
Hydrogen production catalysed by atomically precise metal clusters
Tongxin
Song
,
Xiao
Cai
and
Yan
Zhu
 *
*
School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China
First published on 9th July 2024
Abstract
Atomically precise metal clusters that possess the exact atom number, definitive composition, and tunable geometric and electronic structures have emerged as ideal model catalysts for many important chemical processes. Recently, metal clusters have been widely used as excellent catalysts for hydrogen production to explore the relationship between the structure and catalytic properties at the atomic level. In this review, we systematically summarize the significant developments concerning metal clusters as electrocatalysts and photocatalysts for hydrogen generation. This review also puts forward the challenges and perspectives of atomically precise metal clusters in electrocatalysis and photocatalysis in the hope of providing a valuable reference for the rational design of high-performance catalysts for hydrogen production.
1 Introduction
In recent years, the energy demand has been increasing and traditional fossil fuels have been unable to meet the current needs of society. Therefore, a lot of research studies have been conducted on the development of sustainable and clean energy sources. Hydrogen (H2) energy is a renewable green energy carrier with a high energy density value per unit mass (120–142 MJ kg−1),1,2 which is the best alternative to conventional fossil fuels. Currently, the main methods for H2 production include steam reforming,3,4 coal gasification,5,6 the natural gas method,7,8 industrial by-products for H2 production,9,10 electrocatalytic11–13 and photocatalytic water splitting14,15 and so on. Among these H2 production methods, electrolysis of water utilizes renewable energy to produce H2, with low energy consumption, less greenhouse gas release during the whole preparation process, and the obtained H2 is of high purity and low impurity content.16–18 Photolysis of water is the direct utilization of primary solar energy for splitting water to produce H2 with the assistance of a photocatalyst, which is simple in operation and highly efficient without the waste of energy.19,20 Other H2 generation methods suffer from serious pollution and environmental problems, and the low purity of the obtained H2 makes it difficult to meet the strategic needs of sustainable development.21 Therefore, it is necessary to design and develop high performance catalysts for electrocatalytic and photocatalytic water splitting to produce hydrogen.Atomically precise metal clusters (including Au, Ag, Cu, Ni, etc.) are a class of aggregates consisting of several to hundreds of metal atoms protected by organic ligands, with resolvable geometries and a discrete electronic structure.22,23 Since the dimensions of metal clusters are close to the Fermi energy level of metals, they exhibit molecular-like quantum size effects that are distinguishable from bulk materials.24 With the crystal structure of metal clusters solved, metal clusters become excellent catalysts to study the relationship between structures and catalytic performances.25,26 Although metal clusters for the studies of electrocatalysis and photocatalysis have been reported extensively in recent years,27,28 there are very few systematic summaries based on the application of metal clusters for catalytic H2 production, especially through electrocatalytic and photocatalytic water splitting.
In this review, we systematically summarize the advances in H2 production catalysed by atomically precise metal clusters, including electrochemical H2 evolution and photolysis of water. We highlight the performance of loaded clusters, unloaded pure clusters and theoretical calculations in electrochemistry. We also focus on the cases where the photocatalytic activity has been enhanced through strategies such as binding to metal oxides, surface charge modification and valence bond bridging. It is expected that this minireview will provide valuable information on atomically precise metal clusters for catalytic H2 energy generation.
2 Electrocatalytic H2 production
Metal clusters have been proved to exhibit excellent electrocatalytic properties in various electrochemical reactions.29–33 In this section, we introduce recent progress on the applications of metal clusters as electrocatalysts for the hydrogen evolution reaction (HER). The HER is a half-reaction of electrocatalytic water splitting that occurs at the cathode and which generally follows two possible mechanisms: the Volmer–Tafel mechanism and Volmer–Heyrovsky mechanism.34,35 In the former, a proton (H+) attaches to the active site of the catalysts to generate M–H* and then self-combines to form H2,36 while in the latter M–H* bonds with free H+ to release H2.37 The formation of M–H* is the limiting step for HER, so it is necessary to develop effective catalysts to enhance the HER. Generally, platinum-based nanomaterials are considered to be the most advanced electrocatalysts for HER.38,39 But the high price and rare earth reserves limit their practical applications. It was found that Au, Ag, Cu and other metal clusters with inert H2 evolution also display unique electrocatalytic H2 production activity.40–42 These metal elements feature relatively cheap prices and high earth reserves, making the study of non-platinum metal electrocatalysis even more valuable for HER.Back in 2017, Chen's group reported atomically precise Pd clusters protected by dodecanthiols with the molecular formula Pd6(SC12H25)12 (hereafter Pd6) and investigated the HER catalytic performance of Pd6 loaded onto activated carbon (Pd6/AC) and its counterpart after annealing treatment at 200 °C (Pd6/AC-V) in 0.5 M H2SO4 solution.43Fig. 1a shows the linear sweep voltammetry (LSV) curves of Pd6/AC, Pd6/AC-V and commercial Pt/C catalysts. It can be seen that Pd6/AC exhibited much better catalytic activity than Pt/C, and the onset potential of annealing-treated Pd6/AC-V was as negative as that of Pt/C. At different applied potentials, Pd6/AC-V presented the highest mass activity (Fig. 1b). The authors attributed this to the fact that calcination treatment removed the thiol ligands on the surface of Pd clusters, exposing more catalytic sites. Unfortunately, no explanation was given in the report as to why the thiol-capped clusters exhibited superior HER activity than commercial Pt/C. In addition, Fig. 1c and d show the accelerated durability tests (ADTs) of Pd6/AC-V and Pd6/AC to assess their catalytic stability. The surface-clean Pd6/AC-V displayed superior stability over the fully protected Pd6/AC. Although the catalysts in this study were ligand-free catalysts obtained using Pd6(SC12H25)12 clusters as precursors, it had given rise to a wave of research into the electrocatalytic H2 evolution through water splitting using atomically precise metal clusters as catalysts.44
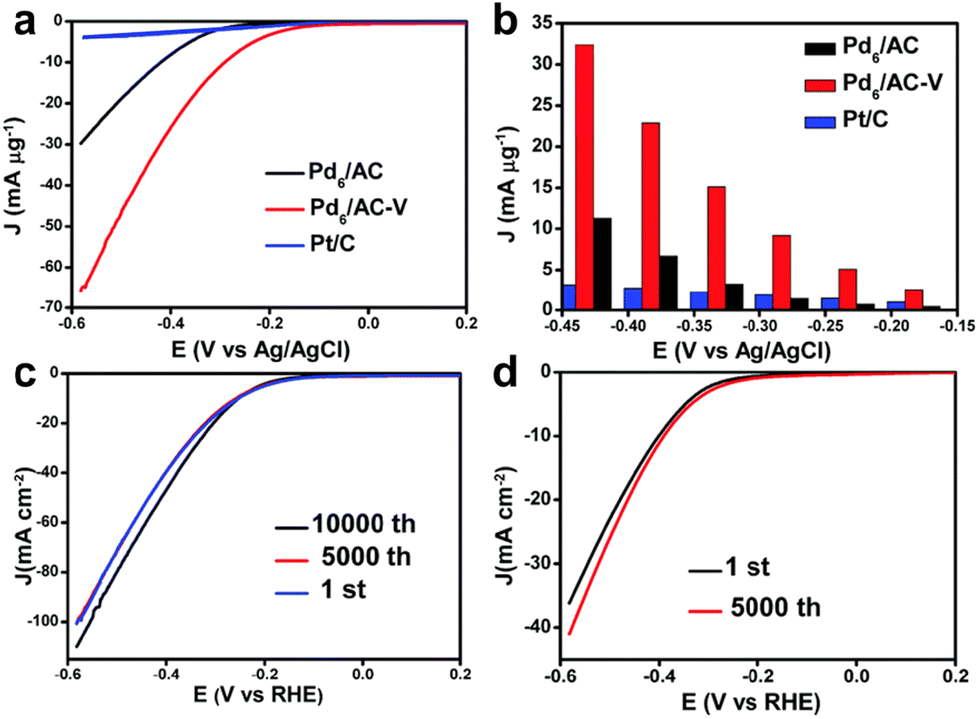 | ||
| Fig. 1 (a) The polarization curves of Pd6/AC, Pd6/AC-V and commercial Pt/C for the HER. (b) The mass activities of the three catalysts at different applied potentials. (c) ADTs of Pd6/AC-V. (d) ADTs of Pd6/AC. All the LSV curves were obtained for a solution of 0.5 M H2SO4 with a scanning rate of 5 mV s−1. Reproduced with permission from ref. 43. Copyright 2017 Royal Society of Chemistry. | ||
Also in 2017, Jin et al. loaded phenylethanethiol (PET) ligand-protected Au25(PET)18 (hereafter Au25) onto layered MoS2,45 and its scanning transmission electron microscopy (STEM) image is shown in Fig. 2a, where the Au25/MoS2 composite exhibited remarkable enhanced HER activity. Fig. 2b compares the polarization curves of a blank electrode, plain MoS2 and Au25/MoS2 with different cluster loadings in acidic electrolyte solution. The introduction of Au25 reduced the onset potential of the Au25/MoS2 composites by 40 mV compared with the pristine MoS2 nanosheets, and the mass activity was more excellent with the increase of Au25 loading. Electrochemical impedance spectroscopy was used to evaluate the charge transfer impedance of the catalysts, and as shown in Fig. 2c, the Au25/MoS2 catalysts exhibited smaller charge transfer impedance, indicating that the introduction of Au25 clusters could effectively promote charge transfer. X-ray photoelectron spectroscopy (XPS) characterization evidenced the electron transfer from Au25 to the MoS2 nanosheets, implying the existence of a pronounced electronic interaction between the clusters and MoS2 (Fig. 2e interface II), which is a major factor in promoting H2 evolution. In addition, the HER performance of electrocatalysts with different ligands of Au25 loaded onto MoS2 was investigated (Fig. 2d), and the catalytic activity of phenylselenol (PhSeH)-capped Au25(SePh)18 was much lower than that of Au25(PET)18. This was attributed to the stronger electronic relay capability of Au25(PET)18, which facilitated the interfacial electronic interactions between metal core and organic ligand (Fig. 2e interface I), thus effectively improving the HER performance. It should be noted that despite the different carbon tails of PET and PhSeH, the catalytic activity of Au25(SePh)18/MoS2 was slightly inferior to that of Au25(PET)18/MoS2, which had better conductivity due to the conjugation effect, suggesting that the interfacial effect, not the conductivity, was the key to the catalytic activity towards the HER. The Au25(PET)18/MoS2 nanocomposite exhibited great catalytic durability in 1000 cycles of ADTs. This finding provided important guidance for subsequent metal cluster electrocatalysis.
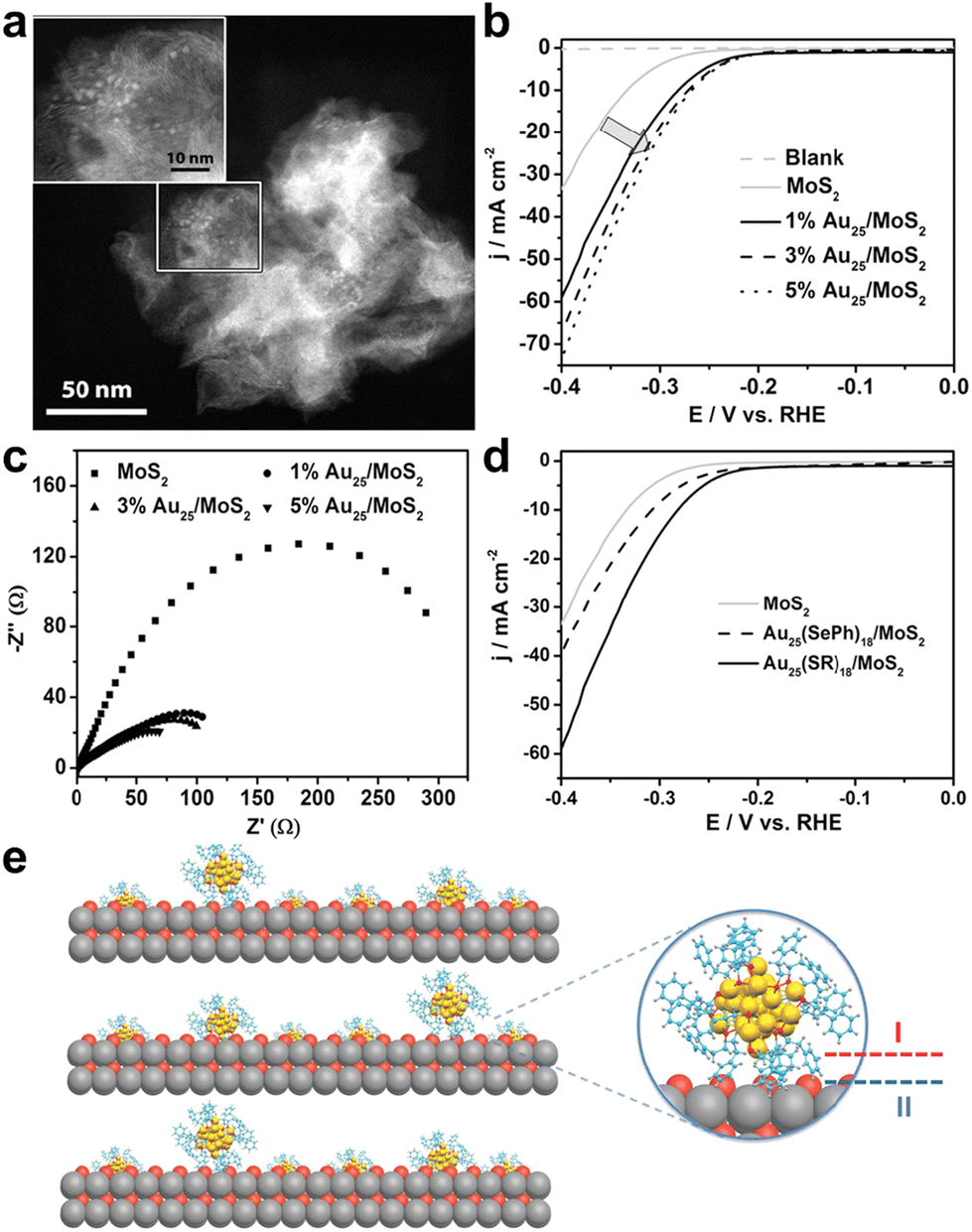 | ||
| Fig. 2 (a) STEM image of Au25(PET)18/MoS2. (b) LSV curves and (c) electrochemical impedance spectroscopy of plain MoS2 and Au25(PET)18/MoS2 with different loadings of composites. (d) The polarization curves of MoS2, 1 wt% Au25(PET)18/MoS2 and 1 wt% Au25(SePh)18/MoS2. All the electrochemical measurements were performed in a solution of 0.5 M H2SO4 with a sweep rate of 5 mV s−1 at a rotation rate of 1600 rpm. (e) Scheme of interfacial effects of composite catalysts. Color codes: yellow = Au, red/orange = S, blue = C and gray = Mo. Reproduced with permission from ref. 45. Copyright 2017 Wiley-VCH. | ||
Gratious et al. used Au11(PPh3)7I3 (PPh3 = triphenylphosphine) as a catalyst supported on MoS2 for electrocatalytic H2 production.46 They found that the interfacial interaction between the cluster and MoS2 was due to the coordination of the metal atoms of clusters with S from MoS2. This interfacial effect also enhanced the structural stability of clusters in supported catalysts. Similarly, Zhu et al. also confirmed this conclusion with the alloy clusters Au2Pd6S4(PPh3)4(SR)6 (SR stands for thiol) and found that the composite catalyst (Au2Pd6S4(PPh3)4(SR)6/MoS2) showed more attractive electrochemical H2 production performance compared to the single-component Au2/MoS2 and Pd3/MoS2 clusters.47 The moderate adsorption behavior of H atom on Au2Pd6S4(PPh3)4(SR)6/MoS2 and the electronic interactions between alloy cluster and MoS2 contributed to the excellent catalytic activity. Other carriers in the favour of the HER had also been reported in the study of electrocatalytic production of H2 over metal cluster-based hybrid catalysts.48 For example, reduced graphene oxide (r-GO) as a support can improve the catalyst stability by increasing electron transfer at the interface.49 Molybdenum selenide (MoSe2) can influence the electronic structure of the catalysts and provide more catalytically active sites.50 We summarized all cases of atomically precise metal clusters employed for electrocatalytic HER in Table 1.
| Catalysts | Supports | Cluster loadings | Electrolyte | E Onset (mV vs. RHE) | η (mV vs. RHE) at 10 mA cm−2 | j (mA cm−2) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pd2Au36(SR)24 | Carbon black | 1 mmol | 1.0 M B–R buffer (pH 3) | 70 | — | 10.9@−0.6 V | — | 40 |
| PtAu24(SR)18 | Carbon black | 1 mmol | 1.0 M B–R buffer (pH 3) | 70 | — | 15.3@−0.6 V | — | 40 |
| Pd6(SR)12 | Active carbon | — | 0.5 M H2SO4 | 73 | — | — | 220 | 43 |
| Au25(SR)18 | MoS2 | 0.2 mg cm−2 | 0.5 M H2SO4 | 200 | 280 | 59.3@−0.4 V | 79.3 | 45 |
| Au11(PPh3)7I3 | MoS2 | 0.1 mg cm−2 | 0.5 M H2SO4 | — | 292 | — | 63 | 46 |
| Au2Pd6S4(PPh3)4(SR)6 | MoS2 | 0.1 mg cm−2 | 0.5 M H2SO4 | 127 | 232 | 91.0@−0.4 V | 67 | 47 |
| Au101(PPh3)21Cl5 | Reduced-graphene oxide | 0.177 mg cm−2 | 0.5 M H2SO4 | — | 360 | 18.9@−0.4 V | 148 | 49 |
| Ni7(SR)7 | MoSe2 | 50 μg cm−2 | 0.5 M H2SO4 | 106 | 170 | 142@−0.35 V | 91 | 50 |
| Au25(SR)18− | — | 2.55 mg cm−2 | 0.2 M HClO4 | 190 | — | 26.0@−0.7 V | — | 51 |
| Au36Ag2(SR)18 | — | 0.2 mg cm−2 | 0.5 M H2SO4 | ∼100 | — | 19.4@−0.3 V | 125 | 52 |
| NiAg24(SR)18 | Carbon black | 2.5 nmol cm−2 | 1.0 M KOH | 50 | 270 | 55.2@−0.5 V | 58 | 53 |
Au24Ag20(tBuPh–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)24Cl2 C)24Cl2 |
Carbon nanotube | 0.2 mg cm−2 | 0.5 M H2SO4 | — | 262 | — | 96 | 54 |
| Au9Ag9(CCArF)18 |
Carbon black | 0.714 mg cm−2 | 0.5 M H2SO4 | 100 | 274 | — | 99 | 55 |
| PdHCu11(SR)6(CCPh)4 |
— | 2.7 nmol cm−2 | 0.5 M H2SO4 | ∼0 | 50 | — | 40 | 56 |
| PtHCu11(SR)6(CCPh)4 |
— | 2.5 nmol cm−2 | 0.5 M H2SO4 | ∼0 | 30 | — | 39 | 57 |
| PtAg28(S2R)12(PPh3)2 | — | 0.2 mg cm−2 | 0.5 M H2SO4 | — | 146 | 63@−0.3 V | 104 | 62 |
| Au15Ag23(tBuCC)18Br6 |
Carbon black | 0.714 mg cm−2 | 0.5 M H2SO4 | — | 125 | — | 121 | 63 |
In addition to composite catalysts, unloaded metal clusters provide an ideal platform for studying the catalytic performance–structure relationship as well as identifying the catalytic active sites due to their precise structures. Recent research studies have also witnessed a great development in this field. For example, Kumar et al. investigated the size effect for HER using Aux(PET)y with different atom numbers.51 As the cluster size decreased, the binding of Au atoms to H+ was gradually enhanced, which facilitated the HER activity. By testing the H2 production activity of Au25 with different ligands, it was found that the length of the ligand created an insulating layer between the electrode and the metal core, which increased the difficulty of electron transfer, and the π–π interactions between ligands containing benzene rings also affected the electron transfer ability, which indirectly impacted the catalytic activity. In addition, the doping effect in electrochemical H2 generation was also studied by modelling Ag-, Cu-, and Pd-doped Au25 as catalysts. The results showed that the doping of heteroatoms can change the electronic structure of the cluster, which affected the adsorption energy of the surface gold atoms with H+.
Jin and colleagues reported that Au36Ag2(SR)18 with low ligand coverage could efficiently catalyse the HER, and identified the H adsorption site through Au25(SR)18− and Au38(SR)24 as counterparts.52Fig. 3a shows the geometries and nuclear structures of three clusters: Au25(SR)18− has an Au@Au12 core structure and exhibits a typical icosahedral configuration; Au38(SR)24 possesses an Au2@Au21 core and shows a dimeric structure of two icosahedra sharing the Au3 plane; Au36Ag2(SR)18 is a trimeric ensemble of three icosahedra sharing the Ag2Au1 plane and the core structure is Au3@Au27Ag2. In the acidic HER reaction, the Au36Ag2(SR)18 cluster exhibited the lowest overpotential and the optimal current density (Fig. 3b). There was no degradation of the activity after 1000 cycles of the LSV test, indicating promising catalytic stability. Fig. 3c shows the plots of the Tafel curves of the three clusters with comparable Tafel slopes indicating their similar surface chemical states. The adsorption sites of H on the three clusters were simulated by theoretical calculation, which were all fully ligand-protected and no ligand detachment occurred. The calculation results showed that the most easily adsorbed H site is the Au atom in the kernel's shell layer, and the optimized adsorption configuration is shown in Fig. 3d. For Au25(SR)18−, H adsorbed on the Au12 shell; for Au38(SR)24, H adhered to the bridge site where the two icosahedra were connected; and for Au36Ag2(SR)18, H could be favourably attached to each exposed Au.
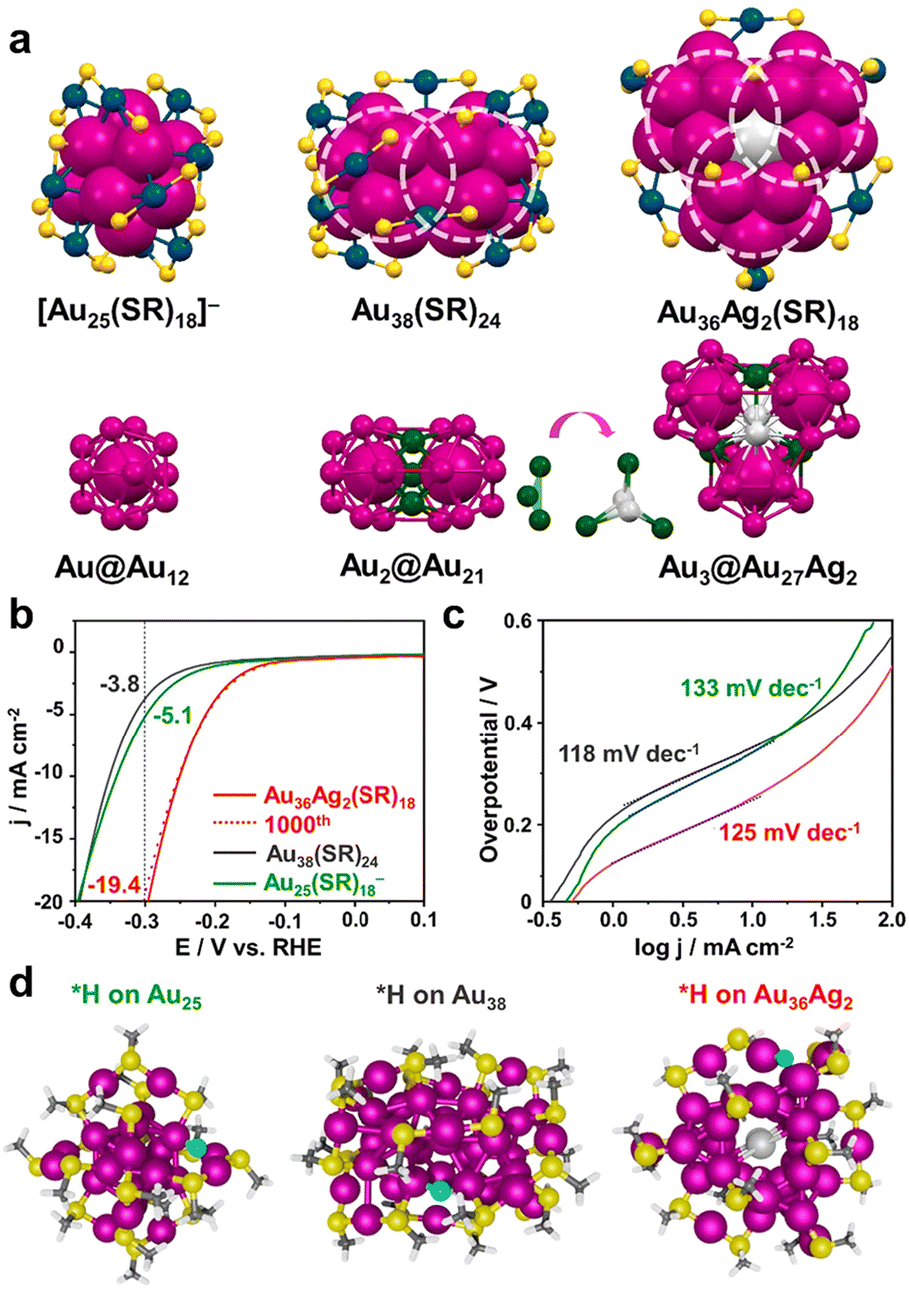 | ||
| Fig. 3 (a) The geometry structures (top) and kernel structures (bottom) of Au25(SR)18−, Au38(SR)24, and Au36Ag2(SR)18, C with H atoms omitted; each circle indicates one icosahedral unit. (b) LSV polarization curves obtained for 0.5 M H2SO4 solution at a sweeping rate of 0.05 V s−1. (c) Tafel plots of the three cluster catalysts. (d) The optimized adsorption structures of H on Au25(SR)18−, Au38(SR)24, and Au36Ag2(SR)18 clusters. Color codes: light gray = Ag, magenta/green/navy = Au, yellow = S, cyan = adsorbed H, gray = C, and white = H. Reproduced with permission from ref. 52. Copyright 2021 American Chemical Society. | ||
Subsequently, a variety of atomically precise alloy clusters have been used for electrochemical hydrogen production, which can significantly modify the electronic structure and reduction potential due to the doping of heteroatoms, thus substantially altering the catalytic activity. For example, Lee et al. synthesized Ni1Ag24(SR)18 clusters, whose HER performance far exceeded that of Ag25(SR)18 clusters.53 Tang's group investigated the catalytic properties of AuAg alloy clusters Au24Ag20(tBuPh–CC)24Cl2 and Au22Ag22(tBuCC)16Br3.28Cl2.72, which were protected with alkyne and halogen, as models for electrochemical H2 evolution.54 In contrast to the Au36Ag2(SR)18 cluster mentioned previously, for the two clusters studied, H preferentially adsorbs on the Au of the staple. And the difference in catalytic performance between the two AuAg clusters originated from the two silver atoms in the core. This group also reported Au9Ag9(CCArF)18 clusters as HER catalysts,55 which displayed a unique ichthyomorphic framework. Compared with alkyne complexes of Au and Ag, the unique geometry of the Au9Ag9(CCArF)18 cluster exposed more catalytic sites and therefore enhanced the catalytic activity.
Recently, H-containing 2-electron PdCu superatomic clusters were reported to offer unexpected catalytic performance for HER, which was the first example of hydride clusters applied to electrochemical H2 production.56 In contrast to PdH2Cu14(SR)6(CCPh)6 and PdHCu12(SR)6(CCPh)4, the PdHCu11(SR)6(CCPh)4 cluster was composed of a central Pd atom and a cuboctahedron Cu11 lacking a vertex with one interstitial hydride. In the case of the two former ones, the complete Cu shell layer surrounded the Pd atom in the centre, as shown in Fig. 4a, and it is worth noting that the H atom was located in the PdCu3 unit. Fig. 4b shows the LSV curves of three Pd–Cu clusters used in the acidic HER, and PdHCu11(SR)6(CCPh)4 only exhibited an overpotential of 50 mV at a current density of 10 mA cm−2. Importantly, PdHCu11(SR)6(CCPh)4 exhibited almost constant HER activity and maintained the structural integrity. Fig. 4c shows the resulting Tafel slope plots, and significantly different Tafel slopes indicated that the reaction pathways are different at the surface of the three cluster catalysts. The rate-determining step for both PdH2Cu14(SR)6(CCPh)6 and PdHCu12(SR)6(CCPh)4 systems was the Volmer pathway, while the rate-limiting step for PdHCu11(SR)6(CCPh)4 was the Heyrovsky reaction, which implied that the open framework exposed Pd site was conducive to the adsorption of H and the promotion of the HER. The group later more strongly demonstrated the conclusion with the Pt-doped Cu–H cluster and that the PtHCu11(SR)6(CCPh)4 cluster had the most outstanding performance among all HER cluster catalysts reported to date.57
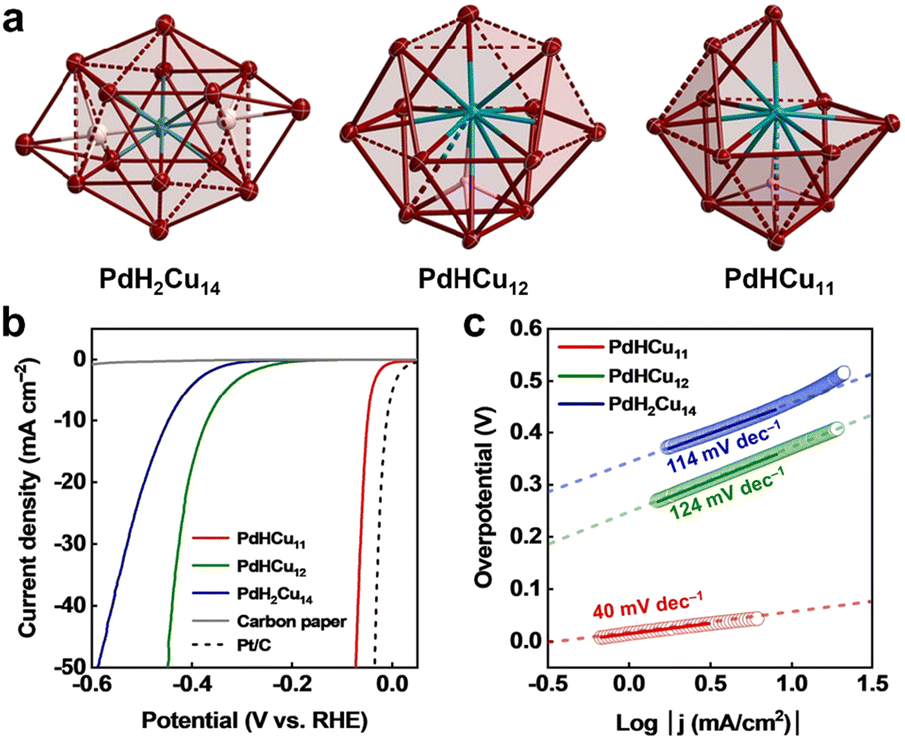 | ||
| Fig. 4 (a) The core structures of PdH2Cu14(SR)6(CCPh)6, PdHCu12(SR)6(CCPh)4, and PdHCu11(SR)6(CCPh)4; ligands are omitted. Color codes: red = Cu, cyan = Pd, pink/white = H. (b) LSV curves for the three catalysts with carbon paper and Pt/C as comparison. (c) Corresponding Tafel plots of the three cluster catalysts. All measurements were conducted in Ar-saturated 0.5 M H2SO4 solution at 1 mV s−1. Reproduced with permission from ref. 56. Copyright 2023 Wiley-VCH. | ||
The clusters can be applied not only for H2 production in aqueous systems, but also in a wide range of organic systems. Kwak et al. catalysed electrochemical H2 evolution in the tetrahydrofuran (THF) system using a centrally doped PtAu24(SR)180 cluster as a homogeneous catalyst in a system also containing 0.1 M tetra-tert-butylammonium hexafluoro-phosphonate (Bu4NPF6) and 1.0 M trifluoroacetic acid (TFA).58 As can be seen from Fig. 5a, Au25(SR)18− exhibited a significant reduction current compared to the blank glassy carbon electrode, and the reduction current of the bimetallic cluster PtAu24(SR)180 was remarkably enhanced over the parent cluster, indicating that Pt doping can dramatically change the reduction potential of PtAu24(SR)180. Fig. 5b shows the polarization curve of 1 mM PtAu24(SR)180 cluster in THF solution with the change of TFA concentration. The two weak reduction peaks corresponded to the reduction potentials of PtAu24(SR)180/1− and PtAu24(SR)181−/2−. When PtAu24(SR)182− was produced, there was a pronounced H2-producing current, which indicated that PtAu24(SR)180 displayed molecular-like properties to transfer individual electrons. Fig. 5c shows the H adsorption configuration and calculated energy in the THF system simulated by theoretical calculation. The protons involved in the HER process were simulated using protons dissolved by two THF molecules, and H was subsequently adsorbed on the surface of the cluster, a step that is thermodynamically advantageous. Step 2a in Fig. 5c is the Tafel pathway, which is endothermic. But process 2b is the Heyrovsky pathway, which is thermodynamically feasible. In addition, it was found that H atoms could spontaneously penetrate through the Au shell layer and adsorb onto the surface of Pt, so Pt was identified as the active site of the HER.
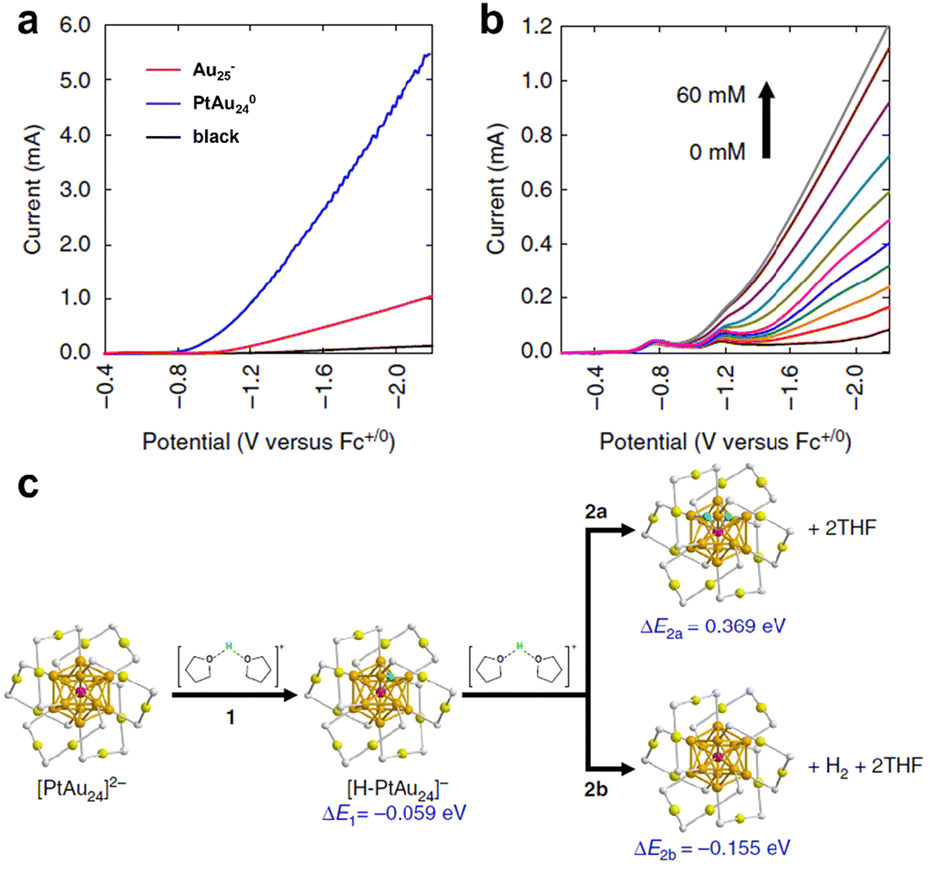 | ||
| Fig. 5 (a) LSV curves of THF containing 0.1 M Bu4NPF6 and 1.0 M TFA solution for Au25(SR)18− and PtAu24(SR)180 clusters. (b) The polarization curves of 1 mM PtAu24(SR)180 in THF with different TFA concentrations. (c) The calculated energy of H2 evolution on PtAu24(SR)180. Reproduced with permission from ref. 58. Copyright 2023 Nature Publishing Group. | ||
With the rapid development of scientific research, density functional theory (DFT) calculations have become an important characterization method to obtain the reaction intermediate states and adsorption energies through configuration optimization and simulation. Studies have also been reported on the adsorption of H atoms on clusters by first-principles DFT calculations.59 In 2017, Jiang's group investigated the interaction of H with Au25(SR)18 and monoatom-doped MAu24(SR)18 (M = Pt, Pd, Ag, Cu, Hg or Cd) clusters by theoretical calculations.60 It was found that the H atom exhibited metallic properties and can contribute 1s electrons to the superatomic free electron number. And when H was adsorbed on the cluster to form an 8e superatom, the binding energy was much stronger.
Subsequently, Hu and the co-workers modelled Au22(L8)6 cluster protected by 1,8-bis(diphenylphosphine)octane (L8) as a ligand, and demonstrated by DFT calculations that this cluster was a promising catalyst for HER.61 The Au22(L8)6 cluster had eight coordinated unsaturated Au atoms at the waist of the cluster, which can serve as active sites for H adsorption. Calculations revealed that up to six of H atoms can be adsorbed onto the Au22(L8)6 cluster, and the optimized H atom adsorption configuration is shown in Fig. 6a. The first four H atoms adsorbed were at the bridge sites of every two ligand-unsaturated Au, the fifth and sixth H atoms bridged on the top and bottom Au, and the latter Au was not coordination unsaturated. Fig. 6b shows the calculated adsorption energy (ΔEH) and adsorption free energy (ΔGH) for each H atom adsorbed on the cluster. It can be seen that the ΔGH of the first six H atoms was close to 0 eV and that the second H was the strongest adsorbed. The seventh H atom can hardly adsorb on the cluster. Fig. 6c shows the Bader charge analysis of the Au22H2(L8)6 cluster with two H atoms adsorbed, and it can be seen that H atoms exhibited hydride-like properties with all negative charge. On the other hand, the Au attached to H showed an obvious lack of electrons. This suggested that the H adsorbed onto the phosphine ligand-capped Au cluster was characterized as a hydride, which differed from the metallic H in the thiol ligand-protected Au clusters.
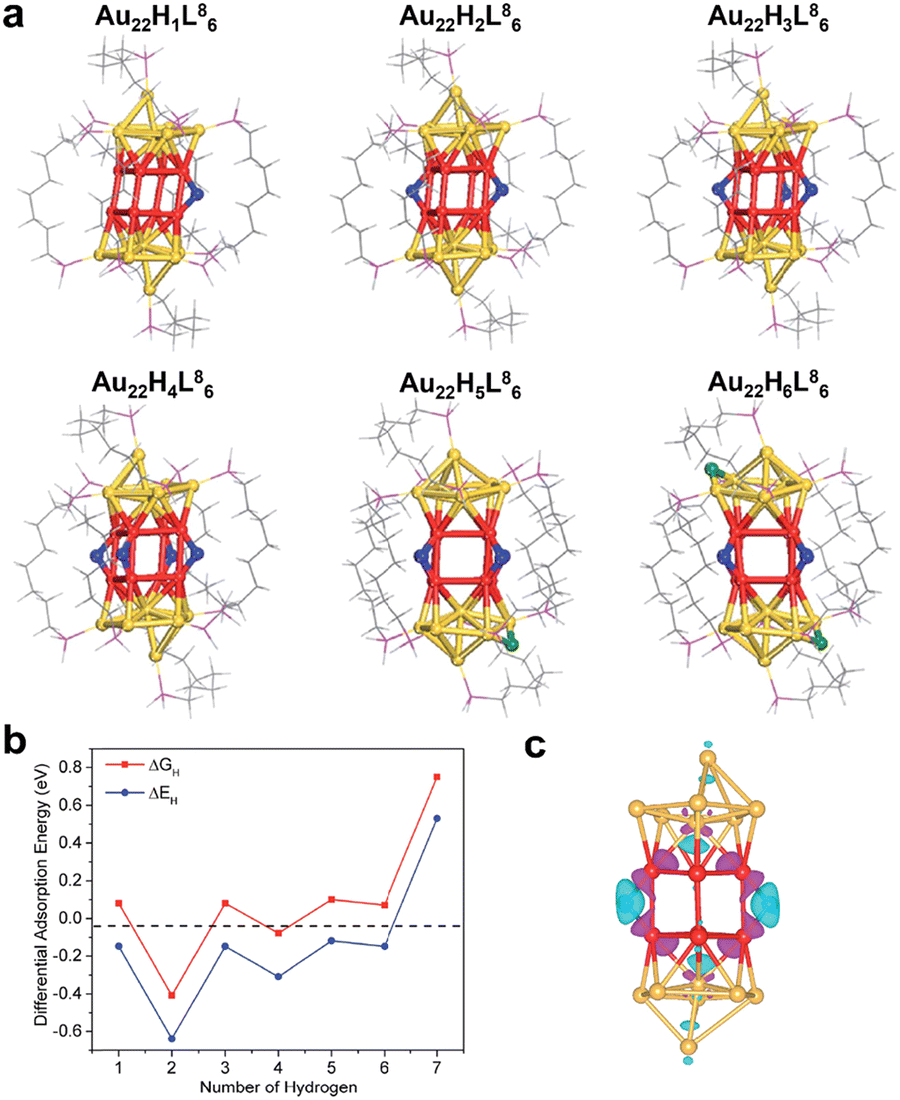 | ||
| Fig. 6 (a) Optimized configurations of different amounts of H atoms adsorbed on Au22(L8)6. (b) Calculated adsorption energies (ΔEH) and adsorption free energies (ΔGH). (c) The Bader charge of the Au22H2(L8)6 cluster with two H atoms adsorbed onto the Au22(L8)6. Color codes: red/yellow = Au, blue/green/light grey = H, magenta = P, grey = C. Reproduced with permission from ref. 61. Copyright 2018 Royal Society of Chemistry. | ||
3 Photocatalytic H2 production
Photocatalytic H2 production can be viewed as the process of catalysing water splitting to produce chemical energy H2 using renewable solar energy assisted by a photocatalyst.64,65 A favourable photocatalyst needs to satisfy the following conditions: a suitable band gap width and band position and effective ability to separate and transport electron–hole pairs.66–68 Atomically precise metal clusters feature diverse energy levels, HOMO–LUMO (highest occupied molecular orbital–lowest unoccupied molecular orbital) transitions and special photo-absorption properties,69,70 which make the clusters excellent for applications in photocatalysis. In recent years, chemists have achieved substantial progress in preparing composite photocatalysts by combining precise metal clusters with conventional semiconductors.71,72 Orlov's group used sub-nm Au particle modification on CdS as a photocatalyst, which displayed amazing photocatalytic H2 production performance.73 However, the sub-nm particle had no resolved crystal structure, and was identified as bare Au9 and Au11 by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry and DFT simulations, which was the first ever demonstration of the potential of sub-nm Au particles for photocatalytic H2 production.As early as 2013, Jin's group demonstrated the high activity of Ni-based clusters as photocatalytic water reduction catalysts by using an Ir complex as a photosensitizer, triethylamine (TEA) as a sacrificial agent, and Ni6(SR)12 clusters as a water reducing catalyst, and the turnover numbers (TONs) and turnover frequencies (TOFs) can reach 3750 and 970 h−1, respectively.74 NiII first undergoes one electron reduction and protonation, possibly forming a hydride intermediate, followed by reduction of another proton to produce H2. This study confirmed the promising potential of Ni-based clusters as catalysts for photocatalytic H2 production from water reduction, which opened up a new mindset for designing photocatalysts.
Chen's group loaded benzyl thiol-protected Ni6(SR)12 cluster onto TiO2.75 This composite catalyst could significantly enhance the photocatalytic H2 production activity under simulated sunlight using methanol as a sacrificial agent, and the molecular structure of the clusters remained unchanged during the catalytic process. Ni6(SR)12 (hereafter Ni6) presented obvious characteristic absorption peaks in the UV-visible region. When Ni6 cluster was loaded onto TiO2, its solid-state UV spectrum exhibited a corresponding photoresponse, which was enhanced with the increase of Ni6 cluster content. Fig. 7a shows the photocatalytic H2 production performance of the catalysts with different Ni6 cluster loadings, and increasing the cluster loading can enhance the photocatalytic H2 generation production activity. The H2 evolution activity of the catalyst only decreased slightly during the 10-cycle stability experiments. Fig. 7b shows the photocurrent density of Ni6/TiO2 and TiO2, which was twice as high as that of pristine TiO2 without the modification of Ni6, implying that Ni6 can improve the separation efficiency of photogenerated electron–hole pairs of TiO2 and reduce the recombination rate. The electron density state indicates that there was an overlap between the electron occupied orbital of Ni6 and the conduction band of TiO2, allowing the photogenerated electrons transfer from the TiO2 conduction band to the occupied orbital of the excited Ni6 cluster (Fig. 7c). Fig. 7d shows a schematic diagram of the mechanism of Ni6 cluster enhancing the photocatalytic performance of TiO2, showing the transfer path of photogenerated electron–hole pairs. It is worth mentioning that the non-bonding weak interaction between the ligands of the clusters and TiO2 played a crucial role in this reaction.
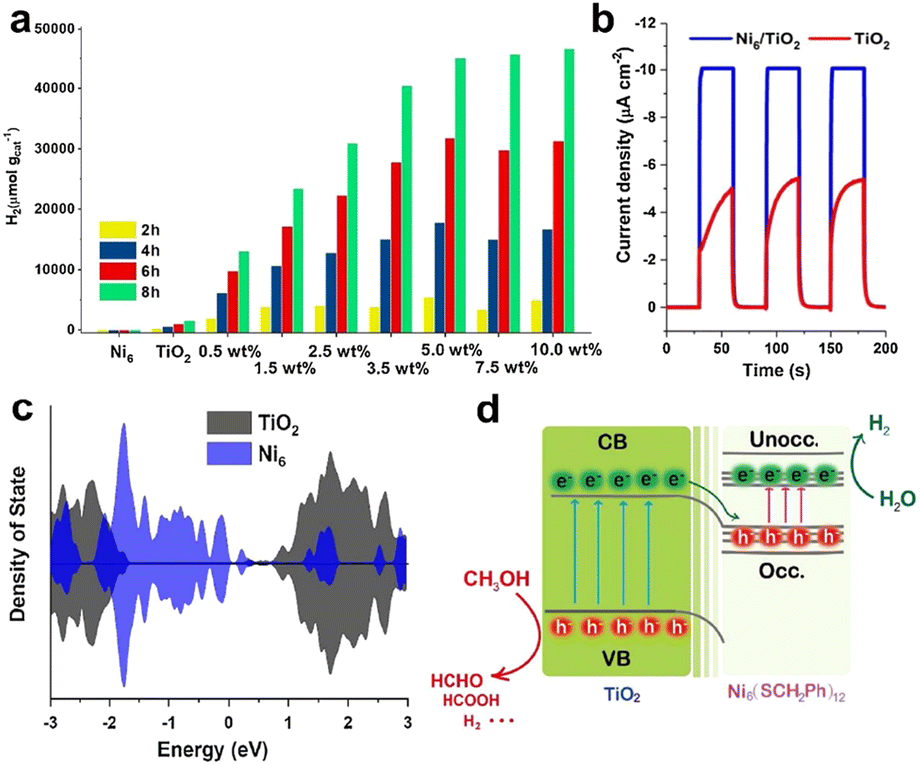 | ||
| Fig. 7 (a) Photocatalytic H2 production performance of catalysts with different cluster loadings in a mixture of distilled water and methanol (v/v = 80 mL/20 mL) with simulated solar light irradiation powered by a 300 W Xe lamp. (b) Photo-response currents of Ni6/TiO2 and TiO2. (c) Electron density states of pristine TiO2 and Ni6 cluster. (d) Schematic diagram of the mechanism of photogenerated electron–hole pair transfer between TiO2 and Ni6 clusters. Reproduced with permission from ref. 75. Copyright 2021 Elsevier. | ||
Subsequently, they investigated the effect of cluster ligands on the photocatalytic performance using graphitic carbon nitride (g-C3N4) with delocalized π bonds as the carrier and Ni12(SR)24 (hereafter Ni12) as the cocatalyst.76 The Ni12 cluster possesses a ring structure, similar to that of a crown, and protected by 4-methylbenzenethiolates as a ligand. The absorption of the cluster in the UV-visible region mainly originated from the electron transition within the Ni–S ring structure of the cluster and the transition from the core structure to the π-bond of the benzene ring in the ligand. When exposed to ultraviolet radiation, electrons are transferred from the ring structure nucleus to the benzene ring ligand. Fig. 8a shows the H2 generation efficiency of the catalysts with different cluster loadings under simulated sunlight radiation. While Ni12 and g-C3N4 alone were not photocatalytically active, Ni12-modified g-C3N4 possessed an extremely high H2 evolution rate, and the catalytic efficiency increased with the increase of cluster loading. Because the clusters featured significant absorption in the visible region, the photocurrent density of the composite catalyst was apparently amplified compared to fresh g-C3N4 (Fig. 8b). The activity of the catalyst was reduced by only 30% in a cycle stability test of greater than 24 hours. The interaction between the Ni12 cluster and g-C3N4 was simulated by molecular dynamics, and it was found that the benzene ring on the cluster ligand was parallel to the g-C3N4 plane, resulting in π–π conjugated interaction, which connected the charge transfer between the cluster and the carrier, and facilitated the effective separation of photogenerated electrons and holes of the composite catalyst to enhance the catalytic activity (benzene rings are shown in green in Fig. 8c). The mechanism of the conjugation interaction to improve the photocatalytic H2 evolution performance is shown in Fig. 8d and marked by red arrows. Under light irradiation, the ground state electrons of Ni12 were excited to produce photogenerated electron–hole pairs. The photogenerated electrons jump to the conduction band of g-C3N4 due to the π–π interactions, and are then captured by the surface to produce H2. This study visualized the weak cluster–support interaction in the composite photocatalysts as the π–π conjugated interaction.
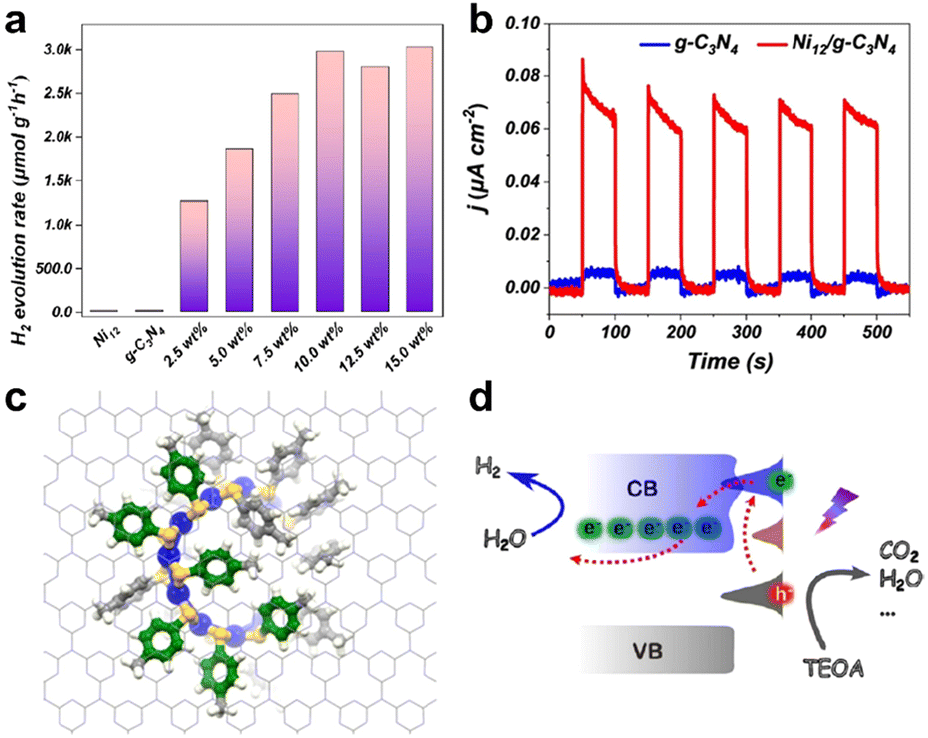 | ||
| Fig. 8 (a) Photocatalytic H2 production efficiency of catalysts with different Ni12 cluster loadings in a mixture of distilled water and methanol (v/v = 80 mL/20 mL) with simulated solar light irradiation powered by a 300 W Xe lamp. (b) Photo-response currents of Ni12/g-C3N4 and g-C3N4. (c) Ab initio molecular dynamic snapshots of Ni12 loaded onto g-C3N4 systems at 10 ps. Color codes: blue = Ni, yellow = S, grey = C, white = H. (d) Schematic diagram of the mechanism of photogenerated electron–hole pair transfer between g-C3N4 and Ni12 clusters. Reproduced with permission from ref. 76. Copyright 2023 American Chemical Society. | ||
In addition to Ni-based clusters, Cu-based clusters have also been shown to be outstanding catalysts for photochemical H2 production. The Cu cluster family has developed gradually, which has more enriched crystallographic structure compared to Ni clusters. Gao's group reported a precise Cu20O1(C20H24O2)12(CH3COO)6 cluster (hereafter UNJ-Cu20) protected by ethinylestradiol (C20H24O2) ligands and acetic acid (CH3COOH) molecules.77 Unlike previously reported Cu clusters, the additional hydroxyl group on C20H24O2 can modulate the hydrophilicity of UNJ-Cu20. Combining UNJ-Cu20 and TiO2 nanosheets (NSs) can form a highly efficient composite H2 evolution photocatalyst. As shown in Fig. 9a, the photocatalytic H2 generation performance of UJN-Cu20@TiO2-NS composites with different loadings of UJN-Cu20 was demonstrated by using a 300 W xenon lamp to simulate sunlight. The original TiO2-NS or UJN-Cu20 could only produce trace amounts of H2. The H2 production activity of UJN-Cu20@TiO2-NS nanocomposites was significantly enhanced, with an optimal H2 production rate of 13 mmol g−1 h−1 at 2% UJN-Cu20 loading. Moreover, UJN-Cu20@TiO2-NS nanocomposite maintained high H2 production activity after 5 cycles. Fig. 9b shows the photocurrent response curve to investigate the photoconversion ability of the catalyst. Both TiO2-NS and UJN-Cu20 exhibited weak photocurrent responses. And the composite catalyst possesses a significantly enhanced photoelectric response, indicating that the synergistic effect of the UJN-Cu20 cluster and TiO2-NS could promote the separation of photoelectrons and holes. As for the interaction between UJN-Cu20 clusters and TiO2-NS, UJN-Cu20 was bound to the surface of TiO2-NS by hydrogen bonding via the hydroxyl group on the ligand (Fig. 9c). Fig. 9d illustrates the relative positions of energy bands of UJN-Cu20 and TiO2-NS as well as the electron transfer mechanism on the composite catalyst. The electrons were excited, when the light energy was larger than the band gap of both. The electrons from the TiO2-NS conduction band combined with the holes in the HOMO level of UJN-Cu20, retaining the electrons from the LUMO level of UJN-Cu20. Triethanolamine (TEOA) could capture the holes with strong oxidation capacity in the TiO2-NS valence band. By constructing a Z-type photocatalytic system, the separation of photoelectrons and holes was significantly promoted.
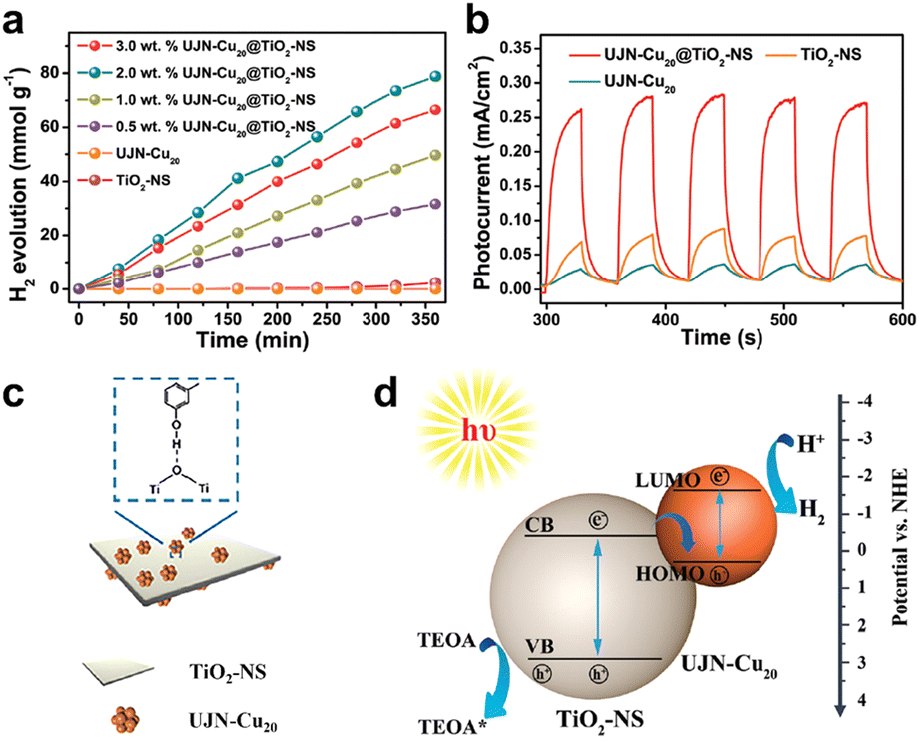 | ||
| Fig. 9 (a) Photocatalytic H2 production rates of composite catalysts with different UNJ-Cu20 cluster loadings using a 300 W xenon lamp as the light source to simulate sunlight. (b) Photo-response currents density of UNJ-Cu20, TiO2-NS and UNJ-Cu20/TiO2-NS. (c) Schematic diagram of the interaction of UNJ-Cu20 with TiO2-NS surface. (d) Schematic illustration of the relative positions of the UNJ-Cu20 and TiO2-NS energy bands and the electron transfer mechanism over the composite catalysts. Reproduced with permission from ref. 77. Copyright 2021 Royal Society of Chemistry. | ||
Loading Cu-based clusters onto semiconductor TiO2 can lead to efficient photocatalysts; on the other hand, self-assembled cluster-based metal organic frameworks (MOFs), a porous network structure with periodicity by connecting cluster nodes with organic ligands, also improved the stability of the clusters. Moreover, the MOFs also could maximize the accessibility of the unsaturated active sites in the spatial separation structure. Xu et al. synthesized a Cu8(SN)4(tBuS)4 (SN = 4-(4-pyridinyl)thiazole-2-thiol) cluster with the precise structure shown in Fig. 10a.78 And the Cu-MOF was obtained by using Cu8(SN)4(tBuS)4 as a metal cluster node, and SN with a double coordination structure as an ideal grafting ligand, with Cu cluster attached at one end and uncoordinated pyridine nitrogen atoms linked to Cu at the other end during the self-assembly process. Fig. 10a shows the mechanism of construction of the Cu cluster-based MOF. When fluorescein (FL) and triethylamine (TEA) were used as photosensitizers and sacrificial agents, the ordered-structured Cu-MOF exhibited excellent photocatalytic H2 production activity (Fig. 10b), and the Cu-MOF displayed the recycling of heterogeneous catalysts. The photo-responsive current densities of Cu8(SN)4(tBuS)4 and Cu-MOF were compared in Fig. 10c, and the introduction of the FL significantly enhanced the photocurrent of Cu-MOF, which indicated that a large number of photogenerated electrons were transferred from the photosensitizer to the Cu-MOF catalyst as well as the efficient separation and transfer of photogenerated electrons. Compared with Cu8(SN)4(tBuS)4 units, the ordered porous frame structure enabled Cu-MOF to carry out charge transfer efficiently, which was the main reason for its enhanced photocatalytic performance. Furthermore, Jiang and his colleagues reported that MAg24(SR)18 (M = Ag, Pd, Pt, and Au) encapsulated in MOFs exhibited outstanding photocatalytic H2 production activity, which was attributed to the fact that the metal clusters generated a charge transfer pathway similar to that of a Z-type heterojunction under visible irradiation, which facilitated charge separation.79
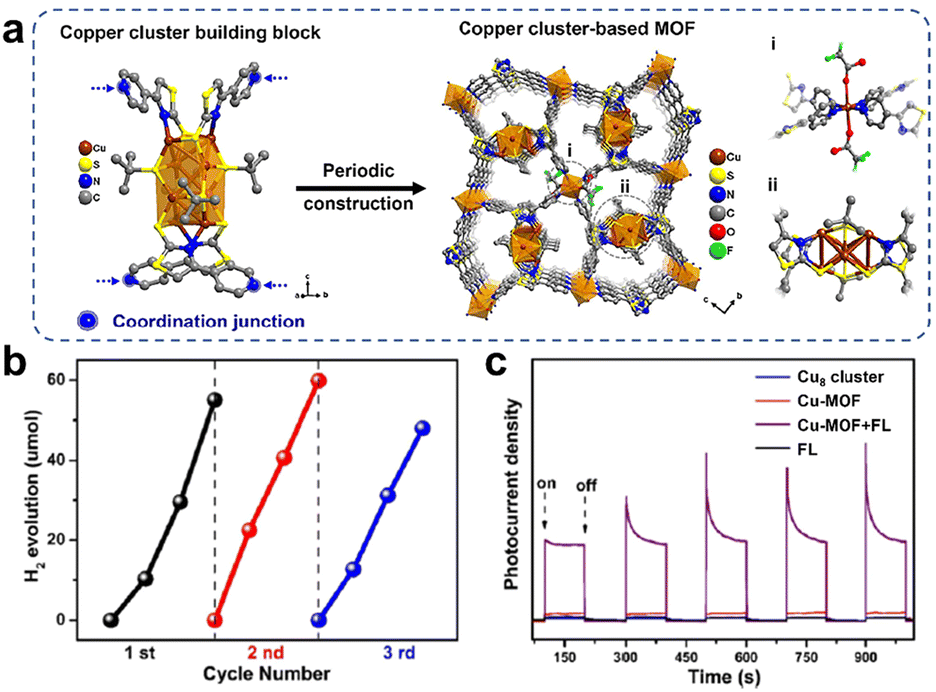 | ||
| Fig. 10 (a) Schematic diagram of the synthesis of Cu8(SN)4(tBuS)4 cluster-based MOFs (Cu-MOF) through self-assembly of copper cluster units. (b) Recyclability of Cu-MOF for photocatalytic H2 production in a mixed solution of ethanol and water (v/v = 1/1) with FL and TEA as the photosensitizer and sacrificial agent. (c) Photo-response currents density of Cu8(SN)4(tBuS)4 and Cu-MOF. Reproduced with permission from ref. 78. Copyright 2023 Royal Society of Chemistry. | ||
In addition to monometallic Cu clusters, Mo–Cu bimetallic clusters have also been reported for photocatalytic H2 production. Zang's group introduced MoOS32− unit into Cu cluster and prepared a series of atomically precise Cu6(MoOS3)2(C7H7S)2(PPh3)4·xCH3CN (Cu6Mo2, C7H7S = benzyl mercaptan).80 The introduction of MoOS32− could improve the structural stability of Cu clusters and introduce new active sites. The composite catalyst loaded with Fe3O4 exhibited excellent catalytic activity and stability in the photocatalytic H2 production reaction due to the electronic effects of ligands that can modify the HOMO–LUMO of the cluster. The electrons on the LUMO of FL are trapped by holes on the HOMO of Cu6Mo2 under light radiation. As a result, the electrons are highly reductive on the LUMO of Cu6Mo2 and thus participate in the generation of H2. This kind of new copper clusters bring new vitality to photocatalytic hydrogen production catalysts.
It is well known that gold bulk is chemically inert. Recently, Xiao's group constructed an interfacial charge-controlled Z-type heterojunction photocatalyst based on Au25(GS)18 (GSH = glutathione) cluster.81Fig. 11a shows a schematic diagram of the preparation of MoSe2/CdSe/Au25 (M/C/A) heterojunction catalyst. First, MoSe2 nanosheets were modified with a layer of mercaptoacetic acid (MAA) to make the surface negatively charged. The surface of CdSe quantum dots was positively charged by surface modified 2-aminoethanethiol (AET). The charge-modified MoSe2 nanosheets and CdSe quantum dots were self-assembled to form the binary heterostructure as shown in Fig. 11b, and the surface of the composite structure was positively charged. GSH-protected Au25 cluster was assembled with a binary heterostructure to form a ternary heterostructure, as shown in Fig. 11c. Fig. 11d shows the photocatalytic H2 production activities of each component. The MoSe2 nanosheets alone were almost inactive, CdSe quantum dots were poorly active, and the catalytic activity of MoSe2/CdSe (M/C) was significantly improved. Fig. 11e compares the effect of the introduction of Au25(GSH)18 on the catalytic performance of MoSe2/CdSe binary heterostructures. The highest catalytic activity was obtained when the content of Au25(GSH)18 was 4 mL (1M/C/4A), which highlighted the great importance of Au25(GSH)18 in the catalysis of ternary heterostructures. Fig. 11f compares the photocatalytic activity of the composite catalyst produced by simple physical mixing without surface charge regulation. The experimental results demonstrated that all of the three components along with charge regulation were indispensable for improving the H2 production activity. The excellent catalytic activity was derived from the fact that MoSe2 can accelerate the migration of photoelectrons from CdSe quantum dots to MoSe2 nanosheets, and reduce the carrier recombination rate of CdSe quantum dots. In addition, the 1M/C/4A catalyst exhibited great cycling stability, with no decay in activity during four cycles of the reaction. Fig. 11g presents a schematic diagram of the electron transfer mechanism of the catalyst. Under visible light irradiation, the HOMO of Au25(GSH)18 produced photogenerated electrons onto the LUMO, and the CdSe quantum dots were also photoexcited to generate charge carriers. The electrons at the LUMO level of Au25(GSH)18 clusters rapidly combine with the holes in the valence band of CdSe quantum dots. At the same time, the electrons in the conduction band of CdSe quantum dots migrated to the neighboring MoSe2 nanosheets to complete the Z-scheme charge transfer. This work inspired the researchers to optimize the catalytic reaction performance by manipulating the surface charge catalyst.
 | ||
| Fig. 11 (a) Schematic diagram of the preparation of MoSe2/CdSe/Au25 (M/C/A) heterojunction catalyst. (b) MoSe2/CdSe heterostructure. (c) MoSe2/CdSe/Au25 ternary heterostructure. (d) Photocatalytic H2 production activities of MoSe2, CdSe and Mx/C (x = 0.1, 1, 3 and 6). (e) Photocatalytic H2 production activities of CdSe, 1M/C and 1M/C/Ax (x = 0.5, 4, and 8). (f) Photocatalytic H2 production activities of 1M/C, 1M/C/4A produced by surface charge regulation and simple physical mixing under visible light irradiation (λ > 420 nm) in Na2SO4 aqueous solution (pH = 6.69). (g) Scheme of the electron transfer mechanism of catalyst. Reproduced with permission from ref. 81. Copyright 2023 Wiley-VCH. | ||
In addition to interfacial charge regulation, Zhu et al. also proposed a covalent bridge strategy connecting atomically precise Au25(L-Cys)18 (L-Cys = L-cysteine) cluster with metal–organic frameworks to enhance the activity and stability of photocatalytic H2 production.82Fig. 12a shows a schematic diagram of –COOH on the Au25(L-Cys)18 ligand bound to –NH2 on UiO-66-NH2 by a dehydration condensation reaction with an amide covalent bond. Through a covalent bridge strategy, the Au25(L-Cys)18 cluster was uniformly and densely embedded throughout UiO-66-NH2. A physical mixture of Au25(PET)18 and UiO-66-NH2 was also prepared for comparison. Fig. 12b compares the photocatalytic H2 production performance of UiO-66-NH2, covalent bonded UiO-66-NH2-Au25(L-Cys)18 and physically mixed UiO-66-NH2/Au25(PET)18. UiO-66-NH2-Au25(L-Cys)18 exhibited the optimal photocatalytic H2 production rate, which was 90 times higher than that of the original UiO-66-NH2. Moreover, the UiO-66-NH2-Au25(L-Cys)18 catalyst linked via covalent bonding displayed outstanding stability (Fig. 12c). The UiO-66-NH2/Au25(PET)18 catalyst by simple mixing in the second cycle showed a significant decrease in catalytic activity. This is attributed to the metal–support interaction created by the covalent bond between UiO-66-NH2 and Au25(L-Cys)18, which can facilitate charge transport and inhibit charge–hole recombination. We also list a comprehensive range of studies of precise metal clusters as photocatalysts in Table 2, including test conditions and catalytic activities, without going into detail.
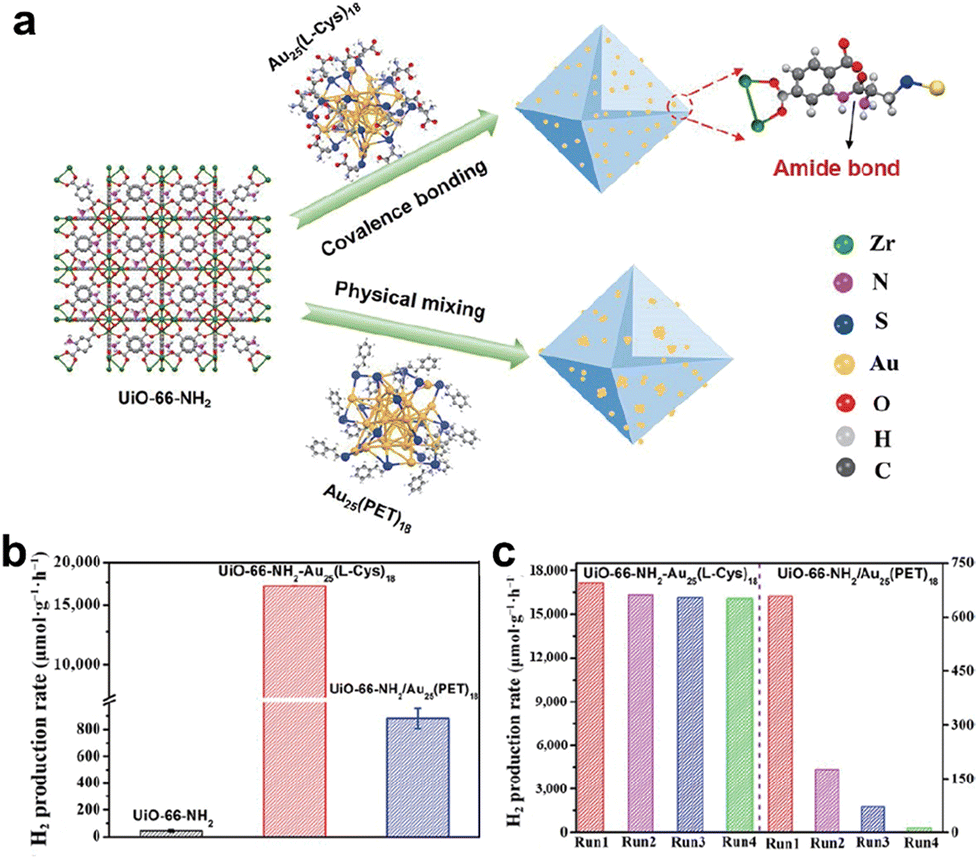 | ||
| Fig. 12 (a) Schematic diagram of Au25(L-Cys)18 bound to UiO-66-NH2 through covalent bridge strategy. (b) Comparison of the photocatalytic H2 production performance of UiO-66-NH2, covalent bonded UiO-66-NH2-Au25(L-Cys)18 and physically mixed UiO-66-NH2/Au25(PET)18 with ErB and TEOA as the photosensitizer and sacrificial agent. The light source was a 300 W xenon lamp with a >420 nm filter. (c) Comparison of cycling stability of UiO-66-NH2-Au25(L-Cys)18 and UiO-66-NH2/Au25(PET)18. Reproduced with permission from ref. 82. Copyright 2023 Springer Nature. | ||
| Catalysts | Co-catalyst | Amount of catalyst (mg) | Light source | Sacrificial agent (vol%) | Activity | Ref. |
|---|---|---|---|---|---|---|
| Ni6(SR)12/TiO2 | Ni6(SR)12 | 20 | 300 W Xe lamp | Methanol (20) | 5600 μmol g−1 h−1 | 75 |
| Ni12(SR)24/C3N4 | Ni12(SR)24 | 10 | 50 W Xe lamp | Triethanolamine (20) | 3000 μmol g−1 h−1 | 76 |
| Cu20@TiO2-NS | Cu20 | — | 300 W Xe lamp (λ > 420 nm) | Triethanolamine (—) | 13 mmol g−1 h−1 | 77 |
| Cu6-MOF | Cu6 | 2 | 300 W Xe lamp (λ > 420 nm) | Triethanolamine (5) | 127.98 mmol per g per 9 h | 78 |
| MoSe2/CdSe/Au25(SR)18 | Au25(SR)18 | 10 | 300 W Xe lamp (λ > 420 nm) | Lactic acid (10) | 140 μmol g−1 h−1 | 81 |
| UiO-66-NH2-Au25(SR)18 | Au25(SR)18 | 5 | 300 W Xe lamp (λ > 420 nm) | Triethanolamine (10) | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 012.045 μmol g−1 h−1 012.045 μmol g−1 h−1 |
82 |
| Au25(SR)18/g-C3N4 | Au25(SR)18 | 20 | 300 W Xe lamp (λ > 420 nm) | Triethanolamine (10) | 320 μmol g−1 h−1 | 83 |
| Au24Pt(SR)18–BaLa4Ti4O15 | Au24Pt(SR)18 | 500 | 400 W high-pressure Hg lamp | Methanol (10) | 5000 μmol h−1 | 84 |
| Au25(SR)18–BaLa4Ti4O15 | Au25(SR)18 | 500 | 400 W high-pressure Hg lamp | — | 500 μmol h−1 | 85 |
| Au101(PPh3)21Cl5–AlSrTiO3–r-GO | Au101(PPh3)21Cl5 | 7 | UV LED (365 nm, 83 mW cm−2) | Methanol (33.3) | 0.5 μmol g−1 h−1 | 86 |
| Au25(SR)18–Cr2O3–BaLa4Ti4O15 | Au25(SR)18 | 500 | 400 W high-pressure Hg lamp | Methanol (10) | 4697 μmol h−1 | 87 |
4 Conclusions
In summary, we have concentrated on summarizing the development of atomically precise metal clusters in electrochemical and photocatalytic hydrogen production in the last few years. Due to the resolvable crystallographic structure of metal clusters, they provide a promising research platform for understanding the relationship between catalytic performance and structure. Many achievements have been witnessed in the application of precise clusters for electrocatalytic and photocatalytic H2 production. With the further development of scientific research, the researchers are more in pursuit of superior catalytic performance, and how to enhance the efficiency of electrocatalysis and photocatalysis remains a challenge for current research. For electrochemical H2 evolution catalysts, understanding the electron transfers between the catalyst and electrode surface as well as the catalyst and reaction substrate is crucial for designing high-performance electrocatalysts. For photocatalytic H2 production, how to effectively promote the separation of photogenerated electron–hole pairs and inhibit their recombination is the key to enhancing the photocatalytic efficiency. Metal clusters with extraordinary optical absorption urgently need to be designed to have the ability to capture light from the visible to the infrared and act as photosensitizers in photocatalytic systems. In addition, ligand-protected metal clusters are fragile in both electrocatalytic and photocatalytic systems, and improving the structural stability of the clusters is conducive to promote catalytic stability. The monitoring of active sites reported so far is usually realized by DFT calculations. Therefore, in situ characterization techniques are of great interest for experimentally detecting active sites and tracking cluster structure evolution. It is expected that the development of atomically precise metal clusters in the field of electrochemical and photocatalytic H2 production will fill the gap between single-atom catalysis and nanoparticle catalysis and bring new opportunities for understanding the catalytic mechanism.Data availability
This is a review, in which the data supporting this article have been reported.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (22125202, 21932004 and 22302091) and the Natural Science Foundation of Jiangsu Province (BK20220033).References
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- K. Perović, S. Morović, A. Jukić and K. Košutić, Materials, 2023, 16, 6319 CrossRef PubMed.
- E. Fernandez, L. Santamaria, M. Artetxe, M. Amutio, A. Arregi, G. Lopez, J. Bilbao and M. Olazar, Fuel, 2022, 312, 122910 CrossRef CAS.
- V. Palma, F. Castaldo, P. Ciambelli and G. Iaquaniello, Appl. Catal., B, 2014, 145, 73–84 CrossRef CAS.
- J. Wang, J. Liu, L. Zhang, S. Dai, A. Li and J. Chen, Fuel, 2022, 327, 125076 CrossRef CAS.
- L. Feng, M. Dong, B. Qin, J. Pang and S. Babaee, Energy, 2024, 291, 130379 CrossRef CAS.
- H. L. Huynh, W. M. Tucho, X. Yu and Z. Yu, J. Cleaner Prod., 2020, 264, 121720 CrossRef CAS.
- X. Hong, V. B. Thaore, S. S. Garud, I. A. Karimi, S. Farooq, X. Wang, A. K. Usadi and B. R. Chapman, Int. J. Hydrogen Energy, 2023, 48, 8743–8755 CrossRef CAS.
- J. Wu, Y. Tao, C. Zhang, Q. Zhu, D. Zhang and G. Li, J. Hazard. Mater., 2023, 443, 130363 CrossRef CAS PubMed.
- M. Zhang, J. Guan, Y. Tu, S. Chen, Y. Wang, S. Wang, L. Yu, C. Ma, D. Deng and X. Bao, Energy Environ. Sci., 2020, 13, 119–126 RSC.
- R. Zhang, Y. Li, X. Zhou, A. Yu, Q. Huang, T. Xu, L. Zhu, P. Peng, S. Song, L. Echegoyen and F. F. Li, Nat. Commun., 2023, 14, 2460 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- X. W. Lv, W. W. Tian and Z. Y. Yuan, Electrochem. Energy Rev., 2023, 6, 23 CrossRef CAS.
- Q. Liu, C. Zeng, Z. Xie, L. Ai, Y. Liu, Q. Zhou, J. Jiang, H. Sun and S. Wang, Appl. Catal., B, 2019, 254, 443–451 CrossRef CAS.
- J. Wu, H. Zhong, Z. F. Huang, J. J. Zou, X. Zhang, Y. C. Zhang and L. Pan, Nanoscale, 2024, 16, 9169–9185 RSC.
- Z. Kang, M. A. Khan, Y. Gong, R. Javed, Y. Xu, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9, 6089–6108 RSC.
- G. Zhao, Y. Jiang, S. X. Dou, W. Sun and H. Pan, Sci. Bull., 2021, 66, 85–96 CrossRef CAS PubMed.
- J. Ding, H. Yang, S. Zhang, Q. Liu, H. Cao, J. Luo and X. Liu, Small, 2022, 18, 2204524 CrossRef CAS PubMed.
- B. J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S. P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- M. U. Shahid, T. Najam, M. H. Helal, I. Hossain, S. M. El-Bahy, Z. M. El-Bahy, A. ur Rehman, S. S. A. Shah and M. A. Nazir, Int. J. Hydrogen Energy, 2024, 62, 1113–1138 CrossRef CAS.
- H. Nazir, C. Louis, S. Jose, J. Prakash, N. Muthuswamy, M. E. M. Buan, C. Flox, S. Chavan, X. Shi, P. Kauranen, T. Kallio, G. Maia, K. Tammeveski, N. Lymperopoulos, E. Carcadea, E. Veziroglu, A. Iranzo and A. M. Kannan, Int. J. Hydrogen Energy, 2020, 45, 13777–13788 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- X. Zou, X. Kang and M. Zhu, Chem. Soc. Rev., 2023, 52, 5892–5967 RSC.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- X. Liu, E. Wang, M. Zhou, Y. Wan, Y. Zhang, H. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202207685 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, 61, e202211771 CrossRef CAS PubMed.
- H. Yan, H. Xiang, J. Liu, R. Cheng, Y. Ye, Y. Han and C. Yao, Small, 2022, 18, 2200812 CrossRef CAS PubMed.
- Y. Sun, X. Cai, W. Hu, X. Liu and Y. Zhu, Sci. China: Chem., 2021, 64, 1065–1075 CrossRef CAS.
- X. Yuan and M. Zhu, Inorg. Chem. Front., 2023, 10, 3995–4007 RSC.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- Z. Song, T. Shen, Y. Hu, G. Liu, S. Bai, X. Sun, S. M. Xu and Y. F. Song, Nanoscale, 2023, 15, 11867–11874 RSC.
- M. Wu, F. Dong, Y. Yang, X. Cui, X. Liu, Y. Zhu, D. Li, S. Omanovic, S. Sun and G. Zhang, Electrochem. Energy Rev., 2024, 7, 10 CrossRef CAS.
- T. H. Chiu, J. H. Liao, R. P. B. Silalahi, M. N. Pillay and C. W. Liu, Nanoscale Horiz., 2024, 9, 675–692 RSC.
- Z. Pu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, S. Mu, W. Zhao, F. Su, G. Zhang, S. Liao and S. Sun, Nano-Micro Lett., 2020, 12, 21 CrossRef CAS PubMed.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- P. Zhu, X. Xiong and D. Wang, Nano Res., 2022, 15, 5792–5815 CrossRef CAS.
- H. Chen, Y. Zhou, W. Guo and B. Y. Xia, Chin. Chem. Lett., 2022, 33, 1831–1840 CrossRef CAS.
- Y. Wang, H. Zhuo, X. Zhang, Y. Li, J. Yang, Y. Liu, X. Dai, M. Li, H. Zhao, M. Cui, H. Wang and J. Li, J. Mater. Chem. A, 2019, 7, 24328–24336 RSC.
- Z. Zhong, Y. Tu, L. Zhang, J. Ke, C. Zhong, W. Tan, L. Wang, J. Zhang, H. Song, L. Du and Z. Cui, ACS Catal., 2024, 14, 2917–2923 CrossRef CAS.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-e. Jiang, J. P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- A. M. Abudayyeh, M. S. Bennington, J. Hamonnet, A. T. Marshall and S. Brooker, Dalton Trans., 2024, 53, 6207–6214 RSC.
- C. R. Ratwani, S. Karunarathne, A. R. Kamali and A. M. Abdelkader, ACS Appl. Mater. Interfaces, 2024, 16, 5847–5856 CrossRef CAS PubMed.
- X. Gao and W. Chen, Chem. Commun., 2017, 53, 9733–9736 RSC.
- K. M. Sithar Shahul, V. Packirisamy and P. Pandurangan, Int. J. Hydrogen Energy, 2024, 62, 1067–1076 CrossRef CAS.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- S. Gratious, A. Karmakar, D. Kumar, S. Kundu, S. Chakraborty and S. Mandal, Nanoscale, 2022, 14, 7919–7926 RSC.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- W. L. Mu, Y. T. Luo, P. K. Xia, Y. L. Jia, P. Wang, Y. Pei and C. Liu, Inorg. Chem., 2024, 63, 6767–6775 CrossRef CAS PubMed.
- H. Mousavi, Y. Yin, S. K. Sharma, C. T. Gibson, V. Golovko, G. G. Andersson, C. J. Shearer and G. F. Metha, J. Phys. Chem. C, 2021, 126, 246–260 CrossRef.
- L. Sahoo, A. Devi and A. Patra, ACS Sustainable Chem. Eng., 2023, 11, 4187–4196 CrossRef CAS.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- Y. Jo, M. Choi, M. Kim, J. S. Yoo, W. Choi and D. Lee, Bull. Korean Chem. Soc., 2021, 42, 1672–1677 CrossRef CAS.
- Y. Tang, F. Sun, X. Ma, L. Qin, G. Ma, Q. Tang and Z. Tang, Dalton Trans., 2022, 51, 7845–7850 RSC.
- G. Ma, Y. Tang, L. Chen, L. Qin, Q. Shen, L. Wang and Z. Tang, Eur. J. Inorg. Chem., 2022, e202200176 CrossRef CAS.
- R. P. Brocha Silalahi, Y. Jo, J. H. Liao, T. H. Chiu, E. Park, W. Choi, H. Liang, S. Kahlal, J. Y. Saillard, D. Lee and C. W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301272 CrossRef CAS PubMed.
- R. P. Brocha Silalahi, H. Liang, Y. Jo, J. H. Liao, T. H. Chiu, Y. Y. Wu, X. Wang, S. Kahlal, Q. Wang, W. Choi, D. Lee, J. Y. Saillard and C. W. Liu, Chem. – Eur. J., 2024, 30, e202303755 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- O. López-Estrada, N. Mammen, L. Laverdure, M. M. Melander, H. Häkkinen and K. Honkala, ACS Catal., 2023, 13, 8997–9006 CrossRef.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D.-e. Jiang, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- G. Hu, Z. Wu and D.-e. Jiang, J. Mater. Chem. A, 2018, 6, 7532–7537 RSC.
- H. Shen, Q. Zhu, J. Xu, K. Ni, X. Wei, Y. Du, S. Gao, X. Kang and M. Zhu, Nanoscale, 2023, 15, 14941–14948 RSC.
- X. Liu, Y. Tang, L. Chen, L. Wang, Y. Liu and Z. Tang, Int. J. Hydrogen Energy, 2024, 53, 300–307 CrossRef CAS.
- S. Tasleem, M. Tahir and W. A. Khalifa, Int. J. Hydrogen Energy, 2021, 46, 14148–14189 CrossRef CAS.
- S. Xiong, R. Tang, D. Gong, Y. Deng, J. Zheng, L. Li, Z. Zhou, L. Yang and L. Su, Chin. J. Catal., 2022, 43, 1719–1748 CrossRef CAS.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- J. Wen, J. Xie, Z. Yang, R. Shen, H. Li, X. Luo, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2017, 5, 2224–2236 CrossRef CAS.
- Y. Yang, C. Zhou, W. Wang, W. Xiong, G. Zeng, D. Huang, C. Zhang, B. Song, W. Xue, X. Li, Z. Wang, D. He, H. Luo and Z. Ouyang, Chem. Eng. J., 2021, 405, 126547 CrossRef CAS.
- Y. Zhao, Z. Niu, J. Zhao, L. Xue, X. Fu and J. Long, Electrochem. Energy Rev., 2023, 6, 14 CrossRef CAS.
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed.
- S. Biswas, S. Das and Y. Negishi, Nanoscale Horiz., 2023, 8, 1509–1522 RSC.
- H. Liang, B. J. Liu, B. Tang, S. C. Zhu, S. Li, X. Z. Ge, J. L. Li, J. R. Zhu and F. X. Xiao, ACS Catal., 2022, 12, 4216–4226 CrossRef CAS.
- P. Shen, S. Zhao, D. Su, Y. Li and A. Orlov, Appl. Catal., B, 2012, 126, 153–160 CrossRef CAS.
- H. N. Kagalwala, E. Gottlieb, G. Li, T. Li, R. Jin and S. Bernhard, Inorg. Chem., 2013, 52, 9094–9101 CrossRef CAS PubMed.
- F. Tian, J. Chen, F. Chen, Y. Liu, Y. Xu and R. Chen, Appl. Catal., B, 2021, 292, 120158 CrossRef CAS.
- F. Tian, X. Huang, W. Li, Y. An, G. Li and R. Chen, ACS Catal., 2023, 13, 12186–12196 CrossRef CAS.
- Y. D. Cao, H. P. Hao, H. S. Liu, D. Yin, M. L. Wang, G. G. Gao, L. L. Fan and H. Liu, Nanoscale, 2021, 13, 16182–16188 RSC.
- Y. Xu, Q. G. Dong, J. P. Dong, H. Zhang, B. Li, R. Wang and S. Q. Zang, Chem. Commun., 2023, 59, 3067–3070 RSC.
- H. Wang, X. Zhang, W. Zhang, M. Zhou and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202401443 CrossRef CAS PubMed.
- Y. D. Cao, D. Yin, S. Li, X. Y. Dong, Y. Feng, H. Liu, L. L. Fan, G. G. Gao and S. Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202307678 CrossRef CAS PubMed.
- X. Yan, X. Y. Fu and F. X. Xiao, Adv. Funct. Mater., 2023, 33, 2303737 CrossRef CAS.
- A. Yao, Y. Du, M. Han, Y. Wang, J. Hu, Q. Zhu, H. Sheng and M. Zhu, Nano Res., 2022, 16, 1527–1532 CrossRef.
- C. Wang, P. Lv, D. Xue, Y. Cai, X. Yan, L. Xu, J. Fang and Y. Yang, ACS Sustainable Chem. Eng., 2018, 6, 8447–8457 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- H. Mousavi, T. D. Small, S. K. Sharma, V. B. Golovko, C. J. Shearer and G. F. Metha, Nanomaterials, 2022, 12, 3638 CrossRef CAS PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |