
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn insight into advances and challenges in the development of potential stearoyl Co-A desaturase 1 inhibitors
Shivani Kirad
a,
Sonakshi Puri
b,
P. R. Deepa
b and
Murugesan Sankaranarayanan
 *a
*a
aMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Pilani-333031, Rajasthan, India. E-mail: murugesan@pilani.bits-pilani.ac.in
bBiochemistry and Enzyme Biotechnology Laboratory, Department of Biological Sciences, Birla Institute of Technology and Science Pilani, Pilani Campus, Pilani-333031, Rajasthan, India
First published on 24th September 2024
Abstract
Stearoyl-CoA desaturase 1 (SCD1) is one of the key enzymes involved in lipid metabolism, plays a vital role in the synthesis of monounsaturated fatty acids (MUFAs) from saturated fatty acids (SFAs). Due to its promising therapeutic potential in treating metabolic disorders, cancers, and skin diseases there is an increasing interest in the development of novel inhibitors against SCD1. This review comprehensively explores the evolution of potential SCD1 inhibitors, focusing on systemic and liver-targeted inhibitors and discusses their structure–activity relationship (SAR) pattern. Among the various small molecules reported, natural products like sterculic acid have emerged as significant SCD1 inhibitors, highlighting the potential of naturally derived compounds in therapeutic development. This review also addresses the challenges in optimizing pharmacokinetic properties and reducing adverse effects, providing insights into the future directions for the development of potential novel SCD1 inhibitors with maximum therapeutic effect and minimum side effects.
 Shivani Kirad | Ms Shivani Kirad completed her MS (Pharm) in Medicinal Chemistry from the National Institute of Pharmaceutical Education and Research, Ahmedabad, India, in 2023. She is pursuing PhD programme in Pharmacy at Birla Institute of Technology and Science (BITS) Pilani, Pilani Campus, India. Her current interest mainly focuses on development of novel molecules for the treatment of obesity, NAFLD and NASH. |
 Sonakshi Puri | Ms Sonakshi Puri completed her MSc in Microbiology from the Institute of Science, Nirma University, Ahmedabad, India, in 2020. She is pursuing PhD programme in Biological Sciences at Birla Institute of Technology and Science (BITS) Pilani, Pilani Campus, India. Her research interest is in a better understanding of disease mechanisms and therapeutic strategies in the area of metabolic syndrome and complications (Obesity, NAFLD). |
 P. R. Deepa | Dr P. R. Deepa is a Professor in the Dept. of Biological Sciences, BITS, Pilani. Her primary research is oriented towards biochemical understanding of disease mechanisms and therapeutic strategies related to metabolic disorders (obesity, NAFLD/NASH, CVDs, and cancers). She is also involved in research efforts in applied nutrition and nutraceuticals. A key thrust area of her research over the past two decades has been to identify and experimentally validate lipid metabolism targets (fatty acid synthase, stearoyl CoA desaturase, PPARs) to treat cancers, obesity and fatty liver disease. Prof. Deepa earned her doctoral degree from the Dept. of Medical Biochemistry, University of Madras. |
 Murugesan Sankaranarayanan | Prof. S. Murugesan is a Professor in the Department of Pharmacy at BITS Pilani. He holds a PhD from BIT, Mesra (2009) in synthetic medicinal chemistry and has over 200 research papers in the journals of international repute and 100 poster presentations at national and international forums. With expertise in Computer Aided Drug Design and Synthetic Medicinal Chemistry, Dr Murugesan is actively involved in the development of non-nucleoside reverse transcriptase inhibitors for combating opportunistic infections like HIV, TB, Malaria, Leishmaniasis as well as addressing rare disorders such as Sickle Cell Disease, Gaucher's disease and lifestyle disorders such as Obesity, NAFLD and NASH. |
1. Introduction
The ongoing search for safe and effective synthetic inhibitors that target SCD1 has been diligently documented over the past decades by an abundance of scientific literature from more than a dozen university and industrial research organisations. Pioneering research highlighting SCD1 being a potential therapeutic target for treating metabolic illnesses prompted this far-reaching venture into the field of chemistry. Although significant progress has been achieved in the identification of potent inhibitors across a range of scaffolds, converting these findings into effective clinical development has proven to be extremely difficult. A partial understanding of the complex biology associated with SCD1 and mechanism-based adverse effects that limit treatment efficacy are among the factors contributing to the obstacles to clinical success.1The focus of recent research efforts has switched to the creation of SCD1 inhibitors that are selective for certain tissues, especially the liver. This strategy is justified by the expectation of increased safety margins regarding mechanism-based side effects seen in animal models. Though they appear safe, these novel compounds have not yet overcome the basic obstacles that come with understanding and treating the intricacies of SCD1 biology. A paradigm change has occurred because of the difficulties in developing effective oral treatments for metabolic illnesses, with a growing focus on the assessment of SCD1 inhibitors in alternative indications.2
The goal of this review of literature is to give a thorough investigation of the structural variety and development of various scaffolds as SCD1 inhibitors, since the first drug-like SCD1 inhibitors were published in the year 2005. The following sections will describe the techniques that have been used as well as the progress that has been made in the ongoing search for new compounds that target SCD1.
Within the lipid homeostasis paradigm, SCD becomes an important participant, with complex interplay between synthesis of fatty acids or lipogenesis and fatty acid metabolism or lipolysis, affecting signalling, cellular and tissue architecture as well as energy storage.3–5 Situated mostly in the endoplasmic reticulum (ER) of the cell, the SCD enzyme selects and reorganises the cis-double bond formation between carbon 9 and 10 in saturated fatty acyl coenzyme-A esters in both regio and stereospecific manner.6 Palmitoyl-CoA (C16:0) and stearoyl-CoA (C18:0), the major substrates were converted into their respective MUFAs, oleoyl-CoA (C16:1) and palmitoleoyl-CoA (C16:1) (C18:1).7
These MUFAs were considered as necessary building blocks of membrane components – phospholipids, cholesterol esters and triglycerides. The highly regulated activity of the SCD enzyme is reliant on iron and co-factors, including cytochrome-b5, nicotinamide adenine dinucleotide (NADH), and flavoprotein cytochrome-b5 reductase.8
The primary human SCD isoform is called hSCD1, but hSCD5, a second SCD gene with possible developmental and protective functions, has also been identified but not been fully explored.9,10
Notwithstanding the difficulties associated with SCD1 biology, the fact that SCD1 is widely expressed in humans—especially in the liver and adipose tissues—has prompted significant efforts to create small-molecule SCD inhibitors as potential treatments for metabolic diseases.11,12 This is corroborated by two clinical trials in patients with non-alcoholic steatohepatitis (NASH) and non-alcoholic fatty liver disease (NAFLD), which showed that Aramchol (Fig. 1A), a partial hepatic SCD1 inhibitor, significantly improved fibrosis, decreased steatohepatitis and reduced liver fat content without causing appreciable adverse drug reactions.13,14 Furthermore, Aramchol showed a decrease in collagen 1 (Col1α1) expression and secretion via decreasing SCD1 expression and boosting peroxisome proliferator-activated receptor γ (PPAR-γ) in hepatic stellate cells (HSCs) in mechanistic research using primary human hepatocytes. It thereby produced an anti-fibrotic effect in people with NASH.15
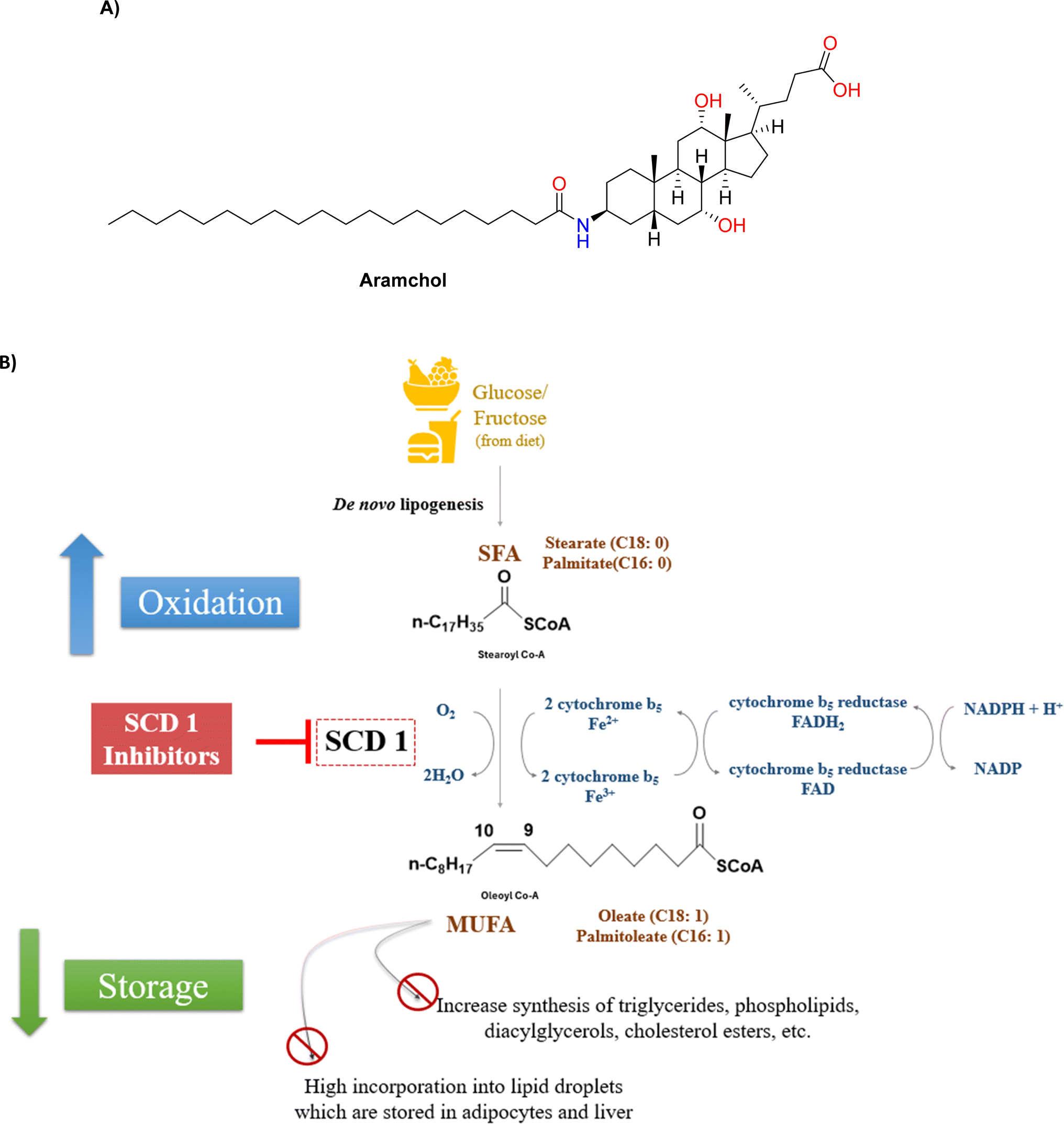 | ||
| Fig. 1 (A) Structure of Aramchol (B) SCD1 converts saturated fatty acids (SFAs) to monounsaturated fatty acids (MUFAs), inhibiting SCD1 results in decreased incorporation of lipid droplets, reduced lipid storage and increased fatty acid oxidation. | ||
In pre-clinical studies, small molecules have shown persistent suppression of SCD to be linked to side effects include baldness, squinting and dry eyes. These occurrences, which are thought to be mechanism-based, highlight a crucial difficulty in creating safe, well-tolerated SCD inhibitors for use in treating humans: striking a compromise between efficacy and a large enough therapeutic margin. Suppression of SCD1 may also trigger inflammation due to accumulation of saturated fatty acids. This may be overcome by co-administration of long chain essential fatty acids.12
It is crucial that we critically analyse the structural variety, evolutionary history, and prospective uses of SCD1 inhibitors outside metabolic illnesses as we make our way through the complex world of these drugs. The complex relationship that exists between the onset of metabolic diseases and lipid dysregulation highlights the therapeutic target SCD1's ongoing relevance and importance. In the hopes that future research projects will not only decipher the complexity of SCD1 biology but also open the door for novel therapeutic treatments in the field of metabolic disorders and beyond, the search for novel, potent and well-tolerated SCD1 inhibitors continues. This thorough study aims to provide an overview of reported SCD1 inhibitors that have been created throughout the years, illuminating the structure–activity relationship, development problems and fresh approaches for ongoing search of novel potent SCD1 inhibitors.
2. SCD1: significance and biological implications
A double bond has been created between carbon 9 and 10 in the fatty acid chain of stearoyl-CoA by the iron-containing fatty acid desaturase SCD1, which is essential for the formation of oleoyl-CoA. Known as Δ9 desaturase (D9D) often, SCD1 exhibits remarkable specificity by desaturating the C-9 position of the acyl chain of, stearic acid (C18:0), resulting in the production of oleic acid (C18:1n9) (Fig. 1B).16SCD functions as an integral membrane desaturase that catalyses the first insertion of a double bond at the delta-9 position in saturated fatty acids with a carbon chain length of 12 to 19. Through an enzymatic mechanism, these fatty acids were changed into monounsaturated fatty acids (MUFAs). Membrane fluidity and cell signalling were significantly impacted by the SCD-regulated modification of the unsaturation level of cellular lipids.17
Furthermore, the monounsaturated products produced by SCD function as the main building blocks for the synthesis of complex lipids, such as wax esters, cholesterol esters, triglycerides (TGs), phospholipids and diacylglycerols. This emphasises the critical function of SCD as a very controlled and evolved conserved enzyme with a variety of isoforms that have different substrate specificities and overlapped but unique tissue distributions.
Similar to other desaturases, SCD is a non-heme Fe-containing enzyme that needs NADH, cytochrome b5, cytochrome b5 reductase or a different transport mechanism of electrons to catalyse reactions. With its N- and C-terminal facing the cytoplasm, the SCD protein binds structurally to the endoplasmic reticulum (ER) membrane via four transmembrane domains. Additionally, the functional integrity of the enzyme is supported by the three catalytically essential and conserved His-box motifs.18
Four recognized isoforms of SCD (SCD1–4) exist in mice, situated within a 200 kilobase region on chromosome. These isoforms encode proteins consisting of 350–360 amino acids, demonstrating identity of over 80% amino acid sequence. Variations in the 5′-flanking regions contribute to tissue specificity differences among the isoforms. SCD2 shares considerable sequence similarity with SCD1 and is expressed everywhere, with elevated levels in the mice brain, especially during the myelination period. In mice, SCD2 plays a crucial developmental role and is essential for establishing an intact skin barrier in neonates. SCD3 is in the Harderian gland and skin of mice, although at considerably lower levels than SCD1, exhibiting a preference for palmitoyl-CoA over stearoyl-CoA. Conversely, SCD4 is predominantly found in the heart and experiences significant induction in the hearts of Scd1−/−mice.19
A similar reaction can be catalysed by SCD1 with substrate-CoAs that are structurally linked to saturated fatty acids. SCD1, for example, can convert palmitate (C16:0) to palmitoleate (C16:1n7), which is also a CoA conjugate. This enzymatic oxidation occurs on the inner face of the endoplasmic reticulum, partnering with NADH-cytochrome B5 reductase and employing linked electron transfers through cytochrome B5, FADH2, and NADH.20 The MUFA products resulting from SCD1 desaturation may undergo further elongation, desaturation within the cell or incorporation into various complex lipids, including triglycerides, phospholipids, wax esters, and other lipid species. Since, SFA substrates are typically abundant through diet and endogenous synthesis, the desaturation reaction catalysed by SCD1 becomes the rate-limiting step in the production and subsequent metabolism MUFAs in the body. Altering lipid composition and the ratio of SFA to MUFA has diverse impacts on cellular functioning. SCD1 enzyme is crucial in determining the balance of saturated and unsaturated lipids within cells, significantly influences membrane fluidity, impacting numerous membrane-bound biological processes and systems. Additionally, SCD1 plays a critical role in energy balance, with SFAs being preferentially utilized or oxidized over unsaturated fatty acids, which are predominantly stored as fat in tissues.
SCD1's central involvement in the de novo lipogenic pathway is evident as it catalyses the further transformation of fats made by fatty acid synthase into derivatives primed for integration into storage lipids, such as TGs. Moreover, its heightened expression under the influence of hormones and transcription factors like insulin and SREBP-1c underscores the crucial role of SCD1 in de novo lipogenesis. Notably, mice lacking SCD1 due to a targeted whole-body deletion of the enzyme exhibit remarkable resistance to obesity and hepatic steatosis induced by a high-carbohydrate diet.21 This resistance is attributed, in significant part, to reduction in hepatic lipogenesis. Liver-specific deletion of SCD1 through conditional knockout model confers the same protection against carbohydrate-induced obesity and hepatic steatosis as observed in whole-body SCD1 deletion.
Mice lacking SCD1 (SCD1−/−) are resistant to obesity and hepatic steatosis, and they are also shielded from the harmful reduction in insulin sensitivity that is frequently linked to obesity and fatty liver.22 SCD1−/− mice show enhanced insulin signalling across a variety of tissues, including skeletal muscle, liver, adipose tissue, and heart, despite having lower fasting plasma insulin levels. This is characterized by increased insulin receptor tyrosine phosphorylation and reduced Ser/Thr phosphorylation of IRS-1. Reduced ceramide formation and greater fat oxidation in SCD1−/− mice may be a factor in their increased insulin sensitivity because ceramides are implicated in mediating lipid-induced alterations in insulin signalling.
SCD1 serves as a metabolic hub, orchestrating various physiological processes including glucose and lipid metabolism, inflammation, autophagy, development, and tumorigenesis by regulating the proportion of MUFAs.1,23 The expression of SCD1 is tightly controlled both transcriptionally and epigenetically, influenced by a myriad of factors such as glucose, insulin, sterol regulatory element binding protein 1c (SREBP-1c), peroxisome proliferator-activated receptor α (PPARα) and liver X receptor (LXR)24–28 which induce its expression, while leptin, estrogen, and polyunsaturated fatty acids (PUFAs) suppress it.29–31
Under pathophysiological conditions, silencing of SCD1 has been shown to promote adipose tissue browning,32 reduce lipid accumulation and enhance lipolysis, offering potential therapeutic benefits for conditions such as obesity and non-alcoholic fatty liver disease (NAFLD). However, this beneficial effect might be offset by adverse impacts on β-cell structure and function, as evidenced by decreased lipid storage capacity and increased susceptibility to lipotoxicity in islet of β-cells. SCD1 plays a complex role in cancer biology,33,34 wherein SCD1 promotes metabolic balance in cancer cells that favours cell proliferation and its survival.35 In cancer cells the lipotoxicity induced by saturated fatty acid accumulation is mitigated by overexpression of SCD1 enzyme. Inhibiting SCD1 would deprive this protection afforded to cancer cells from cytotoxic effects of its saturated fatty acids. Thus, SCD1 inhibitors may be potential anti-cancer agents.36 SCD1 deficiency has also demonstrated cardioprotective effects against high-fat diet (HFD) induced cardiac dysfunction, underscoring its potential in addressing cardiovascular complications associated with metabolic diseases.37 The multifaceted role of SCD1 in various tissues highlights its potential as a therapeutic target for a range of metabolic disorders. Advances in medicinal chemistry and structural biology, including the resolution of the crystal structures of mouse and human SCD1 have provided deeper insights into the enzyme's catalytic mechanisms and have paved the way for the rational design and development of next-generation novel SCD1 inhibitors.
Recent findings indicate that due to wide distribution of SCD1 throughout the body, indiscriminate inhibition of SCD1, on a systemic scale may not be advisable due to the potential heightened susceptibility to cellular inflammation and side effects like alopecia and partial eye closure. While developing safe and effective SCD1 inhibitors, we should consider the metabolic consequences arising from systemic SCD1 inhibition and the indirect impacts of SCD1 inhibition in peripheral tissues such as skin. Taking this into consideration various efforts have been made to design molecules that have preferential liver distribution over other tissues giving liver targeted SCD1 inhibition in order tackle the side effects of systemic SCD1 inhibition. In this context, the availability of various in silico techniques, in vitro models and rodent models provide an immense scope for rational design of potential molecules that can target SCD1 with maximum potency and favourable pharmacokinetic profiles as well as with minimum side effects. Following, we present the reported systemic and liver-targeted SCD1 inhibitor across a variety of scaffolds from the available literature.
3. Small molecules as SCD1 inhibitors
In the present review, the development of SCD1 inhibitors has been broadly categorized into: (A) systemic inhibitors and (B) liver-targeted inhibitors. The scaffold of most of the SCD1 inhibitors exhibited a central ring system that were either (i) monocyclic, (ii) bicyclic or (iii) spirocyclic. These inhibitors were modified on their sidechains in order to enhance their potency. It is emphasized here that the liver targeted SCD inhibitors necessarily bear a tetrazole acetic acid side chain which facilitates directed distribution of the inhibitor molecule to the liver cells (Fig. 2). In the following section, the structural details of the various scaffolds in the reported molecules, along with their activity correlations, have been discussed. | ||
| Fig. 2 General structural representation of SCD1 inhibitors. | ||
3.1 Systemic SCD1 inhibitors
Researchers at Xenon took compound 1 (Fig. 3), a potent hit obtained from library screening and produced compound 2a as a hit-to-lead effort.38 Compound 2a was chosen to start SAR investigation, and the goal was to substitute heterocycles containing nitrogen for the pyridin-2-yl moiety. Analogues of pyrimidine and pyrazine (compound 2b and 2c) exhibited a reduction in potency. In the SCD1 inhibition assay, compound 2d (pyridin-3-yl substitution) maintained its activity but lost some of its potency. In conclusion, it was observed that replacing the pyridine group with pyridazine (compound 2e) increased the potency by almost three-fold. | ||
| Fig. 3 Monocyclic SCD1 inhibitors: piperazinyl – pyridazine based SCD1 inhibitors as reported by Xenon. | ||
With piperazinyl pyridazine as the focal point, further optimization was conducted. On the right (R1), various amines were investigated while keeping the 2-trifluoromethyl benzoyl group constant on the right. SCD1 activity was dependent on the linker lengths of the cycloalkyl and alkyl groups (compounds 3a–f). The cyclopropyl group with ethyl chain showed far more potency than that of methyl group containing analogues. Further with increase in chain length up to butyl group, potency increased whereas with hexyl and pentyl, heptyl it was less than butyl but comparable to that of ethyl group. Groups containing phenyl ring (compounds 3g–3j) showed strong potency and ideal chain lengths. Benzyl group showed good potency compared to directly attached phenyl group, increasing the aliphatic chain attached to the aromatic group decreased the activity. The potency of benzimidazol-2-yl (compound 3k) was preserved. In case of 5-membered heterocycles as side chain, furan and thiazole were active, whereas oxazole and pyrazole (compounds 3l–3o) were not as much as active.
The left-hand side's (R2) SAR study revealed strict tolerance for alterations. For strong SCD1 inhibitory activity, the 2-trifluoromethyl phenyl group (compound 4a) turned out to be the best choice. Electron withdrawing groups showed increased activity, whereas compounds with di-halo substitution (compounds 4b–4g) showed improved potency. Compound 4c (XEN 103) demonstrated good potency (IC50 = 14 nM), strong enzymatic inhibition and good oral bioavailability (F = 49%). These inhibitors were found to be highly potent and orally bioavailable SCD1 inhibitors. Promising in vivo tests using Zucker fatty rats, verified significant decreases in visceral fat pad weight and body weight growth without lowering lean body mass. Subsequent research revealed that it was possible to substitute the piperazine moiety with piperidine and the amide linkage with carbonyl, ether, amino or alkyl without significantly altering the inhibitory potential of SCD1. In 2006, Xenon made these findings public through several patent filings.39–44
Based on these proposed scaffolds using scaffold design strategies, scientists at Abbott developed pyridazine heteroaryl-based potent and orally bioavailable SCD1 inhibitors.45 Replacing pyrazine with piperidine aryl ethers (compounds 5a–b) (Fig. 4), characteristically fluorinated phenoxypiperidine (compound 5b) showed improved potency over un-substituted phenoxy piperidine analogue. Further, using various substituted analogues for SAR exploration, it was observed that either 2 or 2,5-disubstituted phenoxypiperidine were preferred, which has added advantage of 2-substitution to avoid the oxidation of phenoxy ring. Although good potency was achieved, but the molecules failed to show metabolic stability. It was hypothesized that the isoamyl group is responsible for the metabolic instability. Replacing isoamyl amide with methyl amide (compound 6a) improved the metabolic stability in rat liver microsomes. Primary amide substitution (compound 6b) showed similar potency to that of methyl amide. Further SAR was explored using heteroarylmethylamines based pyridazine carboxamides. 3-Pyridinylmethyl (compound 6d) analogue showed good potency against mSCD1 but not human SCD1, this could be recovered by using 3-methyl isoxazole, pyrazole and imidazole amides (compounds 6e–g). But these molecules showed decreased hepatic microsomal stability compared to smaller amides. Further replacement of the amide group was done with different heterocyclic systems (compounds 7a–d). Compound 7b was found as lead, which showed good microsomal stability.
 | ||
| Fig. 4 Monocyclic SCD1 inhibitors: six-membered piperazinyl based SCD1 inhibitors. | ||
Further on exploration, modification of the scaffold was done and series of 1-(4-phenoxypiperidin-1-yl)-2-arylaminoethanone, piperidine aryl urea-based inhibitors were reported.46 It yielded potent molecules like compound 8 but did not improve the potency compared to lead (compound 6a) taken.
Based on the nicotinamide SCD1 inhibitor (compound 2a) researchers at Abbott decided to replace the central aminopyridine with urea group, while conserving its polarity and geometry.47 Although, trans olefin seems to be a suitable linker (compound 9) (Fig. 4), but due to stability problem, benzene was incorporated in the structure. Meta-substituted derivative (compound 10a) showed promising results whereas para-substituted derivative (compound 10b) failed to show any potency. Replacing piperazine benzamide with piperidine ether conserved the potency. It was found that on the phenoxy ring, o-substituted halogen derivative showed improved activity with increase in bulkiness. Improved inhibitory activity was correlated with increased lipophilicity on the phenoxy ring.
The urea containing analogue, compound 11b (A-939572), was an effective and selective SCD1 inhibitor with an IC50 of 4 nM in (mouse SCD1) mSCD1 and 37 nM in hSCD1 enzymatic assays.47 Compound 11b also exhibited good PK profiles, a dose dependent desaturation index reduction in obese mice models and decrease in body weight, plasma insulin, plasma and liver triglycerides and an increase in insulin sensitivity, in a diet-induced obesity model in mice. However, the target-related adverse effects such as alopecia, eye apoptosis and sebaceous gland atrophy were observed 2 weeks of oral administration.46
Further researchers at Daiichi Sankyo replaced the piperazine moiety of the molecules proposed by Xenon using piperidine moiety and obtained a series of potent SCD1 inhibitors.48 It was found that replacing the pyridazine resulted in substantial loss in activity. So, keeping the pyridazine core constant they explored the piperazine fragment. SAR obtained for the benzoyl piperidine based SCD1 inhibitor depicted that electron withdrawing groups (compounds 12a–e) (Fig. 5) showed better activity in both mouse and human cell microsomal SCD1. Similar pattern was observed in the SAR development on the right-hand side aryl moieties. Fluro-phenyl derivatives (compounds 12f–h) showed better potency than pyridine analogue (compound 12i). Fluorine at meta position (compound 12f) did not improve inhibition in humans but at ortho and para (compounds 12g–h), it showed improved results. Replacing phenyl group with bio isostere thiophene (compound 12j) improved the potency. Compound 12k demonstrated plasma triglyceride-lowering effects in Zucker fatty rats in a dose-dependent manner after a 7 days administration. Further, this benzoyl piperidine (compound 12k) was explored by cyclization to form a spiro-piperidine ring. These reported compounds showed excellent potency.49
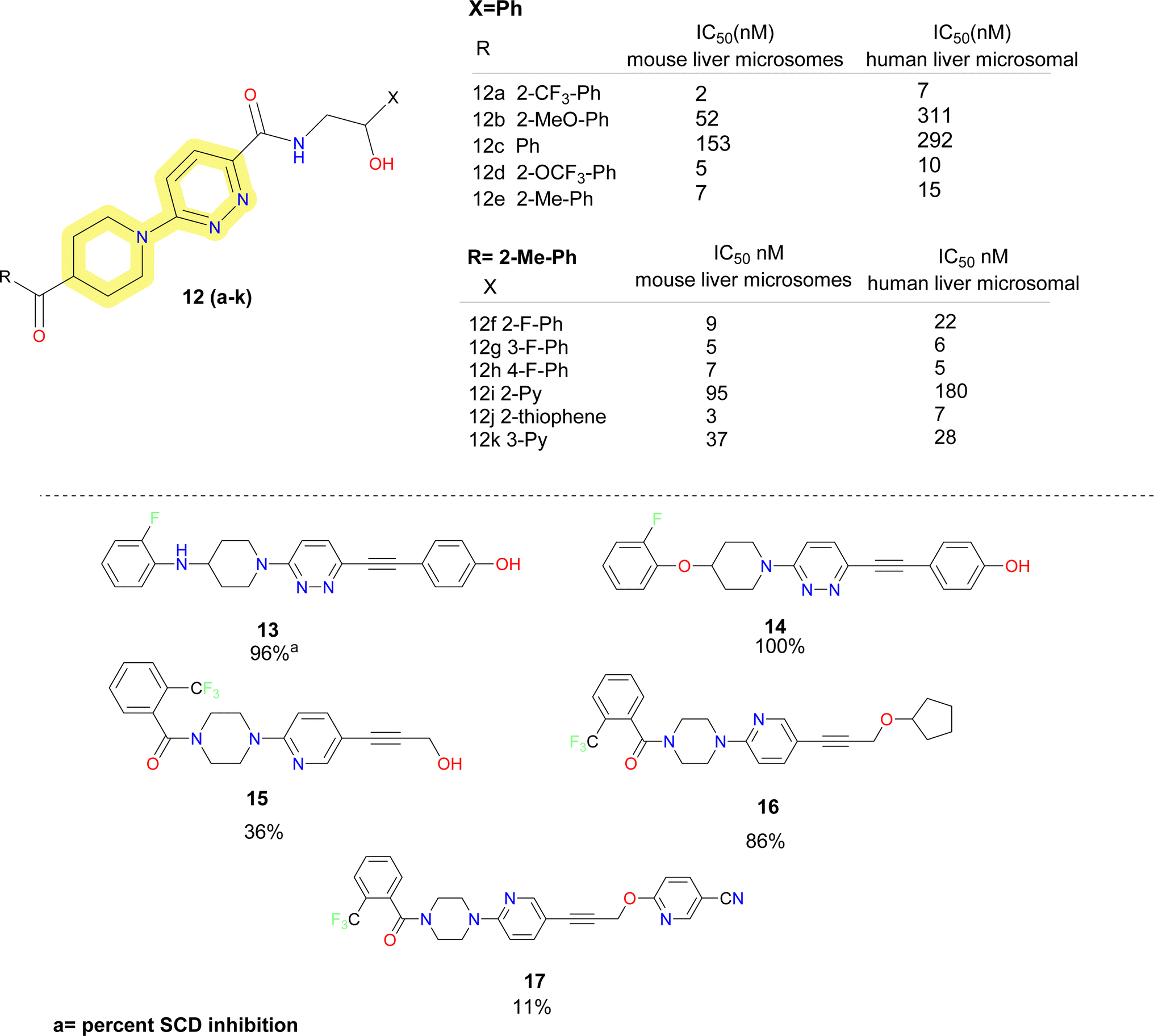 | ||
| Fig. 5 Monocyclic SCD1 inhibitors: pyridazine based carboxamide and acetylene linker containing SCD1 inhibitors. | ||
In the year 2008, researchers at Glenmark reported acetylene derivatives of piperidine–pyridazine based SCD1 inhibitors (compounds 13–17) (Fig. 5).50 Titrated water release assay at 10 μM was performed for the synthesized compounds to determine the percent SCD inhibition. No IC50 values were disclosed. Based on the determined percent inhibition, it was observed that amino and ether linkage showed better inhibition than carbonyl linkage and piperine and azetidine ring are better tolerated than piperazine moiety. Further, they reported acetylene containing spirocyclic SCD1 inhibitors. No biological data was disclosed.51
Researchers at Forest Laboratories also reported various piperazine based SCD1 inhibitors in various patent application, but no biological evaluation data was reported for these compounds.52–55
von Roemeling et al. introduced a pioneering computational-driven strategy for drug discovery, integrating machine learning and functional assessment to devise novel inhibitors of the lipogenic enzyme SCD1.56 This methodology, characterized by the creation of a new compound library based on reported molecules and in house fragments, and the integration of sophisticated in silico scoring techniques encompassing shape analysis, docking simulations and QSAR parameters. This method led to the identification of compound 18 (SSI-4) (Fig. 6), a pyridine–piperidine carboxamide analogue, showcasing potent SCD1 inhibitory property IC50 = 1.9 nM and anti-tumour property.
 | ||
| Fig. 6 Monocyclic SCD1 inhibitors: piperidine based SSI-4 and pyridine based SCD1 inhibitors. | ||
In 2020 Yumanity Therapeutics, Inc. reported a series of pyridazine and pyridine carboxamide molecules (compound 19–26) (Fig. 6) as SCD modulators.57 Compound 19 showed good potency, replacement of the central pyridine with groups like phenyl (compound 20), thiazole (compound 21) reduced the potency. Dihalo-substituted benzyl group (compounds 19, 22) as side chain showed better potency than mono-substituted derivatives. It was observed that pyrimidine (compound 23) and pyridine (compound 26) were well tolerated substituents for the side chain benzyl group. Imidazole (compound 23), pyridine (compound 26) was well tolerated substituent for pyridazinone side chain, whereas replacing it with dihydro imidazopyridine (compound 25) on its place decreased the potency.
Researchers at Takeda Pharmaceutical Company identified a lead compound 27 (Fig. 7), which exhibited high SCD1 binding affinity but lacked favorable PK properties, researchers pursued the design of novel derivatives based on molecular modeling studies.58 Through iterative SAR analysis, they identified compound 28a and subsequently explored various substitutions on the piperidine scaffold to optimize binding affinity and potency. Notably, hydrophilic substitutions like amino (compound 28b), hydroxy (compound 28c) and methoxy (compound 28d) were tolerated in terms of binding affinity, but lipophilic trifluoromethyl group (compounds 28e, 28i) showed a 5-fold increase in potency. Further optimization including substitution at the phenyl ring showed that lipophilic substitutions enhanced potency and regio isomers of the oxadiazole showed that 1,3,4-oxadiazole (compound 29b) and thiadiazole (compound 30b) showed excellent PK profiles and potency. Ultimately, compound 29b (T-3764518) emerged as a promising candidate, demonstrating high binding affinity, GI activity and metabolic stability, warranting further investigation for its therapeutic potential in metabolic disorders.59
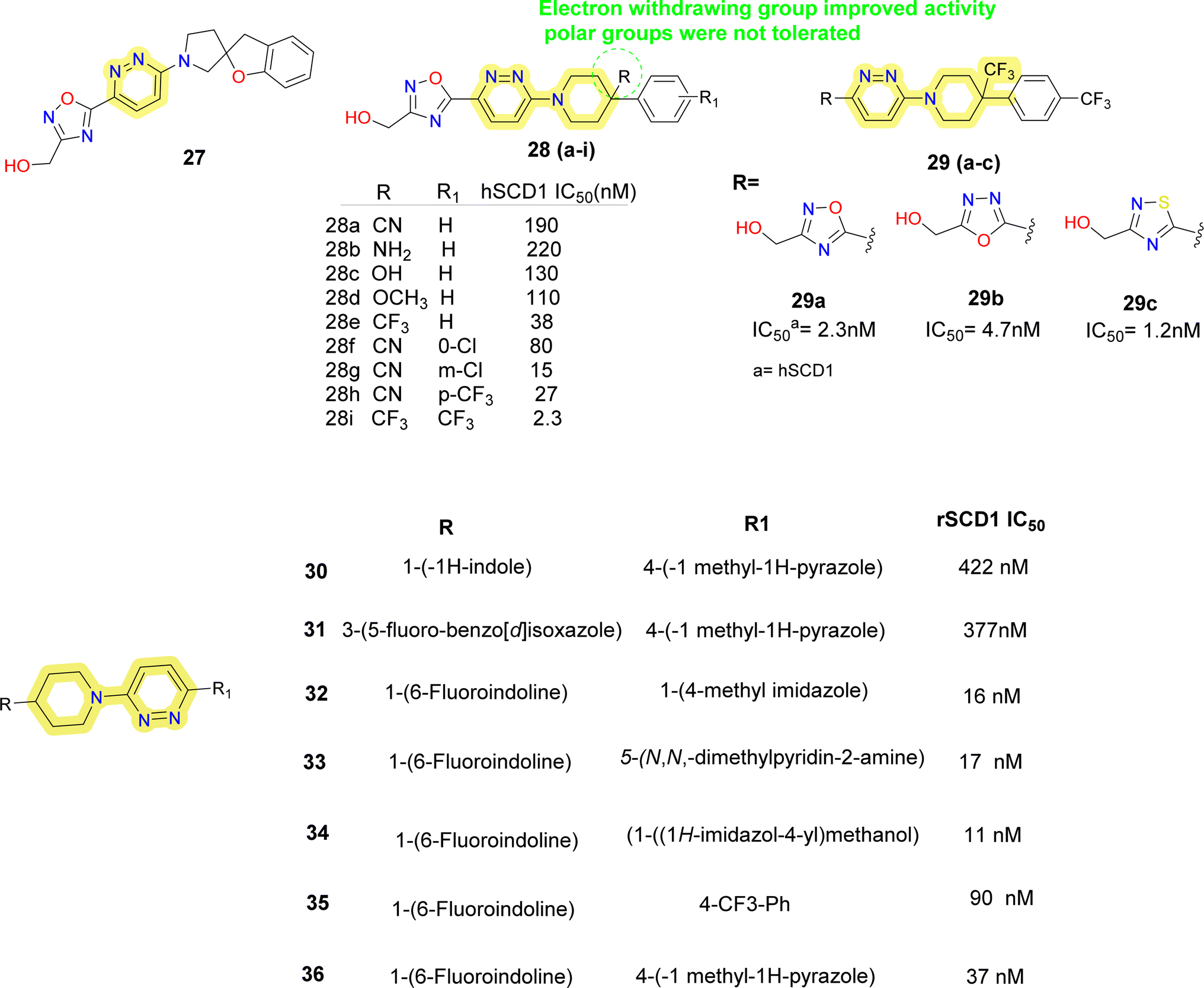 | ||
| Fig. 7 Monocyclic SCD1 inhibitors: pyridazine–piperidine containing SCD1 inhibitors reported by Takeda Pharmaceuticals and Yang et al.60 | ||
Yang et al. discovered 4-bicyclic heteroaryl-piperidine derivatives as potent inhibitors of SCD1 (compounds 30–36) (Fig. 7).60 Initially, compounds with N-methylpyrazole-4-yl substitution were synthesized and tested, revealing that potency was significantly influenced by the type and position of substituents on the bicyclic heteroaryl ring.61 Halogen substitution at the 6-position of indoline rings (compounds 32–36) notably improved potency, while substitutions at other positions decreased activity. Subsequent modifications focused on exploring SAR by varying the pyrazole ring with various substituted 5-membered heteroarenes (compounds 32, 34), revealed promising inhibitory activities. Replacement of the pyrazole ring with para-substituted 6-membered rings such as phenyl (compound 35) or pyridine (compound 33) showed improved inhibitory activity, imidazole with hydroxymethyl substitution (compound 34) also improved the potency. Despite the potency of these inhibitors, some suffered from low microsomal stabilities or significant affinity towards CYP450 inhibition and/or human ether-a-go-go related gene (hERG) channel binding, limiting further development. Notably, compound 36 demonstrated in vivo efficacy by decreasing body weight gain and plasma fatty acid desaturation index (DI) in Zucker fa/fa rats, suggesting its potential as a therapeutic agent. The study highlights the importance of SAR analysis in the rational design of SCD1 inhibitors and suggests that plasma fatty acid DI could serve as a biomarker for SCD1 inhibition, warranting further investigation into the pharmacological effects of these compounds.
Some pyridazine containing SCD1 inhibitors were reported by Xenon (compounds 37–41) (Fig. 8).62 These molecules indicated that sulphide and sulfoxide linkages are tolerated as a substitute to ether linkages and phenyl side chain improves potency rather than an aliphatic side chain.
 | ||
| Fig. 8 Monocyclic SCD1 inhibitors: SCD1 inhibitors reported by Xenon and Merck. | ||
Researchers at Merck reported azetidine containing pyridazine carboxamide SCD1 inhibitors (compounds 42–45) (Fig. 8),63 where dihalo-phenoxy moiety on the azetidine was fixed, on the right hand it is observed that phenyl group substituted with electron donor groups like methoxy (compound 44), alkyl (compounds 42, 45) showed better activity than withdrawing groups like carboxylic acid (compound 43). PF Medicament reported 2H-pyridazin-3-one derivatives as SCD1 inhibitors (compounds 46–50) (Fig. 8).64 Smaller groups like methyl (compounds 46, 47) were tolerated on the pyridine nitrogen whereas bulky groups like benzyl (compound 50) resulted in decrease of potency. Compound 47 was reportedly 100 times more potent than reference sterculic acid (50).
Pfizer researchers took compound 51a(Fig. 9) as the lead molecule and postulated that the benzyl group linked to imidazole was comparable to the halo substituted group by employing Rapid-Fire High Throughput Mass Spectrometry (RF-MS) experiment and acyl guanidine (compound 51a) as the starting point.65 The guanidine moiety was replaced out for the more potent N-methyl pyrazole. Results improved when benzylic tail moieties were substituted for the tail amide region; this was superior to phenyl substitutions. Additionally, a single meta substituent Cl or CF3 on the benzyl moiety was shown to produce ligand-efficient inhibitors.
 | ||
| Fig. 9 Monocyclic SCD1 inhibitors: SCD1 inhibitors containing 5-membered rings as central moiety. | ||
However, this did not help with the objective of in vivo activity because the increase in log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P that accompanied the alteration frequently led to decreased microsomal stability and solubility. Tail substituents were also accepted in certain instances by highly specialised aliphatic groups (compound 52d). For the amide area, similar patterns were seen. The preference was for small, five-membered amino heterocycles (compounds 52a–c). While some six-membered amino heterocycles and aliphatic cyclic (data not shown) amines (compound 52d) were tolerated, their potency was typically lost by one to two orders of magnitude. In rats fed with a low essential fatty acid (LEFA) diet, compound 51b produced a high exposure in vivo to cause dose-dependent alteration of the plasma desaturation index (DI). All dosage groups did, however, see minor to moderate effects on the ocular tissues.
P that accompanied the alteration frequently led to decreased microsomal stability and solubility. Tail substituents were also accepted in certain instances by highly specialised aliphatic groups (compound 52d). For the amide area, similar patterns were seen. The preference was for small, five-membered amino heterocycles (compounds 52a–c). While some six-membered amino heterocycles and aliphatic cyclic (data not shown) amines (compound 52d) were tolerated, their potency was typically lost by one to two orders of magnitude. In rats fed with a low essential fatty acid (LEFA) diet, compound 51b produced a high exposure in vivo to cause dose-dependent alteration of the plasma desaturation index (DI). All dosage groups did, however, see minor to moderate effects on the ocular tissues.
To develop potential SCD1 inhibitors to treat metabolic diseases, researchers from Novartis and Xenon collaborated in the year 2004. By starting with the pyridazine molecule, HTS was carried out to create a novel scaffold.66 Compound 53 (amino thiazole) was obtained as a HIT, following this various patent of thiazole along with imidazolidinones, pyridinones, triazinones, piperidinones and other heteroaryl containing SCD1 inhibitors were reported (compounds 53–64) (Fig. 9).67–71 In thiazolyl imidazolidinone67 molecules reported (compounds 55–59), on the left side of thiazole methyl-pyridinyl and other six membered aromatic moiety were well tolerated, whereas molecules containing un-substituted NH in imidazole (compound 55) were less potent than the molecules where the hydrogen of NH was substituted with benzyl substituents. Electron withdrawing trifluoromethyl phenyl group (compound 58) showed better activity than mono–fluoro substituted phenyl (compound 56) and pyridinyl (compound 57) substituted compounds. Further, replacing imidazole with triazole, thiazole improved the activity.68 Benzyl group on left showed better potency than pyridine substitution and trifluoromethyl phenyl showed better activity than mono-fluoro substituted phenyl analogue.
In 2006 Merck Frosst reported thiazole and thiadiazole analogues as SCD1 inhibitor (compounds 65–68) (Fig. 10).72,73 Compared to oxadiazole and triazoles, thiadiazole demonstrated superior efficacy when substituted for thiazole. Analogs of pyrazine and piperidine with ether and carbonyl linkers (compound 67) shown superior efficacy than those with alkyl and sulfoxide linkers. Whereas urea, thiadiazole and carboxamide reduced the potency, amide and hydroxyl-oxadiazole substitution on the thiazole ring preserved the potency. The potency of compound 68 was 100 times higher than that of compound 67. Even though its potency improved, it had adverse effects on the skin and eyes. Compound 67 was synthesized by substituting the piperazinyl-pyridazine analogues' five-membered thiazole ring for the six-membered pyridazine ring, as previously reported. This drug showed no Δ5 and Δ6 desaturase activity and IC50 values of 0.1 μM against rSCD and 0.3 μM against hSCD1 against SCD1, indicating it's in vitro potency against SCD1. In an acute trial, compound 67 reduced the mouse liver SCD activity index in a dose-dependent manner following oral dosing, with an ED50 of 3.0 mg kg−1. Additionally, compound 67 reduced body weight increase in a 7 weeks chronic mice study by 73%; however, after 14 days of therapy, partial eye closure and progressive baldness has been developed.74 The optimization efforts yielded compound 68, a highly effective SCD inhibitor (HepG2 IC50 = 1 nM). With an ED50 of 0.3 mg kg−1, compound 68 showed a dose-dependent decrease in the liver SCD activity index in mice. In rats given a high-fat diet, compound 68 showed a strong 24% reduction in body weight growth in a 4 weeks chronic testing with oral dosage at 0.2 mg kg−1. Changes in insulin and glucose levels demonstrated an improved metabolic profile along with this outcome, although unfavourable effects on the skin and eyes depending on the mechanism were also noted. Researchers at Glenmark in 2010 reported a series of thiazole containing SCD1 inhibitors.75 Electron withdrawing groups (compound 69a) on the amide showed improved potency compared to electron donating groups such as phenyl (compounds 69b–c) (Fig. 10).
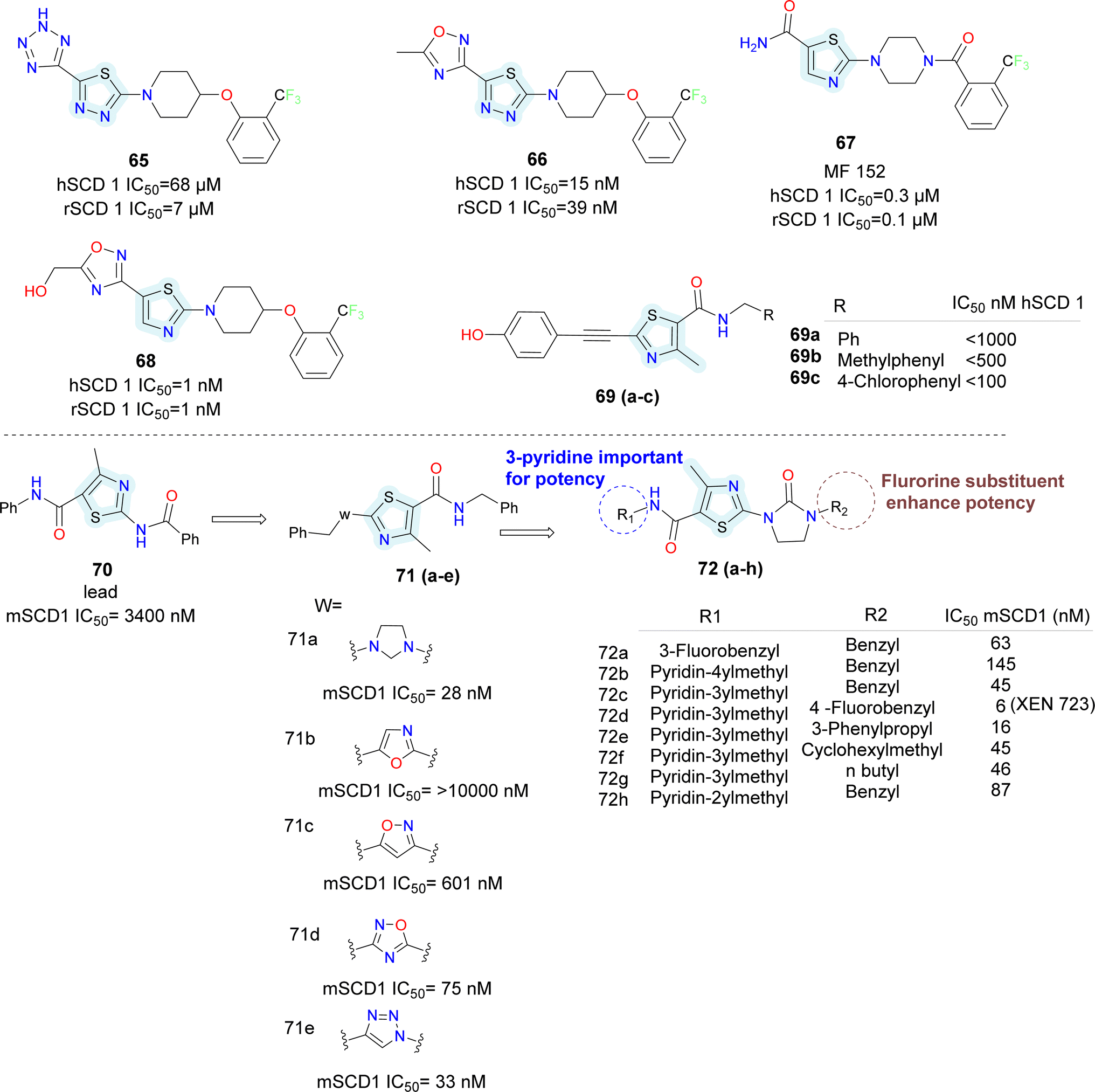 | ||
| Fig. 10 Monocyclic SCD1 inhibitors: thiazole derivatives as SCD1 inhibitors. | ||
Sun et al., utilized bio isostere strategies to replace the amide bond at the C2-position in the lead obtained compound 70, leading to the identification of potent SCD1 inhibitors.76 The replacement of the amide moiety at the C2-position with various heterocycles revealed that SCD1 potency was highly dependent on the nature of the heterocyclic replacements (compounds 71a–e) (Fig. 10). Imidazolidinone analogues emerged as the most potent compound, while other substitutions such as oxadiazole and triazoles, exhibited decreased potency. Halogen substitution at the 6-position of the aromatic ring notably improved the activity. However, substitutions at other positions or with different groups led to decreased potency, emphasizing the importance of substituent type and position. The study also highlighted the significance of thiazolyl imidazolidinone core structure, where analogues (compounds 72a–h) with certain aryl or heteroaryl groups displayed good potency, while longer chain lengths at specific positions led to decreased the activity.
Moreover, the potent and novel (compound 72d) XEN723 was found, exhibiting significant in vivo pharmacodynamic (PD) effects, and demonstrating an enhancement in SCD1 in vitro potency of approximately 560-fold over the original HTS hit compound 70.
While demonstrating potent inhibition and in vivo efficacy, XEN723 was associated with undesirable skin and eye adverse events (AEs), prompting the exploration of alternative chemical space to mitigate these AEs. The synthesis and SAR analysis of a series of triazolone-based SCD1 inhibitors (compounds 73, 74) (Fig. 11) led to the discovery of compound 74a,77 exhibiting improved AEs profile and promising in vivo efficacy. The study systematically modified the chemical structure, particularly focusing on the R2 side chain, to optimize potency and metabolic stability. Replacement of the thiazole core with a pyrazole group in compound 74a resulted in decreased permeability but maintained potency and demonstrated good in vivo efficacy. Furthermore, SAR analysis highlighted the importance of pyridin-3-yl-methyl R1 group for maintaining potency, as modifications to this moiety led to significant loss of activity. Despite challenges in modifying the left-hand side pyridin-3-yl-methyl R1, compound 74a emerged as a potent, orally active SCD1 inhibitor with promising in vivo efficacy with a favorable safety profile, suggesting its potential as a therapeutic agent for metabolic disorders.
 | ||
| Fig. 11 Monocyclic SCD1 inhibitors: five membered heterocycles reported for SCD1 inhibition. | ||
Further Sun et al. discovered novel thiazolylpyridinone-based SCD1 inhibitors by replacing the amide bond with a pyridinone moiety, exemplified by compound 75g(Fig. 11), which showed potent SCD1 inhibition in vitro and in vivo.78 This compound demonstrated a significant increase in potency compared to the original hit, with improved liver distribution and a wide safety margin. Furthermore, compound 75g demonstrated a dose-dependent reduction of plasma desaturation index, indicating potential efficacy in vivo. Additionally, flexible linkers between the pyridinone moiety and the terminal phenyl ring may enhance the potency and cellular activity, warranting further investigation.
Medicis Pharmaceuticals reported a simple phenoxy pyrrolidine potent SCD1 inhibitor compound 76 (Fig. 11),79 with aromatic rings substituted at either end and pyrrolidine as central ring.80
 | ||
| Fig. 12 Bicyclic SCD1 inhibitors: development of SAR 707 and SAR 224. | ||
Hexahydro-pyrrolo-pyrroles were substituted for the piperidine moiety using this data.85,86 SAR studies revealed that substituting aromatic pyridazine for pyridine and using an ether linkage rather than an amide resulted in promising compounds (78a–c). They called this compound 78c as molecule SAR707. Despite demonstrating effectiveness in vitro and in vivo in terms of lowering body weight, improving lipid parameters and lowering serum desaturation index, SAR707 displayed adverse drug reaction (ADR) in the skin when used for an extended period in vivo.
To combat concerns about SAR707's adverse effects, Matter et al. at Sanofi Aventis also carried out ligand-based virtual screening and searched for additional chemotypes. They found that the benzimidazole-carboxamide scaffold was a good place to start and more 2D similarity searches produced compounds 79a–c.87
It was discovered that adding keto linker in place of carboxamide did not increase activity (compounds 79d–e).
After more investigation on SAR, it was found that efficacy was reduced on the left side when benzyl substituents were used in place of thiophene group (compounds 79b–e). Further research revealed that presence of a chloro-substituent on thiophene preserved activity, whereas substituting thiazole on thiophene lowered the activity (compounds 79f–g). Conversely, substituting benzophenone derivatives on biphenyl ether (compound 79g) resulted in finding of SAR224 (compound 79h) (Fig. 12). Serum fatty acid desaturation index significantly decreased in SAR224 in vivo, and its optimal bioavailability and half-life were also demonstrated.
Powell and colleagues at Merck used the competitive rat SCD1 Scintillation Proximity binding Assay (SPA)88 to identify structurally varied inhibitors via HTS, which resulted in the identification of 2-arylbenzimidazole inhibitors with moderate potency.89
Initially taking compound 80a as starting hit, the t-butyl group was replaced with either H or CN (compounds 80b–c) (Fig. 13), which led to a decrease in efficacy. Conversely, analogues of sulfonamide showed comparable activity. The most active replacement among all was methylsulfone (compound 80d). While keeping methylsulfone, several aryl substituents were investigated to substitute the biphenyl group, but none of them showed superior activity. Moreover, these substances reportedly exhibited selectivity for the hSCD1 enzyme in contrast to the hSCD5 isoform.
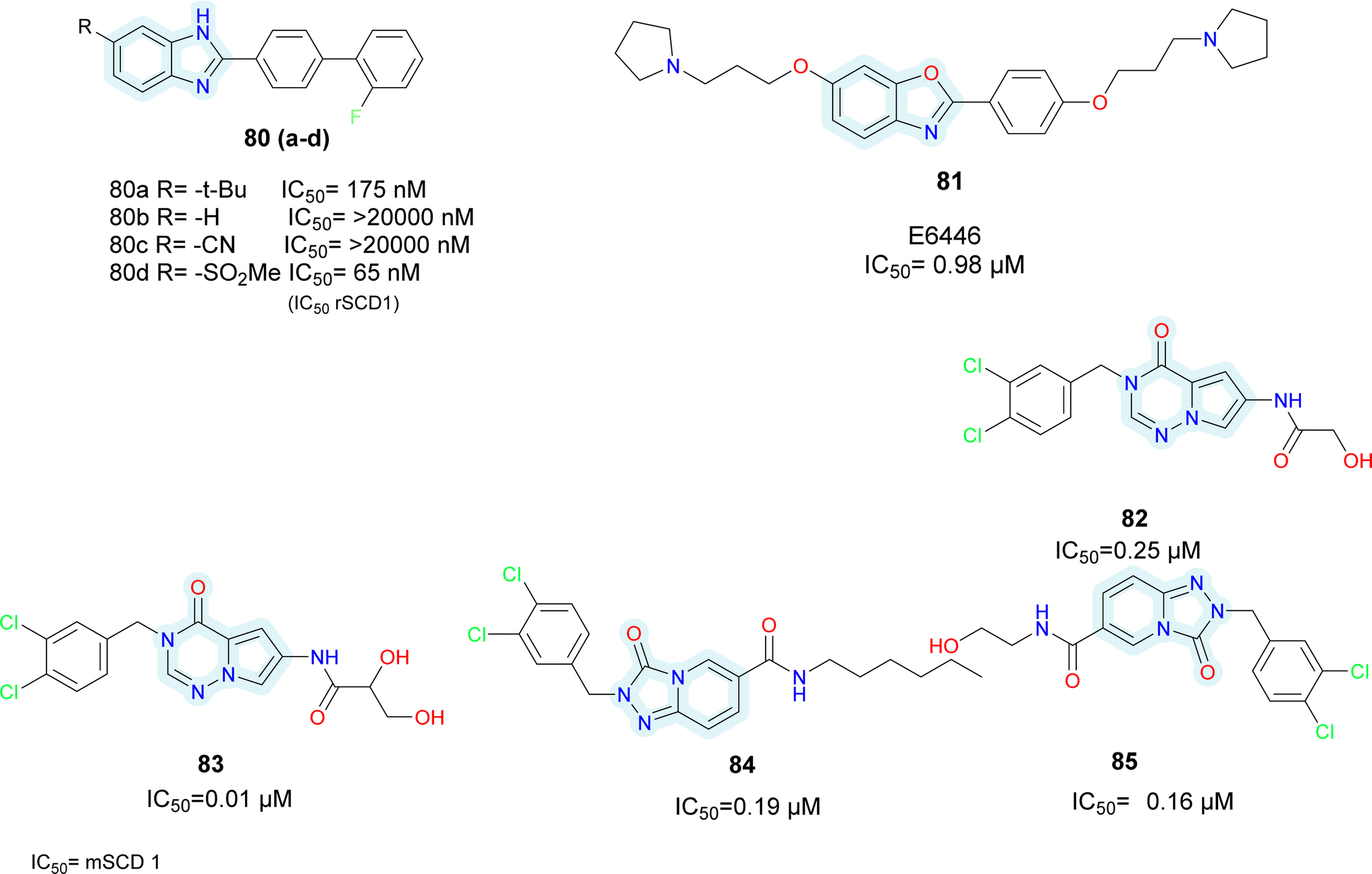 | ||
| Fig. 13 Bicyclic SCD1 inhibitors: various bicyclic heterocycles as potential SCD1 inhibitors. | ||
Wang et al. identified benzo-[d]-oxazole scaffold based E6446 (compound 81) (Fig. 13) using High Throughput Virtual Screening (HTVS).90 E6646 significantly inhibited adipogenic differentiation and hepatic lipogenesis via SCD1-ATF3 signalling. The SPR results showed that E6446 exhibited strong interaction ability with SCD1 (KD:4.61 μM). Additionally, E6646 significantly decreased hepatic steatosis, hepatic lipid droplet accumulation and insulin resistance in high-fat diet (HFD)-fed mice. It also showed excellent potency IC50 = 0.98 μM compared to reference A939572 which showed IC50 = 2.8 μM. Other bicyclic ring based SCD1 inhibitors such as isoquinoline, pyrrolotriazine and triazolopyridinone have been reported in various patents (compounds 82–85) (Fig. 13).91–93
 | ||
| Fig. 14 Spirocyclic SCD1 inhibitors: spiro benzo-oxapine based SCD1 inhibitors. | ||
Various patents for spirocyclic SCD1 inhibitors have been reported (Fig. 15). Xenon reported a series of spiro (chromane-piperidine) analogues (compounds 88a–f).95 Fluorine substitution in the chromane ring (compound 88c) improved potency whereas oxo-chromane (compounds 88a–b) showed reduced potency. Smaller substitution such as methyl on the amide group (compounds 88c–d) showed better activity compared to bulky substitutions such as pyridine and pyrazole (compounds 88e–f). Schering Corp. also reported a series of oxazepine based spiro compounds (compounds 89a–e).96 N-Piperidine substituted with heterocycle (compound 89d) showed better activity than alkyl substitution. Substitutions like CF3 (compound 89d) on the aromatic ring improved the activity rather than alkyl substituents.
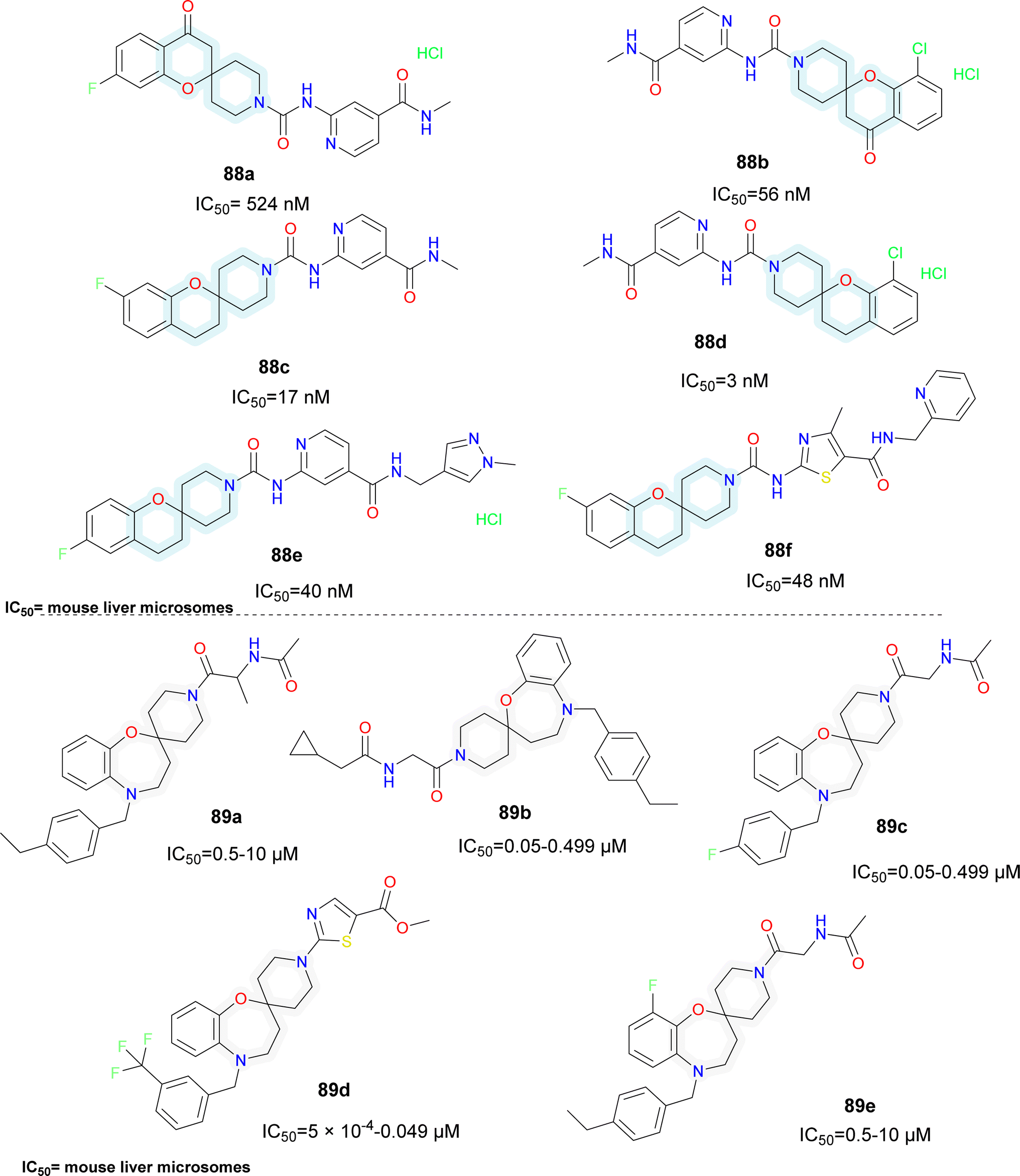 | ||
| Fig. 15 Spirocyclic SCD1 inhibitors: oxazepane-piperidine based spirocyclic systemic SCD1 inhibitors. | ||
3.2 Liver targeted SCD1 inhibitors
Merck researchers in 2011,97 with approach aiming to enhance drug exposure in the liver by utilizing liver-specific organic anion carrying polypeptides to facilitate active transport into hepatocytes (OATPs).98 Their approach consists of the following: (i) using an in vitro rat microsomal enzyme assay to determine whether a compound is a SCD inhibitor; (ii) testing active SCD inhibitors in two cell assays, one containing functional active OATPs (rat hepatocyte) and one lacking (HepG2 cell line); and (iii) conducting mouse tissue distribution studies on those compounds that are at least 4 to 5-fold more potent in the OATP vs. non-OATP cell assays.Acidic moieties were positioned on the left or right side of the molecule, using compound 90 MF-438 (ref. 99) as a reference. It became clear that the molecule could only support acidic moieties on its left side (compound 91b) (Fig. 16). Several acidic moieties were added, and the use of tetrazole caused a loss of efficacy and rendered the compound inert in the HepG2 test. Compound 91b displayed a low level of activity. This proved that an acidic moiety's appendage might actively travel into a hepatocyte while reducing passive cell penetration. SAR experiments were conducted on the right-hand side phenoxy part to boost potency on the rat SCD enzyme assay. As previously reported by them, 2,5-dihalogen substitution resulted in a significant 8-fold improvement in potency.100
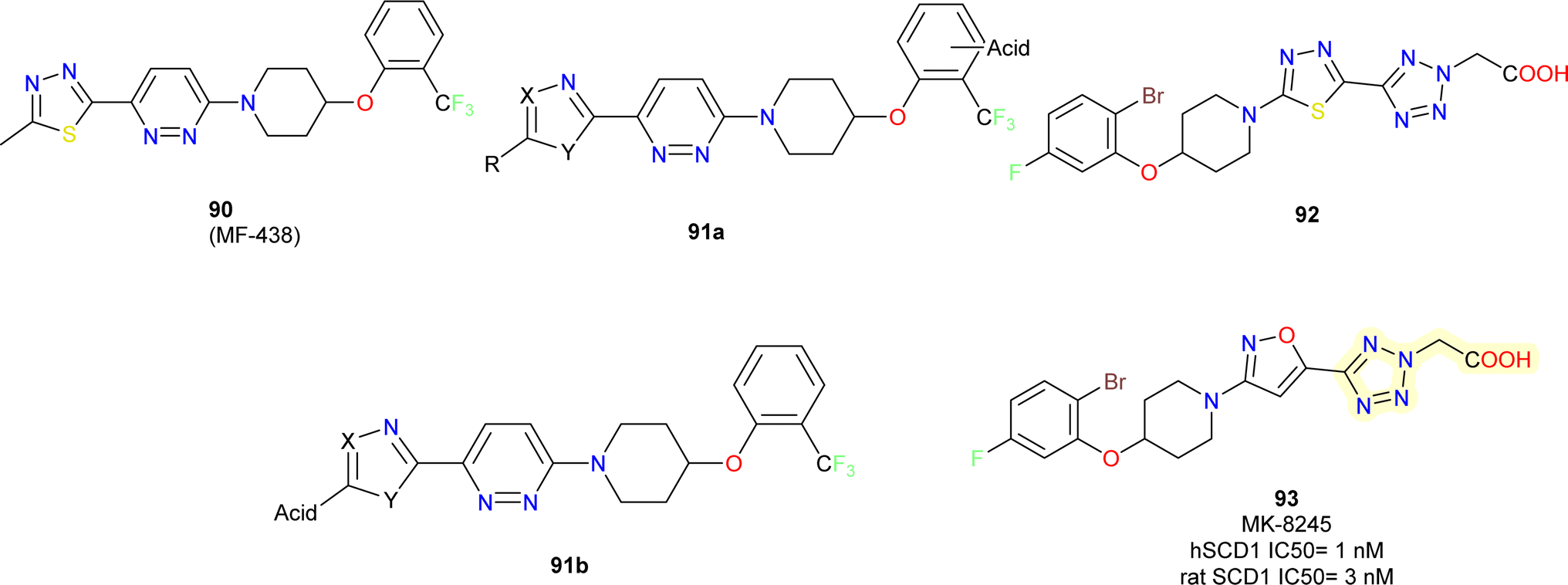 | ||
| Fig. 16 Development of MK-8245 based SCD1 inhibitors. | ||
The tetrazole heterocycle could have its potency increased even further by adding an acetic acid side chain (compound 92). In this instance, a carboxylic acid essentially took the place of the acidic tetrazole molecule. Compound 92 was more potent in the hepatocyte assay and >100-fold shifted in potency in the HepG2 cellular assay compared to molecule without –COOH. Finally, the potency in the rat enzyme and hepatocyte tests was further increased 2-fold while keeping a >300-fold shift in the HepG2 assay by substituting an isoxazole (compound 93) (MK-8245) nucleus instead of the middle thiadiazole ring with excellent potency.101
Compound 93 was able to exhibit maximum suppression of liver SCD while sparing the skin and eye tissues linked to adverse effects due to liver-targeting. At the end, this led to a safer profile for the liver-targeted SCD inhibitor (compound 93) in comparison to the systemically dispersed inhibitor that has been previously described.
Lachance et al.102 looked for structurally diverse liver-targeted SCD1 inhibitors, using MK-8245 as a reference point. While leaving the remainder of MK-8245 unaltered, they chose to replace out the piperidine linker for a bicyclic linker, such as, bis-pyrrolidine moiety. The isoxazole ring was found to be the ideal heterocycle in the bis-pyrrolidine analogues (compounds 94a, b) (Fig. 17), whereas the thiazole ring is quite like the reference. Since, the thiazole is a good substitute for the isoxazole ring and the bis-pyrrolidine linker was the first alternative studied, they conducted more SAR exploration by evaluating a wide variety of heterocyclic linkers (compounds 94d, e).
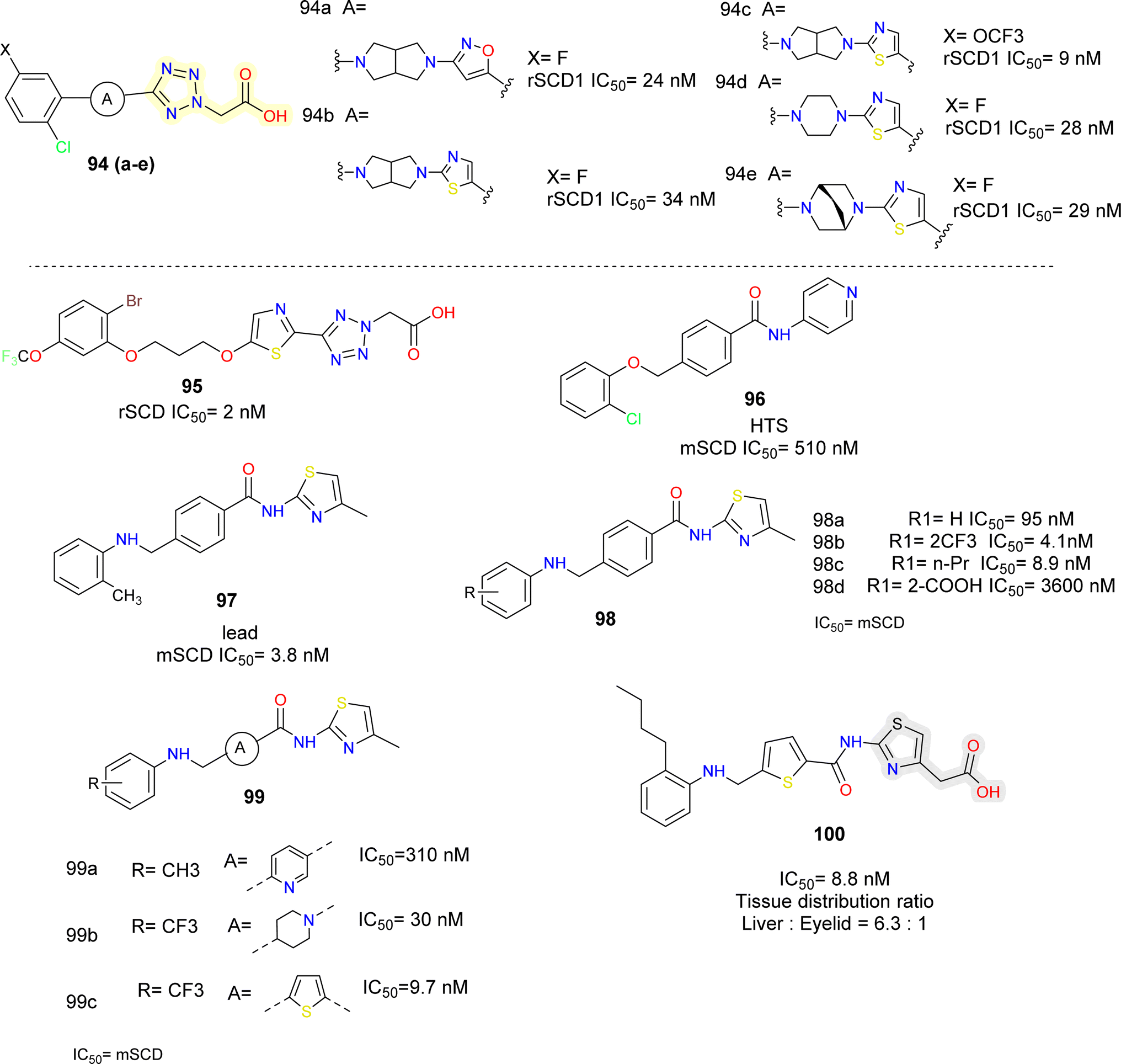 | ||
| Fig. 17 Tetrazole and thiazole acetic acid based SCD1 inhibitors. | ||
Following subsequent in vivo testing, the tissue distribution profile of compounds 94c and 94d found to be liver-selective. When compared to MK-8245, the mouse liver/plasma ratios were marginally lowered up to two folds, and the liver/Harderian glands ratios were improved. Given the link between suppressing SCD in the Harderian glands and AEs in the eyes, the liver organ's good tissue selectivity was desirable.
The moderately rigid heterocycles in these series substitute the piperidine core found in MK-8245 and other widely used SCD-inhibitors by joining the thiazole or isoxazole ring with the phenyl ring via a C–N bond. The thiazole heterocycle and 2-chloro-5-trifluoromethoxyphenyl in compound 94c were identified by SAR around the bis-pyrrolidine core as an excellent combination for both in vitro potency and in vivo efficacy.
In another report103 the core of MK-8245 was replaced using thiazole and an alkyl chain which was connected to the heterocycle using an oxygen atom (compound 95) (Fig. 17). In the metabolic studies conducted, it was observed that fluorine substitution on the phenyl ring led to oxidative defluorination metabolism which was observed in hepatocyte incubations, among different replacements, they found out –OCF3 was more metabolically stable resulting in a compound with good potency.
The researchers obtained compound 96 (Fig. 17), as HTS hit,104 they focused on physiochemical properties such as total polar surface area (tPSA) and partition coefficient (cLogP) to minimize the off-target effects. Further modification by replacing ether linkage with an amine linkage and pyridine with a thiazole group gave a prominent lead (compound 97) with improved potency but no selectivity towards the target. To obtain different compound exposure between liver and eyelids, further lead optimization was done. On the left had phenyl ring with hydrophobic groups such as CF3 (compound 98b), n-propyl (compound 98c) were well tolerated, whereas polar substituents like COOH (compound 98d) were not. It was also found that thiophene (compound 99c) was an equipotent replacement for central phenyl ring. Alcohols with methylene linker were placed at 4th position of the thiazole ring linkers, up to 2 carbon length were tolerated and showed single digit nM potency but further longer chain length decreased the potency. The correlation plots indicated that compounds in the ranges of 3.9 ≤ cLogP ≤5.0 and 90 ≤ tPSA ≤98 were highly selective for the liver targeting. Further optimization led to compound 100 which showed IC50 = 5.4 nM (mouse liver microsomes), was orally available and liver selective.
The phenoxy piperidine isoxazole derivatives of MK-8245 emerged as potent SCD1 inhibitors (compounds 101a–g) (Fig. 18), exhibiting robust enzymatic potency against rat SCD, and demonstrating promising in vivo efficacy in mouse liver pharmacodynamic models without adverse effects. The liver-targeted distribution profile of MK-8245 is attributed to substrate recognition by organic anionic transporter proteins (OATPs) in hepatocytes, primarily facilitated by the tetrazole acetic acid moiety. With a focus on maintaining this liver-targeting functionality while exploring structurally diverse SCD inhibitors, the piperidine core of MK-8245 was replaced with acyclic linkers, leading to the identification of a novel series of SCD1 inhibitors.105 Through SAR and metabolism studies, the optimal combination of a thiazole heterocycle and 2-bromo-5-trifluoromethoxyphenol in compound 101g was delineated for potent in vitro activity and in vivo efficacy. However, further efforts were required to enhance liver-selectivity and mitigate potential adverse effects associated with exposure to the Harderian gland.
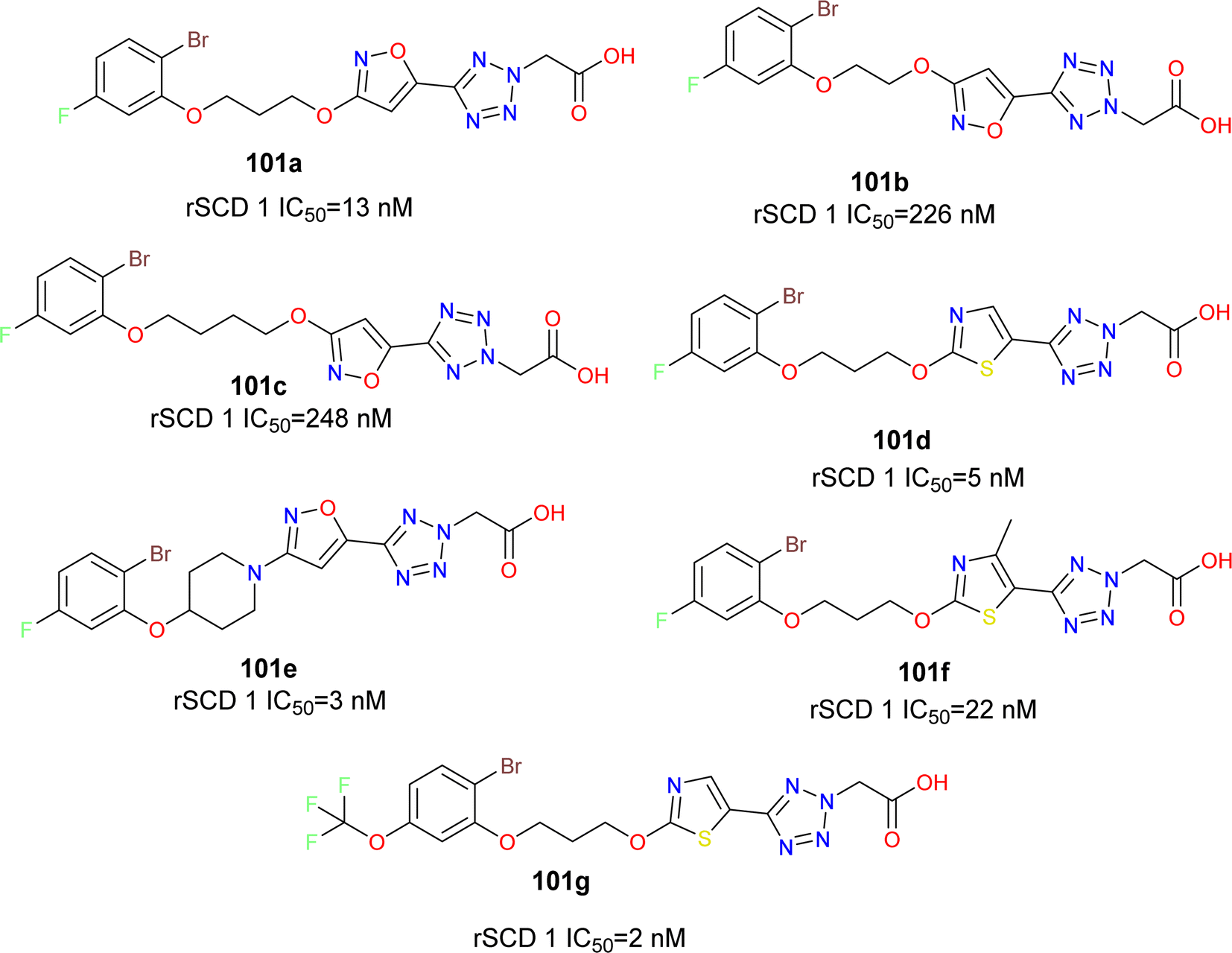 | ||
| Fig. 18 Acyclic linker based liver targeted SCD1 inhibitors. | ||
Developed from a benzo-fused spirocyclic oxazepine scaffold, inhibitors for SCD1 were crafted, demonstrating efficacy in lowering desaturation index in mice (Fig. 19).106 During optimization, preferences emerged for the oxazepine core and benzylic positions, while flexibility in piperidine substitutions allowed for potency and pharmacokinetic adjustments. Investigating SAR at the 5-position highlighted the potency variances with different substituents, particularly noting the influence of electron donating or withdrawing groups (compounds 102a–e). Shifting focus to the 1′-position, incorporation of R1 led to the synthesis of compounds featuring diverse R2 groups, revealing changes in potency. Exploration of heterocycles, especially pyridazine and thiazole, as linker motifs unveiled promising potency and tissue distribution in compound 103d, indicating potential for treating metabolic disorders. Further assessment, in vivo showed a moderate decrease in plasma desaturation index, prompting consideration for enhancing selective liver distribution to minimize side effects and enhance therapeutic efficacy. The SAR analysis focused on optimizing a benzo-fused spirocyclic oxazepine scaffold for SCD1 inhibition, revealing preferences for the oxazepine core and benzylic positions with substituents on the piperidine portion offering flexibility in potency and PK properties. Modifications at the 5-position showed that a benzyl group (compounds 102d–e) has improved the potency, while substitutions on the benzyl group had varying effects, with electron-withdrawing groups enhanced the potency. Introduction of heterocycles, particularly pyridazine (compounds 103a–d), further enhanced the potency, with thiazole derivatives (compounds 103c–d) exhibiting favourable SCD1 inhibition. Compound 103d emerged as a highly potent SCD1 inhibitor with excellent pharmacokinetic profiles and liver tissue distribution, suggesting promise for further development in metabolic disorder treatment. Merck reported a series of chromane-piperidine spiro compounds with various heterocyclic linkers, having tetrazole acetic acid as side chain, thiazole linker showed improved potency than pyridazine linkers. These compounds were reported to be liver selective (compounds 104a–f).107,108
 | ||
| Fig. 19 Spirocyclic liver targeted SCD1 inhibitors. | ||
4. Natural products as SCD1 inhibitors
Among the numerous small molecules reported, various natural products have been reported for SCD1 inhibition and/or by indirect de-regulation of lipid metabolizing molecules (Fig. 20). | ||
| Fig. 20 Natural products based SCD1 inhibitors. | ||
Zhu et al.109 showed that berberine (105) an isoquinoline alkaloid extracted from a Chinese herb, reduced hepatic lipid accumulation along with reduced expression of hepatic SCD1 and other related genes. They also reported that knockdown of SCD1 expression mimicked the effect of berberine decreasing hepatic steatosis. In HepG2 cells and the liver of mice fed with a high-fat diet, berberine enhanced the phosphorylation of AMP-activated protein kinase (AMPK) and sterol regulatory element-binding protein-1c (SREBP-1c). The berberine-induced reduction of SCD1 was caused by activation of the AMPK-SREBP-1c pathway and the sterol regulatory element (SRE) motif in the SCD1 promoter (−920/−550).
Sterculic acid (SA) (50), a natural cyclopropene fatty acid found in plant Sterculia foetida is a natural inhibitor of SCD1.110 Its extremely strained and reactive propene ring as well as the double bond between C9 and C10 in its chemical structure have been suggested as contributing factors to this inhibitory effect. Research conducted on 3T3-L1 adipocyte cells has demonstrated that this inhibition takes place through the down regulation of enzyme activity, independent of SCD mRNA or protein levels.111 Moreover, SA in Toxoplasma gondii does not change SCD's levels of transcription or translation. Therefore, this inhibition may result from SA's conversion to sterculoyl-CoA, which appears to be the enzyme's active form or from the irreversible binding of the enzyme's sulfhydryl groups to cyclopropene groups as a substitute option. However, following a four days injection of sterculic oil (SO), Dallaire et al. observed a 31% rise in SCD1 mRNA levels in mammary cow tissue. According to the authors, this is the result of a drop in SCD product levels, which causes an increase in SCD1 transcription to compensate for the deficit.112,113 In a mouse model of NAFLD, (5R)-5-hydroxytriptolide (LLDT-8) (106), a triptolide analogue, demonstrated hepatoprotective benefits by reducing liver lipid accumulation.114 It reduced lipid synthesis by downregulating the expression of SCD1, which in turn caused the ratios of C16:1/C16:0 and C18:1/C18:0 to decrease. It also up-regulated the genes involved in fatty acid oxidation and promoted lipolysis. It lessened AST, ALT serum markers, hepatic ballooning, and macro vesicular steatosis.
Pédrono et al.115 reported that sciadonic acid, a distinguished fatty acid found in gymnosperm seeds such as pine nuts which has been reported for lowering hepatic and plasma triacylglycerols acts by inhibiting SCD1.116–118 When hepatic cells were treated with sciadonic acid (compound 107), they modify the hepatic acid profile. Monounsaturated fatty acids (18:1n-9, 18:1n-7, 16:1n-7) were all decreased along with decrease in triacylglycerol amounts. Similarly, another seed oil Jacaric acid (compound 108) reportedly decreased the desaturation index, an indicator of SCD1 activity also in further experiments it decreased the expression of SCD1 mRNA.119 Curcumin (compound 109) a dietary polyphenol has been reported to suppress SCD1 regulation in various reports. Curcumin, increases fatty acid-oxidation, and reduces lipid synthesis.120 Li et al. reported that curcumin inhibited SREBP-1c and SCD1, which were enhanced in liver injury and hepatic metabolic disorder.121 Bai et al.122 reported sesamin (compound 110) a lignan from Sesamum indicum, showed anti-hepatic steatosis effects in MUFAs incubated mouse hepatocytes, HepG2 cells and high cholesterol diet induced NASH mice. It was found that sesamin reduced cluster of differentiation 36 (CD36) mediated fatty acid uptake and inhibited lipogenic genes like SCD1.123 Li et al. reported Epigallocatechin-3-gallate (EGCG) (compound 111) which is found in green tea, inhibited the expression of SCD1, in HFD mouse groups. Treatment with EGCG improved lipid characteristic and obesity symptoms.124
Naringin (compound 112), a natural flavone has been reported for inhibiting SCD1 expression along with reducing weight and lipid accumulation.125 Icariin obtained from the herb Epimedium brevicornu reduced lipid accumulation, increased fatty acid oxidation, showing ameliorative effect on hepatic steatosis induced by high fat diet and letrozole. Yang et al. showed that icaritin derivative IC2 (compound 113) illustrated SCD1 inhibitory property in breast cancer cells.126 Miyata et al. reported that taurine (compound 114) a bile acid salt decreased hepatic damage related bio-markers, hepatic triglycerides, non-esterified fatty acids. Expression of SCD1 was significantly decreased in genetic model of hepatic steatosis.127
5. Summary: key SAR insights of SCD1 inhibitors
The development of potential novel SCD1 inhibitor has seen substantial progress, driven by the need to manage metabolic disorders effectively. Table 1 captures some of the significant developments in SCD1 inhibitors during the last decade. Initial efforts focused on identifying potent systemic inhibitors, but the associated adverse effects such as skin and eye reactions, necessitated a shift towards liver-targeted therapies. The liver-specific distribution of inhibitors like MK-8245, facilitated by organic anionic transporter proteins, underscores the importance of targeting to reduce off-target effects and enhance the therapeutic efficacy.| S. no. | Structure | Remarks | Reference |
|---|---|---|---|
| 1 |  |
Pyridazine showed 3-fold improved potency than pyridine. CF3 group increases the potency | 38 |
| XEN 103 showed IC50 = 14 nM, with good potency and oral bioavailability | |||
| 2 |  |
Benzimidazole derivative SAR224 showed decrease in potency compared to the former compound SAR 707 but showed lesser side effects | 87 |
| 3 |  |
Urea based-piperidine SCD inhibitor showed good potency of IC50 = 8 nM | 128 |
| 4 |  |
Benzo-oxazepine based spirocyclic compounds showed excellent activity, pharmacokinetic properties and liver tissue distribution | 106 |
| 5 |  |
Bile acid–arachidonic acid conjugate, has reached phase 3A of clinical trial, with no side effects reported till date for the treatment of NASH. | 14 and 15 |
| 6 |  |
Indoline based bicyclic heteroaryl compounds showed optimum activity but poor microsomal stability, affinity towards CYP450 inhibition | 60 |
| 7 |  |
Imidazolidinone based compound showed 560-fold improved activity than the hit molecule, and better pharmacodynamic properties | 76 |
| 8 |  |
Maintained potency, but lesser side effects than XEN 723 | 77 |
| 9 | 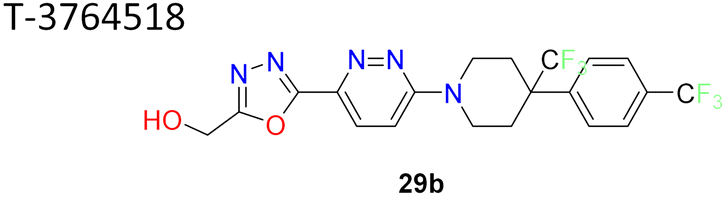 |
1,3,4-Oxadiazole improved pharmacokinetic properties demonstrated high binding affinity, GI activity and metabolic stability | 58 and 59 |
| 10 |  |
Integrated in silico techniques that considered docking score, QSAR, pharmacophore and shape analysis yielded SSI-4 with good potency | 56 |
| 11 |  |
Phenoxy pyrrolidine based SCD1 inhibitor showed excellent potency IC50 hSCD1 = 6.8 nM | 79 |
| 12 |  |
Electron withdrawing group substituted benzyl group on the right-hand side improve the potency | 75 |
| 13 |  |
Pyridine-pyridazinone carboxamide based SCD1 inhibitors showed optimum potency range (IC50 = 40–0.01 μg) | 57 |
| 14 |  |
Icaritin derivative based SCD1 inhibitor | 129 |
| 15 | 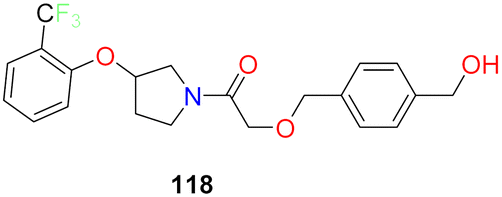 |
Pyrrolidine based SCD1 inhibitor showed excellent potency in human liver microsomes IC50 = 0.9 nM | 130 |
| 16 | 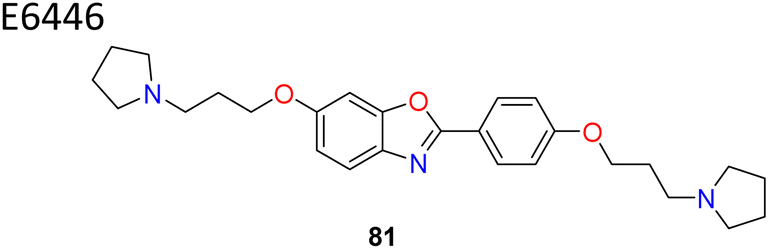 |
Benzo-oxazole based E6446 with flexible side chains showed IC50 = 0.98 μM | 90 |
Initial compounds feature heterocyclic cores such as pyridine, pyrimidine, and pyrazine. Modifications have revealed that pyridazine and piperidine provide three-fold increase in potency compared to pyridine. Flexible linkers between core structures and terminal phenyl ring improves potency and cellular activity. In case of substituent effects, it has been observed that electron withdrawing groups on aromatic rings particularly CF3 in several cases have generally enhanced the activity. Thiazole and pyridazine linkers have been extensively studied. Thiazole derivatives have shown favourable pharmacokinetic characteristics and high potency compared to other heterocycles. Other scaffolds such as imidazolidinone, thiazolylpyridinone were common in certain potent compounds. In cases of six-membered systems like piperazinyl pyridazine linker lengths have shown an influence on potency, for example cyclopropyl group with an ethyl chain showed higher potency. Chain length influenced the potency, with butyl being optimal. Amide linkage replaced with carbonyl, ether, amino or alkyl groups without significantly affecting the potency. Compounds with imidazole and pyrazole rings demonstrated significant SCD1 inhibitory activity. Substitutions at the 5-position of these rings, such as hydroxymethyl groups, enhanced the potency. Imidazole with a hydroxymethyl substitution improved the potency, indicating the importance of specific hydroxyl group positioning for activity. Bicyclic systems, such as those involving isoxazole and thiazole rings, have shown promising results. These linkers, when combined with a phenyl ring via a C–N bond, contributed to in vitro potency and in vivo efficacy. For example, the combination of thiazole and 2-chloro-5-trifluoromethoxyphenyl provided an excellent balance for systemic SCD1 inhibition. Substitutions at specific positions on bicyclic rings like 5-fluorobenzoisoxazole and indoline led to enhanced potency. Fluro-substitution at the 6-position of these rings improved activity, suggesting a key role of fluorine in modulating SCD1 inhibitory effects. Spirocyclic compounds such as 4,5-dihydro-3H-spiro-[benzo-[b][1,4]-oxazepine-2,4′-piperidine] exhibited favourable scaffolds for SCD1 inhibition. Substitutions at the benzyl group's 5-position, such as chlorine and trifluoromethoxy, significantly improved the potency, indicating the importance of specific hydrophobic and electron-withdrawing groups at these positions. When it comes to liver targeted distribution profile, it is attributed to moieties such as tetrazole acetic acid, which facilitate recognition by OATPs. Compounds with carboxylic acid groups demonstrated preferential liver distribution than other organs. Thiazole carboxylic acid derivatives exhibited higher potency and better pharmacokinetic profiles than pyridazine counterparts. Bicyclic linkers like bis-pyrrolidine showed improved liver selectivity.
Despite these advances, challenges remain in balancing potency, selectivity, and safety. The identification of novel scaffolds and the continuous refinement of SAR will be crucial in developing next-generation potential SCD1 inhibitor. Additionally, understanding the metabolic pathways and potential off-target effects are very much essential for optimizing these compounds for clinical use.
6. Conclusion and future prospects
The journey of development of potential SCD1 inhibitor has significantly progressed with the identification of potent systemic and liver-targeted inhibitors, and highlights the intricate balance between efficacy and safety. The structure–activity relationship studies reveal that specific heterocyclic rings, such as five-membered, bicyclic, and spirocyclic systems, play vital roles in determining the potency and selectivity of these inhibitors. Natural products have also contributed valuable scaffolds for SCD1 inhibition, demonstrating the broad potential for chemical diversity in drug design. Continued exploration of novel heterocyclic systems and their substitutions is essential to enhance the efficacy and safety profiles of identified SCD1 inhibitors. Future research canfocus on optimizing liver-targeted delivery mechanisms to minimize systemic side effects. Additionally, the integration of natural product scaffolds with synthetic modifications holds promise for developing more effective and safer SCD1 inhibitor. The future of SCD1 inhibitor development lies in the design of tissue-specific delivery strategies to mitigate systemic side effects. Nanoparticles and lipid nanoparticles targeting the liver, adipose tissue, and skeletal muscle hold promise for effectively delivering SCD1 inhibitors to their intended sites of action. A potential strategy to offset the side effects of systemic SCD1 inhibition can be to develop partial inhibitors of SCD1 enzyme, on the lines of Aramchol. It is envisaged that molecular hybridization by incorporating SCD1 inhibitory pharmacophore with anti-inflammatory pharmacophore may limit the unintended side effects of systemic SCD1 inhibition. Additionally, exploring the lipidomics of SCD1 and identifying new downstream lipid mediators will further elucidate its complex metabolic and signalling networks, offering new avenues for drug development. Understanding the dynamic conformations of SCD1 through computational studies will also enhance the design of novel inhibitors that can more effectively modulate its activity under various physiological and pathological conditions with maximum therapeutic effect and minimum side effects.Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by research grant from the Indian Council of Medical Research (ICMR), New Delhi (Grant Number 52/13/2022-BIO/BMS). SK and SP acknowledge the Institute Fellowship from BITS Pilani. The authors are grateful to the administration of BITS Pilani, Pilani campus, for providing necessary infrastructural support.References
- Q. Sun, X. Xing, H. Wang, K. Wan, R. Fan, C. Liu, Y. Wang, W. Wu, Y. Wang and R. Wang, Biomed. Pharmacother., 2024, 170, 115586 CrossRef CAS PubMed.
- N. Stefan, A. Peter, A. Cegan, H. Staiger, J. Machann, F. Schick, C. D. Claussen, A. Fritsche, H.-U. Häring and E. Schleicher, Diabetologia, 2008, 51, 648–656 CrossRef CAS.
- Y. Zou, Y.-N. Wang, H. Ma, Z.-H. He, Y. Tang, L. Guo, Y. Liu, M. Ding, S.-W. Qian and Q.-Q. Tang, J. Lipid Res., 2020, 61, 1589–1604 CrossRef CAS.
- N. Zhang, Y. Wang, J. Zhang, B. Liu, X. Deng, S. Xin and K. Xu, FASEB J., 2020, 34, 15338–15363 CrossRef CAS PubMed.
- M. W. Hulver, J. R. Berggren, M. J. Carper, M. Miyazaki, J. M. Ntambi, E. P. Hoffman, J. P. Thyfault, R. Stevens, G. L. Dohm, J. A. Houmard and D. M. Muoio, Cell Metab., 2005, 2, 251–261 CrossRef CAS.
- P. Dobrzyn, M. Jazurek and A. Dobrzyn, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1189–1194 CrossRef CAS PubMed.
- A. M. ALJohani, D. N. Syed and J. M. Ntambi, Trends Endocrinol. Metab., 2017, 28, 831–842 CrossRef CAS PubMed.
- U. N. Das, Cell Chem. Biol., 2019, 26, 309–311 CrossRef CAS PubMed.
- J. Wang, L. Yu, R. E. Schmidt, C. Su, X. Huang, K. Gould and G. Cao, Biochem. Biophys. Res. Commun., 2005, 332, 735–742 CrossRef CAS PubMed.
- Y. Bai, J. G. McCoy, E. J. Levin, P. Sobrado, K. R. Rajashankar, B. G. Fox and M. Zhou, Nature, 2015, 524, 252–256 CrossRef CAS PubMed.
- B. Yang, F. Ding, F.-L. Wang, J. Yan, X.-W. Ye, W. Yu and D. Li, Sci. Rep., 2016, 6, 23446 CrossRef CAS PubMed.
- R. Safadi, F. M. Konikoff, M. Mahamid, S. Zelber-Sagi, M. Halpern, T. Gilat, R. Oren, R. Safadi, F. M. Konikoff, A. Hershkovitz, T. Gilat, M. Halpern, Z. Rosenthal-Galili, E. Zuckerman, S. Abu-Mouch, A. Fich, E. Sikuler, A. Issachar, N. Assy, Y. Baruch, Y. Lurie, M. Graif, N. Stern, M. Yaron, A. Blank, D. Ben Bashat, S. Zelber-Sagi, M. Mahamid, M. Mizrahi and R. Oren, Clin. Gastroenterol. Hepatol., 2014, 12, 2085–2091 CrossRef CAS.
- V. Ratziu, L. de Guevara, R. Safadi, F. Poordad, F. Fuster, J. Flores-Figueroa, M. Arrese, A. L. Fracanzani, D. Ben Bashat, K. Lackner, T. Gorfine, S. Kadosh, R. Oren, M. Halperin, L. Hayardeny, R. Loomba, S. Friedman, M. Abdelmalek, F. Angelico, M. Angelico, J. P. Arancibia, E. Bardou-Jacquet, F. Barrera, C. F. Barish, Y. Baruch, Z. Ben-Ari, T. Berg, M. Bourliere, J. Boursier, E. Broide, M. Carmiel, D. S. Denham, L. Di Cesare, D. L. Dumitrascu, A. Francis, S. Gawrieh, M. S. González-Huezo, P. Hillon, A. Iracheta, Z. Kayali, L. Kupcinskas, G. Lau, L. Serfaty, A. Le Cleach, C. Loguercio, M. Manns, B. I. Martinez Saldivar, E. A. Mena, L. A. Morales Garza, J. M. Neutel, L. Nikoleishvili, M. Noureddin, R. Pais, A. H. Paredes, M. Paredes, R. Peters Watkins, A. Picardi, M. Pirisi, G. P. Jofre, L. Preotescu, T. Saadi, D. Samuel, J. F. Sánchez Avila, I. Schiefke, O. Shibolet, M. S. Siddiqui, G. Torres-Mendoza, J. F. Trotter, E. Tsai, E. C. Verna, E. Zuckerman, D. Zur and A. J. Sanyal, Nat. Med., 2021, 27, 1825–1835 CrossRef CAS.
- R. Safadi, F. M. Konikoff, M. Mahamid, S. Zelber-Sagi, M. Halpern, T. Gilat, R. Oren, R. Safadi, F. M. Konikoff, A. Hershkovitz, T. Gilat, M. Halpern, Z. Rosenthal-Galili, E. Zuckerman, S. Abu-Mouch, A. Fich, E. Sikuler, A. Issachar, N. Assy, Y. Baruch, Y. Lurie, M. Graif, N. Stern, M. Yaron, A. Blank, D. Ben Bashat, S. Zelber-Sagi, M. Mahamid, M. Mizrahi and R. Oren, Clin. Gastroenterol. Hepatol., 2014, 12, 2085–2091 CrossRef CAS.
- D. Bhattacharya, B. Basta, J. M. Mato, A. Craig, D. Fernández-Ramos, F. Lopitz-Otsoa, D. Tsvirkun, L. Hayardeny, V. Chandar, R. E. Schwartz, A. Villanueva and S. L. Friedman, JHEP Rep., 2021, 3, 100237 CrossRef.
- C. M. Paton and J. M. Ntambi, Am. J. Physiol.: Endocrinol. Metab., 2009, 297, E28–E37 CrossRef CAS.
- H. Sampath and J. M. Ntambi, Ann. N. Y. Acad. Sci., 2011, 1243, 47–53 CrossRef CAS.
- J. Shen, G. Wu, B. S. Pierce, A.-L. Tsai and M. Zhou, J. Biol. Chem., 2023, 299, 104897 CrossRef CAS.
- L. F. C. Castro, J. M. Wilson, O. Gonçalves, S. Galante-Oliveira, E. Rocha and I. Cunha, BMC Evol. Biol., 2011, 11, 132 CrossRef CAS.
- H. G. Enoch, A. Catalá and P. Strittmatter, J. Biol. Chem., 1976, 251, 5095–5103 CrossRef CAS PubMed.
- Y. Zheng, K. J. Eilertsen, L. Ge, L. Zhang, J. P. Sundberg, S. M. Prouty, K. S. Stenn and S. Parimoo, Nat. Genet., 1999, 23, 268–270 CrossRef CAS PubMed.
- M. Miyazaki, H. Sampath, X. Liu, M. T. Flowers, K. Chu, A. Dobrzyn and J. M. Ntambi, Biochem. Biophys. Res. Commun., 2009, 380, 818–822 CrossRef CAS PubMed.
- M. Minville-Walz, A.-S. Pierre, L. Pichon, S. Bellenger, C. Fèvre, J. Bellenger, C. Tessier, M. Narce and M. Rialland, PLoS One, 2010, 5, e14363 CrossRef CAS.
- H. Sampath, M. T. Flowers, X. Liu, C. M. Paton, R. Sullivan, K. Chu, M. Zhao and J. M. Ntambi, J. Biol. Chem., 2009, 284, 19961–19973 CrossRef CAS.
- H. Sampath and J. M. Ntambi, Curr. Opin. Clin. Nutr. Metab. Care, 2006, 9, 84–88 CrossRef CAS.
- J. M. Ntambi, J. Lipid Res., 1999, 40, 1549–1558 CrossRef CAS.
- J. M. Ntambi, Prog. Lipid Res., 1995, 34, 139–150 CrossRef CAS.
- K. H. Hellemans, J.-C. Hannaert, B. Denys, K. R. Steffensen, C. Raemdonck, G. A. Martens, P. P. Van Veldhoven, J.-Å. Gustafsson and D. Pipeleers, PLoS One, 2009, 4, e7266 CrossRef.
- H. Sampath and J. M. Ntambi, Annu. Rev. Nutr., 2005, 25, 317–340 CrossRef CAS.
- G. Bryzgalova, L. Lundholm, N. Portwood, J.-Å. Gustafsson, A. Khan, S. Efendic and K. Dahlman-Wright, Am. J. Physiol.: Endocrinol. Metab., 2008, 295, E904–E912 CrossRef CAS PubMed.
- S. B. Biddinger, M. Miyazaki, J. Boucher, J. M. Ntambi and C. R. Kahn, Diabetes, 2006, 55, 2032–2041 CrossRef CAS.
- K. Liu, L. Lin, Q. Li, Y. Xue, F. Zheng, G. Wang, C. Zheng, L. Du, M. Hu, Y. Huang, C. Shao, X. Kong, G. Melino, Y. Shi and Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2462–2472 CrossRef CAS.
- Z. Ye, Q. Zhuo, Q. Hu, X. Xu, M. liu, Z. Zhang, W. Xu, W. Liu, G. Fan, Y. Qin, X. Yu and S. Ji, Redox Biol., 2021, 38, 101807 CrossRef CAS.
- E. Piccinin, M. Cariello and A. Moschetta, Mol. Aspects Med., 2021, 78, 100933 CrossRef CAS PubMed.
- A. Noto, S. Raffa, C. De Vitis, G. Roscilli, D. Malpicci, P. Coluccia, A. Di Napoli, A. Ricci, M. R. Giovagnoli, L. Aurisicchio, M. R. Torrisi, G. Ciliberto and R. Mancini, Cell Death Dis., 2013, 4, e947 CrossRef CAS PubMed.
- C. Wang, M. Shi, J. Ji, Q. Cai, Q. Zhao, J. Jiang, J. liu, H. Zhang, Z. Zhu and J. Zhang, Aging, 2020, 12, 15374–15391 CrossRef CAS.
- V. V. Balatskyi and P. Dobrzyn, Int. J. Mol. Sci., 2023, 24, 5531 CrossRef CAS.
- Z. Zhang, S. Sun, V. Kodumuru, D. Hou, S. Liu, N. Chakka, S. Sviridov, S. Chowdhury, D. G. McLaren, L. G. Ratkay, K. Khakh, X. Cheng, H. W. Gschwend, R. Kamboj, J. Fu and M. D. Winther, J. Med. Chem., 2013, 56, 568–583 CrossRef CAS PubMed.
- R. Kamboj, Z. Zhang, J. M. Fu, V. Kodumuru, S. Sviridov, K. Sadalapure, S. Liu, S. Sun, D. Hou, and N. Chakka, Heterocyclic derivatives and their use as therapeutic agents, WO2006/034440A2, 2006.
- R. Kamboj, J. FU, V. Kodumuru, S. Liu, K. Sadalapure, N. Chakka, D. Hou and S. Sun, Bicylic heterocyclic derivatives and their use as Stearoyl-CoA-desaturse (scd), WO2006034312A1, 2006.
- R. Kamboj, Z. Zhang, S. Sviridov, V. Raina, D. Hou, V. Kodumuru and B. M. Seid, Heterocyclic derivatives and their use as stearoyl-CoA desaturase, WO2006034441A1, 2006.
- R. Kamboj, Z. Zaihui, J. Fu, B. M. Seid, S. Sviridov, S. Chowdhury, S. Liu and V. Kodumuru, Heterocyclic derivatives and their use as therapeutic agents, WO2006034279A1, 2006.
- R. Kamboj, Z. Zhang, J. Fu, S. Sviridov, S. Sun, B. M. Seid, B. Raina, D. Hou, S. Chowdhury, S. Liu, V. Kodumuru and N. Chakka, Heterocyclic derivatives and their use as mediators of stearoyl-CoA desaturase, WO2006034338A1, 2006.
- R. Kamboj, Z. Zhang, J. Fu, S. Sviridov, S. Chowdhury and V. Kodumuru, Pyridazine derivatives for inhibiting human stearoyl CoA-desaturase, WO2006034341A3, 2006.
- G. Liu, J. K. Lynch, J. Freeman, B. Liu, Z. Xin, H. Zhao, M. D. Serby, P. R. Kym, T. S. Suhar, H. T. Smith, N. Cao, R. Yang, R. S. Janis, J. A. Krauser, S. P. Cepa, D. W. A. Beno, H. L. Sham, C. A. Collins, T. K. Surowy and H. S. Camp, J. Med. Chem., 2007, 50, 3086–3100 CrossRef CAS.
- H. Zhao, M. D. Serby, H. T. Smith, N. Cao, T. S. Suhar, T. K. Surowy, H. S. Camp, C. A. Collins, H. L. Sham and G. Liu, Bioorg. Med. Chem. Lett., 2007, 17, 3388–3391 CrossRef CAS PubMed.
- Z. Xin, H. Zhao, M. D. Serby, B. Liu, M. Liu, B. G. Szczepankiewicz, L. T. J. Nelson, H. T. Smith, T. S. Suhar, R. S. Janis, N. Cao, H. S. Camp, C. A. Collins, H. L. Sham, T. K. Surowy and G. Liu, Bioorg. Med. Chem. Lett., 2008, 18, 4298–4302 CrossRef CAS PubMed.
- Y. Uto, T. Ogata, Y. Kiyotsuka, Y. Ueno, Y. Miyazawa, H. Kurata, T. Deguchi, N. Watanabe, M. Konishi, R. Okuyama, N. Kurikawa, T. Takagi, S. Wakimoto and J. Ohsumi, Bioorg. Med. Chem. Lett., 2010, 20, 341–345 CrossRef CAS PubMed.
- Y. Uto, Y. Kiyotsuka, Y. Ueno, Y. Miyazawa, H. Kurata, T. Ogata, T. Deguchi, M. Yamada, N. Watanabe, M. Konishi, N. Kurikawa, T. Takagi, S. Wakimoto, K. Kono and J. Ohsumi, Bioorg. Med. Chem. Lett., 2010, 20, 746–754 CrossRef CAS PubMed.
- A. Thomas, V. S. Prasada Rao Lincam, S. K. Phatangare, J. N. Khairatkar, D. M. Shah, D. V. Ukirde, and D. A. More, Acetylene derivatives as stearoyl Co-A desaturase inhibitors, WO2008062276A2, 2008.
- S. S. Chaudhari, A. Thomas, G. B. Gudade, A. Kadam, N. P. Patil, N. Khairatkar-Joshi, D. M. Shah, Spirocyclic compounds as stearoyl Co-A desaturase inhibitors, WO2009037542A2, 2009.
- A. Bischoff, S. Subramanya, S. Kumar, S. Srinivasa, and A. Kumar, Novel piperazine derivatives as inhibitors of stearoyl-Co-A desaturase, WO2008157844A1, 2008.
- A. Bischoff, S. N. Raikar, S. R. Sammeta, G. Prabhu, H. Subramanya, and S. Kumar, Novel piperazine derivatives as inhibitors of stearoyl-Co-A desaturase, WO2009117659A1, 2009.
- A. Bischoff, S. Kumar, A. P. Bala Koteswara, B. Ainan, H. Ayyamperumal, A. R. Girish, S. Tatiparthy, G. Prabhu, and H. Subramanya, Novel piperidine derivatives as inhibitors of stearoyl-Co-A desaturase, WO2009117676A2, 2009.
- A. Bischoff, H. Subramanya, S. Kumar, S. R. Sammeta, and A. K. Vaka, Novel piperazine derivatives as inhibitors of stearoyl-Co-A desaturase, US Pat., US2010160323A1, 2010.
- C. A. von Roemeling, T. R. Caulfield, L. Marlow, I. Bok, J. Wen, J. L. Miller, R. Hughes, L. Hazlehurst, A. B. Pinkerton, D. C. Radisky, H. W. Tun, Y. S. B. Kim, A. L. Lane and J. A. Copland, Oncotarget, 2018, 9, 3–20 CrossRef.
- K. O. Matthew, B. Pandya, D. Tardiff, P. Tivitmahaisoon and I. Wrona, Compounds and uses thereof, WO2020198026A1, 2020.
- K. Imamura, N. Tomita, Y. Kawakita, Y. Ito, K. Ono, N. Nii, T. Miyazaki, K. Yonemori, M. Tawada, H. Sumi, Y. Satoh, Y. Yamamoto, I. Miyahisa, M. Sasaki, Y. Satomi, M. Hirayama, R. Nishigaki and H. Maezaki, Bioorg. Med. Chem., 2017, 25, 3768–3779 CrossRef CAS.
- S. Nishizawa, H. Sumi, Y. Satoh, Y. Yamamoto, S. Kitazawa, K. Honda, H. Araki, K. Kakoi, K. Imamura, M. Sasaki, I. Miyahisa, Y. Satomi, R. Nishigaki, M. Hirayama, K. Aoyama, H. Maezaki and T. Hara, Eur. J. Pharmacol., 2017, 807, 21–31 CrossRef CAS PubMed.
- S.-M. Yang, Y. Tang, T. Rano, H. Lu, G.-H. Kuo, M. D. Gaul, Y. Li, G. Ho, W. Lang, J. G. Conway, Y. Liang, J. M. Lenhard, K. T. Demarest and W. V. Murray, Bioorg. Med. Chem. Lett., 2014, 24, 1437–1441 CrossRef CAS PubMed.
- N. Kurikawa, T. Takagi, S. Wakimoto, Y. Uto, H. Terashima, K. Kono, T. Ogata and J. Ohsumi, Biol. Pharm. Bull., 2013, 36, 259–267 CrossRef CAS.
- H. W. Gschwend, V. Kodumuru, S. Liu, and R. Kamboj, Pyridazine Derivatives and Their Use as Inhibitors of Stearoyl-CoA Desaturase-1 Activity in a Mammal, WO2005011653A2, 2005.
- D. Powell and G. K. Tranmer, Azetidine derivatives as inhibitors of stearoyl-coenzyme-A delta-9 desaturase, WO2010043052A1, 2010.
- E. Dupont-Passelaigue, S. Mialhe, R. Jean-Pierre, J. Didier, and K. Valeille, Derivatives of 2H pyridazin-3-ones, their preparation and their use as SCD1 inhibitors, WO2011015629A1, 2011.
- K. A. Atkinson, E. E. Beretta, J. A. Brown, M. Castrodad, Y. Chen, J. M. Cosgrove, P. Du, J. Litchfield, M. Makowski, K. Martin, T. J. McLellan, C. Neagu, D. A. Perry, D. W. Piotrowski, C. M. Steppan and R. Trilles, Bioorg. Med. Chem. Lett., 2011, 21, 1621–1625 CrossRef CAS PubMed.
- J. Fu, S. Sun, S. Serguei, Z. Zhang, D. Hou, R. Kamboj, V. Kodumuru, N. Pokrovskaia, and V. Raina, Aminothiazole derivatives as human stearoyl-Co-A desaturase inhibitors, WO2007130075A1, 2007.
- N. Davis and Z. Zhang, 2- (pyrazin-2-yl) -thiazole and 2-(1H-pyrazol-3-yl)-thiazole derivatives as well as related compounds as stearoyl-coa desaturase (SCD) inhibitors for the treatment of metabolic, cardiovascular and other disorders, WO2008/024390A2, 2008.
- N. Dales, Z. Zhang, J. Fonarev, J. Fu, R. Kamboj, V. Kodumuru, N. Pokrovskaia and S. Sun, 2-Substituted 5-membered heterocycles as SCD inhibitors, WO2008/074835A1, 2008.
- N. Dales, Z. Zhang, R. Kamboj, J. Fu, S. Sun, N. Pokrovskaia, and S. Sviridov, Heterocyclic organic compounds, WO2008036715A1, 2008.
- S. Chowdhury, N. Dales, J. Fonarev, J. Fu, D. Hou, Q. Jia, V. Kodumuru, N. Pokrovskaia, S. Sun and Z. Zhang, Heterocyclic inhibitors of stearoyl-Co-A desaturase, WO2009103739A1, 2009.
- N. Dales, J. Fonarev, J. Fu, R. Kamboj, V. Kodumuru, S. Liu, N. Pokrovskaia, V. Raina, S. Sun, Z. Zhang, Heterocyclic compounds suitable for the treatment of diseases related to elevated lipid level, WO2008127349A2, 2008.
- C. S. Li, L. Belair, J. Guay, R. Murgasva, W. Sturkenboom, Y. K. Ramtohul, L. Zhang and Z. Huang, Bioorg. Med. Chem. Lett., 2009, 19, 5214–5217 CrossRef CAS.
- Y. K. Ramtohul, C. Black, C.-C. Chan, S. Crane, J. Guay, S. Guiral, Z. Huang, R. Oballa, L.-J. Xu, L. Zhang and C. S. Li, Bioorg. Med. Chem. Lett., 2010, 20, 1593–1597 CrossRef CAS PubMed.
- C. S. Li, L. Belair, J. Guay, R. Murgasva, W. Sturkenboom, Y. K. Ramtohul, L. Zhang and Z. Huang, Bioorg. Med. Chem. Lett., 2009, 19, 5214–5217 CrossRef CAS.
- A. Thomas, S. K. Phatangare, D. M. Shah, D. V. Ukirde, D. A. More, Acetylene derivatives as stearoyl CoA desaturase inhibitors, CN101583600A, 2019.
- S. Sun, Z. Zhang, V. Kodumuru, N. Pokrovskaia, J. Fonarev, Q. Jia, P.-Y. Leung, J. Tran, L. G. Ratkay, D. G. McLaren, C. Radomski, S. Chowdhury, J. Fu, B. Hubbard, M. D. Winther and N. A. Dales, Bioorg. Med. Chem. Lett., 2014, 24, 520–525 CrossRef CAS PubMed.
- S. Sun, Z. Zhang, N. Pokrovskaia, S. Chowdhury, Q. Jia, E. Chang, K. Khakh, R. Kwan, D. G. McLaren, C. C. Radomski, L. G. Ratkay, J. Fu, N. A. Dales and M. D. Winther, Bioorg. Med. Chem., 2015, 23, 455–465 CrossRef CAS.
- S. Sun, Z. Zhang, V. Raina, N. Pokrovskaia, D. Hou, R. Namdari, K. Khakh, L. G. Ratkay, D. G. McLaren, M. Mork, J. Fu, S. Ferreira, B. Hubbard, M. D. Winther and N. Dales, Bioorg. Med. Chem. Lett., 2014, 24, 526–531 CrossRef CAS.
- J. Li, and K. N. Lee, Phenoxy-Pyrrolidine Derivative and Its Use and Compositions, US Pat., US8242286B2, 2018.
- J. Li and K. N. Lee, Preparation of (S)-2-[4-(hydroxymethyl)phenoxy)-1-[3-[2-(trifluoromethyl)phenoxy]pyrrolidin-1-yl]ethanone as a stearoyl-CoA desaturase (SCD1) inhibitor, WO2009019566, 2009.
- M. D. Voss, G. Zoller, H. Matter, A. W. Herling, G. Biemer-Daub, A. Pfenninger, S. Haag-Diergarten, S. Keil, M. Kohlmann and H.-L. Schmidts, Eur. J. Pharmacol., 2013, 707, 140–146 CrossRef CAS.
- G. Liu, J. K. Lynch, J. Freeman, B. Liu, Z. Xin, H. Zhao, M. D. Serby, P. R. Kym, T. S. Suhar, H. T. Smith, N. Cao, R. Yang, R. S. Janis, J. A. Krauser, S. P. Cepa, D. W. A. Beno, H. L. Sham, C. A. Collins, T. K. Surowy and H. S. Camp, J. Med. Chem., 2007, 50, 3086–3100 CrossRef CAS.
- Z. Xin, H. Zhao, M. D. Serby, B. Liu, M. Liu, B. G. Szczepankiewicz, L. T. J. Nelson, H. T. Smith, T. S. Suhar, R. S. Janis, N. Cao, H. S. Camp, C. A. Collins, H. L. Sham, T. K. Surowy and G. Liu, Bioorg. Med. Chem. Lett., 2008, 18, 4298–4302 CrossRef CAS.
- C. Mcmartin and R. S. Bohacek, J. Comput.-Aided Mol. Des., 1997, 11, 333–344 CrossRef CAS.
- G. Zoller, M. Dietrich Voss, S. Keil, A. Herling and H. Matter, 2 -heteroaryl- pyrrolo-[3, 4-c]- pyrrole derivatives and their use as SCD inhibitors, WO2008135141A1, 2008.
- G. Zoller, M. D. Voss, H. Matter and A. Herling, 2-heteroaryl-pyrrolo-[3, 4-c]-pyrrole derivatives and the use thereof as SCD inhibitors, WO2010/028761A1, 2010.
- H. Matter, G. Zoller, A. W. Herling, J.-A. Sanchez-Arias, C. Philippo, C. Namane, M. Kohlmann, A. Pfenninger and M. D. Voss, Bioorg. Med. Chem. Lett., 2013, 23, 1817–1822 CrossRef CAS.
- V. C. Joshi, A. C. Wilson and S. J. Wakil, J. Lipid Res., 1977, 18, 32–36 CrossRef CAS.
- D. A. Powell, Y. Ramtohul, M.-E. Lebrun, R. Oballa, S. Bhat, J.-P. Falgueyret, S. Guiral, Z. Huang, K. Skorey, P. Tawa and L. Zhang, Bioorg. Med. Chem. Lett., 2010, 20, 6366–6369 CrossRef CAS PubMed.
- W. Wang, Y. Kong, X. Wang, Z. Wang, C. Tang, J. Li, Q. Yang, Y. Q. Chen and S. Zhu, Cell Commun. Signaling, 2023, 21, 268 CrossRef CAS.
- D. A. Claude-marie, A. Dean, W. Fillmore, and C. Martin, N-thiazolyl-1, 2, 3, 4-tetrahydro-6-isoquinolinecarboxamide derivatives as inhibitors of stearoyl coenzyme-A desaturase, WO2009150196A1, 2009.
- K. Dmitry, J. Zablocki, and E. Parkhill, Pyrrolotriazinone derivatives for use as stearoyl Co-A desaturase inhibitors, WO2009124259A1, 2009.
- K. Dmitry and Z. Jeff, Triazolopyridinone derivatives for use as stearoyl Co-A desaturase inhibitors, US Pat., US20090253738A1, 2009.
- Y. Deng, Z. Yang, G. W. Shipps, S.-M. Lo, R. West, J. Hwa, S. Zheng, C. Farley, J. Lachowicz, M. van Heek, A. S. Bass, D. P. Sinha, C. R. Mahon and M. E. Cartwright, Bioorg. Med. Chem. Lett., 2013, 23, 791–796 CrossRef CAS PubMed.
- N. Dales, J. Fonarev, J. Fu, and Z. Zhang, Spiro derivatives for the modulation of stearoyl-Co-A desaturase, US Pat., US20120010135A1, 2013.
- J. W. G. Shipps, Z. Yang, and R. West, Spirocyclic oxazepine compounds as stearoyl-Co-A delta-9 desaturase inhibitors, WO2011/011506A1, 2011.
- R. M. Oballa, L. Belair, W. C. Black, K. Bleasby, C. C. Chan, C. Desroches, X. Du, R. Gordon, J. Guay, S. Guiral, M. J. Hafey, E. Hamelin, Z. Huang, B. Kennedy, N. Lachance, F. Landry, C. S. Li, J. Mancini, D. Normandin, A. Pocai, D. A. Powell, Y. K. Ramtohul, K. Skorey, D. Sørensen, W. Sturkenboom, A. Styhler, D. M. Waddleton, H. Wang, S. Wong, L. Xu and L. Zhang, J. Med. Chem., 2011, 54, 5082–5096 CrossRef CAS.
- M. Niemi, Pharmacogenomics, 2007, 8, 787–802 CrossRef CAS.
- S. Léger, W. C. Black, D. Deschenes, S. Dolman, J.-P. Falgueyret, M. Gagnon, S. Guiral, Z. Huang, J. Guay, Y. Leblanc, C.-S. Li, F. Massé, R. Oballa and L. Zhang, Bioorg. Med. Chem. Lett., 2010, 20, 499–502 CrossRef.
- G. Liu, J. K. Lynch, J. Freeman, B. Liu, Z. Xin, H. Zhao, M. D. Serby, P. R. Kym, T. S. Suhar, H. T. Smith, N. Cao, R. Yang, R. S. Janis, J. A. Krauser, S. P. Cepa, D. W. A. Beno, H. L. Sham, C. A. Collins, T. K. Surowy and H. S. Camp, J. Med. Chem., 2007, 50, 3086–3100 CrossRef CAS.
- D. Powell, M. Lebrun, S. Bhat and Y. K. Ramtohul, Novel substituted heteroaromatic compounds as inhibitors of stearoyl-coenzyme-A delta-9 desaturase, WO2009/129625, 2009.
- N. Lachance, Y. Gareau, S. Guiral, Z. Huang, E. Isabel, J.-P. Leclerc, S. Léger, E. Martins, C. Nadeau, R. M. Oballa, S. G. Ouellet, D. A. Powell, Y. K. Ramtohul, G. K. Tranmer, T. Trinh, H. Wang and L. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 980–984 CrossRef CAS.
- N. Lachance, S. Guiral, Z. Huang, J.-P. Leclerc, C. S. Li, R. M. Oballa, Y. K. Ramtohul, H. Wang, J. Wu and L. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 623–627 CrossRef CAS.
- T. Iida, M. Ubukata, I. Mitani, Y. Nakagawa, K. Maeda, H. Imai, Y. Ogoshi, T. Hotta, S. Sakata, R. Sano, H. Morinaga, T. Negoro, S. Oshida, M. Tanaka and T. Inaba, Eur. J. Med. Chem., 2018, 158, 832–852 CrossRef CAS PubMed.
- N. Lachance, S. Guiral, Z. Huang, J.-P. Leclerc, C. S. Li, R. M. Oballa, Y. K. Ramtohul, H. Wang, J. Wu and L. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 623–627 CrossRef CAS PubMed.
- Y. Deng, Z. Yang, G. W. Shipps, S.-M. Lo, R. West, J. Hwa, S. Zheng, C. Farley, J. Lachowicz, M. van Heek, A. S. Bass, D. P. Sinha, C. R. Mahon and M. E. Cartwright, Bioorg. Med. Chem. Lett., 2013, 23, 791–796 CrossRef CAS PubMed.
- L. J. Philippe, C. S. Li, and O. M. Moradei, Novel spiro compounds useful as inhibitors of stearoyl-coenzyme-A delta-9 desaturase, WO2011011872A1, 2011.
- F. Rejean, N. Lachance, C. S. Li, and G. Tranmer, Novel spiro compounds useful as inhibitors of stearoyl-coenzyme-A delta-9 desaturase, WO2011047481A1, 2011.
- X. Zhu, H. Bian, L. Wang, X. Sun, X. Xu, H. Yan, M. Xia, X. Chang, Y. Lu, Y. Li, P. Xia, X. Li and X. Gao, Free Radical Biol. Med., 2019, 141, 192–204 CrossRef CAS.
- L. C. Ortinau, R. T. Pickering, K. J. Nickelson, K. L. Stromsdorfer, C. Y. Naik, R. A. Haynes, D. E. Bauman, R. S. Rector, K. L. Fritsche and J. W. Perfield, ISRN Endocrinol., 2012, 2012, 1–11 CrossRef.
- F. Enrique Gomez, D. E. Bauman, J. M. Ntambi and B. G. Fox, Biochem. Biophys. Res. Commun., 2003, 300, 316–326 CrossRef CAS.
- C. A. Major, K. Ryan, A. J. Bennett, A. L. Lock, D. E. Bauman and A. M. Salter, J. Lipid Res., 2008, 49, 1456–1465 CrossRef CAS.
- M. P. Dallaire, H. Taga, L. Ma, B. A. Corl, R. Gervais, Y. Lebeuf, F. J. Richard and P. Y. Chouinard, J. Dairy Sci., 2014, 97, 6411–6425 CrossRef CAS.
- Y. Dong, H. Lu, Q. Li, X. Qi, Y. Li, Z. Zhang, J. Chen and J. Ren, Life Sci., 2019, 232, 116644 CrossRef CAS PubMed.
- F. Pédrono, N. Boulier-Monthéan, F. Boissel, J. Ossemond and F. Lohézic-Le Dévéhat, Mol. Nutr. Food Res., 2017, 4, 1700567 Search PubMed.
- Y. Endo, Y. Osada, F. Kimura and K. Fujimoto, Nutrition, 2006, 22, 553–558 CrossRef CAS.
- Y. Endo, Y. Osada, F. Kimura, H. Shirakawa and K. Fujimoto, Biosci., Biotechnol., Biochem., 2007, 71, 231–233 CrossRef CAS PubMed.
- Y. Endo, K. Tsunokake and I. Ikeda, Biosci., Biotechnol., Biochem., 2009, 73, 577–581 CrossRef CAS.
- N. Shinohara, J. Ito, T. Tsuduki, T. Honma, R. Kijima, S. Sugawara, T. Arai, M. Yamasaki, A. Ikezaki, M. Yokoyama, K. Nishiyama, K. Nakagawa, T. Miyazawa and I. Ikeda, J. Oleo Sci., 2012, 61, 433–441 CrossRef CAS PubMed.
- R. Taebi, M. R. Mirzaiey, M. Mahmoodi, A. Khoshdel, M. A. Fahmidehkar, M. Mohammad-Sadeghipour and M. R. Hajizadeh, Gene Rep., 2020, 18, 100581 CrossRef.
- N. Lu, X. Li, J. Yu, Y. Li, C. Wang, L. Zhang, T. Wang and X. Zhong, Lipids, 2018, 53, 53–63 CrossRef CAS PubMed.
- Y. Bai, T. Zhang, Z. Hu, Y. Zhang, D. Wang, M. Zhou, Y. Zhang, F. Zhang and X. Kong, Biochem. Pharmacol., 2024, 224, 116240 CrossRef CAS.
- Y. Hai, L. Zuo, M. Wang, R. Zhang, M. Wang, L. Ren, C. Yang and J. Wang, Molecules, 2023, 28, 517 CrossRef CAS PubMed.
- F. Li, C. Gao, P. Yan, M. Zhang, Y. Wang, Y. Hu, X. Wu, X. Wang and J. Sheng, Front. Pharmacol, 2018, 9, 1366 CrossRef CAS.
- H. Mu, Q. Zhou, R. Yang, J. Zeng, X. Li, R. Zhang, W. Tang, H. Li, S. Wang, T. Shen, X. Huang, L. Dou and J. Dong, Front. Microbiol., 2020, 11, 585066 CrossRef.
- C. Yang, Y.-Y. Jin, J. Mei, D. Hu, X. Jiao, H.-L. Che, C.-L. Tang, Y. Zhang and G.-S. Wu, Cancer Cell Int., 2022, 22, 202 CrossRef CAS.
- M. Miyata, A. Funaki, C. Fukuhara, Y. Sumiya and Y. Sugiura, J. Toxicol. Sci., 2020, 45, 87–94 CrossRef CAS.
- S.-M. Yang, Y. Tang, R. Zhang, H. Lu, G.-H. Kuo, M. D. Gaul, Y. Li, G. Ho, J. G. Conway, Y. Liang, J. M. Lenhard, K. T. Demarest and W. V. Murray, Bioorg. Med. Chem. Lett., 2013, 23, 6773–6776 CrossRef CAS PubMed.
- W. Guosheng, Y. Chen, J. Yiyuan, T. Chunlei, C. Yongquan and C. Guozhen, Medicine for inhibiting stearoyl- Co-A desaturase1, CN113057955A, 2021.
- D. A. Bullough, J. D. Roth and J. G. Foulkes, Methods of treating non-alcoholic steatohepatitis with a SCD-1 inhibitor, WO2022178261A1, 2022.
| This journal is © The Royal Society of Chemistry 2024 |