
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnvironmentally persistent free radicals readily form on TiO2 but not on ZnO nanoparticles†
Eric P.
Vejerano
 * and
Jeonghyeon
Ahn
* and
Jeonghyeon
Ahn
Center for Environmental Nanoscience and Risk, Department of Environmental Health Sciences, Arnold School of Public Health, University of South Carolina, 921 Assembly St., Columbia, SC 29208, USA. E-mail: vejerano@mailbox.sc.edu
First published on 24th September 2024
Abstract
Environmentally persistent free radicals (EPFRs) are stable organic radicals and pollutants in atmospheric and soil particles. Knowledge of EPFR formation on pristine, unsupported engineered nanoparticles is limited. This study investigates the potential of TiO2 and ZnO nanoparticles in aiding aromatic precursors in forming EPFRs and is an experimental verification of our earlier prediction on nanoparticles' potential to form EPFRs. TiO2 formed EPFRs regardless of size, while ZnO exhibited size-dependent behavior. Only smaller ZnO particles readily produced EPFRs. Larger ZnO particles only formed EPFRs when pretreated with ethanol. EPFRs formed on TiO2 and ZnO near room temperature, challenging the belief that relatively high temperatures are needed for EPFRs to form. The yields of EPFRs on pristine TiO2 and ZnO were comparable to those found in atmospheric particulate matter. We identified four types of EPFRs: carbon-centered phenoxyl, oxygen-centered phenoxyl, carbon-centered semiquinone, and oxygen-centered semiquinone radicals. Our study suggests the role of band bending in forming EPFRs on TiO2 and ZnO and the adsorption of aromatic precursors acting as electron acceptors or donors. Our findings suggest that EPFRs may form in unexpected places and under conditions where TiO2 and ZnO nanoparticles are present.
Environmental significanceThis study elucidates the interaction of TiO2 and ZnO nanoparticles with environmental systems, particularly their role in forming environmentally persistent free radicals (EPFRs). By demonstrating that EPFRs can form on these nanoparticles at near-room temperatures and near-ambient pressure, our findings challenge the conventional belief that high temperature and low pressure are necessary for EPFR formation. This work highlights the potential of engineered nanoparticles to contribute to atmospheric and soil pollution through EPFR formation. The size-dependent behavior in EPFR formation enhances our understanding of nanomaterial interactions with natural systems. These insights are crucial for assessing the environmental and human health risks associated with the widespread presence of TiO2 and ZnO nanoparticles. |
Introduction
Environmentally persistent free radicals (EPFRs) are a new class of important persistent organic pollutants1 with significant public health impacts.2 EFPRs are stable and persistent, thus capable of being transported over long distances from their sources. EPFRs have been detected in diverse environmental matrices: atmospheric particulate matter,3–13 soils/sediments and biochars,14–25 wood and coal combustion sites,26,27 microplastics28 wildfire charcoals,29 sewage sludge carbonization,30 to name a few. Recently, persistent free radicals are also found in leaves,31 dubbed as biogenic PFRs. Many studies on EPFRs' health effects on murine models demonstrate that EPFRs elicit various health dysfunctions.2,32–34 Despite their low concentration relative to molecular pollutants in particulate matter (PM), EPFRs exert adverse effects because they can catalytically amplify ROS formation in biological systems.5Many studies on EPFRs have focused on the contribution of transition metal oxide NPs that are supported on surfaces of larger particles. This interest is understandable since the major fraction of ambient PM is from combustion sources that contain clusters of metal oxide nanoparticles (NPs) on surfaces of larger particles. However, pristine NPs, specifically those composed of transition metal oxides, are increasingly used in many commercial products.35 TiO2 is among the most widely industrially produced and used NPs.36 Approximately 4 million metric tons of TiO2 NPs will be produced by 2025.37–39 Their eventual disposal warrants investigation if they assist in forming EPFRs from organic components in the waste stream.
Still, questions exist if pristine engineered NPs can form EPFRs under ambient conditions that typify many environmental systems. NPs, when supported on larger particles (e.g., ambient PM), create Schottky contact due to differences in dielectric properties. Hence, they readily form EPFRs. The Schottky contact is absent in pristine NPs. Only limited information exists on the ability of pristine NPs to stabilize and aid EPFRs.
We posited in our earlier paper that pure NPs can aid in forming EPFRs via band bending and that their formation will depend on particle size, chemical composition, and temperature.40 Here, we investigated the following research questions. First, do NPs with slight differences in their band gap energies (energy difference of the valence and conduction bands) drastically differ in their potential to aid EFPR formation? TiO2 and ZnO are good candidates since their band gap energies differ only by 0.12 eV. Second, what is the influence of the NP size in forming EPFRs. Third, what is the effect of temperature since thermal perturbation can increase the propensity of electrons to traverse the band gaps? A previous study has demonstrated that TiO2 can stabilize phenol, forming EPFRs at 220 °C but not at 25 °C.41 Hence, if large-sized NPs stabilize EPFRs, could their smaller counterpart stabilize and form EPFRs at lower temperatures?
The study described here differs from the earlier studies,41–43 which were performed under ultralow pressure (high vacuum, <10−5 Torr). High vacuum can induce a subtle shift in the properties of the material.44 At sufficiently high temperatures, high vacuum conditions can reduce metal oxides.45 These conditions do not mimic those occurring in the post-combustion zone during incineration and thermal processing of waste, which most likely occur at high pressure. The study described in this paper was conducted at higher pressures (∼14–670 Torr).
Materials and methods
NPs
We purchased ZnO (wurtzite, average particle size (APS) 30 nm and 20 nm, 99% purity), TiO2 (mixed anatase and rutile, APS 20 nm, 99% purity), TiO2 (rutile, APS 20 nm, 99.8% purity), TiO2 (anatase, APS 20 nm, 99% purity), and CdO (APS 20–50 nm, 99.8% purity) from Nanostructured & Amorphous Materials, Inc. We used the NPs without any physical or chemical modifications. We used CdO NPs to compare EPFR formation over the ZnO NP surface; both metal oxides are in group 12 (IIb) in the periodic table and have filled d-orbitals. We used pristine TiO2 (ref. 46) and ZnO since they are among the most used and manufactured NPs and to compare our results to the earlier studies.41–43 We are also interested in determining the effect of a transition metal with filled d-orbitals (Zn2+, [Ar] 3d10) and that with multiple unfilled d-orbitals (Ti2+, [Ar] 3d2).Organic precursors for synthesizing EPFRs
We used 2-monochlorophenol (2-MCP, purity ≥99%), phenol (PH, purity ≥99%), and 1,2-dichlorobenzene (1,2-DCB, anhydrous grade, purity ≥99%) as the organic precursors. These precursors are known precursors of EPFRs on metal oxide particles. We purchased all chemicals from Sigma-Aldrich, which we used without further purification.Synthesis of EPFRs
In previous studies, EPFRs were synthesized under a high vacuum of at least 10−6 Torr. Since EPFRs are detected in atmospheric PM, this suggests that they can form at relatively high pressure. Thus, we synthesized the EPFRs at ∼14 Torr and near ambient pressure (670 Torr) to mimic the conditions during combustion and thermal processing. We used a custom-made thermal system (Fig. S1†) for synthesizing EPFRs (details discussed in the ESI†). We placed 100 mg of the metal oxide NPs in each reactor. The reactors were placed inside a ceramic heater and attached to the ports connected to the equilibration chamber. We then pulled a vacuum and slowly opened the valves that connected the reactors to the equilibration chamber to create a final pressure of ∼14 or 670 Torr. We set the temperature to 100 °C and heated the NPs for 1 h to reduce moisture. Water removal is critical since a high-water load will compete with the organic adsorbates during the formation of EPFRs. After cleaning, we set the heater to a reaction temperature of 25, 120, 230, 350, or 400 °C. We closed the vacuum line once the reactors had reached the set temperature. We injected 10 μL of the precursor and allowed the vapors to adsorb/react with the NPs for 5 min. We heated the unwrapped glass regions using a heating gun to avoid cold spots during adsorption/reaction. After the reaction, we again pulled a vacuum (14 or 670 Torr) to remove any excess unreacted precursors. We turned off the heater to cool the reactors to room temperature (22 °C). After cooling, we sealed the samples under vacuum, detached the reactors from the exposure chamber, transferred the sample to the side arm, and measured the signal using an electron spin resonance (ESR) spectrometer (details in the ESI†). At 670 Torr, other molecules in the gas phase can compete with the organic precursor.Calibration curve using DPPH
We used a three-point calibration curve to bracket the expected concentration using DPPH as the standard. We determined the area counts as the double-integrated area of the ESR signal. We generated the calibration curve by plotting the area counts v. the number of molecules of DPPH for each calibration standard. We expressed the [EPFR] in spins per g of NPs. One spin equals one EPFR. We spiked DPPH in silica to obtain a similar matrix with the sample.Decay of EPFRs
We measured the [EPFR] at different time intervals after exposure to air. We monitor the sample at different time intervals ranging from days to years. We repeat the cycle of exposing the sample to air, pulling vacuum from the sample, and measuring the EPFR concentration. We plotted decay of the normalized [EPFR] on a semi-log scale as a function of time. We normalized the concentration to the initial [EPFR]. We determine the pseudo-first order rate constant as the best-fit line. In this case, the 1/e-folding time is the reciprocal of the pseudo-first-order rate constant.Material characterization
Results
In our earlier paper, we posited that EPFRs can form on pristine NPs, invoking band bending. Here, we investigated the effect of chemical composition, size, temperature, and pressure on forming EPFRs. Previous studies have generated EPFRs at extremely high vacuum, with pressure exceeding 10−6 Torr. Our study investigated the formation of EPFRs under ambient conditions that mimic those during thermal processing.Formation of EPFRs on TiO2 NPs at high pressure
First, we measured the formation of EPFRs on a mixture of anatase- and rutile-TiO2 NPs at near ambient pressure (670 Torr) and high temperatures. TiO2 NPs formed EPFRs when dosed with 1,2-DCB and 2-MCP (Fig. 1A and B). To test the formation of EPFRs on TiO2 NPs and to compare the yield of EPFRs, we performed experiments at a low temperature (25 °C) and high temperature (350 °C). Despite the chlorine substituents destabilizing the aromatic ring in 1,2-DCB, we used it as test precursor molecule since it is symmetric and can form 2-chlorophenoxyl and/or o-semiquinone radicals.1 Interestingly, we observed that TiO2 NPs generated EPFRs even at 25 °C and 670 Torr for particles >30 nm but less than 100 nm. However, the [EPFR] was 3–4× lower than those dosed at high temperatures. Fig. 1A and B show representative first-derivative ESR spectra of the EPFRs on TiO2 NPs and 1,2-DCB at 25 and 350 °C. As shown in Fig. 1A and B, the ESR spectra have a single almost symmetric signal at 350 and 450 °C, whereas at lower temperatures, the signals were highly asymmetric. We attributed this difference to the lower [EPFR] at low temperatures and the types of EPFRs formed. The EPFRs had g-values of 2.0035 and 2.0051. Fig. 1B shows the first-derivative ESR spectra of TiO2 NPs and 2-MCP at 25, 250, 350, 450 °C. ESR signals generated at room temperature had a g-value of 1.9983, which is considerably lower for an organic radical (>2.0023). The g-values ranged from ∼2.0041 to 2.0049 for all three high temperatures, suggesting the presence of O-centered EPFRs. The [EPFR] increased with increasing temperatures. At 450 °C, we observed the highest [EPFR] of 1.4 × 1017 spins per g of NPs. The g-values ranged from 1.9983 to 2.0078, and the ΔHp–p ranged from 4 to 9.6, except those prepared at 25° (ΔHp–p ∼ 2.8–26). The ESR spectral characteristics at 350 and 450 °C resemble combustion-generated EPFRs from carbonaceous particles27 with ΔHp–p of 5.3 to 6.8.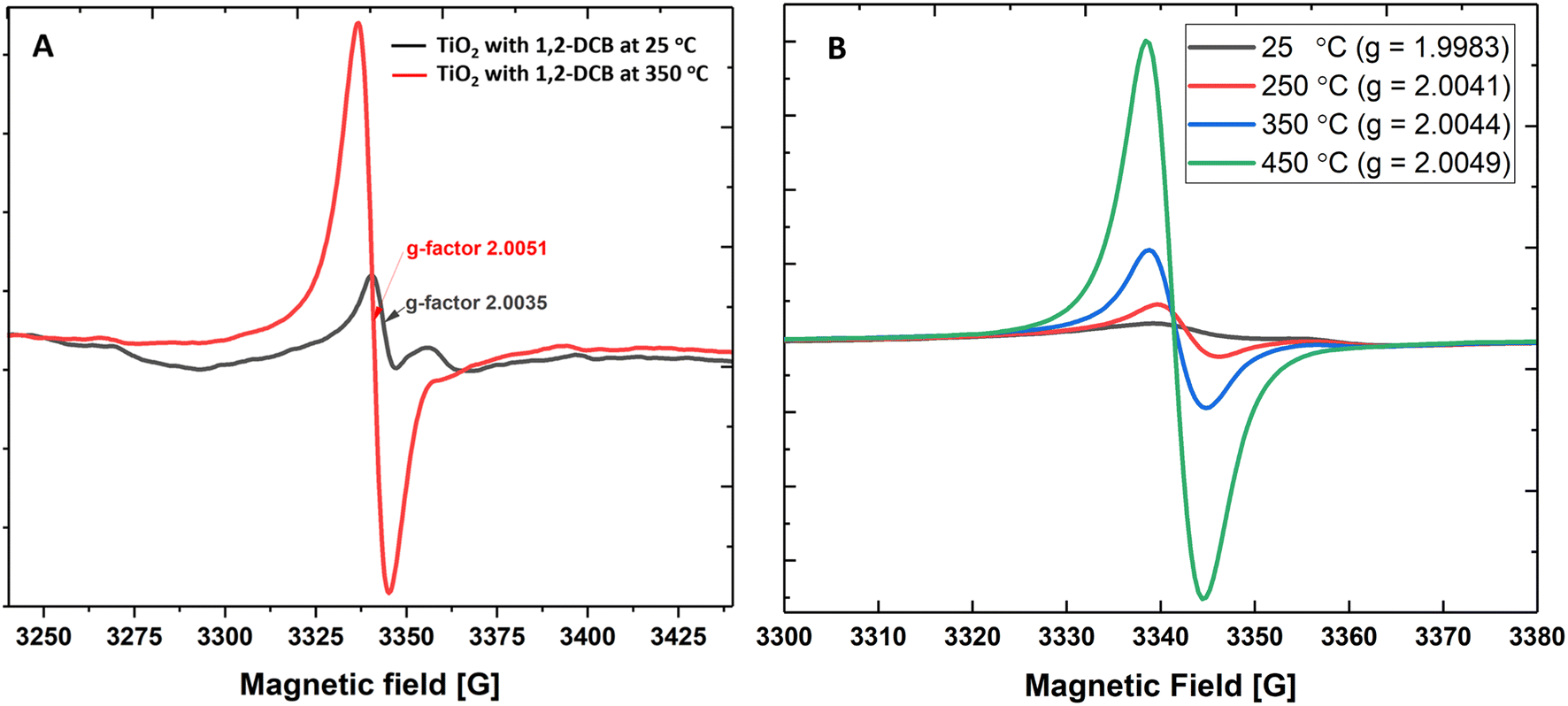 | ||
| Fig. 1 (A) The ESR intensity of TiO2 NPs and 1,2-DCB at low temperature (25 °C) and at high temperature (350 °C) at 670 Torr. (B) The ESR intensity of TiO2 NPs and 2-MCP at 25, 250, 350, and 450 °C at 670 Torr. | ||
Next, we investigated the formation of EPFRs on surfaces of pure rutile-TiO2 NPs to determine if a different crystal structure influences EPFR formation using 2-MCP as the precursor. EPFRs formed on rutile-TiO2 as well. Also, the [EPFR] increased with increasing temperature; the highest [EPFR] of 1.1 × 1018 spins per g of NP was generated at 350 °C. The [EPFR] on rutile-TiO2 was ∼10× higher than that on anatase-TiO2 at 250 °C for 2-MCP and 1,2-DCB (Table S1†).
ZnO forms EFPRs only under unique conditions
While EPFRs readily formed on 30 nm TiO2 NPs and at 670 Torr, they did not form on ZnO (Fig. 2A). We investigated if modifying the ZnO NP surface can aid in forming EPFRs on 30 nm ZnO NPs at high pressure (670 Torr). We dosed ZnO with methanol since it is among the most abundant simple organic alcohols used in combustion systems.47 After treating ZnO NPs with methanol, we dosed them with 2-MCP at 670 Torr and 450 °C. However, we did not detect ESR signals. Fig. 2A depicts the representative ESR spectra on ZnO NPs (≥30 nm) and 2-MCP dosed at 25, 100, 250, and 350 °C, showing the absence of EPFRs. We then tested a slightly longer chain alcohol (ethanol) to pretreat the ZnO NPs (≥30 nm) and dosed them with 2-MCP at 450 °C and 670 Torr. EPFRs formed under these conditions (Fig. 2B). Note that the [EPFR] was much lower than those generated at a similar temperature for similar sized TiO2 NPs. In the absence of 2-MCP, we did not detect any ESR signal for the ZnO NPs pretreated with ethanol. The ESR signal observed at 3400–3450 G (Fig. 2A) corresponds to a g-value range that is not associated with organic radicals, such as EPFRs. Overall, larger NPs only produced EPFRs if we pretreat the surface with ethanol.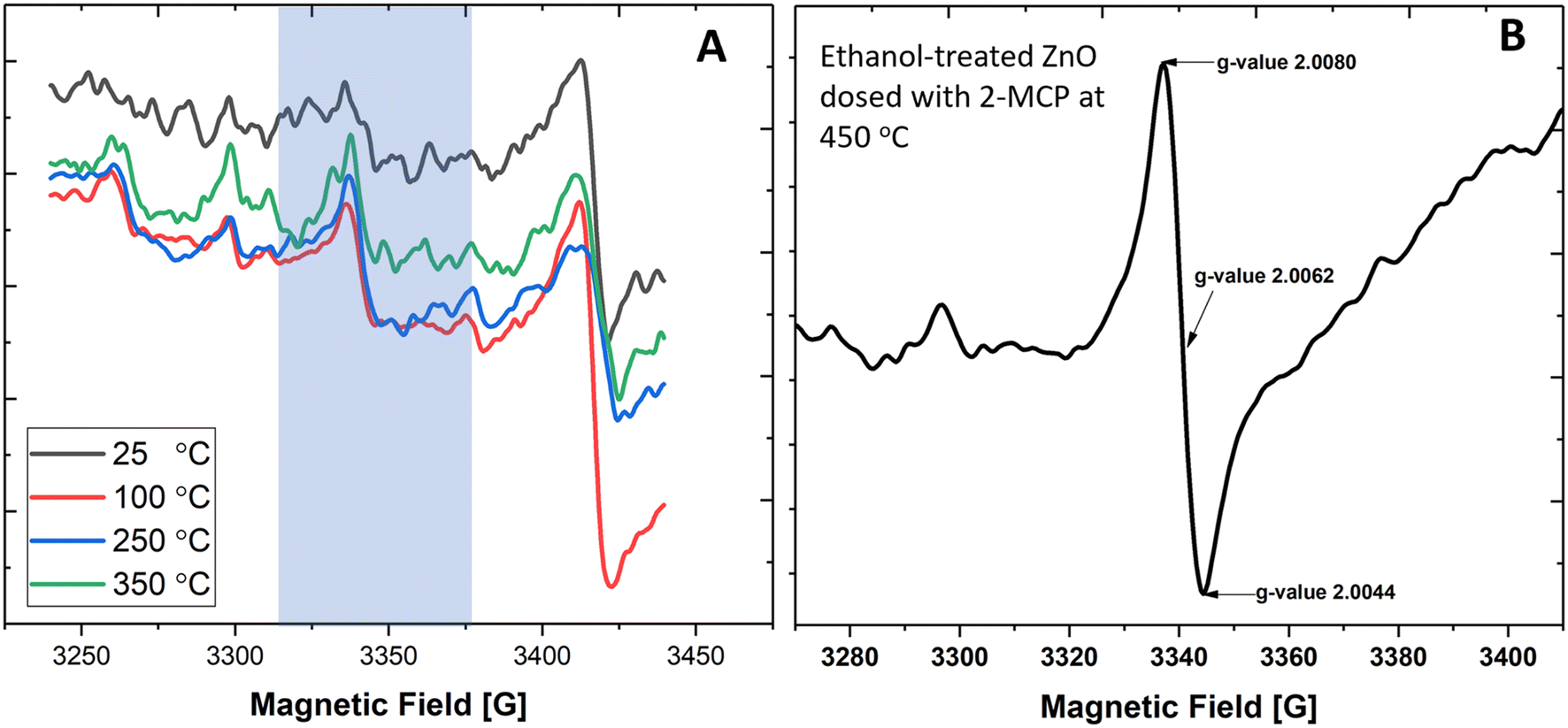 | ||
| Fig. 2 (A) The ESR spectra of ZnO NPs (30 nm) and 2-MCP at 25, 100, 250, and 350 °C at 670 Torr. The shaded region is the magnetic field window for organic radicals (EPFRs). (B) The ESR spectra of ZnO NPs and 2-MCP at 450 °C, pretreated with ethanol at 670 Torr. | ||
Thus, we run a series of experiments to see if EPFRs can form on ZnO under specific conditions (i.e., using smaller ZnO NPs and ethanol treatment). We performed these experiments at pressures of 14 and 670 Torr. Results are summarized in Table 1. Representative ESR spectra are depicted in Fig. 2, 3, and S2.† Hereafter, we will use this notation to describe the different experimental conditions, [organic precursor/NP size/pressure/ethanol dosing]. For example, 2-MCP/30/14T/N means 2-MCP dosed on >30 nm ZnO NPs at 14 Torr without ethanol pretreatment, and Y if the NPs were pretreated with ethanol. Table 1 summarizes the results performed under different conditions. In Table 1, NPs ≥ 30 nm did not form EPFRs (conditions A, 2-MCP/30/670T/N and H, 1,2-DCB/30/670T/N) for 2-MCP and 1,2-DCB at high pressure. Maintaining the dosing at 670 Torr, we investigated whether EPFRs will form on smaller ZnO NPs. Under this condition, we observed that smaller ZnO NPs (<20 nm) formed EPFRs (conditions E, 2-MCP/20/670T/N and M, PH/20/670T/N) but only at high temperatures starting at 350 °C for 2-MCP and 400 °C for PH (Fig. 3A–C) without ethanol treatment. However, under similar conditions (J, 1,2-DCB/20/670T/N) but with 1,2-DCB as the organic precursor, ZnO NPs did not generate EPFRs. The corresponding representative ESR spectra of ZnO NPs under different conditions but at a pressure of 670 Torr are shown in Fig. S2.†
| Organic precursor | Condition | ZnO NP average particle size (nm) | Pressure (Torr) | Ethanol pretreatment | EPFR formation |
|---|---|---|---|---|---|
| 2-MCP | A | >30 | 670 | NO | NO |
| B | >30 | 670 | YES | YES, weak ESR signals | |
| C | >30 | 14 | YES | YES, very strong ESR | |
| D | >30 | 14 | NO | YES, weak ESR | |
| E | <20 | 670 | NO | YES, strong ESR signal at ≥350 °C | |
| F | <20 | 14 | NO | YES, strong ESR signal at ≥250 °C | |
| 1,2-DCB | G | <20 | 14 | NO | EPFRs formed at ≥250 °C |
| H | >30 | 670 | NO | NO | |
| I | >30 | 670 | YES | YES, very strong ESR signal | |
| J | <20 | 670 | NO | NO | |
| K | <20 | 14 | NO | YES, weak ESR signal at ≥350 °C | |
| PH | L | <20 | 14 | NO | EPFRS formed at ≥250 °C |
| M | <20 | 670 | NO | EPFRs formed at ≥400 °C | |
| N | >30 | 14 | NO | NO |
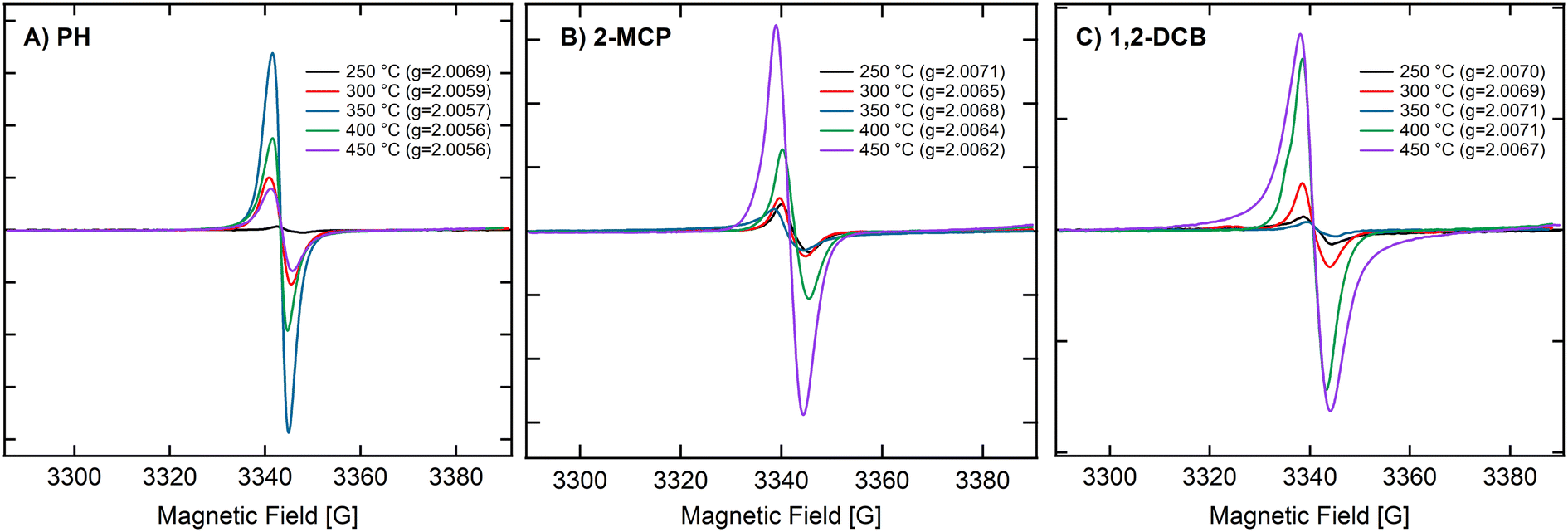 | ||
| Fig. 3 Representative ESR spectra for the formation of EPFRs for three organic precursors on ZnO NPs. (A) The ESR spectra of ZnO NPs (20 nm) and 2-MCP at 250, 300, 350, 400, and 450 °C at 14 Torr. (B) The ESR spectra of ZnO NPs (20 nm) and PH at 250, 300, 350, 400, and 450 °C at 14 Torr. (C) The ESR spectra of ZnO NPs (20 nm) and 1,2-DCB at 250, 300, 350, 400, and 450 °C at 14 Torr. | ||
We investigated the effect of pressure on EPFR formation. While large ZnO NPs (>30 nm) did not form EPFRs at high pressure and without ethanol pretreatment (condition A, 2-MCP/30/670T/N), at low pressure, they yielded low [EPFR] ESR signal (condition D, 2-MCP/30/14T/N). However, despite multiple dosing attempts with PH, condition N, PH/30/14T/N did not yield EPFR and we expected that it would since PH readily forms EPFRs because of the absence of an electron-withdrawing group (e.g., Cl group), unlike 1,2-DCB or 2-MCP. We did not perform EPFR studies for 20 nm ZnO NPs with ethanol since they readily formed EPFRs at 670 Torr. Thus, they will readily form EPFRs at a lower pressure. Since we only observed a weak ESR signal for 2-MCP, we did not expect 1,2-DCB to form EPFRs under this condition [1,2-DCB/30/14T/N] because of its two electron-withdrawing groups. Thus, we did not perform studies under these conditions.
We observed the temperature dependence of ZnO NPs (<20 nm) at 14 Torr with 2-MCP, PH, and 1,2-DCB. Fig. 3A–C depict the ESR spectra of ZnO NPs (<20 nm) and 2-MCP at 250, 300, 350, 400, and 450 °C. The [EPFRs] ranged from 1.4 to 7.2 × 1017 spins per g of NPs. However, the formation of EPFRs was evident for the smaller size, only at temperatures ≥250 °C and at 14 Torr for all organic precursors. With increasing temperatures, we observed a shift in the EPFRs' g-values. The g-values ranged between 2.0056 and 2.0071, suggesting the presence of O-centered EPFRs. We observed the highest [EPFR] of 1.8 × 1017 spins per g of NPs at 350 °C. In general, the [EPFR] followed the order of 350 °C > 400 °C > 450 °C > 300 °C > 250 °C. We also conducted the same set of experiments at 670 Torr (representative spectra depicted in Fig. S2†). Under this condition, only temperatures exceeding 300 °C led to the formation of EPFRs. PH and 1,2-DCB also produced EPFRs at 450 °C only when dosed at 14 Torr. For ZnO, we observed decreasing g-values of the EPFRs with increasing temperatures. The [EPFR] ranged from 1.2 × 1014 to 3.8 × 1018 spins per g of NPs for 2-MCP and 1,2-DCB.
We performed a limited series of experiments with 30 nm CdO to confirm if a filled d-orbital would not readily form EPFRs. Indeed, for similar particle size (large >30 nm) and condition (670 Torr), CdO NPs did not form EPFRs at all temperatures (≤450 °C) for 1,2-DCB and 2-MCP (data not shown).
