 reduction
Photocatalysts play an important role in the field of photocatalytic CO2 reduction because the process of transformation of CO2 into value-added chemicals (such as CO, CH4, HCHO, HCOOH, CH3OH and C2 products) through sunlight is straightforward and involves zero consumption. The materials that can be used as photocatalysts include inorganic semiconductors (ZnO2, TiO2, Fe2O3, CdS, Co3S4, LDHs, etc.),9,124 organic semiconductors (porphyrin, triazine, etc.), and noble metals (Ag nanoparticles (NPs), Au NPs, Pt NPs).28,30 When sunlight irradiates the surface of the photocatalyst, the process of photocatalytic reaction occurs on the surface which is as follows: (1) CO2 molecules are absorbed by the active sites of the photocatalyst; (2) light is harvested by the semiconductor photocatalyst; (3) light energy drives the separation of photogenerated carriers; (4) the photogenerated carriers migrate to the semiconductor's valence (VB) and conduction (CB) bands, respectively; (5) the photogenerated electrons in the CB undergo a reduction reaction with the adsorbed CO2; and (6) product desorption from the photocatalyst surface takes place. This completes the entire catalytic reaction process. In contrast, the photocatalytic activity of noble metal NPs is rooted in the surface plasmon resonance effect (SPR).123,125,126 The evidence presented demonstrates that the condition of photogenerated carriers significantly impacts the performance of photocatalysis.45,112 Porphyrins are highly efficient photo-response molecules, and functionalized porphyrins integrated into MOFs greatly improve the intra-electronic environment of the catalysts, which has led to intensive investigations. Porphyrin-based MOF(Zn) materials have strong photocatalytic activity for photocatalytic CO2 reduction. S. Sharifnia and co-workers synthesized a porphyrin-based metal organic framework (Zn/PMOF), and the ligand in Zn/PMOF is TCPP (Fig. 9a).127 Then, the photocatalytic CO2 reduction behavior of porphyrin-based MOFs was investigated by a Zn/PMOF platform with a gaseous phase reactor, in which vapor acts as a donor. As shown in Fig. 9b, under UV/Visible irradiation, electrons at first are excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) of Zn/PMOF. Subsequently, the interaction between the photogenerated electrons in the CB and the adsorbed CO2, as well as the interaction between the photogenerated holes in the VB and the water molecules, initiates the photocatalytic reaction. In this photocatalytic system, the ultimate product is CH4 with a considerable yield of 8.7 μmol g−1 h−1. In addition, the results of 3-cycling and long-term tests illustrated that the porphyrin-based MOFs possessed excellent stability during the photocatalytic reaction (Fig. 9c and d). | ||
| Fig. 9 (a) Synthetic reaction scheme for Zn/PMOF. (b) Mechanism of photocatalytic reduction of CO2 over Zn/PMOF with H2O vapor as a sacrificial agent. (c) The photocatalytic performance of Zn/PMOF under UV/Visible light. (d) Recycling tests for photocatalytic reduction of CO2 under UV/Visible light over Zn/PMOF. Reproduced from ref. 127. Copyright 2022 Elsevier. | ||
The chemical stability and the selection of the active center of the photocatalyst are key factors for photocatalytic CO2 reduction. Diverse and complex structures of porphyrin-based MOFs demonstrate reliable stability of the photocatalyst closely associated to the suitable synthesis routes and structural designs. Li and his team proposed a strategy wherein a series of BUT-110 with different contents of 4,4′-(porphyrin-5,15-diyl)dibenzolate (DCPP2−) were prepared by in situ substitution of the porphyrin ligand DCPP2− to stabilize and functionalize BUT-109(Zr).128 And the post-synthetic route for DCPP2−-displaced BUT-109(Zr) (BUT-109(Zr)-P) can also be used. The post-synthetic modification would not be applicable for BUT-109(Zr)-P owing to its low porosity resulting from framework interpenetration (Fig. 10a). According to the results of XRD and BET, BUT-109(Zr) and BUT-110 analogs show more chemical stability, which can be reflected by the pH tolerance (pH = 1–10) (Fig. 10b and c). As a comparison, BUT-109(Zr) possesses poor chemical stability which is mainly attributed to the reversible C–N linkage in the imide ligand linker 4,4′-(1,3,6,8-tetraoxobenzo[lmn][3,8]phenanthroline-2,7(1H,3H,6H,8H)-diyl)dibenzoate (NDIDB2−). In addition, a metalloporphyrin, which serves as an excellent active site, was introduced into the most stable BUT-109(Zr)-50% during pre-synthesis to obtain a novel BUT-110-50%-Fe, which was applied in the study of photocatalytic CO2 reduction. Photocatalytic CO2 reduction studies were performed in a reaction chamber charged with CO2 (0.1 MPa) under full-spectrum irradiation; the hydrogen donor and the sacrificial agent were H2O and triethylamine, respectively. As shown in Fig. 10d, the yield of the main product (CO) for BUT-110-50%-Fe was 47.2 μmol g−1 h−1, which demonstrates that the robust porous structure and uniform active site distribution synergistically promote photocatalytic CO2 reduction performance.
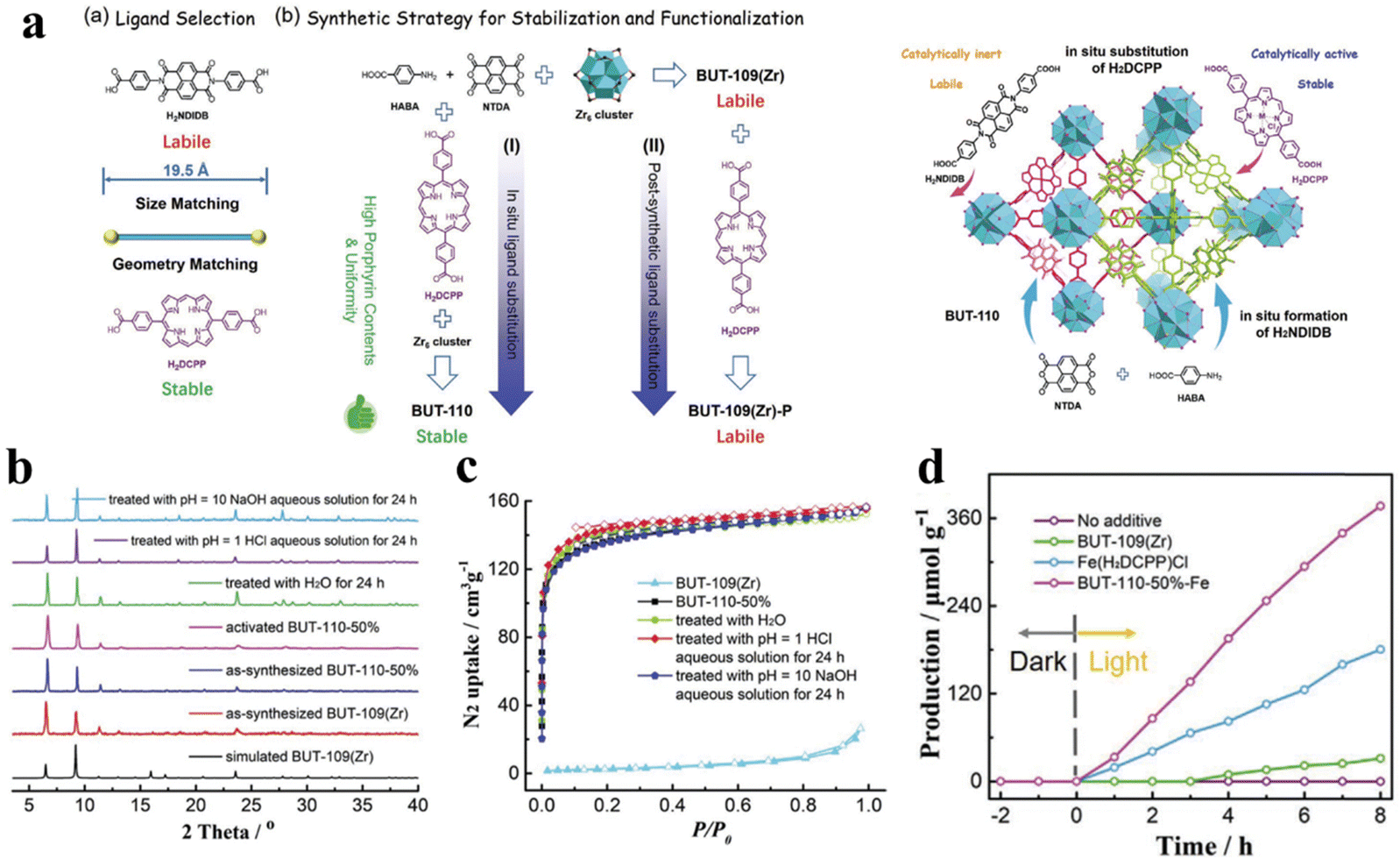 | ||
| Fig. 10 (a) Ligand selection and synthetic strategies of BUT-110 through in situ ligand substitution and post-synthetic ligand substitution, and the schematic illustration of the proposed “in situ ligand substitution” strategy for the construction of BUT-110 from HABD, NTDA, and H2DCPP. (b) Comparison of XRD patterns. (c) N2 adsorption isotherms between BUT-109(Zr) and BUT-110-50% after different treatments. (d) The yield of CO for BUT-110-50%-Fe. Reproduced from ref. 128. Copyright 2022 Wiley. | ||
Porphyrins integrated into MOFs as the functional moiety alter the ligand metal positioned at the porphyrin core towards the modulation of CO2 photocatalysis. Wang and his team synthesized a series of 2-fold interpenetrated indium-porphyrin frameworks In(H2TCPP)(1−n)[M(TCPP)(H2O)](1−n)[DEA](1−n) (In-MnTCPP-MOF) (M = In, Co, Fe), which consisted of H2TCPP (tetrakis(4-benzoic acid)porphyrin) with Fe coordination and DEA (diethylamine) (Fig. 11a).129 The photocatalytic CO2 reduction test was performed in the mixed solution of 20 mg of L-ascorbyl palmitate (L-AP) and 5 mL of ethyl acetate. Herein, high-performance visible-light-driven CO2 to CO conversion resulted by the synergetic effect between porphyrinic rings and Fe reaction active sites, and Fe1.91TCPP-MOF was shown to be the best catalyst for photocatalytic CO2 reduction with a CO yield of up to 3469 μmol g−1 within 24 h with a high selection of 90% (Fig. 11d). According to the results of density functional theory (DFT) (Fig. 11b), COOH− is an important intermediate in the process of photocatalytic CO2 reduction. Free energy changes for the CO2 reduction processes at Fe/Co porphyrins are as follows:
| [Fe0(Por2−)]2− + CO2 + H2CO3 → [FeII(Por2−)(COOH−)]− + HCO3− ΔG = −4.10 kcal mol−1 |
| [Co0(Por2−)]2− + CO2 + H2CO3 → [CoII(Por2−)(COOH−)]− + HCO3− ΔG = −0.25 kcal mol−1 |
 | ||
| Fig. 11 (a) Synthesis and structure of In-Fe/CoTCPP-MOFs. (b) Influence of the electron configurations of the metal centers on metal–carbon interactions in [FeII(Por2−)(COOH−)]− and [CoII(Por2−)(COOH−)]−. (c) Time-resolved PL decays of In-Fe1.91TCPP-MOF, In-Co1.71TCPP-MOF, and In-InTCPP-MOF under excitation at 440 nm. (d) Time profiles for the CO evolution catalyzed by In-Fe1.91TCPP-MOF, In-Co1.71TCPP-MOF, and In-InTCPP-MOF under irradiation with a 300 W xenon light source (>400 nm) in the presence of 0.1 M L-ascorbyl palmitate (L-AP) in ethyl acetate under 1 atm CO2. Reproduced from ref. 129. Copyright 2022 ACS. | ||
[FeII(Por2−)(COOH−)]− originates from [Fe0(Por2−)]2− with a lower free energy compared to that of [CoII(Por2−)(COOH−)]− which transforms from [Co0(Por2−)]2−, which means more stability for [FeII(Por2−)(COOH−)]− compared with [CoII(Por2−)(COOH−)]−. This is attributed to the self-filled dz2 orbital which weakens the coordination of the COOH− moiety from the z-direction for [Co0(Por2−)]2−. However, the empty dz2 orbital of [Fe0(Por2−)]2− can easily accept the long pairs of electrons from COOH−. Thus, the Fe center can lower the negative charge of the COOH− moiety, ultimately, stabilizing the intermediate. Then, the short life-span of Fe1.91TCPP-MOF means the rapid recombination or the fast transportation of photogenerated carriers (Fig. 11c). The electronic properties and CO2 adsorption of Fe1.91TCPP-MOF demonstrate that rapid electron migration may occur on the photocatalyst. In addition, compared with [CoII(Por2−)(COOH−)]−, the Fe active site brings key intermediates closer together (1.9 Å), which means the adsorption state has a low Gibbs free energy.
