
Electronic state modulation of Ag30 nanoclusters within a ring-shaped polyoxometalate†
Daiki
Yanai
a,
Kentaro
Yonesato
 *a,
Soichi
Kikkawa
b,
Seiji
Yamazoe
b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
*a,
Soichi
Kikkawa
b,
Seiji
Yamazoe
b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp; k-yonesato@g.ecc.u-tokyo.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioji, Tokyo 192-0397, Japan
First published on 10th September 2024
Abstract
Atomically precise Ag nanoclusters display distinctive properties that are dictated by their structures and electronic states. However, manipulating the electronic states of Ag nanoclusters is challenging owing to their inherent instability and susceptibility to undesired structural changes, decomposition, and aggregation. Recently, we reported the synthesis of a body-centered cubic {Ag30}22+ nanocluster encapsulated within a ring-shaped polyoxometalate (POM) [P8W48O184]40− by reacting 16 Ag+-containing [P8W48O184]40− with Ag+ using N,N-dimethylformamide (DMF) as a mild reducing agent. This led to a redox-induced structural transformation into a face-centered cubic {Ag30}16+ nanocluster. In this study, we demonstrated the modulation of the electronic states of Ag30 nanoclusters within the ring-shaped POM through two different approaches. A face-centered cubic {Ag30}18+ nanocluster, featuring distinct oxidation states compared to previously reported {Ag30}22+ and {Ag30}16+ nanoclusters, was synthesized using tetra-n-butylammonium borohydride, a stronger reducing agent than DMF, in the reaction of 16 Ag+-containing [P8W48O184]40− and Ag+. Additionally, by leveraging the acid–base properties of POMs, we demonstrated the reversible, stepwise modulation of the charge distribution in the Ag30 nanocluster through controlling protonation states of the ring-shaped POM ligand. These results highlight the potential of engineering POM-stabilized Ag nanoclusters with diverse structures and electronic states, thereby facilitating the exploration of novel properties and applications utilizing the unique characteristics of the POM ligands.
Introduction
Atomically precise metal nanoclusters of group 10 and 11 elements, such as Au, Ag, Cu, Pd, and Pt, have attracted increasing interest in various fields, including catalysis, magnetic devices, pharmaceuticals, optical materials, and sensors.1 This interest stems from their unique properties, which are intrinsically linked to their structures and electronic states. To date, substantial advancements in the synthesis of metal nanoclusters have enabled extensive structural tuning of their constituent metal elements, stabilizing ligands, and sizes, leading to the successful construction of these metal nanoclusters.Recent studies on Au and Au-alloy nanoclusters, particularly [Au25(SR)18]q nanoclusters (where q = −1, 0, and +1),2 have highlighted the profound effect of their oxidation states on the physicochemical properties of metal nanoclusters. These properties include stability,3 magnetic characteristics,4 optical behaviors,5 and catalytic activity.6 Moreover, metal nanoclusters with identical structures and oxidation states exhibit varying physicochemical properties influenced by the electron-donating abilities of stabilizing ligands.7 These studies highlight the importance of controlling metal nanoclusters’ electronic states, including oxidation states and electron donation from stabilizing ligands, to develop novel properties and applications. However, modifying the electronic states of metal nanoclusters is typically difficult, and often leads to undesirable aggregation or decomposition. In particular, Ag nanoclusters are generally unstable and prone to structural changes, limiting the successful modification of their electronic states to only a few reports.8
Polyoxometalates (POMs) are anionic metal oxide clusters known for their diverse, well-defined structures and unique properties, including acid–base, redox, and optical characteristics.9 POMs have been employed as attractive inorganic ligands for Ag nanoclusters owing to their oxygen-enriched molecular surfaces and variable properties.10,11 Recently, we developed a synthetic method for Ag nanoclusters stabilized by lacunary POMs. These act as inorganic multidentate ligands with highly reactive oxygen atoms at vacant sites.12,13 In particular, using a ring-shaped POM [P8W48O184]40− (P8W48), consisting of a tetramer of hexavacant lacunary Dawson-type {P2W12} units,14,15 we synthesized surface-exposed Ag30 nanoclusters via a stepwise reduction method (Fig. 1, and S1a†).13 The reaction involving 16 Ag+-containing P8W48 ([Ag16P8W48O184]24−, denoted as Ag16) and additional Ag+ in N,N-dimethylformamide (DMF), acting as a mild reducing agent, led to the formation of an {Ag30}22+ nanocluster within P8W48 (denoted as Ag30). This {Ag30}22+ nanocluster underwent further reduction upon reaction with tetra-n-butylammonium borohydride (TBABH4), resulting in an {Ag30}16+ nanocluster (denoted as I). Importantly, P8W48 served as an effective stabilizing ligand for Ag nanoclusters with varying structures and oxidation states: the {Ag30}22+ nanocluster featured a body-centered cubic (bcc) atom arrangement. In contrast, the {Ag30}16+ nanocluster exhibited a face-centered cubic (fcc) atom arrangement. Consequently, we expected that P8W48 could facilitate the formation of Ag nanoclusters with diverse electronic states, thus driving the advancement of novel applications for POM-stabilized Ag nanoclusters.
 | ||
| Fig. 1 Diagram illustrating the modulation of electronic states in Ag30 nanoclusters within a ring-shaped POM. (a) Modulation of oxidation states in Ag30 nanoclusters through variations in synthetic conditions. (b) Modulation of charge distribution via protonation and deprotonation of the ring-shaped POM. | ||
In this study, we demonstrated the modulation of electronic states in Ag30 nanoclusters within a ring-shaped POM through two distinct approaches (Fig. 1): (a) oxidation-state modulation by changing the synthetic conditions and (b) charge-distribution modulation by changing the protonation states of P8W48. Specifically, we synthesized an {Ag30}18+ nanocluster (II) within P8W48 using a strong reducing reagent (TBABH4) in a reaction involving Ag16 and Ag+ ions. The {Ag30}18+ nanocluster in II exhibited structural similarity to the {Ag30}16+ nanocluster in I, maintaining an fcc metal arrangement despite the different oxidation states. Subsequently, by leveraging the acid–base properties of POMs, we demonstrated the ability to reversibly and stepwise control the charge distribution of Ag30 nanoclusters in II through the process of protonation and deprotonation of P8W48. These findings demonstrate that the electronic states of POM-stabilized Ag nanoclusters can be manipulated through synthetic conditions and post-synthetic modulation via protonation states. This capability will expedite the advancement of their applications across diverse fields.
Results and discussion
The {Ag30}22+ nanocluster within P8W48 (i.e., Ag30) was synthesized by reacting 16 Ag+-containing P8W48 (i.e., Ag16) with silver acetate in DMF, which acted as a solvent and a mild reducing agent (Fig. 1a).13 As a result, 14 Ag+ were integrated into Ag16 through the partial reduction of Ag+ to Ag0, leading to the creation of the {Ag30}22+ nanocluster. We hypothesized that the electronic state of the Ag nanoclusters within P8W48 could be further modulated using a strong reducing reagent. Upon addition of TBABH4 to the reaction solution of Ag16 and silver acetate (Fig. S1b; see ESI† for details), the color of the solution rapidly shifted from light yellow to dark brown. The UV–vis spectrum of Ag30 in acetonitrile displayed no characteristic absorption in the visible light region. In contrast, the reaction solution exhibited an absorption band at 472 nm (Fig. S2a†), indicating the formation of Ag nanoclusters with distinct structures and/or electronic states, which are different from those of Ag30. The introduction of ethyl acetate into the reaction solution produced dark brown block-shaped single crystals (II). Notably, the UV–vis spectrum of these crystals dissolved in acetonitrile showed no significant difference from that of the reaction solution, suggesting that II was the main product of the reaction (Fig. S2b†).Elemental analysis and acid–base titration (Fig. S3†) revealed that the formula of II was TBA17H5[Ag30P8W48O184], indicating the involvement of 30 Ag atoms within P8W48. Single-crystal X-ray structure analysis of II (Table S1, and Fig. S4†) showed distinct electron densities attributed to Ag atoms confined exclusively within the P8W48 cavity, resulting in the formation of an Ag30 nanocluster (Fig. 2a, b, and S5†). In the crystal structure of II, 26 of the 30 Ag atoms were arranged in the fcc structure, while the remaining 4 Ag atoms were integrated into two hinge sites between adjacent {P2W12} units. The Ag30 nanocluster in II exhibited structural similarity to that of I (Tables S2, S3, and Fig. S5†). The bond valence sum values of P and W indicate the oxidation states of +5 and +6, respectively (Table S4†). The UV–vis spectrum of II did not show strong absorption in the 600–800 nm range associated with W6+/W5+ intervalence charge transfer, indicating that W6+ maintained its oxidation state without being reduced to W5+ (Fig. 2c).
 | ||
| Fig. 2 (a) Crystal structure depicting the anion part of II. Green octahedra, {WO6}; purple tetrahedra, {PO4}; black spheres, Ag atoms; red spheres, O atoms. (b) Structure of an {Ag30}18+ nanocluster in II. (c) UV–vis spectra of I and II in acetonitrile (10 μM, 1 cm quartz cell). (d) XPS spectrum in the Ag 3d region of II (black dots) and the sum of the curve-fitting analysis (orange line) obtained through the linear combination of Ag0 (blue line) and Ag+ (red line) with an area ratio of Ag0/Ag+ = 12/18. | ||
Although I and II exhibited similar structures, the UV–vis spectrum of II significantly differed from that of I (Fig. 2c). Considering that the absorption bands of I in the visible light region were attributed to charge transfer from the {Ag30}16+ nanocluster to the W atoms of P8W48, as well as intra-electron excitation within the {Ag30}16+ nanocluster,13 this result suggests that II possesses a distinct electronic state compared to I. Considering the anion charge of P8W48 ([P8W48O184]40−) and the number of cations (17 TBA+ and 5 H+), the charge of the Ag30 nanocluster in II was determined to be +18. Analysis of the X-ray photoelectron spectroscopy (XPS) spectrum of II in the Ag 3d region revealed two peaks at 367.6 eV (Ag 3d5/2) and 373.6 eV (Ag 3d3/2) (Fig. 2d). Through curve-fitting analysis, these peaks were deconvoluted into Ag+ (3d5/2 367.5 eV; 3d3/2 373.5 eV) and Ag0 (3d5/2 367.9 eV, 3d3/2 373.9 eV) with an area ratio of Ag0/Ag+ = 12/18 (Table S5†). These findings confirm the oxidation state of the {Ag30}18+ nanocluster in II, possessing 12 valence electrons, contrasting with the {Ag30}16+ nanocluster in I, which has 14 valence electrons. To further elucidate the electronic state of the Ag30 nanocluster in II, we conducted solid-state Ag K-edge X-ray absorption fine structure (XAFS) spectroscopy measurements (Fig. 3). The X-ray absorption near-edge structure (XANES) spectrum of II was found to be more similar to that of I ({Ag30}16+) than Ag30 ({Ag30}22+), corroborating the oxidation state of the {Ag30}18+ nanocluster in II (Fig. 3a and b). Additionally, with the introduction of TBABH4 into the acetonitrile solution of II, the UV–vis spectrum showed increased absorption bands around 420 and 380 nm, similar to those of I, suggesting the conversion of II into I through further reduction (Fig. S6†). This finding further validates the successful synthesis of {Ag30}18+ nanoclusters, which possess fewer valence electrons than the {Ag30}16+ nanoclusters in I. Overall, we demonstrated the controllable modulation of the oxidation state and structure of Ag30 nanoclusters within P8W48 by selecting appropriate reducing reagents. Although there have been instances of Au or Au–metal alloy nanoclusters displaying multiple oxidation states,2,16 a pair of Ag nanoclusters sharing similar structures yet differing in oxidation states remains uncommon, largely owing to the inherent instability of Ag nanoclusters, which readily undergo structural change during the modification of oxidation states.17
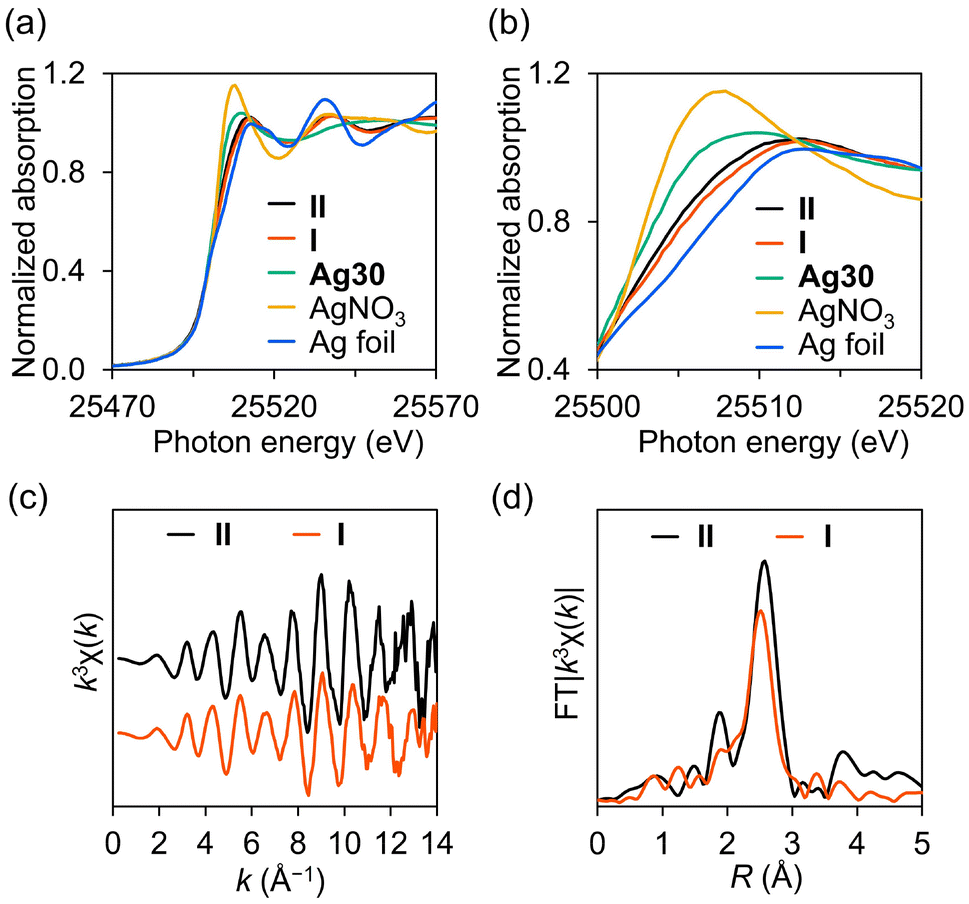 | ||
Fig. 3 Solid-state Ag K-edge XAFS measurements of II and I performed at −263 °C. (a) Wide view and (b) enlarged view around the white line (E = ca. 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 512 eV) of XANES spectra. (c) k3-Weighted k-space EXAFS spectra and (d) Fourier-transformed R-space EXAFS spectra (k range, 3–16 Å). 512 eV) of XANES spectra. (c) k3-Weighted k-space EXAFS spectra and (d) Fourier-transformed R-space EXAFS spectra (k range, 3–16 Å). | ||
The structure of the Ag30 nanocluster in II was further investigated using Ag K-edge extended X-ray absorption fine structure (EXAFS) analysis. The k-space EXAFS of II displayed an oscillation pattern similar to that of I (Fig. 3c), aligned with the crystallographic analysis, which revealed no significant structural disparities between the Ag30 nanoclusters in I and II. Conversely, the Fourier-transformed R-space EXAFS spectrum of II (Fig. 3d) exhibited an intense peak at a slightly longer R value than that of I, underscoring the structural differentiation between I and II. Curve-fitting analysis was conducted for the EXAFS spectra of I and II to elucidate the variance in the Ag–Ag bond length distribution between I and II, stemming from the subtle displacements of the Ag sites. The coordination number (CN) and Ag–Ag distance (R) of II (CN = 4.9 ± 0.3, R = 2.81 ± 0.03 Å) exceeded those of I (CN = 3.9 ± 0.3, R = 2.77 ± 0.03 Å) (Table S6†). This fitting analysis result was consistent with the expected CN and R values derived from crystallographic analysis (CN = 5.0, R = 2.82 Å), calculated based on the count and average bond length of Ag–Ag bonds below 2.88 Å. Conversely, EXAFS analysis did not reveal relatively long Ag–Ag bonds (>2.88 Å), likely owing to interference from a broad bond length distribution. Thus, the fitting analysis of EXAFS, which indicated a larger CN for II than for I, demonstrated that the {Ag30}18+ nanocluster in II had a shorter Ag–Ag bond (<2.88 Å) than the {Ag30}16+ nanocluster in I.
We then attempted to manipulate the charge distribution in the Ag30 nanocluster by protonation and deprotonation of the P8W48 framework. Given the ability of POMs to reversibly store and release multiple protons in their frameworks owing to their unique acid–base properties, the negative charges of POM ligands can be adjusted by varying the number of protons on the POMs. Exploiting this characteristic, we recently demonstrated that electron donation from the [Si2W18O66]16− anion to the {Ag27}17+ nanocluster could be controlled by protonation and deprotonation of the [Si2W18O66]16− anion.12d When p-toluenesulfonic acid (TsOH, 0–4 equivalents relative to II) was added to the acetonitrile solution of II, the UV–vis spectra showed a slight increase in absorbance at λ = 472 nm and decrease at λ = 510 nm (Fig. 4a, and S7†). This result suggests that the electronic state of II can be adjusted by the addition of stoichiometric amounts of TsOH. Moreover, adding TsOH (8–12 equivalents relative to II) reduced the absorbance at λ = 472 nm. After the reaction of II with TsOH, excess diethyl ether was added to isolate the product as a powder, and the peaks assignable to the C–H vibration of TBA cations (2800–3050 cm−1) were significantly decreased in the IR spectrum (Fig. S8†), indicating that the TBA cations of II were exchanged with protons upon the reaction with TsOH. On the other hands, when Na+ ions (sodium trifluoromethanesulfonate) were added to the acetonitrile solution of II instead of protons, no significant change was observed in the UV-vis spectra (Fig. S9†). These findings imply that the structure and/or electronic state of the Ag nanocluster in P8W48 was changed by reacting with TsOH.
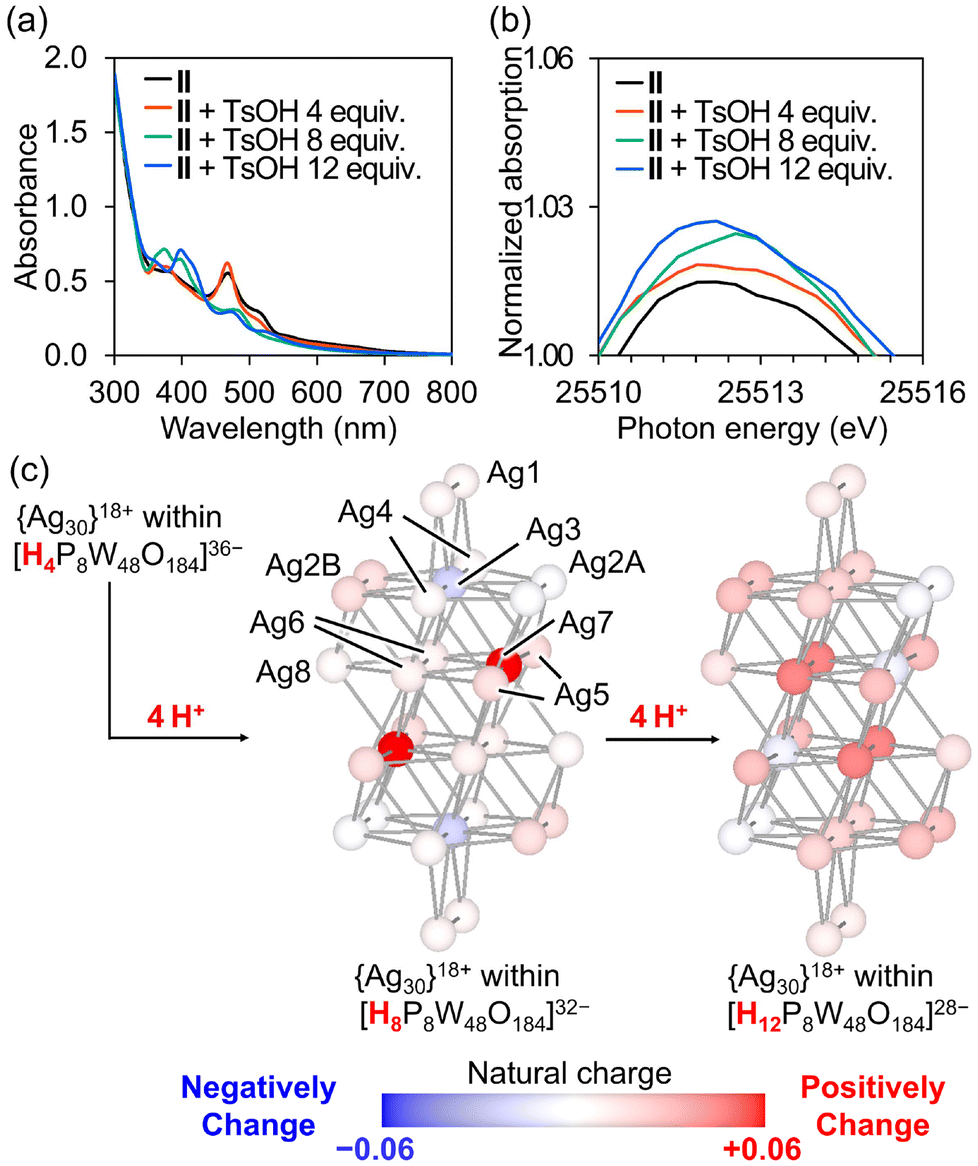 | ||
| Fig. 4 (a) UV–vis spectra of II after adding TsOH (4, 8, and 12 equivalents) in acetonitrile. (b) The enlarged view around the white line (E = ca. 25512 eV) of solution-state Ag K-edge XANES spectra of II upon TsOH addition in acetonitrile. (c) Changes in the natural charge of the {Ag30}18+ nanocluster within a ring-shaped POM upon protonation. Change in the natural charge of each Ag atom after adding protons to a ring-shaped POM framework. Ag atoms are color-coded based on the changes in the natural charges. | ||
To further elucidate the structure and electronic state of the Ag30 nanocluster in II during the reaction with TsOH, solution-state Ag K-edge XAFS studies were performed. The XANES spectra and k-space EXAFS oscillation patterns of II showed no significant difference between the solid-state and acetonitrile solutions, indicating that the structure and electronic state of the Ag30 nanocluster in II remained unchanged in solution (Fig. S10†). Moreover, both the k-space and Fourier-transformed R-space EXAFS spectra of the reaction solution of II and TsOH showed no significant changes, indicating the structural integrity of the Ag30 nanocluster in II during the reaction (Fig. S11 and S12†). However, the XANES spectra of the reaction solution containing II and TsOH revealed an increasing white line (E = ca. 25512 eV) as the amount of TsOH increased (Fig. 4b, S13a, and S13b†). This result suggests that the protonation of II reduces electron donation from the P8W48 ligand to the Ag30 nanocluster, resulting in changes in the UV–vis absorption. Furthermore, upon the addition of an equivalent amount of TBAOH relative to TsOH to the reaction solution of II and TsOH, the UV–vis and XANES spectra reverted to characteristics resembling those of the original II, indicating that the electronic state of the Ag30 nanocluster in II can be controlled by adding stoichiometric amounts of acid and base (Fig. S14, S15a, and S15b†).
Finally, density functional theory calculations were performed to analyze the natural charge population of the {Ag30}18+ nanocluster in II, considering different numbers of protons on the P8W48 ligand (see Experimental details in the ESI; Fig. S16†). The calculation with four protons on the [{Ag30}18+P8W48O184]22− anion showed that the natural charges of the Ag atoms at the center of the Ag30 nanocluster (i.e., Ag4, Ag6, and Ag7; Fig. 4c, S17, and Table S7†) ranged from −0.33 to −0.16, which were more negative than those of the other Ag atoms on the surface or adjacent to the P8W48 ligand (i.e., 0.46–0.81 for Ag1, Ag2, Ag3, Ag5, and Ag8). The total natural charge of the Ag30 nanoclusters within the ring-shaped POM (i.e., [H4P8W48O184]36−) was +11.3, which is more negative than that of the Ag30 nanocluster without the POM ligand (+18.0) (Fig. S18†). This suggests that the POM ligand acts as an electron-donating ligand. The total natural charges of the Ag30 nanocluster with 4, 8, and 12 protons on the P8W48 ligand were +11.3, +11.6, and +11.8, respectively, indicating a decrease in the electron density of the Ag30 nanocluster with an increase in the number of protons on the P8W48 ligand. This observation aligns with the results of the Ag K-edge XANES analysis. With an increase in the number of protons on the P8W48 ligand from 4 to 8 (i.e., [H4P8W48O184]36− to [H8P8W48O184]32−), the natural charge of Ag4 became more negative, whereas those of Ag7 became more positive (Fig. 4c). However, with the addition of 4 more protons (forming [H12P8W48O184]28−), the natural charge of Ag4 became more positive and that of Ag7 became more negative. Additionally, the Ag3, Ag4, and Ag6 sites showed a slight increase in natural charge by 0.02–0.04, indicating a reduction in electron donation from the P8W48 ligand, influencing the electronic state of the Ag30 nanocluster within P8W48. These findings suggest that the charge distribution of Ag nanoclusters can be controlled by the protonation states of the POM ligands. Additionally, given that the changes in the natural charge populations exhibited different trends depending on the number of protons, this computational study aligns with the multistep changes observed in the UV–vis spectra of the reaction solution when II was reacted with increasing amounts of TsOH. These results suggested that the positions of protons added to P8W48 differ every 4 equivalents, resulting in a significant change in the UV-vis spectra of II with the addition of 4, 8, and 12 equivalents of TsOH (Fig. S16†).
Conclusions
In this study, we demonstrated the modulation of the electronic state of Ag30 nanoclusters within a ring-shaped POM ([P8W48O184]40−, denoted as P8W48) using two approaches. An {Ag30}18+ nanocluster in P8W48 was synthesized by adding tetra-n-butylammonium borohydride as a reducing reagent to a reaction solution of 16 Ag+-containing P8W48 and Ag+ ions. The {Ag30}18+ nanocluster in P8W48 possessed an fcc-type structure similar to that of the previously reported {Ag30}16+ nanocluster13 despite the different oxidation states. Moreover, we demonstrated that the charge distribution of the {Ag30}18+ nanocluster could be modulated by leveraging the acid–base properties of the POMs. These findings suggest that POM-stabilized Ag nanoclusters with different structures and electronic states can be obtained by controlling the synthetic conditions and post-synthetic modulation. This capability will accelerate the development of their applications and properties, including their catalytic activity, photochemical properties, adsorption, sensing, and medical applications.Data availability
The data supporting this manuscript is available in the ESI† of and available on request. Crystallographic data for II has been deposited at the CCDC (deposition number 2361048†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST FOREST (JPMJFR213M), JST SPRING (JPMJSP2108), and JSPS KAKENHI (22H04971, 24H02210, 24H02217). XAFS measurements at SPring-8 were conducted with the approval of the Japan Synchrotron Radiation Research Institute (proposal numbers 2023B1651, 2023B2070). Single-crystal X-ray diffraction measurements at SPring-8 were conducted with the approval of the Japan Synchrotron Radiation Research Institute (proposal numbers 2023B1842, 2023A1731). A part of computations was performed using Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C106, 24-IMS-C101).References
- (a) I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed; (b) Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed; (c) G.-G. Luo, Q.-L. Guo, Z. Wang, C.-F. Sun, J.-Q. Lin and D. Sun, Dalton Trans., 2020, 49, 5406–5415 RSC; (d) S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed; (e) M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- (a) Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS PubMed; (b) M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS; (c) M. A. Tofanelli, K. Salorinne, T. W. Ni, S. Malola, B. Newell, B. Phillips, H. Häkkinen and C. J. Ackerson, Chem. Sci., 2016, 7, 1882–1890 RSC.
- (a) M. A. Tofanelli and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 16937–16940 CrossRef CAS PubMed; (b) C. B. Collins, M. A. Tofanelli, M. F. Crook, B. D. Phillips and C. J. Ackerson, RSC Adv., 2017, 7, 45061–45065 RSC.
- (a) M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS PubMed; (b) S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585–15594 CrossRef CAS PubMed.
- (a) R.-J. Mosqueda and G. Mpourmpakis, Phys. Chem. Chem. Phys., 2019, 21, 22272–22282 RSC; (b) K. Kwak, Q. Tang, M. Kim, D.-e. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- (a) D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC; (b) G. Li, W. Hu, Y. Sun, J. Xu, X. Cai, X. Cheng, Y. Zhang, A. Tang, X. Liu, M. Chen, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2020, 59, 21135–21142 CrossRef CAS PubMed; (c) H. Chong, P. Li, S. Wang, F. Fu, J. Xiang, M. Zhu and Y. Li, Sci. Rep., 2013, 3, 3214 CrossRef PubMed.
- (a) Z. Liu, H. Tan, B. Li, Z. Hu, D.-e. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed; (b) Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed; (c) A. Cirri, H. M. Hernández, C. Kmiotek and C. J. Johnson, Angew. Chem., Int. Ed., 2019, 58, 13818–13822 CrossRef CAS PubMed.
- (a) Z.-J. Guan, R.-L. He, S.-F. Yuan, J.-J. Li, F. Hu, C.-Y. Liu and Q.-M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116965 CrossRef CAS PubMed; (b) Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. L. D. M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews, S. Barcikowski, M. M. Kappes, W. J. Parak, C. M. Aikens and I. Chakraborty, J. Am. Chem. Soc., 2021, 143, 9405–9414 CrossRef CAS PubMed.
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef; (b) C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS; (c) H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC; (d) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed; (e) I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680–2717 CrossRef CAS PubMed; (f) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809–10825 CrossRef CAS; (g) N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS; (h) A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS PubMed; (i) J.-H. Kruse, M. Langer, I. Romanenko, I. Trentin, D. Hernández-Castillo, L. González, F. H. Schacher and C. Streb, Adv. Funct. Mater., 2022, 32, 2208428 CrossRef CAS.
- (a) K. Zhou, C. Qin, H.-B. Li, L.-K. Yan, X.-L. Wang, G.-G. Shan, Z.-M. Su, C. Xu and X.-L. Wang, Chem. Commun., 2012, 48, 5844–5846 RSC; (b) Z. Wang, H.-F. Su, C.-H. Tung, D. Sun and L.-S. Zheng, Nat. Commun., 2018, 9, 4407 CrossRef PubMed; (c) H. Liu, C.-Y. Song, R.-W. Huang, Y. Zhang, H. Xu, M.-J. Li, S.-Q. Zang and G.-G. Gao, Angew. Chem., Int. Ed., 2016, 55, 3699–3703 CrossRef CAS PubMed; (d) Y.-M. Su, Z. Wang, G.-L. Zhuang, Q.-Q. Zhao, X.-P. Wang, C.-H. Tung and D. Sun, Chem. Sci., 2019, 10, 564–568 RSC; (e) Z. Zhao, M. Zhao, L. Deng, Q. Li, J. Zhang, H. Su, H. Lv and G.-Y. Yang, Chem. Commun., 2024, 60, 5415–5418 RSC.
- (a) K.-G. Liu, X.-Y. Liu, Z.-J. Guan, K. Shi, Y.-M. Lin and Q.-M. Wang, Chem. Commun., 2016, 52, 3801–3804 RSC; (b) S. Tamari, K. Ono, M. Hashimoto and T. Ozeki, Dalton Trans., 2015, 44, 19056–19058 RSC; (c) F. Gruber and M. Jansen, Angew. Chem., Int. Ed., 2010, 49, 4924–4926 CrossRef CAS PubMed; (d) G.-G. Gao, P.-S. Cheng and T. C. W. Mak, J. Am. Chem. Soc., 2009, 131, 18257–18259 CrossRef CAS PubMed; (e) X. Fan, F. Yuan, D. Li, S. Chen, Z. Cheng, Z. Zhang, S. Xiang, S.-Q. Zang, J. Zhang and L. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12949–12954 CrossRef CAS PubMed; (f) X. Fan, S. Chen, L. Zhang and J. Zhang, Chem. – Eur. J., 2021, 27, 15563–15570 CrossRef CAS PubMed.
- (a) K. Yonesato, H. Ito, H. Itakura, D. Yokogawa, T. Kikuchi, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 19550–19554 CrossRef CAS PubMed; (b) K. Yonesato, H. Ito, D. Yokogawa, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 16361–16365 CrossRef CAS PubMed; (c) K. Yonesato, S. Yamazoe, D. Yokogawa, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2021, 60, 16994–16998 CrossRef CAS PubMed; (d) K. Yonesato, S. Yamazoe, S. Kikkawa, D. Yokogawa, K. Yamaguchi and K. Suzuki, Chem. Sci., 2022, 13, 5557–5561 RSC.
- K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940–947 CrossRef CAS PubMed.
- (a) R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS; (b) S. Sasaki, K. Yonesato, N. Mizuno, K. Yamaguchi and K. Suzuki, Inorg. Chem., 2019, 58, 7722–7729 CrossRef CAS PubMed.
- (a) S. S. Mal and U. Kortz, Angew. Chem., Int. Ed., 2005, 44, 3777–3780 CrossRef CAS PubMed; (b) S. S. Mal, M. H. Dickman, U. Kortz, A. M. Todea, A. Merca, H. Bögge, T. Glaser, A. Müller, S. Nellutla, N. Kaur, J. van Tol, N. S. Dalal, B. Keita and L. Nadjo, Chem. – Eur. J., 2008, 14, 1186–1195 CrossRef CAS PubMed; (c) P. Yang, M. Alsufyani, A.-H. Emwas, C. Chen and N. M. Khashab, Angew. Chem., Int. Ed., 2018, 57, 13046–13051 CrossRef CAS PubMed; (d) Y. Koizumi, K. Yonesato, S. Kikkawa, S. Yamazoe, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 14610–14619 CrossRef CAS PubMed.
- (a) M. Suyama, S. Takano, T. Nakamura and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 14048–14051 CrossRef CAS PubMed; (b) Q. Li, J. Chai, S. Yang, Y. Song, T. Chen, C. Chen, H. Zhang, H. Yu and M. Zhu, Small, 2021, 17, 1907114 CrossRef CAS PubMed; (c) Y. Song, S. Jin, X. Kang, J. Xiang, H. Deng, H. Yu and M. Zhu, Chem. Mater., 2016, 28, 2609–2617 CrossRef CAS.
- (a) R. S. Dhayal, Y.-R. Lin, J.-H. Liao, Y.-J. Chen, Y.-C. Liu, M.-H. Chiang, S. Kahlal, J.-Y. Saillard and C. W. Liu, Chem. – Eur. J., 2016, 22, 9943–9947 CrossRef CAS PubMed; (b) W.-J. Yen, J.-H. Liao, T.-H. Chiu, J.-Y. Chen, Y. J. Chen, S. Kahlal, J.-Y. Saillard and C. W. Liu, Nanoscale, 2024, 16, 7011–7018 RSC; (c) P.-K. Liao, K.-G. Liu, C.-S. Fang, Y.-Y. Wu and C. W. Liu, J. Cluster Sci., 2019, 30, 1185–1193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Tables S1–S7 and Fig. S1–S18. CCDC 2361048. For ESI and crystallographic data in CIF, see DOI: https://doi.org/10.1039/d4nr02547d |
| This journal is © The Royal Society of Chemistry 2024 |