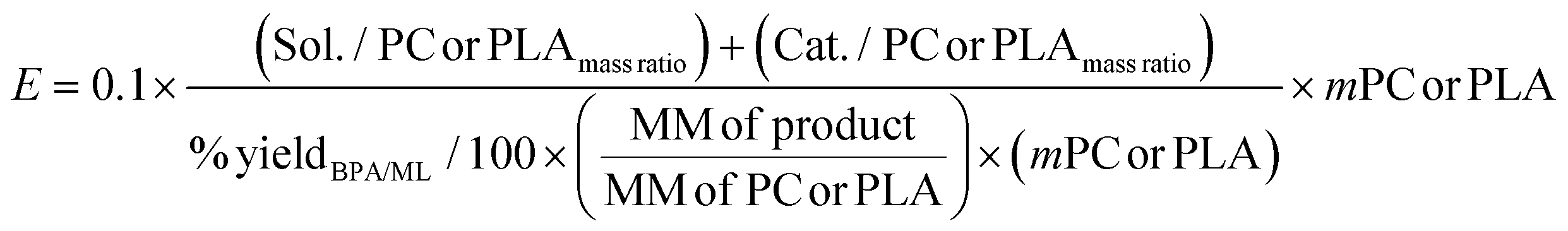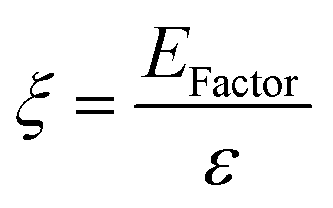DOI:
10.1039/D3GC04907H
(Paper)
Green Chem., 2024,
26, 3814-3831
Solvent-free chemical upcycling of poly(bisphenol A carbonate) and poly(lactic acid) plastic waste using SBA-15-functionalized basic ionic liquids†
Received
13th December 2023
, Accepted 6th February 2024
First published on 7th February 2024
Abstract
Chemical upcycling of plastic waste has garnered global attention due to its sustainable approach to addressing the growing plastic waste accumulation problem and facilitating the establishment of a circular plastic economy. Methanolysis is a chemical upcycling process for the depolymerization of post-consumer polycarbonates and polyesters into their monomeric feedstock, which generally requires an excess amount of co-solvents and homogeneous inorganic salts. Herein, a solvent-free heterogeneous catalytic chemical upcycling of poly(bisphenol A carbonate) (BPA-PC) and poly(lactic Acid) (PLA) is proposed for the production of bisphenol A (BPA) and methyl lactate (ML) with a high yield using SBA-15 functionalized basic ionic liquid catalysts. Among all the synthesized catalysts, SBA-15-Pr-MIM-OH exhibited the highest basicity and demonstrated the best performance for depolymerization of PC and PLA at 120 °C, completing the reaction in 1 h and 4 h, respectively, with a complete conversion and a monomer yield of >98%. The reaction condition was optimized to get the best catalytic performance and product selectivity. Furthermore, the “one-pot” depolymerization strategy was applied for the chemical upcycling of mixed plastic waste (BPA-PC/and PLA) to their monomers. A detailed depolymerization pathway is provided, supported by FT-IR spectroscopy, 1H NMR spectroscopy, and TGA. The parameters for green chemistry metrics were evaluated to show the efficiency and sustainability of the proposed system, opening doors for the industrial upscaling of plastic depolymerization.
1. Introduction
Plastics have been widely used since the 1950s and are ubiquitous in our daily lives. The present assessment suggests that global plastic production stands at approximately 385 million tons annually, with a projected surge to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 million tons by 2050, generating a massive amount of plastic waste annually, which is hard to handle and manage. More than 300 million tons of plastic waste are taken to landfills and incinerated every year.1,2 In 2018, 79% of plastic waste was accumulated in landfills and 12% was incinerated, whereas only 9% was recycled. The accumulated plastic waste in the natural environment by landfilling causes several harmful effects by introducing microplastics into the ecosystem.3 In addition, the incineration of plastic waste leads to vast amounts of CO2 emission, accelerating environmental threats. In contrast to mechanical recycling, chemical upcycling of plastic waste has emerged as a viable alternative to foster a waste-free, carbon-neutral society.4
000 million tons by 2050, generating a massive amount of plastic waste annually, which is hard to handle and manage. More than 300 million tons of plastic waste are taken to landfills and incinerated every year.1,2 In 2018, 79% of plastic waste was accumulated in landfills and 12% was incinerated, whereas only 9% was recycled. The accumulated plastic waste in the natural environment by landfilling causes several harmful effects by introducing microplastics into the ecosystem.3 In addition, the incineration of plastic waste leads to vast amounts of CO2 emission, accelerating environmental threats. In contrast to mechanical recycling, chemical upcycling of plastic waste has emerged as a viable alternative to foster a waste-free, carbon-neutral society.4
Polycarbonate (PC) is a rapidly growing engineering plastic with diverse potential applications ranging from electronics to medical fields, attributed to its exceptional mechanical strength, thermal stability, and durability.5,6 Nonetheless, recycling PC presents challenges. Similarly, bio-based plastics like poly(lactic acid) (PLA) offer an eco-friendly alternative to counter the environmental impact of petroleum-based plastic accumulation.7 However, PLA is relatively costly, and its natural degradation is sluggish. Global PLA production exceeded 800000 tons by 2020. Additionally, microorganisms capable of digesting PLA are not prevalent. They can only break down to low molecular weight PLA (below 10000 Da).8–10 Conventional mechanical recycling transforms plastic waste into lower-grade products. Over time, recycled plastic becomes unsuitable and is eventually discarded in landfills or incinerated. Chemical recycling has garnered significant attention due to its lower energy demand and the potential to produce materials for making fresh virgin plastic.11,12 It also enables the stepwise degradation of mixed plastic waste, bypassing the need for sorting, and offers the ability to process traditionally non-recyclable polymers.13 Given the escalating global production and use of PC and PLA, adopting a more environmentally conscious, sustainable, and cost-effective chemical recycling approach has become imperative for waste management.
Numerous chemical upcycling methodologies, including pyrolysis, hydrogenolysis, cracking, and solvolysis, have been developed to valorize plastic waste.14,15 Pyrolysis typically yields a liquid oil blend of hydrocarbons, emitting greenhouse gases and displaying low product selectivity.16,17 Approaches like hydrogenolysis and hydrocracking, employed for generating value-added chemicals and fuels, usually need high temperatures, pressure, and catalysts based on transition metals.18,19 Solvolysis, including hydrolysis, alcoholysis, aminolysis, and glycolysis, stands as an alternate sustainable technique wherein plastic waste is converted to its monomers, fostering circular plastic production within a circular economy framework.20 Among these, alcoholysis emerges as an energy-efficient and sustainable process, enabling recycling and acquiring essential chemicals. Methanolysis of PC and PLA can produce BPA and ML under mild reaction conditions. Prior studies highlighted the demand for substantial quantities of potent bases or superbases in alcoholysis.21 Additionally, ionic liquids (ILs) and deep eutectic solvents were employed for the same transformation. Initially, [Bmim][Cl] and [Bmim][Ac] were used as catalyst–solvent hybrids for PC methanolysis under mild conditions. Quaranta et al. reported the alcoholysis of PC over a highly active 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) organocatalyst.22 However, the use of homogeneous DBU had some problems; for example, it was difficult to reuse and unpleasant during the operation, which limited its industrial application. Various solid catalysts, including ZnO-NPs/NBu4Cl nanoparticles (Iannone et al., 2017),23 CeO2 (Taguchi et al., 2016),24 CaO(SrO,BaO)/SBA-15 (Zhao et al., 2017),25 and CaO–CeO2/SBA-15 (Yang et al., 2019),26 have demonstrated the ability to catalyze the reaction. However, these catalysts often required substantial amounts of solvent (THF), leading to cumbersome operations and increased waste generation. The studies have explored the methanolysis of PC using a solid base CaO(SrO,BaO)-modified SBA-15 catalyst at higher temperatures (130 °C) and longer durations (3 h) with a higher ratio of n(CH3OH):n(PC) = 8:1. However only a few works demonstrated the solvent-free protocol of depolymerization of PC under mild conditions. Huang W. et al. reported an efficient Mg/Al layer double hydroxide catalyst for the methanolysis of PC.40 A metal-based catalyst was used for this transformation, which used the higher mass of methanol (m(CH3OH)/m(PC) = 2.5) and also needed a longer reaction time (2 h). Furthermore, Mengshuai Liu et al. explored various ILs as catalysts for metal and solvent-free methanolysis of PC under mild conditions,27 and the catalyst [HDBU][LAc] displayed efficient depolymerization of PC. However, the other catalysts posed challenges such as poor reusability, operational inconveniences, and adduct formation with the monomer, hampering their industrial viability.28–32 These studies lack a detailed exploration of the PC depolymerization pathway, and the catalysts were employed in a limited scope. Additionally, the catalyst was not utilized for authentic PC waste depolymerization, and a study on product separation methodology was absent.
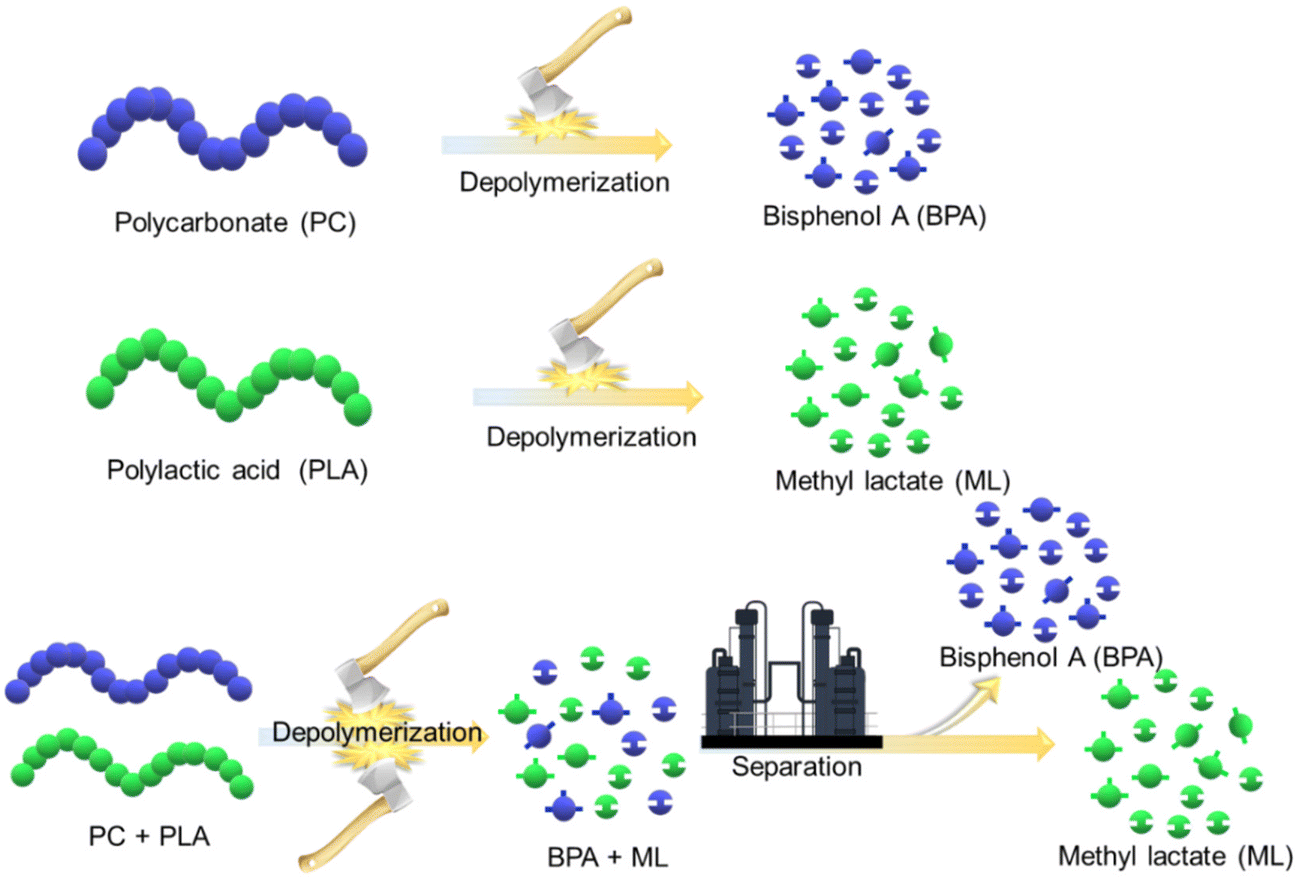
A simple approach involving heterogeneously catalyzed, solvent-free depolymerization of PC and PLA plastic waste is presented to address the aforementioned challenges. This study showcases significant advancements in user-friendly catalyst synthesis, operational ease, and environmental compatibility. In this work, basic ionic liquids-functionalized SBA-15 were prepared and employed for depolymerizing PC and PLA waste, yielding their respective monomers, such as BPA and ML. The catalysts were systematically evaluated, and the impact of the reaction parameters on their efficacy was thoroughly investigated. The evolution of the reactions and the depolymerization pathways for PC and PLA were elucidated through progressive 1H NMR analysis at varying time intervals. Subsequently, the practical utility of the process was explored using authentic sources of PC and PLA waste, thereby addressing real-world plastic waste challenges. Furthermore, a unified approach for depolymerizing mixed PC and PLA plastic waste and separating the resultant products was proposed to deal with real industrial scenarios involving diverse plastic compositions (Scheme 1). A plausible mechanistic insight into the methanolysis of PC and PLA, catalyzed by SBA-15-Pr-MIM-OH, was established through FT-IR, 1H NMR, and TGA–DSC analysis. These highly efficient and environmentally friendly solvent-free heterogeneous catalytic processes exhibit substantial potential for recycling PC and PLA waste, contributing to a diminished environmental footprint.