The lifespan of photogenerated carriers is a significant influence for photocatalytic CO2 reduction, which can be regulated through modifying the microstructure and permanent channels of porphyrin-based MOFs. Deria and co-workers report topological control over the photophysical properties of MOFs via modular interchromophoric electronic coupling to manifest different fluorescence lifetimes.130 There are four target samples for lifetime research, NU-902(H2) and MOF-525(H2), consisting of free-base TCPP(H2), and NU-902(Zn) and MOF-525(Zn), consisting of zinc(II)-metallated TCPP(Zn) linkers. In addition, NU-902 with a topological network of ftw and MOF-525 with a topological network of scu manifest the smallest porphyrin–porphyrin torsional angle of 90° and 60°, respectively (Fig. 12a–c). According to Fig. 12d and e, the order of fluorescence lifetime for the corresponding Zn(II)-based porphyrinic MOFs is TCPPOMe(Zn) > MOF-525(Zn) > NU-902(Zn) and the same trend appears on TCPPOMe(H2) > MOF-525(H2) > NU-902(H2). Combined topological structures (ftw and scu) of porphyrin-based MOFs and fluorescence lifetime spectra indicate that stronger interchromophoric interaction occurs on NU-902 and leads to more red shifted spectra with faster emissive state radiative decay. Therefore, the idea is potential to be applied in the investigation of photocatalytic CO2 reduction.
 | ||
| Fig. 12 (a) Molecular structures of porphyrin-based MOFs NU-902 and MOF-525 (M = H2 or Zn) and their corresponding building blocks. (b) and (c) DFT optimized structures of closely positioned porphyrin dimers in MOF-525(H2) [90°, dM–M = 13.5 Å] and NU-902(H2) [60°, dM–M = 10.5 Å]. (d) and I Transient emission decay profile for free-base and zinc(II)-metallated NU-902 and MOF-525. Reproduced from ref. 130. Copyright 2022 RSC. | ||
The single-atom strategy has been widely employed in the field of photocatalytic CO2 reduction. The addition of single atoms enables a better dispersion of the reactive sites, while optimizing the photoelectronic properties of the photocatalyst. Ye et al. incorporated coordinatively unsaturated Co single atoms into a porphyrin-based MOF, denoted as MOF-525-Co, which consists of Zr6 clusters and porphyrin-based molecular units (TCPP), as shown in Fig. 13a.131 In this photocatalytic system, single-atom Co is identified as the active site, as shown in Fig. 13b. This active site enhances the absorption of CO2 by increasing the concentration of localized electrons in the near-surface region and inducing surface corrugation. The introduction of electrons to the Co active sites lowers the reaction activation energy of CO2 (reducing the energy barrier (EB) from 4.13 to 3.08 eV), suggesting that photo-excited electrons from porphyrin migrate to CO2 molecules through the Co active site (Fig. 13c and d). Ultimately, the optimal photocatalyst presented up to 3.13-fold improvement in the CO evolution rate (200.6 μmol g−1 h−1) and a 5.93-fold enhancement in the CH4 generation rate (36.67 μmol g−1 h−1) compared to the primitive porphyrin-based MOF (Fig. 12e).
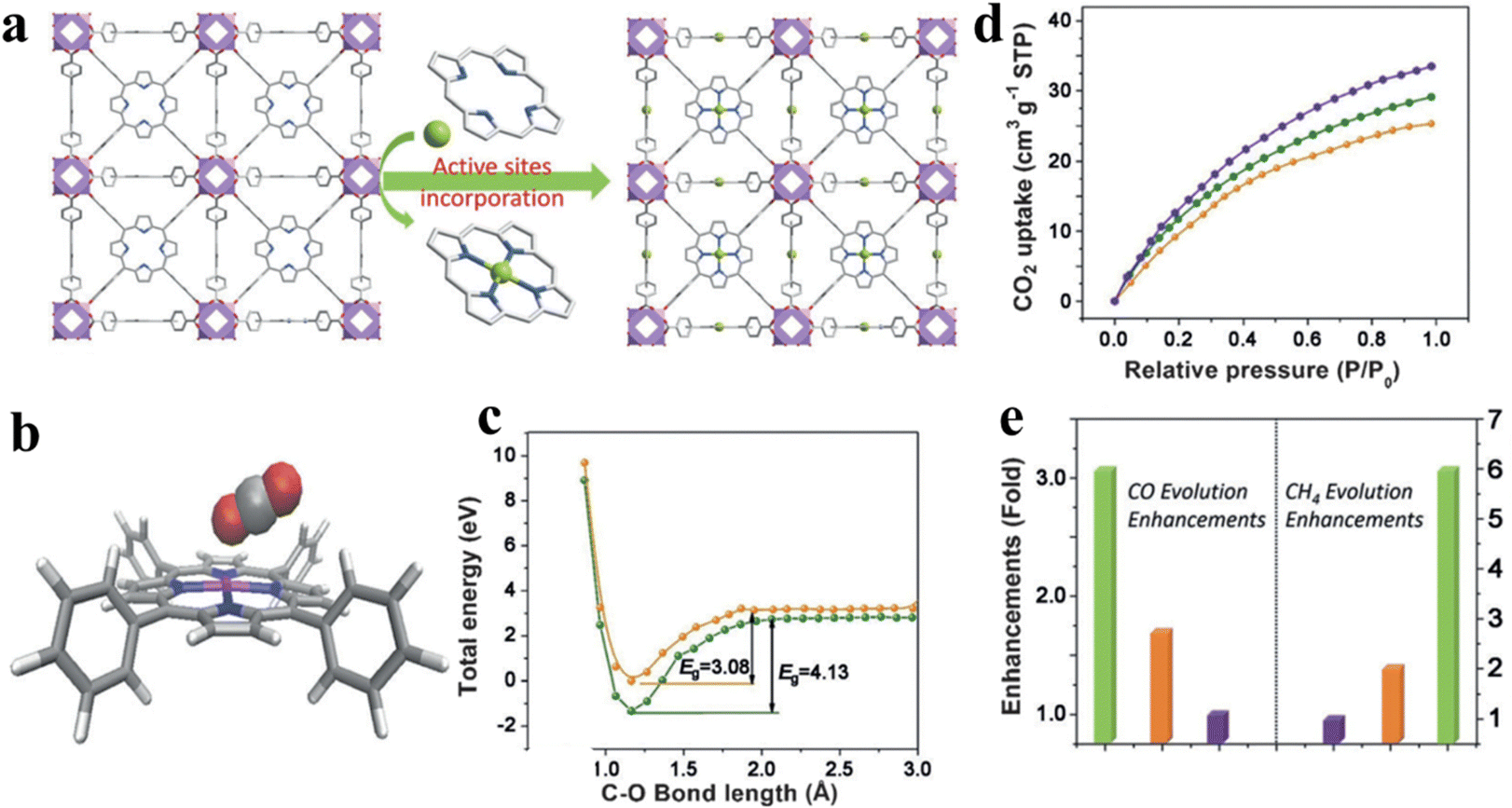 | ||
| Fig. 13 (a) View of the 3D network of MOF-525-Co featuring a highly porous framework and incorporated active sites. (b) The optimized structure for CO2 adsorption on a porphyrin-Co unit. (c) The O–C bond length-dependent CO2 activation energy barrier, charged with one electron (orange) and neutral state (green). (d) CO2 adsorption behaviors for MOF-525 (orange) as well as MOF-525-Co (purple) and MOF-525-Zn (green) implanted with single atomI(e) Enhancement of production over MOF-525-Co (green), MOF-525-Zn (orange), and MOF-525-Zn (purple). Reproduced from ref. 131. Copyright 2022 Wiley. | ||
A heterojunction comprises dissimilar semiconductors with distinct electronic properties, most notably differing band gaps (Eg). The band gap represents the energy difference between the valence band (VB) and the conduction band (CB), determining the minimum energy required for an electron to transition from the VB to the CB. In a heterojunction system, the semiconductors may have different chemical compositions, crystal structures, or dopant concentrations, resulting in varying Eg values and band edge positions. Its impact on photocatalysis is characterized by several key aspects: efficient charge separation, broadened light absorption, improved charge transport, and interface catalytic effects. Ye and his team prepared a Z-scheme photocatalyst consisting of PCN-224(Cu) and TiO2 nanoparticles to improve the photocatalytic performance in CO2 reduction. As shown in Fig. 14a, TiO2 and PCN-224(Cu) are intimately connected and generate a built-in electric field, under which photogenerated electrons migrate from the oxidizing side TiO2 to the reducing side PCN-224(Cu).64 The advanced structure of the hybrid photocatalyst retains the original redox strength of the semiconductor and enhances the photoelectric absorption capability. As shown in Fig. 14b and c, the light harvesting and light response of the hybrid photocatalyst 15%P(Cu)/TiO2 are obviously improved. The photocatalytic CO2 reduction test was carried out in a gas phase system without any sacrificial agent and photosensitizer. And the integration is capable of significantly triggering the improvement in photocatalytic CO2 reduction into CO, with an evolution rate of 37.21 μmol g−1 h−1, which is 10 times and 45.5 times higher than that of pristine PCN-224(Cu) and pure TiO2, respectively (Fig. 14d).
