
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic and surface properties of titanium dioxide nanoparticles in soil solutions†
Karolina
Solymos
*ab,
Izabella
Babcsányi
b,
Badam
Ariya
 a,
Tamás
Gyulavári
a,
Áron
Ágoston
ac,
Ákos
Szamosvölgyi
a,
Ákos
Kukovecz
a,
Zoltán
Kónya
a,
Andrea
Farsang‡
b and
Zsolt
Pap
*ade
a,
Tamás
Gyulavári
a,
Áron
Ágoston
ac,
Ákos
Szamosvölgyi
a,
Ákos
Kukovecz
a,
Zoltán
Kónya
a,
Andrea
Farsang‡
b and
Zsolt
Pap
*ade
aDepartment of Applied and Environmental Chemistry, University of Szeged, Szeged, Hungary. E-mail: pzsolt@chem.u-szeged.hu
bDepartment of Geoinformatics, Physical and Environmental Geography, University of Szeged, Szeged, Hungary
cDepartment of Physical Chemistry and Materials Sciences, University of Szeged, Szeged, Hungary
dNanostructured Materials and Bio-Nano-Interfaces Center, Institute for Interdisciplinary Research on Bio-Nano-Sciences, Babeş–Bolyai University, Cluj-Napoca, Romania
eInstitute of Research-Development-Innovation in Applied Natural Sciences, Babeş–Bolyai University, Cluj-Napoca, Romania
First published on 11th January 2024
Abstract
Nanotechnology has emerged as a rapidly growing scientific field with diverse applications. Titanium dioxide nanoparticles are among the most commonly used nanoparticles due to their unique properties. However, their release into the environment poses potential ecological risks to the soil, groundwater, and aquatic systems. Therefore, this study explores the behavior of anatase and rutile nanoparticles in Chernozem soil solutions, focusing on their optical, surface, and photocatalytic properties. After immersion in soil solutions, both anatase and rutile nanoparticles exhibited optical changes, while their semiconductive properties, as indicated by the band gap, remained unaltered. Rutile nanoparticles displayed a decreased isoelectric point after interaction with soil solutions, probably due to deprotonation based on infrared spectroscopy results. Photocatalytic activity assessments revealed a slowdown in the case of anatase by 14% and rutile by 27% after exposure to soil solutions. However, after a three-hour degradation process, the titanias regained their initial levels of activity. The reduction in photoactivity was attributed to adsorption onto the surface of nanoparticles of Zn2+, Ca2+, PO43− organic molecules (such as amines, alkenes, and carboxyl groups) present in the soil solution, hindering catalytic reactions. Anatase nanoparticles exhibited superior performance compared to rutile, which can be attributed to their larger specific surface area and higher hydrophilicity, resulting in the enhanced generation of reactive species. This study provides valuable insights into the complex interactions between nanoparticles and soil solutions, shedding light on their optical properties, surface characteristics, and catalytic activity.
Environmental significanceNanotechnology shows excellent potential across various sectors with numerous applications. However, concerns arise due to releasing nanomaterials, such as titanium dioxide, into the environment. With extensive use in sunscreens, cosmetics, paints, and food additives, accumulated titanium dioxide nanoparticles pose potential risks, demanding a thorough environmental assessment. These materials are already present in soil, groundwater, and aquatic media, raising questions about their long-term impacts. Understanding their behavior and properties changes is critical in determining their ecological impact and interactions with organisms and environmental components. Research on titanium dioxide nanoparticles in the environment plays a huge role in ensuring their safe and sustainable use. It guides the development of regulations, contributing to environmental and human health protection. |
1. Introduction
Nanotechnology and photocatalysis are some of the fastest-growing industry-leading research areas of the 21st century.1 Nanoscale materials have new physical, chemical, and electrical properties compared to bulk materials due to their size, shape, and surface chemistry.2There are many photocatalysts, including ZnO, Fe2O3, CuO, and CdS, that are already widely applied.3–5 However, titanium dioxide (TiO2) stands out due to its distinct characteristics and applications, both as a semiconducting material and an efficient photocatalyst.6 There are several properties of TiO2 nanoparticles (NPs), which is why they are versatile materials, including high UV-absorbing effect7 and chemical stability.8
TiO2 is widely used as a white pigment in various products, including paints, plastics, and sunscreens.9 Their semiconductive properties are also utilized in various electronic applications, including solar cells, sensors, and electronic devices.10 Last, TiO2 NPs can be applied as catalysts11 due to the presence of active centers on their surface, which can enable chemical reactions.12 This makes them suitable for the preparation of self-cleaning surfaces by breaking down organic pollutants and removing dirt and grime upon UV light irradiation.13 Additionally, TiO2 coatings often have hydrophilic properties, promoting the formation of a water film that carries away broken-down particles. This reduces the need for manual cleaning and maintenance, which is especially beneficial in outdoor applications.14,15 Common applications include self-cleaning glasses, automotive coatings, and outdoor signage.16
NPs show increased reactivity and interact with the environment to a greater extend compared to bulk materials.17 Therefore, the environmental and health risks they pose are higher than those of their macroscopic counterparts.18 They may have deleterious effects19 such as phytotoxicity and bioaccumulation in plants, cause acute and chronic diseases in humans and animals,20 and alter the ecological niches of microbes.21 The smallest NPs can even pass through the alveolar membranes of the lungs and enter the bloodstream.2 Several studies have shown that NPs present ecotoxicological concerns due to their widespread use and ubiquitous presence in environments affected by human activity.22–24 The International Agency for Research on Cancer (IARC) has classified TiO2 as a potential human carcinogen (class 2B) because of the results on animals.25
TiO2 NPs can be released into the soil through several pathways: during production, use, waste treatment, waste disposal, air pollution, and fertilizer application (sewage sludge).26 NPs released into the air during various industrial processes (e.g., combustion and manufacturing) can settle down by precipitation and mobilisating into the soil.2,27 In the early 2010s, it was observed that TiO2 NPs can also originate from textiles and painted surfaces due to washing and the weather. This resulted in the accumulation of titania in sewage sludge reaching an approximate concentration of 2 g kg−1.28 Several models were set to predict the fate of TiO2 NPs in the environment29,30 and the results show that their concentration is continuously increasing. Thus, it is crucial to investigate their behavior in the soil and the aquatic environment.29,31
TiO2 NPs released into the soil undergo transformation that ultimately change their mobility, bioavailability, and toxicity.32 The behavior of TiO2 NPs in soil depends on various factors, including the type of soil (pH, cation exchange capacity, salt content, dissolved organic matter)33 or the size, shape, and surface properties of the NPs.34 These properties also strongly influence the various ways NPs can enter soil solutions. The term “soil solution” refers to the liquid component in the soil, containing dissolved substances like mineral salts and organic matter. In the context of TiO2 NPs, they can be present in this soil solution, contributing to the dynamic nature of the soil's chemical composition.35,36 Rainfall is a key player in this process, interacting with the soil and washing off TiO2 NPs from surfaces,37–39 NPs can enter soil solutions is through leaching. In this process, the NPs are transferred into soil pores by water, which can occur if the NPs are not strongly adsorbed onto soil particles.40
It is important to note that the behavior of TiO2 NPs in the soil is not well understood. Most of the available research studied the behaviors of NPs in aquatic environments. Although these matrices are closely intertwined, the information on how these particles can change or behave in soil solutions is limited. Thus, the main objective of this study is to investigate the interaction of TiO2 NPs in different soil solutions. For this purpose, soil solutions were prepared from Chernozem soils, which account for 1.8% of the total continental land area on Earth41 and represent one of the most fertile arable lands in Europe. Then, the effect of pure anatase and pure rutile TiO2 polymorphs was investigated on the as-obtained Chernozem soil solutions. We investigated the potential changes in morphology, crystal structure, surface chemistry, optical properties, and photocatalytic activity of the NPs.
2. Materials and methods
2.1. Materials and sample preparation
The following TiO2 NPs were used during the experiments: Sigma Aldrich Anatase (AA) and Sigma Aldrich Rutile (AR) with 98% purity. Ultrapure Millipore Milli-Q water was applied in all cases (18.2 MΩ cm−1). To determine photocatalytic activities, we used phenol (VWR, >99%) as a model pollutant.2.2. Soil sampling and analysis
Ten topsoil samples were collected from an agricultural area near Szeged, Hungary (Fig. 1) and used for the preparation of the soil solutions. The exact GPS-coordinates of the soil samples are attached in Table S1.† Based on the World Reference Base of Soil, the collected samples can be classified as the Chernozem soil type. | ||
| Fig. 1 Location of the sampling area. | ||
The soil samples were taken from a depth of 0–20 cm in September 2021. The sampling was performed by mixing topsoil samples from the 0–10 cm and 10–20 cm layers. The soil samples were air-dried for 10 days and sieved through a 2 mm sieve. The pH, electrical conductivity (Ec), total salt content, texture, organic matter content, and total heavy metal concentration of the soil samples were determined following the Hungarian Standards (MSZ), and the results are included in the ESI† material.
2.3. Soil solution extraction and analysis
The soil solutions were prepared based on the methodology developed by Klitzke et al.,35 and Qiu et al.36 Briefly, the soil solutions were prepared using Milli-Q water with a solid-to-water volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5. The suspension was shaken (40 rpm) for 18 h, centrifuged at 3700 rpm for 30 min, and subsequently filtered through a 0.45 μm cellulose nitrate membrane (Ahlstrom-Munksjö GmbH). The pH was measured using a digital pH meter (Inolab pH 720).42 The total organic carbon content of the soil solutions (TOC) was measured using Analytik Jena – Multi N/C 2100 S.
:2.5. The suspension was shaken (40 rpm) for 18 h, centrifuged at 3700 rpm for 30 min, and subsequently filtered through a 0.45 μm cellulose nitrate membrane (Ahlstrom-Munksjö GmbH). The pH was measured using a digital pH meter (Inolab pH 720).42 The total organic carbon content of the soil solutions (TOC) was measured using Analytik Jena – Multi N/C 2100 S.
The electrical conductivity (Ec) and total salt content were analyzed using an Orion 3-Star (Thermo Electron Corporation) conductivity meter. The ionic strength was calculated from the Ec results (eqn (1)):36
| IS = Ec·0.0127 | (1) |
• Ec – electrical conductivity (mS cm−1).
The concentration of major elements (Na, K, Ca, Mg, Al, Fe, Mn) and trace elements (Cu, Zn, Ni, Co, As) was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES), during which Ar was used as the carrier gas.
For flow injection analysis (FIA), a Foss FIA Star 5000 spectrometer was applied, using an NH4Cl buffer (pH = 8.5) to determine NO3− and NO2− concentrations. For PO43− determination, SnCl2 and (NH4)6Mo7O24 were used as reagents. F−, Cl−, SO42−, HCO3− andNH4+ concentrations were determined using ion chromatography (IC) with a Dionex ICS-1600 ion chromatograph, applying an eluent composition of 0.5 M Na2CO3 and NaHCO3.
2.4. Soil solution experiments with TiO2 NPs
The 10 prepared soil solutions were combined, and throughout the experiments, a single representative homogenous sample was utilized. The stock suspension of anatase and rutile NPs was prepared with Milli-Q water using ultrasonication. The final concentration of the TiO2 NPs was 1 g L−1 in the soil solutions following mixing. Following the addition of the TiO2 stock solution to the soil solution, the resulting suspension was mixed using a magnetic stirrer while being protected from light. After 4 hours of interaction between TiO2 NPs with the soil solutions, the suspensions were centrifuged for 10 min at 3700 RPM (1531 RCF).2.5. Characterization of the nanomaterials
The X-ray diffractogram (XRD) patterns were registered using a Rigaku Miniflex II diffractometer with Cu-Kα radiation (λ = 1.5406 Å) equipped with a graphite monochromator. Data points were taken in the 2θ = 20–40° range at a scan speed of 1 min−1 and with a scan step of 0.02°. The primary crystallite size of the TiO2 samples was calculated using the Scherrer equation.43 The morphology of the samples was investigated by a Hitachi S-4700 Type II scanning electron microscope (SEM). Infrared (IR) spectroscopy was used to analyze the surface of various samples (Bruker Equinox 55 spectrometer). The samples were prepared with KBr powder pressed into pellets, and the IR spectra were recorded with a resolution of 2 cm−1. A JASCO-V650 spectrophotometer with an integration sphere (ILV-724) was used to measure the diffuse reflectance spectra (DRS) of the samples (λ = 220–800 nm). BaSO4 was used as a reference, and band gaps were obtained using Tauc plots following the Kubelka–Munk transformation.44The zeta potential (ZP) of the NPs was determined using a Horiba SZ-100 Nanoparticle Analyzer (Retsch Technology GmbH, Germany). The measurements were performed in a cell with a carbon electrode. The values were determined using the Smoluchowski model. The measured dispersion concentration was 0.001 w/v%.
For the X-ray photoelectron spectroscopy (XPS) A Specs XPS instrument equipped with an XR50 dual anode X-ray source and a Phoibos 150 hemispherical electron analyzer was used for data acquisition. The Al Kα source was operated with 150 W power. During X-ray irradiation an electron flood gun was used to negate sample charging. The survey spectra were collected with a pass energy of 100 eV while the high resolution spectra were collected with a pass energy of 20 eV. The evaluation was done with CasaXPS software version 2.3.25PR1.0.45 The aliphatic component of the C1s spectrum was used as an inner reference @ 284.8 eV binding energy. The high resolution spectra were corrected with a Shirley background and all peaks were fit with a Gauss–Lorentz product function with 30% Lorentzian contribution. Elements were quantified based on their peak area on the survey spectrum, corrected with the relative sensitivity factors based on Scofield cross sections.
The investigation of photoluminescence (PL) in the samples utilized a Horiba Jobin Yvon Fluoromax-4 spectrofluorometer (Horiba, Kyoto, Japan) with an excitation wavelength of 300 nm.
Photocurrent measurements were conducted using a Metrohm Autolab PGSTAT302n potentiostat/galvanostat in an aqueous medium with a classical three-electrode system. For the fluorine-doped tin oxide (FTO)/photocatalyst systems, 1 × 2.5 cm FTO plates were cut and coated with 100 mg cm−2 TiO2 samples over a 1 cm2 surface using the spray coating method. The working electrode, counter electrode, and reference electrode were FTO/photocatalysts, a platinum wire, and a Ag/AgCl wire (in 3 mol L−1 NaCl), respectively. All currents were normalized to the geometric surface area of the electrodes. The measurements were conducted in a 0.5 mol L−1 Na2SO3 solution, which facilitated conduction and served as a hole scavenger. The incoming light had an intensity of 100 mW cm−2, and current density was recorded in mA cm−2. Photocurrents were measured following the linear sweep voltammetry (LSV) method within the potential range of −1 V to 0.3 V at a sweeping rate of 2 mV s−1.
The photocatalytic activity of TiO2 NPs was evaluated by phenol degradation. The model pollutant (c0,phenol = 0.1 mM) was added to the TiO2 suspensions (1 g L−1 concentration), which were then sonicated (Ultrapulse, Digitial Ultrasonic Cleaner) for 10 min. The experiment was carried out in a double-walled glass vessel (V = 100 mL) surrounded by six fluorescent tubes (Vilber-Lourmat T-6L UV-A, 6W). The suspensions were stirred in the dark for 10 min46 before switching the lights on to achieve the adsorption/desorption equilibrium. The phenol concentration changes were followed by a Hitachi high-performance liquid chromatograph (HPLC) consisting of a Merck-Hitachi L-4250 UV-vis detector and a Merck Hitachi L-7100 low-pressure pump (0.7 cm3 min−1 flow rate, detection at 210 nm).
3. Results and discussion
3.1. Soil solution properties
The physicochemical properties and the ionic content of the Chernozem soil solutions are presented in Tables 1 and 2. The samples were named “Csn”, where “Cs” stands for “Chernozem” and n corresponds to the number associated with the soil solution samples, CsALL, is a homogeneous sample that was formed by combining the 10 samples.| Samples | pH | IS (mmol L−1) | TOC (mg L−1) |
|---|---|---|---|
| Cs1 | 7.76 ± 0.1 | 3.38 ± 0.1 | 37.19 ± 1.2 |
| Cs2 | 7.72 ± 0.3 | 3.39 ± 0.1 | 48.37 ± 0.5 |
| Cs3 | 7.71 ± 0.1 | 4.12 ± 0.1 | 49.61 ± 2.1 |
| Cs4 | 7.79 ± 0.4 | 4.59 ± 0.1 | 41.11 ± 1.3 |
| Cs5 | 7.69 ± 0.1 | 3.86 ± 0.1 | 46.74 ± 2 |
| Cs6 | 7.72± 0.1 | 3.19 ± 0.08 | 35.14 ± 0.7 |
| Cs7 | 7.75 ± 0.2 | 4.15 ± 0.1 | 54.05 ± 2.3 |
| Cs8 | 7.74 ± 0.2 | 3.42 ± 0.05 | 58.84 ± 2.6 |
| Cs9 | 7.81 ± 0.5 | 4.62 ± 0.09 | 39.62 ± 0.8 |
| Cs10 | 7.73 ± 0.2 | 4.84 ± 0.1 | 40.87 ± 1.1 |
| CsALL | 7.71 ± 0.2 | 4.32 ± 0.14 | 43.19 ± 0.4 |
| Samples | mg L−1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Na+ | K+ | Ca2+ | Mg2+ | F− | Cl− | NO3− | NO2− | PO43− | SO42− | HCO3− | NH4+ | |
| Cs1 | 5.91 ± 0.2 | 5.69 ± 0.1 | 52.6 ± 1.2 | 5.85 ± 0.3 | 0.70 ± 0.02 | 7.56 ± 0.1 | 44.26 ± 0.5 | 1.18 ± 0.04 | 1.07 ± 0.02 | 4.82 ± 0.09 | 24.19 ± 0.3 | 0.51 ± 0.02 |
| Cs2 | 7.56 ± 0.1 | 5.33 ± 0.4 | 53.92 ± 0.7 | 6.22 ± 0.2 | 0.69 ± 0.01 | 10 ± 0.3 | 18.98 ± 0.3 | 2.52 ± 0.1 | 1.25 ± 0.11 | 4.11 ± 0.04 | 21.32 ± 0.18 | 0.30 ± 0.02 |
| Cs3 | 6.73 ± 0.1 | 4.03 ± 0.1 | 60.42 ± 1.1. | 6.53 ± 0.3 | 0.56 ± 0.04 | 4.9 ± 0.1 | 93.51 ± 1.4 | 4.51 ± 0.3 | 0.71 ± 0.06 | 2.36 ± 0.01 | 8.13 ± 0.3 | 0.34 ± 0.01 |
| Cs4 | 13.94 ± 0.5 | 6.54 ± 0.2 | 60.6 ± 0.5 | 7.46 ± 0.1 | 0.61 ± 0.08 | 19 ± 0.5 | 48.49 ± 0.3 | 0.31 ± 0.08 | 0.99 ± 0.05 | 3.57 ± 0.04 | 16.71 ± 0.09 | 0.41 ± 0.02 |
| Cs5 | 7.79 ± 0.3 | 5.4 ± 0.1 | 58.7 ± 1 | 6.99 ± 0.1 | 0.60 ± 0.02 | 5.8 ± 0.2 | 78.51 ± 1.1 | 5.1 ± 0.5 | 0.98 ± 0.14 | 3.43 ± 0.06 | 9.85 ± 0.06 | 0.36 ± 0.01 |
| Cs6 | 8.82 ± 0.4 | 3.87 ± 0.1 | 47.26 ± 0.3 | 5.30 ± 0.4 | 0.75 ± 0.05 | 7.85 ± 0.1 | 19.27 ± 0.4 | 2.5 ± 0.02 | 0.82 ± 0.08 | 5.76 ± 0.08 | 27.86 ± 0.1 | 0.32 ± 0.01 |
| Cs7 | 8.03 ± 0.3 | 4.49 ± 0.3 | 49.96 ± 0.7 | 5.45 ± 0.2 | 0.6 ± 0.01 | 10.77 ± 0.4 | 55.46 ± 0.6 | 4.07 ± 0.1 | 0.85 ± 0.03 | 2.70 ± 0.1 | 9.57 ± 0.14 | 0.22 ± 0.01 |
| Cs8 | 7.38 ± 0.1 | 6.01 ± 0.2 | 50.97 ± 1.1 | 6.34 ± 0.1 | 0.7 ± 0.03 | 7.55 ± 0.3 | 31.19 ± 0.2 | 1.13 ± 0.09 | 1.27 ± 0.01 | 3.82 ± 0.06 | 8.99 ± 0.12 | 0.26 ± 0.01 |
| Cs9 | 10.29 ± 0.1 | 5.47 ± 0.1 | 71.22 ± 1.4 | 7.73 ± 0.3 | 0.44 ± 0.01 | 14.09 ± 0.1 | 187.21 ± 2.5 | 3.21 ± 0.2 | 0.69 ± 0.04 | 2.65 ± 0.17 | 10.14 ± 0.17 | 0.22 ± 0.01 |
| Cs10 | 6.71 ± 0.1 | 9.00 ± 0.4 | 70.47 ± 0.9 | 8.11 ± 0.2 | 0.54 ± 0.02 | 9.05 ± 0.3 | 169.23 ± 1.9 | 3.90 ± 0.5 | 1.23 ± 0.1 | 2.34 ± 0.09 | 12.76 ± 0.2 | 0.31 ± 0.06 |
| CsALL | 8.85 ± 0.3 | 5.59 ± 0.1 | 57.61 ± 0.3 | 6.61 ± 0.1 | 0.58 ± 0.04 | 9.48 ± 0.3 | 78.14 ± 0.8 | 2.25 ± 0.1 | 0.98 ± 0.09 | 3.2 ± 0.15 | 18.53 ± 0.2 | 0.33 ± 0.04 |
The pH of CsALL was sample 7.71, which is slightly alkaline. The IS of the sample was 4.32 mmol L−1, considered low from a hydrochemical point of view.47 At this pH and IS value, the ZP of TiO2 is typically negative, which can affect its behavior in soil solutions in several ways. Particles with a negative ZP tend to repel each other, which can influence their aggregation and dispersion,48 and they are more stable than particles with a positive ZP.48,49
The TOC concentration is 43.19 mg L−1. Although these values are relatively small,50 they can still affect the properties of TiO2. TOC can also influence the stability of particles, which may undergo aggregation along with a reduction in the active sites.51,52 Accordingly, the dissolved organic matter can behave as a reactant for the TiO2 photocatalyst.53
The effect of hydrochemical parameters on the behavior of TiO2 NPs in the soil solution can be complex and may also depend on the different ion contents present in the soil solution. The concentration of Na+ (8.85 mg L−1) and Ca2+ (57.61 mg L−1) was found to be outlier. At similar Na+ and Ca2+ concentrations, it was observed that they increased the sedimentation and aggregation of TiO2 NPs in water,54 and inhibited their photocatalytic activity.55 The NO3− concentration in soil solution was 78.14 mg L−1. A previous study shows56 that 50 mg L−1 NO3− concentration decreased the ZP of TiO2 NPs and increased the size of the aggregates. The concentration of PO43− was 0.98 mg L−1. This ion can strongly adsorb on the surface of titania, potentially affecting the photoactivity of TiO2 even in the millimolar concentration region.57 Finally, considerations were given to F−, Cl−, and SO42− ions. Noteworthy distinctions were identified for the first two ions, whereas the variance in the case of SO42− was less pronounced. These ions have the capacity to attach to the active sites of catalysts, impeding the accessibility for reactant molecules and consequently obstructing photocatalytic reactions.58–60
Besides the parameters mentioned above, the behavior and photocatalytic activity of TiO2 NPs in soil solutions can be influenced by the particle size, morphology, surface chemistry, and crystal structure of NPs. These issues are discussed in the next section.
3.2. Characteristics of TiO2 NPs before immersion in the soil solution
Before immersing the TiO2 NPs in the soil solutions, we characterized them by XRD, SEM, DRS and photocatalytic activity measurements. Two TiO2 samples with different crystal structures have been chosen: Aldrich Anatase (AA) and Aldrich Rutile (AR). Both materials are used as photocatalysts.61 At the same time, Evonik Aeroxide P25 contains anatase and rutile crystal phases.62 However, for our measurements, not the well-known P25 TiO2 was considered because we wanted to investigate pure crystal phases without considering any possible synergistic effects.Based on the SEM micrographs, AA and AR have a polycrystalline structure, i.e., they do not contain particles of different shapes, and there is no change in orientation, (Fig. 2a and b). Their crystalline structure was investigated based on their XRD patterns (Fig. 2c). The characteristic diffraction signals of AA were located at 25.2° (101), 36.9° (103), 37.8° (004), and 38.5° (112), for AR at 25.2° (101), 27.4° (101), 36° (101), and 39.1° (200). The primary particle sizes were ∼85 nm for AA and ∼315 nm for AR. However, it is important to point out that with such a large crystal size, the uncertainty in the calculation could be large.
 | ||
| Fig. 2 Characterization of TiO2 NPs: a) SEM-micrographs of Aldrich Anatase and b) Aldrich Rutile; c) XRD-patterns of Aldrich Anatase and Aldrich Rutile; d) band gap of Aldrich Anatase and Aldrich Rutile; e) phenol degradation performance. | ||
As mentioned above, AA and AR are also used as photocatalysts; therefore, their band gap was also considered. Based on the literature, the band gaps for AA and AR are 3.2 and 3.0 eV, respectively.61 In this study, we measured 3.19 eV for AA and 2.96 eV for AR (Fig. 2d). Their photocatalytic activity (before immersion in the soil solution) was also investigated by the degradation of phenol. The phenol degradation process63 required 120 minutes for AA and 180 minutes for AR (Fig. 2e). Throughout this photocatalytic degradation, reactive oxygen species play a crucial role. Notably, hydroxyl radicals (·OH), formed through the combination of photogenerated electrons and holes, are pivotal in breaking down phenol molecules. Simultaneously, photogenerated holes (h+) directly oxidize phenol, contributing significantly to its degradation. Furthermore, superoxide radicals (·O2−), generated by the reaction of electrons with oxygen molecules, also actively participate in the degradation process.63–65
AA is usually considered a better photocatalyst than AR due to higher specific surface area and adsorption capacity, moreover, the presence of oxygen vacancies and surface-anchored hydroxyl groups also contributes to the increase in activity.66
3.3. Morphology, crystal structure, and optical properties of TiO2 NPs after immersion in the soil solution
Indeed, the properties of TiO2 NPs, including particle size, morphology, surface chemistry, and crystal structure, play a significant role in determining their behavior and photocatalytic activity in soil solutions. Investigating these properties helps in understanding how the NPs interact with soil components, how their structure may change, and how these changes can influence their photocatalytic efficiency.The following abbreviations have been used consistently throughout the study: AA_REF: pure/reference Aldrich Anatase, AA_AP: Aldrich Anatase after phenol degradation, AA_SS_AP: Aldrich Anatase after interaction with soil solution and phenol degradation, AA_SS: Aldrich Anatase after interaction with soil solution; AR_REF: pure/reference Aldrich Rutile, AR_AP: Aldrich Rutile after phenol degradation, AR_SS_AP: Aldrich Rutile after interaction with soil solution and phenol degradation, AR_SS: Aldrich Rutile after interaction with soil solution.
The XRD patterns revealed that there were no significant changes in the crystal structure and the particle size of TiO2 NPs after immersion in the soil solutions (Fig. 3). This indicates that the structure of the NPs remained intact, suggesting that the AA and AR can be considered stable withing such a time range after their release into the environment. Possible differences in morphology were also studied to determine how different soil solution parameters affect the shape and structure of the NPs. We found that, there is no changes were observed in the morphology of the TiO2 NPs after their interaction with the soil solutions. This means that the NPs maintained their original shape and structure, further proving their stability.
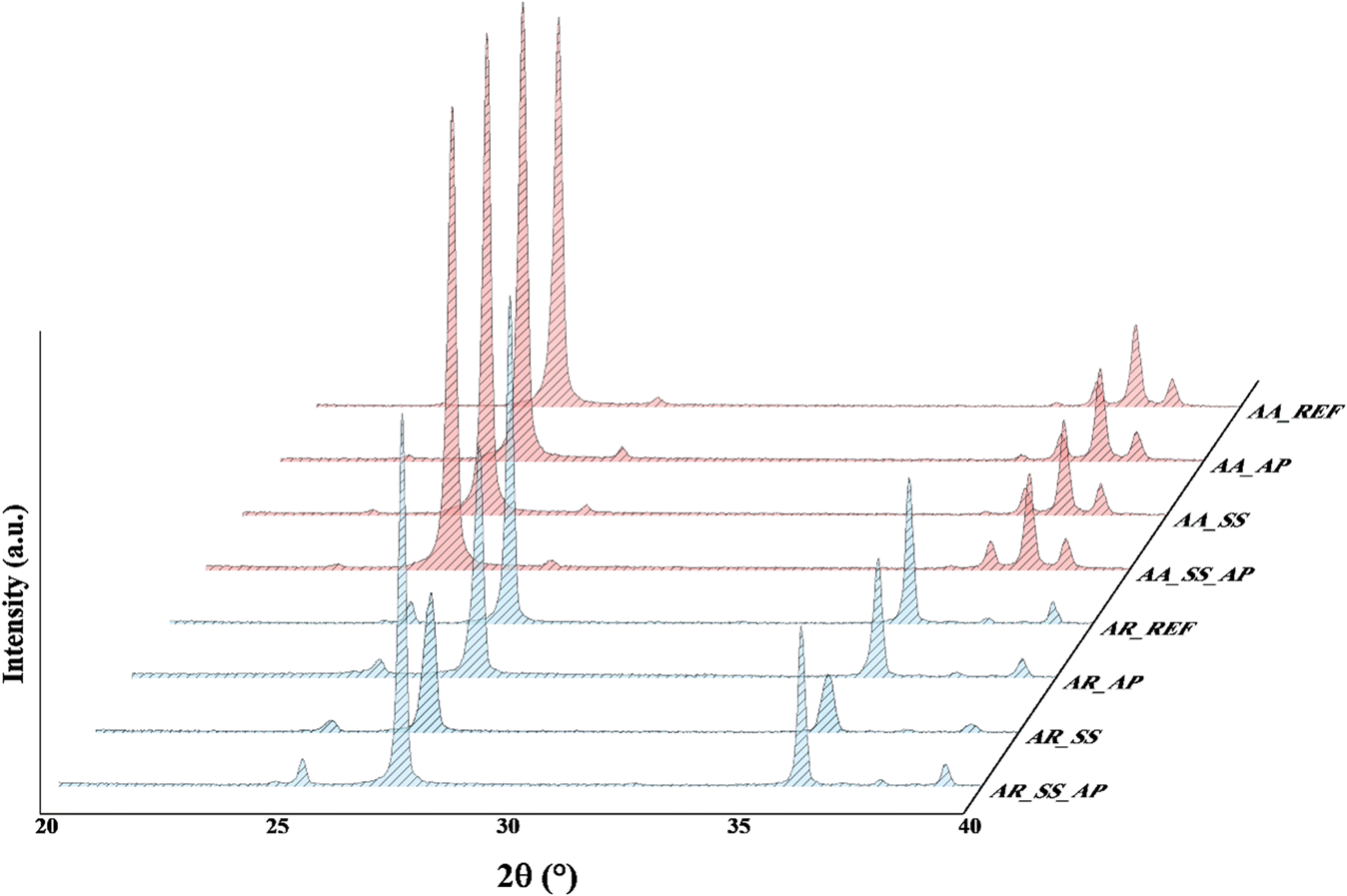 | ||
| Fig. 3 XRD patterns of TiO2 NPs. | ||
Optical properties directly relate to the excitation and activation of the materials; thus, DRS measurements were carried out (Fig. 4a and b). The color of the samples changed after their interaction with the soil solutions, as shown in Fig. 4c. However, the color changes for AA were not as significant as for AR. The band gaps of AR and AA remained at 2.95 and 3.19 eV, respectively, even after phenol degradation and the interaction with the soil solution. Thus, it can be ascertained that even if a material changes color, it does not necessarily lead to changes in band gap67 (Fig. 4). Consequently, the color difference can be attributed to the presence of organic matter on the surface. Based on these results, we investigated the chemical properties of the surface of the samples including surface charge, and adsorption of organic compounds in the next section.
 | ||
| Fig. 4 a) DR spectra of AA, b) DR spectra of AR, and c) the color change of the titania samples (before and after interaction with soil solution, and after phenol degradations). | ||
3.4. Surface properties of TiO2 NPs
ZP can play a crucial role in the way particles interact with each other and with other species in an aquatic environment. For this reason, the ZP and isoelectric point (IEP) of AA and AR were measured in the soil solutions and also in distilled water as a reference.Based on literature data68 the IEP of AA is around pH 3, which agrees with our measurements: in distilled water, the IEP of AA was in the pH 3 approximately and the same value was observed in the soil solution. These results were supported by the DRS measurements as no significant change was observed in the band gap of AA (Fig. 4). It can be concluded that no significant changes occur when AA interacts with the soil solution, so the degree of aggregation did not change compared to the reference sample, either. Hence, only functional groups adsorbed on the surface could have caused the change in its optical properties.
The IEP of AR in distilled water was at pH 4, which is also in accord with the literature.69 However, the IEP of AR immersed in soil solution was measured to be pH 3. This may suggest that various dissolved ions such as cations (Na+ or Ca2+) or organic species have shifted the IEP of AR in the soil solution. Indeed, these ions can adsorb onto the surface of AR particles and alter their surface charge. However, it is also possible that a functional group adsorbed on the surface of AR particles caused deprotonation (e.g., carboxyl groups from humic acids). The reason that the IEP only changed in AR could be because AA and AR have different crystal structures, resulting in differences in their atomic arrangements and surface energy. These structural variations can influence the adsorption of ions and molecules on the surfaces of AA and AR, leading to the observed differences in the IEP values.69
During the photocatalytic activity measurements, the initial pH was also measured. No significant difference was observed between the pH values measured for the AA_REF (pH 6.56) and for the AA_SS (pH 6.65). At this pH range, the NPs have a negative ZP, which can help to repel other particles and maintain the stability of the suspension. In contrast, the initial pH of the AR immersed in the soil solution was 6.95, compared to 6.50 measured for the reference AR (Fig. 5).
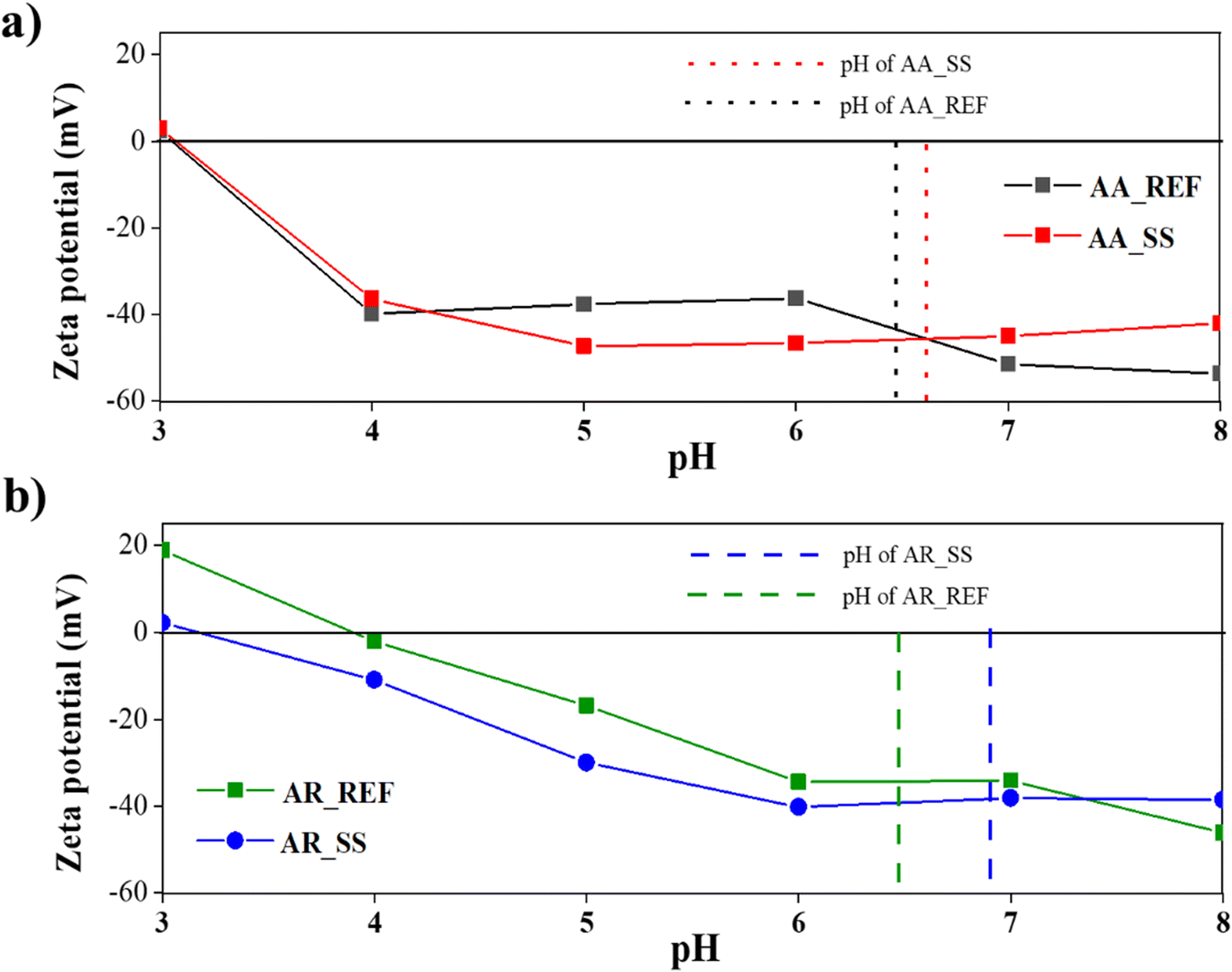 | ||
| Fig. 5 ZP and IEP of TiO2 NPs: a) AA_REF: reference Aldrich Anatase, AA_SS: Aldrich Anatase in soil solution; b) AR_REF: reference Aldrich Rutile, AR_SS: Aldrich Rutile in soil solution. | ||
Specific vibrational modes corresponding to different functional groups can be identified based on the IR spectra, providing information about the chemical composition and surface chemistry of the NPs. Such results can contribute to understanding the nature of surface interactions, such as the adsorption of organic compounds or the formation of surface complexes, which can have implications for the photoactivity and behavior of the NPs in the soil solution. The IR spectra of the samples are shown in Fig. 6.
 | ||
| Fig. 6 IR spectra of the AA and AR samples in the 900–3500 cm−1 region. | ||
The bands at 500–800 cm−1 correspond to metal oxide (Ti–O) vibrations, which are the major components of the samples, and there has been no change in this region. Because metal oxides have the potential to oxidize, but TiO2 turns blue upon reduction in the presence of powerful reductants, such as NaBH4,70 which would have been visible in the DR spectrum (Fig. 4). For this reason, the spectra were normalized to this region to interpret the spectral features of the other regions.
Vibrations associated with CO2 were identified at 2350 cm−1 in all samples. These features were expected to appear due to the adsorption of CO2 from the air and its presence in the optical path of the IR source.
The 1650 cm−1 region shows whether the surface of a semiconductor contains O–H groups or not (hydrophilicity).71 This region should vary parallel with the band associated with water (3000–4000 cm−1). If they do not change together, then the surface contains species with extra O–H groups. For AA_AP and AA_SS_AP, a significant increase was observed for both bands, which can be attributed to the intermediates of phenol formed in the degradation process, such as hydroquinone, resorcinol, pyrocatechol.65,72 For AA_SS, the band at 3000–4000 cm−1 was less intense. The same trend can be observed for AR: both O–H bands in this region had a high intensity in AR_AP and AR_SS_AP, but a low intensity in AR_SS (although the intensity of the band associated with water increased in the 3000–4000 cm−1 region and the one at 1650 cm−1 did not change). The intensity of the band associated with water increased after immersing AA in the soil solution and phenol solution (during the degradation experiments). This increase, which was also observed in AA_AP, could be due to the intermediates of phenol degradation (surface O–H groups originating from hydroquinone, resorcinol, and pyrocatechol). For the AR samples, this band increased in each case except in the AR_REF sample.
Additionally, after evaluating the M–O and –OH/H2O regions, the other regions in the IR spectra were also considered to investigate the effect of interactions with soil and phenol solutions. In the AA_SS and AA_SS_AP samples, the observed bands were in the same regions with different intensities. Between 1080 and 1170 cm−1, C–O vibrations were observed, which could be attributed to the adsorption of long-chain carboxylic acids from the soil solution (e.g., from humic acids). In the 1250–1342 cm−1 region, vibrations of C–N bonds were observed, which may refer to the presence of amines and amides.73 The signals between 1370–1470 cm−1 indicate the presence of C–H bonds in lignin, which may originate from the decomposition of plants in the soil.74 Similarly, both samples display C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds at 1600–1655 cm−1, probably indicating the presence of alkenes. They are commonly found in soil environments originating from various sources, mainly from the decomposition of organic matter, such as plant material and microbial biomass.75 At the 1680–1725 cm−1 region, the vibrations of CO bonds were identified, which can be attributed to the presence of amides. Amides can be derived from organic matter in soil: from carboxylic acids that have an amine group (–NH2) attached to the carbonyl C atom. Between 3000 and 2840 cm−1, C–H vibrational bonds were detected, referring to alkenes, alkanes, and aldehydes. They can be primarily derived from the decomposition of plant residues and are also products of the soil's microbiological activity.76
C bonds at 1600–1655 cm−1, probably indicating the presence of alkenes. They are commonly found in soil environments originating from various sources, mainly from the decomposition of organic matter, such as plant material and microbial biomass.75 At the 1680–1725 cm−1 region, the vibrations of CO bonds were identified, which can be attributed to the presence of amides. Amides can be derived from organic matter in soil: from carboxylic acids that have an amine group (–NH2) attached to the carbonyl C atom. Between 3000 and 2840 cm−1, C–H vibrational bonds were detected, referring to alkenes, alkanes, and aldehydes. They can be primarily derived from the decomposition of plant residues and are also products of the soil's microbiological activity.76
AR_SS also shows these bands (at 1080–1170, 1100, 1250–1342, 1370–1470, and 1600–1655 cm−1 corresponding to C–O, Si–O, C–N, C–H, and CC vibrations, respectively), but with very low intensities. In these regions, intensive bands were observed in AR_SS_AP (Fig. 6). Again, these bands either originate from the soil solution or the phenol degradation (i.e., bands at 1120–1270, 1390–1490, 1550–1650, 1728, and 2960 cm−1 correlate to C–O (ether, ester, carboxylic acid), O–H (phenol), CC, CO (carboxylic acid), and C–H (aldehyde) vibrations, respectively).
XPS is utilized to examine the surface composition of photocatalysts after they undergo immersion in a soil solution. The analysis encompasses two key aspects: elemental analysis, which quantifies the atom % concentrations of detected elements on the surface (indicating the percentage of atoms out of 100 for each element), and component composition analysis, which identifies the various chemical species of these elements.
The findings, presented in Table 3, highlight the elements distinctly identified on the photocatalyst's surface. Ti was detectable in all samples; however, as shown in the table, it is present in varying amounts. The reference samples (AA_REF and AR_REF), which were not immersed in the soil solution, clearly exhibit higher concentrations. In contrast, the samples immersed in the soil solution show lower Ti amounts, as other elements from the solution displace it from the material's surface. The trend is comparable for O, observed at higher concentrations in the reference samples. However, unlike Ti, the O concentrations in both the reference and post-interaction samples with soil solutions do not significantly differ in terms of proportions. This lack of distinction could be attributed to the adsorption of various organic functional groups from the soil solution onto the surface, as indicated by IR measurements, which also include O. C was detectable in all samples, but it was higher in AA_SS, AA_SS_AP, AR_REF, and AR_REF_AP. This elevation may be attributed to the adsorption of various organic matter from soil solution and carbon from phenol decomposition on the surface.
| Sample name | Elemental composition (a.m. %) | |||||||
|---|---|---|---|---|---|---|---|---|
| O | C | N | Zn | Ti | Si | Ca | P | |
| a n.d. – not detected. | ||||||||
| AA_REF | 38.0 | 48.2 | n.d.a | n.d. | 11.2 | n.d. | n.d. | n.d. |
| AA_SS | 35.3 | 48.8 | 1.0 | 0.18 | 7.5 | 3.5 | 1.3 | 1 |
| AA_SS_AP | 33.7 | 53 | 0.4 | 0.2 | 9.2 | 1.3 | 1.1 | 0.7 |
| AR_REF | 36.2 | 51.5 | n.d. | n.d. | 11.9 | n.d. | n.d. | n.d. |
| AR_SS | 30.1 | 60.1 | 0.3 | 0.2 | 6.8 | n.d. | 1.7 | 0.4 |
| AR_SS_AP | 29.1 | 61.8 | 0.6 | 0.2 | 5.9 | n.d. | 1.1 | 0.7 |
After interacting with the soil solutions, both AA and AR samples (AA_SS, AR_SS, AA_SS_AP, AR_SS_AP) exhibit the presence of N. The peak labeled N 1s (∼399.9 eV) most likely corresponds to –NH2 groups, which consistent with the detection IR results. In the same samples, Zn, Ca, and P were detected, and these elements were likewise found in the soil solution (with P measured as PO43− in the solution) (Table 2). Notably, Si was detected in samples AA_SS and AA_SS_AP, as confirmed by IR measurements. While it remains a possibility that various elements could have adsorbed to the surface from the soil solutions, it is important to note that only those outlined in Table 3 fell within the detection limit.
3.5. Photoluminescence and photocurrent measurements
PL offer valuable insights into the recombination of charge carriers after excitation, allowing for the comparison of samples based on energy consumption from photons. These spectra enable the identification of new energy levels and lattice defects. Direct recombination, observable in emission curves, occurs when electrons return from the conduction band to the valence band, emitting photons. When photons of varying energies overlap, the intensity at the specified wavelength increases.77,78Fig. 7 displays the PL spectra of anatase and rutile as reference, both before and after interaction with a soil solution. It's crucial to note that the complexity of the surface chemistry hinders a clear deconvolution of the samples.
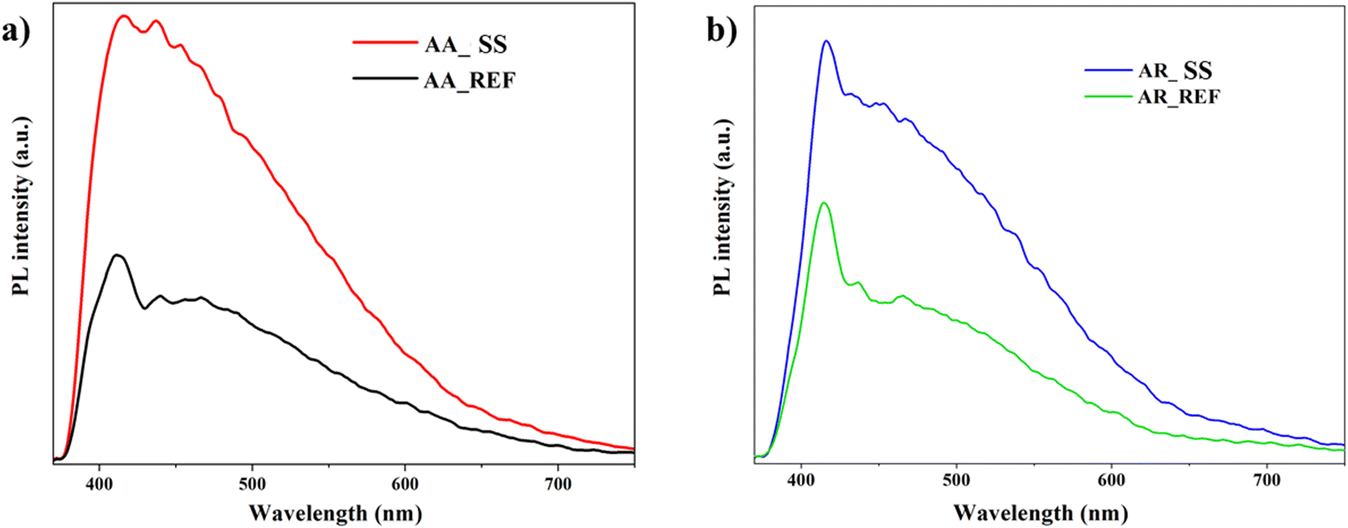 | ||
| Fig. 7 Photoluminescence spectra of TiO2 NPs: a) AA before and after immersion of soil solution; b) AR before and after immersion of soil solution. | ||
In both cases, at λ = 411 nm in the spectra, direct recombination is evident, while emission at λ = 465 nm suggests minimal TiO2 presence.79 The higher photoluminescence intensity after interaction with the soil solution indicates charge recombination due to insufficient separation. This is attributed to various components from the solution adsorbing onto the material's surface, impeding efficient charge carrier utilization. Briefly, the automatic increase in photoluminescence suggests inhibited activity, revealing ineffective utilization of charge carriers.
Photocurrent is also a suitable method to investigate the efficiency of charge carriers within a given catalyst. The measurement is based on the principle of generating a flow of electric current in a material when it absorbs light and generates charge carriers. In the context of a photocatalyst, when photons of sufficient energy are absorbed, electron–hole pairs are created, leading to a flow of current. As can be seen in Fig. 8, the current density of both materials decreased after the immersion of soil solution. It's due to the various adsorbed species onto the surface of the photocatalyst which is from the soil solution. This can create additional recombination centers, hindering the effective separation of photoinduced charge carriers and reducing the overall photocurrent.
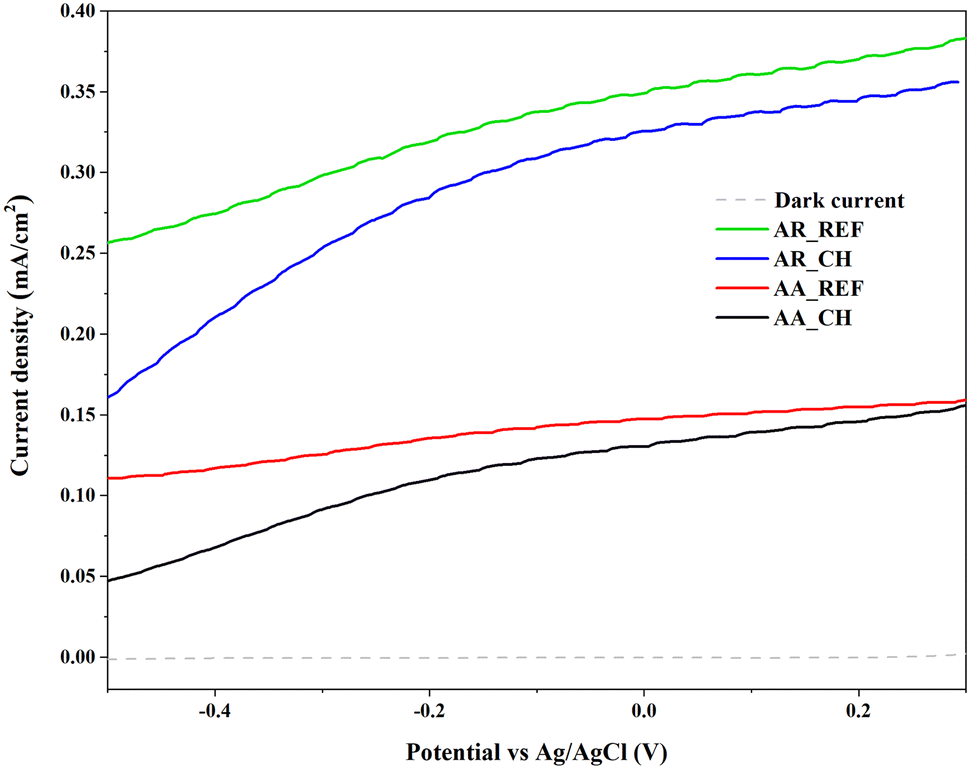 | ||
| Fig. 8 Photovoltammograms of AA and AR samples before and after immersion of soil solution. | ||
3.6. Evaluation of the photocatalytic activity of TiO2 NPs after immersion in the soil solution
The effect of soil solution on the photoactivity of AA and AR was investigated by phenol degradation measurements. Previous adsorption studies indicated that the adsorption of phenol on TiO2 is negligible (around 2–4%).46,80–82 Hence, only a 10 minute dark period was implemented to ensure that concentration changes occur due to catalytic degradation rather than adsorption.AA_REF exhibited the complete degradation of phenol after approximately 120 min of UV-A irradiation. However, for AA_SS, 180 min was required to completely degrade the model pollutant. AR_REF was less efficient than AA_REF: it took 180 min to entirely degrade phenol by the former catalyst. AR_SS was ∼25–27% less efficient than AR_REF. This tendency shows that the presence of the soil solution negatively affected the photocatalytic performance in both cases.
Fig. 9 demonstrates that the activity of AA is superior to that of AR, both as a reference and after interaction with the soil solution. This can be attributed to the higher surface area of AA compared to AR.83 The larger surface area allows AA to degrade the molecules adsorbed on the surface more effectively, including those derived from the soil solution. The degradation curve illustrates that AA is capable of virtually “cleaning” its surface during the irradiation period. Furthermore, AA has higher hydrophilicity compared to AR, primarily due to its higher surface area and more accessible crystal structure.84 The surface of AA tends to possess a higher abundance of surface hydroxyl groups, which serve as active sites for the generation of reactive species such as hydroxyl radicals (·OH) during photocatalysis. These reactive species play a critical role in the degradation of organic pollutants. Thus, the higher concentration of hydroxyl groups on the hydrophilic surface of AA promotes an increased generation of reactive species and enhances its photocatalytic activity.85
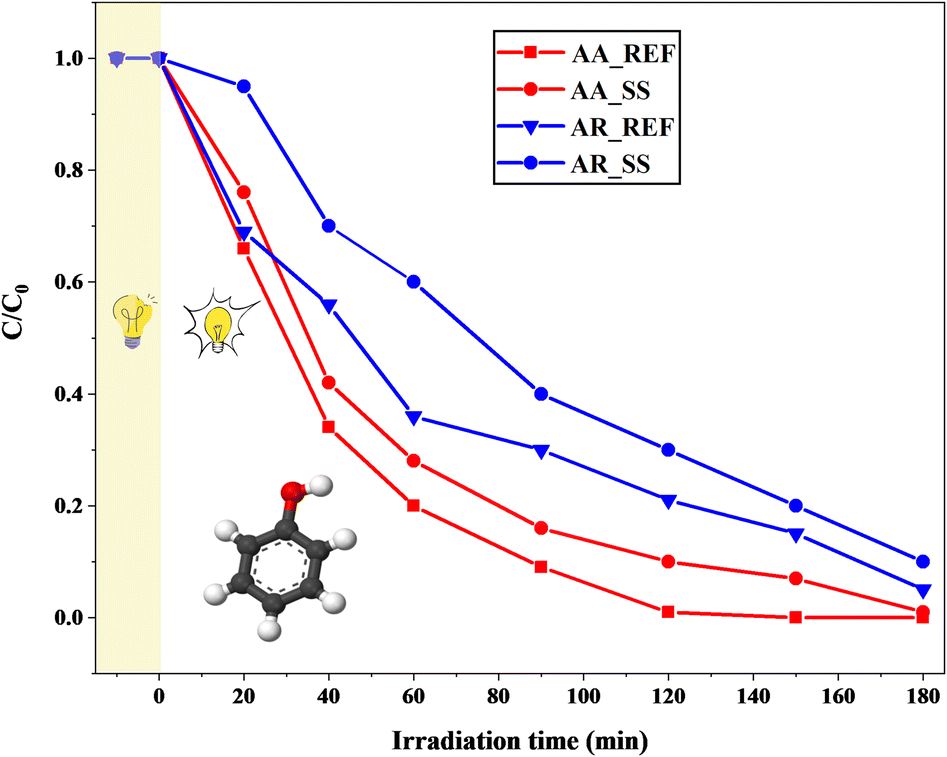 | ||
| Fig. 9 Phenol degradation curves using Aldrich Anatase and Aldrich Rutile as references before and after interactions with the soil solution. | ||
The decrease in photoactivity observed in both AA and AR can be attributed to the presence of chemical components in the soil solution. As discussed earlier, organic molecules with different functional groups can have inhibitory effects on photocatalytic activity when adsorbed on the surface of photocatalysts. This inhibition occurs through multiple mechanisms. Firstly, organic matter acts as a physical barrier, occupying the active sites and limiting the available surface area for pollutant degradation. Secondly, the adsorption of organic matter on the surface can create electron traps, reducing the overall efficiency of photocatalysis. This effect is more pronounced for polar compounds such as O–H and carboxyl groups, which preferentially adsorb onto the AR and AA surfaces.
The XPS spectra revealed the adsorption of Ca2+, Zn2+, P (PO43−), C, and N on the material surface after immersion in the soil solution, potentially influencing catalytic activity in various ways. Ca2+ has the ability to enhance the sedimentation and aggregation of TiO2 NPs, adversely affecting photocatalytic activity.54,86 PO43− ions tend to form surface complexes with TiO2, resulting in surface passivation and a reduction in reactive sites and surface area for adsorption, leading to a decline in photocatalytic activity and potential strong adsorption on the titania surface.57
Moreover, the increased presence of C on the material surface indicates that the surface is actively degrading organic matter but not phenol. As the surface becomes “cleaner” the photo-oxidation efficiency also increases. Therefore, the soil solution does not act as a “poison” for the catalyst. This finding is supported by PL, where the heightened PL intensity in AR_SS and AR samples indicates inefficient utilization of charge carriers due to adsorbed substances on the surface.
Additionally, it is noteworthy that the soil solution contains Zn2+ ions (Table S4†), which may positively impact titania photoactivity. This occurs by promoting the adsorption of reactive molecules on the catalyst surface and facilitating the availability of active sites for catalytic reactions.87
4. Summary and conclusions
The behavior of anatase (AA) and rutile (AR) NPs in a soil solution has been investigated, shedding light on their interactions and properties. The optical properties of AA and AR exhibited changes following their interaction with the soil solution. However, it was observed that their band gap remained unchanged. This suggested that while they may undergo some structural modifications, their fundamental electronic characteristics remain intact. Moreover, the IEP of AR was found to decrease after interaction with the soil solution. This decrease can be attributed to the deprotonation of the NPs, which was supported by IR measurements. Various organic molecules present in the soil solution were found to adsorb onto the surfaces of AA and AR NPs, further confirming the complex nature of their interactions with the soil. Their photocatalytic activity decreased after exposure to the soil solution. This decrease can be attributed to the presence of chemical components in the soil solution, which may hinder catalytic reactions and reduce the efficiency of the NPs as photocatalysts. Overall, the findings of this study highlight the importance of understanding the behavior of NPs in soil environments. The interactions between NPs and soil solutions can lead to changes in their optical properties, surface characteristics, and photocatalytic activity. These findings have implications for the application of NPs in environmental remediation, agriculture, and other fields, where their behavior in the soil is of significance. Further research is needed to explore the specific mechanisms underlying these interactions and develop strategies to optimize the performance of NPs in soil environments.Author contributions
K. S. conceptualization, investigation, writing, editing; I. B. conceptualization, supervision; B. A. investigation; T. G. investigation, writing, editing; A. A. investigation; A. Sz. investigation; Z. K. resources, funding acquisition; Á. K. resources, funding acquisition; A. F. resources, supervision; Z. P. conceptualization, supervision, writing, editing, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude for the financial support provided by the 2019-2.1.13-TÉT_IN-2020-00015 project. T. Gyulavári is grateful for the financial support of the NKFI-PD-138248 project and the Bolyai János scholarship provided by the Hungarian Academy of Sciences (BO/00447/23). TKP2021-NVA-19 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NVA funding scheme.References
- A. A. Keller, S. McFerran, A. Lazareva and S. Suh, J. Nanopart. Res., 2013, 15, 1692 CrossRef
.
- M. F. Hochella, D. W. Mogk, J. Ranville, I. C. Allen, G. W. Luther, L. C. Marr, B. P. McGrail, M. Murayama, N. P. Qafoku, K. M. Rosso, N. Sahai, P. A. Schroeder, P. Vikesland, P. Westerhoff and Y. Yang, Science, 2019, 363, eaau8299 CrossRef PubMed
- F. Hasanvandian, A. Shokri, M. Moradi, B. Kakavandi and S. Rahman Setayesh, J. Hazard. Mater., 2022, 423, 127090 CrossRef CAS PubMed
- A. Payan, A. Akbar Isari and N. Gholizade, Chem. Eng. J., 2019, 361, 1121–1141 CrossRef CAS
- V. Rajeswari, R. Jayavel and A. Clara Dhanemozhi, Mater. Today: Proc., 2017, 4, 645–652 Search PubMed
- T. Gyulavári, K. Kovács, Z. Kovács, E. Bárdos, G. Kovács, K. Baán, K. Magyari, G. Veréb, Z. Pap and K. Hernadi, Appl. Surf. Sci., 2020, 534, 147327 CrossRef
- H. El-Hosainy, S. Mine, T. Toyao, K.-I. Shimizu, N. Tsunoji, M. Esmat, E. Doustkhah, M. El-Kemary and Y. Ide, Mater. Today Nano, 2022, 19, 100227 CrossRef CAS
- F. Yurt, M. Ince, S. G. Colak, K. Ocakoglu, O. Er, H. M. Soylu, C. Gunduz, C. B. Avci and C. C. Kurt, Int. J. Pharm., 2017, 524, 467–474 CrossRef CAS PubMed
- A. Weir, P. Westerhoff, L. Fabricius, K. Hristovski and N. von Goetz, Environ. Sci. Technol., 2012, 46, 2242–2250 CrossRef CAS PubMed
- T. Archana, K. Vijayakumar, M. Arivanandhan and R. Jayavel, Surf. Interfaces, 2019, 17, 100350 CrossRef CAS
- R. Li, T. Li and Q. Zhou, Catalysts, 2020, 10, 804 CrossRef CAS
- L. Collado, P. Reñones, J. Fermoso, F. Fresno, L. Garrido, V. Pérez-Dieste, C. Escudero, M. D. Hernández-Alonso, J. M. Coronado, D. P. Serrano and V. A. de la Peña O'Shea, Appl. Catal., B, 2022, 303, 120931 CrossRef CAS
- S. Mehregan, F. Hayati, M. Mehregan, A. A. Isari, A. Jonidi Jafari, S. Giannakis and B. Kakavandi, Environ. Sci. Pollut. Res., 2022, 29, 74951–74966 CrossRef CAS PubMed
- F. Hasanvandian, M. Zehtab Salmasi, M. Moradi, S. Farshineh Saei, B. Kakavandi and S. Rahman Setayesh, Chem. Eng. J., 2022, 444, 136493 CrossRef CAS
- B. Kakavandi, E. Dehghanifard, P. Gholami, M. Noorisepehr and B. MirzaHedayat, Appl. Surf. Sci., 2021, 570, 151145 CrossRef CAS
- P. Shahzadi, S. R. Gilani, B. B. Rana, A. Ghaffar and A. Munir, Sci. Rep., 2021, 11, 20743 CrossRef CAS PubMed
- E. Kabir, V. Kumar, K. H. Kim, A. C. K. Yip and J. R. Sohn, J. Environ. Manage., 2018, 225, 261–271 CrossRef CAS PubMed
- S. Maiti, I. Fournier, S. K. Brar, M. Cledon and R. Y. Surampalli, J. Hazard., Toxic Radioact. Waste, 2016, 20, B4014004 CrossRef
- N. Jones, B. Ray, K. T. Ranjit and A. C. Manna, FEMS Microbiol. Lett., 2008, 279, 71–76 CrossRef CAS PubMed
- H. Chen, Chem. Speciation Bioavailability, 2018, 30, 123–134 CrossRef CAS
- L. Nyberg, R. F. Turco and L. Nies, Environ. Sci. Technol., 2008, 42, 1938–1943 CrossRef CAS PubMed
- W. Jiang, H. Mashayekhi and B. Xing, Environ. Pollut., 2009, 157, 1619–1625 CrossRef CAS PubMed
- C. O. Dimkpa, A. Calder, D. W. Britt, J. E. McLean and A. J. Anderson, Environ. Pollut., 2011, 159, 1749–1756 CrossRef CAS PubMed
- V. Aruoja, S. Pokhrel, M. Sihtmäe, M. Mortimer, L. Mädler and A. Kahru, Environ. Sci.: Nano, 2015, 2, 630–644 RSC
- S. Nakamura, M. Sato, Y. Sato, N. Ando, T. Takayama, M. Fujita and M. Ishihara, Int. J. Mol. Sci., 2019, 20(15), 3620 CrossRef CAS PubMed
- B. Nowack, J. F. Ranville, S. Diamond, J. A. Gallego-Urrea, C. Metcalfe, J. Rose, N. Horne, A. A. Koelmans and S. J. Klaine, Environ. Toxicol. Chem., 2012, 31, 50–59 CrossRef CAS PubMed
- A. A. Keller and A. Lazareva, Environ. Sci. Technol. Lett., 2014, 1, 65–70 CrossRef CAS
- L. Windler, C. Lorenz, N. von Goetz, K. Hungerbühler, M. Amberg, M. Heuberger and B. Nowack, Environ. Sci. Technol., 2012, 46, 8181–8188 CrossRef CAS PubMed
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed
- S. Heilgeist, R. Sekine, O. Sahin and R. A. Stewart, Water, 2021, 13, 734 CrossRef CAS
- M. Bundschuh, J. Filser, S. Lüderwald, M. S. McKee, G. Metreveli, G. E. Schaumann, R. Schulz and S. Wagner, Environ. Sci. Eur., 2018, 30, 6 CrossRef PubMed
- Q. Abbas, B. Yousaf, A. Amina, M. U. Ali, M. A. M. Munir, A. El-Naggar, J. Rinklebe and M. Naushad, Environ. Int., 2020, 138, 105646 CrossRef CAS PubMed
- W. Tan, J. R. Peralta-Videa and J. L. Gardea-Torresdey, Environ. Sci.: Nano, 2018, 5, 257–278 RSC
- M. R. Wiesner, G. V. Lowry, K. L. Jones, J. M. F. Hochella, R. T. Di Giulio, E. Casman and E. S. Bernhardt, Environ. Sci. Technol., 2009, 43, 6458–6462 CrossRef CAS PubMed
- S. Klitzke, G. Metreveli, A. Peters, G. E. Schaumann and F. Lang, Sci. Total Environ., 2015, 535, 54–60 CrossRef CAS PubMed
- Y. Qiu, Z. Mu, N. Wang, X. Wang, M. Xu and H. Li, Sci. Total Environ., 2020, 731, 139215 CrossRef CAS PubMed
- M. M. Nabi, J. Wang and M. Baalousha, Chemosphere, 2021, 263, 128261 CrossRef CAS PubMed
- J. Makselon, N. Siebers, F. Meier, H. Vereecken and E. Klumpp, Environ. Pollut., 2018, 238, 1027–1034 CrossRef CAS PubMed
- S. V. Gudkov, G. A. Shafeev, A. P. Glinushkin, A. V. Shkirin, E. V. Barmina, I. I. Rakov, A. V. Simakin, A. V. Kislov, M. E. Astashev, V. A. Vodeneev and V. P. Kalinitchenko, ACS Omega, 2020, 5, 17767–17774 CrossRef CAS PubMed
- M. Simonin, J. M. F. Martins, G. Uzu, L. Spadini, A. Navel and A. Richaume, Sci. Total Environ., 2021, 783, 146952 CrossRef CAS PubMed
- E. Eckmeier, R. Gerlach, E. Gehrt and M. W. I. Schmidt, Geoderma, 2007, 139, 288–299 CrossRef CAS
- MSZ-08–0206–2, Evaluation of Some Chemical Properties of the Soil. Laboratory Tests. (pH Value, Phenolphtaleine Alkalinity Expressed in Soda, All Water Soluble Salts, Hydrolite (y1-Value) and Exchanging Acidity (y2-Value)), Hungarian Standard Association, Budapest (in Hungarian), 1978.
- V. Uvarov and I. Popov, Mater. Charact., 2007, 58, 883–891 CrossRef CAS
- I. Tunc, M. Bruns, H. Gliemann, M. Grunze and P. Koelsch, Surf. Interface Anal., 2010, 42, 835–841 CrossRef CAS
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef
- E. Szabó, Z. Pap, G. Simon, A. Dombi, L. Baia and K. Hernádi, Water, 2016, 8, 21 CrossRef
- S. Dahaan, N. Al-Ansari and S. Knutsson, Engineering, 2016, 8, 823–830 CrossRef
- P. Han, X. Wang, L. Cai, M. Tong and H. Kim, Colloids Surf., A, 2014, 454, 119–127 CrossRef CAS
- D. Lin, S. D. Story, S. L. Walker, Q. Huang, W. Liang and P. Cai, Environ. Pollut., 2017, 228, 35–42 CrossRef CAS PubMed
- WHO-EM/CEH/142/E, A compendium of standards for wastewater reuse in the Eastern Mediterranean Region, World Health Organization, Regional Office for the Eastern Mediterranean, Regional Centre for Environmental Health Activities (CEHA), 2006.
- M. Zhu, H. Wang, A. A. Keller, T. Wang and F. Li, Sci. Total Environ., 2014, 487, 375–380 CrossRef CAS PubMed
- L. Haroune, M. Salaun, A. Ménard, C. Y. Legault and J.-P. Bellenger, Sci. Total Environ., 2014, 475, 16–22 CrossRef CAS PubMed
- R. A. Mathew, G. Wu, Y. Zhang, S. Shakiba, Y. Yao, A.-L. Tsai and S. M. Louie, Environ. Sci.: Nano, 2021, 8, 2165–2176 RSC
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS PubMed
- M. L. Tran, C.-C. Fu and R.-S. Juang, J. Mol. Liq., 2019, 276, 32–38 CrossRef CAS
- H. He, Y. Cheng, C. Yang, G. Zeng, C. Zhu and Z. Yan, J. Environ. Sci., 2017, 54, 135–141 CrossRef CAS PubMed
- D. Zhao, C. Chen, Y. Wang, H. Ji, W. Ma, L. Zang and J. Zhao, J. Phys. Chem. C, 2008, 112, 5993–6001 CrossRef CAS
- M. Delarmelina, M. W. Dlamini, S. Pattisson, P. R. Davies, G. J. Hutchings and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2023, 25, 4161–4176 RSC
- J. Zhao, S. Liu, X. Zhang and Y. Xu, Catal. Sci. Technol., 2020, 10, 6552–6561 RSC
- J. Zhang, X. Wang, J. Wang, J. Wang and Z. Ji, Chem. Phys. Lett., 2016, 643, 53–60 CrossRef CAS
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC
- B. Ohtani, O. O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS
- P. Qiu, B. Park, J. Choi, B. Thokchom, A. B. Pandit and J. Khim, Ultrason. Sonochem., 2018, 45, 29–49 CrossRef CAS PubMed
- F. Hayati, M. R. Khodabakhshi, A. A. Isari, S. Moradi and B. Kakavandi, J. Water Proc.engineering, 2020, 38, 101693 CrossRef
- T. Gyulavári, G. Veréb, Z. Pap, B. Réti, K. Baan, M. Todea, K. Magyari, I. M. Szilágyi and K. Hernadi, Materials, 2019, 12, 2537 CrossRef PubMed
- H. Cheng and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 092101 CrossRef
- Z. Pap, K. Mogyorósi, G. Veréb, A. Dombi, K. Hernádi, V. Danciu and L. Baia, J. Mol. Struct., 2014, 1073, 157–163 CrossRef CAS
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Nanoscale Res. Lett., 2010, 6, 27 CrossRef PubMed
- J. W. Bullard and M. J. Cima, Langmuir, 2006, 22, 10264–10271 CrossRef CAS PubMed
- P. Wen, Y. Zhang, G. Xu, D. Ma, P. Qiu and X. Zhao, J. Materiomics, 2019, 5, 696–701 CrossRef
- T. Bezrodna, G. Puchkovska, V. Shymanovska, J. Baran and H. Ratajczak, J. Mol. Struct., 2004, 700, 175–181 CrossRef CAS
- G. Vardar and T. K. Wood, Appl. Environ. Microbiol., 2004, 70, 3253–3262 CrossRef CAS PubMed
- M. S. Demyan, F. Rasche, E. Schulz, M. Breulmann, T. Müller and G. Cadisch, Eur. J. Soil Sci., 2012, 63, 189–199 CrossRef CAS
- A. P. d. Silva, L. C. Babujia, J. C. Franchini, R. Ralisch, M. Hungria and M. d. F. Guimarães, Soil Tillage Res., 2014, 142, 42–53 CrossRef
- C. M. Monreal, Y. Sultan and M. Schnitzer, Geoderma, 2010, 159, 237–242 CrossRef CAS
- L. Zehlike, A. Peters, R. H. Ellerbrock, L. Degenkolb and S. Klitzke, Sci. Total Environ., 2019, 688, 288–298 CrossRef CAS PubMed
- S. Wang, S. Tang, H. Gao, X. Chen, H. Liu, C. Yu, Z. Yin, X. Zhao, X. Pan and H. Yang, Opt. Mater., 2021, 118, 111273 CrossRef CAS
- B. Liu, X. Zhao and L. Wen, Mater. Sci. Eng., B, 2006, 134, 27–31 CrossRef CAS
- A. Galdámez-Martinez, G. Santana, F. Güell, P. R. Martínez-Alanis and A. Dutt, Nanomaterials, 2020, 10, 857 CrossRef PubMed
- G. Veréb, Z. Ambrus, Z. Pap, Á. Kmetykó, A. Dombi, V. Danciu, A. Cheesman and K. Mogyorósi, Appl. Catal., A, 2012, 417–418, 26–36 CrossRef
- N. Balázs, K. Mogyorósi, D. F. Srankó, A. Pallagi, T. Alapi, A. Oszkó, A. Dombi and P. Sipos, Appl. Catal., B, 2008, 84, 356–362 CrossRef
- N. Balázs, D. F. Srankó, A. Dombi, P. Sipos and K. Mogyorósi, Appl. Catal., B, 2010, 96, 569–576 CrossRef
- W.-K. Jo and T. S. Natarajan, Chem. Eng. J., 2015, 281, 549–565 CrossRef CAS
- R. Kaplan, B. Erjavec and A. Pintar, Appl. Catal., A, 2015, 489, 51–60 CrossRef CAS
- J. Du, Q. Wu, S. Zhong, X. Gu, J. Liu, H. Guo, W. Zhang, H. Peng and J. Zou, J. Rare Earths, 2015, 33, 148–153 CrossRef CAS
- M. Abdullah, G. K. C. Low and R. W. Matthews, J. Phys. Chem., 1990, 94, 6820–6825 CrossRef CAS
- G. L. M. Léonard, C. A. Pàez, A. E. Ramírez, J. G. Mahy and B. Heinrichs, J. Photochem. Photobiol., A, 2018, 350, 32–43 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3en00622k |
| ‡ We sincerely honor the memory of Professor Andrea Farsang and her remarkable achievements with the article. Her spirit and unwavering passion for knowledge will be cherished and remembered. |
| This journal is © The Royal Society of Chemistry 2024 |