
Advances and challenges in MXene-based electrocatalysts: unlocking the potential for sustainable energy conversion
Lei
He†
ac,
Haizheng
Zhuang†
c,
Qi
Fan†
cd,
Ping
Yu
b,
Shengchao
Wang
cd,
Yifan
Pang
e,
Ke
Chen
 cf and
Kun
Liang
*cf
cf and
Kun
Liang
*cf
aSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang 315211, China
bSchool of Electronic and Information Engineering, Ningbo University of Technology, Ningbo 315211, China
cZhejiang Key Laboratory of Data-Driven High-Safety Energy Materials and Applications, Ningbo Key Laboratory of Special Energy Materials and Chemistry, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: kliang@nimte.ac.cn
dUniversity of Chinese Academy of Sciences, 19 A Yuquan Rd, Shijingshan District, Beijing 100049, China
eDepartment of Materials Science and Engineering, the Ohio State University, Columbus, OH 43210, USA
fQianwan Institute of CNITECH, Ningbo 315336, China
First published on 22nd August 2024
Abstract
MXenes, a novel class of two-dimensional materials, have garnered significant attention for their promising electrocatalytic properties in various energy conversion applications such as water splitting, fuel cells, metal–air batteries, and nitrogen reduction reactions. Their excellent electrical conductivity, high specific surface area, and versatile surface chemistry enable exceptional catalytic performance. This review highlights recent advancements in the design and application strategies of MXenes as electrocatalysts, focusing on key reactions including hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), and nitrogen reduction reaction (NRR). We discuss the tunability of MXenes’ layered structures and surface properties through surface modification, MXene lattice substitution, defect and morphology engineering, and heterostructure construction. Despite the considerable progress, MXenes face challenges such as restacking during catalysis, stability issues, and difficulties in large-scale production. Addressing these challenges through innovative engineering approaches and advancing industrial synthesis techniques is crucial for the broader application of MXene-based materials. Our review underscores the potential of MXenes in transforming electrocatalytic processes and highlights future research directions to optimize their catalytic efficiency and stability.
 Lei He | Lei He is a master candidate in Prof. Kun Liang's research group at the Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. His research interest focuses on the preparation and catalytic properties of LDH/MXene composites. He received his bachelor's degree in polymer materials and engineering from Hunan University of Technology in 2022. |
 Haizheng Zhuang | Haizheng Zhuang is a postdoctoral research fellow at Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. He received his PhD degree in Materials Science and Technology from New Jersey Institute of Technology in 2022. His research interests focus on two-dimensional transition metal carbide and/or nitrides materials, polymeric nitrogen materials, and their potential applications in energy storage and electrocatalysis. |
 Qi Fan | Qi Fan is a PhD candidate in Prof. Kun Liang's research group at the Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. Her research interests focus on the structure editing and applications of MXenes materials based on the intercalation chemistry. She received MS degree in physical chemistry from Shandong University in 2019. |
 Ping Yu | Ping Yu is an associate professor at Ningbo University of Technology. He received his PhD degree in Microelectronics from Zhejiang University, Hangzhou, China, in 2013. His current research interests include the design, modeling, and fabrication of nanophotonic materials & devices. |
 Shengchao Wang | Shengchao Wang is a PhD candidate in Prof. Kun Liang's group at the Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. His research interests focus on the structure editing and applications of new MAX and MXene materials. He received his bachelor's degree from Hebei University of Technology in 2023. |
 Yifan Pang | Yifan Pang is an undergraduate student at Department of Materials Science and Engineering, the Ohio State University. He is a research assistant at Prof. Alan Luo's group. He conducts summer research internship in Prof. Kun Liang's research group at the Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. |
 Ke Chen | Ke Chen is an associate professor at Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. After receiving his master's degree in materials physics and chemistry from the University of Chinese Academy of Sciences in 2015. He studied for a doctorate in materials science and engineering at Seoul National University, South Korea. He returned to Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, to carry out research work in 2018 and was promoted to associate professor in 2021. His research interests focus on the structural editing of MAX and MXenes. |
 Kun Liang | Kun Liang is a professor at Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences. After receiving his PhD degree in Materials Science and Technology from University of Electronic Science and Technology of China in 2015, he moved to the University of Central Florida and Tulane University (U.S.A.) as a postdoctoral research fellow. His research interests focus on the structural editing of two-dimensional transition metal carbide and/or nitrides materials, functional control and integration, and potentials in energy storage, electrocatalysis, and flexible devices etc. |
Wider impactOur manuscript presents significant advancements in MXene-based electrocatalysts for key electrochemical reactions, including hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), and nitrogen reduction reaction (NRR). By leveraging innovative strategies such as surface modification, defect engineering, and the creation of hybrid structures, we significantly enhance the catalytic efficiency and stability of MXenes. These advancements have profound implications for sustainable energy and chemical processing technologies, offering potential solutions for renewable energy conversion, green ammonia synthesis, and advanced manufacturing. Our research bridges the gap between fundamental science and practical applications, aligning with the mission of Materials Horizons to deliver impactful, high-quality scientific contributions that address global challenges and benefit society at large. |
1. Introduction
Two-dimensional materials have attracted a lot of attention due to their unique physical, chemical, and electronic properties and have a broad potential for applications in electrocatalysis.1–3 Typically, two-dimensional materials contain several atomic layers that exhibit strong covalent in-plane bonding and weak interlayer van der Waals interactions, and the weak interlayer bonding allows for the exfoliation into thinner nanosheets consisting of several layers or monolayers.4–6 Two-dimensional materials can maximize the use of the surface area, thus increasing the catalytic active sites of electrocatalysts. Moreover, their electronic structure has a special energy band structure that enables the adsorption and activation of reactants, thus increasing the electrocatalytic efficiency.7–10Two-dimensional transition metal carbides, nitrides, and carbonitrides (MXene) are emerging two-dimensional materials obtained by etching a certain atomic layer in a layered parent, which is mostly in the MAX phase.11,12 Yury Gogotsi et al.13 produced the first MXene by selectively etching the Ti3AlC2 MAX phase's middle layer Al. The general molecular formula of the MAX phase is Mn+1AXn, where n can vary between 1 and 4; M is an early transition metal element; A is mainly a group IIIA–IVA element; X is a carbon, nitrogen or a mixture of both elements.14 The A–M interaction force in the MAX phase is weaker than that between M and X. This is because the X atoms are located in the octahedron of M, which can be seen as an alternating stack of Mn+1Xn layers with A layers. Therefore, the elements of the A-layer in the MAX phase can be selectively etched away to obtain graphene-like 2D materials. The general formula of MXene is Mn+1XnTx, where Tx represents the terminal group. The terminal group depends on the etching environment, where the anions (–OH, –O, –F, –CI, etc.) are adsorbed as terminals on the surface of the Mn+1Xn layer, reducing the surface energy.15 In the last few years, MXene has emerged in the field of electrocatalysis due to its large specific surface area, hydrophilicity, good electrical conductivity, and designability.16–18
Herein, we discuss the optimization strategies and design ideas of MXene electrocatalytic activity in recent years, including surface modification, MXene lattice substitution (MX substitution), defect engineering, morphology engineering, and heterostructure engineering (Fig. 1). In addition, representative applications of MXene-based electrocatalysts, including HER, OER, ORR, and NRR, are systematically introduced. Finally, the prospects of MXene-based electrocatalysts are proposed. We hope that this review article will provide theoretical and experimental guidance for future research on the design of various MXene-based electrocatalysts and promote the application and development of MXene-based electrocatalysts.
 | ||
| Fig. 1 Overview of design strategies and representative applications of MXene-based electrocatalysts. | ||
2. A brief overview of MXene
2.1 MXene synthesis
Recently, a gas-phase selective etching with a simple, versatile process and controllable surface terminals has been reported to utilize the strong oxidizing power of halogen and hydrogen halide gases to etch the A-layer elements of the MAX phase.28 Meanwhile, due to the low boiling point of the by-products, the separation of MXene and by-products can be realized, thus eliminating the subsequent processing steps. This strategy is expected to produce MXene on a large scale, and conducive to promoting the application range of MXene.
2.2 MXene structure
MXene inherits the hexagonal close-packed structure of the MAX phase, where X atoms are filled in the octahedral interstitial sites, and the adjacent layers are connected by van der Waals force.19,29,30Fig. 2(a) and (c) show four common MXene structures and their composition based on the periodic table of elements. However, M2X has an HCP sequence (ABABAB) for its M atom, while M3X2 and M4X3 have an FCC sequence (ABCABC).31 Corresponding to the precursor 211, 312, 413 MAX phase, there are two M6X layers to separate the A layer in the 211 structure, while in the 312 structure, the number of M6X layers is three, and in the 413 structure is four. Meanwhile, the 211 phase has a single polymorph, while the 312 phase has two (α and β), and the 413 phase has three (α, β, and γ).32 According to the M element, MXene can be divided into single transition metal and two or more transition metal doping. In the latter two elemental distributions are found: disordered solid solution and ordered diatomic structure.33 The ordered MXenes and solid solutions are produced by changing the M in the lattice of MXene structure. For the disordered solid solution, the multiple elements are randomly distributed within the M-layer. The ordered diatomic structure has two distributions: out-of-plane ordered and in-plane ordered. The properties of structure and component are favorable to regulate the catalytic active site and design ordered vacancy distribution, providing a prospect for optimizing its electrocatalytic performance.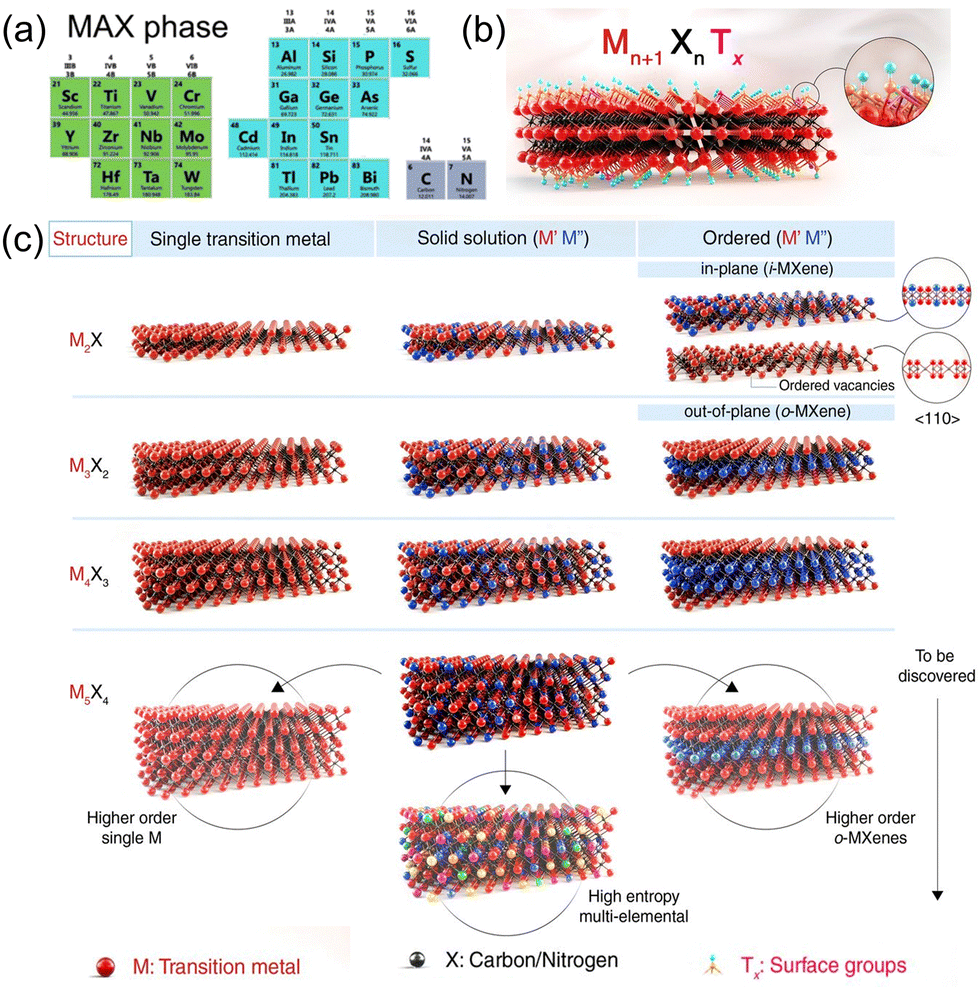 | ||
| Fig. 2 (a) Composition of various types of MAX.30 Copyright 2023, Elsevier. (b) and (c) Schematic illustration of the MXene structures.29 Copyright 2021, AAAS. | ||
2.3 Electronic properties
The electronic properties of MXene depend mainly on M, X, and surface functional groups. Most MXenes have metal-like electron transport, and the outer transition metal type and surface functional groups can cause MXene to exhibit semiconductor behavior (Fig. 2(b)).34,35 For example, Han et al.36 studied the electronic conductivity of 16 MXenes with different compositions and structures, finding that Ti-based and V-based MXenes have great conductivity whereas MXenes containing Mo or Nb have decreased conductivity. Additionally, compared to the –F and –OH terminals, –O would require 1 extra electron from the MXene surface to achieve stability, which would have bigger effects on the metallic behavior of MXenes.37 Theoretical calculations have demonstrated that,38 the terminals of MXene are closely related to work function (WF): –OH hinders WF, –O increases WF, and the trend of –F is usually between the two. Therefore, changing the type and content of transition metals and functional groups of MXene can modulate the electronic properties to obtain the best catalytic activity.2.4 Stability
The lack of stability of MXene limits the application of MXene-based materials of practical importance. In the field of electrocatalysis, the rapid oxidation of MXene affects its electrochemical properties and leads to catalytic deactivation. Zhang et al.39 demonstrated that the oxidation of Ti3C2Tx starts from the edges and proceeds inward, and its kinetics follow a single-exponential decay. Also, the oxidation process is size-dependent, with smaller-sized lamellar structures being less stable. Meanwhile, this work argues that dissolved oxygen in water is the main cause of the oxidation of MXene flakes. On the other hand, Huang et al.40 emphasized that it was water rather than dissolved oxygen that played a key role in causing MXene oxidation. Further studies have shown,41 that different MXene in their conversion to oxides in water, carbide MXene formed CH4, while Ti3CN MXene formed CH4 and NH3. The actual oxidation mechanism of MXene is still controversial and the interaction of water and dissolved oxygen with MXene needs to be further investigated.3. Design strategy of MXene-based electrocatalysts
The catalytic activity of MXene varies largely for different chemical reactions. For instance, the original MXene has a good performance for HER but has little catalytic activity for ORR/OER.42,43 Therefore, to expand the electrocatalytic application of MXene, there are many specific modifications and designs of MXene. In general, the intrinsic catalytic activity of the original MXene can be improved by surface terminals, defects, heteroatom incorporation, etc., while the number and accessibility of active sites can be increased by the morphology transformation or hybridization with other catalytic active materials. The intrinsic and hybrid strategies of MXene electrocatalytic activity, including surface modification, MX substitution, defect engineering, morphology engineering, and extremely specialized heterostructure engineering in hybridization, are discussed in detail in the following chapters.3.1 Surface modification
Surface modification of MXene is a broad strategy that can change the types, content, and distribution of functional groups, all of which will affect the electronic structure of MXene as well as the catalytic active site.The types of functional groups has a significant effect on the electrocatalytic activity of MXene, and the most common terminal species are –O, –OH, and –F. Gao et al.44 computationally studied the HER electrocatalytic activity of MXene with the –O terminal. As shown in the volcano diagram in Fig. 3(a), the interaction of bare MXene with H* is strong due to the large surface energy, which is not favorable for the release of H*. When –O terminals are added, the interaction of H* with MXene is weakened, and Ti2CO2's hydrogen adsorption Gibbs free energy subsequently approaches the optimum value. In Fig. 3(b) and (c), where the –F content varied with different etching conditions, Handoko et al.45 investigated the content effect of –F in Ti-based and Mo-based MXene on HER theoretically and experimentally. It was found that increasing the –F terminal will reduce the HER activity. This is because the –F terminal on the surface does not adsorb hydrogen, but releases hydrogen molecules by stripping hydrogen from the neighboring hydroxyl groups.46 It follows that the surface –O terminal theoretically favors the HER activity of MXene, while the –F terminal group is the opposite. The MXene obtained using fluorine-containing etchants is inevitably functionalized by functional groups such as –F, –O, and –OH on its surface, and obviously the electrocatalytic activity of such MXene needs adjustment. Therefore, Jiang et al.47 took advantage of the fact that the –OH terminal of Ti3C2Tx is less thermally stable than the –O terminal and allowed the conversion of the –OH terminal to the –O terminal through a dehydration reaction. Specifically, the layered Ti3C2Tx was immersed in KOH solution to convert Ti3C2Tx to Ti3C2(OH)x and calcined in an argon atmosphere to obtain Ti3C2Ox. As shown in Fig. 3(d), the characteristic peak of –OH at 3460 cm−1 gradually diminished with increasing calcination temperature and disappeared at about 450 °C, which verifies the transformation of Ti3C2(OH)x to Ti3C2Ox. The HER polarization curves show that Ti3C2Ox has a lower overpotential compared to Ti3C2Tx-450 and Ti3C2(OH)x (Fig. 3(e)). In addition, the effect of terminal groups on catalysis is not limited to HER, according to the theoretical calculation of DFT, and Mo2C capped by –O and –H also facilitates the NRR-catalyzed process.48
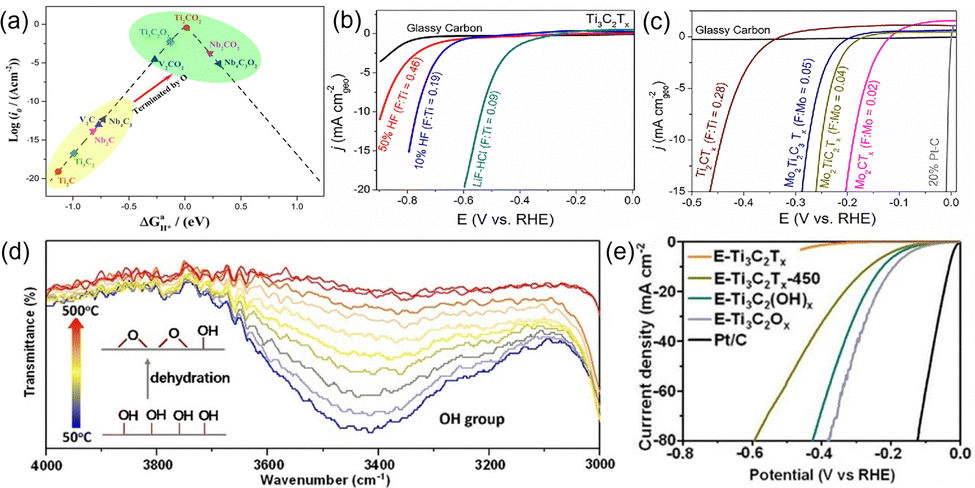 | ||
| Fig. 3 (a) Volcano curve of exchange current as a function of the average Gibbs free energy of hydrogen adsorption.44 Copyright 2017, American Chemical Society. LSVs of (b) Ti3C2Tx and (c) Mo2CTx in 0.5 M H2SO4 electrolyte.45 Copyright 2018, American Chemical Society. (d) Magnified in situ temperature-dependent FTIR spectra of the as-synthesized E-Ti3C2(OH)x in Ar atmosphere; (e) polarization curves of various MXene.47 Copyright 2019, Wiley. | ||
Alternation of the functional group on MXene represents a straightforward approach to enhance its electrocatalytic activity for HER. However, in order to pursue higher catalytic activity and application range, this is not enough. For example, MXene is not suitable for ORR/OER due to the weak interaction of –O or –F capped MXene with the intermediate, which is difficult to bind to the reactant or provide electrons.49 Although Nb2C(OH)2 exhibits high catalytic activity for ORR, the transition from –OH to –O is irreversible.49 Therefore, in addition to the adjustment of the common functional groups of MXene (–OH, –F, –O), other elements can also be introduced. Based on this, Yoon et al.46 used NaNH2 to nitride the Ti2CTx surface to obtain an efficient HER electrocatalytic material (Fig. 4(a)). NaNH2 decomposes into Na, N, and H2 at high temperature, and thus surface terminal groups of MXene will be removed by Na and H2, while Ti will be irreversibly bonded with N.50 This strategy can be extended to the electrocatalytic modification of other MXene. To eliminate inactive groups on NRR, Guo et al.51 prepared Fe-capped MXene, as shown in Fig. 4(c). The Fe terminals replace the inactive groups and reduce the surface work function while exposing more transition metal active sites for NRR catalysis.
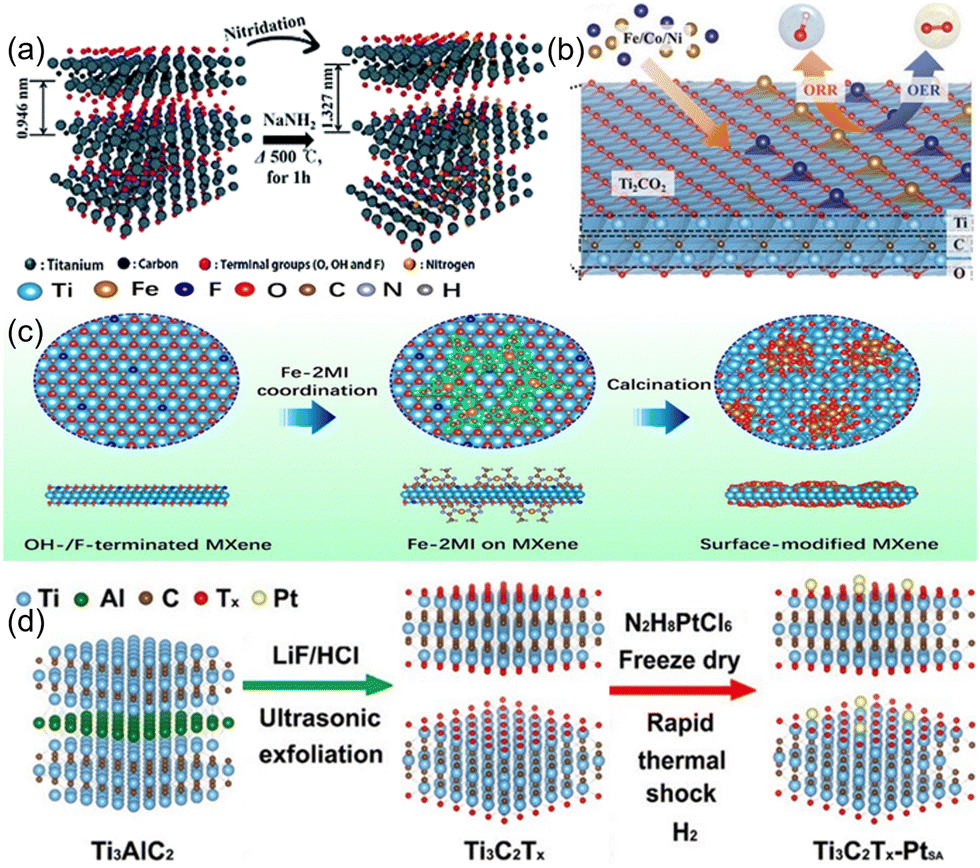 | ||
| Fig. 4 (a) Synthesis procedures for the N–Ti2CTx nanosheets and their dispersion.46 Copyright 2018, Royal Society of Chemistry. (b) Mechanisms for the conformation of Fe/Co/Ni-DACs based on Ti2CO2 and for the electrocatalytic reactions in aqueous electrolytes.52 Copyright 2021, Wiley. (c) Schematic illustration for the preparation procedure of the surface-tuned MXene/TiFeOx nanosheets.51 Copyright 2020, American Chemical Society. (d) Schematic illustration of constructing Pt single atoms on monolayer Ti3C2Tx.53 Copyright 2022, American Chemical Society. | ||
Additionally, single-atom catalysts are particularly special. By introducing metal atoms through surface doping or functional group substitution, single-atom catalysts can be synthesized on MXene surfaces, enabling electronic structure modulation and the creation of novel catalytic active sites, resulting in high atom utilization efficiency and superior catalytic activity.54 For example, He et al.55 prepared efficient Ru single-atom catalysts for HER with N and S-doped Ti3C2Tx. The present study reveals the occurrence of a robust electronic coupling between individual monatomic Ru and Ti3C2Tx MXene carriers, which is facilitated by the involvement of N and S atoms. The differential binding affinity of N and S atoms towards the Ru species contributes significantly to the observed augmentation in the electrocatalytic activity for HER. This is a good research direction to enhance the interaction of metal atoms with MXene by using heterogeneous atoms as intermediates. In addition, oxygen vacancies can also be used as a bridge to connect metal atoms with MXene. Gong et al.53 used the oxygen vacancies of Ti3C2Tx to capture the monatomic Pt to form Ti–Pt bonds by a fast thermal shock strategy (Fig. 4(d)). The produced monatomic catalysts exhibited excellent HER performance, good stability, high-quality activity, and large conversion frequency. Compared with monoatomic catalysts, diatomic catalysts have higher metal loading and more complex and flexible active sites.56 Zhang et al.52 achieved bifunctional and efficient catalysis of ORR/OER by introducing non-precious metals (e.g., Fe/Co/Ni) on the surface as metal atom sites for oxygen-capped MXene to facilitate the catalytic reaction (Fig. 4(b)). The bimetallic atoms are adsorbed on the –O terminal surface or when two oxygen vacancies are formed, the bimetals adsorb on the face-centered cubic sites of the vacancies to form bimetallic atom catalysts.
3.2 MX substitution
The influence of the M and X components, as well as surface terminations Tx, on the electrical characteristics of MXene is significant. Furthermore, distinct early-transition metal layers on MXene exhibit visible variation in their electrocatalytic influence. MX substitution is the replacement of sites or vacancies in a specific lattice structure by elemental doping, which leads to significant changes in electronic properties and modulates the active sites.Single-atom catalysts with high atomic utilization, as described above, can occupy M-atom vacancies in MXene in addition to replacing surface functional groups. Wang et al.57 used electrochemical exfoliation to prepare Mo2TiC2Tx MXene containing a large number of Mo vacancies, where the Mo vacancies are used to immobilize Pt by forming covalent Pt–C bonds with the surrounding C atoms. This single-atom catalyst has Pt-like catalytic kinetics for HER in acidic and neutral solutions. Peng et al.58 modified the Mo2CTx surface with single-atom Ru, Mo vacancies. The negatively charged functional groups are introduced during Mo2Ga2C etching and the functional groups contribute to anchoring Ru3+, while the Mo vacancies reduce and stabilize Ru3+, forming Ru-doped single-atom NRR catalysts. Therefore, single atoms can occupy the vacancies (M vacancies and oxygen vacancies) on the MXene carrier, and can also use surface terminals or heteroatoms as bridges. The vacancy-anchored single atom forms a chemical bond with the surrounding atoms and becomes a part of the lattice, which has high stability. Moreover, M vacancy can be directly used to anchor metal atoms without using reducing agents due to its high reducibility. However, to ensure the stability of the lattice structure, the type and loading of metal atoms need to be reasonably selected. At the same time, heteroatoms can be used to realize the strong interaction between single atoms and MXene, and heteroatoms can further regulate the electronic structure of metal atoms.
Moreover, alloying is also a way to optimize the electrocatalytic performance of MXene. Alloying includes multi-metal MXene and alloy-embedded MXene. To predict ordered binary alloy MXene with high HER activity, Su et al.59 computationally investigate the effects of alloys on electrical and geometric properties. Potential catalysts were screened out, which have thermal stability and excellent HER activity beyond noble metal platinum. The multi-metal MXene catalysts are still at the theoretical stage and need to be further investigated. In contrast, alloy-embedded MXene has been demonstrated experimentally. For example, Yan et al.60 doped Ti3C2Tx with Nb and modified the Nb–Ti3C2Tx with Ni/Co alloy, which exhibited excellent HER activity in an alkaline solution. The Nb doping, instead of Ti, could shift the Fermi energy level up to the conduction band, while the Ni/Co alloy modification could modulate the M–H affinity.
In addition to the M element in MXene, the X element species is also important. Theoretically, nitrogen has fewer empty electron orbitals than carbon and exhibits higher catalytic activity.61 Nitrogen-doped MXene, generally in three forms, adsorbing on the surface groups, replacing the surface functional groups, and replacing C in carbon-based MXene. For Example, Lee et al.62 replaced C in Ti3C2Tx with N by annealing treatment under an ammonia atmosphere to form O–Ti–N (Fig. 5(a)). It is easier to introduce N atoms by substitution at the carbon sites or at the terminal functional group sites at high temperature, which is favorable to form O–Ti–N. However, too high temperature (800 °C) will decrease the O–Ti–N content, since oxygen becomes unstable and mostly removed. Meanwhile, –OH is replaced by N-related groups (Fig. 5(c)). To investigate the doping mechanism and doping sites, Shi and Liu63 investigated N-doped Ti3C2Tx using a two-way isotope labeling technique, where N substitutes C in the lattice structure, expanding the layer spacing and exposing more surface active sites. Meanwhile, the doped N changes the electron density of Ti3+ by filling incomplete orbitals, decreasing the orbital overlap between N2 and Ti3+, and decreasing the energy of the valence electron system. This will facilitate the adsorption and desorption of NH3 in NRR and improve the catalytic efficiency (Fig. 5(b)).
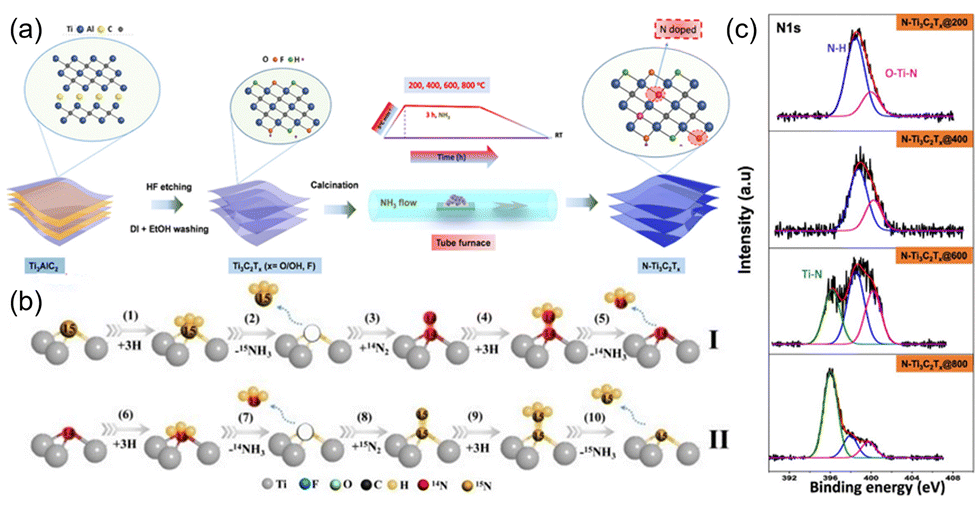 | ||
| Fig. 5 (a) Illustration of synthesis of N-doped MXene from the Ti3AlC2 MAX phase.62 Copyright 2019, American Chemical Society. (b) The proposed N dopants in 15N doped-TiV-Ti3-xC2Ty-1.2 MXene catalysts act as an active catalytic site in ENRR.63 Copyright 2021, Elsevier. (c) Core-level XPS of the N 1s of N-doped MXene samples annealed at various temperatures.63 Copyright 2021, Elsevier. | ||
3.3 Defect engineering
During the etching process, MXene inherits all existing vacancies from the precursor MAX phase and may further develop new vacancies due to direct contact between the top and bottom atomic layers and the etchant solution.64 The presence of these defects leads to changes in the surface electronic structure, which consequently influences the electrocatalytic activity. Furthermore, these defects can serve as anchoring sites for catalyst loading and surface modification purposes. It is important to note that defects have a negative impact on the quality and stability of MXene. Ti3C2Tx, for example, defect sites and edge sites are preferred sites for electron–hole accumulation, which can lead to the oxidation of Ti and C.65 The oxidation products inhibit electron transport and hinder the electrocatalytic kinetics of MXene.Using etching conditions to introduce M/X vacancies in MXene, the density and size of defects can be simply but imprecisely controlled by adjusting etching concentration, time, and temperature.66 For example, Ti3C2Tx was synthesized under varying concentrations of HF etchant to achieve samples with minimal Ti vacancies, single Ti vacancies, and Ti vacancy clusters.67 These Ti3C2Tx samples were subsequently utilized as supports for loading precious metal atom clusters. Compared with the former two, Ti vacancy clusters are used to induce superb metal-carrier interactions due to size expansion that exposes more lattice C ligands and is used to anchor metal clusters. The surface functional group of MXene also affects defect formation. Ti vacancies will be readily present on bare or –OH functionalized surfaces but not commonly present on –O functionalized ones, while C and N vacancies are more common in –O functionalized surfaces.68 Furthermore, the selective removal of specific transition metal atoms in the A and M layers can be achieved in quaternary or higher-order meta-MAX phases by leveraging the distinctive reactivity of various transition metals. For example, J. Rosen et al.69 obtained Nb1.33CTx with a large number of metal vacancies by selective etching of Sc and Al of quaternary MAX phase (Nb2/3Sc1/3)2AlC. After that, the catalytic activity of vacancy-ordered MXene (Nb1.33C, Mo1.33C, and W1.33C) on HER was further investigated experimentally and theoretically, where W1.33C showed the best performance.70
Compared to the atomic vacancies, edge defects will produce abundant electrocatalytic active sites. The edge sites of MXene are theoretically identified as a source of highly reactive HER due to their low coordination, which gives them weak hydrogen binding energy and low water dissociation potential.71 To increase the catalytic activity, the edge sites need to be exposed as many as possible. This can be done by cutting 2D MXene into low-dimensional structures that include 1D nanowires or nanotubes and 0D nanodots, or by allowing the generation of porous structures in MXene to increase the edge active sites. For example, Kou et al.72 sheared MXene (M–Ti3C2, M–V2C, S–Nb2C, M–Nb2C) into 0D quantum dots by Li embedding method. This method not only enriches the active edge sites, but also optimizes the density of states of active M atoms and lowers the HER energy barrier. Interestingly, the metal atoms at the edges of MXene quantum dots are more catalytically active on HER than the metal atoms at the basal plane. Lee et al.73 prepared highly porous structured layered Ti3C2Tx by wet chemistry to support the main catalytically active center IrCo. The low coordination elements at the edges and defective sites of Ti3C2 can be used as anchor points for growing the active phase, while the porous and edge-defective sites on the substrate surface enhance the ion/mass transport kinetics by shortening the diffusion pathways of charge/gas/reactant-related species and inhibit the 2D MXene self-stacking. In conclusion, the defects are diverse and complex, and the simultaneous presence of multiple types of vacancies and edge defects in MXene makes the conformational relationships between various types of defective MXene-based electrocatalysts and their intrinsic catalytic properties still lack deep understanding.
3.4 Morphology engineering
It is well known that, like other 2D materials, MXene tends to accumulate during electrode preparation due to van der Waals force attraction and hydrogen bonding interactions, which leads to a decrease in the utilization of active sites. Adjusting the MXene morphology is beneficial for the exposure of active sites.As previously described, decreasing the dimensionality of MXene increases the surface area and exposure of active sites, with an important contribution to enhanced electrocatalytic activity. In addition to 0D MXene quantum dots, 0D MXene nanoparticles can also be prepared. Zhang et al.74 prepared Ti3C2Tx MXene nanoparticles using ultrasonic cell crushing and used them to enhance the electrocatalytic activity of g-C3N4 towards ORR. The high electrical conductivity of MXene nanoparticles and the high O2 adsorption due to the coupling effect between MXene and g-C3N4 enhanced the electrocatalytic activity of ORR. 1D MXene (T3C2Tx) nanofibers were prepared by KOH solution for hydrolysis of MAX phase (T3AlC2) into nanofibers followed by simple HF selective etching (Fig. 6(a)).75 Further, it was investigated that hydrolysis of Nb2AlC with KOH followed by electrochemical etching without HF in 4 h synthetically prepared 1D Nb2CTx.76 In addition to the alkali treatment of the MAX phase, 1D nanostructures can also be obtained by treating MXene in an alkali solution. Wei et al.77 showed a significant increase in N2 immobilization efficiency by converting MXene nanosheets into nanoribbons, which was due to the large exposure of Ti–OH at the active site (Fig. 6(b)).
 | ||
| Fig. 6 (a) TEM image of Ti3C2 NFs.75 Copyright 2018, American Chemical Society. (b) HRTEM image of Ti3C2 MNRs, a single MNR with a width of ∼16 nm, taken from A position A.77 Copyright 2022, Wiley. (c) Cross-sectional SEM images of the 3D macroporous Ti3C2Tx film.78 Copyright 2017, Wiley. (d) and (e) SEM images of 3D Ti3C2 MXene architectures.79 Copyright 2018, American Chemical Society. (f) SEM images of Pt/MXene.80 Copyright 2022, Wiley. | ||
In addition, 2D MXene can be constructed into 3D structures. For example, 2D Ti3C2Tx MXene was self-assembled into hollow spheres and 3D macroporous structural frameworks on the surface by the template method (Fig. 6(c)).78 The hollow shell layer of MXene effectively suppressed interlayer aggregation and improved ion accessibility. However, this method is still very challenging to synthesize in practice. Hence, a nanoflower-like 3D MXene material with layered layers was successfully synthesized via ultrasonic aerosol spray drying of MXene colloids, which underwent self-assembly under the influence of capillary forces (Fig. 6(d) and (e)).79 The resulting material was employed as an effective host for loading CoP as a bifunctional electrocatalytic hydrolysis catalyst, exhibiting excellent catalytic performance. With its anti-agglomeration properties and porous structure, this 3D MXene framework enhances the transport of ions and diffusion of reaction-related species at the three-phase interface of the electrode, electrolyte, and gas. Additionally, the conductive network provided by the 3D MXene backbone structure facilitates rapid electron transfer. Mai et al.80 used a fast and continuous spray drying method to prepare 3D folded Ti3C2Tx MXene loaded with sub-nanometer Pt clusters to construct HER electrocatalysts (Fig. 6(f)). 3D folded structures hindered the re-stacking of 2D MXene nanosheets, allowing complete exposure of Pt catalysts, and HER catalytic activity exceeded that of commercial Pt/C catalysts.
3.5 Heterostructure engineering
Elaborately engineered structured electrocatalysts can expose a greater number of catalytically active central sites that exhibit speedy charge and mass transport kinetics, which can be achieved through intricate morphology and defect engineering techniques applied to MXene. However, the intrinsic catalytic activity of MXene is not high, which requires hybridization with other active substances, which has been shown in some previous strategies.The heterostructure modulates the electronic structure by inducing spontaneous electron transfer at the interface and generating more active sites by expanding the exposed edges, which can combine the structural advantages of each component and thus achieve significant synergistic coupling effects.81,82 Moreover, due to the diversity of the components of the heterostructure, the composition, morphology, structure, and electronic state of the heterostructure can be tuned for efficient electrocatalysis and stability. Notably, MXene is prone to oxidative deactivation, which can significantly limit its electrocatalytic applications. For example, during hydrothermal etching of Ti3C2, dissolved oxygen in an aqueous solution combines with the most active edges of MXene to generate TiO2, leading to structural changes and degradation of electrochemical properties.39 Therefore, the construction of heterojunctions of MXene with thermodynamically stable active phases is an effective strategy to explore stable and efficient electrocatalysts.
Briefly, there is self-assembly and in situ synthesis methods for the construction of MXene-based heterostructures. Self-assembly usually relies on the electrostatic interaction between the negative charge of the base surface of MXene nanosheets and the positive charge of the active phase. Chen et al.83 self-assembled MOFs on 2D nitrogen-doped MXene by electrostatic interaction, and then prepared 0D bimetallic selenide particles (CoZn–Se) by in situ selenization strategy to form amphiphilic lithium–sulfur bond binding sites, which can effectively immobilize and catalyze the conversion of LiPS intermediates. This 0D–2D heterostructured catalyst has a hierarchical porous structure, which not only has a large active area but also synergistically prevents the aggregation of CoZn–Se nanoparticles and the restacking of N–MXene nanosheets. In situ synthesis is the direct synthesis of another material on the MXene substrate with high interfacial binding strength. Zou et al.84 synthesized MXene–TiN ultrathin heterojunctions by controlled nitridation of MXene nanosheets and in situ growth of a layer of TiN on the MXene surface and constructed hollow spherical structures after removing the template. This 2D heterostructure exhibited good adsorption capacity and high catalytic activity by forming many interfacial regions through the generated bonds in close contact between Ti3C2Tx MXene and TiN.
Depending on the type, morphology, and mode of action of MXene with another material, the specific heterostructures are constructed in a variety of ways. For example, in the work of Chen and Zou,83,84 2D MXene was used as a carrier and acted synergistically as a catalyst. By contrast, MXene can also serve as the main active center. For instance, Chu et al.85 combined 0D Ti3C2Tx–MXene quantum dots (MQDs) with porous Cu nanosheets, which are densely and uniformly covered with a large number of MQDs on the surface. According to DFT theory calculations, the synergistic effect of MQDs and Cu makes the interfacial Cu–Ti dimer a double active center, which greatly improves the NRR catalytic performance. In addition, hydroxyl oxides as promising catalysts for OER, Li et al.86 showed Ru/Rh-doped FeOOH nanoarray structures with abundant oxygen vacancies grown in situ on Ti3C2Tx MXene. The Ti3C2Tx MXene matrix provided a good substrate for the ordered growth of FeOOH nanoarrays and provided open spaces with abundant edge activity. Interestingly, Sun et al.87 prepared heterostructures of Ti3C2Tx/MAX phase in situ by controlled etching conditions. The center of the heterostructure is the MAX phase, which is supported between the MXene sheets and forms a heterostructure interface. Especially after enlargement, the heterostructure shows a unique “flared” multilayer structure, which makes it exhibit a good nano-limited effect and reflects a unique catalytic mechanism. Therefore, the construction of heterostructures of MXenes is very effective and brings more possibilities for electrochemical catalysis.
In conclusion, there are many MXene-based electrocatalytic design strategies, and the easy idea is to adjust the composition of MXene. This gives rise to surface modification and MX substitution. Moreover, defects (vacancies and edge defects) can be incorporated into the MXenes catalytic system by rational design, which can adjust the electronic structure and catalytic sites. In addition to the treatment for MXenes at the atomic level, the morphological engineering of MXenes is also fascinatingly attractive. By downscaling or upscaling, the original 2D MXene nanosheets, such as 0D quantum dots or nanoparticles, 1D nanowires or nanoribbons, 3D is much more constructible, which will all have a great impact on the active sites and accessibility of MXene. The electrocatalytic performance of MXene itself is not enough to meet the requirements, especially in OER, ORR, NRR, etc. MXene has some reducibility and negatively charged surface functional groups that can anchor the nanoparticles well. The introduction of other active phases is feasible and indispensable, which is also shown in surface modification and MX substitution. The heterostructure is particularly unique in hybridization with other materials, allowing MXenes to form electron–hole transport interfaces with other materials. This fosters simultaneous modulation of the electronic state and physicochemical characteristics of the material, harnessing the interfacial component coupling effect to effectively enhance its electrocatalytic performance. These strategies are not independent of each other, and each strategy has its own concerns and shortcomings (Table 1). For example, decreasing the MXene dimension increases the catalytically active surface area, which naturally also increases the degree of defects, and brings about antioxidant performance concerns. To design more efficient MXene-based electrocatalysts, more comprehensive considerations are required when modifying MXenes electrocatalytic properties. For example, in the construction of 3D MXene, it is certainly to increase the exposure of active sites but also to enhance the resistance of MXenes to aggregation. Notably, MXenes are prone to oxidation, which eventually destroys the non-metallic carbon layer and the intact metal phase inside MXenes, hindering electron transfer and charge exchange, which has a significant impact on electrocatalysis.
| Design strategy | Advantage | Shortage |
|---|---|---|
| Surface modification | Modulation of MXene surface active sites and electronic structure | High requirements for MXene surface terminals; easy inactivation |
| MX substitution | Improved MXene intrinsic catalysis | Insufficient catalytic applicability |
| Defect engineering | Modulation of electronic structure; subsequent load | Easily oxidized |
| Morphology engineering | Increase the number of active site exposures | Limited catalytic activity |
| Heterostructure engineering | Wide range of active materials; suitable for different catalytic areas | Complex interfacial mechanism; complex preparation |
4. Representative electrocatalytic applications of MXene-based materials
The electrochemical active sites of MXene-based materials can be designed and tuned by surface modification, MX substitution, defect engineering, morphology engineering and heterostructure engineering to facilitate their applications in many electrocatalytic fields. In this section, MXene-based electrocatalysts in the fields of HER, OER, ORR, and NRR will be discussed.4.1 HER
Compared to fossil fuels, hydrogen is a renewable and clean energy source with high energy density and excellent storage capacity, obtained by the electrocatalytic decomposition of water into hydrogen and oxygen, which can be reacted with oxygen in a fuel cell to produce electricity and water without producing pollutants.88,89 Therefore, ideal HER catalysts are essential for efficient and low-consumption hydrogen production. MXene itself has some HER electrocatalytic activity, but compared to Pt, it is also necessary to adjust its electronic structure and catalytic sites to enhance HER catalytic activity (Table 2).| HER catalyst | Strategy | Electrolyte | Overpotential η10 [mV] | Tafel slope [mV dec−1] | Ref. |
|---|---|---|---|---|---|
| 11N-Ti2CTx | Surface modification | 0.5 M H2SO4 | 215 | 67 | 46 |
| Ti3C2Ox | Surface modification | 0.5 M H2SO4 | 190 | 60.7 | 47 |
| RuSA–N–S–Ti3C2Tx | Surface modification | 0.5 M H2SO4 | 76 | 90 | 55 |
| Ti3C2Tx–PtSA | Surface modification | 0.5 M H2SO4 | 38 | 32 | 53 |
| Pd/Ti3C2Tx–CNT | Surface modification | 0.1 M KOH | 158 | 50 | 90 |
| Mo2TiC2Tx–PtSA | MX substitution | 0.5 M H2SO4 | 30 | 30 | 57 |
| Ni0.9Co0.1@Ti3C2Tx | MX substitution | 1.0 M KOH | 43.4 | 116 | 60 |
| N–Ti3C2Tx–600 | MX substitution | 0.5 M H2SO4 | 198 | 92 | 62 |
| NiNPs@N–Nb2CTx | MX substitution/surface modification | 1.0 M HClO4 | 383 (η500) | 88 | 91 |
| Ru@Ti3C2Tx-Vc | Defect/surface modification | 1.0 M KOH | 35 | 32 | 67 |
| M–Ti3C2 QDs | Defect/morphology | 1.0 M KOH | 201 | 106 | 72 |
| M–V2C QDs | Defect/morphology | 1.0 M KOH | 244 | 157 | 72 |
| M–Nb2C QDs | Defect/morphology | 1.0 M KOH | 271 | 137 | 72 |
| CoP@3D Ti3C2–Mxene | Morphology/surface modification | 1.0 M KOH | 168 | 58 | 79 |
| Pt/Ti3C2Tx | Morphology/surface modification | 0.5 M H2SO4 | 34 | 29.7 | 80 |
| GQDs (5%)/Ti3C2Tx | Heterostructure | 0.5 M H2SO4 | 260 | 89 | 82 |
| MoS2/Nb2C hybrids | Heterostructure | 1.0 M KOH | 117 | 65.1 | 92 |
| Rh–CoNi LDH/MXene | Heterostructure | 1.0 M KOH | 74.6 | 43.9 | 93 |
| Ti3CN(OH)x@MoS2 | Heterostructure | 0.5 M H2SO4 | 120 | 64 | 94 |
The key to the HER reaction depends on the free energy of hydrogen adsorption, which should not be either too strong or too weakly bound to the catalyst. If the binding of hydrogen to the surface is too weak, the adsorption step will limit the overall reaction rate (Volmer step), and if the binding is too strong, the desorption step will limit the rate (Heyrovsky or Tafel steps). For example, adjusting the MXene functional group species and content can promote hydrogen adsorption and desorption, and the surface –O terminal theoretically favors the HER activity of MXene, while the –F terminal does the opposite.44 Previous theoretical calculations have considered the case of complete substitution; in fact, the surface of MXene is the coexistence of multiple terminal groups. To a certain degree, –F groups are beneficial in regulating hydrogen bond strength, and the presence of –F groups is not detrimental when the –O coverage is 2/3.95 In addition to tuning the functional groups, both non-metallic and transition metal atom doping can be effective in improving HER catalytic activity, either by surface modification or MX substitution depending on the doping site differences. Meanwhile, defect engineering can be used to provide anchor points for heterogeneous atoms. He et al.55 prepared RuSA–N–S–Ti3C2Tx catalysts that utilized heteroatoms to enhance the coupling and synergize the Ru monoatomic with the carrier Ti3C2Tx, achieving an overpotential of 76 mV at a current density of 10 mA cm−2. The addition of this catalyst to the n+np+–Si photocathode significantly increased the photocurrent density (37.6 mA cm−2) in the photoelectrochemical (PEC) water splitting. Compared to the Mo vacancy anchored Pt with Mo2TiC2Tx (η10 of 30 mV),57 Gong et al.53 achieved the immobilization of single-atom Pt on the oxygen vacancy of Ti3C2Tx with an overpotential of 38 mV at j of 10 mA cm−2, exhibiting excellent HER catalytic performance. Lee et al.62 controlled the appropriate amount of N at a suitable calcination temperature to facilitate the shift of adsorption Gibbs free energy towards 0 eV, thus promoting hydrogen adsorption. Nitrogen appeared in the lattice sites at temperatures higher than 600 °C, with better HER performance compared to not being replaced by the lattice. This approach demonstrates a judicious manipulation of experimental parameters to optimize adsorption energetics. Compared with loading on the surface of MXene, Huang et al.91 first N-doped Nb2CTx at 600 °C and then encapsulated Ni nanoparticles from the surface embedded into the interlayer edges of N–Nb2CTx by an electrochemical process. It exhibited an overpotential of 383 mV under acidic conditions and 500 mA cm−2, which is much higher than that of N–Nb2CTx loaded Ni (642 mV).
Besides MXene itself, the inherent HER catalytic activity of MXene is further enhanced through hybridization with other active materials. It not only introduces additional catalytically active sites within the active phase but also serves to mitigate the negative surface charge of the MXene material, thereby precluding aggregation and enabling more cohesive catalytic performance, such as carbon nanotubes,90 layered double hydroxides,93 MoS2,94etc. The morphology of the components, as well as the construction of heterostructures, can be targeted. For example, Huang et al.82 devised a method for fabricating tunable 0D/2D heterostructures composed of graphene quantum dots (GQDs) and MXene nanosheets, which exhibit a charge transfer resistance of approximately 4.1 Ω during the hydrogen evolution reaction. By contrast, GQDs alone evince a markedly higher resistance value of 174.7 Ω. This discrepancy underscores the capacity of the 0D/2D architecture to optimize electron transfer kinetics in the context of HER. In addition, Liu et al.93 used in situ synthesis method to grow LDH with rich oxygen vacancies and Rh doping on the surface of MXene, resulting in a low overpotential (74.6 mV). The design not only includes 2D/2D nanoarray structure, but also includes Rh and oxygen vacancies to regulate the electronic behavior and structure of CoNi-LDH, thereby increasing the active sites and optimizing the adsorption/desorption of intermediate products. Zhu et al.92 employed a hydrothermal technique to grow MoS2 on the 3D petal-like Nb2C MXene surface, creating a heterostructural material with high lattice adaptation. The resulting conductive network, established by the 3D flower-like Nb2C–MXene framework, significantly boosted electron transport capacity and improved the stability of the MoS2/Nb2C catalyst.
In short, MXene surface terminals have a significant effect on the HER kinetic properties, yet the types and ratios of MXene surface terminals obtained by conventional etching are uncontrollable. Therefore, it is necessary to develop new etching methods and then further modulate the end groups. Some noble metal-loaded MXene showed excellent HER performance, outperforming commercial Pt/C catalysts in acidic electrolytes. However, most of the studies focused on short-term stability tests and could not guarantee long-term cycling operation. Therefore, the durability of MXene-based catalysts needs to be tested over a long period of time.
4.2 OER
OER is a key step in many clean energy storage and conversion processes (hydrolysis or metal–air batteries). OER involves multiple electron transfer processes, slow kinetic reactions, and high overpotential, which are bottlenecks limiting hydrolysis and metal–air batteries.96 Unlike HER, pristine MXene exhibited a very limited OER activity. Therefore, MXene needs to be hybridized with other active substances to achieve charge transfer in the OER process and improve the catalytic activity (Table 3).| OER catalyst | Strategy | Overpotential η10 [mV] | Tafel slope [mV dec−1] | Ref. |
|---|---|---|---|---|
| IrCo@ac-Ti3C2 | Defect/morphology | 220 | 60 | 73 |
| CoP@3D Ti3C2-Mxene | Morphology/surface modification | 298 | 51 | 79 |
| Ru–FeOOH@ Ti3C2Tx | Heterostructure | 223 | 63.6 | 86 |
| Rh–FeOOH@Ti3C2Tx | Heterostructure | 230 | 67.7 | 86 |
| 0D Co3O4/2D Ti3C2 | Heterostructure | 300 | 118 | 97 |
| H2PO2-/FeNi-LDH-V2C | Heterostructure | 250 | 64.5 | 98 |
| Cr–FeNi LDH/MXene | Heterostructure | 232 | 54.4 | 99 |
| Ni0.7Fe0.3PS3@MXene | Heterostructure | 282 | 36.5 | 100 |
Transition metal oxides (TMOs) have emerged as a highly promising class of materials that hold great potential for replacing current state-of-the-art OER catalysts, while simultaneously enhancing the OER performance of initial metal oxides through the transition of oxidation states within metallic oxides.101 The hybridization of MXene with TMO, especially the construction of heterostructures, can improve the electrical conductivity, increase the number of active sites and shorten the charge transfer pathway to achieve effective OER catalytic activity over a single component. For example, Yang et al.97 constructed 0D/2D heterostructures by combining in situ electrostatic assembly with a solvothermal approach to load 0D Co3O4 onto an exfoliated small amount of layered Ti3C2Tx. In the latest study, Daire Tyndall et al.102 explored the mechanism that TMOs/MXene hybrids exhibit better OER activity comparing to single-component materials. 1% MXene hybridization with TMOs not only allows the liganded Co metal to reach a higher oxidation state at a lower input potential, but also results in a robust observable film after electrochemical cycling. The high oxidation state of Co at a lower overpotential on the catalyst surface stimulates the reactivity and the films formed enhance the mechanical properties of the hybrid catalyst. Unfortunately, although higher content of MXene will have lower overpotential, it will degrade into insulator TiO2 in the subsequent long-term OER process, which will seriously hinder electron transfer.
In addition, layered double hydroxide (LDH) is considered as a promising electrocatalyst for oxygen precipitation.103 The LDH has a layered structure, tunability, and strong coupling with the MXene interface, which will act as an anti-agglomeration and enhanced conductivity. For instance, Shi et al.98 prepared H2PO2−/FeNi-LDH-V2C electrocatalysts by in situ assembly of layer less V2C nanosheets and FeNi-LDH nanosheets by a simple hydrothermal method, and by embedding H2PO2− into the LDH layer due to the tunable properties of the interlayer anion of LDH. Strong electronic interactions and synergistic interactions between FeNi LDHs and V2C MXene ensured the balance of significant charge transfer and the adsorption and desorption of O2. The catalyst has good durability in rechargeable zinc–air batteries compared to conventional Pt/C + RuO2 air cathodes, and in practical applications, powering light-emitting diodes (2–2.2 V) for 36 h with no degradation in brightness. The heteroatom doping of LDH/MXene also changes the catalytic structure. For example, the generation of vacancies in the LDH layer and Cr doping,99 or S and P doping100 that have a synergistic effect on the surface electronic structure of the central metal atom.
In short, if these MXene-based catalysts are to be used in electrolytic systems, they must perform reliably at high current densities. Due to the severe oxidizing environment present in the system during OER, there is instability in the MXene-based catalysts, leading to the decomposition of MXene into TiO2. For example, in a 2000 cycle test of Co3O4/Ti3C2Tx hybrids, a gradual decrease in the activity of the hybrids was observed.97 Therefore, the strategy of constructing MXene-based catalysts should also pay attention to improving the stability of MXene, such as covering the edges of MXene by some active materials to hinder the further oxidation of MXene.
4.3 ORR
ORR is fundamentally the inverse reaction of OER and one of the key reactions for electrochemical reduction in the electrochemical energy conversion systems of fuel cells and metal–air batteries.104 However, the sluggish ORR kinetics and the dominant energy conversion require carefully designed stable and efficient electrocatalysts to facilitate the ORR process. Although the intrinsic ORR performance of MXene is insufficient,105 MXene can be combined with other ORR actives to achieve synergistic promotion, showing the potential to replace precious metal ORR catalysts (Table 4).| ORR catalyst | Strategy | E on-set (V vs. RHE) | E 1/2 (V vs. RHE) | Tafel slope [mV dec−1] | Ref. |
|---|---|---|---|---|---|
| Nb2CTx | Surface modification | 0.77 | — | 69.93 | 105 |
| g-C3N4/Ti3C2-NP | Heterostructure | 0.924 | 0.79 | — | 74 |
| Fe–N–C/Ti3C2Tx | Heterostructure | 1.00 | 0.814 | 30 | 106 |
| 2D/2D Fe–N–C/Ti3C2Tx | Heterostructure | 0.92 | 0.84 | — | 107 |
| Co–CNT/Ti3C2-60 | Heterostructure | — | 0.82 | 63 | 108 |
| N–CoSe2/3D–Ti3C2Tx | Heterostructure | 0.95 | 0.79 | 87 | 109 |
| MoS2QDs@ Ti3C2Tx QDs@MWCNTs-2 | Heterostructure | 0.87 | 0.75 | 92 | 110 |
Recently, metal–N–C materials, especially Fe–N–C materials, have been extensively studied in non-precious metal ORR catalysts. The central structure of Fe–N–C (Fe–N4), stabilized by four Fe–N bonds, has been identified as one of the most active catalytic centers.111,112 Moreover, carbon carriers with porous structures play a positive role in promoting electron and mass transfer. For example, iron phthalocyanine (FePc), with an active Fe–N–C structure, is well suited for ORR. Strong interactions between FePc and Ti3C2Tx MXene lead to significant 3d electron delocalization and spin conformational changes of Fe, which makes the active FeN4 sites more susceptible to oxygen adsorption and reduction.113 In contrast to the usual Fe–N–C cations bound to Ti3C2Tx anions under electrostatic forces,106 the Fe–N–C was enabled to form 2D/2D superlattice like heterostructures with MXene nanosheets by Fe-clustering assistance in van der Waals or electrostatic forces, showing a positive onset potential of 0.92 V, a four-electron transfer pathway and 20 h durability under alkaline conditions in electrocatalytic ORR applications.107 In addition, the hybridization of Co–N–C with MXene also showed good performance in ORR electrocatalysis. Zhang et al.108 grew CNT directly perpendicular to the MXene surface by pyrolysis of ZIF-67, which exhibited good ORR activity with abundant ∼10 nm diameter cobalt nanoparticles embedded at the tip of the CNT. Direct growth of CNT on MXene ensures strong interactions between CNT and MXene, promotes fast electron transfer, and exposes more active sites that avoid the aggregation of CNTs.
Notably, morphological engineering will allow the advantages of the hybridization of MXene with other active materials to be fully exploited. The 3D structure of carrier-type MXene is characterized by its increased surface area and porous structure. Therefore, it is endowed with the ability to anchor a large number of active sites and plays an effective role in dispersing the carrier. For example, Zeng et al.109 synthesized CoSe2–MXene sheets by in situ method, freeze-dried CoSe2–Ti3C2Tx sheets using polyvinyl alcohol and finally calcined in ammonia to form porous 3D structures. 3D MXene structures can anchor a large number of CoSe2 nanocrystals, thus preventing stacking between MXene sheets. Strong interactions between MXene and CoSe2 drive electron transfer from MXene to CoSe2, which improves the ORR catalytic activity of the Co site. When MXene acts as the ORR active center, 0D quantum dots have stronger size-dependent quantum-limited domains, higher specific surface area, and more edge sites. For example, 0D Ti3C2Tx quantum dots and 0D MoS2 quantum dots loaded on multi-walled CNTs exhibit excellent ORR catalytic activity.110,114 The high surface activity of quantum dots easily binds to other atoms, 0D Ti3C2Tx quantum dots facilitate the electron transfer for oxygen reduction, and 0D MoS2 quantum dots provide active sites for oxygen adsorption. The MXene-based ORR catalyst design strategy tends to achieve heterostructures with other active materials, thereby improving the original deficiencies of the active material. Therefore, the terminals design of MXene is also a top priority for the design of heterostructures.
In short, the use of MXene as a support for the active materials enabled the composites to exhibit excellent ORR catalytic activity, approaching or even exceeding that of commercially available Pt/C catalysts. However, the stability of MXene-loaded electrocatalysts under fuel cell operating conditions is still unsatisfactory and needs to be further improved when used as cathode catalysts in fuel cells. For example, the cathode catalyst, Pt/CNT–Ti3C2Tx, was employed in a proton exchange membrane fuel cell, achieving a maximum power density of 138 W and sustaining a consistent current density at 0.30 V for a 360 min operational period.115
4.4 NRR
Ammonia (NH3) is an important chemical raw material that is essential to produce nitrogen fertilizers and is one of the most common chemicals.114,116 Compared to the conventional Haber–Bosch process, ammonia synthesis using electrocatalytic nitrogen reduction reaction does not require high-temperature and high-pressure conditions and would be a promising green alternative.117 Theoretical calculations indicate that MXene is a promising material for N2 capture and has a thermodynamic advantage over the capture of CO2 and H2O.118 Therefore, MXenes are widely used as potential catalyst components for NRR enhancement due to their unique properties (Table 5).| NRR catalyst | Strategy | Electrolyte | NH3 yield | FE (%@V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| Ti3C2Tx-medium F | Surface modification | 0.01 M Na2SO4 | 2.81 × 10−5 μmol (cm−2 s−1) | 7.40@−0.7 | 119 |
Ti3C2Tx (T![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OH) O and OH) |
Surface modification | 0.1 M KCI | 36.9 μg (mg h)−1 | 9.10@−0.3 | 120 |
| Ti3C2Tx/TiFeOx-700 | Surface modification | 0.05 M H2SO4 | 2.19 μg (cm2 h)−1 | 25.44@−0.2 | 51 |
| Ru–Mo2CTx | MX substitution | 0.5 M K2SO4 | 40.77 μg (mg h)−1 | 25.77@−0.3 | 58 |
| Ti3C2OH QDs | Morphology | 0.1 M HCI | 62.94 μg (mg h)−1 | 13.30@−0.5 | 121 |
| MQDs/Cu | Heterostructure | 0.5 M LiClO4 | 78.5 μg (mg h)−1 | 21.30@−0.4 | 85 |
| Ti3C2 MXene/MAX | Heterostructure | 0.1 M Na2SO4 | 2.73 μg (cm2 h)−1 | 36.90@−0.5 | 87 |
| BNQDs/Ti3C2Tx | Heterostructure | 0.5 M LiClO4 | 52.8 ± 3.3 μg (mg h)−1 | 19.1 ± 1.6@−0.4 | 122 |
| COF–Fe/Mxene | Heterostructure | 0.1 M Na2SO4 | 41.8 μg (mg h)−1 | 43.1@−0.5 | 123 |
The NRR catalytic activity can be influenced by adjusting the MXene surface functional groups. Ti3C2Tx with a medium density of –F functional groups exhibits the best NRR catalytic activity, which is because excessive –F density limits the adsorption of H+, while too low (–O terminals become more numerous) allows the highly active HER to compete with NRR.119 In addition, the H atoms in –OH can be trapped by the intermediate and participate in NRR, which helps the NRR process to form a higher degree of *N(H)NH reduction in the first step of hydrogenation and reduce the overpotential.120 For example, Ti3C2Tx (–O and –OH) nanosheets (∼50–100 nm) can effectively reduce N2 to NH3 with higher NRR catalytic activity than fluorine-capped Ti3C2Tx.121 This is due to the higher electrical conductivity of Ti and –OH functional groups present at the edges, which facilitates the adsorption and activation of N2, thus improving the catalytic performance. Therefore, the synthesis of MXene with more edge sites is considered as one of the most feasible methods to improve the catalytic efficiency of NRR. Xu et al.124 synthesized hydroxyl-rich Ti3C2Tx quantum dots by alkalinization and intercalation. The –OH and edge defects led to a significant increase in NRR catalytic activity (NH3 yield of 62.94 μg (mg h)−1 and FE of 13.30% in 0.1 M HCl electrolyte medium and at a potential of −0.50 V). The tuning of surface terminals and the exposure of edge defects were beneficial for the enhancement of NRR catalytic activity. The introduction of other active substances is a common strategy for MXene-based NRR catalysts, in which single atoms hosted by MXene are one of the candidates for NRR catalysts. For example, Zhang et al.125 calculated 18 single-atom catalysts on S-functionalized MXene for NRR activity and selectivity by DFT theory, among which Mo@MXene, Nb@MXene, and V@MXene are suitable candidates. Meanwhile, MXene heterostructures have been widely used in NRR. For example, the boron carbide pair was made into self-assembled quantum dots on MXene (BNQDs/Ti3C2Tx).122 And the electronic interaction between the two can enhance the electron-donating ability of N2 activation and protonation at the interface B. Meanwhile, the BNQDs can block the HER active site of MXene, which makes the catalyst have high NRR selectivity. In addition, it is possible to actively modify the MXene surface to achieve a better coupling effect. By amino-functionalizing the MXene surface, COFs are homogeneously and tightly bound to the aminated MXene to form a heterostructure (COF/MXene) by covalent coupling, which is then post-treated to obtain hydrophobic COF–Fe/MXene.123 This NRR catalyst showed an NH3 yield of 41.8 μg (mg h)−1 with a FE of 43.1% in 0.1 M Na2SO4 aqueous solution and at −0.5 V relative to the RHE. The confinement effect is also particularly attractive in catalysis. Sun et al.87 constructed interesting MXene/MAX heterostructures, which exhibited unique and efficient NRR catalysis due to the domain-limiting effect. The reaction intermediates alternately diffuse, collide, adsorb, and reduce in the narrow confinement space constituted by the MAX/MXene surface, thus achieving efficient NRR electrocatalysis.
It is possible that MXene may exhibit greater activity towards NRR than towards HER, due to the high surface charge density of these materials. Modulation of the end groups on the surface of MXene is of great significance, as the –O end group results in weaker binding to N2, which in turn leads to competitive HER. Furthermore, MXene can serve as an excellent carrier for the reduction of N2 to NH3, and displays excellent NRR catalytic properties in combination with the active substance. Nevertheless, MXene-based NRR catalysts are currently the focus of laboratory research and are still a considerable distance from achieving industrial commercialization. This is because the low selectivity and reaction rate of the MXene-based NRR catalyst still hinder the practical application of the NRR process (Table 5).
In general, modifying and constructing heterostructures for MXene electrocatalytic properties can modulate the electronic structure, increase the surface area, and provide more active sites, making MXene-based materials promising for electrocatalytic applications.
5. Summary and outlook
In summary, MXenes have emerged as a highly promising class of materials for electrocatalysis due to their excellent electrical conductivity, high specific surface area, and versatile surface chemistry. This review has detailed the advancements in MXene-based catalysts for key reactions such as HER, OER, ORR, and NRR, highlighting the tunability of their layered structures and surface properties through surface modification, MX substitution, defect engineering, morphology control, and heterostructure construction.Despite the promising potential and significant advancements, several challenges remain:
Restacking during synthesis and catalysis: the high surface energy of MXenes can cause nanosheets to restack, reducing the exposure of active sites. Constructing intercalation and 3D structures using organic macromolecules or integrating other materials can mitigate restacking. However, further studies are needed to understand the host–guest interlayer chemistry and various forms of 3D frameworks.
Stability issues: MXenes can undergo irreversible oxidation, which compromises their unique properties like metallic conductivity and layered structure. Optimizing the synthesis of the MAX phase to produce low-defect and complete lattice precursors, along with constructing stable heterostructures, can enhance the stability and activity of MXene-based catalysts.
Scalable production: the large-scale production of high-quality MXene materials is currently limited by existing synthesis methods, which often involve fluorine-containing etching not conducive to industrialization. Developing scalable, industrial synthesis techniques is essential for broader application.
Exploring MXene diversity: most research has focused on specific MXenes like Mo2C and Ti3C2, while other variants, such as double transition metal MXenes and V-based MXenes, require more attention. Moreover, controlling the content and positioning of single atoms anchored in MXene vacancies necessitates further studies to generate ordered vacancy defects.
Addressing these challenges will require innovative engineering solutions and the development of robust industrial synthesis techniques. Furthermore, exploring the diverse forms of MXenes and optimizing their atomic structures and surface end-groups can unlock further potential in electrocatalytic applications.
With continued research and novel approaches, MXenes can play a pivotal role in advancing sustainable energy conversion technologies, offering improved catalytic efficiency and stability. The future of MXene-based electrocatalysts appears bright, with the potential to make significant contributions to the field of green chemistry and renewable energy.
Data availability
The data that support the findings of this review are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ten-Thousand Talents Plan of Zhejiang Province (no. 2022R51007), Ningbo Top-talent Team Program, National Natural Science Foundation of China (U23A2093). KL gratefully acknowledges financial support from Anglo American Resources Trading (China) Co., Ltd.References
- X. Wu, S. Xiao, Y. Long, T. Ma, W. Shao, S. Cao, X. Xiang, L. Ma, L. Qiu, C. Cheng and C. Zhao, Small, 2022, 18, 2105831 CrossRef CAS PubMed.
- P.-F. Guo, Y. Yang, B. Zhu, Q.-N. Yang, Y. Jia, W.-T. Wang, Z.-T. Liu, S.-Q. Zhao and X. Cui, Carbon Energy, 2024, 6, e532 CrossRef.
- H. Su, X. Pan, S. Li, H. Zhang and R. Zou, Carbon Energy, 2023, 5, e296 CrossRef.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef.
- J. Ren, Y. Huang, H. Zhu, B. Zhang, H. Zhu, S. Shen, G. Tan, F. Wu, H. He, S. Lan, X. Xia and Q. Liu, Carbon Energy, 2020, 2, 176–202 CrossRef.
- Z. Zheng, D. Wu, G. Chen, N. Zhang, H. Wan, X. Liu and R. Ma, Carbon Energy, 2022, 4, 901–913 CrossRef CAS.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS.
- H. M. Barkholtz and D.-J. Liu, Mater. Horiz., 2017, 4, 20–37 RSC.
- J. Hao, K. Wu, C. Lyu, Y. Yang, H. Wu, J. Liu, N. Liu, W.-M. Lau and J. Zheng, Mater. Horiz., 2023, 10, 2312–2342 RSC.
- C. Chen, S. Li, X. Zhu, S. Bo, K. Cheng, N. He, M. Qiu, C. Xie, D. Song, Y. Liu, W. Chen, Y. Li, Q. Liu, C. Li and S. Wang, Carbon Energy, 2023, 5, e345 CrossRef CAS.
- W. Eom, H. Shin, W. Jeong, R. B. Ambade, H. Lee and T. H. Han, Mater. Horiz., 2023, 10, 4892–4902 RSC.
- H. Ye, M.-B. Wu, Q.-H. Ye, R.-M. Wen, Z.-T. Hu, J. Yao and C. Zhang, Mater. Horiz., 2024, 11, 2685–2693 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef PubMed.
- A. Zhou, Y. Liu, S. Li, X. Wang, G. Ying, Q. Xia and P. Zhang, J. Adv. Ceram., 2021, 10, 1194–1242 CrossRef.
- R. Ding, J. Xiong, Q. Yan, Z. Chen, Z. Liu, X. Zhao, Q. Peng and X. He, Mater. Horiz., 2023, 10, 2262–2270 RSC.
- Y. Chen, Y. Dai, S. C. Bodepudi, X. Liu, Y. Ma, S. Xing, D. Di, F. Tian, X. Ming, Y. Liu, K. Pang, F. Xue, Y. Zhang, Z. Yu, Y. Dan, O. V. Penkov, Y. Zhang, D. Qi, W. Fang, Y. Xu and C. Gao, InfoMat, 2024, 4, e12596 CrossRef.
- C. Zhang, L. Cui, S. Abdolhosseinzadeh and J. Heier, InfoMat, 2020, 2, 613–638 CrossRef CAS.
- X. Cheng, R. Guan, Z. Wu, Y. Sun, W. Che and Q. Shang, InfoMat, 2024, 6, e12535 CrossRef CAS.
- S. Jin, Y. Guo, F. Wang and A. Zhou, MRS Bull., 2023, 48, 245–252 CrossRef CAS.
- K. Matthews, T. Zhang, C. E. Shuck, A. VahidMohammadi and Y. Gogotsi, Chem. Mater., 2022, 34, 499–509 CrossRef CAS.
- M. Anayee, N. Kurra, M. Alhabeb, M. Seredych, M. N. Hedhili, A.-H. Emwas, H. N. Alshareef, B. Anasori and Y. Gogotsi, Chem. Commun., 2020, 56, 6090–6093 RSC.
- M. Alhabeb, K. Maleski, T. S. Mathis, A. Sarycheva, C. B. Hatter, S. Uzun, A. Levitt and Y. Gogotsi, Angew. Chem., Int. Ed., 2018, 57, 5444–5448 CrossRef CAS PubMed.
- F. Liu, A. Zhou, J. Chen, J. Jia, W. Zhou, L. Wang and Q. Hu, Appl. Surf. Sci., 2017, 416, 781–789 CrossRef CAS.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L.-Å. Näslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374–2381 CrossRef CAS PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Å. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X.-H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Piñero, P.-L. Taberna, P. Simon and Q. Huang, Nat. Mater., 2020, 19, 894–899 CrossRef CAS PubMed.
- W. Sun, S. A. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. J. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC.
- T. Li, L. Yao, Q. Liu, J. Gu, R. Luo, J. Li, X. Yan, W. Wang, P. Liu, B. Chen, W. Zhang, W. Abbas, R. Naz and D. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6115–6119 CrossRef CAS PubMed.
- J. Zhu, S. Zhu, Z. Cui, Z. Li, S. Wu, W. Xu, T. Ba, Y. Liang and H. Jiang, Energy Storage Mater., 2024, 70, 103503 CrossRef.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- C. Lamiel, I. Hussain, J. H. Warner and K. Zhang, Mater. Today, 2023, 63, 313–338 CrossRef CAS.
- O. Salim, K. A. Mahmoud, K. K. Pant and R. K. Joshi, Mater. Today Chem., 2019, 14, 100191 CrossRef CAS.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, Trends Chem., 2019, 1, 210–223 CrossRef CAS.
- Z. Li and Y. Wu, Small, 2019, 15, 1804736 CrossRef PubMed.
- B. Anasori, C. Shi, E. J. Moon, Y. Xie, C. A. Voigt, P. R. C. Kent, S. J. May, S. J. L. Billinge, M. W. Barsoum and Y. Gogotsi, Nanoscale Horiz., 2016, 1, 227–234 RSC.
- K. Hantanasirisakul and Y. Gogotsi, Adv. Mater., 2018, 30, 1804779 CrossRef PubMed.
- M. Han, C. E. Shuck, R. Rakhmanov, D. Parchment, B. Anasori, C. M. Koo, G. Friedman and Y. Gogotsi, ACS Nano, 2020, 14, 5008–5016 CrossRef CAS PubMed.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- Y. Liu, H. Xiao and W. A. Goddard, III, J. Am. Chem. Soc., 2016, 138, 15853–15856 CrossRef CAS.
- C. J. Zhang, S. Pinilla, N. McEvoy, C. P. Cullen, B. Anasori, E. Long, S.-H. Park, A. Seral-Ascaso, A. Shmeliov, D. Krishnan, C. Morant, X. Liu, G. S. Duesberg, Y. Gogotsi and V. Nicolosi, Chem. Mater., 2017, 29, 4848–4856 CrossRef CAS.
- S. Huang and V. N. Mochalin, Inorg. Chem., 2019, 58, 1958–1966 CrossRef CAS.
- S. Huang and V. N. Mochalin, ACS Nano, 2020, 14, 10251–10257 CrossRef CAS PubMed.
- H. Lei, S. Tan, L. Ma, Y. Liu, Y. Liang, M. S. Javed, Z. Wang, Z. Zhu and W. Mai, ACS Appl. Mater. Interfaces, 2020, 12, 44639–44647 CrossRef CAS PubMed.
- J. Xu, X. Zhong, X. Wu, Y. Wang and S. Feng, J. Energy Chem., 2022, 71, 129–140 CrossRef CAS.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2018, 1, 173–180 CrossRef.
- Y. Yoon, A. P. Tiwari, M. Lee, M. Choi, W. Song, J. Im, T. Zyung, H.-K. Jung, S. S. Lee, S. Jeon and K.-S. An, J. Mater. Chem. A, 2018, 6, 20869–20877 RSC.
- Y. Jiang, T. Sun, X. Xie, W. Jiang, J. Li, B. Tian and C. Su, ChemSusChem, 2019, 12, 1368–1373 CrossRef PubMed.
- L. R. Johnson, S. Sridhar, L. Zhang, K. D. Fredrickson, A. S. Raman, J. Jang, C. Leach, A. Padmanabhan, C. C. Price, N. C. Frey, A. Raizada, V. Rajaraman, S. A. Saiprasad, X. Tang and A. Vojvodic, ACS Catal., 2020, 10, 253–264 CrossRef CAS.
- D. Kan, D. Wang, X. Zhang, R. Lian, J. Xu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 3097–3108 RSC.
- Q. Guo, Y. Xie, X. Wang, S. Lv, T. Hou and C. Bai, J. Am. Ceram. Soc., 2005, 88, 249–251 CrossRef CAS.
- Y. Guo, T. Wang, Q. Yang, X. Li, H. Li, Y. Wang, T. Jiao, Z. Huang, B. Dong, W. Zhang, J. Fan and C. Zhi, ACS Nano, 2020, 14, 9089–9097 CrossRef CAS.
- B. Wei, Z. Fu, D. Legut, T. C. Germann, S. Du, H. Zhang, J. S. Francisco and R. Zhang, Adv. Mater., 2021, 33, 2102595 CrossRef CAS.
- J. Zhang, E. Wang, S. Cui, S. Yang, X. Zou and Y. Gong, Nano Lett., 2022, 22, 1398–1405 CrossRef CAS.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS.
- V. Ramalingam, P. Varadhan, H.-C. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma, Y. Wang, H. N. Alshareef and J.-H. He, Adv. Mater., 2019, 31, 1903841 CrossRef CAS.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C. L. Dong, R. S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T.-S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- X. Wang, C. Wang, S. Ci, Y. Ma, T. Liu, L. Gao, P. Qian, C. Ji and Y. Su, J. Mater. Chem. A, 2020, 8, 23488–23497 RSC.
- C.-F. Du, X. Sun, H. Yu, Q. Liang, K. N. Dinh, Y. Zheng, Y. Luo, Z. Wang and Q. Yan, Adv. Sci., 2019, 6, 1900116 CrossRef.
- D. Kan, R. Lian, D. Wang, X. Zhang, J. Xu, X. Gao, Y. Yu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 17065–17077 RSC.
- T. A. Le, Q. V. Bui, N. Q. Tran, Y. Cho, Y. Hong, Y. Kawazoe and H. Lee, ACS Sustainable Chem. Eng., 2019, 7, 16879–16888 CrossRef CAS.
- Y. Shi and Y. Liu, Appl. Catal., B, 2021, 297, 120482 CrossRef CAS.
- X. Sang, Y. Xie, M.-W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS PubMed.
- F. Xia, J. Lao, R. Yu, X. Sang, J. Luo, Y. Li and J. Wu, Nanoscale, 2019, 11, 23330–23337 RSC.
- K. Rajavel, X. Yu, P. Zhu, Y. Hu, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2020, 12, 49737–49747 CrossRef CAS.
- X. Wang, J. Ding, W. Song, X. Yang, T. Zhang, Z. Huang, H. Wang, X. Han and W. Hu, Adv. Energy Mater., 2023, 13, 2300148 CrossRef CAS.
- R. Ibragimova, P. Rinke and H.-P. Komsa, Chem. Mater., 2022, 34, 2896–2906 CrossRef.
- J. Halim, J. Palisaitis, J. Lu, J. Thörnberg, E. Moon, M. Precner, P. Eklund, P. Persson, M. Barsoum and J. Rosen, ACS Appl. Nano Mater., 2018, 1, 2455–2460 CrossRef.
- H. Lind, B. Wickman, J. Halim, G. Montserrat-Sisó, A. Hellman and J. Rosen, Adv. Sustainable Syst., 2021, 5, 2000158 CrossRef.
- X. Yang, N. Gao, S. Zhou and J. Zhao, Phys. Chem. Chem. Phys., 2018, 20, 19390–19397 RSC.
- B. Jiang, T. Yang, T. Wang, C. Chen, M. Yang, X. Yang, J. Zhang and Z. Kou, Chem. Eng. J., 2022, 442, 136119 CrossRef CAS.
- T. A. Le, N. Q. Tran, Y. Hong, M. Kim and H. Lee, ChemSusChem, 2020, 13, 945–955 CrossRef CAS PubMed.
- X. Yu, W. Yin, T. Wang and Y. Zhang, Langmuir, 2019, 35, 2909–2916 CrossRef CAS PubMed.
- W. Yuan, L. Cheng, Y. An, H. Wu, N. Yao, X. Fan and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 8976–8982 CrossRef CAS.
- S.-Y. Pang, W.-F. Io, L.-W. Wong, J. Zhao and J. Hao, Adv. Sci., 2020, 7, 1903680 CrossRef CAS.
- H. Wei, Q. Jiang, C. Ampelli, S. Chen, S. Perathoner, Y. Liu and G. Centi, ChemSusChem, 2020, 13, 5614–5619 CrossRef CAS.
- M.-Q. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef.
- L. Xiu, Z. Wang, M. Yu, X. Wu and J. Qiu, ACS Nano, 2018, 128, 8017–8028 CrossRef PubMed.
- Y. Wu, W. Wei, R. Yu, L. Xia, X. Hong, J. Zhu, J. Li, L. Lv, W. Chen, Y. Zhao, L. Zhou and L. Mai, Adv. Funct. Mater., 2022, 32, 2110910 CrossRef CAS.
- S. Jin, W. Shao, S. Chen, L. Li, S. Shang, Y. Zhao, X. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202113411 CrossRef CAS.
- Y. Xue, Y. Xie, C. Xu, H. He, Q. Jiang, G. Ying and H. Huang, Surf. Interfaces, 2022, 30, 101907 CrossRef CAS.
- Z. Ye, Y. Jiang, L. Li, F. Wu and R. Chen, Adv. Mater., 2021, 33, 2101204 CrossRef CAS.
- H. Wang, Z. Cui, S.-A. He, J. Zhu, W. Luo, Q. Liu and R. Zou, Nano-Micro Lett., 2022, 14, 189 CrossRef CAS PubMed.
- Y. Cheng, X. Li, P. Shen, Y. Guo and K. Chu, Energy Environ. Mater., 2023, 6, e12268 CrossRef CAS.
- B. Zhang, J. Shan, X. Wang, Y. Hu and Y. Li, Small, 2022, 18, 2200173 CrossRef CAS.
- K. Ba, D. Pu, X. Yang, T. Ye, J. Chen, X. Wang, T. Xiao, T. Duan, Y. Sun, B. Ge, P. Zhang, Z. Liang and Z. Sun, Appl. Catal., B, 2022, 317, 121755 CrossRef CAS.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S.-Z. Qiao, Adv. Mater., 2021, 33, 2007508 CrossRef CAS PubMed.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef.
- P. Zhang, R. Wang, T. Xiao, Z. Chang, Z. Fang, Z. Zhu, C. Xu, L. Wang and J. Cheng, Energy Technol., 2020, 8, 2000306 CrossRef.
- J. Huang, M. Feng, Y. Peng, C. Huang, X. Yue and S. Huang, Small, 2023, 19, 2206098 CrossRef PubMed.
- H. Zong, L. Hu, S. Gong, K. Yu and Z. Zhu, Electrochim. Acta, 2022, 404, 139781 CrossRef.
- L. Yan, D. Song, J. Liang, X. Li, H. Li and Q. Liu, J. Colloid Interface Sci., 2023, 640, 338–347 CrossRef CAS PubMed.
- J. Jiang, F. Li, S. Bai, Y. Wang, K. Xiang, H. Wang, J. Zou and J.-P. Hsu, Nano Res., 2023, 16, 4656–4663 CrossRef CAS.
- L. Meng, L.-K. Yan, F. Viñes and F. Illas, J. Mater. Chem. A, 2023, 11, 6886–6900 RSC.
- Y. He, F. Yan, B. Geng, C. Zhu, X. Zhang, X. Zhang and Y. Chen, J. Colloid Interface Sci., 2022, 619, 148–157 CrossRef CAS.
- Y. Lu, D. Fan, Z. Chen, W. Xiao, C. Cao and X. Yang, Sci. Bull., 2020, 65, 460–466 CrossRef CAS PubMed.
- Y. Chen, H. Yao, F. Kong, H. Tian, G. Meng, S. Wang, X. Mao, X. Cui, X. Hou and J. Shi, Appl. Catal., B, 2021, 297, 120474 CrossRef CAS.
- L. Yan, Z. Du, X. Lai, J. Lan, X. Liu, J. Liao, Y. Feng and H. Li, Int. J. Hydrogen Energy, 2023, 48, 1892–1903 CrossRef CAS.
- C.-F. Du, K. N. Dinh, Q. Liang, Y. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127 CrossRef.
- Y. Zhang, Q. Fu, B. Song and P. Xu, Acc. Mater. Res., 2022, 3, 1088–1100 CrossRef CAS.
- D. Tyndall, L. Gannon, L. Hughes, J. Carolan, S. Pinilla, S. Jaśkaniec, D. Spurling, O. Ronan, C. McGuinness, N. McEvoy, V. Nicolosi and M. P. Browne, npj 2D Mater. Appl., 2023, 7, 15 CrossRef CAS PubMed.
- L. Huang, Y. Zou, D. Chen and S. Wang, Chin. J. Catal., 2019, 40, 1822–1840 CrossRef CAS.
- J. Zhao, J. Lian, Z. Zhao, X. Wang and J. Zhang, Nano-Micro Lett., 2022, 15, 19 CrossRef PubMed.
- X. Huang, M. Song, J. Zhang, J. Zhang, W. Liu, C. Zhang, W. Zhang and D. Wang, Nano Res., 2022, 15, 3927–3932 CrossRef CAS.
- Y. Wen, C. Ma, Z. Wei, X. Zhu and Z. Li, RSC Adv., 2019, 9, 13424–13430 RSC.
- L. Jiang, J. Duan, J. Zhu, S. Chen and M. Antonietti, ACS Nano, 2020, 14, 2436–2444 CrossRef CAS.
- J. Chen, X. Yuan, F. Lyu, Q. Zhong, H. Hu, Q. Pan and Q. Zhang, J. Mater. Chem. A, 2019, 7, 1281–1286 RSC.
- Z. Zeng, G. Fu, H. B. Yang, Y. Yan, J. Chen, Z. Yu, J. Gao, L. Y. Gan, B. Liu and P. Chen, ACS Mater. Lett., 2019, 1, 432–439 CrossRef CAS.
- X. Yang, Q. Jia, F. Duan, B. Hu, M. Wang, L. He, Y. Song and Z. Zhang, Appl. Surf. Sci., 2019, 464, 78–87 CrossRef CAS.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 30, 1803220 CrossRef PubMed.
- G. F. Chen, Y. F. Yuan, H. F. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. P. Wu, J. Lu and H. H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef.
- C. Xu, C. Fan, X. Zhang, H. Chen, X. Liu, Z. Fu, R. Wang, T. Hong and J. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 19539–19546 CrossRef CAS PubMed.
- W. Qiu, N. Yang, D. Luo, J. Wang, L. Zheng, Y. Zhu, E. M. Akinoglu, Q. Huang, L. Shui, R. Wang, G. Zhou, X. Wang and Z. Chen, Appl. Catal., B, 2021, 293, 120216 CrossRef CAS.
- M. Jin, X. Zhang, X. Zhang, H. Zhou, M. Han, Y. Zhang, G. Wang and H. Zhang, Nano Res., 2022, 15, 8826–8835 CrossRef CAS.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- Y. Ding, J. Zhang, A. Guan, Q. Wang, S. Li, A. M. Al-Enizi, L. Qian, L. Zhang and G. Zheng, Nano Convergence, 2021, 8, 14 CrossRef CAS.
- X. Lv, L. Kou and T. Frauenheim, ACS Appl. Mater. Interfaces, 2021, 13, 14283–14290 CrossRef CAS.
- T. Li, X. Yan, L. Huang, J. Li, L. Yao, Q. Zhu, W. Wang, W. Abbas, R. Naz, J. Gu, Q. Liu, W. Zhang and D. Zhang, J. Mater. Chem. A, 2019, 7, 14462–14465 RSC.
- K. Chu, X. Li, Y. Tian, Q. Li and Y. Guo, Energy Environ. Mater., 2022, 5, 1303–1309 CrossRef.
- H. He, H.-M. Wen, H.-K. Li, P. Li, J. Wang, Y. Yang, C.-P. Li, Z. Zhang and M. Du, Adv. Sci., 2023, 10, 2206933 CrossRef PubMed.
- Z. Jin, C. Liu, Z. Liu, J. Han, Y. Fang, Y. Han, Y. Niu, Y. Wu, C. Sun and Y. Xu, Adv. Energy Mater., 2020, 10, 2000797 CrossRef.
- B. Huang, J. Yang, G. Ren, Y. Qian and Y.-W. Zhang, Appl. Catal., A, 2022, 646, 118886 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |