
Ditopic ligand effects on solution structure and redox chemistry in discrete [Cu12S6] clusters with labile Cu–S bonds†
Michael J.
Trenerry
 and
Gwendolyn A.
Bailey
*
and
Gwendolyn A.
Bailey
*
University of Minnesota – Twin Cities, Minneapolis, MN 55455, USA. E-mail: gbailey@umn.edu
First published on 23rd July 2024
Abstract
Copper chalcogenide nanoclusters (Cu–S/Se/Te NCs) are a broad and diverse class of atomically precise nanomaterials that have historically been studied for potential applications in luminescent devices and sensors, and for their beautiful, mineral-like crystal structures. By the “cluster-surface” analogy, Cu–S/Se NCs are prime candidates for the development of nanoscale multimetallic catalysts with atomic precision. However, the majority of studies conducted to date have focused exclusively on their solid-state structures and physical properties, leaving open questions as to their solution stability, dynamics, and reactivity. Herein, we report the first detailed interrogation of solution structure, dynamics, electrochemistry, and decomposition of Cu–S NCs. Specifically, we report the detailed NMR spectroscopy, diffusion-ordered spectroscopy, MALDI mass spectrometry, electrochemical and stoichiometric redox reactivity studies, and DFT studies of a series of [Cu12S6] clusters with labile Cu–S bonds supported by monodentate phosphines and ditopic bis(diphenylphosphino)alkane ligands PPh2R (R = Et, –(CH2)5–, –(CH2)8–). We find that the ligand binding topology dictates the extent of speciation in solution, with complete stability being afforded by the longer octane chelate in dppo (1,8-bis(diphenylphosphino)octane) according to 1H and DOSY NMR and MALDI-MS studies. Furthermore, a combined electrochemical and computational investigation of [Cu12S6(dppo)4] reveals that the intact [Cu12S6] core undergoes a quasireversible one-electron oxidation at mild applied potentials ([Cu12S6]0/+: −0.50 V vs. Fc0/+). In contrast, prolonged air exposure or treatment with chemical oxidants results in cluster degradation with S atom extrusion as phosphine sulfide byproducts. This work adds critical new dimensions to the stabilization and study of atomically precise metal chalcogenide NCs with labile M–S/Se bonds, and demonstrates both progress and challenges in controlling the solution behaviour and redox chemistry of phosphine-supported copper chalcogenide nanoclusters.
Introduction
Base metal sulfides are some of Nature's most ancient and ubiquitous catalysts capable of cooperatively activating and converting small, traditionally inert molecules into valuable products.1–5 Understanding and controlling the interaction of small molecules at the surface is important for tailoring the reactivity of a system, e.g. towards improved selectivity, turnover, and robustness.6,7 Unfortunately, the factors governing activity and selectivity are often elusive and complex, sometimes varying according to subtle differences in material preparation, crystallinity, or composition.8,9In this context, metal chalcogenide nanoclusters (NCs) have shown significant promise for development of earth-abundant, multimetallic catalyst systems with atomistic control.10–12 These NCs capture the essential features of bulk metal chalcogenides, including their highly delocalized electronic structures and flexible configuration of multimetallic active sites.13–25 Additionally, their discrete, atomically precise structures confer some distinct advantages. First, NCs feature well-defined arrangements of surface ligands that can be tuned to manipulate active site steric environments, secondary coordination sphere interactions, and NC electronic character. Additionally, the configuration and number of core atoms can be known precisely and varied systematically to improve catalyst performance.9,26–29 Finally, NCs incorporate a higher density of active surface sites, leading to higher turnover frequencies relative to analogous surfaces. For example, Besenbacher and coworkers showed that [Mo3S13]2− NCs supported on highly ordered pyrolytic graphite catalyzed the hydrogen evolution reaction with roughly an order of magnitude greater turnover frequency than either amorphous or nanocrystalline MoS2.9 Similarly, Zang and coworkers evaluated the electrocatalytic CO2 reduction activity of a thiolate-supported ditetrahedron-shaped Cu8 cluster, finding that the Faradaic efficiencies for generation of formate exceeded that of an isomeric cube-shaped cluster by up to 1.5-fold.28 In related studies, the photocatalytic activity of thiopyrimidine-supported Cu6 clusters for CO2 reduction to CO was enhanced by the presence of the thiopyrimidine group, which acted as a proton relay.30 Clearly, there is enormous potential to develop multimetallic catalysts with unprecedented control via changes to the NC core composition, size, morphology, active site structure, ligand configuration, and ligand structure.
While extensive research has focused on understanding and developing catalytically active NCs of M–S/Se where M = Fe, Mo, W, and others,3,9,31–38 examples of Cu–S/Se NCs remain rare.39–42 This is despite recent compelling evidence that S/Se-modified Cu phases are highly active for electrocatalytic CO2 reduction (eCO2R) to high-value liquid products.43–51 According to these reports, the incorporation of chalcogenide improves product selectivity by shutting down the formation of H2 and boosting the production of formate, methane, acetic acid, and C2–C3 alcohols. Still, the origins of product selectivity, including the mechanistic details of catalyst turnover and structural requirements for selective turnover, are under debate.44–46,49–53 Moreover, facile and dynamic changes to the surface structure under catalytic conditions further complicate tuning by modification of catalyst structure.46,49,50,53 Discrete NCs of Cu–S/Se have the potential to offer improved understanding and rational control over multimetallic architectures, and they are accessible in a wide range of core configurations and sizes that rival the structural complexity of the bulk solids.54–56 While such flexibility facilitates the identification of specific, discrete active sites, it also presents a challenge due to the inherent Cu–S/Se bond lability and hence cluster instability.57–60 Indeed, most studies of Cu–S/Se NCs have focused exclusively on their synthesis and solid-state properties, including their crystallographic structures and photoluminescence.54–56,61,62 Better understanding of the solution behavior and redox properties of discrete Cu–S/Se NCs is needed to inform the development of robust molecular congeners of catalytically active bulk surfaces.
To this end, the [Cu12S6] core configuration presents an interesting test case. One of the earliest derivatives, [Cu12S6(PEt3)8] (1·PEt3), was reported to decompose in solution at −20 °C as evidenced by the brown colour of diethyl ether solutions from which violet crystals of 1·PEt3 were isolated.57 In contrast, ditopic bis(diphenylphosphino)alkane ligands such as dppo and dppt (1,8-(bis(diphenylphosphino)octane) and 1,5-bis(diphenylphosphino)pentane, respectively) appear to impart some stability, according to rare electronic absorption and emission studies in THF.58,59 For [Cu12S6(dppo)4] (1·dppo, Fig. 1), bright red phosphorescence was observed with emission maxima centered at 640 and 655 nm (λex = 350 and 455 nm, respectively), consistent with an intact [Cu12S6] core.59 For [Cu12S6(dppt)4] (1·dppt), however, the solution spectrum exhibited an additional emission maximum centered at 750 nm (λex = 425 nm) not present in solid-state spectra, consistent with at least partial speciation. While suggestive, these studies can only confirm the presence of intact clusters and cannot necessarily detect other species that may have formed in solution. Moreover, the details of the solution structure, dynamics, and origin of improved stability for 1·dppo remain unclear.
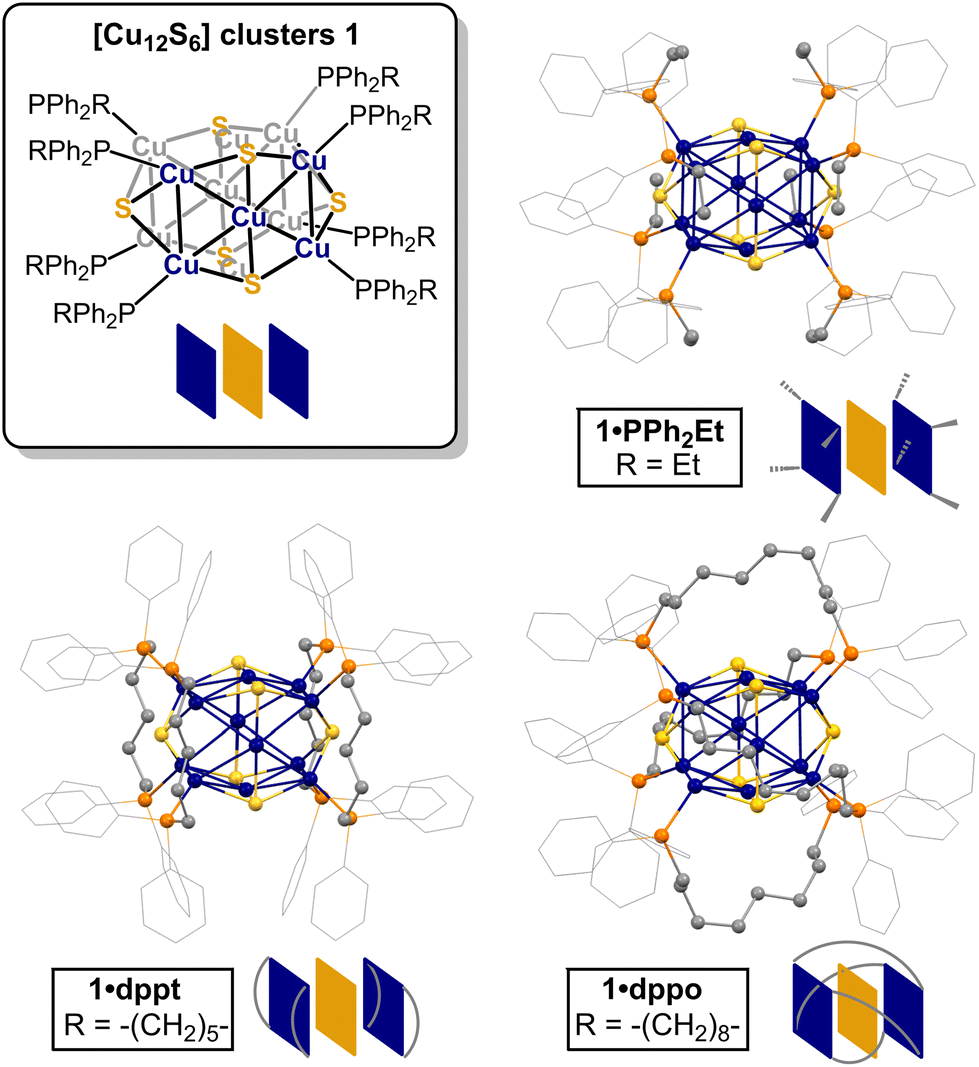 | ||
| Fig. 1 Structures of [Cu12S6] clusters 1 supported by alkyl diphenylphosphine ligands: ball and stick diagrams and cartoon representations of 1·PPh2Et (top right), 1·dppt (bottom left), and 1·dppo (bottom right) showing ditopic ligand binding mode (adapted from reported crystallographic data59,61). Colour scheme: grey (C); orange (P); blue (Cu); yellow (S). H atoms omitted for clarity. PPh2Et: diphenylethylphosphine; dppt: 1,5-bis(diphenylphosphino)pentane; dppo: 1,8-bis(diphenylphosphino)octane. | ||
Herein, we present a systematic study of solution structure, dynamics, and redox properties of [Cu12S6] clusters 1via1H/31P{1H} NMR and diffusion-ordered spectroscopy, MALDI-MS, and electrochemistry. These studies unequivocally confirm that 1·dppo remains fully intact in THF-d8 solutions and reveal previously unrecognized subtleties in the stabilization of [Cu12S6] by dppo, including the retention of a rigid, conformationally locked ligand configuration in solution and the potential role of bridging CH⋯S interactions in stabilizing the ligands in place. In addition, combined electrochemical, computational (DFT), and stoichiometric oxidation studies reveal that 1·dppo can support a chemically reversible [Cu12S6]0/+ redox couple—an important prerequisite for electrocatalytic studies. However, chemical oxidation rapidly triggers intramolecular S atom transfer and elimination of the supporting dppo ligands as phosphine sulfide products (dppoS and/or dppoS2) alongside cluster decomposition. Combined, these studies shed light on the structural vulnerabilities of the [Cu12S6] core and emergent design principles of Cu–S/Se NCs as discrete molecular analogues of some of the leading multimetallic catalysts for eCO2R to valuable products.
Results and discussion
Solution structure of [Cu12S6] clusters 1
According to the crystallographic metrics, the [Cu12S6] core of clusters 1 consists of a stretched S6 octahedron interpenetrated within a Cu12 cuboctahedron, where the cuboctahedron is axially elongated due to phosphine coordination at the eight Cu edge sites.61 The result is an overall prolate core configuration with two axial Cu4S pyramids (blue squares, Fig. 1) separated by a central Cu4S4 equator (yellow squares). In the solid state, dppo and dppt bind to the [Cu12S6] core in one of two preferential coordination modes: in 1·dppo, the longer octane linker bridges diagonally across each Cu5S2 face (avg. dCu–Cu = 5.06 Å), while in 1·dppt, the pentane linker is too short to bridge in this coordination mode and is thus confined to binding adjacent Cu sites within the axial Cu4S pyramid substructures (avg. dCu–Cu = 2.86 Å).59Prior solution characterization of clusters 1 is confined to the room-temperature electronic absorption and photoluminescence excitation/emission spectra for 1·dppt and 1·dppo in THF, as noted above.59 To better understand the unusual stability of 1·dppo, we performed detailed 1H, 31P{1H}, and diffusion-ordered spectroscopy (DOSY) NMR studies in THF-d8. Remarkably, the NMR data for 1·dppo not only confirms that the [Cu12S6] core is conserved in solution but indicates a highly symmetrical, locked ligand configuration. Thus, the 31P{1H} and 1H NMR spectra of 1·dppo reveal a single set of dppo resonances distinct from those of the free ligand, consistent with ligand binding in a C4 symmetry about the cluster core. Notably, each pair of geminal protons within the octyl chain of dppo is split into distinct diastereotopic signals CHaHb (Fig. 2a; bottom), suggesting constrained ligand rotation upon binding. This effect is most pronounced for the α and β methylene protons, which are more rigorously locked into different chemical environments facing towards or away from the [Cu12S6] core.
 | ||
| Fig. 2 Top: (a) 1H NMR spectrum (THF-d8, 400 MHz) of 1·dppo showing a conformationally locked ligand configuration about [Cu12S6] (‡THF solvent, *residual pentane). Ph o/m/p-CH signals belonging to cisoid versus transoid Ph rings are denoted with c and t subscripts, respectively. Bottom: truncated crystallographic structures of 1·dppo showing CH⋯S interactions and representative distances with (b) Ph o-CH; (c) α-CHa. Colour scheme: dark grey (cisoid Ph–C); light grey (transoid Ph–C); orange (P); blue (Cu); yellow (S); white (H). H atoms are omitted except cisoid Ph o-CH and octane α-CHa, which are shown interacting with S. | ||
Likewise, the two Ph rings in each PPh2R group are rendered chemically inequivalent, resulting in two distinct sets of o, m, and p-CH signals (Fig. 2a; top). Of note, one set of o-CH signals is shifted significantly downfield at 8.45 ppm (vs. 7.45 ppm in the free ligand), suggesting that these protons may be directly interacting with the core. However, we see no evidence of close CH⋯Cu interactions in the crystallographic metrics, suggesting that the Cu is too far to participate in an anagostic interaction63 (CH⋯Cu: 3.10–3.27 Å for one set of o-CH). In contrast, the o-CH⋯S contacts between the cisoid Ph pointed towards the cluster and apical Cu4S are short (CH⋯S: 2.78 ± 0.20 Å; ∠142–164°;59Fig. 2b). These distances fall in the lower range of CH⋯S contacts found in the crystal structure database for metal complexes of group 11 and may reflect an attractive CH⋯S interaction (typical CH⋯S: 2.84–3.40 Å; ∠144–180° & 117–135°).64 Short CH⋯S contacts are also observed in the crystal structures of 1·dppt and 1·PPh2Et (1·dppt: 2.92 ± 0.21 Å; 1·PPh2Et: 2.87 ± 0.12 Å), although these clusters do not remain intact in solution (vide infra).
Similarly, one diastereotopic α-CHaHb signal is shifted downfield at 2.71 ppm (vs. 2.02 ppm in free dppo), consistent with weaker but still appreciable CH⋯S interactions with the central sulfides, as reflected in the crystallographic metrics (CH⋯S: 3.18 ± 0.23 Å, ∠117 ± 4°; Fig. 2c). By contrast, the β-CHb signal is shifted substantially upfield at 0.15 ppm (versus 2.02 ppm in the free ligand), suggesting a possible CH⋯Cu agostic interaction. However, close CH⋯Cu contacts were not observed with any of the neighbouring Cu in the crystallographic coordinates (average CH⋯Cu: 3.12 ± 0.18 Å; range 2.77–3.84 Å), perhaps reflecting the impact of crystal packing and/or solvent effects on the interaction of β-CHb with the cluster core.
In comparison, the solution 1H NMR signals for samples of 1·dppt and 1·PPh2Et are broadened and shifted slightly upfield, but otherwise show little change relative to the respective free ligands, consistent with fluxional coordination to electron-rich Cu(I) centers (ω1/2 21.3 Hz and 17.3 Hz, respectively, for the most downfield signal; Fig. 3b). Solution-phase equilibria with smaller clusters and/or monometallic species is confirmed by the 1H DOSY NMR spectra, which reveal significantly larger diffusion coefficients than expected for the intact clusters, indicating a smaller radius on average for these fluxional species (e.g.1·PPh2Et, Fig. 3b: D = 11.0 × 10−10 m2 s−1; rDOSY = 4.3 Å; cf. rXRD = 8.8 Å estimated from the XRD structure; see the ESI† for details). These results contrast the 1H DOSY NMR spectrum of 1·dppo, which exhibits a single uniform and exceptionally small diffusion coefficient for all non-solvent 1H NMR signals in close agreement with the expected value for an intact cluster (Fig. 3a: D = 5.1 × 10−10 m2 s−1; rDOSY = 9.4 Å; cf. rXRD ≈ 9.4 Å).59 Further evidence for the intact [Cu12S6] core was obtained by MALDI-MS analysis, in which samples were dropcast from THF solutions and cocrystallized with the charge-transfer matrix anthracene (base peak: [Cu12S6(dppo)2]˙+m/z 1919.5 Da, expected 1919.4 Da; see Fig. S1 and S2† for details).
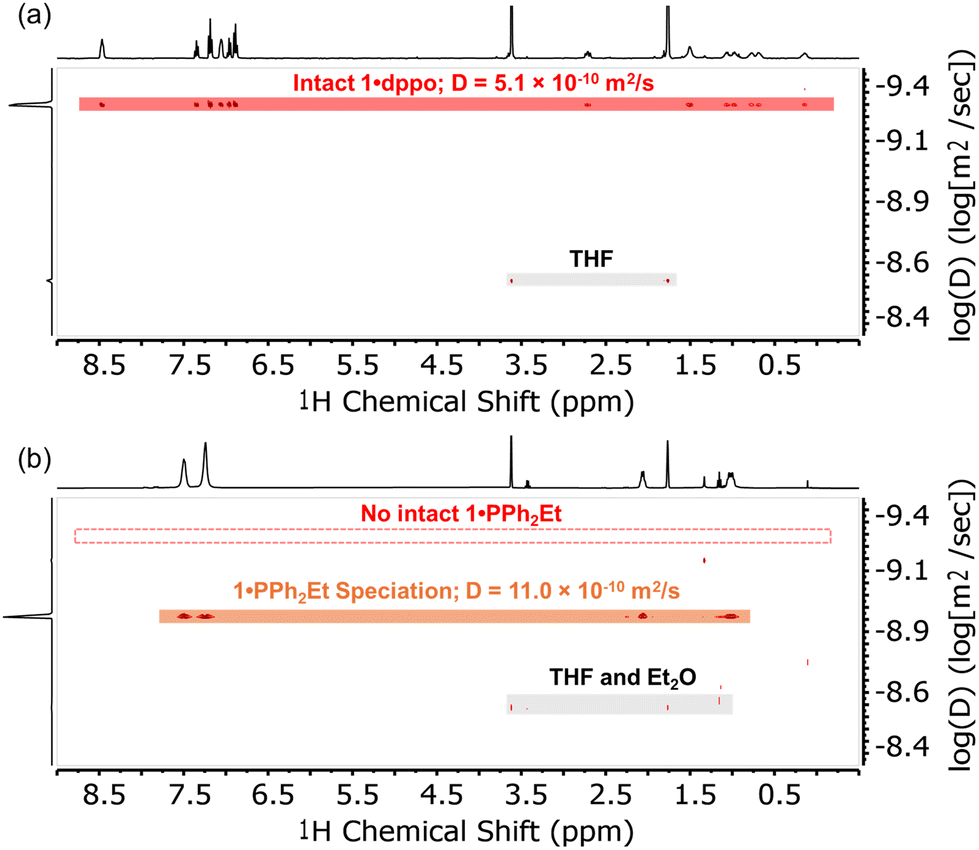 | ||
| Fig. 3 1H DOSY NMR spectra (THF-d8, 400 MHz) of (a) 1·dppo and (b) 1·PPh2Et showing the unique ability of the dppo ligand to arrest speciation of the [Cu12S6] cluster core. Dashed line in (b) indicates the calculated diffusion coefficient for intact 1·PPh2Et based on the crystallographic structure.61 | ||
Combined, these studies shed light on the unique ability of the dppo ligand to stabilize and enforce rigidity of the [Cu12S6] core. Clusters 1 are inherently unstable structures that speciate spontaneously in solution due to the lability of core Cu–S bonds. By contrast, the rigid, conformationally locked ligand configuration in 1·dppo is likely responsible for its improved core stability. The dppo ligand spans diagonally across each [Cu5S2] cluster face of 1·dppo and in doing so, supports the cluster along all 3 axes (Fig. 1 above). This includes stabilizing the weaker Cu–S linkages that extend between the axial Cu and equatorial S, which are notably elongated in the crystallographic coordinates (Cu–S: 2.36–2.40 Å, vs. 2.14–2.27 Å in the apical and equatorial linkages, see ESI† for details).59,61 In contrast, the shorter pentyl linker of dppt permits binding at only adjacent apical Cu sites, leaving the vulnerable Cu–S bonds liable for breakage. These studies emphasize the importance of long, ditopic linkers in buttressing the vulnerable axes of labile Cu–S/Se NCs and ultimately ensuring solution stability. Moreover, they demonstrate that while CH⋯S interactions may play a stabilizing role in locking the dppo ligand in place, they are likely not the primary cause of the stability of 1·dppo.
Solution electrochemistry studies
The distinct stability of 1·dppo presents an opportunity to study the solution-phase electrochemistry of the [Cu12S6] core. Thus, 1·dppo exhibits an irreversible reduction with cathodic peak potential at −3.2 V vs. Fc0/+, suggesting that partial Cu(0) character may be electrochemically accessible though transient in the [Cu12S6] core (red trace, Fig. 4). We also observe a quasireversible redox couple at −0.50 V vs. Fc0/+, which we assign to the one-electron oxidation of the [Cu12S6] core ([1·dppo]0/+; red trace, Fig. 4). Further, irreversible oxidation events occur at potentials more positive than −0.25 V vs. Fc0/+ (dotted pink trace). Scanning beyond −0.25 V also causes the cathodic return wave of the [1·dppo]0/+ redox couple to become diminished, indicating that 1·dppo is highly sensitive to oxidizing conditions and likely degrades following loss of more than one electron. A similar phenomenon was recently reported for the carbene-stabilized Cu4S cluster, [Cu4S(L)2]2+, which is structurally analogous to the apical Cu4S pyramids in 1·dppo (E1/2 [Cu4S(L)2]2+/3+: −0.65 V vs. Fc0/+; L = a tetracarbene ligand).65 The free dppo ligand similarly undergoes irreversible oxidation at mildly anodic potentials (blue trace), with a smaller pre-wave feature beginning at −0.2 V vs. Fc0/+ that aligns with the onset of irreversible oxidations in 1·dppo. However, a sharp increase in current is only observed at potentials of 0.5 V and higher, whereas in 1·dppo the irreversible features are cathodically shifted by ca. 250 mV. We thus conclude that the degradation of 1·dppo ultimately stems from oxidation of the composite cluster, rather than its supporting ligands exclusively, at distinctly milder conditions. | ||
| Fig. 4 Cyclic voltammograms (top) and differential pulse voltammograms (bottom) of 1·dppo (1 mM concentration, red and dotted pink traces) and free dppo ligand (5 mM concentration, blue trace). Collected at 20 mV s−1 in THF with 100 mM tetrabutylammonium hexafluorophosphate ([Bu4N][PF6]) electrolyte. | ||
We next targeted the putative oxidized cluster [1·dppo]+via chemical oxidation using [Cp*2Fe][PF6] (Cp* = pentamethylcyclopentadienyl; E1/2: −0.44 V in THF). [Cp*2Fe]+ was chosen as a mild oxidant to avoid over-oxidation of the [Cu12S6] core and ensuing decomposition. Nevertheless, by 1H and 31P{1H} NMR we observe the complete disappearance of 1·dppo only upon the addition of two equivalents of oxidant (Fig. 5a). This change is accompanied by the growth of new, diagnostic 31P NMR signals at ca. 41.8 and 42.0 ppm and a downfield 1H NMR signal at 7.95 ppm. Assignment of these spectral features as 1,8-bis(diphenylphosphino)octane sulfides (Ph2RP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S: dppoS and/or dppoS2), presumably originating from oxidation of the parent dppo ligand, is confirmed by independent synthesis (see the ESI† for details). A broad 31P NMR signal ranging from −7 to −9 ppm and a 1H NMR multiplet at ∼7.4 ppm also grow in with additional equivalents of oxidant. We assign these signals to remaining dppo ligands bound to the Cu sites of cluster degradation products.
S: dppoS and/or dppoS2), presumably originating from oxidation of the parent dppo ligand, is confirmed by independent synthesis (see the ESI† for details). A broad 31P NMR signal ranging from −7 to −9 ppm and a 1H NMR multiplet at ∼7.4 ppm also grow in with additional equivalents of oxidant. We assign these signals to remaining dppo ligands bound to the Cu sites of cluster degradation products.
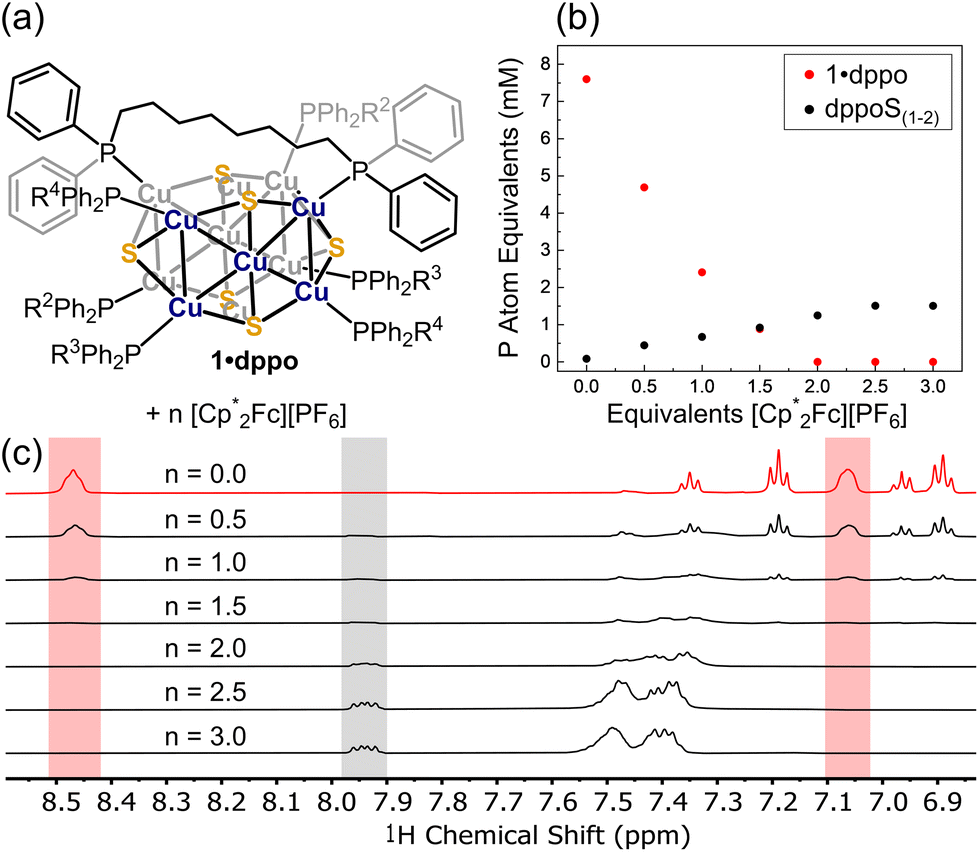 | ||
| Fig. 5 (a) Reaction of 1·dppo with [Cp*2Fe][PF6] (b) concentrations of 1·dppo (red trace) and phosphine sulfide products dppoS1–2 (black trace), expressed in terms of phosphorus atom equivalents, are plotted versus equivalents of [Cp*2Fe][PF6] added. (c) 1H NMR spectra (THF-d8, 400 MHz) collected for 1·dppo with n equivalents of [Cp*2Fe][PF6] added. Diagnostic o-CH proton signals used to construct the plot in part (b) are highlighted for both 1·dppo (red) and phosphine sulfides dppoS1–2 (grey). | ||
By quantitative 1H NMR measurements (Fig. 5b), we find that the oxidation of 1·dppo with two equiv. [Cp*2Fe]+ yields ca. 1 phosphine sulfide group per starting cluster. Importantly, the conversion of phosphine to phosphine sulfide is a two-electron oxidation and is thus consistent with the complete disappearance of 1·dppo after the addition of two equivalents of oxidant, as well as the electrochemical measurements above indicating loss of reversibility following oxidation by more than one electron. Overoxidation in the presence of the comparatively mild oxidant [Cp*2Fe]+ likely reflects spontaneous disproportionation of the putative one-electron oxidized species [1·dppo]+. Minor growth of phosphine sulfide continues following the addition of >2 equivalents of [Cp*2Fe][PF6], suggesting a reaction between the remaining sulfides of cluster decomposition products and residual dppo and/or dppoS. Interestingly, phosphine sulfide is also observed as a byproduct following air oxidation of solution samples of 1·dppo, indicating that this decomposition pathway is also active under aerobic conditions. 1H DOSY analysis of the air-oxidized species reveals that the products formed on air oxidation correspond to species that are smaller than 1·dppo itself (Fig. S21 and S22†).
Electronic structure calculations
Geometry optimization and electronic structure calculations were carried out using density functional theory (DFT) to probe the nature of redox events and degradation under oxidizing conditions. We performed these calculations on the model species [1′·PPh2Et]0/+/2+ in which the dppo ligands are truncated as separate PPh2Et ligands for the sake of computational cost. Importantly, the relative orientations of phosphines are preserved from 1·dppo, making the structure of 1′·PPh2Et distinct from 1·PPh2Et. The highest occupied molecular orbitals of 1′·PPh2Et span a narrow energetic window and primarily depict antibonding interactions between Cu sites and neighboring atoms (Fig. 6). These orbitals are relatively delocalized across the [Cu12S6] core but are best categorized as having predominantly Cu–P σ*, equatorial S3p, and apical S3p character. Importantly, the HOMO and HOMO−1 of 1′·PPh2Et comprise a pseudo-degenerate pair featuring prominent Cu–P σ* interactions between apical Cu sites and binding phosphines, as well as minor Cu–Cu σ* interactions between apical and equatorial Cu sites. Minor electron density is also placed on apical sulfides, which are out of phase with neighboring Cu and P sites.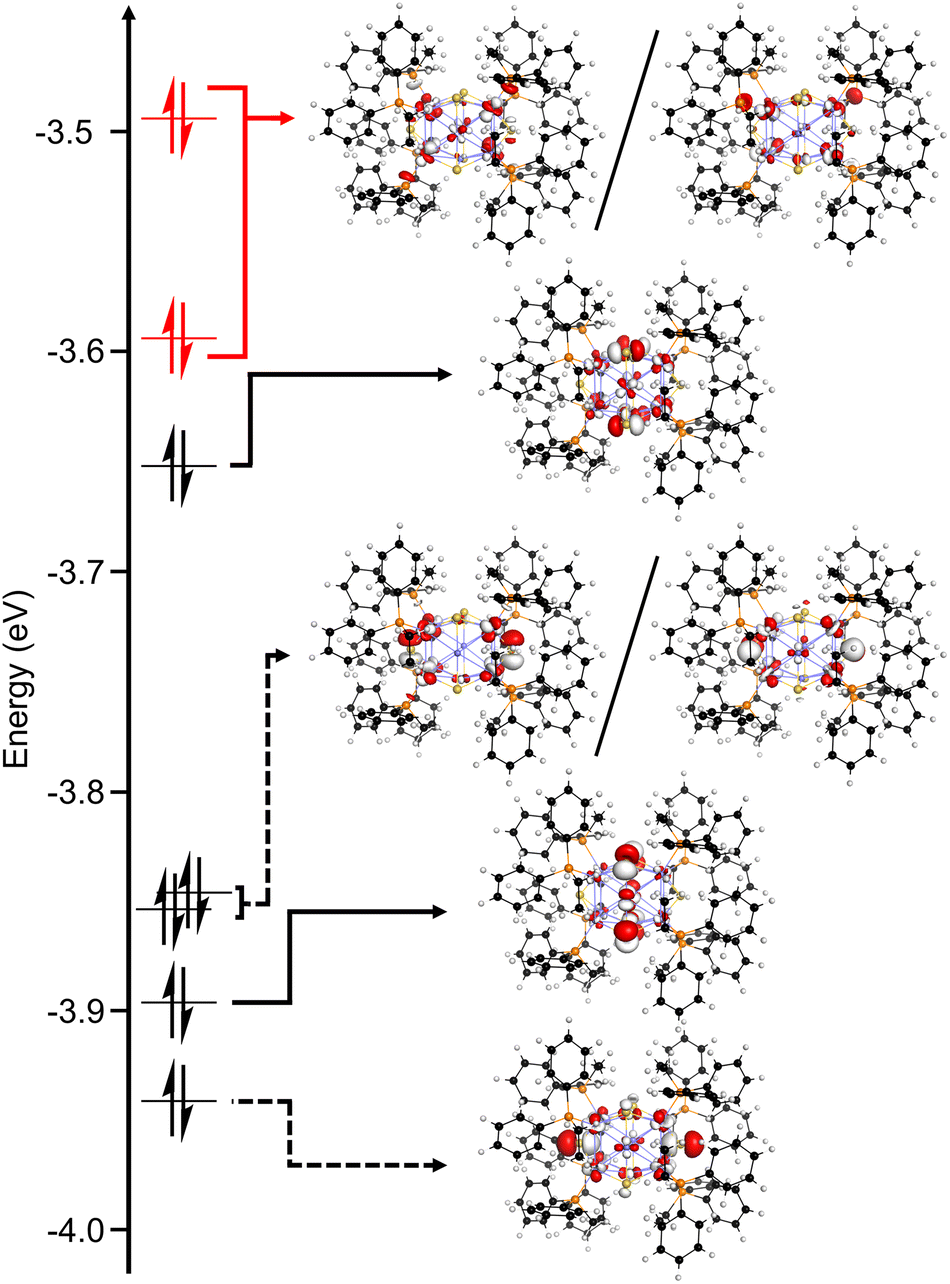 | ||
| Fig. 6 Calculated molecular orbital diagram (DFT: BP86/def2-SVP) for the truncated model complex [1′·PPh2Et] showing the highest occupied molecular orbitals. | ||
Electronic structure calculations of the oxidized clusters [1′·PPh2Et]+ and [1′·PPh2Et]2+ reveal broadly similar trends, with predominantly Cu–P and Cu–Cu σ* character in the frontier orbitals. Accordingly, oxidation constricts the [Cu12S6] core in [1′·PPh2Et]+ and [1′·PPh2Et]2+, giving overall shorter Cu–Cu and Cu–S bond lengths and conferring substantial partial positive charge to the P atoms (Table S6†). Collectively, these effects prime the phosphines for interaction with nearby bridging sulfides, which we infer to be strongly nucleophilic based on the prominent S3p character of the next highest energy-occupied molecular orbitals (e.g., HOMO−1 and HOMO−2 of [1′·PPh2Et]2+; Fig. 7). This results in facile nucleophilic attack of a nearby sulfide, culminating in net loss of sulfur and phosphine as a phosphine sulfide and decomposition of the sulfur-deficient [Cu12S5]2+ core.
 | ||
| Fig. 7 (a) Proposed pathway for decomposition of [Cu12S6] via oxidative PS bond formation. (b) Calculated HOMO−1 and HOMO−2 (DFT: BP86/def2-SVP) of [1′·PPh2Et]2+ showing significant S3p orbital character on nearby bridging sulfides. | ||
The oxidative decomposition behavior of 1·dppo is reminiscent of a similar pathway observed for the phosphine-supported copper hydride cluster [(PPh3)6Cu6H6], which also exhibits a reversible one-electron oxidation (E1/2 = 1.01 V vs. Fc0/+) and an irreversible second oxidation event (Epa = −0.57 V vs. Fc0/+).66 Treatment of [(PPh3)6Cu6H6] with two equivalents of [Cp2*Fe]+ results in net hydride loss to yield [(PPh3)6Cu6H5]+, presumably via deprotonation of the putative dicationic intermediate [(PPh3)6Cu6H6]2+.67 In both [(PPh3)6Cu6H6] and 1·dppo, structural changes to the cluster core occur only following two-electron oxidation and ensuing group transfer, either of a sulfur atom or a proton. These results are also in line with recent studies of the highly oxidized, all-ferric iron–sulfur clusters [Fe4S4(SAr)4]0, which to date have been only isolated in the presence of bulky thiolate ligands that presumably protect the core from decomposition via bimolecular pathways (e.g., Ar = 2,6-bis(mesityl)phenyl; E1/2 [Fe4S4(SAr)4]1−/0: −0.19 V vs. Fc0/+).68,69 Thus, the oxidative sensitivity of metal–sulfur clusters is likely a more general phenomenon that necessitates judicious application of ligands that modulate the electronic structure towards greater inherent robustness of oxidized states. For example, Mankad and coworkers have employed electron-rich, ditopic formamidinate and triazenide ligands to support reduction and disproportionation of N2O by [Cu4S] clusters via a [Cu4S]3+/4+ redox couple (E1/2 = −1.28 V vs. Fc0/+).23,40,42 [Cu4S]2+ clusters supported by ditopic bis(diphenylphosphino)amine ligands also promote reduction of N2O to N2, but require a protic methanol solvent for activation.41 Similar strategies are likely to be effective for higher nuclearity copper–sulfur clusters such as [Cu12S6] and are currently being explored in our laboratories.
Conclusions
We report herein the first detailed study of solution structure, dynamics, and electrochemistry of [Cu12S6] NCs with varied monotopic and ditopic supporting phosphine (PPh2R) ligands. Importantly, these clusters bear labile Cu–S bonds and thus present a stringent test case for the ability of ditopic ligands to stabilize vulnerable cluster configurations. Our studies rigorously demonstrate that the longer octyl linker in [Cu12S6(dppo)4] 1·dppo results in striking robustness as demonstrated by solution 1H, 31P{1H}, and DOSY NMR studies in THF-d8, in contrast to shorter linkers or monotopic phosphines in other structurally related clusters. In addition, while the 1H NMR and crystallographic data indicate the presence of relatively strong CH⋯S interactions in 1·dppo, these interactions do not appear to be solely responsible for the unique stability of 1·dppo and instead may assist by enforcing a rigid ligand conformation about [Cu12S6]. These results are important considering recent evidence supporting the significance of CH⋯SR interactions (R = alkyl, aryl; S = monovalent or divalent sulfur) in controlling the behaviour of enzymes, metal complexes, and supramolecular assemblies.64 In contrast, the role of CH⋯S interactions in stabilizing cluster assemblies bearing bridging S2− ligands has been studied in far less detail.64Moreover, our in-depth electrochemical investigations have demonstrated the [Cu12S6] core of 1·dppo to be redox-active, exhibiting a quasireversible [Cu12S6]0/+ redox couple at mildly oxidizing potentials (−0.50 V vs. Fc0/+). Unfortunately, attempts at isolating this putative oxidized species resulted in facile S atom transfer and elimination of dppo supporting ligands as phosphine sulfides (Ph2RPS) with ultimate decomposition of the cluster. Collectively, these results underscore both challenges and opportunities in leveraging Cu–S/Se NCs for electrocatalytic studies. First, labile ligands such as phosphines must be reinforced by multitopic binding, ideally across multiple axes of the core. Second, while one- and two-electron oxidations are electrochemically accessible at mild potentials, the oxidized clusters are unstable entities that decompose via extrusion of Ph2RPS. This inherent limitation of phosphine-supported Cu–S/Se NCs highlights the importance of new cluster discovery towards extending truly functional, catalytic behavior to the atomically precise nanoscale. Specifically, there is a need to explore alternative, tailor-designed ligand sets that resist both speciation and decomposition via S-atom extrusion by leveraging the effects of specific ditopic ligand coordination modes explored in this work in combination with other features, such as stronger ligand binding to Cu sites and local steric environments that favor cluster core morphologies with different arrangements of bridging sulfides.
Author contributions
M. J. T. and G. A. B. conceived and planned the experiments. M. J. T. performed the synthesis of all compounds, collected experimental data, and performed DFT calculations. M. J. T. and G. A. B. jointly wrote the manuscript.Data availability
Data for this article, including 1H, 31P{1H}, and DOSY NMR spectra, MALDI mass spectra, electrochemical data, powder X-ray diffraction data, computational details, and Cartesian coordinates of DFT-optimized structures, have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by startup funds through the University of Minnesota – Twin Cities. Powder X-ray diffraction experiments were carried out in the Characterization Facility at the University of Minnesota, which receives partial support from the NSF through the MRSEC (#DMR-2011401) and the NNCI (#ECCS-2025124) programs. NMR and MS experiments were carried out in the LeClaire–Dow Instrumentation Facility at UMN.References
- C. Huber and G. Wächtershäuser, Activated Acetic Acid by Carbon Fixation on (Fe,Ni)S Under Primordial Conditions, Science, 1997, 276(5310), 245–247, DOI:10.1126/science.276.5310.245.
- C. Huber and G. Wächtershäuser, Peptides by Activation of Amino Acids with CO on (Ni,Fe)S Surfaces: Implications for the Origin of Life, Science, 1998, 281(5377), 670–672, DOI:10.1126/science.281.5377.670.
- K. Tanifuji and Y. Ohki, Metal–Sulfur Compounds in N2 Reduction and Nitrogenase-Related Chemistry, Chem. Rev., 2020, 120(12), 5194–5251, DOI:10.1021/acs.chemrev.9b00544.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper Active Sites in Biology, Chem. Rev., 2014, 114(7), 3659–3853, DOI:10.1021/cr400327t.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase, Chem. Rev., 2014, 114(8), 4149–4174, DOI:10.1021/cr400461p.
- J. T. Perryman and J. M. Velázquez, Design Principles for Multinary Metal Chalcogenides: Toward Programmable Reactivity in Energy Conversion, Chem. Mater., 2021, 33(18), 7133–7147, DOI:10.1021/acs.chemmater.1c01983.
- M. Nath, H. Singh and A. Saxena, Progress of Transition Metal Chalcogenides as Efficient Electrocatalysts for Energy Conversion, Curr. Opin. Electrochem., 2022, 34, 100993, DOI:10.1016/j.coelec.2022.100993.
- M. A. Reynolds, Minireview on the Evolution of Tetrathiometallate Salts as Protean Building Blocks of Catalysts and Materials for the Energy Transition: Recent Advances and Future Perspectives, Energy Fuels, 2021, 35(19), 15285–15300, DOI:10.1021/acs.energyfuels.1c02039.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Building an Appropriate Active-Site Motif into a Hydrogen-Evolution Catalyst with Thiomolybdate [Mo3S13]2− Clusters, Nat. Chem., 2014, 6(3), 248–253, DOI:10.1038/nchem.1853.
- C. Dong, S. Nematulloev, P. Yuan and O. M. Bakr, Atomically Precise Copper Nanoclusters, in Atomically Precise Nanochemistry, 2023, pp. 257–283. DOI:10.1002/9781119788676.ch8.
- X. Liu and D. Astruc, Atomically Precise Copper Nanoclusters and Their Applications, Coord. Chem. Rev., 2018, 359, 112–126, DOI:10.1016/j.ccr.2018.01.001.
- T. W. Hayton, Atomically Precise Nanoclusters of Iron, Cobalt, and Nickel, in Atomically Precise Nanochemistry, 2023, pp. 285–307. DOI:10.1002/9781119788676.ch9.
- R. Maity, B. S. Birenheide, F. Breher and B. Sarkar, Cooperative Effects in Multimetallic Complexes Applied in Catalysis, ChemCatChem, 2021, 13(10), 2337–2370, DOI:10.1002/cctc.202001951.
- T. Jia, Y.-X. Li, X.-H. Ma, M.-M. Zhang, X.-Y. Dong, J. Ai and S.-Q. Zang, Atomically Precise Ultrasmall Copper Cluster for Room-Temperature Highly Regioselective Dehydrogenative Coupling, Nat. Commun., 2023, 14(1), 6877, DOI:10.1038/s41467-023-42688-3.
- A. W. Beamer and J. A. Buss, Synthesis, Structural Characterization, and, CO2 Reactivity of a Constitutionally Analogous Series of Tricopper Mono-, Di-, and Trihydrides, J. Am. Chem. Soc., 2023, 145(23), 12911–12919, DOI:10.1021/jacs.3c04170.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, An Organometallic Cu20 Nanocluster: Synthesis, Characterization, Immobilization on Silica, and “Click” Chemistry, J. Am. Chem. Soc., 2018, 140(1), 394–400, DOI:10.1021/jacs.7b10960.
- B. S. Mitchell, A. Chirila, J. A. Kephart, A. C. Boggiano, S. M. Krajewski, D. Rogers, W. Kaminsky and A. Velian, Metal–Support Interactions in Molecular Single-Site Cluster Catalysts, J. Am. Chem. Soc., 2022, 144(40), 18459–18469, DOI:10.1021/jacs.2c07033.
- L. N. V. Le, J. P. Joyce, P. H. Oyala, S. DeBeer and T. Agapie, Highly Activated Terminal Carbon Monoxide Ligand in an Iron–Sulfur Cluster Model of FeMco with Intermediate Local Spin State at Fe, J. Am. Chem. Soc., 2024, 146(8), 5045–5050, DOI:10.1021/jacs.3c12025.
- A. G. Scott, D. Alves Galico, I. Bogacz, P. H. Oyala, J. Yano, E. A. Suturina, M. Murugesu and T. Agapie, High-Spin and Reactive Fe13 Cluster with Exposed Metal Sites, Angew. Chem., Int. Ed., 2023, 62(49), e202313880, DOI:10.1002/anie.202313880.
- C. E. Juda, R. Handford, A. K. Bartholomew, T. Powers, N. X. Gu, E. Meyers, N. Roth, Y.-S. Chen, S.-L. Zheng and T. Betley, Cluster Dynamics of Heterometallic Trinuclear Clusters during Ligand Substitution, Redox Chemistry, and Group Transfer Processes, Chem. Sci., 2024, 15, 8242–8248, 10.1039/D3SC03606E.
- A. A. Fertig, W. W. Brennessel, J. R. McKone and E. M. Matson, Concerted Multiproton–Multielectron Transfer for the Reduction of O2 to H2O with a Polyoxovanadate Cluster, J. Am. Chem. Soc., 2021, 143(38), 15756–15768, DOI:10.1021/jacs.1c07076.
- A. C. Brown, N. B. Thompson and D. L. M. Suess, Evidence for Low-Valent Electronic Configurations in Iron–Sulfur Clusters, J. Am. Chem. Soc., 2022, 144(20), 9066–9073, DOI:10.1021/jacs.2c01872.
- S. C. Rathnayaka, S. M. Islam, I. M. DiMucci, S. N. MacMillan, K. M. Lancaster and N. P. Mankad, Probing the Electronic and Mechanistic Roles of the u4-Sulfur Atom in a Synthetic CuZ Model System, Chem. Sci., 2020, 11(13), 3441–3447, 10.1039/C9SC06251C.
- S. Pattanayak and L. A. Berben, Pre-Equilibrium Reaction Mechanism as a Strategy to Enhance Rate and Lower Overpotential in Electrocatalysis, J. Am. Chem. Soc., 2023, 145(6), 3419–3426, DOI:10.1021/jacs.2c10942.
- M. C. Eaton, V. J. Catalano, J. Shearer and L. J. Murray, Dinitrogen Insertion and Cleavage by a Metal–Metal Bonded Tricobalt(I) Cluster, J. Am. Chem. Soc., 2021, 143(15), 5649–5653, DOI:10.1021/jacs.1c01840.
- S. Zhao, N. Austin, M. Li, Y. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. Jin, Influence of Atomic-Level Morphology on Catalysis: The Case of Sphere and Rod-Like Gold Nanoclusters for CO2 Electroreduction, ACS Catal., 2018, 8(6), 4996–5001, DOI:10.1021/acscatal.8b00365.
- S. Li, X. Du, Z. Liu, Y. Li, Y. Shao and R. Jin, Size Effects of Atomically Precise Gold Nanoclusters in Catalysis, Precis. Chem., 2023, 1(1), 14–28, DOI:10.1021/prechem.3c00008.
- L.-J. Liu, Z.-Y. Wang, Z.-Y. Wang, R. Wang, S.-Q. Zang and T. C. W. Mak, Mediating CO2 Electroreduction Activity and Selectivity over Atomically Precise Copper Clusters, Angew. Chem., Int. Ed., 2022, 61(35), e202205626, DOI:10.1002/anie.202205626.
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Dimeric [Mo2S12]2− Cluster: A Molecular Analogue of MoS2 Edges for Superior Hydrogen-Evolution Electrocatalysis, Angew. Chem., Int. Ed., 2015, 54(50), 15181–15185, DOI:10.1002/anie.201507529.
- J.-P. Dong, Y. Xu, X.-G. Zhang, H. Zhang, L. Yao, R. Wang and S.-Q. Zang, Copper-Sulfur-Nitrogen Cluster Providing a Local Proton for Efficient Carbon Dioxide Photoreduction, Angew. Chem., Int. Ed., 2023, 62(48), e202313648, DOI:10.1002/anie.202313648.
- M. R. DuBois, Catalytic Applications of Transition-Metal Complexes with Sulfide Ligands, Chem. Rev., 1989, 89(1), 1–9, DOI:10.1021/cr00091a001.
- S. C. Lee, W. Lo and R. H. Holm, Developments in the Biomimetic Chemistry of Cubane-Type and Higher Nuclearity Iron–Sulfur Clusters, Chem. Rev., 2014, 114(7), 3579–3600, DOI:10.1021/cr4004067.
- Y. Ohki, K. Uchida, M. Tada, R. E. Cramer, T. Ogura and T. Ohta, N2 Activation on a Molybdenum–Titanium–Sulfur Cluster, Nat. Commun., 2018, 9(1), 3200, DOI:10.1038/s41467-018-05630-6.
- J. A. Kephart, B. S. Mitchell, A. Chirila, K. J. Anderton, D. Rogers, W. Kaminsky and A. Velian, Atomically Defined Nanopropeller Fe3Co6Se8(Ph2PNTol)6: Functional Model for the Electronic Metal–Support Interaction Effect and High Catalytic Activity for Carbodiimide Formation, J. Am. Chem. Soc., 2019, 141(50), 19605–19610, DOI:10.1021/jacs.9b12473.
- A. L. Gushchin, Y. A. Laricheva, M. N. Sokolov and R. Llusar, Tri- and Tetranuclear Molybdenum and Tungsten Chalcogenide Clusters: On the Way to New Materials and Catalysts *, Russ. Chem. Rev., 2018, 87(7), 670, DOI:10.1070/RCR4800.
- S. Shitaya, K. Nomura and A. Inagaki, Light-Driven Catalytic Hydrogenation of Carbon Dioxide at Low-Pressure by a Trinuclear Iridium Polyhydride Complex, Chem. Commun., 2019, 55(35), 5087–5090, 10.1039/C9CC00916G.
- Y. Ohki, K. Munakata, Y. Matsuoka, R. Hara, M. Kachi, K. Uchida, M. Tada, R. E. Cramer, W. M. C. Sameera, T. Takayama, Y. Sakai, S. Kuriyama, Y. Nishibayashi and K. Tanifuji, Nitrogen Reduction by the Fe Sites of Synthetic [Mo3S4Fe] Cubes, Nature, 2022, 607(7917), 86–90, DOI:10.1038/s41586-022-04848-1.
- K. Patra, W. W. Brennessel and E. M. Matson, Molybdenum Sulphide Clusters as Redox-Active Supports for Low-Valent Uranium, Chem. Commun., 2024, 60(5), 530–533, 10.1039/D3CC05561B.
- M. V. L. Ocampo and L. J. Murray, Metal-Tuned Ligand Reactivity Enables CX2 (X = O, S) Homocoupling with Spectator Cu Centers, J. Am. Chem. Soc., 2024, 146(1), 1019–1025, DOI:10.1021/jacs.3c11928.
- B. J. Johnson, W. E. Antholine, S. V. Lindeman, M. J. Graham and N. P. Mankad, A One-Hole Cu4S Cluster with N2O Reductase Activity: A Structural and Functional Model for CuZ*, J. Am. Chem. Soc., 2016, 138(40), 13107–13110, DOI:10.1021/jacs.6b05480.
- C.-W. Hsu, S. C. Rathnayaka, S. M. Islam, S. N. MacMillan and N. P. Mankad, N2O Reductase Activity of a [Cu4S] Cluster in the 4CuI Redox State Modulated by Hydrogen Bond Donors and Proton Relays in the Secondary Coordination Sphere, Angew. Chem., Int. Ed., 2020, 59(2), 627–631, DOI:10.1002/anie.201906327.
- N. P. Mankad, Triazenide-Supported [Cu4S] Structural Mimics of CuZ That Mediate N2O Disproportionation Rather than Reduction, Chem. Sci., 2024, 15(5), 1820–1828, 10.1039/D3SC05451A.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Recent Advances in the Utilization of Copper Sulfide Compounds for Electrochemical CO2 Reduction, Nano Mater. Sci., 2020, 2(3), 235–247, DOI:10.1016/j.nanoms.2019.10.006.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Steering Post-C–C Coupling Selectivity Enables High Efficiency Electroreduction of Carbon Dioxide to Multi-Carbon Alcohols, Nat. Catal., 2018, 1(6), 421–428, DOI:10.1038/s41929-018-0084-7.
- A. Saxena, W. Liyanage, J. Masud, S. Kapila and M. Nath, Selective Electroreduction of CO2 to Carbon-Rich Products with a Simple Binary Copper Selenide Electrocatalyst, J. Mater. Chem. A, 2021, 9(11), 7150–7161, 10.1039/D0TA11518E.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, Sulfur-Modified Copper Catalysts for the Electrochemical Reduction of Carbon Dioxide to Formate, ACS Catal., 2018, 8(2), 837–844, DOI:10.1021/acscatal.7b03161.
- P. Kar, S. Farsinezhad, X. Zhang and K. Shankar, Anodic Cu2S and CuS Nanorod and Nanowall Arrays: Preparation, Properties and Application in CO2 Photoreduction, Nanoscale, 2014, 6(23), 14305–14318, 10.1039/C4NR05371K.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, Efficient and Stable Electroreduction of CO2 to CH4 on CuS Nanosheet Arrays, J. Mater. Chem. A, 2017, 5(38), 20239–20243, 10.1039/C7TA05507B.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, Sulfide-Derived Copper for Electrochemical Conversion of CO2 to Formic Acid, J. Phys. Chem. Lett., 2018, 9(15), 4407–4412, DOI:10.1021/acs.jpclett.8b01601.
- Y. Huang, Y. Deng, A. D. Handoko, G. K. L. Goh and B. S. Yeo, Rational Design of Sulfur-Doped Copper Catalysts for the Selective Electroreduction of Carbon Dioxide to Formate, ChemSusChem, 2018, 11(1), 320–326, DOI:10.1002/cssc.201701314.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Double Sulfur Vacancies by Lithium Tuning Enhance CO2 Electroreduction to n-Propanol, Nat. Commun., 2021, 12(1), 1580, DOI:10.1038/s41467-021-21901-1.
- Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, On the Role of Sulfur for the Selective Electrochemical Reduction of CO2 to Formate on CuSx Catalysts, ACS Appl. Mater. Interfaces, 2018, 10(34), 28572–28581, DOI:10.1021/acsami.8b08428.
- R. García-Muelas, F. Dattila, T. Shinagawa, A. J. Martín, J. Pérez-Ramírez and N. López, Origin of the Selective Electroreduction of Carbon Dioxide to Formate by Chalcogen Modified Copper, J. Phys. Chem. Lett., 2018, 9(24), 7153–7159, DOI:10.1021/acs.jpclett.8b03212.
- S. Dehnen, A. Eichhöfer and D. Fenske, Chalcogen-Bridged Copper Clusters, Eur. J. Inorg. Chem., 2002, 2002(2), 279–317, DOI:10.1002/1099-0682(20022)2002:2<279::AID-EJIC279>3.0.CO;2-H.
- O. Fuhr, S. Dehnen and D. Fenske, Chalcogenide Clusters of Copper and Silver from Silylated Chalcogenide Sources, Chem. Soc. Rev., 2013, 42(4), 1871–1906, 10.1039/C2CS35252D.
- J. F. Corrigan, O. Fuhr and D. Fenske, Metal Chalcogenide Clusters on the Border between Molecules and Materials, Adv. Mater., 2009, 21(18), 1867–1871, DOI:10.1002/adma.200802897.
- S. Dehnen, A. Schäfer, D. Fenske and R. Ahlrichs, New Sulfur- and Selenium-Bridged Copper Clusters; Ab Initio Calculations on [Cu2nSen(PH3)m] Clusters, Angew. Chem., Int. Ed. Engl., 1994, 33(7), 746–749, DOI:10.1002/anie.199407461.
- C. B. Khadka, B. K. Najafabadi, M. Hesari, M. S. Workentin and J. F. Corrigan, Copper Chalcogenide Clusters Stabilized with Ferrocene-Based Diphosphine Ligands, Inorg. Chem., 2013, 52(12), 6798–6805, DOI:10.1021/ic3021854.
- X.-X. Yang, I. Issac, S. Lebedkin, M. Kühn, F. Weigend, D. Fenske, O. Fuhr and A. Eichhöfer, Red-Luminescent Biphosphine Stabilized ‘Cu12S6’ Cluster Molecules, Chem. Commun., 2014, 50(75), 11043–11045, 10.1039/C4CC04702H.
- A. M. Polgar, F. Weigend, A. Zhang, M. J. Stillman and J. F. Corrigan, A N-Heterocyclic Carbene-Stabilized Coinage Metal-Chalcogenide Framework with Tunable Optical Properties, J. Am. Chem. Soc., 2017, 139(40), 14045–14048, DOI:10.1021/jacs.7b09025.
- A. Eichhöfer, G. Buth, S. Lebedkin, M. Kühn and F. Weigend, Luminescence in Phosphine-Stabilized Copper Chalcogenide Cluster Molecules—A Comparative Study, Inorg. Chem., 2015, 54(19), 9413–9422, DOI:10.1021/acs.inorgchem.5b01146.
- O. Veselska and A. Demessence, d10 Coinage Metal Organic Chalcogenolates: From Oligomers to Coordination Polymers, Coord. Chem. Rev., 2018, 355, 240–270, DOI:10.1016/j.ccr.2017.08.014.
- M. Brookhart, M. L. H. Green and G. Parkin, Agostic Interactions in Transition Metal Compounds, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(17), 6908–6914, DOI:10.1073/pnas.0610747104.
- H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, C–H⋯S Hydrogen Bonding Interactions, Chem. Soc. Rev., 2022, 51(4), 1454–1469, 10.1039/D1CS00838B.
- Y. Liu, S. Chatterjee, G. E. Cutsail III, S. Peredkov, S. K. Gupta, S. Dechert, S. DeBeer and F. Meyer, Cu4S Cluster in “0-Hole” and “1-Hole” States: Geometric and Electronic Structure Variations for the Active CuZ* Site of N2O Reductase, J. Am. Chem. Soc., 2023, 145, 18477–18486, DOI:10.1021/jacs.3c04893.
- M. S. Eberhart, J. R. Norton, A. Zuzek, W. Sattler and S. Ruccolo, Electron Transfer from Hexameric Copper Hydrides, J. Am. Chem. Soc., 2013, 135(46), 17262–17265, DOI:10.1021/ja408925m.
- S. Liu, M. S. Eberhart, J. R. Norton, X. Yin, M. C. Neary and D. W. Paley, Cationic Copper Hydride Clusters Arising from Oxidation of (Ph3P)6Cu6H6, J. Am. Chem. Soc., 2017, 139(23), 7685–7688, DOI:10.1021/jacs.7b02183.
- L. Grunwald, M. Clémancey, D. Klose, L. Dubois, S. Gambarelli, G. Jeschke, M. Wörle, G. Blondin and V. Mougel, A Complete Biomimetic Iron-Sulfur Cubane Redox Series, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(31), e2122677119, DOI:10.1073/pnas.2122677119.
- G. Moula, T. Matsumoto, M. E. Miehlich, K. Meyer and K. Tatsumi, Synthesis of an All-Ferric Cuboidal Iron–Sulfur Cluster [Fe4S4(SAr)4], Angew. Chem., Int. Ed., 2018, 57(36), 11594–11597, DOI:10.1002/anie.201803679.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02615b |
| This journal is © The Royal Society of Chemistry 2024 |