Lifetime and decay of EPFRs
We investigated and quantified the initial concentration of EPFRs from TiO2 NPs and studied their decay rates. Fig. S3† depicts the [EPFR] decay patterns of TiO2 NPs (>30 nm) dosed with 2-MCP at 25, 250, and 450 °C. Table S1† shows the ESR characteristics and [EPFR]. The initial [EPFR] values were 2.33 × 1016, 4.63 × 1016, and 1.43 × 1017 spins per g of NPs at 25, 250, and 450 °C, respectively. For EPFRs formed at 250 and 450 °C, the [EPFR] decreased substantially but remained stable after that. Overall, the EPFRs on TiO2 had a 1/e-folding time of at least 112 days for those formed at 25 °C. Even after more than a year, the [EPFR] remained at ∼1015 spin per g of NP. Also, EPFRs on ZnO NP (20 nm, dosed with 2-MCP at 350 °C) had a longer lifetime (Fig. S4†) than those on TiO2 under the same conditions. The normalized [EPFR] only decayed to 0.167 after 230 days, thus, decaying only by one order of magnitude from its initial [EPFR].Effects of heating and EPFR formation on the TiO2 crystal structure
Since TiO2 can form EPFRs from low to high temperatures, we investigated if the formation of EPFRs induced the reorientation of the crystal structure. Fig. S5† shows the electron diffraction patterns and fringe lines of the pristine (untreated) and treated TiO2. Fig. S5a, c, and e† show the electron diffraction patterns, and Fig. S5b, d, and f† show fringe lines. Fig. S5a, c, and e† depict diffraction ring distances of pure rutile NPs, which is like the heat-treated TiO2 and those that formed EPFRs. We observed a fringe distance of 0.331–0.332 nm for all three samples, which suggests that thermal treatment does not change the crystallographic properties of TiO2 under the conditions we experimented with. We did not acquire TEM images for anatase since the transformation of anatase to rutile (phase change) in the presence of air occurs only at temperatures >500 °C,48 which are above our highest reaction temperature of 450 °C. We did not acquire the electron diffraction and fringe line pattern for ZnO because it is not expected to undergo phase changes at our dosing temperature.49Discussion
The formation of EPFRs during combustion/thermal processes relies on two components: a surface (e.g., transition metal oxide surface) and an aromatic molecule as a free radical precursor. The single-electron transfer (SET) mechanism is the most invoked conceptual model of EPFR formation.40 The SET mechanism assumes a one-electron reduction transfer. SET successfully explains the formation of EPFRs on some metal oxides but not on ZnO since it would become Zn+, a nonexistent oxidation state.50The idea that electrons can move in both directions (semiconductor-to-organics and vice versa) has important consequences. In our earlier paper,40 we predicted that band bending (VBB) is a potential mechanism in forming EPFRs on pristine NPs using the equation developed by Albery,51
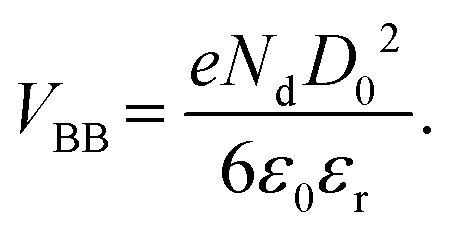 | (1) |
In this equation, VBB is the magnitude of band bending, e is the electronic charge, Nd is the dopant concentration,52–55D is the depletion layer (absence of charge carriers), εr is the relative dielectric constant,56,57 and εo is the permittivity of vacuum. For a small particle, 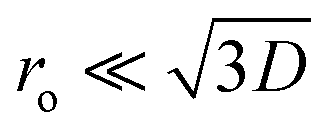 , where ro is the particle radius.52,58 Such an equation has an important consequence in forming EPFRs due to differences in chemistry (εr), size (ro), and condition of the physicochemical environment that influences charge transfer (i.e., temperature)59 during the reaction of an adsorbate on the NP surface. Of the three variables (Nd, D, and εr), εr is modulated by temperature. εr decreases with increasing temperature.60,61 Also, εr varies with particle size depending on the nanomaterials.62–67
, where ro is the particle radius.52,58 Such an equation has an important consequence in forming EPFRs due to differences in chemistry (εr), size (ro), and condition of the physicochemical environment that influences charge transfer (i.e., temperature)59 during the reaction of an adsorbate on the NP surface. Of the three variables (Nd, D, and εr), εr is modulated by temperature. εr decreases with increasing temperature.60,61 Also, εr varies with particle size depending on the nanomaterials.62–67
Thus, NPs unsupported on an insulating or a semiconducting layer (i.e., no Schottky contact) can form EPFRs. We posited that EPFRs can form on pure or pristine NPs, depending on their chemical composition and conditions that differ from those in the post-combustion zone below 450 °C.
NP size
From eqn (1), we predicted that small NPs will induce small or negligible VBB compared to larger NPs since VBB is proportional to ro,59 For pristine NPs (i.e., NPs with a single chemical composition), this approximation indicates that VBB for sufficiently small particles depends on ro and εr. Using eqn (1), assuming an Nd of 1024 m−3, the 30 nm ZnO NPs have a VBB of 0.077 V (Fig. 4Bi and iii). However, the 30 nm TiO2 NPs have a VBB of only 0.007 V (Fig. 4Ai and iii). Such low VBB values facilitate the easy formation of EPFRs on TiO2 NPs, consistent with our results. Even the 100 nm TiO2 NPs have only a VBB of 0.075 V, which is lower than the VBB of the 30 nm ZnO NPs (Fig. 4Ai and 4Bi). Smaller ZnO NPs (20 nm), which produced EPFRs, have a VBB of 0.034 V. This VBB is lower than the VBB of the 30 nm ZnO NPs. For this reason, small particles produced EPFRs more easily than their larger counterpart, as we confirmed in our study. This size is equivalent to large TiO2 NPs (∼60 nm), which, by their VBB, would still form EPFRs. The graph in Fig. 4A and B depict the calculated VBB as a function of the size of TiO2 and ZnO NPs, respectively. Small particles have no depletion layer; thus, charge carriers are available on the surface.59 Thus, in the presence of an organic adsorbate, small NPs easily induce band bending to form EPFRs, as in the case of <20 nm ZnO NPs.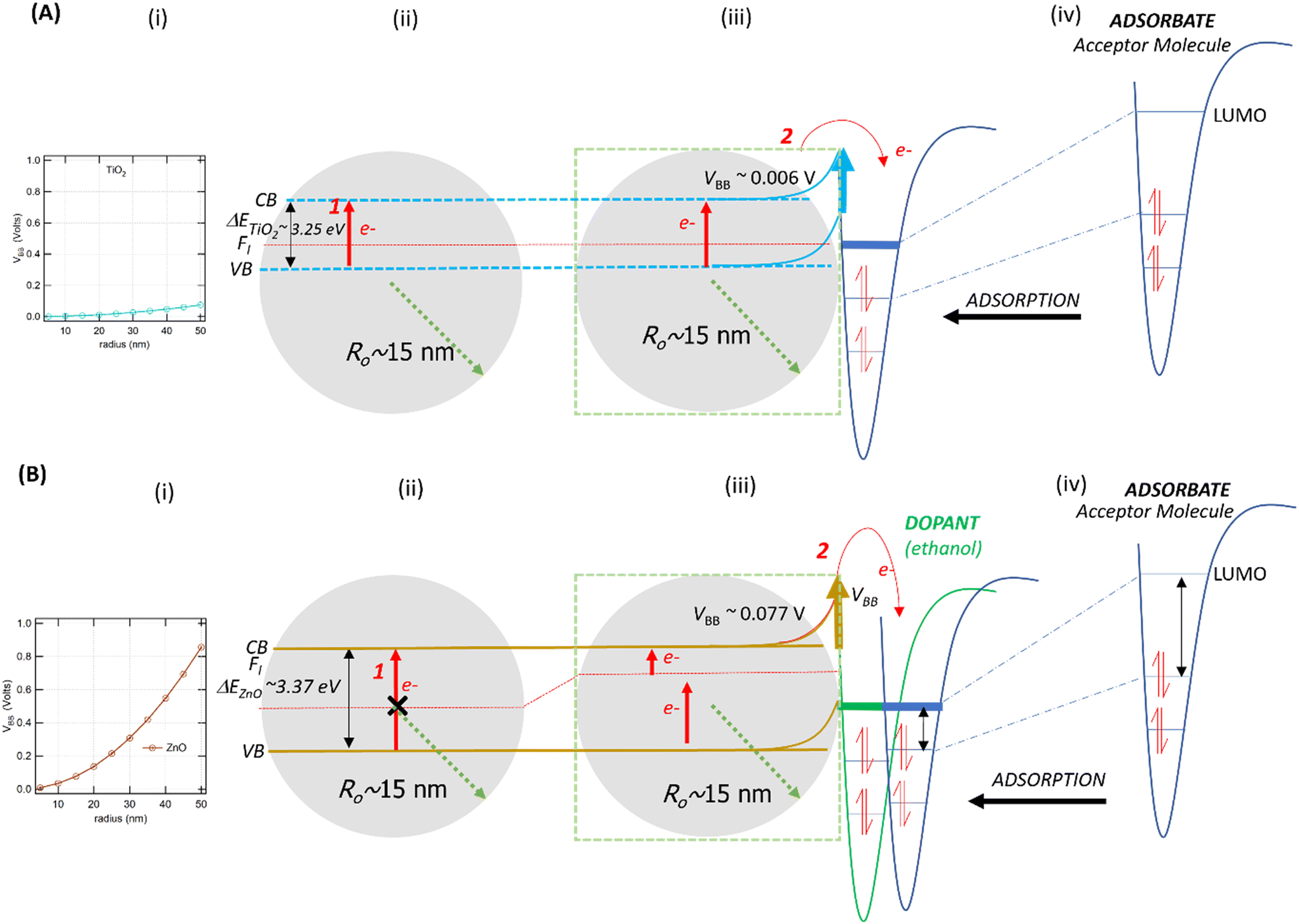 | ||
| Fig. 4 Schematic of the band bending mechanism and electron movement on large (A) TiO2 NPs and (B) ZnO NPs. Fl is the Fermi level. CB and VB are the conduction and valence bands, respectively. (i): Plot of variation in band bending (VBB) as a function of particle size (r) for TiO2 (A) and ZnO (B). (ii): Energy band diagram of TiO2 (A) and ZnO (B), showing the CB, VB, and Fl. (iii) Schematic of the VBB for both TiO2 (A) and ZnO (B). In TiO2, VBB is ∼0.006 V, while in ZnO, it is much higher (∼0.077 V). For ZnO, the presence of a dopant (e.g., ethanol) is needed for electron transfer to occur. (iv): Illustration of the electron transfer process from the semiconductor surface to the acceptor molecule. | ||
Effect of the material physicochemical properties
For metal oxide NPs, the interfacial contact between the metal and semiconductor produces an energy barrier known as the Schottky barrier (SB). For this reason, the presence of the SB facilitates the formation of EPFRs on metal oxide NPs supported on large-size silica or alumina particles. Charge carriers in NPs (e.g., electrons) cross the SB to promote electron transfer between the NP and the organic precursor to form EPFRs. However, the SB is absent in pristine NPs. We posited that NPs may form EPFRs whose properties depart from those observed on NPs that are supported on a surface depending on their chemical composition (different εr). Using eqn (1), the VBB of ZnO is ∼10× higher than that of TiO2, assuming all other parameters are equal. We predicted that ZnO will not easily form EPFRs because it has a higher VBB than TiO2.2 The calculated VBB for ZnO is higher because of its lower εr (∼8.8)57 than that for TiO2 (εr ∼ 100).59 Indeed, we have experimentally verified that such is the case for larger (>30 nm) ZnO NPs even at low pressure, even more so under high pressure (670 Torr), where they compete with other gas phase molecules. Thus, no EPFRs were generated under these conditions. Only when we reduced the size that ZnO NPs formed EPFRs with or without pre-adsorption of a small organic compound, as discussed later.Although oxygen vacancies (surface defects) are not a prerequisite for generating EPFRs, when present, they enhance EPFR formation.68,69 The oxygen vacancy can increase the binding energy of both the core level and the valence band electrons.70 On the ZnO surface, it could enhance band bending in the presence of an organic precursor. Another explanation for why larger 30 nm ZnO NPs do not produce EPFRs is the presence of a few surface-level defects. The ideality factor for spherical shaped ZnO NPs is 1.63,71 suggesting a low number of surface defects. Consequently, the surface electrons on pristine spherical ZnO NPs are less mobile.71 This phenomenon may contribute to the difficulty in forming EPFRs on pristine spherical ZnO NPs. We observed that 20 nm ZnO NPs turned yellow, suggesting that oxygen defects formed.72 Such defects may have contributed to the formation of EPFRs.
Another prediction due to the dielectric constant is the higher relative lifetime of free radicals on ZnO compared to TiO2 NPs of similar sizes.56 The lifetimes of EPFRs on TiO2 and ZnO NPs are comparable to those in other matrices, such as 5–60 days in soils contaminated with polycyclic aromatic hydrocarbons depending on anoxic conditions or the presence of light irradiation,21 0.3–40 days in organic matter of atmospheric particles,11 57–59 days in atmospheric haze,9 160–401 days in hydrothermal processing of sewage sludge.30 The [EPFR] on ZnO shifted only by one order of magnitude compared to that on TiO2 generated under similar conditions. The lifetime for ZnO NPs was much higher than that for ZnO (ref. 50) supported on silica particles. The lower εr for ZnO may explain the longer lifetimes compared to the EPFRs on TiO2, consistent with our prediction.40
Effect of temperature
The band gap energy for semiconductors decreases with increasing temperature, as does the VBB. Increasing temperature enhances lattice vibrations73 and generates a higher charge-carrier density on the surface.74 Thermal perturbation can enhance electron transfer, for instance, an electron traversing a band gap and an electron moving between the energy levels of molecular orbitals of a donor/acceptor molecule. ZnO and TiO2 are wide-band gap materials; their band gap energies are similar to semiconductors (<4 eV).75 Thus, the formation of EPFRs may not require the relatively elevated temperature of the post-combustion zone. Such is the case for most EPFRs from various matrices, which often are generated during combustion and thermal processes. Thus, EPFRs can form at room temperature. Indeed, such is the case for TiO2 in which even under near ambient pressure conditions and 25 °C, EPFRs easily formed. Note that the yield was lower relative to those formed at higher temperatures. However, the signal was strong, as evidenced by the low-noise ESR spectra (Fig. 1A). An earlier study demonstrated that EPFRs form on ZnO at room temperature.76 However, this study used a base pressure below ∼10−5 Torr and heated the ZnO particles at 250 °C before dosing them with the adsorbate at room temperature.76 As mentioned earlier, at sufficiently high temperatures,45 high vacuum can modify the surfaces of metal oxides.44 While such a study is important from a mechanistic standpoint, it does not mimic the conditions at which EPFRs potentially form in the environment.If there is no Schottky contact, electrons on the NPs are depleted, creating a depletion layer. Under this condition, the energy needed to traverse the band gap becomes larger. This phenomenon is not an issue for TiO2 since the band gap is only ∼3.2 eV (Fig. 4A), easily allowing electrons to flow to the valence band. It is difficult for electrons to flow from the conduction band to the valence band. The thermal energy required for an electron to cross a band gap of 3.2 eV can be estimated using the Boltzmann constant and requires high temperature. Although temperature alone is not a viable mechanism for significant electronic conduction in many materials, increasing temperature increases electronic conduction. For ZnO, in which the band is 0.12 eV higher than that for TiO2, the electrons can only be promoted to the conduction band by thermally activating them at high temperature. Indeed, this hypothesis is consistent with the results that only elevated temperature resulted in higher EPFR yields for the 30 nm ZnO NPs. Thus, doping with an organic precursor, high temperature, and small particle size are needed to generate EPFRs on ZnO NPs.
Evidence supporting band bending as the likely mechanism for EPFR formation on pristine NPs
Band bending in solid-state materials can be induced by multiple modes.77 In our study, band bending occurred by two modes: first, the movement of electrons from the valence band to the conduction band of the NPs (Fig. 4A and B) and second, the adsorption of organic molecules acting as acceptors or donors of electrons on surfaces of the NPs, which act as semiconductors (Fig. 4Aiv and Biv). NPs easily sorb volatile organics (e.g., 1,2-DCB, PH, and 2-MCP) to minimize their solid–gas interface's free energy.78,79 From our XPS analysis, we did not observe Ti4+ being reduced to Ti3+ (Fig. S6†). This evidence strengthens the band bending mechanism for TiO2 NPs as the potential mechanism for EPFR formation rather than the SET mechanism, which should be evident if Ti4+ was reduced.Previous studies conducted with phenol adsorption on TiO2 NPs,41,42 where an organic precursor molecule interacts with an NP, indicated the effect of band bending under ultra-high vacuum conditions. We have shown in this study that such a case also happens under low vacuum (14 Torr) and nearly ambient pressure (670 Torr). We demonstrate that TiO2 NPs can generate EPFRs not only at ≥150 °C but also at 25 °C. The differences in EPFR formation in TiO2 NPs at various temperatures are evident from their g-values, a property of an unpaired electron that reveals chemical information of radicals in any specific environment. Reported g-values are <2.0030 for C-centered radicals and >2.0040 for O-centered radicals.80 At high temperatures, thermal excitation and band bending facilitate the easy formation of EPFRs81 on TiO2 from the adsorption of phenol at >220 °C at ≥10−3 Torr. But band bending could also be a viable mechanism near room temperature and high pressure (670 Torr) for the sorption of 1,2-DCB on TiO2. Laser-induced band bending at room temperature has been observed in some material under ultrahigh vacuum82 and is reported that small nanoparticles (5–32 nm) have low band bending potential.77,83–85 Future studies should elucidate the detailed mechanism of EPFR formation on TiO2 at room temperature, which is beyond the scope of the current paper.
As illustrated in Fig. 4 and 5, two electron-transfer processes must occur to generate EPFRs: first, electron transfer in the band gap (Fig. 4i and 5i, red arrows) and second, electron transfer from the semiconductor to the aromatic adsorbate (Fig. 4Aiii and Biii). For TiO2, the movement of electrons from the valence band to the conduction band readily happens due to its relatively lower band gap energy, as discussed above. Additionally, TiO2 lowers and broadens an organic molecule's lowest unoccupied orbital (LUMO) as it adsorbs on its surface.59 Adsorption induces electron flow from the TiO2 to the adsorbate (upward band bending) (Fig. 4Aiii and iv), forming EPFRs. PH accepts electrons during band bending,76 and most likely its substituted analogs, 2-MCP and 1,2-DCB, suggesting upward band bending.
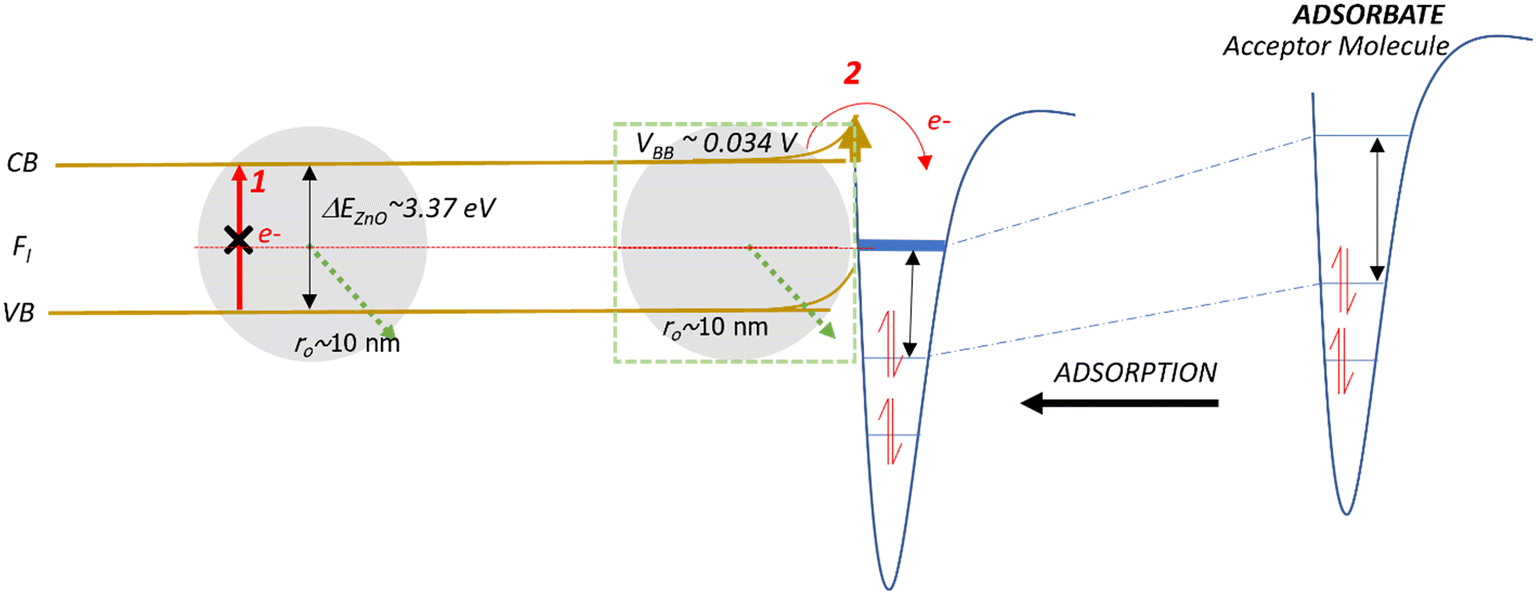 | ||
| Fig. 5 Schematic of the band bending mechanism and electron movement on a small ZnO NP. Fl is the Fermi level. CB and VB are the conduction and valence bands, respectively. | ||
ZnO has a wide bandgap energy. A highly pure ZnO NP has an energy gap of 3.31 to 3.49 eV.86 This band gap does not change beyond 0.1 eV for a size between 0.15 and 80 nm.87 For ZnO NPs, the promotion of electrons from the valence band to the conduction band does not readily occur because of the 0.12 eV increase in the band gap energy (Fig. 4Bii), especially for larger particles (>30 nm). This slight increase in energy prevents EPFRs from readily forming. However, when doped, materials lower their band gap energies.88
In the case of larger ZnO NPs, they produce EPFRs only in the presence of a primary adsorbate (ethanol, Fig. 4Biii). We hypothesize that ethanol unpins the Fermi level of the semiconductor (Fig. 4Biii). This unpinning increases the population of electrons at the Fermi level. Consequently, these electrons easily move to the conduction band of ZnO (red arrows, Fig. 4Biii). However, such unpinning does not immediately result in electron flow from the ZnO to ethanol (i.e., the absence of upward band bending). It is plausible that the molecular structure of ethanol could not stabilize the donated electron, but only a larger aromatic molecule can stabilize the donated electron (Fig. 4Biv). The result that ethanol dosed on larger ZnO NPs facilitating the formation of EPFRs is consistent with our earlier studies89 and that simple acidic/basic organics remove the activation barrier by acting as a bridge between the surface and the aromatic adsorbate to produce EPFRs.89
The SET mechanism is also unlikely for several reasons. Zn2+(II) is ZnO's only known and confirmed stable valence state. Unlike other metal oxide NPs (Fe2O3, NiO, CuO),1,90,91 ZnO does not act as an electron acceptor while forming EPFRs. Only a few studies have mentioned that the charge transfers from ZnO to the phenol adsorbate, revealing a mechanism different from SET.43,92 However, the SET mechanism is not operative in forming EPFRs on pristine ZnO surfaces. The reduction of Zn2+ to Zn0 or its oxidation to Zn3+ states is still debatable. Our XPS analysis for ZnO (Fig. S6†) supports this explanation. A lone-oxidized Zn3+ cannot be created thermodynamically;93 its DFT calculations are heavily contested.93,94 Zn+ exists only under rare conditions when metal–metal bonds dominate,95,96 which is unlikely in our case. In such a case, only an oxidizing Zn surface is predicted to proceed via SET by transferring electrons to the organic precursor, forming EPFRs.76
Conclusions
The insights into the processes of EPFR formation on NP surfaces have important implications. Given that NPs like TiO2 (ref. 46) and ZnO (ref. 78) are heavily used in various industrial processes, engineering applications, food processing,97,98 and consumer products,99 understanding their ability to generate EPFRs is critical. One important finding of our study is that EPFRs can form on NPs even at room temperature under ambient conditions. Our result challenges existing notions that EPFR formation requires elevated temperatures. Our findings indicate that TiO2 NPs readily form EPFRs regardless of size, whereas ZnO nanoparticles exhibit size-dependent behavior. The ease with which TiO2 forms EPFRs raises concerns about its widespread use,46 especially because they are abundant in air, soil, and water.100–104 Additionally, TiO2 NPs are released into the environment more than any other NPs.105 Our study highlights the difficulty of predicting the environmental fate and toxicity of NPs. For instance, the fact that large ZnO NPs only form EPFRs when dosed with organic molecules adds a layer of complexity in predicting EPFR formation on NPs. This finding suggests that slight modifications in NP characteristics can yield significantly different behaviors on their ability to form EPFRs. Large ZnO NPs do not easily form EPFRs unless pretreated with small organic molecules. However, organic molecules abound in nature and in consumer products such as sunscreens. The heavy use of ZnO NPs in sunscreen necessitates research on their potential to form EPFRs under ambient condition and via photo-driven reactions during their use. Thus, EPFRs may form via a light-driven process, which for some reactions is more efficient than thermally activated reactions.106 Experiments are underway to investigate this hypothesis. The generation and toxicity of EPFRs on transition metal-oxide NPs like TiO2 and ZnO and their complexes with organic radicals are yet to be fully characterized. Our findings, however, offer a starting point for identifying which NPs may be suitable for specific applications, considering their environmental and health impacts. Given the widespread use of these materials, further research is urgently needed to inform the safer and more sustainable practices when using nanomaterials on their potential to form EPFRs.Data availability
The data for this study are available in the article and its ESI.†Conflicts of interest
We declare we have no competing interests.Acknowledgements
The National Science Foundation supported this work through grants 1834638 (CBET) and 1738337 (EPSCoR RII-Track 4).References
- S. Lomnicki, H. Truong, E. Vejerano and B. Dellinger, Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter, Environ. Sci. Technol., 2008, 42(13), 4982–4988 CrossRef CAS PubMed.
- T. R. Dugas, S. Lomnicki, S. A. Cormier, B. Dellinger and M. Reams, Addressing Emerging Risks: Scientific and Regulatory Challenges Associated with Environmentally Persistent Free Radicals, Int. J. Environ. Res. Public Health, 2016, 13(6), 573 CrossRef PubMed.
- A. Valavanidis, K. Fiotakis, E. Bakeas and T. Vlahogianni, Electron paramagnetic resonance study of the generation of reactive oxygen species catalysed by transition metals and quinoid redox cycling by inhalable ambient particulate matter, Redox Rep., 2005, 10(1), 37–51 CrossRef CAS PubMed.
- A. Valavanidis, N. Iliopoulos, G. Gotsis and K. Fiotakis, Persistent free radicals, heavy metals and PAHs generated in particulate soot emissions and residue ash from controlled combustion of common types of plastic, J. Hazard. Mater., 2008, 156(1), 277–284 CrossRef CAS PubMed.
- M. A. Kelley, V. Y. Hebert, T. M. Thibeaux, M. A. Orchard, F. Hasan and S. A. Cormier, et al., Model Combustion-Generated Particulate Matter Containing Persistent Free Radicals Redox Cycle to Produce Reactive Oxygen Species, Chem. Res. Toxicol., 2013, 26(12), 1862–1871 Search PubMed.
- W. Gehling, L. Khachatryan and B. Dellinger, Hydroxyl Radical Generation from Environmentally Persistent Free Radicals (EPFRs) in PM2.5, Environ. Sci. Technol., 2014, 48(8), 4266–4272 CrossRef CAS.
- L. W. Kiruri, L. Khachatryan, B. Dellinger and S. Lomnicki, Effect of Copper Oxide Concentration on the Formation and Persistency of Environmentally Persistent Free Radicals (EPFRs) in Particulates, Environ. Sci. Technol., 2014, 48(4), 2212–2217 CrossRef CAS.
- C. K. Borrowman, S. Zhou, T. E. Burrow and J. P. D. Abbatt, Formation of environmentally persistent free radicals from the heterogeneous reaction of ozone and polycyclic aromatic compounds, Phys. Chem. Chem. Phys., 2015, 18(1), 205–212 RSC.
- L. Yang, G. Liu, M. Zheng, R. Jin, Q. Zhu and Y. Zhao, et al., Highly Elevated Levels and Particle-Size Distributions of Environmentally Persistent Free Radicals in Haze-Associated Atmosphere, Environ. Sci. Technol., 2017, 51(14), 7936–7944 CrossRef CAS PubMed.
- H. Tong, P. S. J. Lakey, A. M. Arangio, J. Socorro, F. Shen and K. Lucas, et al., Reactive Oxygen Species Formed by Secondary Organic Aerosols in Water and Surrogate Lung Fluid, Environ. Sci. Technol., 2018, 52(20), 11642–11651 CAS.
- Q. Chen, H. Sun, M. Wang, Y. Wang, L. Zhang and Y. Han, Environmentally Persistent Free Radical (EPFR) Formation by Visible-Light Illumination of the Organic Matter in Atmospheric Particles, Environ. Sci. Technol., 2019, 53(17), 10053–10061 CrossRef CAS PubMed.
- Y. Wang, S. Li, M. Wang, H. Sun, Z. Mu and L. Zhang, et al., Source apportionment of environmentally persistent free radicals (EPFRs) in PM2.5 over Xi'an, China, Sci. Total Environ., 2019, 689, 193–202 CrossRef.
- Q. Chen, H. Sun, W. Song, F. Cao, C. Tian and Y. L. Zhang, Size-resolved exposure risk of persistent free radicals (PFRs) in atmospheric aerosols and their potential sources, Atmos. Chem. Phys., 2020, 20(22), 14407–14417 CrossRef.
- E. S. Odinga, M. G. Waigi, F. O. Gudda, J. Wang, B. Yang and X. Hu, et al., Occurrence, formation, environmental fate and risks of environmentally persistent free radicals in biochars, Environ. Int., 2020, 134, 105172 CrossRef.
- A. L. N. dela Cruz, R. L. Cook, B. Dellinger, S. M. Lomnicki, K. C. Donnelly and M. A. Kelley, et al., Assessment of Environmentally Persistent Free Radicals in Soils and Sediments from Three Superfund Sites, Environ. Sci.: Processes Impacts, 2014, 16(1), 44–52 RSC.
- J. Yang, B. Pan, H. Li, S. Liao, D. Zhang and M. Wu, et al., Degradation of p-Nitrophenol on Biochars: Role of Persistent Free Radicals, Environ. Sci. Technol., 2016, 50(2), 694–700 CrossRef.
- U. G. Nwosu, L. Khachatryan, S. G. Youm, A. Roy, A. L. N. dela Cruz and E. E. Nesterov, et al., Model system study of environmentally persistent free radicals formation in a semiconducting polymer modified copper clay system at ambient temperature, RSC Adv., 2016, 6(49), 43453–43462 RSC.
- U. G. Nwosu, A. Roy, A. L. N. dela Cruz, B. Dellinger and R. Cook, Formation of environmentally persistent free radical (EPFR) in iron(III) cation-exchanged smectite clay, Environ. Sci.: Processes Impacts, 2016, 18(1), 42–50 RSC.
- H. Jia, G. Nulaji, H. Gao, F. Wang, Y. Zhu and C. Wang, Formation and Stabilization of Environmentally Persistent Free Radicals Induced by the Interaction of Anthracene with Fe(III)-Modified Clays, Environ. Sci. Technol., 2016, 50(12), 6310–6319 CrossRef CAS.
- H. Jia, S. Zhao, G. Nulaji, K. Tao, F. Wang and V. K. Sharma, et al., Environmentally Persistent Free Radicals in Soils of Past Coking Sites: Distribution and Stabilization, Environ. Sci. Technol., 2017, 51(11), 6000–6008 CrossRef CAS PubMed.
- H. Jia, S. Zhao, Y. Shi, X. Fan and T. Wang, Formation of environmentally persistent free radicals during the transformation of anthracene in different soils: Roles of soil characteristics and ambient conditions, J. Hazard. Mater., 2019, 362, 214–223 CrossRef CAS.
- K. Zhang, P. Sun and Y. Zhang, Decontamination of Cr(VI) facilitated formation of persistent free radicals on rice husk derived biochar, Front. Environ. Sci. Eng., 2019, 13(2), 22 CrossRef.
- G. Fang, J. Gao, C. Liu, D. D. Dionysiou, Y. Wang and D. Zhou, Key Role of Persistent Free Radicals in Hydrogen Peroxide Activation by Biochar: Implications to Organic Contaminant Degradation, Environ. Sci. Technol., 2014, 48(3), 1902–1910 CrossRef CAS PubMed.
- G. Fang, C. Liu, J. Gao, D. D. Dionysiou and D. Zhou, Manipulation of Persistent Free Radicals in Biochar To Activate Persulfate for Contaminant Degradation, Environ. Sci. Technol., 2015, 49(9), 5645–5653 CrossRef CAS PubMed.
- T. Lieke, X. Zhang, C. E. W. Steinberg and B. Pan, Overlooked Risks of Biochars: Persistent Free Radicals trigger Neurotoxicity in Caenorhabditis elegans, Environ. Sci. Technol., 2018, 52(14), 7981–7987 CrossRef CAS.
- A. L. N. dela Cruz, W. Gehling, S. Lomnicki, R. Cook and B. Dellinger, Detection of Environmentally Persistent Free Radicals at a Superfund Wood Treating Site, Environ. Sci. Technol., 2011, 45(15), 6356–6365 CrossRef CAS.
- H. Jia, S. Li, L. Wu, S. Li, V. K. Sharma and B. Yan, Cytotoxic Free Radicals on Air-Borne Soot Particles Generated by Burning Wood or Low-Maturity Coals, Environ. Sci. Technol., 2020, 54(9), 5608–5618 CrossRef CAS.
- K. Zhu, H. Jia, S. Zhao, T. Xia, X. Guo and T. Wang, et al., Formation of Environmentally Persistent Free Radicals on Microplastics under Light Irradiation, Environ. Sci. Technol., 2019, 53(14), 8177–8186 CrossRef CAS PubMed.
- G. Sigmund, C. Santín, M. Pignitter, N. Tepe, S. H. Doerr and T. Hofmann, Environmentally persistent free radicals are ubiquitous in wildfire charcoals and remain stable for years, Commun. Earth Environ., 2021, 2(1), 1–6 CrossRef.
- Y. Zhu, J. Wei, Y. Liu, X. Liu, J. Li and J. Zhang, Assessing the effect on the generation of environmentally persistent free radicals in hydrothermal carbonization of sewage sludge, Sci. Rep., 2019, 9(1), 1–10 CrossRef.
- E. P. Vejerano and J. Ahn, Leaves are a Source of Biogenic Persistent Free Radicals, Environ. Sci. Technol. Lett., 2023, 10(8), 662–667 CrossRef CAS PubMed.
- B. R. Burn and K. J. Varner, Environmentally persistent free radicals compromise left ventricular function during ischemia/reperfusion injury, Am. J. Physiol., 2015, 308(9), H998–H1006 CAS.
- S. Balakrishna, S. Lomnicki, K. M. McAvey, R. B. Cole, B. Dellinger and S. A. Cormier, Environmentally persistent free radicals amplify ultrafine particle mediated cellular oxidative stress and cytotoxicity, Part. Fibre Toxicol., 2009, 6, 11 CrossRef.
- S. Balakrishna, J. Saravia, P. Thevenot, T. Ahlert, S. Lominiki and B. Dellinger, et al., Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs, Part. Fibre Toxicol., 2011, 8, 11 CrossRef PubMed.
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, M. F. Hochella and D. Rejeski, et al., Nanotechnology in the real world: Redeveloping the nanomaterial consumer products inventory, Beilstein J. Nanotechnol., 2015, 6, 1769–1780 CrossRef.
- C. J. Dedman, A. M. King, J. A. Christie-Oleza and G. L. Davies, Environmentally relevant concentrations of titanium dioxide nanoparticles pose negligible risk to marine microbes, Environ. Sci.: Nano, 2021, 8(5), 1236–1255 RSC.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Modeled Environmental Concentrations of Engineered Nanomaterials (TiO2, ZnO, Ag, CNT, Fullerenes) for Different Regions, Environ. Sci. Technol., 2009, 43(24), 9216–9222 CrossRef PubMed.
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Estimates of Upper Bounds and Trends in Nano-TiO2 Production As a Basis for Exposure Assessment, Environ. Sci. Technol., 2009, 43(12), 4227–4233 CrossRef.
- A. Weir, P. Westerhoff, L. Fabricius, K. Hristovski and N. von Goetz, Titanium Dioxide Nanoparticles in Food and Personal Care Products, Environ. Sci. Technol., 2012, 46(4), 2242–2250 CrossRef PubMed.
- E. P. Vejerano, G. Rao, L. Khachatryan, S. A. Cormier and S. Lomnicki, Environmentally Persistent Free Radicals: Insights on a New Class of Pollutants, Environ. Sci. Technol., 2018, 52(5), 2468–2481 CrossRef CAS.
- M. C. Patterson, C. A. Thibodeaux, O. Kizilkaya, R. L. Kurtz, E. D. Poliakoff and P. T. Sprunger, Electronic Signatures of a Model Pollutant–Particle System: Chemisorbed Phenol on TiO2(110), Langmuir, 2015, 31(13), 3869–3875 CrossRef CAS PubMed.
- M. C. Patterson, N. D. Keilbart, L. W. Kiruri, C. A. Thibodeaux, S. Lomnicki and R. L. Kurtz, et al., EPFR Formation from Phenol adsorption on Al2O3 and TiO2: EPR and EELS studies, Chem. Phys., 2013, 422, 277–282 CrossRef CAS PubMed.
- M. C. Patterson, M. F. DiTusa, C. A. McFerrin, R. L. Kurtz, R. W. Hall and E. D. Poliakoff, et al., Formation of environmentally persistent free radicals (EPFRs) on ZnO at room temperature: Implications for the fundamental model of EPFR generation, Chem. Phys. Lett., 2017, 670(Supplement C), 5–10 CrossRef CAS PubMed.
- H. E. Frankel, Effect of vacuum on materials, 1969, Jan [cited 2023 Sep 27]. Report No.: NASA-TM-X-61789. Available from: https://ntrs.nasa.gov/citations/19690026573.
- I. Vishnevetsky and M. Epstein, Metal oxides reduction in vacuum: setup development and first experimental results, Proc. ScolarPaces, 2011 Search PubMed.
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Estimates of Upper Bounds and Trends in Nano-TiO2 Production As a Basis for Exposure Assessment, Environ. Sci. Technol., 2009, 43(12), 4227–4233 CrossRef CAS PubMed.
- S. Verhelst, J. W. Turner, L. Sileghem and J. Vancoillie, Methanol as a fuel for internal combustion engines, Prog. Energy Combust. Sci., 2019, 70, 43–88 CrossRef.
- M. Jarosz, K. Syrek, J. Kapusta-Kołodziej, J. Mech, K. Małek and K. Hnida, et al., Heat Treatment Effect on Crystalline Structure and Photoelectrochemical Properties of Anodic TiO2 Nanotube Arrays Formed in Ethylene Glycol and Glycerol Based Electrolytes, J. Phys. Chem. C, 2015, 119(42), 24182–24191 CrossRef CAS.
- P. S. Sokolov, A. N. Baranov and V. L. Solozhenko, Phase stability and thermal expansion of ZnO solid solutions with 3d transition metal oxides synthesized at high pressure, J. Phys. Chem. Solids, 2023, 180, 111437 CrossRef CAS.
- E. Vejerano, S. Lomnicki and B. Dellinger, Lifetime of combustion-generated environmentally persistent free radicals on Zn(II)O and other transition metal oxides, J. Environ. Monit., 2012, 14(10), 2803–2806 RSC.
- W. J. Albery, The Transport and Kinetics of Photogenerated Carriers in Colloidal Semiconductor Electrode Particles, J. Electrochem. Soc., 1984, 131(2), 315 CrossRef CAS.
- Z. Zhang, C. C. Wang, R. Zakaria and J. Y. Ying, Role of Particle Size in Nanocrystalline TiO2-Based Photocatalysts, J. Phys. Chem. B, 1998, 102(52), 10871–10878 CrossRef CAS.
- Y. Wang, H. Cheng, Y. Hao, J. Ma, W. Li and S. Cai, Photoelectrochemical properties of metal-ion-doped TiO2 nanocrystalline electrodes, Thin Solid Films, 1999, 349(1), 120–125 CrossRef CAS.
- A. W. Xu, Y. Gao and H. Q. Liu, The Preparation, Characterization, and their Photocatalytic Activities of Rare-Earth-Doped TiO2 Nanoparticles, J. Catal., 2002, 207(2), 151–157 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Photoinduced reactivity of titanium dioxide, Prog. Solid State Chem., 2004, 32(1), 33–177 CrossRef CAS.
- M. Quintana, T. Edvinsson, A. Hagfeldt and G. Boschloo, Comparison of Dye-Sensitized ZnO and TiO2 Solar Cells: Studies of Charge Transport and Carrier Lifetime, J. Phys. Chem. C, 2007, 111(2), 1035–1041 CrossRef CAS.
- Ü. Özgür, Ya. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov and S. Doğan, et al., A comprehensive review of ZnO materials and devices, J. Appl. Phys., 2005, 98(4), 041301 CrossRef.
- J. Balajka, U. Aschauer, S. F. L. Mertens, A. Selloni, M. Schmid and U. Diebold, Surface Structure of TiO2 Rutile (011) Exposed to Liquid Water, J. Phys. Chem. C, 2017, 121(47), 26424–26431 CrossRef PubMed.
- Z. Zhang and J. T. Yates, Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces, Chem. Rev., 2012, 112(10), 5520–5551 CrossRef.
- G. A. Samara, Temperature and Pressure Dependence of the Dielectric Constants of the Thallous Halides, Phys. Rev., 1968, 165(3), 959–969 CrossRef.
- G. A. Samara, Temperature and pressure dependences of the dielectric constants of semiconductors, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27(6), 3494–3505 CrossRef.
- M. Karmaoui, E. V. Ramana, D. M. Tobaldi, L. Lajaunie, M. P. Graça and R. Arenal, et al., High dielectric constant and capacitance in ultrasmall (2.5 nm) SrHfO3 perovskite nanoparticles produced in a low temperature non-aqueous sol–gel route, RSC Adv., 2016, 6(57), 51493–51502 RSC.
- M. Descoteaux, J. P. Sunnerberg and C. Staii, Quantitative characterization of dielectric properties of nanoparticles using electrostatic force microscopy, AIP Adv., 2020, 10(11), 115118 CrossRef.
- H. A. Abid and S. N. T. Al-Rashid, Study of the effect of nanoparticle size on the dielectric constant and concentration of charge carriers of Si and CdS materials, Chalcogenide Lett., 2020, 17(12), 623–629 CrossRef CAS.
- A. Wypych, I. Bobowska, M. Tracz, A. Opasinska, S. Kadlubowski and A. Krzywania-Kaliszewska, et al., Dielectric Properties and Characterisation of Titanium Dioxide Obtained by Different Chemistry Methods, J. Nanomater., 2014, 2014(1), 124814 CrossRef.
- M. D. Parvez Ahmad, A. Venkateswara Rao, K. Suresh Babu and G. Narsinga Rao, Particle size effect on the dielectric properties of ZnO nanoparticles, Mater. Chem. Phys., 2019, 224, 79–84 CrossRef CAS.
- M. Shoeb, S. Ahmad, F. Mashkoor, M. N. Khan, I. Hasan and B. R. Singh, et al., Investigating the size-dependent structural, optical, dielectric, and photocatalytic properties of benign-synthesized ZnO nanoparticles, J. Phys. Chem. Solids, 2024, 184, 111707 CrossRef CAS.
- W. Wang, Z. Liu, Y. Li, W. Wang, Q. Zhang and Q. Wang, Heterogeneous formation of EPFRs from aromatic adsorbates on the carbonaceous particulate matter, Appl. Surf. Sci., 2022, 602, 154316 CrossRef CAS.
- W. Wang, R. Zhang, Z. Liu, W. Wang, Q. Zhang and Q. Wang, Periodic DFT calculation for the formation of EPFRs from phenol on γ-Al2O3 (110): Site-dependent mechanism and the role of ambient water, J. Environ. Chem. Eng., 2022, 10(5), 108386 CrossRef CAS.
- Y. Y. Tay, T. T. Tan, M. H. Liang, F. Boey and S. Li, Specific defects, surface band bending and characteristic green emissions of ZnO, Phys. Chem. Chem. Phys., 2010, 12(23), 6008–6013 RSC.
- S. Middya, A. Layek, A. Dey, J. Datta, M. Das and C. Banerjee, et al., Role of zinc oxide nanomorphology on Schottky diode properties, Chem. Phys. Lett., 2014, 610–611, 39–44 CrossRef CAS.
- F. Hai-Bo, Y. Shao-Yan, Z. Pan-Feng, W. Hong-Yuan, L. Xiang-Lin and J. Chun-Mei, et al., Investigation of Oxygen Vacancy and Interstitial Oxygen Defects in ZnO Films by Photoluminescence and X-Ray Photoelectron Spectroscopy, Chin. Phys. Lett., 2007, 24(7), 2108–2111 CrossRef.
- P. G. Klemens, Thermal conductivity and lattice vibrational modes, Solid State Phys., 1958, 7, 1–98 CAS.
- G. Li, X. Zhou, C. Hao, Q. Lei and Y. Wei, Temperature and electric field dependence of charge conduction and accumulation in XLPE based on polarization and depolarization current, AIP Adv., 2019, 9(1), 015109 CrossRef.
- R. Koole, E. Groeneveld, D. Vanmaekelbergh, A. Meijerink and C. de Mello Donegá, Size effects on semiconductor nanoparticles, Nanoparticles: Workhorses of Nanoscience, Springer, 2014, pp. 13–51 Search PubMed.
- M. C. Patterson, M. F. DiTusa, C. A. McFerrin, R. L. Kurtz, R. W. Hall and E. D. Poliakoff, et al., Formation of environmentally persistent free radicals (EPFRs) on ZnO at room temperature: Implications for the fundamental model of EPFR generation, Chem. Phys. Lett., 2017, 670, 5–10 CrossRef CAS PubMed.
- Z. Zhang and J. T. Yates, Band bending in semiconductors: Chemical and physical consequences at surfaces and interfaces, Chem. Rev., 2012, 112(10), 5520–5551 CrossRef PubMed.
- V. Bolis, Fundamentals in Adsorption at the Solid-Gas Interface. Concepts and Thermodynamics, Calorimetry and Thermal Methods in Catalysis, Springer, 2013, pp. 3–50 Search PubMed.
- L. Lin, Y. Chai, B. Zhao, W. Wei, D. He, B. He and Q. Tang, Photocatalytic oxidation for degradation of VOCs, Open J. Inorg. Chem., 2013, 3 DOI:10.4236/ojic.2013.31003.
- B. Dellinger, S. Lomnicki, L. Khachatryan, Z. Maskos, R. W. Hall and J. Adounkpe, et al., Formation and stabilization of persistent free radicals, Proc. Combust. Inst., 2007, 31(1), 521–528 CrossRef PubMed.
- M. C. Patterson, N. D. Keilbart, L. W. Kiruri, C. A. Thibodeaux, S. Lomnicki and R. L. Kurtz, et al., EPFR formation from phenol adsorption on Al2O3 and TiO2: EPR and EELS studies, Chem. Phys., 2013, 422, 277–282 CrossRef.
- A. Cricenti and B. A. Orlowski, Laser-induced band-bending variation on room-temperature CdTe(110)1×1 surfaces observed in photoemission and through the Franz-Keldish effect in surface differential reflectivity, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51(4), 2322–2325 CrossRef.
- M. E. Franke, T. J. Koplin and U. Simon, Metal and Metal Oxide Nanoparticles in Chemiresistors: Does the Nanoscale Matter?, Small, 2006, 2(1), 36–50 CrossRef PubMed.
- J. Tamaki, Z. Zhang, K. Fujimori, M. Akiyama, T. Harada and N. Miura, et al., Grain-Size Effects in Tungsten Oxide-Based Sensor for Nitrogen Oxides, J. Electrochem. Soc., 1994, 141(8), 2207 CrossRef CAS.
- C. Xu, J. Tamaki, N. Miura and N. Yamazoe, Grain size effects on gas sensitivity of porous SnO2-based elements, Sens. Actuators, B, 1991, 3(2), 147–155 CrossRef.
- V. R. Venu Gopal and S. Kamila, Effect of temperature on the morphology of ZnO nanoparticles: a comparative study, Appl. Nanosci., 2017, 7(3), 75–82 CrossRef.
- D. T. Donia, E. M. Bauer, M. Missori, L. Roselli, D. Cecchetti and P. Tagliatesta, et al., Room Temperature Syntheses of ZnO and Their Structures, Symmetry, 2021, 13(4), 733 CrossRef.
- J. D. Hwang, F. H. Wang, C. Y. Kung and M. C. Chan, Using the Surface Plasmon Resonance of Au Nanoparticles to Enhance Ultraviolet Response of ZnO Nanorods-Based Schottky-Barrier Photodetectors, IEEE Trans. Nanotechnol., 2015, 14(2), 318–321 Search PubMed.
- L. Wang, D. Liang, J. Liu, L. Du, E. Vejerano and X. Zhang, Unexpected catalytic influence of atmospheric pollutants on the formation of environmentally persistent free radicals, Chemosphere, 2022, 303(Pt 1), 134854 CrossRef PubMed.
- E. Vejerano, S. Lomnicki and B. Dellinger, Formation and stabilization of combustion-generated environmentally persistent free radicals on an Fe(III)2O3/silica surface, Environ. Sci. Technol., 2010, 45(2), 589–594 CrossRef PubMed.
- E. Vejerano, S. Lomnicki and B. Dellinger, Formation and stabilization of combustion-generated environmentally persistent free radicals on an Fe(III)2O3/silica surface, Environ. Sci. Technol., 2011, 45(2), 589–594 CrossRef.
- C. A. Thibodeaux, E. D. Poliakoff, O. Kizilkaya, M. C. Patterson, M. F. DiTusa and R. L. Kurtz, et al., Probing environmentally significant surface radicals: Crystallographic and temperature dependent adsorption of phenol on ZnO, Chem. Phys. Lett., 2015, 638(Supplement C), 56–60 CrossRef.
- T. Schlöder, M. Kaupp and S. Riedel, Can Zinc Really Exist in Its Oxidation State +III?, J. Am. Chem. Soc., 2012, 134(29), 11977–11979 CrossRef.
- D. Samanta and P. Jena, Zn in the +III Oxidation State, J. Am. Chem. Soc., 2012, 134(20), 8400–8403 CrossRef PubMed.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Decamethyldizincocene, a Stable Compound of Zn(I) with a Zn-Zn Bond, Science, 2004, 305(5687), 1136–1138 CrossRef.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez and E. Gutierrez-Puebla, et al., Zinc−Zinc Bonded Zincocene Structures. Synthesis and Characterization of Zn2(η5-C5Me5)2 and Zn2(η5-C5Me4Et)2, J. Am. Chem. Soc., 2007, 129(3), 693–703 CrossRef.
- K. Rebecca, Engineered Nanoparticles in Consumer Products: Understanding a New Ingredient, Environ. Health Perspect., 2011, 119(3), A120–A125 Search PubMed.
- D. J. McClements and H. Xiao, Is nano safe in foods? Establishing the factors impacting the gastrointestinal fate and toxicity of organic and inorganic food-grade nanoparticles, npj Sci. Food, 2017, 1(1), 1–13 CrossRef PubMed.
- T. G. Smijs and S. Pavel, Titanium dioxide and zinc oxide nanoparticles in sunscreens: focus on their safety and effectiveness, Nanotechnol., Sci. Appl., 2011, 4, 95–112 CrossRef.
- A. R. Deline, W. M. Young and J. A. Nason, Gold core-labeled TiO2 nanoparticles for tracking behavior in complex matrices: synthesis, characterization, and demonstration, Environ. Sci.: Nano, 2018, 5(4), 956–968 RSC.
- S. Ottofuelling, F. Von Der Kammer and T. Hofmann, Commercial Titanium Dioxide Nanoparticles in Both Natural and Synthetic Water: Comprehensive Multidimensional Testing and Prediction of Aggregation Behavior, Environ. Sci. Technol., 2011, 45(23), 10045–10052 CrossRef PubMed.
- A. J. Koivisto, K. I. Kling, A. S. Fonseca, A. B. Bluhme, M. Moreman and M. Yu, et al., Dip coating of air purifier ceramic honeycombs with photocatalytic TiO2 nanoparticles: A case study for occupational exposure, Sci. Total Environ., 2018, 630, 1283–1291 CrossRef.
- P. Sanderson, J. M. Delgado-Saborit and R. M. Harrison, A review of chemical and physical characterisation of atmospheric metallic nanoparticles, Atmos. Environ., 2014, 94(Supplement C), 353–365 CrossRef.
- C. Zhang, J. Lohwacharin and S. Takizawa, Properties of residual titanium dioxide nanoparticles after extended periods of mixing and settling in synthetic and natural waters, Sci. Rep., 2017, 7(1), 1–11 CrossRef.
- A. A. Keller and A. Lazareva, Predicted Releases of Engineered Nanomaterials: From Global to Regional to Local, Environ. Sci. Technol. Lett., 2014, 1(1), 65–70 CrossRef.
- T. Stensitzki, Y. Yang, V. Kozich, A. A. Ahmed, F. Kössl and O. Kühn, et al., Acceleration of a ground-state reaction by selective femtosecond-infrared-laser-pulse excitation, Nat. Chem., 2018, 10(2), 126–131 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00500g |
| This journal is © The Royal Society of Chemistry 2024 |