 | ||
| Fig. 14 (a) Direct Z-scheme photocatalytic mechanism. (b) Energy band gaps (Eg) of TiO2, PCN-224(Cu), and PCN-224(Cu)/TiO2. (c) Photocurrent–time (I–t) curves of TiO2, PCN-224(Cu), and 15%P(Cu)/TiO2 photoelectrodes at 0.6 V vs. REH bias potential in 0.5 M Na2SO4 (pH ∼7.35). (d) CO and CH4 generation rate over PCN-224(Cu), 6%P(Cu)/TiO2, 7.5%P(Cu)/TiO2, 10%P(Cu)/TiO2, 15%P(Cu)/TiO2, 30%P(Cu)/TiO2, and TiO2 under 300 W Xe lamps. Reproduced from ref. 64. Copyright 2022 ACS. | ||
The photocatalytic reduction of CO2 offers a promising avenue for harnessing sunlight to convert the abundant greenhouse gas into valuable chemical commodities and fuels, while operating under mild and energy-efficient conditions. This environmentally benign and potentially transformative technology has attracted significant attention, prompting numerous research endeavors aimed at enhancing the photocatalytic performance of porphyrin-based MOFs. These efforts are geared towards expediting the practical deployment of porphyrin MOFs as efficient photocatalysts for CO2 reduction. This section provides an overview of how various synthetic strategies, metal active sites, configurations, single-atom doping, and heterojunction designs influence the chemical stability, product selectivity, photocatalytic activity, and photoelectrochemical properties of porphyrin-based MOFs. Subsequently, recent advances in applying these materials to the photocatalytic reduction of CO2 are summarized in Table 1. IHEP-22(Co) shows the best performance of photocatalytic reduction of CO2 to CO, with a yield of 350.9 μmol h−1 g−1. The metal center not only serves as a pathway for the photogenerated electrons from porphyrin to transfer to CO2 molecules, but also, through spatial regulation, it has been observed that the stacking method, spatial angle, and distance of porphyrin in MOFs greatly influence the photocatalytic conversion of CO2. The high modifiability of porphyrin-based MOFs allows for significant potential for future research exploring the correlation between spatial structure and catalytic activity.
| Samples | Porphyrins | Products | Performance (μmol g−1 h−1) | Selectivity | Ref. |
|---|---|---|---|---|---|
| PCN-224(Cu)/TiO2 | CuTCPP | CO/CH4 | 37.21/0.22 | 99.4% | 64 |
| In-Fe1.91TCPP-MOF | FeTCPP | CO/CH4 | 144/0.72 | 99.5% | 129 |
| In-Co1.71TCPP-MOF | CoTCPP | CO | 38.1 | — | 129 |
| In-InTCPP-MOF | InTCPP | CO | 551 | — | 129 |
| IHEP-22(Co) | CoTCPP | CO/CH4 | 350.9/8.9 | 97.5% | 132 |
| IHEP-22(Cu) | CuTCPP | CO/CH4 | 230/19.45 | 92.2% | 132 |
| MOF-525-Co | CoTCPP | CO/CH4 | 200.6/36.8 | 84.5% | 131 |
| g-C3N4/Cu-CuTCPP MOF | CuTCPP | CO/CH4 | 11.6/18.5 | 71% | 133 |
| Zn/PMOF | ZnTCPP | CH4 | 8.7 | — | 127 |
| BUT-109(Zr) | DCPP | CO | 22.6 | — | 128 |
| BUT-110-50%-Co | DCPP | CO/CH4 | 64/11.3 | 85% | 128 |
| MOF 1(monolayer) | TPP | CO | 60.9 | 100% | 134 |
| MOF 2 (bilayer) | TPP | CO | 46.14 | 100% | 134 |
| g-CNQDs/PMOF | TCPP | CO/CH4 | 16.1/6.8 | 70.1% | 135 |
| Cu-PMOF | CuTCPP | HCOOH | 130.6 | — | 136 |
5. Porphyrin-based MOFs for electrocatalytic CO2 reduction
CO2RR is typically conducted in a saturated aqueous sodium carbonate solution, organic solution, or ionic solution, under milder conditions compared to the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and oxygen reduction reaction (ORR) conducted in strong acidic and alkaline systems.78,137,138 The integration of porphyrin-based MOFs has allowed for the transition of porphyrins from original homogeneous catalytic systems to heterogeneous catalytic systems.127,139,140 Common products resulting from heterogeneous catalytic electrochemical CO2RR include CO, CH4, HCOO−, and C2H4, among others.38,75,141 However, research on the electrochemical CO2RR of porphyrin-based MOFs is limited due to challenges such as the competitive hydrogen precipitation reaction, low reaction efficiency, and poor selectivity.142–144 Gu et al. prepared copper(II) paddle wheel cluster-based porphyrinic metal–organic framework (MOF) nanosheets for electrochemical CO2RR. The discussion further examines the outstanding product selectivity and structural modifications of Cu2(CuTCPP) nanosheets.145 As shown in Fig. 15a, Cu-MOF nanosheets exhibit significant activity for formate production with a faradaic efficiency (FE) of 68.4% and acetate production with a FE of 16.8% under CO2-saturated CH3CN solution with 1 M H2O and 0.5 M EMIMBF4. Based on the analysis of linear sweep voltammetry (LSV) curves of Cu2(CuTCPP), a significant amount of hydrogen was generated as a byproduct of the electrocatalytic process, and the changing trend of current density over time indicates the possibility of chemical restructuring of Cu2(CuTCPP) occurring after the first hour. Thus, the pre-electrolyzed Cu2(CuTCPP) nanosheets were further used to eliminate the side reactions during the initial process and further confirm the activity of the final cathodized catalyst. At the appropriate working potential (−1.55 V), the total faradaic efficiency of formate and acetate formation reached 85.2% (Fig. 15b–e). The excellent performance is mainly attributed to synergy between Cu2(CuTCPP) nanosheets and CuO, Cu2O and Cu4O3. In addition, the energy-state change after 5 h reaction for Cu species is reflected through XPS spectra showing the peaks at 569.5 eV and 572.5 eV, which are ascribed to the kinetic energy of Cu+ and Cu2+, respectively (Fig. 15f and g). The results indicate the instability of the Cu2(CuTCPP) catalyst in the electrocatalytic CO2RR process. However, the substitution of the catalyst resulted in an unforeseen impact on the catalytic process. | ||
| Fig. 15 (a) Crystal structure of Cu2(CuTCPP) nanosheets along the c axis. Red is O, blue is N, grey is C and cyan is Cu and the CO2 electrochemical reduction system with Cu2(CuTCPP) nanosheets as the catalyst. (b) Faradaic efficiencies of Cu2(CuTCPP) nanosheets. (c) Faradaic efficiencies of Cu2(CuTCPP) nanosheets at different times. (d) Faradaic efficiencies of pre-electrolyzed Cu2(CuTCPP) nanosheets. (e) Total and partial current densities for CO2RR products on pre-electrolyzed Cu2(CuTCPP). (f) Cu 2p XPS spectra and g. O 1s XPS spectra of Cu2(CuTCPP) on FTO before and after 5 h reaction. All potentials were set at −1.55 V vs. Ag/Ag+. Reproduced from ref. 145. Copyright 2022 RSC. | ||
The manipulation and alteration of linkers in porphyrin-based metal–organic frameworks (MOFs) represent a strategic approach to tuning their electrical conductivity, thereby enhancing their electrochemical performance.53,91,126 This is achieved through the introduction or substitution of functional linkers that facilitate charge transport while maintaining the inherent porphyrin topology.146 Huang and his team integrated viologen groups into cobalt porphyrin (Por(Co))-based MOFs by a self-assembly method for the first time to act as electron-transfer mediators (ETMs) to enhance the electron-transfer capacity and thus improve the activity of the CO2RR (Fig. 16a).147 Interestingly, both Vg and Por(Co) possess a similar length (19.65 Å and 19.62 Å, respectively) and the same end functional group, which is beneficial to form coordination bonds with the Hf6O8 (Zr6O8) cluster. And the more positive onset potential and higher current densities in the CO2-saturated 0.5 M KHCO3 solution demonstrate outstanding reaction activity and feasibility of the CO2RR, according to the LSV curves (Fig. 16b and c). Vg-Por(Co)-MOF (n: 1) (n = 1, 3, 5, 9) showed lower potential under a current density of 10 mA g−1 indicating the more electron transfer efficiency than pure Vg-MOF and Por(Co)-MOF. And compared with other components, Vg-Por(Co)-MOF (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) exhibited the best activity in the CO2RR with a jCO of 24.3 mA cm−2 at −1.0 V and a high FECO of 93.7% at −0.6 V (Fig. 16d). Finally, a long term electrocatalytic test at a fixed potential of −0.6 V indicated that Vg-Por(Co)-MOF (9:1) has electrochemical stability for CO2RR (Fig. 16e).
