
Tunable nanofluidic device for digital nucleic acid analysis†
Imman I.
Hosseini
 a,
Seyed Vahid
Hamidi
a,
Xavier
Capaldi
b,
Zezhou
Liu
b,
Matheus Azevedo
Silva Pessoa
b,
Sara
Mahshid
*a and
Walter
Reisner
*b
a,
Seyed Vahid
Hamidi
a,
Xavier
Capaldi
b,
Zezhou
Liu
b,
Matheus Azevedo
Silva Pessoa
b,
Sara
Mahshid
*a and
Walter
Reisner
*b
aDepartment of Biomedical Engineering, McGill University, 3775 Rue University, Montreal, Quebec H3A 2B4, Canada. E-mail: sara.mahshid@mcgill.ca; Tel: +1 (514) 398-8964
bDepartment of Physics, McGill University, 3600 Rue University, Montreal, Quebec H3A 2T8, Canada. E-mail: walter.reisner@mcgill.ca; Tel: +1 (514) 398-8964
First published on 8th March 2024
Abstract
Nano/microfluidic-based nucleic acid tests have been proposed as a rapid and reliable diagnostic technology. Two key steps for many of these tests are target nucleic acid (NA) immobilization followed by an enzymatic reaction on the captured NAs to detect the presence of a disease-associated sequence. NA capture within a geometrically confined volume is an attractive alternative to NA surface immobilization that eliminates the need for sample pre-treatment (e.g. label-based methods such as lateral flow assays) or use of external actuators (e.g. dielectrophoresis) that are required for most nano/microfluidic-based NA tests. However, geometrically confined spaces hinder sample loading while making it challenging to capture, subsequently, retain and simultaneously expose target NAs to required enzymes. Here, using a nanofluidic device that features real-time confinement control via pneumatic actuation of a thin membrane lid, we demonstrate the loading of digital nanocavities by target NAs and exposure of target NAs to required enzymes/co-factors while the NAs are retained. In particular, as proof of principle, we amplified single-stranded DNAs (M13mp18 plasmid vector) in an array of nanocavities via two isothermal amplification approaches (loop-mediated isothermal amplification and rolling circle amplification).
The Nucleic Acid Test (NAT) is a reliable method for early disease detection. For example, polymerase chain reaction (PCR) has been used to detect viral infections such as SARS-CoV-2,1 and next-generation sequencing (NGS) has been used for whole genome sequencing (WGS) to detect cancer associated single nucleotide variants (SNVs).2 In NA amplification, detection specificity can be significantly improved by using designed primers to enable the detection of single nucleotide variants.3 In addition, partitioning the sample, i.e. dividing the sample into small volumes either in micro wells4 or droplets,5 can increase detection specificity and sensitivity. Sample partitioning prior to amplification enhances the detection specificity by reducing cross-contamination between the samples in each partition.5 Partitioning can also enhance detection sensitivity for low-abundant target molecule samples.6 In this case, certain partitions may contain one or more target molecules, although many other partitions may be devoid of the target molecule. Consequently, detecting the target molecule is more probable within partitions enriched with the target molecule. Furthermore, there is a higher likelihood of detecting a response signal within each positive partition compared to traditional methods where the detection signal is diluted throughout the entire sample. Moreover, partitioning can provide accurate quantitative measurement of the target molecule concentration by mitigating quantitative biases found in traditional quantitative NAT methods. These include biases arising from target molecule properties (such as GC contents or secondary structures such as hairpins or stem-loops) or inhibiting effects (caused by DNA binding proteins or amplification contamination such as nucleases and salts).5
Digital PCR (dPCR) is the gold standard approach for partitioned NA amplification.7 A key requirement of dPCR is thermal cycling. In thermal cycling the temperature is increased to around 95 °C for DNA denaturation, reduced to 50 °C for primer annealing, and then increased to 65 °C for new strand synthesis. Isothermal amplification methods have been proposed as alternatives to dPCR to eliminate the need for the complex setup required for thermal cycling and increase detection sensitivity and specificity. Loop-mediated isothermal AMPlificaiton (LAMP) method uses primers targeting multiple regions of the target molecule to decrease the non-specific amplification8 at 65 °C. The polymerase used in LAMP (Bst 2.0) is a high-speed strand-displacing DNA polymerase with a high tolerance to inhibitors. Digital LAMP has been proposed to further increase assay sensitivity.9,10 Although LAMP has been reported as a powerful technique for point-of-care testing due to its robustness in DNA amplification and high selectivity via the use of three primer pairs, the high-temperature activity of Bst DNA polymerase (65 °C) makes it challenging to couple LAMP with other platforms. Rolling Circle Amplification (RCA) is another isothermal amplification method that can work at room temperature due to the room-temperature activity of phi29 polymerase.11 So far RCA has been integrated with microfluidic platforms,12 and combined with multiple read-outs including surface plasmon resonance (SPR),13 and electrochemistry.14 Compared with RCA, LAMP assays are more likely to generate unwanted non-specific amplification products due to primer dimerization and mismatched hybridization, arising from the fact that in LAMP reactions about six primers at high concentrations (3.6–4.4 μM) are needed in comparison with RCA reactions where two primers at lower concentrations (0.4–1.0 μM) are employed.15,16
Nanofluidic devices can be used for controlled digitization of NAT samples. Using nanofluidic devices, a larger number of partitions can be accommodated in a given field of view, consequently enhancing the sensitivity and specificity of detection by mitigating the influence of quantitative biases and inhibitors. Also, the sample partitioning can be performed very accurately using nanofluidic devices, reducing the biases introduced by sample loading inside the partitions. The primary step in nanofluidic based NATs is molecule capture. A number of approaches have been demonstrated for NA capture, including immunoaffinity17,18 and using external forces such as dielectrophoresis19 and optical trapping.20
Nucleic Acids can also be captured by confinement in nanofluidic compartments (e.g., nano-wells or nanochannels) without external forces or treatment of the nanofluidic device for immunoaffinity immobilization. In confinement-based capture, NAs are loaded in a slit with a nanoscale (~100nm) height dimension containing the compartments as embedded features. The embedded compartments act as entropic traps for the NAs while accessibility to reagents required for biochemical processing of the NAs is ensured by leaving a fixed nanoscale gap between the bonding lid and the slit floor.21,22 For example, Marie et al. confined lambda DNA in an array of nanopits and showed that phi29-driven amplification can be achieved while the DNA is immobilized in the nanopits with a slit gap.23 Öz et al. stretched lambda DNAs in nanochannels and loaded nucleolytic enzyme DNase I to digest the stretched DNA while the molecules were retained in the nanochannels.24 Two main obstacles exist in this approach. First, the target NA size must be significantly larger than the enzyme and primers; otherwise, the target NA can escape from the compartments during the buffer exchanges required to introduce processing reagants. Secondly, a device design with a fixed slit height can only work with a limited range of DNA sizes. At high slit heights, only large DNA molecules can be loaded and confined in the nanofluidic compartments, as small molecules tend to escape quickly. Conversely, only small DNA molecules can be loaded and confined for low slit heights, while the loading of large molecules is hindered by the narrowness of the slit.
One solution is to dynamically adjust the slit height so that the optimum degree of confinement can be achieved for a given assay step. Here we harness a coupled-membrane nanofluidic device featuring tunable confinement to confine and partition nucleic acids inside an array of nanocavities and then amplify the target nucleic acids using isothermal amplification methods including RCA and LAMP. In our previous work,25–27 we introduced a nanofluidic system that exploits pneumatic pressure applied to a flexible free-standing SiNx membrane lid to adjust in situ the degree of confinement experienced by nanoparticles and single molecule analytes in nanoconfined volumes. Pneumatic pressure applied from above deflects the lid downwards varying the degree of confinement; at the highest degree of deflection, the lid seals off nanocavity structures embedded in a nanoslit. Here we have enhanced our previously developed nanofluidic device design to facilitate performing an amplification reaction on cavity-confined target species. Amplifying the cavity confined target species requires the capability of introducing chemical reagents into the nanocavities, such as DNA polymerase, amplification buffer and fluorescent dye while the DNA targets are retained. This can be achieved using our ability to adjust the degree of membrane deflection with nm precision, creating gaps sufficiently small around the nanocavity to prevent the escape of target DNA while allowing the introduction of amplification reagents. Reagents are introduced via a set of parallel nanochannels located sufficiently close (500 nm) to the nanocavities for diffusive transport through the thin access gap to be efficient and a uniform distribution of reagents achieved within and across the cavities. The nanochannels are in turn interfaced with side microchannels with separate inlets for loading of target molecules and amplification reagents. The use of separate loading inlets prevents unwanted amplification in the microchannels and constrains the amplification to take place only at the channel intersection where the cavity array is located.
By employing dynamic adjustments to the slit height, we demonstrated the staining of unlabeled NAs within nanocavities after concentrating the NAs inside the nanocavity array (see ESI section V†). To accurately assess the extent of NA retention within the nanocavities, we observed stable retention of NAs while allowing small oligonucleotides (20 bps) to be loaded and washed out from the cavities. Additionally, we demonstrated precise size dependent particle retention in the cavities by quantifying the diffusive escape of fluorescently labelled gold nanoparticles from the cavities as the slit height is varied. Following comprehensive control tests, we successfully amplified nanocavity confined M13Mp18 plasmid vector using Loop-mediated isothermal AMPlification (LAMP) and Rolling Circle Amplification (RCA).
Our adjustable confinement device is fabricated using the procedure described in Capaldi et al.28 The device consists of a borosilicate substrate that incorporates two intersecting nanoslits, namely the target-loading channel and the reagent-loading channel (Fig. 1a). The nanoslit depth is 420 nm and each slit is connected to two loading reservoirs (refer to Fig. 1a and ESI section I†). An array of circular nanocavities (with a radius Rc = 1000 nm and a depth of 300 nm) are embedded at the intersection of the microchannels (Fig. 1b and c). Nanochannels (with a width of 100 nm) are positioned a distance of 500 nm from the nanocavities so that they are in close proximity. Above the nanocavities, there is a flexible lid formed from a free-standing silicon nitride membrane (200 nm thick). When pneumatic pressure is applied the membrane deflects, resulting in a reduction of the vertical gap (slit height). As we have previously shown,25 the slit height as a function of applied pressure can be calibrated by measuring the intensity of fluorescent dye introduced into the nanoslit see (Fig. 1d and ESI section III†).
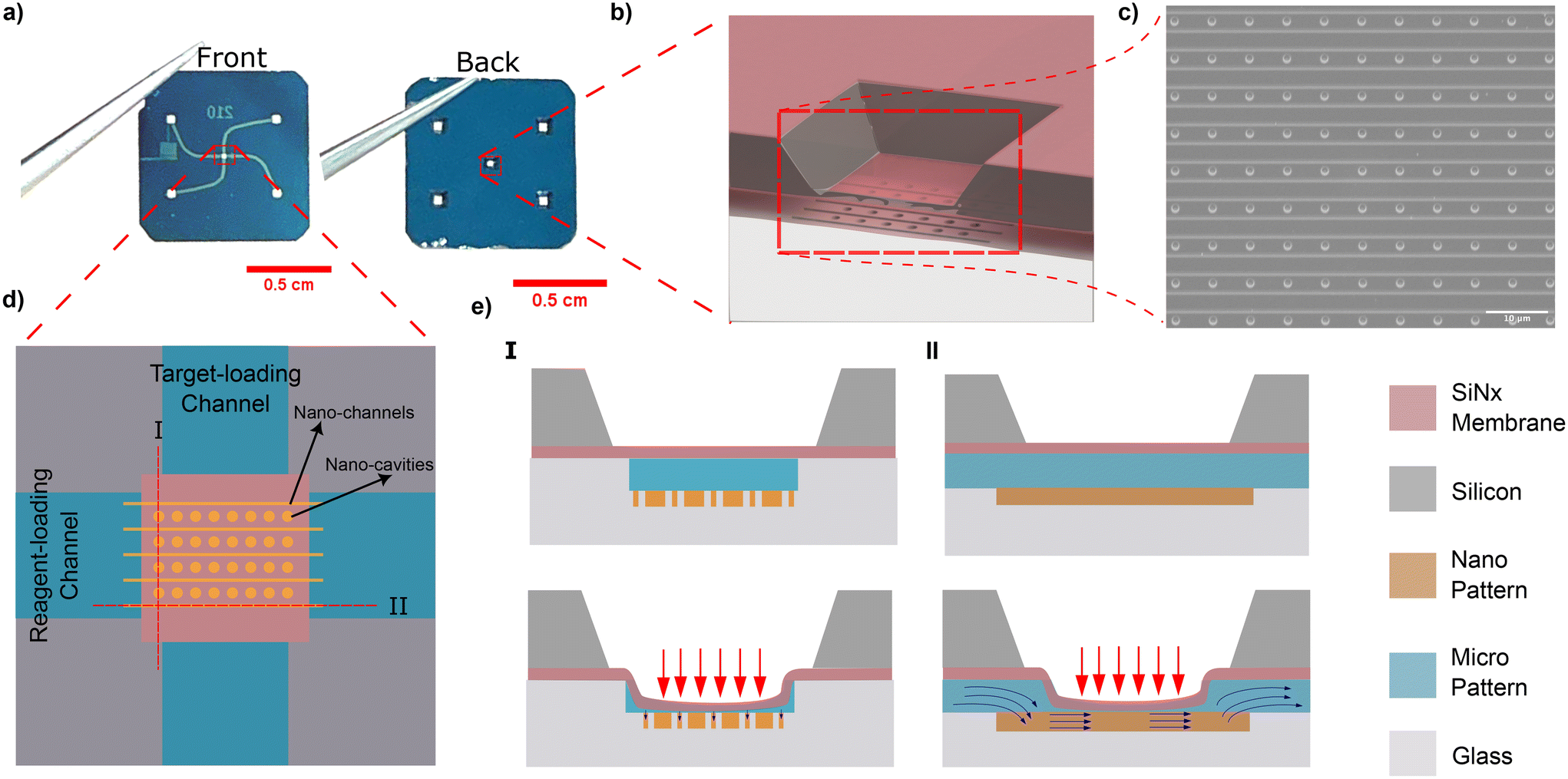 | ||
| Fig. 1 Nanofluidic device and its working principle. (a) Front and back views of the nanofluidic device (b) 3D schematic of the deformable membrane. (c) SEM of nanocavity array after the membrane is removed. (d) The device has two crossed microchannels (target-loading channel and reagent-loading channel) which intersect at the membrane position. (e) (I) Cut-out view of the nanofluidic device at the reagent-loading channel. When the membrane is pushed down, the membrane prevents the reagents from flowing from the nanochannels to the nanocavities. (e) (II) Cut-out view of the nanofluidic device at the target loading channel. When the membrane is deflected, the reagents can enter the nanochannels as the nanochannels are connected to the reagent-loading channel external to the membrane area. | ||
Digital DNA amplification can be divided into two main steps. The first step is the confinement and concentration of DNA molecules in the nanocavities (Fig. 2a). The second step is DNA analysis, including removing extra molecules from the chamber via introducing a washing buffer (e.g., 1× TE buffer), fluorescent staining, and DNA polymerization. In the first step, the DNA is loaded from the target-loading channel into the membrane region by constant pressure applied at the inlet (Fig. 2b-I). During this step no pneumatic pressure is applied to the membrane so the slit height is at its maximum value (420 nm). The membrane is then deflected to drive the DNA inside the cavities. When sufficient applied pressure is applied, the membrane will reach its maximum deflection at which the nanocavities are completely sealed (Fig. 2b-I).
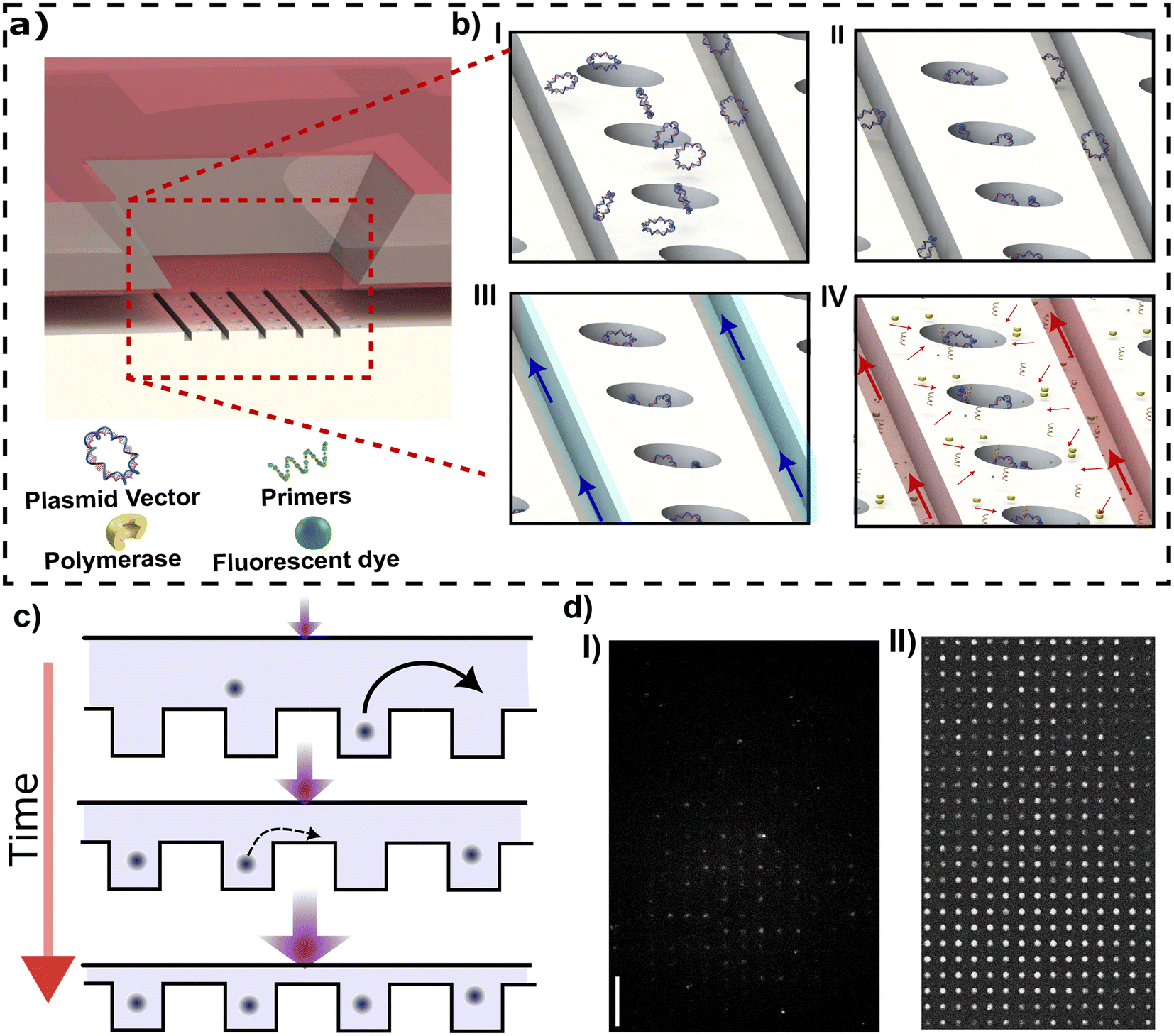 | ||
| Fig. 2 (a) Schematic image of the membrane region. (b) Amplification steps of DNA inside cavities; (b-I) loading of target DNAs from target loading channel; (b-II) DNA confinement via membrane deflection; (b-III) washing out extra DNAs in side nanochannels and reagent-loading channels; (b-IV) adding amplification reagents from nanochannels while the target DNAs remain confined in nanocavities. (c) Cartoon showing molecule concentration in cavities as the membrane lid is lowered. (d) Fluorescent images showing the confinement of YOYO-1 labelled-pBR322 plasmid vectors in nanocavities (I) without and (II) with the concentrating procedure. | ||
In the second step, the detection reagents are loaded from the reagent-loading channel. First, NA molecules present in the side nanochannels are removed via a washing buffer introduced in the reagent-loading channel (Fig. 2b-III). Note that the membrane comes into contact with the bottom of the slit channel during the washing step. This contact prevents any particles from jumping or transferring from the nanochannel to the nanocavities. Then, the amplification reagents including polymerase, dNTPs, and fluorescent dyes are loaded from the reagent-loading channel (Fig. 2b-IV). The pneumatic pressure is reduced to create a narrow gap between the membrane and nanocavities which lets the amplification reagents diffuse through the gap into the nanocavity while the large target NAs stay confined in the nanocavities.
In this approach, particle confinement in cavities is size-dependent. Whether particles can migrate between cavities is determined by their size relative to the gap between the membrane and substrate. If the size of particles exceeds this gap, they cannot cross over to adjacent cavities, thus preventing false positives. Notably, amplicons generated through the RCA method are inherently larger than the target molecule, ensuring their retention within the gap created between the membrane and substrate. Note that the gap spacing can be precisely controlled, with spacings below 60 nm being maintained with a precision of 8 nm (as depicted in ESI section III†).
While membrane deflection will confine molecules inside the nanocavities, confinement alone does not lead to DNA concentration (see Fig. 2d-I). Additional concentration can be necessary if the sample contains a low concentration of target NAs. Combined concentration and confinement can be achieved by a protocol combining membrane deflection with fluid flow. When target NAs are driven over the embedded nanocavity array, the NAs will accumulate inside the cavities row by row.23 This arises as induced flow, increasing the molecule throughput, increases the filling rate of cavities from molecules passing by in the slit that can then enter a cavity. At the same time, molecules that are captured in a cavity remain trapped, even in the presence of the flow, due to the high entropic barrier created by the slit. A high cavity filling rate combined with a very small escape rate leads to NA concentration within the cavities. As molecules are repelled from cavities that are already filled due to the strong excluded-volume interactions with molecules present in the filled cavities, cavity rows will tend to fill sequentially. Therefore, an intermediate flow should be adopted to obtain a high occupancy; however, a long waiting time is required to obtain full occupancy at an intermediate flow.
The entropic barrier can be increased by decreasing the slit height, allowing for using higher flow to achieve a higher occupancy in a shorter time. However, using a very thin slit height hinders the loading and washing of target NAs from cavities. The ability to adjust confinement in real time allows us to separate the particle loading and washing steps from the concentration step. Consequently, a narrow slit height can be employed specifically for the concentration step without any concerns regarding the loading and washing steps. Also, in our method, since the occupancy of one cavity is independent of the occupancy of all other cavities, a defect in one cavity does not affect the occupancy of other cavities, as is the case for pure flow-based loading without confinement modulation.
To demonstrate combined loading and concentration, YOYO-1 stained pBR322 vector plasmid with 1 μg mL−1 concentration is loaded into the membrane area by applying pressure at the reagent-loading inlet. The membrane is then gradually depressed from an initial slit height of 197 ± 6 nm (25 mbar) to a final height of 113 ± 6 nm (35 mbar). The membrane is lowered in small increments, with 0.5 mbar pressure increase applied per increment each increment taking 5 s; the overall process then takes 20 such steps. Following this procedure we observed that more than 95% of cavities are occupied with the vector plasmid in 100 seconds.
It is critical to ensure the retention of target DNAs in nanocavities during washing and reagent loading steps. To quantify the degree of retention, first our confinement and concentration protocol was used to introduce YOYO-3 labeled pBR322 plasmid vectors into the nanocavity arrays with the lid maximally deflected to ensure complete nanocavity sealing. Then, 20 bp primers tagged with FAM are loaded from the reagent-loading channel. The primers’ radius of gyration is estimated to be 1.9 nm (see ESI section VII†). After the primers reach the membrane region, a 5.4 ± 2.2 nm gap is introduced so that the primers can enter the cavities. Primer loading in the nanocavities leads to an increase in a FAM-associated signal at the nanocavity location. While the FAM signal increases, the YOYO-3 signal associated with target DNAs stays stable. The observation suggests that we can keep the target NAs fully confined while primers are loaded (Fig. 3a).
 | ||
| Fig. 3 (a) Schematic showing target molecule retention inside cavities while small nucleic acids are loaded and then washed away from the cavities. (b) Retention of YOYO-3 labelled-pBR322 plasmid vectors in nanocavities during the introduction of FAM-tagged primers (20 bp). The inset shows the relative intensity, the intensity normalized to the intensity before loading primers; this reaches zero when the cavities are completely empty of primers. | ||
To perform sequential analysis, we need to remove from the nanocavities the molecules used in the previous round of analysis; their retention in subsequent analysis cycles might introduce noise or errors. For example, in sequential multiplex amplification, it is possible to quench fluorescent dye and perform another amplification round. However, the sequential amplification needs the reaction chamber to be emptied of the primers and reagents used in the previous analysis round (Fig. 3b). We demonstrate that we can remove the primers from the nanocavities while the target DNA molecules are stably retained. To achieve this, the reagent-loading inlet is flushed with washing buffer. When the washing buffer reaches the membrane region, the FAM channel intensity drops indicating primers have been removed from the nanocavities. The cavities have been completely emptied of primers when the relative intensity, the intensity normalized to the intensity before loading primers, reaches zero (Fig. 3b-inset). During the washing step, the YOYO-3 signal remains constant indicating the retention of the target DNA inside the cavities.
To demonstrate our ability to retain small particles, we examined the retention of Cy3 labelled-gold nanoparticles with mean diameters of 15 and 50 nm (Nanocs Inc.) at different slit heights. The particles are introduced from the target-loading channel and confined in the nanocavities by lowering the membrane to ensure complete sealing. Free fluorescent dye and extra gold nanoparticles in the side nanochannels are removed by introducing washing buffer from the reagent-loading channel. A very low flow (10 μm s−1) is continuously applied from the reagent-loading chamber to wash out the side channels. Then, the applied membrane pressure is decreased to create a gap between the membrane and cavities allowing nanoparticles to leave the cavities. The average fluorescent signal of six nanocavities in the middle of the membrane region is recorded for two minutes for a range of slit heights (Fig. 4). Background intensity is removed by capturing a reference frame before nanoparticle loading. Linear photobleaching correction is applied to the time-dependent fluorescence intensity by measuring fluorescence depletion outside of the nanofluidic device.
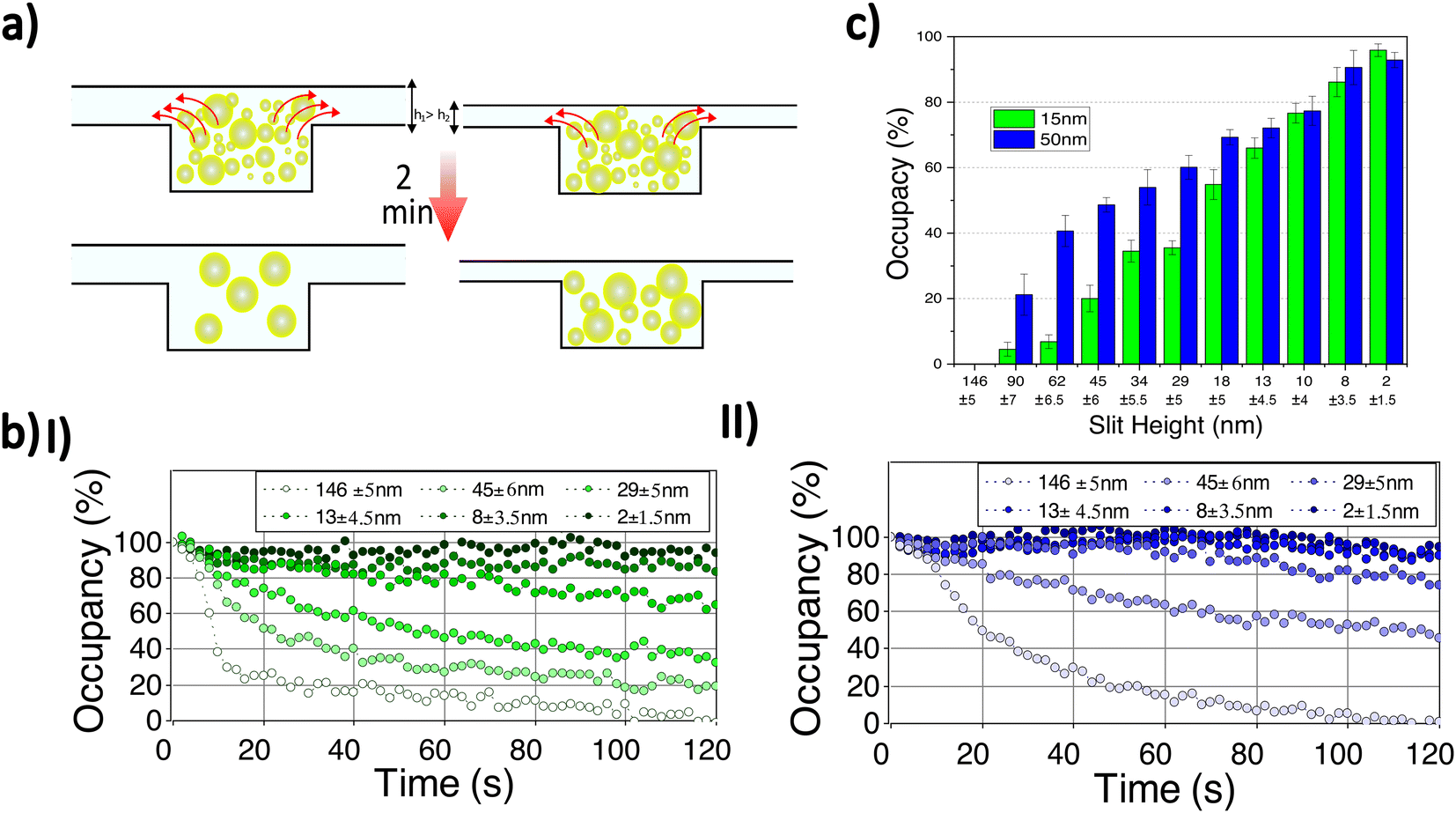 | ||
| Fig. 4 (a) Schematic images of particle retention at two slit heights. (b) Time-dependence of percent occupancy for (b-I) 15 nm and (b-II) 50 nm particles retained in nanocavities for a range of slit heights. Percent occupancy refers to percentage of particles that remain in cavity at a given time (as inferred from particle fluorescence). (c) Percent occupancy for 15 and 50 nm nanoparticles retained in cavities for different slit heights after 2 min have elapsed. | ||
When a small gap is introduced, the fluorescence intensity due to the confined nanoparticles decreases, indicating depletion of nanoparticles from the nanocavities (Fig. 4a, b). By increasing the slit height still further, particles escape more rapidly and a lower concentration is reached. At a very high slit height (≈146 ± 5 nm), the intensity reaches zero in two minutes while particles are retained for lower slit heights (Fig. 4b). Note the samples of gold nanoparticles used have a fairly broad size distribution with a ratio of standard-deviation to mean diameter of 15% (Nanocs Inc.), indicating that samples contain nanoparticles with a diameter varying considerably about the mean. For instance, in the case of the sample with a 50 nm mean diameter, there are nanoparticles that can pass through a slit with a height below 50 nm because their diameter is smaller than 50 nm.
The nanoparticles with a mean diameter of 15 nm leave the nanocavities more rapidly compared to nanoparticles with a 50 nm mean diameter at the same slit height (Fig. 4b,c). Initially, the nanocavities are occupied with maximum concentration (100% occupancy) and are empty before loading (0% occupancy). Assuming that the fluorescence signal is proportional to the number of particles retained in the cavity, the signal at maximum concentration and empty state can be used, for any given intermediate signal level, to determine the percent occupancy, the percentage of particles that are retained in the cavity at a given time. At very high slit heights (146 ± 5 nm) all particles exit the nanocavities. By reducing the slit height (45 ± 6 nm), although more than 50% of nanoparticles with 50 nm mean diameter remain confined, most of the nanoparticles (≈80%) with 15 nm mean diameter leave the nanocavities. Finally, at a very small gap height (2 ± 1.5 nm), only a tiny reduction (less than 10%) in final occupancy is observed for both samples (Fig. 4c).
We performed amplification of single-stranded DNA (M13mp18 plasmid vector) in nanocavities using two isothermal amplification methods, namely LAMP and RCA. Once the cavities are loaded with target NAs the amplification assay can be implemented. LAMP forms loop structures on the target NA, resembling dumbbells, containing open single-stranded sequences below the denaturation temperature. Primers can hybridize to the open single-stranded sequences without needing to increase the solution temperature up to the denaturation temperature (95 °C). The formation of the loop structures involves two primers (Forward Inner Primer (FIP) and F3). FIP consists of two main sequences: F2 sequences which are complementary to the target DNA sequences F2C, and F1C sequences positioned before F2. F1C is identical to the target DNA sequences located after F2C. The polymerase (usually BST 2.0) in the amplification reagents (LAMP 1X Master Mix, NewEngland Biolabs) initiates synthesis along the FIP primer and generates F1C complementary sequences (F1). Consequently, the synthesized strand along the FIP primer contains F1C–F2–F1 sequences. Secondly, F3 primer binds to the complementary sequences in the target DNA (F3C) which is positioned before F2C. A new polymerase starts synthesizing along F3 primer and detaches the synthesized strand initiated by the FIP primer. Finally, the detached strand forms the loop structure by hybridization between F1C and F1 sequences. After loop formation, a Loop Forward (LF) primer hybridizes with the formed strand containing the open single-stranded sequences in the loop structure and initiates the amplification. While FIP can also trigger amplification (by attaching to the F2C sequences of the formed strand with F2 Sequence), it may not offer the highest stability due to limitations in choosing the optimal position for loop structure formation, dependent on F1 and F2 affinities. LAMP produces double-stranded amplicons that can be detected by an intercalating fluorescent dye (1X LAMP, NewEngland Biolabs). The LAMP primers sequences (ThermoFisher) are provided in ESI section VII.†
In the RCA method, the target molecule is a circular DNA molecule. When a primer binds to the target molecule, the polymerase (Phi29, NewEngland Biolabs) initiates the synthesis from 3′ of the attached primer. As the polymerase completes a whole circle and reaches the 5′ end of the primer, it opens up the synthesized circle and continues synthesizing DNA. This process continues until the polymerase reaches its processivity limit, which is the number of nucleotide polymerizations it can perform before dissociating from the target molecule. For Phi29 polymerase, the processivity is approximately 70 kilobase pairs (kbp).29 RCA primers (ThermoFisher) are discussed in the ESI section VII.† Similar to LAMP method, the RCA products are double-stranded DNA which are detected by intercalating fluorescent dye (1X LAMP, NewEngland Biolab).
In the LAMP experiment, the reagents are loaded at the reagent-loading inlet. The target DNAs are protected from contamination via loading in a separate channel (regent-loading channel). Therefore, several consecutive experiments can be performed in one device, i.e., after testing a batch of confined DNAs, the tested DNAs are released, washed away, and fresh intact DNA is loaded. The device is heated to 65 °C using the method discussed in ESI section II.† Then, the reagents are introduced inside the nanochannels. The largest reagent molecule is Bst 2.0 polymerase (WarmStart) with ≈2.7 nm radius of gyration and primers with a maximum ≈3.0 nm radius of gyration (see ESI section VII†). Therefore, by creating about 5–10 nm gap between the membrane and nanocavities, the reagent molecules can diffuse from the side nanochannels into the nanocavities. The fluorescent dye (NEB LAMP fluorescent dye) has minimal interaction with the single-stranded M13mp18 and the signal only arises upon dye intercalation with the synthesized dsDNA. We observed an increase in fluorescent signal at ≈120 seconds at which the signal reaches its maximum value. In the LAMP method, the amplicons contain molecules with a large range of sizes, from oligonucleotide size (<100 bp) to very large molecules (>10 kbp) (see gel electrophoresis results in ESI section VI†). After saturation of cavities with amplicons, the small molecules can diffuse out from the cavities. The diffused DNAs to the nanochannels can react with the amplification reagents and, by producing very large amplicons in the nanochannels, the nanochannels can be clotted with small defects that lead to particle accumulation (see Fig. 5a ESI Video II†).
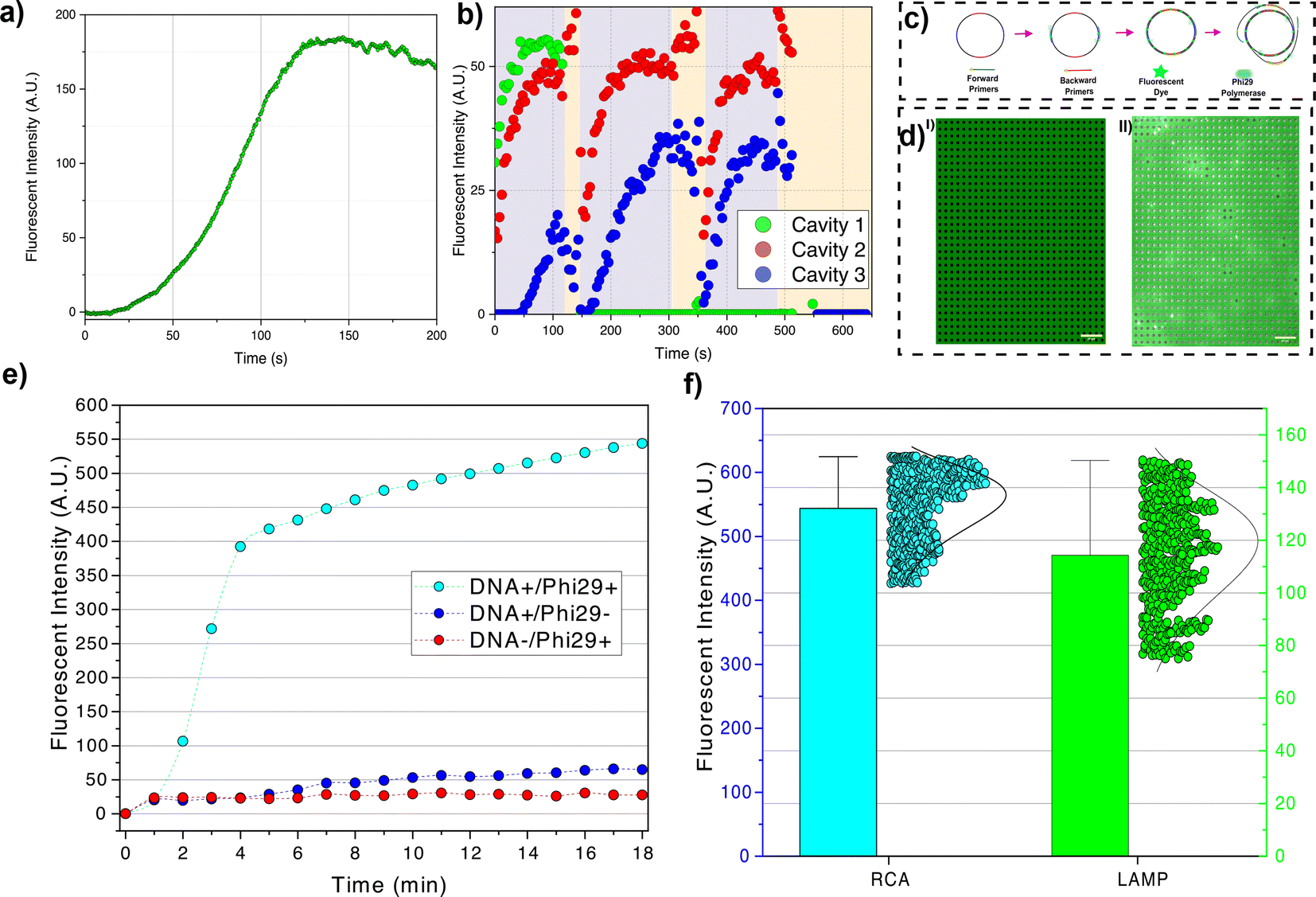 | ||
| Fig. 5 Digital amplification of M13mp18 in nanocavities. (a) Real-time nanocavity fluorescence intensity arising from LAMP averaged over 440 cavities. (b) Sequential retention, amplification and release of amplified molecules. The fluorescence signal in three selected cavities is shown, with shaded regions indicating the raising of the lid to introduce additional amplification reagents. (c) Four primer RCA amplification procedure. (d) Fluorescent images before and 18 minutes after loading the RCA amplification reagents. (e) Real-time nanocavity fluorescence intensity arising from RCA averaged over 924 cavities compared to controls where Phi29 was not included (DNA+/Phi29−) or target DNA was not included (DNA-/Phi29+). (f) fluorescence intensity distribution over cavities for the two isothermal amplification methods used (RCA and LAMP). | ||
We employed the LAMP method to demonstrate that we can retain molecules following amplification, perform additional amplification cycles on the retained molecules, and then release all cavity-confined amplicons (Fig. 5b). Following DNA (M13mp18) capture in cavities, we perform an initial amplification round by introducing amplification reagents from the reagent loading chamber. Approximately 140 seconds later, the membrane is raised to around 170 nm (25 mbar pressure). As the LAMP-generated amplicons cover a broad size range, only those smaller than 170 nm can escape through the gap formed between the membrane and the substrate; the larger amplicons are retained. The amplification rate, and consequently the fluorescence signal in the cavities, is influenced by the size and number of retained amplicons inside the cavities. Due to the variability in the size of generated amplicons, the second round of amplification may yield a higher, lower, or zero signal (indicating the removal of all amplicons from the cavity). Following the third round of amplification, the membrane is fully released, resulting in the washing out of all amplicons from the cavities (reducing all signals to zero).
In the LAMP method, a single amplicon generates several amplicons after forming a loop. The polymerase can bind to the newly generated amplicons and perform several amplifications in parallel (similar to PCR30). The parallel amplification and the high strand displacement rate of Bst 2.0 WarmStart polymerase give rise to a rapid increase in flouorescence signal. Amplicons generated using the LAMP method can migrate to other cavities which can cause contamination of other cavities and consequently give rise to false positives. In our digital amplification approach in which the confinement of particles depends on the size of the target molecules, the small amplicons can escape from their cavities. The migration of amplicons from one cavity to another has undesirable effects on applications like multiplexing which requires that targets should be kept isolated from other molecules. Therefore, a single-molecule amplification method in which the amplification product is one single large molecule instead of several small amplicons like RCA is needed for digital amplification purposes.
In the RCA method, a single polymerase attaches to a DNA and synthesizes the complementary sequences. This gives rise to particles that will always be bigger than the target molecule (see ESI section VI†); therefore, no amplicon can leave the cavity. Given a single path of amplification (i.e., not being parallel like LAMP), the fluorescence signal increases slowly. To reduce the effect of a slow amplification rate, we used four primers (two forward and two backward) instead of a single forward primer. The real-time PCR results for single primer and four primers amplification are presented in the ESI section VI† which shows the effectiveness of the proposed four primers method. We also took images with longer exposure time (2 s) and longer time-laps (1 min) to increase the signal and reduce bleaching, respectively. The two forward primers are placed on two opposite sides of the DNA, so, instead of one polymerase, two polymerases initiate the synthesis. The fluorescence intensity sharply increases in 2–3 min. This sharp increase might represent the termination of one complete circle amplification. After the full circle completion, the polymerase opens up the synthesized double strand and generates ssDNA tails. The two backward primers which were placed on two opposite sides of the vector, attach to the ssDNA tails and another Phi29 polymerase starts to synthesize the complementary part of the tails (Fig. 5c and d). The generated double-stranded tail results in an increase in fluorescence intensity with a lower slope (Fig. 5e).
We also compared the distribution of final fluorescence signals for both isothermal amplification methods. In the LAMP method, the final signal intensity correlates with the size of large particles generated during amplification (as the smaller particles leave the cavity). Given that the size distribution of generated particles is random, there is a relatively wide distribution in final fluorescence intensity (Fig. 5f). In the RCA method, we observed most cavities are in the same range of final intensity after 18 minutes which shows most DNAs follow the same amplification procedure (Fig. 5f).
In RCA method, it is possible that only one of the forward primers continues the amplification. If the polymerase starts the amplification by one of the forward primers, and the second primer cannot find the target while the first polymerase syntheses the half circle of M13mp18 vector (given the fast strand displacement speed of Phi29, 50–200 bases per second (ref. 31)), the second primer can no longer attach to the target; therefore, we expect a slower rate of fluorescence intensity increase.
In this study, we have introduced a novel nanofluidic coupled-membrane device designed for the digital amplification of nucleic acids (NA) within a nanocavity array. The device employs a flexible membrane that can be deflected to physically confine the NA within the nanocavities. By utilizing this membrane, we are able to retain the NA inside the nanocavities while allowing the loading of amplification reagents into the cavities through a narrow gap created between the membrane and nanocavities. For the amplification process, we employed two isothermal amplification methods: Loop-mediated Isothermal Amplification (LAMP) and Rolling Circle Amplification (RCA). These methods facilitate the amplification of the NAs directly within the confined nanocavities. By implementing this nanofluidic coupled-membrane device and utilizing LAMP and RCA techniques, we successfully achieved the amplification of nucleic acids within the nanocavities, providing a platform for digital amplification in a controlled and confined environment.
In the future, we propose to extend the device to applications where amplification is multiplexed. Multiplexing can be achieved by designing padlock probes (commonly used for amplification barcoding32). Multiple DNA sequences can be detected by barcoding the padlock probe. Multiplexing digital amplification is important for single particle analysis such as single EV analysis. Following our previous study, in future, a single EV can be confined in the nanocavities. We should be able to lyse the EV and then extract the vesicle's encapsulated mRNA cargo using our device while the mRNA is retained in the cavities. We can then detect multiple mRNA-associated biomarkers via sequential amplification rounds applied to the extracted cavity confined mRNA.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Canadian Cancer Society (255878 CCSRI), Natural Science and Engineering Research Council of Canada (NSERC RGPIN-2018-06232 and RGPIN-2018-06125), New Frontiers for Research Fundexploration (250326 NFRFE) and Canada Foundation for Innovation (CFI, G248924) for financial support. The authors acknowledge Nanotools-Microfab at McGill University and INRS Varennes. I. I. M. acknowledge Faculty of Engineering at McGill University for MEDA award and CMC for the financial support.References
- P. S. Wikramaratna, R. S. Paton, M. Ghafari and J. Lourenço, Estimating the false-negative test probability of SARS-CoV-2 by RT-PCR, Eurosurveillance, 2020, 25, 2000568 CrossRef CAS PubMed.
- R. Goya, M. G. Sun, R. D. Morin, G. Leung, G. Ha, K. C. Wiegand, J. Senz, A. Crisan, M. A. Marra and M. Hirst, et al., SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors, Bioinformatics, 2010, 26, 730–736 CrossRef CAS PubMed.
- L. Miotke, B. T. Lau, R. T. Rumma and H. P. Ji, High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR, Anal. Chem., 2014, 86, 2618–2624 CrossRef CAS PubMed.
- F. Shen, W. Du, J. E. Kreutz, A. Fok and R. F. Ismagilov, Digital PCR on a SlipChip, Lab Chip, 2010, 10, 2666–2672 RSC.
- G. Perkins, H. Lu, F. Garlan and V. Taly, Droplet-based digital PCR: application in cancer research, Adv. Clin. Chem., 2017, 79, 43–91 CAS.
- K. A. Heyries, C. Tropini, M. VanInsberghe, C. Doolin, O. I. Petriv, A. Singhal, K. Leung, C. B. Hughesman and C. L. Hansen, Megapixel digital PCR, Nat. Methods, 2011, 8, 649–651 CrossRef CAS PubMed.
- A. A. Kojabad, M. Farzanehpour, H. E. G. Galeh, R. Dorostkar, A. Jafarpour, M. Bolandian and M. M. Nodooshan, Droplet digital PCR of viral‎ DNA/RNA, current progress, challenges, and future perspectives, J. Med. Virol., 2021, 93, 4182–4197 CrossRef CAS PubMed.
- L. Becherer, N. Borst, M. Bakheit, S. Frischmann, R. Zengerle and F. von Stetten, Loop-mediated isothermal amplification (LAMP)–review and classification of methods for sequence-specific detection, Anal. Methods, 2020, 12, 717–746 RSC.
- Y.-L. Tan, H. Chen, Z.-K. Wu, J. He and J.-H. Jiang, Digital loop-mediated isothermal amplification-based absolute methylation quantification revealed hypermethylated DAPK1 in cervical cancer patients, Anal. Chem., 2021, 93, 8077–8083 CrossRef CAS PubMed.
- X. Lin, X. Huang, K. Urmann, X. Xie and M. R. Hoffmann, Digital loop-mediated isothermal amplification on a commercial membrane, ACS Sens., 2019, 4, 242–249 CrossRef CAS PubMed.
- L. Xu, J. Duan, J. Chen, S. Ding and W. Cheng, Recent advances in rolling circle amplification-based biosensing strategies-A review, Anal. Chim. Acta, 2021, 1148, 238187 CrossRef CAS PubMed.
- T. Konry, I. Smolina, J. M. Yarmush, D. Irimia and M. L. Yarmush, Ultrasensitive detection of low-abundance surface-marker protein using isothermal rolling circle amplification in a microfluidic nanoliter platform, Small, 2011, 7, 395–400 CrossRef CAS PubMed.
- D. Shi, J. Huang, Z. Chuai, D. Chen, X. Zhu, H. Wang, J. Peng, H. Wu, Q. Huang and W. Fu, Isothermal and rapid detection of pathogenic microorganisms using a nano-rolling circle amplification-surface plasmon resonance biosensor, Biosens. Bioelectron., 2014, 62, 280–287 CrossRef CAS PubMed.
- C. Feng, X. Mao, Y. Yang, X. Zhu, Y. Yin and G. Li, Rolling circle amplification in electrochemical biosensor with biomedical applications, J. Electroanal. Chem., 2016, 781, 223–232 CrossRef CAS.
- Q. Lin, X. Ye, Z. Huang, B. Yang, X. Fang, H. Chen and J. Kong, Graphene oxide-based suppression of nonspecificity in loop-mediated isothermal amplification enabling the sensitive detection of cyclooxygenase-2 mRNA in colorectal cancer, Anal. Chem., 2019, 91, 15694–15702 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, J. Qin, J. Wei, W. Wu and Y. Zhao, Engineered Janus probes modulate nucleic acid amplification to expand the dynamic range for direct detection of viral genomes in one microliter crude serum samples, Chem. Sci., 2018, 9, 392–397 RSC.
- J. H. Monserud and D. K. Schwartz, Mechanisms of surface-mediated DNA hybridization, ACS Nano, 2014, 8, 4488–4499 CrossRef CAS PubMed.
- G. B. Aktas, A. Ribera, V. Skouridou and L. Masip, DNA immobilization and detection using DNA binding proteins, Anal. Bioanal. Chem., 2021, 413, 1929–1939 CrossRef CAS PubMed.
- S. Mahshid, J. Lu, A. A. Abidi, R. Sladek, W. W. Reisner and M. J. Ahamed, Transverse dielectrophoretic-based DNA nanoscale confinement, Sci. Rep., 2018, 8, 1–12 CAS.
- C. J. Bustamante, Y. R. Chemla, S. Liu and M. D. Wang, Optical tweezers in single-molecule biophysics, Nat. Rev. Methods Primers, 2021, 1, 1–29 CrossRef.
- F. Persson, P. Utko, W. Reisner, N. B. Larsen and A. Kristensen, Confinement spectroscopy: probing single DNA molecules with tapered nanochannels, Nano Lett., 2009, 9, 1382–1385 CrossRef CAS PubMed.
- F. Ruggeri, F. Zosel, N. Mutter, M. Różycka, M. Wojtas, A. Ożyhar, B. Schuler and M. Krishnan, Single-molecule electrometry, Nat. Nanotechnol., 2017, 12, 488–495 CrossRef CAS PubMed.
- R. Marie, J. N. Pedersen, K. U. Mir, B. Bilenberg and A. Kristensen, Concentrating and labeling genomic DNA in a nanofluidic array, Nanoscale, 2018, 10, 1376–1382 RSC.
- R. Öz, K. Sriram and F. Westerlund, A nanofluidic device for real-time visualization of DNA–protein interactions on the single DNA molecule level, Nanoscale, 2019, 11, 2071–2078 RSC.
- I. I. Hosseini, Z. Liu, X. Capaldi, T. AbdelFatah, L. Montermini, J. Rak, W. Reisner and S. Mahshid, Nanofluidics for Simultaneous Size and Charge Profiling of Extracellular Vesicles, Nano Lett., 2021, 21, 4895–4902 CrossRef CAS PubMed.
- X. Capaldi, Z. Liu, Y. Zhang, L. Zeng, R. Reyes-Lamothe and W. Reisner, Probing the organization and dynamics of two DNA chains trapped in a nanofluidic cavity, Soft Matter, 2018, 14, 8455–8465 RSC.
- Z. Liu, X. Capaldi, L. Zeng, Y. Zhang, R. Reyes-Lamothe and W. Reisner, Confinement anisotropy drives polar organization of two DNA molecules interacting in a nanoscale cavity, Nat. Commun., 2022, 13, 1–12 Search PubMed.
- X. Capaldi, Z. Liu, Y. Zhang, L. Zeng, R. Reyes-Lamothe and W. Reisner, Probing the organization and dynamics of two DNA chains trapped in a nanofluidic cavity, Soft Matter, 2018, 14, 8455–8465 RSC.
- S. Kamtekar, A. J. Berman, J. Wang, J. M. Lázaro, M. de Vega, L. Blanco, M. Salas and T. A. Steitz, Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage φ29, Mol. Cell, 2004, 16, 609–618 CrossRef CAS PubMed.
- C. R. Newton, A. Graham and J. S. Ellison, PcR, BIOS Scientific Publishers Oxford, UK, 1997 Search PubMed.
- H. Takahashi, H. Yamazaki, S. Akanuma, H. Kanahara, T. Saito, T. Chimuro, T. Kobayashi, T. Ohtani, K. Yamamoto and S. Sugiyama, et al., Preparation of Phi29 DNA polymerase free of amplifiable DNA using ethidium monoazide, an ultraviolet-free light-emitting diode lamp and trehalose, PLoS One, 2014, 9, e82624 CrossRef PubMed.
- M. Szemes, P. Bonants, M. de Weerdt, J. Baner, U. Landegren and C. D. Schoen, Diagnostic application of padlock probes—multiplex detection of plant pathogens using universal microarrays, Nucleic Acids Res., 2005, 33, e70 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Device fabrication and operation; temperature controller; slit height calibration; flow measurement; gel electrophoresis and real-time PCR; particle size measurement; primer design. Also, Supporting Movie (MP4): staining of fully confined pBR322 plasmid vectors inside cavities; amplification of fully confined M13Mp18 using loop-mediated isothermal amplification method; amplification of fully confined M13Mp18 using rolling circle amplification method. See DOI: https://doi.org/10.1039/d3nr05553a |
| This journal is © The Royal Society of Chemistry 2024 |