
Atomically precise alkynyl-protected Ag19Cu2 nanoclusters: synthesis, structure analysis, and electrocatalytic CO2 reduction application†
Xin
Zhu‡
a,
Pan
Zhu‡
b,
Xuzi
Cong‡
c,
Guanyu
Ma
a,
Qing
Tang
 *b,
Likai
Wang
*c and
Zhenghua
Tang
*ad
*b,
Likai
Wang
*c and
Zhenghua
Tang
*ad
aNew Energy Research Institute, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Center, Guangzhou, 510006, China. E-mail: zhht@scut.edu.cn
bChongqing Key Laboratory of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 401331, China. E-mail: qingtang@cqu.edu.cn
cSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, 255049, Shandong, China. E-mail: lkwangchem@sdut.edu.cn
dKey Laboratory of Functional Inorganic Materials Chemistry, Ministry of Education, Heilongjiang University, Harbin, 150001, China
First published on 15th August 2024
Abstract
We report the synthesis, structure analysis, and electrocatalytic CO2 reduction application of Ag19Cu2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CArF)12(PPh3)6Cl6 (abbreviated as Ag19Cu2, CCArF: 3,5-bis(trifluoromethyl)phenylacetylene) nanoclusters. Ag19Cu2 has characteristic absorbance features and is a superatomic cluster with 2 free valence electrons. Single-crystal X-ray diffraction (SC-XRD) revealed that the metal core of Ag19Cu2 is composed of an Ag11Cu2 icosahedron connected by two Ag4 tetrahedra at the two terminals of the Cu–Ag–Cu axis. Notably, Ag19Cu2 exhibited excellent catalytic performance in the electrochemical CO2 reduction reaction (eCO2RR), manifested by a high CO faradaic efficiency of 95.26% and a large CO current density of 257.2 mA cm−2 at −1.3 V. In addition. Ag19Cu2 showed robust long-term stability, with no significant drop in current density and FECO after 14 h of continuous operation. Density functional theory (DFT) calculations disclosed that the high selectivity of Ag19Cu2 for CO in the eCO2RR process is due to the shedding of the –CCArF ligand from the Ag atom at the very center of the Ag4 unit, exposing the active site. This study enriches the potpourri of alkynyl-protected bimetallic nanoclusters and also highlights the great advantages of using atomically precise metal nanoclusters to probe the atomic-level structure–performance relationship in the catalytic field.
CArF)12(PPh3)6Cl6 (abbreviated as Ag19Cu2, CCArF: 3,5-bis(trifluoromethyl)phenylacetylene) nanoclusters. Ag19Cu2 has characteristic absorbance features and is a superatomic cluster with 2 free valence electrons. Single-crystal X-ray diffraction (SC-XRD) revealed that the metal core of Ag19Cu2 is composed of an Ag11Cu2 icosahedron connected by two Ag4 tetrahedra at the two terminals of the Cu–Ag–Cu axis. Notably, Ag19Cu2 exhibited excellent catalytic performance in the electrochemical CO2 reduction reaction (eCO2RR), manifested by a high CO faradaic efficiency of 95.26% and a large CO current density of 257.2 mA cm−2 at −1.3 V. In addition. Ag19Cu2 showed robust long-term stability, with no significant drop in current density and FECO after 14 h of continuous operation. Density functional theory (DFT) calculations disclosed that the high selectivity of Ag19Cu2 for CO in the eCO2RR process is due to the shedding of the –CCArF ligand from the Ag atom at the very center of the Ag4 unit, exposing the active site. This study enriches the potpourri of alkynyl-protected bimetallic nanoclusters and also highlights the great advantages of using atomically precise metal nanoclusters to probe the atomic-level structure–performance relationship in the catalytic field.
1. Introduction
Thanks to the alloying effect, bimetallic nanoclusters usually display different physicochemical properties and functionalities from their monometallic counterparts.1–5 Specifically, in the catalytic field, upon introducing another metal atom into the core, on the one hand, the geometric configuration of the metal nanocluster is changed and new catalytically active sites can be generated, and on the other hand, it can modulate the electronic structure of the metal nanocluster hence tuning the adsorption capacities to the reaction substrates and intermediates. For instance, the Jiang and Lee group reported that the Au24Pt(SC6H13)18 cluster possesses much higher hydrogen evolution reaction (HER) activity than Au25(SC6H13)18, as the central Pt atom can more easily form an M–H chemical bond to lower the energy barrier.6 In another study, Deng et al. found that Cu doping can significantly boost the electrochemical CO2 reduction reaction (eCO2RR) of atomically precise Au nanoclusters. In a gas diffusion electrode-based membrane electrode assembly cell, the Au15Cu4 nanocluster exhibited a high CO faradaic efficiency (over 90%), markedly larger than that of the Au18 cluster (∼60%). Density functional theory (DFT) calculations disclosed that Cu doping induced catalytic synergistic effects, where the exposed pair of AuCu dual sites can accelerate the eCO2RR process.7 As seen above, atomically precise bimetallic nanoclusters have good catalytic activity and selectivity for the eCO2RR, and more importantly, the atomically precise structure of bimetallic nanoclusters is conducive to the in-depth analysis of the reaction mechanism and the elucidation of the catalyst's structure–performance relationship at the atomic level.To prevent the bimetallic core from aggregation, there are various types of small molecules that can be used as capping agents or ligands for preparing bimetallic nanoclusters.8 The ligand molecules include but are not limited to thiolate molecules,9 N-containing molecules,10 carbenes,11 hydrides,12 alkynyl molecules,13–15 and so on. The ligand molecules can not only tune the physical and chemical properties of the metal nanoclusters,16 but also affect the catalytic performance drastically.17–19 The most widely employed ligand is thiolate molecules; however, alkynyl molecules as a protecting ligand have been gaining more and more research attention.13–15 In a quite recent study, the Wang and Zhou groups reported the photophysics of Au22(tBuPhCC)18 and Au16Cu6(tBuPhCC)18, where the latter exhibited >99% photoluminescence quantum yield (PLQY) in de-aerated solution at room temperature with an emission maximum at 720 nm tailing to 950 nm and 61% PLQY in an oxygen-saturated solution.20 This is due to the fact that the Cu doping suppressed the non-radiative decay (∼60-fold less) and promoted the intersystem crossing rate. Moreover, in the catalytic regime, alkynyl ligands can perturb the electronic structure of metal nanoclusters by forming σ and/or π bonds, thereby promoting the catalytic performance of some specific reactions.20 In 2017, the Wang group fabricated two isostructural [Au38L20(Ph3P)4]2+ (L = alkynyl or thiolate) clusters with the ligand being the only variable.21 The alkynyl-protected Au38 cluster is very active in the semi-hydrogenation of alkynes with the substrate conversion ratio over 97% while the thiolate one possesses negligible activity (less than 2%). In another study, our group disclosed that, in the electrochemical CO2 reduction reaction (eCO2RR), alkynyl-protected Ag32 clusters can outcompete the phosphine and thiolate co-protected Ag32 clusters in terms of FECO, as the former has a much lower energy barrier for forming the key intermediate *COOH.22 Similar phenomena have also been documented for alkynyl-protected Au25 nanoclusters to catalyze the HER,23 and alkynyl-protected Au28 nanoclusters supported by NiFe-LDH for catalyzing the oxygen evolution reaction.24
So far, significant progress has been achieved on the synthesis, electronic properties, luminescent behaviours, and catalytic applications of alkynyl-protected bimetallic nanoclusters, yet the family or potpourri of alkynyl-protected bimetallic nanoclusters is still limited. For AgCu bimetallic nanoclusters, molecules with atomically precise structures and alkynyl protection only include Ag9Cu6,25 Ag20Cu12,26 Ag15Cu6,27 Ag22Cu7,28 Ag14Cu2,29 and so on. Inspired by the above studies, herein, we report the synthesis, structural analysis, and eCO2RR application of Ag19Cu2(CCArF)12(PPh3)6Cl6 nanoclusters (hereafter referred to as Ag19Cu2, CCArF: 3,5-bis(trifluoromethyl)phenylacetylene). Ag19Cu2 has characteristic absorbance features, and its overall structure was analyzed by SC-XRD. Ag19Cu2 exhibited excellent catalytic performance, evidenced by a high CO faradaic efficiency (FECO) of 95.26%, a high current density of 257.2 mA cm−2 at −1.3 V, and robust stability with no significant current decay during 14 h of continuous operation.
2. Experimental section
See all the experimental details in the ESI.†3. Results and discussion
The UV-visible absorption spectrum of Ag19Cu2 is shown in Fig. 1a, and two characteristic absorbance peaks at 498 nm and 703 nm can be easily identified. The overall structure of Ag19Cu2 was then analyzed by single-crystal X-ray diffraction (Fig. 1b). Specifically, the Ag19Cu2 nanocluster is positively charged, with BF4− as the counter ion. Ag19Cu2 crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) space group. Ag19Cu2 has 21 metal atoms in the core, which is protected by twelve –CC–ArF ligands, six PPh3 molecules and six chlorine ions. The calculated free valence electron number is 2, indicating that it is a superatomic cluster with a closed-shell electronic structure. In fact, after being stored at room temperature for 3 months, the absorbance features of the Ag19Cu2 clusters remained almost unchanged (Fig. S5†), confirming the excellent chemical stability of Ag19Cu2 under ambient conditions.
(2) space group. Ag19Cu2 has 21 metal atoms in the core, which is protected by twelve –CC–ArF ligands, six PPh3 molecules and six chlorine ions. The calculated free valence electron number is 2, indicating that it is a superatomic cluster with a closed-shell electronic structure. In fact, after being stored at room temperature for 3 months, the absorbance features of the Ag19Cu2 clusters remained almost unchanged (Fig. S5†), confirming the excellent chemical stability of Ag19Cu2 under ambient conditions.
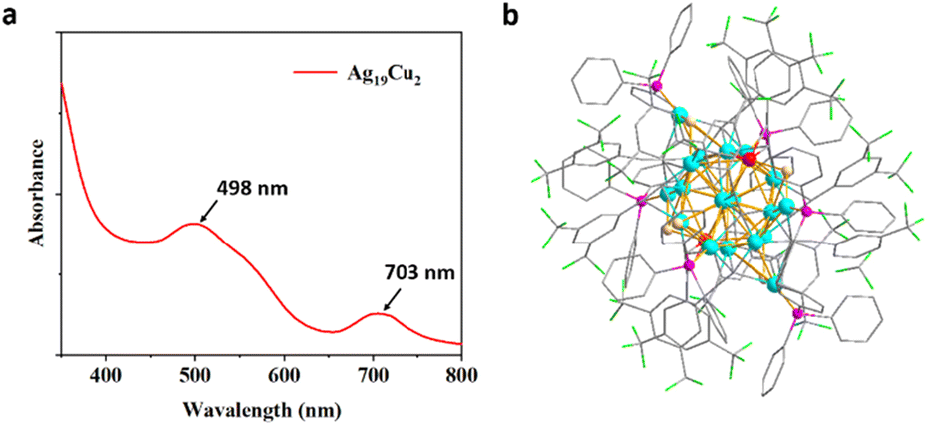 | ||
| Fig. 1 (a) Absorbance spectra of the Ag19Cu2 nanocluster in CH2Cl2. (b) The overall structure of Ag19Cu2. Color legend: red, Cu; blue, Ag; green, Cl; purple, P; laurel green, F; white, H; gray, C. | ||
Subsequently, the metal core configuration of Ag19Cu2 was detailedly examined. As illustrated in Fig. 2a, Ag11Cu2 forms an icosahedron with two Cu atoms on two diagonal points, and two Ag4 tetrahedra are connected with two faces of the two opposite sides in the Ag11Cu2 icosahedron to form the Ag19Cu2 core. Apparently, the Ag19Cu2 core is centrosymmetric. Interestingly, the average Ag–Ag bonding length in the Ag11Cu2 kernel is 2.9300 Å, slightly longer than that in the Ag4 tetrahedra (see Fig. S2†). Next, the metal–ligand coordination mode between Ag/Cu and the alkynyl molecule was analyzed and is summarized in Fig. 2b. Basically, there are four types of coordination modes: Motif A, B, C, and D. In Motif A, the alkynyl molecule binds with three Ag atoms with σ bonding and one Ag atom with both σ and π bonding; hence Motif A adopts a μ4–η1, η1, η1, η2 coordination mode. In Motif B, the alkynyl molecule binds with four Ag atoms with σ bonding; hence the coordination mode is μ4–η1, η1, η1, η1. In Motif C, the alkynyl molecule binds with two Ag atoms with σ bonding and one Cu atom with both σ and π bonding; hence Motif C adopts a μ3–η1, η1, η2 coordination mode. Motif D has a similar coordination pattern to Motif C, and the alkynyl molecule binding with both σ and π bonding is one Ag atom in Motif D but one Cu atom in Motif C; therefore, Motif D adopts a μ3–η1, η2, η3 coordination mode. There are twelve alkynyl molecules capping the metal core, and the total number of Motif A, B, C, and D is 4, 4, 3, and 3, respectively. In addition, six PPh3 ligands are coordinated with six Ag atoms on two sides in the cluster (Fig. S3†), and six Cl atoms are also coordinated with Ag11Cu2 by σ bonds in the cluster (Fig. S4†). Four Cl atoms are connected to three Ag atoms and two Cl atoms are connected to one Cu atom and two Ag atoms. All the detailed structure parameters are summarized in Table S1.†
 | ||
| Fig. 2 (a) The structural anatomy of the Ag19Cu2 core. (b) Four different coordination modes between Ag/Cu and one alkynyl molecule. Color legend: red, Cu; blue, Ag; green, Cl; purple, P; laurel green, F; white, H; gray, C. | ||
The elemental composition and charge states of Ag19Cu2 were subsequently analyzed using X-ray photoelectron spectroscopy (XPS). The survey scan spectra shown in Fig. S6a† confirmed the presence of Ag, Cu, C, F, and P atoms. The high resolution XPS spectra of the Ag 3d and Cu 2p electrons in Ag19Cu2 can be found in Fig. S6b and S6c,† respectively. Notably, the binding energy of the Ag 3d5/2 electrons is 368.7 eV, suggesting that the valence state of Ag is between (0) and (I). Such a value is also in good agreement with the previously reported Ag15Cu6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and Ag20Cu1226 clusters, whose Ag 3d5/2 binding energies are 368.1 eV and 368.84 eV, respectively. Meanwhile, the binding energy of the Cu 2p3/2 electrons is 933.34 eV, indicating that the valence state of Cu is (0, 1). The XPS results further validate that the superatomic cluster of Ag19Cu2 is successfully obtained.
27 and Ag20Cu1226 clusters, whose Ag 3d5/2 binding energies are 368.1 eV and 368.84 eV, respectively. Meanwhile, the binding energy of the Cu 2p3/2 electrons is 933.34 eV, indicating that the valence state of Cu is (0, 1). The XPS results further validate that the superatomic cluster of Ag19Cu2 is successfully obtained.
The Ag19Cu2 nanoclusters were then loaded onto carbon nanotubes (CNTs) to investigate the eCO2RR catalytic performance. As shown in Fig. 3a, in N2 saturated 1 M KOH, both the CNTs and Ag19Cu2/CNTs exhibited no activity, while in CO2 saturated 1 M KOH, both displayed effective activity. At the same applied potential, Ag19Cu2/CNTs has a much higher current density than CNTs, indicating superior catalytic activity. In the eCO2RR, the main product is CO, along with H2 as the side product. Faradaic efficiency (FE) is defined as the amount of product collected relative to the amount that can be produced by the total charge passing through, expressed as a fraction or percentage. In electrochemical reactions, a higher FE means that the actual power of the reaction is closer to the theoretical power and the efficiency of the reaction is higher. The FECO for Ag19Cu2/CNTs and CNTs is presented in Fig. 3b. The FECO for CNTs is nearly zero in the whole tested potential window, but for Ag19Cu2/CNTs, when the applied potential becomes more negative, the FECO first increases and then decreases, and the highest FECO is achieved at −1.37 V with a value of 95.26%. When the potential becomes quite negative, the hydrogen evolution reaction starts to dominate, which is the main reason why FECO decreases. Meanwhile, Ag19Cu2/CNTs displays a strong capability to suppress hydrogen evolution, as the FEH2 is below 5% in the whole potential window. However, the HER process dominates the reaction for CNTs, as the FEH2 increases from ∼51% to nearly 100% (Fig. 3c). To further evaluate the catalytic performance of the Ag19Cu2/CNT catalyst, we also calculated the energy efficiency (EE), which is defined as the conversion efficiency of the chemical energy from the input electrical energy to the final product, and a higher EE means a higher yield of the reaction product and less energy wasted. From −1.47 V to −0.77 V, the EE first increased and then decreased, reaching a maximal value of 39.76% at an EE of −0.97 V (Fig. 3d). In addition, the single-pass conversion efficiency (SPCE) of the Ag19Cu2/CNT catalyst increased when the potential changed from −0.77 V to −1.47 V, achieving a maximal value of 7.46% at −1.47 V. Furthermore, long-term stability is another important criterion to further assess the intrinsic catalytic properties of a catalyst. As demonstrated in Fig. S7,† after continuous operation for 14 h, the current density and FECO of the Ag19Cu2/CNT catalyst remained almost unchanged, suggesting excellent long-term stability.
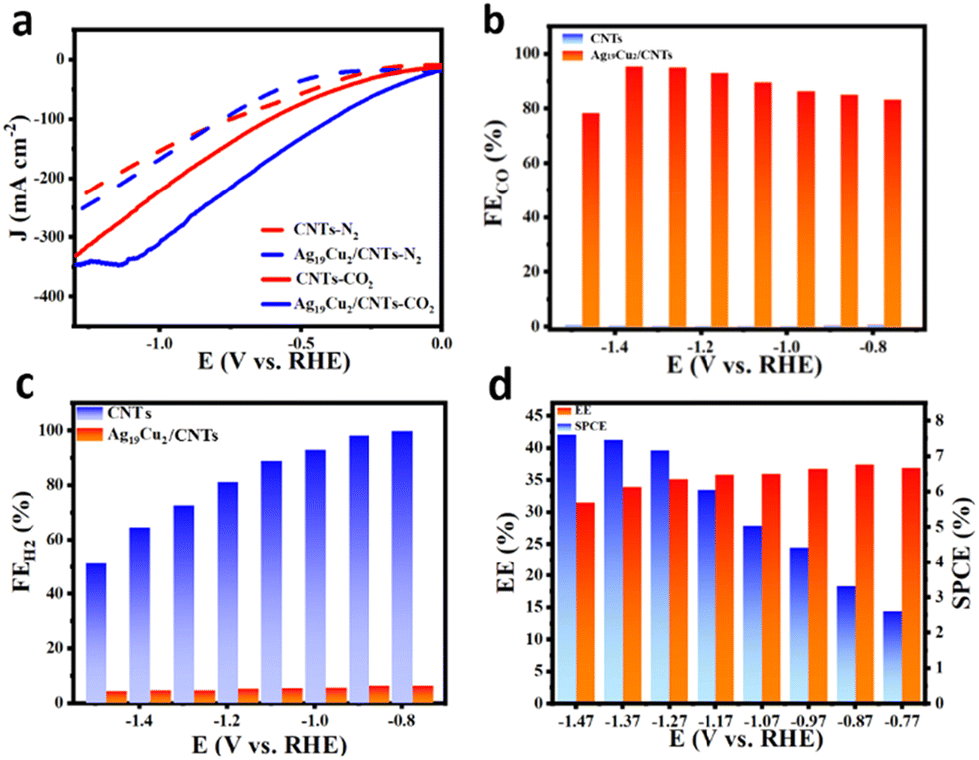 | ||
| Fig. 3 (a) Polarization curve of CNTs and Ag19Cu2/CNTs in N2/CO2 saturated 1 M KOH. (b) FECO of CNTs and Ag19Cu2/CNTs. (c) FEH2 of CNTs and Ag19Cu2/CNTs. (d) EE and SPCE of the eCO2RR. | ||
The spin-polarized density functional theory (DFT) calculations were subsequently carried out to investigate the electrocatalytic activity and selectivity of Ag19Cu2 nanoclusters by using the Vienna ab initio simulation package (VASP5.4.4).30 In order to save the computational cost, we simplified the –CCArF groups to –CCR (R = 3,5-C6H3F2) and the –PPh3 groups to –P(CH3)3, respectively. The nanoclusters were placed in a cubic box (26 Å × 26 Å × 26 Å), and their structures were optimized. The interactions of electron exchange–correlation were represented by the Perdew–Burke–Ernzerhof (PBE) functional form of the generalized gradient approximation (GGA).31 The projector augmented-wave (PAW) method was utilized to describe the ion–electron interactions,32 and the wave functions of all the computations were extended by a plane-wave cutoff energy of 400 eV. Only the Gamma point was used to sample the Brillouin zone, and the convergence criteria for energy and force were set as 10−4 eV and −0.05 eV Å−1, respectively. In addition, considering the non-negligible van der Waals interactions between ligands, we used the empirical density functional dispersion (DFT-D3) method.33
Based on the computational hydrogen electrode (CHE) model,34 the change in Gibbs free energy (ΔG) for each elementary step of the CO2 reduction reaction and hydrogen evolution reaction can be calculated as follows:
| ΔG = ΔE + ΔZPE − TΔS |
To further understand the high eCO2RR activity and selectivity of Ag19Cu2 nanoclusters observed in the experiment, DFT calculations were carried out to identify the catalytically active sites for the CO2RR. To simplify the calculations, we replaced the –CCArF groups with –CCR (R = 3,5-C6H3F2) groups and the –P(Ph)3 groups with –P(CH3)3 groups, using the [Ag19Cu2(CCR)12(P(CH3)3)6Cl6]+ cluster as the computational model. Here, we consider all cases where the removal of a single ligand exposes under-coordinated metal active sites. We found that after the removal of the –Cl ligand, the spatial accessibility around the under-coordinated metal site (highlighted by red circles, Fig. S8†) is too small to act as a catalytic site; thus its catalytic activity is negligible. We therefore focused on the catalytic performance of Ag19Cu2 clusters after the removal of a single –CCR or –P(CH3)3 group. For the intact Ag19Cu2 cluster, –P(CH3)3 ligands and part of the –CCR ligands are attached to the Ag4 unit of the Ag19Cu2 cluster, and the remaining –CCR ligands are connected with the Ag11Cu2 unit of the Ag19Cu2 cluster. We first consider the case of the removal of a single –P(CH3)3 ligand from the Ag4 unit, as predicted by the free energy profile shown in Fig. 4a, where the formation of *COOH is the potential-limiting step (PDS) of the CO2RR with a high reaction energy up to 1.82 eV. With the removal of a –CCR ligand from the Ag11Cu2 unit to form Ag19Cu2(CCR)11(P(CH3)3)6Cl6+-(1) clusters, the formation of the *COOH intermediate remains the most uphill step with a relatively high energy barrier of 1.53 eV. It is noteworthy that when a –CCR ligand is removed from the Ag4 unit, the reaction energy for COOH* formation (PDS) on the Ag19Cu2(CCR)11(P(CH3)3)6Cl6+-(2) cluster is lower (1.09 eV). In comparison, the Ag19Cu2(CCR)11(P(CH3)3)6Cl6+-(2) cluster has a better catalytic activity for the CO2RR. The optimized structures of all substrates and intermediates are depicted in Fig. S9 and S10† and Fig. 4c. We found that for the Ag19Cu2(CCR)11(P(CH3)3)6Cl6+-(2) cluster, the under-coordinated central Ag atom in the Ag4 unit prefers to act as a catalytically active site for the CO2RR, with both COOH* and CO* adsorbed to the central Ag atom.
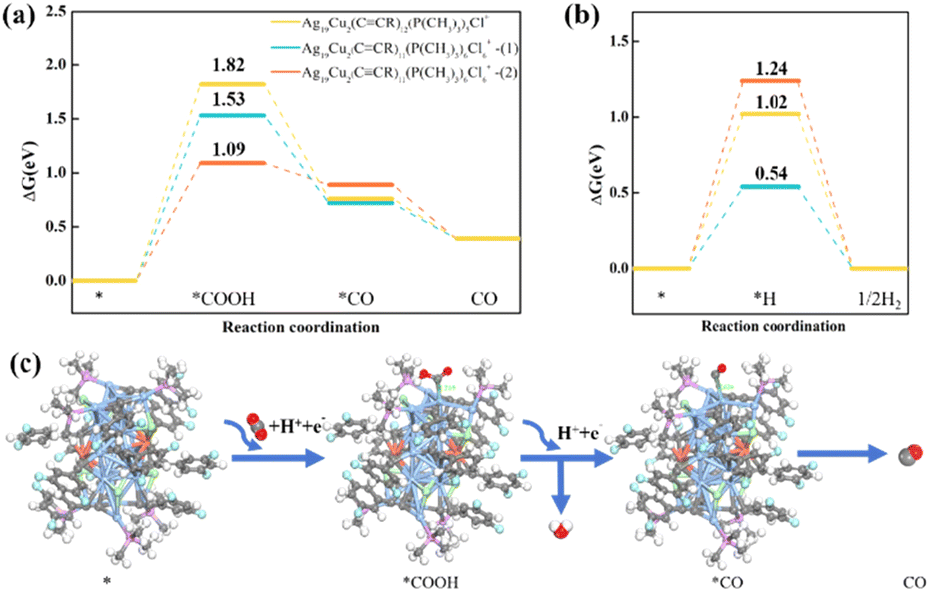 | ||
| Fig. 4 Free energy diagrams (ΔG) for CO2 reduction (a) and hydrogen evolution (b) on Ag19Cu2 clusters with different ligands removed at zero potential. (c) Schematic presentation of the electrocatalytic CO2 reduction process on the [Ag19Cu2(CCR)11(P(CH3)3)6Cl6]+-(2) cluster. Colour legend: Ag, baby blue; Cu, dark orange; C, grey; Cl, light green; F, cyan; P, purple; H, white; O, red. | ||
Furthermore, we analyzed the competitive HER process on the surface of [Ag19Cu2(CCR)12(P(CH3)3)5Cl6]+, [Ag19Cu2(CCR)11(P(CH3)3)6Cl6]+-(1) and (2), and the free energy diagrams are shown in Fig. 4b. Our calculations show that the hydrogen-adsorption step is the potential-determining step (PDS) for the HER on the three NCs, with limiting potentials calculated to be 1.02, 0.54 and 1.24 eV respectively. All schematic presentations of the adsorption structures of intermediates are shown in Fig. S11.† Compared with the result of the electroreduction of CO2 to CO, we found a contrasting product selectivity of Ag19Cu2 clusters with the removal of different ligands. Ag19Cu2 clusters after the removal of a single –P(CH3)3 group from the Ag4 unit or a single –CCR group from the Ag11Cu2 unit are more favourable to undergo the HER process. However, the limiting potential of the HER (1.24 eV) significantly exceeds that of the CO2RR (1.09 eV) on the [Ag19Cu2(CCR)11(P(CH3)3)6Cl6]+-(2) cluster, fully supporting the experimentally observed high CO selectivity. Overall, the DFT results suggest that the high CO selectivity of Ag19Cu2 clusters is probably due to the removal of the –CCR group from the Ag4 unit, exposing the under-coordinated Ag site as a catalytically active centre to promote electrochemical CO2 reduction.
4. Conclusions
In summary, we successfully synthesized an atomically precise nanocluster of Ag19Cu2(CCArF)12(PPh3)6Cl6, which possesses two valence electrons and characteristic absorbance features. The overall structure of Ag19Cu2 was analyzed by SC-XRD, and it has a novel Ag19Cu2 core composed of a Ag11Cu2 kernel connected by two Ag4 units and various metal–ligand binding coordination modes. When loading into carbon nanotubes, it exhibited excellent catalytic performance, manifested by a high FECO value of 95.26%, a large CO current density of 257.2 mA cm−2 at −1.3 V, and robust long-term stability for a continuous 14 h test. DFT calculations revealed that the high CO selectivity is due to the shedding of the –CCR group from the Ag4 unit during the catalytic process, which exposes the undercoordinated Ag atom as the catalytically active site. This study enriches the family of alkynyl-protected bimetallic nanoclusters, and also provides atomic-level mechanistic insights into employing bimetallic nanoclusters as catalysts for complex electrochemical reactions.
Data availability
All data supporting the findings of this study are available within the paper and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. T. acknowledges the funding from the Guangdong Natural Science Funds (No. 2023A0505050107). L. W. acknowledges the financial support from the National Natural Science Foundation of China (No. 21805170). Q. T. acknowledges the funding by the Chongqing Science and Technology Commission (cstc2020jcyj-msxmX0382).References
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- X.-M. Luo, Y.-K. Li, X.-Y. Dong and S.-Q. Zang, Chem. Soc. Rev., 2023, 52, 383–444 RSC.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- C. Sun, B. K. Teo, C. Deng, J. Lin, G.-G. Luo, C.-H. Tung and D. Sun, Coord. Chem. Rev., 2021, 427, 213576 CrossRef CAS.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- G. Deng, H. Yun, M. S. Bootharaju, F. Sun, K. Lee, X. Liu, S. Yoo, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2023, 145, 27407–27414 CrossRef CAS PubMed.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2022, 123, 5948–6002 CrossRef PubMed.
- Y. Li, M. Zhou and R. Jin, Adv. Mater., 2021, 33, 2006591 CrossRef CAS PubMed.
- S. F. Yuan, W. D. Liu, C. Y. Liu, Z. J. Guan and Q. M. Wang, Chem. – Eur. J., 2022, 28, e202104445 CrossRef CAS PubMed.
- H. Shen, G. Tian, Z. Xu, L. Wang, Q. Wu, Y. Zhang, B. K. Teo and N. Zheng, Coord. Chem. Rev., 2022, 458, 214425 CrossRef CAS.
- Y. Lv, T. Jiang, Q. Zhang, H. Yu and M. Zhu, Polvoxometalates, 2024, 3, 9140050 CrossRef.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- X. Ma, Y. Tang, G. Ma, L. Qin and Z. Tang, Nanoscale, 2021, 13, 602–614 RSC.
- L. Chen, L. Wang, Q. Shen, Y. Liu and Z. Tang, Mater. Chem. Front., 2023, 7, 1482–1495 RSC.
- Y. Wang, Z. Liu, A. Mazumder, C. G. Gianopoulos, K. Kirschbaum, L. A. Peteanu and R. Jin, J. Am. Chem. Soc., 2023, 145, 26328–26338 CrossRef CAS PubMed.
- Q. J. Wu, D. H. Si, P. P. Sun, Y. L. Dong, S. Zheng, Q. Chen, S. H. Ye, D. Sun, R. Cao and Y. B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306822 CrossRef CAS PubMed.
- H. Shan, J. Shi, T. Chen, Y. Cao, Q. Yao, H. An, Z. Yang, Z. Wu, Z. Jiang and J. Xie, ACS Nano, 2023, 17, 2368–2377 CrossRef CAS PubMed.
- Z. Liu, H. Tan, B. Li, Z. Hu, D.-e. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed.
- W.-Q. Shi, L. Zeng, R.-L. He, X.-S. Han, Z.-J. Guan, M. Zhou and Q.-M. Wang, Science, 2024, 383, 326–330 CrossRef CAS PubMed.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- L. Chen, F. Sun, Q. Shen, L. Qin, Y. Liu, L. Qiao, Q. Tang, L. Wang and Z. Tang, Nano Res., 2022, 15, 8908–8913 CrossRef CAS.
- X. Li, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2021, 125, 23226–23230 CrossRef CAS.
- Q.-L. Shen, L.-Y. Shen, L.-Y. Chen, L.-B. Qin, Y.-G. Liu, N. M. Bedford, F. Ciucci and Z.-H. Tang, Rare Met., 2023, 42, 4029–4038 CrossRef CAS.
- X. Ma, F. Sun, L. Qin, Y. Liu, X. Kang, L. Wang, D.-e. Jiang, Q. Tang and Z. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC.
- G. Ma, F. Sun, L. Qiao, Q. Shen, L. Wang, Q. Tang and Z. Tang, Nano Res., 2023, 16, 10867–10872 CrossRef CAS.
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2022, 145, 3401–3407 CrossRef PubMed.
- G. Deng, K. Lee, H. Deng, S. Malola, M. S. Bootharaju, H. Häkkinen, N. Zheng and T. Hyeon, Angew. Chem., Int. Ed., 2023, 62, e202217483 CrossRef CAS PubMed.
- Q. Shen, X. Cong, L. Chen, L. Wang, Y. Liu, L. Wang and Z. Tang, Dalton Trans., 2023, 52, 16812–16818 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- I. L. Garzón and A. Posada-Amarillas, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11796–11802 CrossRef PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. W. Liu, D. Lee and D.-e. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supporting figures and tables, and more calculation results. CCDC 2366717. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr02702g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |