
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive review of enhanced CO2 capture using activated carbon derived from biomass feedstock
Shreyase Kundu†
a,
Tasmina Khandaker†b,
Md Al-Amin Mia Anik†a,
Md. Kamrul Hasana,
Palash Kumar Dhara,
Sagar Kumar Dutta a,
M. Abdul Latifc and
Muhammad Sarwar Hossain*a
a,
M. Abdul Latifc and
Muhammad Sarwar Hossain*a
aChemistry Discipline, Khulna University, Khulna-9208, Bangladesh. E-mail: sarwar@chem.ku.ac.bd
bDepartment of Chemistry, Bangladesh Army University of Engineering & Technology (BAUET), Qadirabad Cantonment, Natore-6431, Bangladesh
cDepartment of Chemistry, Begum Rokeya University, Rangpur-5404, Bangladesh
First published on 18th September 2024
Abstract
The increasing level of atmospheric CO2 requires the urgent development of effective capture technologies. This comprehensive review thoroughly examines various methods for the synthesis of carbon materials, modification techniques for converting biomass feedstock into carbon materials and pivotal factors impacting their properties. The novel aspect of this review is its in-depth comparison of how these modifications specifically affect the pore structure and surface area together with the exploration of the mechanism underlying the enhancement of CO2 adsorption performance. Additionally, this review addresses research gaps and provides recommendations for future studies concerning the advantages and drawbacks of CO2 adsorbents and their prospects for commercialization and economic feasibility. This article revealed that among the various strategies, template carbonization offers a viable option for providing control of the material pore diameter and structure without additional modification treatments. Optimizing the pore structure of activated carbons, particularly those activated with agents such as KOH and ZnCl2, together with synthesizing hybrid activated carbons using multiple activating agents, is crucial for enhancing their CO2 capture performance. Cost-benefit analysis suggests that biomass-derived activated carbons can significantly meet the escalating demand for CO2 capture materials, offering economic advantages and supporting sustainable waste management.
1. Introduction
In the last 20 years, the amount of atmospheric carbon dioxide (CO2), a potent greenhouse gas, has exhibited a steady increase, as documented by Goel (2021).1 This increase can be attributed to multiple factors, including the repercussions of the industrial revolution, characterized by an annual increment of 0.17 °C, and burning of fossil fuels. Industries such as those involved in energy production from coal, oil, and natural gases; cement manufacturing; chemical synthesis; metal production; and refineries significantly contribute to CO2 emissions. Consequently, the atmospheric CO2 concentration reached 423 parts per million (ppm) in 2023, surpassing the anticipated level of around 400 ppm or 0.04% of CO2 in the air by volume.2 This surge represents an approximately 50% increase since the pre-industrial era and an additional 13% since the turn of the millennium. The escalating rate of CO2 emissions raises concerns on various fronts, primarily because of its pivotal role in climate change and its adverse impact on human health. Manifestations of these impacts include respiratory complications, elevated blood pressure, and accelerated heart rates among individuals. Thus, to lower CO2 emissions, several carbon capture systems have been developed. Numerous initiatives focus on mitigating climate change by capturing and separating CO2, particularly from large power plants and the atmosphere, using different technologies such as membranes, absorption, microbial methods, cryogenics, and chemical looping. However, these technologies often face challenges such as high operational costs and substantial energy consumption. Fortunately, physical processes such as adsorption offer a cost-effective solution and have been widely used to capture various contaminants, including CO2.3 Despite the initial skepticism, the need to develop green technologies has become increasingly evident, given the incontrovertible link between CO2 emissions, climate change, and the resulting health risks. Therefore, the development and adoption of effective and socially acceptable solutions are imperative. Addressing the challenge of CO2 emissions necessitates innovative approaches and concerted efforts to safeguard both the environment and human well-being.Accordingly, adsorption has emerged as a viable and cost-effective approach for the sequestration and storage of carbon. Among the many absorbents used, those that stand out include polymers, carbon compounds, metal oxides, amine-based materials, zeolites, alumina, silica, and metal–organic frameworks (MOFs). In this case, zeolites exhibit sensitivity to moisture, adsorption temperature, and pressure, whereas carbon materials offer notable advantages. Carbon materials, including hydrocarbons, charcoal, carbon nanotubes, graphite, graphene, fullerene, and activated carbon, are simple to prepare and control. Efficient CO2 capture is possible due to their well-developed pore architectures and high specific surface area. Unlike MOFs, carbon materials are not moisture sensitive, making them more reliable for industrial applications.4 Therefore, it is necessary to develop carbon materials with remarkable capabilities for capturing CO2, given their abundant and economical sources. The process for the preparation of carbon materials is straightforward and easily manageable. Their substantial specific surface area and well-formed pore structures significantly enhance their capacity for capturing CO2 effectively. Furthermore, carbon materials exhibit resistance to moisture, unlike their alternatives. Additionally, activated carbon demonstrates the ability to function across a broad temperature range.
Activated carbons (AC) have emerged as incredibly encouraging adsorbents for CO2 capture due to their significantly enhanced physiochemical, mechanical, thermal, and chemical stability, as depicted in Fig. 1. Although numerous porous carbon-based materials exist, porous carbonaceous materials derived from biomass hold particular appeal as CO2 adsorbents. The preference for biomass as a material is driven by its excellent textural characteristics, high CO2 adsorption capacity, and ease of synthesis, combined with its cost-effectiveness and the renewable nature of biomass resources.5 Biomass, which is derived from both plant and animal origins, captures solar energy through photosynthesis, making it a sustainable energy source that can be used for heating or transformed into liquid and gaseous fuels using various methods. Among the diverse biomass sources, traditional options such as bagasse, algae, municipal waste, wet waste, forestry residues, agricultural crop residues, and wood processing by-products stand out due to their potential to reduce costs. Pyrolysis stands out as the predominant thermochemical technology for biomass conversion, wherein biomass undergoes decomposition into biochar, tar, and gas. Traditionally, biomass is subjected to pyrolysis, a thermal treatment conducted at moderate temperatures without oxygen, resulting in the production of biochar.6 Pyrolysis methods include different approaches such as slow or fast pyrolysis, low or high-temperature conditions, and dry or wet pyrolysis.7 A variety of biomass sources, including wood, non-wood materials, and agricultural or fruit residues, has been utilized for the production of charcoal. The creation of activated porous carbons, similar to biochar, involves the addition of an activating agent either before or after the biochar is produced. An efficient technique for producing activated porous carbon has been developed using Arundo donax biomass, which was activated in a single step with KOH.8 This innovative approach simplifies the conventional two-step activation process, thereby reducing the time constraints. Various activating agents, whether physical or chemical, are utilized in the production of these carbons, including steam, CO2, KOH, ZnCl2, H3PO4, HNO3, and H2O2.9 Changes in the pyrolysis conditions affect the yield, specific surface area, and surface chemistry of biochar. For instance, Ighalo et al. conducted pyrolysis at temperatures ranging from 250 °C to 650 °C, demonstrating an increase in specific surface area of the biochar with an increase in temperature, together with a decrease in its yield, as evidenced by BET data.10 The composition and quantity of functional groups on the surface of carbon materials are influenced by their preparation techniques and the characteristics of the employed biomass sources. Fig. 2 illustrates the formation of carbon fibers, activated porous carbons, and other byproducts that arise from the combustion of various biomass types, together with their potential applications across different industries.11
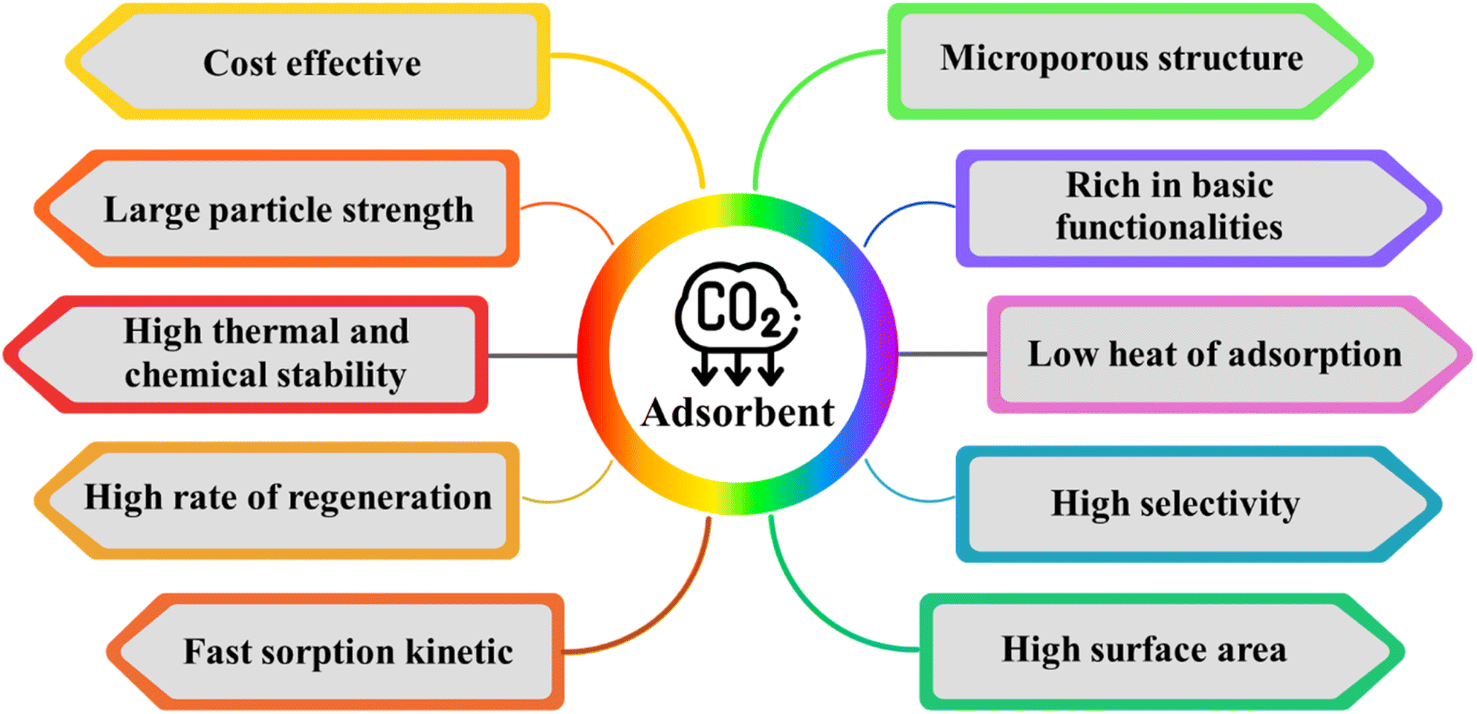 | ||
| Fig. 1 Properties of activated carbon for carbon dioxide capture. | ||
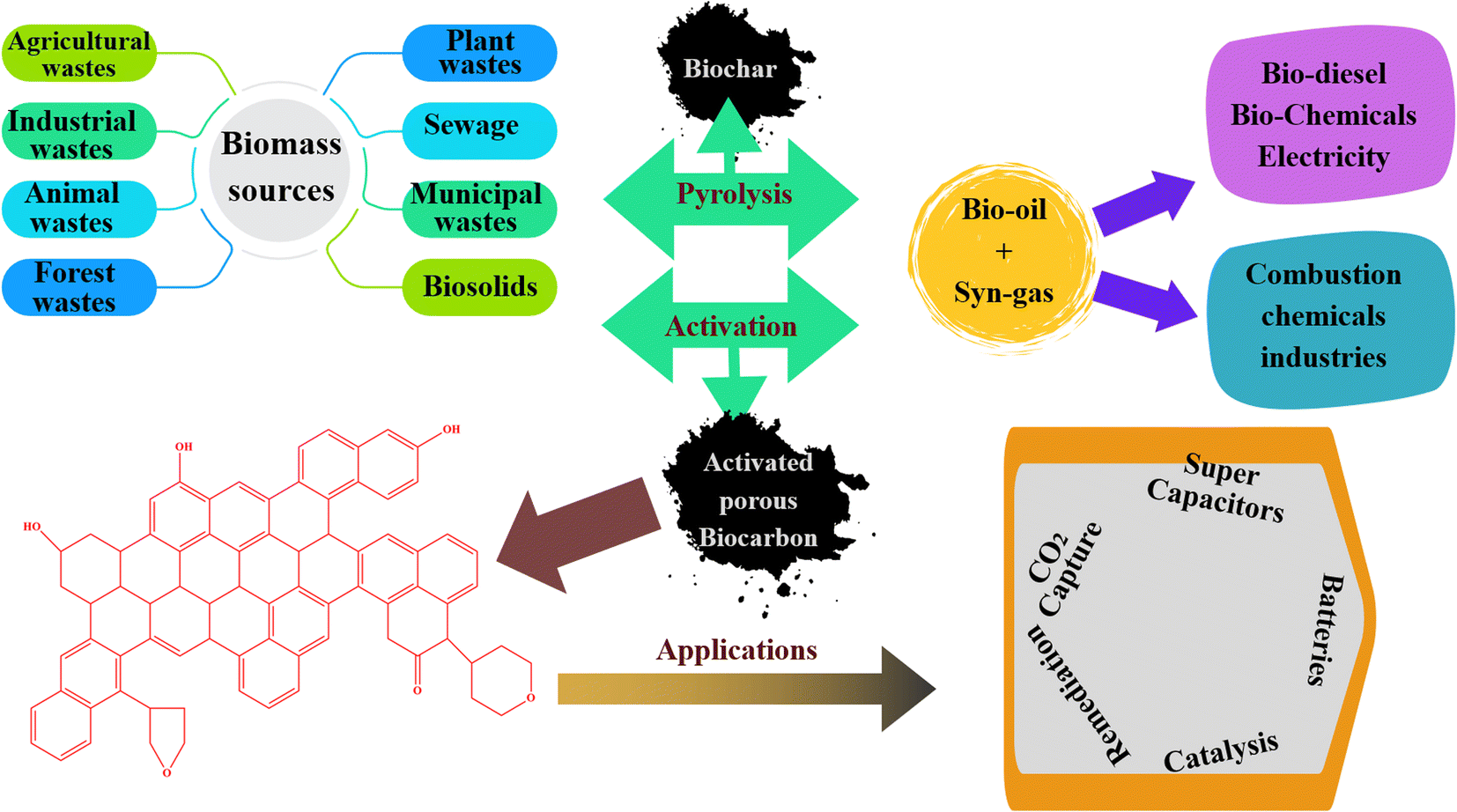 | ||
| Fig. 2 Illustration of the process of producing biochar and activated carbons from biomass together with their potential applications across different industries. Reproduced from ref. 11. | ||
Several studies have shown that unmodified biochar has limited effectiveness in absorbing contaminants, leading to the investigation of various modification techniques, including reactions with steam, acid or alkaline treatments, and doping with heteroatoms.12 To significantly improve the ability of carbon sorbents to capture CO2, it is essential to introduce basic sites and increase the porosity of the material. Nitrogen doping, a prevalent modification technique, involves the incorporation of nitrogen-containing chemicals such as polyethyleneimide (PEI) and triethylenephosphoramide (TEPA) into carbon materials, resulting in the introduction of pyridinic N or pyrrolic N groups, which both play pivotal roles in CO2 capture.13,14 For example, incorporating chitosan into carbonized materials has been shown to significantly improve their CO2 adsorption capacity, achieving 5.83 mmol g−1 at 0 °C.15 Additionally, metal oxides or salts are used to modify carbon materials, allowing the introduction of alkaline metal oxides or hydroxides. This modification increases the alkalinity of carbon materials, thereby enhancing their ability to adsorb acidic CO2. Research has demonstrated that biochar derived from walnut shells and impregnated with metal nitrates, particularly magnesium–biochar combinations, exhibits superior CO2 adsorption capabilities compared to other metal–biochar composites (such as that with aluminum, iron, nickel, calcium, or sodium).1,16 However, despite the progress in modification techniques and their application in CO2 capture, there is still a lack of comprehensive reviews on the methods for the preparation and modification of biomass-based carbon materials. Thus, this review provides a critical analysis of the preparation technologies and conversion conditions that influence the properties of carbon materials derived from biomass. Also, the emerging techniques for modifying these carbon materials, their applications in CO2 capture, and the underlying capture mechanisms are explored. Finally, the potential future directions and challenges for research in this field are highlighted.
2. Navigating thermochemical conversion technologies for biomass
A wide range of thermochemical conversion methods designed for utilizing biomass has been investigated, including pyrolysis, gasification, and hydrothermal processes. This study assesses their effectiveness in converting biomass into valuable energy resources, with a focus on environmental sustainability and resource optimization.2.1 Conventional pyrolysis
Pyrolysis, a thermochemical process devoid of oxygen, is instrumental in converting biomass into valuable products such as char, oil, and syngas through high-temperature heating.17 This process is highly versatile, encompassing variations such as fast, flash, and slow pyrolysis, which are categorized based on the temperature and heating rate. Typically, natural biomass derived from plants primarily consists of three key biopolymers, i.e., cellulose (making up 35–50% of the biomass), hemicellulose (comprising 25–30%), and lignin (constituting 10–30%), with a small fraction of mineralized inorganic components.18,19 These components differ in their chemical structures and undergo distinct, complex reactions during pyrolysis. Among the various pyrolytic conditions, temperature is regarded as the most critical factor, given that it greatly impacts the final textural properties and surface functionalities of biochar. Fig. 3 depicts the thermal decomposition behavior of these components over a wide temperature range, highlighting the specific temperature intervals at which each component undergoes thermal breakdown. The intricate mechanism of pyrolysis involves sequential decomposition stages of biomass constituents. Initially, water evaporation occurs, followed by the degradation of hemicellulose at around 200–350 °C. Subsequently, cellulose undergoes decomposition with an increase in temperature, while lignin is decomposed at temperatures exceeding 400 °C. These temperature-dependent reactions are pivotal in determining the final properties and functionalities of the resulting biochar. Among the pyrolytic conditions, temperature stands out as a critical parameter influencing the texture and surface characteristics of biochar. Slow pyrolysis, which operates at temperatures between 300–600 °C and with low heating rates of 0.1–1 °C s−1, primarily produces biochar, with the typical yields in the range of 30–50%.20 In contrast, fast and flash pyrolysis methods are designed to efficiently produce bio-oil. These methods are characterized by extremely high heating rates, such as 1000 °C s−1, and very short residence times.21 The pyrolysis cycle can be comprehensively understood through four distinct stages, as elucidated by the thermogravimetric analysis (TGA) of biomass. These stages involve moisture removal, hemicellulose degradation, cellulose decomposition, and lignin degradation, occurring in specific temperature ranges. Thus, a comprehensive understanding of these stages is crucial for optimizing the properties of biochar and activated porous carbons. This knowledge allows better control of the product selectivity, particularly in balancing the production of char, bio-oil, and syngas.22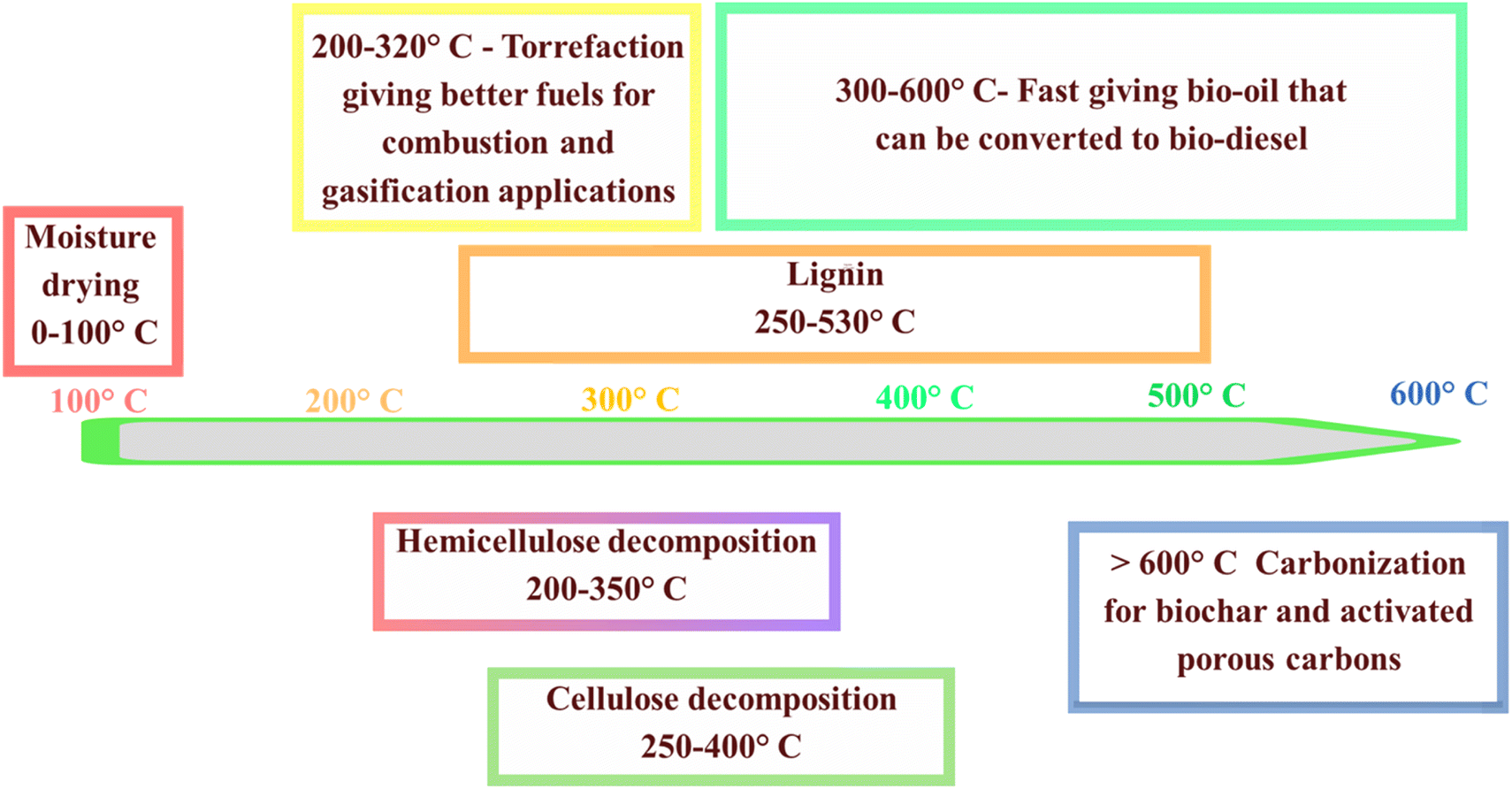 | ||
| Fig. 3 Decomposition temperatures for lignin, cellulose, and hemicellulose in biomass during thermal processing. | ||
The initial phase of pyrolysis involves removing moisture entirely at temperatures up to 220 °C, followed by the disintegration of hemicellulose in the temperature range of 220–315 °C. Subsequently, cellulose decomposes in the range of 315–400 °C, and lignin degradation occurs beyond 400 °C.23 Understanding the transformations these biopolymers undergo during pyrolysis is crucial for gaining deep insight into the overall mechanism. This deep understanding not only enables an improvement in the inherent properties of biochar and precise control of the selectivity for products such as char, bio-oil, and syngas, as well as activated porous carbons, but it also aids in selecting the most suitable biomass sources from a diverse range of naturally occurring feedstocks.24 Fig. 4 illustrates the quantities of biochar, syngas, and bio-oil produced under various pyrolysis conditions. Fast and flash pyrolysis techniques yield significantly more bio-oil than slow pyrolysis, mainly because the shorter residence time minimizes secondary cracking reactions, thereby increasing the bio-oil yield. Maintaining the optimal pyrolysis temperature is crucial for maximizing bio-oil production, given that higher temperatures typically favor its generation. However, conventional pyrolysis methods have inherent drawbacks, such as low heat transfer efficiency and prolonged heating times, resulting in high costs. In contrast, microwave pyrolysis offers rapid heating rates and shorter reaction times, thereby enhancing the productivity and reducing expenses. Additionally, water, a strong absorber of microwaves, enables the direct microwave-pyrolysis of high-moisture biomass without pretreatment. Comparative studies indicate that microwave pyrolysis results in biochar with a greater specific surface area and higher porosity compared to conventional pyrolysis under similar temperature conditions. However, conventional pyrolysis tends to produce a higher yield of char. This is because conventional methods often lead to reduced yields of biochar and bio-oil due to the increased occurrence of biomass cracking and gas formation.
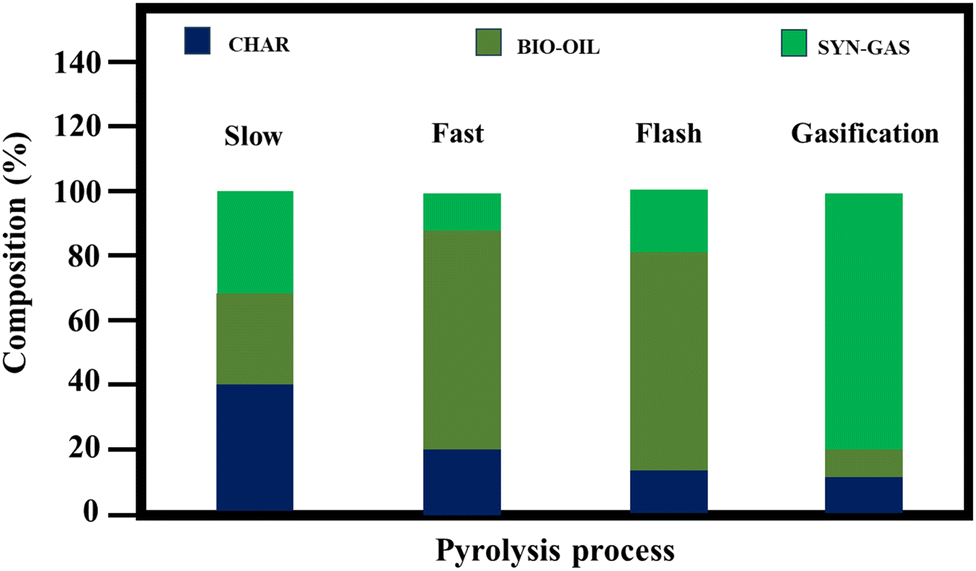 | ||
| Fig. 4 Proportional breakdown of char, bio-oil, and syngas produced through various pyrolysis methods. | ||
Table 1 shows the influence of the pyrolysis parameters on the generation of biochar, bio-oil, and syngas. Additionally, these insights will aid in identifying suitable biomass sources from the diverse array of naturally occurring biomass feedstocks. In summary, elucidating the mechanism of pyrolysis and its intricate stages not only improves the understanding of biochar production but also facilitates the development of efficient thermochemical conversion processes for the utilization of biomass, contributing to sustainable resource management and energy production.
| Classification of pyrolysis | Biochar/bio-oil/syngas% | Temperature range (°C) | Heating rate (°C min−1) | Holding time (s) |
|---|---|---|---|---|
| Flash | ∼10/65–70/20–25 | <650 | ∼1000 | <1 |
| Fast | ∼10–12/70–75/13–20 | 400–600 | >10 | 0.5–5 |
| Slow | ∼35/30/35 | 300–800 | <10 | min–h |
| Gasification | ∼10/5/85 | 700–1500 | ∼1000 | s |
2.2 Microwave pyrolysis
Microwave pyrolysis has emerged as a promising strategy for carbonizing raw materials, offering solutions to the challenges associated with conventional pyrolysis methods. Analogous to commercial microwave applications in daily life, this approach transfers electromagnetic energy to the precursor material, effecting rapid and targeted heating. Microwave pyrolysis offers distinct advantages compared to traditional furnaces, primarily due to its capability for precise and rapid heating, which results in a highly energy-efficient system. Additionally, carbon-based compounds with high microwave absorbance enable the selective absorption of microwave radiation, allowing the targeted heating of the reactants and promoting specific chemical reactions. Consequently, microwave pyrolysis is expected to result in higher char yields and maintain a more stable temperature profile.25 These attributes contribute to enhanced production rates and optimal production costs, underscoring the potential of microwave pyrolysis as a transformative approach in biomass conversion processes. Table 2 summarizes the key differences between conventional and microwave pyrolysis based on insights gathered from studies conducted by various research groups.26,27 The key advantage of microwave pyrolysis is its unique heating mechanism. Unlike traditional methods, which rely on convection and conduction through heated coils to transfer heat to materials, often facing limitations due to the thermal properties and surface temperature of materials, microwave pyrolysis uses the interaction between microwave energy and the dipoles in the target material to generate heat internally. This internal heating process makes the system more efficient than traditional surface heating, given that it facilitates uniform heat distribution and consistent heating throughout the material. Consequently, microwave pyrolysis is recognized for its energy efficiency, rapid processing, and time-saving procedures, facilitating high heating rates and temperatures.28 These advantages contribute to the superior reputation of microwave pyrolysis compared to traditional pyrolysis methods.| Physical parameters | Conventional pyrolysis | Microwave pyrolysis |
|---|---|---|
| Heating procedure | Inconsistent surface heating | Internal and bulk heating |
| Physical activation temperature (°C) | 750–1000 | 600–900 |
| Chemical activation temperature (°C) | 400–700 | 300–650 |
| Physical activation time (min) | 30–300 | 15–210 |
| Chemical activation time (min) | 30–120 | 5–20 |
| Process flexibility | Less flexible | Flexible |
| Heat control | Not as exact | Accurate and regulated heating |
| Char yield | 15–20% | 20–35% |
| Gas yield | 10–15% | 30–60% |
| Bio-oil yield | 65–75% | 30–60% |
| Capital plant expenses, USD | ∼16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
∼20000 |
| Complexity | Low | High |
Pecan nutshells were used as precursors for producing activated carbon through a one-step process. The nutshells were treated and mixed with two different chemical agents, potassium carbonate and potassium hydroxide, in a 1:1 ratio. Subsequently, this mixture was subjected to microwave pyrolysis at power levels of 300 W and 400 W for periods ranging from 2 to 6 min. Notably, microwave pyrolysis significantly improved the formation of microporous structures, especially in the samples activated with potassium hydroxide. For example, the samples pyrolyzed at 300 W for 6 min exhibited more than 73% ultra-microporosity. Additionally, these samples demonstrated an excellent CO2 adsorption performance, achieving 5.3 mmol g−1 at 1 bar and 0 °C. This level of performance was comparable to that of the activated carbon synthesized through conventional methods reported in the literature, underscoring the significant potential of microwave pyrolysis for future application in the production of activated carbon. Vairaja et al. investigated the performance disparities between conventional and microwave pyrolysis in producing AC.29 Both methods were employed to fabricate carbon activated from the husk of sunflower seeds. Following washing and drying, the precursor material was impregnated with varying concentrations of ZnCl2 and subjected to microwave carbonization at power levels ranging from 200 to 1000 W for durations spanning 5 to 40 min. Subsequently, the samples underwent conventional treatment. The results indicated that the highest the activated carbon surface area was achieved at 600 W microwave power. In a separate study, Ali and Idris examined the influence of activation processes on the characteristics of synthesized activated carbon.30 Pistachio shells were combined with different concentrations of K2CO3, KOH, ZnCl2, and H2SO4, and the impact of the microwave power level in the range of 450 to 800 W was assessed. The optimal yields were obtained using a microwave power of 600 W with 15 min of irradiation when K2CO3 was used as the activation agent. Additionally, the surface area of the activated carbon increased by nearly 600% when the power was increased from 450 W to 600 W. However, this increase was followed by a decline of 27% and 58% in surface area as the power was further increased to 700 W and 800 W, respectively. Microwave pyrolysis shows promising results in biochar production due to its high yield, reduced energy requirements, and shorter activation times. Despite these advantages, the use of microwave technology for the synthesis of activated carbon is still limited to laboratory-scale applications, largely due to the technical challenges associated with scaling up to industrial levels.
2.3 Mechanism of pyrolysis
In elucidating the mechanism of pyrolysis, it is crucial to delineate the sequential transformations that three key biopolymers undergo throughout the pyrolysis cycle. Understanding the intricate processes involved not only sheds light on the fundamental principles governing pyrolysis but also aids in identifying the optimal biomass sources for extraction. By comprehending the underlying mechanisms, researchers can discern the most suitable biomass substrates from the vast array of naturally occurring sources, thereby facilitating the efficient conversion of biomass into valuable products. This elucidation of the pyrolysis mechanisms not only enhances our understanding of bioenergy production but also contributes to sustainable resource utilization.The presence of hydroxyl groups (–OH) plays a pivotal role in the formation of biochar, particularly influencing its hydrophilic surface properties, which are crucial for facilitating the dispersion of metal species. This investigation specifically addresses the relationship between the functionalities of biochar and its polarities by subjecting cellulose to pyrolysis at temperatures in the range of 200 °C to 700 °C, and subsequently characterizing the resulting biochar. The findings revealed that –OH, rather than the carbon–oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carbon–oxygen–carbon (C–O–C) functionalities, significantly contribute to the formation of biochar with a hydrophilic surface. Furthermore, this study identified the temperature of 440 °C as crucial, marking the peak abundance of hydroxyl groups (–OH) and aliphatic carbon–hydrogen (C–H) bonds. Beyond this temperature threshold in the range of 460 °C to 700 °C, a notable transition from oxygen-rich functionalities to carbon-rich functionalities occurs, with the dominant aromatization process leading to a substantial increase in the hydrophobicity of the biochar. The hydrophilic nature of the biochar surface is vital in its utility as a support material for enhancing the dispersion of metals, such as copper (Cu), as demonstrated by the creation of bonding sites conducive to chelating with Cu2+ (Fig. 5).34
O) and carbon–oxygen–carbon (C–O–C) functionalities, significantly contribute to the formation of biochar with a hydrophilic surface. Furthermore, this study identified the temperature of 440 °C as crucial, marking the peak abundance of hydroxyl groups (–OH) and aliphatic carbon–hydrogen (C–H) bonds. Beyond this temperature threshold in the range of 460 °C to 700 °C, a notable transition from oxygen-rich functionalities to carbon-rich functionalities occurs, with the dominant aromatization process leading to a substantial increase in the hydrophobicity of the biochar. The hydrophilic nature of the biochar surface is vital in its utility as a support material for enhancing the dispersion of metals, such as copper (Cu), as demonstrated by the creation of bonding sites conducive to chelating with Cu2+ (Fig. 5).34
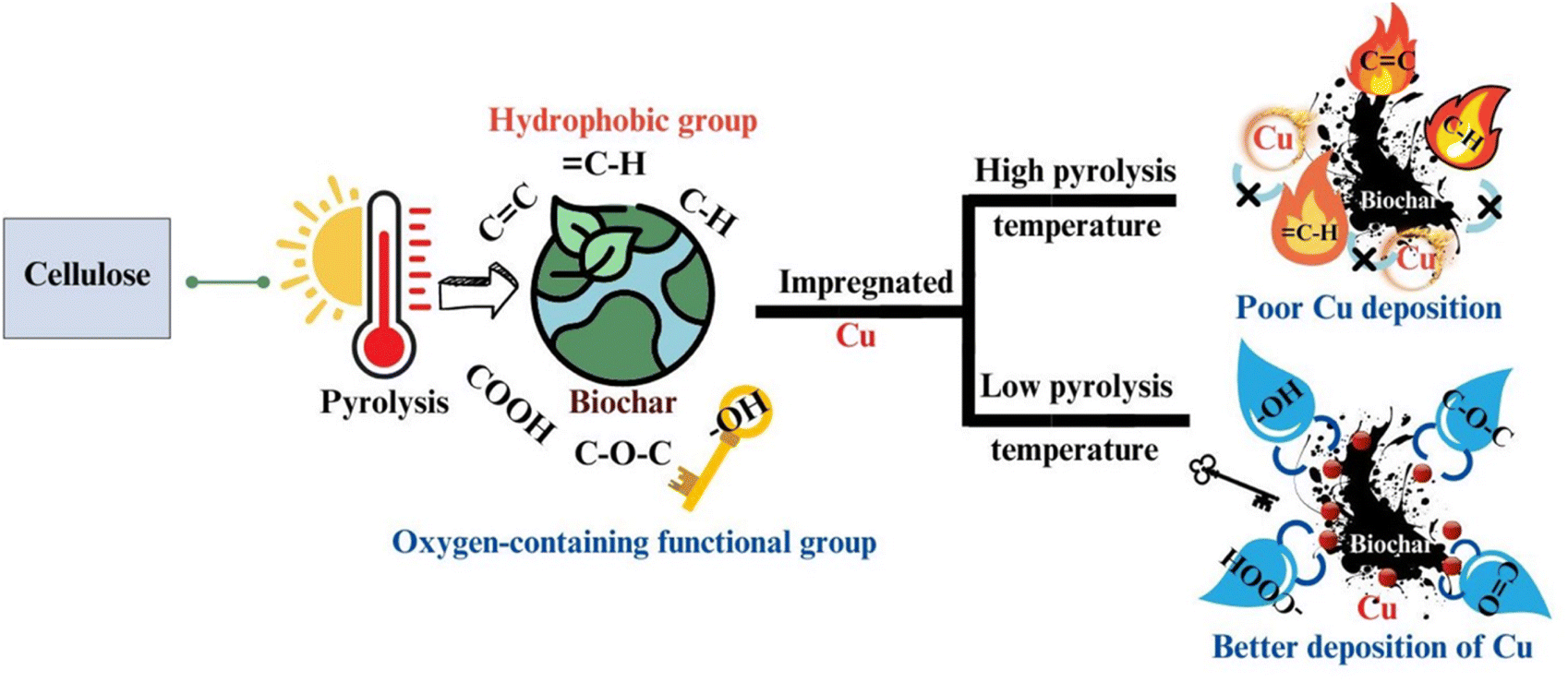 | ||
| Fig. 5 Schematic showing the relationship between hydrophilicity and functional development of biochar. Reproduced from ref. 34. | ||
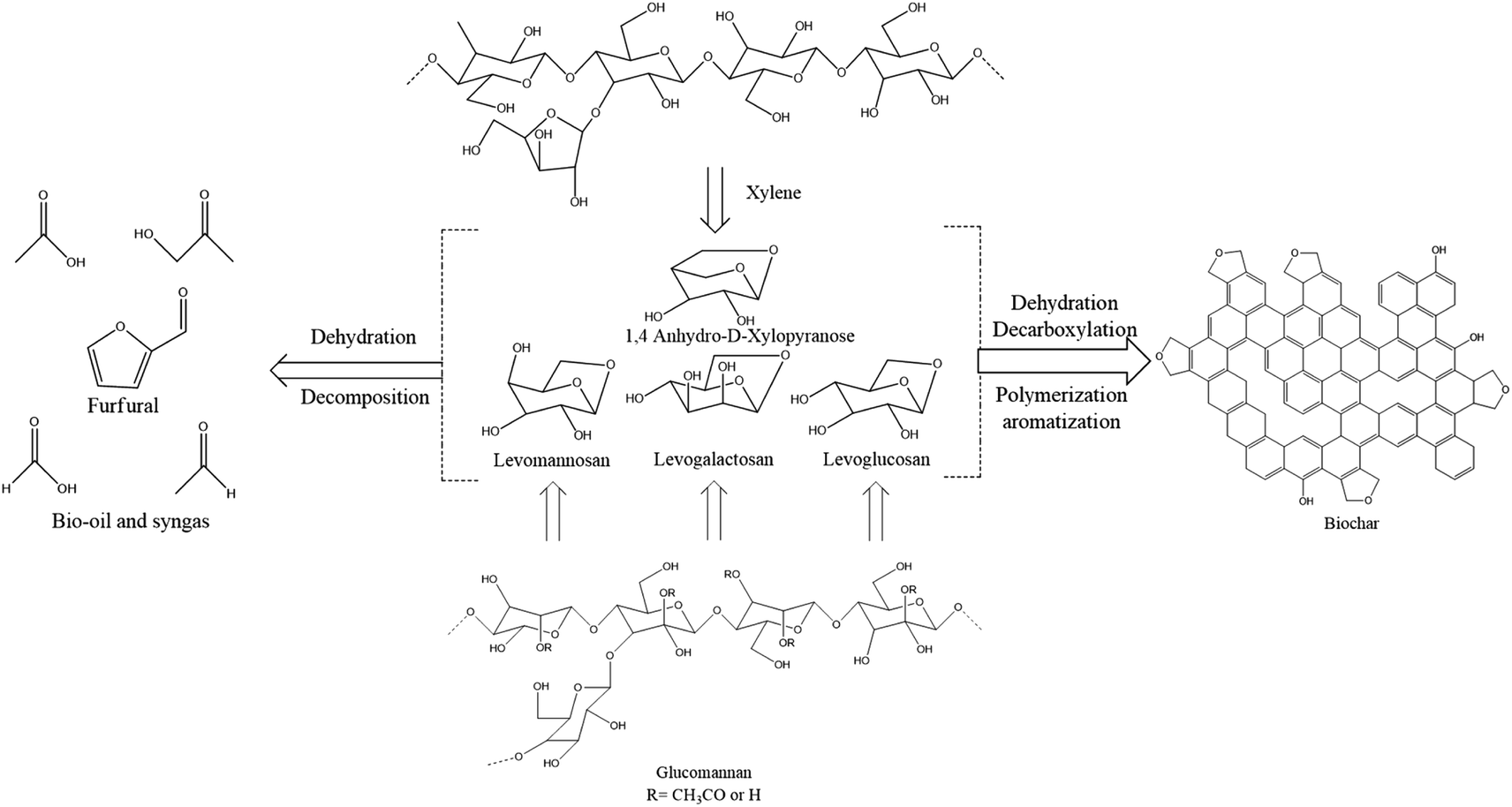 | ||
| Fig. 6 Illustration of the breakdown mechanism of hemicellulose during pyrolysis and subsequent biochar formation. | ||
O confer high polarity to lignin molecules. The pyrolysis of lignin is a complex process occurring over a wide temperature range owing to its diverse structure. The majority of interconnections in lignin involve β-O-4 linkages, constituting about 60% of its structure.38 Upon pyrolysis, these linkages undergo cleavage, generating free radical species. Subsequent radical reactions lead to chain polymerization, ultimately leading to the production of a biochar network.39 Moreover, methane gas is released during pyrolysis when the methoxy groups are broken. The intricate nature of biomass, combined with the distinct behavior of its constituent biopolymers at varying temperatures during pyrolysis, poses a significant challenge in elucidating the precise mechanism of the entire process. Furthermore, understanding the process by which activated porous carbons are derived from biomass presents an even greater challenge. Despite these complexities, previous review articles have attempted to describe the pyrolysis process, highlighting the ongoing efforts to unravel the intricate mechanisms underlying biomass conversion and biochar formation.362.4 Hydrothermal carbonization
Hydrothermal carbonization (HTC), recognized as hydrous pyrolysis, stands out as cost-effective and sustainable thermochemical conversion technology. It operates through exothermic reactions, primarily involving dehydration and decarboxylation reactions aimed at reducing the oxygen (O) and hydrogen (H) content in biomass. In a sealed reactor system using water as the reaction solvent, biomass undergoes a sequence of chemical transformations, including hydrolysis, dehydration, decarboxylation, polymerization, and aromatization. HTC is executed in the temperature range of 180 °C to 260 °C, under pressures of approximately 2–6 MPa, and for durations spanning 5–240 min.40 This process encompasses two main techniques, i.e., hydrothermal vaporization and liquefaction, which occur at temperatures exceeding 260 °C, leading to the substitution of gaseous and liquid fuel generation for solid fuel.41 A distinctive advantage of HTC lies in its ability to process high moisture content feedstock and wet biomasses, eliminating the need for pre-drying as required in traditional pyrolysis methods. This not only streamlines the process but also significantly reduces the energy consumption, thereby enhancing the overall efficiency.42 The resulting products from HTC include hydrochar (solid carbon matter), bio-oil (liquid), and small amounts of gases, with their properties and percentage distribution influenced by the process conditions.43Hydrocar has emerged as a crucial product due to its versatile applications, displaying properties such as hydrophobicity and low ash content, which improve the mass, energy density, and combustion performance compared to the original feedstock.44 Alternatively, biochar offers advantages in terms of porosity and surface area, making it suitable for applications such as adsorption and soil conditioning. HTC involves hydrothermal carbonization at both high and low temperatures (>220 °C, HHTC and ≤220 °C, LHTC, respectively). Although HHTC produces materials similar to coal under natural formation processes, LHTC, conducted under milder conditions, is more commonly employed in laboratories.45 Notably, current LHTC technology has been employed for the production of useful carbon compounds in a variety of shapes, including honeycomb and fibrous structures.46 HTC has significant utility in treating high-water-content biomass, similar to that seen in municipal sludge, resulting in considerable energy savings and positioning HTC as technology for green thermochemical conversion.
The primary byproducts of HTC treatment are gas, bio-oil, and hydrochar, which exhibit distinct characteristics. Hydrochar is comprised of matrix and coke microparticles, with the matrix formed through condensation, dehydration, and decarboxylation of unhydrolyzed biomass. In contrast, the coke microparticles originate from the aggregation of organic molecules and are rich in functional groups such as –OH, CO, and –COOH, facilitating their reactivity with organic molecules in the liquid phase. Overall, HTC represents a promising avenue for biomass conversion, offering a range of valuable products with diverse applications and demonstrating potential for both laboratory-scale research and industrial implementation (Table 3).71
| Material | Conditions | Product yield (%) | Ref. | ||
|---|---|---|---|---|---|
| Temperature (°C) | Residence time (h) | Activation | |||
| Apple pomace | 300 | 0.5 | Air | 40.5 | 47 |
| Prosopis juliflora | 300–700 | 1–4 | KOH | 62 | 47 |
| Yerba mate twig (YMT) | 400–600 | — | H3PO4 | 63 | 48 |
| Date palm waste | 500 | 2 | — | 75.4 | 49 |
| Cassava peel | 500 | 0.5 | — | 55 | 50 |
| Palm kernel shell | 450 | 1.5 | KOH/carbon = 2 at 750 °C | 57 | 51 |
| Rambutan peel | 600 | 3 | KOH/HC = 2 at 850 °C | 122 | 52 |
| Corn stover | 250 | 10 | KOH | 88 | 53 |
| Bamboo waste | 700 | 3.0 | Alkali metal salt | 70 | 54 |
| Cellulose | 450 | 1 | Alkali metal salt | 28 | 55 |
| Bamboo waste | 400–600 | 2–3 | Steam | 40 | 56 |
| Sugarcane bagasse | 450–600 | 1–3 | KOH | 55 | 57 |
| Plastic waste | 300 | — | KOH | 17 | 58 |
| Birch bark | 750 | 2 | KOH | 57 | 59 |
| Wood chips | 500 | 2 | — | 25 | 60 |
| Loblolly pine | 240 | — | — | 48.54 | 61 |
| Tobacco stalk | 180 | 2 | — | 80 | 62 |
| Rice husk | 400–700 | 1–2 | CO2 | 60 | 63 |
| Eucalyptus bark | 220 | 2 | — | 46.4 | 64 |
| Sweet potato waste | 180 | 1 | — | 63.18 | 65 |
| Lignocellulose | 200 | — | — | 50 | 66 |
| Holocellulose | 220 | 4 | — | 66.7 | 67 |
| Lignin | 300–390 | — | KOH/carbon = 2, 600 °C | 35.6 | 68 |
| Lignin | 300–390 | — | KOH/carbon = 2, 700 °C | 27.0 | 68 |
| D-Glucose with urea | 180 | 12 | KOH/carbon = 1–3, 600–700 °C | — | 69 |
| Food waste | 200 | 1 | — | 7 | 70 |
3. Key factors impacting the characteristics of carbon materials
Considering that pyrolysis stands out as the dominant method for biomass conversion, this section delves deeply into the diverse array of factors that influence the initiation and characteristics of carbon materials during pyrolysis. Besides the inherent qualities of biomass itself, significant emphasis is placed on manipulating the variables involved in the reaction. Parameters such as pyrolysis temperature, duration of residence, rate of heating, and ambient conditions all play pivotal roles in determining the outcomes of carbon materials. These variables not only govern the yield of char but also intricately mold its properties, encompassing aspects such as surface area and pH levels. Given the profound interplay between adsorption capacity and the attributes of sorbents, it is essential to meticulously scrutinize the full spectrum of factors that impact carbon materials.3.1 Biomass-derived feedstocks for the production of carbon materials
Exploring the diverse array of biomass-derived feedstocks for the production of carbon materials reveals a promising avenue for sustainable resource utilization. The categorization of feedstock for AC production plays a pivotal role in evaluating the nutrient content and determining appropriate conversion techniques. Carbon-rich biomass, derived from both plant and animal sources, stands as the primary feedstock for AC production, with the objective of maximizing the carbon content in solid form.72 Activated carbon encompasses carbon materials with a robust carbon framework and a highly developed internal pore structure and surface area. The properties of activated carbon are influenced by the structural polymers found in biomass, including cellulose, hemicelluloses, and lignin. A higher cellulose and hemicellulose content contributes to increased volatile vapor production, while lignin serves as the principal precursor for the synthesis of AC.73 The characteristics of biomass, including mineral matter content, significantly impact the pyrolysis properties and resultant product yield.74 Classification based on moisture content delineates between wet (above 30%) and dry (below 30%) biomass, aiding in the selection of suitable conversion methods.75 Biomass can be further categorized as ‘purpose-grown’ and ‘waste biomass’. Purpose-grown biomass, cultivated specifically for AC production, exhibits a low moisture content (<5%), high yield, and energy density, finding applications in the energy and biofuel sectors. Conversely, waste biomass, lacking economic value, serves as an alternative feedstock for char production, often undergoing hydrothermal carbonization due to its elevated moisture content.76 Biomass sources encompass a wide range, including animal sources (such as pig dung, pigskin, wool, and fishbone), plant sources (such as straw, rapeseed, sawdust, and peanut shell), and municipal waste (such as sludge).77 The main elements in biomass are carbon, hydrogen, and oxygen, with minor amounts of nitrogen and sulfur. The appropriate treatment of biomass enables its conversion into carbon materials with commendable adsorption capabilities. However, certain types of biomass, such as rice husk, which contains high levels of silicon, are commonly used in the production of silicon carbide (SiC).78 Thus, the selection of suitable biomass constitutes the initial step in preparing high-quality carbon-based adsorbents. This chapter primarily reviews the biomass thermochemical conversion technologies, including pyrolysis, gasification, and hydrothermal carbonization, together with the factors influencing the properties of carbon materials such as the feedstock, temperature, residence time, and heating.3.2 Pressure
In the domain of carbon material science, the influence of pressure stands as a pivotal yet often underestimated determinant. Pressure serves as a dynamic force that profoundly impacts the synthesis, structure, and properties of carbon materials across various manufacturing processes. Whether in the realm of crafting carbon nanomaterials or producing activated carbons, the application of varying pressure levels plays a central role in shaping the final product. Under high-pressure conditions, structural transformations are induced, porosity is altered, and mechanical properties are enhanced, thereby offering avenues for precise material tailoring. Conversely, low-pressure environments may foster the formation of specific carbon allotropes or facilitate unique chemical transformations, adding to the intricacy of material design. The significance of pressure is particularly notable in the production of biochar and hydrochar. The reactions occurring within the reaction chamber adhere to Le Chatelier's principle, highlighting the critical role of pressure modulation. Increasing the pressure within the chamber, either through the introduction of non-reactive gases or elevating reaction temperatures, leads to notable enhancements in the yield and quality of both hydrochar and biochar.45 This improvement is attributed to the accelerated decomposition of biomass, resulting in a superior end-product. In processes such as HTC, surpassing the critical pressure threshold enhances both the hydrolysis process and biomass decomposition, underscoring the multifaceted impact of pressure.79 Although the physical structure of hydrochar remains relatively unchanged under increasing pressure, finer and more uniformly distributed particles emerge, accentuating the role of pressure in morphology control. Similarly, in pyrolysis, an increase in pressure prolongs the residence time, facilitating the formation of secondary carbon and augmenting the char quality. Moreover, pressure exerts a tangible influence on the carbon content and energy density of biochar, with high-pressure pyrolysis yielding superior outcomes in these aspects.80 Understanding the subtle interplay between pressure and carbon materials is paramount for optimizing their performance across an extensive array of applications, spanning from energy conversion and storage to environmental remediation and beyond.3.3 Temperature
Temperature stands as a pivotal determinant in shaping the characteristics of carbon materials, exerting a profound influence on their synthesis, structure, and intrinsic properties. Varied temperature regimes dictate the ultimate outcomes of carbon materials, exerting notable effects on parameters such as porosity, surface area, and chemical reactivity. In addition to feedstock considerations, the significance of operating conditions cannot be overstated. Among these factors, the synthesis temperature holds particular prominence. As the pyrolysis temperature increases, discernible trends emerge, where the specific surface area and pH levels tend to increase, while the char yield experiences a decline. For instance, investigations by Singh et al. illustrated a notable reduction in biochar yield together with an increase in pH and surface area across the temperature range of 250 °C to 650 °C during the pyrolysis of rice husk.11 However, excessive temperatures can lead to detrimental effects, such as the destruction of surface and pore structures due to melting and collapse. This phenomenon can be ascribed to the high temperature-induced disintegration and reorganization of the biomass components. Moreover, pH levels exhibit a positive correlation with temperature due to the enrichment of alkaline minerals such as sodium and potassium under high-temperature pyrolysis conditions. Elevated pyrolysis temperatures also facilitate the expulsion of volatile substances, thereby contributing to the formation of more developed pore structures. The research by Lahijani et al. underscored the impact of pyrolysis temperature on the CO2 capture efficiency, revealing that the biochar produced at higher temperatures exhibited enhanced CO2 adsorption capabilities.81 This enhancement can be attributed to the fragmentation of the carbon particles, reduction in average pore size, and formation of micropores induced by increasing the carbonization temperatures. The presence of a microporous structure significantly augments the adsorption of CO2, underscoring the critical role of temperature in tailoring carbon materials for diverse applications, including environmental remediation and carbon capture technologies.3.4 Residence time
Residence time, which denotes the duration in which the feedstock undergoes thermal decomposition in a reactor at a predefined temperature, has emerged as a critical parameter influencing the characteristics of carbon materials. Particularly in pyrolysis processes, the residence time exerts a substantial influence on both the yield and physicochemical attributes of biochar, primarily by fostering pore formation and structural refinement.82 Prolonged reaction durations often yield positive outcomes, enhancing the energy density and facilitating the production of secondary hydrochar, distinguished by its polyaromatic structure. Conversely, shorter residence times tend to yield a higher hydrochar content, a phenomenon notably observed in HTC processes.83 Manipulation of the residence time induces the release of volatile compounds, consequently affecting the composition of the resulting materials, with a lower soil product content observed under increased residence times.84 Additionally, the residence time significantly impacts the texture of hydrochar, modulating processes such as polymerization and hydrolysis, and influencing the micropore diameter. Co-pyrolysis experiments involving agricultural residues and sewage sludge further elucidated the role played by the residence time, demonstrating a pronounced decrease in biochar yield with extended durations, suggesting the completion of organic compound degradation within 150 min.85 Correspondingly, surface area exhibits a notable increase with longer residence times, which is attributed to the enhanced thermal decomposition and subsequent porous structure development, potentially augmenting the CO2 adsorption capacity.86 However, a further increase beyond 150 min resulted in a decline in surface area due to structural collapse. These insights underscore the intricate relationship between residence time and properties of carbon materials, emphasizing the need for meticulous control and optimization to tailor the characteristics of materials for diverse applications, including carbon sequestration and environmental remediation.3.5 Heating rate
The rate at which heating occurs plays a crucial role in shaping the characteristics of carbon materials, influencing parameters such as carbonization efficiency, surface morphology, and chemical composition. Typically, accelerated heating rates result in higher carbonization temperatures, fostering rapid pyrolysis and potentially inducing structural alterations, whereas slower rates afford a more controlled decomposition process, yielding materials with distinctive surface functionalities. This aspect of the heating rate significantly impacts the pyrolysis carbonization mechanism and the characteristics of the resultant biochar. Notably, an increase in heating rate is often correlated with enhanced biomass weight loss. For instance, the research conducted by Li et al. scrutinized the pyrolysis of lignin at varying heating rates, revealing a decrease in biochar yield from 55.5% to 50.1% as the heating rate increased from 5 to 20 °C min−1, together with a concomitant rise in gas yield from 18.2% to 22.2%.87 Moreover, increased heating rates facilitate the cracking of organic constituents and volatiles within biochar, expediting the generation of gas. Despite the prevailing assumptions that higher heating rates uniformly augment the specific surface area of biochar, contradictory findings have been documented. Hu et al. observed a decrease in specific surface area from 362 to 146 m2 g−1 with an increase in the heating rate from 2 to 15 °C min−1, followed by an increase to 327 m2 g−1 at 25 °C min−1.88 This phenomenon is attributed to the intense decomposition and volatile diffusion occurring at steep heating rates, underscoring the interplay between the heating rate and biochar characteristics.4. Activated carbon
Activated carbon, commonly known as AC, encompasses carbonaceous materials featuring a sturdy carbon framework together with a fully developed surface area and interior pore structure. Various organic materials possess the potential for conversion into activated carbon, with petroleum coke, peat, and wood being the common precursors utilized in its development.89 Therefore, comprehending the composition and structure of the precursor material is imperative in determining the most suitable activating agent for AC production. The constituent structural polymers within biomass, including cellulose, hemicelluloses, and lignin, significantly influence the characteristics of the resulting AC. Typically, biomass is comprised of approximately 40% cellulose, 30% hemicellulose, and 30% lignin, with lignin requiring higher temperatures for complete decomposition compared to cellulose.90 Consequently, materials rich in lignin content tend to yield AC with a lower surface area at lower activation temperatures.91 Additionally, non-structural components such as proteins, ash, waxes, pectin, tannins, and chlorophylls contribute to the raw material composition.92 Micropores, among the various pore structures present, play a pivotal role in adsorption due to their small void space, facilitating the trapping of organic molecules. The micropores formed during the activation of carbon are crucial for the adsorption of carbon dioxide, given that they allow localized van der Waals forces to overlap, promoting strong electrostatic attraction and physisorption.93 A pore size of less than 1.0 nm is deemed effective for CO2 capture due to the small molecular size of CO2.94 The adsorption process exhibits low activation energy, enabling reversible adsorption and the potential for multiple-layer CO2 adsorption on AC, thereby enhancing the adsorption capacity. Thus, optimizing the synthesis process is paramount for producing AC with a high surface area, which is conducive to efficient CO2 adsorption. However, despite its stability and superior surface area, activated carbon faces challenges regarding economic and environmental viability.95 Recent advancements highlight the equivalent CO2 adsorption capabilities of activated carbon derived from various waste material precursors compared to their commercial counterparts, offering promising avenues for the sustainable production of AC.964.1 Carbon material modification techniques
In the field of carbon materials, investigating techniques for their modification is a crucial area of study driving research and innovation forward. This review offers a comprehensive examination of the various methodologies intended to customize the properties and functions of carbon materials, elucidating a wide array of modification techniques and their applications across different fields. Carbon materials derived from biomass in their original state exhibit inherent limitations in chemical composition and pore structure, restricting their ability to adsorb CO2 effectively. Therefore, modifying carbon materials becomes exceedingly significant, particularly in the realm of CO2 capture. The two primary approaches for modifying carbon materials are outlined, as illustrated in Fig. 7, i.e., pre-treatment, which involves directly mixing raw biomass with acids or metal salts before carbonization, and post-treatment, modifying carbonized biomass through activation with substances such as acids, bases, metal salts, and gases. Notably, it is imperative to treat raw biomass with diluted acid prior to its modification to remove its ash content, thus averting pore blockage during the carbonization process. This review thoroughly explores the modification methods, which are classified into three main categories of physical methods (steam/air/CO2 activation), chemical processes (acid/alkaline activation, metal or metal oxide impregnation, and heteroatom doping), and supplementary techniques (ball milling, microwave, ultrasound, and plasma treatment). Through a detailed analysis of these techniques, this review seeks to offer valuable insights into the latest advancements and future trajectories in the modification of carbon materials, aiming to enhance their performance and widen their range of applications. | ||
| Fig. 7 Schematic illustrating the two primary approaches for modifying carbon materials: (a) pre-treatment and (b) post-treatment methods. | ||
4.1.1.1 Steam activating methods. Steam activation is a significant technique for improving the textural characteristics of carbon materials by facilitating the creation of additional pores.97 This process involves the diffusion of steam into the carbon material, which promotes the formation of pores. The underlying mechanisms of steam activation encompass various reactions, including the interaction between carbon and water vapor to generate carbon monoxide and hydrogen, together with the production of methane and other gases.
| C + H2O → C(O) + H2 | (1) |
| C(O) → CO + C | (2) |
| CO + H2O → CO2 + H2 | (3) |
| C + 2H2O → CO2 + 2H2 | (4) |
| C + CO2 → 2CO | (5) |
| C + 2H2 → CH4 | (6) |
| CH4 + H2O → CO + 3H2 | (7) |
Observations demonstrate a notable enhancement in specific surface area and pore volume of biochar following its steam activation. For instance, Xie et al. employed steam activation to modify biochar derived from tea waste, resulting in a significant increase in specific surface area from 342 to 576 m2 g−1 and enhancement in pore volume from 0.022 to 0.109 cm3 g−1.98 The enhancement in pore structure resulting from steam activation can be attributed to the elimination of pore-clogging tar, the creation of new micropores, and the enlargement of existing pores. Different steam activation conditions, such as temperature and reaction time, have varying effects on the activation results. Other studies using cellulose fibers as a raw material revealed that steam activation facilitated the formation of new pores and influenced the expansion of the pore sizes, leading to a remarkable CO2 adsorption capacity of 3.78 mmol g−1.99 Furthermore, the steam activation of carbon derived from cellulose fibers resulted in the expansion of existing ultra-micropores and the generation of additional ultra-micropores (Fig. 8).100 This activation process disrupted the graphitic structure, resulting in pore sizes exceeding 2 nm and a significant increases in specific surface area and pore volume. Additionally, Chen et al. demonstrated that steam activation of biochar produced from pine sawdust not only enhanced its textural properties but also enriched its surface functional groups.101 The specific surface area increased from 316 to 582 m2 g−1, and the pore volume increased from 0.17 to 0.25 cm3 g−1. XPS analysis indicated an increase in the oxygen content of biochar from 9.16% to 11.05% following steam activation, which was attributed to the increase in the content of C–O, C–OH, and O–CO functional groups. These findings underscore the efficacy of steam activation in improving the textural properties and surface chemistry of carbon materials, thereby expanding their potential applications across various fields.
 | ||
| Fig. 8 Diagram illustrating the process of activating the surface of carbon with steam. | ||
4.1.1.2 Air activating methods. Air activation has emerged as a notable strategy for altering carbon materials, offering an environmentally friendly alternative compared to chemical activation techniques. This method involves utilizing air as the activating agent, eliminating the need for additional chemicals. Oxygen, known for its potent oxidizing properties, plays a crucial role in the activation process. However, due to the prohibitive cost of pure oxygen, air is commonly utilized for activation purposes. The underlying mechanism for air activation can be understood through various reactions,102 as follows:
| 2C + O2 (g) → 2CO | (8) |
| C + O2 (g) → CO2 | (9) |
| C(O) + O2 (g) → CO (g) + CO2 | (10) |
Air oxidation has the potential to introduce new functional groups into biochar. However, this oxidation process may lead to a slight decrease in surface area due to the new functional groups obstructing the pores. Kim et al. observed that air oxidation enriched the micropores of biochar and introduced O-groups, albeit resulting in reduced specific surface area.103 Furthermore, carbon materials produced at lower temperatures are more susceptible to oxidation, leading to the introduction of additional –COOH and CO groups. In comparison with steam activation, air oxidation tends to introduce O-containing groups more readily. Another research group reported differing outcomes between steam activation and air oxidation, with steam activation resulting in an increased surface area but decreased functional group concentration, while air oxidation led to a higher functional group concentration in biochar.104 Lee et al. conducted air activation on biochar at various temperatures and noted a significant enhancement in surface groups such as –OH, CO, and C–O.105 This enhancement is attributed to the reactions between O2 and the lignocellulosic components or aromatic substances within the biochar. Additionally, they examined the impact of activation time on the process, observing an initial increase in the micropores within the first 15 min, followed by an increase in the content of mesopores. However, a prolonged activation time could potentially lead to micropore destruction, highlighting the importance of selecting an appropriate activation duration. For carbon materials intended for CO2 capture applications, determining the optimal oxygen activation treatment time is essential to maximize their efficacy.
4.1.1.3 CO2 activating methods. CO2 activation has emerged as a significant technique for modifying carbon materials, providing an eco-friendly avenue to enhance their properties. This method involves employing CO2 as the activating agent, leading to tailored advancements in the pore structures and surface functionalities within carbon materials. Its adoption is gaining recognition due to its environmentally benign nature and the abundance of CO2 as a readily accessible resource. CO2 activation can be carried out either simultaneously with biomass pyrolysis or as post-pyrolysis treatment. Studies suggest that pyrolysis conducted in a CO2 atmosphere is advantageous for preserving C and N in biomass. Moreover, this activation approach significantly improves the porosity and specific surface area of carbon materials. For instance, Tomin et al. illustrated that CO2 activation predominantly yields micropores in porous carbon fibers, resulting in a specific surface area of up to 1012 m2 g−1.106 However, the development of mesoporous structures may be observed in certain carbon materials following CO2 activation, which is influenced by factors such as the type of pristine materials and activation conditions. Activation of various biochars with CO2, including wheat straw, softwood, and peach stones, resulted in a more pronounced development of mesoporous structure in wheat straw and softwood biochar, while peach stone biochar exhibited some enhancement in microporosity.107 CO2 activation involves the Boudouard reaction, an endothermic process, and thus higher temperatures favor CO2 activation. For instance, CO2 activation of biochar conducted at temperatures in the range of 800 °C to 950 °C showed an escalation in pore improvement with an increase in the activation temperature.108 Specifically, the specific surface area of biochar increased from 392 m2 g−1 to 506, 854, 1059, and 2186 m2 g−1, respectively, with micropores less than 2 nm dominating the pore structures for all the samples. These findings underscore the efficacy of CO2 activation in enhancing the porosity and specific surface area of carbon materials, offering promising avenues for their utilization in various applications.
4.1.2.1 Alkaline activating methods. Alkaline activation is a crucial method for modifying carbon materials, offering a flexible approach to customize their properties. This process involves the use of alkaline agents such as KOH and NaOH to activate the carbon precursors, leading to the formation of tailored pore structures and surface functionalities. This method is particularly advantageous for enhancing the CO2 adsorption capacity by promoting pore development and incorporating alkaline groups. The common alkaline activators include KOH, NaOH, and amino (–NH2) groups. Among them, KOH is widely recognized for its effectiveness as a metal hydroxide in activating carbon materials. Typically, the activation process begins by thoroughly mixing KOH with the carbon precursor. The redox reaction between KOH and the carbon precursor starts at around 400 °C, producing K2CO3. As the temperature increases to about 600 °C, KOH is fully converted into K2CO3. At temperatures above 700 °C, K2CO3 begins to decompose into CO2 and potassium oxide (K2O). Additionally, at even higher temperatures, both K2CO3 and K2O can be reduced to potassium (K), which infiltrates the carbon lattice in vapor form, assisting the formation of micropores and enhancing the porous structure.
The reaction sequence for KOH activation is outlined as follows:108
| 2C + 6KOH → 2K + 2K2CO3 + 3H2 | (11) |
| K2CO3 → K2O + CO2 | (12) |
| C + CO2 → 2CO | (13) |
| 2C + K2CO3 → 2K + 3CO | (14) |
| C + K2O → 2K + CO | (15) |
KOH activation enhances the surface functionality of carbon materials by introducing various groups such as –OH, CO, C–C, and = C–H. However, as the synthesis temperature increases, the O-containing groups on the surface of the carbon material decompose, resulting in the production of CO and CO2.109 Due to the acidic nature of carbon dioxide, incorporating Lewis bases on the surface of carbon materials has been proven to be advantageous for CO2 capture. An investigation into biochar derived from a mixture of food and wood waste demonstrated that although KOH activation led to superior microporous structures and enhanced CO2 adsorption capacity, activation with KOH–CO2 did not yield further improvements.110 Researchers explored the impact of KOH activation on the production of porous carbon from biomass for supercapacitors, noting the significant influence of KOH on the microstructure of the resulting char. The optimized carbon materials exhibited a specific surface area of up to 2790 m2 g−1, with a graded pore structure enhancing the rate performance and achieving a maximum specific capacitance of 327 F g−1.
Alternatively, NaOH activation offers stronger oxidizing properties and is more environmentally friendly. Studies indicate that the redox reaction between NaOH and carbon contributes to forming both micropores and mesopores in carbon materials. During activation, NaOH interacts with carbon materials through reactions as follows:111
| 2C + 6NaOH → 2Na + 2Na2CO3 + 3H2 | (16) |
| 2C + Na2CO3 → 2Na + 3CO | (17) |
| 2Na + CO2 → Na2O + CO | (18) |
Sodium hydroxide (NaOH) activation of carbon generates active intermediates such as CO, CO2, and H2, simultaneously facilitating the infiltration of Na and Na2CO3 into the carbon layer, which expands its atomic structure and enhances the specific surface area and pore diameter of the resulting carbon materials. However, excessive NaOH concentrations can intensify gasification reactions, ultimately reducing the effective area. Elevated NaOH concentrations may also induce a series of reactions (e.g., C–NaOH, C–Na2CO3, C–Na2O, C–Na, C–CO2, and C–CO), leading to the rupture of the C–C and C–O–C bonds and diminishing the yield of carbon materials.112 Studies by Shin et al. demonstrated the synthesis of carbon materials from macroalgae using NaOH activation, yielding materials with a significant surface area of 1238 m2 g−1. Another investigation highlighted the enhancement of aromatic properties in biochar through NaOH activation, promoting π–π interactions during pollutant adsorption.113
Incorporating amino groups into carbon materials also significantly enhances their CO2 capture capacity. NH3 treatment, typically performed at high temperatures, introduces basic functional groups and removes acidic groups from the surface of carbon. This treatment results in the formation of amides, lactams, imides, and heterocyclic compounds such as pyrrole and pyridine.114 Additionally, NH3 activation moderately improves the pore size and specific surface area. Studies by Wu et al. demonstrated an increase in the specific surface area of biochar from coconut fiber from 4, 7, and 541 m2 g−1 to 9, 24, and 553 m2 g−1, respectively, after NH3·H2O modification.115 Liu et al. synthesized microporous N-doped carbon materials using NH3 as the activator, highlighting the synergistic effects of pores and N-groups in determining high CO2 capture capacity and selectivity over N2.116
4.1.2.2 Acid activating methods. Acid activation serves as a crucial technique for tailoring the characteristics of carbon materials, significantly enhancing their surface area, pore structure, and functional groups, thereby improving their adsorption capabilities. Typically, this process involves treating carbonaceous materials, such as activated carbon, biochar, and carbon fibers, with strong acids such as H3PO4, H2SO4, and HNO3. The acid activation process not only creates new pores but also widens the existing pores, leading to a well-developed pore structure, which is essential for various adsorption applications.
H3PO4 has emerged as the predominant acid activator in the production of activated carbon for carbon capture applications, undergoing transformation into polyphosphate during the activation process.117
| 2P2O5 + 5C → P4 + 5CO2 | (19) |
| 2H2P2O72− → P4 + 6O2 + 2H2O | (20) |
Activation with H3PO4 not only stimulates the development of pores but also introduces novel functional groups into the structure of carbon. For instance, the immersion of Lantana camara biomass in 85 wt% H3PO4 solution led to a significant increase in the specific surface area of the activated biochar from 117 to 1177 m2 g−1 and a 20-fold increase in pore volume compared to the untreated biochar.118 Furthermore, the oxygen content of the biochar increased, with the XPS analysis confirming the presence of surface groups such as C–PO3, C–P2O6, and C–P2O5. Similarly, Bonga et al. observed surface groups such as C–O–PO3 and C–PO3 in biochar produced through phosphoric acid activation in the temperature range of 400–600 °C.119 However, it is notable that the acidic PxOy groups resulting from H3PO4 activation have been found to hinder CO2 chemisorption in carbon materials. In this case, fine-tuning of the surface chemistry and porous structures of carbon materials can be achieved by adjusting the concentration of H3PO4 and the treatment temperature.120 Although a porous structure plays a crucial role in CO2 capture at lower temperatures, surface chemistry, including the presence of functional groups, becomes increasingly influential at higher temperatures during adsorption. Thus, meticulous attention is warranted in both the preparation and application of H3PO4-activated carbon.
Sulfuric acid is an extremely reactive substance, which can interact with organic compounds, such as carbohydrates, to extract water and break down organic precursors into carbon, according to the following reaction:121
| CnH2nOn + H2SO4 → nC + H2SO4·nH2O | (21) |
Additionally, sulfuric acid reacts with the mineral content in lignocellulosic materials, serving as an effective cleaning and de-ashing agent in the preparation of activated carbon. This makes H2SO4 a cost-effective option for carbonization processes, offering both economic and functional advantages.121 Furthermore, the activation process using sulfuric acid is known to enhance the porous structure of the resulting activated carbon. Notably, the porous carbon treated with H2SO4 exhibited a commendable CO2 adsorption capacity of 3.60 mmol g−1, marking a 39.5% improvement compared to the untreated sample.122 The acid penetrates the material, creating a surface with a combination of large and medium-sized pores, which is beneficial for a variety of applications.121,123
Additionally, nitric acid activation is primarily used to introduce a variety of oxygen-containing functional groups, such as carboxyl, hydroxyl, and carbonyl groups, on the surface of carbon. This treatment is usually conducted at room temperature to 100 °C. Nitric acid oxidizes the surface of carbon, creating defects and functional groups, which enhance the adsorption capacity for polar molecules, including heavy metals and organic pollutants. The reaction mechanism is as follows:124
| HNO3 → NO2 + O2 + H2O | (22) |
| 2NO2 + C → C(O) + 2NO | (23) |
| C(O) + H2O → COOH | (24) |
| C + HNO3 → C–OH + 2NO2 | (25) |
|
C + O2 → CO
| (26) |
The oxidation process increases the surface acidity and introduces active sites, which improve the interaction of carbon materials with the adsorbate, enhancing their performance in various applications.
4.1.2.3 Salt activating method. Salt activation is a crucial method for tailoring the properties of carbon materials, offering a versatile approach to enhance their surface area, pore structure, and adsorption capabilities. This technique employs various salts, such as potassium carbonate (K2CO3), sodium carbonate (Na2CO3), zinc chloride (ZnCl2), and ferric chloride (FeCl3), to activate the carbonaceous precursors through thermal treatment. Unlike traditional alkaline activators such as KOH and NaOH, which are strong bases, salts such as K2CO3 and Na2CO3 also contribute to activation by providing different chemical environments. These salts activate the carbonaceous precursors through thermal treatment, promoting the development of porous structures via specific reaction mechanisms.125
K2CO3 is a common activating agent, which upon heating typically between 700 °C and 900 °C, decomposes to release CO2 and K2O. The CO2 generated in this reaction acts as a gasifying agent, creating pores within the carbon matrix. Additionally, K2O can further react with the carbon structure to form metallic potassium, which intercalates into the carbon lattice, enhancing its porosity, as follows:
| K2CO3 → K2O + CO2 | (27) |
| K2O + C → 2K + CO | (28) |
This process leads to a significant increase in surface area and pore volume, making K2CO3-activated carbons highly effective for adsorption applications.126
Na2CO3 follows a similar activation pathway to K2CO3. During thermal treatment, Na2CO3 decomposes into sodium oxide (Na2O) and (CO2). CO2 aids in the development of a porous structure by gasifying the carbon material. Na2O further reacts with carbon to produce elemental sodium, which intercalates into the carbon structure, enhancing the formation of pores, as follows:127
| Na2CO3 → Na2O + CO2 | (29) |
| Na2O + C → 2Na + CO | (30) |
Carbon materials activated by Na2CO3 exhibit a high surface area and improved adsorption properties, making them suitable for various environmental applications, including pollutant capture and energy storage.
ZnCl2 is widely recognized as an effective activation agent, particularly due to its strong dehydrating properties. The activation process with ZnCl2 typically involves three stages during pyrolysis. Initially, biomass decomposes into small particles and volatiles as the temperature increases. In the subsequent stage, ZnCl2 interacts with these particles, producing char, tar, and volatiles through the following reaction:128
| ZnCl2 + C → Zn + Cl2 + CO | (31) |
In the final stage, the char undergoes further reaction with ZnCl2, leading to significant pore development. Carbon materials activated with ZnCl2 are noted for their increased prevalence of mesopores and macropores, which is attributed to the deep penetration of molten ZnCl2 into the carbon lattice, which expands the pore structure. Co-activation with CO2 further enhances the specific surface area, total pore volume, and even generates ultra-fine pores.129,130 Conversely, activation with ferric chloride (FeCl3) is more effective in promoting micropore formation, as demonstrated by Elewa et al., who achieved a micropore volume of 0.468 cm3 g−1 using FeCl3.131 However, the environmental implications of ZnCl2 activation warrant attention. During this process, some ZnCl2 can be converted to ZnO, which may adsorb on the activated carbon, posing the risk of soil and groundwater contamination. Considering these concerns, the EU proposed restrictions on high concentrations of ZnO since 2017, with similar measures initiated by China in 2018.132
FeCl3 is another effective activating agent, particularly favoring micropore formation. In the activation process, FeCl3 acts as a catalyst, promoting the removal of volatile compounds and facilitating the development of a highly porous structure, as follows:
| 2FeCl3 + 3C → 2Fe + 3Cl2 + 3CO | (32) |
This reaction results in carbon materials with a high micropore volume, which is beneficial for applications such as gas storage and separation.133
4.1.2.4 Metal doping. The impregnation of biomass-derived carbon materials with metals such as sodium, calcium, magnesium, aluminum, nickel, and iron has attracted significant attention as a strategy to enhance their CO2 capture capabilities. Biomass-derived carbon materials, including biochar and activated carbon, are attractive due to their sustainable and eco-friendly nature, offering a renewable alternative by recycling agricultural and forestry waste. These materials naturally possess high surface areas and porous structures, making them highly effective for gas adsorption. Furthermore, the introduction of metal species into these carbon matrices can enhance their CO2 adsorption capacities and selectivity, making them more efficient and versatile for carbon capture applications.134
Sodium and calcium, both alkaline metals, have been shown to significantly enhance CO2 capture through chemisorption mechanisms. When impregnated into biomass-derived carbon materials, these metals form stable carbonates, such as Na2CO3 and CaCO3, respectively, which effectively bind CO2 molecules. This chemisorptive process not only increases the efficiency of CO2 capture but also reduces the likelihood of desorption, making these materials more reliable for long-term CO2 storage.135 The stability of these carbonates ensures that the captured CO2 remains securely bound, reducing the risk of its release back into the atmosphere. Magnesium, another alkaline earth metal, also enhances the capture of CO2 when incorporated into biomass-derived carbon materials. The formation of MgCO3 within the carbon matrix, combined with the high surface area of the material, creates an effective platform for CO2 adsorption. The presence of MgCO3 not only increases the affinity of the material for CO2 but also contributes to the overall stability of the adsorbed CO2.136 Aluminum impregnation introduces Lewis acid sites on the surface of carbon through the formation of alumina (Al2O3). These acid sites interact strongly with CO2 molecules, significantly enhancing both the adsorption capacity and selectivity of the material.13 The strong interaction between CO2 and the aluminum-impregnated carbon material increases the efficiency of CO2 capture, making it particularly suitable for selective CO2 adsorption in mixed-gas streams.
Transition metals such as iron and nickel play crucial roles in improving CO2 capture. Iron, forms iron oxides, such as Fe2O3 and Fe3O4, on the surface of carbon. These oxides facilitate both physisorption and chemisorption mechanisms, significantly enhancing the overall CO2 capture efficiency.137 Alternatively, Ni doping is a promising strategy to enhance their properties for various applications, particularly in CO2 adsorption and electrochemical energy storage. Nickel doping involves impregnating carbon materials derived from biomass with a nickel precursor such as nickel nitrate (Ni(NO3)2) or nickel chloride (NiCl2). This process begins with the pyrolysis of biomass to produce a porous carbon framework. Subsequently, this carbon material is impregnated with a nickel salt solution, allowing nickel ions to diffuse and become uniformly distributed within the carbon matrix. After impregnation, the material undergoes thermal activation, typically at temperatures in the range of 300 °C to 900 °C in an inert atmosphere. This thermal treatment reduces the nickel salts to metallic nickel, which is embedded within the carbon structure, forming nickel oxides and carbonates, serving as active sites for CO2 interaction.138
The incorporation of nickel into biomass-derived carbons significantly enhances their CO2 adsorption capacity and selectivity. Nickel carbonates and oxides provide additional active sites, which promote stronger interactions with CO2 molecules, thereby increasing the amount of CO2 that can be adsorbed. This is particularly advantageous for applications requiring selective adsorption, such as carbon capture and storage.102 The presence of nickel also improves the selectivity of materials for CO2 over other gases, making it more efficient in separating CO2 from gas mixtures. This enhanced selectivity is due to the specific interactions between CO2 molecules and the nickel-based active sites, which are less favorable for other gases. In addition to gas adsorption, nickel doping significantly improves the electrochemical properties of biomass-derived carbons. In applications such as fuel cells, batteries, and supercapacitors, nickel acts as a catalyst, which enhances the electrocatalytic activity and stability of the carbon material. Also, the presence of nickel increases the electrical conductivity and facilitates efficient charge transfer, which are crucial for high-performance energy storage and conversion devices.139
4.1.2.5 Heteroatom doping. Heteroatom doping provides a promising pathway for tailoring the characteristics of carbon materials by deliberately introducing elements such as nitrogen, sulfur, and boron into the carbon framework. This approach alters the electronic structure and surface chemistry of carbon, thereby improving its conductivity, catalytic activity, and ability to adsorb various substances. Nitrogen doping has attracted considerable interest due to its capacity to create active sites and enhance the electrochemical performance of carbon-based materials. Similarly, sulfur and boron doping have been explored for their potential to augment the adsorption capacity and stability of carbon materials across diverse applications. Although micropores play a pivotal role in carbon dioxide capture, surface functional groups also significantly contribute to the adsorption process, accounting for about 38%. Surface alkalinity plays a crucial role in CO2 adsorption, and heteroatom doping has been demonstrated to enhance the surface alkalinity, thereby improving the CO2 uptake.140 Nitrogen doping, being the most extensively researched method, not only regulates the pore structure of carbon materials but also enriches them with various forms of N-containing groups such as pyridone, cyanide, pyrrole, pyridine, amine, quaternary, and pyridine-N-oxide. Compounds such as PEI, TEPA, and DETA have been shown to be effective as nitrogen dopants for CO2 capture.129 However, the additional cost associated with nitrogen doping underscores the importance of identifying nitrogen-rich biomass sources for the synthesis of carbon materials. However, although biomass with a high nitrogen content tends to yield nitrogenous biochar, not all nitrogen in biomass can be converted to N in carbon materials, given that some may be transformed into NH3 or other gases during the process. Beyond single nitrogen doping, other elements such as sulfur and co-doping have been proven to be effective. For instance, researchers synthesized sulfur-doped porous carbon with a CO2 adsorption capacity of 4.5 mmol g−1, surpassing similar materials doped with nitrogen, which is possibly due to the higher binding energy of CO2–S compared to CO2–N, resulting in more stable adsorption and higher capacity.141 Guo et al. utilized poplar sawdust as a raw material and sulfur-containing wastewater as a modifier to co-load nitrogen, sulfur, and oxygen on activated carbon, yielding porous carbon with a CO2 adsorption capacity of 5.61 mmol g−1 at 0 °C.142
4.1.2.6 Other modification methods. Besides the primary modification methods discussed previously, supplementary approaches such as ball milling, microwave irradiation, ultrasound treatment, and plasma modification show potential for further improving carbon materials. Ball-milling modification utilizes the kinetic energy produced during the movement of balls in machinery to diminish the particle size of materials or disrupt their chemical bonds, thereby modifying their pore structures. An examination of crayfish shell biochar pre- and post-ball milling unveiled notable enhancements in its specific surface area and micropore volume, escalating from 128 to 290 m2 g−1 and 0.028 to 0.061 cm3 g−1, respectively.143 Additionally, the comparison among pristine biochar, ball-milled biochar, and ball-milled-N-doped biochar demonstrated superior pore structures in the modified biochar, coupled with increased CO2 adsorption capacities.144 Microwave radiation, an emerging heating method, presents advantages such as swift heating, convenient regulation, and uniform heat dispersion. Given their elevated microwave absorption capacity, carbon materials can be transformed under microwave heating, generating new carbons with tailored characteristics.145 The rapid heating facilitated by microwave radiation may prompt the formation of new pores and modify the pore structures and surface functional groups in carbon materials. Studies suggested an increase in the number of functional groups on the molecular bonds of graphene materials after microwave treatment, providing additional adsorption sites for the adsorption process.146
Ultrasonic treatment has emerged as a novel method for altering carbon materials. By exploiting cavitation and microjet effects, ultrasonic treatment effectively eradicates the lumps formed during the pyrolysis of biomass, thus preventing pore blockage and facilitating the creation of additional microporous structures.134 Typically, ultrasonic treatment is paired with chemical modification techniques to optimize its effectiveness. For example, in the study by Cao et al., biochar was synthesized through ultrasonic treatment followed by amination at room temperature. Preceding amination, ultrasonic treatment of biochar induced the detachment of the graphite layer, resulting in the formation of new pores.147 The resultant exfoliated biochar exhibited an abundance of functional groups such as –COOH, –OH, and –CH(O)CH–, which facilitated amine grafting. As a result, the biochar treated with ultrasound and amines exhibited a nine-times greater CO2 adsorption capacity than that of the untreated biochar. Plasma, consisting of cations, particles, and free electrons, serves as a neutral substance for modifying the surface characteristics of materials, which is typically generated through corona, glow, or microwave discharge methods. During the modification process, interactions between plasma and the material surface facilitate the formation of new groups. In the study by Khan et al., they utilized atmospheric pressure plasma to modify activated carbon, observing improvements in its surface area and pore volume together with the enrichment of surface oxygen-containing functional groups.148
Furthermore, the choice of chemical treatment is pivotal in determining the optimal activation temperature. Various activating agents interact differently with the carbonaceous components in biomass, leading to diverse outcomes in terms of pore structure and surface characteristics. Studies conducted by numerous researchers have underscored the significance of selecting the appropriate chemical treatment based on the reactivity with the precursor material and its specific chemical properties.149–151 Factors such as boiling and melting points, as well as the ability to mobilize metallic ions, further influence the effectiveness of the activation process. Understanding these intricate relationships among chemical treatment, activation temperature, and the resultant properties of activated carbon is essential for tailoring materials to meet the diverse needs of industrial, environmental, and technological applications.
4.1.3.1 Optimum KOH activation temperature. Determining the optimal activation temperature for KOH is paramount for the production of activated carbon, given that it profoundly influences the properties and performance of materials. This review summarizes a vast body of literature dedicated to pinpointing the ideal activation temperature using KOH as the activating agent. The activation temperature serves as a critical parameter in modulating the pore structure, surface area, and adsorption properties of the resulting activated carbon. By collating insights from various studies, this review endeavors to elucidate the multifaceted factors shaping the selection of the optimum activation temperature for KOH, considering variables such as precursor material, activation duration, and intended applications. Dewi asserted that the most effective activation temperature for KOH treatment is in the range of 700 °C to 800 °C.152 Correspondingly, Williams et al. observed a significant enhancement in surface area and micropore volume with an increase in activation temperature from 700 °C to 800 °C.153 The surface area experienced a remarkable 33% enhancement, increasing from 1955 m2 g−1 to 2600 m2 g−1, in contrast to the less than 10% improvement witnessed when the temperature was increased from 600 °C to 700 °C. This trend was corroborated by a group of researchers, further demonstrating that elevating the activation temperature from 700 °C to 800 °C led to a substantial increase in surface area, reaching up to 3228 m2 g−1, albeit at the expense of carbon yield and surface area when the temperature exceeded 850 °C.154–156 Additionally, Ma et al. observed a progressive widening of the average pore width from 0.520 nm at 500 °C to 0.573 nm at 800 °C.156 This exponential enhancement can be attributed to the formation and mobilization of metallic potassium at around 700 °C, facilitating pore development. Furthermore, the interaction among potassium, carbon and water yields potassium carbonate, which mitigates precursor burn-off, contributing to enhanced pore formation. Understanding these intricate temperature-dependent mechanisms is indispensable for optimizing KOH activation processes and tailoring activated carbon for diverse applications.
4.1.3.2 Optimum H3PO4 activation temperature. Determining the most effective activation temperature for H3PO4 is crucial in producing activated carbon, given its significant influence on the characteristics and performance of materials. This review presents an in-depth exploration of the extensive research dedicated to identifying the optimal activation temperature using phosphoric acid as the activating agent. The activation temperature plays a pivotal role in shaping the pore structure, surface area, and adsorption capabilities of the resulting activated carbon. By summarizing the insights from various studies, this review aims to highlight the factors influencing the selection of the best temperature for H3PO4 activation, considering variables such as precursor material, activation duration, and intended applications. Notably, empirical studies suggest that the most suitable activation temperature for H3PO4-treated precursors is typically 500 °C or lower, regardless of the impregnation ratio or precursor source.152,157 For instance, researchers treated Paulownia wood with H3PO4 at a ratio of 4 and subjected it to carbonization in the temperature range of 300 °C to 600 °C.158 The results indicated that higher temperatures were associated with a decrease in the activated carbon yield. Additionally, a notable trend emerged, wherein the micropore volume and BET surface area exhibited trend for activated carbon carbonized in the range of 300 °C to 400 °C, followed by a decline as the carbonization temperature increased from 400 °C to 600 °C.152,157 This phenomenon was attributed to the contraction of the carbon structure on Paulownia wood at elevated temperatures, resulting in a diminished void space. Moreover, the activated carbon produced in these experiments demonstrated a dual-porosity nature, comprising micropores and mesopores, which is consistent with findings from prior studies utilizing alternative precursor materials such as date pits.
The observed trends in activation behavior can be elucidated by considering the chemical properties and organic chemistry of phosphoric acid. In the temperature range of 300 °C to 400 °C, phosphoric acid not only acts as a heat facilitator but also as a dehydrating agent, capturing minerals to inhibit ash formation, and thereby promoting pore development. Additionally, H3PO4 forms a protective layer on carbon in liquid materials and various chemical structures such as acetic acid, methanol, and polyphosphates, which reinforce the pore stability against temperature-induced collapse. However, at higher temperatures, these supporting structures, particularly acetic acid, encounter resistance limits. Beyond 440 °C, acetic acid decomposes into a mixture of carbon dioxide and methane or water and ketene, resulting in compromised pore stability and subsequent surface area reduction. Thus, understanding these intricate temperature-dependent mechanisms is vital for optimizing the H3PO4 activation processes and tailoring activated carbon for diverse applications in industries, environmental remediation, and emerging technologies.
4.1.3.3 Optimum ZnCl2 activation temperature. Determining the optimal activation temperature for ZnCl2 is a critical factor in the production of activated carbon, given its significant impact on the properties and performance of materials. This review aims to delve into the extensive research dedicated to identifying the ideal activation temperature using zinc chloride as the activating agent. The activation temperature plays a pivotal role in molding the pore structure, surface area, and adsorption properties of the resulting activated carbon. By summarizing the insights from various studies, this review endeavors to elucidate the factors influencing the selection of the optimum temperature for ZnCl2 activation, considering variables such as precursor material, activation duration, and intended applications. According to the study conducted by a research group, the optimal activation temperature for ZnCl2 treatment was suggested to be 500 °C, which is attributed to the higher surface area and micropore volume reported for activated carbon precursor cherry stones.159 This finding was corroborated by other researchers, where an increase in temperature from 500 °C to 800 °C resulted in a decrease in surface area.151 Additionally, Nurdin et al. reported the highest surface area of activated carbon precursor oil palm shell at 1429.71 m2 g−1 when activated at 500 °C, with a high micropore volume also observed.149 However, an increase in activation temperature from 500 °C to 800 °C led to a decrease in specific surface area and micropore volume, coupled with an increase in average pore size distribution, as supported by similar research utilizing different precursors.152 Furthermore, in the adsorption-based study by Njeh et al., the highest iodine and methylene blue adsorption capacities were recorded for sawdust activated at 500 °C.160 It was noted that ZnCl2 can vaporize at temperatures above 400 °C, potentially leading to rapid carbon burnout due to the loss of protective carbon elements, according to Wei et al.161 However, Huang disagreed these claims, suggesting that the optimum activation temperature for ZnCl2 treatment is around 700 °C.162 Though scarce, this assertion is reinforced by similar findings, where the highest micropore formation upon activation up to 800 °C is possibly linked to the physical properties of ZnCl2, with a boiling point of 732 °C.151,163 In summary, higher temperatures generally result in increased pore formation, but each activating agent exhibits a specific range of optimum temperatures, depending on their chemical and physical properties such as boiling point. According to the literature, the suggested activation temperatures for precursors incorporating KOH, H3PO4, and ZnCl2 are 700–800 °C, 500 °C, and 500 °C, respectively.
In the case of biomass-derived materials, the impregnation ratio is particularly important, influencing their structural and functional characteristics. Several studies conducted by researchers underscore the significance of the impregnation ratio in shaping the pore structures.151,164,165 The typical impregnation ratios for biomass are in the range of 1:1 to 3:1, depending on the type of biomass and the specific requirements of the end product.166 Studies have highlighted the effect of the impregnation ratio on the structural properties of activated carbons. For example, increasing the impregnation ratio has been shown to improve the porosity and surface functionality of activated carbon from coconut shells, enhancing its adsorption capabilities.167 Additionally, optimizing the impregnation ratio is crucial for achieving a well-balanced pore structure and thermal stability, as seen in biochar produced from rice husks. Research also indicates that an impregnation ratio of 1:1 can result in activated carbons with higher surface areas and larger pore volumes compared to lower ratios.168 Increasing the ratio of phosphoric acid from 1:1 to 2:1 was found to significantly increase the micropore volume in activated carbons.169 However, it is important to note that excessively high impregnation ratios can lead to the overactivation of carbon materials, compromising their structural integrity. Therefore, it is essential to carefully optimize the impregnation ratio to achieve the desired balance among surface area, pore structure, and mechanical stability, tailoring the properties of activated carbon to meet specific application needs.
In essence, the impregnation ratio plays a critical role in the production of activated carbon, shaping its pore structure and surface area, and consequently affecting its adsorption capacity and performance. The optimal adjustment of this ratio is indispensable for tailoring activated carbon to specific applications, harmonizing diverse factors such as porosity, yield, and cost-effectiveness. A profound comprehension of the intricate interplay between the impregnation ratio and properties of activated carbon is pivotal for refining production processes and devising activated carbon materials endowed with enhanced performance attributes.
Research indicates that the temperature at which carbonization occurs and the flow rate of the inert gas, typically nitrogen are critical factors in determining the porous architecture of carbon materials. Studies have shown that the optimal conditions for achieving the maximum specific surface area, pore volume, and micropore content are carbonization temperature in the range of 800–850 °C with a nitrogen flow rate of 7.5–15 dm3 min−1. These parameters help in effectively removing volatile components from the precursor material, promoting the formation of a well-developed porous structure and increasing the surface area.171
Inert gases such as nitrogen play multiple roles during the carbonization process. Primarily, they prevent the oxidation of the carbon material by displacing oxygen and other reactive gases, thus maintaining the integrity of the carbon structure. Additionally, the flow rate of nitrogen influences how quickly volatile compounds are expelled from the carbon matrix. A higher nitrogen flow rate facilitates the faster removal of volatiles, enhancing the development of a more extensive pore network and higher surface areas. Alternatively, a lower flow rate can lead to a less porous structure due to the slower release of volatiles and potential re-deposition of carbonaceous residues within the pores.96,171
Furthermore, the interaction between the carbonization temperature and inert gas flow rate is vital in achieving the desired textural properties. Higher temperatures enhance the decomposition of the precursor material, and when combined with a controlled nitrogen flow, can result in a uniform and highly porous structure. However, if the temperature is too high or the nitrogen flow is inadequate, the pore structure may collapse or unwanted tar formation may occur, clogging the pores and reducing the overall effectiveness of the material.172
Overall, the use of inert gases such as nitrogen during carbonization is critical for regulating the textural characteristics of activated carbon. By optimizing the carbonization temperature and gas flow rate, it is possible to fine-tune the pore structure and surface area, thereby enhancing the adsorption capacity and overall performance of the material.
4.2 Utilizing waste-derived activated carbon for CO2 capture
The widespread application of activated carbon spans multiple sectors, encompassing gas and water purification, gold processing, wastewater treatment, metal extraction, decaffeination, and pharmaceuticals. The rapid expansion of the AC market is underscored by the significant rise in its annual demand. According to Market Data Forecast, the global AC market is projected to grow from $9.17 billion in 2023 to $11.46 billion by 2029, indicating robust growth.96 Thus, to meet escalating demands and address cost considerations, researchers are actively exploring alternative pathways for the production of AC, with a specific emphasis on low-cost or waste materials. The utilization of waste materials not only addresses environmental and waste management challenges but also unlocks lucrative opportunities for the business sector. In parallel, biomass can be broadly classified into two main categories, conventional waste and non-conventional waste. Conventional waste is comprised of biomasses sourced from the agricultural and wood industries, such as coconut shells, rice straws, sawdust, and wood, while non-conventional wastes include materials such as plastic, sugarcane bagasse, citrus peel, and tires, originating from municipal and industrial sources. Coconut shells, in particular, have attracted significant attention as a frequently studied precursor due to their elevated carbon content and robust mechanical properties, as evidenced by the compositional analysis in Fig. 9. Comparative studies conducted by several researchers have focused on determining the adsorption capacity and practical feasibility of coconut shell-derived AC precursors.173–176 The literature underscores the importance of understanding the carbon content and compositions of potential raw materials, given that these factors profoundly influence the characteristics of the resulting AC. Crucially, parameters such as fixed carbon and volatile matter levels play pivotal roles in evaluating the effects of carbonization and pore formation processes, with carbonization notably enhancing pore development by augmenting the mobility and reducing the volatile matter content. | ||
| Fig. 9 Process for the synthesis of activated carbon. | ||
The analysis presented in Table 4 highlights that nutshell, coconut shell, algae, and sea mango possess a carbon content exceeding the average of 44.95 wt%. Other studies suggest that utilizing a concentrated carbon precursor can potentially enhance the yield of AC by leveraging its higher carbon content, thereby facilitating the formation of microporous structures.215 Moreover, gaining a thorough understanding of the structural intricacies of various waste materials, influenced by variations in lignocellulose compositions, is considered crucial. Comparative studies indicate that precursors abundant in lignin tend to yield activated carbon with spherical porous structures, while that rich in cellulose tends to produce cylindrical pore structures. These observations emphasize the pivotal role of precursor type in determining the texture of AC.177,216 However, achieving the optimal activated carbon for CO2 adsorption poses significant challenges due to the diverse composition properties of waste materials from different sources. Notably, it can be observed in Table 4 that algae, walnut shell, and waste palm shell exhibit elevated nitrogen levels compared to other waste samples. With nitrogen levels of 7.20 wt%, 3.20 wt%, and 3.00 wt%, respectively, surpassing the average nitrogen content of 1.67 wt% among the samples. Despite the significance of the carbon content in precursor selection, this indicates that a higher nitrogen concentration in the precursor enhances both the carbon dioxide adsorption performance and CO2 selectivity, irrespective of the carbon composition.178,217 Treeweranuwat et al. emphasized that a low carbon content in the precursor leads to a reduction in the surface area of the resulting activated carbon. However, incorporating heteroatom-rich materials during the carbonization and activation of shrimp-derived AC not only increased its surface area but also boosted the carbon yield.218 Furthermore, the addition of melamine, sodium thiosulfate, and KOH was shown to double the AC yield due to the enhanced thermal stability.
| Precursor materials | Elemental composition (weight%) | Ref. | ||||
|---|---|---|---|---|---|---|
| Carbon | Oxygen | Hydrogen | Nitrogen | Ash | ||
| Corn stover | 60.50 | 24.30 | 5.60 | 1.20 | 7.40 | 53 |
| Miscanthus | 74.60 | 17.42 | 3.38 | 0.45 | 3.41 | 147 |
| Miscanthus | 84.47 | 7.61 | 2.28 | 0.30 | 5.02 | 147 |
| Switchgrass | 78.64 | 4.88 | 1.46 | 0.79 | 13.94 | 147 |
| Sugarcane bagasse | 59.00 | 25.00 | 7.00 | 1.30 | 8.70 | 150 |
| Walnut shell | 84.86 | 13.63 | 1.16 | 0.34 | — | 177 |
| Shrimp shell | 42.07 | — | 5.74 | 24.81 | — | 178 |
| Crab shell | 10.23 | — | 2.82 | 3.94 | — | 179 |
| Algae | 50.20 | 35.00 | 6.80 | 7.20 | 6.70 | 180 |
| Bituminous coal | 85.09 | 5.05 | 3.13 | 1.50 | 5.23 | 181 |
| Bituminous coal | 83.26 | 12.20 | 3.44 | 0.77 | — | 182 |
| Anthracite coal | 90.02 | 3.15 | 3.50 | 1.25 | 2.08 | 181 |
| Apple tree branches | 80.01 | 6.59 | 2.72 | 1.28 | 9.40 | 183 |
| Torrefied cornstalk | 35.63 | 32.31 | 4.48 | 1.04 | — | 184 |
| Raw cornstalk | 39.57 | 40.05 | 5.61 | 0.52 | — | 184 |
| Lignite coal | 52.54 | 17.94 | 2.88 | 0.77 | — | 184 |
| Palm kernel shell | 45.40 | 41.92 | 6.10 | 0.70 | 5.00 | 185 |
| Cotton stalk | 38.96 | 56.47 | 3.56 | 1.01 | 6.03 | 186 |
| Bamboo waste | 65.00 | 20.01 | 6.99 | 0.90 | 7.10 | 187 |
| Rambutan peel | 76.36 | 19.22 | 2.90 | 1.35 | — | 188 |
| Rambutan peel | 83.38 | 14.71 | 0.90 | 0.77 | — | 188 |
| Buckwheat husks | 78.40 | 15.50 | 3.10 | 1.30 | — | 189 |
| Sewage sludge | 12.37 | 8.94 | 0.83 | 0.95 | — | 190 |
| Olive stone | 47.10 | 46.46 | 6.23 | 0.21 | 0.45 | 191 |
| Microalgae | 54.28 | 31.45 | 9.12 | 4.74 | — | 192 |
| Apricot stone | 48.45 | 45.08 | 6.03 | 0.44 | 1.68 | 193 |
| Date stone | 48.43 | 44.46 | 6.44 | 0.67 | 1.58 | 194 |
| Cherry stone | 49.90 | 44.15 | 6.37 | 0.24 | 0.24 | 195 |
| Almond shell | 51.40 | 41.60 | 6.10 | 0.30 | 1.30 | 196 |
| Peanut shell | 46.82 | 37.64 | 6.58 | 0.80 | 8.61 | 197 |
| Macadamia nut shell | 53.19 | 40.68 | 5.78 | 0.29 | 0.30 | 198 |
| Pecan shell | 47.53 | 45.97 | 5.53 | 0.33 | — | 199 |
| Rice husk | 36.52 | 41.10 | 4.82 | 0.86 | 16.70 | 200 |
| Sugarcane bagasse | 41.55 | 52.86 | 5.55 | 0.03 | 6.20 | 200 |
| Coconut shell | 49.62 | 42.75 | 7.31 | 0.22 | 0.80 | 201 |
| Coconut shell | 40.33 | 51.57 | 2.78 | 0.72 | 4.18 | 202 |
| Hemp straw | 48.40 | 44.90 | 6.40 | 0.30 | 1.80 | 203 |
| Wheat straw | 39.90 | 41.97 | 5.75 | 0.65 | 12.30 | 204 |
| Soybean straw | 41.50 | 41.39 | 5.52 | 0.28 | 8.87 | 205 |
| Wheat straw | 41.52 | 36.24 | 5.76 | 0.61 | — | 206 |
| Rice straw | 40.67 | 32.30 | 5.73 | 0.80 | — | 206 |
| Waste tea | 48.47 | 37.60 | 6.40 | 0.32 | 3.37 | 207 |
| Douglas fir | 47.90 | 45.57 | 6.55 | 0.08 | 0.21 | 208 |
| Pinewood sawdust | 49.09 | 44.53 | 6.05 | 0.33 | — | 209 |
| Citrus peel | 41.87 | 51.62 | 5.49 | 0.58 | 2.86 | 210 |
| Cassava peel | 47.21 | 43.70 | 7.74 | 1.35 | 1.92 | 211 |
| Lemon peel | 38.48 | — | 4.98 | 1.21 | 3.68 | 212 |
| Cassava peel | 38.34 | 38.55 | 6.13 | 2.08 | 4.32 | 213 |
| Waste palm peel | 51.00 | 39.00 | 7.00 | 3.00 | 4.00 | 214 |
An essential criterion for an effective precursor in the production of adsorbents is a minimal ash content.219 Douglas fir stands out with its notably low recorded ash content of 0.21 wt% (as indicated in Table 4), which is significantly lower than the average of 4.12 wt%. In contrast, algae and rice husk exhibit a higher ash content of 6.70 wt% and 16.70 wt%, respectively. The reduced ash content in Douglas fir suggests its potential as a superior adsorbent precursor. Although the ash content does not directly influence the development of porosity, it impacts the adsorption properties due to the formation of inert sites. Understanding the prevalent lignocellulose structure is crucial due to the diverse compositions of waste materials. Comparative analyses indicate that precursors rich in lignin tend to yield activated carbon with spherical porous structures, while that with a higher cellulose content tends to yield cylindrical pore structures.177,180 The varying composition properties of waste materials pose challenges in achieving the optimal activated carbon for CO2 adsorption. Agricultural residues, such as palm kernel shells, rice husks, coconut shells, and bagasse, are extensively utilized for producing activated carbon due to their abundance, high carbon content, and relatively low ash levels. Conducting a comprehensive precursor analysis is essential to facilitate the conversion of AC into a suitable CO2 adsorbent. Besides possessing a substantial surface area and high microporosity concentration, waste-derived synthesized AC materials must be economically feasible, possess basic functionalities, exhibit low heat absorption, high CO2 selectivity, and be suitable for industrial-scale production. Also, the composition of waste products plays a crucial role in determining the quality of the resulting activated carbon.
:1 ratio. This study revealed that coconut shells exhibited a higher yield of activated carbon precursors compared to palm kernel shells, positioning them as promising candidates for raw material utilization in carbon capture applications.Similarly, another research group investigated the efficacy of utilizing bagasse and rice husk as precursor materials for the production of activated carbon.200 Employing ZnCl2 as an activator at a temperature of 500 °C for 60 min, this study demonstrated that bagasse-derived activated carbon displayed superior adsorption capabilities over its rice husk-derived counterpart across various adsorption temperatures. These findings underscore the potential of leveraging readily available agricultural waste streams for the scalable and economically viable production of activated carbon, thereby facilitating the transition towards a more sustainable waste management paradigm. However, despite the notable advancements in CO2 adsorption performance observed in these studies, challenges persist in ensuring the thermal stability of the synthesized activated carbon for sustained CO2 capture efficacy. Particularly, the decline in CO2 adsorption performance at elevated temperatures underscores the need for targeted modifications to enhance the thermal stability of activated carbon materials tailored for CO2 capture applications. According to the findings gleaned from extensive literature studies, coconut shell and palm kernel shells emerge as the most promising agricultural waste precursors for the production of activated carbon, owing to their high carbon content and low ash composition.176,219 This ranking underscores the critical importance of selecting appropriate precursor materials to optimize the performance and sustainability of activated carbon production processes.
:1 activating agent to carbon, and subjecting it to activation at 850 °C for 4 h.223 Similarly, Jia et al. investigated shrimp shells as activated carbon precursors, leveraging their high nitrogen content to enhance the formation of basic functional groups in the synthesized activated carbon.217 In another study, coffee grounds were utilized as the raw material and activated with KOH at 400 °C, yielding activated carbon with an average CO2 adsorption capacity of 0.27 cm3 g−1 at 0 °C.224 Moreover, Wen et al. demonstrated the potential of beer waste as an activated carbon precursor, employing hydrothermal carbonization followed by activation at 800 °C for 2 h, resulting in activated carbon with high CO2 selectivity compared to other studied precursors.225 Comprehending the structural and elemental makeup of waste materials is crucial for confirming their viability as precursors for activated carbon in CO2 capture. Although agricultural residues are favored due to their abundance, high carbon content, and low ash content, food waste shows promise despite its lower average carbon content, which is attributed to its high nitrogen heteroatoms. Furthermore, industrial residues often possess elevated concentrations of functional groups, potentially enhancing the carbon capture capabilities. Consideration of factors such as volatile matter fraction, ash content, moisture content, and compositional complexity of biomass is crucial to ensure the uniform performance of the produced activated carbon, particularly for commercial applications where a high variation in carbon dioxide adsorption capacity is intolerable. Agricultural wastes are favored due to their abundance, ease of management, and the ability to categorize the raw materials based on species, age, and origin. By repurposing food waste streams, this approach not only mitigates food waste disposal challenges but also offers an environmentally friendly solution for producing activated carbon with diverse applications in water treatment, air purification, and energy storage.5. Application
5.1 CO2 capture with carbon materials
Carbon materials are increasingly recognized for their potential in industrial applications, particularly due to their cost-effectiveness, efficient regeneration, and exceptional ability to capture substantial amounts of CO2. These materials play a crucial role in addressing the growing need for CO2 capture from various emission sources, leveraging their high surface area and adaptable pore structures to serve as effective adsorbents for CO2 molecules. The versatility of carbon materials extends beyond CO2 capture, where they are also employed in pollutant adsorption, catalysis, and energy storage, making them vital in confronting environmental challenges and promoting sustainable technologies. Recent research has focused on the use of carbon materials for CO2 capture, largely driven by the increasing release of carbon dioxide from industrial activities, which exacerbates the global warming crisis. In this case, biomass, rich in carbon content, is emerging as a valuable resource for producing biochar or hydrochar, contributing to efforts in reducing CO2 emissions. However, pristine carbon materials face limitations in their textural structure and chemical composition, which restricts their effectiveness in CO2 capture. Thus, to overcome these challenges, pre-capture modifications are necessary to enhance the properties of carbon materials and improve their CO2 capture efficiency. This review critically examines the reported studies on biomass-derived carbon materials for CO2 capture, exploring the underlying adsorption mechanisms and identifying promising avenues for future research and practical application. By understanding these mechanisms and advancing the development of modified carbon materials, it is possible to optimize the CO2 capture processes, thereby contributing to the global effort to mitigate the impacts of climate change and move towards a more sustainable future.Numerous adsorbents have been proposed for CO2 capture, each possessing unique characteristics that contribute to their effectiveness in this application. As shown in Fig. 10, an optimal CO2 adsorbent should exhibit high adsorptive capacity, selectivity for CO2 in gaseous mixtures, rapid sorption kinetics, a microporous structure, and morphological and chemical stability, while also offering a low heat of adsorption, high regeneration rate, and cost-effectiveness.235 The following table presents a comprehensive overview of various AC and carbon materials derived from different precursors, including details on their synthesis and activation methods. It includes information on the precursor materials, synthesis techniques, activation agents, and the ratio of precursor to activation agent used. Additionally, this table summarizes key textural properties such as surface area, pore size or volume, and pore size distribution (PSD), as well as the surface functionalities of each material. This comparative analysis provides valuable insights into the performance and characteristics of different carbon materials for applications such as CO2 capture (Table 5).
 | ||
| Fig. 10 Characteristic properties required for an ideal adsorbent for CO2 capture. | ||
| Precursor | Synthesis method | Activation method/agent | Ratio (precursor) | Surface area (m2 g−1) | Pore size (nm) | Pore volume (cm3 g−1) | Surface functionalities | Ref. |
|---|---|---|---|---|---|---|---|---|
| Olive stones | Hydrothermal carbonization | KOH solution | 1:1 |
969 | — | 0.443 | Functional groups: –OH, CO |
191 |
| Olive stones | Hydrothermal carbonization | Bioorganic solution | 1:1 |
915 | — | 0.444 | Functional groups: –OH, CO |
191 |
| Olive stones | Hydrothermal carbonization | H3PO4 | 1:1 |
849 | — | 0.339 | Functional groups: –OH, CO |
191 |
| Rice husk | Carbonization | Potassium acetate | 1:1.5 |
80 | 3.0 | 0.58 | Carboxylic and –OH | 236 |
| Rice husk | Carbonization | H3PO4 | 1:1 |
1500 | 2.5 | 0.8 | High oxygen content | 237 |
| Coconut shell | Physical activation (steam) | CO2/KOH | 1:2 |
2300 | 2.8 | 1.12 | High carbon content, OH, CO |
238 |
| Coconut shell | Pyrolysis | Potassium citrate | 1:2 |
1050 | 2.5 | 0.62 | High oxygen content, acetic groups | 239 |
| Citric acid residue | Hydrothermal carbonization | H3PO4 | 1:4 |
1100 | 2.0 | 0.7 | Carboxylic and phenolic groups | 240 |
| Corncob | Pyrolysis | Bioorganic K2CO3 | 1:2 |
1500 | 1.8 | 0.758 | High oxygen content, carboxyl, hydroxyl, and lactone groups | 241 |
| Cashew nut shell | Hydrothermal carbonization | K2CO3 | 1:1 |
1693 | 1.63 | 0.839 | Functional groups: –OH, CO, COOH |
242 |
| Sugarcane bagasse | Sol–gel method | Potassium oxalate | 1:2 |
1250 | 2.7 | 0.78 | Carboxyl, phosphate, lactone group | 216 |
| Guhanshan coal | Direct carbonization | KOH | 1:2 |
1300 | 1.5 | 1.0 | Nitrogen-doped groups | 7 |
| Gulfweed | Chemical activation | KOH | 1:1 |
2500 | 2.8 | 1.2 | High surface oxygen groups | 243 |
| Rambutan peel | Hydrothermal carbonization | KOH | 1:1 |
2500 | 2.8 | 0.313 | High oxygen content | 52 |
| Pine cone | Hydrothermal carbonization | KOH | 1:3 |
2100 | 2.3 | 0.87 | Functional groups: –OH, CO |
244 |
| Pineapple peel | Wet impregnation | K2CO3 | 1:1.25 |
1006 | 0.59 | 0.28 | Functional groups: –OH, CO |
245 |
| Lotus leaf | Hydrothermal carbonization | KOH | 1:4 |
2020 | 3.4 | 0.92 | Functional groups: –OH, CO, COOH |
246 |
| N-doped carbon | Carbonization | KOH | 1:3 |
1800 | 3.5 | 1.0 | Pyridinic-N, OH, CO |
247 |
| Sunflower seed hull | Wet impregnation | ZnCl2 | 1:4 |
1956 | 1.12 | 0.28 | Functional groups: –OH, CO |
248 |
| Sawdust | N & C doping carbonization | KOH | 1:4 |
1600 | 3.2 | 1.1 | Functional groups: –OH, CO, SOx |
156 |
| N-rich organic compound | One-pot synthesis | KOH or ZnCl2 | 1:3 |
2000 | 3.0 | 0.8 | Pyridinic-N, OH, CO |
249 |
| Bamboo shoot shell | Carbonization | KOH (heteroatom doping) | 1:3 |
1850 | 1.2 | 0.75 | Pyridinic-N, OH, CO |
250 |
| Agricultural residue | Hydrothermal carbonization | KOH | 1:3 |
1800 | 3.0 | 1.2 | Functional groups: –OH, CO |
251 |
| Oak wood | Wet impregnation | H3PO4 | 1:1 |
2553 | 1.61 | 0.94 | Functional groups: –OH, CO |
96 |
| Organic polymer-rich biomass | Carbonization | KOH | 1:3 |
1900 | 3.5 | 1.1 | Functional groups: thiols, sulfides, –OH, CO |
252 |
| Lignin | Carbonization | KOH (N, S-doping) | 1:4 |
2200 | 3.0 | 1.2 | Pyridinic-N, thiols, OH, CO |
253 |
| Argan paste cake waste | Carbonization | KOH | 1:4 |
1500 | 3.0 | 1.0 | High carbon content, OH, CO |
254 |
| PET bottles | Carbonization | KOH + urea | 1:2:1 |
1165 | 0.47 | 0.47 | Pyridinic-N, OH, CO |
255 |
| Chestnut | Soft-templating method | KOH | 1:2 |
747 | 4.0 | 0.8 | High carbon content, OH, CO |
256 |
| Fox nut shell | Chemical activation | H3PO4 | 1:3 |
2000 | 1.2 | 1.1 | Functional groups: –OH, CO |
257 |
| Crustacean shell | Carbonization | KOH | 1:4 |
2000 | 4.0 | 1.2 | High carbon content, OH, CO |
178 |
| Hazelnut shell | Chemical activation | KOH | 1:1 |
1696 | 3.5 | 0.7 | Pyridinic-N, OH, CO |
258 |
| Celtuce leaves | Carbonization | KOH | 1:4 |
3404 | 3.5 | 1.2 | High carbon content | 259 |
Recent studies on CO2 capture, both pre- and post-combustion, have investigated various techniques, including adsorption on functionalized porous materials, absorption in liquid amine solutions, membrane separation, and cryogenic separation at ultra-low temperatures.260 However, each method has its strengths and limitations, such as the use of harsh chemicals, equipment wear and tear, disposal challenges, performance inconsistencies, and high associated costs. In contrast, activated porous carbons derived from biomass have emerged as promising and economically viable options for addressing the elevated CO2 levels in the atmosphere. These carbons produced via biomass pyrolysis offer a sustainable approach by converting unwanted biomass materials into efficient adsorbents suitable for large-scale CO2 capture.261 Porous carbon materials offer a triple advantage in CO2 emission reduction, as follows: firstly, by mitigating CO2 emissions from decomposing biomass; secondly, by amending soils with porous carbon to diminish CO2 generation; and thirdly, by utilizing porous carbons for CO2 capture in flue gas streams during both pre- and post-combustion processes. This section delves into the potential applications of biochar and activated porous carbons derived from various biomass precursors for CO2 capture. Although pristine biochar may possess limited porosity and efficacy for CO2 capture, activation procedures can enhance its surface area and porosity, making it suitable for this purpose. Researchers have explored diverse biomass sources and experimental parameters to develop various types of activated porous carbons, aiming to optimize their CO2 capture efficiency.
| Precursor | Modification | Sample name | Adsorption condition | SBET (m2 g−1) | CO2 capture (mmol g−1) | Ref. |
|---|---|---|---|---|---|---|
| Tobacco stem | KOH (500 °C 1 h) | OC500 | 0 °C, 1 bar | 786 | 4.76 | 262 |
| KOH (600 °C 1 h) | OC600 | 1086 | 6.32 | |||
| KOH (700 °C 1 h) | OC700 | 1922 | 7.98 | |||
| KOH (800 °C 1 h) | OC800 | 2399 | 6.60 | |||
| Coconut shell | 500 °C 2 h | C-500 | 0 °C, 1 bar | 21 | 2.0 | 263 |
| (NH2)2CO (600 °C 2 h) | C-600 | 1023 | 5.6 | |||
| KOH (650 °C 1 h) | A-650 | 1535 | 7.0 | |||
| Olive stones | H3PO4 | AC-H3PO4 | 30 °C, 1 bar | 1178 | 10.9 | 264 |
| CO2 | AC-CO2 | 757 | 5.89 | |||
| H2O (g) | AC-H2O | 754 | 7.97 | |||
| Black locust | — | AC | 0 °C, 1 bar | 1175 | 2.79 | 265 |
| KOH | AC-KOH | 2064 | 5.86 | |||
| NH3 | AC-KOH-N | 2511 | 7.19 | |||
| Walnut shells | Urea/carbon 1:3 |
KNWS-13 | 25 °C, 10 bar | 1047 | 5.72 | 266 |
| Urea/carbon 2:3 |
KNWS-23 | 2707 | 10.06 | |||
| Urea/carbon 3:3 |
KNWS-33 | 2461 | 11.02 | |||
| Persian ironwood | H3PO4 | HP5 | 30 °C, 1 bar | 1802 | 3.02 | 267 |
| NiO | HP5/Ni3-1 | 1945 | 6.48 | |||
| CuO | HP5/Cu3-1 | 1954 | 6.78 | |||
| Coffee grounds | — | HC | 35 °C, 1 bar | 34 | 0.14 | 268 |
| Melamine | MHC | 402 | 0.85 | |||
| Melamine, KOH | KMHC | 990 | 2.67 |
5.1.2.1 Pristine carbon materials. Pristine carbon materials, in their natural state, serve as the fundamental building blocks for various applications, including the capture of CO2. However, despite possessing inherent carbonaceous properties such as surface area and porosity, pristine carbon materials often have limited CO2 adsorption capabilities due to their underdeveloped pore structures and surface chemistry. These materials are directly derived from the thermochemical conversion of biomass, such as biochar or hydrochar, well-known for their environmentally friendly characteristics and wide availability from diverse biomass sources. Despite their initial limitations, pristine carbon materials provide the foundation for subsequent modifications aimed at enhancing their CO2 capture efficiency. Through the implementation of various activation methods and doping techniques, pristine carbon materials can be customized to meet specific requirements for CO2 capture, making them a versatile platform for developing effective CO2 adsorbents to counteract the rising atmospheric CO2 levels. Numerous studies have highlighted the CO2 adsorption potential of biochar, which is attributed to its polarity, high specific surface area, and functional groups.270 For instance, composite biochar synthesized from sewage sludge and Leucaena wood in different proportions showed superior CO2 uptake compared to pure sewage sludge biochar. Increasing the Leucaena wood biochar content resulted in a higher carbon content, and subsequently enhanced CO2 adsorption capacity.271 Similarly, date palm leaf biochar produced at varying temperatures exhibited a corresponding increase in CO2 adsorption capacity with higher preparation temperatures, owing to the increased carbon content of the biochar.272 These findings underscore the potential of pristine carbon materials, particularly biochar, as effective CO2 adsorbents and emphasize the significance of optimizing preparation conditions to maximize their CO2 capture efficiency.
5.1.2.2 Activated porous carbon. Activated porous carbon (APC) has emerged as a pivotal material for CO2 capture due to its unique properties, including highly porous structure and large surface area, which enhance its adsorption capabilities. Traditionally, APC has been extensively utilized in various applications such as pollutant removal, industrial purification, and air filtration. Its versatility is attributed to its ability to be tailored through different activation methods, both physical and chemical, to achieve the desired porosity and surface characteristics. Biomass provides a distinctive opportunity for creating activated porous carbons with beneficial structural characteristics. This approach is considered more advantageous than using traditional fossil fuel-based precursors for the production of activated carbons due to low cost and abundant availability of biomass and the environmentally friendly nature of the pyrolysis process used. The process of converting biomass into APC involves pyrolysis, followed by activation using agents such as steam, CO2, and chemical reagents such as KOH or ZnCl2. This process results in a material with a high surface area and tunable pore size distribution, making it highly effective for capturing CO2.273
Physical activation, such as CO2 or steam activation, plays a crucial role in enhancing the microporosity of the carbon structure, thereby increasing its specific surface area and improving CO2 adsorption. The choice of biomass precursor and the conditions of the pyrolysis process, including temperature and residence time, significantly influence the final properties of the APC. For instance, CO2 activation is known to produce microporous carbons with uniform pore structures, which are ideal for gas adsorption, while steam activation can introduce mesopores and macropores, potentially enhancing the diffusion of CO2 molecules into the carbon matrix.274 Alternatively, chemical activation can further enhance the porosity and surface functionality of APCs, making them even more effective for CO2 capture. Activating agents such as KOH and ZnCl2 facilitate the development of a hierarchical pore structure and the introduction of functional groups that can interact with CO2 molecules, improving the adsorption efficiency.226,275 The ability to engineer APCs with specific surface properties through controlled activation processes makes them highly promising materials for addressing the growing challenge of CO2 emissions and climate change.
Fig. 11 illustrates the process of CO2 capture using activated porous carbon derived from biomass, which was chemically activated with potassium hydroxide (KOH). The process begins with the raw biomass of Jujun grass, which undergoes KOH activation to create a highly porous carbon material. Subsequently, this activated porous carbon is used for CO2 adsorption, effectively capturing CO2 molecules at 25 °C as a function of pressure.276 This highlights the potential of KOH-activated porous carbon as a highly efficient material for CO2 capture, which is crucial for mitigating the impact of greenhouse gases on the environment.
 | ||
| Fig. 11 CO2 adsorption performance of KOH-activated biomass-derived porous carbon.276 | ||
5.1.2.3 Non-functionalized activated carbon. Non-functionalized activated carbon derived from various precursor materials undergoes activation processes without additional chemical modifications to its surface, preserving its inherent physical and chemical attributes. In contrast to functionalized activated carbon, which incorporates specific functional groups to target adsorption, non-functionalized activated carbon relies solely on its porous structure and high surface area for adsorption. Generally, activated carbons demonstrate superior CO2 adsorption capacities compared to biochar due to their improved textural properties and surface chemistry resulting from activation treatments. CO2 capture mechanisms involve both physical and chemical processes. Physical adsorption arises from van der Waals forces between CO2 molecules and the carbon surface, with surface area and pore size playing critical roles in the adsorption efficiency. Conversely, chemical adsorption is attributed to the alkali functional groups on the carbon surface, enhancing the interaction and uptake of CO2. Activation significantly enhances the textural properties of carbon materials, thereby boosting their CO2 adsorption performance.
Highly microporous KOH-activated carbons derived from Jujun grass (JG) and Camellia japonica (CJ) biomass exhibited significant CO2 capture capabilities in both pre- and post-combustion scenarios.277 The activated carbons produced at 700 °C with a KOH impregnation ratio of 2 demonstrated the highest CO2 adsorption at 1 bar (4.9–5.0 mmol g−1) under room temperature conditions. Conversely, that prepared at 800 °C with a higher KOH impregnation ratio of 4 showed the highest CO2 adsorption at 20 bar (21.1 mmol g−1). Mesquite wood, when transformed into carbon-rich material through single-step KOH activation at 800 °C, achieved a high surface area of 3167 m2 g−1 and CO2 adsorption of 26.0 mmol g−1 at 23 °C and 30 bar.261 Similarly, microporous activated carbons synthesized from Arundo donax at 600 °C displayed an excellent CO2 adsorption capacity of 15.4 mmol g−1 at 30 bar, with a surface area of 1122 m2 g−1. ZnCl2 activation of Arundo donax resulted in an even higher surface area of 3298 m2 g−1 and CO2 adsorption of 30.2 mmol g−1 at 30 bar.278 These results emphasize the critical role of surface area and porosity in enhancing the CO2 adsorption by activated porous carbons.
The microporous content of activated carbon significantly influences CO2 adsorption, with a higher micropore content correlated with increased CO2 adsorption.279 Research on KOH-activated rice husk char showed that lower activation temperatures (640–710 °C), reduced KOH impregnation ratios (1:1), and elevated nitrogen content resulted in the high CO2 uptake of 2.11 mmol g−1 at 0.1 bar. A narrow pore size distribution and micropores less than 0.7 nm were identified as crucial for the enhanced CO2 adsorption at sub atmospheric pressures. These activated carbons also exhibited high selectivity (19.9) for CO2 over N2. Another study highlighted the importance of ultra-micropores in CO2 capture using KOH-activated bamboo porous carbons, reporting a notable CO2 adsorption value of 7.0 mmol g−1 at 0 °C and 1 bar, which was attributed to the micropores of 0.55 nm.280 However, wider pores resulting from increased KOH impregnation amounts led to reduced CO2 adsorption due to the limited pore diffusion.
Likewise, Patel et al. highlighted the importance of narrow micropores (<1 nm) in CO2 capture by activated porous carbons produced through the KOH activation of sawdust.281 Mild KOH treatment resulted in narrow micropores with lower surface areas compared to severe treatment, yielding high CO2 uptake (4.8 mmol g−1) at room temperature. The presence of a microporous structure, hydroxyl groups, and heteroatoms such as nitrogen and oxygen collectively regulated the CO2 adsorption on activated porous carbons derived from KOH activation of gelatin and starch biomasses, resulting in substantial CO2 adsorption (7.49 mmol g−1) at 1 bar and 0 °C, with favorable selectivity for CO2 over N2 (52–98%).282 The isosteric heats of adsorption values (21.7–62.9 kJ mol−1) suggest the involvement of both chemical and physical adsorption mechanisms. Additionally, wood ash-based activated porous carbon has been proposed for CO2 capture, primarily via chemical sorption.283 Analysis pre- and post-CO2 adsorption revealed the participation of alkali and alkaline earth metals in wood ash, facilitating chemical reactions with CO2 and H2O and resulting in the formation of diverse carbonate products.
5.1.2.4 Heteroatom-doped activated porous carbon. Doping activated porous carbon with heteroatoms such as nitrogen, oxygen, and sulfur during activation modifies its electronic properties and surface chemistry, enhancing its adsorption capabilities and selectivity, particularly for CO2. Although micropores are crucial for CO2 capture, they also promote the adsorption of other gases such as CH4 and N2, leading to reduced CO2 adsorption capacity due to competitive adsorption. However, heteroatom doping addresses this issue by introducing functional groups that exclusively interact with CO2, boosting the CO2 selectivity. Incorporating basic or electron-rich heteroatoms such as nitrogen into the carbon framework further enhances the CO2 absorption capacity, given that the abundant basic sites on the surface act as anchors for capturing the weakly acidic CO2 molecules. For example, Nazir et al. added nitrogen-containing functional groups to activated porous carbons for CO2 extraction during carbonization using an NH3 solution.265 This three-step process resulted in a final carbon material with a high nitrogen content (7.21%) incorporated as functional groups, exhibiting a higher surface area (2511 m2 g−1) compared to the carbon prepared using KOH activation alone (2064 m2 g−1). The enhanced surface area and basic sites facilitated significant CO2 adsorption at both 0 °C (7.19 mmol g−1) and 25 °C (5.5 mmol g−1), with high ultra-microporosity aiding in physical and chemical CO2 adsorption.284 Additionally, the sample demonstrated high selectivity for CO2 over N2 (30.75%), rapid kinetics, and efficient regeneration, presenting a promising approach for CO2 capture using biomass-based materials.
In a study conducted by a research group, activated carbon was produced from husk using carbonization and KOH activation, with chitosan introduced during its activation.200 The resultant doped activated carbon displayed an enhanced CO2 adsorption capacity, reaching 5.83 mmol g−1 at 0 °C. The XPS analysis detected pyridine-N and pyrrole-N on the surface of the doped activated carbon, enhancing its surface alkalinity and facilitating CO2 uptake. Similarly, another research team enhanced the CO2 capture performance of activated carbon by modifying it with ammonium sulfate.285 The presence of sulfur functional groups enabled interaction with CO2 molecules, thereby improving their adsorption. Nitrogen doping plays a vital role in heteroatom doping to boost the CO2 capture efficiency. Nazir and Wang et al. employed various amines (TEPA, MEA, DEA, PEI, and DETA) to modify waste wood ashes for CO2 capture. Among them, the TEPA-sorbent exhibited a superior CO2 capture performance, with a capacity of 1.76 mmol g−1 and an impressive regeneration rate of 92.6% (Fig. 12).265,286 Despite the DEA-sorbents having the highest actual nitrogen loading, their adsorption capacity was lower than that of TEPA, highlighting the multifaceted nature of CO2 adsorption, where the nitrogen content alone does not determine the capacity. The significance of micropores in CO2 adsorption was demonstrated by the optimized loading of TEPA (45 wt%), which yielded a superior CO2 capture performance (2.02 mmol g−1). Furthermore, the type of nitrogen-containing functional groups significantly impacts the CO2 capture efficiency. Wang et al. investigated the roles of various nitrogen functional groups in the CO2 adsorption process, with pyridine-N showing the highest CO2 adsorption energy (−21.4 kJ mol−1) due to its robust dipole–dipole interactions with CO2, similar to H+ and O2−.269,287 Thus, understanding these mechanisms is crucial for tailoring adsorbents for the optimal CO2 capture performance.
 | ||
| Fig. 12 Heteroatom-doped carbons for CO2 capture and CO2 adsorption performance of wood ash (WA-1) activated with various polyamines, showing (a) CO2 adsorption breakthrough profiles, and (b) adsorption capacity along with amine efficiency. Regeneration performance of WA-1 activated with different polyamines: (c) CO2 desorption curves, and (d) adsorbent regeneration potential and conversion efficiency. Reprinted with the permission.265,286 | ||
5.1.2.5 Metal/metal oxide-embedded activated porous carbon. Metal or metal oxide-embedded activated porous carbons stand out as a versatile and highly effective class of carbon-based materials, which are characterized by the integration of metal or metal oxide nanoparticles into their porous structure. These materials are synthesized using a variety of methods, such as impregnation, co-precipitation, and in situ growth, resulting in enhanced adsorption properties and catalytic activity owing to the incorporation of metal or metal oxide nanoparticles into the carbon matrix. Another strategy to boost the CO2 adsorption capacity involves functionalizing activated porous carbons with metal atoms to create additional surface-active sites. Numerous studies have explored the impact of metal functionalization on CO2 uptake, with alkaline metals proving particularly effective in adsorbing acidic carbon dioxide. In the adsorption process, CO2 serves as an electron acceptor, capable of receiving electrons from metal oxides possessing basic characteristics.
The adsorption mechanism of CO2 onto metal-impregnated AC is depicted in the Fig. 13. Metal oxides are typically incorporated in the surface of AC using the wet impregnation method. This involves mixing the carbon precursor with metal oxide solutions before subjecting it to a high-temperature carbonization process, or alternatively, mixing the already prepared AC with metal oxide solutions followed by calcination at elevated temperatures.13 For instance, Botomé et al. produced AC from wood treated with chromated copper arsenate (CCA), which was pyrolyzed at 700 °C, and the resulting material demonstrated an adsorption capacity of 1.88 mmol g−1 for CO2.288 The metal oxide impregnation on AC significantly impacts both the structural characteristics and CO2 adsorption capacity, depending on the metal loading. A low metal loading may not enhance the adsorption capacity, while an excessively high metal loading can cause pore clogging, which reduces the surface area and pore volume, thereby diminishing the adsorption performance.
 | ||
| Fig. 13 Mechanism of impregnating activated carbon with metal oxides. | ||
Recent studies have focused on enhancing CO2 capture using carbon composites derived from walnut shells and doped with metals such as Mg and Mg–Al. Although metal loading led to reduced specific surface areas due to pore clogging, the CO2 capture performance of these composites improved significantly.289 Notably, the composites with 10% Mg and 5% Al achieved a CO2 uptake of 4.5 mmol g−1. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) confirmed the effective distribution of Mg and Al on the carbon surface, while Fourier-transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD) revealed weakened metal oxide peaks after CO2 adsorption, indicating enhanced chemisorption. This improved performance is attributed to the combined effects of physical and chemical adsorption mechanisms in the metal-doped carbon materials.290
In parallel, biochar modified with AlCl3 demonstrated superior CO2 uptake compared to unmodified biochar. This was due to the primary role of surface adsorption facilitated by the metallic oxides present. Additionally, activated porous carbons derived from pine cones and treated with KOH, incorporating nitrogen and metals, showed high CO2 adsorption capacities.291 For instance, the samples prepared at 700 °C with a KOH ratio of two, containing 0.5% nitrogen and calcium, achieved a CO2 uptake of 4.7 mmol g−1.292 These materials also exhibited an excellent performance in the oxygen reduction reaction (ORR), suggesting their potential in various applications beyond CO2 capture. Table 7 provides a comparative overview of various types of sorbents, highlighting their physical and chemical properties together with their CO2 adsorption capacities.
| Sorbent type | Physical properties | Chemical properties | CO2 adsorption capacity | Ref. |
|---|---|---|---|---|
| Non-modified carbon | High surface area, microporous structure | Primarily carbon, with surface oxides | 30.2 mmol g−1 at 25 °C, 30 bar | 278 |
| Nitrogen-doped carbon | Moderately high surface area, mesoporous | Nitrogen groups improve basicity | 5.83 mmol g−1 at 0 °C, 1 bar | 285 |
| Sulfur-doped carbon | Microporous and mesoporous structure | Sulfur groups increase binding sites | ∼2.8–3.8 mmol g−1 at 25 °C, 1 bar | 293 |
| Metal-modified carbon | Surface area depends on metal type, porosity varies | Enhanced by metal oxides (e.g., Al, Cu, Fe) | 4.5 mmol g−1 at 25 °C, 1 bar | 290 |
| Metal oxide-embedded AC | High surface area (∼600–1200 m2 g−1), microporous/mesoporous | Embedded metal oxides (e.g., Fe3O4), surface oxygen groups | 4.7 mmol g−1 at 25 °C, 1 bar | 292 |
Furthermore, the incorporation of metal oxides such as MgO, BaO, and Fe2O3 into activated carbon has been shown to enhance its CO2 adsorption capacity. Basic oxides, in particular, are more effective than acidic or neutral oxides. For example, MgO significantly improved the CO2 capture at higher temperatures, and BaO provided the greatest enhancement due to its high electronegativity.294 These findings underscore the potential of metal-functionalized activated carbons in optimizing CO2 capture and other environmental applications.
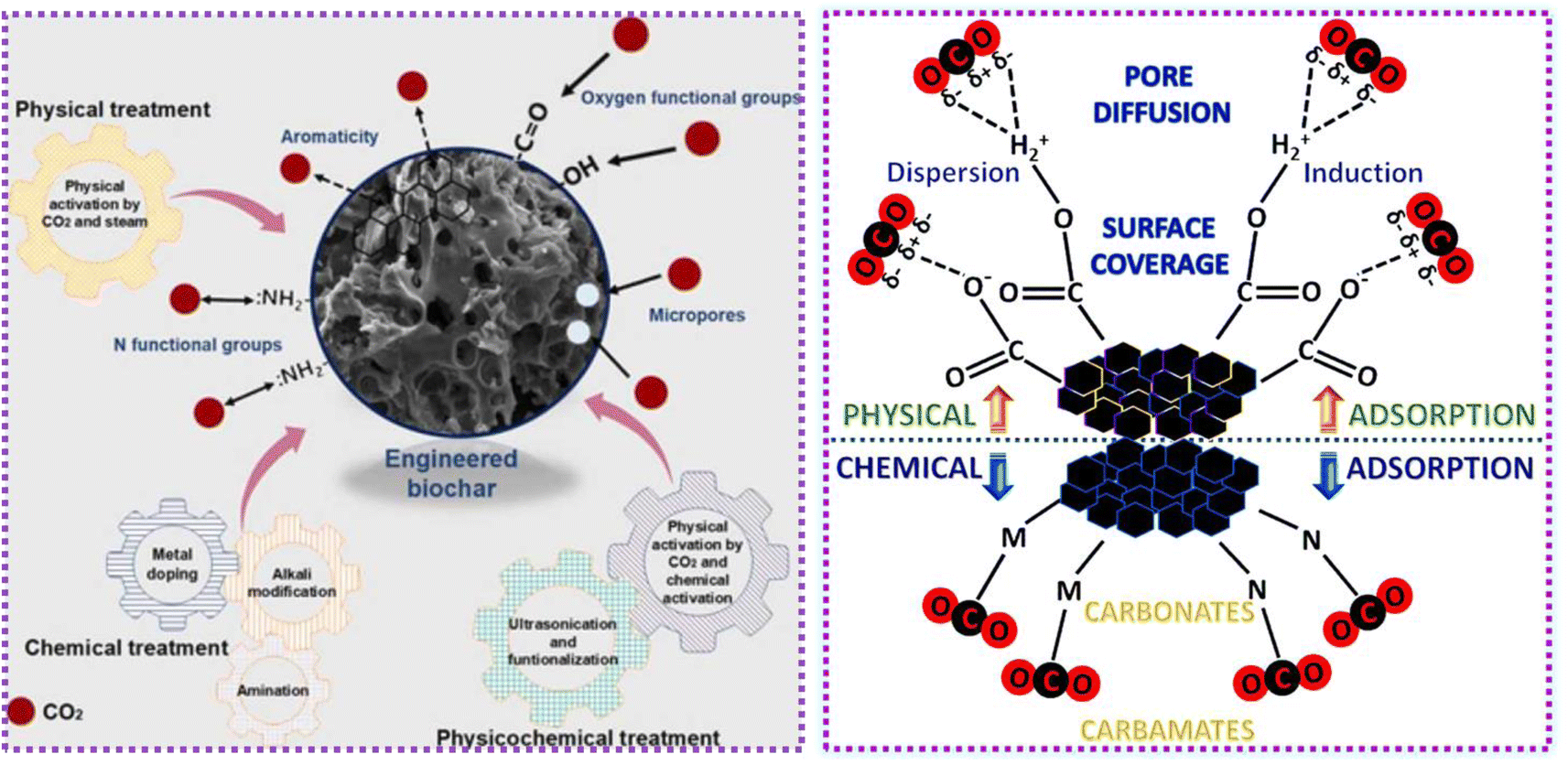 | ||
| Fig. 14 Processes involved in CO2 capture by carbon materials. Reprinted with permission.269,295 | ||
Physisorption, a reversible process driven by van der Waals forces and electrostatic forces, is primarily governed by the textual properties of carbon materials, including pore structure and surface area. Although the large specific surface area and porosity of carbon materials enhance CO2 capture, their pore size is more crucial than specific surface area and total pore volume for CO2 adsorption. Micropores significantly contribute to CO2 adsorption, particularly at low adsorption pressures. Notably, studies have identified narrow micropores with sizes in the range of 0.5 to 0.7 nm as critical for enhancing the CO2 adsorption capacity.295 Under higher adsorption pressures, adsorption occurs through surface coverage, where the specific surface area plays a pivotal role. Adsorption temperature also influences physical adsorption, with higher temperatures promoting CO2 diffusion and weakening CO2–carbon material interactions. Chemisorption, an irreversible chemical reaction process, involves the formation of new chemical bonds between adsorbents and adsorbates, facilitated by surface functional groups and alkali ions. Hydrogen bonds and Lewis acid–base interactions play crucial roles in chemisorption. Studies have highlighted the significance of hydroxyl groups in enhancing CO2 capture, given that CO2 molecules can form hydrogen bonds with –OH, promoting CO2 adsorption. Nitrogen-containing polymers have been employed to modify carbon materials, enhancing their alkalinity and electron density to strengthen the Lewis acid–base interaction. Moreover, the covalent coordination bonds formed between CO2 and the adsorbent surface contribute to chemisorption. Researchers have developed novel N, P co-doped porous carbon materials with high specific surface areas and rich mesopores, micro pores, and N, P, resulting in enhanced CO2 uptake capacity.296 The presence of N groups in porous carbon derived from lotus stalks has been linked to excellent CO2 adsorption capacity, with the N groups playing a dominant role in CO2 adsorption at higher temperatures and lower pressures. However, the excessive loading of modifying agents such as melamine can clog the pores and reduce the specific surface area of carbon materials, thus impacting their CO2 capture performance. Thus, achieving a balance between loading amount and porosity changes is essential to realize the optimal CO2 capture efficiency. Studies have demonstrated an enhancement in surface area through heat treatment after melamine impregnation, highlighting the importance of carefully managing the loading amounts to maximize the capture performance. These findings underscore the multifaceted mechanisms underlying CO2 capture by carbon materials and emphasize the need for tailored material design to optimize the efficiency in various applications.
| Sorbent | Characteristics/processes/advantages | Drawbacks/disadvantages | Ref. |
|---|---|---|---|
| Metal oxides | Both low toxicity and abundant availability | High energy needs for regeneration | 297 and 298 |
| Waste products | Renewable sources, low cost and abundant | CO2 adsorption capacity varies significantly based on the chemical, physical, and structural properties of the raw material, as well as its preparation history and treatment conditions | 299–301 |
| Commercial activated carbon | The best adsorbent available | Initial cost of the carbon and non-renewable resources | 302 |
| Activated alumina | Relatively well-known and commercially available | High cost of adsorbent | 303–305 |
| Zeolites | Large surface area, stable temperature, and less heat needed for regeneration | Performance decrease in the presence of moisture and impurities in gas feed | 306 |
| MOF | High surface area and physiochemical stability | Costly and difficult to produce for industrial scale | 307–309 |
| Silica | Large surface area, strong thermal and mechanical stability | High regeneration energy requirements | 310 and 311 |
| Agricultural wastes | Low cost and abundant | Performance varies depending on the precursor | 312 |
| Industrial by products | Highly available and low-cost precursor | Varies adsorption strength depending on the precursor | 313 |
| Shrimp shells/chitosan | Plentiful, renewable, biodegradable, and environmentally friendly resource | Low surface area | 314 and 315 |
| Peat | A cheap, plentiful, and easily accessible biosorbent | Low mechanical strength | 316 |
| Biomass | Plenty resources with high adsorption capacity, low cost and effective technology | In study/experimental stage | 222 and 225 |
| Biochar | Many resources with a large capacity for adsorption, inexpensive costs, and efficient technology | Low CO2 uptake compared to AC | 317 |
Activated carbon fibers (ACFs) were developed from mechanically recycled acrylic (PAN), cotton (CO), and their blends (PAN/CO) to enhance CO2 capture.320 The production process involved stabilization, carbonization, and chemical activation. However, although PAN is effective for ACF production, it is costly and has significant environmental impacts due to its energy-intensive production. Thus, to address these issues, recycled or renewable materials were utilized to reduce both costs and environmental footprint. PAN fibers were stabilized using oxidation and phosphoric acid treatment, followed by carbonization at a temperature of 800 °C or 900 °C under nitrogen, and activation with various KOH ratios at 700 °C. The stabilization process preserved the fiber structure and improved the heat resistance.321 Generally, the PAN fibers maintained their integrity after carbonization and activation, although some samples showed rough surfaces due to the incomplete KOH removal.
The CO2 adsorption capacity of ACFs made from recycled fibers was notably high, with higher KOH ratios enhancing their surface area and pore volume, which in turn improved their CO2 capture performance. These recycled ACFs showed comparable or superior CO2 adsorption capacities to commercial ACFs, highlighting their potential for sustainable CO2 capture. Additionally, this study evaluated the mass yield and elemental composition of PAN and PAN/cellulose (PAN/cell) nanofibers in producing activated carbon nanofibers (ACnFs). Oxidative stabilization reduced the mass of PAN/cell fibers more than PAN fibers. Carbonization and activation caused significant mass loss in the PAN/cell nanofibers due to their lower carbon content and thermal stability, although the PAN/cell fibers had a higher nitrogen content. The SEM and FTIR analyses confirmed the changes in the fiber diameter and chemical structure. Fig. 15a shows SEM images of biomass fibers before and after grafting with poly(acrylic acid) (PAAc), highlighting the increase in surface roughness.322 Fig. 15b presents the SEM images of the PAN fibers at various stages, showing changes in surface texture and porosity with treatment. Fig. 15c shows a comparison of the untreated PAN fibers with that carbonized at different temperatures, revealing significant structural transformations. The PAN-based ACnFs carbonized at 800 °C exhibited higher CO2 adsorption compared to that processed at 900 °C, emphasizing the impact of the production conditions on CO2 capture efficiency.324 Recent evaluations of the CO2 adsorption capacities for various the carbon-based adsorbents at 1 bar and 25 °C highlight their effectiveness in CO2 capture. This study shows that recycled ACFs derived from acrylic carbon show a comparable performance or surpass that of many commercial carbon adsorbents.323
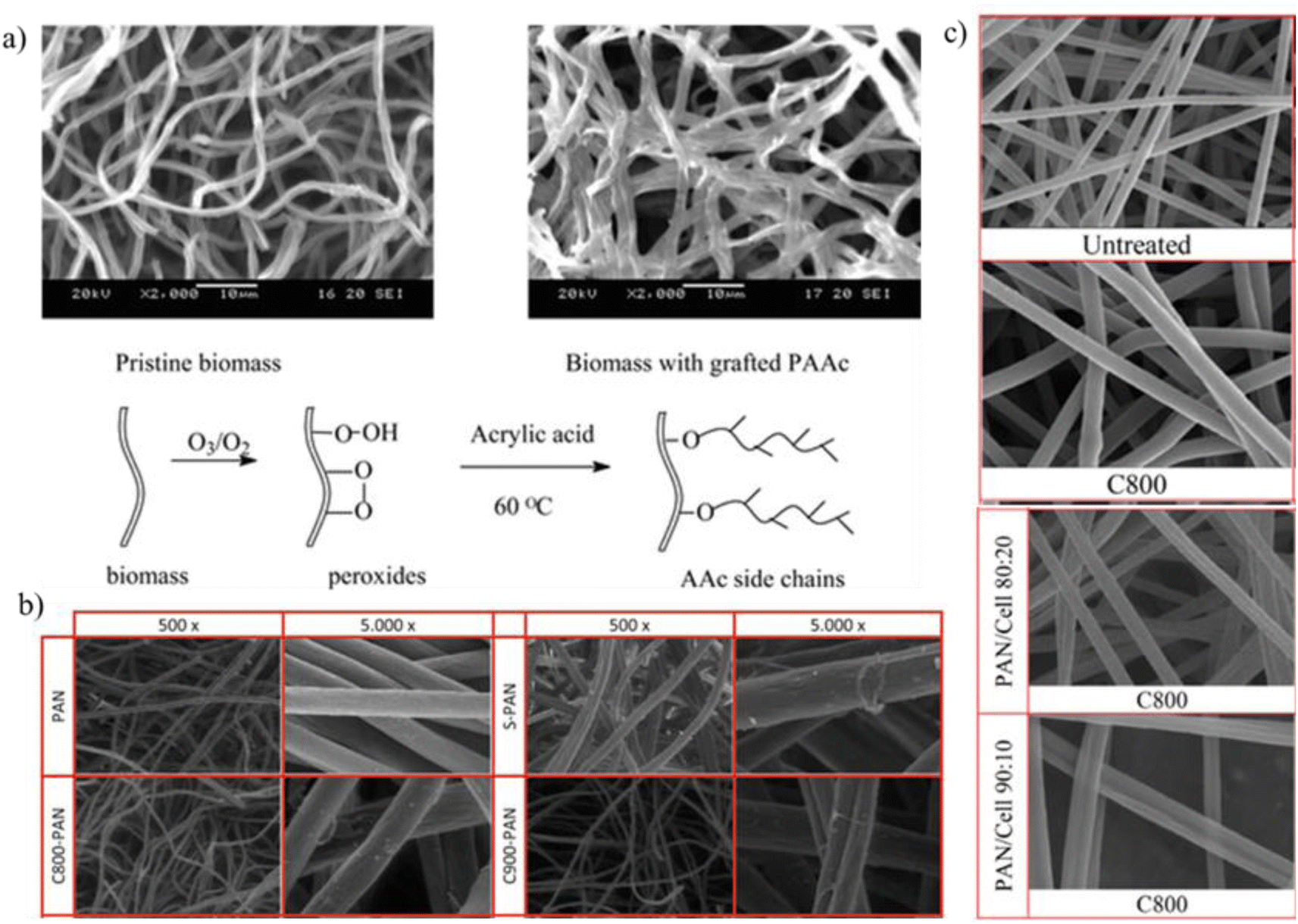 | ||
| Fig. 15 Surface morphology of biomass and polyacrylonitrile (PAN)-based nanofibers for enhanced CO2 capture: (a) pristine vs. PAAc-grafted biomass, (b) SEM comparison of untreated PAN, C800, and PANC/cell composites, and (c) PAN nanofiber morphology under varying treatments and magnifications. Reproduced from ref. 322 and 323. | ||
Furthermore, doping acrylic carbon with metal nanoparticles, such as alkali and alkaline earth metals, has shown to boost the CO2 adsorption significantly. Metals such as sodium, calcium, and magnesium interact synergistically with CO2, combining the benefits of both physisorption and chemisorption. Metal-doped acrylic carbon adsorbents exhibit higher CO2 capture capacities and improved selectivity, given that the metal sites provide additional reactive centers for CO2 binding.265 These modifications not only enhance the CO2 capture performance but also improve the stability and recyclability of the adsorbent, making them viable for long-term industrial applications.
The application of acrylic carbon for CO2 adsorption holds promise across various sectors, including flue gas treatment, air purification, and carbon capture and storage technologies. The versatility in its synthesis and the ability to finely tune its structural and chemical properties make acrylic carbon a highly adaptable material for CO2 capture. Recent advancements focus on optimizing the synthesis routes to improve its pore structure and surface functionality, thereby maximizing its adsorption efficiency under different operational conditions.325 Continued research is essential to fully understand the adsorption mechanisms at the molecular level, which will aid in designing more efficient and cost-effective acrylic carbon-based adsorbents. Moreover, scaling up the production of these materials, while maintaining their performance and stability is crucial for practical applications. As global efforts to mitigate climate change intensify, the development of advanced materials such as acrylic carbon for CO2 capture is becoming increasingly critical. The integration of these materials into existing CO2 capture frameworks can significantly enhance their efficiency and contribute to reducing atmospheric CO2 levels.326
5.2 Prospects for commercialization
The potential for commercializing a product or technology relies on diverse factors such as market demand, technological feasibility, scalability, regulatory compliance, and economic viability. In the domain of CO2 adsorbents, the prospects for their commercialization depend on their capacity to meet or exceed industry-defined performance standards, while remaining cost-effective and environmentally sustainable. Additionally, considerations such as the availability of raw materials, manufacturing simplicity, and compatibility with existing infrastructure play pivotal roles in determining the feasibility of their large-scale production and deployment. With various synthesis strategies for biomass-derived porous carbon materials, the transition from laboratory-scale or pilot plant development to industrial-scale production capacity becomes crucial. Achieving this transition requires integrating chemistry and chemical engineering technologies. Nonetheless, several challenges need to be addressed. Firstly, scaling up synthesis protocols from gram to kilo batch sizes poses a significant hurdle, compounded by uncertainties in biomass supply and associated logistical costs. Establishing local biomass resources, enhancing annual yields, and devising new logistics routes can offer sustainable supply chains from field to processing plant. Subsequently, for improved qualities, micro and mesoporous carbon powder materials must be transformed into granules, pellets, beads, or extrudates with or without binders. Granulation techniques sometimes result in the loss of specific surface area, despite the fact that granular materials offer higher mechanical strength, decreased attrition, and lower resistance in the adsorbent bed. However, studies suggest that incorporating biomass-based pore-forming materials can enhance the sorption properties.327 Once the characteristics of materials are understood, finalizing the design of CO2 capture plants becomes essential. Strategies may involve temperature swing adsorption (TSA), pressure swing adsorption (PSA), vacuum swing adsorption (VSA), or combinations thereof. These next-generation carbon materials outperform traditional sorbents such as zeolites due to their inherent hydrophobic properties, low isosteric heat of adsorption, high CO2 adsorption capacities, and long-term stability across multiple cycles. Fig. 16 schematically illustrates the transition from porous carbon powder to plant-level deployment. Further material and design considerations should be made based on the specific CO2 removal application, whether for pre- or post-combustion scenarios requiring high CO2 concentration removal or for confined environments such as cockpits and underground spaces with lower CO2 concentrations. In these cases, high-performance materials such as carbon with strong interactions at low surface coverage, requiring minimal heat for regeneration, are essential. Additionally, anticipating the adsorption of CO2 under humid conditions underscores the need for porous carbons capable of efficient operation in these circumstances. | ||
| Fig. 16 Diagram illustrating the complete CO2 capture process, starting from powder form to palletization, and depicting the sorption cycle for real-world application. | ||
5.3 Cost assessment framework
Cost assessment frameworks for producing activated porous carbons, especially for CO2 capture, play a pivotal role in determining the economic feasibility of transitioning from laboratory-scale experiments to full-scale industrial production. Producing activated carbons from biomass encompasses various cost components, which can be broadly categorized into fixed and variable costs. Typically, fixed costs involve capital investments in infrastructure, equipment, and facilities necessary for the initial setup of the production plant. Alternatively, variable costs include raw materials, energy consumption, labor, maintenance, and other operational expenses. The interplay among these costs significantly impacts the overall financial outlook of producing activated carbon on an industrial scale.Establishing a production plant to meet industrial demand involves substantial capital investments, which are proportional to the plant size and the scale of equipment required for different production stages, from raw material preparation to the final collection of activated carbon. For instance, producing around 450 tonnes of physically activated carbon from biomass annually necessitates an initial capital investment of approximately $2.12 million, coupled with annual operational costs of about $1.22 million. In contrast, using a chemical activation method, such as with H3PO4, for similar production volumes requires a total cost investment of approximately $4.25 million, with market prices ranging from $2.7 kg−1 to $2.9 kg−1. These figures underscore the significant financial commitment required for large-scale production, emphasizing the importance of precise cost assessment frameworks.328 The cost dynamics of producing activated carbon from biomass are complex and multifaceted, as shown by recent studies. Skoczko et al. estimated that physical activation processes generally cost between $1.5 and $2.0 per kilogram, with variations influenced by factors such as the scale of operation and the type of biomass feedstock used.329 Chemical activation, particularly with H3PO4, incurs significantly higher costs, with Raja et al. reporting costs ranging from $3.0 to $3.5 per kilogram.330 This difference underscores the importance of selecting the appropriate activation method, given that it plays a crucial role in the overall production costs.
The feedstock choice is another critical factor affecting the cost structure. Agricultural residues and forestry by-products are often more affordable options, but chemical activation methods tend to drive up costs due to the expensive reagents required. For instance, Ng et al. found that producing activated carbon from pecan shells costs between $2.60 and $2.76 per kilogram, depending on the activation method.331 Similarly, Lima et al. achieved a competitive cost of $1.38 per kilogram for activated carbon derived from organic waste using physical activation, assuming no raw material expenses.332 In contrast, Stavropoulos et al. provided a comparative analysis of the production costs for activated carbon from various materials, revealing significantly higher costs for non-biomass sources such as used tires ($10.88 kg−1), wood ($6.09 kg−1), and lignite ($5.13 kg−1).333 Serafin et al. further illustrated the variability in production costs by estimating that producing 50000 kg of activated carbon from fern leaves will cost approximately $2.72 per kilogram based on an annual production cost of $135.690.334,335 Additionally, energy consumption remains a key cost component due to the energy-intensive nature of the carbonization process, with market prices for activated carbon ranging from $2.7 to $3.3 per kilogram, depending on its quality and application.336
According to the comparison of these market prices with the estimated production costs, biomass-derived activated carbons show a competitive edge, particularly when considering the environmental benefits of using renewable resources and minimizing waste. Looking at the commercial landscape, the global production of activated carbon reached USD 7.2 billion in 2023 and is projected to grow by 12.42%, reaching USD 14.50 billion by 2031. This surge in demand for activated carbon can be attributed to various factors, including population growth, extensive consumption across industries, and stringent environmental regulations governing air and water purification systems. Currently, the commercial-scale production of activated carbon predominantly relies on coconut shell and coal-based precursors. However, these raw materials alone may not be sufficient to meet the escalating demand across diverse sectors. Therefore, exploring alternative precursors, particularly biomass, can offer a cost-effective solution to meet the projected demands. Globally, approximately 105 billion metric tonnes of biomass, sourced from both land and oceans, is available annually.337
Refining the cost assessment frameworks involves a comprehensive analysis of the production costs, market trends, and operational efficiencies. As technological advancements and process optimizations continue to lower production costs, the economic feasibility of large-scale biomass-based activated carbon production is improving, making it an increasingly attractive option for applications such as CO2 capture. Ongoing research and development will be crucial in further reducing costs and enhancing the viability of this sustainable production pathway.
6. Research gap
Despite the considerable volume of research conducted on the potential of waste products as precursors for AC, a significant research gap exists concerning the cost-effectiveness and scalability of the process. Many studies lack detailed procedures during the synthesis stage, resulting in inconsistent characteristics and adsorption properties of the manufactured product. To commercialize this type of activated carbon, there is a need for a thorough investigation into the continuity of waste supply for precursors, given that abundant resources are essential. Although the average biochar yield is around 20 wt% compared to the raw materials, detailed statistics on precursor availability are necessary to ensure long-term sustainability. Additionally, more research effort is required to explore regeneration methods for activated carbon, given that current technologies often necessitate high heat, which may not be feasible in the long run. Furthermore, there is a lack of studies on the synthesis of hybrid activated carbons incorporating heteroatoms and metal oxides. These hybrid materials have the potential to exhibit enhanced adsorption properties and stability. Moreover, there is a dearth of research focusing on the detailed understanding of CO2 adsorption at the quantum level. These insights are crucial for predicting the behavior and performance of activated carbons under various operating conditions accurately. Comprehensive studies in this area will significantly contribute to advancing the understanding of the CO2 capture mechanisms and optimizing the performance of activated carbon-based adsorbents for practical applications.7. Summary and prospects for future investigations
This review article provided a comprehensive examination of the synthesis and modification techniques for biomass-derived carbon materials intended for CO2 capture. It delved into the fundamental methodologies for converting biomass into carbon materials and the critical factors influencing their properties. The importance of selecting appropriate biomass sources and processing parameters to tailor the structure and chemical composition of carbon materials was emphasized. Various modification approaches, both physical and chemical, were outlined, highlighting their role in modifying pore structures or introducing functional groups or metals to enhance the CO2 capture efficiency. However, despite the advancements, challenges and research opportunities persist, particularly in understanding the relationship between pore structure and factors such as biomass characteristics, pyrolytic conditions, and activation methods. Optimizing the pore structure of activated carbons, especially that activated with agents such as KOH and ZnCl2, is crucial. Additionally, synthesizing hybrid activated carbons using multiple activating agents holds promise for improving the CO2 capture performance through hybrid porous features.Moreover, although renewable biomass serves as a cost-effective precursor for carbon capture materials, pristine carbon materials have inherent limitations in terms of pore structure and surface chemistry, constraining their adsorption capacities. Although various modification methods have emerged to address these limitations, some processes are complex and costly, hindering their scalability for industrial applications. Template carbonization offers a viable alternative, providing control of the material pore diameter and structure without additional modification treatments. However, much of the research on CO2 adsorption by carbon materials remains confined to laboratory settings, overlooking real-world challenges such as moisture and corrosion. Future research should focus on enhancing the mechanical properties of carbon materials to withstand harsh environments and assessing their suitability for industrial applications. Furthermore, it is necessary to explore competitive adsorption mechanisms among different gases and simulate the carbon material adsorption performance in complex gas compositions. Additionally, research efforts should extend beyond CO2 capture to encompass CO2 conversion, leveraging the catalytic properties of biomass-based carbon materials. The in situ conversion of adsorbed CO2 presents an intriguing avenue for sustainable CO2 recovery. However, challenges related to regenerating CO2-saturated carbon materials and mitigating the effects of contaminants require further investigation. Understanding the impact of contaminants on carbon materials and developing strategies to enhance their resistance can enhance their longevity and performance in CO2 capture applications.
Transitioning from laboratory-scale studies to pilot-scale investigations is crucial to evaluate material performance under realistic industrial conditions. Furthermore, gaining deeper insights into the underlying mechanisms for the formation of activated porous carbons from biomass and their regeneration processes can facilitate the design of advanced carbon materials for various applications. Addressing these research gaps and challenges is essential to harness the full potential of biomass-derived activated porous carbons for CO2 capture, water decontamination, and energy storage, thereby advancing carbon capture technologies and promoting environmental sustainability.
Data availability
The data supporting the findings of this review can be obtained from the corresponding author upon request. Due to privacy concerns and other restrictions, the data are not publicly accessible.Author contributions
Shreyase Kundu, Tasmina Khandaker & Md Al-Amin Mia Anik: writing – original draft, review and editing; Md. Kamrul Hasan, Palash Kumar Dhar, Sagar Kumar Dutta & M. Abdul Latif: proof reading and editing; Muhammad Sarwar Hossain: formal analysis, methodology, resources, validation, writing and editing, supervision and project administration.Conflicts of interest
The authors declare that they have no known financial or personal conflicts of interest that could have influenced the work reported in this paper.Acknowledgements
The authors express their deep gratitude to the Chemistry Discipline at Khulna University for offering a conducive research environment and necessary facilities. This project was funded by the faculty budget and research grant of Khulna University, Bangladesh.References
- B. Dziejarski, J. Serafin, K. Andersson and R. Krzyżyńska, Mater. Today Sustain., 2023, 100483 Search PubMed.
- G. Polya, WMO Warning: 1.5 Degree C Warming Breach Very Soon & With Increasing Frequency. Act Now!, Climate Change, 2023 Search PubMed.
- T. Khandaker, M. S. Hossain, P. K. Dhar, M. S. Rahman, M. A. Hossain and M. B. Ahmed, Processes, 2020, 8, 654 CrossRef CAS.
- V. Ntouros, I. Kousis, A. L. Pisello and M. N. Assimakopoulos, Energies, 2022, 15, 1489 CrossRef CAS.
- J. Serafin, B. Dziejarski, X. Vendrell, K. Kiełbasa and B. Michalkiewicz, Biomass Bioenergy, 2023, 175, 106880 CrossRef CAS.
- Y. Liu, Z. Weng, B. Han, Z. Guo, H. Tian, Y. Tang, Y. Cai and Z. Yang, J. Cleaner Prod., 2023, 138495 CrossRef CAS.
- Y. Zhang, J. Jia, Y. Sun, B. Xu, Z. Jiang, X. Qu and C. Zhang, Nanomaterials, 2023, 13, 2909 CrossRef CAS.
- G. Singh, A. M. Ruban, X. Geng and A. Vinu, Chem. Eng. J., 2023, 451, 139045 CrossRef CAS.
- K. H. H. Aziz, F. S. Mustafa, K. M. Omer, S. Hama, R. F. Hamarawf and K. O. Rahman, RSC Adv., 2023, 13, 17595–17610 RSC.
- J. O. Ighalo, J. Conradie, C. R. Ohoro, J. F. Amaku, K. O. Oyedotun, N. W. Maxakato, K. G. Akpomie, E. S. Okeke, C. Olisah and A. Malloum, Ind. Crops Prod., 2023, 204, 117300 CrossRef CAS.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef CAS.
- S. Ha, S. G. Jeong, S. Myeong and Y.-S. Lee, J. CO2 Util., 2023, 76, 102589 CrossRef CAS.
- K. Malini, D. Selvakumar and N. Kumar, J. CO2 Util., 2023, 67, 102318 CrossRef CAS.
- C. Deng, L. Xu, K. Hu, X. Chen, R. Gao, L. Zhang, L. Wang and C. Zhang, Atmosphere, 2023, 14, 1510 CrossRef CAS.
- Z. Nezafat, M. Nasrollahzadeh, S. Javanshir, T. Baran and Y. Dong, Green Chem., 2023, 25, 9603–9643 RSC.
- J. Gopalan, A. Buthiyappan and A. A. A. Raman, J. Ind. Eng. Chem., 2022, 113, 72–95 CrossRef CAS.
- W. A. Rasaq, C. O. R. Okpala, C. A. Igwegbe and A. Białowiec, Materials, 2024, 17, 725 CrossRef CAS PubMed.
- A. Devi, S. Bajar, H. Kour, R. Kothari, D. Pant and A. Singh, BioEnergy Res., 2022, 15, 1820–1841 CrossRef CAS PubMed.
- K. S. Khoo, I. Ahmad, K. W. Chew, K. Iwamoto, A. Bhatnagar and P. L. Show, Prog. Energy Combust. Sci., 2023, 96, 101071 CrossRef.
- T. Sharma, I. G. Hakeem, A. B. Gupta, J. Joshi, K. Shah, A. K. Vuppaladadiyam and A. Sharma, J. Energy Inst., 2024, 101559 CrossRef CAS.
- A. K. Vuppaladadiyam, S. S. V. Vuppaladadiyam, A. Awasthi, A. Sahoo, S. Rehman, K. K. Pant, S. Murugavelh, Q. Huang, E. Anthony and P. Fennel, Bioresour. Technol., 2022, 364, 128087 CrossRef CAS PubMed.
- X. Peng, Y. Jiang, Z. Chen, A. I. Osman, M. Farghali, D. W. Rooney and P.-S. Yap, Environ. Chem. Lett., 2023, 21, 765–801 CrossRef CAS.
- P. Manasa, S. Sambasivam and F. Ran, J. Energy Storage, 2022, 54, 105290 CrossRef.
- K. Yogalakshmi, P. Sivashanmugam, S. Kavitha, Y. Kannah, S. Varjani, S. AdishKumar and G. Kumar, Chemosphere, 2022, 286, 131824 CrossRef PubMed.
- M. Xu, Z. Shi, X. Zhu, Y. Lai, A. Xia, Y. Huang, X. Jiang, J. He, M. Zhou and X. Zhu, Int. J. Hydrogen Energy, 2024, 52, 83–96 CrossRef CAS.
- R. D. L. C. Iturbides, L. Ubiera, U. J. Haza and I. Polaert, React. Chem. Eng., 2024, 9, 1235–1250 RSC.
- N. A. Binti Mohd, Conventional and microwave pyrolysis of empty fruit bunch and rice husk pellets, PhD thesis, University of Sheffield, 2017.
- W. Song, Y. Zhang, C. H. Tran, H. K. Choi, D.-G. Yu and I. Kim, Prog. Polym. Sci., 2023, 142, 101691 CrossRef CAS.
- N. Vasiraja, R. S. S. Prabhahar and A. Joshua, J. Cleaner Prod., 2023, 397, 136579 CrossRef CAS.
- A. Ali and R. Idris, Adv. Mater. Lett., 2017, 8, 70–76 CrossRef CAS.
- A. A. Adeleke, P. P. Ikubanni, S. S. Emmanuel, M. O. Fajobi, P. Nwachukwu, A. A. Adesibikan, J. K. Odusote, E. O. Adeyemi, O. M. Abioye and J. A. Okolie, Renewable Sustainable Energy Rev., 2024, 199, 114502 CrossRef CAS.
- M. N. Romanias, M. M. Coggon, F. Al Ali, J. B. Burkholder, P. Dagaut, Z. Decker, C. Warneke, C. E. Stockwell, J. M. Roberts and A. Tomas, ACS Earth Space Chem., 2024, 8, 857–899 CrossRef CAS.
- S. P. S. Yadav, S. Bhandari, D. Bhatta, A. Poudel, S. Bhattarai, P. Yadav, N. Ghimire, P. Paudel, P. Paudel and J. Shrestha, J. Agric. Food Res., 2023, 11, 100498 Search PubMed.
- M. Fan, C. Li, Y. Shao, S. Zhang, M. Gholizadeh and X. Hu, Sci. Total Environ., 2022, 825, 153959 CrossRef CAS.
- N. I. W. Azelee, H. I. Mahdi, Y.-S. Cheng, N. Nordin, R. M. Illias, R. A. Rahman, S. M. Shaarani, P. Bhatt, S. Yadav and S. W. Chang, Fuel, 2023, 339, 126982 CrossRef.
- J. Liu, X. Chen, W. Chen, M. Xia, Y. Chen, H. Chen, K. Zeng and H. Yang, Proc. Combust. Inst., 2023, 39, 3157–3181 CrossRef CAS.
- A. Shafizadeh, H. Rastegari, H. Shahbeik, H. Mobli, J. Pan, W. Peng, G. Li, M. Tabatabaei and M. Aghbashlo, J. Cleaner Prod., 2023, 400, 136705 CrossRef CAS.
- W.-l. Xie, B. Hu, Y. Liu, H. Fu, J. Liu, B. Zhang and Q. Lu, Proc. Combust. Inst., 2023, 39, 3303–3311 CrossRef CAS.
- Y. Fan, Z. Zhang, Z. Wang, H. Yu, X. Kong, P. Li, M. Li, R. Xiao and C. Liu, Bioresour. Technol., 2022, 360, 127648 CrossRef CAS PubMed.
- A. L. Pauline and K. Joseph, J. Hazard., Toxic Radioact. Waste, 2024, 28, 04024025 CrossRef.
- R. H. Teoh, A. S. Mahajan, S. R. Moharir, N. A. Manaf, S. Shi and S. Thangalazhy-Gopakumar, Energy Nexus, 2024, 100301 CrossRef CAS.
- X. Zhuang, J. Liu, Q. Zhang, C. Wang, H. Zhan and L. Ma, Renewable Sustainable Energy Rev., 2022, 154, 111877 CrossRef CAS.
- T.-D. Hoang, L. Yan and T. M. Le, Sustainability, 2024, 16, 5495 CrossRef.
- C. Goel, S. Mohan, P. Dinesha and M. A. Rosen, Chem. Pap., 2024, 78, 3845–3856 CrossRef CAS.
- M. Cavali, N. L. Junior, J. D. de Sena, A. L. Woiciechowski, C. R. Soccol, P. Belli Filho, R. Bayard, H. Benbelkacem and A. B. de Castilhos Junior, Sci. Total Environ., 2023, 857, 159627 CrossRef CAS PubMed.
- T. Bi, H. Chen, J. Li, X. Zhang and Q. Lin, Electrochim. Acta, 2022, 433, 141266 CrossRef CAS.
- P. Baghel, A. K. Sakhiya and P. Kaushal, Renewable energy, 2022, 185, 538–551 CrossRef CAS.
- J. J. Alvear-Daza, A. Sosa, D. M. Ruiz, G. A. Pasquale, J. A. Rengifo-Herrera, G. P. Romanelli and L. R. Pizzio, Catalysts, 2024, 14, 522 CrossRef CAS.
- Y. Liu, J. Pan, J. Wang, X. Yang, W. Zhang, K. H. D. Tang, H. Wang, X. Zhang, R. Gao and G. Yang, Environ. Sci. Pollut. Res., 2024, 1–12 Search PubMed.
- J. O. Ighalo and A. G. Adeniyi, Environ. Processes, 2021, 8, 267–285 CrossRef CAS.
- M. Pahnila, A. Koskela, P. Sulasalmi and T. Fabritius, Energies, 2023, 16, 6936 CrossRef CAS.
- N. A. Zubbri, A. R. Mohamed, P. Lahijani and M. Mohammadi, J. Environ. Chem. Eng., 2021, 9, 105074 CrossRef CAS.
- A. Soomro, S. Chen, M. H. Mallah, I. A. Samo, A. A. Siyal, B. Ali, K. A. Samo, S. Ma, K. C. Mukwana and W. Xiang, BioEnergy Res., 2024, 1–12 Search PubMed.
- D. Aboelela, H. Saleh, A. M. Attia, Y. Elhenawy, T. Majozi and M. Bassyouni, Sustainability, 2023, 15, 11238 CrossRef CAS.
- W. Chen, X. Tao, X. Shi, W. Guo, Y. Wang, B. Liu and H. Yang, npj Mater. Sustain., 2024, 2, 25 CrossRef.
- A. A. Ahmad, A. Alwahbi, L. A. Al Khatib and H. Dammag, in From Biomass to Biobased Products, IntechOpen, 2024 Search PubMed.
- S. Pathak, A. K. Sakhiya and P. Kaushal, in Thermochemical and Catalytic Conversion Technologies for Future Biorefineries, Springer, 2022, vol. 1, pp. 205–239 Search PubMed.
- R. Blanchard and T. H. Mekonnen, RSC appl. polym., 2024, 2, 557–582 RSC.
- Q. Ma, W. Chen, Z. Jin, L. Chen, Q. Zhou and X. Jiang, Fuel, 2021, 289, 119932 CrossRef CAS.
- R. Muzyka, E. Misztal, J. Hrabak, S. W. Banks and M. Sajdak, Energy, 2023, 263, 126128 CrossRef CAS.
- Q. Wu, S. Yu, N. Hao, T. Wells Jr, X. Meng, M. Li, Y. Pu, S. Liu and A. J. Ragauskas, Bioresour. Technol., 2017, 244, 78–83 CrossRef CAS.
- M. J. Ahmed and B. H. Hameed, Environ. Res., 2024, 118203 CrossRef CAS.
- A. N. Amenaghawon, C. L. Anyalewechi, C. O. Okieimen and H. S. Kusuma, Environ. Dev. Sustain., 2021, 1–55 Search PubMed.
- Z.-H. Chen, X.-L. Du, J.-B. He, F. Li, Y. Wang, Y.-L. Li, B. Li and S. Xin, ACS Appl. Mater. Interfaces, 2017, 9, 33855–33862 CrossRef CAS PubMed.
- X. Chen, X. Ma, X. Peng, Y. Lin and Z. Yao, Bioresour. Technol., 2018, 249, 900–907 CrossRef CAS PubMed.
- H. Su, X. Zhou, R. Zheng, Z. Zhou, Y. Zhang, G. Zhu, C. Yu, D. Hantoko and M. Yan, Sci. Total Environ., 2021, 754, 142192 CrossRef CAS PubMed.
- F. Liu, R. Yu, X. Ji and M. Guo, Bioresour. Technol., 2018, 263, 508–516 CrossRef CAS PubMed.
- W. Sangchoom and R. Mokaya, ACS Sustain. Chem. Eng., 2015, 3, 1658–1667 CrossRef CAS.
- L. Rao, R. Ma, S. Liu, L. Wang, Z. Wu, J. Yang and X. Hu, Chem. Eng. J., 2019, 362, 794–801 CrossRef CAS.
- N. U. Saqib, S. Baroutian and A. K. Sarmah, Bioresour. Technol., 2018, 266, 357–363 CrossRef CAS PubMed.
- L. Wang, G. Yin, Y. Chang and S. Qiao, Waste Manage., 2024, 177, 182–195 CrossRef CAS PubMed.
- Q. Wang, S. Wu, D. Cui, H. Zhou, D. Wu, S. Pan, F. Xu and Z. Wang, Sci. Total Environ., 2022, 850, 158034 CrossRef CAS PubMed.
- S. Ullah, S. S. A. Shah, M. Altaf, I. Hossain, M. E. El Sayed, M. Kallel, Z. M. El-Bahy, A. ur Rehman, T. Najam and M. A. Nazir, J. Anal. Appl. Pyrolysis, 2024, 106480 CrossRef CAS.
- N. Rathnayake, S. Patel, I. G. Hakeem, J. Pazferreiro, A. Sharma, R. Gupta, C. Rees, D. Bergmann, J. Blackbeard and A. Surapaneni, Process Saf. Environ. Prot., 2023, 173, 75–87 CrossRef CAS.
- W.-H. Chen, B.-J. Lin, Y.-Y. Lin, Y.-S. Chu, A. T. Ubando, P. L. Show, H. C. Ong, J.-S. Chang, S.-H. Ho and A. B. Culaba, Prog. Energy Combust. Sci., 2021, 82, 100887 CrossRef.
- K. Van der Cruijsen, M. Al Hassan, G. van Erven, O. Dolstra and L. M. Trindade, Molecules, 2021, 26, 254 CrossRef CAS PubMed.
- H. Sun, D. Feng, S. Sun, Y. Zhao, L. Zhang, G. Chang, Q. Guo, J. Wu and Y. Qin, J. Anal. Appl. Pyrolysis, 2021, 156, 105128 CrossRef CAS.
- F. Narciso-Romero and F. Rodriguez-Reinoso, J. Mater. Sci., 1996, 31, 779–784 CrossRef CAS.
- Y. X. Seow, Y. H. Tan, N. Mubarak, J. Kansedo, M. Khalid, M. L. Ibrahim and M. Ghasemi, J. Environ. Chem. Eng., 2022, 10, 107017 CrossRef CAS.
- M. Rasul, M. Hazrat, M. Sattar, M. Jahirul and M. Shearer, Energy Convers. Manage., 2022, 272, 116326 CrossRef CAS.
- P. Lahijani, M. Mohammadi and A. R. Mohamed, J. CO2 Util., 2018, 26, 281–293 CrossRef CAS.
- M. Ahmed and B. Hameed, J. Cleaner Prod., 2020, 265, 121762 CrossRef CAS.
- M. Siddique, S. Akram, Z. Liaqat and M. Mushtaq, Sewage and Biomass from Wastewater to Energy, 2024, pp. 1–41 Search PubMed.
- D. Lachos-Perez, P. C. Torres-Mayanga, E. R. Abaide, G. L. Zabot and F. De Castilhos, Bioresour. Technol., 2022, 343, 126084 CrossRef CAS PubMed.
- X. Li, K. Cen, L. Wang, D. Jia, X. Zhu and D. Chen, Ind. Crops Prod., 2024, 219, 119071 CrossRef CAS.
- Y. Li, H. Yu, L. Liu and H. Yu, J. Hazard. Mater., 2021, 420, 126655 CrossRef CAS PubMed.
- C. Li, J.-i. Hayashi, Y. Sun, L. Zhang, S. Zhang, S. Wang and X. Hu, J. Anal. Appl. Pyrolysis, 2021, 155, 105031 CrossRef CAS.
- Q. Hu, W. Cheng, Q. Mao, J. Hu, H. Yang and H. Chen, Fuel, 2022, 314, 122789 CrossRef CAS.
- K. Rasouli, J. Rasouli, M. S. Mohtaram, S. Sabbaghi, H. Kamyab, H. Moradi and S. Chelliapan, J. Cleaner Prod., 2023, 138181 CrossRef CAS.
- M. Gayathiri, T. Pulingam, K. Lee and K. Sudesh, Chemosphere, 2022, 294, 133764 CrossRef CAS PubMed.
- P. B. n. Ramos, A. Mamaní, M. Erans, F. Jerez, M. F. Ponce, M. F. Sardella, A. Arencibia, M. A. Bavio, E. S. Sanz-Pérez and R. l. Sanz, Energy Fuels, 2024, 38, 6102–6115 CrossRef CAS.
- M. Mujtaba, L. F. Fraceto, M. Fazeli, S. Mukherjee, S. M. Savassa, G. A. de Medeiros, A. d. E. Santo Pereira, S. D. Mancini, J. Lipponen and F. Vilaplana, J. Cleaner Prod., 2023, 402, 136815 CrossRef CAS.
- S. Purayil, M. O. Hamdan, S. Al-Omari, M. Selim and E. Elnajjar, Energy Rep., 2023, 9, 4547–4573 CrossRef.
- Q. Pu, J. Zou, J. Wang, S. Lu, P. Ning, L. Huang and Q. Wang, J. Alloys Compd., 2021, 887, 161406 CrossRef CAS.
- Y. Li, L. Lu, S. Lyu, H. Xu, X. Ren and Y. A. Levendis, J. Anal. Appl. Pyrolysis, 2021, 156, 105137 CrossRef CAS.
- J. Serafin and B. Dziejarski, Environ. Sci. Pollut. Res., 2024, 31, 40008–40062 CrossRef CAS PubMed.
- J. Fu, J. Zhang, C. Jin, Z. Wang, T. Wang, X. Cheng and C. Ma, Bioresour. Technol., 2020, 310, 123413 CrossRef CAS PubMed.
- Y. Xie, L. Wang, H. Li, L. J. Westholm, L. Carvalho, E. Thorin, Z. Yu, X. Yu and Ø. Skreiberg, J. Anal. Appl. Pyrolysis, 2022, 161, 105405 CrossRef CAS.
- A. Rehman, G. Nazir, K. Y. Rhee and S.-J. Park, Chem. Eng. J., 2021, 420, 130421 CrossRef CAS.
- W. Suliman, J. B. Harsh, N. I. Abu-Lail, A.-M. Fortuna, I. Dallmeyer and M. Garcia-Perez, Biomass Bioenergy, 2016, 85, 1–11 CrossRef CAS.
- L. Chen, W. Fang, J. Liang, M. Nabi, Y. Cai, Q. Wang, P. Zhang and G. Zhang, Resour., Conserv. Recycl., 2023, 188, 106720 CrossRef CAS.
- B. Petrovic, M. Gorbounov and S. M. Soltani, Microporous Mesoporous Mater., 2021, 312, 110751 CrossRef CAS.
- D. Kim and S. Hadigheh, Renewable Energy, 2024, 224, 120106 CrossRef CAS.
- Q. Qin, F. Zhong, T. Song, Z. Yang, P. Zhang, H. Cao, W. Niu and Z. Yao, Chem. Eng. J., 2023, 477, 146763 CrossRef CAS.
- J. H. Lee, S. J. Sim, J. H. Kang and S. S. Choi, Chem. Phys. Lett., 2021, 780, 138962 CrossRef CAS.
- O. Tomin, R. Vahala and M. R. Yazdani, Heliyon, 2024, 10, e24722 CrossRef CAS PubMed.
- W. K. Tong, C. Dai, C. Jia, J. Hu, M.-t. Gao, J. Li, J. B. Zhang, H. Tang, Y. Liang and W. Teng, Chem. Eng. J., 2024, 481, 148422 CrossRef CAS.
- J. Ma, L. Zheng and F. Yu, Desalination, 2024, 117597 CrossRef CAS.
- F. Yang, Z. He, F. Yu, S. Zhou and X. Zhu, Sci. Total Environ., 2023, 867, 161425 CrossRef CAS PubMed.
- L. Cao, X. Zhang, Y. Xu, W. Xiang, R. Wang, F. Ding, P. Hong and B. Gao, Sep. Purif. Technol., 2022, 287, 120592 CrossRef CAS.
- M. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41, 267–275 CrossRef.
- A. El Hadrami, S. Ojala and R. Brahmi, Surf. Interfaces, 2022, 30, 101849 CrossRef.
- J. Shin, J. Kwak, Y.-G. Lee, S. Kim, M. Choi, S. Bae, S.-H. Lee, Y. Park and K. Chon, Environ. Pollut., 2021, 270, 116244 CrossRef CAS.
- F. Ma, H. Zhao, X. Zheng, B. Zhao, J. Diao and Y. Jiang, J. Environ. Chem. Eng., 2023, 11, 109747 CrossRef CAS.
- J. Li, L. Zheng, S.-L. Wang, Z. Wu, W. Wu, N. K. Niazi, S. M. Shaheen, J. Rinklebe, N. Bolan and Y. S. Ok, Sci. Total Environ., 2019, 672, 572–582 CrossRef CAS PubMed.
- B. Liu, R. Shi, X. Ma, R. Chen, K. Zhou, X. Xu, P. Sheng, Z. Zeng and L. Li, Carbon, 2021, 181, 270–279 CrossRef CAS.
- M. Olivares-Marín, C. Fernández-González, A. Macías-García and V. Gómez-Serrano, Carbon, 2006, 44, 2347–2350 CrossRef.
- K. Chen, D. Ma, H. Yu, S. Zhang, B. C. Seyler, Z. Chai and S. Peng, Chemosphere, 2022, 291, 132721 CrossRef CAS.
- C. Bonga, L. Y. Lima, C. T. Leea, P. Y. Ongb, J. Jaromír, C. L. Klemešc and Y. Gaod, Chem. Eng., 2020, 81, 427–432 Search PubMed.
- X. Ma, C. Su, B. Liu, Q. Wu, K. Zhou, Z. Zeng and L. Li, Sep. Purif. Technol., 2021, 259, 118065 CrossRef CAS.
- M. Olivares-Marín, C. Fernández-González, A. Macías-García and V. Gómez-Serrano, J. Anal. Appl. Pyrolysis, 2012, 94, 131–137 CrossRef.
- D. Li, W. Li, J. Shi and F. Xin, RSC Adv., 2016, 6, 50138–50143 RSC.
- C. Cheng, H. Liu, P. Dai, X. Shen, J. Zhang, T. Zhao and Z. Zhu, J. Taiwan Inst. Chem. Eng., 2016, 67, 532–537 CrossRef CAS.
- E. Wolak and A. Orzechowska-Zięba, Adsorption, 2024, 30, 121–128 CrossRef CAS.
- J. M. Illingworth, B. Rand and P. T. Williams, Fuel Process. Technol., 2022, 235, 107348 CrossRef CAS.
- Z. Liu, W. Fan and Y. Xu, Environ. Sci. Pollut. Res., 2024, 1–12 Search PubMed.
- A. Komaei, M. Yadollah-Roudbari and S. M. Fattahi, Cleaner Mater., 2024, 100264 CrossRef CAS.
- J. O. Velasco, Biomass to chemicals and fuels using advanced thermochemical concepts, PhD thesis, University of Groningen, 2024.
- W. Shi, J. Yu, H. Liu, D. Gao, A. Yuan and B. Chang, ACS Appl. Nano Mater., 2023, 6, 7887–7900 CrossRef CAS.
- M. S. Khosrowshahi, H. Mashhadimoslem, H. Shayesteh, G. Singh, E. Khakpour, X. Guan, M. Rahimi, F. Maleki, P. Kumar and A. Vinu, Advanced Science, 2023, 10, 2304289 CrossRef CAS PubMed.
- A. M. Elewa, A. A. Amer, M. F. Attallah, H. A. Gad, Z. A. M. Al-Ahmed and I. A. Ahmed, Materials, 2023, 16, 1251 CrossRef CAS PubMed.
- D. Hong, J. Zhou, C. Hu, Q. Zhou, J. Mao and Q. Qin, Fuel, 2019, 235, 326–335 CrossRef CAS.
- M. Kwiatkowski, C. Belver and J. Bedia, Sci. Rep., 2024, 14, 2266 CrossRef CAS.
- N. M. Amer, P. Lahijani, M. Mohammadi and A. R. Mohamed, Biomass Convers. Biorefin., 2024, 14, 7401–7448 CrossRef CAS.
- X. Duan, G. Song, G. Lu, Y. Wang, J. Sun, A. Chen and X. Xie, Mater. Today Sustain., 2023, 23, 100453 Search PubMed.
- A. U. Khan, O. Samuel, M. H. D. Othman, M. Younas, R. Kamaludin, M. H. Puteh, T. A. Kurniawan, K. Y. Wong, F. Kadirkhan and N. Yoshida, Sep. Purif. Technol., 2024, 347, 127511 CrossRef.
- Y. Guo, G. Wang, J. Yu, P. Huang, J. Sun, R. Wang, T. Wang and C. Zhao, Sep. Purif. Technol., 2023, 305, 122455 CrossRef CAS.
- S. Park, J. Kim and K. Kwon, Chem. Eng. J., 2022, 446, 137116 CrossRef CAS.
- X. Ma, X. Li, H. Cui, W. Zhang, Z. Cheng and Z. Zhou, AIChE J., 2023, 69, e17520 CrossRef CAS.
- L. Shao, L. Wang, J. Wang, Z. Liu, Z. Wu, P. Zhan, L. Zhang, X. Ma and J. Huang, Sep. Purif. Technol., 2023, 313, 123440 CrossRef CAS.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30, 1909265 CrossRef CAS.
- X. Guo, G. Zhang, C. Wu, J. Liu, G. Li, Y. Zhao, Y. Wang and Y. Xu, J. Environ. Chem. Eng., 2021, 9, 105165 CrossRef CAS.
- D. Zhang, Q. He, X. Hu, K. Zhang, C. Chen and Y. Xue, Colloids Surf., A, 2021, 615, 126254 CrossRef CAS.
- Y. Qiao and C. Wu, Carbon Capture Sci. Technol., 2022, 2, 100018 CrossRef CAS.
- A. D. Kute, R. P. Gaikwad, I. R. Warkad and M. B. Gawande, Green Chem., 2022, 24, 3502–3573 RSC.
- P. Pareek, H. Dave and L. Ledwani, Mater. Today: Proc., 2021, 43, 3277–3285 CAS.
- W. Cao, H. Xu, X. Zhang, W. Xiang, G. Qi, L. Wan and B. Gao, Chem. Eng. J., 2023, 471, 144523 CrossRef CAS.
- A. A. Khan, Y. S. Ling and Z. Z. Chowdhury, Eur. Phys. J. Plus, 2024, 139, 1–11 CrossRef.
- A. Nurdin, I. Iriani, H. Harahap and A. Fahmi, Indones. J. Chem. Res., 2022, 10, 8–13 CrossRef CAS.
- A. El Nemr, R. M. Aboughaly, A. El Sikaily, M. S. Masoud, M. S. Ramadan and S. Ragab, Carbon Lett., 2022, 32, 229–249 CrossRef.
- M. N. Islam, J. Sarker, A. Khatton, S. M. Hossain, H. A. Sikder, R. Ahmed and A. S. Chowdhury, Sch. Int. J. Chem. Mater. Sci., 2022, 5, 33–39 CrossRef.
- R. Dewi, A. Azhari and I. Nofriadi, J. Teknol. Kim. Unimal, 2021, 9, 12–22 CrossRef.
- N. E. Williams, O. A. Oba and N. P. Aydinlik, ChemBioEng Rev., 2022, 9, 164–189 CrossRef CAS.
- S. Li, K. Han, J. Li, M. Li and C. Lu, Microporous Mesoporous Mater., 2017, 243, 291–300 CrossRef CAS.
- Z. Gu, X. Wang, P. Huang, Y. Huang, X. He, X. Wei, J. Yue, J. Jiang and C. Zhao, Process Saf. Environ. Prot., 2022, 160, 573–583 CrossRef CAS.
- C. Ma, T. Lu, J. Shao, J. Huang, X. Hu and L. Wang, Sep. Purif. Technol., 2022, 281, 119899 CrossRef CAS.
- Y. Cao, L. Yang, F. Liu and Q. Yu, Biomass Bioenergy, 2024, 182, 107074 CrossRef CAS.
- Y. Chen, C. Zhou, X. Xing, L. Chen, B. Yao, L. Chao, Y. Zhang, J. Wang, J. Dong and C. Liu, Chem. Eng. J., 2024, 495, 153579 CrossRef CAS.
- S. Sangon, K. Kotebantao, T. Suyala, Y. Ngernyen, A. J. Hunt and N. Supanchaiyamat, Environ. Sci.: Water Res. Technol., 2024, 10, 1389–1405 RSC.
- F. Njeh, M. Hamza, I. Bouaziz, R. Abdelhedi and M. Abdelmouleh, Korean J. Chem. Eng., 2022, 1–15 Search PubMed.
- S. Wei, Q. Qin and Z. Liu, J. Anal. Appl. Pyrolysis, 2024, 179, 106500 CrossRef CAS.
- B. Huang, W. Li, Z. Shi, L. Yang, Z. Wen, G. Zi and L. Luo, Clean: Soil, Air, Water, 2024, 52, 2300148 CAS.
- W. Li and J. Shi, Front. Bioeng. Biotechnol., 2023, 11, 1121027 CrossRef PubMed.
- P. M. Yeletsky, M. V. Lebedeva and V. A. Yakovlev, J. Energy Storage, 2022, 50, 104225 CrossRef.
- P. Ozpinar, C. Dogan, H. Demiral, U. Morali, S. Erol, C. Samdan, D. Yildiz and I. Demiral, Renewable Energy, 2022, 189, 535–548 CrossRef CAS.
- B. S. Rathi and R. V, Environ. Qual. Manag., 2024, 33, 907–928 CrossRef.
- A. A. Chan, A. A. A. Raman, W. L. Chong and A. Buthiyappan, J. Cleaner Prod., 2024, 437, 140655 CrossRef CAS.
- Z. Baassou, F. Benmahdi, A. Reffas and A. Benhaya, Biomass Convers. Biorefin., 2024, 1–14 Search PubMed.
- J. Liang, C. Li, S. Zhang, S. Wang and X. Hu, Ind. Crops Prod., 2024, 221, 119387 CrossRef CAS.
- A. Martusevich, A. Surovegina, A. Popovicheva, N. Didenko, M. Artamonov and V. Nazarov, BioMed Res. Int., 2022, 2022, 5857979 Search PubMed.
- J. Serafin, K. Kiełbasa and B. Michalkiewicz, Chem. Eng. J., 2022, 429, 131751 CrossRef CAS.
- X. Wang, H. Cheng, G. Ye, J. Fan, F. Yao, Y. Wang, Y. Jiao, W. Zhu, H. Huang and D. Ye, Chemosphere, 2022, 287, 131995 CrossRef CAS PubMed.
- N. Karić, A. S. Maia, A. Teodorović, N. Atanasova, G. Langergraber, G. Crini, A. R. Ribeiro and M. Đolić, Chem. Eng. J. Adv., 2022, 9, 100239 CrossRef.
- S. Das, Microbial Biodegradation and Bioremediation, Elsevier, 2014 Search PubMed.
- İ. Kılınçer, L. Khanyile, K. Gürcan, Ö. Şimşek, A. Uzun and A. Nikbakht-Dehkordi, Genet. Resour. Crop Evol., 2024, 71, 2229–2246 CrossRef.
- P. Chaudhary, S. Bansal, B. B. Sharma, S. Saini and A. Joshi, J. Energy Storage, 2024, 78, 109996 CrossRef.
- H. Ma, W. Xu, P. Wang, Y. Ding and S. Zhou, Environ. Geochem. Health, 2024, 46, 1–20 CrossRef PubMed.
- S. Shao, Y. Wang, L. Ma, Z. Huang and X. Li, Fuel, 2024, 355, 129287 CrossRef CAS.
- D. Pereira, M. Ilkaeva, F. Vicente, R. Vieira, M. Sardo, M. A. Lourenço, A. Silvestre, I. Marin-Montesinos and L. Mafra, ACS Omega, 2024, 9, 17956–17965 CrossRef CAS PubMed.
- S. Amreen, S. K. Dash, B. Naik, A. K. Dash, A. K. Verma and A. Pradhan, Environ. Qual. Manag., 2024, 34, e22213 CrossRef.
- R. Ganjoo, S. Sharma, A. Kumar and M. Daouda, Royal Society of Chemistry Books, 2023, p. 450 Search PubMed.
- M. An, T. Guo and Q. Guo, Carbon Resour. Convers., 2024, 7, 100205 CrossRef CAS.
- S.-X. Zhao, N. Ta and X.-D. Wang, Energies, 2017, 10, 1293 CrossRef.
- Y. Liu, W. Tan, S. Liang, X. Bi, R. Sun and X. Pan, Biomass Convers. Biorefin., 2024, 14, 781–793 CrossRef CAS.
- U. Onochie, A. Obanor, S. Aliu and O. Igbodaro, Niger. J. Technol., 2017, 36, 987–990 CrossRef.
- P. Pramanik, H. Patel, S. Charola, S. Neogi and S. Maiti, J. CO2 Util., 2021, 45, 101450 CrossRef CAS.
- Z. Wu, Y. Liu, Y. Xu, W. Zhan, Y. Liu, D. Zhang and Y. Xu, Biomass Convers. Biorefin., 2024, 1–13 Search PubMed.
- H. Li, M. Tang, X. Huang, L. Wang, Q. Liu and S. Lu, Chem. Eng. J., 2023, 466, 143095 CrossRef CAS.
- J. Kawalerczyk, J. Walkiewicz, J. Sedliačik, D. Dukarska, M. Woźniak and R. Mirski, Polymers, 2024, 16, 1350 CrossRef CAS PubMed.
- Y.-j. Wang, Y. Yu, H.-j. Huang, C.-l. Yu, H.-s. Fang, C.-h. Zhou, X. Yin, W.-h. Chen and X.-c. Guo, Sci. Total Environ., 2022, 803, 149874 CrossRef CAS PubMed.
- J. Serafin, B. Dziejarski and J. Sreńscek-Nazzal, Sustainable Mater. Technol., 2023, 38, e00717 CrossRef CAS.
- J. de Siqueira Castro, P. P. Assemany, A. C. de Oliveira Carneiro, J. Ferreira, M. M. de Jesus Júnior, F. de Ávila Rodrigues and M. L. Calijuri, Sci. Total Environ., 2021, 768, 144480 CrossRef PubMed.
- M. Farnane, A. Machrouhi, M. Abdennouri, H. Tounsadi, Z. Rais, S. Qourzal and N. Barka, Biointerface Res. Appl. Chem., 2021, 12, 5941–5952 Search PubMed.
- A. B. Fadhil and B. A. Kareem, J. Anal. Appl. Pyrolysis, 2021, 158, 105249 CrossRef CAS.
- J. M. González-Domínguez, C. Fernández-González, M. Alexandre-Franco and V. Gómez-Serrano, Processes, 2024, 12, 149 CrossRef.
- P. Riquelme-García, J. Chaparro-Garnica, M. Navlani-García and D. Cazorla-Amorós, ChemCatChem, 2024, e202400160 CrossRef.
- X. Li, H. Tian, S. Yan, H. Shi, J. Wu, Y. Sun, Y. Xing, H. Bai and H. Zhang, Int. J. Hydrogen Energy, 2024, 50, 324–336 CrossRef CAS.
- K. Suwannahong, S. Wongcharee, T. Kreetachart, C. Sirilamduan, J. Rioyo and A. Wongphat, Appl. Sci., 2021, 11, 7432 CrossRef CAS.
- M. Hamid, S. A. Amaturrahim, I. B. Dalimunthe and A. Daulay, Mater. Sci. Energy Technol., 2023, 6, 429–436 CAS.
- S. He, G. Chen, H. Xiao, G. Shi, C. Ruan, Y. Ma, H. Dai, B. Yuan, X. Chen and X. Yang, J. Colloid Interface Sci., 2021, 582, 90–101 CrossRef CAS.
- S. Omokafe, A. Adeniyi, E. Igbafen, S. Oke and P. Olubambi, Int. J. Electrochem. Sci., 2020, 15, 10854–10865 CrossRef CAS.
- J. Liu, X. Yang, H. Liu, X. Jia and Y. Bao, Chemosphere, 2021, 282, 131116 CrossRef CAS PubMed.
- L. Manjakkal, A. Jain, S. Nandy, S. Goswami, J. T. Carvalho, L. Pereira, C. H. See, S. C. Pillai and R. A. Hogg, Chem. Eng. J., 2023, 465, 142845 CrossRef CAS.
- M. V. Penzik, A. N. Kozlov, S. Zhang, V. V. Badenko, I. K. Sosnovsky and V. A. Shamansky, Thermochim. Acta, 2022, 711, 179209 CrossRef CAS.
- S. Leng, W. Li, C. Han, L. Chen, J. Chen, L. Fan, Q. Lu, J. Li, L. Leng and W. Zhou, Bioresour. Technol., 2020, 298, 122502 CrossRef CAS PubMed.
- H. Wang, H. Teng, X. Wang, J. Xu and L. Sheng, J. Environ. Manage., 2022, 310, 114758 CrossRef CAS PubMed.
- M. A. A. Mariah, K. Rovina, J. M. Vonnie and K. H. Erna, S. Afr. J. Chem. Eng., 2023, 44, 113–122 Search PubMed.
- E. Huo, D. Duan, H. Lei, C. Liu, Y. Zhang, J. Wu, Y. Zhao, Z. Huang, M. Qian and Q. Zhang, Energy, 2020, 199, 117459 CrossRef CAS.
- C. H. Pimentel, M. S. Freire, D. Gómez-Díaz and J. González-Álvarez, Appl. Sci., 2024, 14, 841 CrossRef CAS.
- J. Weng, M.-M. Wei, S.-J. Wu, Y.-Q. Liu, S.-R. Li, Y.-Y. Ye, M. Wang and D. Wang, BioResources, 2019, 14, 3899–3913 CAS.
- V. T. W. Thuppahige, L. Moghaddam, Z. G. Welsh, T. Wang, H.-W. Xiao and A. Karim, LWT-Food Sci. Technol., 2023, 182, 114787 CrossRef.
- A. Herrera-Barros, N. Bitar-Castro, Á. Villabona-Ortíz, C. Tejada-Tovar and Á. D. González-Delgado, Sustainable Chem. Pharm., 2020, 17, 100299 CrossRef.
- G. Cruz, A. d. L. P. Rodrigues, D. F. da Silva and W. C. Gomes, J. Therm. Anal. Calorim., 2021, 143, 3611–3622 CrossRef CAS.
- M. L. Yeboah, Int. J. Environ. Anal. Chem., 2023, 103, 4411–4429 CrossRef CAS.
- R. I. Ismail, K. C. Yee, A. R. Shaari, A. R. Mohamed, M. H. B. A. Halim, A. A. A. Rahman, L. Y. Leng, N. L. Makhtar and N. A. Razak, AIP Conf. Proc., 2023, 2703, 130001 CrossRef CAS.
- I. K. Tetteh, I. Issahaku and A. Y. Tetteh, Carbon Trends, 2024, 100328 CrossRef CAS.
- W. Jia, S. Li, J. Wang, J. T. Lee, C. S. K. Lin, O. Mašek, H. Zhang and X. Yuan, Green Chem., 2024, 26, 1790–1805 RSC.
- P. Treeweranuwat, P. Boonyoung, M. Chareonpanich and K. Nueangnoraj, ACS Omega, 2020, 5, 1911–1918 CrossRef CAS PubMed.
- A. Hidayu and N. Muda, Procedia Eng., 2016, 148, 106–113 CrossRef CAS.
- C. Wu, Q. Huang, Z. Xu, A. T. Sipra, N. Gao, L. P. de Souza Vandenberghe, S. Vieira, C. R. Soccol, R. Zhao and S. Deng, Carbon Capture Sci. Technol., 2024, 11, 100178 CrossRef CAS.
- C. Ma, J. Bai, X. Hu, Z. Jiang and L. Wang, J. Environ. Sci., 2023, 125, 533–543 CrossRef CAS.
- S. Song, Z. Liu, G. Liu, X. Cui and J. Sun, Constr. Build. Mater., 2023, 405, 133373 CrossRef CAS.
- A. Zaker, S. ben Hammouda, J. Sun, X. Wang, X. Li and Z. Chen, J. Environ. Chem. Eng., 2023, 11, 109741 CrossRef CAS.
- X. Xiong, Recycling food waste into value-added chemicals over carbon-based catalysts, PhD thesis, Hong Kong Polytechnic University, 2021.
- C. Wen, T. Liu, D. Wang, Y. Wang, H. Chen, G. Luo, Z. Zhou, C. Li and M. Xu, Prog. Energy Combust. Sci., 2023, 99, 101098 CrossRef.
- O. H. Gunawardene, C. A. Gunathilake, K. Vikrant and S. M. Amaraweera, Atmosphere, 2022, 13, 397 CrossRef CAS.
- A. Mukhtar, S. Ullah, A. Inayat, S. Saqib, N. B. Mellon, M. A. Assiri, A. G. Al-Sehemi, M. B. K. Niazi, Z. Jahan and M. A. Bustam, Energy, 2021, 216, 119230 CrossRef CAS.
- S. Kamalakannan, K. R. Maiyelvaganan, K. Palanisamy, A. Thomas, R. B. Said, M. Prakash and M. Hochlaf, Chemosphere, 2022, 286, 131612 CrossRef CAS PubMed.
- N. Abuelnoor, A. AlHajaj, M. Khaleel, L. F. Vega and M. R. Abu-Zahra, Chemosphere, 2021, 282, 131111 CrossRef CAS PubMed.
- Y. Han, G. Bai, J. Yang, J. Huang, Z. Fei, Q. Liu, Z. Zhang, X. Chen, J. Tang and M. Cui, J. Solid State Chem., 2020, 290, 121531 CrossRef CAS.
- V. Patel, A. Baskar, S. Tiburcius, B. Morrison, B. Mod, P. S. Tanwar, P. Kumar, A. Karakoti, G. Singh and A. Vinu, Adv. Sens. Res., 2023, 2, 2300024 CrossRef CAS.
- Y. Ye, Y. Qu and J. Sun, Catal. Rev., 2023, 1–56 CrossRef CAS.
- C. Zhou, L. Xu, H. Song, J. Feng, Z. Hu, M.-J. Yang, P. Shi, Y.-R. Li, Y.-J. Guo and H.-Z. Li, Aquaculture, 2023, 563, 738916 CrossRef CAS.
- Z. Chen, S. Deng, H. Wei, B. Wang, J. Huang and G. Yu, Front. Environ. Sci. Eng., 2013, 7, 326–340 CrossRef CAS.
- H. Liu, H. Lu and H. Hu, Renewable Sustainable Energy Rev., 2024, 189, 113908 CrossRef CAS.
- M. M. Alam, M. A. Hossain, M. D. Hossain, M. Johir, J. Hossen, M. S. Rahman, J. L. Zhou, A. K. Hasan, A. K. Karmakar and M. B. Ahmed, Processes, 2020, 8, 203 CrossRef CAS.
- N. A. Rashidi, Y. H. Chai, I. S. Ismail, M. F. H. Othman and S. Yusup, Biomass Convers. Biorefin., 2022, 1–15 Search PubMed.
- J. Bai, J. Huang, Q. Yu, M. Demir, E. Akgul, B. N. Altay, X. Hu and L. Wang, Front. Chem. Sci. Eng., 2023, 17, 1122–1130 CrossRef CAS.
- X. Gao, S. Yang, L. Hu, S. Cai, L. Wu and S. Kawi, Carbon Capture Sci. Technol., 2022, 3, 100039 CrossRef CAS.
- M. P. Maniscalco, M. Volpe and A. Messineo, Energies, 2020, 13, 4098 CrossRef CAS.
- L. Zhu, N. Zhao, L. Tong and Y. Lv, RSC Adv., 2018, 8, 21012–21019 RSC.
- Ó. J. Fonseca-Bermúdez, L. Giraldo, R. Sierra-Ramírez, J. Serafin, B. Dziejarski, M. G. Bonillo, G. Farid and J. C. Moreno-Piraján, J. CO2 Util., 2024, 83, 102799 CrossRef.
- V. Thithai, X. Jin, M. Ajaz Ahmed and J.-W. Choi, Energies, 2021, 14, 3002 CrossRef CAS.
- N. Kaya and Z. Y. Uzun, Biomass Convers. Biorefin., 2021, 11, 1067–1083 CrossRef CAS.
- K. Foo and B. Hameed, Microporous Mesoporous Mater., 2012, 148, 191–195 CrossRef CAS.
- Q. Li, S. Liu, L. Wang, F. Chen, J. Shao and X. Hu, J. Environ. Sci., 2021, 103, 268–278 CrossRef CAS.
- J. Shao, J. Wang, Q. Yu, F. Yang, M. Demir, O. C. Altinci, A. Umay, L. Wang and X. Hu, Sep. Purif. Technol., 2024, 333, 125891 CrossRef CAS.
- M. M. Hassan and C. M. Carr, Chemosphere, 2021, 265, 129087 CrossRef CAS.
- Q. Yu, J. Bai, J. Huang, M. Demir, B. N. Altay, X. Hu and L. Wang, Molecules, 2022, 27, 6816 CrossRef CAS.
- W. Wu, C. Wu, J. Liu, H. Yan, G. Li, Y. Zhao, K. Bei and G. Zhang, Sep. Purif. Technol., 2024, 339, 126690 CrossRef CAS.
- M. Vega, E. Díaz-Faes and C. Barriocanal, J. CO2 Util., 2024, 81, 102716 CrossRef CAS.
- C. Liu, Y. Zhi, Q. Yu, L. Tian, M. Demir, S. G. Colak, A. A. Farghaly, L. Wang and X. Hu, ACS Appl. Nano Mater., 2024, 7, 5434–5441 CrossRef CAS.
- Y. Bai, Y. Li, M. Li, X. Lin, C. Cai, H. He and S. Liu, Ind. Crops Prod., 2024, 220, 119183 CrossRef CAS.
- J. Serafin, M. C. Román-Martínez, M. Saidi, A. R. Gallego, S. Atlas and M. Ouzzine, J. Anal. Appl. Pyrolysis, 2024, 181, 106637 CrossRef.
- X. Yuan, S. Li, S. Jeon, S. Deng, L. Zhao and K. B. Lee, J. Hazard. Mater., 2020, 399, 123010 CrossRef CAS PubMed.
- K. M. Nelson, S. M. Mahurin, R. T. Mayes, B. Williamson, C. M. Teague, A. J. Binder, L. Baggetto, G. M. Veith and S. Dai, Microporous Mesoporous Mater., 2016, 222, 94–103 CrossRef CAS.
- A. Kumar and H. M. Jena, Results Phys., 2016, 6, 651–658 CrossRef.
- R. Pang, T. Lu, J. Shao, L. Wang, X. Wu, X. Qian and X. Hu, Energy Fuels, 2020, 35, 1620–1628 CrossRef.
- R. Wang, P. Wang, X. Yan, J. Lang, C. Peng and Q. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 5800–5806 CrossRef CAS PubMed.
- Y.-Y. Hao, M.-X. Xiao, G.-C. Mao, J.-P. Wang, Z.-K. Guo, B.-X. Dong and Y.-L. Teng, J. Environ. Manage., 2024, 359, 120782 CrossRef CAS PubMed.
- M. S. Ribeiro, M. Zanatta and M. C. Corvo, Fuel, 2022, 327, 125164 CrossRef.
- M. S. Khosrowshahi, M. A. Abdol, H. Mashhadimoslem, E. Khakpour, H. B. M. Emrooz, S. Sadeghzadeh and A. Ghaemi, Sci. Rep., 2022, 12, 8917 CrossRef CAS PubMed.
- J. Chen, J. Yang, G. Hu, X. Hu, Z. Li, S. Shen, M. Radosz and M. Fan, ACS Sustain. Chem. Eng., 2016, 4, 1439–1445 CrossRef CAS.
- B. N. Ho, D. Pino-Perez, C. M. Ghimbeu, J. Diaz, D. Peredo-Mancilla, C. Hort and D. Bessieres, J. Nat. Gas Sci. Eng., 2021, 95, 104124 CrossRef CAS.
- G. Nazir, A. Rehman, S. Hussain, Q. Mahmood, M. Fteiti, K. Heo, M. Ikram and M. A. U. Din, Green Chem., 2023, 25, 4941–4980 RSC.
- O. Hakami, Surf. Interfaces, 2023, 37, 102668 CrossRef CAS.
- A. Ghaemi, H. Mashhadimoslem and P. Zohourian Izadpanah, Int. J. Environ. Sci. Technol., 2022, 19, 727–746 CrossRef CAS.
- A. Mukherjee, Generation of Activated Carbon from Spent Coffee Grounds: Process Optimization, Kinetics and CO2 Capture, PhD thesis, University of Saskatchewan, 2022.
- C. Quan, Y. Zhou, J. Wang, C. Wu and N. Gao, J. CO2 Util., 2023, 68, 102373 CrossRef CAS.
- X.-F. Tan, S.-S. Zhu, R.-P. Wang, Y.-D. Chen, P.-L. Show, F.-F. Zhang and S.-H. Ho, Chin. Chem. Lett., 2021, 32, 2939–2946 CrossRef CAS.
- Y.-F. Huang, P.-T. Chiueh and S.-L. Lo, Environ. Sci. Pollut. Res., 2023, 30, 22211–22221 CrossRef CAS PubMed.
- C. Zhang, S. Sun, S. Xu and C. Wu, Biomass Bioenergy, 2022, 166, 106608 CrossRef CAS.
- S. K. Guchhait, S. Khatana, R. K. Saini, A. D. Singh and A. K. Sarma, Appl. Catal. O, 2024, 206924 Search PubMed.
- M. Vafaeinia, M. S. Khosrowshahi, H. Mashhadimoslem, H. B. M. Emrooz and A. Ghaemi, RSC Adv., 2022, 12, 546–560 RSC.
- H. Zhang, Z. Wei, D. Xiong, Y. Wu, M. Tong, H. Su, Z. Zhang and J. Liao, Materials, 2024, 17, 942 CrossRef CAS PubMed.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280–289 RSC.
- M. T. Islam, A. I. Sultana, C. Chambers, S. Saha, N. Saha, K. Kirtania and M. T. Reza, Energies, 2022, 15, 9340 CrossRef CAS.
- N. F. Attia, S. E. Elashery, A. A. Galhoum, H. Oh and I. E. T. El Sayed, Sustainable Solutions for Environmental Pollution: Air, Water and Soil Reclamation, 2022, pp. 503–531 Search PubMed.
- J. Serafin, B. Dziejarski, O. F. C. Junior and J. Sreńscek-Nazzal, Carbon, 2023, 201, 633–647 CrossRef CAS.
- S. Heo, W. Kim, Y. Jo and A. A. Adelodun, Sustainability, 2024, 16, 1634 CrossRef CAS.
- H. Patel, H. Weldekidan, A. Mohanty and M. Misra, Carbon Capture Sci. Technol., 2023, 8, 100128 CrossRef CAS.
- M. Karimi, L. F. Zafanelli, J. P. Almeida, G. R. Ströher, A. E. Rodrigues and J. A. Silva, J. Environ. Chem. Eng., 2020, 8, 104069 CrossRef CAS.
- W. Y. Wu, M. Zhang, C. Wang, L. Tao, J. Bu and Q. Zhu, Chem.–Asian J., 2024, 19, e202400180 CrossRef CAS PubMed.
- B. Petrovic, M. Gorbounov and S. M. Soltani, Carbon Capture Sci. Technol., 2022, 3, 100045 CrossRef CAS.
- S. Biti, A. J. McCue, D. Dionisi, I. Graça and C. F. Martín, Fuel, 2024, 358, 130246 CrossRef CAS.
- P. Wang, Y. Guo, C. Zhao, J. Yan and P. Lu, Appl. Energy, 2017, 201, 34–44 CrossRef CAS.
- G. A. R. Bari, H.-J. Kang, T.-G. Lee, H. J. Hwang, B.-H. An, H.-W. Seo, C. H. Ko, W. H. Hong and Y.-S. Jun, Carbon Lett., 2023, 33, 811–822 CrossRef.
- M. L. Botomé, P. Poletto, J. Junges, D. Perondi, A. Dettmer and M. Godinho, Chem. Eng. J., 2017, 321, 614–621 CrossRef.
- Q. Pu, Y. Wang, X. Wang, Z. Shao, S. Wen, J. Wang, P. Ning, S. Lu, L. Huang and Q. Wang, J. CO2 Util., 2021, 54, 101756 CrossRef CAS.
- L. Zhao, Z.-F. Sun, X.-W. Pan, J.-Y. Tan, S.-S. Yang, J.-T. Wu, C. Chen, Y. Yuan and N.-Q. Ren, Water Res.: X, 2023, 18, 100167 CAS.
- Y. Shen, Fuel Process. Technol., 2022, 236, 107437 CrossRef CAS.
- B. Igliński, W. Kujawski and U. Kiełkowska, Energies, 2023, 16, 1829 CrossRef.
- A. Allangawi, E. F. Alzaimoor, H. H. Shanaah, H. A. Mohammed, H. Saqer, A. A. El-Fattah and A. H. Kamel, C, 2023, 9, 17 CAS.
- F. Nawaz, M. Ali, S. Ahmad, Y. Yong, S. Rahman, M. Naseem, S. Hussain, A. Razzaq, A. Khan and F. Ali, Chemosphere, 2024, 143014 CrossRef CAS PubMed.
- A. N. Shafawi, A. R. Mohamed, P. Lahijani and M. Mohammadi, J. Environ. Chem. Eng., 2021, 9, 106869 CrossRef CAS.
- J. Serafin and O. F. Cruz Jr, J. Environ. Chem. Eng., 2022, 10, 107642 CrossRef CAS.
- L. Fu, Z. Ren, W. Si, Q. Ma, W. Huang, K. Liao, Z. Huang, Y. Wang, J. Li and P. Xu, J. CO2 Util., 2022, 66, 102260 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- K. Azam, N. Shezad, I. Shafiq, P. Akhter, F. Akhtar, F. Jamil, S. Shafique, Y.-K. Park and M. Hussain, Chemosphere, 2022, 306, 135566 CrossRef CAS PubMed.
- S. K. Jena, Min. Metall. Explor., 2021, 38, 47–68 Search PubMed.
- M. H. Khan, N. M. Akash, S. Akter, M. Rukh, C. Nzediegwu and M. S. Islam, J. Environ. Manage., 2023, 338, 117825 CrossRef CAS PubMed.
- G. Ramalingam, A. Priya, L. Gnanasekaran, S. Rajendran and T. K. Hoang, Fuel, 2024, 355, 129490 CrossRef CAS.
- C. Gao, J. Feng, W. Mo, W. Guo, S. Ma, X. Su, J. Yang, D. Wang, W. Sun and H. Jia, Sep. Purif. Technol., 2024, 348, 127756 CrossRef CAS.
- A. Shokri and A. Ghaemi, Case Stud. Chem. Environ. Eng., 2024, 9, 100725 CrossRef CAS.
- F. Bahmanzadegan, M. A. Pordsari and A. Ghaemi, Sci. Rep., 2023, 13, 12533 CrossRef CAS PubMed.
- E. Pérez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695 CrossRef PubMed.
- L. Santamaría, S. Korili and A. Gil, Chem. Eng. J., 2023, 455, 140551 CrossRef.
- V. C. Nguyen, H. T. Nguyen, H. D. Tran, T. N. Tu, W.-S. Kim and J. Kim, Microporous Mesoporous Mater., 2023, 360, 112716 CrossRef.
- R. Li, Z. Wang, Z. Yuan, C. Van Horne, V. Freger, M. Lin, R. Cai and J. Chen, Crit. Rev. Environ. Sci. Technol., 2022, 52, 4038–4071 CrossRef.
- F. Bai, X. Liu, S. Sani, Y. Liu, W. Guo and C. Sun, Sep. Purif. Technol., 2022, 299, 121539 CrossRef CAS.
- H. Wang, Y. Huang, S. Liu, C. Meng, X. Cheng and Y. Gao, Eur. Polym. J., 2024, 113367 CrossRef.
- J. Zhang, C. Duan, X. Huang, M. Meng, Y. Li, H. Huang, H. Wang, M. Yan and X. Tang, J. Mater. Sci., 2024, 59, 5271–5292 CrossRef CAS.
- C. Han, J. Yang, S. Dong, L. Ma, Q. Dai and J. Guo, Sep. Purif. Technol., 2024, 128957 Search PubMed.
- E. Priya, S. Kumar, C. Verma, S. Sarkar and P. K. Maji, J. Water Proc. Eng., 2022, 49, 103159 CrossRef.
- M. M. Rahman and M. Maniruzzaman, S. Afr. J. Chem. Eng., 2024, 47, 300–311 Search PubMed.
- A. Singh and J. G. Vijayan, in Application of Nanotechnology for Resource Recovery from Wastewater, CRC Press, 2024, pp. 254–294 Search PubMed.
- M. H. Ali and A. H. Fahmi, IOP Conf. Ser.: Earth Environ. Sci., 2024, 1371, 082029 CrossRef.
- R. Singh, L. Wang, K. Ostrikov and J. Huang, Adv. Mater. Interfaces, 2024, 11, 2202290 CrossRef CAS.
- T.-D. Hoang, S. A. Bandh, F. A. Malla, I. Qayoom, S. Bashir, S. B. Peer and A. Halog, Recycling, 2023, 8, 53 CrossRef.
- A. C. Pina, N. Tancredi, C. O. Ania and A. Amaya, J. Mater. Sci. Eng. B, 2021, 268, 115115 CrossRef CAS.
- S. Dubal, S. Chavan and P. Jadhav, J. Inst. Eng. (India): C, 2022, 103, 1415–1422 Search PubMed.
- S. Deng and Y. P. Ting, Langmuir, 2005, 21, 5940–5948 CrossRef CAS PubMed.
- S. Kocak, C. Akduman, J. Yanik, E. P. A. Kumbasar and A. Cay, Polym. Eng. Sci., 2024, 64, 1355–1364 CrossRef CAS.
- T. Töngüç Yalçınkaya, A. Çay, Ç. Akduman, E. P. Akçakoca Kumbasar and J. Yanık, J. Appl. Polym. Sci., 2024, 141, e55475 CrossRef.
- Y. Fei and Y. H. Hu, J. Mater. Chem. A, 2022, 10, 1047–1085 RSC.
- F. Raganati and P. Ammendola, Energy Fuels, 2024, 38, 13858–13905 CrossRef CAS.
- L. Jiang, Y. Zhang, P. Kong, L. Cheng, G. Liu and J. Sun, Carbon Capture Sci. Technol., 2024, 10, 100155 CrossRef CAS.
- J. He, Z. Jin, F. Gan, L. Xie, J. Guo, S. Zhang, C. Q. Jia, D. Ma, Z. Dai and X. Jiang, Green Chem., 2022, 24, 3376–3415 RSC.
- I. Skoczko and R. Guminski, J. Ecol. Eng., 2024, 25(1), 285–302 CrossRef.
- V. Raja, S. Dutta, P. Murugesan, J. Moses and C. Anandharamakrishnan, Environ. Chem. Lett., 2023, 21, 839–864 CrossRef CAS.
- C. Ng, W. E. Marshall, R. M. Rao, R. R. Bansode and J. N. Losso, Ind. Crop. Prod., 2003, 17, 209–217 CrossRef CAS.
- I. M. Lima, A. McAloon and A. A. Boateng, Biomass Bioenergy, 2008, 32, 568–572 CrossRef CAS.
- G. Stavropoulos and A. Zabaniotou, Fuel Process. Technol., 2009, 90, 952–957 CrossRef CAS.
- J. Serafin, B. Dziejarski, P. Rodríguez-Estupiñán, V. B. Fernández, L. Giraldo, J. Sreńscek-Nazzal, B. Michalkiewicz and J. C. Moreno-Piraján, Fuel, 2024, 374, 132462 CrossRef CAS.
- J. Serafin, J. Sreńscek-Nazzal, A. Kamińska, O. Paszkiewicz and B. Michalkiewicz, J. CO2 Util., 2022, 59, 101970 CrossRef CAS PubMed.
- C. A. Toles, W. E. Marshall, M. M. Johns, L. H. Wartelle and A. McAloon, Bioresour. Technol., 2000, 71, 87–92 CrossRef CAS.
- T. Kalak, Energies, 2023, 16, 1783 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |