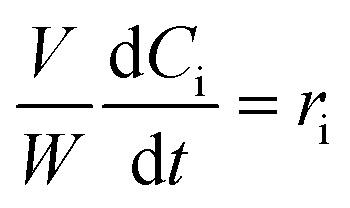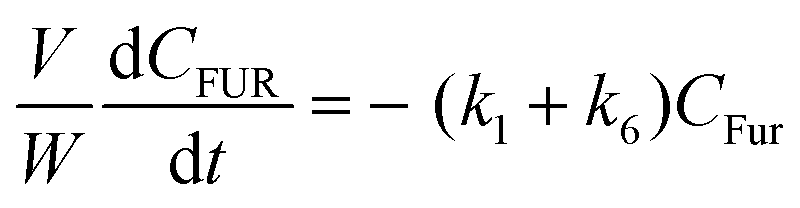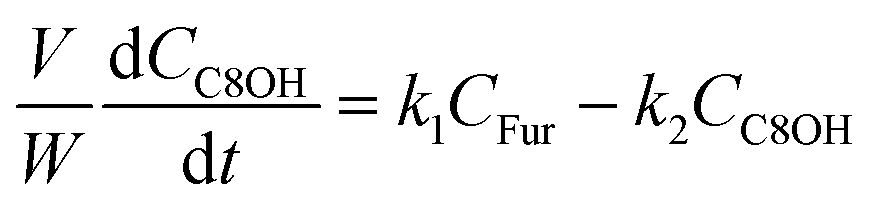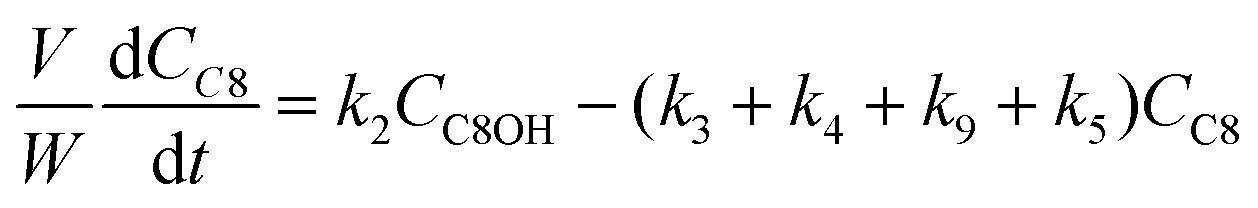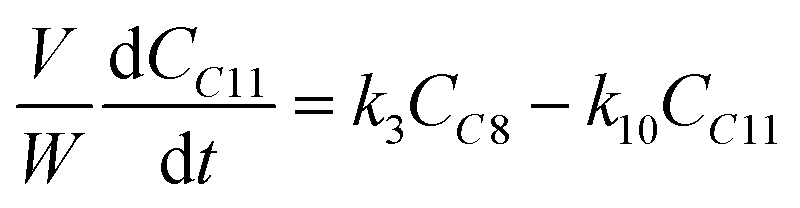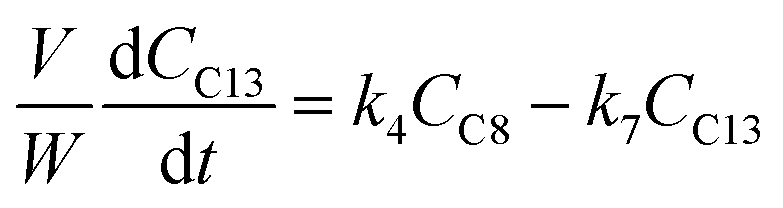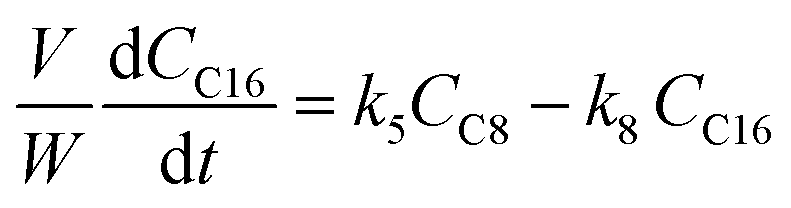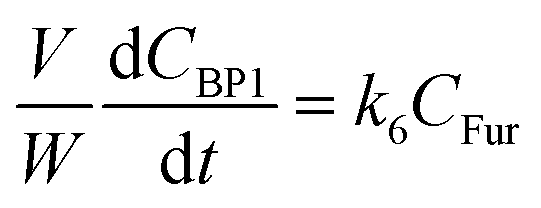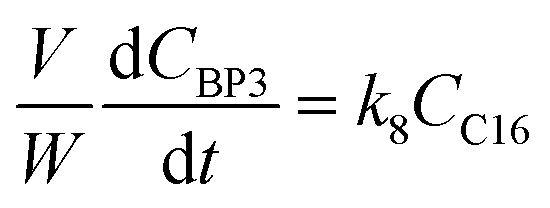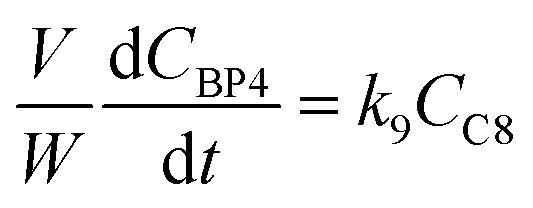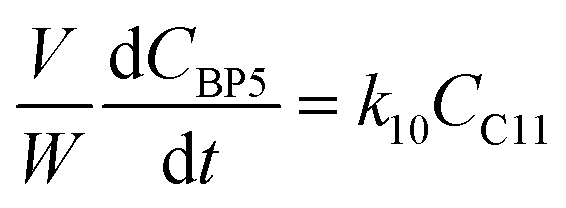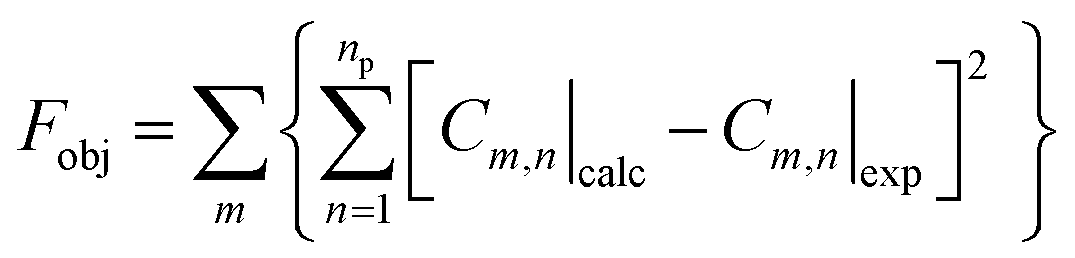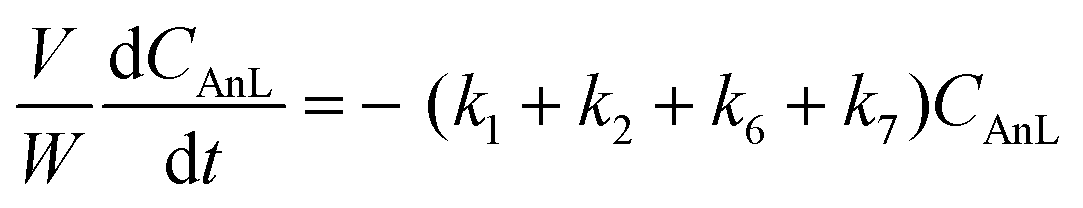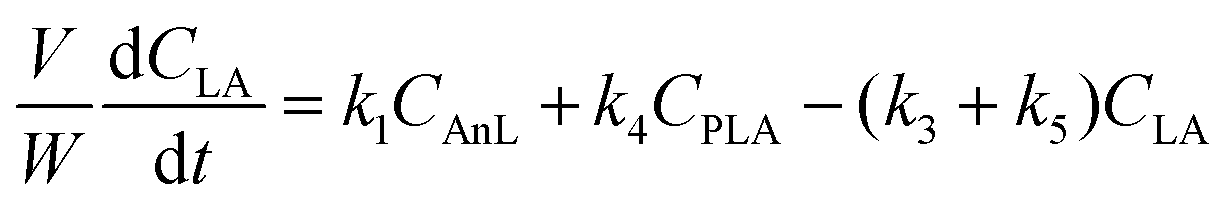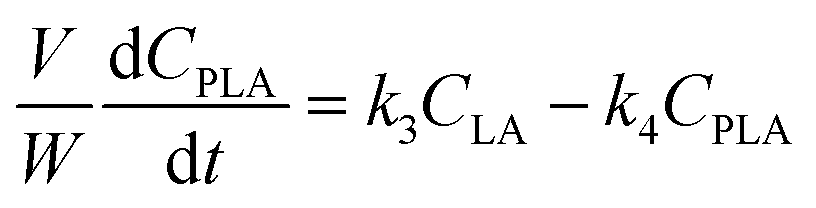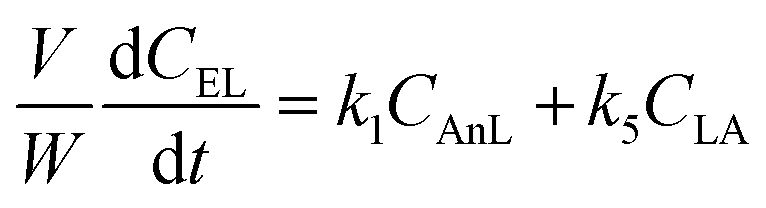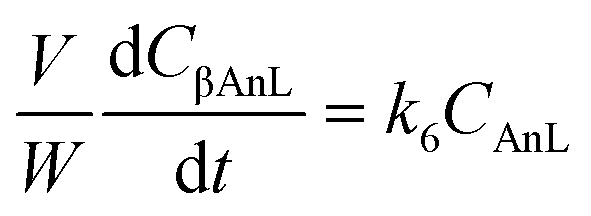DOI:
10.1039/D4GC00207E
(Paper)
Green Chem., 2024,
26, 4820-4833
Chemical valorisation of biomass derived furanics and carboxylic acids over niobium-based catalysts†
Received
13th January 2024
, Accepted 11th March 2024
First published on 12th March 2024
Abstract
Furfural is an industrial renewable platform chemical, which can be converted to useful furanics such as α-angelica lactone, carboxylic acids such as levulinic acid and valeric acid, or to higher carbon content products via condensation routes for producing drop-in fuel replacements and chemicals with diverse applications. These important conversion processes may be carried out in selective fashions, although they require adequate catalysts. They were successfully carried out using versatile, stable silica-wrapped niobium oxide nanostructured catalysts. For example, α-angelica lactone was converted in an integrated fashion to ethyl levulinate in 90% yield, and the esterification of levulinic and valeric acids gave ethyl levulinate in quantitative yield and ethyl valerate in 90% yield, respectively, at 140 °C. Catalytic, mechanistic and kinetic modelling studies shed light on the influence of the materials properties on the catalytic performances. These catalysts outperformed pure Nb2O5, as well as hydrothermally synthesized composites consisting of Nb2O5 nanoparticles embedded in a mesoporous siliceous matrix.
Introduction
The continued use of fossil fuels and the increase of anthropogenic CO2 emissions can lead to irreparable damage such as decline in agriculture, increase of natural disasters, and extreme temperatures. Hence, it is important to search for sustainable green alternatives to respond to the increasing energy demands. Vegetable biomass is a renewable source of organic carbon, and its main components include carbohydrates, which can be found in forestry/agricultural residues/surpluses and industrial byproduct streams (e.g., those from sugarcane processing, cellulosic fiber production, etc.) and converted to useful biobased products.
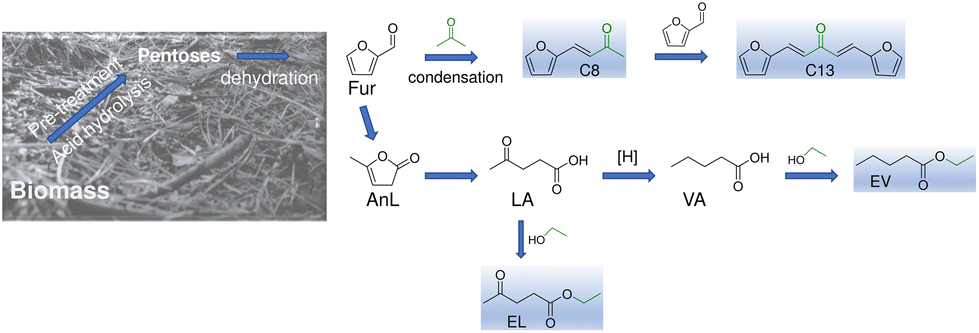
For over a century, one of the most important and versatile platform chemicals produced from carbohydrate matter is furfural (Fur),1 specifically via aqueous phase, acid-catalysed reactions of hemicelluloses.2,3 Fur is used as solvent (e.g., in petroleum refining) or upgraded for different end use industries such as agriculture, paints/coatings, drugs, food/beverages, etc.,4–6 and for synthesizing drop-in fuels.7
The C/O atomic ratio of Fur can be enhanced by chemical routes, such as condensation with ketones, to produce fuel grade alkanes.8–11 Aiming at fully integrated biobased Fur conversion processes, ketones such as acetone are attractive for Fur condensation since they may be produced via the Acetone–Butanol–Ethanol (ABE) fermentation of biomass.12 The condensation of Fur with acetone gives 4-(furan-2-yl)but-3-en-2-one (C8) and 1,5-di(furan-2-yl)penta-1,4-dien-3-one (C13) (Scheme 1), which are interesting intermediates for producing drop-in fuels, avoiding engine adjustments, etc.11 C13 is also an attractive intermediate for jet fuel range hydrocarbons.13 In the search for improved solutions and efficient catalysts to enhance productivity, different catalytic approaches have been reported to produce condensation products from Fur/acetone using acid, base or acid–base catalysts.8,14 The C8 and C13 products were produced using solid base catalysts, most of which possessed magnesium, such as for C8, Mg-containing metal oxides,15–22 hydrotalcites (HTs) or layered double hydroxides (LDHs),14,23,24–31,32,33 carbons,34,35 and mineral meixnerite;14 and for C13, metal oxides36–39 and carbons.40 In general, the reported Mg-containing catalysts were effective for the condensation reactions, but several studies reported the occurrence of catalyst deactivation18,27,28,34,36 or did not report catalyst recyclability,14,23,25,30,41 which represents an important knowledge gap because catalyst stability is fundamental in the development of sustainable heterogeneous catalytic processes. The use of solid acids for the target reaction systems instead of solid base catalysts advantageously avoids intermediate neutralization processes between the upstream acid-catalysed production of Fur and the catalytic upgrading of Fur. Different types of solid acid catalysts have been reported for C8 production such as zeotypes/zeolites,42–45 metal organic frameworks46 and organic frameworks.47
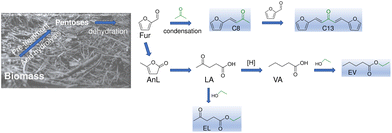 |
| | Scheme 1 Catalytic upgrading of biobased furfural to bioproducts with higher number of carbon atoms such as 4-(furan-2-yl)but-3-en-2-one (C8), ethyl levulinate (EL) and ethyl valerate (EV). | |
Water, besides being present from upstream Fur production, is a co-product of important biomass conversion processes. In this sense, niobium based oxides may be promising acid catalysts since they may be relatively water-tolerant48,49 for various biomass conversion routes50 involving reactions such as dehydration,49,51–53 etherification, alcoholysis,54 acetalization,55 hydrolysis and esterification.56,57 According to the literature, niobium oxides may possess Brønsted and Lewis acidity capable of activating carbon–oxygen bonds and oxygen-containing functional groups,58 accounting for superior performances compared to those of other metal oxides such as MgO, CaO, MgAl hydrotalcites, Al2O3, and ZrO2 in Fur/4-heptanone condensation.59 Zeolites were modified with niobium in order to trigger Fur condensation,60 and a preliminary study showed good catalytic potential of niobium oxide nanoparticles wrapped within a mesoporous silica component.61
In this work, various reaction systems involving acid catalysis were studied for converting biobased furanics and carboxylic acids to renewable products (Scheme 1), which are of interest in the context of an integrated biorefinery for the production and upgrading of the important industrial platform chemical, Fur. Specifically, Fur condensation, the one-pot conversion of α-angelic lactone (AnL) to ethyl levulinate, and the esterification of important biobased carboxylic acids, namely levulinic acid (LA) and valeric acid (VA), are reported. These are important reactions since, for example, EL has a growing market for food additives, fragrances, and drug intermediates,62 and is an attractive fuel blend for decreased emissions and improved flow properties.63–66 Moreover, EL and EV have higher energy density than ethanol, making them interesting fuel additives, besides being useful to different industrial sectors (perfumes, cosmetics, flavours, lubricants, plasticizers, etc.).67,68 The development of catalysts which are versatile, i.e., capable of promoting the different reaction processes within an integrated biorefinery, can be economically attractive by advantageously avoiding additional catalytic material production units, several specific catalyst treatment processes, etc. In this challenging context, the present study discusses the potential of silica–niobia nanostructures as versatile catalysts (sol–gel synthesis using acetophenone) for the various abovementioned processes involving acid catalysis (Fur, AnL, LA, and VA), relevant for Fur upgrading. Mechanistic and kinetic modelling studies contributed to the understanding of the influence of the materials properties on the performances of the versatile catalysts.
Experimental
Catalytic materials
The silica–niobia nanostructured catalysts and Nb2O5 nanoparticles were obtained as described in the literature, via a fast non-hydrolytic sol–gel method. Simply, acetophenone as the reaction solvent plus the metal precursors (silicon tetrachloride and niobium(V) chloride) in the desired amounts, were subjected to microwave heating at 220 °C for 20 min.61 The materials were used in the catalytic reactions without calcination (only washing with acetone and ethanol, and drying at 65 °C; samples denoted as SiNbx-NC) or after calcination at 400 °C (air, 5 h, 1 °C min−1; samples denoted as SiNbx).61 In the sample names, x is the at.% of niobium relative to silicon in the materials.
For comparative studies, a composite consisting of the Nb2O5 nanoparticles embedded (ca. 20 wt% loading) in a mesoporous siliceous TUD-1 type matrix was synthesized without using any surfactant or expensive polymeric templating agents, adapting known procedures.69,70 Specifically, tetraethylorthosilicate (TEOS, Sigma-Aldrich, 98%; 2.19 g, 10.5 mmol) was added dropwise with stirring at 200 rpm and at room temperature to a suspension of Nb2O5 (350 mg) in a mixture of triethanolamine (TEA, Fluka, 97%, 1.57 g, 10.5 mmol) and water (1.22 g, 67.8 mmol). Then tetraethylammonium hydroxide (TEAOH, Sigma-Aldrich, 35 wt% in water, 0.50 g, 3.36 mmol) was added, and the stirring was continued for 2 h. The gel (with a molar composition of TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEA:0.32TEAOH:6.46 H2O and a molar ratio 80 Si:20 Nb) was then aged at room temperature for 24 h, followed by drying at 100 °C for another 24 h. The obtained solid was gently ground in an agate mortar with pestle, transferred to a Teflon-lined autoclave and heated at 180 °C under static conditions for 8 h. After being cooled to room temperature, the recovered product was washed with water until neutral pH, dried at 60 °C for 24 h, and finally calcined at 600 °C (heating rate of 1 °C min−1) for 10 h, in air flow (20 mL min−1). Pristine silica TUD-1 was prepared in a similar fashion, but without the Nb2O5 nanoparticles, as described in the literature.71
:TEA:0.32TEAOH:6.46 H2O and a molar ratio 80 Si:20 Nb) was then aged at room temperature for 24 h, followed by drying at 100 °C for another 24 h. The obtained solid was gently ground in an agate mortar with pestle, transferred to a Teflon-lined autoclave and heated at 180 °C under static conditions for 8 h. After being cooled to room temperature, the recovered product was washed with water until neutral pH, dried at 60 °C for 24 h, and finally calcined at 600 °C (heating rate of 1 °C min−1) for 10 h, in air flow (20 mL min−1). Pristine silica TUD-1 was prepared in a similar fashion, but without the Nb2O5 nanoparticles, as described in the literature.71
Materials characterisation
The PXRD data were collected on an Empyrean PANalytical diffractometer (Cu-Kα X-radiation, K = 1.5 4060 Å) in a Bragg–Brentano para-focusing optical configuration (45 kV, 40 mA) at ambient temperature. The sample was prepared in a spinning flat plate sample holder and step-scanned in the 2θ range from 3° to 70° with steps of 0.026°. A PIXEL linear detector with an active area of 3.3473° was used with a scan speed of 0.0515° per second. Low angle (0.5–6° 2θ) PXRD data were collected using the transmission mode, and with the samples deposited between Mylar foils. The samples were step-scanned in 0.013° 2θ steps with a counting time of 100 s per linear detector active area of 1.01°.
Scanning electron microscopy (SEM) images, elemental mapping images (Si and Nb) and energy dispersive X-ray spectroscopy (EDS) images were obtained on a Hitachi SU-70 SEM microscope with a Bruker Quantax 400 detector operating at 15 kV. Samples were prepared by deposition on aluminium sample holders followed by carbon coating using an Emitech K 950 carbon evaporator. Scanning transmission electron microscopy (STEM) (mapping) images (Si and Nb) were collected using a Hitachi HD2700 scanning electron microscope, equipped with a Bruker EDS detector; a drop of an ethanol suspension of the composite was added to a holey amorphous carbon-film-coated 400 mesh copper grid (Agar Scientific). Elemental analysis for C was performed on a Leco TruSpec 630–200–200 analyzer.
The N2 sorption isotherms were measured at −196 °C, using a QuantaChrome Autosorb iQ2 Automated Gas Sorption analyser. The sample was pre-treated at 250 °C for 3 h, under vacuum (<4 × 10−3 bar). The specific surface area was calculated using the Brunauer, Emmett, Teller equation (SBET) and mesopore sizes (dp) were calculated from the adsorption branch using the BJH method.
The characterisation results of the Nb2O5/TUD-1 composite and the pristine mesoporous silica TUD-1 (three-dimensional, sponge-like structure) are included in the ESI.† Briefly, Nb2O5/TUD-1 possessed mesopore widths in the range 4–10 nm, SBET = 413 m2 g−1 and ca. 18 at% Nb relative to Si, and the composite particles were irregular in size and shape. The textural properties were somewhat comparable to those of the mesoporous silica TUD-1 (synthesized in a similar fashion, but without Nb2O5) which possessed SBET = 471 m2 g−1 and dp = 8.5 nm.71
The SiNbx materials consisted of niobium oxide nanoparticles (ca. 5 nm) uniformly distributed within mesoporous silica, with Nb at.% relative to Si of 7, 20, 42, 65 and 82; the specific surface area (SBET) of the SiNbx was in the range 224–913 m2 g−1 and the average mesopore sizes were in the range 7–27 nm (Table S4†).61 SiNbx contained 138–267 μmolTAS g−1 of total amount of Lewis plus Brønsted acid sites (TAS) and a Lewis/Brønsted molar ratio (L/B) of 1.8–3.7.61 Increasing x led to increasing L/B ratio, but the relation between x and TAS was not linear: for x = 7 (L/B = 1.8, TAS = 138 μmol g−1); x = 20 (L/B = 2.4, TAS = 195 μmol g−1); x = 42 (L/B = 2.9, TAS = 267 μmol g−1); x = 65 (L/B = 3.1, TAS = 252 μmol g−1); and x = 82 (L/B = 3.7, TAS = 156 μmol g−1) (Table S4†).61 Pure Nb2O5 (1.5–5 nm particle size) synthesized in a similar fashion to SiNbx, but without silicon, possessed lower SBET (161 m2 g−1) and TAS (150 μmol g−1, L/B = 4.0) than SiNbx with x in the range 20–82.61
Catalytic tests
The catalytic tests at temperatures of up to 140 °C were carried out using borosilicate batch reactors, equipped with a PTFE valve for purging and a PTFE-coated magnetic stirring bar (800 rpm, avoiding mass transfer limitations). Catalytic tests at a higher temperature of 160 °C were carried out using a PTFE-lined stainless-steel autoclave with 5 mL capacity, equipped with a PTFE-coated magnetic stirring bar. For the Fur/acetone system, the reactor was loaded with a solution of 1.6 M furfural (Fur) in acetone (initial mole ratio of acetone:Fur ≅ 7.6) and 29.7 gcatalyst L−1. For the reactions of α-angelic lactone (AnL), levulinic acid (LA) and valeric acid (VA), the reactors were loaded with a solution of 0.35 M substrate in ethanol and 10 gcatalyst L−1 and heated at the desired reaction temperature (typically 140 °C). The initial activity (mmol gcat−1 h−1) was based on substrate conversion at 1 h.
The evolution of the reactions was monitored by gas chromatography (GC), using a Thermo Scientific Trace 1300 Series GC equipped with an Agilent Technologies, Inc. capillary column (DB-5, 30 m × 0.32 mm × 0.25 μm; He as the carrier gas) and a flame ionization detector. Exceptionally, Fur was quantified by liquid chromatography (HPLC), using a Knauer Smartline HPLC Pump 100 and a Shodex SH1011 H+ 300 mm × 8 mm (i.d.) ion exchange column (Showa Denko America, Inc., New York), coupled to a Knauer Smartline UV detector 2520 (210 nm). The mobile phase was 0.005 M aq. H2SO4 at a flow rate of 0.8 mL min−1 and column temperature of 35 °C. Calibration curves with internal standards were measured for quantifications. For each experimental point, separate experiments were carried out (the presented results are the mean values of at least two replicates (error < 5%)). The products (Tables S1 and S2†) were identified by GC-MS using a Shimadzu QP 2010 ultra-GC-MS equipped with a HT-5 GC column (25 m × 0.32 mm × 0.10 μm; He as the carrier gas).
Catalyst stability
The catalyst stability studies involved catalyst reuse at 140 °C, contact tests (CT) and characterisation of the used catalysts (with Fur as the substrate). Between runs, the used catalyst was separated from the reaction mixture by centrifugation (10000 rpm), thoroughly washed with acetone, dried at 85 °C overnight, and calcined at 400 °C for 5 h (1 °C min−1). The contact test (CT) consisted of contacting the fresh catalyst with acetone for 5 h at 140 °C, under the same conditions to those used for a normal catalytic test, but without Fur. Afterwards, the solid was separated from the liquid phase by centrifugation for 5 min at 10000 rpm; this liquid phase was passed through a 220 nm pore size PTFE membrane, giving the solution LP. Subsequently, the substrate (Fur) was added to the LP solution (to give an initial Fur concentration of 1.6 M), and left to react for 5 h, at 140 °C. This homogeneous mixture was analysed by chromatography and the results were compared to those for a normal catalytic test (with solid catalyst) and the blank test (without adding catalyst).
Kinetic modelling for Fur condensation
The kinetic modelling was carried out in Matlab, considering a batch reactor that is perfectly stirred and isothermal, for which the material balances are expressed according to eqn (1):| | 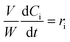 | (1) |
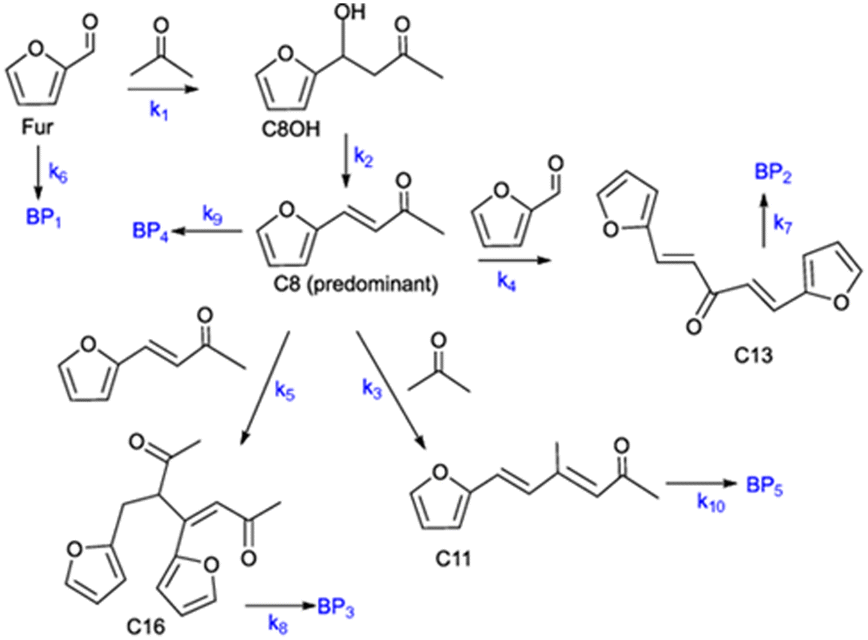
where V is the volume of the reaction mixture (L), W is the mass of catalyst (g), Ci is the molar concentration of species i (M), t is the reaction time (h), and ri is the rate of the reaction of species i (mol gcat−1 h−1); C8 = 4-(furan-2-yl)but-3-en-2-one, C8OH = 4-(furan-2-yl)-4-hydroxybutan-2-one, C11 = 6-(furan-2-yl)-4-methylhexa-3,5-dien-2-one, C13 = 1,5-di(furan-2-yl)penta-1,4-dien-3-one, C16 = 4-(furan-2-yl)-5-(furan-2-ylmethyl)hept-3-ene-2,6-dione (Table S1† and Scheme 2). The ratio W/V was maintained constant in all experiments (29.7 gcat−1 L−1).
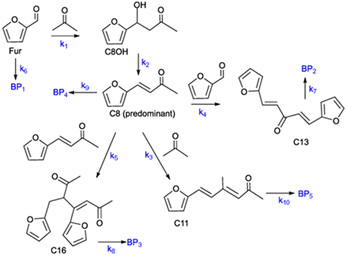 |
| | Scheme 2 Reaction mechanism of the Fur/acetone reaction system (ki are the kinetic constants used for kinetic modelling and BPi are possible byproducts). | |
Based on the mechanism proposed for Fur conversion (Scheme 2), a pseudo-homogeneous kinetic model was developed, considering irreversible, first order reactions for all steps (eqn (2)–(12)):
| | 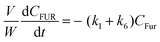 | (2) |
| | 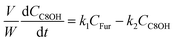 | (3) |
| | 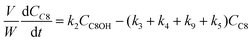 | (4) |
| | 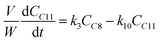 | (5) |
| | 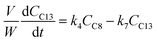 | (6) |
| | 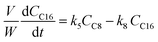 | (7) |
| | 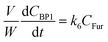 | (8) |
| | 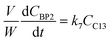 | (9) |
| | 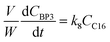 | (10) |
| | 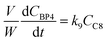 | (11) |
| | 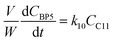 | (12) |
where
kj (
j = 1–10) are the kinetic constants (L g
cat−1 h
−1) of step
j at constant temperature. The formation of byproducts (denoted BP
i for the decomposition of species
i) was considered for closing the material balances.
The equation system was solved by numerical integration, using appropriate initial conditions (at t = 0), and the solution was refined by minimizing the objective function (Fobj) according to eqn (13), giving the values of the kinetic constants (ki, L gcat−1 h−1) by fitting the model to the experimental data:
| | 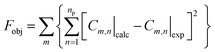 | (13) |
where
Cm,n|
calc and
Cm,n|
exp are the calculated (predicted by the model) and experimental concentrations, respectively, at each instant of time
n, and for each species
m. The ESI
† presents a similar kinetic model (model B) but considering second order reactions for the steps regarding
k4 and
k5, which gave very similar results (Table S5 and Fig. S6, S7
†) to the above model.
Kinetic modelling for the integrated conversion of AnL
A kinetic model was developed in a similar fashion to that described for Fur, but considering the mechanism discussed for the integrated conversion of AnL, using eqn (1) and (13)–(19):| | 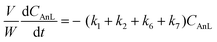 | (14) |
| | 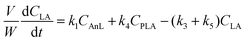 | (15) |
| | 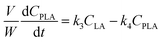 | (16) |
| | 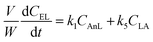 | (17) |
| | 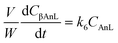 | (18) |
| | 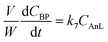 | (19) |
Results and discussion
Furfural–acetone condensation
General considerations.
The Fur condensation system was investigated over silica–niobia catalysts for several reasons including the following: (1) aldol/ketol condensation routes lead to products possessing lower O/C atom ratio and increased number of carbon atoms, being important for biomass valorisation; (2) Fur2 and acetone12 may be derived from vegetable biomass, making them interesting feedstocks for producing useful biobased products; (3) acid-catalyzed condensation routes are compatible with upstream Fur production routes,9 since the latter are also based on acid catalysis;2 and (4) the pore sizes of the silica–niobia materials are greater (7–27 nm mesopore sizes, Table S4†)61 than the approximate molecular dimensions of the Fur/acetone condensation products (up to 1.6 nm, Table S3†), which may advantageously favour mass transfer.
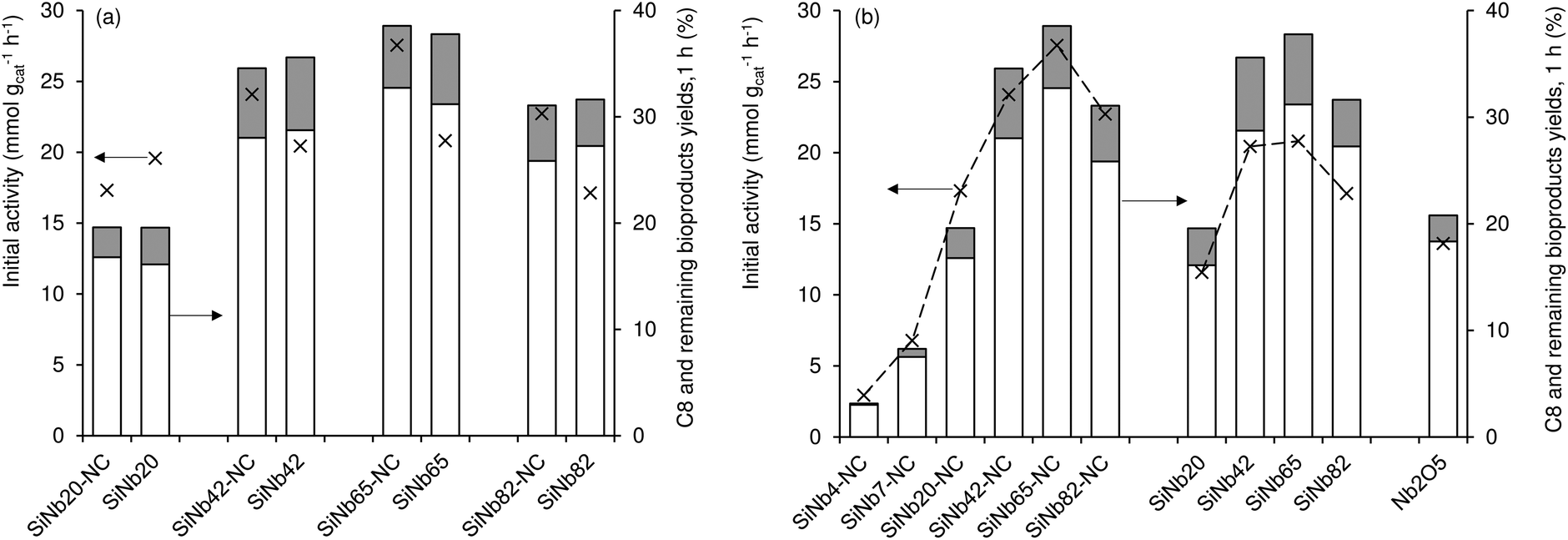
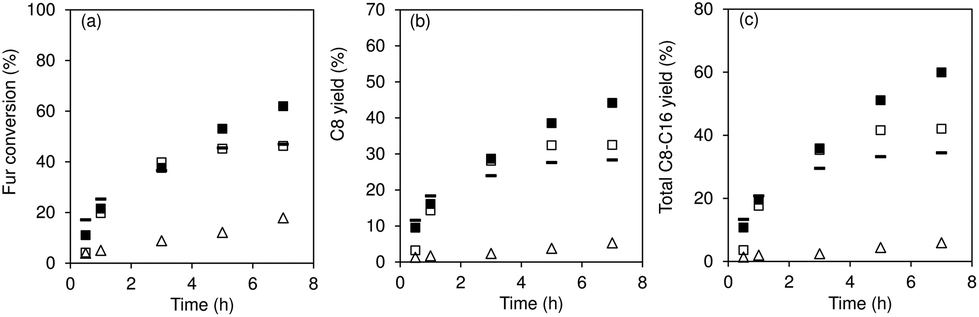
The Fur reaction with acetone gave predominantly 4-(furan-2-yl)but-3-en-2-one (C8) (up to 62% yield, at 140 °C) (Fig. 1). Other bioproducts included 6-(furan-2-yl)-4-methylhexa-3,5-dien-2-one (C11; ca. 1% maximum yield), 1,5-di(furan-2-yl)penta-1,4-dien-3-one (C13, 10% maximum yield) and 4-(furan-2-yl)-5-(furan-2-ylmethyl)hept-3-ene-2,6-dione (C16; 5% maximum yield) (Table S1†). Adding a cosolvent to the Fur/acetone system, such as toluene (acetone:toluene = 0.07 v/v, initial Fur concentration = 0.43 M), did not favour the catalytic reaction: conversion at 5 h decreased from 75% (without toluene) to 67% (with toluene), and C8 yield at 5 h decreased from 62 to 52%. On the other hand, a comparative study of SiNbx synthesized using the highest Nb content (x = 82) and pure niobia nanoparticles (Nb2O5) indicated that the former was more active and led to higher bioproduct yields. Moreover, the material balances (considering Fur and C8-C16 products, at 7 h/140 °C) reached 90–98% for the SiNbx catalysts, which was far superior to that for Nb2O5 (reached 77%, under similar conditions, Fig. S5†). On the other hand, commercial niobium oxide (Nb2O5-com) led to very sluggish results: 2% Fur conversion, <1% C8 yield, at 5 h/140 °C. A comparative study between Nb2O5 nanoparticles and Nb2O5-com suggested that decreasing the particle size to the nanoscale and enhancing the specific surface area (SBET) may considerably improve the catalytic performance; specifically, Nb2O5-com was microcrystalline (ca. 5 μm average particle size, Fig. S4†) with a low SBET of 20 m2 g−1, whereas Nb2O5 consisted of very small nanoparticles (1.5–5 nm) with a significant SBET of 161 m2 g−1.61
 |
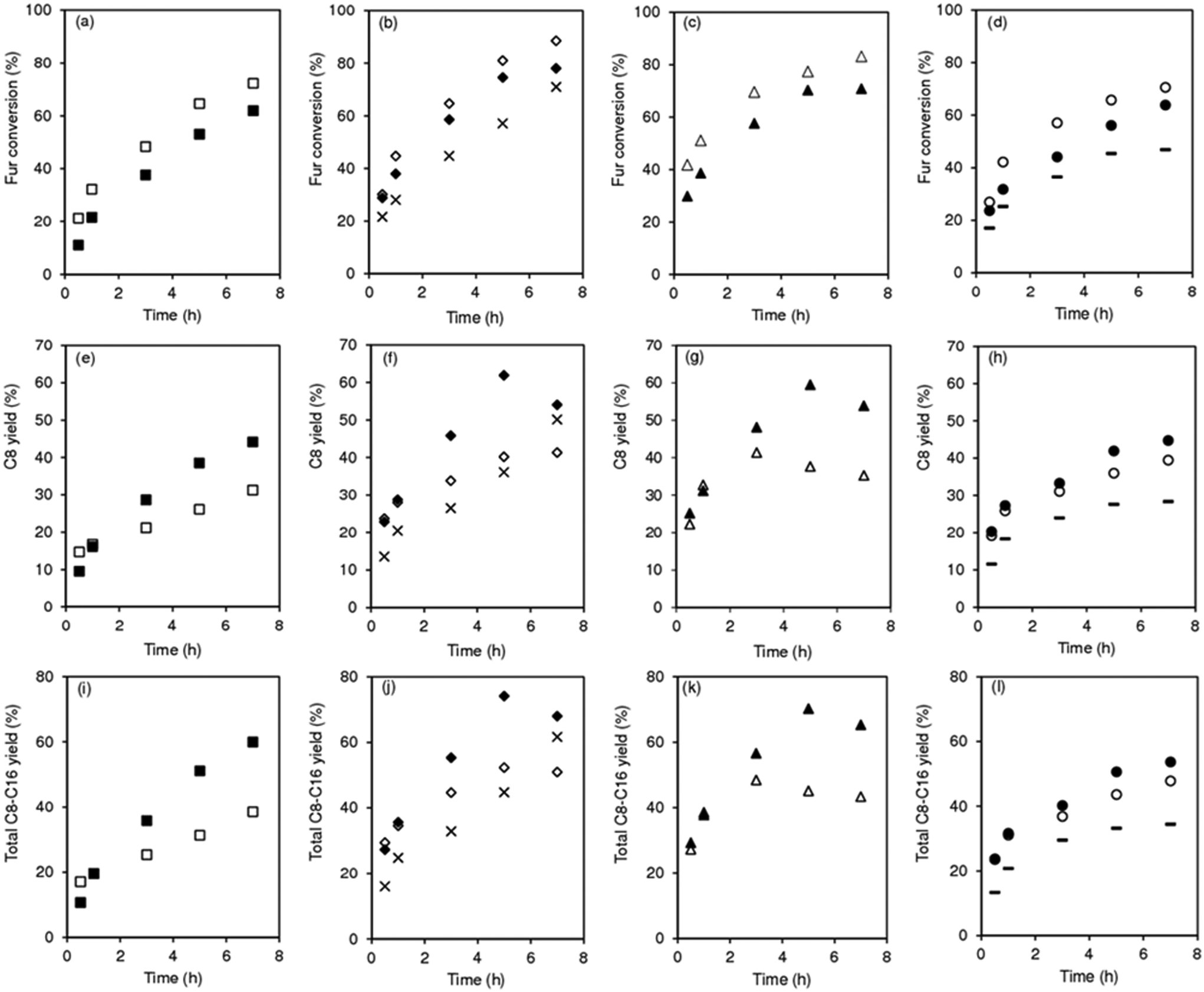
| | Fig. 1 Comparison of the catalytic performances of the as-synthesized (SiNbx-NC) and calcined (SiNbx) materials (a) possessing different Nb contents (b), and of the nanomaterial niobia (Nb2O5 in (b)); initial activity for Fur conversion (×) and yields of C8 (white bars) and remaining bioproducts (C11, C13 plus C16; grey bars). Reaction conditions: 1.6 M Fur in acetone, 29.7 gcat L−1, 1 h, 140 °C. | |
Influence of calcination of the materials.
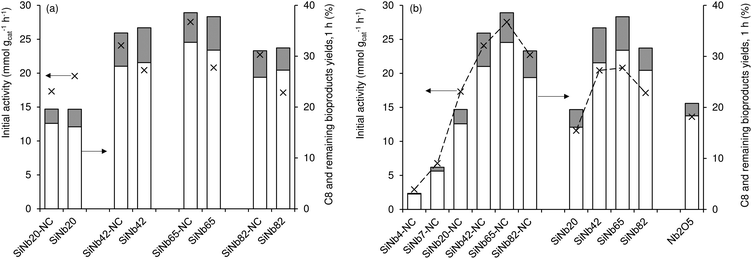
The influence of calcination of materials prior to catalytic application was investigated (Fig. 1). The as-synthesized (non-calcined) materials (SiNbx-NC) were more active than the respective calcined ones (SiNbx) (Fig. 1a) and led to roughly comparable C8 and total bioproduct yields at 1 h (Fig. 1a). However, for longer reaction times, SiNbx led to higher product yields than SiNbx-NC. Moreover, the carbon balance decreased with increasing Fur conversion, which was (disadvantageously) more pronounced for SiNbx-NC than for SiNbx (Fig. S5†), i.e., the overall selectivity was greater for SiNbx. Higher selectivity is important from a practical point of view, since it may avoid downstream separation/purification (energy intensive) processes and waste production, which negatively impact the process economics and green chemistry metrics (e.g., environmental (E) factor (mass of waste/mass of target product)). Increasing the calcination temperature from 400 to 500 °C did not result in a gain of the catalytic performance (kinetic curves in Fig. 2b, f and j for x = 42).
 |
| | Fig. 2 Fur/acetone condensation in the presence of SiNbx-NC (open symbols) and SiNbx (solid symbols) catalysts with x = 20 (a, e and i; squares), 42 (b, f and j; diamonds), 65 (c, g and k; triangles) and 82 (d, h and l; circles). For comparison, the results for the material possessing x = 42, calcined at 500 °C (×) are given in (b,f,j). The results for the nanomaterial Nb2O5 are given in (d,h,l: (−)). Reaction conditions: 1.6 M Fur in acetone, 29.7 gcat L−1, 140 °C. | |
Mechanistic and kinetic studies and influence of materials properties.
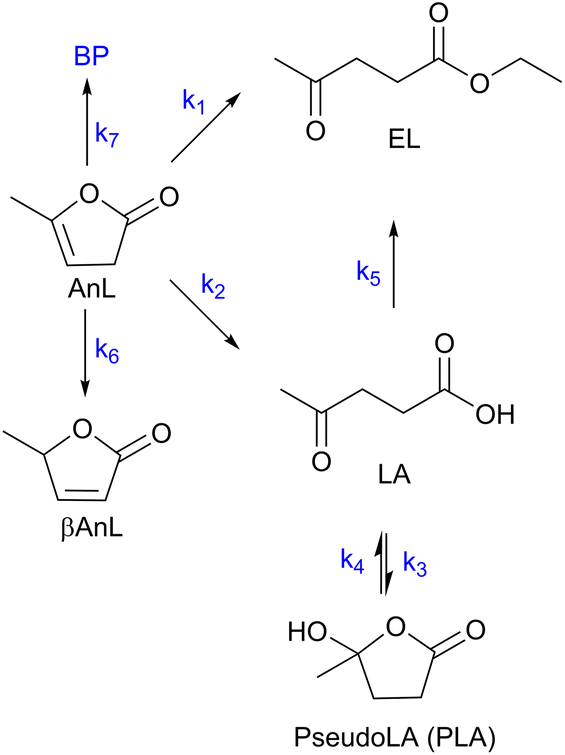
C8 is formed via Fur/acetone condensation and may be subsequently converted to other products. To gain mechanistic insights, catalytic tests were performed using C8 as the substrate, specifically, (i) the reaction of C8 with acetone (without Fur) and (ii) the reaction of C8 with Fur (without acetone) (initial C8 concentration ≅ 1.6 M, SiNb42, 140 °C). Test (i) gave mainly C11 and C16 (13 and 3% yield, respectively, at 46% C8 conversion, 5 h), and test (ii) gave C13 (19% yield at 33% C8 conversion, 5 h). Hence, in the integrated reaction system, C8 may react with acetone giving C11, or with Fur giving C13, or may undergo self-condensation to C16. The fact that the total selectivity was lower than 100% for the two tests suggested that C8 and/or the remaining products may be partly converted to byproducts. Based on these results and supportive literature data,20,44–46,72 the overall mechanistic proposal for Fur conversion is presented in Scheme 2. According to the literature, C8 may be formed via dehydration of the intermediate 4-(furan-2-yl)-4-hydroxybutan-2-one (C8OH),22,43,72–74 and, on the other hand, C13 may be formed via the dehydration of the intermediate 1,5-di(furan-2-yl)-5-hydroxypent-1-en-3-one (C13OH).16,22,23,43 C8OH is relatively unstable (may rapidly dehydrate to the α,β-unsaturated ketone C8),73 which may partly explain the fact that C8OH was always present in very small amounts (<3% yield). The intermediate C13OH was not detected in measurable amounts, parallel to other literature studies.22,23,43 The presence of some Brønsted acidity in the catalysts may promote these dehydration steps.75 According to the literature, considerably enhancing Brønsted acidity may promote undesirable reactions.60
Based on the mechanistic proposal (Scheme 2), kinetic modelling studies were carried out for the SiNbx and Nb2O5 catalysts. The kinetic constants ki are given in Table 1 and the calculated kinetic curves are given in Fig. S7.† For all catalysts, the fastest step predicted by the model was the conversion of C8OH to C8 (k2, Table 1), consistent with the very small amounts of C8OH present in the reaction media. Accordingly, the conversion of Fur to C8 was governed by the primary Fur conversion (because k2 ≫ k1). In general, Fur conversion (k1) was faster than the subsequent conversion of C8 to the bioproducts C11, C13 and C16 (k3, k4, k5, respectively, Table 1), contributing to the high C8 yields. Somewhat consistently, a comparative study of the catalytic tests (i) and (ii) (mentioned above), suggested that the C8/acetone reaction was slower (46% conversion at 140 °C/5 h) than the Fur/acetone reaction (75% conversion at 140 °C/5 h).
Table 1 Kinetic modelling results (kinetic constants and objective function (Fobj)) for Fur condensation, in the presence of the niobium-based nanocatalysts, at 140 °C
|
k
i
|
Step |
SiNb20 |
SiNb42 |
SiNb65 |
SiNb82 |
Nb2O5 |
|
Kinetic constant (L gcat−1 h−1).
|
|
k
1
|
Fur-C8OH |
5.110 × 10−3 |
1.059 × 10−2 |
1.017 × 10−2 |
6.449 × 10−3 |
4.600 × 10−3 |
|
k
2
|
C8OH-C8 |
1.442 × 103 |
1.452 × 103 |
1.452 × 103 |
1.442 × 103 |
1.442 × 103 |
|
k
3
|
C8-C11 |
2.686 × 10−6 |
1.910 × 10−5 |
1.540 × 10−6 |
3.545 × 10−7 |
3.672 × 10−7 |
|
k
4
|
C8-C13 |
1.951 × 10−3 |
2.950 × 10−3 |
3.463 × 10−3 |
3.947 × 10−3 |
5.047 × 10−3 |
|
k
5
|
C8-C16 |
1.606 × 10−3 |
6.255 × 10−4 |
2.523 × 10−4 |
4.265 × 10−4 |
2.542 × 10−3 |
|
k
6
|
Fur-BP1 |
1.584 × 10−7 |
1.044 × 10−5 |
2.163 × 10−8 |
1.753 × 10−9 |
1.808 × 10−9 |
|
k
7
|
C13-BP2 |
3.737 × 10−3 |
4.327 × 10−3 |
7.268 × 10−3 |
1.504 × 10−2 |
5.029 × 10−2 |
|
k
8
|
C16-BP3 |
9.035 × 10−3 |
1.219 × 10−2 |
1.296 × 10−2 |
1.437 × 10−2 |
3.605 × 10−1 |
|
k
9
|
C8-BP4 |
2.686 × 10−6 |
1.910 × 10−5 |
1.540 × 10−6 |
3.545 × 10−7 |
3.672 × 10−7 |
|
k
10
|
C11-BP5 |
6.786 × 103 |
6.788 × 103 |
6.788 × 103 |
6.786 × 103 |
6.786 × 103 |
|
F
obj
|
|
0.0283 |
0.1853 |
0.2469 |
0.2091 |
0.1085 |
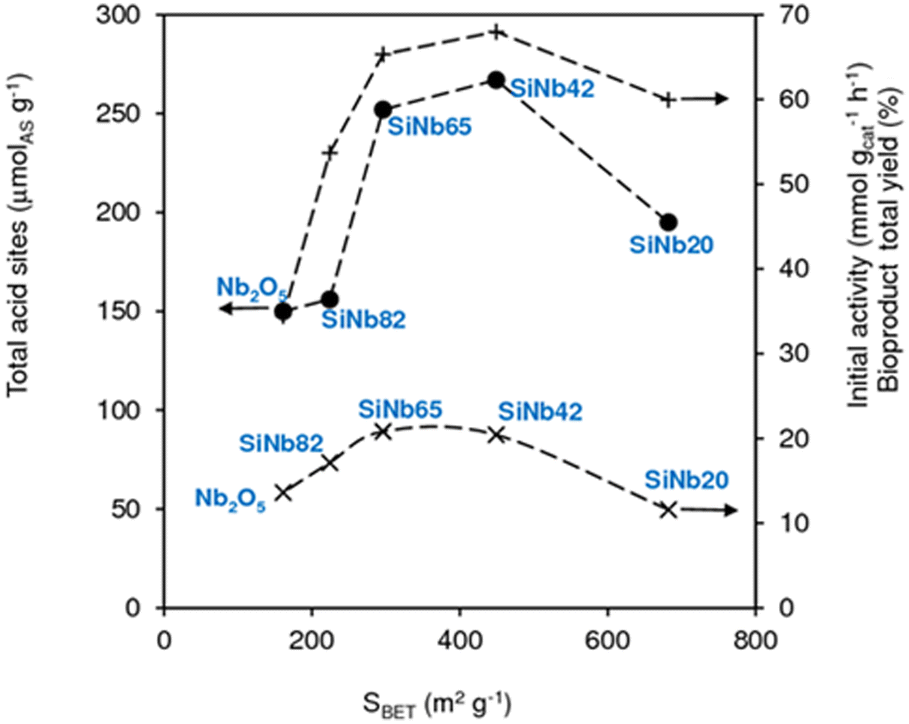
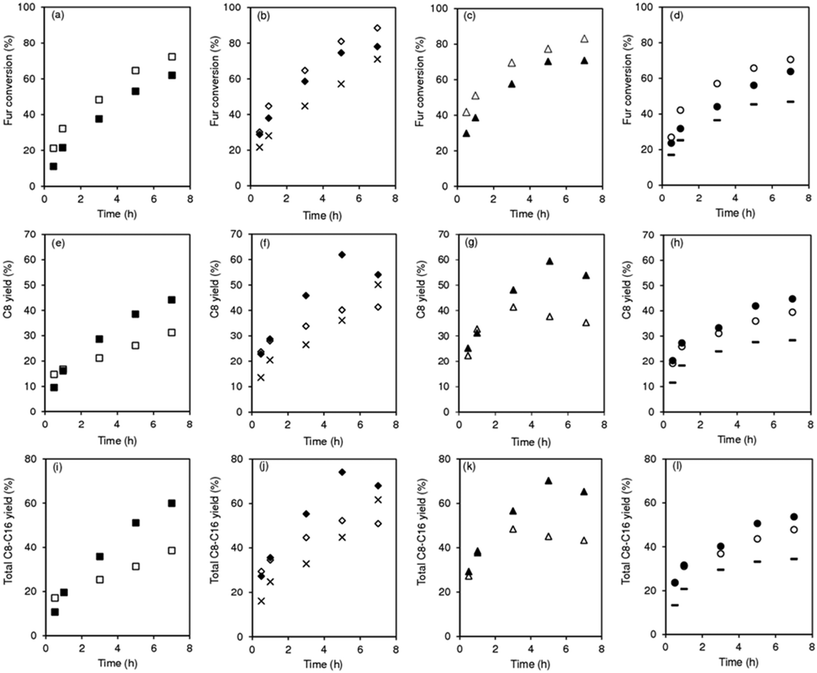
According to the literature, the condensation reactions may be promoted by Lewis acid sites (L) which may activate the carbonyl groups.43,60,75,76 Brønsted acid sites (B) may also promote condensation reactions77 and dimerization29,44 of C8 to C16. The degree of participation of the B and L acid sites in the overall mechanism is difficult to determine unambiguously.46 The SiNbx materials are essentially Lewis acid catalysts and possess little B acidity (Table S4†). Besides acidity, enhanced specific surface area may favour the catalytic reaction by increasing the amount of effective (accessible) active sites. Fig. 4 shows the relation between initial activity and these materials properties,61 specifically the amount of total acid sites (TAS = L + B) and SBET (Fig. 3). TAS versus SBET presented a maximum for SiNbx with x = 42 and 65, which were the catalysts with the highest initial activity (20–21 mmol gcat−1 h−1) and led to the highest total bioproduct yields at 7 h (65–68%, Fig. 3). Pure Nb2O5 possessed lower TAS and SBET, and thus lower initial activity (9 mmol gcat h−1).
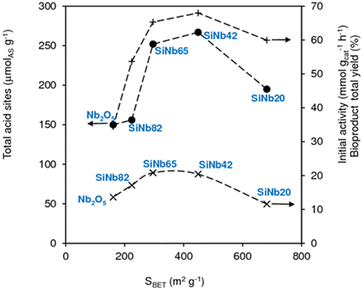 |
| | Fig. 3 Amount of total (L + B) acid sites (●), initial activity (×) and yields of total bioproducts (C8, C11, C13, C16) at 140 °C/7 h (+), versus SBET of SiNbx and Nb2O5. | |
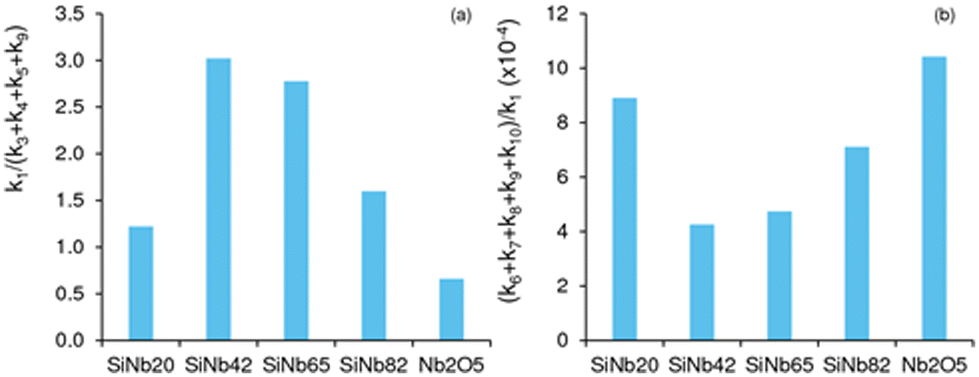
Fig. 4a compares the ratio of kinetic constants k1/(k3 + k4 + k5 + k9) (ki values are based on catalyst mass, L gcat−1 h−1); it is worth mentioning that the ki values expressed per active site ((AS), L molAS−1 h−1) followed a similar trend (Fig. S8†) to that observed in Fig. 4. A higher ratio somewhat reflects favourable C8 formation (please note that, since k2 ≫ k1, the kinetics of C8 formation may be evaluated based on k1). The highest ratio was verified for x = 42 and 65, i.e., these catalysts were more effective for targeting C8. Regarding the undesirable (unproductive) decomposition paths that lead to (unknown) byproducts (BPi), the slowest decompositions were those of Fur and C8 (k6 and k9, respectively), which explains the enhanced selectivity towards C8 for SiNbx and Nb2O5. Nevertheless, a comparative study of the ratio (k6 + k7 + k8 + k9 + k10)/k1 (a higher ratio reflects enhanced decomposition paths to BPi), suggested that undesirable pathways were more pronounced for Nb2O5 than for SiNbx (especially compared to SiNb42 and SiNb65) (Fig. 4b).
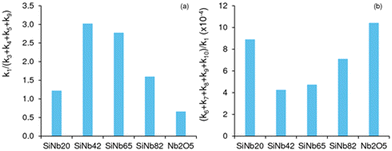 |
| | Fig. 4 Ratio of rate constants (a) reflecting favourable C8 formation (higher ratio k1/(k3 + k4 + k5 + k9)), or (b) enhanced decomposition paths to byproducts (higher ratio (k6 + k7 + k8 + k9 + k10)/k1). Reaction conditions: 1.6 M Fur in acetone, 140 °C, 29.7 gcat L−1. | |
Reaction temperature effect.
Fig. S9† shows the influence of the reaction temperature (60–140 °C) on the catalytic performance for Fur conversion (SiNb42). The carbon balance reached at least 88% in the studied temperature range. The initial activity (mmol gcat h−1) increased with temperature in the order 1.4 (60 °C) < 5.6 (100 °C) < 20.4 (140 °C). Based on the linearized Arrhenius equation, the apparent activation energy (Ea) was ca. 27 kJ mol−1 (R2 = 0.9996), which is intermediate of that reported in the literature for Cu/Al-MCM-41 (15.5 kJ mol−1, 150–200 °C),78 Mg–Al mixed oxides (24.8 kJ mol−1, 30–90 °C25 or 50.67 kJ mol−1, 130–160 °C15), 2.6%Nb-MF (52.9 kJ mol−1, 100–160 °C),60 and Sn-MFI and Sn-Beta (59 and 44 kJ mol−1, respectively, 100–160 °C).43
At 140 °C, not only was the Fur reaction faster, but also the maximum yield of C8 (62% at 5 h) was almost double that achieved at 100 °C (33%) and much higher than that at 60 °C (15%) (Fig. S9†). The bioproducts C11, C13 and C16 were formed in increasing total yields as the temperature increased (ca. 0, 4 and 10% yield at 60, 100 and 140 °C, respectively, at 5 h), and C13 was predominant (0%, 3% and 8% at 60, 100 and 140 °C, respectively, at 5 h). Increasing the reaction temperature from 140 to 160 °C, enhanced C8 and C13 yields (71 and 18%, respectively, at 100% conversion and 5 h; C11 and C16 were formed in a total yield < 3%), but the carbon balance reached a lower value of 93% compared to >99% for the reactions carried out at 100 or 140 °C.
Acetone efficiency.
An important factor to consider in the evaluation of new catalysts for the target reaction, concerns the “non-productive” decomposition of acetone, which should be avoided to enhance the process sustainability, e.g., reduce the E factor. The self-condensation of acetone was reported in the literature in the presence of acid and base catalysts, leading to products such as 4-hydroxy-4-methylpentan-2-one (MPOH) which may dehydrate to 4-methylpent-3-en-2-one (MP) (Scheme S1†).33,36–38,42,44–47,79 For SiNbx, the total yield of the acetone decomposition products was less than 1% at 140 °C/7 h, which were essentially MP and residual amounts of MPOH, 2,6-dimethylhepta-2,5-dien-4-one, 3,5,5-trimethylcyclohex-2-en-1-one and mesitylene. According to the literature, Brønsted acidity may favour acetone self-condensation.42 Accordingly, the fact that the Lewis acidity predominates over the Brønsted acidity for SiNbx may partly explain the good acetone consumption efficiency.
Catalyst stability.
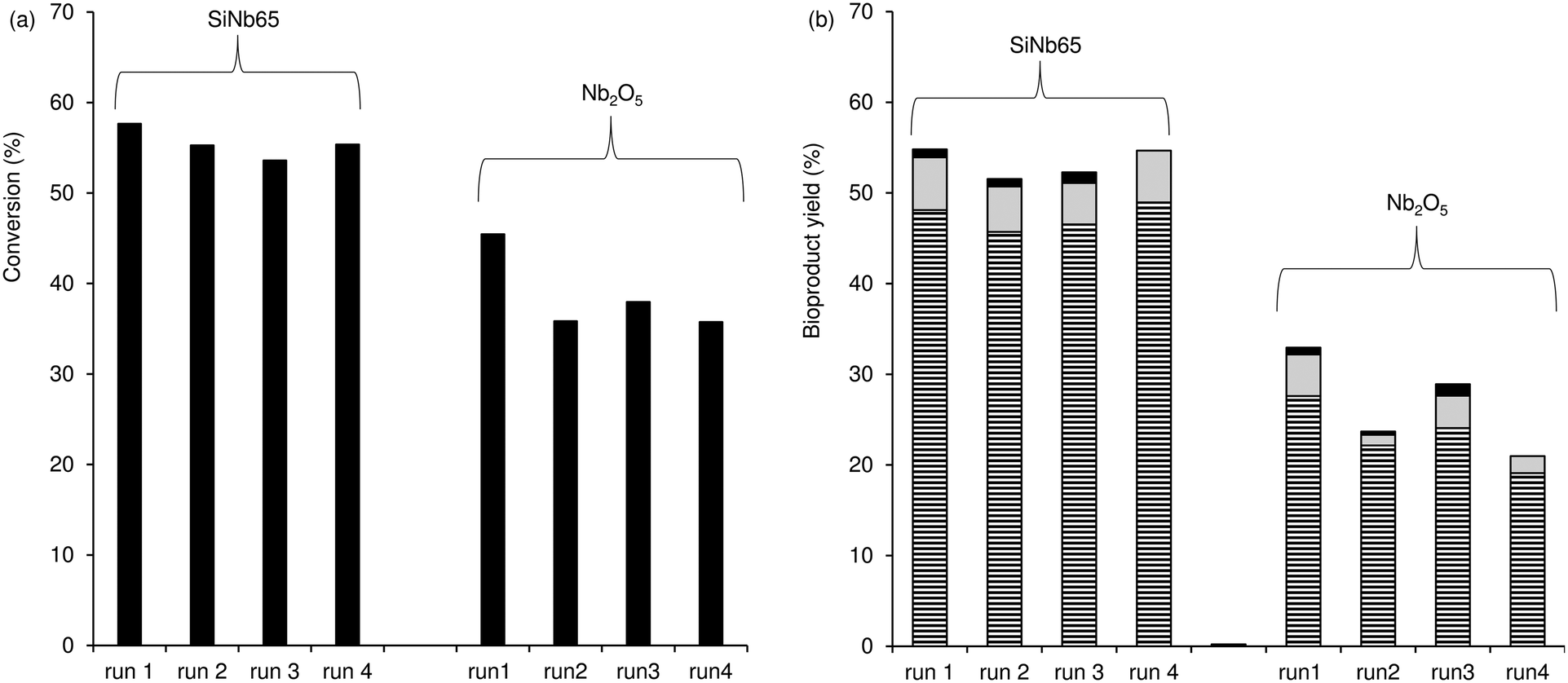
The used catalysts (at 140 °C) were dark brownish in colour, suggesting the presence of carbonaceous organic matter (e.g., ca. 12 wt% C for used SiNb65, based on elemental analysis). These results are somewhat consistent with those in the literature in that humins may be formed over solid catalysts in the Fur condensation process.80 Hence, it is desirable that the catalysts developed for these reaction systems are thermally resistant to withstand calcination. SiNb65, one of the best catalysts, was regenerated at 400 °C prior to reuse (the solid turned white in colour, like the original catalyst). The catalyst performed steadily in consecutive 3 h-batch runs at 140 °C (Fig. 5). The characterisation studies of the fresh and used solids indicated no significant alterations in the PXRD patterns (Fig. S10†), specific surface area (SBET of the original and used solids were approximately the same) and chemical composition ((Si/Nb of the used catalyst)/(Si/Nb of the original catalyst) = 1.02). Moreover, a contact test was carried out for SiNb65 (in acetone at 140 °C; details in the Experimental section) to check the heterogeneous nature of the catalytic reaction. After separating the solid from the reaction mixture, Fur was added to the filtrate and left to react further at 140 °C. This led to ca. 5% Fur conversion at 5 h, whereas a normal catalytic test (in the presence of SiNb65) led to 70% conversion. These results, together with the fact that without a catalyst, Fur conversion was 5%, further supported that the catalytic process was heterogeneous.
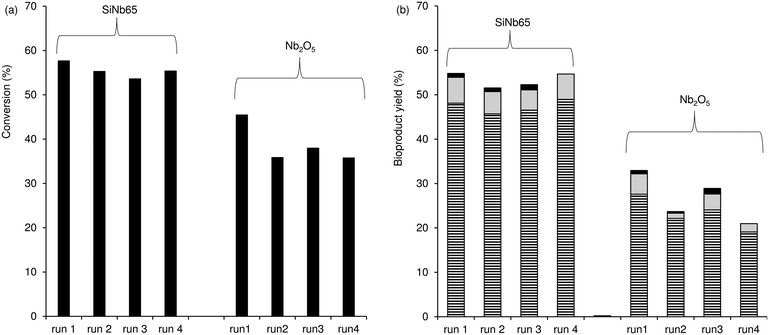 |
| | Fig. 5 Consecutive batch runs of Fur condensation in the presence of SiNb65 or Nb2O5; (a) Fur conversion and (b) yield of C8 (stripped bars), C13 (grey bars) and C16 (black bars); C8OH and C11 yields were always less than 2%. Reaction conditions: 1.6 M Fur in acetone, 140 °C, 29.7 gcat L−1, 3 h. | |
Benchmarking with the Nb2O5/TUD-1 composite.
For comparative studies, the catalytic reaction of Fur was carried out in the presence of hydrothermally synthesized Nb2O5/TUD-1 consisting of Nb2O5 nanoparticles embedded on mesoporous siliceous TUD-1 type matrix, possessing ca. 18 at% Nb relative to Si, which is comparable to SiNb20 (ca. 20 at% Nb).
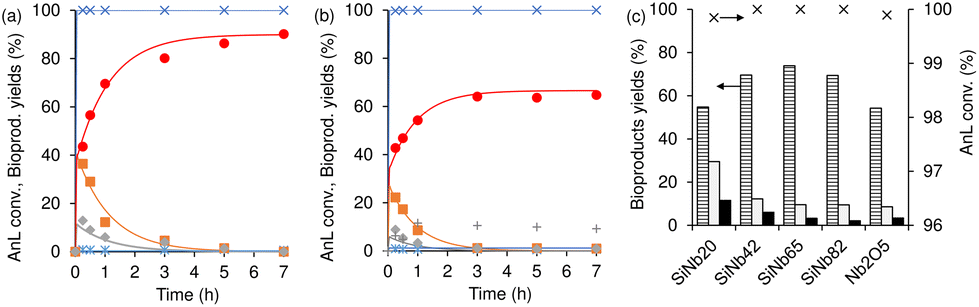
Fig. 6 compares the catalytic results for Nb2O5/TUD-1, SiNb20 and Nb2O5 using the same mass of catalyst, or based on a comparable molar amount of niobium (in the catalyst that was added to the reactor). A comparison of Nb2O5/TUD-1 versus Nb2O5 based on the same mass of catalyst, indicated that the former was more selective to C8 at similar Fur conversion (ca. 67% C8 selectivity at ca. 50% conversion, and ca. 52% selectivity at 55% conversion, respectively). Using a comparable amount of niobium added to the reactor, Nb2O5/TUD-1 led to faster reaction than Nb2O5 (45 and 12% conversion, respectively, at 5 h). Moreover, Nb2O5/TUD-1 led to 45% conversion and 32% C8 yield at 5 h, whereas Nb2O5 led to only 12% conversion and 4% C8 yield (Fig. 6a and b). Pristine silica TUD-1 led to sluggish Fur reaction (<1% total bioproducts yield at 140 °C, 5 h). Hence, the composite Nb2O5/TUD-1 performed superiorly to its individual (pure) components. The above results somewhat parallel those verified for the SiNbx family versus Nb2O5; e.g., SiNb20 led to 73% C8 selectivity at 53% Fur conversion, versus 60% C8 selectivity at 47% conversion for Nb2O5 using the same mass of catalyst (or 29% C8 selectivity at 18% Fur conversion for Nb2O5 used in a comparable amount of niobium). The combination of Nb2O5 nanoparticles with silica matrices seems to advantageously result in composites with distributed nanoparticles and enhanced specific surface areas. Nevertheless, the Nb2O5/TUD-1 composite performed inferiorly to SiNb20. Specifically, the former led to 45%/46% Fur conversion at 5 h/7 h (versus 53%/62% conversion for SiNb20), and 40% total bioproduct yield between 5 and 7 h (whereas SiNb20 led to 51%/60% yield at 5 h/7 h). Moreover, catalyst stability studies indicated partial loss of activity of the reused Nb2O5/TUD-1 and Nb2O5 solids (please see the ESI† (Fig. S3) for details).
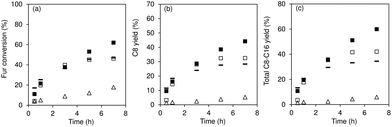 |
| | Fig. 6 Conversion (a) and product yields (b and c) for Fur condensation at 140 °C, in the presence of SiNb20 (solid squares), Nb2O5/TUD-1 (open squares) and Nb2O5 on the basis of the same mass of catalyst (−) or comparable amount of niobium (open triangle). Reaction conditions: 1.6 M Fur in acetone, 29.7 gcat L−1 for the composite and SiNb20, and ca. 9.5 gcat L−1 for Nb2O5 (open triangles). | |
SiNbx versus literature data for SiOx-based catalysts.
Table S6† compares the results for SiNb42 to literature data for different silica based solid catalysts tested for the target reaction.41,42,43–45,60,73,76–78,80–82 Fair, clear comparisons are difficult to establish due to the different reaction conditions used in the different studies, and sometimes catalytic stability tests were not reported. Hence, this section is not intended to rank catalysts, but instead to give a summarized overview. The types of SiOx-based catalysts reported were essentially fully inorganic zeolites/zeotypes (FAU, BEA, MFI, MWW, LTL topologies) and organic–inorganic hybrids (e.g., organo-functionalised silicas), and were mostly base catalysts possessing alkali or alkaline earth metals, or organic base groups (which may present drawbacks, such as the possible poisoning by impurities coming from the upstream acid-catalysed industrial production of Fur, insufficient stability towards the thermal regeneration of the catalyst and/or poisoning by atmospheric CO2). The C8 yields ranged from 14% at 100 °C (entry 14)77 to 95% at 160 °C (entries 15 and 16).42 The results and reaction conditions of entries 15 and 16 are identical, and regard zeolites treated with KNO3 (catalyst:furfural (Cat:Fur) mass ratio = 0.31, initial molar concentration of Fur ([Fur]0) = 1.21 M and 160 °C/1 h), which led to at least 95% C8 yield. For SiNb42, using a lower Cat:Fur ratio and higher [Fur]0 (i.e., more demanding conditions), the C8 yield reached 71% at 160 °C/5 h (entry 4). An aluminosilicate MFI zeolite possessing NbOx clusters led to 80% C8 yield at 87% Fur conversion, 160 °C/2 h (entry 5); in relation to the conditions used for SiNb42, the Cat:Fur ratio was higher and [Fur]0 was lower (kinetically favourable) for the zeotypes.60 Besides catalytic activity and product yields, the stability of the catalysts is important from a practical point of view. The SiNbx type materials were stable whereas, for example, Ca-ZSM-5 (entry 12),81 1.1 K-N-BEA (entry 13)81 and 20K2O/12wt% MgAl-SBA-15 (entry 19)82 suffered relatively rapid deactivation,80 and, on the other hand, no catalytic stability tests were reported for MCM-22 (entry 11).45
Conversion routes to biobased esters
The catalytic performances were further explored for different reaction routes of Fur derivatives (Scheme 1), namely, the esterification of levulinic acid (LA) and valeric acid (VA) in ethanol (derivable from carbohydrate biomass, in a fully integrated biobased approach) to produce ethyl levulinate (EL) and ethyl valerate (EV), respectively, and, on the other hand, the integrated conversion of α-angelica lactone (AnL) to LA and EL (SiNbx with x in the range 20–82, at 140 °C). Ethanol acts as the solvent and reactant in these systems; residual amounts of ethanol degradation products were formed, namely 1,1-diethoxyethane and 1,1,1-triethoxyethane in a total yield of less than 0.18% for the AnL reaction, and less than 0.23% for the LA and VA reactions, at 140 °C (at 100% substrate conversion, catalyst SiNb42), suggesting very good ethanol consumption efficiency.
Integrated conversion of α-angelica lactone.
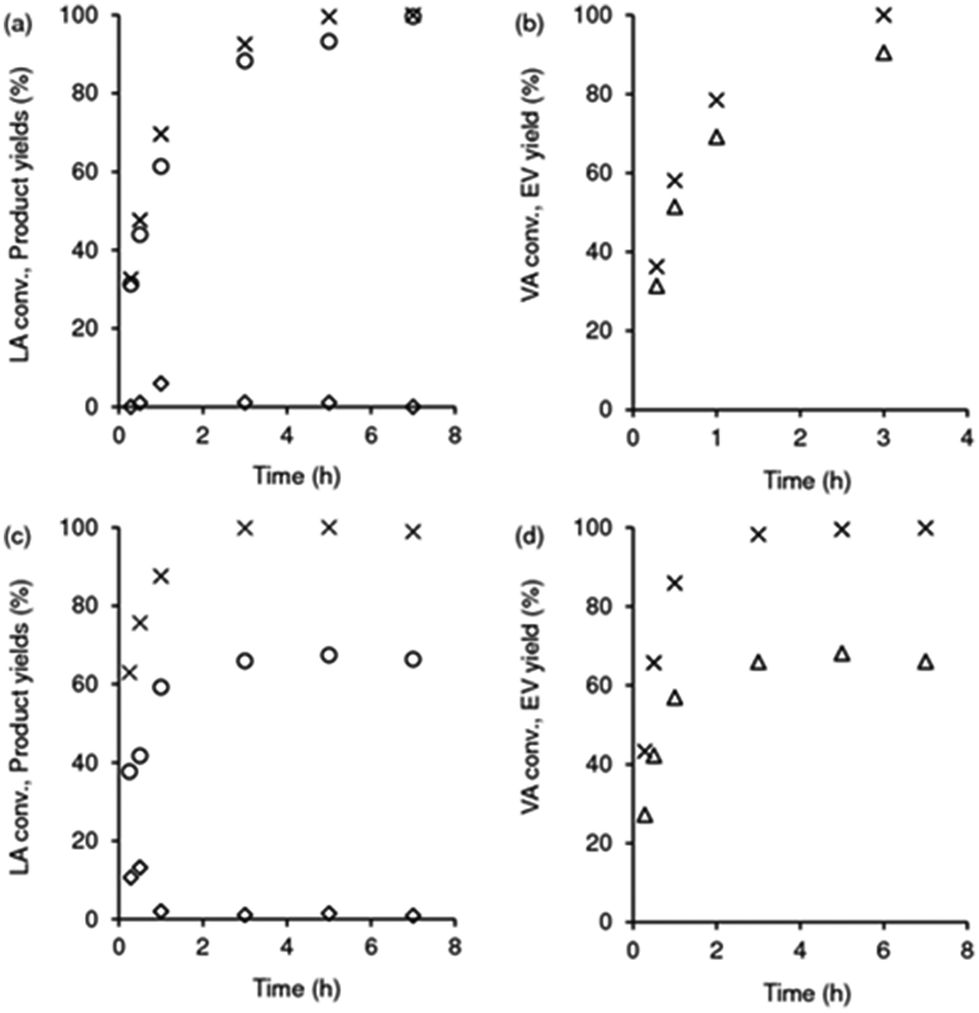
The integrated conversion of AnL over SiNbx, at 140 °C, gave mainly LA, pseudoLA and EL (Fig. 7c and Scheme 3). β-Angelica lactone (bAnL), an isomer of AnL, was present in minor amounts (<1% yield). The kinetic profiles (for SiNb42 and Nb2O5 in Fig. 7a and b, respectively) showed that LA and pseudoLA were initially formed in a total yield of 49% at 15 min, which was higher than that of EL (44% yield at 15 min). Over time, the total yield of LA plus pseudoLA decreased (reaching 0% at 7 h), which was accompanied by increasing EL yield (reached 90% at 7 h). These results suggested that AnL was, to a certain extent, converted to EL via the intermediate formation of LA. This was somewhat supported by the kinetic model developed based on the mechanistic proposal in Scheme 3, which fitted the experimental results reasonably well (Fig. 7a and b for SiNb42 and Nb2O5; kinetic constants in Table 2). The model predicted that the conversion of AnL to LA (k1) was faster than AnL to EL (k2). On the other hand, EL formation from AnL (k2; possibly via alcoholysis83) was more favourable than via the esterification of LA (k5);84–87 the latter was the slowest step. For bulk Nb2O5 the kinetic constant of AnL decomposition (k7) was comparable to that of AnL to EL (k2), whereas for SiNb42 it was much lower. Hence, Nb2O5 performed inferiorly to SiNb42 (Fig. 7a), in parallel to that verified with Fur.
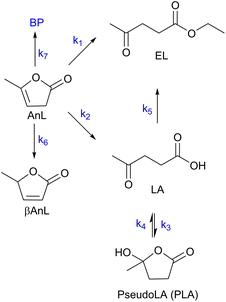 |
| | Scheme 3 Mechanistic proposal for the integrated reaction of AnL (BP are byproducts of the decomposition of AnL). | |
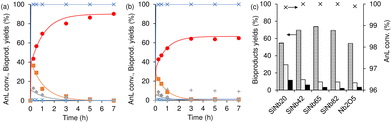 |
| | Fig. 7 Kinetic profiles (a, b) of the integrated reaction of AnL, in the presence of SiNb42 (a) or Nb2O5 (b): AnL conversion (×), and yields of EL (●), LA (■), pseudoLA (◆) and βAnL (*). In (a) and (b), the lines are the calculated kinetic curves based on the kinetic model. (c) AnL conversion (x) and bioproduct distributions at 1 h for the SiNbx and Nb2O5 catalysts: yields of EL (stripped bars) and LA (light grey bars), pseudoLA (black bars). Reaction conditions: 0.35 M AnL in ethanol, 10 gcat L−1, 140 °C. | |
Table 2 Kinetic modelling results (kinetic constants and objective function (Fobj)) for the integrated conversion of AnL, in the presence of SiNb42 or Nb2O5, at 140 °C
| Kinetic constant |
Step |
SiNb42 ki (L gcat−1 h−1) |
Nb2O5ki (L gcat−1 h−1) |
|
k
1
|
AnL-LA |
252.1501 |
103.5654 |
|
k
2
|
AnL-EL |
170.7505 |
96.8608 |
|
k
3
|
LA-PLA |
28.0045 |
17.3278 |
|
k
4
|
PLA-LA |
99.5136 |
80.7796 |
|
k
5
|
LA-EL |
0.1129 |
0.1289 |
|
k
6
|
AnL-βAnL |
1.7176 |
3.5907 |
|
k
7
|
AnL-BP |
45.2952 |
96.8126 |
|
F
obj
|
— |
0.0018 |
0.0012 |
The influence of the Nb content (x) on the AnL reaction (Fig. 7c) presented some similarities to that verified with Fur as the substrate (Fig. 1). Specifically, for x in the range 20–82, 100% AnL conversion was reached within 1 h, although the EL yield increased with increasing x up to 65 (55% EL yield (x = 20) < 70% (x = 42) < 74% (x = 65)) and then decreased for x = 82 (69%), being lowest for Nb2O5 (54%).
The catalyst stability was also checked for AnL conversion (SiNb42, Fig. S11†). The catalytic results were somewhat comparable for four consecutive batch runs, suggesting relatively good catalyst stability. As mentioned in the Introduction, water is likely present in biomass conversion processes. In the condensation and esterification systems, water is a coproduct which did not seem to affect catalytic stability (Fig. 5 and Fig. S11†). To further check the effect of water, a catalytic test was carried out in which 5 mol% water (relative to ethanol) was added to the reaction mixture (Fig. S11†). Additionally, the catalyst was consecutively reused under these conditions (SiNb42, 140 °C). On the one hand, a comparative study for the reaction without and with the addition of water indicated a slight increase in LA yield when water was added (from 5% to 12% yield, at 3 h). Water seemed to favour the kinetics of the AnL-to-LA step, which agrees with the literature; e.g., it was reported for an acid ion exchange resin catalyst, that the addition of 5 mol% water (relative to AnL) favoured LA formation.84 On the other hand, the consecutive batch runs with the addition of water led to similar results (73–78% EL yield and 12–15% LA yield, 3 h; Fig. S11†), suggesting that the presence of water was not detrimental to catalytic stability.
To the best of our knowledge, SiNbx are the first Si,Nb-oxide catalysts without other metals, studied for the integrated conversion of AnL to alkyl levulinates, in alcohol media. The only two literature studies of fully inorganic SiOx based acid catalysts tested for the AnL/ethanol reaction concerned perchloric acid supported on silica gel (HClO4–SiO2) and H-beta-14.5 (entries 3, 4, Table S7†).87,88 Although HClO4–SiO2 led to a high EL yield of 91% at 90 °C/2 h, the catalyst stability was not reported.88 On the other hand, H-Beta-14.5 (initial cat:AnL mass ratio = 0.4; [AnL]0 = 0.28 M, entry 4) led to 10% EL yield and 22% total yield of LA plus pseudoLA, at 95% conversion, 110 °C/1.5 h.87 For comparison, SiNb42 was tested at 110 °C using (more demanding) lower catalyst load and higher initial concentration of AnL (cat:AnL mass ratio = 0.28, [AnL]0 = 0.35 M), and led to superior results (58% EL yield and 34% total yield of LA plus pseudoLA, at 96% conversion, reached within 1 h).
Esterification of biobased carboxylic acids.
As referred above, besides the Fur and AnL systems, the LA and VA esterification are also relevant processes involving acid catalysis, in the context of a furfural integrated biorefinery concept. The reaction of LA/ethanol gave EL in approximately quantitative yield within 7 h, at 140 °C (Scheme 1 and Fig. 8a). The bioproduct 5-hydroxy-5-methyldihydrofuran-2(3H)-one (pseudoLA, which is an isomer of LA) was formed in <1% yield. In relation to SiNb42, pure Nb2O5 performed inferiorly, leading to 66% EL yield at 100% LA conversion, 3 h (Fig. 8c); increasing the reaction time up to 7 h did not alter the catalytic results (i.e., EL was stable) and the material balance closed in ca. 69%.
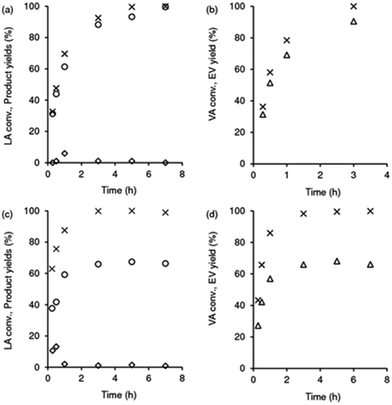 |
| | Fig. 8 Esterification of levulinic acid (LA; a and c) and valeric acid (VA; b and d), in the presence of SiNb42 (a and b) or Nb2O5 (c and d): (a and c) LA conversion (×), yields of EL (o) and pseudoLA (◊); (b and d) VA conversion (×), EV yield (Δ). Reaction conditions: 0.35 M substrate in ethanol, 10 gcat L−1, 140 °C. | |
Fig. 9a shows the influence of the Nb content (x) of the SiNbx catalysts on the LA reaction (Fig. 9a), which indicated that higher LA conversions and EL yields were reached for x = 42 and 65; initial activity was 24–25 mmol gcat−1 h−1, and EL yield reached 61–68% at 70–71% conversion, 1 h/140 °C. The catalysts with x = 20 and 82 performed inferiorly to SiNb42 and SiNb65, which correlated with the lower amount of total acid sites of the former (156–195 μmol g−1) in relation to the latter (252–267 μmol g−1, Table S4†). The addition of 5 mol% H2O to the LA system with SiNb42 as catalyst did not considerably affect the esterification reaction kinetics (Fig. S12a†).
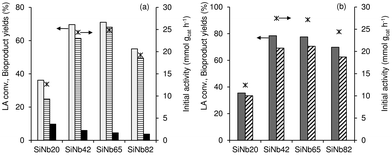 |
| | Fig. 9 Influence of the Nb content (×) of the SiNbx catalysts on the performances for (a) LA and (b) VA esterification: (a) initial activity (*), LA conversion (light grey bars), yields of EL (stripped bars) and pseudoLA (black bars); (b) initial activity (*), VA conversion (dark grey bars), EV yield (stripped bars). Reaction conditions: 0.35 M substrate in ethanol, 10 gcat L−1, 1 h, 140 °C. | |
To the best of our knowledge, these are the first fully inorganic Si,Nb oxide catalysts tested for LA/ethanol esterification. Table S8† presents the literature data for fully inorganic silica-based (e.g., modified SiO2, SBA-15, MCM-41, KIL-2) catalysts tested for LA/ethanol conversion. Different reaction conditions were used in the different studies, which makes clear comparisons difficult. Nevertheless, based on EL yields reported, SiNb42 seems a promising catalyst; EL yields reaching 92–98% were reported for WO3-SBA-1689 (entry 6), HClO4–SiO2 (entry 11)90 and silicotungstic acid on silica gel spheres (catalyst stability was not reported, entry 16),91 in the temperature range of 85–250 °C.
With VA as the substrate, ethyl valerate (EV) was formed in 90% yield at 100% conversion, 140 °C/3 h (Fig. 8b). Nb2O5 performed inferiorly, leading to a lower EV yield of 63% at 100% conversion, 3 h (Fig. 8d); no other products were formed in significant amounts and increasing the reaction time to 7 h did not influence the EV yield (i.e., the byproducts do not seem to be formed from EV). Hence, SiNb42 was a more selective catalyst than Nb2O5.
In parallel to that observed for the remaining substrates in the presence of SiNbx, the catalysts with x = 42 and 65 possessed higher initial activity for VA conversion to EV than those with x = 20 and 82 (Fig. 9b), correlating with the higher amount of total acid sites of the former two catalysts. On the other hand, the addition of 5 mol% H2O to the VA system did not considerably affect the esterification reaction kinetics (Fig. S12b†).
To the best of our knowledge, there is only one study for VA esterification using a Si,Nb-oxide catalyst, but it also possessed aluminium, namely a mesoporous niobium aluminosilicate of the type Nb,Al-SBA-15, which led to up to 35% VA conversion at 150 °C (cat:VA mole ratio = 0.54, [VA]0 = 0.43 M, microwave heating 300 W for 15 min; entry 4, Table S9†); for these materials, higher VA conversions were reached by introducing 10 wt% fluoride (using NH4F) which led to up to ca. 55% conversion at 120 °C, under microwave heating (300 W for 15 min; entry 5, Table S9†).92 Overall, the superior performance of SiNb42 and SiNb65 in relation to Nb2O5 for the reactions of LA and VA to bioesters, parallels that verified for the other reaction systems of Fur and AnL, and these materials may be promising for broader applications.
Conclusions
Catalytic, mechanistic and kinetic modelling studies of the conversion of vegetable biomass derived furanic compounds (furfural (Fur) and α-angelica lactone (AnL)) and carboxylic acids (levulinic acid (LA) and valeric acid (VA)), via different routes which involve aldol condensation, esterification and furan ring-opening reactions, were successfully carried out in the presence of versatile silica-niobia nanostructured catalysts SiNbx (obtainable via sol–gel synthesis without templates). These catalysts performed superiorly to bulk Nb2O5 and the composite consisting of Nb2O5 nanoparticles embedded in a mesoporous siliceous TUD-1 type matrix (synthesised without surfactants or expensive polymeric templates). The calcined SiNbx generally performed superiorly to the respective non-calcined materials, and, on the other hand, an intermediate at.% of Nb relative to Si (x value) seemed favourable for enhancing acidity and improving the catalytic performances. The catalytic Fur condensation route was selective towards 4-(furan-2-yl)but-3-en-2-one (C8), with high acetone consumption efficiency.
SiNbx are, to the best of our knowledge, the first fully inorganic Si,Nb oxide catalysts reported for the conversion systems AnL/alcohol, VA/alcohol and LA/ethanol, leading to biobased esters with promising results and good ethanol consumption efficiency: up to 90% yield of ethyl levulinate (EL) was reached at 100% AnL conversion, 140 °C/7 h; 100% EL yield at 100% LA conversion, 140 °C/7 h; and 90% yield of ethyl valerate at 100% VA conversion, 140 °C/3 h.
Overall, the results show the potential of the SiNbx materials as versatile catalysts for various processes involving acid catalysis, of relevance within a furfural integrated biorefinery concept. These types of nanostructured catalytic materials are obtainable via a fast, versatile, one-pot synthesis (e.g., different metals may be incorporated, with different compositions). It is envisaged that the reaction and substrate scopes may be considerably broadened in the context of the chemical valorisation of biomass using these types of materials.
Author contributions
Margarida M. Antunes: investigation, methodology, formal analysis, conceptualization, writing – original draft, review and editing, and validation. Pedro S. Cabanelas: calculation and formal analysis. Kai Skrodczky: investigation. Nicola Pinna: conceptualization, project administration, and validation. Patrícia A. Russo: conceptualization, project administration, supervision, and validation. Anabela A. Valente: conceptualization, resources, project administration, supervision, formal analysis, validation, and writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MCTES (PIDDAC). The position held by M. M. A. was funded by national funds (OE), through FCT, I. P., in the scope of the framework contract envisaged in the numbers 4, 5 and 6 of article 23 of the Decree-Law 57/2016 of 29 August, changed by Law 57/2017 of 19 July (https://doi.org/10.54499/DL57/2016/CP1482/CT0062).
References
-
A. K. Mathew, A. Abraham, K. K. Mallapureddy and R. K. Sukumarn, in Waste Biorefinery, Potential and Perspectives, ed. T. Bhaskar, A. Pandey, S. V. Mohan, D.-J. Lee and S. K. Khanal, Elsevier B. V., 1st edn, 2018, pp. 267–297 Search PubMed
 .
.
-
K. J. Zeitsch, The chemistry and technology of furfural and its many by-products, Elsevier B.V., Amsterdam, 1st edn, vol. 4, 2000 Search PubMed .
- S. Global, Furfural: Chemical Economics Handbook, https://www.spglobal.com/commodityinsights/en/ci/products/furfural-chemical-economics-handbook.html, (accessed 7 November 2023).
-
M. P. Alphy, P. A. Balakumaran, R. Sindhu, A. Pandey and P. Binod, in Biomass, Biofuels, Biochemicals- Circular Bioeconomy; Technologies for Biofuels and Biochemicals, ed. S. Varjani, T. Bhaskar, D. C. W. Tsang, P. Ashok; and S. V. Mohan, Elsevier B.V., 2022, pp. 163–187 Search PubMed .
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sábada and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC .
-
C. C. Truong, V. K. Verma, P. Mishra, Y.-W. Suh and D. K. Mishra, in Biomass, Biofuels, Biochemicals: Biochemicals and Materials Production from Sustainable Biomass Resources, ed. H. Li, A. Pandey, S. Saravanamurugan and S. Elumalai, Elsevier B.V., Amesterdam, Netherlands, 2022, pp. 673–720 Search PubMed .
- B. L. Wegenhart, L. Yang, S. C. Kwan, R. Harris, H. I. Kenttämaa and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2742–2747 CrossRef CAS PubMed .
- A. Bohre, S. Dutta, B. Saha and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2015, 3, 1263–1277 CrossRef CAS .
- J. He, Q. Qiang, S. Liu, K. Song, X. Zhou, J. Guo, B. Zhang and C. Li, Fuel, 2021, 306, 121765 CrossRef CAS .
- H. Zang, K. Wang, M. Zhang, R. Xie, L. Wang and E. Y.-X. Chen, Catal. Sci. Technol., 2018, 8, 1777–1798 RSC .
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed .
- E. Ketabchi, L. Pastor-Pérez, T. R. Reina and H. Arellano-García, Renewable Energy, 2020, 156, 1065–1075 CrossRef CAS .
- A. Zitouni, R. Bachir, W. Bendedouche and S. Bedrane, Fuel, 2021, 297, 120783 CrossRef CAS .
- A. Tampieri, K. Föttinger, N. Barrabés and F. Medina, Appl. Catal., B, 2022, 319, 121889 CrossRef CAS .
- D. S. Desai and G. D. Yadav, Ind. Eng. Chem. Res., 2019, 58, 16096–16105 CrossRef CAS .
- R. Huang, J. Chang, H. Choi, J. M. Vohs and R. J. Gorte, Catal. Lett., 2022, 152, 3833–3842 CrossRef CAS .
- A. Bohre, M. I. Alam, K. Avasthi, F. Ruiz-Zepeda and B. Likozar, Appl. Catal., B, 2020, 276, 119069 CrossRef CAS .
- I. Sábada, M. Ojeda, R. Mariscal, J. L. G. Fierro and M. L. Granados, Appl. Catal., B, 2011, 101, 638–648 CrossRef .
- O. Kikhtyanin, V. Korolova, A. Spencer, L. Dubnová, B. Shumeiko and D. Kubička, Catal. Today, 2021, 367, 248–257 CrossRef CAS .
- A. Parejas, D. Cosano, J. Hidalgo-Carrillo, J. R. Ruiz, A. Marinas, C. Jiménez-Sanchidrián and F. J. Urbano, Catalysts, 2019, 9, 203 CrossRef .
- O. Kikhtyanin, Z. Tišler, R. Velvarská and D. Kubička, Appl. Catal., A, 2017, 536, 85–96 CrossRef CAS .
- J. Kocík, K. Frolich, P. Ilona; and J. Horácek, J. Chem. Technol. Biotechnol., 2019, 94, 435–445 CrossRef .
- A. Tampieri, M. Lilic, M. Constantí and F. Medina, Crystals, 2020, 10, 833 CrossRef CAS .
- N. Sosa, N. Chanlek and J. Wittayakun, Ultrason. Sonochem., 2020, 62, 104857 CrossRef CAS PubMed .
- S. Arhzaf, M. N. Bennani, S. Abouarnadasse, J. Houssaini, H. Ziyat and O. Qabaqous, Moroccan J. Chem., 2021, 9, 614–627 CAS .
- V. Korolova, O. Kikhtyanin, M. Veselý, D. Vrtiška, I. Paterová, V. Fíla, L. Čapek and D. Kubička, Catalysts, 2021, 11, 992 CrossRef CAS .
- O. Kikhtyanin, L. Hora and D. Kubicka, Catal. Commun., 2015, 58, 89–92 CrossRef CAS .
- H. Liu, W. Xu, X. Liu, Y. Guo, Y. Guo, G. Lu and Y. Wang, Kinet. Catal., 2010, 51, 75–80 CrossRef CAS .
- L. Smoláková, K. Frolich, J. Kocík, O. Kikhtyanin and L. Čapek, Ind. Eng. Chem. Res., 2017, 56, 4638–4648 CrossRef .
- K. Jaroslav, K. Jiří, A. Uliana and T. Zdeněk, Catalysts, 2020, 10, 1484 CrossRef CAS .
- L. Hora, O. Kikhtyanin, L. Čapek, O. Bortnovskiy and D. Kubička, Catal. Today, 2015, 241, 221–230 CrossRef CAS .
- P. Hájková and Z. Tišler, Catal. Lett., 2017, 147, 374–382 CrossRef .
- L. Hora, K. Vendula, O. Kikhtyanin, O. Bortnovskiy and D. Kubicka, Catal. Today, 2014, 223, 138–147 CrossRef CAS .
- M. Li, X. Xu, Y. Gong, Z. Wei, Z. Hou, H. Li and Y. Wang, Green Chem., 2014, 16, 4371–4377 RSC .
- P. A. Zapata, J. Faria, M. P. Ruiz and D. E. Resasco, Top. Catal., 2012, 55, 38–52 CrossRef CAS .
- L. Faba, E. Díaz and S. Ordóñez, Biomass Bioenergy, 2013, 56, 592–599 CrossRef CAS .
- L. Faba, E. Díaz and S. Ordóñez, Appl. Catal., B, 2012, 113–114, 201–211 CrossRef CAS .
- S. Ordóñez, E. Díaz, M. León and L. Faba, Catal. Today, 2011, 167, 71–76 CrossRef .
- W. Shen, G. A. Tompsett, K. D. Hammond, R. Xing, D. Fulya, C. P. Grey, W. C. Conner Jr., S. M. Auerbach and G. W. Huber, Appl. Catal., A, 2011, 392, 57–68 CrossRef CAS .
- C. Ramirez-Barria, A. Guerrero-Ruiz, E. Castillejos-López, I. Rodríguez-Ramos, J. Durand, J. Volkman and P. Serp, RSC Adv., 2016, 6, 54293–54298 RSC .
- T. Kondratowicz, S. Slang, L. Dubnová, O. Kikhtyanin, P. Belina and L. Capek, Appl. Clay Sci., 2022, 216, 106365 CrossRef CAS .
- A. Al-Ani, C. Freitas and V. Zholobenko, Microporous Mesoporous Mater., 2016, 293, 109805 CrossRef .
- M. Su, W. Li, T. Zhang, H. S. Xin, S. Li, W. Fan and L. Ma, Catal. Sci. Technol., 2017, 7, 3555–3561 RSC .
- O. Kikhtyanin, V. Kelbichová, D. Vitvarová, M. Kubů and D. Kubička, Catal. Today, 2014, 227, 154–162 CrossRef CAS .
- O. Kikhtyanin, P. Chlubná, T. Jindrová and D. Kubicka, Dalton Trans., 2014, 43, 10628–10641 RSC .
- O. Kikhtyanin, D. Kubička and J. Čejka, Catal. Today, 2015, 243, 158–162 CrossRef CAS .
- M. Y. Talanova, V. A. Yarchak and E. A. Karakhanov, Russ. J. Appl. Chem., 2019, 92, 857–864 CrossRef CAS .
- M. Hara, Bull. Chem. Soc. Jpn., 2014, 87, 931–941 CrossRef CAS .
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed .
- K. Tanabe and S. Okazaki, Appl. Catal., A, 1995, 133, 191–218 CrossRef CAS .
- E. I. García-López, F. R. Pomilla, B. Megna, M. L. Testa, L. F. Liotta and G. Marcì, Nanomaterials, 2021, 11, 1821 CrossRef PubMed .
- C. García-Sancho, J. M. Rubio-Caballero, J. M. Mérida-Robles, R. Moreno-Tost, J. Santamaría-González and P. Maireles-Torres, Catal. Today, 2014, 234, 119–124 CrossRef .
- J. L. Vieira, M. Almeida-Trapp, A. Mithöfer, W. Plass and J. M. R. Gallo, Catal. Today, 2020, 344, 92–101 CrossRef CAS .
- K. Skrodczky, M. M. Antunes, X. Han, S. Santangelo, G. Scholz, A. A. Valente, N. Pinna and P. A. Russo, Commun. Chem., 2019, 2, 129 CrossRef CAS .
- L. C. Kao, W. C. Kan, R. M. Martin-Aranda, M. O. Guerrero-Perez, M. Bañares and S. Y. H. Liou, Catal. Today, 2020, 356, 80–87 CrossRef CAS .
- S. Kang, R. Miao, J. Guo and J. Fu, Catal. Today, 2021, 374, 61–76 CrossRef CAS .
- L. Oliveira, M. Pereira, A. P. Heitman, J. Filho, C. Oliveira and M. Ziolek, Molecules, 2023, 28, 1527 CrossRef CAS PubMed .
-
Q. Xia and Y. Wang, in Nanoporous Catalysts for Biomass Conversion, ed. C. V. Stevens, F.-S. Xiao and L. Wang, John Wiley, 2017, pp. 253–281 Search PubMed .
- Y. Jing, Y. Xin, Y. Guo, X. Liu and Y. Wang, Chin. J. Catal., 2019, 40, 1168–1177 CrossRef CAS .
- E. Yuan, W. Dai, G. Wu, N. Guan and L. Li, Microporous Mesoporous Mater., 2020, 305, 110361 CrossRef CAS .
- K. Skrodczky, M. M. Antunes, Q. Zhu, A. A. Valente, N. Pinna and P. A. Russo, Nanomaterials, 2023, 13, 3046 CrossRef CAS PubMed .
- Expert Market Research (EMR), Global Ethyl Levuliante Market to Reach USD 14 Million by 2026. Aided by Their Rising Demand From the Fragrance Industry, https://www.expertmarketresearch.com/reports/ethyl-levulinate-market, (accessed 9 December 2023).
- D. D. M. Di Bucchianico, Y. Wang, J.-C. Buvat, Y. Pan, V. C. Moreno and S. Leveneur, Green Chem., 2022, 24, 614–646 RSC .
- Y. Tian, F. Zhang, J. Wang, L. Cao and Q. Han, Bioresour. Technol., 2021, 342, 125977 CrossRef CAS PubMed .
- J. F. L. Silva, R. Grekin, A. P. Mariano and R. M. Filho, Energy Technol., 2018, 6, 613–639 CrossRef .
- E. Ahmad, M. I. Alam, K. K. Pant and M. A. Haider, Green Chem., 2016, 18, 4804–4823 RSC .
- R. Pothu, R. Gundeboyina, R. Boddula, V. Perugopu and J. Ma, New J. Chem., 2022, 46, 5907–5921 RSC .
- M. Sajid, U. Farooq, G. Bary, M. M. Azim and X. Zhao, Green Chem., 2021, 23, 9198–9238 RSC .
- M. M. Antunes, P. Neves, A. Fernandes, S. Lima, A. F. Silva, M. F. Ribeiro, C. M. Silva, M. Pillinger and A. A. Valente, Catal. Sci. Technol., 2016, 6, 7812–7829 RSC .
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141–148 CrossRef CAS .
- D. M. Gomes, P. Neves, M. M. Antunes, A. J. S. Fernandes, M. Pillinger and A. A. Valente, Catalysts, 2022, 12, 12 CrossRef .
- M. Xu, S. Célerier, J.-D. Comparot, J. Rousseau, M. Corbet, F. Richard and J.-M. Clacens, Catal. Sci. Technol., 2019, 9, 5793–5802 RSC .
- W. Li, M. Su, T. Zhang, Q. Ma and W. Fan, Fuel, 2019, 237, 1281–1290 CrossRef CAS .
- L.-P. Li, Z. Fang, X. Kong and W. J. Cong, Mol. Catal., 2021, 515, 111893 CrossRef CAS .
- A. Kumar, R. Bal and R. Srivastava, Energy Fuels, 2021, 35, 11366–11381 CrossRef CAS .
- R. Balaga, P. Yan, K. Ramineni, H. Du, Z. Xia, M. R. Marri and Z. C. Zhang, Appl. Catal., A, 2022, 648, 118901 CrossRef CAS .
- O. Kikhtyanin, Y. Ganjkhanlou, D. Kubička, R. Bulánek and J. Čejka, Appl. Catal., A, 2018, 549, 8–18 CrossRef CAS .
- P. Gandhi, B. Saha, S. Vedachalam and A. K. Dalai, Sustainable Energy Fuels, 2023, 4260–4272 RSC .
- L. Faba, E. Díaz and S. Ordõñez, ChemSusChem, 2013, 6, 463–473 CrossRef CAS PubMed .
- O. Kikhtyanin, R. Bulánek, K. Frolich, J. Cejka and D. Kubička, J. Mol. Catal. A: Chem., 2016, 424, 358–368 CrossRef CAS .
- X. Fang, Z. Wang, W. Song and S. Li, J. Taiwan Inst. Chem. Eng., 2020, 108, 16–22 CrossRef CAS .
- M. Arumugam, O. Kikhtyanin, A. Osatiashtiani, V. Kyselová, V. Fila, I. Paterova, K. L. Wong and D. Kubička, Sustainable Energy Fuels, 2023, 7, 3047–3059 RSC .
- P. Latos, A. Szelwicka, S. Boncel, S. Jurczyk, M. Swadźba-Kwaśny and A. Chrobok, ACS Sustainable Chem. Eng., 2019, 7, 5184–5191 CrossRef CAS .
- M. G. Al-Shaal, W. Ciptonugroho, F. J. Holzhäuser, J. B. Mensah, P. J. C. Hausoul and R. Palkovits, Catal. Sci. Technol., 2015, 5, 5168–5173 RSC .
- M. M. Antunes, S. Lima, A. Fernandes, A. L. Magalhães, P. Neves, C. M. Silva, M. F. Ribeiro, D. Chadwick, K. Hellgardt, M. Pillinger and A. A. Valente, ChemCatChem, 2017, 9, 2747–2759 CrossRef CAS .
- W. Ciptonugroho, J. B. Mensah, G. Al-Shaal and R. Palkovits, Chem. Pap., 2023, 77, 3769–3778 CrossRef CAS .
- M. Paniagua, J. A. Melero, J. Iglesias, G. Morales, B. Hernándes and C. López-Aguado, Appl. Catal., A, 2017, 537, 74–82 CrossRef CAS .
- S. B. Onkarappa, N. S. Bhat and S. Dutta, Biomass Convers. Biorefin., 2020, 10, 849–856 CrossRef CAS .
- F. Ciardelli, M. Michelotti, A. Altomare and E. Roland, J. Mol. Catal. A: Chem., 2017, 426, 30–38 CrossRef .
- F. Yang and J. Tang, ChemistrySelect, 2019, 4, 1403–1409 CrossRef CAS .
- X. Kong, S. Wu, X. Li and J. Liu, Energy Fuels, 2016, 30, 6500–6504 CrossRef .
- M. Blanco-Sánchez, E. Pfab, N. Lázaro, A. M. Balu, R. Luque and A. Pineda, Front. Chem., 2020, 8, 42 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: chemical structure, abbreviations and dimensions, carbon balances, characterisation of Nb2O5-com, kinetic models (kinetic constants, calculated curves), acetone decomposition products, PXRD of the used catalyst, Reaction temperature effect, catalytic stability (AnL), tables comparing literature catalytic data for the reactions of Fur, AnL, LA and VA. See DOI: https://doi.org/10.1039/d4gc00207e |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Kai
Skrodczky
b,
Pedro S.
Cabanelas
a,
Nicola
Pinna
*a,
Kai
Skrodczky
b,
Pedro S.
Cabanelas
a,
Nicola
Pinna