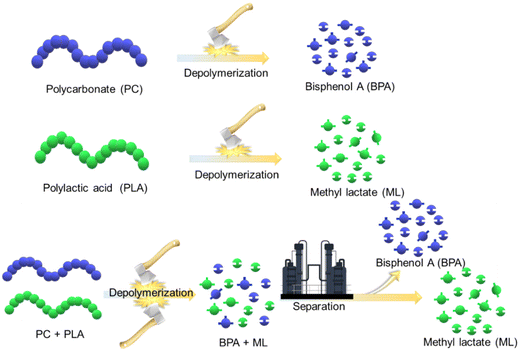 |
| | Scheme 1 Strategies adopted for the depolymerization of single and mixed plastic waste. | |
2. Results and discussion
2.1 Catalysts characterization
SBA-15 functionalized ionic liquids were prepared and used for PC and PLA plastic waste depolymerization reactions (Scheme 2).33 Comprehensive characterization techniques were applied to systematically examine the inherent structural properties of SBA-15 and its functionalized basic ionic liquid catalysts.
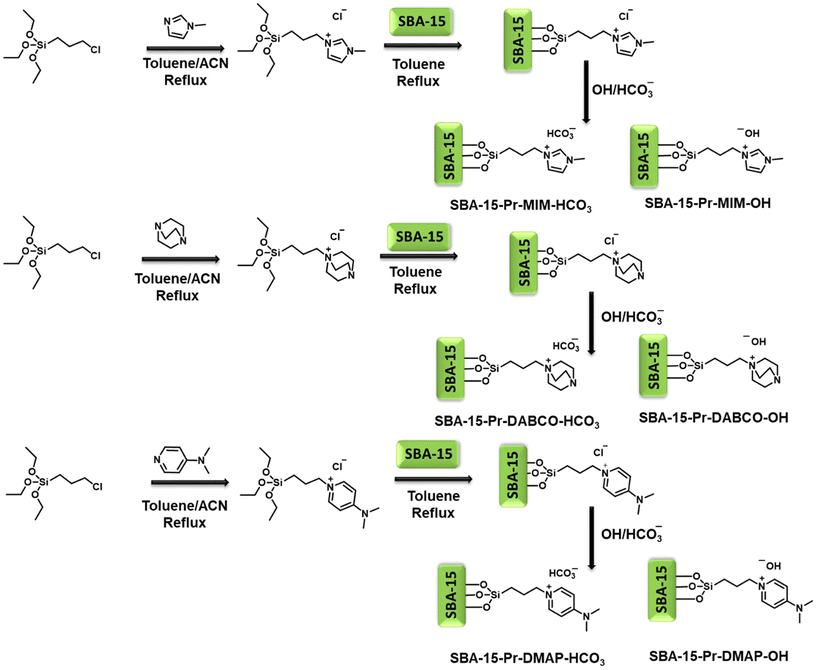 |
| | Scheme 2 Schematic representation of the synthetic process for synthesizing basic ionic-liquid-functionalized SBA-15 catalysts. | |
The X-ray diffraction (XRD) patterns of all synthesized catalysts are displayed in Fig. S1 and S2.† Fig. S1† shows that the SBA-15 and functionalized SBA-15 exhibit a low angle powder XRD diffraction peak at 1.0° (2Θ), indicating a d100 spacing of 8.8 nm that corresponds to the SBA-15 hexagonal structure.34 The basic ionic liquid-functionalized SBA-15 showed a similar low-angle XRD pattern to that of the parent SBA-15 with a marginal shift towards a lower 2Θ value, indicating that the incorporation of organic groups did not disturb the low-range ordering of the SBA-15 material. Furthermore, the wide angle PXRD pattern of the synthesized catalyst was recorded in the 2Θ range of 5–80° (Fig. S2†). The diffraction peak at the 2Θ range of 20–30° corresponds to the amorphous mesoporous silica material. The distinctive peaks observed in the wide-angle ranges for the SBA-15 functionalized with basic ionic liquids closely resembled those of the parent SBA-15, with no additional peaks detected. The absence of any diffraction peak in the wide range XRD pattern indicates the persistence of a stable amorphous mesoporous structure in SBA-15 after the functionalization. The surface area and pore size distribution of unmodified SBA-15 and its ionic liquid-functionalized catalysts were determined through N2 sorption isotherm analyses employing the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively, and the obtained results are summarized in Table 1 and Fig. S3.† Fig. S3† illustrates that SBA-15 and functionalized SBA-15 exhibited type IV isotherm with an H1 hysteresis loop with a capillary condensation occurring in the P/P0 range of 0.65–0.9. After the functionalization, a shift in the capillary condensation to a lower P/P0 range (0.6–0.8) and a marginal change in the isotherm, hysteresis, and adsorbed volume were observed (Fig. S3†). The decrease in textural properties, such as surface area and pore volume of the ionic liquid-functionalized SBA-15, is due to the functionalization of an organic group onto the surface and into the mesopores of SBA-15 (Table 1).
Table 1 Physicochemical properties of SBA-15 and ionic liquid-functionalized SBA-15
| S. no. |
Catalyst |
S
BET
(m2 g−1) |
V
total
(cm3 g−1) |
Avg. pore sizec (nm) |
Amount of functionalized organic moietyd (mmol g−1) |
Basicitye (mmol g−1) |
|
Calculated by the BET equation.
Using the BJH method.
Using the BJH method.
Using TGA analysis.
Using the acid–base titration method.
Recycled catalyst.
Regenerated catalyst.
|
| 1 |
SBA-15 |
545 |
0.92 |
7.6 |
— |
— |
| 2 |
SBA-15-Pr-MIM-Cl |
— |
— |
— |
1.25 |
0.52 |
| 3 |
SBA-15-Pr-MIM-OH |
322 |
0.70 |
5.8 |
1.21 |
0.98 |
| 4 |
SBA-15-Pr-DMAP-OH |
362 |
0.74 |
5.9 |
1.12 |
0.84 |
| 5 |
SBA-15-Pr-DABCO-OH |
316 |
0.68 |
5.5 |
1.19 |
0.86 |
| 6 |
SBA-15-Pr-MIM-OHf |
301 |
0.68 |
5.6 |
— |
0.82 |
| 7 |
SBA-15-Pr-MIM-OHg |
— |
— |
— |
— |
0.96 |
FT-IR was employed to characterize the organic functional groups in the catalytic materials. The FT-IR spectra of SBA-15 exhibited distinct peaks, including a narrow band at 1640 cm−1 and a broad band at 3460 cm−1, which correspond to the bending and stretching vibrations of water or hydroxyl groups located on the surface of SBA-15, respectively (Fig. S4†). Additionally, asymmetric stretching modes of Si–O–Si were identified in the broadband range from 1250 cm−1 to 1000 cm−1, whereas the bending mode and symmetric stretching mode of Si–O–Si were observed at 793 cm−1 (Fig. S4†).35 The stretching vibration of –C–N overlapped with the asymmetric Si–O–Si vibrations.33 When functionalized with organic groups such as SBA-15-Pr-MIM-OH, SBA-15-Pr-DMAP-OH, and SBA-15-Pr-DABCO-OH, these materials displayed peaks consistent with the parent SBA-15. A peak at 1497 cm−1 is attributed to –CH2 bending vibrations, while peaks at 1645 cm−1 and 1655 cm−1 are associated with the –C–C– stretching vibration of imidazole and pyridine rings in SBA-15-Pr-MIM-OH and SBA-15-Pr-DMAP-OH, respectively.33,36 The –C–N stretching vibration mode specific to the SBA-15 functionalized catalysts is associated with a characteristic peak at 1566 cm−1 (Fig. S4†).37
The morphological study of the SBA-15 and SBA-15-Pr-MIM-OH catalysts was conducted with electron microscopy (Fig. S5†). The SEM images of SBA-15 display an aggregated morphology composed of crystals with uniform sizes, and the functionalization with ionic liquid (–Pr-MIM-OH) did not influence the surface morphologies of the parent SBA-15. Additionally, the ‘N’ presence was confirmed by the energy dispersive X-ray (EDAX) and elemental mapping (Fig. S5d–i†). The elemental mapping images of SBA-15-Pr-MIM-OH were recorded at a scale of 5 μm. The elemental mapping images (Fig. S5g–i†) reveal abundant ‘Si’ and ‘O’ atoms within the catalyst framework, with a uniform distribution of ‘N’ throughout the mesoporous silica structure. TEM images of SBA-15 and functionalized SBA-15 (Fig. S4j–l†) reveal the high-order porous structure of the material. The estimated pore size, derived from the intensity profiles of TEM images of SBA-15 and SBA-15-Pr-MIM-OH, was 7.5 nm and 5.9 nm, respectively, suggesting the successful functionalization of the organic moiety. These results are consistent with the results obtained from the N2-sorption study.
Thermogravimetric analysis (TGA) was conducted to assess the thermal stability of SBA-15 functionalized with ionic liquids (ILs) and to quantify the amount of functionalized organic moiety in the SBA-15 material. The thermograms of the functionally modified specimens reveal distinct stages of mass loss, each corresponding to the desorption of water molecules and the thermal degradation of organic constituents present in the sample (Fig. S6†). Utilizing the data obtained from the mass loss profiles, it becomes feasible to compute the quantity of functionalized organic ILs (Table 1). The basicity of catalysts was calculated by performing an acid–base titration method (a detailed procedure is mentioned in ESI†),38 and the corresponding results are depicted in Table 1. Among all catalysts, SBA-15-Pr-MIM-OH was highly basic, with a basicity of 0.98 mmol g−1, which is attributed to its high aromatic character and the conjugation of imidazolium nitrogen atoms. The SBA-15-Pr-MIM-OH shows almost two times higher basicity than SBA-15-Pr-MIM-Cl due to the presence of –MIM and –OH functional groups in the catalyst. The other catalysts exhibited lower basicity than SBA-15-Pr-MIM-OH (Table 1).
2.2 Methanolysis of poly(bisphenol A carbonate)
PC has become the fastest-growing engineering plastic. Its monomer, bisphenol A, leaches out from waste during traditional waste treatment methods, and has diverse effects on human health and the environment.39 Therefore, chemical recycling is particularly important. In this work, the SBA-15 functionalized quaternary ammonium salt-based imidazole, DMAP, and DABCO were synthesized to study the influence of the basic ionic liquids. Cl− was replaced with OH− and HCO3−, and were evaluated for the methanolysis of PC without any external solvent. Reactions were conducted employing commercial PC granules (Mw ∼ 58000) and various sources of PC plastic waste (derived from municipal waste) to systematically evaluate the catalytic depolymerization under optimized conditions (Fig. S8–S12†). Methanol was used as a transesterification alcohol, and additional solvents such as THF, 2-MTHF, and DCM were avoided. Moderately polar solvents like THF and 2-MTHF have shown favorable effects on PC alcoholysis, potentially augmenting the rate of PC depolymerization.24–26 In contrast, some recent research has indicated that insufficient THF could restrict the reaction occurring at the PC surface, leading to reduced activity and product selectivity.40 Furthermore, adopting THF as a solvent can lead to intricate procedural steps, resource wastage, and challenges in separation. The utilization of chlorinated solvents was avoided to enhance the environmentally friendly aspects of the process.41 In this work, PC (1.0 g) and catalysts (0.03 g) were mixed with methanol, and the reactions were carried out at an optimized temperature. The PC conversion and yield of BPA were calculated using eqn (S1)–(S4) (ESI†). The products obtained by methanolysis of PC were analyzed by 1H NMR spectroscopy (CDCl3). A virtual concentration of the BPA functional group was also measured as a relative percentage. In Fig. 1, the aromatic and methyl protons of BPA were considered to be in a different environment: bisphenol A (BPA) (7.0–6.6 ppm – aromatic protons) & (1.48–1.37 ppm – methyl protons), monocarbonated bisphenol A (oligomer) (7.2–6.9 ppm – aromatic protons) & (1.58–1.48 ppm – methyl protons), and polycarbonate (PC) (7.4–7.2 ppm – aromatic protons) & (1.65–1.58 ppm – methyl protons).42
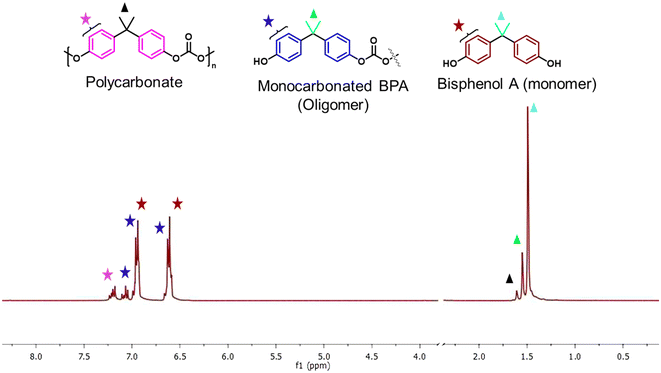 |
| | Fig. 1
1H NMR assignment for PC depolymerization products, such as polycarbonate (PC), monocarbonated BPA (oligomer), and BPA (monomer). | |
The results corresponding to PC methanolysis over all synthesized catalysts are depicted in Table 2. The catalytic activity exhibited by SBA-15-Pr-MIM-Cl highlights the significance of quaternary ammonium salts for this transformation. Without a catalyst, the reaction did not occur. The support SBA-15 was also inactive for this transformation due to the absence of active sites, indicating the critical role of the functionalized quaternary ammonium salt. Although the activity of SBA-15-Pr-MIM-Cl (Table 2, entry 3) was significantly higher than that of SBA-15, it was far lower than that required for its commercial application. [Bmim][Cl] was also subjected to identical reaction conditions for the comparison. The [Bmim][Cl] catalyst demonstrated the capacity to achieve full depolymerization of PC. However, the separation of the targeted product posed challenges due to complex separation processes, and the recycling of the catalyst proved unachievable due to the emergence of BPA-[Bmim][Cl] adduct formation within the system.43 It may be noted that the low efficiency of SBA-15-Pr-MIM-Cl towards PC methanolysis could be due to poor basicity, causing lower efficiency in the adsorption and activation of methanol. Therefore, to further enhance the catalytic efficiency, the chlorine ion (Cl−) was replaced with OH− and HCO3− from SBA-15-Pr-MIM-Cl to prepare SBA-15 supported basic ILs. The order of catalytic reactivity towards PC methanolysis follows the following trend: OH− > HCO3− > Cl−. Results demonstrate that SBA-15-Pr-MIM-OH exhibited 99.9% conversion of PC and ∼99% selectivity of BPA (Table 2, entry 4). The excellent activity of SBA-15-Pr-MIM-OH agreed with the basicity and textural properties of the material. The external surface area and silanol groups on the surface of SBA-15 played a pivotal role in anchoring highly dispersed surface-functionalized –Pr-MIM-OH groups that exhibited superior basicity of the SBA-15-Pr-MIM-OH.33 The higher basicity of the catalyst facilitates the attraction of the methanol proton, resulting in the formation of an oxyanion intermediate. This intermediate undergoes a nucleophilic attack on the ester groups in PC, facilitating the reaction and leading to the formation of BPA. Thus, FT-IR and 1H NMR studies were performed to demonstrate the interaction between methanol and SBA-15-Pr-MIM-OH (Fig. S13 and S14†). With an increase in the catalyst amount from 0.01 g to 0.03 g, the –OH vibration band became broader and shifted from 3326 cm−1 to 3225 cm−1 (Fig. S13†). The red shifts demonstrate the strong adsorption and activation of methanol by forming an oxyanion with the catalyst SBA-15-Pr-MIM-OH.27 Moreover, the interaction between the methanol and SBA-15-Pr-MIM-OH was also proved by 1H NMR spectra (Fig. S14†). Fig. S14† illustrates a downfield shift in the methanol –OH proton signal in the presence of SBA-15-Pr-MIM-OH. The shift signifies the activation of methanol by SBA-15-Pr-MIM-OH, and the findings align with the FT-IR and experimental results.
Table 2 Catalytic methanolysis of polycarbonate over different catalysts
| S. no. |
Catalyst |
m(Cat.):m(PC) |
m(CH3OH):m(PC) |
C
PC
(%) |
S
BPA
(%) |
S
others (%) |
Y
BPA
(%) |
| The effect of catalysts on the catalytic depolymerization of PC. Reaction conditions: PC (3.93 mmol = 1 g), methanol (46 mmol = 1.5 g), catalyst (0.03 g), 120 °C, and 1 h. Calculated using eqn (S1).† Calculated using eqn (S2).† Calculated using eqn (S4).† |
| 1 |
None |
0.03:1 |
1.5:1 |
0 |
— |
— |
— |
| 2 |
SBA-15 |
0.03:1 |
1.5:1 |
0 |
— |
— |
— |
| 3 |
SBA-15-Pr-MIM-Cl |
0.03:1 |
1.5:1 |
79.3 |
63.8 |
36.0 |
55.3 |
| 4 |
SBA-15-Pr-MIM-OH |
0.03:1 |
1.5:1 |
99.9 |
99.2 |
— |
99.0 |
| 5 |
SBA-15-Pr-MIM-HCO3 |
0.03:1 |
1.5:1 |
94.2 |
92.3 |
7.5 |
87.0 |
| 6 |
SBA-15-Pr-DMAP-OH |
0.03:1 |
1.5:1 |
90.6 |
96.8 |
3.1 |
87.7 |
| 7 |
SBA-15-Pr-DABCO-OH |
0.03:1 |
1.5:1 |
88.7 |
96.4 |
3.5 |
85.5 |
| 8 |
SBA-15-Pr-MIM-OH |
0.01:1 |
1.5:1 |
72.4 |
83.9 |
15.8 |
60.7 |
| 9 |
SBA-15-Pr-MIM-OH |
0.05:1 |
1.5:1 |
99 |
98.5 |
1.5 |
99.0 |
| 10 |
SBA-15-Pr-MIM-OH |
0.03:1 |
0.75:1 |
59.2 |
80.6 |
19.3 |
47.7 |
| 11 |
SBA-15-Pr-MIM-OH |
0.03:1 |
2.0:1 |
99.9 |
98 |
2.9 |
98.0 |
| 12 |
SBA-15-Pr-MIM-OH |
0.03:1 |
3.0:1 |
99.9 |
98.5 |
1.5 |
98.5 |
2.3 Effect of the reaction parameters
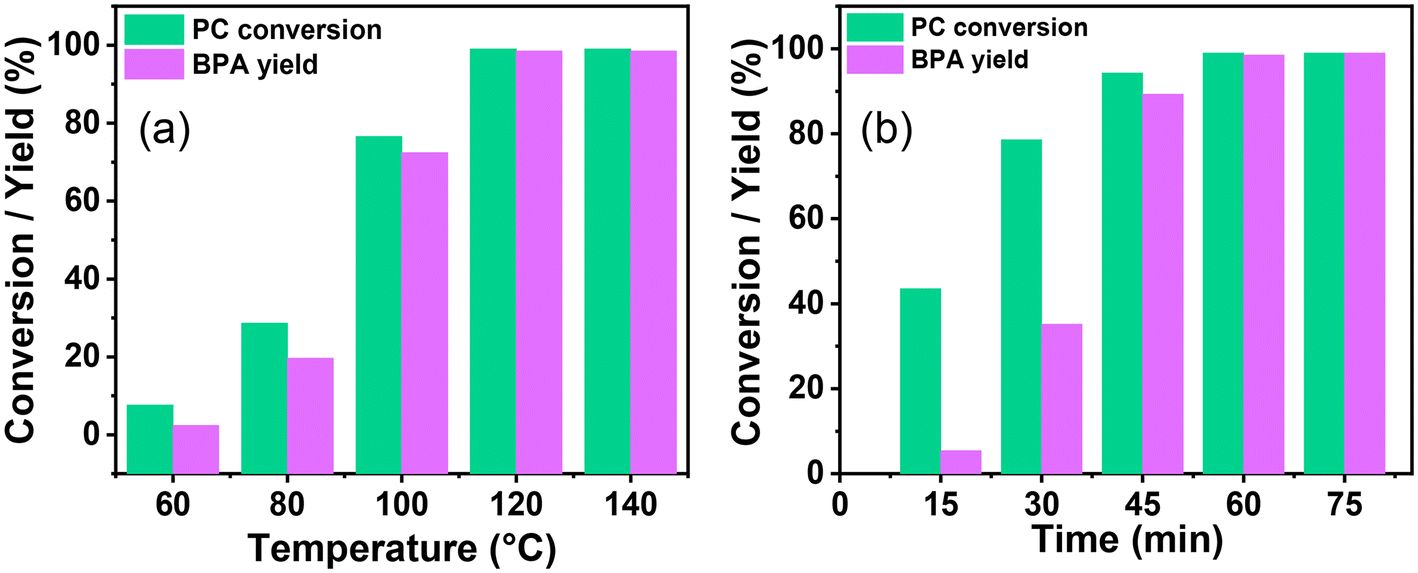
The highly active catalyst SBA-15-Pr-MIM-OH was chosen to study the influence of reaction parameters such as temperature, catalyst amount, methanol concentration, and time. Several studies reveal that the dissolution or swelling of PC is a rate-determining step for PC depolymerization, and temperature plays a crucial role.44Fig. 2 displays the influence of temperature on the catalytic activity. The temperature was increased from 60 °C to 140 °C at a catalyst-to-substrate mass ratio of 0.03:1, CH3OH:PC mass ratio (1.5:1), for 1 h. At a temperature of 60 °C, only 7.6% conversion of PC and 31.5% selectivity of BPA was observed (Fig. 2 and Fig. S7†), indicating that the dissolution of rigid PC was not possible at lower temperatures. After increasing the temperature from 60 °C to 120 °C, the PC conversion increased from 7.6% to 99.9%, the BPA selectivity increased from 31.5% to 99%, and the oligomer concentration decreased from 68.4% to <1.0%. Upon further increasing the temperature from 120 °C to 140 °C, no obvious changes were detected in the catalytic activity. Thus, 120 °C was selected for the next optimization.
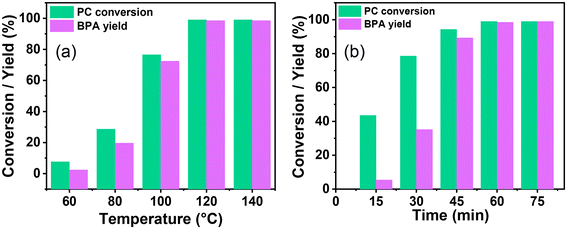 |
| | Fig. 2 The effect of the reaction condition on the catalytic depolymerization of PC. (a) Reaction temperature, reaction conditions: PC (3.93 mmol = 1 g), methanol (46 mmol = 1.5 g), catalyst (0.03 g), and 1 h. (b) Reaction time, reaction condition: PC (3.93 mmol = 1 g), methanol (46 mmol = 1.5 g), catalyst (0.03 g), and temperature 120 °C. A comparative analysis of data, including the PC conversion and BPA selectivity in relation to variations in temperature and time, is given in Fig. S7.† | |
The influence of the methanol concentration and the formation of a corresponding BPA monomer was examined by taking the CH3OH:PC mass ratio from 0.75:1 to 2.0:1. When the mass ratio of CH3OH:PC was 0.75:1, a moderate PC conversion (59.2%) and BPA yield (47.7%) were obtained over SBA-15-Pr-MIM-OH (Table 2). The lower catalytic activity could be obtained due to a lower concentration of CH3OH, leading to the formation of partial transesterification products such as oligomers. It was observed that the depolymerization of PC and corresponding BPA yield increased to >99% PC conversion and ∼99% BPA yield when the CH3OH:PC mass ratio increased from 0.75:1 to 1.5:1. However, the complete PC depolymerization was obtained when the CH3OH:PC mass ratio was 1:1, while the BPA yield was low (84.6%). A nearly complete PC depolymerization (99.9%) and the highest BPA yield (99%) were obtained at the mass ratio of 1.5:1 (CH3OH:PC). Furthermore, no further enhancement for the BPA yield was observed when the mass ratio of CH3OH:PC was increased from 1.5:1 to 2.0:1. To achieve a satisfactory BPA yield, the optimized molar ratio of 1.5:1 for CH3OH:PC was chosen for the depolymerization of PC.
The effect of catalyst amount on PC depolymerization and BPA yield was explored to get higher catalytic activity and to set the process economy. At a lower catalyst amount, the catalyst:PC mass ratio was 0.01:1. Moderate PC depolymerization (72.4%) could be afforded under these optimized reaction parameters. When the catalyst:PC mass ratio was increased from 0.01:1 to 0.03:1, the PC was completely converted and a 99% BPA yield was observed (Table 2). Higher catalytic activity could be obtained due to the faster swelling of PC with increasing catalyst amounts and the resulting facile PC activation with catalyst. When the catalyst:PC mass ratio was further increased from 0.03:1 to 0.05:1, no obvious change was observed for the BPA yield. Thus, a catalyst:PC mass ratio of 0.03:1 was selected for further optimization.
The depolymerization route of PC and its progress with time was examined, and are displayed in Fig. 2b and Fig. S7b,† respectively. The PC conversion and corresponding BPA yield were examined with time from 15 min to 75 min. At 15 min, 43.5% PC conversion and 12.4% BPA selectivity were observed. 1H NMR was conducted with time to investigate the progress of PC depolymerization into its oligomer and monomer (Fig. 3). The higher selectivity of the oligomer and poor selectivity of BPA at lower time indicate that the first depolymerization of PC into oligomers is more facile over SBA-15-Pr-MIM-OH. The further conversion of the oligomer into monomer BPA could be the rate-determining step under the selected reaction conditions. When increasing the reaction time from 15 min to 60 min, enhancement for PC conversion and BPA yield was observed, indicating that the depolymerization of PC progressed significantly over time. The PC was completely converted to give a 99% BPA yield within 60 min (Fig. 2b and 3). Upon further increasing the time, no significant changes were observed in the BPA selectivity. Thus, 60 min was selected for the higher catalytic activity.
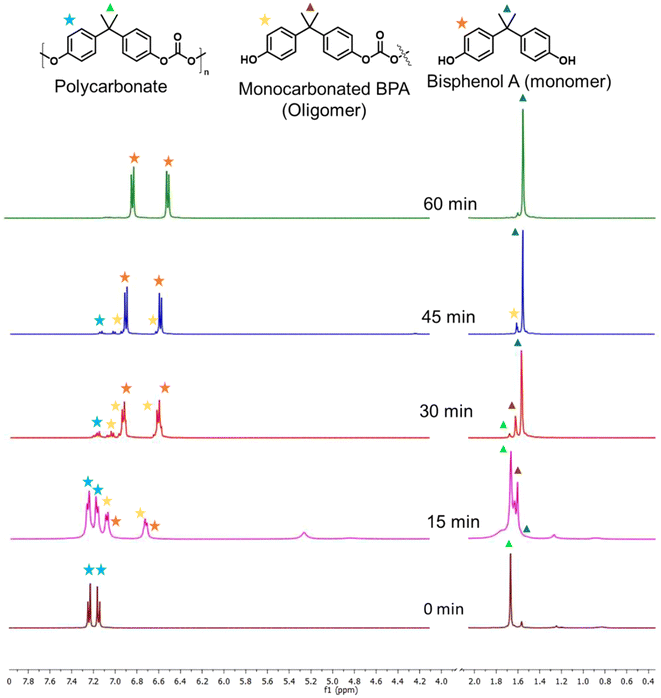 |
| | Fig. 3
1H NMR spectra of the PC depolymerisation reaction collected with time to reveal the progress of the reaction. | |
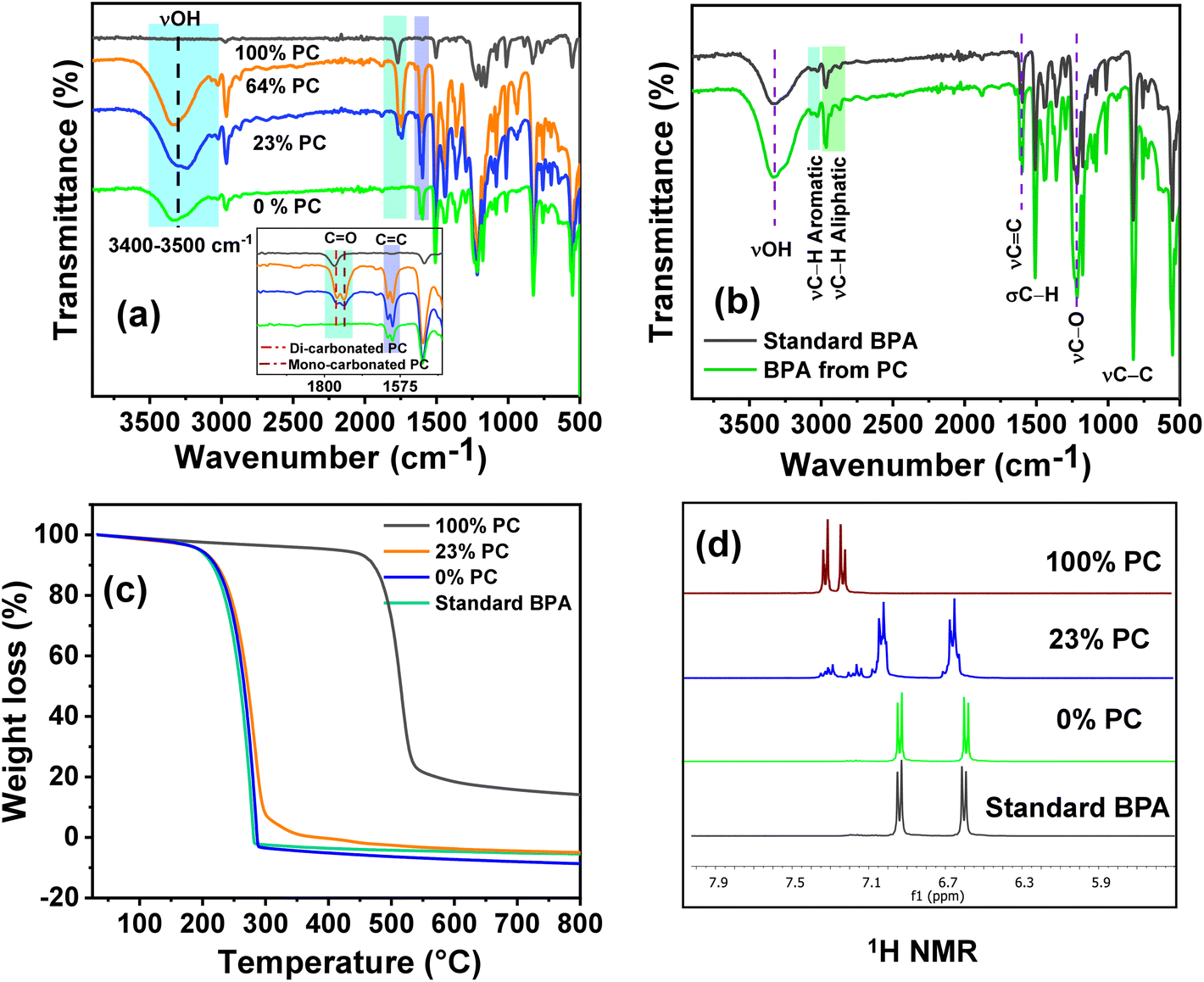
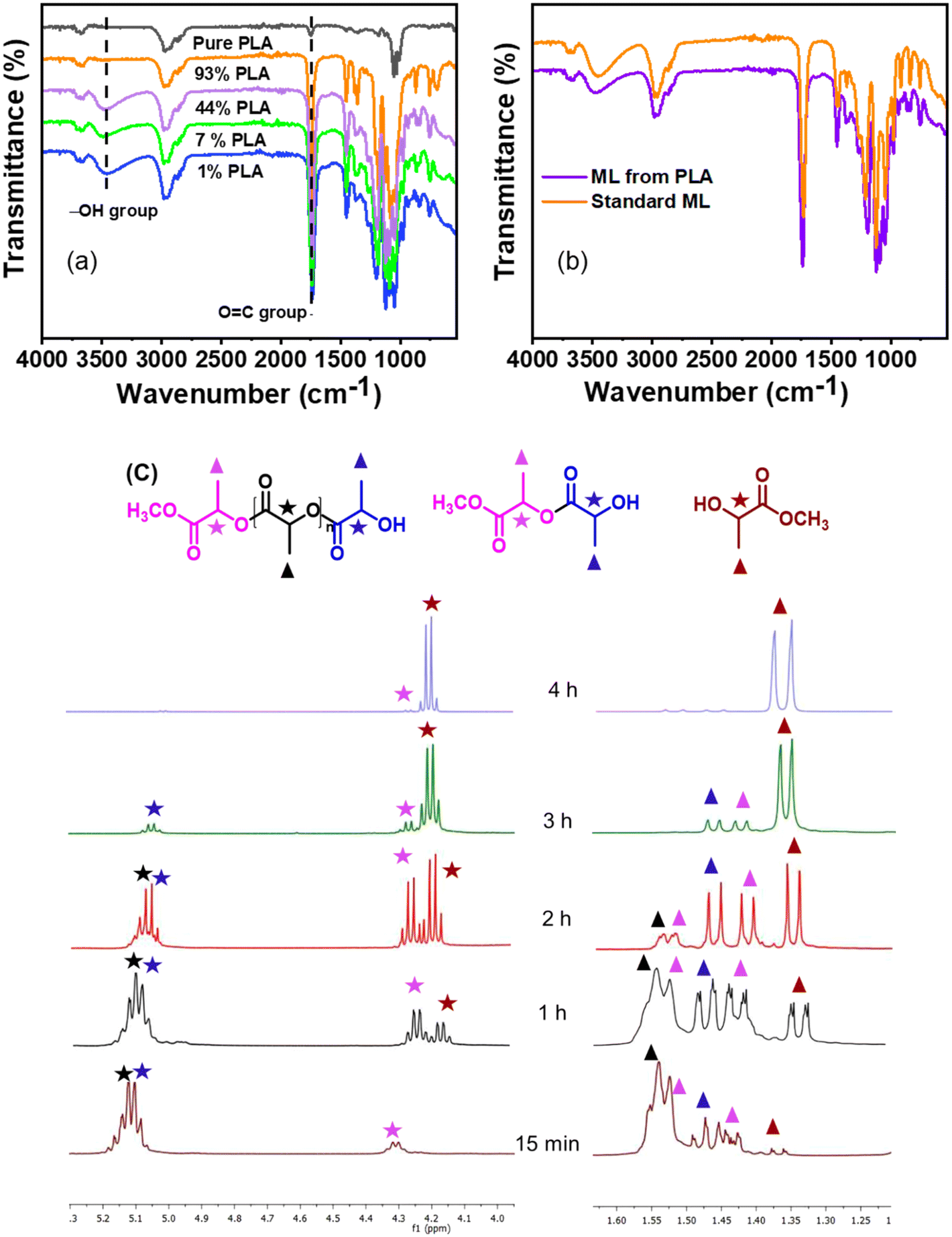
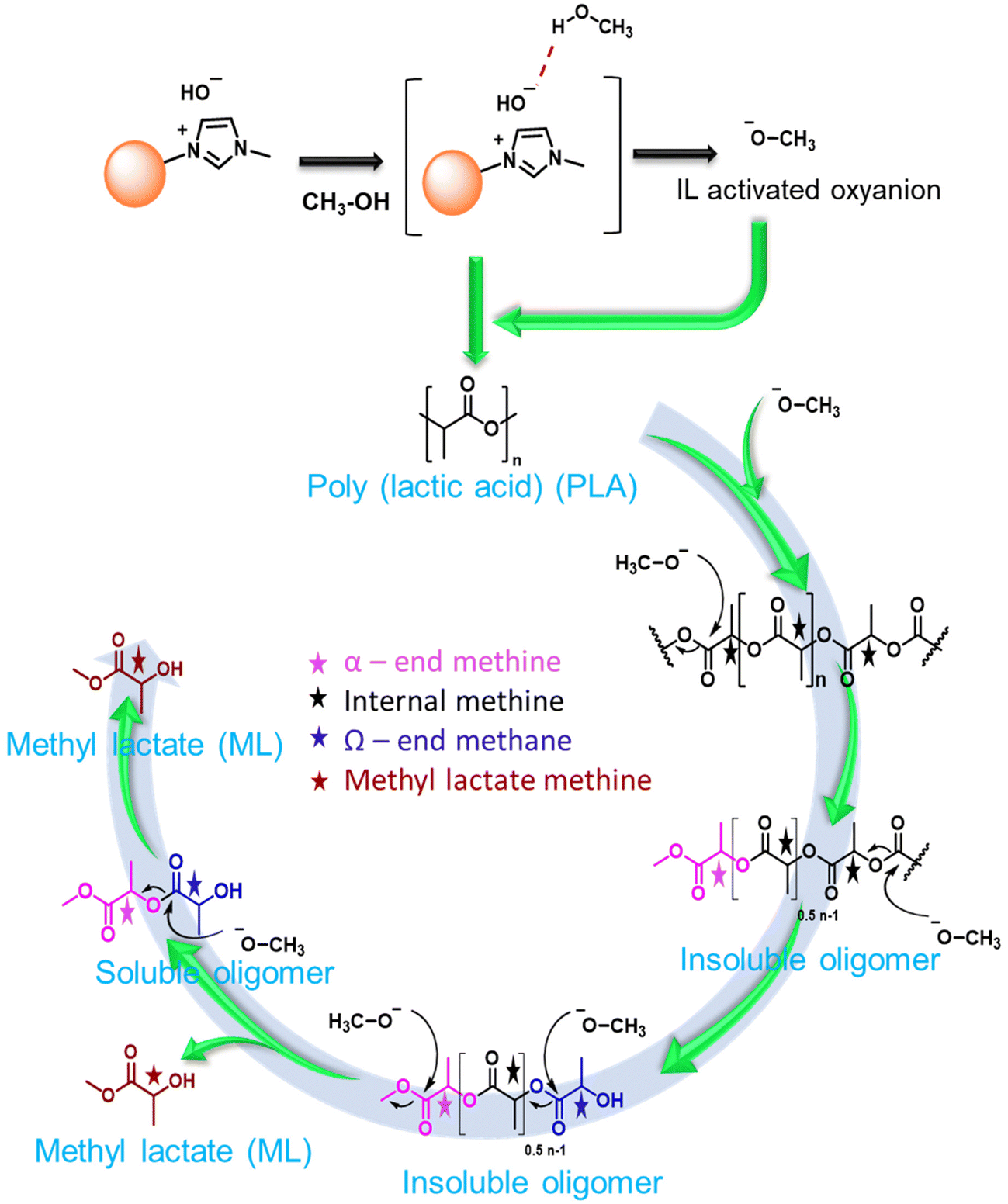
To understand the reaction mechanism for the depolymerization of PC, different structure characterizations such as 1H NMR, FT-IR, TGA, and DSC analysis were performed at different PC conversion and product yields (Fig. 4). At the lower PC conversion, the residue was almost identical to pure PC, which indicates the minimal presence of oligomers and monomers in the residual material. FT-IR spectra show that the broad peak at 3400–3500 cm−1 corresponding to the hydroxyl group gradually appeared and increased with increasing PC conversion. Similarly, the sharp peak at 1760 cm−1 corresponds to carbonyl, increasing and slightly shifting towards a lower wavenumber. The red-shift of the carbonyl peak shows the doublet, which indicates that the PC is partially depolymerized to form a mono-carbonated PC as an oligomer (Fig. 4a).27,45 The oligomers formed during the reaction are attributed to the fragmentation of partially soluble PC through the nucleophilic attack of methanol in the presence of a base catalyst. However, after the complete conversion of PC, the carbonyl peak completely disappeared, and the pure form of BPA was observed (Fig. 4a). The FT-IR spectra of commercial BPA and the BPA produced from PC are displayed in Fig. 4b. Furthermore, 1H NMR was conducted to understand the progress of the reaction and path of oligomer formation. Fig. 3 and 4d show that PC was partially depolymerized at the initial stage, and no BPA was detected. With time, oligomers were converted into BPA monomers. The DSC profile of commercial BPA and the BPA obtained from PC are compared, which shows a sharp peak at ∼115 °C corresponding to the melting temperature, confirming the absence of soluble oligomer in the product (Fig. S10†). The slight increase in decomposition temperature in the TGA profile is due to the presence of some soluble oligomer. After completion of the reaction, no soluble oligomers were detected, and pure BPA was observed, which was confirmed by FTIR, 1H-NMR, and TGA analysis. In accordance with the obtained results and existing literature, a plausible mechanism catalyzed by SBA-15-Pr-MIM-OH is elucidated (Scheme 3). Initially, PC undergoes activation through dissolution or swelling within the catalytic process. Simultaneously, electrostatic interactions occur between the anions of SBA-15-Pr-MIM-OH and the methanol protons, adhering to the principles of conjugated acid–base theory. Additionally, hydrogen bonds are formed between the O atom of SBA-15-Pr-MIM-OH and the –OH group in methanol, leading to the activation of methanol and the generation of oxyanion intermediates. Such activation was not observed in the absence of catalysts, which was confirmed through FT-IR and 1H NMR analyses (Fig. S13 and S14†). Subsequently, the activated methanol, in the form of oxyanion intermediates, undergoes nucleophilic attack on the ester groups in PC, forming insoluble oligomers and smaller soluble oligomeric species. These oligomers react with the oxyanion, yielding the selective monomer BPA.
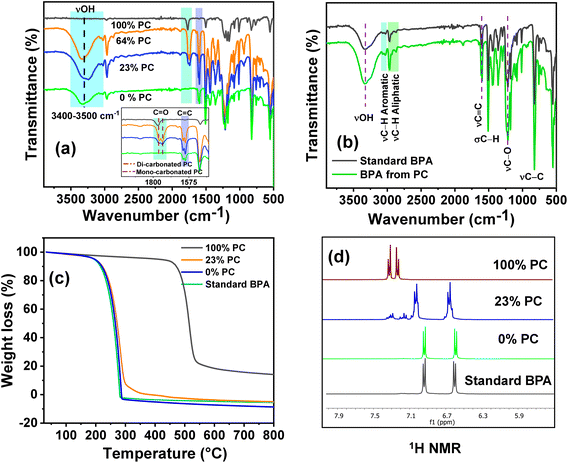 |
| | Fig. 4 Investigating and understanding of the PC depolymerization pathway through various techniques. (a) FT-IR spectra at different PC concentrations and BPA yields; (b) comparative FT-IR spectra of standard BPA and BPA obtained from PC; (c) TGA profiles of PC at different PC conversions; and (d) 1H NMR spectra of the aromatic region of PC at different PC conversions. | |
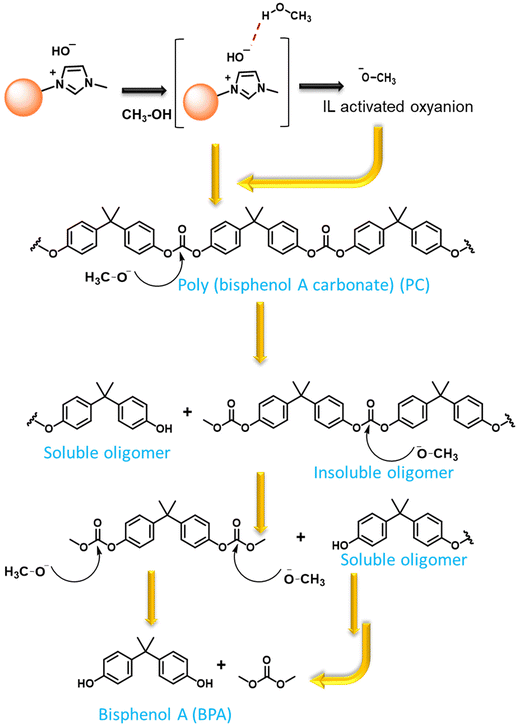 |
| | Scheme 3 Plausible depolymerisation pathways for the conversion of PC to BPA. | |
2.4 Methanolysis of poly(lactic acid)
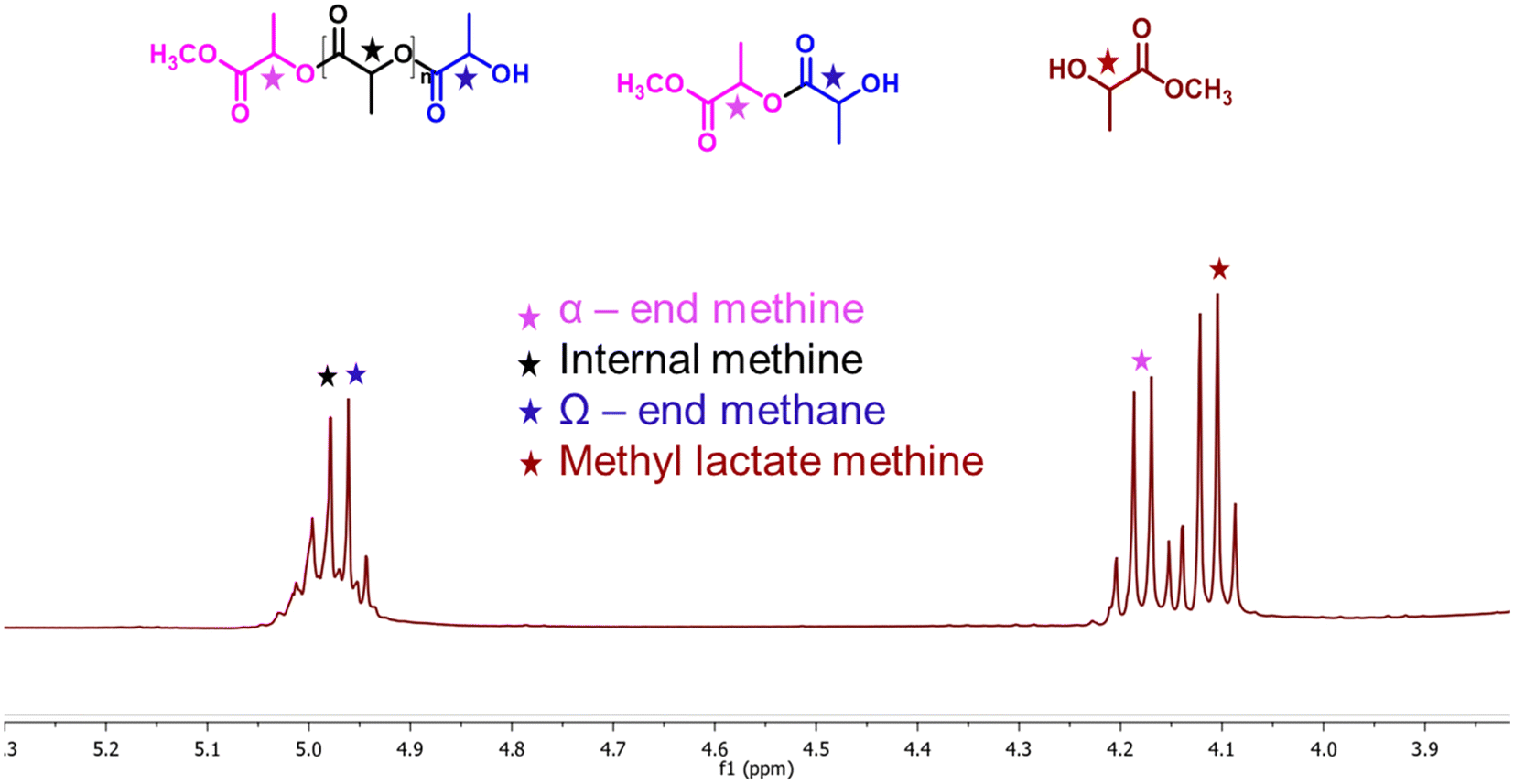
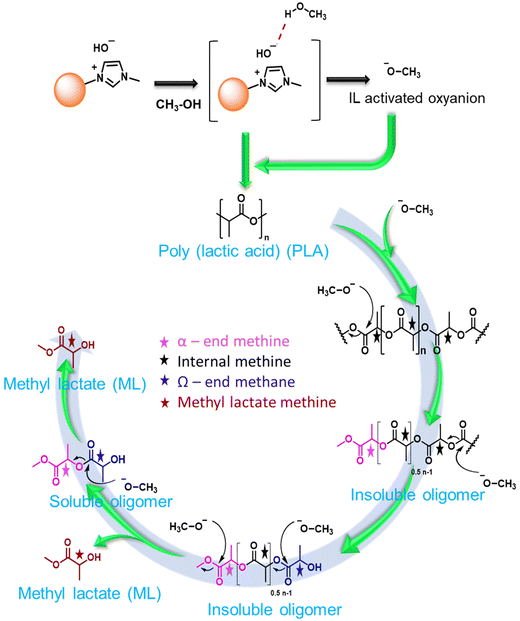
Biodegradable plastic such as PLA is comparatively more expensive, and its inherent biodegradation rate is relatively slow. Opting for upcycling rather than conventional disposal methods would be the more favorable approach for enhancing sustainability.46 The depolymerization of PLA is commonly catalyzed by Lewis acid and basic conditions. Herein, a synthesized solid ionic liquid catalyst is employed for the depolymerization of PLA (LX-175, 3.0 mm × 2.5 mm pallets, Mw ∼ 163000, composition L-lactide = 95.7%, D-lactide = 4.1%, meso-lactide = 0.2%) using methanol as a transesterification solvent. As discussed in the polycarbonate section, solvents such as DCM, THF, and MTHF were avoided due to their limitations. All catalysts synthesized in this study were evaluated for catalytic depolymerization using PLA (1 g), catalyst (0.03 g), methanol (2.0 g), and 120 °C for 4.0 h, and summarized in Table 3. In the absence of a catalyst, PLA depolymerization did not occur. Moreover, SBA-15 showed no activity for this transformation due to the absence of any active sites. In contrast, the remarkable PLA depolymerization activity was noted over the SBA-15 functionalized quaternary ammonium salt based on imidazole, DMAP, and DABCO. The yield to the formation of methyl lactate (ML) followed the order SBA-15-Pr-MIM-Cl > SBA-15-Pr-DABCO-Cl > SBA-Pr-DMAP-Cl (Table 3). The catalysts SBA-15-Pr-MIM-Cl exhibited 63.4% PLA conversion and 42.6% ML yield, which was not satisfactory from a commercial point of view (Table 3, entry 3). Therefore, the Cl−-exchanged OH− and HCO3− were evaluated for the same reaction to get a higher ML yield. The results demonstrated that SBA-15-Pr-MIM-OH exhibited complete PLA depolymerization and >98% yield of ML, and it followed the activity trend: OH− > HCO3− > Cl− for high ML yield. The higher basicity of SBA-15-Pr-MIM-OH could activate methanol, and facilitate the rate of transesterification and depolymerization of PLA to the corresponding ML. The enantiomeric purity of the obtained ML was assessed through 31P NMR spectroscopy,47 and the observed spectrum indicates that ∼88% of ML was in the L-form (Fig. S15†). At the lower conversion of PLA, the progress of depolymerization was examined by 1H NMR analysis. The quantification of methine functional groups was determined by assessing their relative percentages of NMR peak. The methine protons were found to be within three distinct chemical environments; namely, methyl lactate (ML), chain end methine, and internal methine, with chemical shifts in the range of 4.05–4.15 ppm, 4.15–4.24 ppm/4.93–4.96 ppm, and 4.98–5.4 ppm, respectively (Fig. 5).48,49
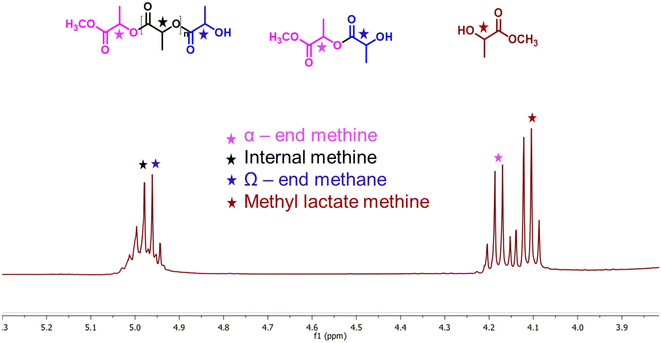 |
| | Fig. 5
1H NMR assignment of the PLA depolymerisation products (methyl lactate, chain-end, and internal methine groups). | |
Table 3 Catalytic methanolysis of poly(lactic acid) over different catalysts
| S. no. |
Catalyst |
m(Catal.):m(PLA) |
m(CH3OH):m(PLA) |
C
PLA
(%) |
S
ML
(%) |
S
others (%) |
Y
ML
(%) |
| The effect of catalysts on the catalytic depolymerization of PC. Reaction condition: PLA (1.0 g), methanol (2.0 g), catalyst (0.03 g), 120 °C, and 4 h. Calculated using eqn (S1).† Calculated using eqn (S4).† Calculated using eqn (S3).† |
| 1 |
None |
0.03:1 |
2.0:1 |
— |
— |
— |
— |
| 2 |
SBA-15 |
0.03:1 |
2.0:1 |
— |
— |
— |
— |
| 3 |
SBA-15-Pr-MIM-Cl |
0.03:1 |
2.0:1 |
63.4 |
67.2 |
32.8 |
42.6 |
| 4 |
SBA-15-Pr-MIM-OH |
0.03:1 |
2.0:1 |
99.9 |
99.0 |
0.9 |
99.0 |
| 5 |
SBA-15-Pr-MIM-HCO3 |
0.03:1 |
2.0:1 |
96.5 |
95.7 |
4.3 |
92.4 |
| 6 |
SBA-15-Pr-DMAP-OH |
0.03:1 |
2.0:1 |
90.8 |
95.6 |
4.4 |
86.8 |
| 7 |
SBA-15-Pr-DABCO-OH |
0.03:1 |
2.0:1 |
93.1 |
95.0 |
5.0 |
88.5 |
| 8 |
SBA-15-Pr-MIM-OH |
0.01:1 |
2.0:1 |
49.6 |
89.3 |
10.7 |
44.3 |
| 9 |
SBA-15-Pr-MIM-OH |
0.05:1 |
2.0:1 |
99.9 |
99.0 |
1.0 |
99.0 |
| 10 |
SBA-15-Pr-MIM-OH |
0.03:1 |
1.0:1 |
72.6 |
82.0 |
18.0 |
59.6 |
| 11 |
SBA-15-Pr-MIM-OH |
0.03:1 |
1.5:1 |
84.6 |
85.5 |
14.5 |
72.4 |
| 12 |
SBA-15-Pr-MIM-OH |
0.03:1 |
3.0:1 |
98.3 |
96.0 |
4.0 |
94.6 |
| 13 |
SBA-15-Pr-MIM-OH |
0.03:1 |
5.0:1 |
89.3 |
92.0 |
8.0 |
82.0 |
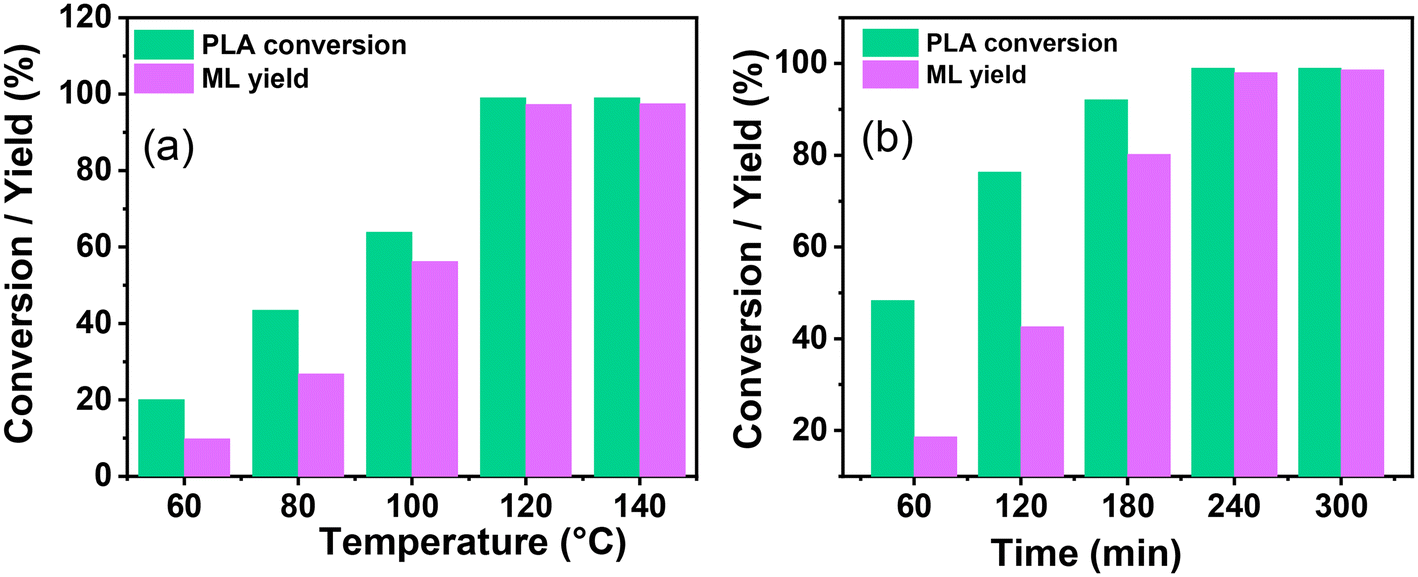
The influence of different reaction parameters such as temperature, CH3OH concentration, catalyst loading, and reaction time was examined to develop a sustainable and economical selective process of PLA depolymerization and ML yield. Fig. 6a depicts the influence of the reaction temperature on the catalytic depolymerization of PLA. The influence of temperature was evaluated between 60–140 °C, and it was observed that the conversion of PLA and ML yield increased with temperature (Fig. 6a). The swelling of PLA was favored at higher temperatures. The complete PLA depolymerization (100%) and the highest ML yield (99%) were attributed at 120 °C, and no further significant changes were observed in catalytic activity. Therefore, considering the practical standpoint of energy conservation, the temperature of 120 °C was selected for further optimization.
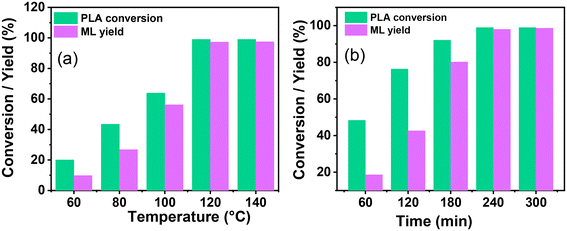 |
| | Fig. 6 The effect of the reaction condition on the catalytic depolymerization of PLA: (a) reaction temperature, reaction condition: PLA (1.0 g), methanol (2.0 g), catalyst (0.03 g), and 4 h. (b) Reaction time, reaction condition: PLA (1.0 g), methanol (2.0 g), catalyst (0.03 g), and temperature 120 °C. Comparative analysis of the data, including the PLA conversion and ML selectivity, in relation to variations in temperature and time is given in Fig. S7c and d.† | |
During the reaction, it was noted that the concentration of CH3OH plays a crucial role in the depolymerization of PC and the selective production of BPA. The mass ratio of CH3OH:PLA was kept at 1.5:1.0, which provided a good PLA depolymerization (84.6%) and ML yield (72.4%) over SBA-15-Pr-MIM-OH at 120 °C for 4.0 h. However, this was not satisfactory (Table 3, entry 11). Thus, the mass ratio of CH3OH:PLA was examined, and a summary of its influence is displayed in Table 3. The results depict that it constantly increased and reached 99% ML yield when the CH3OH:PLA mass ratio was increased to 2.0:1. Surprisingly, the PLA conversion and ML yield were decreased by a further increase in the mass ratio from 2.0:1.0 to 5.0:1.0. This decrease in catalytic activity may be attributed to the decrease in catalytic active sites in the mixture with an increase in methanol dosages and saturation of excess methanol on the catalyst surface. A higher PLA conversion (99.9%) and ML yield (99%) were obtained when the CH3OH:PLA mass ratio was 2.0:1.0. Therefore, the mass ratio 2.0:1.0 was selected as desirable.
The influence of catalyst loading was examined by taking the catalyst:PLA mass ratio between 0.01:1.0 to 0.05:1.0. When the mass ratio increases from 0.01:1.0 to 0.03:1.0, the PLA conversion and ML yield increased from 49.6% to 99.9% and 44.3% to 99%, respectively (Table 3). The results indicate that the higher catalyst amount provided higher basic sites, and this is ascribed to the facile swelling of PLA via activation of methanol by the catalytic system and enhanced catalytic activity. Furthermore, when the catalyst:PLA mass ratio to 0.03:0.05, a similar catalytic activity was observed. Therefore, a catalyst:PLA mass ratio of 0.03:1.0 was chosen for further optimization.
The reaction progress was monitored over time by keeping all parameters constant. With time, PLA depolymerized into its oligomers, and then the oligomers were converted into monomers, ML. At the lower time, the main selectivity of the oligomer and poor selectivity of ML were attributed to PLA first depolymerizing into the Ω chain end and α chain end methine oligomers, which were then further converted into monomers such as ML under the selected reaction conditions. The depolymerization process of PLA exhibited notable progression over time. With an increasing reaction time from 60 min to 240 min, the PLA conversion and ML selectivity was increased from 48.3% to 99% and 38.5% to 99%, respectively (Fig. 6b and 7c). PLA was efficiently depolymerized and selectively converted into ML (99% yield) in 4 h. After extending the reaction time beyond 4 h, there was no significant change observed. In addition, the FT-IR spectra of PLA and ML at different conversions and yields were conducted to investigate the depolymerization route of PLA to ML. Fig. 7 shows that the peak at 3500–3600 cm−1 corresponding to the hydroxyl group is absent in the PLA at 0% conversion, and the peak becomes intense and broad with increasing conversion and ML selectivity. The increase in intensity and the broadening of the –OH peak indicate that PLA is partially depolymerized from the end chain side to form soluble and insoluble oligomers, and then it is slowly converted into their monomer ML.50 Moreover, Fig. 7b shows similar FT-IR spectra of commercial ML and ML produced from PLA, indicating its purity. In addition, 1H NMR spectra were obtained to understand the depolymerization pathway and the nature of the soluble and insoluble oligomers and monomers. Fig. 7c depicts the distinct 1H NMR chemical shifts of the methine protons within each group in DMSO-d6, enabling the monitoring of the relative concentrations of each chemical species through NMR analysis. It indicates that PLA was completely depolymerized in 3.0 h, and extra time was required to convert the chain end methine oligomer into ML. 1H NMR spectra at different reaction times revealed that PLA first swelled in the reaction mixture and depolymerized to the internal methine in the Ω chain end methine intermediate, which is an insoluble oligomer (Fig. 7c). With time, the insoluble Ω chain end methine oligomer was converted into a soluble α chain end methine oligomer. Then, the soluble α chain end methine oligomer was converted into ML through the transesterification process. After completion of the reaction, the pure and soluble α chain end methine oligomer-free ML was obtained, which was confirmed by 1H NMR and FT-IR analysis. These observations conclude that after the completion of the reaction, no soluble oligomers were detected. Pure ML was observed, which was confirmed by 1H NMR, FT-IR, and TGA analysis. Based on these results, a plausible reaction mechanism is proposed (Scheme 4), which follows a similar pathway to that explained for PC.
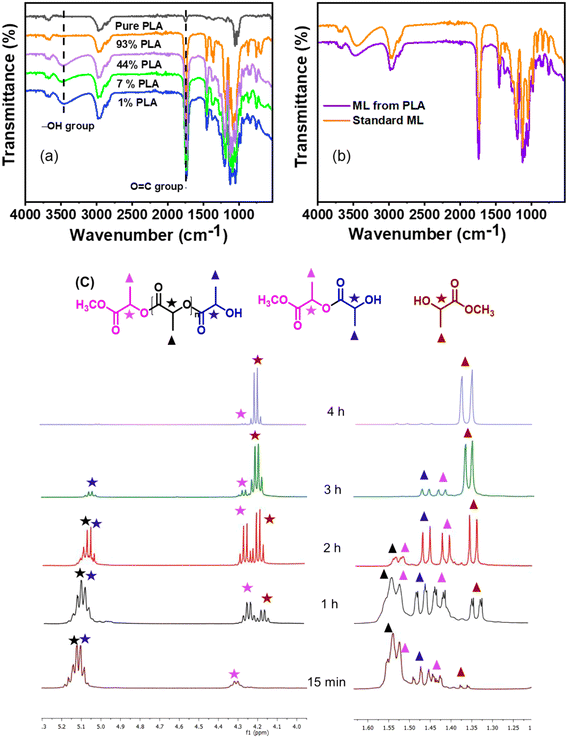 |
| | Fig. 7 Investigating and understanding of the PLA depolymerization pathway through various techniques; (a) FT-IR spectra at different PLA conversions and ML yields; (b) comparative FT-IR spectra of commercial ML and ML obtained from PLA; and (c) 1H NMR spectra of the reaction mixture at different conversions with respect to time. | |
 |
| | Scheme 4 Plausible depolymerisation route for PLA to methyl lactate. | |
3. Methanolysis of real plastic waste
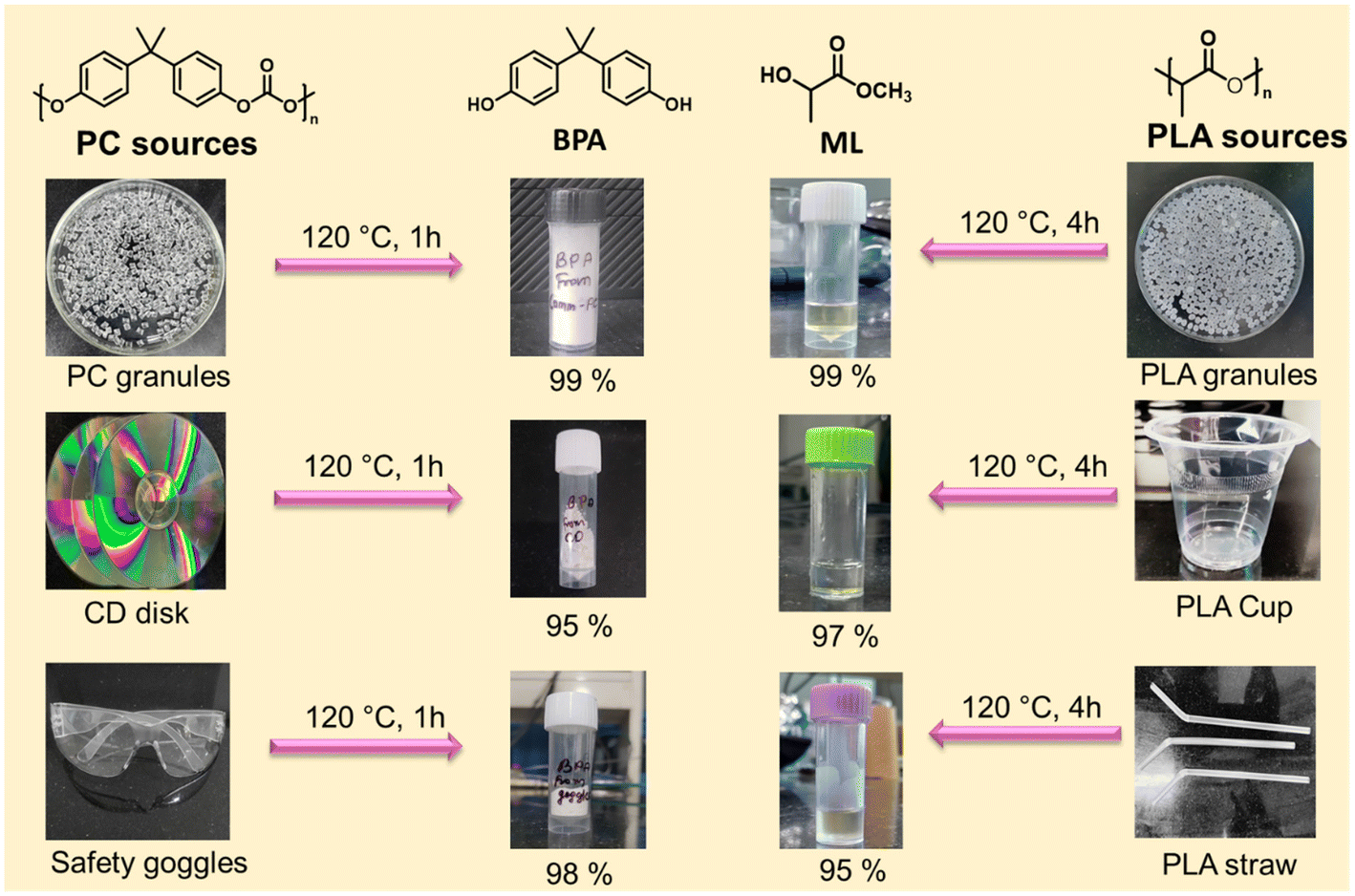
The application of the developed catalytic system was further expanded, and the practicability of the catalyst was examined by taking various plastic waste sources for depolymerization reactions. Depolymerization of discarded PC and PLA plastic waste such as CD disks, safety goggles, transparent PLA cups, and PLA-based straws was conducted in this study (Fig. 8). 1H NMR measurements were taken after methanolysis of PC/PLA waste at the evaluated temperature. The results show that the PC/PLA wastes were completely converted into the product BPA/ML. The distillation method was used to recover BPA/ML with a yield of >95%. Real plastic waste may contain additional additives that barely impact the depolymerization reaction. The aforementioned series of examples proved the viability of the presented catalytic system for the degradation of commonly used plastic waste to promote a resource-efficient circular economy. Firstly, the catalyst and reaction parameters were optimized for commercial PC granules, resulting in 99% PC depolymerization and 99% BPA yield at 120 °C, CH3OH:PC (1.5:1.0), and catalyst:PC (0.03:1.0) for 1.0 h. Furthermore, the same reaction parameters were used for real PC plastic waste depolymerization. The PC waste, such as CDs and safety goggles, was chopped into small pieces, washed with ethanol, and directly introduced for catalytic depolymerization under optimized conditions (detailed procedure provided in ESI†). Similarly, the PLA plastic waste, such as transparent cups and straws, was chopped into small pieces and directly used for the depolymerization reaction under optimized reaction conditions. After completing the reaction, the reaction mixture was filtered, the residue was separated, and the conversion and yield were calculated using eqn (S1)–(S3).† BPA/ML was separated by vacuum distillation. It was observed that the PC/PLA plastic wastes were completely depolymerized after the desired reaction time, and an excellent yield was achieved (Fig. 8). These results suggest that the catalytic system presented herein holds significant promise for effectively recycling actual plastic waste, thereby mitigating its environmental impact.
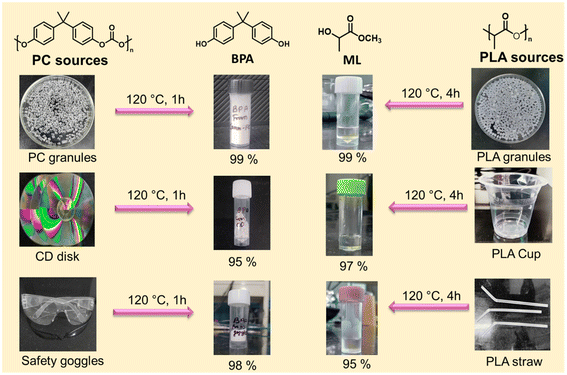 |
| | Fig. 8 Methanolysis of various PC and PLA waste sources to the corresponding monomer using the SBA-15-Pr-MIM-OH catalyst. | |
4. “One-pot” depolymerization of a mixture of PC and PLA plastic waste
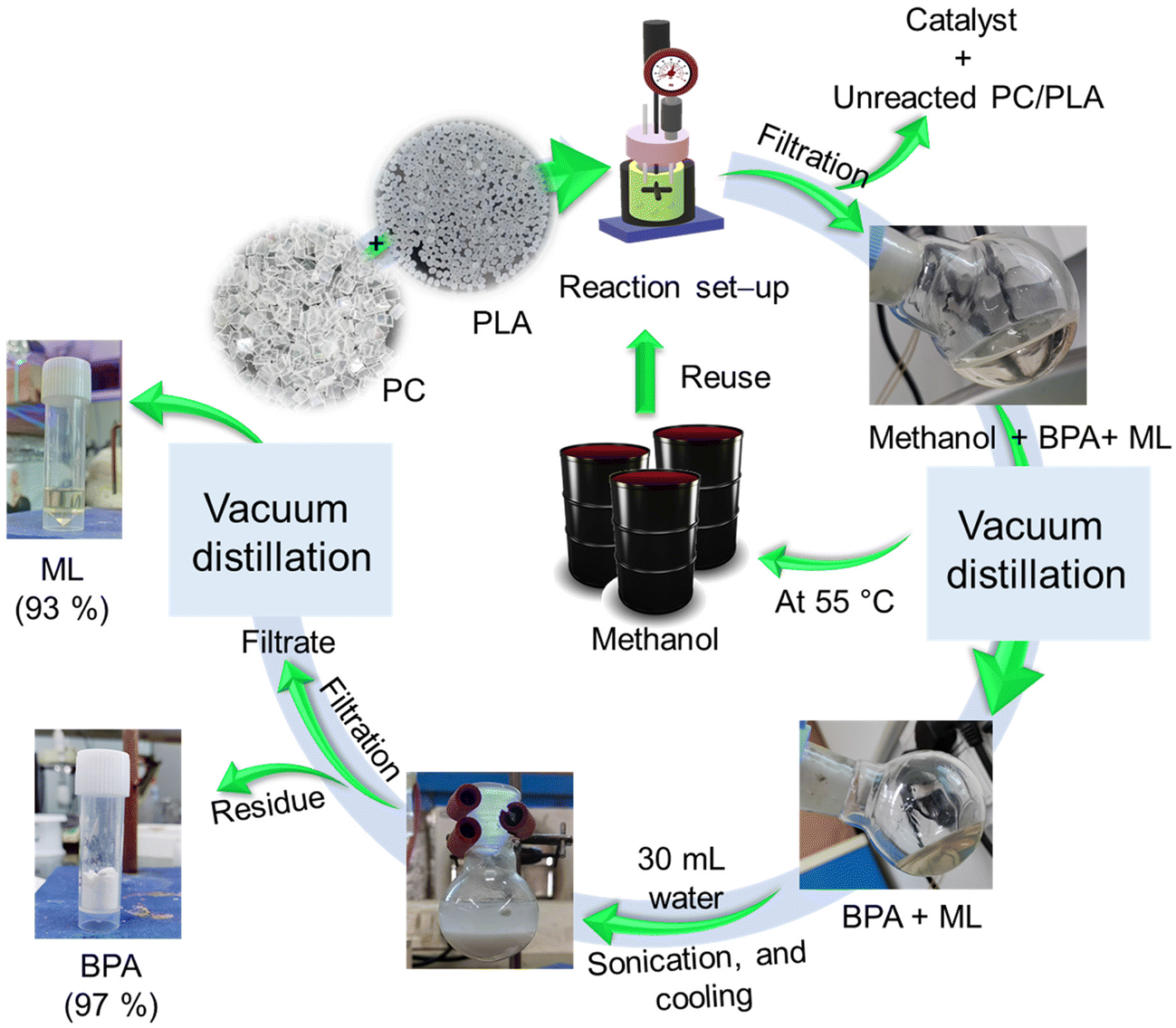
In practical production and application scenarios, combining two or more plastic waste types is frequently required to achieve enhanced comprehensive properties through complementary interactions. For this purpose, the depolymerization of a PC and PLA plastic waste mixture was explored in a single reaction using an SBA-15-Pr-MIM-OH catalyst, and degradation was carried out at an evaluated temperature (120 °C) for 6 h. After 6 h of reaction time, the reactor was cooled to ambient temperature, and the reaction mixture was filtered to remove the catalyst and unreacted PC/PLA using the vacuum filtration method (Scheme 5). The combined PC and PLA conversion was calculated according to eqn (S1).† PC and PLA particles disappeared after the completion of the reaction, and BPA and ML were formed, respectively. The filtrate containing methanol, BPA, and ML was collected. The methanol was first separated by distillation under reduced pressure (40 °C, 600 mbar). The residue containing BPA and ML was mixed with 30 mL of water, and the mixture was sonicated for 10 min followed by cooling under ice-cold conditions for 30 min. Then, the BPA precipitate was filtered, and the residue of BPA was separated. The mother liquor containing the ML was separated by a vacuum distillation process (Scheme 5). In addition, ∼86% solvent (methanol) was recovered from the reaction mixture. The yield of BPA and ML was calculated, and it was found to be 97% and 93%, respectively. The FT-IR spectra of the separated BPA and ML completely matched with those of commercial BPA and ML. Furthermore, the purity of the product was confirmed by NMR and GC-MS analysis (Fig. S11 and S12†).
 |
| | Scheme 5 Schematic representation of the “one-pot” depolymerisation of PC and PLA mixed plastic waste and separation process of BPA and ML. | |
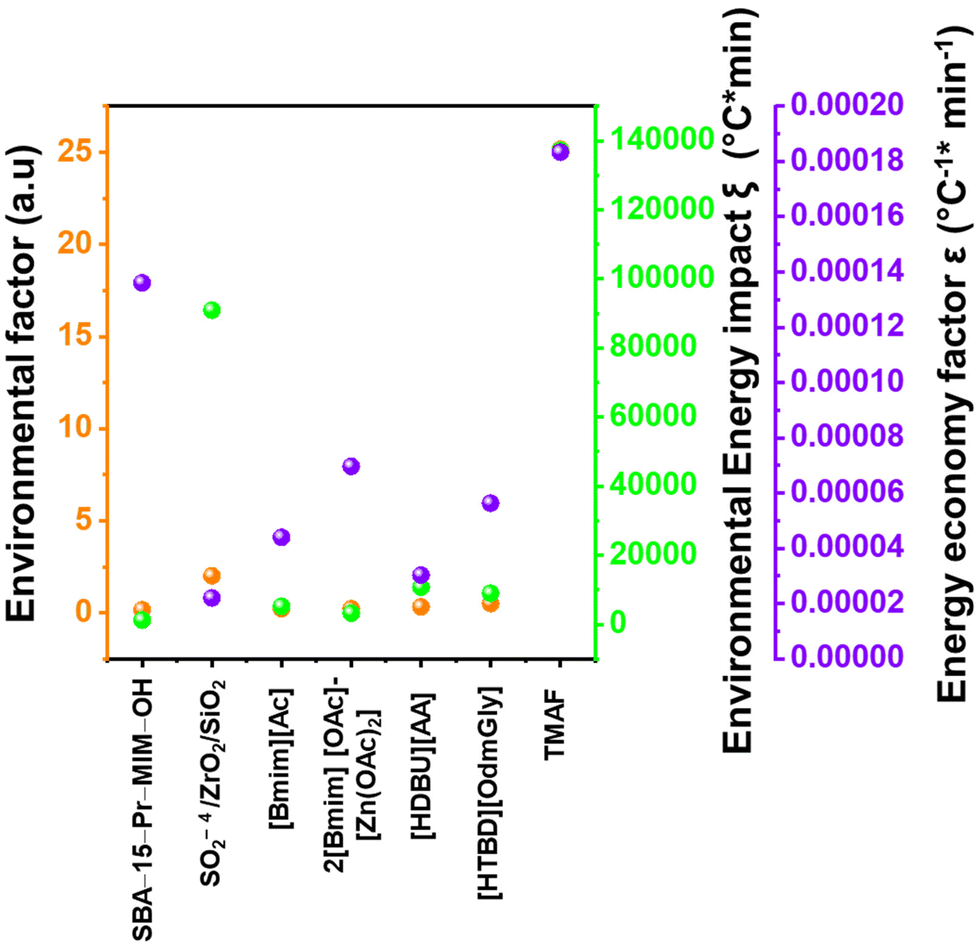
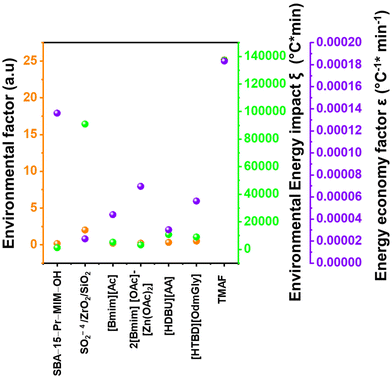
5. Comparative assessment of green chemistry matrix
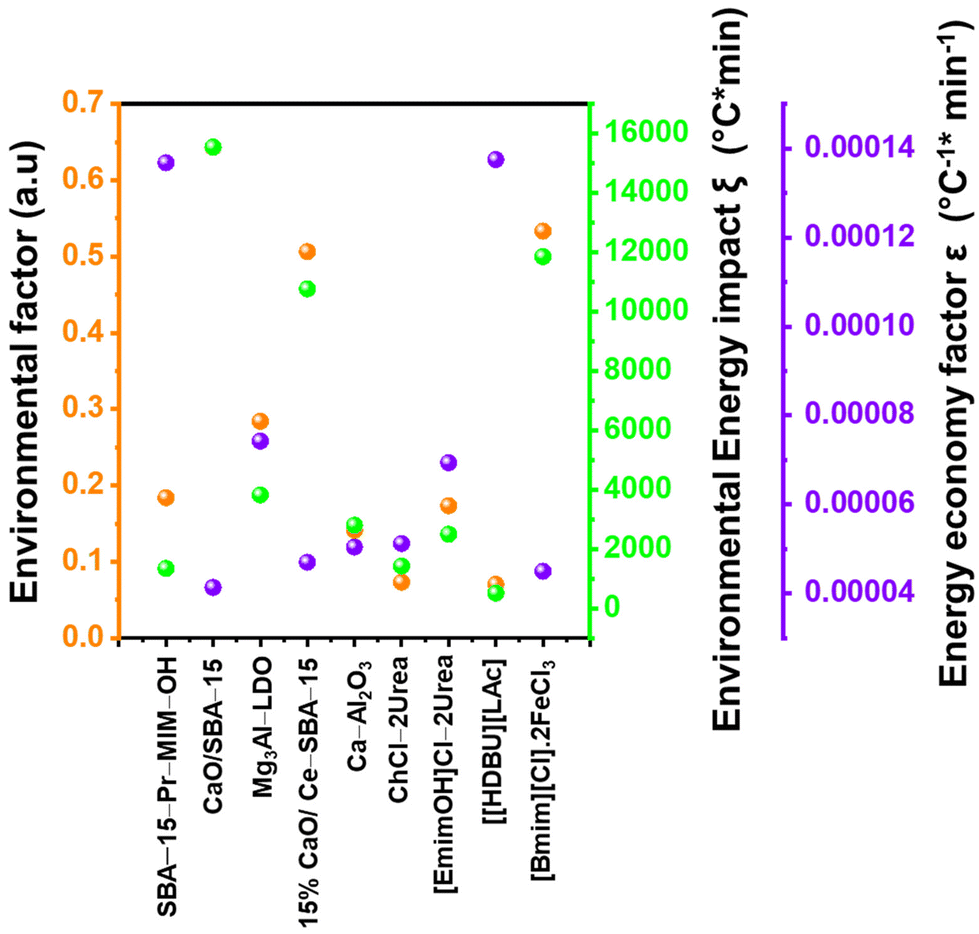
To make a quantitative comparison among different studies, energy and environmental impact parameters were calculated to demonstrate the applicability and scalability of the reported polycarbonate and polyester depolymerization process (Fig. 9, 10 and Tables S1, S2†). First, the energy economy coefficient (ε) was calculated. The ε coefficient relates the monomer yield produced from the depolymerization process temperature and time required for the reaction. The higher value of ε suggests a milder and more economical process. Furthermore, the green chemistry matrix was compared by calculating the environmental factor (E) by considering the mass of waste generated in obtaining a certain amount of monomer. It comprehensively incorporates considerations such as the product yield, encompassing reagents, process aids, solvent losses, and energy consumption. A higher E value corresponds to increased wastage, thereby indicating a greater environmental impact. The ε coefficient provides information related to the energy consumption of the depolymerization process and E compares the different mass inputs separately. Therefore, by combining these two factors, the environmental energy impact factor (ξ) was calculated to assess the performance of the reported processes and to identify the economic and environmental impact, thus enhancing their capacity for widespread industrial deployment.20,51| | 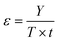 | (1) |
| |  | (2) |
| | 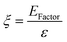 | (3) |
where Y, T, and t represent the yield of the product, temperature (°C), and time (min), respectively. Moreover, MM and m represent the molar mass (g mol−1) and mass (g) of the polymers, respectively. The high values of ε and low values of E and ξ suggest the best catalytic process for depolymerization, thereby facilitating their potential for large-scale industrial implementation. The E and ξ values of the reaction catalyzed by SBA-15-Pr-MIM-OH were much lower than those of other reported heterogeneous catalysts, demonstrating a preeminent role among the reported heterogeneous catalysts for the PC25–27,44,45,52–54 and PLA29,55–59 methanolysis process. The improvement contributes to an enhanced ε, indicating a higher yield under milder conditions. In addition, the E of the presented study shows that less waste was generated during the process than that generated by the reported catalysts (Fig. 9, 10 and Tables S1, S2†).
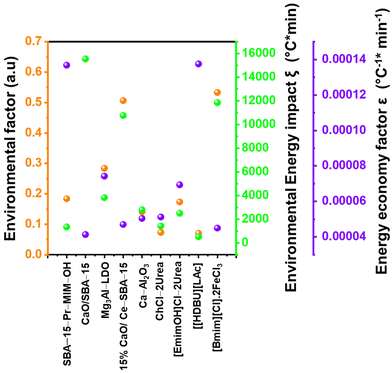 |
| | Fig. 9 Comparison of the performance of SBA-15-Pr-MIM-OH with reported catalysts for PC methanolysis by calculating the green chemistry parameters. | |
 |
| | Fig. 10 Comparison of the performance of the SBA-15-Pr-MIM-OH catalysts with the reported catalysts for PLA methanolysis by calculating green chemistry parameters. | |
6. Recycling and regeneration of catalyst
The catalyst stability and reusability were examined by performing the depolymerization reaction of PC and PLA under optimized reaction conditions using SBA-15-Pr-MIM-OH (Fig. S16†). The catalyst recycling experiments were performed by taking 2.5 times the catalyst, substrate, and methanol contents. After each catalytic cycle, the catalyst was separated, washed with water and ethanol three times, dried, and reused for the next cycle. After five cycles, the PC depolymerization activity dropped from 99.9% to 78.4% (Fig. S16a†). Similarly, the PLA conversion was also decreased from 99.9% to 72.1% after five cycles. This study on recyclability demonstrates that the catalyst deactivation may be attributed to the slight loss of basic sites during the reaction. The basicity of the recycled catalyst was found to be 0.83 mmol g−1, which was lower than that of the fresh catalyst (the basicity of the fresh catalyst was 0.98 mmol g−1) (Table 1). Other structural and textual features of the spent SBA-15-Pr-MIM-OH were similar after the recycling study, which was confirmed using FT-IR, N2-sorption, TGA, and SEM-EDAX analysis (Fig. S18–S20†). Furthermore, to enhance the catalytic activity, the catalyst was mixed with 0.2 M methanol solution of TMAOH (25%) and stirred at room temperature for 4 h. The basicity of the regeneration catalyst was 0.96 mmol g−1, which was very close to that of the fresh catalyst. After the regeneration of SBA-15-Pr-MIM-OH, the catalyst was examined for the depolymerization of PC and PLA. It was observed that the catalyst regained its activity after regeneration and exhibited 99.9% and 98.2% PC and PLA conversions, respectively, suggesting that the catalyst was recyclable and the catalytic activity can regained just by a simple TPAOH treatment. Furthermore, the heterogeneity of the catalyst was confirmed by performing a catalyst leaching test (Fig. S17†). After the desired time (0.25 h for PC and 1 h for PLA), the catalyst was separated from the reaction mixture, and the reaction proceeded without a catalyst. There were slight changes observed in the PC conversion (2.4%) and PLA conversion (1.8%) even after the completion of the desired reaction time, confirming that the catalyst plays a critical role in the catalytic transformation and the process was heterogeneous.
7. Conclusions
A solvent-free chemical upcycling of polycarbonate and polylactic acid waste was successfully demonstrated using a heterogeneous catalyst. SBA-15 functionalized basic ionic liquids were prepared and examined for the methanolysis of PC and PLA waste to BPA and ML, respectively. The proposed protocol allowed the complete conversion of PC and PLA with excellent selectivity and yield of corresponding monomers such as BPA and ML, starting from commercial PC/PLA and PC/PLA municipal plastic waste. Among all the synthesized catalysts, SBA-15-Pr-MIM-OH demonstrated higher basicity (0.98 mmol g−1) and delivered excellent performance in the depolymerization of PC and PLA at a relatively low temperature of 120 °C. SBA-15-Pr-MIM-OH achieved complete conversion within 1 h and 4 h, and exhibited >98% yield of BPA and ML, respectively. Reaction parameters such as the temperature, time, methanol amount, and catalyst dosage on the PC and PLA methanolysis were investigated. A comprehensive examination of the depolymerization process, employing FT-IR, 1H NMR, and TGA analysis, elucidated the mechanistic insights into the methanolysis of PC and PLA. The catalyst SBA-15-Pr-MIM-OH exhibited remarkable activity toward the transformation of diverse commercial plastic substrates into pristine monomeric compounds under mild reaction conditions with commendable yields. The “one-pot” depolymerization of mixed PC & PLA and separation of the resulting depolymerized products were achieved under similar conditions. The catalyst was efficiently recycled after a simple regeneration process. The process exhibited a low environmental energy footprint. Overall, this work introduced the synthesis of heterogeneous basic ionic liquid-functionalized catalysts for the depolymerization of single and mixed PC/PLA plastic waste with a reduced environmental energy footprint.
Author contributions
Arjun K. Manal: conceptualization, methodology development, data acquisition, manuscript writing; Garima Saini: preliminary experiments; Rajendra Srivastava: main project investigator, conceptualization, supervision, writing – review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
AKM is grateful to PMRF for the fellowship (PMRF-ID, 2902500). RS thanks the Research and Innovation award from IIT Ropar.
References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3(7), e170078 CrossRef PubMed.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, Sustainable Mater. Technol., 2020, 25, e00188 CrossRef CAS.
- O. Eriksson and G. Finnveden, Energy Environ. Sci., 2009, 2, 907 RSC.
- C.-H. Wu, L.-Y. Chen, R.-J. Jeng and S. A. Dai, ACS Sustainable Chem. Eng., 2018, 6, 8964–8975 CrossRef CAS.
- T.-W. Jiang, K. S. K. Reddy, Y.-C. Chen, M.-W. Wang, H.-C. Chang, M. M. Abu-Omar and C.-H. Lin, ACS Sustainable Chem. Eng., 2022, 10, 2429–2440 CrossRef CAS.
- L. Ranakoti, B. Gangil, S. K. Mishra, T. Singh, S. Sharma, R. A. Ilyas and S. El-Khatib, Materials, 2022, 15, 4312 CrossRef CAS PubMed.
- H. Tsuji and Y. Ikada, J. Appl. Polym. Sci., 1997, 63, 855–863 CrossRef CAS.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- X. Qi, Y. Ren and X. Wang, Int. Biodeterior. Biodegrad., 2017, 117, 215–223 CrossRef CAS.
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860–9871 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed.
- R. Mishra, A. Kumar, E. Singh and S. Kumar, ACS Sustainable Chem. Eng., 2023, 11, 2033–2049 CrossRef CAS.
- D. Yao, H. Yang, H. Chen and P. T. Williams, Appl. Catal., B, 2018, 239, 565–577 CrossRef CAS.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- A. K. Manal, G. V. Shanbhag and R. Srivastava, Appl. Catal., B, 2023, 338, 123021 CrossRef CAS.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, RSC Adv., 2022, 12, 31564–31576 RSC.
- E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC.
- F. Iannone, M. Casiello, N. Monopoli, M. M. Dell’Anna and A. Nacci, J. Mol. Catal. A: Chem., 2017, 426, 107–116 CrossRef CAS.
- M. Taguchi, Y. Ishikawa, S. Kataoka, T. Naka and T. Funazukuri, Catal. Commun., 2016, 84, 9397 CrossRef.
- Y. Zhao, X. Zhang, X. Song and F. Liu, Catal. Lett., 2017, 147, 2940–2949 CrossRef CAS.
- Y. Yang, C. Wang, F. Liu, X. Sun, G. Qin, Y. Liu and J. Gao, J. Mater. Sci., 2019, 54, 9442–9455 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu and S. Yu, ACS Sustainable Chem. Eng., 2018, 6, 13114–13121 CrossRef CAS.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- S. Xie, Z. Sun, T. Liu, J. Zhang, T. Li, X. Ouyang, X. Qiu, S. Luo, W. Fan and H. Lin, J. Catal., 2021, 402, 61–71 CrossRef CAS.
- L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- M. Hofmann, C. Alberti, F. Scheliga, R. R. Meißner and S. Enthaler, Polym. Chem., 2020, 11, 2625–2629 RSC.
- B. Sarmah and R. Srivastava, Ind. Eng. Chem. Res., 2017, 56, 8202–8215 CrossRef CAS.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448–2459 CrossRef CAS.
- A. K. Manal and R. Srivastava, Appl. Catal., A, 2023, 650, 119018 CrossRef CAS.
- A. Pourjavadi, S. H. Hosseini, M. Doulabi, S. M. Fakoorpoor and F. Seidi, ACS Catal., 2012, 2, 1259–1266 CrossRef CAS.
- B. Sarmah, R. Kore and R. Srivastava, Inorg. Chem. Front., 2018, 5, 1609–1621 RSC.
- A. K. Singh and S. D. Fernando, Energy Fuels, 2008, 22, 2067–2069 CrossRef CAS.
- A. A. Adeyi and B. A. Babalola, Sci. Rep., 2019, 9, 17458 CrossRef PubMed.
- W. Huang, H. Wang, X. Zhu, D. Yang, S. Yu, F. Liu and X. Song, Appl. Clay Sci., 2021, 202, 105986 CrossRef CAS.
- K. Wolf, A. Yazdani and P. Yates, J. Air Waste Manage. Assoc., 1991, 41, 1055–1061 CrossRef CAS PubMed.
- Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529–4537 CrossRef CAS.
- F. Liu, Z. Li, S. Yu, X. Cui and X. Ge, J. Hazard. Mater., 2010, 174, 872–875 CrossRef CAS PubMed.
- F. Liu, L. Li, S. Yu, Z. Lv and X. Ge, J. Hazard. Mater., 2011, 189, 249–254 CrossRef CAS PubMed.
- Y.-Y. Liu, G.-H. Qin, X.-Y. Song, J.-W. Ding, F.-S. Liu, S.-T. Yu and X.-P. Ge, J. Taiwan Inst. Chem. Eng., 2018, 86, 222–229 CrossRef CAS.
- L. A. Román-Ramírez, P. McKeown, C. Shah, J. Abraham, M. D. Jones and J. Wood, Ind. Eng. Chem. Res., 2020, 59, 11149–11156 CrossRef PubMed.
- P. Majgaonkar, R. Hanich, F. Malz and R. Brüll, J. Chem. Eng., 2021, 423, 129952 CrossRef CAS.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347–18353 CrossRef CAS.
- C. Eang, B. Nim, P. Sreearunothai, A. Petchsuk and P. Opaprakasit, New J. Chem., 2022, 46, 14933–14943 RSC.
- H. Liu, R. Zhao, X. Song, F. Liu, S. Yu, S. Liu and X. Ge, Catal. Lett., 2017, 147, 2298–2305 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- W. Huang, H. Wang, W. Hu, D. Yang, S. Yu, F. Liu and X. Song, RSC Adv., 2021, 11, 1595–1604 RSC.
- X. Song, W. Hu, W. Huang, H. Wang, S. Yan, S. Yu and F. Liu, Chem. Eng. J., 2020, 388, 124324 CrossRef.
- J. Guo, M. Liu, Y. Gu, Y. Wang, J. Gao and F. Liu, Ind. Eng. Chem. Res., 2018, 57, 10915–10921 CrossRef CAS.
- B. Ye, R. Zhou, C. Wang, Z. Wang, Z. Zhong and Z. Hou, Appl. Catal., A, 2023, 649, 118936 CrossRef CAS.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- X. Song, Z. Bian, Y. Hui, H. Wang, F. Liu and S. Yu, Polym. Degrad. Stab., 2019, 168, 108937 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst synthesis, characterization details, procedures for the depolymerization of PC and PLA, purification process of PC, characterization catalysts, catalytic activity data, product purity data, mechanistic investigation, recycling data, and comparison of the literature report. See DOI: https://doi.org/10.1039/d3gc04907h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Rajendra
Srivastava
and
Rajendra
Srivastava