:1) exhibited the best activity in the CO2RR with a jCO of 24.3 mA cm−2 at −1.0 V and a high FECO of 93.7% at −0.6 V (Fig. 16d). Finally, a long term electrocatalytic test at a fixed potential of −0.6 V indicated that Vg-Por(Co)-MOF (9:1) has electrochemical stability for CO2RR (Fig. 16e).
 | ||
| Fig. 16 (a) Schematic of the synthesis of Vg-MOF, Por(Co)-MOF, and Vg-Por(Co)-MOF by the solvothermal method. (b) LSV curves of Vg-Por(Co)-MOF (9:1) in CO2- and Ar-saturated 0.5 M KHCO3. (c) LSV curves of Vg-MOF, Por(Co)-MOF and Vg-Por(Co)-MOF (n: 1) (n = 1, 2, 5, 9) in a 0.5 M KHCO3 electrolyte under CO2. (d) Faradaic efficiencies of CO for Vg-MOF, Por(Co)-MOF and Vg-Por(Co)-MOF (n: 1) (n = 1, 3, 5, 9). (e) Stability of Vg-Por(Co)-MOF (9:1) at a potential of −0.6 V versus RHE for 10 h. Reproduced from ref. 147. Copyright 2022 RSC. | ||
Selectivity is indeed a critical challenge in the electrocatalytic reduction of carbon dioxide (CO2RR) as it determines the efficiency of converting CO2 into valuable chemical feedstocks and fuels.21,41,44,49 A novel strategy to enhance the selectivity of porphyrin-based MOFs for CO2RR involves the careful modification of cationic groups within the MOF structure to stabilize specific reaction intermediates. This approach capitalizes on the intrinsic properties of porphyrin MOFs and tailors their electrochemical environment to favor desired product formation.42 Hod et al. prepared Fe-porphyrin (Hemin)-modified Zr6-oxo based 2D-MOF (Zr-BTB@Hemin) for electrocatalytic CO2RR, owing to the porous, robust structure and mass transport.78 However, unsatisfactory catalytic activity and product selectivity are the drawbacks of Zr-BTB@Hemin to be applied in CO2RR. In this case, the authors tethered a cationic functional group, (3-carboxypropyl)trimethylammonium (TMA), proximal to the Fe-porphyrin active site via post-synthesis (Zr-BTB@Hemin-TMA). The details of the synthesis process and catalyst's structure can be found in Fig. 17a. As shown in Fig. 17c and d, Zr-BTB@Hemin-TMA exhibits higher electrochemical activity under a CO2 atmosphere and more outstanding CO product selectivity (about 92%, at the potential of 1.2 V vs. NHE) than that without the TMA group, which is mainly attributed to electrostatic stabilization of a weakly bound CO intermediate, which accelerated its release as a catalytic product. In order to obtain detailed insights into the mechanisms governing the improved electrocatalytic CO2-to-CO performance of Zr-BTB@Hemin-TMA compared to Zr-BTB@Hemin, Raman spectroscopy was carried out which revealed the possible electronic communication between MOF-installed Hemin and TMA ligands. The peaks of Zr-BTB@Hemin at 1367 cm−1 and 1562 cm−1 shift to 1369 cm−1 and 1568 cm−1 after being modified by TMA, which means the conversion of low-spin Fe3+-Hemin into a high-spin species (Fig. 17g and h). Then, in situ Raman measurements were performed at a set of applied potentials (1.1 to 1.6 V vs. NHE) under CO2 reduction conditions. Compared to Zr-BTB@Hemin, the intensity of the 2060 cm−1 peak of Zr-BTB@Hemin-TMA is larger than that of the one at 1840 cm−1, which means stability of the intermediate CO with weak bonds, allowing for swift release of the CO product (Fig. 17e and f). The conclusion is reinforced by the bonding mode illustrated by the peak of 2060 cm−1 and 1840 cm−1 in Fig. 17b. Thus, electrostatic secondary-sphere functionalities enable substantial improvement of CO2-to-CO conversion activity and selectivity.
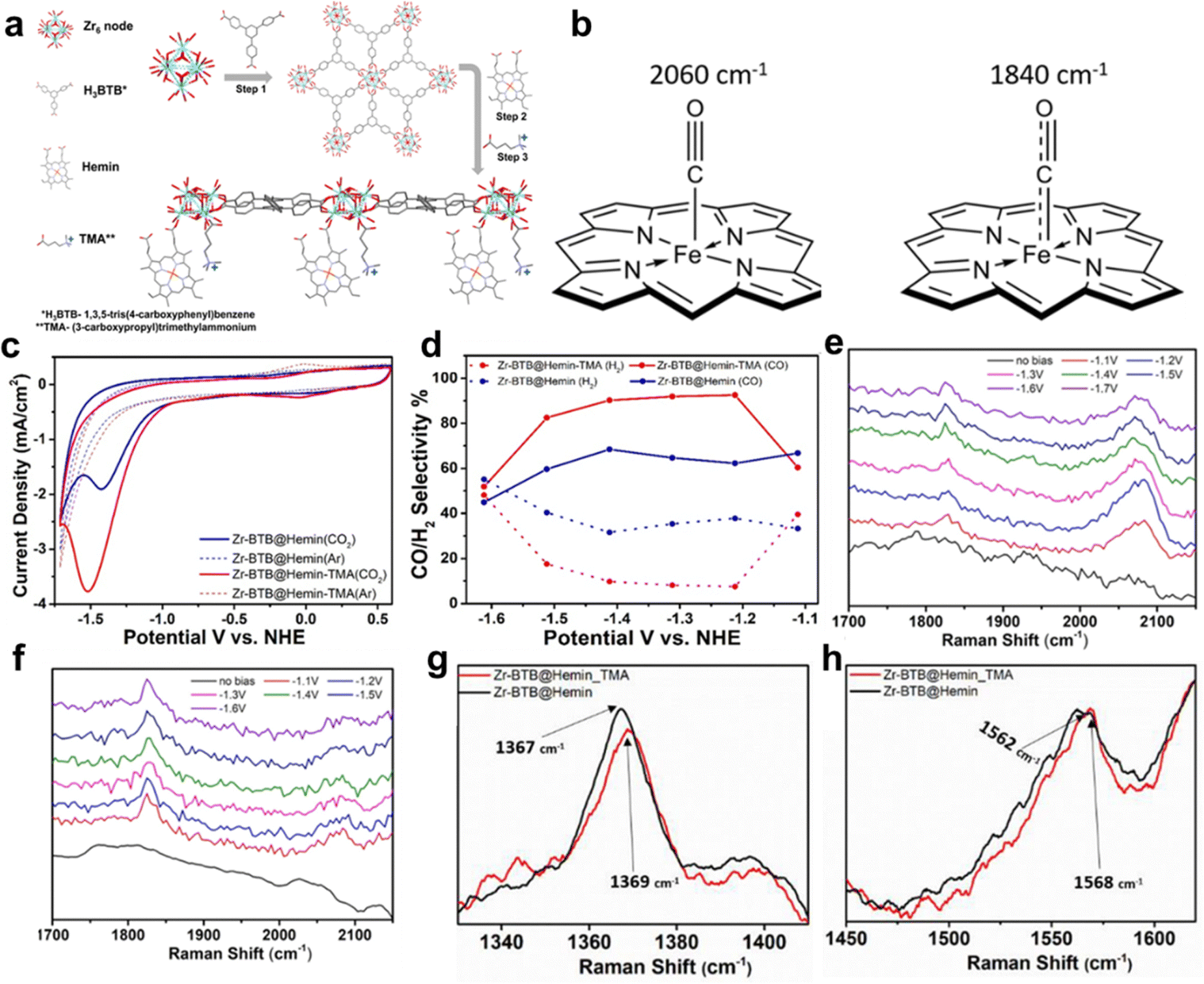 | ||
| Fig. 17 (a) Schematic illustration of Zr-BTB@Hemin-TMA synthesis. Step 1, preparation of the Zr-BTB nanosheet. Step 2, post-synthetic modification with Hemin (molecular catalyst). Step 3, post-synthetic modification with TMA (electrostatic secondary-sphere ligand). (b) Schematic illustration showing the different C–O and Fe–C bond orders for the 2 Hemin-bound CO intermediates detected using in situ Raman spectroscopy. (c) Cyclic voltammetry measurements comparing Zr-BTB@Hemin (blue) and Zr-BTB@Hemin-TMA (red) in both Ar (dashed line) and CO2 environments (solid line). (d) Comparison of catalytic selectivity towards CO (solid line) and H2 (dashed line) for Zr-BTB@Hemin (blue) and Zr-BTB@Hemin-TMA (red). (e) and (f) In situ Raman spectroscopy measurements conducted under working electrocatalytic conditions: the Raman spectra of Zr-BTB@Hemin (black) and Zr-BTB@Hemin_TMA (red). (g) Expansion of the region showing the ν4 peak of the pyrrole half-ring stretching. (h) Expansion of the region showing the ν2 peak region of the pyrrole half-ring stretching. Reproduced from ref. 78. Copyright 2022 Wiley. | ||
The porphyrin moiety in porphyrin-based MOFs is characterized by a highly stable, planar, aromatic ring structure adorned with 18 electron π-bonds.41,56,60,148 Despite the inherent stability and unique electronic properties of porphyrins, the majority of research efforts have focused on utilizing composite catalyst systems to enhance the electrical conductivity and cyclic stability of these MOFs.149 This is often achieved by introducing additional components that can establish weak π–π interactions with the porphyrin rings.76 While such interactions can marginally improve the electrical performance of the hybrid material, they fall short of fully exploiting the potential electrical conductivity offered by the porphyrin-based MOF. To truly unlock the full photoelectric potential of these complexes, direct coordination of the porphyrin core with suitable ligands or dopants becomes essential. Mu and her team created a porphyrin-based Cu MOF (Cu-(Co + Cu)PMOF/CNT-COOH hybrid) by integrating a two-dimensional bimetallic porphyrin-based Cu-MOF with carboxyl-modified carbon nanotubes (Fig. 18a and b).150 When compared with (Co + Cu)PMOF/CNT, which is formed through weak π–π interactions between (Co + Cu)PMOF and carbon nanotubes (CNT), Cu-(Co + Cu)PMOF/CNT-COOH shows superior stability, additional exposed active sites, and a higher electron transfer rate from CNT-COOH to Cu-(Co + Cu)PMOF (Fig. 18c) and achieves a CO faradaic efficiency of 95.98% with an impressive current density of −3.48 mA cm−2 at an overpotential of −0.9 V vs. RHE. Then, a stability test of 40000 s further demonstrates the superstability of Cu-(Co + Cu)PMOF/CNT-COOH (Fig. 18d–f). As we know, C2+ products, derived predominantly from petroleum, are fundamental chemicals widely used in industries such as plastics, synthetic fibers, and solvents.151 However, their production via electrocatalytic CO2 reduction is hampered by kinetic barriers and inefficiencies in proton/electron transfer, particularly concerning the challenging C–C coupling reactions.152 Porphyrin-based Cu MOFs have garnered significant attention in the field of electrocatalytic CO2-to-C2+ products, primarily due to the unique advantage that the Cu coordination environment can be finely tuned through adjusting Cu–N interactions. This precise modulation of the electronic properties of the Cu active site enables porphyrin-based Cu MOFs to exhibit enhanced selectivity towards C2+ products. Nonetheless, sluggish kinetics and inefficient mass transport remain as formidable challenges impeding the full realization of their potential in this application.153 Yang and his team successfully synthesized porphyrin-based Cu MOFs comprising metalloporphyrin Cu centers and impregnated Au nanoneedles by exploiting the ligand carboxylates as the reducing agent (AuNN@PCN-222(Cu)).154 AuNN@PCN-222(Cu) demonstrated significantly improved ethylene production up to an FE of 52.5%. The satisfactory FE was mainly attributed to the Cu–N4 motif which undergoes an out-of-plane displacement toward the Au plane, freeing the Cu center to accept another Au-generated CO affording the co-adsorption of *CO and *CHO with an exothermic energy difference of −0.08 eV. Subsequently, C–C coupling occurs by bridging the two intermediates to form *CO-CHO with a surmountable energy of 0.44 eV. The presence of co-adsorption sites has been demonstrated to lower the energy barrier for C–C coupling while concurrently enhancing mass transfer efficiency, thereby capitalizing on copper's versatile valence properties. This strategic approach holds instructive value for future investigations into electrocatalytic CO2-to-C2+ products, guiding the design of more efficient and selective catalyst systems.
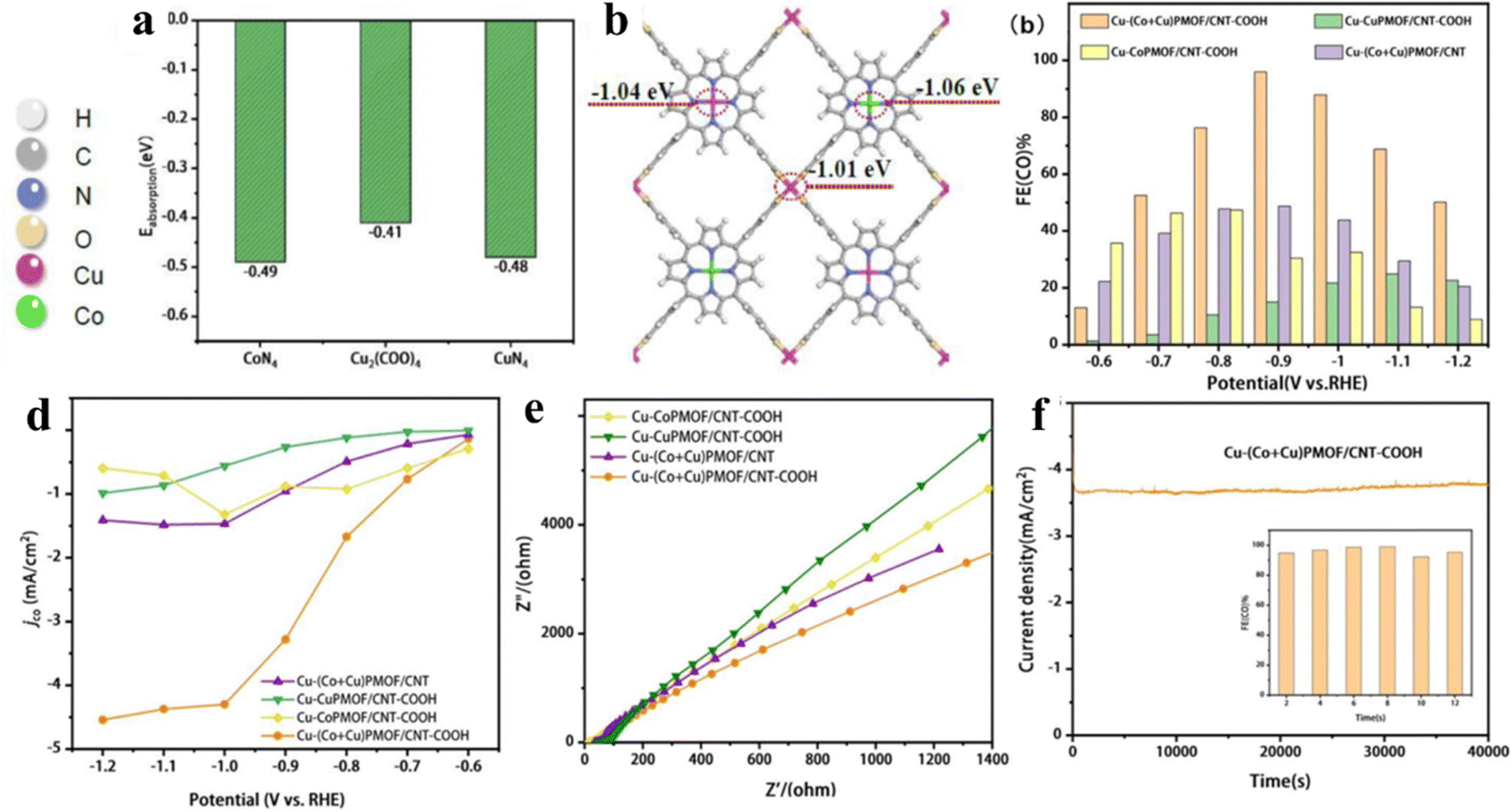 | ||
| Fig. 18 (a) Adsorption energies of CO2 on Cu2(COO)4, CoN4 and CuN4 sites. (b) Bader charge of Cu2(COO)4, CuN4 and CoN4 sites on Cu-(Cu + Co)PMOF. (c) Faradaic efficiency for CO and (d) CO partial current density of Cu-CoPMOF/CNT-COOH, Cu-CuPMOF/CNT-COOH, Cu-(Co + Cu)PMOF/CNT and Cu-(Co + Cu)PMOF/CNT-COOH at different potentials (−0.6 to −1.2 V vs. RHE). (e) Nyquist plots of Cu-CuPMOF/CNT-COOH, Cu-CoPMOF/CNT-COOH, Cu-(Co + Cu)PMOF/CNT-COOH and Cu-(Co + Cu). (f) Long-term stability test of Cu-(Co + Cu)PMOF/CNT-COOH at -0.9 V for 12 h. Reproduced from ref. 150. Copyright 2022 Elsevier. | ||
Electrocatalytic CO2RR holds great promise in transforming the greenhouse gas CO2 into valuable chemicals and fuels, contributing to both environmental sustainability and resource utilization. Porphyrin-based metal–organic frameworks (MOFs) have emerged as intriguing candidates for this purpose, given their unique structural versatility, tunable porosity, and rich redox chemistry.44,61,148 Nevertheless, the practical implementation of porphyrin MOFs as efficient electrocatalysts for CO2RR necessitates improvements in several critical aspects, including product selectivity, cycle stability, conductivity, and catalytic activity. Recognizing the potential of these materials, substantial research efforts have been dedicated to optimizing porphyrin MOFs for high-value conversion of CO2. This section elucidates how the stability, selectivity, and conductivity of porphyrin-based electrocatalysts can be meticulously tailored through judicious manipulation of the surrounding groups, porphyrin metal centers, and diverse porphyrin linkers. Finally, the recent advances in applying porphyrin-based MOFs to the electrocatalytic CO2 reduction are summarized in Table 2. Notably, MOF-NS-Co demonstrates remarkable activity in electrocatalytic CO2 reduction, with the CO faradaic efficiency of MOF-NS-Co surpassing 90% across a broad potential range of −0.5 to −1.0 V, peaking at 98.7% with a mere 100 mV increment. This outstanding performance can be attributed to the unique Kagome-type layered pillar structure of the porphyrin-based MOFs, which exposes a high density of active metal sites and lowers the energy barrier for the generation and conversion of intermediates in the CO2 reaction.
| Samples | Porphyrins | Products | Electrolyte | FE and Eon set (V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| Cu-porphyrin MOF | CuTCPP | C2H5OH/C2H4/CH4/CO | 0.1 M KHCO3 | 11.9% to 41.1% at −1.4 V | 155 |
| g-C3N4@Co-(Co + Cu)PMOF-50% | TCPP | CO | 0.1 M KHCO3 | 97.8% at −1.4 V | 156 |
| MOF-525 | CoTCPP | CH4/CO | 0.1 M KHCO3 | 87% at −0.89 V | 150 |
| MOF-NS-Co | TCPP | CO | 0.1 M KHCO3 | 98.7% at −0.6 V | 157 |
| Al2(OH)2TCPP-Co | CoTCPP | CO | 0.5 M KHCO3 | 76% at −0.7 V | 158 |
| TCPP(Co)/Zr-BTB-PSABA | CoTCPP | CO | 0.5 M KHCO3 | 85.1% at −0.77 V | 87 |
| 2D Cu-MOF | CuTCPP | CH3COOH/HCOOH/CO/CH4 | 1 M H2O and 0.5 M EMIMBF4 | 16.8% and 68.4% at −1.55 VAg/Ag+ | 159 |
| PPy@MOF-545-Co | CoTCPP | CO | 0.5 M KHCO3 | 98% at −0.8 V | 96 |
| PPy@MOF-545-Fe | FeTCPP | CO | 0.5 M KHCO3 | 89.2% at −0.7 V | 96 |
| PPy@MOF-545-Ni | NiTCPP | CO | 0.5 M KHCO3 | 87.8% at −0.9 V | 96 |
| Fe-Porphyrin (Hemin)-based MOF | Hemin | CO | 0.5 M KHCO3 | 92% at −1.2 V | 78 |
| PCN-222(Fe)/C | FeTCPP | CO | 0.5 M KHCO3 | 80.4% at −0.6 V | 41 |
| Vg-Por(Co)-MOF(9:1) |
DPDBP | CO | 0.5 M KHCO3 | 93.8% at 2.3 V | 44 |
6. Conclusions and outlook
Porphyrin-based MOFs have recently emerged as promising candidates for photo(electro)catalytic CO2 conversion reactions due to their straightforward synthesis, high chemical stability, abundant metallic active sites, tunable crystalline structure, and high specific surface area. This review comprehensively discusses the fundamentals of porphyrin-based MOFs and their derivative catalysts for valuable CO2 conversion, covering topics such as synthesis, catalytic mechanisms, and strategies for enhancing photo(electro)catalytic CO2 reduction. The potential of porphyrin-based MOFs in this field is clearly demonstrated. Despite significant progress in recent years, further research is needed to fully explore the capabilities of porphyrin-based MOFs in photo(electro)catalytic CO2 reduction. Several challenges and bottlenecks remain to be addressed.(i) One major obstacle in photocatalytic and electrocatalytic reduction of CO2 is the low selectivity towards desired products, as HER also occurs simultaneously. While the Fischer–Tropsch process can potentially convert CO2 and H2 into hydrocarbons, it does not address the selectivity problem and adds an energy-intensive conversion step. Thus, improving the direct selectivity of CO2 reduction processes is essential for their practical implementation.
(ii) The pursuit of a cost-effective and efficient porphyrin-based MOF catalyst as an alternative to noble metal catalysts in CO2 reduction encounters various challenges. Additionally, the practical application of photo(electro)catalytic CO2RR is impeded by the intricate processes involved in their synthesis, processing, and less than ideal photo(electro)chemical properties. These obstacles necessitate extensive research and development endeavors in this field, which are both technically complex and financially taxing.
(iii) Long-term stable operation is a crucial requirement for any practical catalyst. Porphyrin-based MOFs are susceptible to structural degradation, loss of active components, pore blockage, and other issues during actual operation, which can impact their catalytic efficiency and lifespan. It is essential to develop new MOF materials with superior corrosion resistance and structural stability, along with researching suitable packaging techniques and anti-aging methods, to enhance their long-term stability in real-world applications.
(iv) Although porphyrin-based MOFs have shown promising CO2RR performance in laboratory settings, challenges such as high production costs, limited feedstock availability, and process complexity hinder their commercialization. To overcome these barriers, it is crucial to focus on streamlining synthetic routes, utilizing cost-effective feedstocks, implementing continuous production methods, and enhancing recycling strategies. These efforts are key to reducing costs, enhancing resource efficiency, and facilitating industrial-scale production.
Despite the development bottlenecks mentioned above, we will find appropriate strategies to address these issues as we continue to explore them.
(i) The reaction pathway for CO2 catalysis is complicated, and the catalytic selectivity of porphyrin-based MOFs can be improved by precisely controlling the catalytic environment. Firstly, the presence of a substantial quantity of evenly distributed metal-active centers in porphyrin-based MOFs is a significant factor in the absorption and stabilization of CO2 intermediates, as well as the selective desorption of products. Secondly, porphyrin ligands are functionalized molecules. Choosing the suitable porphyrin ligands and integrating them into MOFs can greatly enhance selectivity. Finally, the configuration of the pore structure plays a major role in stabilizing reaction intermediates and facilitating their specific passage.
(ii) The porphyrin organic linkers’ interaction with the metal nodes of porphyrin-based MOFs significantly affects their electrical conductivity. In porphyrin-based MOFs, metal nodes typically function as electron acceptors, while porphyrin ligands act as electron donors. Therefore, introducing a guest molecule to regulate the band gap structure formed between the electron donor and acceptor greatly impacts the photoelectrochemical performance.
(iii) Some common noble metal industrial catalysts remain active for at least 2400 h reducing production costs. Although most of the current porphyrin-based MOF catalysts demonstrate high CO2 conversion efficiency, their stability issues necessitate extensive efforts. Therefore, a long-term stability test is crucial. Current approaches to tackling the catalytic stability issues of porphyrin-based MOFs primarily include introducing robust linkers to enhance structural integrity, incorporating protective groups to shield sensitive sites against degradation, and designing hybrid structures that synergistically merge the advantages of both organic and inorganic components. Furthermore, the dynamic restructuring of these catalysts during the catalytic cycle has been shown to significantly augment their longevity, thereby holding promise for achieving industrially relevant operational lifetimes in the future. The superb catalytic stability means that porphyrin-based MOFs will be more cost-effective for future applications in photocatalytic CO2 reduction.
(iv) The adsorption capacity of porphyrin-based MOFs can effectively enhance the catalytic conversion efficiency of CO2 within the catalytic system. For instance, increasing the local CO2 concentration in the catalytic active center can improve the reaction yield and selectivity. Furthermore, porphyrin-based MOFs with high CO2 adsorption selectivity can exhibit high catalytic activity in mixed gas or low CO2 concentration or partial pressure environments.
In light of current research on porphyrin-based MOFs, this study suggests key areas for future exploration in the field of porphyrin-based MOFs for photo(electro)catalytic CO2 reduction. Specifically, investigating the potential enhancement of photocatalytic CO2 activity through photoelectric synergy and understanding its underlying mechanisms are essential. The modifiable nature of porphyrin MOFs presents an opportunity to explore how steric structures can impact product selectivity. Furthermore, exploring various forms of metal active sites (such as single atoms, multi-metal atoms, and central coordination) in conjunction with steric effects could lead to the production of complex multi-carbon products. This area of research is poised to become a significant focus in the future. In conclusion, porphyrin-based MOFs show great potential as a photo(electro)catalytic material with a wide range of applications in CO2 reduction. Continuous research and innovation in this field will undoubtedly contribute significantly towards resolving global energy and environmental challenges.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52203110 and 32371978) and the Natural Science Foundation of Fujian Province (Grant No. 2023J05052). This work is dedicated to Professor Geoffrey Ozin on the occasion of his 80th birthday.References
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- G. Ding, C. Li, Y. Ni, L. Chen, L. Shuai and G. Liao, EES Catal., 2023, 1, 369–391 RSC.
- F. Tian, X. Wu, J. Chen, X. Sun, X. Yan and G. Liao, Dalton Trans., 2023, 52, 11934–11940 RSC.
- G. Ding, Z. Wang, J. Zhang, P. Wang, L. Chen and G. Liao, EcoEnergy, 2024, 2, 22–44 CrossRef.
- G. Liao, Y. He, H. Wang, B. Fang, N. Tsubaki and C. Li, Device, 2023, 1, 100173 CrossRef.
- L. Tan, S. M. Xu, Z. Wang, Y. Xu, X. Wang, X. Hao, S. Bai, C. Ning, Y. Wang, W. Zhang, Y. K. Jo, S. J. Hwang, X. Cao, X. Zheng, H. Yan, Y. Zhao, H. Duan and Y. F. Song, Angew. Chem., Int. Ed., 2019, 58, 11860–11867 CrossRef CAS PubMed.
- L. Tan, S. M. Xu, Z. Wang, Y. Xu, X. Wang, X. Hao, S. Bai, C. Ning, Y. Wang, W. Zhang, Y. K. Jo, S. J. Hwang, X. Cao, X. Zheng, H. Yan, Y. Zhao, H. Duan and Y. F. Song, Angew. Chem., Int. Ed., 2019, 58, 11860–11867 CrossRef CAS.
- G. Liao, G. Ding, B. Yang and C. Li, Precis. Chem., 2024, 2, 49–56 CrossRef CAS.
- R. R. Ikreedeegh and M. Tahir, J. CO2 Util., 2021, 43, 101381 CrossRef CAS.
- Q. Zhang, G. Liao, B. Yang, Y. Zhang, G. Ge, A. Lipovka, J. Liu, R. D. Rodriguez, X. Yang and X. Jia, Appl. Surf. Sci., 2023, 638, 157989 CrossRef CAS.
- X. Wang, S. Sahoo, J. Gascon, M. Bragin, F. Liu, J. Olchowski, S. Rothfarb, Y. Huang, W. Xiang, P.-X. Gao, S. P. Alpay and B. Li, Energy Environ. Sci., 2023, 16, 4388–4403 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- T. Yan, X. Chen, L. Kumari, J. Lin, M. Li, Q. Fan, H. Chi, T. J. Meyer, S. Zhang and X. Ma, Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS PubMed.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- Y. Zhang, B. Xia, J. Ran, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 1903879 CrossRef CAS.
- A. Kumar Singh, C. Das and A. Indra, Coord. Chem. Rev., 2022, 465, 214516 CrossRef CAS.
- Z. Wang, G. Ding, J. Zhang, X. Lv, P. Wang, L. Shuai, C. Li, Y. Ni and G. Liao, Chem. Commun., 2024, 60, 204–207 RSC.
- B. Yang, X. Li, Q. Zhang, X. Yang, J. Wan, G. Liao, J. Zhao, R. Wang, J. Liu, R. D. Rodriguez and X. Jia, Appl. Catal., B, 2022, 314, 121521 CrossRef CAS.
- C. Li, B. Cheng, H. Lu, G. Ding, Z. Jiang and G. Liao, Inorg. Chem., 2023, 62, 6843–6850 CrossRef CAS PubMed.
- C. Li, X. Liu, G. Ding, P. Huo, Y. Yan, Y. Yan and G. Liao, Inorg. Chem., 2022, 61, 4681–4689 CrossRef CAS PubMed.
- S. Bai, C. Ning, H. Wang, G. Liu, L. Zheng and Y. F. Song, Small, 2022, 18, e2203787 CrossRef PubMed.
- B. Yang, Z. Wang, J. Zhao, X. Sun, R. Wang, G. Liao and X. Jia, Int. J. Hydrogen Energy, 2021, 46, 25436–25447 CrossRef CAS.
- H. Zhu, C. Zhang, K. Xie, X. Li and G. Liao, Chem. Eng. J., 2023, 453, 139775 CrossRef CAS.
- G. Liao, C. Li, S.-Y. Liu, B. Fang and H. Yang, Phys. Rep., 2022, 983, 1–41 CrossRef CAS.
- C. Li, H. Lu, G. Ding, Q. Li and G. Liao, Catal. Sci. Technol., 2023, 13, 2877–2898 RSC.
- G. Liao, C. Li, S.-Y. Liu, B. Fang and H. Yang, Trends Chem., 2022, 4, 111–127 CrossRef CAS.
- H. Zhu, L. Gou, C. Li, X. Fu, Y. Weng, L. Chen, B. Fang, L. Shuai and G. Liao, Device, 2024, 2, 100283 CrossRef.
- Y. Bo, P. Du, H. Li, R. Liu, C. Wang, H. Liu, D. Liu, T. Kong, Z. Lu, C. Gao and Y. Xiong, Appl. Catal., B, 2023, 330, 122667 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- M. Li, Z. Zuo and S. Zhang, ACS Catal., 2023, 13, 11815–11824 CrossRef CAS.
- G. Liao, L. Zhang, C. Li, S.-Y. Liu, B. Fang and H. Yang, Matter, 2022, 5, 3341–3374 CrossRef CAS.
- M. Yang, P. Wang, Y. Li, S. Tang, X. Lin, H. Zhang, Z. Zhu and F. Chen, Appl. Catal., B, 2022, 306, 121065 CrossRef CAS.
- G. Marcandalli, K. Boterman and M. T. M. Koper, J. Catal., 2022, 405, 346–354 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef CAS.
- C. I. Ezugwu, S. Liu, C. Li, S. Zhuiykov, S. Roy and F. Verpoort, Coord. Chem. Rev., 2022, 450, 214245 CrossRef CAS.
- B. Zhu, Q. Xu, X. Bao, H. Yin, Y. Qin and X.-C. Shen, Chem. Eng. J., 2022, 429, 132284 CrossRef CAS.
- C. Li, G. Ding, X. Liu, P. Huo, Y. Yan, Y. Yan and G. Liao, Chem. Eng. J., 2022, 435, 134740 CrossRef CAS.
- C. Li, X. Liu, P. Huo, Y. Yan, G. Liao, G. Ding and C. Liu, Small, 2021, 17, 2102539 CrossRef CAS.
- B.-X. Dong, S.-L. Qian, F.-Y. Bu, Y.-C. Wu, L.-G. Feng, Y.-L. Teng, W.-L. Liu and Z.-W. Li, ACS Appl. Energy Mater., 2018, 1, 4662–4669 CrossRef CAS.
- Y. Chen, D. Wang, X. Deng and Z. Li, Catal. Sci. Technol., 2017, 7, 4893–4904 RSC.
- Y. Qiao, C. Sun, J. Jian, T. Zhou, X. Xue, J. Shi, L. Zhao and G. Liao, Inorg. Chem., 2024, 63, 2060–2071 CrossRef CAS PubMed.
- Y.-L. Dong, Z.-Y. Jing, Q.-J. Wu, Z.-A. Chen, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2023, 11, 8739–8746 RSC.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef.
- X. J. Kong, T. He, J. Zhou, C. Zhao, T. C. Li, X. Q. Wu, K. Wang and J. R. Li, Small, 2021, 17, e2005357 CrossRef.
- C. Li, W. Qiu, W. Long, F. Deng, G. Bai, G. Zhang, X. Zi and H. He, J. Mol. Catal. A: Chem., 2014, 393, 166–170 CrossRef CAS.
- R. J. Li, M. Li, X. P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- K. Epp, B. Bueken, B. J. Hofmann, M. Cokoja, K. Hemmer, D. De Vos and R. A. Fischer, Catal. Sci. Technol., 2019, 9, 6452–6459 RSC.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 CrossRef CAS PubMed.
- X. Li and Q.-L. Zhu, EnergyChem, 2020, 2, 100033 CrossRef.
- Y. Liu, Y. Yang, Q. Sun, Z. Wang, B. Huang, Y. Dai, X. Qin and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CrossRef CAS PubMed.
- E. G. Percástegui and V. Jancik, Coord. Chem. Rev., 2020, 407, 213165 CrossRef.
- D. Yang, S. Zuo, H. Yang, Y. Zhou, Q. Lu and X. Wang, Adv. Mater., 2022, 34, e2107293 CrossRef PubMed.
- X. Chen, D. Wang, Z. Wang, Y. Li, H. Zhu, X. Lu, W. Chen, H. Qiu and Q. Zhang, Chem. Eng. J., 2021, 424 Search PubMed.
- J. Gu, Y. Peng, T. Zhou, J. Ma, H. Pang and Y. Yamauchi, Nano Res. Energy, 2022, 1, e9120009 CrossRef.
- W. Han, X. Ma, J. Wang, F. Leng, C. Xie and H. L. Jiang, J. Am. Chem. Soc., 2023, 145, 9665–9671 CrossRef CAS PubMed.
- B. J. Burnett, P. M. Barron and W. Choe, CrystEngComm, 2012, 14, 3839 RSC.
- D. Chen, Z. Guo, B. Li and H. Xing, New J. Chem., 2022, 46, 16297–16302 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- Y. T. Guntern, J. R. Pankhurst, J. Vavra, M. Mensi, V. Mantella, P. Schouwink and R. Buonsanti, Angew. Chem., Int. Ed., 2019, 58, 12632–12639 CrossRef CAS PubMed.
- Z. W. Huang, K. Q. Hu, X. B. Li, Z. N. Bin, Q. Y. Wu, Z. H. Zhang, Z. J. Guo, W. S. Wu, Z. F. Chai, L. Mei and W. Q. Shi, J. Am. Chem. Soc., 2023, 145, 18148–18159 CrossRef CAS.
- L. Wang, H. Fan and F. Bai, MRS Bull., 2020, 45, 49–56 CrossRef.
- L. Wang, P. Jin, J. Huang, H. She and Q. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15660–15670 CrossRef CAS.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS.
- Z. Wang, Q. Sun, B. Liu, Y. Kuang, A. Gulzar, F. He, S. Gai, P. Yang and J. Lin, Coord. Chem. Rev., 2021, 439, 213945 CrossRef CAS.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- S. Mehrzad Sajjadinezhad, L. Boivin, K. Bouarab and P. D. Harvey, Coord. Chem. Rev., 2024, 510, 215794 CrossRef CAS.
- F. Liu, I. Rincón, H. G. Baldoví, A. Dhakshinamoorthy, P. Horcajada, S. Rojas, S. Navalón and A. Fateeva, Inorg. Chem. Front., 2024, 11, 2212–2245 RSC.
- Y. G. Gorbunova, Y. Y. Enakieva, M. V. Volostnykh, A. A. Sinelshchikova, I. A. Abdulaeva, K. P. Birin and A. Y. Tsivadze, Russ. Chem. Rev., 2022, 91, RCR5038 CrossRef CAS.
- P. D. Harvey, J. Mater. Chem. C, 2021, 9, 16885–16910 RSC.
- Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2020, 7, 300–339 RSC.
- T. Xia, Y. Lin, W. Li and M. Ju, Chinese Chem. Lett., 2021, 32, 2975–2984 CrossRef CAS.
- Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi, J. Ge and J. Tang, Small, 2021, 17, e2006590 CrossRef PubMed.
- F. ZareKarizi, M. Joharian and A. Morsali, J. Mater. Chem. A, 2018, 6, 19288–19329 RSC.
- S. Yuan, J. S. Qin, L. Zou, Y. P. Chen, X. Wang, Q. Zhang and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636–6642 CrossRef CAS.
- R. Shimoni, Z. Shi, S. Binyamin, Y. Yang, I. Liberman, R. Ifraemov, S. Mukhopadhyay, L. Zhang and I. Hod, Angew. Chem., Int. Ed., 2022, 61, e202206085 CrossRef CAS.
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 3501–3510 RSC.
- J. Wang, Y. Zhang, Y. Ma, J. Yin, Y. Wang and Z. Fan, ACS Mater. Lett., 2022, 4, 2058–2079 CrossRef CAS.
- P. Deria, J. Yu, R. P. Balaraman, J. Mashni and S. N. White, Chem. Commun., 2016, 52, 13031–13034 RSC.
- B. Karadeniz, D. Zilic, I. Huskic, L. S. Germann, A. M. Fidelli, S. Muratovic, I. Loncaric, M. Etter, R. E. Dinnebier, D. Barisic, N. Cindro, T. Islamoglu, O. K. Farha, T. Friscic and K. Uzarevic, J. Am. Chem. Soc., 2019, 141, 19214–19220 CrossRef CAS PubMed.
- Y. Zhao, L. Jiang, L. Shangguan, L. Mi, A. Liu and S. Liu, J. Mater. Chem. A, 2018, 6, 2828–2833 RSC.
- S. Ali Akbar Razavi and A. Morsali, Coord. Chem. Rev., 2019, 399, 213023 CrossRef CAS.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- T.-F. Liu, D. Feng, Y.-P. Chen, L. Zou, M. Bosch, S. Yuan, Z. Wei, S. Fordham, K. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2014, 137, 413–419 CrossRef PubMed.
- X. D. Zhang, S. Z. Hou, J. X. Wu and Z. Y. Gu, Chem. – Eur. J., 2020, 26, 1604–1611 CrossRef CAS.
- L. Yang, P. Cai, L. Zhang, X. Xu, A. A. Yakovenko, Q. Wang, J. Pang, S. Yuan, X. Zou, N. Huang, Z. Huang and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 12129–12137 CrossRef CAS PubMed.
- D.-J. Li, Y.-B. Tian, Q. Lin, J. Zhang and Z.-G. Gu, ACS Appl. Mater. Interfaces, 2022, 14, 33548–33554 CrossRef CAS PubMed.
- K. Nisa, M. Saxena, I. A. Lone and R. Kumar, Sustain. Energy Fuels, 2023, 7, 2774–2801 RSC.
- J. Park, M. Xu, F. Li and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 5493–5499 CrossRef CAS PubMed.
- J. Zheng, M. Wu, F. Jiang, W. Su and M. Hong, Chem. Sci., 2015, 6, 3466–3470 RSC.
- Q. Chen, S. Xian, X. Dong, Y. Liu, H. Wang, D. H. Olson, L. J. Williams, Y. Han, X. H. Bu and J. Li, Chem. Commun., 2021, 60, 10593–10597 CAS.
- Z. Zhu, Z. Wang, Q. H. Li, Z. Ma, F. Wang and J. Zhang, Dalton Trans., 2023, 52, 4309–4314 RSC.
- C. Xia, R. T. Guo, Z. X. Bi, Z. R. Zhang, C. F. Li and W. G. Pan, Dalton Trans., 2023, 52, 12742–12754 RSC.
- Z. Xin, J. Liu, X. Wang, K. Shen, Z. Yuan, Y. Chen and Y. Q. Lan, ACS Appl. Mater. Interfaces, 2021, 13, 54959–54966 CrossRef CAS PubMed.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, L. Zou, Y.-P. Chen, X. Wang, Q. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636–6642 CrossRef CAS PubMed.
- Z. Wang, Q. Sun, B. Liu, Y. Kuang, A. Gulzar, F. He, S. Gai, P. Yang and J. Lin, Coord. Chem. Rev., 2021, 439, 213945 CrossRef CAS.
- Y. Wang, L. Feng, J. Pang, J. Li, N. Huang, G. S. Day, L. Cheng, H. F. Drake, Y. Wang, C. Lollar, J. Qin, Z. Gu, T. Lu, S. Yuan and H. C. Zhou, Adv. Sci., 2019, 6 Search PubMed.
- S. Kumar, M. A. Isaacs, R. Trofimovaite, L. Durndell, C. M. A. Parlett, R. E. Douthwaite, B. Coulson, M. C. R. Cockett, K. Wilson and A. F. Lee, Appl. Catal., B, 2017, 209, 394–404 CrossRef CAS.
- T. Lai, J. Wang, W. Xiong, H. Wang, M. Yang, T. Li, X. Kong, X. Zou, Y. Zhao, D. O’Hare and Y.-F. Song, Chem. Eng. Sci., 2022, 257, 117704 CrossRef CAS.
- L. Cheng, X. Yue, L. Wang, D. Zhang, P. Zhang, J. Fan and Q. Xiang, Adv. Mater., 2021, 33, e2105135 CrossRef PubMed.
- A. A. Zhang, D. Si, H. Huang, L. Xie, Z. B. Fang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202203955 CrossRef CAS PubMed.
- N. Heidary, M. Morency, D. Chartrand, K. H. Ly, R. Iftimie and N. Kornienko, J. Am. Chem. Soc., 2020, 142, 12382–12393 CrossRef CAS PubMed.
- B. Petrovic, M. Gorbounov and S. Masoudi Soltani, Micropor. Mesopor. Mater., 2021, 312, 110751 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- M. Cheng, P. Yan, X. Zheng, B. Gao, X. Yan, G. Zhang, X. Cui and Q. Xu, Chem. – Eur. J., 2023, 29, e202302395 CrossRef CAS PubMed.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- J.-S. Qin, S. Yuan, Q. Wang, A. Alsalme and H.-C. Zhou, J. Mater. Chem. A, 2017, 5, 4280–4291 RSC.
- Q. Wu, J. Wang, Z. Wang, Y. Xu, Z. Xing, X. Zhang, Y. Guan, G. Liao and X. Li, J. Mater. Chem. A, 2020, 8, 13685–13693 RSC.
- K. Yu, D.-I. Won, W. I. Lee and W.-S. Ahn, Korean J. Chem. Eng., 2021, 38, 653–673 CrossRef CAS.
- Z.-B. Fang, T.-T. Liu, J. Liu, S. Jin, X.-P. Wu, X.-Q. Gong, K. Wang, Q. Yin, T.-F. Liu, R. Cao and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- M. Cheng, B. Gao, X. Zheng, W. Wu, W. Kong, P. Yan, Z. Wang, B. An, Y. Zhang, Q. Li and Q. Xu, Appl. Catal., B, 2024, 353, 124097 CrossRef CAS.
- M. R. Smith, C. B. Martin, S. Arumuganainar, A. Gilman, B. E. Koel and M. L. Sarazen, Angew. Chem., Int. Ed., 2023, 62, e202218208 CrossRef CAS PubMed.
- S. M. Pratik, L. Gagliardi and C. J. Cramer, J. Phys. Chem. C, 2019, 124, 1878–1887 CrossRef.
- P. J. Chmielewski and L. Latos-Grażyński, Coord. Chem. Rev., 2005, 249, 2510–2533 CrossRef CAS.
- C. Wang, X.-M. Liu, M. Zhang, Y. Geng, L. Zhao, Y.-G. Li and Z.-M. Su, ACS Sustainable Chem. Eng., 2019, 7, 14102–14110 CrossRef CAS.
- S. Xie, C. Deng, Q. Huang, C. Zhang, C. Chen, J. Zhao and H. Sheng, Angew. Chem., Int. Ed., 2023, 62, e202216717 CrossRef CAS PubMed.
- M. Zhou, Z. Qu, J. Zhang, H. Jiang, Z. Tang and R. Chen, Chem. Commun., 2024, 60, 3170–3173 RSC.
- H. Che, C. Liu, G. Che, G. Liao, H. Dong, C. Li, N. Song and C. Li, Nano Energy, 2020, 67, 104273 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- Z. Jiang, Y. Liu, T. Jing, B. Huang, Z. Wang, X. Zhang, X. Qin and Y. Dai, Appl. Catal., B, 2017, 200, 230–236 CrossRef CAS.
- R.-t Guo, C. Xia and W.-g Pan, J. Chem. Educ., 2023, 100, 1621–1626 CrossRef CAS.
- H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao and S. Z. Qiao, J. Am. Chem. Soc., 2023, 145, 14335–14344 CrossRef CAS PubMed.
- N. Sadeghi, S. Sharifnia and M. Sheikh Arabi, J. CO2 Util., 2016, 16, 450–457 CrossRef CAS.
- X. J. Kong, T. He, J. Zhou, C. Zhao, T. C. Li, X. Q. Wu, K. Wang and J. R. Li, Small, 2021, 17 Search PubMed.
- S. Bai, Z. Wang, L. Tan, G. I. N. Waterhouse, Y. Zhao and Y.-F. Song, Ind. Eng. Chem. Res., 2020, 59, 5848–5857 CrossRef CAS.
- P. Deria, J. Yu, R. P. Balaraman, J. Mashni and S. N. White, Chem. Commun., 2016, 52, 13031–13034 RSC.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- Z.-W. Huang, K.-Q. Hu, X.-B. Li, Z.-N. Bin, Q.-Y. Wu, Z.-H. Zhang, Z.-J. Guo, W.-S. Wu, Z.-F. Chai, L. Mei and W.-Q. Shi, J. Am. Chem. Soc., 2023, 145, 18148–18159 CrossRef CAS PubMed.
- S. Xie, Y. Li, B. Sheng, W. Zhang, W. Wang, C. Chen, J. Li, H. Sheng and J. Zhao, Appl. Catal., B, 2022, 310, 121320 CrossRef CAS.
- J. Liang, H. Yu, J. Shi, B. Li, L. Wu and M. Wang, Adv. Mater., 2023, 35, 2209814 CrossRef CAS PubMed.
- C. Zheng, X. Qiu, J. Han, Y. Wu and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 42243–42249 CrossRef CAS PubMed.
- L. Tayebi, R. Rahimi and A. R. Akbarzadeh, ACS Omega, 2022, 7, 40869–40881 CrossRef CAS PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- K. Liu, Y. Liao, P. Wang, X. Fang, J. Zhu, G. Liao and X. Xu, Nanoscale, 2024, 16, 11096–11108 RSC.
- T. Rhauderwiek, S. Waitschat, S. Wuttke, H. Reinsch, T. Bein and N. Stock, Inorg. Chem., 2016, 55, 5312–5319 CrossRef CAS PubMed.
- S. Senthilkumar, R. Goswami, N. L. Obasi and S. Neogi, ACS Sustainable Chem. Eng., 2017, 5, 11307–11315 CrossRef CAS.
- G. Zhou, Y. Wang and Z. Huang, Chem. Catal., 2022, 2, 3304–3319 CrossRef CAS.
- J. Liang, H. Yu, J. Shi, B. Li, L. Wu and M. Wang, Adv. Mater., 2023, 35, e2209814 CrossRef PubMed.
- Z. Liang, H. Y. Wang, H. Zheng, W. Zhang and R. Cao, Chem. Soc. Rev., 2021, 50, 2540–2581 RSC.
- J. Liu, Y.-Z. Fan, X. Li, Z. Wei, Y.-W. Xu, L. Zhang and C.-Y. Su, Appl. Catal., B, 2018, 231, 173–181 CrossRef CAS.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- X. Li, L. Wang, Y. Xing and W. Su, Sustain. Energy Fuels, 2022, 6, 3516–3526 RSC.
- Y.-L. Dong, Z.-Y. Jing, Q.-J. Wu, Z.-A. Chen, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2023, 11, 8739–8746 RSC.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, e1705512 CrossRef PubMed.
- C. Ma, Z. Wang, J. Li, M. Mu and X. Yin, Electrochim. Acta, 2023, 463, 142774 CrossRef CAS.
- D. Yang, S. Zuo, H. Yang, Y. Zhou, Q. Lu and X. Wang, Adv. Mater., 2022, 34, 2107293 CrossRef CAS PubMed.
- H.-L. Zhu, H.-Y. Chen, Y.-X. Han, Z.-H. Zhao, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2022, 144, 13319–13326 CrossRef CAS PubMed.
- H. Yang, S. Li and Q. Xu, Chinese J. Catal., 2023, 48, 32–65 CrossRef CAS.
- X. Xie, X. Zhang, M. Xie, L. Xiong, H. Sun, Y. Lu, Q. Mu, M. H. Rummeli, J. Xu, S. Li, J. Zhong, Z. Deng, B. Ma, T. Cheng, W. A. Goddard and Y. Peng, Nat. Commun., 2022, 13, 63 CrossRef CAS PubMed.
- S. Xie, Y. Li, B. Sheng, W. Zhang, W. Wang, C. Chen, J. Li, H. Sheng and J. Zhao, Appl. Catal., B, 2022, 310, 121320 CrossRef CAS.
- J. Li, H. Shen, C. Ma, H. Zhang, P. Luo, J. Chen, M. Mu and X. Yin, Electrochim. Acta, 2023, 443, 123684 CrossRef.
- Y. Zhou, L. Zheng, D. Yang, H. Yang, Q. Lu, Q. Zhang, L. Gu and X. Wang, Small Methods, 2021, 5, e2000991 CrossRef PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- J. X. Wu, S. Z. Hou, X. D. Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |