
Chemical activation of atom-precise Pd3 nanoclusters on γ-Al2O3 supports for transfer hydrogenation reactions†
Siddhant
Singh
a,
Dami
Wi
a,
Kholoud E.
Salem
b,
Drew
Higgins
b and
Robert W. J.
Scott
 *a
*a
aDepartment of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada. E-mail: robert.scott@usask.ca
bDepartment of Chemical Engineering, McMaster University, 1280 Main St W, Hamilton, Ontario L8S 4L7, Canada
First published on 30th September 2024
Abstract
Deposition of atom-precise nanoclusters onto solid supports is a promising route to synthesize model heterogeneous catalysts. However, to enhance nanocluster-support interactions, activation of the nanoclusters by removal of surface ligands is necessary. Thermal treatment to remove surface ligands from supported metal nanoclusters can yield highly active heterogeneous catalysts, however, the high temperatures employed can lead to poor control over the final size and speciation of the nanoclusters. As an alternative to high-temperature thermal treatments, chemical activation of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]+ (Pd3) nanoclusters on γ-Al2O3 supports under mild reaction conditions has been demonstrated in this work. Hydride-based reducing agents such as NaBH4, LiBH4, and LiAlH4 have been examined for the activation of the Pd3 nanoclusters. The structural evolution and speciation of the nanoclusters after activation have been monitored using a combination of XAS, XPS, STEM-EDX mapping, and solid-state NMR techniques. The results indicate that treatment with borohydride reducing agents successfully removed surface phosphine and chloride ligands, and the extent of size growth of the nanoclusters during activation is directly correlated with the amount of borohydride used. Borate side products remain on the γ-Al2O3 surface after activation; moreover, exposure to high amounts of NaBH4 resulted in the incorporation of B atoms inside the lattice of the activated Pd nanoclusters. LiAlH4 treatment, on the other hand, led to no significant size growth of the nanoclusters and resulted in a mixture of Pd single-atom sites and activated nanoclusters on the γ-Al2O3 surface. Finally, the catalytic potential of the activated nanoclusters has been tested in the transfer hydrogenation of trans-cinnamaldehyde, using sodium formate/formic acid as the hydrogen donor. The catalytic results showed that smaller Pd nanoclusters are much more selective for hydrogenating trans-cinnamaldehyde to hydrocinnamaldehyde, but overall have lower activity compared to larger Pd nanoparticles. Overall, this study showcases chemical activation routes as an alternative to traditional thermal activation routes for activating supported nanoclusters by offering improved speciation and size control.
Introduction
Solid-supported nanoclusters (NCs) of Pd are highly selective and active catalysts for essential catalytic processes such as the selective hydrogenation of alkynes (e.g., acetylene), CO oxidation, C–C coupling, and dehydrogenation reactions.1–6 The high catalytic selectivity and activity of NCs can be attributed to their large surface area and the size-dependent arrangement of surface atoms.3–5 One common strategy to obtain solid-supported NCs is the deposition of pre-synthesized ligand-protected atom-precise NCs onto a support material followed by activation (i.e., removal of surface ligands).5,7,8 Bottom-up, surface ligand-assisted synthetic approaches have enabled structural control over atom-precise NCs which contain well-defined metal cores with surface ligands coating the metal core.9 However, upon immobilization of NCs on solid supports (i.e., solid-supported NCs) for heterogeneous catalytic processes, their surface ligands can prevent strong support–NC interactions and also act as a barrier for substrates to access catalytically active metal surfaces during catalytic processes.8,10 Therefore, atom-precise NCs are first deposited on a suitable solid support followed by removing the surface ligands from the NCs in a subsequent activation step.7,8,10 A central question in the field is how to effectively remove the ligands from the surface of pre-fabricated NCs with minimum distortion in the structure and size of the NCs.10–12[Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]+ (abbreviated hereafter as Pd3) NCs have shown promising catalytic activity and selectivity in cross-coupling reactions when used without immobilization on solid supports.13,14 However, a recent study by Fairlamb and coworkers suggested that non-immobilized Pd3 NCs are less stable during cross-coupling reactions compared to resin-immobilized Pd3 NCs.6 Yun et al. demonstrated that non-immobilized Pd3 NCs are almost inactive catalysts for the oxidation of benzyl alcohol, however, upon immobilizing the Pd3 NCs on titanate nanotubes, a remarkable increase in their activity was observed due to metal–support interactions.15 These studies suggest that immobilizing Pd3 NCs onto solid supports not only stabilizes the NCs but can also significantly enhance their catalytic potential.6,15 Our group previously reported the immobilization of Pd3 NCs onto activated carbon supports for hydrogenation and cross-coupling reactions, and found that Pd3 NCs adsorbed on activated carbon supports do not behave as true heterogeneous catalysts, as intact Pd3 NCs leached into the reaction mixture during cross-coupling reactions.16 Thus the presence of phosphine ligands on the Pd3 NCs hindered strong interactions between NCs and the support. However, thermal activation of Pd3 NCs in the air led to the complete removal of surface ligands, resulting in catalysts that exhibited true heterogeneous behavior.16 While the thermal activation of NCs is a straightforward strategy, drawbacks associated with this strategy include uncontrolled sintering of NCs to larger nanoparticles (NPs), particularly at higher activation temperatures.16–20 Additionally, many metals, including Pd, tend to oxidize and form metal oxides at moderate temperatures of thermal activation.16,20 Therefore due to the poor speciation and morphology control over NCs during thermal activation, the catalytic potential of the NCs can be adversely affected.10,16
An alternative method to activate NCs is through chemical activation, where ligands are removed from the surface of the NCs following chemical treatment.10,21 One of the most promising strategies for chemically activating NCs involves treating them with reducing agents.10 This process includes the reduction of surface ligands, which are subsequently removed as free ligands.10 Collins et al. have shown a comparison between the thermal and reductive chemical activation of oleylamine, dodecanethiol, and polyvinylpyrrolidone-stabilized Pd NPs.22 They found that the chemical activation of Pd NPs using NaBH4 was less effective in completely removing strongly binding ligands such as dodecanethiol from the surfaces of NPs than thermal activation routes.22 However, the chemically activated NPs were found to be more catalytically active in cross-coupling reactions compared to both non-activated and thermally activated NPs.22 Several groups, including our own, have previously shown that thiolate monolayer-protected Au NCs can be reductively activated in the presence of excessive borohydride reducing agents, and the post-activation size growth of NCs directly depends upon both the concentrations and strength of reducing agents used for the activation.23–25 For example, the activation of γ-Al2O3-supported Au25(SC8H9)18 NCs with an excess of NaBH4 led to the partial removal of thiolates from the Au NCs and resulted in minimal size growth of Au NCs.25 Zhang and co-workers noted that hydrides exhibit a higher binding affinity to the surface of Au compared to organothiolate ligands, and consequently, an excess of hydride species can displace surface ligands from precious metals.26 To the best of our knowledge, reductive chemical routes toward the activation of Pd NCs have not yet been explored and require further investigation. Chemical activation pathways have the potential to act as an efficient way to activate Pd NCs while avoiding the sintering and oxidation of Pd NCs seen during thermal activation in air.16
In this study, the chemical activation of γ-Al2O3-supported atomically precise Pd3 NCs using hydride-based reducing agents i.e., NaBH4, LiBH4, and LiAlH4, has been investigated. Various loadings of these reducing agents were examined to explore their impact on the activation process and subsequent structural changes of the NCs. The findings from this work indicate that all the selected reducing agents effectively removed phosphine and chloride ligands from the Pd3 NC surfaces. In the case of NaBH4 and LiBH4, ligand removal was accompanied by very mild post-activation growth of the Pd3 NCs at lower reducing agent amounts, while higher amounts of NaBH4 during activation promoted the transformation of Pd3 NCs into larger Pd NPs and led to the incorporation of B into the NPs. In contrast, LiAlH4 treatment removed ligands from the Pd3 NCs without any noticeable size growth, facilitating the formation of a mix of activated NCs and single-atom Pd sites on the γ-Al2O3 surface. The structural evolution of Pd3 NCs during activation directly impacted the catalytic activity of the NCs, as demonstrated in their use as catalysts for the transfer hydrogenation of trans-cinnamaldehyde using sodium formate as the hydrogen donor. Smaller NCs exhibited high selectivity for hydrocinnamaldehyde formation, albeit with lower activity compared to larger Pd NPs. This work demonstrates the facile removal of phosphine and chloride ligands from Pd3 NC surfaces via chemical means, highlighting how the choice and amount of the reducing agent used can lead to distinct structural transformations during activation.
Results and discussion
The synthesis, purification, and characterization of Pd3 NCs were carried out following protocols established in the literature and our previous work.16,27 Upon immobilization of the Pd3 NCs on the surface of γ-Al2O3, the resulting composite has been denoted as Pd3/Al2O3. Elemental analysis of the Pd3/Al2O3 by atomic absorption spectroscopy showed a 0.5 wt% loading of the Pd on the γ-Al2O3 support. For chemical activations, three amounts of NaBH4, LiBH4, and LiAlH4 have been used (as shown in Table S1†). Based on the reducing agents and their concentrations, the activated samples have been labelled as Pd/Al2O3-RA(x) (where RA is the used reducing agent) (i.e., NaBH4, LiBH4 or LiAlH4) and x is the molar equivalents of the reducing agent used with respect to the Pd (labelled A, B, or C (Table S1†)). For the NaBH4 reducing agent, 300 (A), 600 (B), or 1200 (C) mole equivalents were used for activation. Meanwhile, for LiBH4 and LiAlH4 reducing agents, 10 (A), 50 (B), and 100 (C) mole equivalents of the reducing agent were used for activation. The amounts of NaBH4 used were larger because our previous work demonstrated that higher concentrations of aqueous NaBH4 were necessary to activate thiolate-protected Au NCs compared to LiBH4.25To investigate the speciation and bonding environment of the NCs after chemical activation, XAS analysis has been performed. Pd K edge XANES spectra of all the activated samples have been provided in Fig. 1(a–c), along with Pd, PdO, and Pd(OH)2 standards. Upon treatment with reducing agents, small changes in the shape of the white lines compared to the initial Pd3/Al2O3 were observed. These changes indicate a modification in the speciation and coordination environment of the supported Pd NCs due to activation. In the case of Pd3/Al2O3–NaBH4(A) and Pd3/Al2O3–LiAlH4(A/B/C), the feature above the white line appeared slightly more intense compared to Pd3/Al2O3, suggesting an increase in Pd(II) species. In contrast, the intensity of the sharp feature above the white line in the remaining NaBH4 (Pd3/Al2O3–NaBH4(B/C)) and LiBH4 (Pd3/Al2O3–LiBH4(A/B/C)) treated samples were found to be relatively lower, suggesting more Pd in the zerovalent state. However, the small XANES changes observed at the edge after activation suggest that XANES data does not offer detailed insights into the speciation changes within these systems. In contrast, changes detected by EXAFS (discussed below) are more informative.
 | ||
| Fig. 1 Pd K-edge XANES spectra of the Pd3/Al2O3 activated via treatment with different amounts of (a) NaBH4, (b) LiBH4 and (c) LiAlH4 along with some standards. Pd K-edge Fourier transformed EXAFS spectra in R-space for Pd3/Al2O3 activated via treatment with different amounts of (d) NaBH4, (e) LiBH4 and (f) LiAlH4. All EXAFS spectra are shown without phase shift corrections. | ||
To investigate the coordination environment of the activated Pd3 NCs, EXAFS analysis at the Pd K edge was performed. The Fourier-transformed (FT) EXAFS spectra of activated samples along with the spectra of Pd3/Al2O3 are provided in Fig. 1(d–f). In the spectra of Pd3/Al2O3, a broad peak at around 1.6–1.8 Å can be seen due to the Pd–P and Pd–Cl interactions of the Pd core with the phosphine and chloride ligands. The small feature at around 2.7 Å can be assigned to Pd–Pd bonding in the triangular metal core of Pd3 NCs. Notably, the Pd–Pd bond distance in Pd3/Al2O3 was found to be slightly longer than that in fcc Pd, which is also the case in the reported single-crystal structure of Pd3 NCs.27 In the EXAFS spectra of all the activated NCs, an intense peak can be observed at 1.5 Å, which is shifted to the left in comparison to the Pd–P and Pd–Cl peak of the non-activated Pd3/Al2O3 (at 1.6–1.8 Å). This suggests a complete removal of phosphines and chloride surface ligands from the metal core of the Pd3 NCs upon activation; the peak at 1.5 Å in the Pd K edge EXAFS spectra is likely indicative of new Pd–O interaction. The appearance of this peak in activated samples indicates the interaction of Pd NCs with the basic surface oxygen sites of γ-Al2O3 after the removal of surface ligands from the Pd3 NCs.
In the case of Pd3/Al2O3–NaBH4(A), the peak pertaining to the Pd–Pd interaction (originally at 2.7 Å) became less prominent in comparison to the Pd–Pd peak of Pd3/Al2O3. This is likely due to the partial collapse of the triangular core of the Pd3 NCs after the removal of surface ligands. Similar behaviour was previously observed with the carbon-supported Pd3 NCs during their mild thermal activation at 150 °C.16 With the use of higher amounts of NaBH4 (i.e., Pd3/Al2O3–NaBH4(B/C)) for activation, the peak for the Pd–Pd interactions around 2.6 Å became higher in intensity than seen for un-activated Pd3/Al2O3, indicating an increase in the size of the Pd NCs during the activation. Meanwhile, the intensity of the Pd–O peak at around 1.5 Å in the spectra of Pd3/Al2O3–NaBH4(B/C) became lower in intensity compared to the corresponding Pd3/Al2O3–NaBH4(A). This implies the reduction in the Pd–O interaction between Pd and the support due to the growth in the size of the Pd NPs. Furthermore, the peak shapes in the 1.5–2.0 Å region in the FT EXAFS spectra of the Pd3/Al2O3–NaBH4(B/C) samples differ significantly from those observed in the Pd3/Al2O3–NaBH4(A) sample. However, confirming whether this change in peak shape is attributable to the presence of a second scatterer cannot be achieved by solely examining the FT EXAFS spectra. To further investigate the possibility of the presence of a second weak scatterer, wavelet transformed (WT) EXAFS spectra were generated.28 In the WT EXAFS spectra of Pd3/Al2O3–NaBH4(A), a single peak can be seen at R ∼ 1.5 Å (Fig. S1(c and d)†) across all the k range, which reflects the presence of a single scatter i.e., O of the γ-Al2O3 support. However, in the cases of Pd3/Al2O3–NaBH4(B/C), significantly intense peaks around R ∼ 2.6 Å from Pd–Pd interactions can be seen in the WT EXAFS data (Fig. S1(e and g)†). Furthermore, in the region between R-values of 1.5 to 2.0 Å in the FT EXAFS spectra, two distinct peaks can be observed in different k-regions (0.5 to 8.5 and 8.5 to 12 Å−1) of the WT EXAFS spectra, suggesting the probable existence of two scattering atoms (Fig. S1(f and h)†). Based on STEM-EDX mapping, solid-state NMR, and XPS analyses indicating the presence of B (with the B source being the borohydride-reducing agent) in these samples (vide infra), it is likely that the two scatterers are O and B.
In the FT EXAFS spectra of LiBH4-treated samples (Pd3/Al2O3–LiBH4(A/B/C)) (Fig. 1e), the peaks of Pd–Pd interaction around 2.6 Å appeared to be higher in the intensity in comparison to Pd3/Al2O3, reflecting a growth in the size of the Pd3 NCs upon activation. The growth in the Pd–Pd peak became larger with an increase in the amount of LiBH4 for activation (shown in Fig. 1e) suggesting the formation of larger Pd NCs with higher amounts of LiBH4. All the WT EXAFS spectra of the LiBH4-activated NCs (i.e., Pd3/Al2O3–LiBH4(A/B/C)) (Fig. S2†) showed the presence of a single scatter for the first peak of the FT EXAFS around R ∼ 1.5 Å. This affirms that Pd atoms in these systems are attached only to the O atoms of the surface of γ-Al2O3 after the activation, and no secondary scatterers are present. Moreover, another peak for the Pd–Pd interactions at around R ∼ 2.6 Å in the WT EXAFS spectra can also be seen, which became more prominent with a further increase in the amount of the reducing agent. Finally, for Pd3/Al2O3–LiAlH4(A/B/C) (Fig. 1f), the FT EXAFS spectra showed that the peak corresponding to Pd–Pd interactions at 2.6–2.7 Å is nearly completely absent. This indicates that the triangular metal core of Pd3 NCs has been broken after activation using LiAlH4 without any significant size growth. Similar to the LiBH4 treated samples, the LiAlH4 treated samples have shown the presence of only one scatterer for the first peak at R ∼ 1.5 Å in the WT EXAFS spectra (Fig. S3†), indicating the presence of Pd–O interactions. In addition, a very weak peak can be seen at around R ∼ 2.6 Å for the Pd–Pd bonds.
EXAFS data modelling for these samples have been performed to get further insight into the structural evolution of the Pd3 NCs upon activation. The k-space EXAFS fits for Pd3/Al2O3 activated via different NaBH4, LiBH4, and LiAlH4 amounts are shown in Fig. S4–S6,† respectively. The corresponding fitting parameters from the EXAFS fits can be found in Table 1. The fitting of the non-activated Pd3/Al2O3 sample was carried out using Pd–P, Pd–Cl, and Pd–Pd interactions based on the structure of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]+.27 EXAFS fitting of the Pd3/Al2O3 data provided coordination number (CN) values of 2.9 ± 0.3, 0.7 ± 0.3, and 1.8 ± 0.1 for Pd–P, Pd–Cl, and Pd–Pd bonds, respectively, which is quite consistent with the crystal structure of Pd3 NCs.27 The fitting of Pd3/Al2O3–NaBH4(A) suggested the presence of Pd–O contributions at a bond distance of 2.009 ± 0.001 Å with a CN value of 3.3 ± 0.1, likely due to metal–support interactions. Compared to Pd3/Al2O3, a drop in the CN of Pd–Pd for this sample can be due to the distortion in the triangular core of the Pd3 NCs upon activation. Since the WT EXAFS spectra of Pd3/Al2O3–NaBH4(B) and Pd3/Al2O3–NaBH4(C) (Fig. S1†) indicated the presence of two scatterers in the first coordination shell of Pd, and solid-state NMR and XPS also confirmed the presence of B in these two samples (vide infra), a Pd–B model has been chosen for fitting along with the Pd–O model for the peak present at R ∼ 1.5 Å in the FT EXAFS of these two samples. B atoms are small enough to be intercalated inside the interstitial sites of the Pd fcc lattice.29–31 In the literature, the use of borohydride reducing agents such as (CH3)2NH·BH3,29 NaBH4,30 and C4H8O·BH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 during the synthesis of Pd NPs has been reported to show the incorporation of B in the lattice of Pd NPs. Chen and co-workers have found that the B content in the Pd lattice can be directly correlated with the concentration of NaBH4 used during the synthesis of Pd NPs.30 From the EXAFS modelling, the CNs for Pd–O, Pd–B, and Pd–Pd interactions in Pd3/Al2O3–NaBH4(B) were 2.8 ± 0.1, 2.3 ± 0.6, and 2.4 ± 0.3, respectively. Bond lengths for the new Pd–B interactions were fitted at 1.87(3) Å and 1.98(2) Å, respectively, for the Pd3/Al2O3–NaBH4(B) and Pd3/Al2O3–NaBH4(C) samples. These Pd–B bond lengths are consistent with Pd–B bond lengths seen for PdBx solid-state solutions, molecular complexes, and B-doped Pd NPs in the literature (ca. 1.9–2.2 Å).32–34 In Pd3/Al2O3–NaBH4(C), the fitted CN values for Pd–B (3.3 ± 0.1) and Pd–Pd (4.0 ± 0.5) interactions are slightly higher than in the previous sample, while the CN for Pd–O has decreased significantly to 1.4 ± 0.3. This implies that as the amount of NaBH4 used for activation increased, the size of the final Pd NPs also increased, along with the incorporation of B in the lattice of Pd NPs. The decrease in Pd–O interactions can be attributed to reduced metal–support interactions as the size of the supported NPs increased.
31 during the synthesis of Pd NPs has been reported to show the incorporation of B in the lattice of Pd NPs. Chen and co-workers have found that the B content in the Pd lattice can be directly correlated with the concentration of NaBH4 used during the synthesis of Pd NPs.30 From the EXAFS modelling, the CNs for Pd–O, Pd–B, and Pd–Pd interactions in Pd3/Al2O3–NaBH4(B) were 2.8 ± 0.1, 2.3 ± 0.6, and 2.4 ± 0.3, respectively. Bond lengths for the new Pd–B interactions were fitted at 1.87(3) Å and 1.98(2) Å, respectively, for the Pd3/Al2O3–NaBH4(B) and Pd3/Al2O3–NaBH4(C) samples. These Pd–B bond lengths are consistent with Pd–B bond lengths seen for PdBx solid-state solutions, molecular complexes, and B-doped Pd NPs in the literature (ca. 1.9–2.2 Å).32–34 In Pd3/Al2O3–NaBH4(C), the fitted CN values for Pd–B (3.3 ± 0.1) and Pd–Pd (4.0 ± 0.5) interactions are slightly higher than in the previous sample, while the CN for Pd–O has decreased significantly to 1.4 ± 0.3. This implies that as the amount of NaBH4 used for activation increased, the size of the final Pd NPs also increased, along with the incorporation of B in the lattice of Pd NPs. The decrease in Pd–O interactions can be attributed to reduced metal–support interactions as the size of the supported NPs increased.
| Catalyst | Component | CN | R (Å) | σ 2/Å2 | E 0 shift (eV) | R-factor |
|---|---|---|---|---|---|---|
| Pd3/Al2O3 | Pd–P | 2.9 (3) | 2.250 (2) | 0.0063 (1) | 1.1 (2) | 0.019 |
| Pd–Cl | 0.7 (3) | 2.312 (3) | 0.0058 (3) | |||
| Pd–Pd | 1.8 (1) | 2.98 (1) | 0.0101 (5) | |||
| Pd3/Al2O3–NaBH4(A) | Pd–O | 3.3 (1) | 2.009 (1) | 0.0028 (2) | 5.3 (2) | 0.004 |
| Pd–Pd | 1.1 (1) | 2.65 (1) | 0.010 (2) | |||
| Pd3/Al2O3–NaBH4(B) | Pd–O | 2.8 (1) | 1.99 (1) | 0.0043 (2) | 9.2 (7) | 0.003 |
| Pd–B | 2.3 (6) | 1.87 (3) | 0.005 (4) | |||
| Pd–Pd | 2.4 (3) | 2.762 (7) | 0.010 (1) | |||
| Pd3/Al2O3–NaBH4(C) | Pd–O | 1.4 (3) | 2.01 (3) | 0.0048 (2) | 9.6 (5) | 0.014 |
| Pd–B | 3.3 (1) | 1.98 (2) | 0.006 (2) | |||
| Pd–Pd | 4.0 (5) | 2.781 (3) | 0.009 (3) | |||
| Pd3/Al2O3–LiBH4(A) | Pd–O | 2.3 (1) | 2.014 (4) | 0.0021 (6) | 2.4 (8) | 0.016 |
| Pd–Pd | 2.0 (5) | 2.74 (1) | 0.012 (9) | |||
| Pd3/Al2O3–LiBH4(B) | Pd–O | 2.7 (1) | 2.006 (2) | 0.0022 (3) | −2.5 (2) | 0.007 |
| Pd–Pd | 2.3 (1) | 2.727 (4) | 0.0075 (4) | |||
| Pd3/Al2O3–LiBH4(C) | Pd–O | 2.4 (1) | 2.000 (3) | 0.0027 (5) | 3.2 (3) | 0.012 |
| Pd–Pd | 2.5 (1) | 2.725 (5) | 0.0073 (6) | |||
| Pd3/Al2O3–LiAlH4(A) | Pd–O | 2.5 (1) | 2.017 (3) | 0.0025 (5) | 4.7 (4) | 0.017 |
| Pd–Pd | 0.5 (2) | 2.68 (3) | 0.011 (4) | |||
| Pd3/Al2O3–LiAlH4(B) | Pd–O | 2.8 (1) | 2.008 (3) | 0.0024 (4) | 4.6 (4) | 0.019 |
| Pd–Pd | 0.6 (1) | 2.69 (2) | 0.007 (2) | |||
| Pd3/Al2O3–LiAlH4(C) | Pd–O | 2.5 (1) | 2.013 (2) | 0.0024 (3) | 4.1 (4) | 0.022 |
| Pd–Pd | 0.5 (1) | 2.69 (2) | 0.008 (2) |
For the LiBH4-activated samples (Pd3/Al2O3–LiBH4(A/B/C)), the CN for the Pd–O interaction was in the 2.3–2.7 range for the three samples, while the CN for Pd–Pd interactions was 2.0 ± 0.5, 2.3 ± 0.1, and 2.5 ± 0.1 for Pd3/Al2O3–LiBH4(A), Pd3/Al2O3–LiBH4(B), and Pd3/Al2O3–LiBH4(C), respectively. This suggests a very slight growth of NCs after LiBH4 treatment. Similar to LiBH4-activated samples, the CN of Pd–O in samples activated by LiAlH4 (Pd3/Al2O3–LiAlH4(A/B/C)) was found to be around 2.5–2.8 for the three samples. However, the Pd–Pd interactions showed a significantly lower CN of approximately 0.5 in the fitting of these samples. This indicates a substantial disruption of Pd–Pd bonds in the triangular metal core of Pd3 NCs upon LiAlH4 activation. This is consistent with the possibility of a mixture of Pd NCs and single-atom sites in the LiAlH4-activated samples. Unlike the NaBH4 and LiBH4-treated samples, no correlation between the loadings of the reducing agent and the resulting Pd–Pd CN of the activated NCs has been observed in the case of samples activated via LiAlH4. Overall, the XAS results showed that all the reducing agents can activate Pd3 NCs by removing phosphine and chloride ligands from their surfaces. After activation, the Pd atoms interact with oxygen atoms on the surface of γ-Al2O3. In the case of NaBH4 and LiBH4-activated samples, the structure and morphology of the NCs upon activation depend on the amount of reducing agent used.
XPS analysis was conducted on activated Pd3/Al2O3 samples to investigate the speciation of Pd and validate the presence of B atoms in the samples. The Pd 3d region of XPS spectra for Pd3/Al2O3, Pd3/Al2O3–NaBH4(A/B/C), and Pd3/Al2O3–LiAlH4(A/B/C), along with the Cl 2p, B 1s, and P 2s regions of Pd3/Al2O3–NaBH4(A/B/C) have been shown in Fig. 2. The Pd 3d XPS spectra of the Pd3/Al2O3 showed peaks corresponding to two components of Pd, with 3d5/2 peaks at approximately 336.1 and 338.0 eV and an area ratio close to 1:2, consistent with XPS results reported in the literature (Fig. 2a).16 In the case of the samples activated using NaBH4 (Pd3/Al2O3–NaBH4(A/B/C)), two species of Pd can be seen in the XPS spectra i.e., Pd(0) and Pd(II) with Pd 3d5/2 peaks at around 335.3 and 337.2 eV respectively (Fig. 2b). The fraction of Pd(0) relative to Pd(II) can be seen increasing with the amount of NaBH4 used for the activation progressing from Pd3/Al2O3–NaBH4(A) to Pd3/Al2O3–NaBH4(C), which can be due to reduction of Pd(II) to Pd(0) upon exposure to NaBH4. For Pd3/Al2O3–NaBH4(C), the XPS spectra only showed Pd(0), with the Pd(II) fraction completely absent. This observation is consistent with the XAS analysis, which indicated that the size of the Pd NPs increased at higher NaBH4 loadings, while the interaction of Pd atoms with the O atoms of the γ-Al2O3 decreased. The interaction of Pd with surface O of γ-Al2O3 likely accounts for the presence of the high-valent Pd(II) species. The absence of high valent Pd(II) species in the XPS data for the Pd3/Al2O3–NaBH4(C) sample may be due to the presence of buried Pd–O interfaces for the larger Pd NPs (as EXAFS data shows a Pd–O CN of 1.4(3) for this sample).
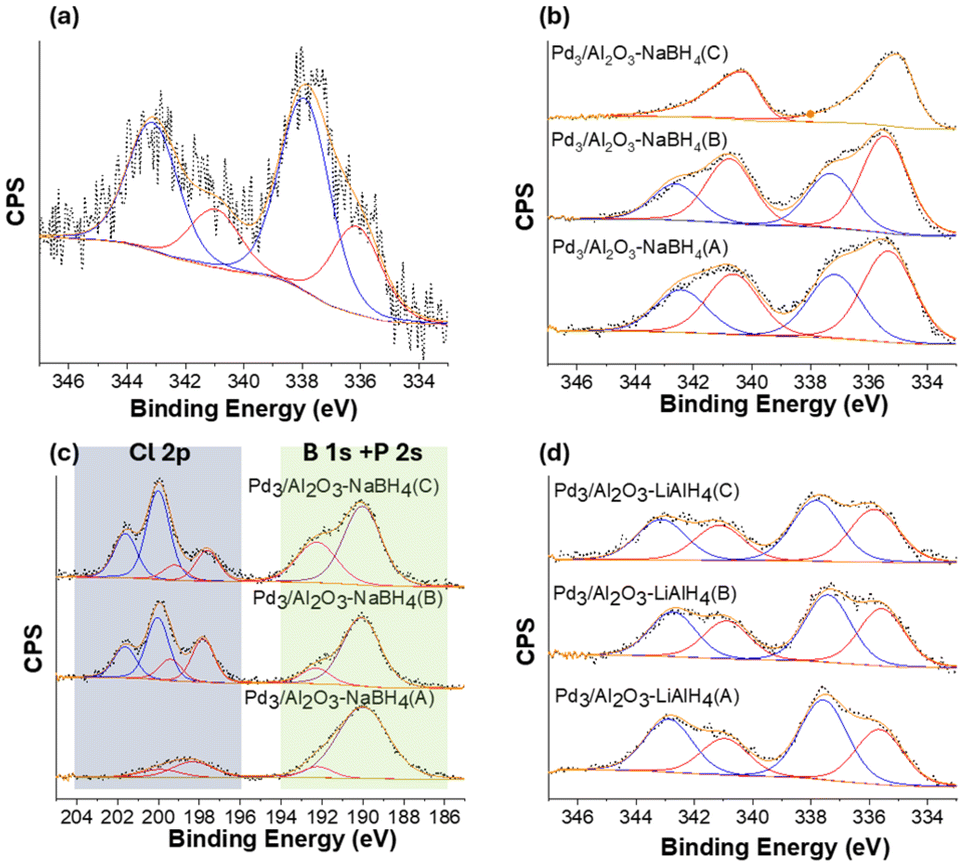 | ||
| Fig. 2 (a) XPS Pd 3d spectra of Pd3/Al2O3, XPS Pd 3d spectra of Pd3/Al2O3 catalysts treated with (b) NaBH4, and (d) LiAlH4; black dots and orange solid lines represent the experiment data and fit respectively; red lines represent the 3d5/2 (right) and 3d3/2 (left) regions for Pd(0); blue lines represent the 3d5/2 (right) and 3d3/2 (left) regions for Pd(II). (c) XPS Cl 2p + B 1s + P 2s spectra of Pd3/Al2O3 catalysts treated with NaBH4; black dots and orange solid lines represent the experiment data and fit respectively; red lines represent the 2p3/2 (right) and 2p1/2 (left) regions for the first Cl component; blue lines represent the 2p3/2 (right) and 2p1/2 (left) regions for the second Cl component; purple lines represent the P 2s region and pink lines represent the B 1s region. | ||
While Pd–P and Pd–Cl interactions were not present in the EXAFS spectra of the Pd3/Al2O3–NaBH4(A/B/C), in the XPS analysis, peaks for phosphine and chloride species can be observed for these samples (Fig. 2c). Most likely, these ligands were adsorbed onto the surface of γ-Al2O3 after being removed from the metal core of Pd3 NCs during the activation. In the literature, thiolate-protected Au25(SR)18 and Ag25(SR)18 NCs have been reported to exhibit similar behavior during thermal activation on a solid support.18,35 A very weak feature in the Cl 2p region of Pd3/Al2O3–NaBH4(A) can be seen with the Cl 2p3/2 peak at 198.3 eV. With further increase in the concentration of the NaBH4 for the activation i.e., Pd3/Al2O3–NaBH4(B/C), peaks for two distinct components of Cl can be observed with Cl 2p3/2 peak position at 197.6 and 200.0 eV. The peak at around 197.6 eV can be assigned to NaCl and the peak at around 200.0 eV is likely due to the interaction between Al and Cl.36,37 A strong P 2s signal in the samples can be seen at ca. 190 eV. The B 1s region in the XPS is very close to the P 2s region. In the case of Pd3/Al2O3–NaBH4(A/B/C), a B 1s component in the XPS spectra at around 192.3 eV (Fig. 2c) has been fitted. The hydrolysis of NaBH4 typically produces borates as side products,38 and the location of the B 1s peak in the spectra of the activated samples is close to that of the reported shift of sodium tetraborate (i.e., 192.6 eV).36 Therefore this peak can be assigned to tetraborate side products adsorbed on the γ-Al2O3 support. The relative peak area of this 192.3 eV B 1s peak increased with higher amounts of NaBH4 used for activation. This suggests that higher amounts of the reducing agent resulted in greater adsorption of tetraborate species on the surface of the γ-Al2O3. While XAS studies indicated that B also begins to intercalate into Pd NPs at large NaBH4 excesses, a separate B 1s peak for this species can not be assigned, which is likely due to overlap with the large P 2s signal present at 190 eV.
Similar to Pd3/Al2O3–NaBH4(A/B), the Pd 3d XPS spectrum of Pd3/Al2O3–LiBH4(B) (Fig. S7a†) showed the presence of two Pd components with Pd 3d5/2 peaks at 335.3 eV for Pd(0) and 337.1 eV for Pd(II). The peak area for the Pd(II) component was found to be slightly lower than that of Pd(0). Conversely, in the case of the LiAlH4-treated samples i.e., Pd3/Al2O3–LiAlH4(A/B/C), the peak area for the Pd(II) component has been found higher than that of Pd(0), indicating that much of the Pd is present in a higher-valent state after activation (Fig. 2d). Moreover, the relative peak areas of Pd 3d regions for Pd3/Al2O3–LiAlH4(A/B/C) did not vary much from each other, suggesting identical speciation of Pd, which is consistent with EXAFS analysis. The Al 2p XPS spectra for the Pd3/Al2O3–NaBH4(A/B/C) and Pd3/Al2O3–LiAlH4(A/B/C) are shown in Fig. S7b and S7c,† respectively. The peak in the Al 2p region at approximately 74.1 eV is consistent with the reported binding energy value of γ-Al2O3.39 The Al 2p region did not show the appearance of any new peaks after treatment with the reducing agents, suggesting that the surface γ-Al2O3 remained unchanged during the reducing agent treatments.
To validate the presence of adsorbed side products of B in the NaBH4-and LiBH4-activated samples, solid-state 11B magic angle spinning (MAS) NMR analysis was performed on Pd3/Al2O3–NaBH4(A/B/C) and Pd3/Al2O3–LiBH4(B) samples (Fig. 3). The 11B MAS NMR spectra of all the activated NCs showed three peaks at 15.6, 10.5, and 1.3 ppm. The positions and shapes of these peaks are in good agreement with those of sodium tetraborate reported in the literature.40 This indicates that during the activation of Pd3 NCs, NaBH4 and LiBH4 formed tetraborate (or related species) as a side product and remained adsorbed onto the γ-Al2O3 surface even after several washing cycles. Similar peaks were seen for a control sample in which γ-Al2O3 was treated with NaBH4 in the absence of Pd3 NCs (Fig. 3a). For the Pd3/Al2O3–NaBH4(B) sample, a new small peak was found around 5.8–5.9 ppm in 11B MAS NMR, which increased in intensity further for the Pd3/Al2O3–NaBH4(C) sample. Chan et al. reported a similar shift for the peaks of intercalated or surface B in fcc Pd.31 Therefore, the peak at 5.9 ppm in 11B MAS NMR was attributed to the intercalated B atoms inside the Pd lattice, which is consistent with EXAFS data showing Pd–B interactions in these samples. However, the new peak for intercalated B was not seen for the LiBH4-treated samples, showing that there was no B incorporation in the Pd NCs, likely because of the smaller concentrations of LiBH4 used for the activation compared to NaBH4. These results are consistent with the absence of a Pd–B interaction in the EXAFS data for the LiBH4-activated samples. Finally, 31P MAS NMR of these samples also indicated the complete removal of phosphine ligands from the Pd3 NCs, however, peaks for remaining phosphine impurities on the surface of the γ-Al2O3 after activation were present (Fig. S8†).
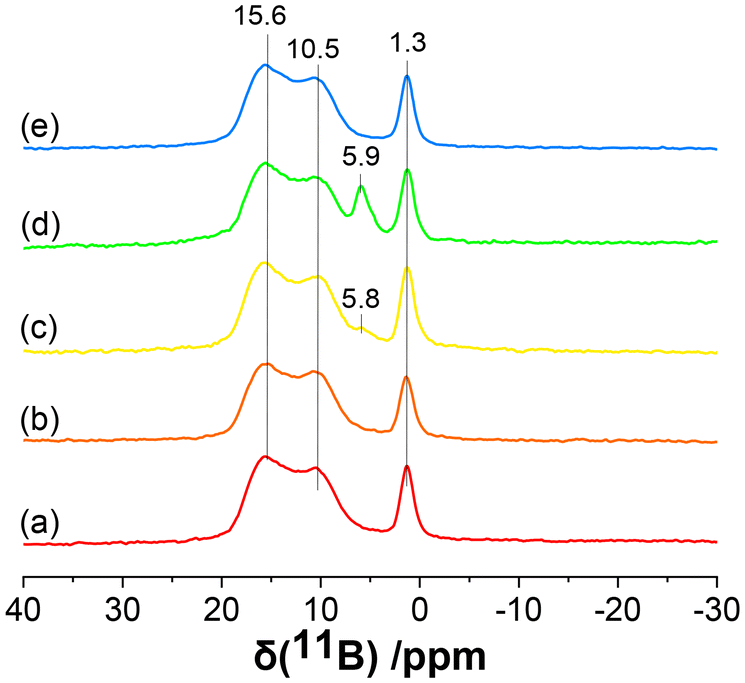 | ||
| Fig. 3 11B MAS NMR of (a) Al2O3–NaBH4(C) control, (b) Pd3/Al2O3–NaBH4(A), (c) Pd3/Al2O3–NaBH4(B), (d) Pd3/Al2O3–NaBH4(C) and (e) Pd3/Al2O3–LiBH4(B). | ||
HAADF-STEM analyses of the activated samples were performed to investigate the size, shape, and distribution of the NCs upon activation (Fig. 4). The Pd3 NCs appeared to be well-dispersed on the surface of the γ-Al2O3 (Fig. 4a), with an average NC size of 0.9 ± 0.3 nm. Upon activation with low amounts of NaBH4, the average NC size was 0.8 ± 0.4 nm for Pd3/Al2O3–NaBH4(A) (Fig. 4b). In contrast, for Pd3/Al2O3–NaBH4(B), an increase in Pd particle size was observed after activation, and Pd NPs with an average size of 3.4 ± 0.8 nm are apparent in both HR-TEM (Fig. S9a†) and HAADF-STEM (Fig. 4c) images. The HAADF-STEM image of Pd3/Al2O3–LiBH4(A) revealed the formation of NCs with an average size of 1.7 ± 0.5 nm (Fig. 4d), while activation with a higher concentration of LiBH4 in Pd3/Al2O3–LiBH4(B) resulted in slightly larger NCs (2.5 ± 0.6 nm) (Fig. S9b† and Fig. 4e). In the HAADF-STEM images of Pd3/Al2O3–LiAlH4(A) (Fig. 4f) and Pd3/Al2O3–LiAlH4(B) (Fig. S10†), Pd NCs with average sizes of approximately 1.1 ± 0.5 nm and 1.5 ± 0.4 nm are apparent. However, it should be noted that the EXAFS fitting revealed a very low Pd–Pd CN of around 0.5 for these samples, which would suggest that the sample contains both NCs and single-atom sites γ-Al2O3 surface, the latter of which could not be imaged.
 | ||
| Fig. 4 HAADF-STEM images of (a) Pd3/Al2O3 (avg. Pd size: 0.9 ± 0.3 nm), (b) Pd3/Al2O3–NaBH4(A) (avg. Pd size: 0.8 ± 0.4 nm), (c) Pd3/Al2O3–NaBH4(B) (avg. Pd size: 3.4 ± 0.8 nm), (d) Pd3/Al2O3–LiBH4(A) (avg. Pd size: 1.7 ± 0.5 nm), (e) Pd3/Al2O3–LiBH4(B) (avg. Pd size: 2.5 ± 0.6 nm) and (f) Pd3/Al2O3–LiAlH4(A) (avg. Pd size: 1.1 ± 0.5 nm). | ||
To visualize the dispersion of the Pd particles and borates on the γ-Al2O3 support, STEM-EDX elemental mapping analysis of the activated samples was performed. STEM-EDX images of Pd3/Al2O3–NaBH4(A) (Fig. S11†) showed a homogenous distribution of Pd over the surface of the γ-Al2O3 support. In the case of Pd3/Al2O3–NaBH4(B), some larger Pd particles can be observed on the γ-Al2O3 support in the STEM-EDX images (Fig. S12†). However, in both of these cases (i.e., for Pd3/Al2O3–NaBH4(A/B)), the B density did not seem to be localized near the position of Pd (Fig. S11 and S12†). In the case of Pd3/Al2O3–NaBH4(B), the atomic fraction of B in the provided STEM-EDX image was found to be around 0.23% (Table S2†). We believe the large amount of borates still present on the γ-Al2O3 support makes it very difficult to visualize small amounts of B incorporated into the Pd NPs (as evidence by EXAFS and NMR). For Pd3/Al2O3–LiBH4(B), the STEM-EDX maps (Fig. S13†) indicated the random distribution of large Pd NCs with an average size of around 2.6 nm on the γ-Al2O3 surface. However, no B signal was detected from this LiBH4-treated sample, likely due to the low concentration of tetraborate species on the γ-Al2O3 support (Table S2†). In the case of Pd3/Al2O3–LiAlH4(B), the STEM-EDX maps (Fig. S14†) showed a fairly homogeneous distribution of Pd over the γ-Al2O3 surface with a 0.35% Pd loading (Table S2†).
Overall, the results from the combination of the aforementioned techniques indicated that all the used reducing agents are capable of activating the Pd3 NCs on the γ-Al2O3 surface by removing phosphine and chloride ligands from the NC surfaces. Treatment with a low amount of NaBH4 in Pd3/Al2O3–NaBH4(A) led to the activation of Pd3 NCs without noticeable size growth, while treatment with higher amounts of NaBH4 in Pd3/Al2O3–NaBH4(A/B) resulted in the formation of larger Pd NPs with intercalated B atoms. In the case of LiBH4-treated samples, i.e., Pd3/Al2O3–LiBH4(A/B/C), slightly larger NCs were formed depending on the amount used for activation, and no intercalated B species were seen in the resulting Pd NCs. Finally, activation with LiAlH4 in Pd3/Al2O3–LiAlH4 (A/B/C) resulted in the formation of a mixture of Pd single-atom sites and very small NCs, and the size and distribution of the Pd particles were found to be independent of the amounts of the LiAlH4 used for the activation.
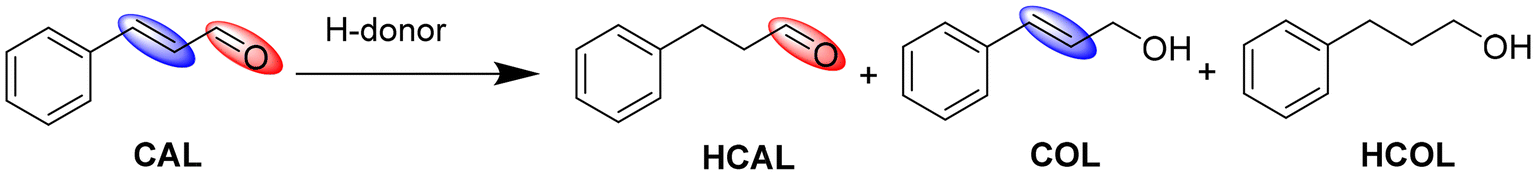
To measure the effect of activation of the Pd3 NCs on their catalytic activity, the transfer hydrogenation of an α,β-unsaturated aldehyde, trans-cinnamaldehyde (CAL), was performed. Selective partial hydrogenation of CAL can yield hydrocinnamaldehyde (HCAL) or cinnamic alcohol (COL), depending on whether the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or CO bonds are hydrogenated, respectively.41 However, upon complete hydrogenation of both CC and CO bonds, hydrocinnamic alcohol (HCOL) can be obtained.41 The products of this reaction, i.e., HCAL and COL have various applications in the pharmaceutical and fragrance fields.41 Thermodynamically, hydrogenation of the CC bond is favoured over the CO bond due to its lower bond energy.42 For this transfer hydrogenation reaction, formic acid and its formates have been selected as the hydrogen donors. Due to increased sustainability and cost-effectiveness, biomass-derived hydrogen donors such as formic acid and related formates have attracted research attention in the field of transfer hydrogenation.43 Formic acid has a very high volumetric capacity for hydrogen storage which can be released by either dehydrogenation or dehydration pathways.44 However, one major challenge in using formic acid in high concentrations at the industrial level is the corrosion of the reactors; thus, using neutralized acids (e.g., formates) instead of formic acid is more favourable.43
C or CO bonds are hydrogenated, respectively.41 However, upon complete hydrogenation of both CC and CO bonds, hydrocinnamic alcohol (HCOL) can be obtained.41 The products of this reaction, i.e., HCAL and COL have various applications in the pharmaceutical and fragrance fields.41 Thermodynamically, hydrogenation of the CC bond is favoured over the CO bond due to its lower bond energy.42 For this transfer hydrogenation reaction, formic acid and its formates have been selected as the hydrogen donors. Due to increased sustainability and cost-effectiveness, biomass-derived hydrogen donors such as formic acid and related formates have attracted research attention in the field of transfer hydrogenation.43 Formic acid has a very high volumetric capacity for hydrogen storage which can be released by either dehydrogenation or dehydration pathways.44 However, one major challenge in using formic acid in high concentrations at the industrial level is the corrosion of the reactors; thus, using neutralized acids (e.g., formates) instead of formic acid is more favourable.43
The reaction conditions for the transfer hydrogenation of CAL were optimized by using Pd3/Al2O3–NaBH4(A) as a model catalyst at 70 °C for 3 hours (Table 2). The reaction was first examined with formic acid as a hydrogen source and in the absence of a base, which resulted in a very low conversion, i.e., 12.2% (Table 2, entry 1). Upon adding 5 mmol of KOH to the reaction, the conversion increased to 63.4% (Table 2, entry 2). With a decrease in the amount of KOH from 5 mmol to 1 mmol, the conversion dropped by a factor of 2 (Table 2, entry 3). Previously, Lee et al. have shown that the decomposition of formic acid over Pd involves the formation and interaction of HCOO− with the metal center followed by C–H activation via β-hydride elimination reaction (Scheme S1†).45 The use of strong bases as the additive can make this process thermodynamically more favourable as the base can facilitate the formation of HCOO− species.44 Changing the solvent from MeOH to EtOH did not have much effect on the conversion (Table 2, entry 4). However, upon using water as a solvent, the conversion dropped significantly to 28.9% (Table 2, entry 5). The main reason behind this large drop is likely the poor solubility of CAL in water. With the absence of the base, the catalysts did not show any significant activity in water (Table 2, entry 6). Upon using the NaOH as a base in water, the catalyst showed 25.7% conversion (Table 2, entry 7). However, upon using a comparatively milder base, i.e., K2CO3, the conversion further dropped to 11.4% (Table 2, entry 8). At room temperature, no conversion was obtained in water with formic acid as the hydrogen donor and KOH as the base (Table 2, entry 9). Conversely, sodium formate as a hydrogen donor in the absence of a base provided 35.4% conversion in water (Table 2, entry 10), which further increased to 62.8% and 60.8% upon using EtOH and MeOH as the solvent, respectively (Table 2, entries 11 and 12). The reaction did not show any conversion when toluene was used as the solvent, probably due to the poor solubility of sodium formate in toluene (Table 2, entry 13). The best result, i.e., 68.7% conversion, was obtained using a mixture of water and EtOH as the solvent (Table 2, entry 14). However, at room temperature, the same conditions provided only a 3.3% conversion (Table 2, entry 15). This is likely due to the unfavorable conditions for C–H activation via β-hydride elimination at low temperatures (Scheme S1†). Doubling the amount of sodium formate resulted in a slight increase in conversion (Table 2, entry 16). Conversely, reducing the sodium formate to 1 mmol led to a conversion of only 21% (Table 2, entry 17). Finally, ammonium formate as the hydrogen donor gave a conversion rate similar to sodium formate (Table 2, entry 18). Notably, in all of these optimization reactions, no formation of COL was observed, and the selectivity towards HCAL remained consistently high, ranging from 85% to 93% (with HCOL as the other minor product). This suggests that the reaction selectivity remains largely unaffected by varying reaction conditions and is primarily governed by catalyst properties.
|
|
||||
|---|---|---|---|---|
| Entry | Hydrogen source/amount (mmol) | Solvent | Additive/amount (mmol) | Conversion% |
| Reaction conditions: 1.0 mmol of CAL, 4.0 ml of solvent, 25.0 mg of Pd3/Al2O3–NaBH4(A) (Pd mol% = 0.11%) at 70 °C for 3 hours.a Reaction was carried out at room temperature. The selectivity for HCAL in all these reactions is between 85–93% with no traceable formation of COL. | ||||
| 1 | HCOOH/5 | MeOH | — | 12.2% |
| 2 | HCOOH/5 | MeOH | KOH/5 | 63.4% |
| 3 | HCOOH/5 | MeOH | KOH/1 | 31.1% |
| 4 | HCOOH/5 | EtOH | KOH/5 | 68.3% |
| 5 | HCOOH/5 | H2O | KOH/5 | 28.9% |
| 6 | HCOOH/5 | H2O | — | 5.3% |
| 7 | HCOOH/5 | H2O | NaOH/5 | 25.7% |
| 8 | HCOOH/5 | H2O | K2CO3/5 | 11.4% |
| 9a | HCOOH/5 | H2O | KOH/5 | 0% |
| 10 | HCOONa/5 | H2O | — | 35.4% |
| 11 | HCOONa/5 | EtOH | — | 62.8% |
| 12 | HCOONa/5 | MeOH | — | 60.8% |
| 13 | HCOONa/5 | Toluene | — | 0% |
| 14 | HCOONa/5 | EtOH:H2O (1:1) |
— | 68.7% |
| 15a | HCOONa/5 | EtOH:H2O (1:1) |
— | 3.3% |
| 16 | HCOONa/10 | EtOH:H2O (1:1) |
— | 73.2% |
| 17 | HCOONa/1 | EtOH:H2O (1:1) |
— | 21.0% |
| 18 | HCOONH4/5 | EtOH:H2O (1:1) |
— | 63.9% |
After identifying the optimum conditions, non-activated Pd3/Al2O3 and all the activated NCs were employed as catalysts in the transfer hydrogenation of CAL using sodium formate as the hydrogen donor in a 1:1 H2O:EtOH solvent (Table 3). Non-activated Pd3/Al2O3 showed a low conversion of 38.3% with a selectivity of around 78.1% towards HCAL. The activated Pd3/Al2O3–NaBH4(A) catalysts showed improved activity, achieving a conversion rate of 68.7% and an improved selectivity towards HCAL of 90.5%. In the case of Pd3/Al2O3–NaBH4(B) and Pd3/Al2O3–NaBH4(C), the obtained conversions were very high (98.3% and 95.0%, respectively), however, these catalysts showed much lower selectivity towards HCAL and a higher proportion of the completely hydrogenated products, i.e., HCOL, formed. This result is consistent with the larger size of the Pd NPs under these activation conditions, and the higher activity may also be enhanced due to B-incorporation into the Pd NPs. Previously, it has been shown in the literature that the intercalation of B inside the Pd lattice can boost their efficiency in hydrogen generation from formic acid.46–48 As these samples also contained the B intercalated Pd particles, their higher catalytic activity for the transfer hydrogenation of CAL is likely associated with their enhanced capability of extracting hydrogen from formate species during catalysis. Pd3/Al2O3–LiBH4(A/B/C) catalysts demonstrated reasonable conversions between 63–73% coupled with high selectivity of around 90% towards HCAL. This is consistent with the moderate NC sizes seen after LiBH4 activation (1.7–2.5 nm) as evidenced by EXAFS and HAADF-STEM analyses. In contrast, Pd3/Al2O3–LiAlH4(A/B/C) catalysts showed slightly lower conversions, ranging from 49.6–61.1%, with selectivities towards HCAL exceeding 90%. Based on EXAFS and HAADF-STEM results, these catalysts consist of a combination of single-atom sites and small NCs ranging in size from 1.1 to 1.5 nm. Consequently, due to the small size of the Pd NCs, these catalysts exhibited lower catalytic activity but higher selectivity towards HCAL. To compare the catalytic performance of the chemically activated NCs with thermally activated NCs, Pd3/Al2O3 was treated at 150 °C for 3 hours to obtain Pd3/Al2O3-150.16 Previously, it has been found that after thermal activation at 150 °C, phosphines on the Pd3 NCs can be effectively removed without any increase in NC size on activated carbon supports; but the resulting Pd3 NCs have significant Pd–Cl interactions.16 In the hydrogenation of CAL, Pd3/Al2O3-150 exhibited a relatively lower conversion of 37.2% along with a very high selectivity towards HCAL. The lower activity of the thermally activated sample may be due to the presence of Cl ligands on the surface of NCs after thermal treatment.16
|
|
||||
|---|---|---|---|---|
| Catalyst | Conversion, % | Selectivity, % | ||
| HCAL | COL | HCOL | ||
| Reaction conditions: 1.0 mmol of CAL, 5.0 mmol of HCOONa, 4 ml of solvent (1:1 H2O:EtOH), 25.0 mg of catalyst (Pd mol% = 0.11%) at 70 °C for 3 hours. |
||||
| Pd3/Al2O3 | 38.3% | 78.1% | ≤0.1% | 21.9% |
| Pd3/Al2O3–NaBH4(A) | 68.7% | 90.5% | ≤0.1% | 9.4% |
| Pd3/Al2O3–NaBH4(B) | 98.3% | 71.2% | ≤0.1% | 28.7% |
| Pd3/Al2O3–NaBH4(C) | 95.0% | 67.1% | ≤0.1% | 32.9% |
| Pd3/Al2O3–LiBH4(A) | 69.3% | 91.2% | ≤0.1% | 8.7% |
| Pd3/Al2O3–LiBH4(B) | 72.3% | 89.9% | ≤0.1% | 9.9% |
| Pd3/Al2O3–LiBH4(C) | 63.3% | 93.2% | ≤0.1% | 6.8% |
| Pd3/Al2O3–LiAlH4(A) | 57.3% | 91.3% | ≤0.1% | 8.6% |
| Pd3/Al2O3–LiAlH4(B) | 49.6% | 90.0% | ≤0.1% | 9.9% |
| Pd3/Al2O3–LiAlH4(C) | 61.1% | 94.0% | ≤0.1% | 5.8% |
| Pd3/Al2O3-150 | 37.2% | 96.0% | ≤0.1% | 4.0% |

None of the catalysts exhibited significant formation of COL, consistent with previous work on Pd-based nano-sized catalysts.41,49,50 In the hydrogenation of CAL, the Pd-based catalysts are mainly selective towards HCAL, however, other noble metals such as Pt, Au, and Ir are more selective towards the formation of COL.41,49,50 This implies that the η4 adsorption mode of CAL is preferred over the Pd surface of these catalysts, which leads to the hydrogenation of the CC bond followed by the hydrogenation of the CO bond.41 Previously, Liu and co-workers demonstrated through DFT calculations that the adsorption energy of the CC bond on Pd4 NCs is lower than that of the CO bond, whereas, larger Pd particles with Pd(111) surfaces exhibit stronger CO bond adsorption.51 Thus, smaller Pd NCs are more selective for the conversion of CAL to HCAL. In recent work on di-1-adamantylphosphine-stablized Pdn (n = 2–5) NCs supported on carbon,52 Tang et al. noted that the Pd5 NCs exhibited high selectivity (95.3%) towards HCAL formation at a high conversion in the hydrogenation of CAL. Conversely, Pd2 NCs analogs showed significantly lower activity but maintained a high selectivity of 99.9% towards HCAL, while Pd3 NCs achieved complete conversion but a lower selectivity (80.5%) towards HCAL.52 These results indicated that changes in both the particle size and electronic structure of the Pdn NCs significantly influenced their activity and selectivity.51,52 Recent work by Li et al. showed that the carbon-nitride-nanosheet-supported Pd NCs along with Pd single-atom sites led to a complete conversion in the hydrogenation of CAL with a 97.3% selectivity towards HCAL.53 Using DFT calculations, a synergistic effect between single atom sites and NCs was found, where the CAL is preferentially adsorbed on Pd single-atom sites and hydrogen adsorbed on NCs.53 In the case of Pd3/Al2O3–LiAlH4(A/B/C), a mix of NCs and atomically dispersed Pd sites were observed, however, the conversions for these catalysts were somewhat lower compared to the reported results for this similar system.53 This may be due to a non-uniform distribution of the Pd sites on the γ-Al2O3 support in comparison to the homogenous distribution of Pd sites on g-C3N4.53 Overall the catalytic results in this manuscript showed that the post-activation evolution of NCs has a direct effect on the activity and selectivity of the catalysts. Larger Pd NPs have shown higher activity for the transfer hydrogenation reactions, likely due to the presence of well-faceted Pd(111) surfaces and the presence of intercalated B species in the Pd NPs. However, the presence of Pd(111) surfaces also led to strong adsorption of the substrate on the metal surface resulting in the increased formation of a completely hydrogenated product.54 However, in the case of small-size NCs and single-atom sites, the activity has been found lower due to weaker interactions of the catalyst with the substrates, which leads to higher selectivity toward the partially hydrogenated product, HCAL.
Conclusions
This work demonstrates the chemical activation of the γ-Al2O3-supported Pd3 NCs using various hydride-based reducing agents for transfer hydrogenation reactions. Different amounts of NaBH4, LiBH4, and LiAlH4 were used for activation, and the post-activation structural evolution of the NCs was monitored using a combination of various techniques. XAS results indicate that all reducing agents effectively removed surface phosphine and chloride ligands from Pd3 NCs. Both XAS and HAADF-STEM analyses showed that lower amounts of NaBH4 activated the Pd3 NCs without any noticeable size growth, while higher NaBH4 amounts caused a transformation of Pd NCs into Pd NPs. XPS, solid-state NMR, and STEM-EDX analyses revealed the presence of tetraborate and phosphine impurities on the surface of γ-Al2O3, even after post-activation washings in the NaBH4-treated samples. Additionally, solid-state NMR and EXAFS analysis suggested the intercalation of B atoms within the lattice of formed Pd NPs for samples activated with high NaBH4 loadings. Activation with LiBH4 resulted in a slight increase in the size of the Pd NCs, producing NCs ranging from 1.7 to 2.5 nm in size. Activation with LiAlH4 converted Pd3 NCs into a mixture of single-atom sites and small NCs around 1.4 nm in size. The post-activation evolution of Pd3 NCs directly impacted their catalytic behavior in the transfer hydrogenation of CAL. Smaller Pd NCs were highly selective towards the formation of HCAL, the partial hydrogenation product, whereas larger Pd NPs were more active catalysts, but also increased the complete hydrogenation of CAL to HCOL. Overall, our results demonstrate that immobilizing atom-precise Pd3 NCs onto an γ-Al2O3 support followed by chemical activation is an effective method to obtain supported Pd NCs for heterogeneous catalysis. This approach offers greater control over the structure and speciation of the Pd NCs compared to traditional thermal activation methods.Author contributions
All authors have approved the final version of the manuscript. S. S. and R. W. J. S. contributed to the initial concept development, experimental design, characterization of catalysts, analysis of results, and writing of the manuscript. D. W. carried out catalytic studies under the supervision of S. S. HR-TEM, HAADF-STEM and STEM-EDX analysis has been carried out by K. E. S. under the supervision of D. H.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial assistance from the National Sciences and Engineering Research Council of Canada. We thank Ning Chen and Weifeng Chen at the HXMA beamline (Canadian Light Source) for their assistance in XAS measurements. XAS experiments were performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation, the National Sciences and Engineering Research Council of Canada, the National Research Council, the Canadian Institutes of Health Research, the Government of Saskatchewan, and the University of Saskatchewan. The Saskatchewan Structural Sciences Centre (SSSC) is acknowledged for providing NMR and XPS facilities to conduct this research. Sarah Purdy and Jianfeng Zhu at SSSC are acknowledged for their assistance with XPS and solid-state NMR analysis, respectively. Funding from the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, and the University of Saskatchewan support research at the SSSC. Electron microscopy was performed at the Canadian Centre for Electron Microscopy at McMaster University.References
- C. Dong, Z. Gao, Y. Li, M. Peng, M. Wang, Y. Xu, C. Li, M. Xu, Y. Deng, X. Qin and F. Huang, Nat. Catal., 2022, 5, 485–493 CrossRef CAS
.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen, N. Wang and H. Liu, J. Am. Chem. Soc., 2018, 140, 13142–13146 CrossRef CAS PubMed
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y. G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS
- I. Cano, A. Weilhard, C. Martin, J. Pinto, R. W. Lodge, A. R. Santos, G. A. Rance, E. H. Åhlgren, E. Jónsson, J. Yuan and Z. Y. Li, Nat. Commun., 2021, 12, 4965 CrossRef CAS PubMed
- X. Li, S. Mitchell, Y. Fang, J. Li, J. Perez-Ramirez and J. Lu, Nat. Rev. Chem., 2023, 7, 754–767 CrossRef CAS PubMed
- N. Jeddi, N. W. J. Scott, T. Tanner, S. K. Beaumont and I. J. S. Fairlamb, Chem. Sci., 2024, 15, 2763–2777 RSC
- T. I. Levchenko, H. Yi, M. D. Aloisio, N. K. Dang, G. Gao, S. Sharma, C. Dinh and C. M. Crudden, ACS Catal., 2024, 14, 4155–4163 CrossRef CAS
- D. Yazaki, T. Kawawaki, D. Hirayama, M. Kawachi, K. Kato, S. Oguchi, Y. Yamaguchi, S. Kikkawa, Y. Ueki, S. Hossain, D. J. Osborn, F. Ozaki, S. Tanaka, J. Yoshinobu, G. F. Metha, S. Yamazoe, A. Kudo, A. Yamakata and Y. Negishi, Small, 2023, 19, 2208287 CrossRef CAS PubMed
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed
- V. Sudheeshkumar, K. O. Sulaiman and R. W. J. Scott, Nanoscale Adv., 2020, 2, 55–69 RSC
- K. K. Li, C. Wu, T. Yang, D. Qin and Y. Xia, Acc. Chem. Res., 2023, 56, 1517–1527 CrossRef PubMed
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682–9693 CrossRef CAS PubMed
- N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, Chem. Sci., 2019, 10, 7898–7906 RSC
- Y. Yun, H. Sheng, J. Yu, L. Bao, Y. Du, F. Xu, H. Yu, P. Li and M. Zhu, Adv. Synth. Catal., 2018, 360, 4731–4743 CrossRef CAS
- S. Singh, K. O. Sulaiman, Mahwar and R. W. J. Scott, Catal. Sci. Technol., 2024, 14, 322–333 RSC
- K. O. Sulaiman, M. Zubair, G. King, N. Bedford and R. W. J. Scott, Phys. Chem. Chem. Phys., 2022, 24, 24834–24844 RSC
- K. O. Sulaiman, V. Sudheeshkumar and R. W. J. Scott, RSC Adv., 2019, 9, 28019–28027 RSC
- Y. Liu, H. Tsunoyama, T. Akitab and T. Tsukuda, Chem. Commun., 2010, 46, 550–552 RSC
- V. Sudheeshkumar, M. Lavier, B. Lukan, J. Shen, N. Semagina and R. W. J. Scott, Catal. Today, 2023, 407, 223–229 CrossRef CAS
- J. Kilmartin, R. Sarip, R. Grau-Crespo, D. D. Tommaso, G. Hogarth, C. Prestipino and G. Sankar, ACS Catal., 2012, 2, 957–963 CrossRef CAS
- G. Collins, F. Davitt, C. Dwyer and J. D. Holmes, ACS Appl. Nano Mater., 2018, 1, 7129–7138 CrossRef CAS
- S. Das, A. Goswami, M. Hesari, J. F. Al-Sharab, E. Mikmeková, F. Maran and T. Asefa, Small, 2014, 10, 1473–1478 CrossRef CAS PubMed
- M. Dasog, W. Hou and R. W. J. Scott, Chem. Commun., 2011, 47, 8569–8571 RSC
- A. Shivhare and R. W. J. Scott, RSC Adv., 2016, 6, 62579–62584 RSC
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed
- F. Fu, J. Xiang, H. Cheng, L. Cheng, H. Chong, S. Wang, P. Li, S. Wei, M. Zhu and Y. Li, ACS Catal., 2017, 7, 1860–1867 CrossRef CAS
- T. J. Penfold, I. Tavernelli, C. J. Milne, M. Reinhard, A. E. Nahhas, R. Abela, U. Rothlisberger and M. Chergui, J. Chem. Phys., 2013, 138, 014104 CrossRef CAS PubMed
- J. Wang, Y. Kang, H. Yang and W. Cai, J. Phys. Chem. C, 2009, 113, 8366–8372 CrossRef CAS
- J. Li, J. Chen, Q. Wang, W. Cai and S. Chen, Chem. Mater., 2017, 29(23), 10060–10067 CrossRef CAS
- C. W. A. Chan, A. H. Mahadi, M. M. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed
- A. Leineweber, T. G. Berger, A. Udyansky and V. N. Bugaev, Z. Kristallogr. – Cryst. Mater., 2014, 229, 353–367 CrossRef CAS
- T. T. Zhuang, D. H. Nam, Z. Wang, H. H. Li, C. M. Gabardo, Y. Li, Z. Q. Liang, J. Li, X. J. Liu, B. Chen and W. R. Leow, Nat. Commun., 2019, 10, 4807 CrossRef PubMed
- A. P. Deziel, S. Gahlawat, N. Hazari, K. H. Hopmann and B. Q. Mercado, Chem. Sci., 2023, 14, 8164–8179 RSC
- B. Zhang, A. Sels, G. Salassa, S. Pollitt, V. Truttmann, C. Rameshan, J. Llorca, W. Olszewski, G. Rupprechter, T. Bürgi and N. Barrabés, ChemCatChem, 2018, 10, 5372–5376 CrossRef CAS PubMed
-
C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr., NIST Standard Reference Database 20, Version 3.4 (web version), 2003 (https://srdata.nist.gov/xps/) Search PubMed
- X. Yang, D. Kim, D. Um, G. Kim and C. Kim, J. Vac. Sci. Technol., A, 2009, 27, 821–825 CrossRef CAS
- C. Su, M. Lu, S. Wang and Y. Huang, RSC Adv., 2012, 2, 2073–2079 RSC
- B. R. Strohmeier, Surf. Sci. Spectra, 1994, 3, 135–140 CrossRef CAS
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef PubMed
- X. Wang, X. Liang, P. Geng and Q. Li, ACS Catal., 2020, 10, 2395–2412 CrossRef CAS
- P. Gallezot and D. Richard, Catal. Rev.: Sci. Eng., 1998, 40, 81–126 CrossRef CAS
- R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang, X. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC
- W. J. Lee, Y. J. Hwang, J. Kim, H. Jeong and C. W. Yoon, ChemPhysChem, 2019, 20, 1382–1391 CrossRef CAS PubMed
- Z. Li, J. Xu, F. Meng, K. Yang and D. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 30735–30745 CrossRef CAS PubMed
- J. S. Yoo, Z. Zhao, J. K. Nørskov and F. Studt, ACS Catal., 2015, 5, 6579–6586 CrossRef CAS
- K. Jiang, K. Xu, S. Zou and W. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed
- S. Fujiwara, N. Takanashi, R. Nishiyabua and Y. Kubo, Green Chem., 2014, 16, 3230–3236 RSC
- Y. Zhao, M. Liu, B. Fan, Y. Chen, W. Lv, N. Lu and R. Li, Catal. Commun., 2014, 57, 119–123 CrossRef CAS
- F. Jiang, J. Cai, B. Liu, Y. Xu and X. Liu, RSC Adv., 2016, 6, 75541–75551 RSC
- J. Tang, T. Ge, W. Wang, C. Liu and J. Huang, Chem. Commun., 2023, 59, 8854–8857 RSC
- X. Li, J. Liu, J. Wu, L. Zhang, D. Cao and D. Cheng, ACS Catal., 2024, 14, 2369–2379 CrossRef CAS
- X. Zhao, Y. Chang, W. Chen, Q. Wu, X. Pan, K. Chen and B. Weng, ACS Omega, 2022, 7, 17–31 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03364g |
| This journal is © The Royal Society of Chemistry 2024 |