
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring heterocyclic scaffolds in carbonic anhydrase inhibition: a decade of structural and therapeutic insights
Nafeesa Naeem a,
Amina Sadiqb,
Gehan Ahmed Othmanc,
Habab M. Yassinc and
Ehsan Ullah Mughal
*a
a,
Amina Sadiqb,
Gehan Ahmed Othmanc,
Habab M. Yassinc and
Ehsan Ullah Mughal
*a
aDepartment of Chemistry, University of Gujrat, Gujrat 50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
bDepartment of Chemistry, Govt. College Women University, Sialkot 51300, Pakistan
cBiology Department, College of Science, King Khalid University, Abha, 61421, Saudi Arabia
First published on 12th November 2024
Abstract
Heterocyclic compounds represent a prominent class of molecules with diverse pharmacological activities. Among their therapeutic applications, they have gained significant attention as carbonic anhydrase (CA) inhibitors, owing to their potential in the treatment of various diseases such as epilepsy, cancer and glaucoma. CA is a widely distributed zinc metalloenzyme that facilitates the reversible interconversion of carbon dioxide and bicarbonate. This reaction is essential for numerous physiological and pathological processes. In humans, CA exists in sixteen different isoforms, labeled hCA-I to hCA-XV, each distributed across various tissues and organs and involved in crucial physiological functions. Clinically utilized CA inhibitors, such as brinzolamide, dorzolamide and acetazolamide, exhibit poor selectivity, leading to undesirable side effects. A significant challenge in designing effective CA inhibitors is achieving balanced isoform selectivity, prompting the exploration of new chemotypes. This review compiles recent strategies employed by various researchers in developing CAIs across different structural classes, including pyrazoline, quinoline, imidazole, oxadiazole, pyrimidine, coumarin, chalcone, rhodanine, phthalazine, triazole, isatin, and indole. Additionally, the review summarizes structure–activity relationship (SAR) analyses, isoform selectivity evaluations, along with mechanistic and in silico investigations. Insights derived from SAR studies provide crucial directions for the rational design of next-generation heterocyclic CA inhibitors, with improved therapeutic efficacy and reduced side effects. To the best of our knowledge, for the first time, we have comprehensively summarized all known isoforms of CA in relation to various heterocyclic motifs. This review examines the use of different heterocycles as CA inhibitors, drawing on research published over the past 11 years. It offers a valuable resource for early-career researchers, encouraging further exploration of synthetic heterocycles in the development of CA inhibitors.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal obtained his PhD degree in Organic Chemistry from Bielefeld University, Germany in 2013. Currently, he is working as Tenured Associate Professor in the Department of Chemistry, University of Gujrat, Pakistan. He was a post-doc fellow at Max-Planck Institute for Polymer Research, Mainz, Germany from 2013 to 2014. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metal-based terpyridine complexes and their uses in the fabrication of DSSCs and as efficient photocatalysts. His research in the field has resulted in more than 97 published articles (research papers and review papers) in reputable high impact journals. His publications have made a great impact in the field by receiving 2059 citations with h-index of 28. |
1. Introduction
Heterocycles represent a class of molecules present in numerous biological compounds within the human body, characterized by the inclusion of heteroatoms such as sulfur, nitrogen and oxygen in a ring structure.1 Their natural occurrence, intricate structure, and propensity for hydrogen bonding render heterocycles highly coveted for their potential application as pharmaceutical agents.2 Medicinal chemists strategically design drug molecules that mimic the structure of endogenous heterocycles, aiming to either enhance or inhibit their physiological functions.3,4 Recent advancements in synthetic methodologies have facilitated the creation of diverse heterocyclic scaffolds, thereby streamlining the process of drug discovery.5,6 These compounds have attained considerable attention in recent years owing to their ability to interact with enzymes and modulate their activity.7,8 Enzymes, vital biological molecules that catalyze various chemical reactions, play pivotal roles in cellular metabolism and signaling pathways, rendering them attractive targets for drug intervention.9–11 Heterocyclic compounds can effectively engage with enzymes by binding to their active sites, consequently altering their activity and exerting inhibitory effects.12 Particularly, the exploration of heterocyclic compounds as inhibitors of CA activity represents a promising avenue for therapeutic intervention.Carbonic anhydrases (CAs, EC 4.2.1.1) represent a widely distributed superfamily of zinc-metalloenzymes.13 Their primary function involves catalyzing the reversible hydration of carbon dioxide to bicarbonate and a proton (CO2 + H2O ↔ HCO3− + H+).14,15 This catalytic activity is integral to numerous biochemical and physiological processes across both prokaryotic and eukaryotic organisms.16–18 They are involved in various physiological processes such as pH regulation, electrolyte transport, bone resorption, and biosynthetic reactions.19,20 The varied functions of CA render them appealing therapeutic targets for numerous diseases, including cancer, epilepsy, obesity, glaucoma and several neurological disorders.21–24 Furthermore, CA isoenzymes emerge as promising targets for Alzheimer's disease (AD) treatment.25 These enzymes are extensively distributed throughout nature and are currently categorized into eight genetically unrelated families: the α-, β-, γ-, δ-, η-, ζ-, θ-, and ι-classes.26,27 To date, nature has revealed eight genetically distinct CA families (a–i).28 Among these, only the alpha class of CA (a-CAs) are exclusively found in higher vertebrates.29 In Homo sapiens, only fifteen isoforms of have been identified, each differing in catalytic activity, tissue/subcellular localization, and physiological role.30,31 In the lungs, CA in red blood cells catalyzes the conversion of HCO3− back into CO2, which is then exhaled. In the human enzyme, zinc serves as an essential metal ion component.32 The zinc ion is coordinated by three nitrogen atoms from three histidine residues within the protein33 (Fig. 1).
 | ||
| Fig. 1 The role of CA in erythrocytes involves facilitating the hydration of CO2 in tissues and the dehydration of HCO3− in erythrocytes within the lungs. | ||
The human alpha CAs comprise 15 distinct isoforms (I–XIV), with 12 demonstrating catalytic activity (Fig. 2).34 These isoforms differ in their catalytic activity, cellular localization, and distribution throughout various organs and tissues. Specifically, hCA-VIII, X, and XI are classified as non-catalytically active CA-related proteins.35 CAs are categorized into four groups based on their localization: cytosolic isoforms (hCAs-I–III, VII, and XIII), transmembrane isoforms (hCAs-IV, IX, XII, and XIV), mitochondrial isoforms (hCAs-VA and VB), and the secreted isoenzyme hCA-VI, found in saliva and milk. Dysregulation of carbonic anhydrase isoforms is associated with various diseases, such as glaucoma (hCAs-I, II, IV, and XII), epilepsy (hCAs-VII and XIV), cerebral and retinal edema (hCAs-I and II), and different cancers (hCAs-IX and XII). Among the α-CA superfamily, CA-IX and CA-XII are notably significant due to their roles in the survival of hypoxic tumors.36,37 These cancer-associated CAs are normally expressed at low levels in healthy tissues but are often overexpressed in a range of hypoxic solid tumors.38–41 Such overexpression patterns are often attributed to the robust transcriptional activation of the hypoxia-inducible transcription factor 1 (HIF1), through which they exert influence over critical processes like cell proliferation, malignant cell invasion and adhesion.42 The Table 1 outlines the distribution of various α-CAs across different organs/tissues and their involvement in various diseases.43,44
 | ||
| Fig. 2 Various isoforms of human carbonic anhydrase. | ||
| CA type | Tissue/organ distribution | Associated diseases |
|---|---|---|
| CA-I | Erythrocytes, kidneys, liver, muscle, and brain | Glaucoma, epilepsy, osteoporosis |
| CA-II | Widespread distribution, especially in erythrocytes, kidneys, and eyes | Osteopetrosis, renal tubular acidosis, retinitis pigmentosa |
| CA-III | Skeletal muscle, liver, adipose tissue, and heart | Obesity, insulin resistance |
| CA-IV | Plasma membrane of various tissues including erythrocytes, kidney, and eye | Renal tubular acidosis, retinitis pigmentosa |
| CA-VA | Mitochondria of liver and adipose tissue | Obesity, metabolic disorders |
| CA-VB | Mitochondria of liver and adipose tissue | Obesity, metabolic disorders |
| CA-VI | Saliva, milk, and cerebrospinal fluid | Dental caries, Sjögren's syndrome |
| CA-VII | Brain, pancreas, and testes | Epilepsy, ataxia |
| CA-IX | Tumor cells, especially in hypoxic conditions | Cancer progression, metastasis |
| CA-XII | Plasma membrane of various tissues including kidney, colon, and pancreas | Cancer progression, metastasis |
| CA-XIII | Brain, gut, kidney, lung, reproductive tract | Sterility |
| CA-XIV | Heart, skeletal muscle, and brain | Epilepsy, retinopathies |
Over the years, extensive research has been conducted to develop small molecule inhibitors targeting CAs, with the aim of modulating their activity for therapeutic benefit. Among the various classes of CA inhibitors (CAIs), heterocyclic scaffolds have emerged as particularly promising candidates due to their structural diversity, synthetic accessibility, and ability to interact with the active site of CAs with high affinity and specificity.33,45
This systematic review aims to provide a comprehensive overview of the recent advancements in the design and development of heterocyclic scaffold-based CAIs for drug discovery. We will explore the diverse array of heterocyclic scaffolds employed, their structural features, synthetic strategies, and their inhibitory activities against different isoforms of CAs. Furthermore, we will discuss the structure–activity relationships (SAR) governing the interaction between heterocyclic scaffold-based CAIs and their target enzymes, highlighting key molecular determinants for potency, selectivity, and pharmacokinetic properties.
The significance of this review lies in its potential to offer insights into the design principles for developing novel CAIs with improved therapeutic efficacy and reduced off-target effects. By examining the latest advancements and trends in the field, we aim to provide researchers and medicinal chemists with valuable guidance for the rational design of heterocyclic scaffold-based CAIs, ultimately facilitating the discovery of next-generation therapeutics for various diseases associated with dysregulated CA activity.
Additionally, we will discuss the therapeutic potential of heterocyclic scaffold-based CAIs in the context of different disease indications, along with the challenges and future perspectives in this exciting area of drug discovery.
2. hCA structure and mechanism/structural overview of carbonic anhydrases
X-ray crystallographic studies have offered valuable insights into the three-dimensional structures of catalytically active hCAs, aiding in the understanding of the active site architecture, amino acid residues, and ligand interactions crucial for drug development.38,46,47 However, it's worth noting that the hCA-VA and VB are murine enzymes and hence are exceptions to these studies.48The catalytic activity of hCAs relies on the presence of the essential zinc ion (Zn2+), as corroborated by numerous investigations. The metal ion is located approximately 15 Å deep within the active site cleft and is coordinated by three conserved histidine residues: His94, His96, and His119, along with a water molecule or hydroxide ion. The Zn2+-bound water molecule or hydroxide forms hydrogen bonds with nearby amino acids, including the conserved threonine residue Thr199 and the glutamic acid residue Glu106, within the binding pocket. This interaction enhances the nucleophilicity of the Zn2+-bound water molecule (Fig. 3).49,50 This results in the enzyme adopting a Zn2+-bound water molecule or hydroxide state, representing the catalytically inactive or resting state, from which the enzyme can initiate another catalytic cycle by attacking a new CO2 molecule.51–53
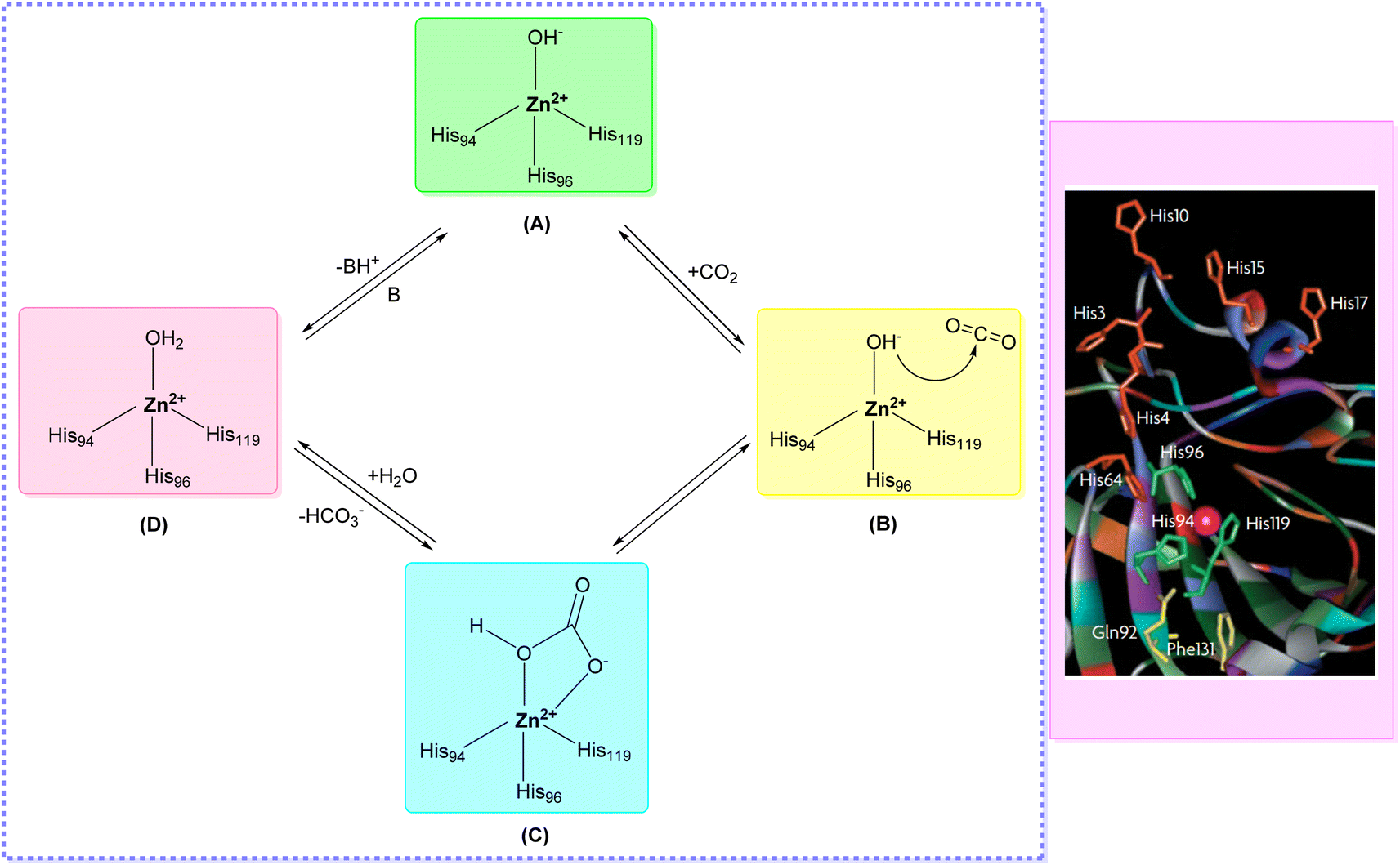 | ||
| Fig. 3 Catalytic mechanism of hCAs entails the reversible hydration and dehydration reactions (A–D) between CO2 with HCO3−. | ||
3. Importance of carbonic anhydrase inhibition
3.1. Therapeutic applications of CAIs
CAIs have demonstrated significant therapeutic potential in treating a wide range of diseases (Fig. 4), including: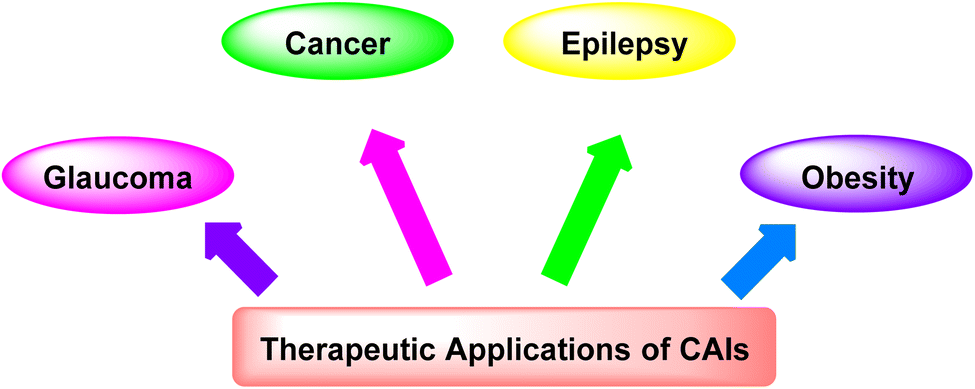 | ||
| Fig. 4 Therapeutic applications of CAIs. | ||
(i) Glaucoma: CAIs reduce intraocular pressure by inhibiting CA in the ciliary processes of the eye, thereby decreasing the production of aqueous humor and alleviating symptoms associated with glaucoma.54
(ii) Cancer: CAIs have emerged as promising agents for cancer therapy due to their ability to disrupt tumor pH regulation, inhibit tumor cell proliferation, and enhance the effectiveness of chemotherapeutic agents.55
(iii) Epilepsy: CAIs have been explored as adjunctive therapy for epilepsy, as they modulate neuronal excitability by altering pH levels and bicarbonate concentrations in the brain, potentially reducing the frequency and severity of seizures.56
(iv) Obesity: CAIs have shown potential in managing obesity by targeting CA isoforms involved in lipid metabolism and appetite regulation, offering a novel approach for weight management.57,58
3.2. Challenges and limitations associated with current CAIs
Despite their therapeutic promise, current CAIs face several challenges and limitations, including:(i) Side effects: some CAIs can cause adverse effects such as metabolic acidosis, electrolyte imbalances, renal dysfunction, and ocular irritation, limiting their clinical utility and patient compliance.59
(ii) Isoform selectivity: achieving selectivity for specific CA isoforms is challenging, as many CAIs exhibit broad-spectrum activity, leading to off-target effects and potential toxicity.60
(iii) Blood–brain barrier penetration: limited penetration of CAIs across the blood–brain barrier hinders their efficacy in neurological disorders such as epilepsy, where precise targeting within the central nervous system is essential.61
(iv) Resistance development: prolonged use of CAIs can lead to the development of drug resistance in certain disease contexts, necessitating the exploration of alternative treatment strategies and novel CAIs with improved efficacy profiles.62
Addressing these challenges through innovative drug design strategies, such as enhancing isoform selectivity, improving pharmacokinetic properties, and minimizing off-target effects, holds promise for advancing the clinical utility of CAIs in the treatment of various diseases.
4. Classification of carbonic anhydrase inhibitors based on various diseases
4.1. CAIs as diuretics
CAIs have been utilized as diuretics due to their ability to modulate renal physiology and fluid balance.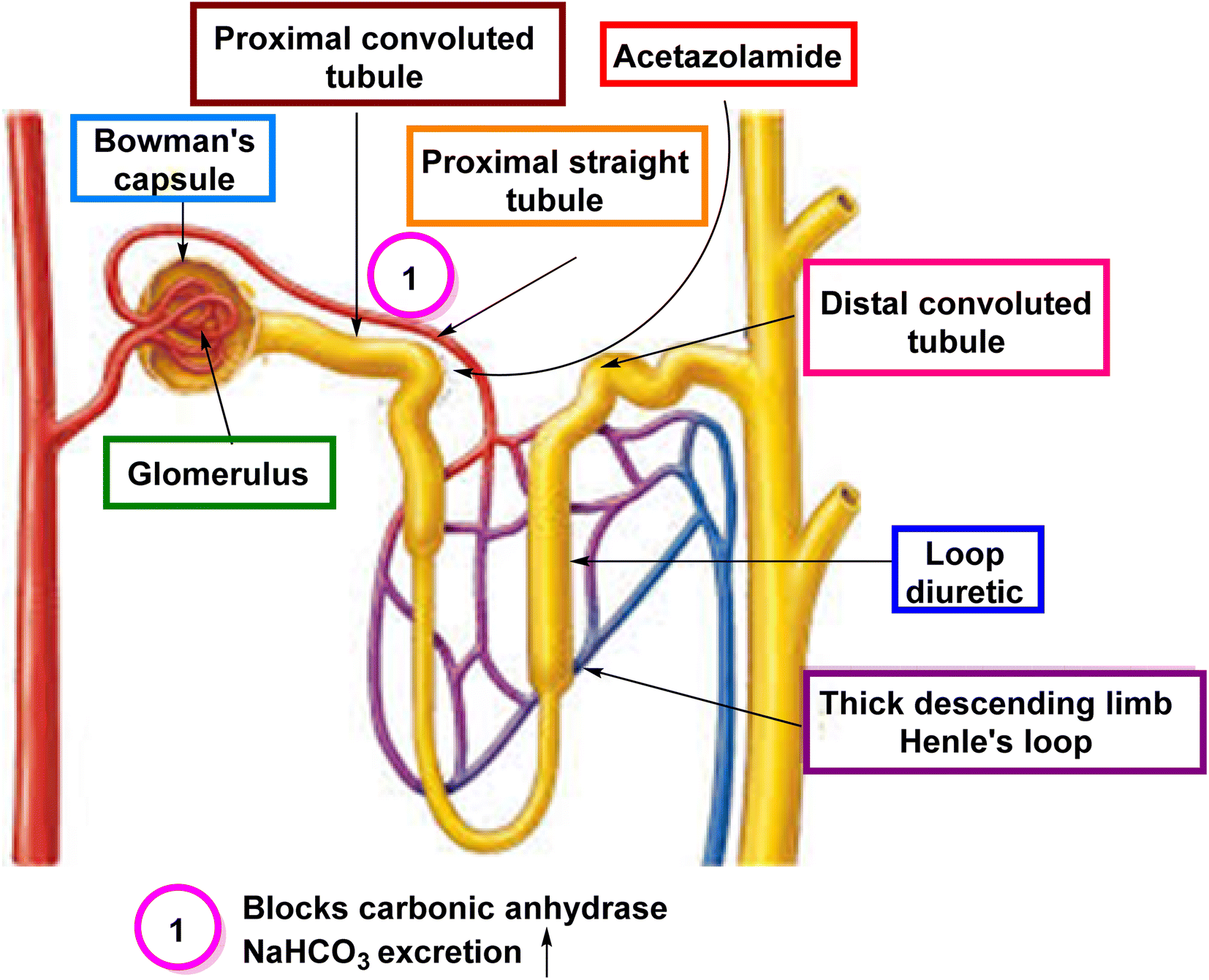 | ||
| Fig. 5 Diuretic therapy with CAIs. | ||
4.2. CAIs as drugs for eye disorders
CAIs have found significant therapeutic applications in the treatment of various eye disorders.4.3. CAIs as potential anti-obesity drugs
CAIs have emerged as potential candidates for the treatment of obesity due to their involvement in metabolic pathways and physiological processes related to energy balance.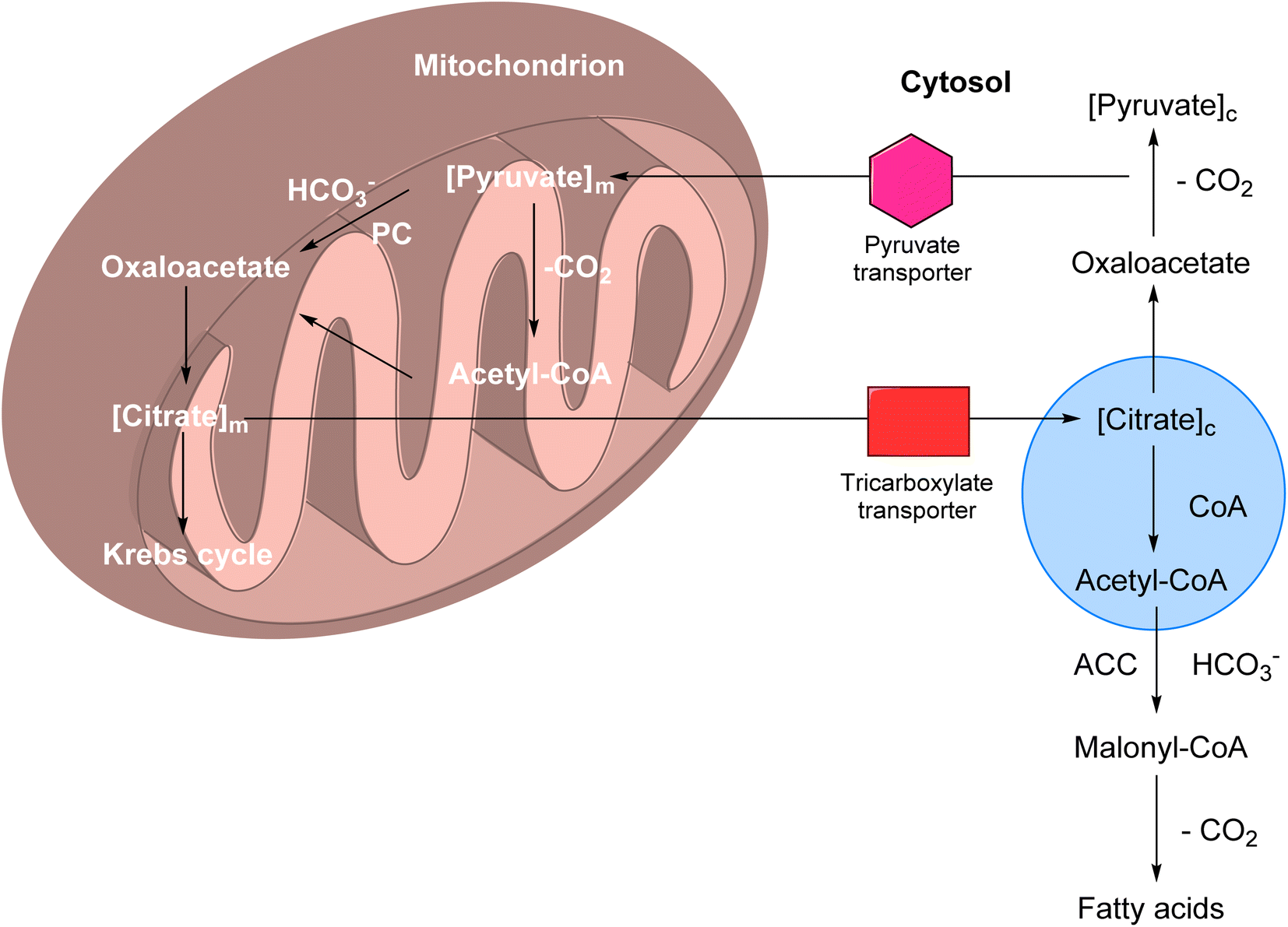 | ||
| Fig. 6 Role of CA-VA, VB and CA-II in the de novo lipogenesis. | ||
4.4. Anticancer potential of CAIs
CAIs have gained significant attention in cancer research due to their potential as anticancer agents.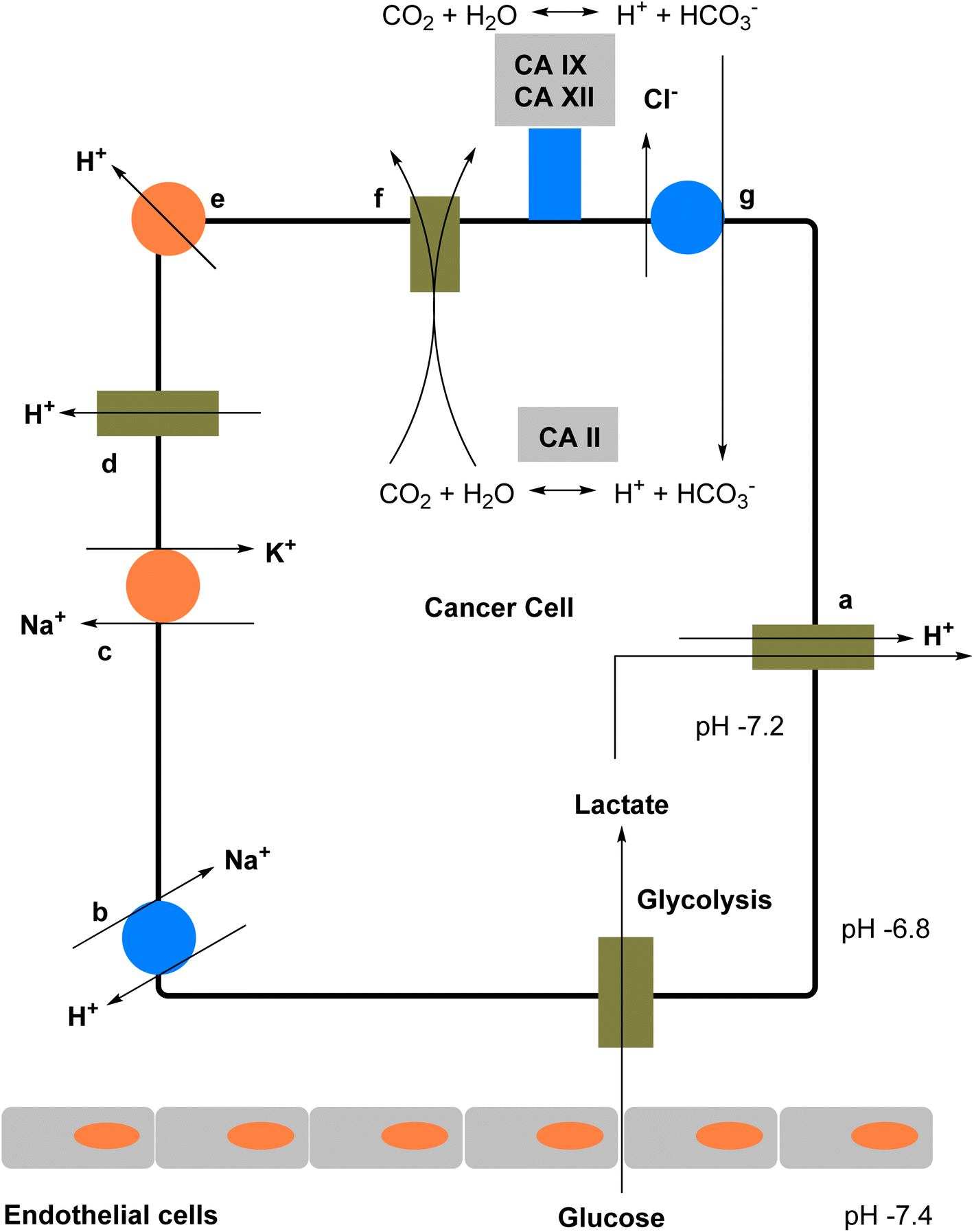 | ||
| Fig. 7 Schematic representation illustrating the role of hCA-IX in regulating pH in hypoxic tumor cells. | ||
4.5. CAIs as potential therapeutics for osteoporosis
Osteoporosis is a common skeletal condition marked by reduced bone strength and an elevated risk of fractures. Conventional therapies for osteoporosis primarily target bone resorption through antiresorptive agents or promote bone formation using anabolic agents. However, these treatments have limitations, underscoring the need for alternative therapeutic strategies. CAIs have emerged as potential candidates for osteoporosis therapy due to their involvement in bone metabolism and remodeling processes.805. Carbonic anhydrase inhibitors
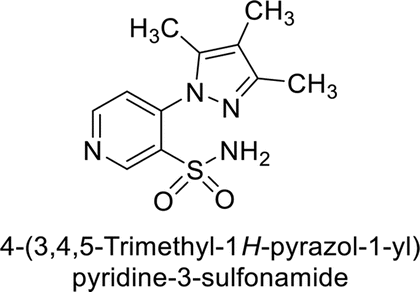
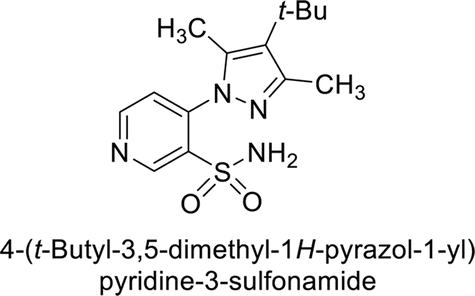
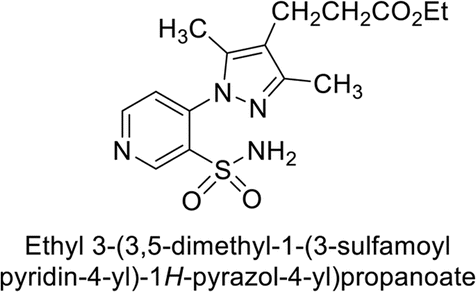
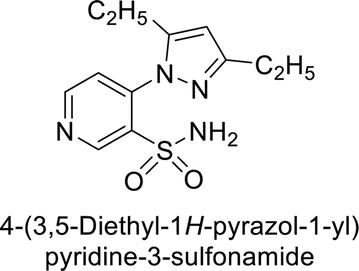
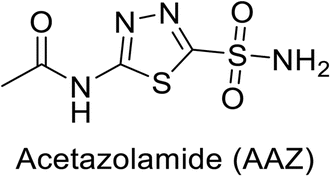
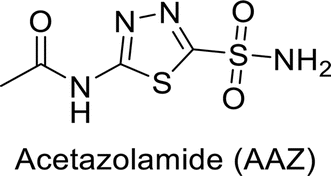
Sławiński et al. (2013) synthesized a series of heterocyclic pyridine-3-sulfonamides and evaluated them against four isoforms of CA. The newly synthesized compounds displayed Ki values against hCA-I ranging from 169–5400 nM, against hCA-II ranging from 58.5–1238 nM, against hCA-IX ranging from 19.5–652 nM, and against hCA-XII ranging from 16.8–768 nM. Particularly, compounds 1–5, which are pyrazole substituted pyridinesulfonamides, exhibited significant inhibitory efficacy against hCA-IX, with Ki values ranging from 19.5–48.6 nM, comparable to the clinically used sulfonamides such as acetazolamide (AAZ), methazolamide (MZA), ethoxazolamide (EZA) and indisulam (IND) (with Ki values ranging from 24–50 nM)84 (Table 2). Future studies exploring the SAR of these compounds, particularly focusing on the impact of substituents on their inhibitory potency and selectivity, would provide valuable insights for optimizing their pharmacological properties and therapeutic utility in treating diseases associated with CA dysregulation.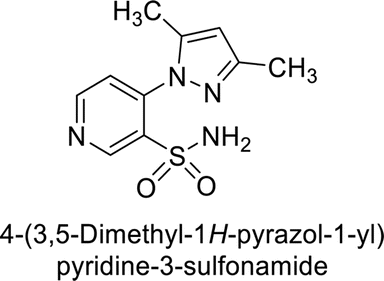
Supuran et al. (2013) synthesized a series of sulfamides containing the dopamine motif. These sulfamides were evaluated for their inhibitory effects on six α-CAs (CA-I, II, VA, IX, XII, and XIV). Compounds 6–10 showed excellent Ki values as compared to the standard AAZ (Table 3). These synthesized sulfamides exhibit potential for targeting medically relevant CA isoforms, suggesting promising applications in the treatment of epilepsy, obesity, tumors, or infections.85
| Compound no. | Chemical structures & IUPAC names | Ki | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| CA-I (μM) | CA-II (μM) | CA-V (μM) | CA-IX (μM) | CA-XII (μM) | CA-XIV (μM) | |||
| 6 | 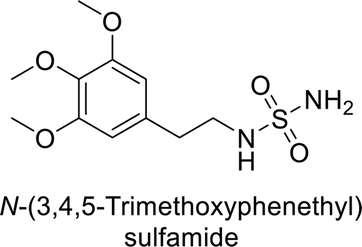 |
1.82 | 2.03 | >50 | 0.221 | 1.52 | 0.22 | 85 |
| 7 | 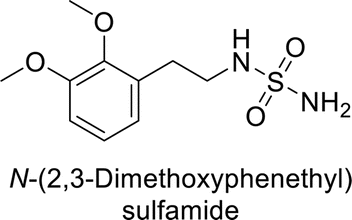 |
0.25 | 2.95 | >50 | >50 | 0.047 | 0.18 | 85 |
| 8 | 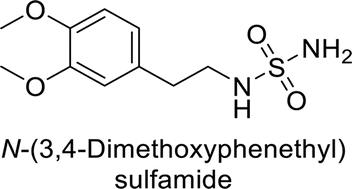 |
0.28 | 2.40 | 2.28 | 0.092 | 0.044 | 0.031 | 85 |
| 9 | 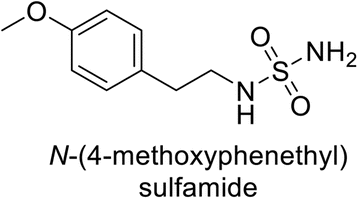 |
0.061 | 1.47 | 3.25 | 0.376 | 0.021 | 0.007 | 85 |
| 10 | 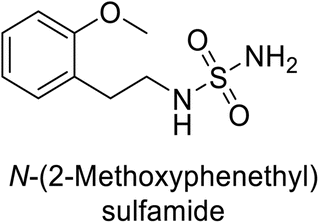 |
0.25 | 2.04 | 3.34 | 0.041 | 0.416 | 0.16 | 85 |
| Standard | 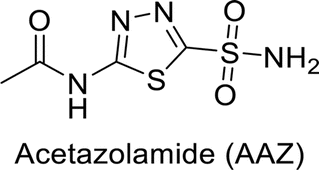 |
0.250 | 12 | 0.063 | 0.025 | 0.006 | 0.041 | 85 |
Gencer et al. (2013) prepared a series of urea and thiourea derivatives, substituted with phthalazine and tested for their inhibitory effects on purified hCA-I and II. Compound 11 demonstrated the most potent inhibition (IC50 = 6.40 μM for hCA-I and 6.13 μM for hCA-II) (Fig. 8). This compound acts as a competitive inhibitor. The bulky nature of the synthesized compounds suggests a probable binding mode similar to coumarin derivatives rather than directly near the zinc ion.86
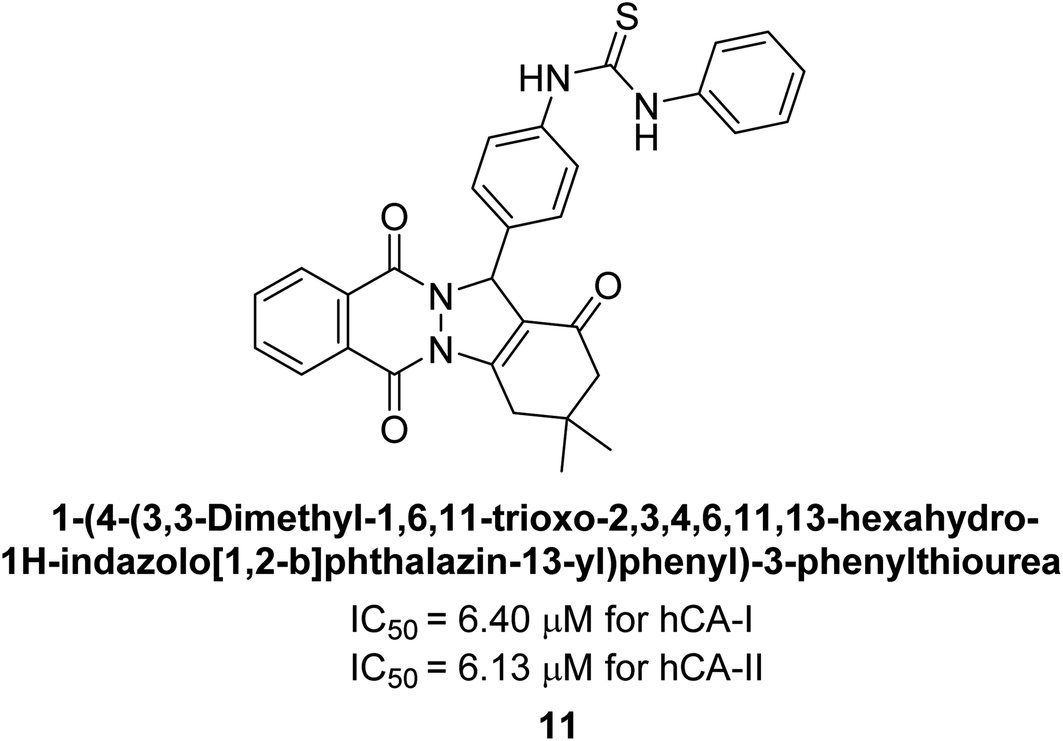 | ||
| Fig. 8 Chemical structure of compound 11 and its hCA-I and II inhibition. | ||
Altuntas et al. (2013) designed a series of 2-amino-3-cyanopyridine compounds and evaluated their inhibitory effects on hCA-I and II. Compounds 12 and 13 displayed the most promising Ki against hCA-I, with IC50 values of 33 and 34 μM and Ki values of 23.8 and 31 μM, respectively. Compound 12 exhibited the highest activity against hCA-II, showing an IC50 value of 56 μM and a Ki value of 41 μM, compared to the standard sulfanilamide, which has Ki values of 25.5 μM and 0.023 μM for hCA-I and II, respectively (Fig. 9). The target compounds act as a competitive inhibitors.87
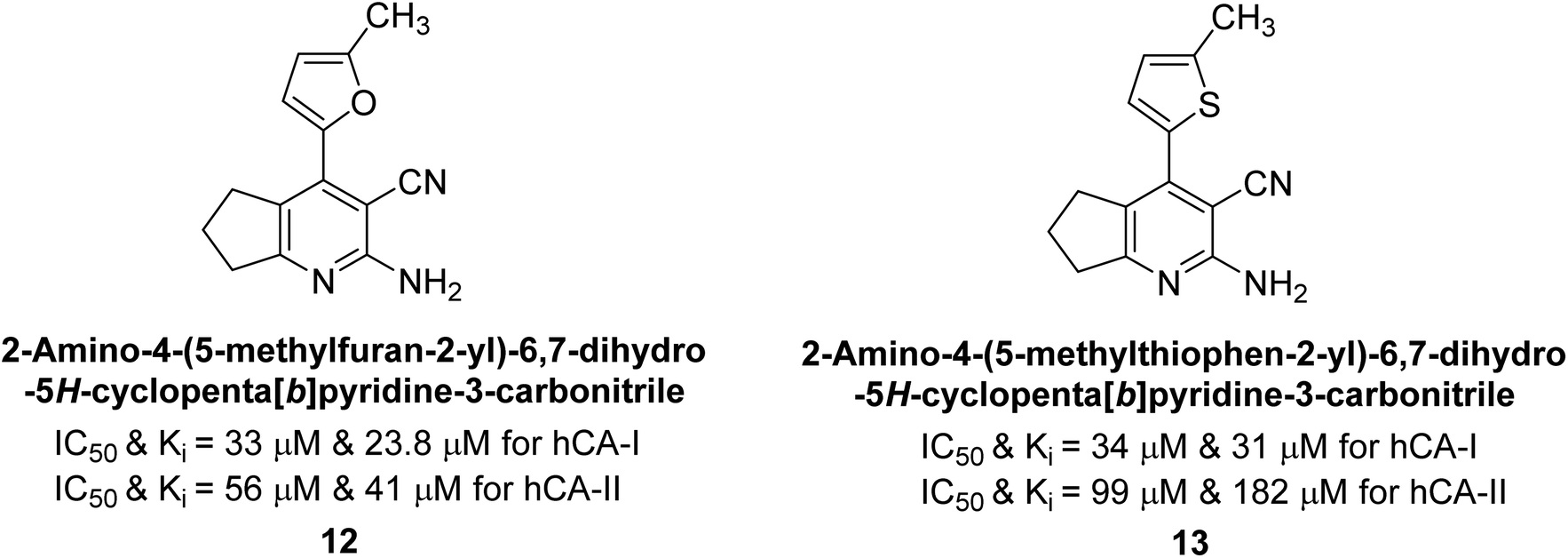 | ||
| Fig. 9 Chemical structures of compounds 12 and 13 and their IC50 and Ki values against hCA-I and II. | ||
Zhu et al. (2013) synthesized a series of sulfonamides incorporating coumarin moieties and determined their inhibitory effects on two CAs. These compounds were tested against the hCA-II and IX. Among them, compound 14 demonstrated the maximum potency against hCA-II with an IC50 = 23 nM, while compound 15 showed the strongest inhibition of hCA-IX with an IC50 = 24 nM as compared to standards AAZ and SA. These sulfonamides, bearing coumarin moieties, hold promise as lead candidates for targeting tumor-associated CA isozymes (Fig. 10).88
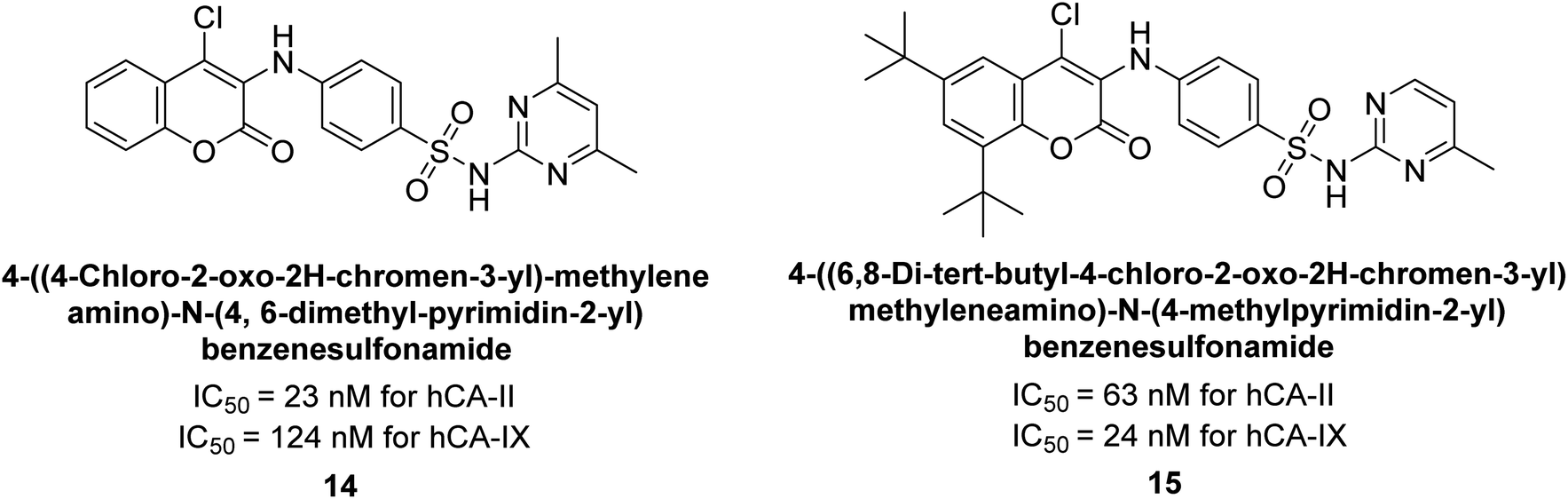 | ||
| Fig. 10 Chemical structures of compounds 14 and 15 and their IC50 values against hCA-II and IX. | ||
Büyükkıdan et al. (2013) developed Co(II) complexes with pyrazole and evaluated their carbonic anhydrase inhibitory activity. For hCA-I, compounds 16 and 17 had IC50 values of 0.473 μM and 0.065 μM, respectively, while for hCA-II, the IC50 values were 0.213 μM and 0.833 μM. In esterase activity assays, IC50 values were 0.058 μM and 0.297 μM for hCA-I, and 0.110 μM and 0.052 μM for hCA-II. The Ki for esterase activity were 0.039 μM and 0.247 μM for hCA-I, and 0.078 μM and 0.363 μM for hCA-II (Fig. 11).89
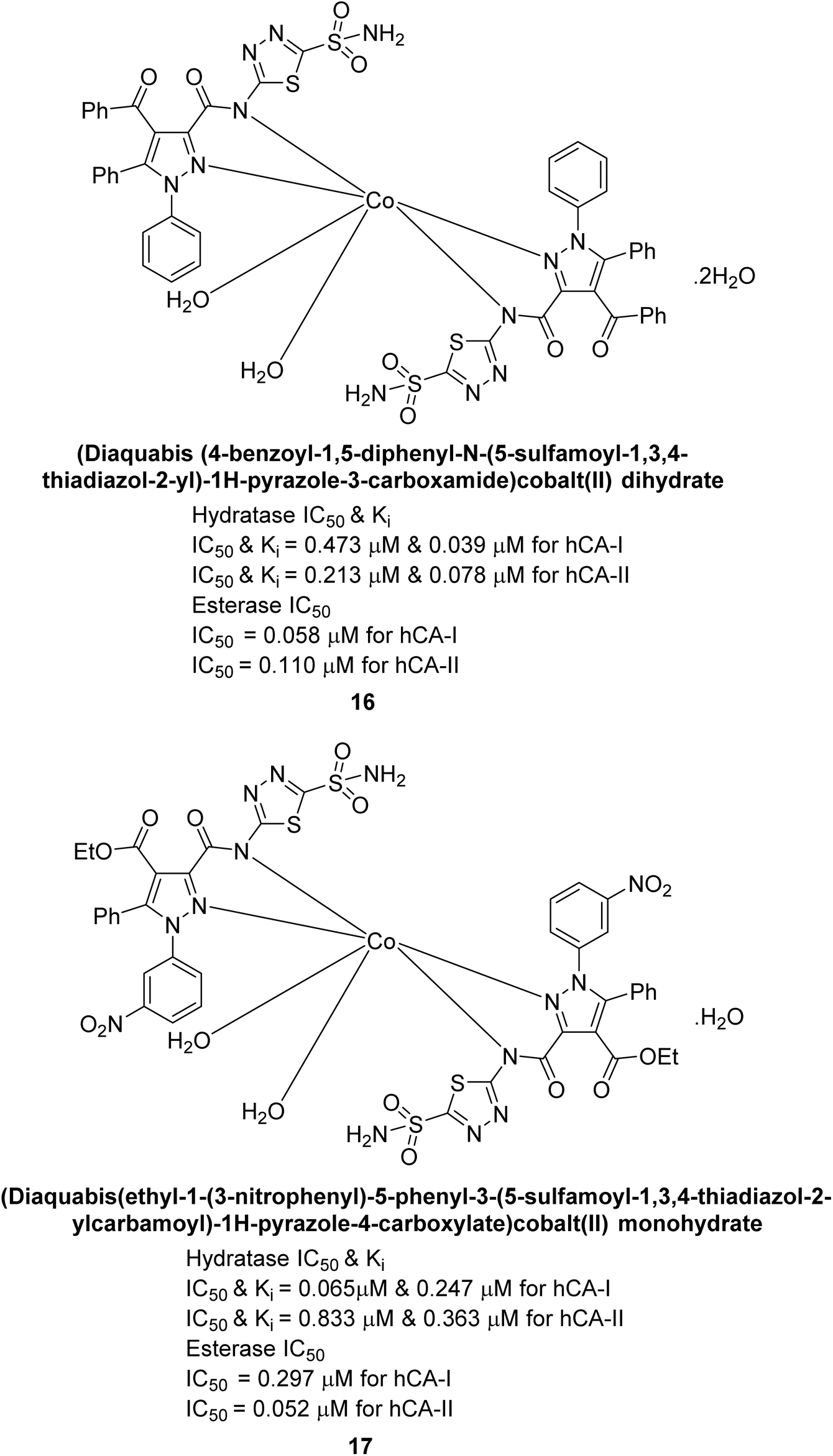 | ||
| Fig. 11 Chemical structures of compounds 16 and 17 and their IC50 and Ki values against hCA-I and II. | ||
Gençer et al. (2013) designed and synthesized a series of water-soluble benzimidazolium chloride salts, bearing chromone moiety and evaluated them against hCA-I and II. Results demonstrated inhibition of hCA isoenzyme activity by all synthesized compounds. Notably, compounds 18 and 19 demonstrated the highest activity against hCA-I and II, with IC50 values of 22.09 μM and 20.33 μM, respectively (Fig. 12).90
 | ||
| Fig. 12 Chemical structures of compounds 18 and 19 and their IC50 values against hCA-I and II. | ||
Ok et al. (2013) designed and synthesized a new series of pyrazole derivatives and evaluated against hCA enzymes. Remarkably, compounds 20 (Ki = 0.108 μM for hCA-I and 0.055 μM for hCA-II) and 21 (Ki = 0.129 μM for hCA-I and 0.064 μM for hCA-II) exhibited the maximum inhibition of both hCA-I and II isozymes (Fig. 13). Both derivatives act as a non-competitive inhibitor. Furthermore, the presence of resonance structures in the molecules reduces their inhibitory effect. The findings suggest that minor alterations in the –R moiety (substituents) significantly impact inhibitory activity. Additionally, the compounds' hydrogen bonding capability and hydrophilic/hydrophobic properties may influence their inhibition behavior. Overall, the study provides insights into the potential of pyrazole carboxamide derivatives as inhibitors of hCA enzymes.91
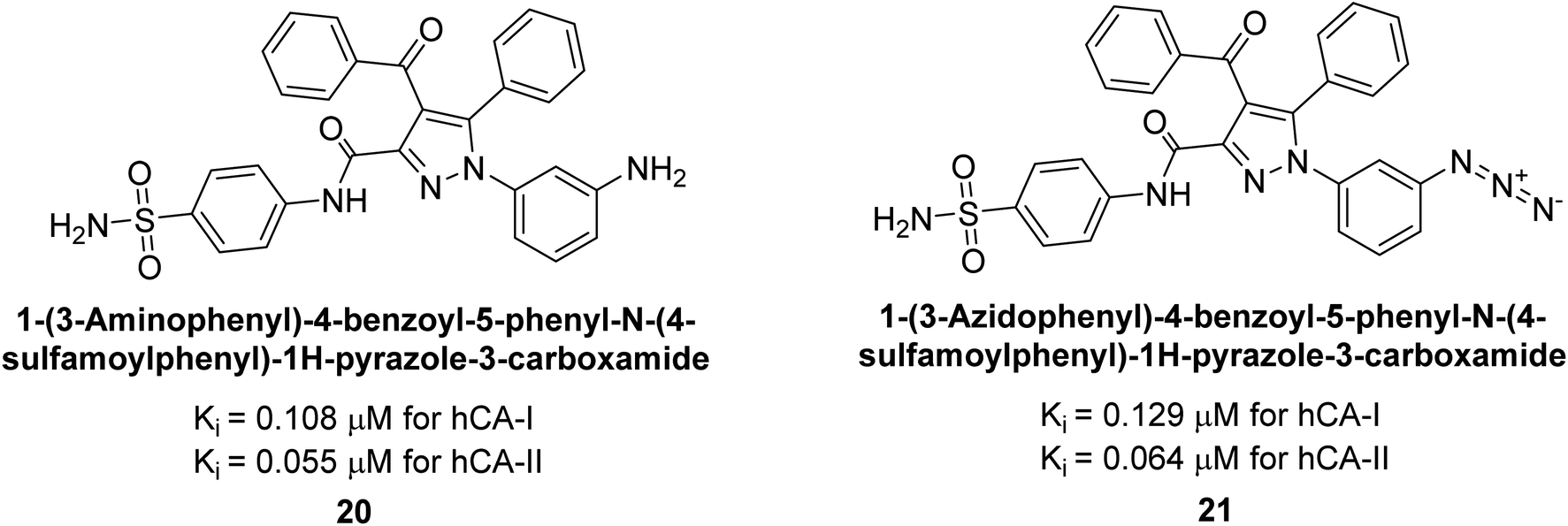 | ||
| Fig. 13 Chemical structures of compounds 20 and 21 and their Ki values against hCA-I and II. | ||
Suthar et al. (2013) designed sulfonamide derivatives as selective inhibitors of CA-IX for potential anti-cancer applications. Among the compounds tested, 22 emerged as the most potent CA-IX inhibitor with Ki value of 2.2 nM as compared to the standard AAZ (Ki = 25.2 nM). Moreover, studies revealed that 22 induced apoptosis in COLO-205 cells, characterized by cell shrinkage and nuclear fragmentation. In a solid tumor model, 22 reduced tumor volume by 64.83% with minimal increase in body weight, indicating its potential as an effective anticancer agent. Among the compounds screened virtually, compound 22 exhibited the lowest estimated free energy of binding at −7.15 kcal mol−1. Molecular docking analysis indicated that compound 22, the most potent inhibitor, could interact with the catalytic Zn(II) ion, which may account for its higher binding affinity for CA-IX (Fig. 14).92
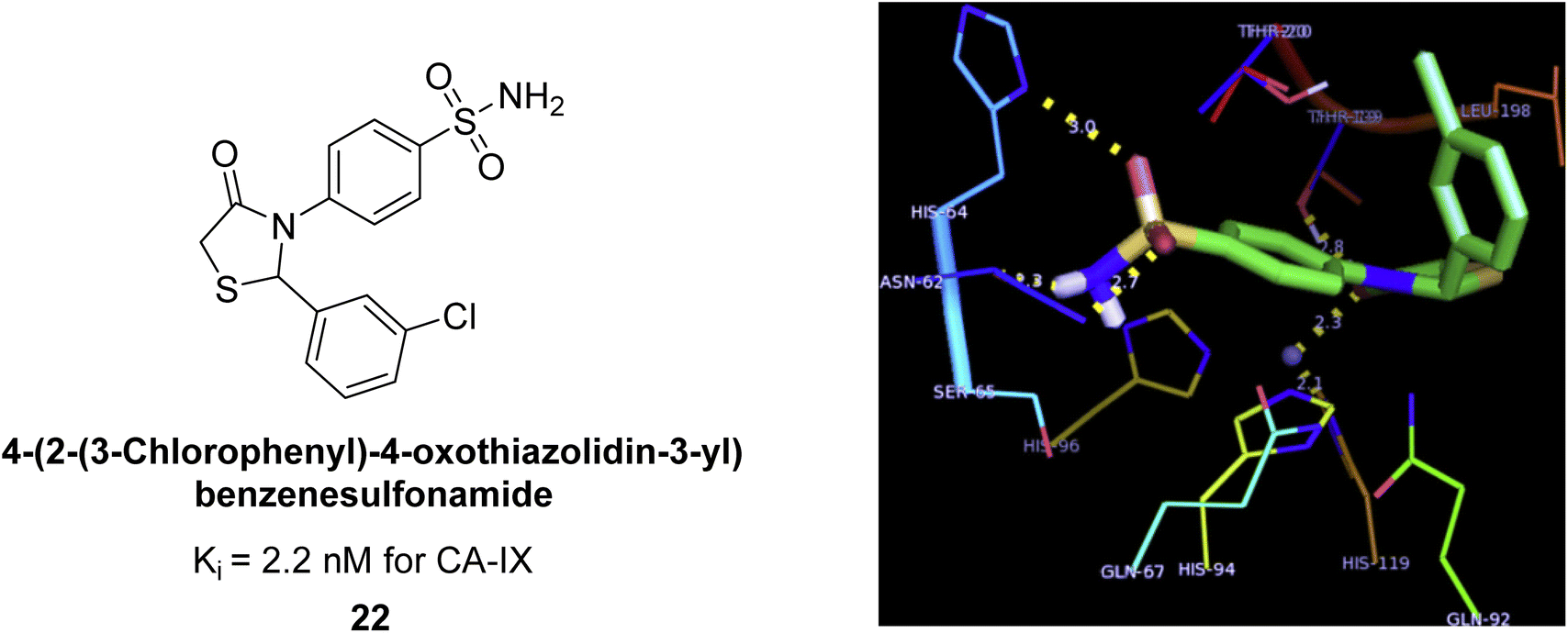 | ||
| Fig. 14 Chemical structure and Ki value of compound 22 and its docking image inside the active pocket of CA-IX enzyme. | ||
Gençer et al. (2013) synthesized a series of phenylurea/phenylthiourea-substituted chalcones and investigated them against CA enzymes. Compounds 23 and 24 exhibited the highest activity against hCA-I with IC50 values of 25.4 μM and 23.1 μM, respectively. Compound 25 demonstrated the maximum activity against hCA-II with an IC50 value of 14.4 μM (Fig. 15).93
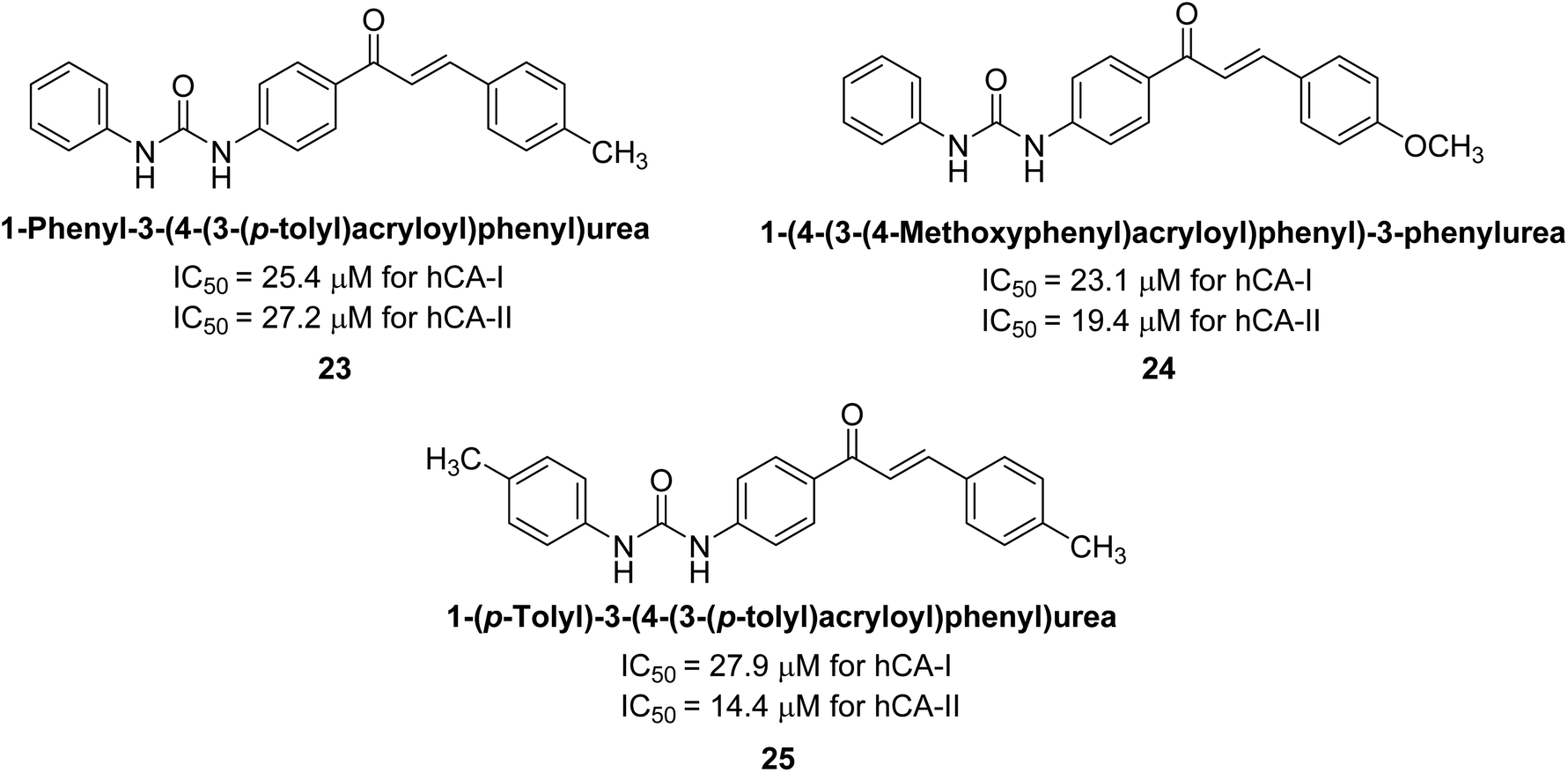 | ||
| Fig. 15 Chemical structures of compounds 23, 24 and 25 and their IC50 values against hCA-I and II. | ||
Xu et al. (2013) synthesized a series of new heterocyclic substituted benzene sulfonamides compound 26 emerged as the most potent and selective, inhibiting CA-IX at subnanomolar levels (IC50 of 0.48 nM), representing a 40-fold increase in potency compared to the lead compound 27 (IC50 = 18.5 nM). Furthermore, it exhibited over 103-fold selectivity over CA-I and CA-II (Fig. 16). Compound 26 exhibits a similar interaction profile: the sulfonamide nitrogen forms a hydrogen bond with the hydroxyl group of Thr199, and one of the sulfonamide oxygens interacts with the backbone amide of Thr199. The benzene rings engage with Val121, Val143, and Leu198 in the hydrophobic region, where methyl groups enhance the affinity. Introducing a methyl group at the o-position of the left benzenesulfonamide produced two highly effective CA-IX inhibitors. In the hydrophilic pocket, the linker NH forms hydrogen bonds with His64 and Asn62, which contribute to the high binding affinities for CA-IX (Fig. 16).94
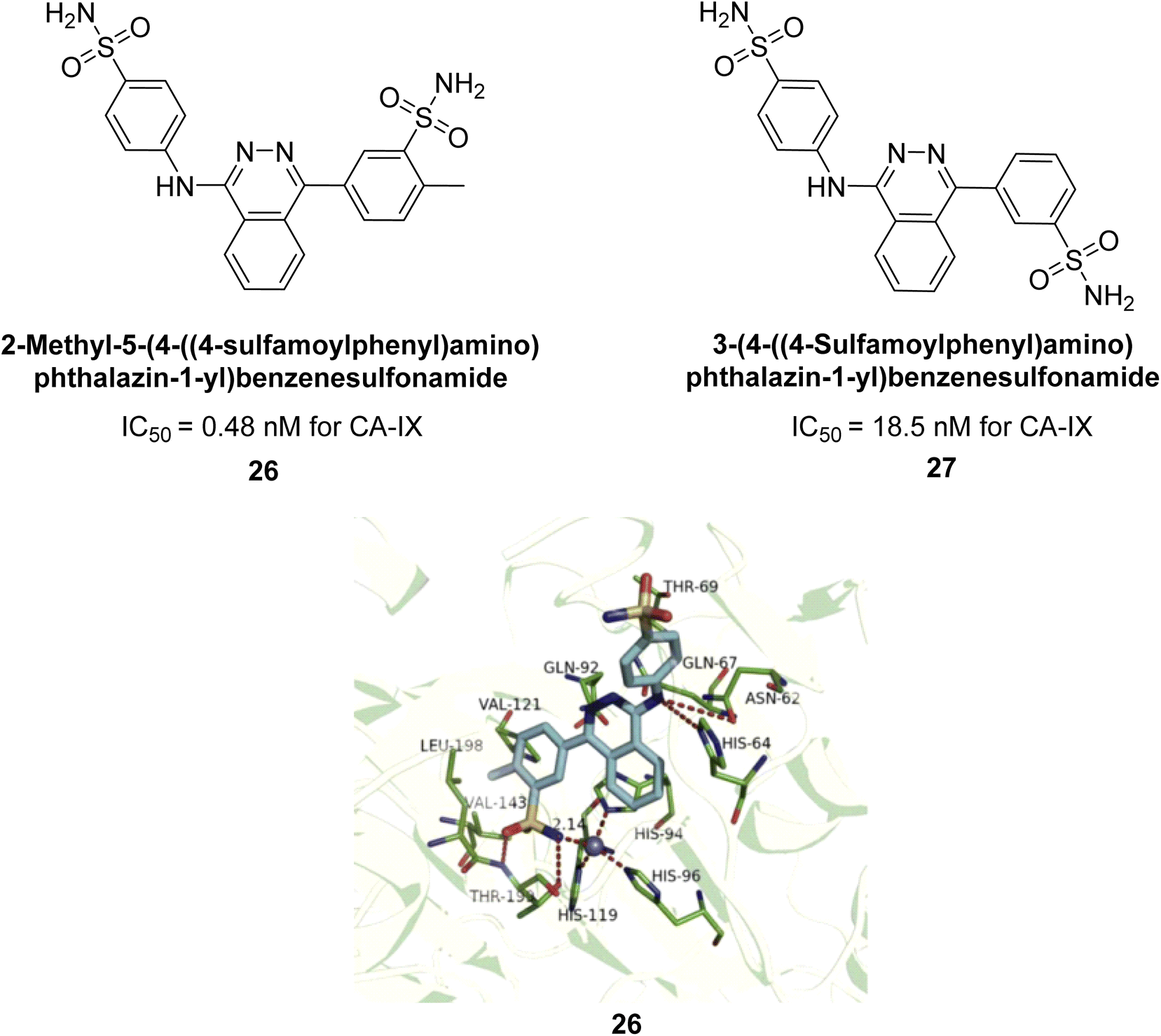 | ||
| Fig. 16 Chemical structures of compounds 26 and 27 and their IC50 values against hCA-IX and docking image of compound 26 against CA-IX (PDB ID: 3IAI). | ||
Allouche et al. (2013) designed and synthesized a series of aminocyanopyrazole derivatives and evaluated their inhibitory effects on hCA-IX and XII. Compound 28 exhibited stronger inhibition against the tumor-associated transmembrane isoforms hCA-IX (Ki = 0.15 μM) and hCA-XII (Ki = 0.47 μM) compared to the cytosolic ones (Fig. 17).95
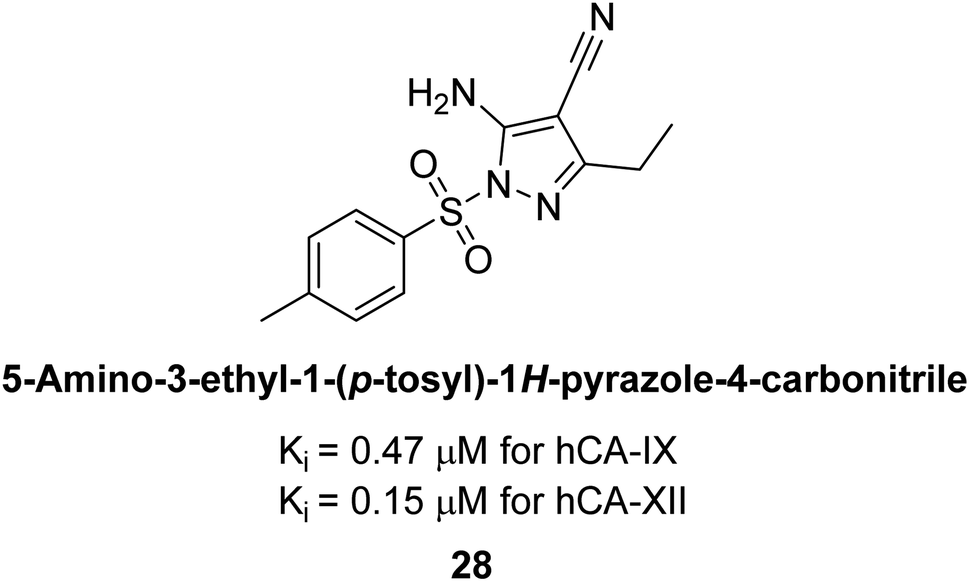 | ||
| Fig. 17 Chemical structure of compound 28 and its Ki values against hCA-IX and XII. | ||
Supuran et al. (2014) synthesized and evaluated 1,3-diarylpyrazoles. Inhibition assays against hCA-II revealed that all synthesized compounds displayed varying degrees of inhibition, ranging from excellent to moderate. Specifically, compounds 29, 30, 31, 32, 33, and 34 exhibited Ki values less than 12 nM, comparable to the Ki value of AAZ. Most compounds exhibited moderate inhibition of hCA-IX, except compound 32, which displayed nearly 9 times greater potency compared to AAZ. In the case of hCA-XII, compounds 32, 35, 36, and 37 displayed Ki values comparable to that of AAZ (Table 4).96
| Compound no. | Chemical structures & IUPAC names | Ki (nM) | Ref. | |||
|---|---|---|---|---|---|---|
| CA-I | CA-II | CA-IX | CA-XII | |||
| 29 | 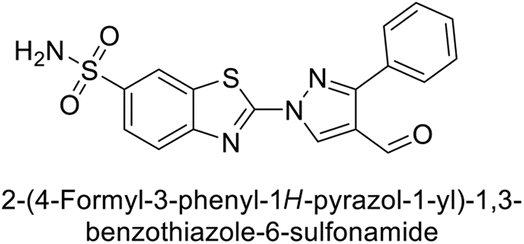 |
8806 | 10.9 | 1747 | 214 | 96 |
| 30 | 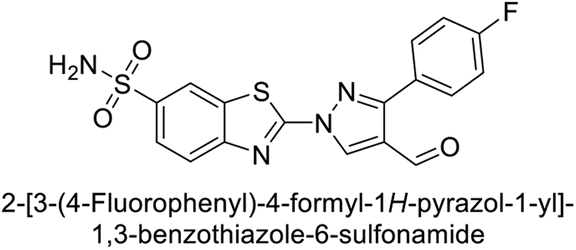 |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 690 690 |
10.4 | 1022 | >10000 |
96 |
| 31 | 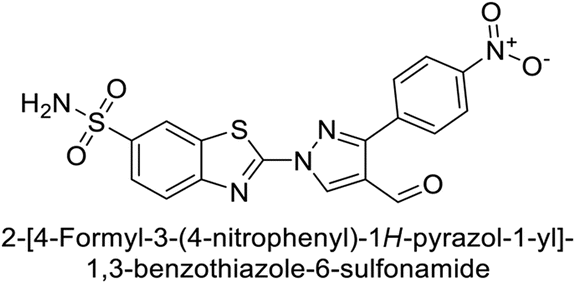 |
2331 | 3.8 | 2109 | 74.5 | 96 |
| 32 | 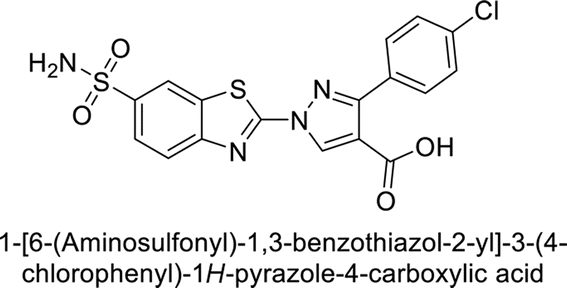 |
407 | 7.3 | 2.8 | 5.5 | 96 |
| 33 |  |
7161 | 8.5 | 224 | 83.8 | 96 |
| 34 | 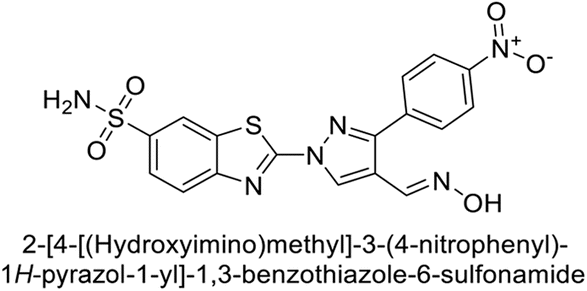 |
23102 |
11.6 | 119 | 96.8 | 96 |
| 35 | 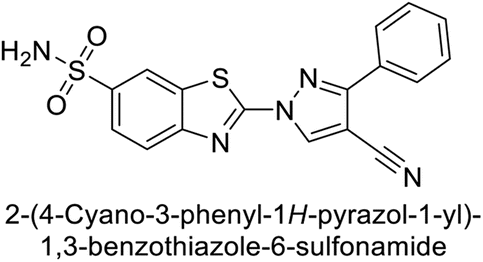 |
9819 | 139 | 81.6 | 6.6 | 96 |
| 36 | 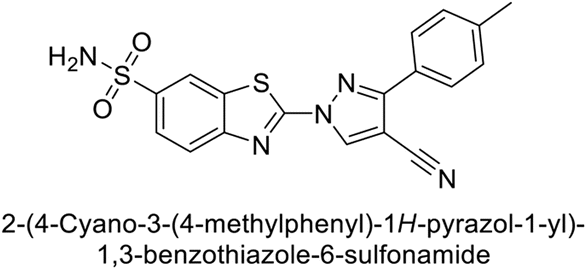 |
>50000 |
48.1 | 186 | 7.2 | 96 |
| 37 |  |
12108 |
27 | 739 | 7.4 | 96 |
| Standard | 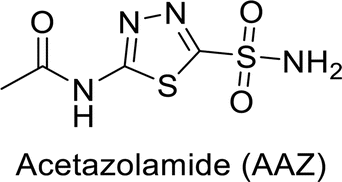 |
250 | 12 | 25 | 5.7 | 96 |
Ghorab et al. (2014) synthesized benzenesulfonamides incorporating various heterocyclic moieties exhibit promising inhibitory activity against hCA-I, II, IX and XII. Among the investigated compounds, derivatives 38, 39, and 40–44 demonstrated notable potency as inhibitors of hCA-IX and XII (Table 5). Their inhibitory activity was particularly significant, falling within the nanomolar and subnanomolar ranges, indicating high efficacy. Moreover, these compounds exhibited considerable selectivity towards the targeted isoforms. Overall, the findings of this study highlight the importance of rational design and synthesis in the development of novel CAIs with potential therapeutic applications.97
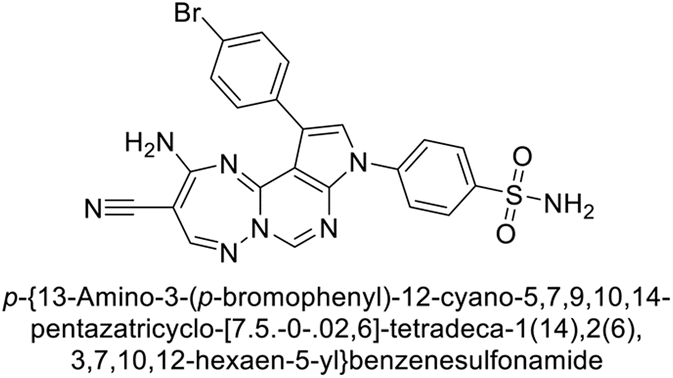
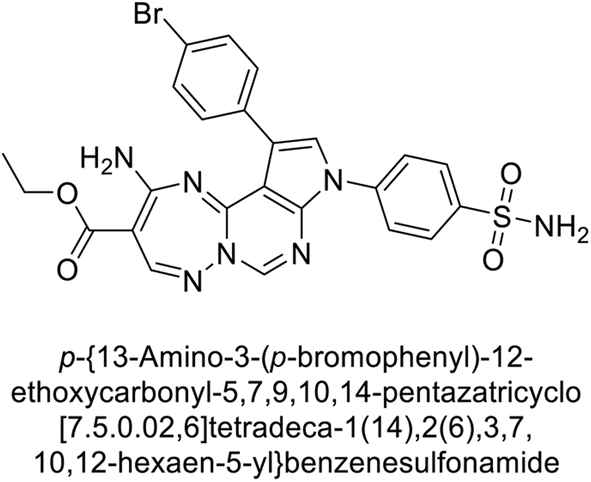
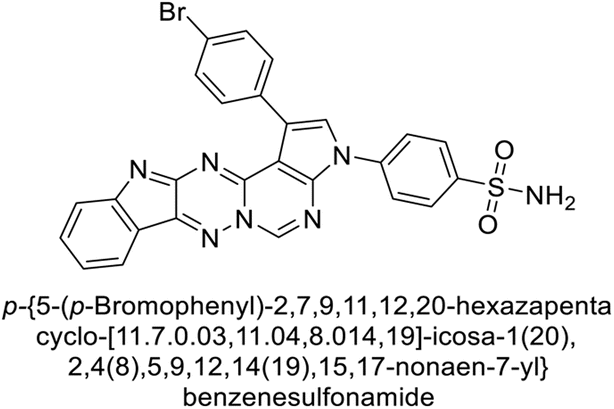
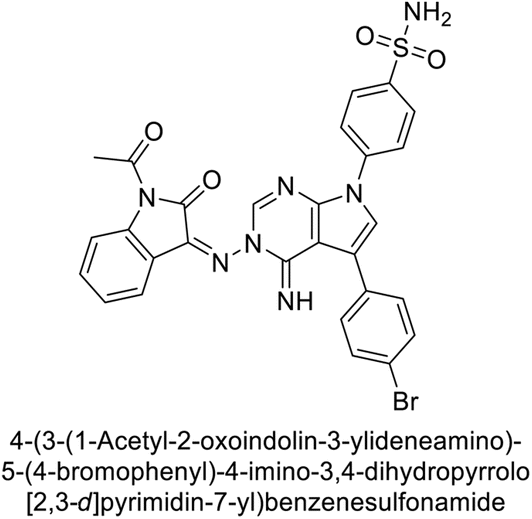
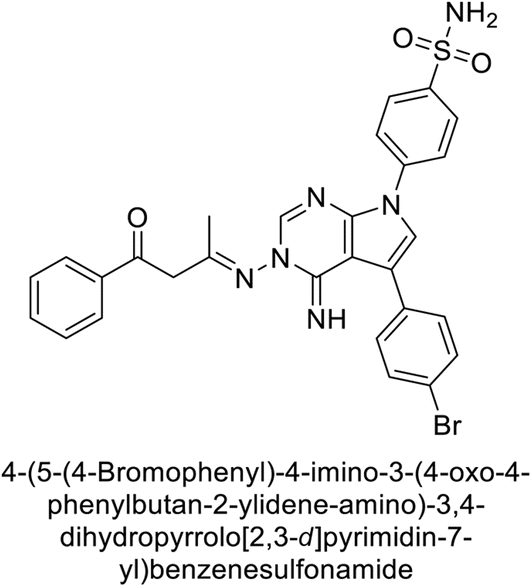
Mojzych et al. (2014) reported pyrazolo-triazine sulfonamides have emerged as a class of compounds with promising CA inhibitory activity. The pyrazolo-triazine scaffold offers structural diversity and flexibility, making it an attractive platform for the synthesis of novel hCAIs. Compound 45 showed good to moderate inhibitory activity against the isoforms of hCA. The hCA-I and hCA-II showed Ki = 270 μM and 8 μM, respectively (Fig. 18). The newly synthesized compounds showed negligible activity as inhibitors of hCA-I and displayed weak inhibition against hCA-II. Henceforth, compound 45 exhibited enhanced efficacy against the hCA-IX (Ki = 43.8 μM) and XII (Ki = 7.9 μM).98
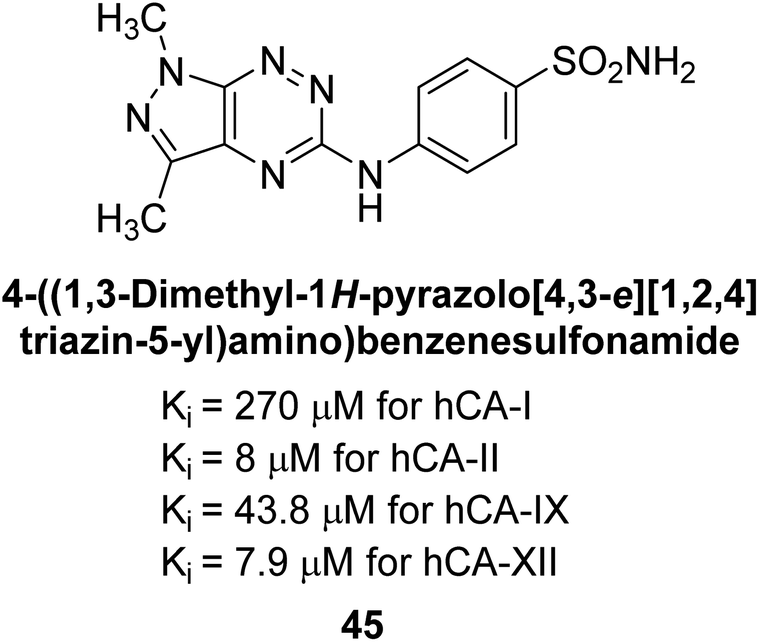 | ||
| Fig. 18 Chemical structure of compound 45 and its Ki values against hCA-I, II, IX, XII. | ||
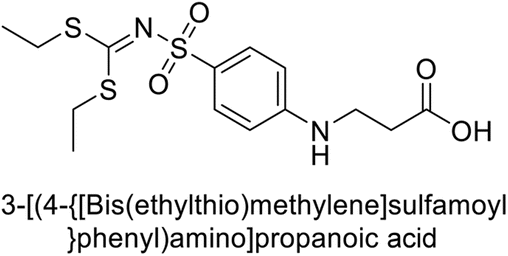
Matulis et al. (2014) investigated the inhibitory potential of 4-amino-substituted benzenesulfonamides on hCAs. The majority of N-aryl-β-alanine derivatives exhibited enhanced binding affinity for hCA-II, whereas diazobenzenesulfonamides demonstrated nanomolar affinities specifically towards the hCA-I isozyme. Most N-aryl-β-alanine derivatives exhibited higher affinities for hCA-II compared to other tested CA isoforms. Notably, compound 46, containing the tertiary sulfonamide group, exclusively bound to hCA-II (Kd = 40 μM). Compound 47 demonstrated increased binding potency towards hCA-XII (Kd = 1.85 μM) relative to other isoforms and standard AAZ (Table 6). Moreover, SAR analysis reveals key structural features that contribute to enhanced inhibition. Further investigations, including in vivo studies and optimization of pharmacokinetic properties, are warranted to advance these compounds toward clinical applications in diseases where hCA inhibition is therapeutically beneficial.99
Sławiński et al. (2014) synthesized and evaluated a series of novel N4-substituted 4-(2-aminoethyl)benzenesulfonamides as inhibitors against four isoforms of hCAs. Particularly, excellent inhibitory activity was observed against hCA-IX and XII. Among the target analogues, noteworthy Ki against hCA-IX (Ki = 5.9–10.7 nM) was observed, surpassing clinically utilized CAIs such as AAZ, DCP, EZA, MZA and IND (24–50 nM). Compounds 48 and 49 emerged as the excellent inhibitors against hCA-IX and hCA-XII (Ki = 5.9 and 6.2 nM for hCA-IX; 4.3 and 4.0 nM for hCA-XII, respectively), characterized by their cationic nature and significant affinity for hCA-IX and XII rather than the hCA-I and II (Fig. 19).100
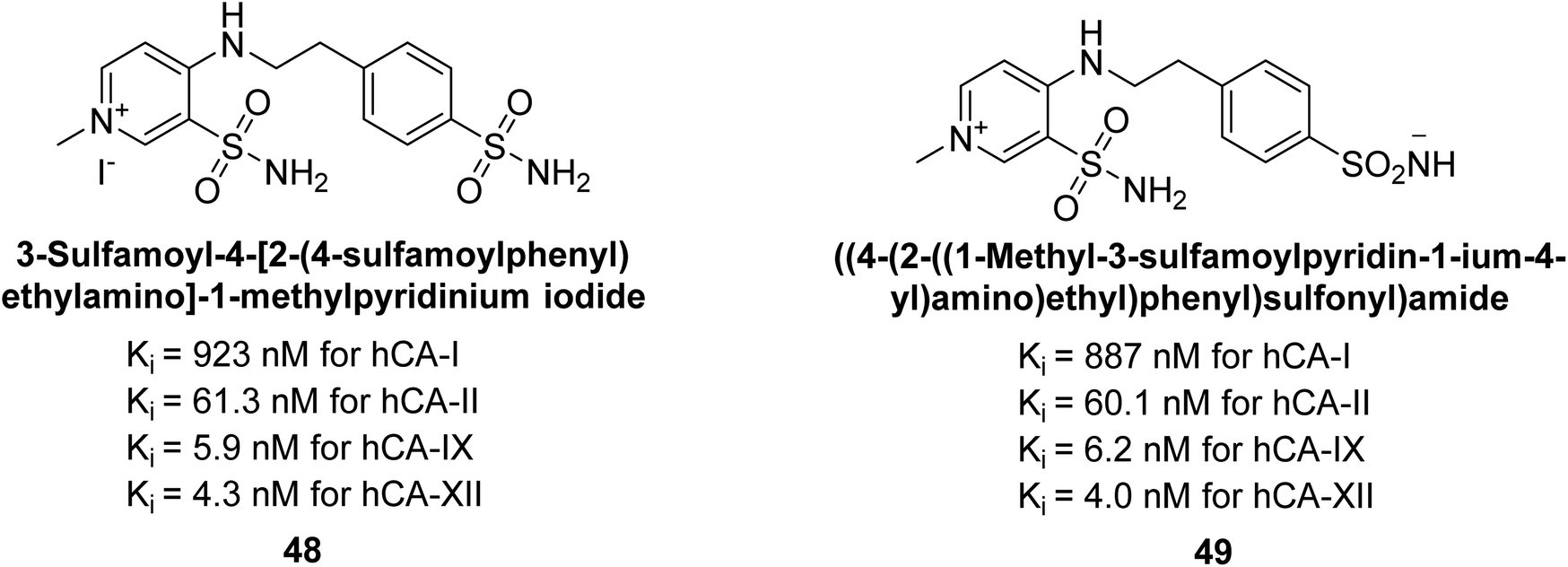 | ||
| Fig. 19 Chemical structure of compounds 48 and 49 and their Ki values against hCA isoforms. | ||
Matulis et al. (2014) synthesized fluorinated benzenesulfonamides, featuring various substituents on the aryl ring. Several of these compounds exhibited strong and selective inhibition against CA-IX. The introduction of three fluorine atoms especially enhanced affinity by electron withdrawal, thereby lowering the pKa of the benzenesulfonamide group. Compound 50 (Kd = 1.1 nM), which features bulky o-substituents, was observed to fit most effectively into the hydrophobic pocket of the hCA-IX active site but not in hCA-II, as demonstrated by co-crystal structure analysis with chimeric CA-IX. The most potent inhibitor compound 51 achieved an affinity of 0.050 nM for the catalytic domain of recombinant human CA-IX in human cells (Fig. 20). However, this heightened affinity compromised selectivity. The inhibitors identified in this study lay the groundwork for the development of novel anticancer therapeutics targeting hCA-IX.101
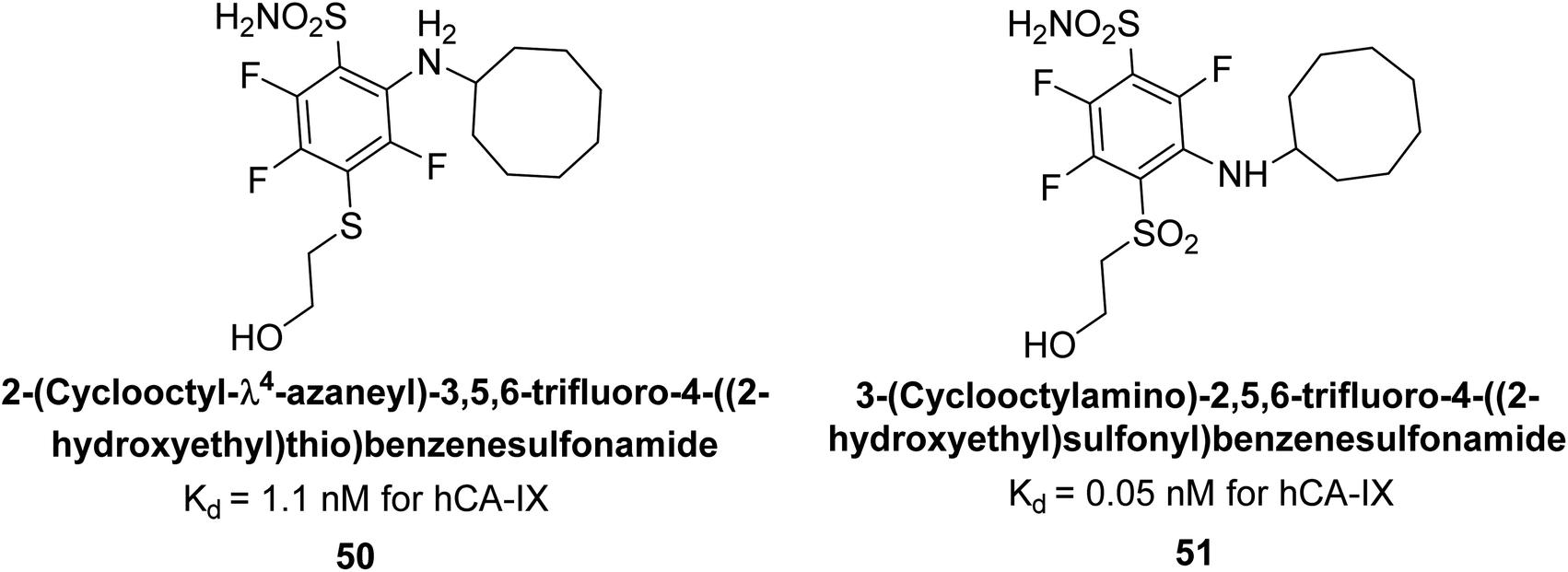 | ||
| Fig. 20 Chemical structure of compounds 50 and 51 and their Kd values against hCA isoforms. | ||
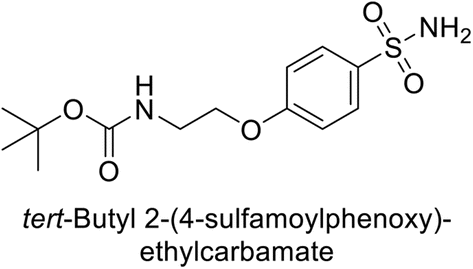
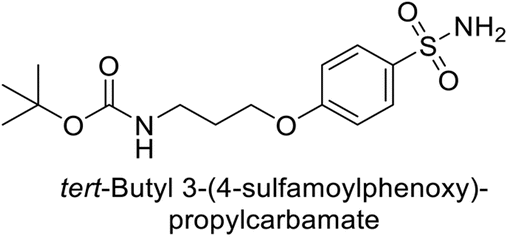
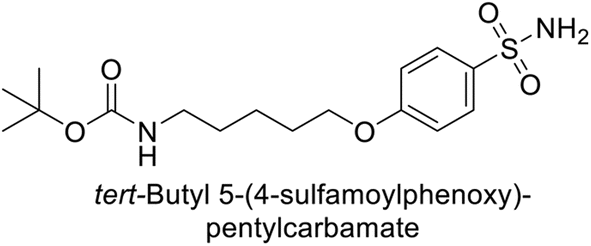
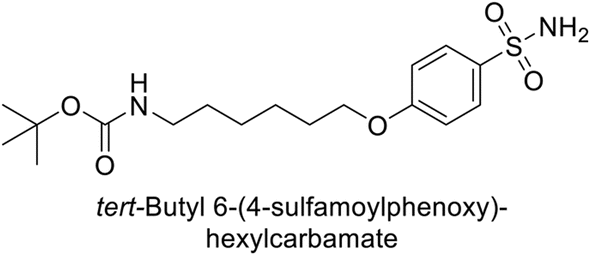
Supuran et al. (2014) synthesized a variety of fluorescent sulfonamides and demonstrated intriguing inhibitory properties against hCA-IX and XII. All derivatives 52–57 reported in this study exhibited potent inhibition against hCA-IX, with Ki values ranging from 5.8 to 32.6 nM. For instance, Boc-protected derivatives 52–55 displayed minimal variation in inhibition constants, ranging from 5.8 to 8.3 nM, while fluorescent sulfonamides 56 and 57 demonstrated consistent inhibitory activity (Table 7).102
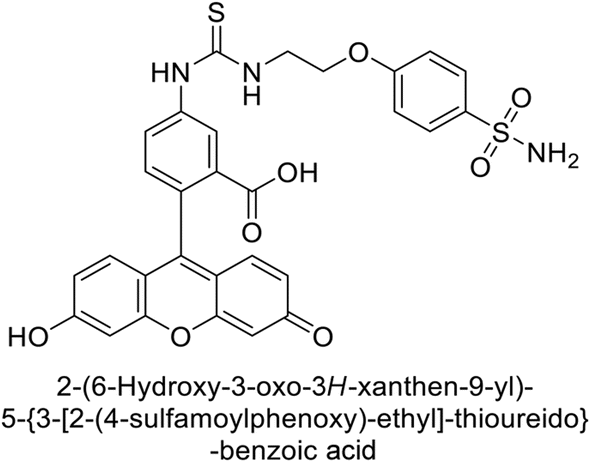
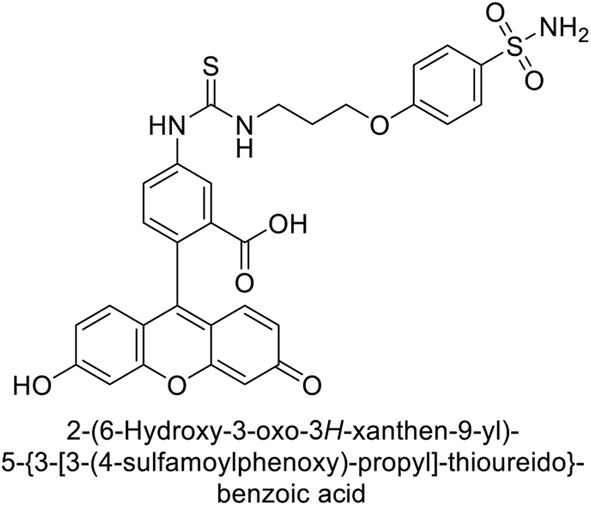
Göksu et al. (2014) synthesized sulfonamides and assessed their inhibitory effects on hCA-I and II. Among the synthesized analogs, compound 58 showed the most potent inhibition of hCA-I, with a Ki value of 46 μM and an r2 of 0.978. In contrast, compound 59 demonstrated the highest inhibition of hCA-II, with a Ki value of 94 μM and an r2 of 0.982 (Fig. 21). Further research and optimization of these compounds could lead to the development of new pharmaceutical agents with improved selectivity and efficacy for CA inhibition.103
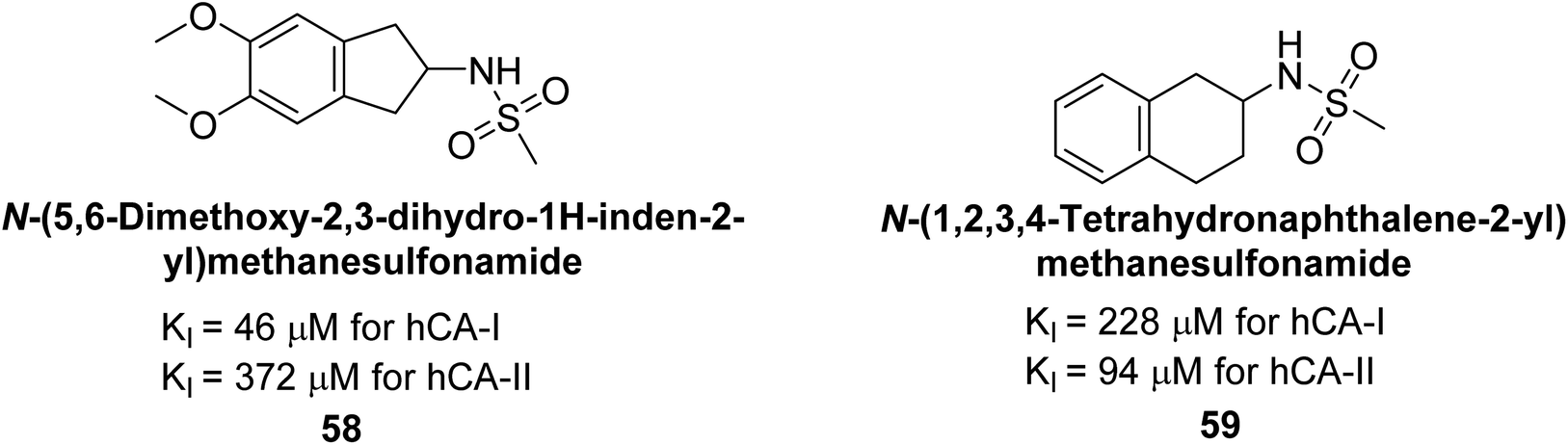 | ||
| Fig. 21 Chemical structure of compounds 58 and 59 and their Kd values against hCA-I and II. | ||
Sharma et al. (2014) designed, synthesized and evaluated a library of 4-functionalized 1,3-diarylpyrazoles against hCA-I, II, IX and XII. Remarkably, compounds 60–65 exhibited low Ki < 5 nM for hCA-IX, while compounds 60, 61, 62, 63, 66 and 67 displayed Ki < 10 nM against hCA-XII (Fig. 22). Furthermore, these derivatives acted as selective hCAIs for isoforms IX and XII over the I and II and as compared to standard AAZ. Additionally, the compounds described in this study demonstrated noteworthy antitumor efficacy, indicating the need for further exploration to expand the understanding of the SAR within this intriguing class of sulfonamide CAIs.104
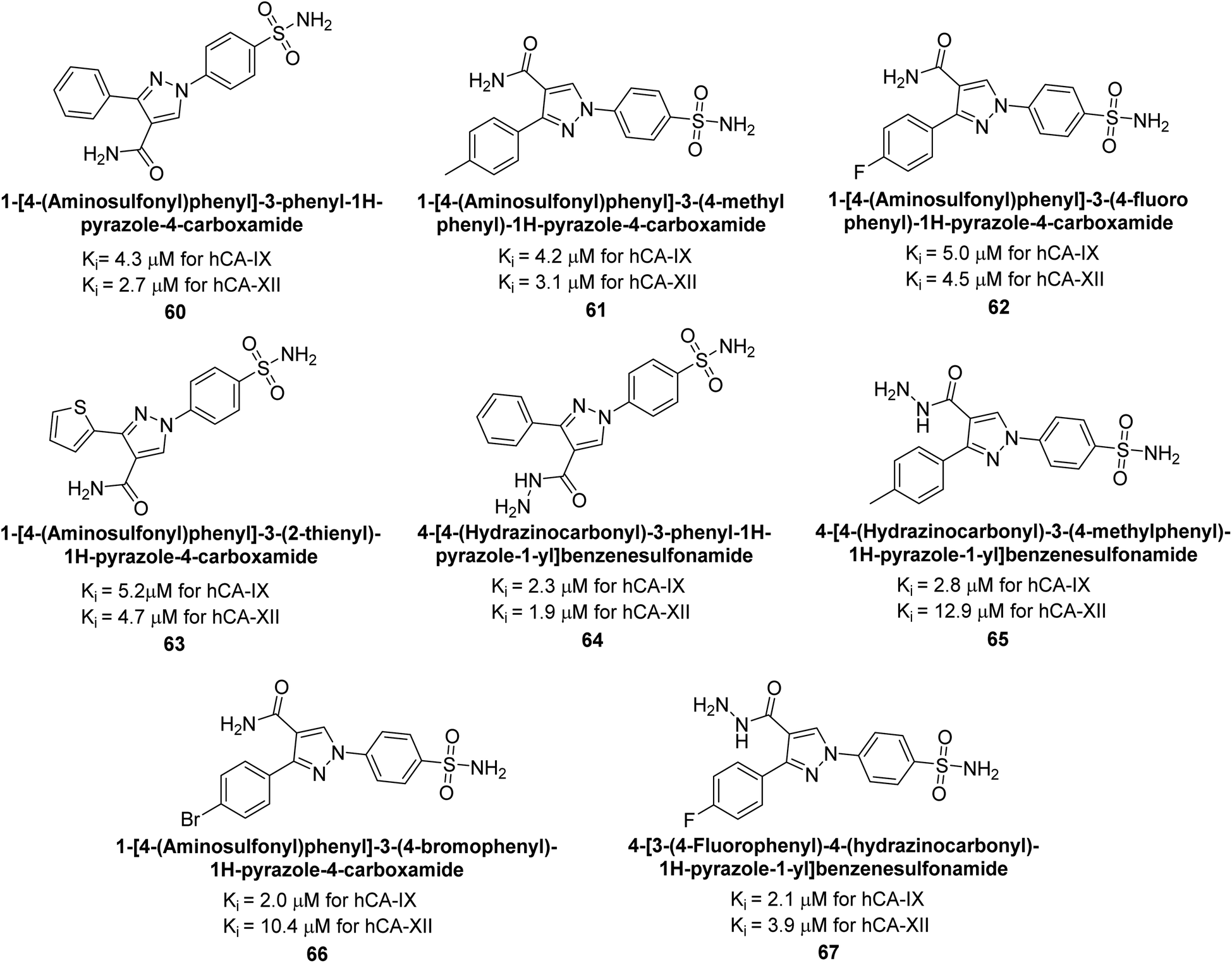 | ||
| Fig. 22 Chemical structure of compounds 60–67 and their Ki values against hCA-IX and XII. | ||
Gençer et al. (2014) investigated saccharin derivatives on purified hCA-I and II using CO2 as a substrate. The findings revealed that all tested analogues exhibited inhibitory effects on both hCA-I and II enzyme activities. Particularly, compound 68 displayed the highest activity against hCA-I (IC50 = 13.67 μM), while compound 69 exhibited the greatest activity against hCA-II (IC50 = 6.54 μM) (Fig. 23). SAR analysis indicated that, generally, thiourea derivatives displayed greater inhibition of both hCA-I and II compared to urea derivatives. Based on the findings, it is suggested that saccharin derivatives hold a promising potential for the treatment of glaucoma.105
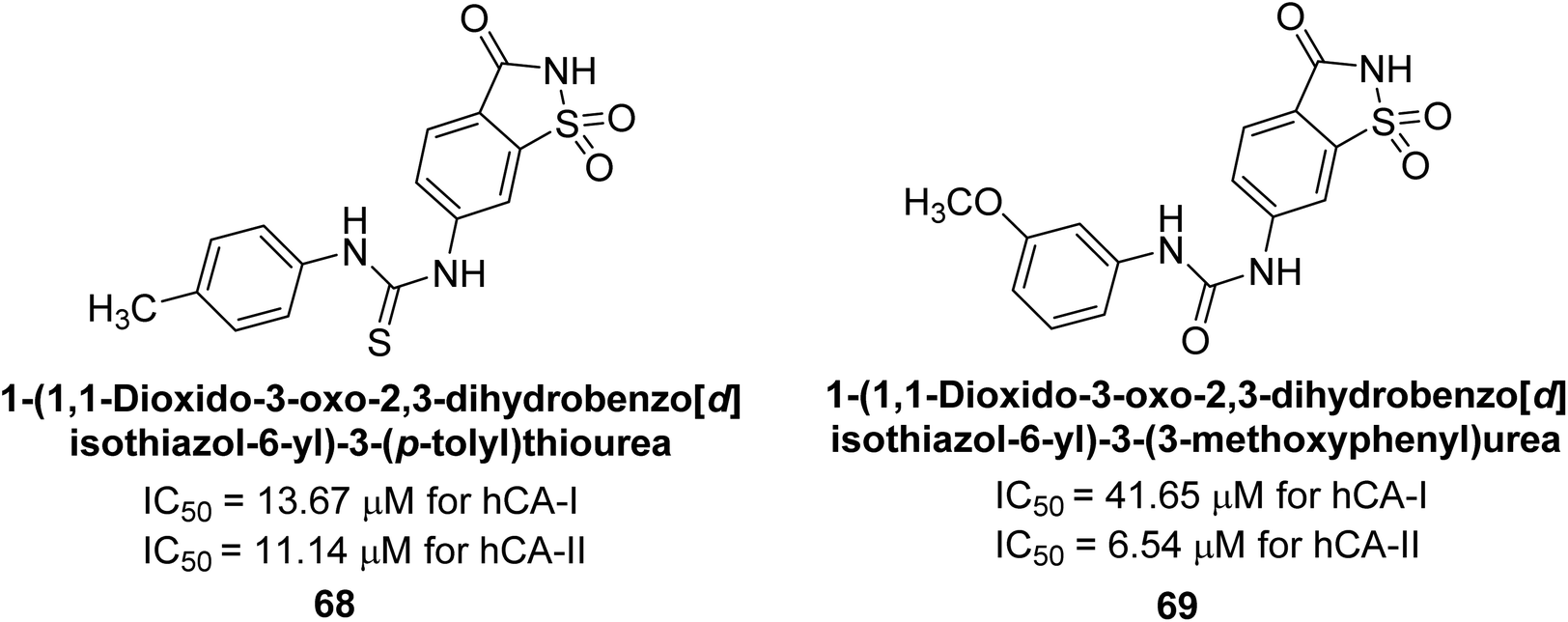 | ||
| Fig. 23 Chemical structure of compounds 68 and 69 and their IC50 values against hCA-I and II. | ||
Sharma et al. (2014) designed triazole-based benzenesulfonamides and evaluated their inhibitory effects against hCA-IX and XII. All target derivatives showed moderate to good inhibitory potential, with Ki values ranging from 2.8 to 431 nM and 1.3 to 63 nM, against hCA-IX and XII, respectively. Compounds 70 and 71, identified as the most potent inhibitors of hCA-IX and XII, exhibited significantly higher IC50 as compared to the standard AAZ (Fig. 24). Additionally, the activity profile revealed that both the nuclear triazole scaffold and triazole fused with thiadiazines serve as highly effective inhibitors of hCA-IX and XII.106
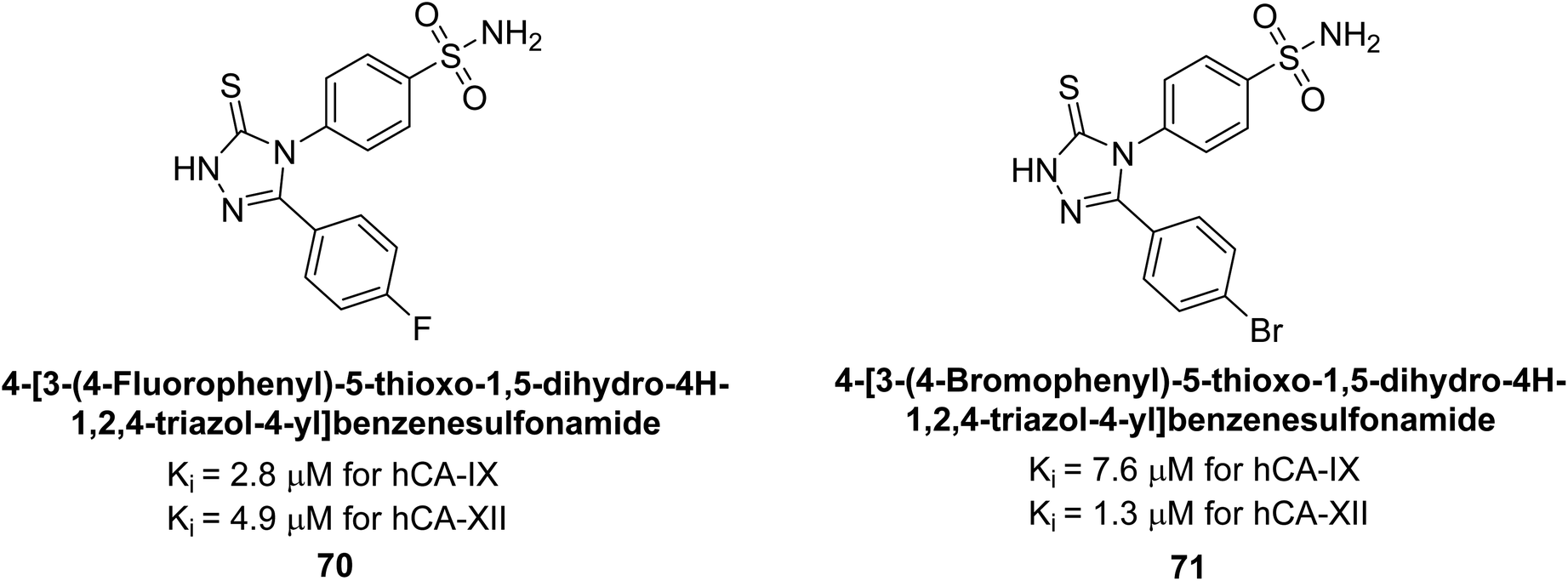 | ||
| Fig. 24 Chemical structure of compounds 70 and 71 and their Ki values against hCA-XI and XII. | ||
Gençer et al. (2014) investigated the inhibitory effects of indolylchalcones and pyrimidines on hCA-I and II. Among the synthesized derivatives, 72 and 73 exhibited the highest inhibitory activities against hCA-I (IC50 = 6.79 μM) and hCA-II (IC50 = 7.22 μM), respectively (Fig. 25). SAR analysis indicated that chalcones exhibited greater IC50 values than pyrimidine derivatives against both hCA-I and II. Furthermore, the presence of a methyl group bonded to the uracil ring increased inhibitory activities against both hCA-I and II. Hence, the compounds examined in this study are promising candidates for drug development and further assessment in in vivo studies.107
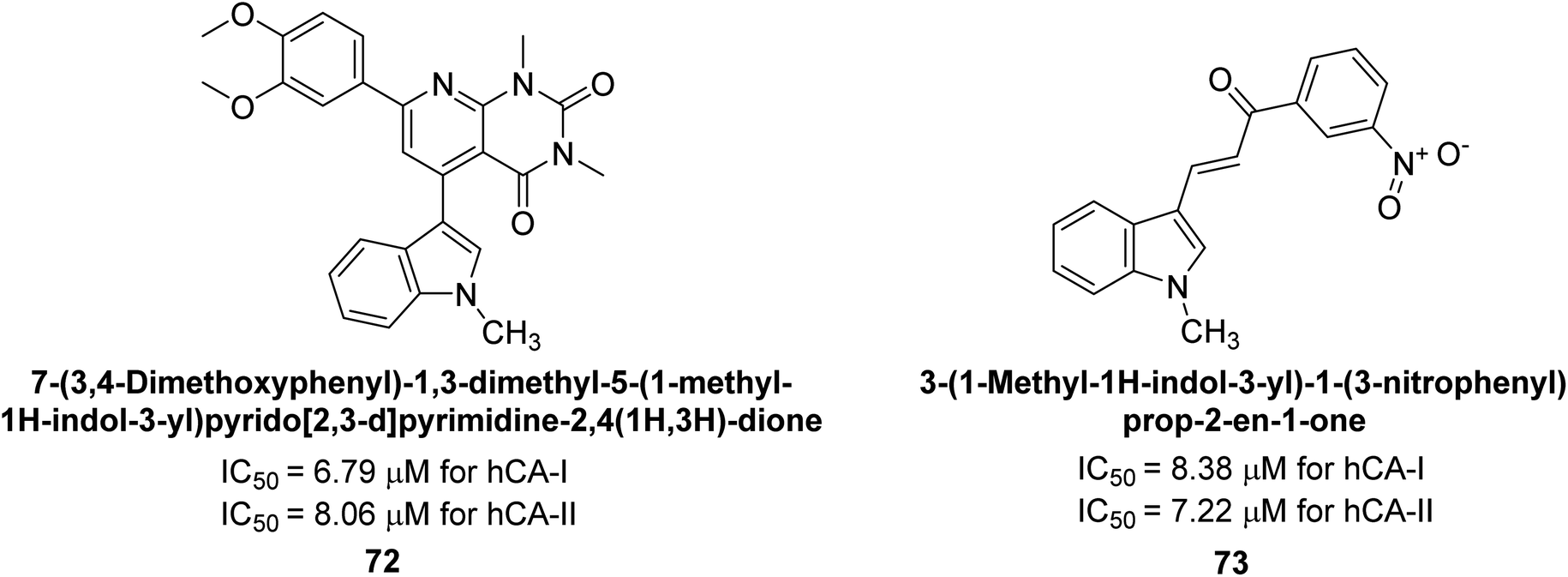 | ||
| Fig. 25 Chemical structure of compounds 72 and 73 and their IC50 values against hCA-I and II. | ||
Iqbal et al. (2014) synthesized a series of thioureas-based sulfonamides, and assessed for their IC50 on bovine CA (bCA-II). These compounds demonstrated considerable activity, with the most potent inhibitor in the series 74 exhibiting an IC50 = 0.261 μM. Both molecular docking and SAR analyses indicate that ortho substitution with bulky electronegative groups (e.g., –NO2) enhances the inhibitory activity against CA. The increased inhibition observed for compound 74 is attributed to the enhanced stability of the enzyme–inhibitor complex, which is facilitated by Thr200 forming two hydrogen bonds: one with the nitrogen atom of the amide group and the other with the oxygen atom of the nitro group (Fig. 26).108
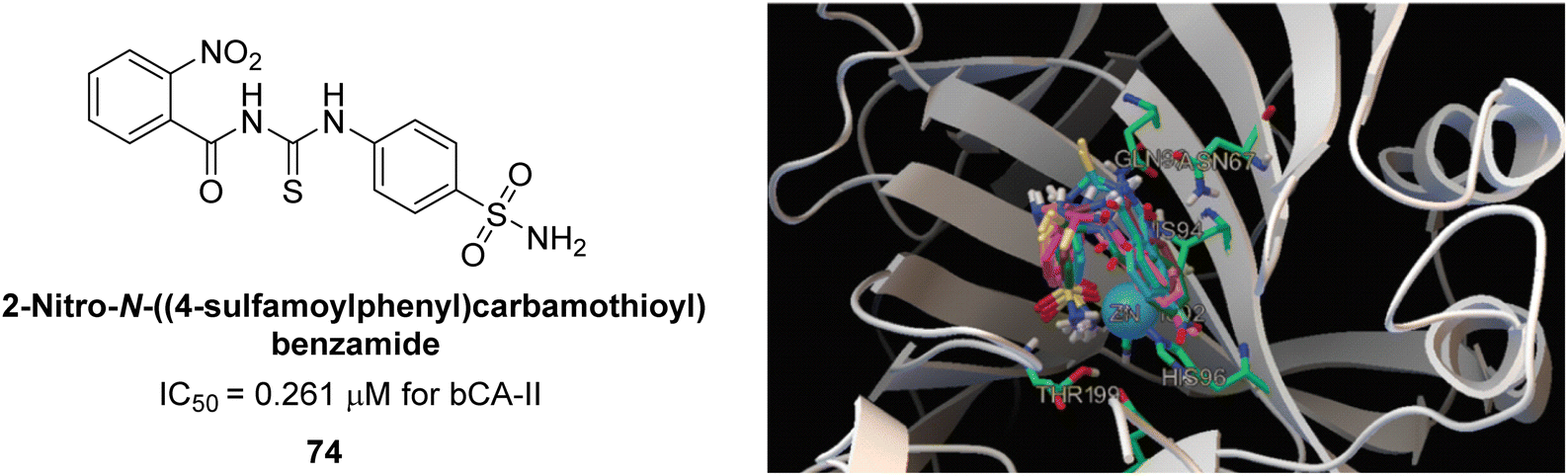 | ||
| Fig. 26 Chemical structure of compound 74 and its IC50 value against bCA-II along with its docking image. | ||
Maraş et al. (2014) synthesized and evaluated sulfamides and demonstrated competitive inhibition of hCA-I and II at nanomolar concentrations. Among the synthesized compounds, derivative 75 was the most effective inhibitor of hCA-I (Ki = 153.88 nM), while derivative 76 was the most potent against hCA-II (Ki = 117.80 nM) (Fig. 27). These findings indicate that the synthesized sulfamide carbamates and sulfamides have the potential to serve as potent inhibitors of hCA isoenzymes, making them promising candidates for the treatment of conditions such as obesity, glaucoma and epilepsy.109
 | ||
| Fig. 27 Chemical structure of compounds 75 and 76 and their Ki values against hCA-I and II. | ||
Sechi et al. (2014) synthesized benzene- and tetrafluorobenzene-sulfonamides using click chemistry and evaluated against hCA-IX and XII isoforms. Remarkably, only two compounds 77 and 78, featuring an electron-withdrawing carboxymethyl group, exhibited reduced activity against hCA-IX and XII as compared to the standard AAZ (Fig. 28). Compound 77, featuring a nonpolar, puckered cyclohexyl ring, was located in the hydrophobic pocket formed by residues L204, P202, V135 and F131. In contrast, the tail of compound 78 was oriented toward the bulk solvent and did not engage in polar or hydrophobic interactions with the protein surface (Fig. 28).110
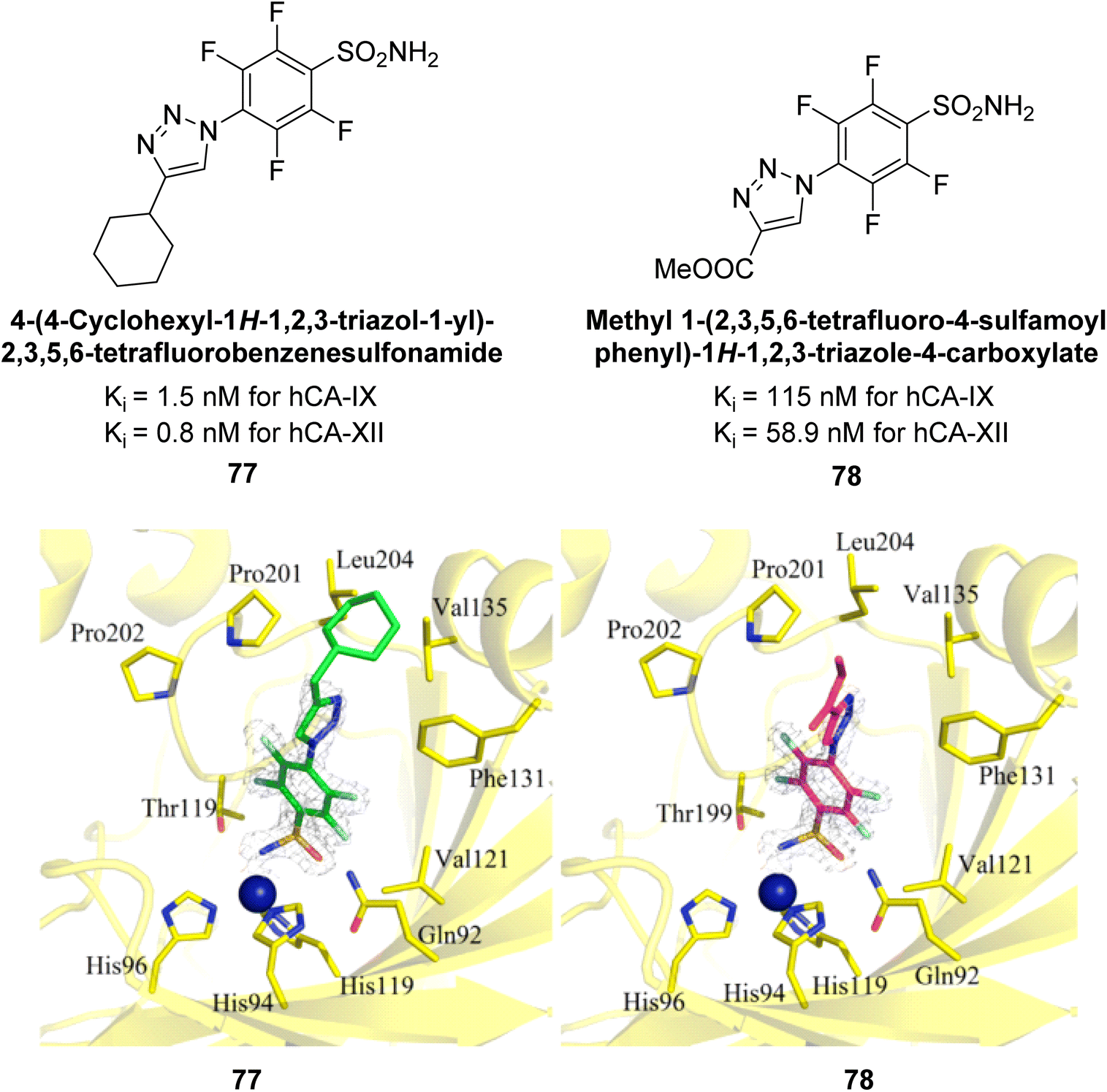 | ||
| Fig. 28 Chemical structure of compounds 77 and 78 and their Ki values against hCA-IX and XII. | ||
Sławiński et al. (2014) synthesized a series of substituted guanidines and evaluated them against two isoforms CA-IX and XII. Interestingly, certain compounds exhibited potent inhibitory activity against hCA-IX, with Ki values ranging from 4.7 to 21 nM, surpassing the reference sulfonamides AAZ, MZA, EZA, DCP, and IND, which had Ki values of 24 to 50 nM. Among these, compound 79 emerged as the most potent inhibitor of hCA-IX (Ki = 4.7 nM), and hCA-XII (Ki = 0.96 nM) (Fig. 29). Future perspectives could involve further optimization of these compounds to enhance their selectivity and efficacy as potential anticancer agents, as well as exploring their therapeutic potential in preclinical and clinical studies. Additionally, structural modifications could be explored to improve pharmacokinetic properties and reduce off-target effects, ultimately advancing their utility in cancer therapy.111
 | ||
| Fig. 29 Chemical structure of compound 79 its Ki values against hCA-IX and XII. | ||
Gençer et al. (2014) designed, synthesized and assessed hCA-I properties of 1,4-dihydropyrimidinone compounds. The compounds were synthesized using a multicomponent Biginelli reaction. Among the synthesized compounds, compound 80 (IC50 = 0.0547 μM) showed excellent inhibitory activity (Fig. 30). These findings underscore the potential of substituted 1,4-dihydropyrimidinone compounds as promising candidates for further development as CA inhibitors with potential implications in cancer therapy.112
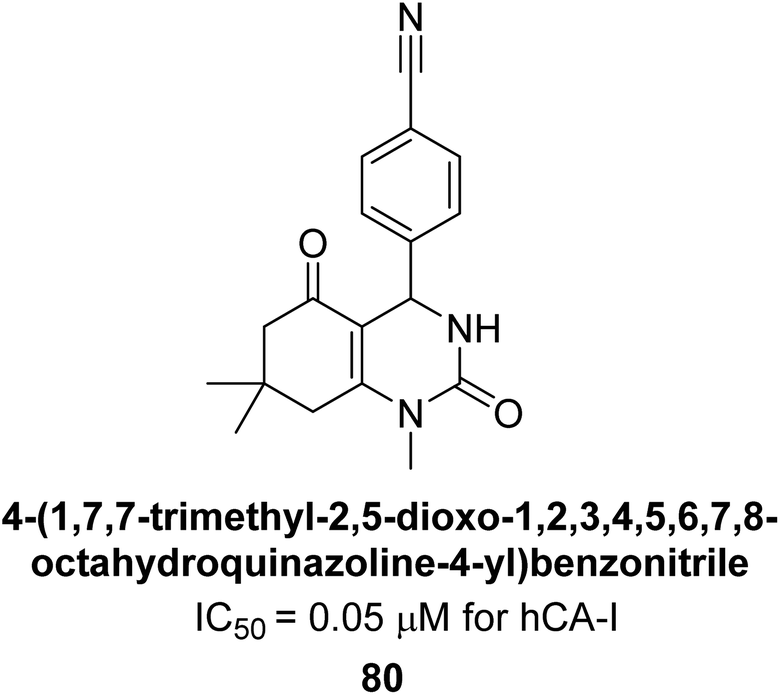 | ||
| Fig. 30 Chemical structure of compound 80 and its IC50 value against hCA-I. | ||
Gençer et al. (2014) synthesized 1,3-dicarbonyl derivatives of substituted sulfonamides and assessed their inhibitory effects on hCA-I and II. The compounds showed effective inhibition of both isoforms, with compounds 81 and 82 demonstrating the highest activity due to methyl substitutions, achieving IC50 values of 2.12 μM for hCA-I and 2.52 μM for hCA-II (Fig. 31). These sulfonamide derivatives are promising for glaucoma treatment and allow further in vivo evaluation.113
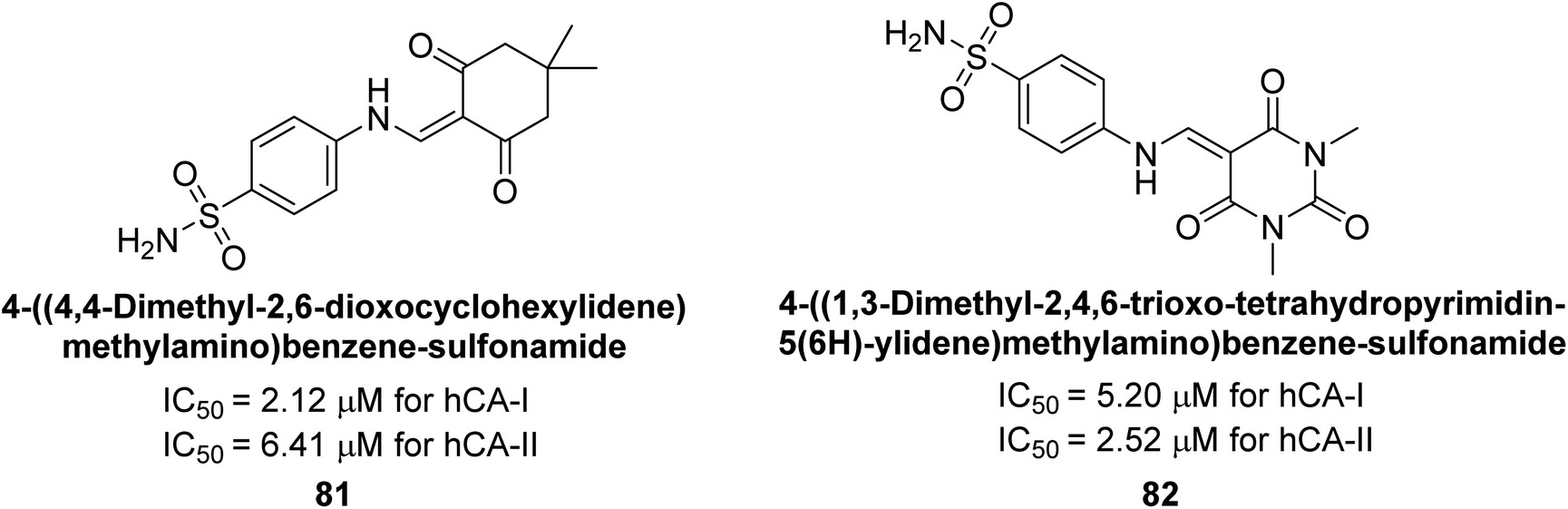 | ||
| Fig. 31 Chemical structures of compounds 81 and 82 and their IC50 values against hCA-I and II. | ||
Gülçin et al. (2014) designed and synthesized sulphamides and sulfonamides and assessed heir inhibitory activity against hCA-I and II. Among the tested derivatives, compounds 83 and 84 displayed a particularly potent inhibitory effect on hCA, with Ki = 0.71 μM and 1.56 μM (hCA-I) and Ki = 0.69 μM and 0.39 μM (hCA-II), respectively (Fig. 32), against the reference standard AAZ. These findings suggest that the target compounds hold as potent inhibitors of CA, highlighting their potential for further exploration as therapeutic agents.114
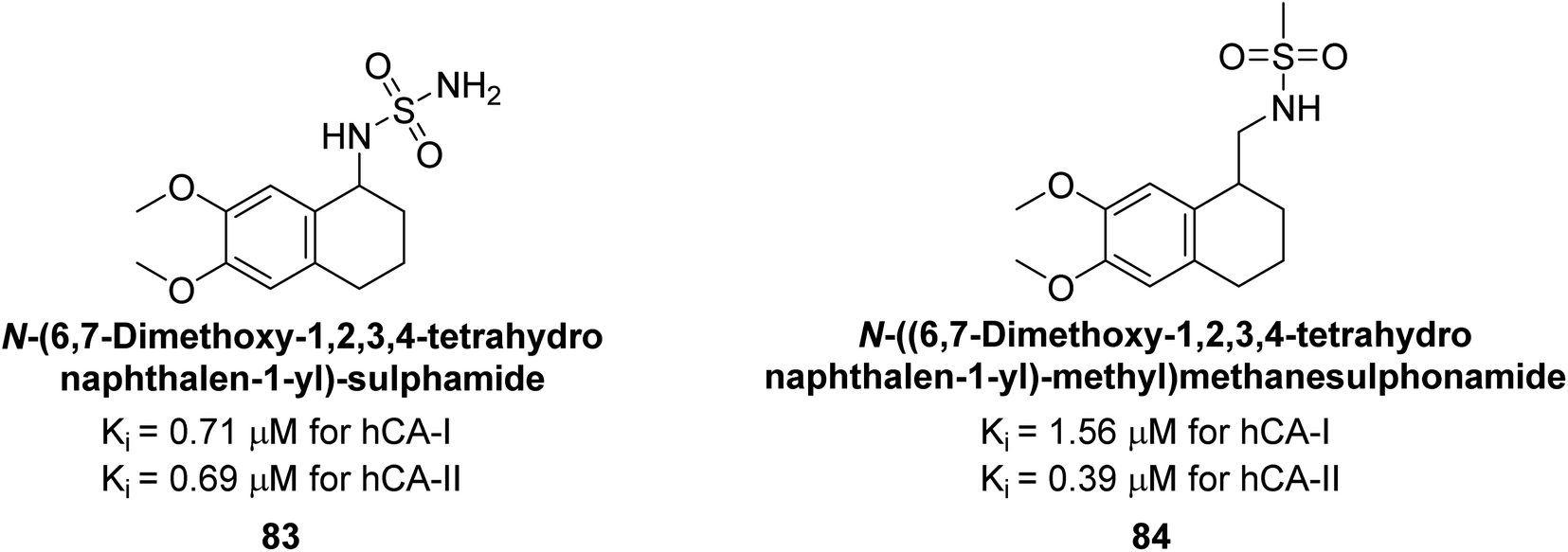 | ||
| Fig. 32 Chemical structure of compounds 83 and 84 and their Ki values against hCA-I and II. | ||
Saeed et al. (2014) synthesized sulfonamide-thiourea and evaluated their inhibitory activity against bCA-II with inhibition constants falling within the range of 0.011 to 17.1 μM. Remarkably, among the compounds evaluated, compound 85 and 86 exhibited the highest potency as inhibitors, demonstrating IC50 values of 0.052 μM and 0.011 μM, respectively (Fig. 33). These analyses aimed to elucidate the structural interactions and dynamics governing the inhibitory properties observed, providing valuable insights for further development of potential therapeutic agents targeting CA enzymes.115
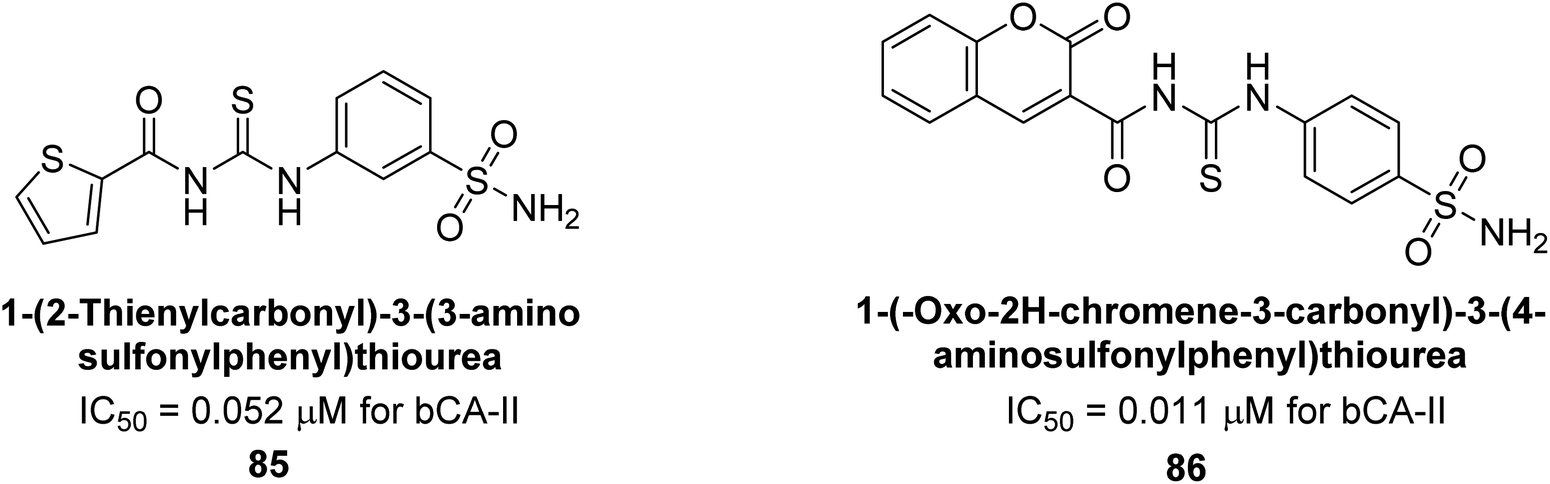 | ||
| Fig. 33 Chemical structure of compounds 85 and 86 and their IC50 values against bCA-II. | ||
Sławiński et al. (2014) synthesized substituted sulfonamides and assessed for their inhibitory activity against hCA-I, II, IX and XII. The investigated compounds exhibited varying inhibitory potencies against these isoforms. Remarkably, the compounds demonstrated excellent inhibitory activity against hCA-IX, with synthesized compounds showing potent Ki values ranging from 2.8 to 21.7 nM compared to clinically used CA inhibitors with Ki values between 24 and 50 nM. Particularly, compound 87 emerged as the most potent hCA-IX inhibitor with a Ki = 2.8 nM, which was 8.5-fold stronger than IND (Ki = 24 nM). Furthermore, compounds 88 and 89 demonstrated superior inhibitory potency against hCA-XII with Ki values of 2.7 and 2.8 nM, respectively, surpassing the reference sulfonamides MZA and IND (Ki = 3.4 nM) (Fig. 34).116
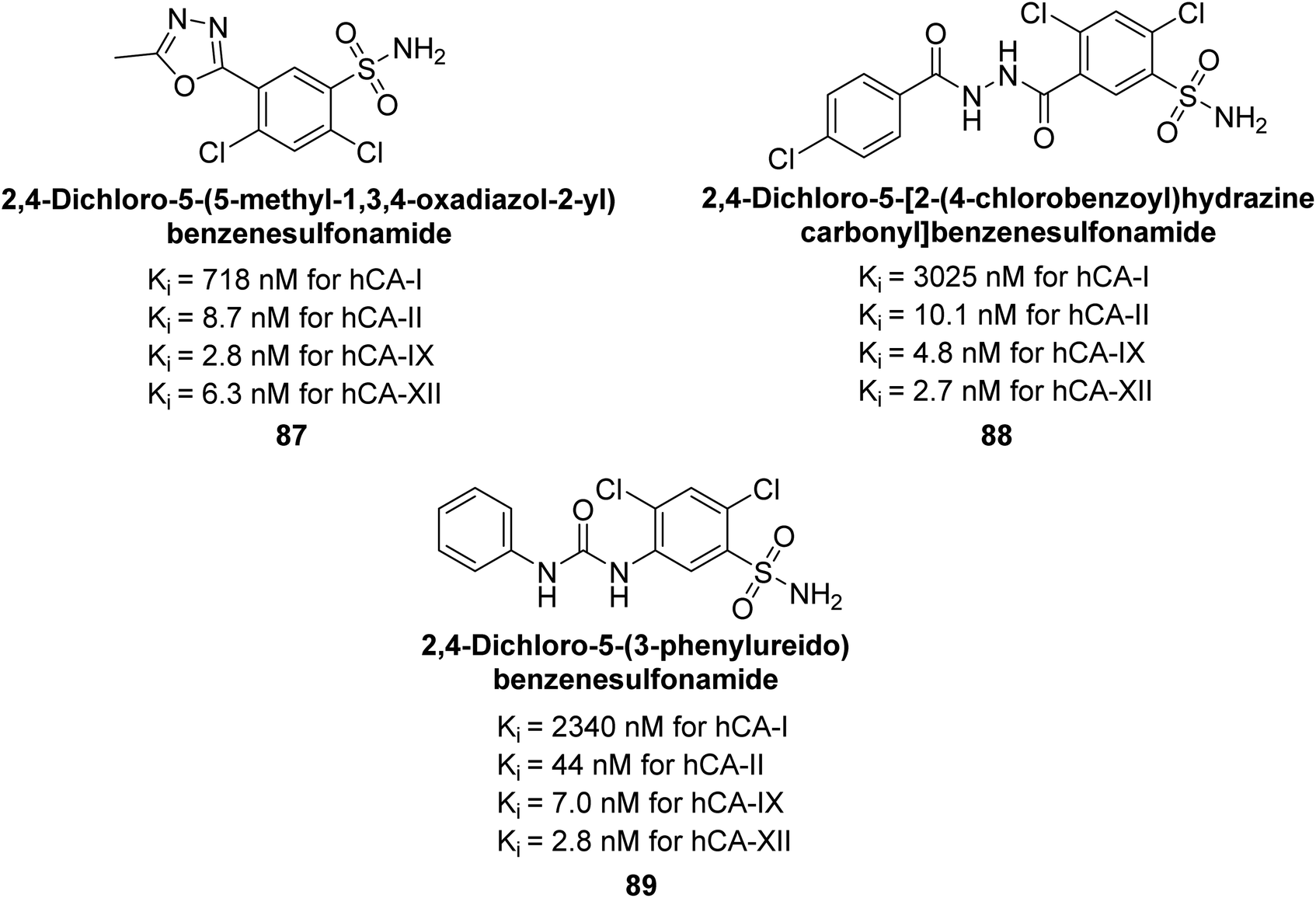 | ||
| Fig. 34 Chemical structure of compounds 87, 88 and 89 and their Ki values against hCA-I, II, IX and XII. | ||
Zhu et al. (2014) designed and synthesized metronidazole-sulfonamides and evaluated for their inhibitory effects on hCA-II and IX. Many of these compounds demonstrated significant inhibition against hCA-II and IX, with IC50 values falling within the ranges of 16–137 nM and 38–169 nM, respectively. Of all the compounds tested, compounds 90 (IC50 = 16 nM) and 91 (IC50 = 38 nM) emerged as the most potent inhibitors against hCA-II and IX, respectively (Fig. 35).117
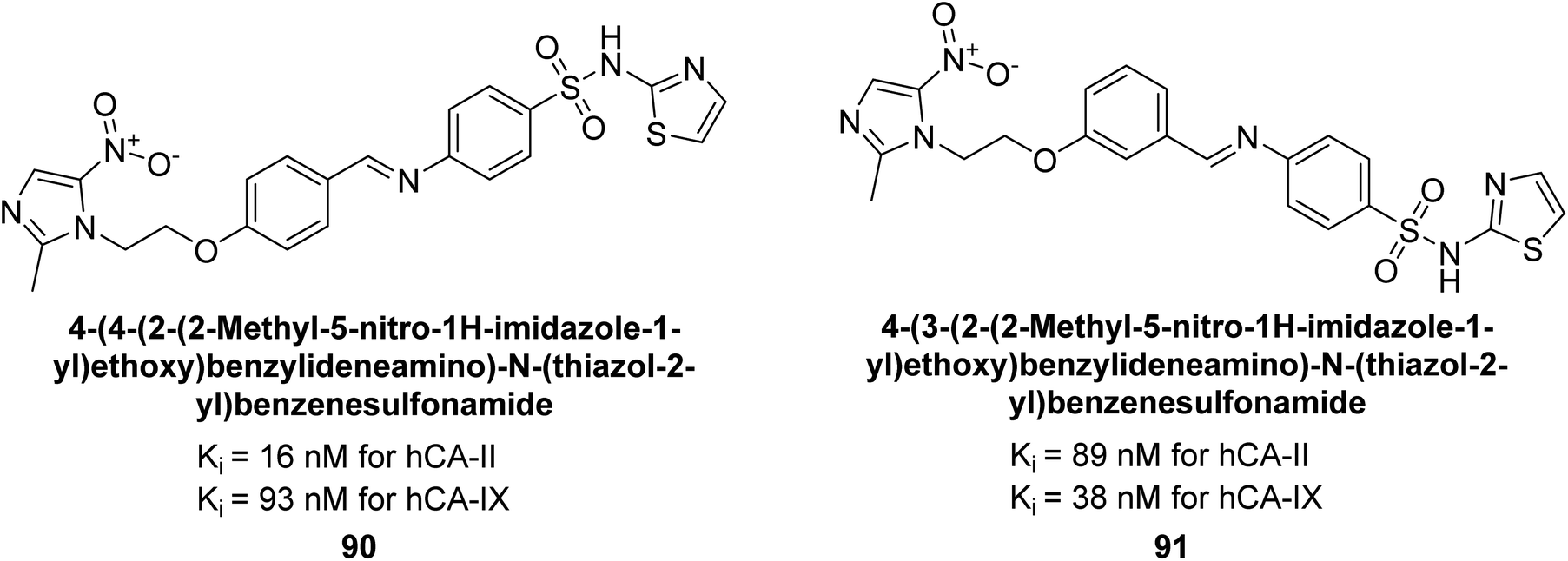 | ||
| Fig. 35 Chemical structure of compounds 90 and 91 and their Ki values against hCA-I, II, IX and XII. | ||
Supuran et al. (2014) synthesized sulfonyl semicarbazides and measured for their inhibitory effects on hCA. These compounds were evaluated against a panel of hCA isoforms including I, II, IX, and XII, with AAZ serving as the standard reference. The 4-nitro substituted derivative 92 was identified as the most potent inhibitor, with Ki values of 6.0 nM for hCA-II and 0.79 nM for hCA-IX (Fig. 36). The study highlighted the importance of p-substitution on the sulfonyl-substituted aromatic ring in influencing both potency and selectivity for specific hCA isoforms. Overall, the research developed highly potent and selective hCA inhibitors with promising therapeutic potential.118
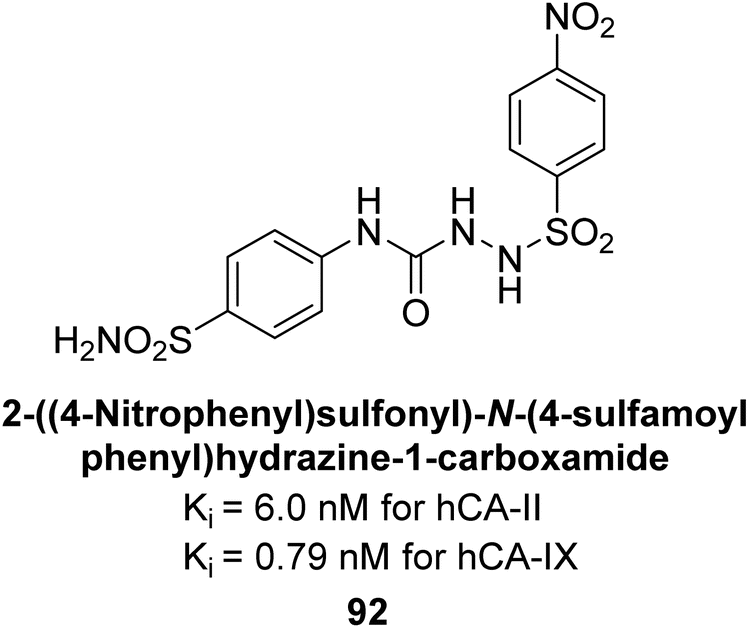 | ||
| Fig. 36 Chemical structure of compound 92 and their Ki values against hCA-II and IX. | ||
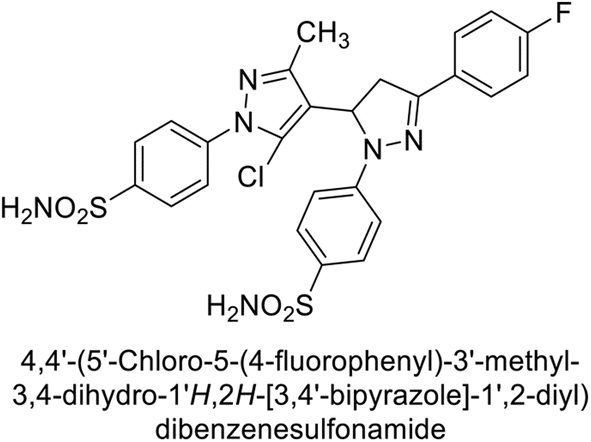
Supuran et al. (2015) synthesized pyrazolines and assessed for their inhibitory activity against CA enzymes. Particularly, compounds 93, 94, and 95–100 exhibited superior inhibition against hCA-XII (Ki = 0.47–5.1 nM) compared to AAZ (Ki = 5.7 nM), with compounds 95, 97, 98 and 101 showing nearly a 10-fold improvement over the reference drug. Against hCA-II, all tested compounds surpassed the standard drug, particularly compounds 93, 97, 98 and 102 (Ki = 1.1–1.7 nM), which were significantly more potent inhibitors than AAZ (Ki = 12.1 nM) (Table 8).119
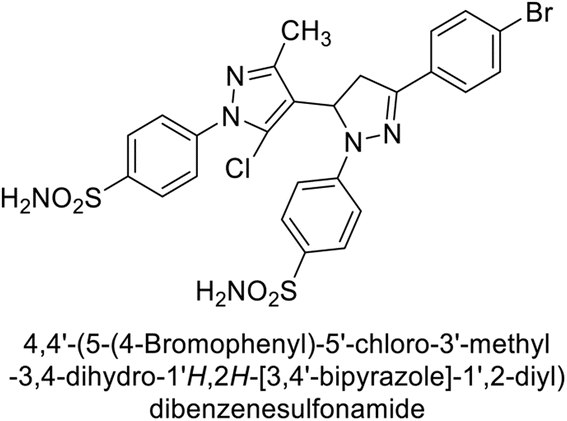
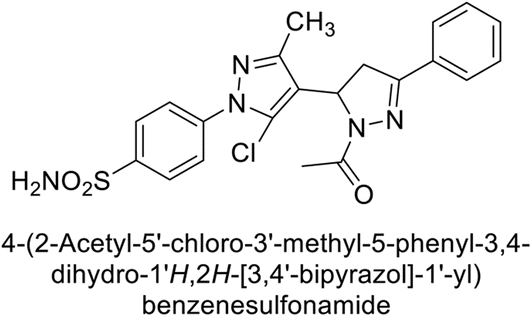
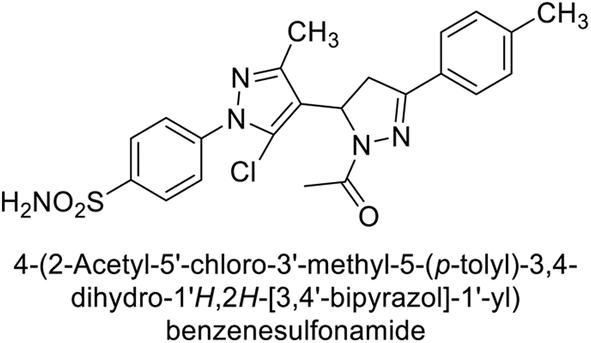
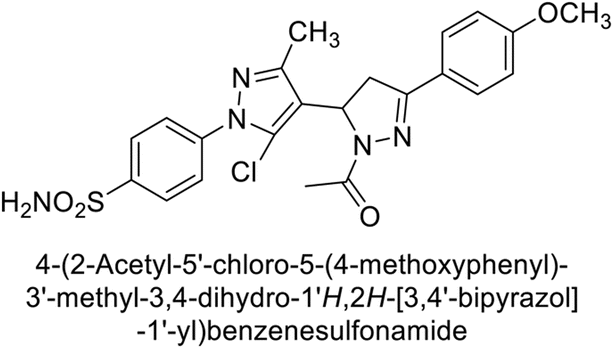
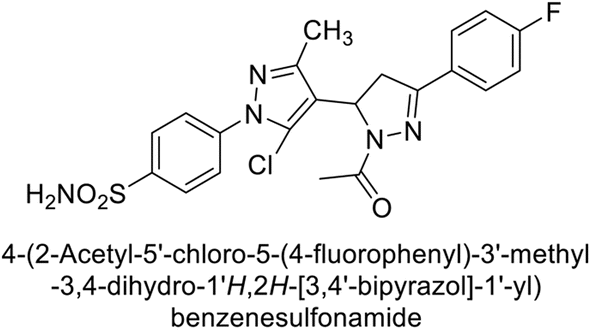
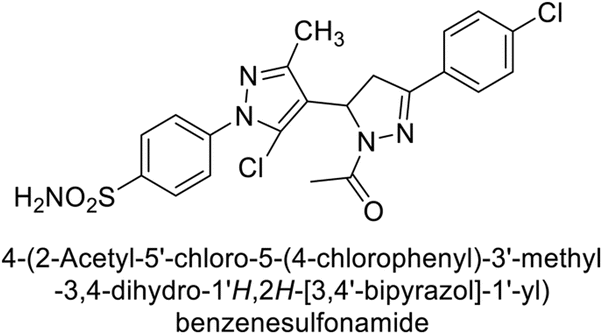
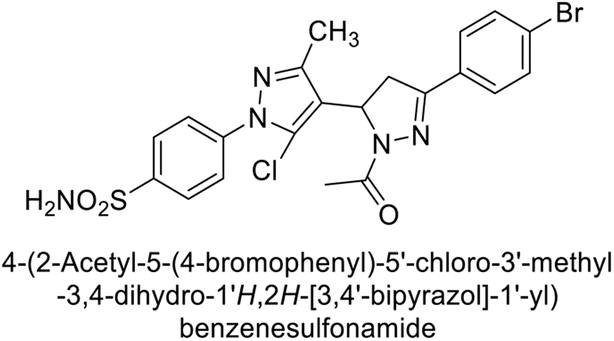
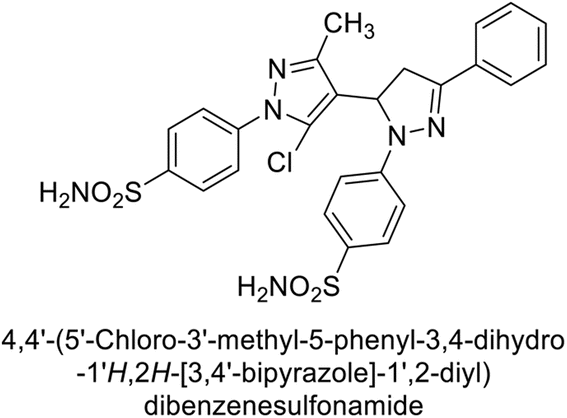
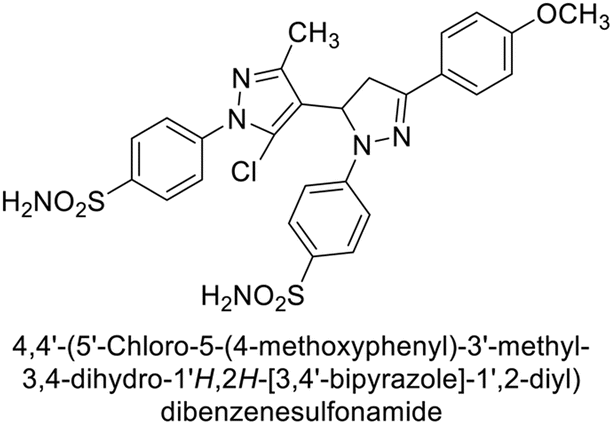
Ismail et al. (2015) designed and synthesized bisindolylmethanes and assessed for their potential inhibitory activity against hCA-II. Particularly, bisindoles with halogen substituents at the fifth position demonstrated enhanced inhibitory activity compared to their unsubstituted counterparts. Moreover, docking investigations of the active compound 103 (IC50 = 42.9 μM) indicated that the p-positioned nitro (NO2) substituent is adept at fitting into the core of the active site and interacting with the Zn ion. Compounds containing a NO2 substituent exhibited close interaction with Zn ion, His94, His119, Thr199 and His96 disrupting the Zn–OH– Glu106–Thr199 hydrogen bond network (Fig. 37). Despite the bulky nature of the indole ring, it was unable to directly interact with the active site. However, the NH group of the indole ring was observed to form a hydrogen bond with Glu69, contributing to the stabilization of the ligand–receptor complex. These newly discovered compounds demonstrate inhibitory activity worthy of consideration for future development.120
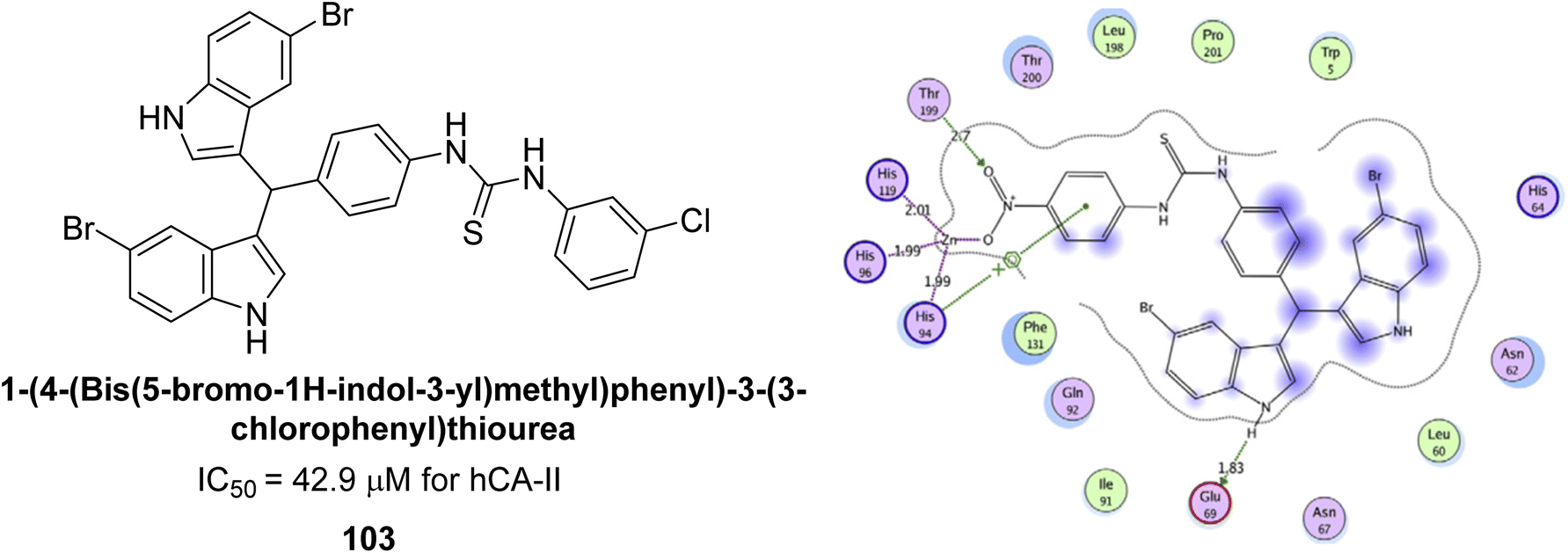 | ||
| Fig. 37 Chemical structure of compound 103 and its IC50 value against hCA-II along with its docking image. | ||
Gitto et al. (2015) synthesized a series of substituted benzenesulfonamides and assessed as inhibitors of hCAs. These novel sulfonamide derivatives underwent screening against hCA-IX, with AAZ and topiramate (TPM). Notably, compounds 104–109 exhibited low nanomolar inhibition of the hCA-IX isoform (Ki values < 10 nM) (Fig. 38). These findings contribute to the development of targeted therapies for conditions associated with dysregulated CA activity, particularly in cancer.121
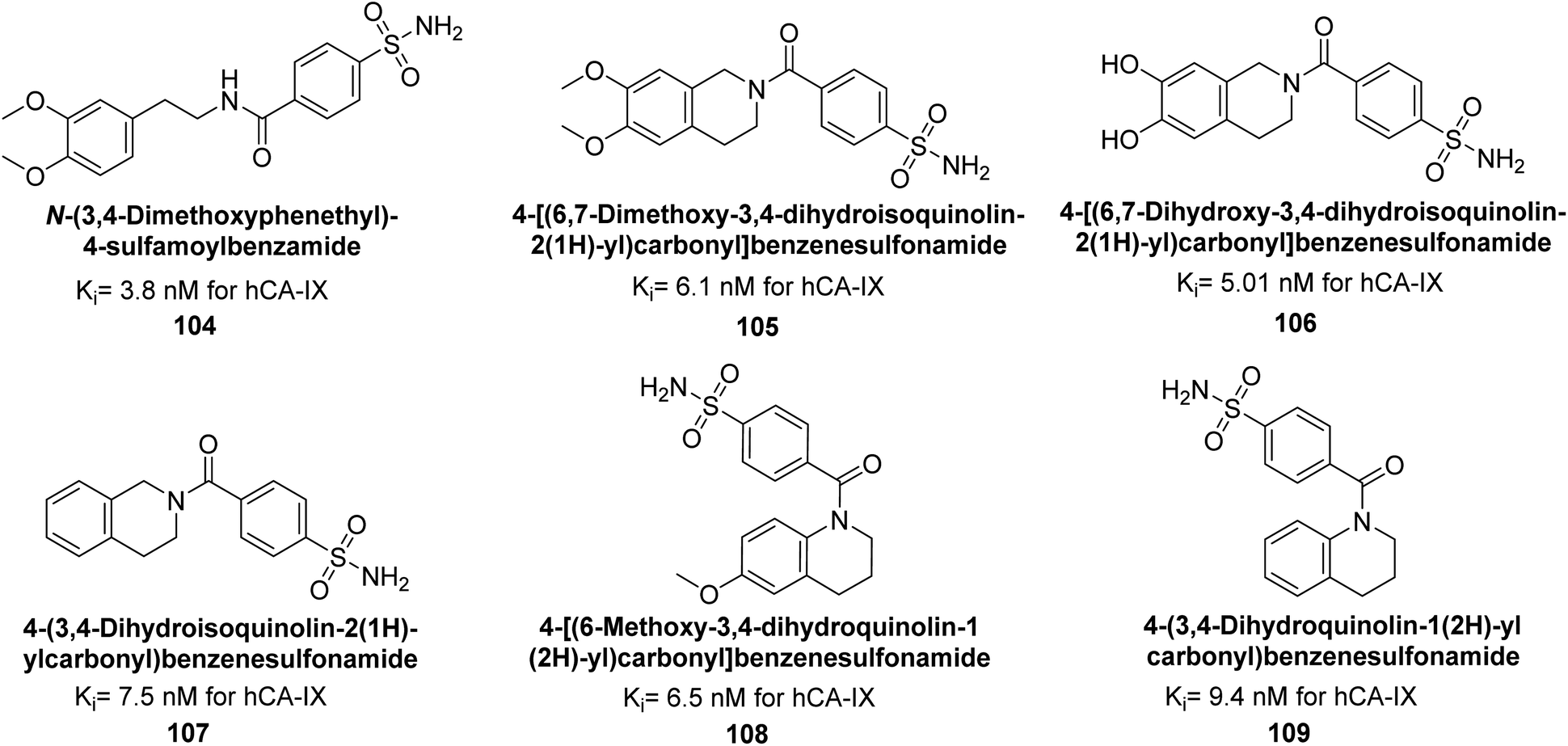 | ||
| Fig. 38 Chemical structure of compounds 104–109 and their Ki values against hCA-IX. | ||
Supuran et al. (2015) synthesized a series of benzenesulfonamide derivatives containing pyrazole and isatin functionalities based on celecoxib as the lead compound. The biological activity of these derivatives was evaluated against hCA-IX and XII. The majority of the tested compounds demonstrated significant inhibition of hCA-IX with Ki as low as 2.5 × 102 nM, surpassing the potency of the standard drug AAZ. Specifically, compounds 110–113 displayed notable inhibitory activity against hCA-XII, with Ki values ranging from 3.7–7.2 nM (Fig. 39). Compounds 110 and 112, featuring a 5-nitro substitution on the isatin moiety, exhibited selectivity towards inhibiting hCA-XII. Molecular docking studies revealed that the NO2 group in these compounds interacted with Asp132 in the active site of hCA-IX and with Asp130 and Lys67 in hCA-XII (Fig. 39).122
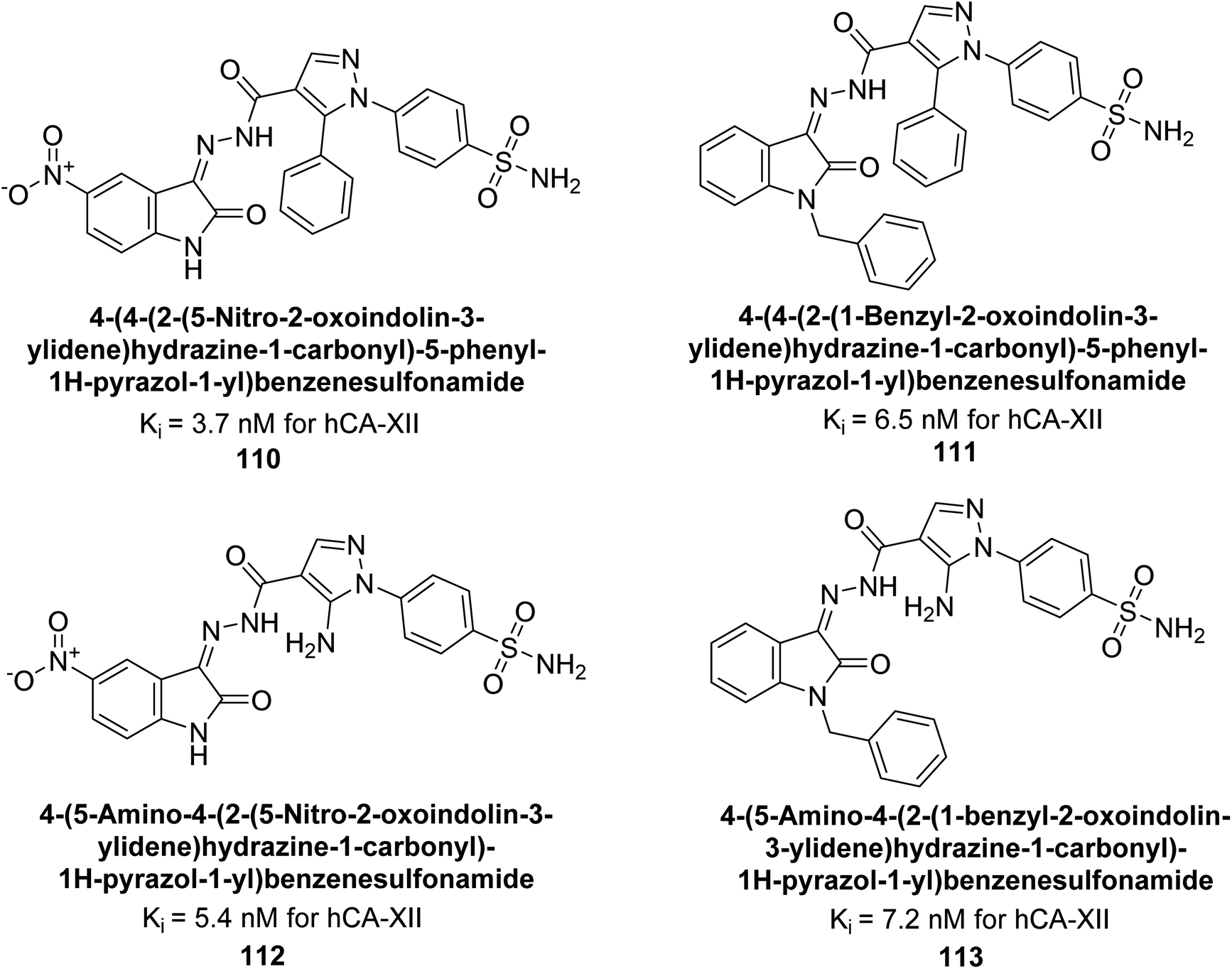 | ||
| Fig. 39 Chemical structure of compounds 110–113 and their Ki values against hCA-XII. | ||
Gençer et al. (2015) synthesized a series of substituted β-lactam derivatives and assessed their inhibitory effects on the activity of hCA-I and II. Remarkably, the compound 114 exhibited the most potent inhibitory effect, with IC50 values of 6.97 μM for hCA-I and 8.48 μM for hCA-II (Fig. 40). The bulky phthalazine group influences the inhibitory effects of these compounds. They likely bind similarly to coumarin derivatives. Additionally, the interaction involves the enzyme's active site and the β-lactam ring.123
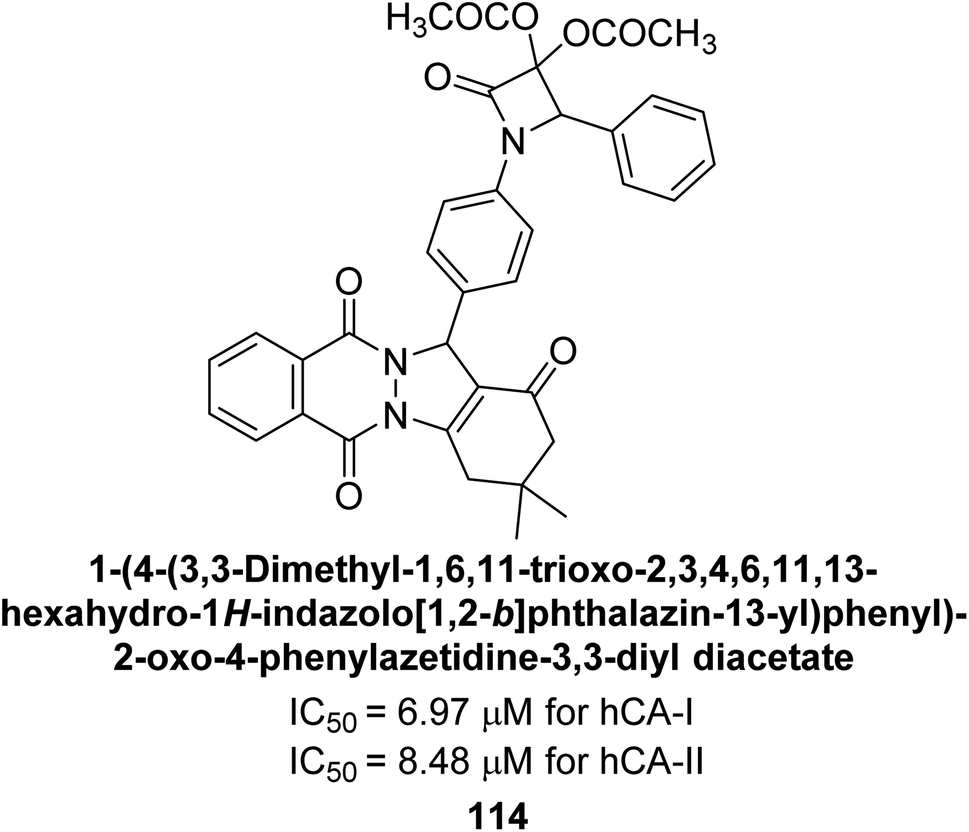 | ||
| Fig. 40 Chemical structure of compound 114 and its IC50 values against hCA-I and II. | ||
Toraskar et al. (2015) synthesized and evaluated Schiff bases from sulfanilamide semicarbazone and various heterocyclic aldehydes and evaluated against different CA isoforms. Compounds 115, 116, 117 and 118 demonstrated hCA-I inhibition (IC50 = 71.2–95.2 nM), surpassing the standard AAZ. Only 119 (2,4-dichlorophenyl) and 120 (3-nitrophenyl) were moderately effective hCA-II inhibitors, with Ki of 51.6–75.0 nM. For CA-IX, only compound 121 (4-fluorophenyl) showed comparable activity to AAZ (Ki = 27.3 nM). Compounds 118 and 122 (with 2-thienyl and 9-anthranyl moieties, respectively) were effective hCA-XII inhibitors (Ki = 21.5–23.8 nM) (Table 9).124
| Compound no. | Chemical structures & IUPAC names | Ki (nM) | Ref. | |||
|---|---|---|---|---|---|---|
| CA-I | CA-II | CA-IX | CA-XII | |||
| 115 | 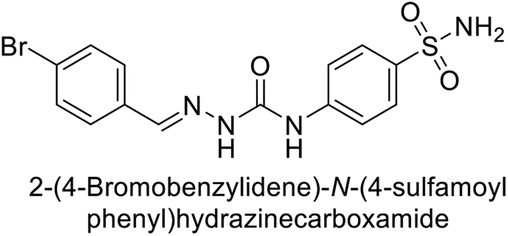 |
95.2 | 6.2 | 90.4 | 92.3 | 124 |
| 116 | 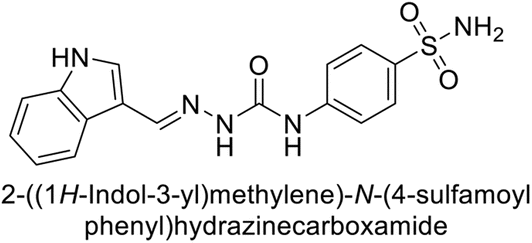 |
71.2 | 4.0 | 103 | 63.1 | 124 |
| 117 | 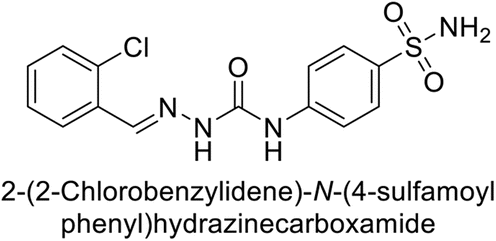 |
77.6 | 4.2 | 213 | 42.3 | 124 |
| 118 | 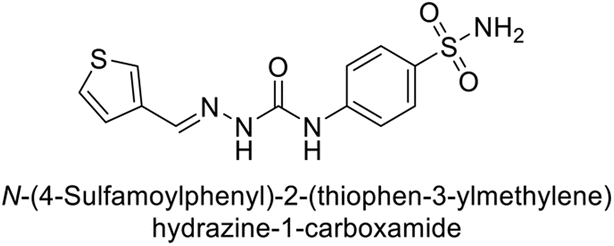 |
79.3 | 4.6 | 395 | 23.8 | 124 |
| 119 | 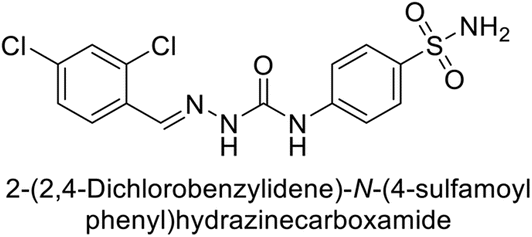 |
6530 | 75.0 | 98.1 | 90.7 | 124 |
| 120 | 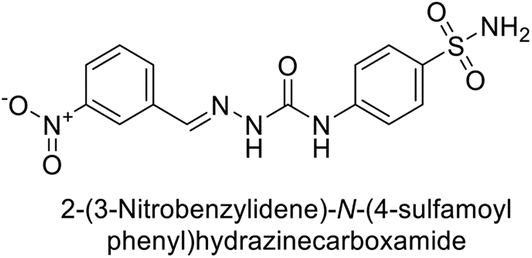 |
4930 | 51.6 | 415 | 83.9 | 124 |
| 121 |  |
675 | 5.6 | 27.3 | 95.8 | 124 |
| 122 |  |
1640 | 19.5 | 381 | 21.5 | 124 |
| Standard |  |
250 | 12 | 25 | 5.8 | 124 |
Khan et al. (2015) synthesized quinazoline derivatives and tested for their inhibitory activity against bCA-II. Compounds 123 (IC50 = 61.33 μM), 124 (IC50 = 108.30 μM), 125 (IC50 = 191.93 μM) and 126 (IC50 = 20.94 μM) exhibited selective inhibition of bCA-II, compared to the standard AAZ (IC50 = 0.12 μM) for bCA-II. The inhibitory activity of these derivatives is attributed to the different substituents (–NO2, –OH, –OMe) the motif (Fig. 41).125 Further refinement of these lead molecules, through kinetic and in vivo studies, may yield potent inhibitors for both enzymes.
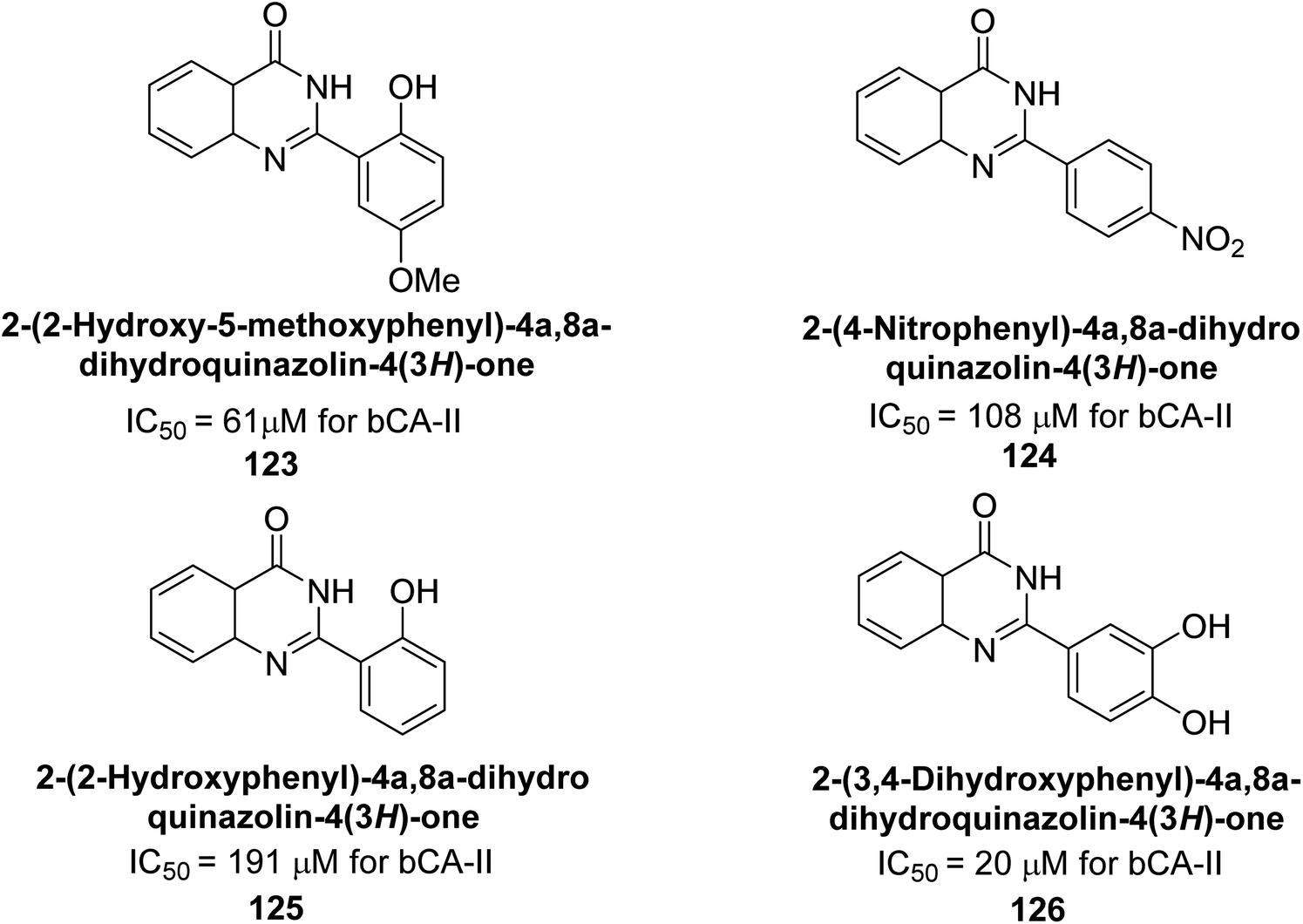 | ||
| Fig. 41 Chemical structure of compounds 123–126 and their IC50 values against bCA-II. | ||
Supuran et al. (2015) synthesized 2-aminobenzothiazole derivatives and evaluated as inhibitors of hCA-XII. Compound 127 (Ki = 9.5 nM) demonstrated selective inhibition of hCA-XII, indicating that certain heterocyclic moieties can be utilized to design effective and selective inhibitors for hCA-XII as compared to AAZ (Ki = 22 nM). Compound 127 primarily binds through coordination of the deprotonated primary sulfonamide group with the active site zinc ion. Secondary interactions involve the benzothiazole ring occupying regions more than 5 Å from the zinc, enhancing binding. The amide linker between the heterocycles facilitates favorable conformations and interactions with a solvent-exposed region defined by Ser130, Arg60, Gln89 and Asn62 (Fig. 42).126
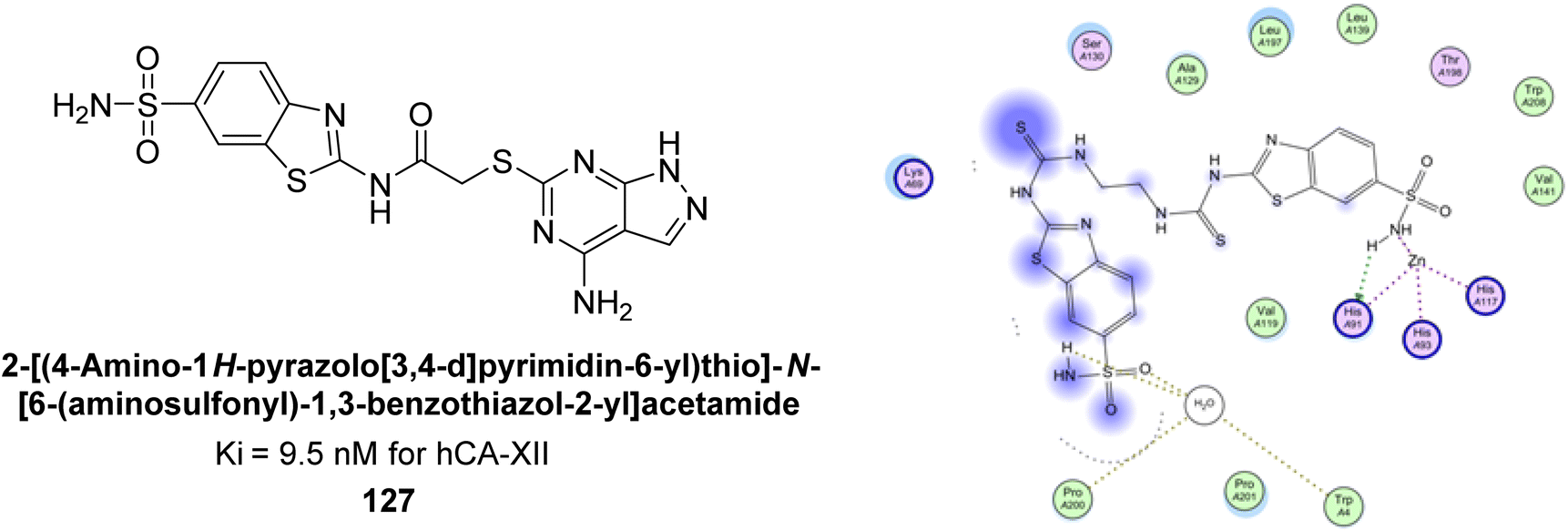 | ||
| Fig. 42 Chemical structure of compound 127 and its Ki value against hCA-XII along with its docking image. | ||
Supuran et al. (2016) synthesized substituted benzenesulfonamides and evaluated their inhibitory effects on various hCA isoforms. Remarkably, compound 128 showed moderate inhibition with Ki values of 73.7 μM for hCA-I and 85.8 μM for hCA-VII. Compounds 129 and 130 inhibited hCA-II with Ki values of 96.0 μM and 87.8 μM, respectively (Fig. 43). The SAR study indicated that modifications to the phenyl ring generally did not significantly affect inhibition, except for the 4-methoxy substitution.127
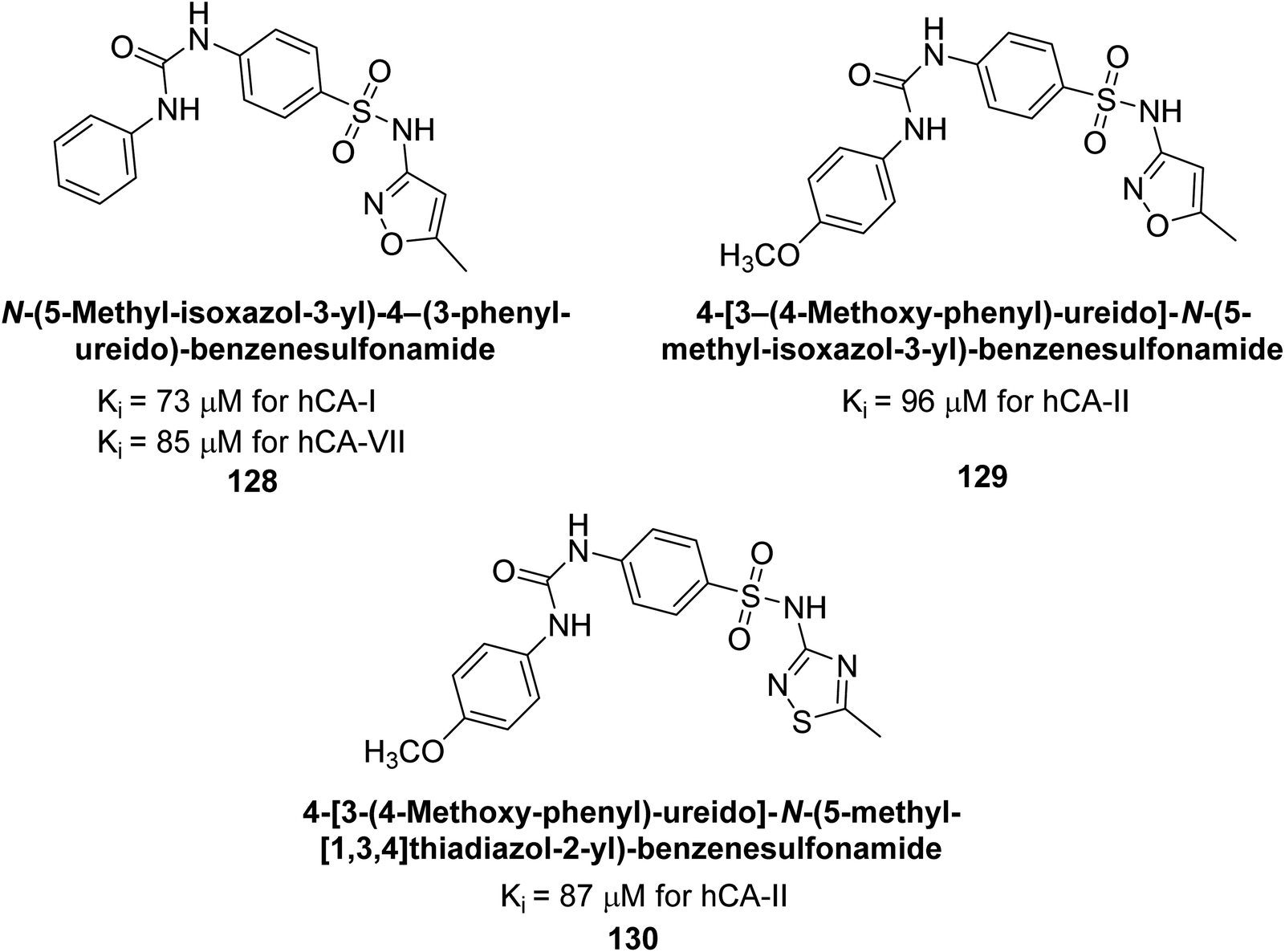 | ||
| Fig. 43 Chemical structures of compounds 128–130 and their Ki values against hCA-I, II and VII. | ||
Gülçin et al. (2016) synthesized a series of urea and phenolic derivatives and evaluated their Ki values against hCA-I and II. The compounds effectively inhibited hCA-I and II, with Ki values ranging from 0.307 to 0.432 nM for hCA-I and 0.149 to 0.278 nM for hCA-II. In comparison, AAZ, a clinically established CA inhibitor, had a Ki value of 49.60 nM for hCA-II. Compound 131, with a phenolic hydroxyl group, showed the highest potency against hCA-II, with a Ki value of 0.307 nM (Fig. 44). The inhibition of hCA-II is attributed to the compound's ability to bind to the catalytic Zn2+ ion in the enzyme's active site, mimicking the tetrahedral transition state.128
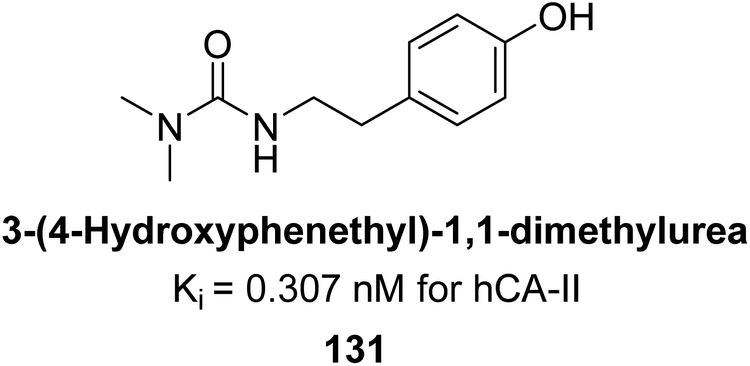 | ||
| Fig. 44 Chemical structure of compound 131 and its Ki value against hCA-II. | ||
Gülçin et al. (2016) synthesized β-lactams and assessed their Ki values against hCA-I and II. The β-lactams demonstrated inhibitory activity, with Ki values ranging from 0.44 to 6.29 nM for hCA-I and 0.93 to 8.34 nM for hCA-II. Notably, β-lactam derivative 132 showed the highest potency against hCA-I with a Ki value of 0.35 nM, while derivative 133 exhibited the strongest inhibition of hCA-II with a Ki value of 0.93 nM (Fig. 45). These findings indicate that β-lactams, with various functional groups, inhibit hCA-I and II in the nanomolar range and could serve as novel CAIs, complementing existing sulfonamides.129
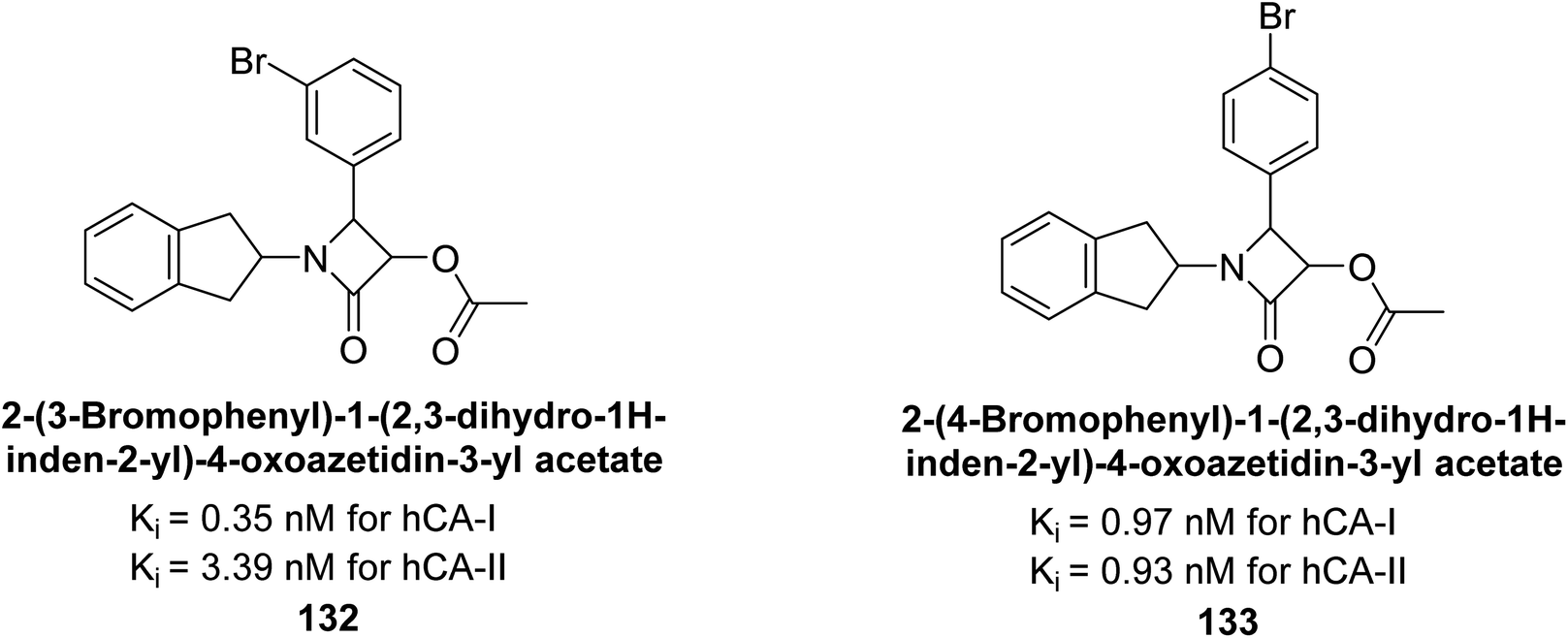 | ||
| Fig. 45 Chemical structures of compounds 132 and 133, and their Ki value against hCA-I and II. | ||
Beydemir et al. (2016) synthesized a series of pyrazole-3,4-dicarboxamides and assessed their inhibitory effects on hCA-I and II. The Ki values ranged from 0.119 to 3.999 μM for hCA-I and from 0.084 to 0.878 μM for hCA-II. Among the tested compounds, compound 134 (–Me and –Ph substituents) exhibited the strongest inhibitory effect (Ki = 0.119 μM) on hCA-I, while compound 135 (–Pr and –OEt substituents) showed the highest inhibition (Ki = 0.084 μM) on hCA-II (Fig. 46).130
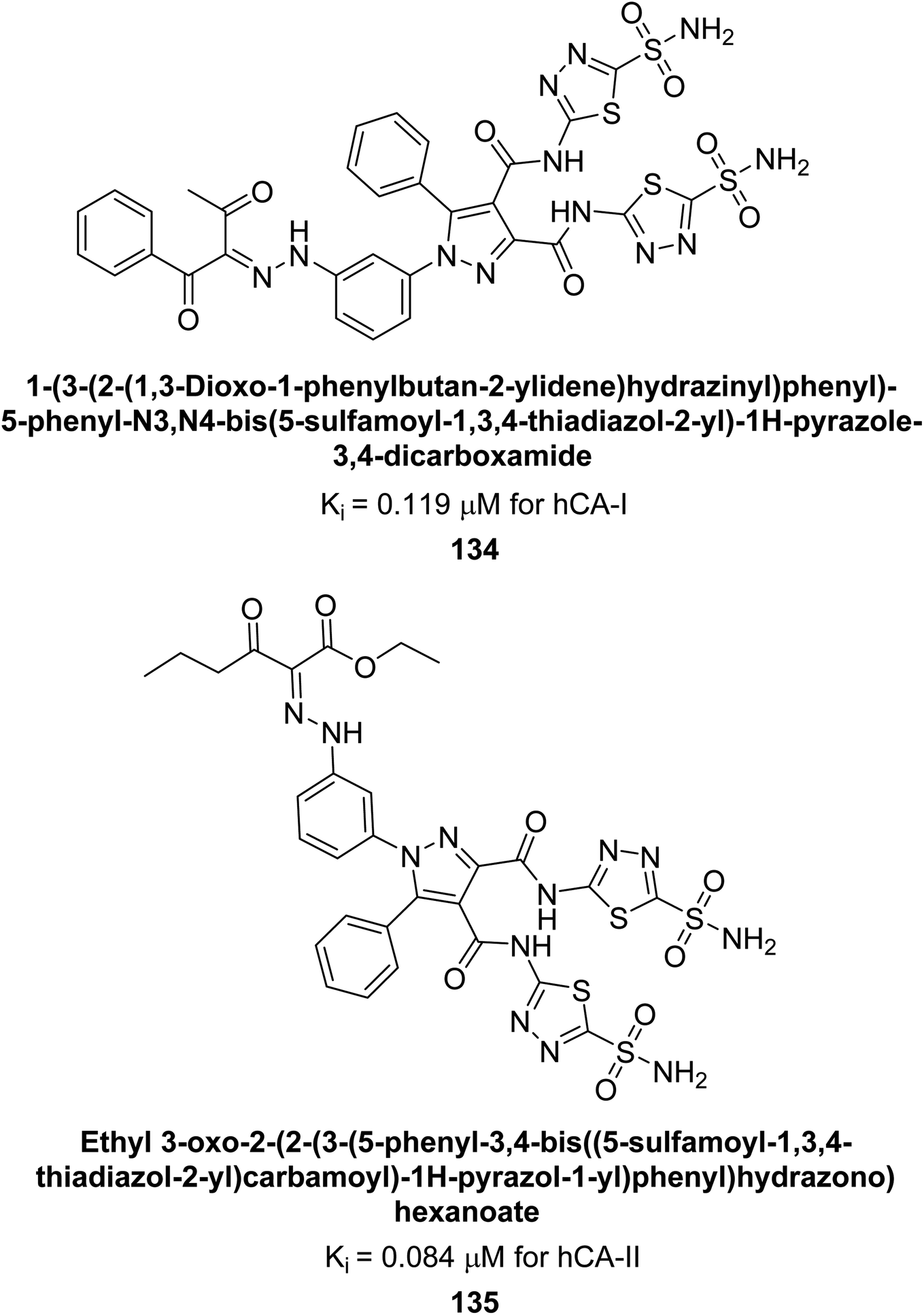 | ||
| Fig. 46 Chemical structures of compounds 134 and 135, and their Ki value against hCA-I and II. | ||
Gülçin et al. (2016) synthesized tetrahydropyrimidinethiones and evaluated their inhibitory effects on hCA-I and II. The compounds showed strong inhibition, with Ki values ranging from 47.40 to 76.06 nM for hCA-I and from 30.63 to 76.06 nM for hCA-II. Compound 136 was the most potent inhibitor of hCA-I with a Ki of 47.40 nM, surpassing AAZ, a clinically used CA inhibitor. Compound 137 exhibited the highest inhibition of hCA-II, with a Ki value of 30.63 nM (Fig. 47). These cyclic thioureas effectively interacted with both hydrophilic and hydrophobic regions of the hCA-II active site.131
 | ||
| Fig. 47 Chemical structures of compounds 136 and 137, and their Ki value against hCA-I and II. | ||
In another study, Gülçin et al. (2016) synthesized tetrahydropyrimidine thiones and assessed for their inhibition of hCA-I and II. These new thiones effectively inhibited both isoforms, with Ki values ranging from 218.5–261.0 pM for hCA-I and from 181.8 to 273.6 pM for hCA-II. In contrast, AAZ, a moderate hCA-I inhibitor, exhibited a Ki of 369.4 pM. The most potent hCA-I inhibition was achieved with compound 138, showing a Ki value of 218.5 pM. Additionally, the strongest hCA-II inhibition was also observed with compound 138, which had a Ki value of 155.4 pM (Fig. 48). This relatively unexplored class of derivatives could offer valuable insights into the field of non-sulfonamide CA inhibitors.132
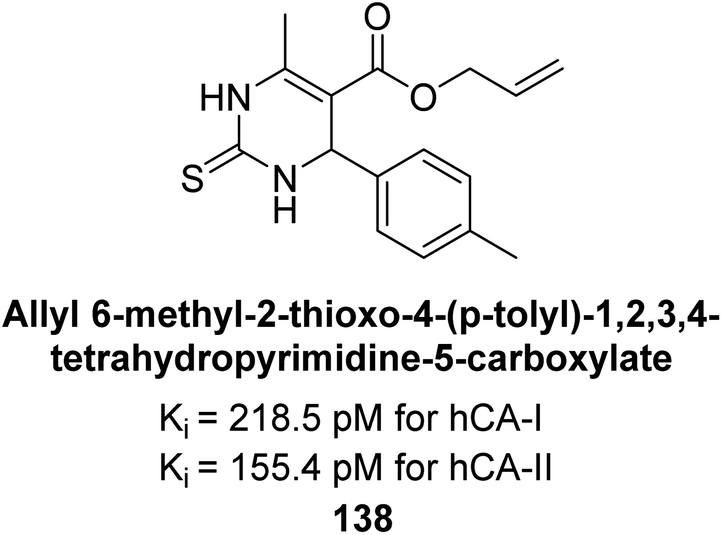 | ||
| Fig. 48 Chemical structure of compound 138 and its Ki value against hCA-I and II. | ||
Amin et al. (2016) synthesized substituted benzenesulfonamides and evaluated for their Ki on hCA-I and IX. These new sulfonamides demonstrated excellent inhibition across all three isoforms, with Ki values ranging from 0.84 to 702 nM for hCA-I and 5.6 to 29.2 nM for hCA-IX. The most effective inhibitor for hCA-I is compound 139, which features a sulfaguanidine moiety and has a Ki of 0.85 nM. For hCA-IX, compound 140 displayed excellent inhibition with a Ki of 29.2 nM, demonstrating potency comparable to that of AAZ (Fig. 49).133
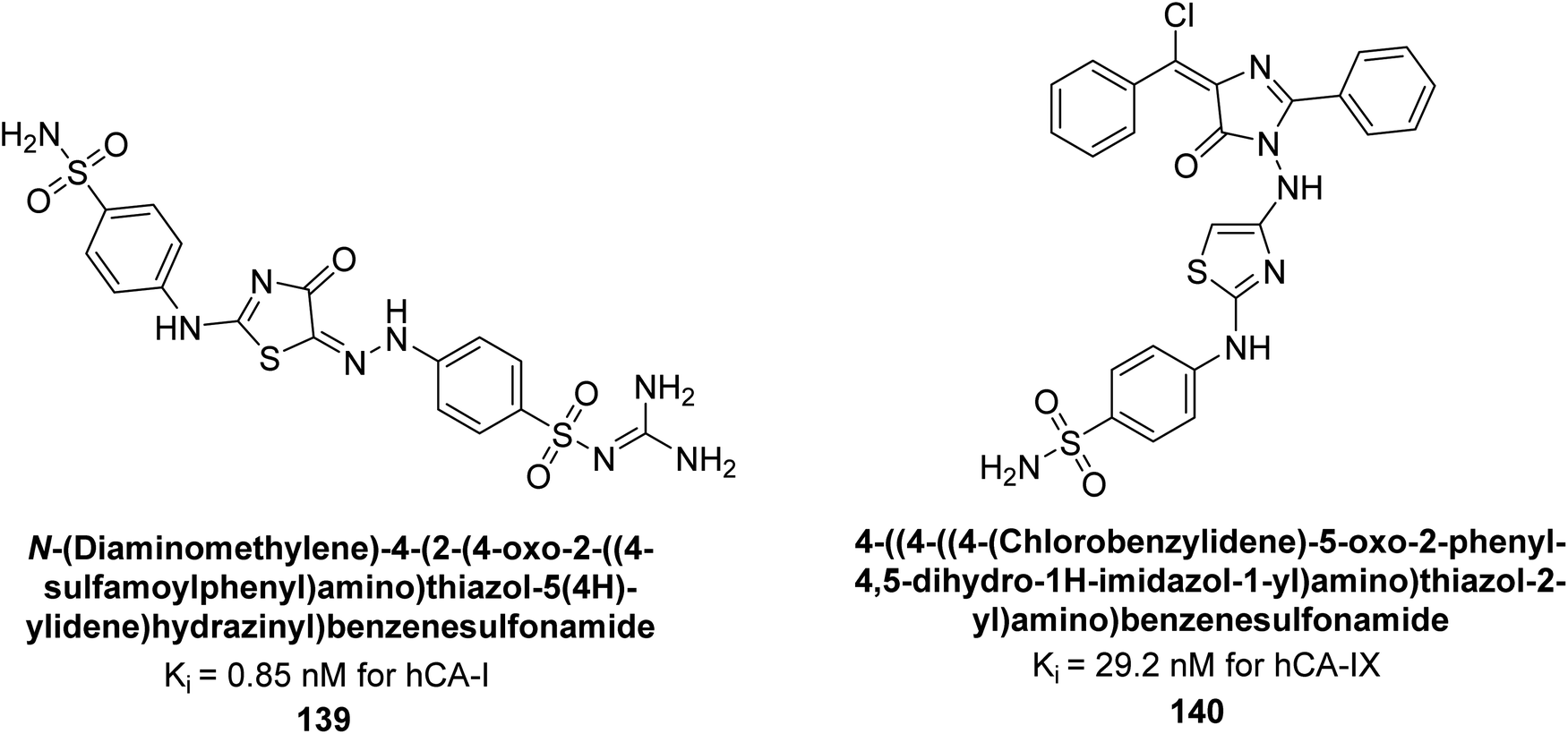 | ||
| Fig. 49 Chemical structures of compounds 139 and 140 and their Ki value against hCA-I and IX. | ||
Kucukoglu et al. (2016) synthesized a series of pyrazoline benzenesulfonamides and assessed for their inhibitory activity on hCA-I and II. All synthesized compounds exhibited superior CA inhibitory activity compared to the reference compound AAZ, with Ki ranging from 26.5 to 55.5 nM for hCA-I and from 18.9 to 28.8 nM for hCA-II. The compounds 141–146 [141 (30.1 nM), 142 (40.8 nM), 143 (46.2 nM), 144 (38.5 nM), 145 (49.2 nM), 146 (36.5 nM)] were found to be 5.9–9.7 times more potent inhibitors of hCA-I compared to AAZ. For hCA-II, the IC50 values of the compounds ranged from 23.8 to 30.1 nM, whereas AAZ had an IC50 value of 146.5 nM. This indicates that compounds 141–146 [141 (24.7 nM), 142 (30.1 nM), 143 (26.7 nM), 144 (23.8 nM), 145 (25.7 nM), 146 (23.9 nM)] were 4.8–6.3 times more potent inhibitors of hCA-II than AAZ (Table 10).134
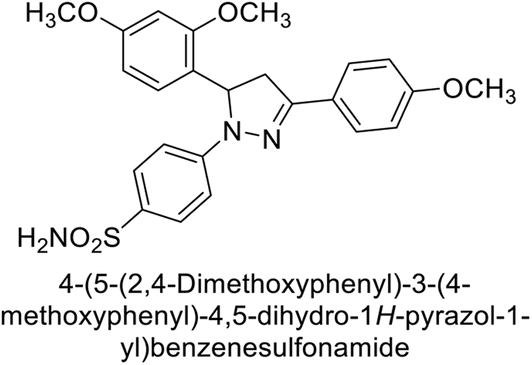
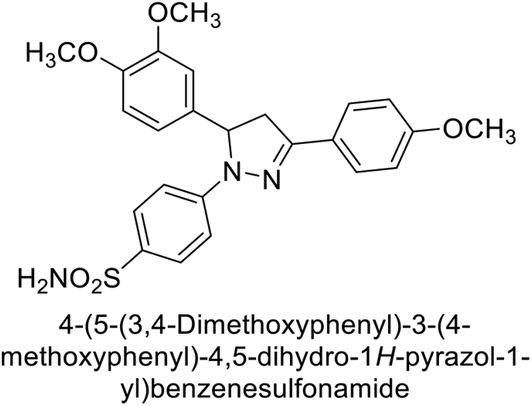
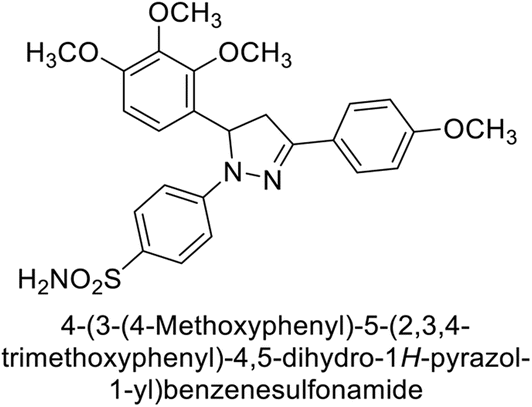
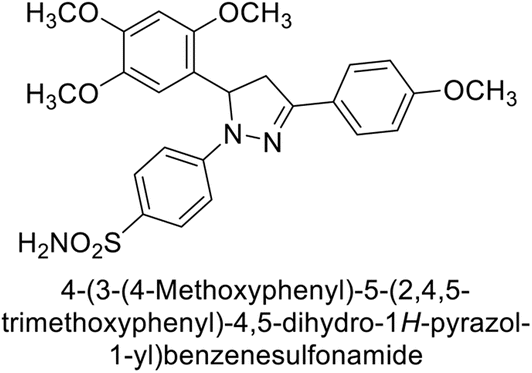
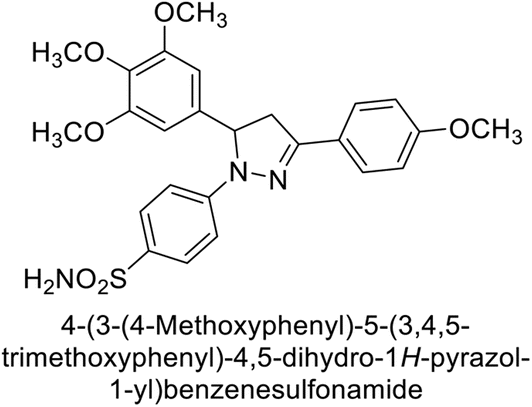
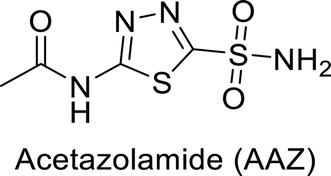
Eldehna et al. (2016) synthesized substituted isatin-based benzenesulfonamides and evaluated for their inhibitory activity against hCA-I, II, IX and XII. The Ki for hCA-I ranged from 7.9–894 nM, for hCA-II from 7.5–1645 nM, for hCA-IX from 5.0–240 nM, and for hCA-XII from 0.47–2.83 nM. Sulfonamides 147, 148, 149, 150, 151 and 152 exhibited exceptional potency against hCA-XII, with Ki between 0.47 and 0.71 nM (Fig. 50). Given the involvement of these isoforms in various pathologies where their inhibition could have therapeutic potential, the synthesized derivatives may represent valuable additions to the repertoire of sulfonamide-based CAI.135
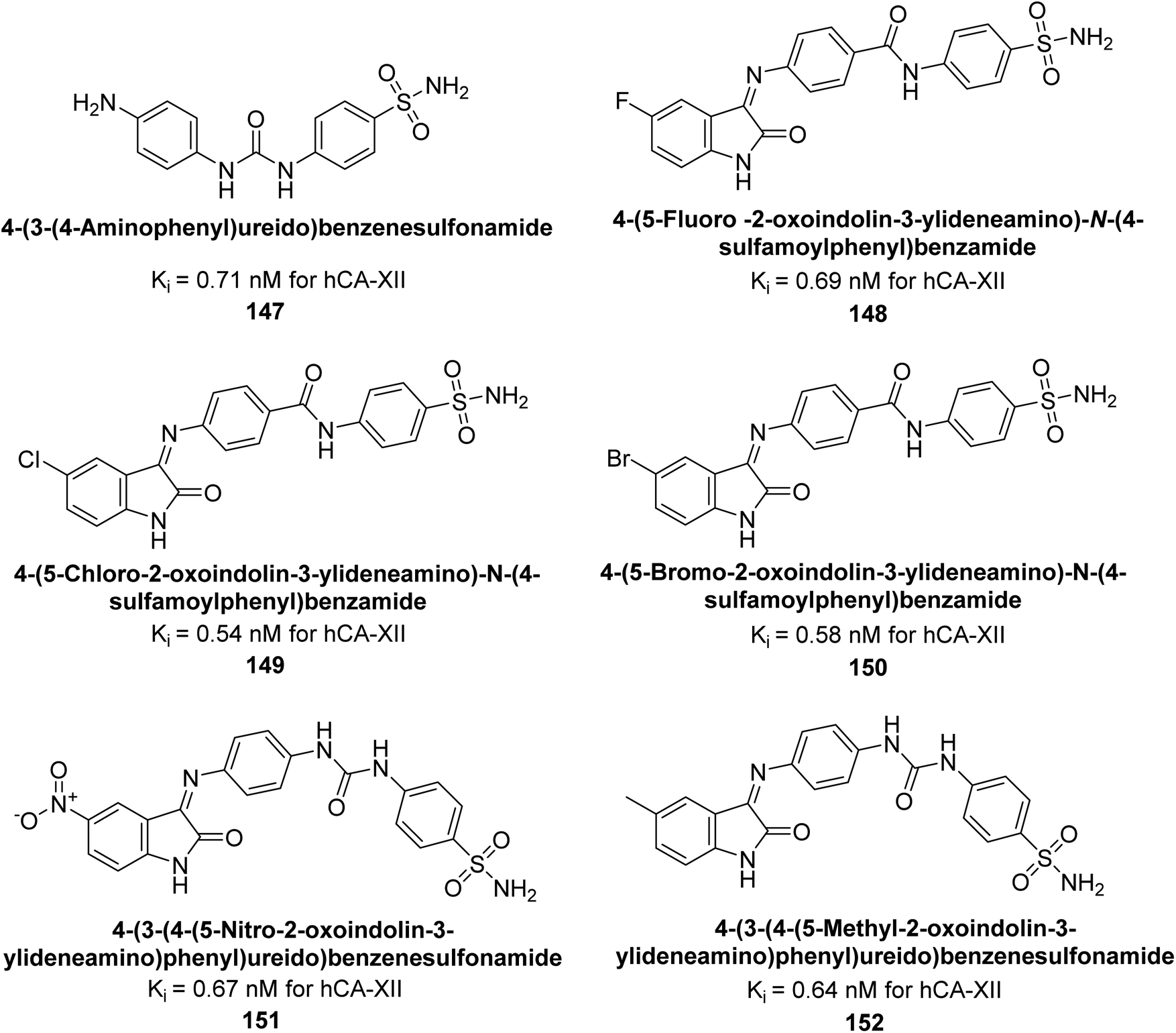 | ||
| Fig. 50 Chemical structures of compounds 147–152 and their Ki value against hCA-XII. | ||
Saeed et al. (2017) synthesized a series of benzyledinyl–hydrazinyl substituted thiazole derivatives and assessed for their inhibitory potential against bCA-II. The majority of these compounds exhibited excellent inhibitory activity against inhibitory potency against bCA-II, with compound 153 showing the highest activity (IC50 = 1.26 μM) (Fig. 51). The high potency of compound 153 is attributed to the presence of ortho-positioned halogen atoms (–F and –Cl), which activate the thiourea nitrogen through a negative inductive effect. Molecular docking studies of these potent dual inhibitors provided insights into their binding site interactions.136
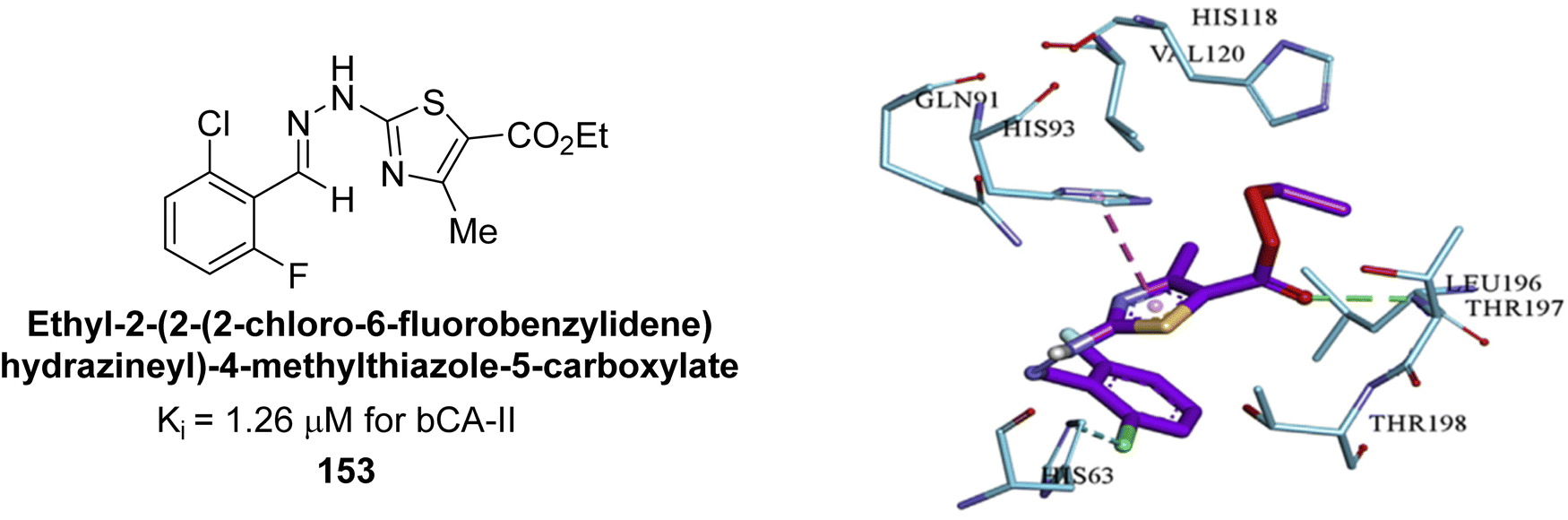 | ||
| Fig. 51 Chemical structure of compound 153 and its Ki value against bCA-II along with its docking image. | ||
Khan et al. (2017) synthesized a series of carbohydrazones and assessed their in vitro inhibitory activity against bCA. Among these, compounds 154 (IC50 = 1.33 μM), 155 (IC50 = 1.85 μM), 156 (IC50 = 1.37 μM), and 157 (IC50 = 1.46 μM) demonstrated superior inhibition compared to the standard reference drug zonisamide (IC50 = 1.86 μM) (Fig. 52). Compound 154, featuring a nitro group at the p-position of the phenyl ring, exhibited the strongest enzyme inhibition with an IC50 = 1.33 μM. Compound 155, with a nitro group at the o-position, showed slightly lower activity (IC50 = 1.85 μM). Compound 157, with 2,6-dichloro substituents, had significant inhibitory potential (IC50 = 1.46 μM), suggesting increased potency with chloro groups at both ortho positions, but decreased effectiveness with ortho and para substitution (2,4-dichloro). Cheminformatic analysis indicated that compounds 154 and 155 possess properties consistent with lead-like candidates. The figure presents a 2D interaction map of compounds 154 and 157, illustrating their binding modes with the active site residues of the enzyme (Fig. 52).137
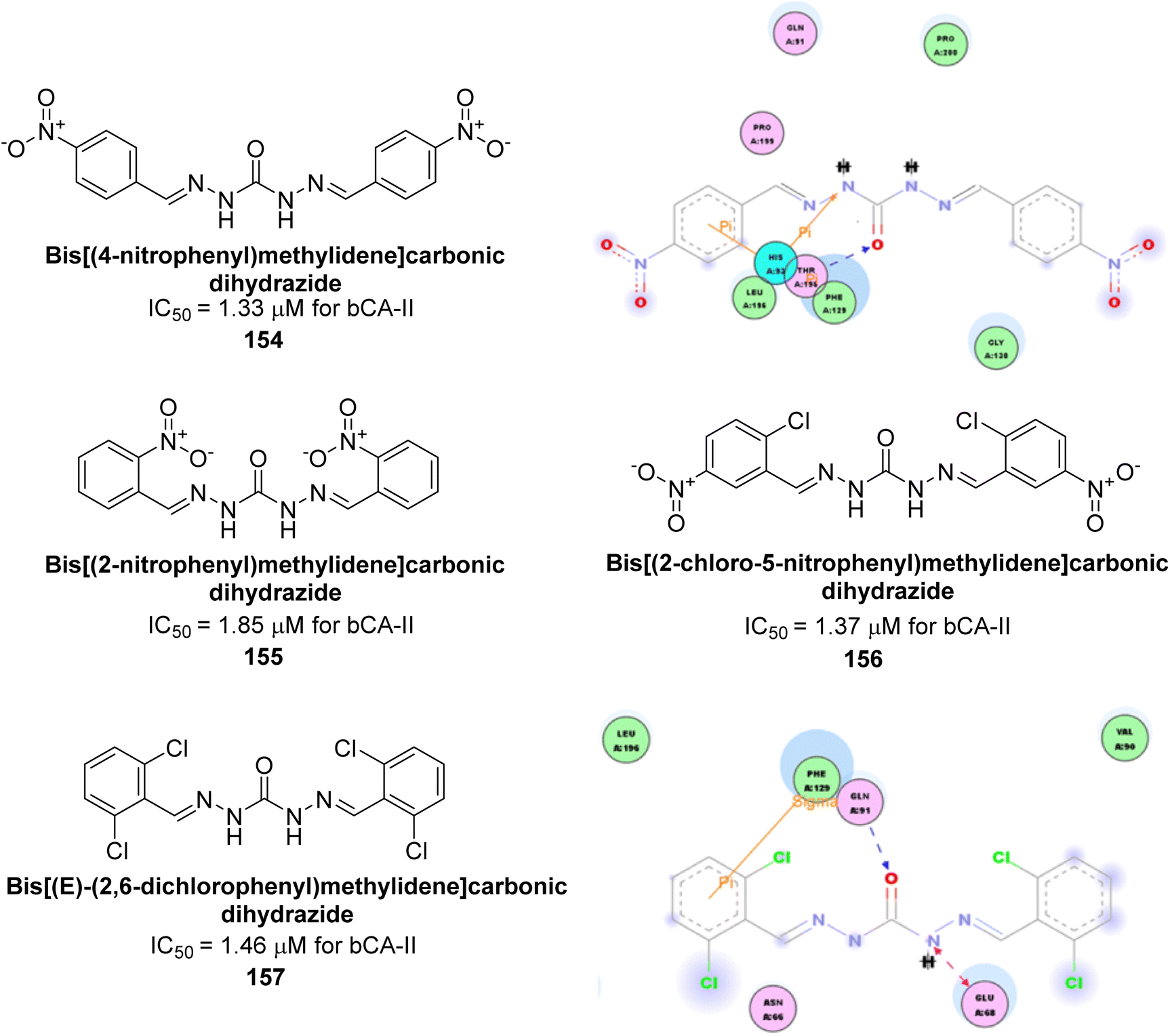 | ||
| Fig. 52 Chemical structures of compounds 154–157 and their IC50 value against bCA-II along with docking images of 154 and 157. | ||
Gencer et al. (2017) synthesized spiroindoline-substituted sulphonamide compounds and assessed their inhibitory effects on hCA-I and II. Remarkably, compound 158 demonstrated the highest inhibition of hCA-I with a Ki value of 0.042 μM, while compound 159 showed the greatest inhibition of hCA-II with a Ki value of 0.151 μM (Fig. 53).138 The findings suggest that these compounds could have potential medical applications. Consequently, they warrant further evaluation through in vivo studies.
 | ||
| Fig. 53 Chemical structures of compounds 158 and 159 and their Ki values against hCA-I and II. | ||
Supuran et al. (2017) synthesized substituted 3,4-dihydroquinolin-2(1H)-one, and evaluated for their inhibitory effects on four CA isoforms: hCA-I, II, IV and IX. The most effective hCA-IX inhibitors were compounds 160 and 161, with Ki ranging from 243.6 to 292.8 nM. These compounds are notably different: compound 160 is a urea derivative containing a pentafluorophenyl group, whereas compound 161 features a secondary sulfonamide functionality (Fig. 54).139
 | ||
| Fig. 54 Chemical structures of compounds 160 and 161 and their Ki values against hCA-IX. | ||
Arifuddin et al. (2017) synthesized substituted indazole-3-carboxamide hybrids and evaluated as inhibitors of hCA-IX. Specifically, compounds 162, 163, and 164 demonstrated potent inhibition of the tumor-associated, hypoxia-induced hCA-IX, with Ki values of 1.8, 2.3, and 2.0 nM, respectively (Fig. 55). Compounds 162, 163, and 164 exhibited 11- to 14-fold greater potency due to the alkyl substituents (isopropyl, ethyl and methyl, respectively) compared to the sulfonamide standard AAZ (Ki = 25 nM).140
 | ||
| Fig. 55 Chemical structures of compounds 162, 163 and 164 and their Ki values against hCA-IX. | ||
Kamal et al. (2017) synthesized sulfonamide derivatives inspired by curcumin and evaluated them against four isoforms: hCA-I, II, IX, and XII. The new compounds showed moderate inhibition of hCA-I (Ki = 191.8–904.2 nM), high inhibition of hCA-II (Ki = 0.75–8.8 nM), potent inhibition of hCA-IX (Ki = 2.3–87.3 nM), and inhibition of hCA-XII (Ki = 6.1–71.8 nM). Markedly, compound 165 displayed nearly 9-fold selectivity for hCA-II (Ki = 0.89 nM) over hCA-IX and XII, while compound 166 showed 3-fold and 70-fold selectivity for hCA-II (Ki = 0.75 nM) over hCA-IX and XII, respectively (Fig. 56). This selectivity difference is attributed to the varying placement of methoxy groups on the second aryl ring derived from chalcone.141
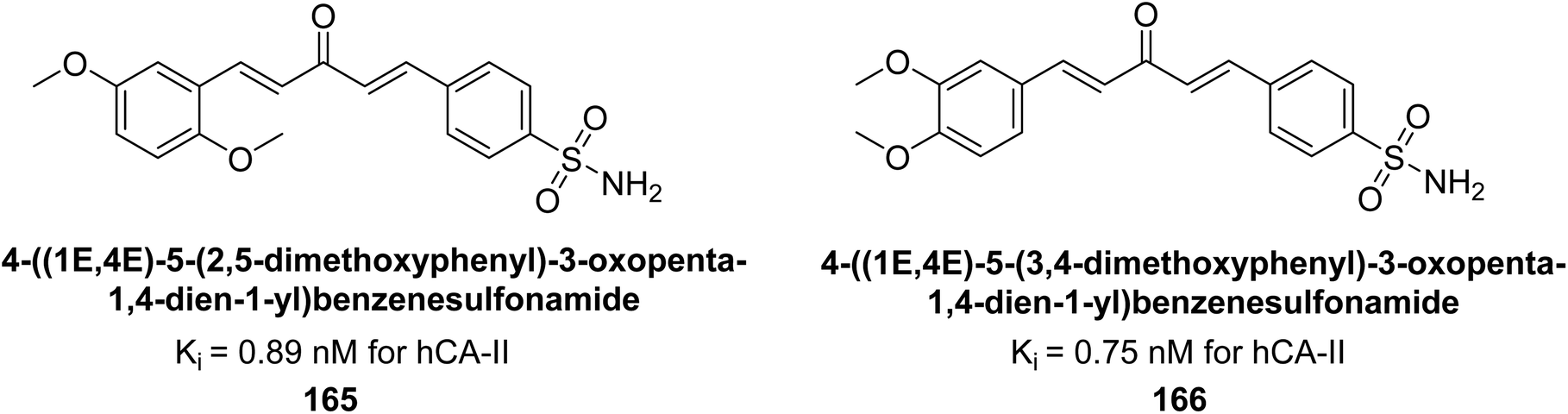 | ||
| Fig. 56 Chemical structures of compounds 165 and 166 and their Ki values against hCA-II. | ||
Gülçin et al. (2017) synthesized novel 2-amino-3-cyanopyridines and assessed their inhibitory effects on hCA-I and II. Compound 167 demonstrated the highest inhibition of hCA-I with a Ki = 2.84 μM, while compound 168 was the most potent inhibitor of hCA-II with a Ki = 2.56 μM (Fig. 57). These compounds are promising candidates for future drug development.142
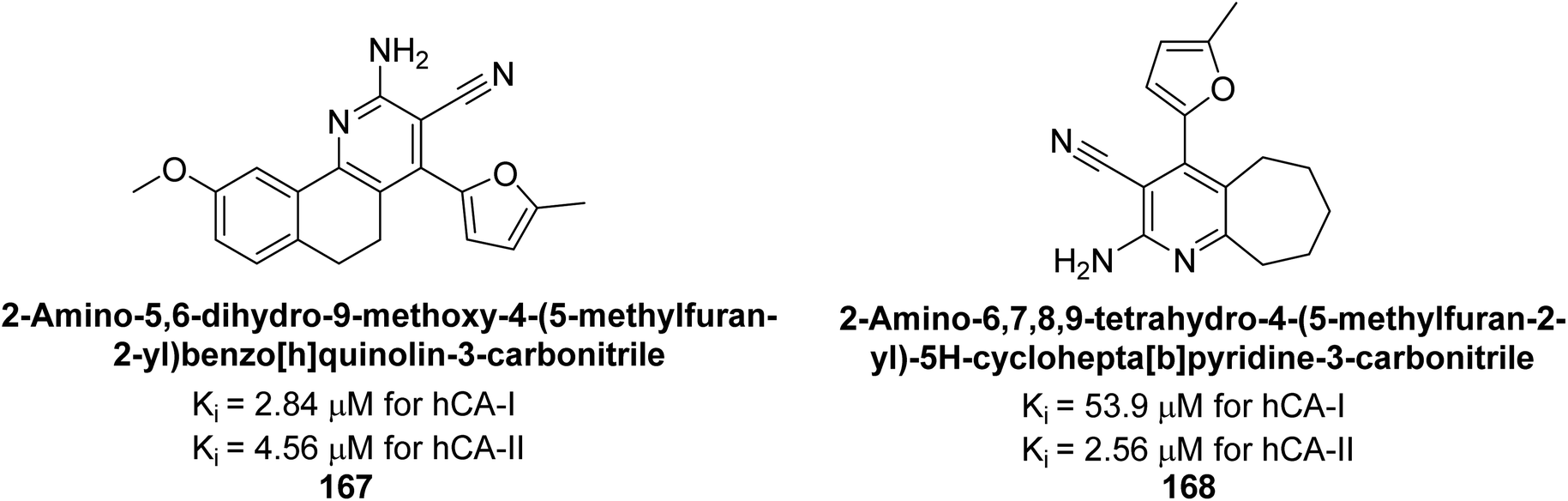 | ||
| Fig. 57 Chemical structures of compounds 167 and 168 and their Ki values against hCA-I & II. | ||
Gülçin et al. (2017) synthesized amides and thiazolidine-4-ones and accessed their hCA activity. These compounds demonstrated effective inhibition profiles with Ki values ranging from 23.76 to 102.75 nM against hCA-I and 58.92 to 136.64 nM against hCA-II. In comparison, AAZ (standard) exhibited Ki values of 482.63 nM against hCA-I and 1019.60 nM against hCA-II. Among the inhibitor compounds, compound 169 exhibited the highest inhibition of hCA-I with a Ki value of 23.76 nM. Additionally, the compound 170 demonstrated the most potent inhibition of hCA-II with a Ki of 58.92 nM (Fig. 58).143
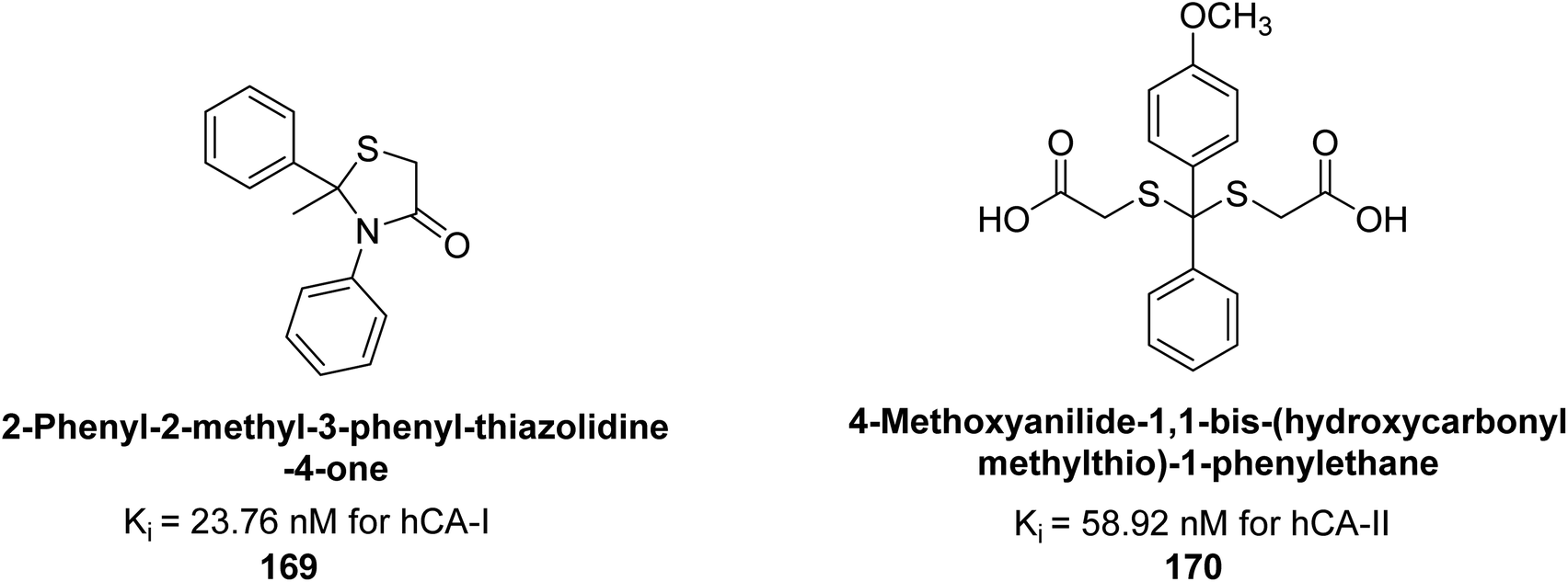 | ||
| Fig. 58 Chemical structures of compounds 169 and 170 and their Ki values against hCA-I & II. | ||
Gülçin et al. (2017) synthesized pyrrolidine-2,5-diones derivatives in good yields. Compound 171, featuring two carbonyl groups and one chlorine group, demonstrated potent inhibition of hCA-I with a Ki of 23.27 nmol L−1. Furthermore, compound 172, which also includes two carbonyl groups and a chlorine group, proved to be the most effective inhibitor of hCA-II with a Ki of 10.64 nmol L−1 (Fig. 59). It is widely recognized that compounds containing carbonyl (–CO) and chlorine (–Cl) groups exhibit effective CAI properties.144
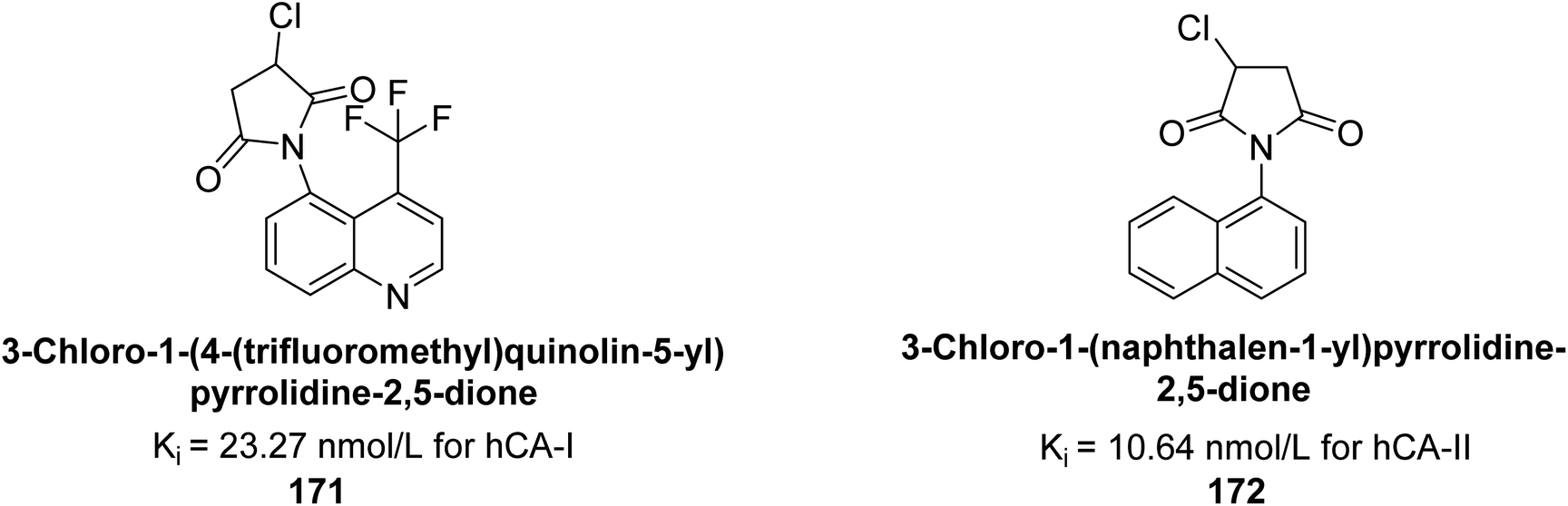 | ||
| Fig. 59 Chemical structures of compounds 171 and 172 and their Ki values against hCA-I & II. | ||
Durdagi et al. (2017) synthesized coumaryl-carboxamide derivatives and evaluated for their inhibitory activity against hCA-I, II, VII, and IX. Specifically, compound 173 selectively inhibited hCA-IX with a Ki of 107.9 nM (Fig. 60). Compound 173 displayed the highest activity against hCA-IX, with superior docking scores for both hydrolyzed (−9.006 kcal mol−1) and non-hydrolyzed (−8.429 kcal mol−1) forms compared to hCA-I, II, and VII. Docking studies showed that the hydrolyzed form of 173 interacted with His96, His64, His94, and Gln92 at the hCA-IX active site, while the non-hydrolyzed form interacted with His64, His94, and His119 (Fig. 60).145
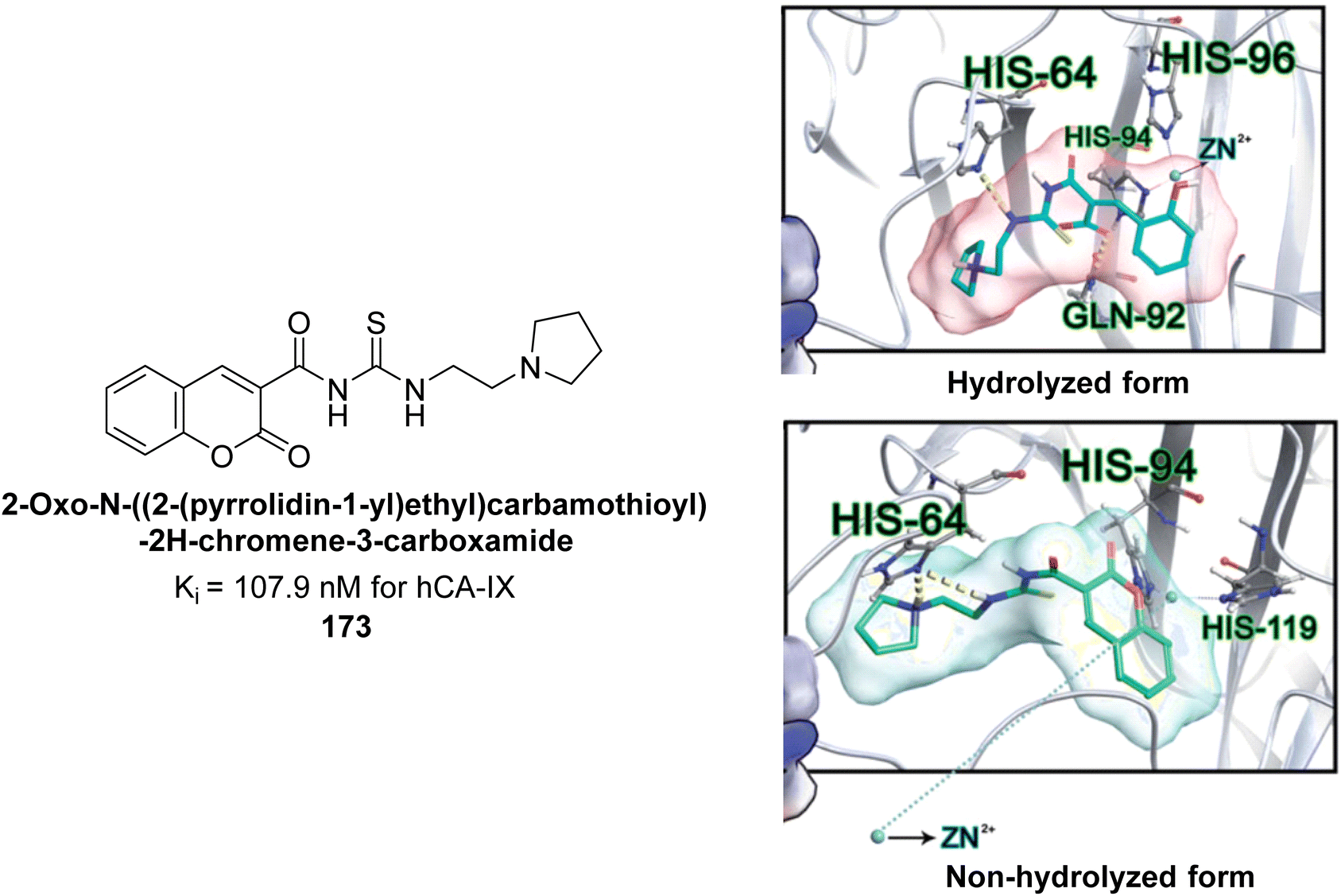 | ||
| Fig. 60 Chemical structure of compound 173 and its Ki value against hCA-IX along with its docking images in hydrolyzed and non-hydrolyzed forms. | ||
Supuran et al. (2017) synthesized different alkoxymethyl derivatives of mercaptobenzoxazole and 2-aminothiazole. This study investigated the inhibitory effects of these molecules on hCA-I and II. The newly synthesized molecules significantly inhibited both hCA isoenzymes, with Ki values ranging from 58 to 157 nM for hCA-I and 76 to 215 nM for hCA-II. In particular, compound 174 (Ki = 58 nM) and compound 175 (Ki = 70 nM) exhibited stronger inhibition of the hCA-I isoform compared to the standard compound AAZ (Ki = 333 nM). For hCA-II, the most significant inhibition was observed with compound 176 (Ki = 76 nM) (Fig. 61).146
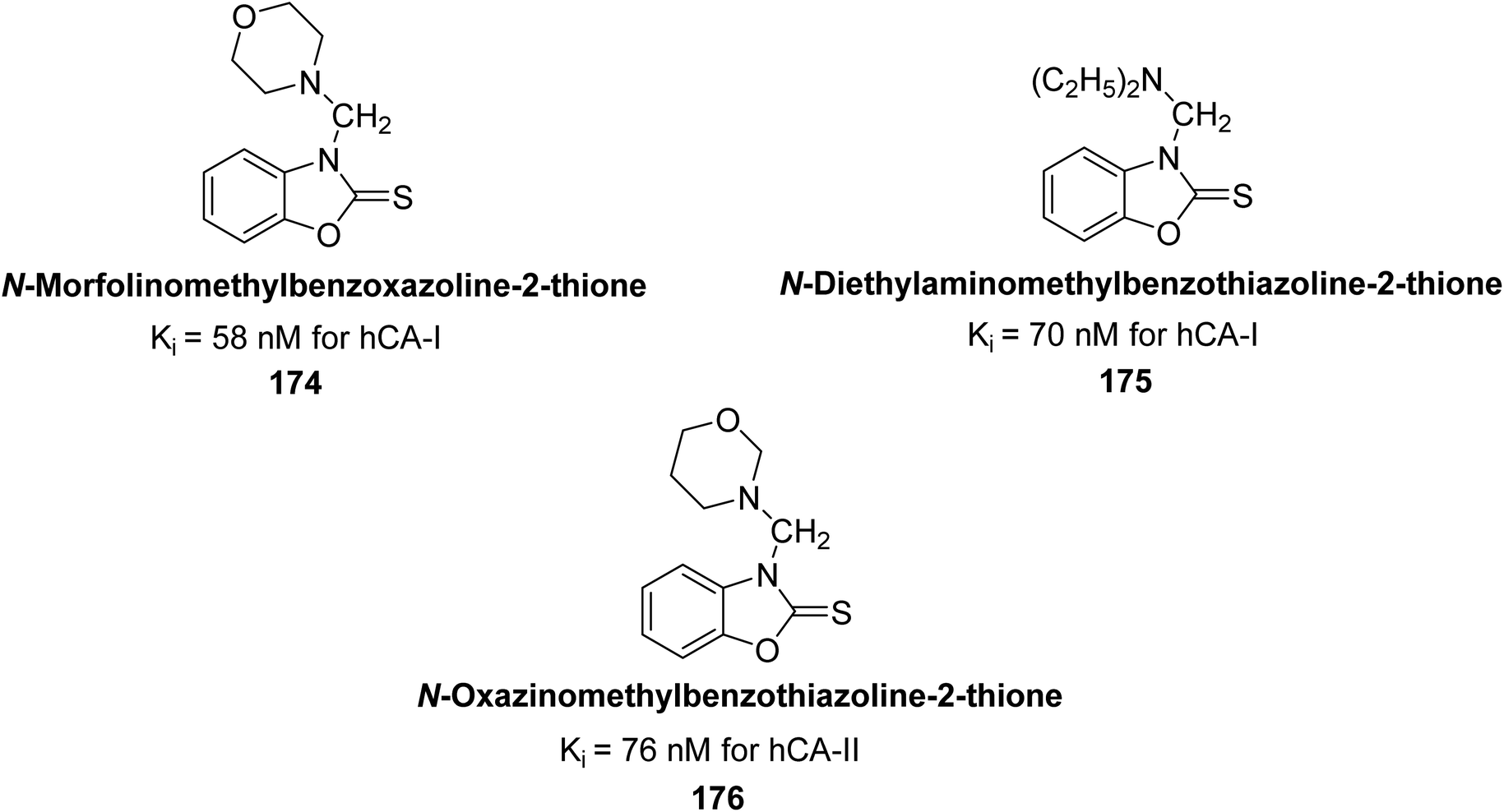 | ||
| Fig. 61 Chemical structures of compounds 174, 175 and 176 and their Ki values against hCA-I & II. | ||
Gülçin et al. (2018) synthesized a series of N-heterocyclic carbene precursors with 2-hydroxyethyl substitutions. These novel NHC precursor derivatives exhibited significant inhibitory activity against hCA-I and II. The Ki were found to range from 13.90 to 41.46 nM for hCA-I and 12.82 to 49.95 nM for hCA-II, respectively. Compound 177 exhibited the most potent inhibition of the hCA-I isoform, with a Ki value of 13.90 nM and 177 exhibited a Ki value of 12.82 nM against hCA-II (Fig. 62).147
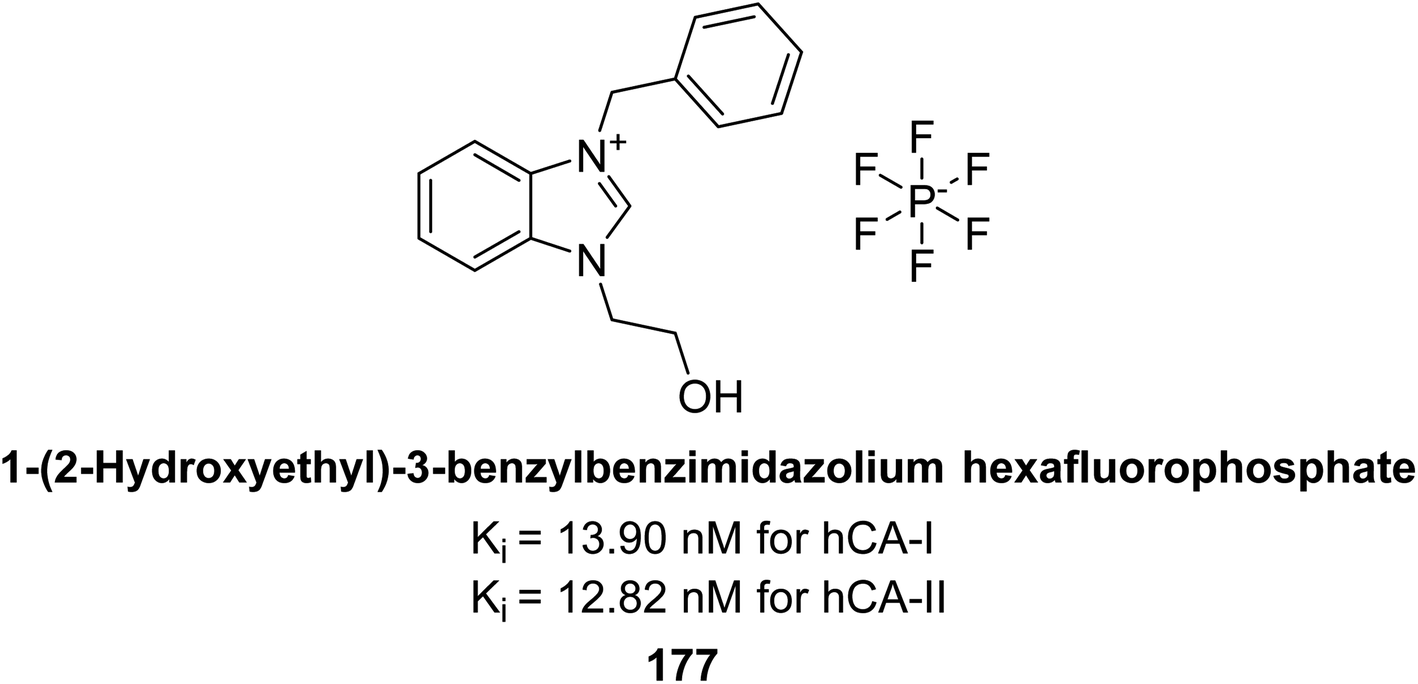 | ||
| Fig. 62 Chemical structure of compound 177 and its Ki values against hCA-I & II. | ||
Gülçin et al. (2018) synthesized a series of benzimidazolium salts evaluated their hCA-I and II activities. Compound 178 showed the highest inhibition of hCA-I with a Ki value of 17.33 nM, while compound 179 demonstrated excellent inhibition of hCA-II with a Ki value of 33.98 nM (Fig. 63). Compared to the standard drug AAZ, which has Ki values of 132.55 nM and 176.15 nM for hCA-I and II, respectively, these compounds exhibited superior inhibitory profiles.148
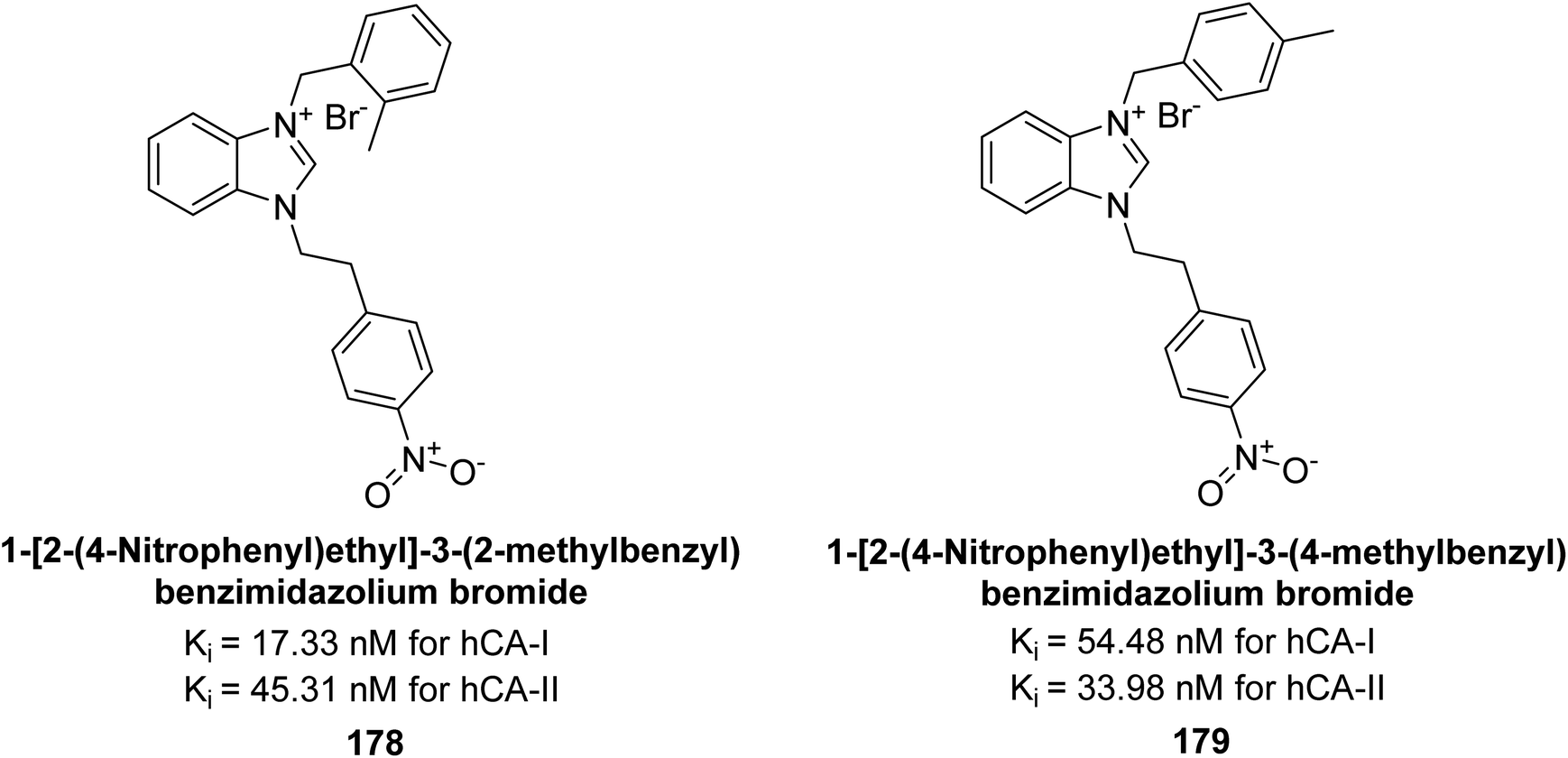 | ||
| Fig. 63 Chemical structures of compounds 178 and 179 and their Ki values against hCA-I & II. | ||
Ahmed et al. (2018) synthesized a series of sulfonamides based on a curcumin scaffold and assessed their inhibitory activity against hCA-I and bCA-II isoforms. Compound 180 showed the highest inhibition of hCA-I with a Ki value of 0.99 μM. For bCA-II, compounds 180, 181, and 182 had Ki values of 0.71, 0.67, and 0.71 μM, respectively (Fig. 64). These compounds exhibited inhibitory activities comparable to the standard drug AAZ, and molecular docking studies suggested they bind to hCA-I in a similar manner (Fig. 64).149
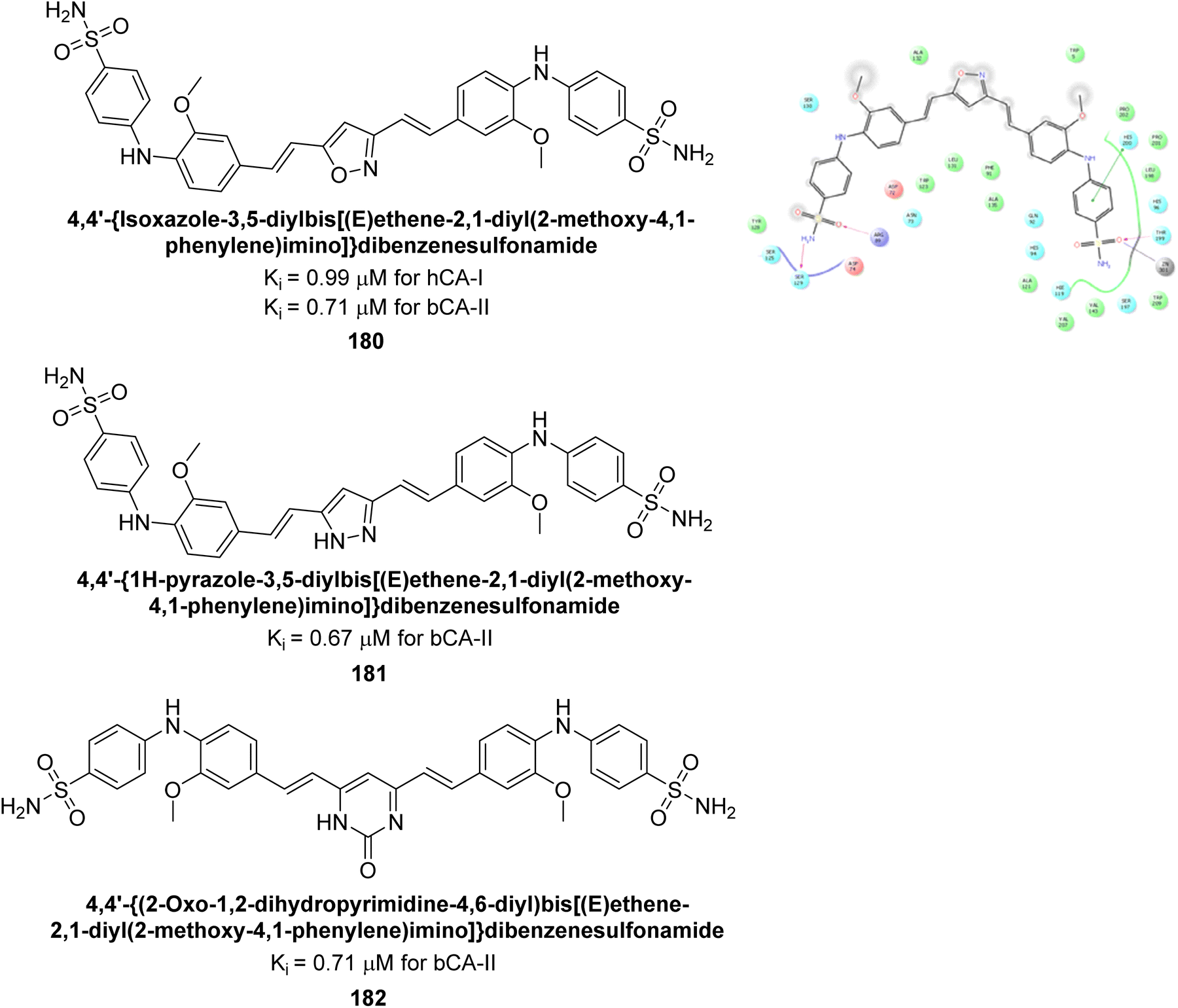 | ||
| Fig. 64 Chemical structures of compounds 180, 181 and 182 and their Ki values against hCA-I & bCA-II along with docking image of compound 180. | ||
Azam et al. (2018) synthesized pyridinethiazolidinone derivatives and tested them as inhibitors of CA-IX. Compounds 183 and 184 demonstrated strong inhibition with IC50 values of 1.61 μM and 1.84 μM, respectively (Fig. 65). Their binding affinities for CA-IX were high, with KD values of 11.21 μM for 183 and 2.32 μM for 184. Docking studies showed that both compounds effectively bind to the CA-IX active site, forming multiple hydrogen bonds and van der Waals interactions with key residues.150
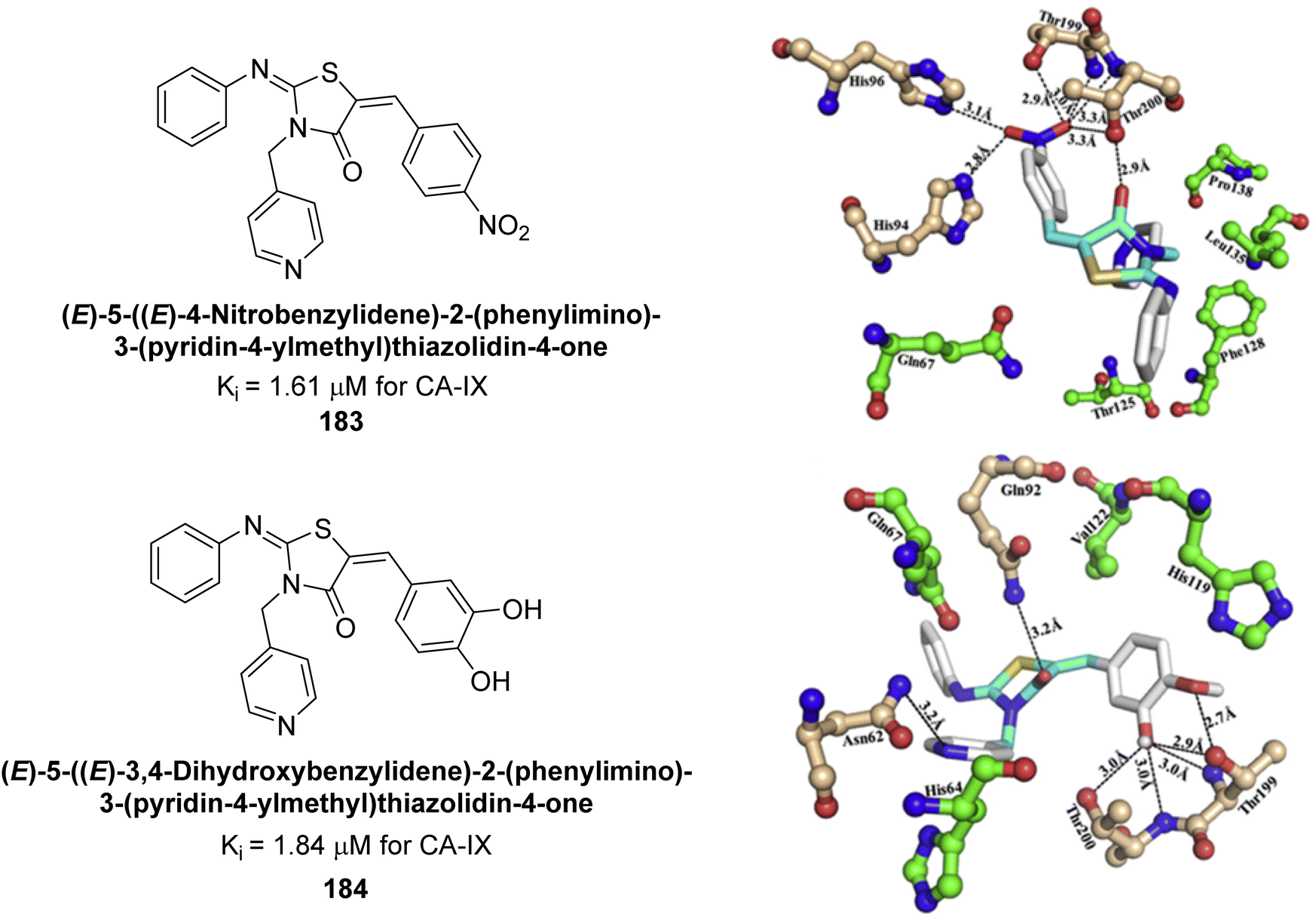 | ||
| Fig. 65 Chemical structures of compounds 183 and 184 and their Ki values against hCA-IX along with their docking images. | ||
Sharma et al. (2018) synthesized a library of substituted 1,2,3-triazole carboxylates and assessed for their inhibitory activity against hCA-I, II, IV, and IX which are established drug targets. Carboxylic acid hydrazide 185 showed the highest inhibition potency against hCA-II with a Ki of 27.2 nM, while derivative 186 demonstrated superior inhibition of hCA-IX with a Ki of 14.3 nM, outperforming the reference drug AAZ (Ki = 25.8 nM) (Fig. 66).151
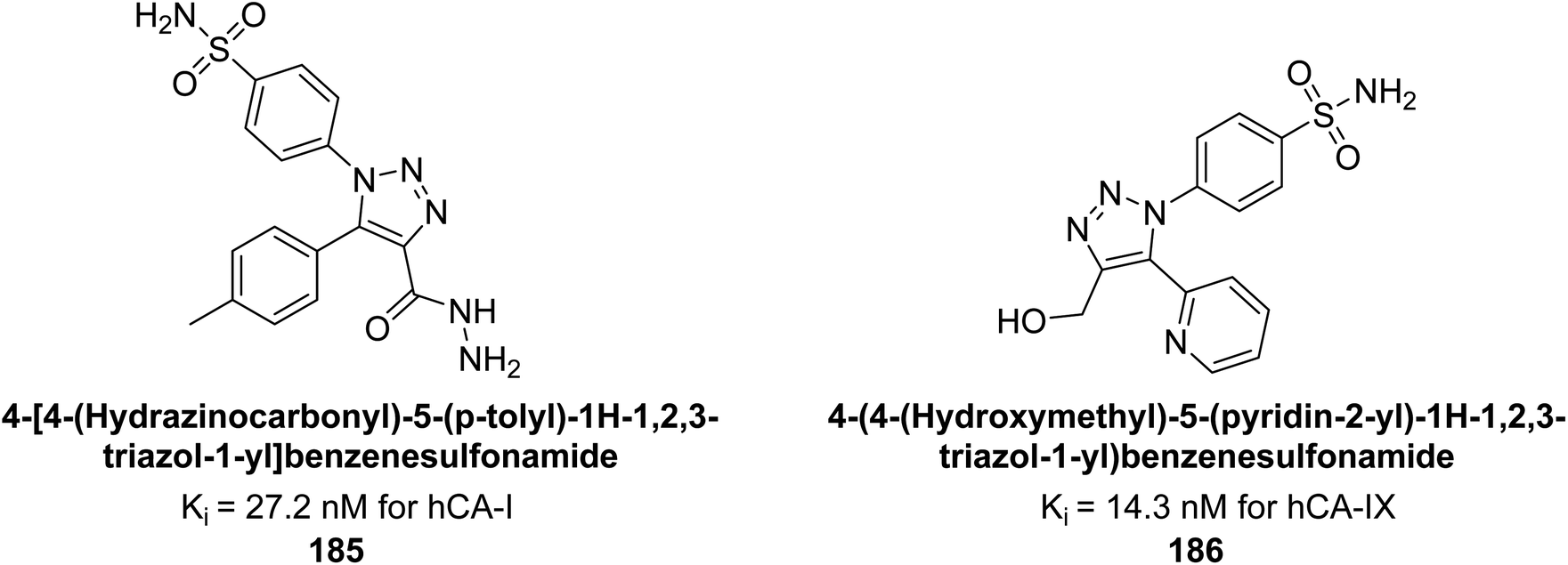 | ||
| Fig. 66 Chemical structures of compounds 185 and 186 and their Ki values against hCA-I and IX. | ||
Supuran et al. (2018) synthesized a series of benzenesulfonamide bearing 1,2,3-triazole ring, and evaluated against hCA-I, II, IV, and IX. All newly synthesized compounds demonstrated excellent inhibition of the hCA-I, with Ki ranging from 30.1 to 86.8 nM, significantly lower than the standard drug AAZ, which has a Ki of 250 nM. For the hCA-IV, compound 187 showed superior inhibition (Ki = 52.4 nM) compared to AAZ (Ki = 74 nM) (Fig. 67), while for the hCA-IX, all compounds exhibited moderate inhibition potential. This study contributes to the ongoing exploration of the 1,2,3-triazole moiety in medicinal chemistry.152
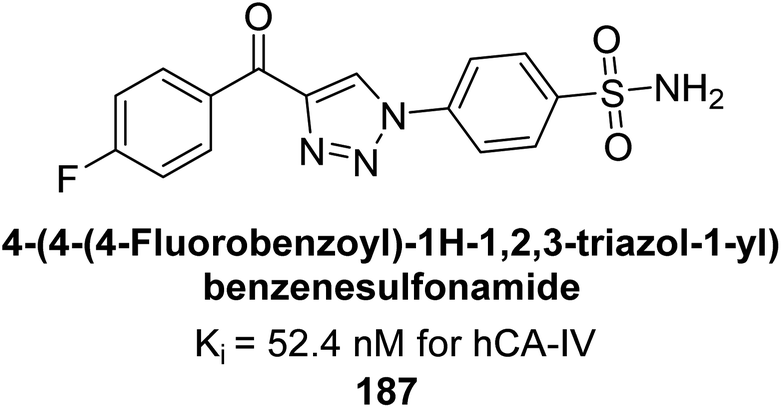 | ||
| Fig. 67 Chemical structure of compound 187 and its Ki value against hCA-IV. | ||
Supuran et al. (2018) synthesized and characterized triazole benzenesulfonamides and assessed against four isozymes: hCA-I, II, IV and IX. The results indicated that the hCA-IX was the most susceptible to inhibition by these derivatives. Especially, compound 188 (Ki = 46.4 nM), 189 (Ki = 42.6 nM) and 190 (Ki = 35.1 nM) emerged as the most potent CA inhibitors (Fig. 68). p-Substitution on benzenesulfonamide with a triazolopyridine moiety was found to be highly effective for CA inhibition. Adding an aryl group at the 5-position of triazolopyridine increased affinity for hCA-IX. Particularly, the N-dimethyl derivative 190 demonstrated the highest potency in this study.153
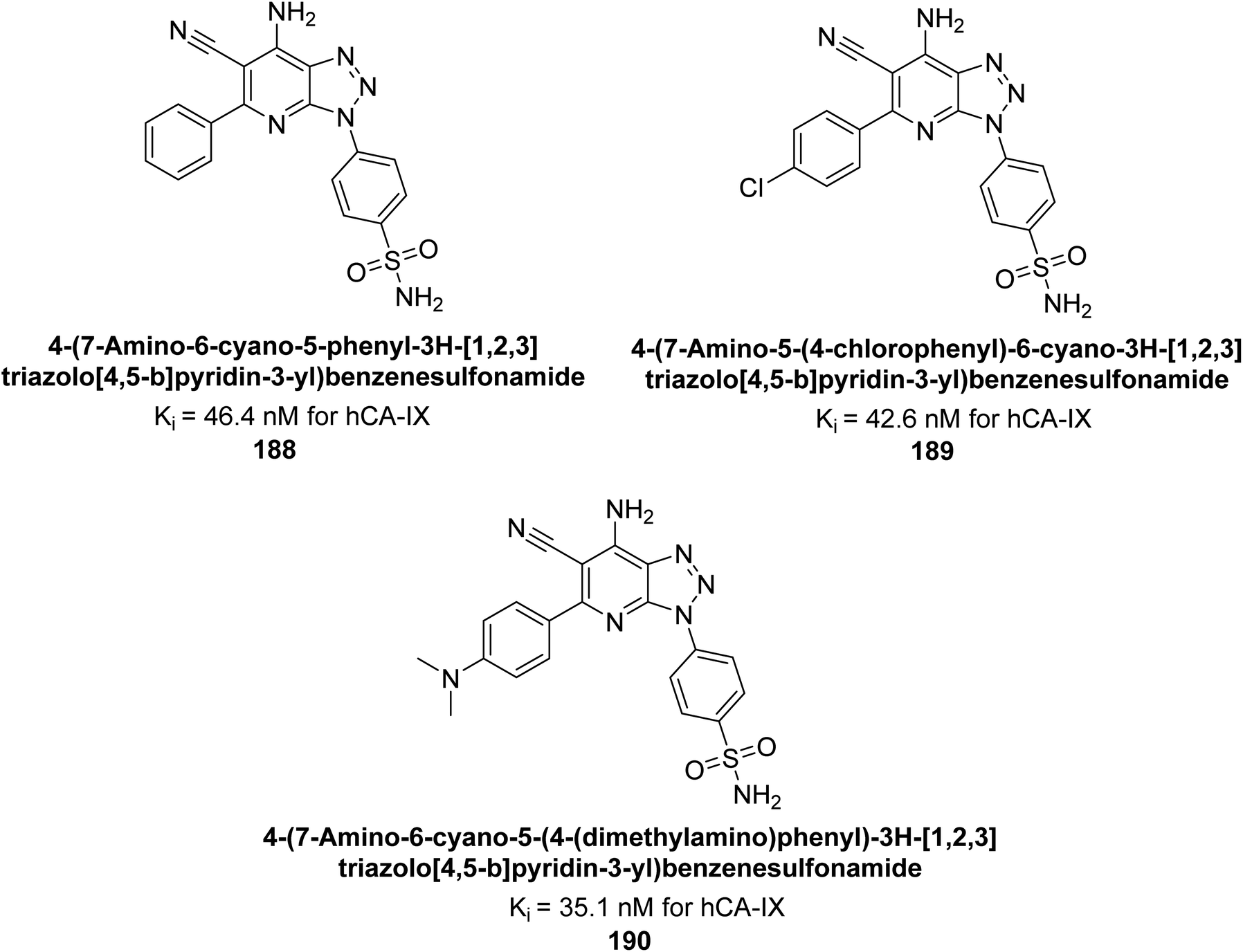 | ||
| Fig. 68 Chemical structures of compounds 188, 189 and 190 and their Ki values against hCA-IX. | ||
Ökten et al. (2018) designed and synthesized bromoindenoquinolines and evaluated them against hCA-I and II. Indenoquinolines with Br substituents at the C-3 and C-8 positions showed notable inhibition of hCA-I and II, outperforming other derivatives and AAZ. Among all the synthesized derivatives, only compound 191 exhibited significant inhibitory activity, with IC50 values of 120.9 nM and 267.5 nM, compared to AAZ (Fig. 69). This study suggests that indenoquinoline amine derivatives substituted with bromine and phenyl groups have potential as promising inhibitors of hCA-I and II.154
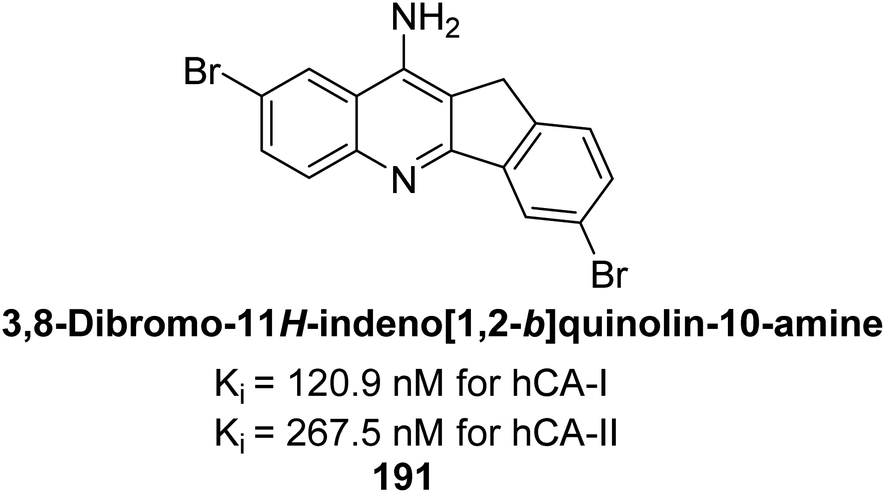 | ||
| Fig. 69 Chemical structure of compound 191 and its Ki values against hCA-I and II. | ||
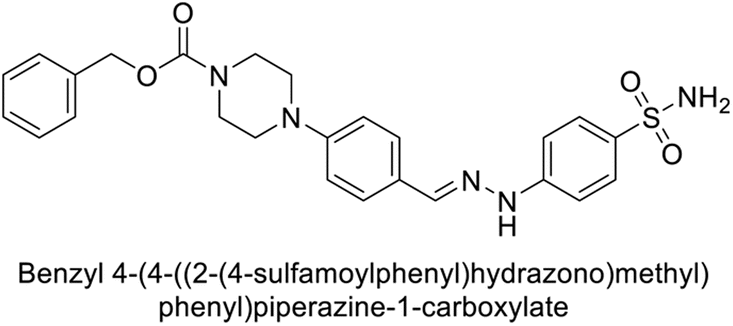
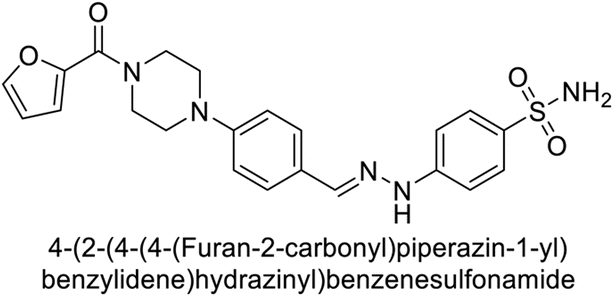
Supuran et al. (2018) synthesized novel benzenesulfonamide derivatives and evaluated against hCA-I, II, VII, and IX. The target compounds exhibited strong inhibition of hCA-VII, with less effectiveness against other hCA isoforms. The benzyl carbamate piperazine derivative 192 had a Ki = 11.4 nM, while the furoyl piperazine derivative 193 showed an even lower Ki = 6.2 nM. Among the benzylidenehydrazine carbonyl series, the o,p-dimethyl piperazine derivative 194 was the most selective hCA-VII inhibitor, with a Ki of 17.8 nM. The findings indicate that compounds 192 and 193 are potent inhibitors of hCA-II and VII, while compound 194 is a highly selective inhibitor of hCA-VII with low nanomolar inhibition (Table 11). These three compounds were therefore chosen for further investigation of their anticonvulsant effects in preclinical animal models of epilepsy.155
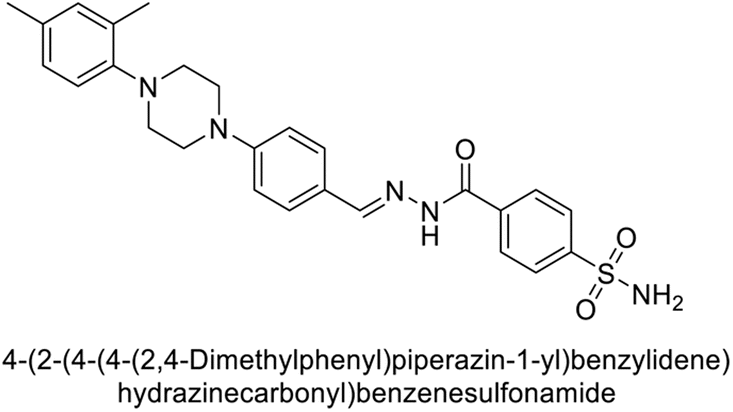
Ashraf et al. (2018) synthesized 1,3-oxazine derivatives and evaluated their inhibitory activity against bCA-II. Compound 195, featuring a 4-methoxy phenyl group, was the most potent, with an IC50 = 0.144 μM, outperforming the standard AAZ (IC50 = 0.997 μM). Molecular docking studies showed that compound 195 binds effectively to the enzyme's active site, interacting with key residues including His2, Lys168, His3, Ser1, Trp4, Asn10, Gly5, Gly63, His9, Leu 237, Phe229 and His63. This compound is suggested as a promising lead for developing more effective CAIs (Fig. 70).156
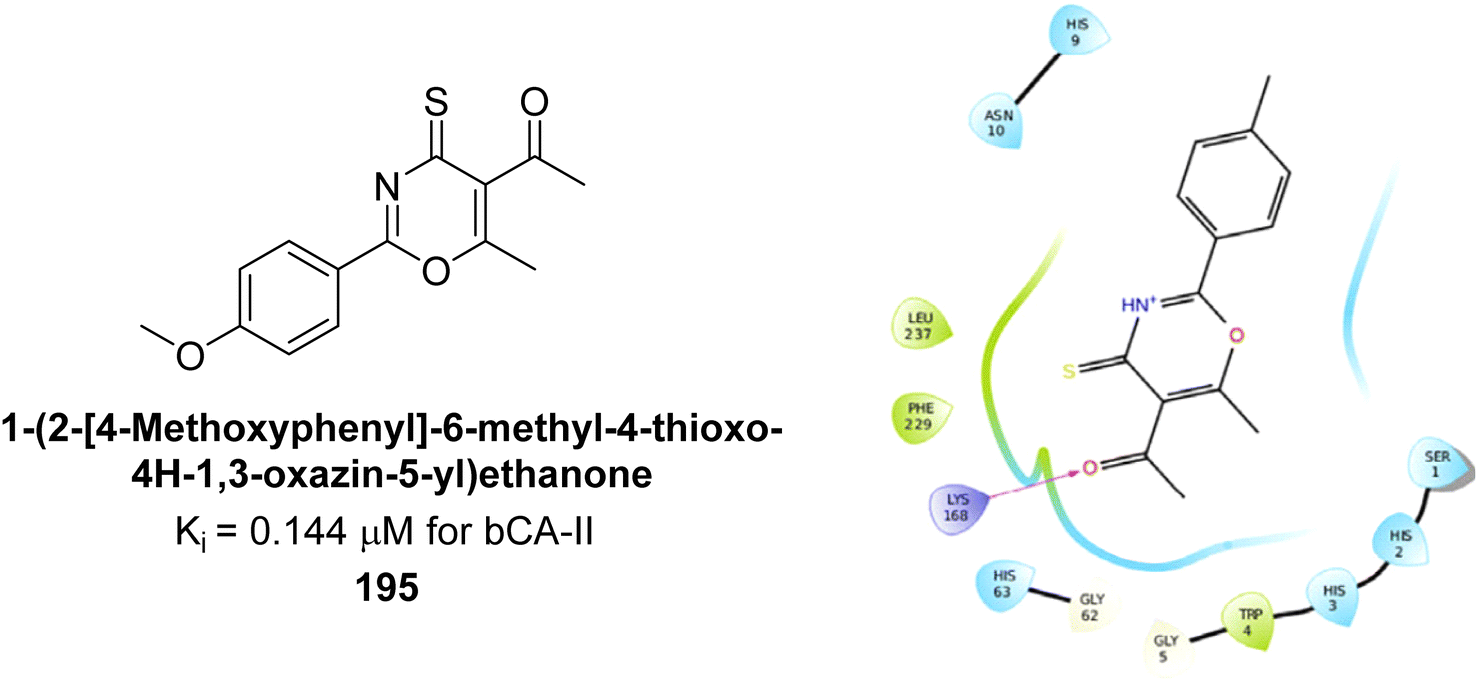 | ||
| Fig. 70 Chemical structure of compound 195 and its Ki value against bCA-II along with docking image of compound 195. | ||
Sağlık et al. (2019) synthesized new sulfonamide-hydrazone derivatives and evaluated their inhibitory effects on hCA-I and II. Compound 196 demonstrated the highest inhibition of hCA-I, with a Ki = 0.1676 μM, while compound 197 was most effective against hCA-II, with a Ki = 0.2880 μM. Docking studies showed that compound 196 interacts with hCA-I through a hydrogen bond with Gln92, a π–π interaction with His200, and a salt bridge with Lys57. Compound 197 binds to hCA-II via three hydrogen bonds: the amide carbonyl group with Asn62 and Asn67, and the pyridine nitrogen with Trp5 (Fig. 71).157
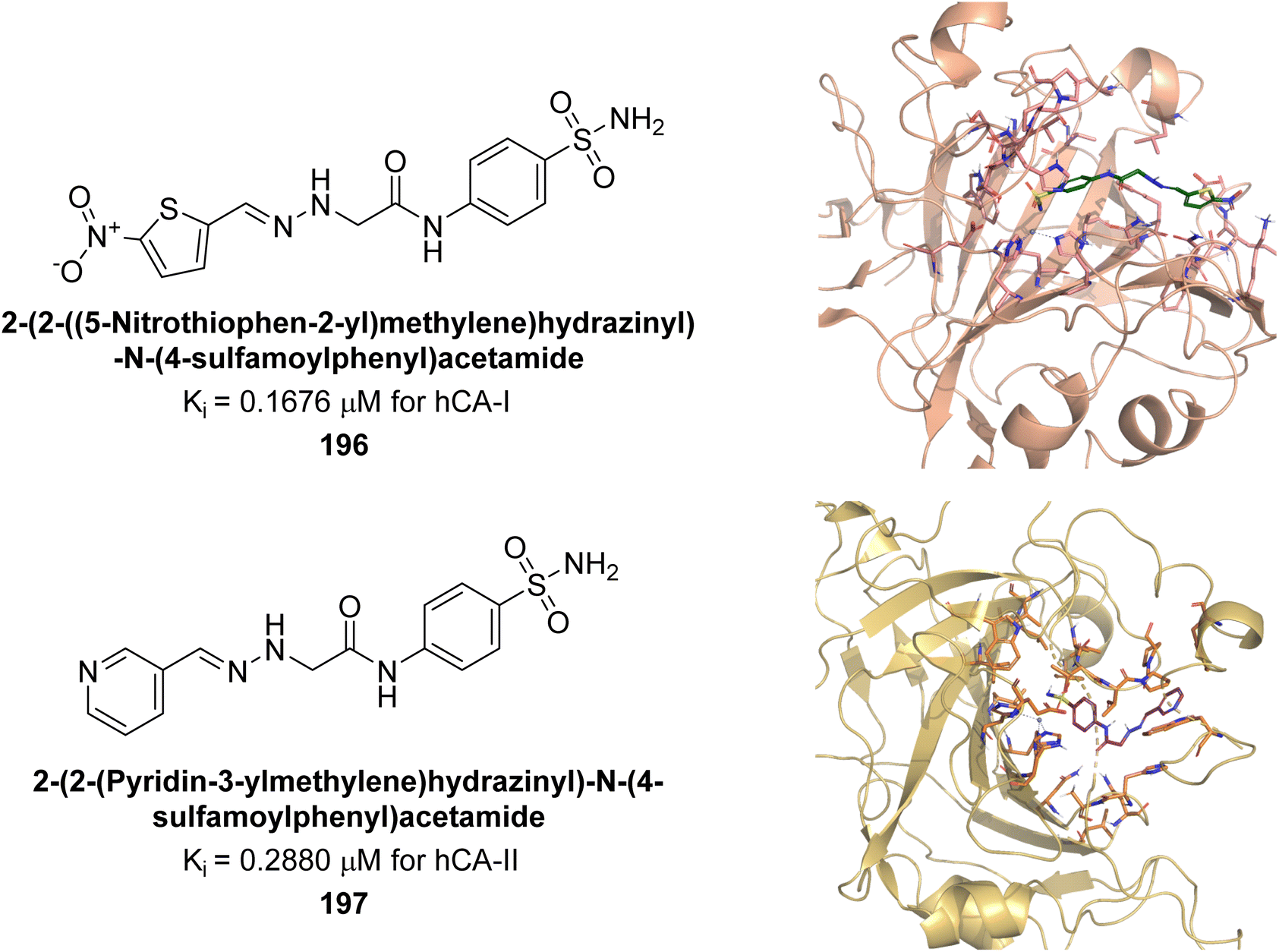 | ||
| Fig. 71 Chemical structures of compounds 196 and 197; their Ki values against hCA-I and II along with docking images of compounds 196 and 197. | ||
Supuran et al. (2019) synthesized pyrazoline-linked benzenesulfonamides and evaluated against hCA-I, IX, and XII. All the compounds demonstrated potent inhibition of hCA-I, with Ki ranging from 87.8 to 244.1 nM, beating AAZ (Ki = 250.0 nM). Compounds 198, 199, 200, 201, 202, and 203 showed effective inhibition of hCA-IX, with Ki values between 5.5 and 37.0 nM, either surpassing or matching the efficacy of AAZ. Additionally, compounds 199, 200, 202 and 204 exhibited strong inhibitory activity against hCA-XII, with Ki values ranging from 7.1–10.1 nM, compared to AAZ (Ki = 5.7 nM) (Table 12).158
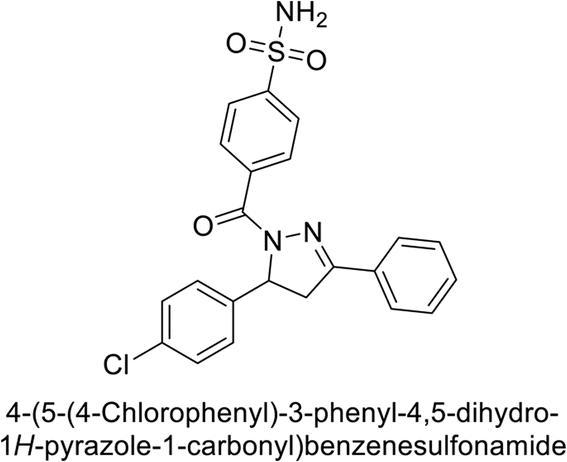
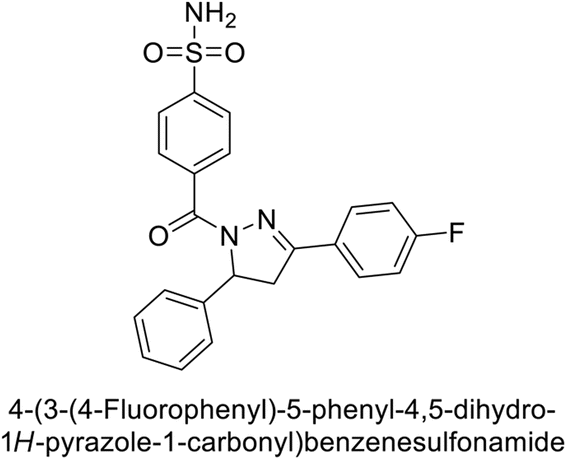
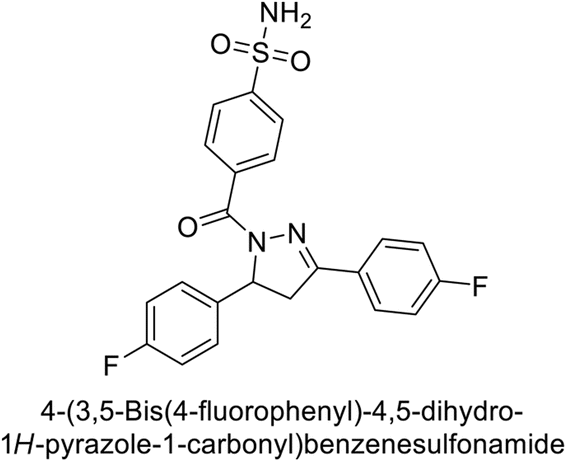
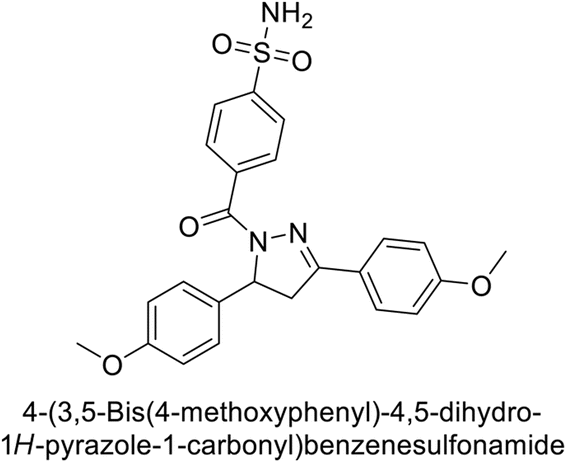
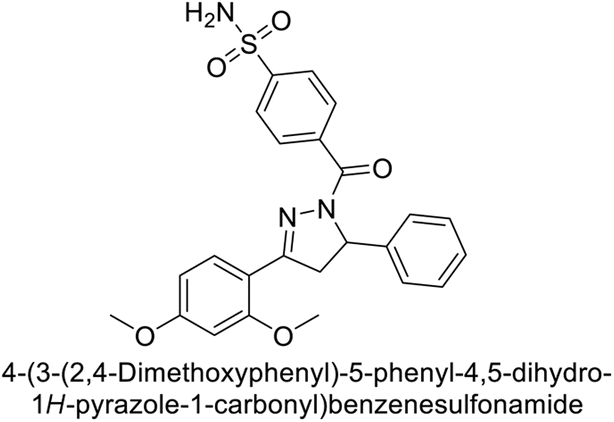
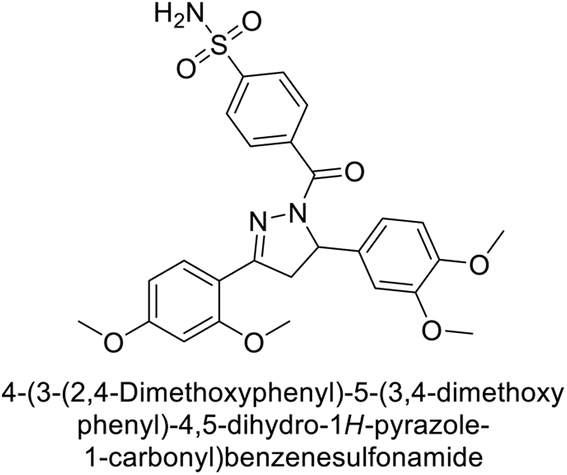
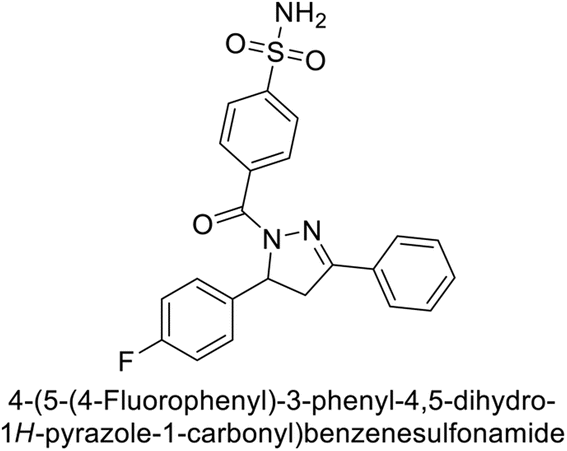
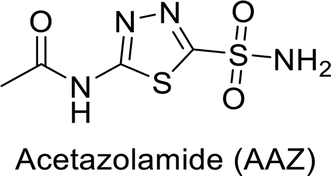
Larik et al. (2019) synthesized a novel silyl-yne chalcone derivatives and the derivatives were tested for inhibition of bCA-II. All compounds showed better inhibition than the standard AAZ. Among them, compound 205 emerged as the most potent, with an IC50 value of 0.054 μM. Binding analysis indicated that compound 205 binds to the active site of bCA-II, with a single π–π stacking interaction observed between the ligand's ring structure and Phe129, with a bond length of 4.90 Å (Fig. 72).159
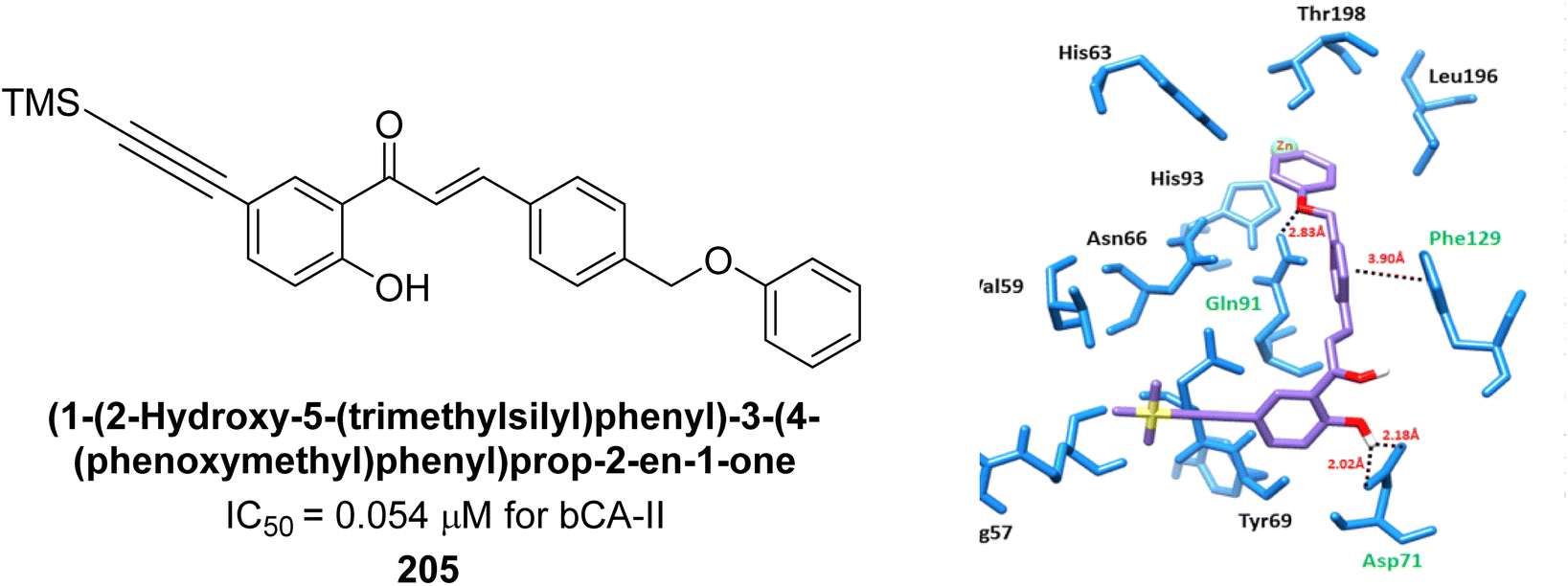 | ||
| Fig. 72 Chemical structure of compound 205; its IC50 value against bCA-II along with docking image of compound 205. | ||
Taslimi et al. (2019) synthesized substituted pyrazole compounds and assessed their inhibition of hCA-I and II. The Ki values ranged from 1.03–22.65 μM for hCA-I and from 1.82–27.94 μM for hCA-II. Remarkably, compounds 206 (Ki = 1.03 nM for hCA-I and 1.82 nM for hCA-II) and 207 (Ki = 6.58 nM for hCA-I and 8.93 nM for hCA-II) were the most active. Compound 207 interacts with Phe297 and Trp286 via π–π interactions and forms hydrogen bonds with Phe295 and Tyr72, with additional π–π interactions involving the aromatic ring (Fig. 73). These findings highlight the potential of these compounds in drug development for anticholinergic and antiepileptic therapies.160
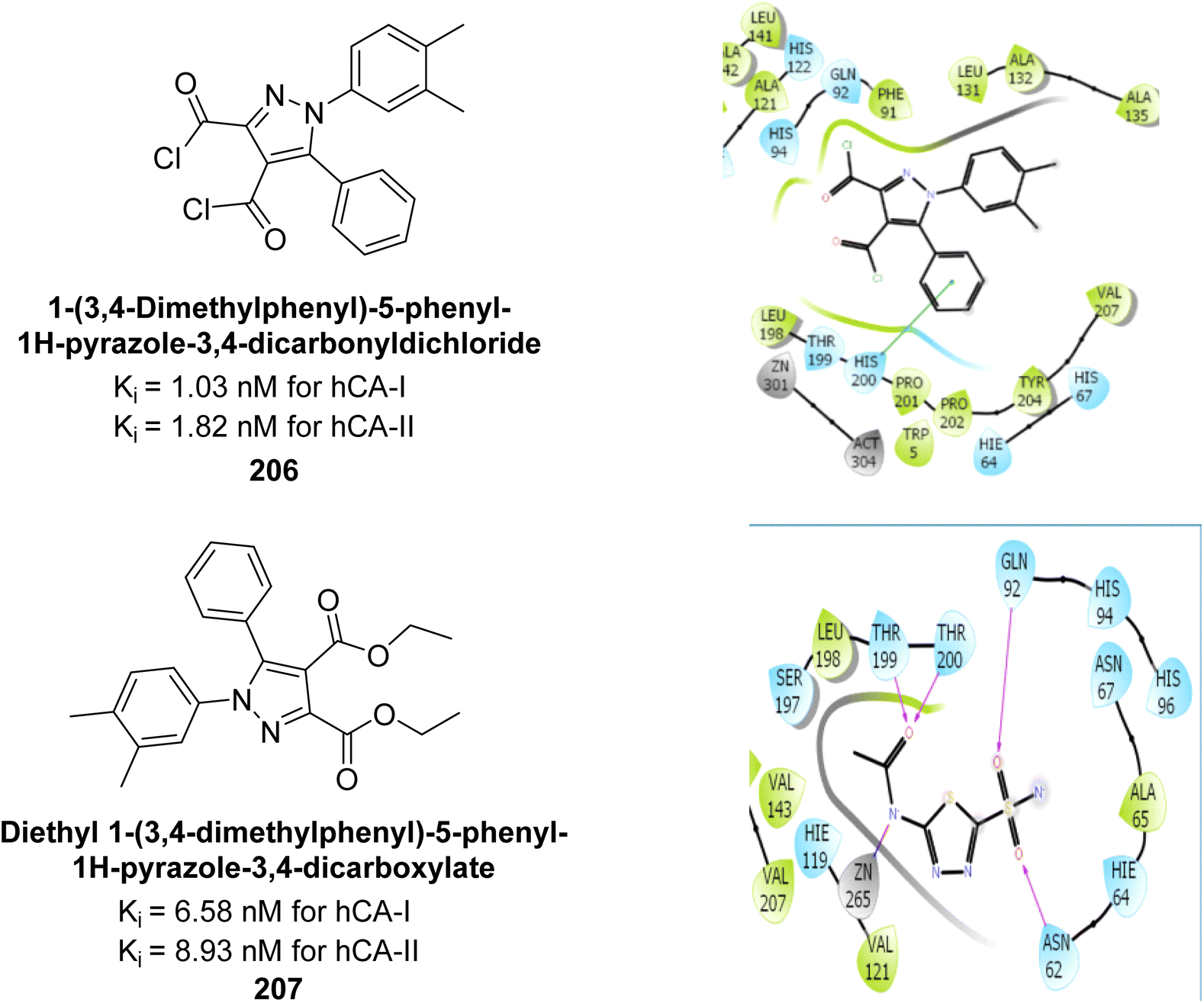 | ||
| Fig. 73 Chemical structures of compounds 206 and 207; their Ki values against hCA-I and II along with docking images of compound 206 and 207. | ||
Arifuddin et al. (2019) synthesized novel 7-hydroxycoumarin-3-carboxamides and evaluated their inhibition of hCA-IX and XII. Compound 208 exhibited the highest efficacy, with a Ki = 0.2 μM against both isoforms. Docking studies revealed that compound 208 binds effectively in the catalytic clefts of hCA-IX and XII, interacting with key residues such as Thr199, Zn262 and Gln92 in hCA-IX, and Zn301, Asn64 and Thr198 in hCA-XII (Fig. 74). These interactions contribute to its potent sub-micromolar inhibition and suggest its potential as a lead for anticancer therapy, particularly for metastatic hypoxic tumors.161
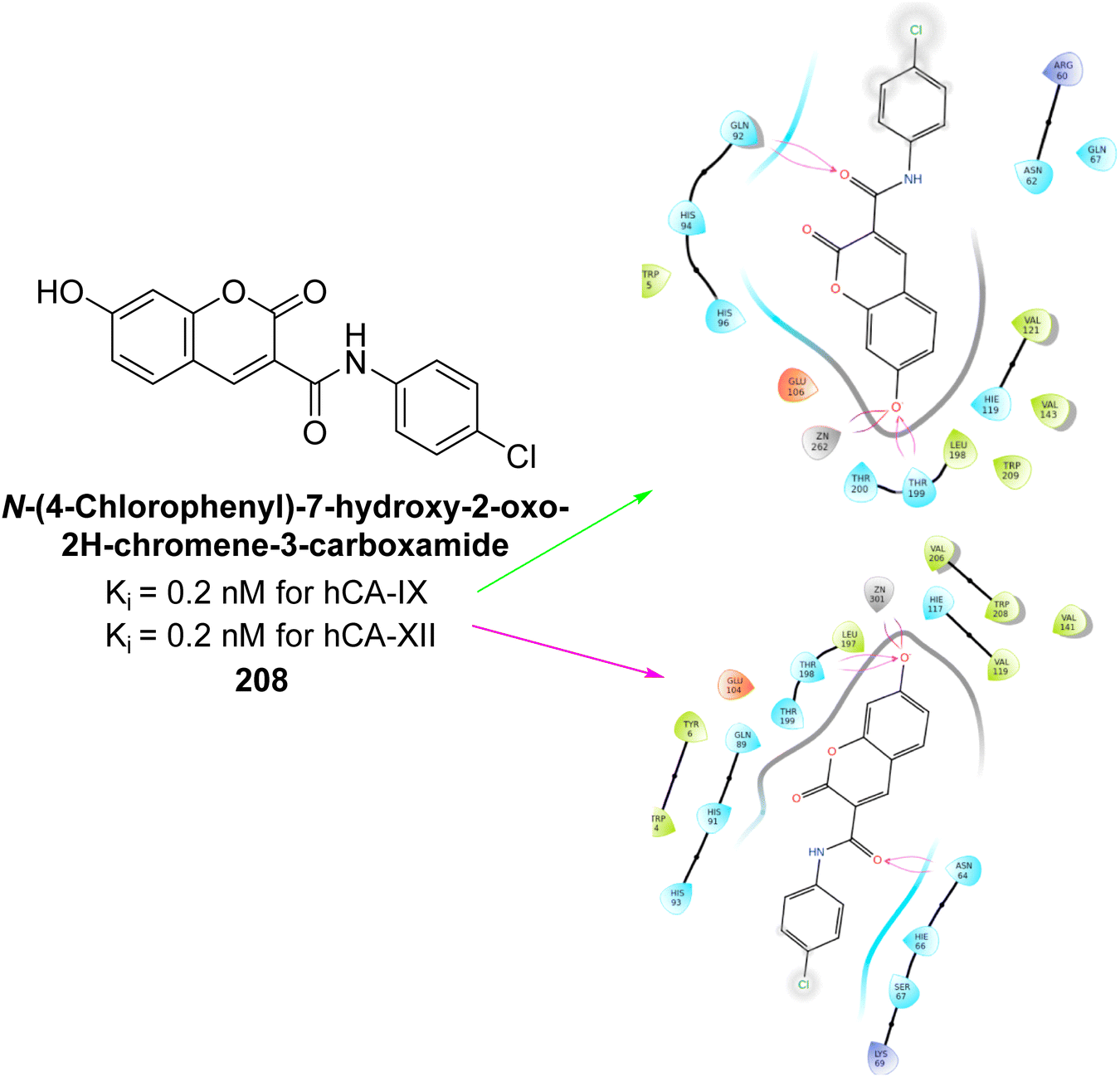 | ||
| Fig. 74 Chemical structure of compound 208; its Ki value against hCA-IX and XII along with docking images of compound 208. | ||
Durdagi et al. (2019) synthesized novel bis-coumarin derivatives linked by a triazole moiety and assessed their inhibition of hCA-I, II, IX, and XII. Compound 209 demonstrated the strongest inhibition of hCA-IX with a Ki = 144.6 nM, while compound 210 had the highest inhibition of hCA-XII with a Ki = 71.5 nM. Although no direct correlation between alkyl chain length and inhibition was observed, compounds with longer alkyl chains (10 and 12 carbons) were more effective. This suggests that longer chains provide greater molecular flexibility, allowing the coumarin moieties to better interact with the enzyme's active sites (Fig. 75).162
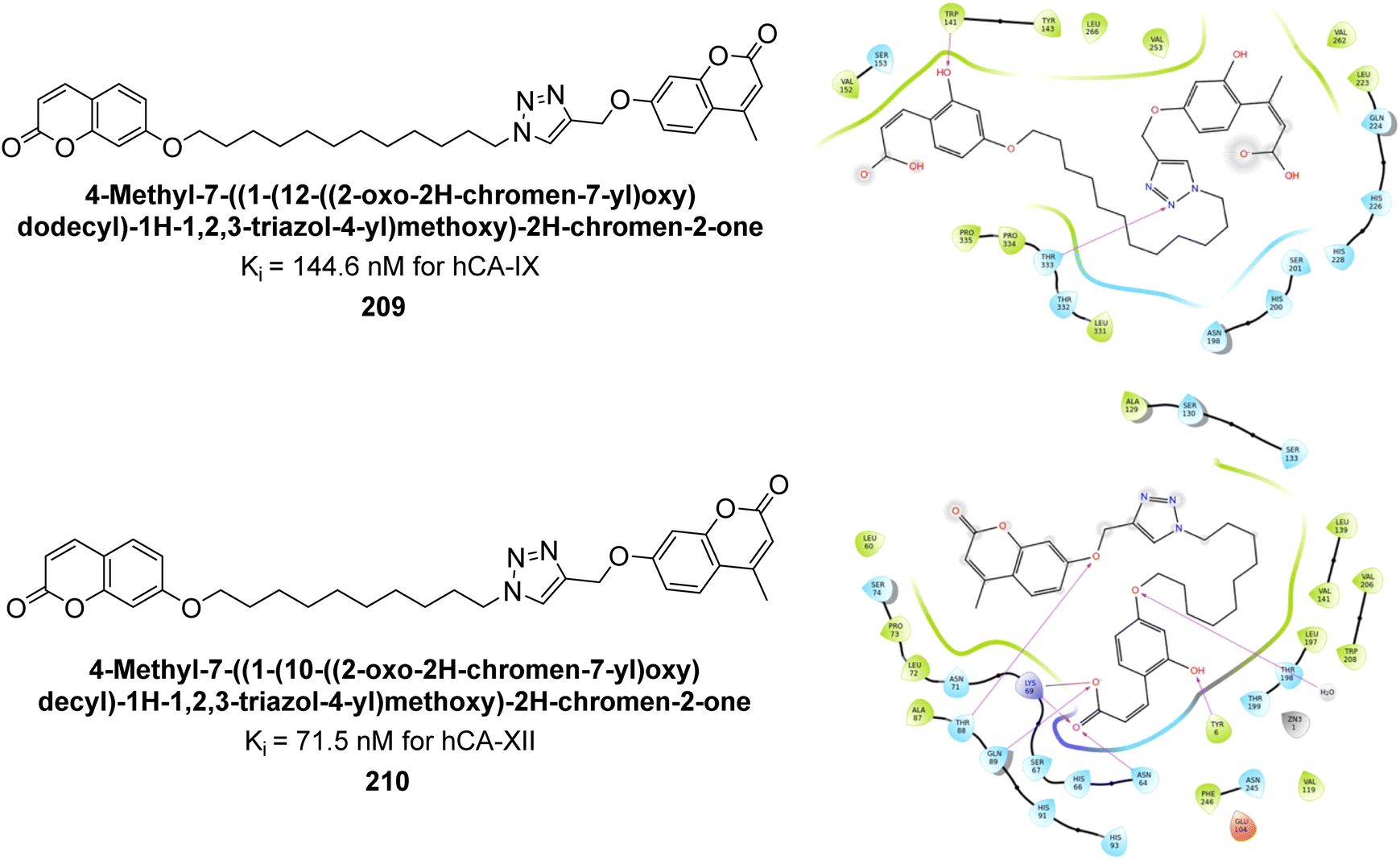 | ||
| Fig. 75 Chemical structures of compounds 209 and 210; their Ki values against hCA-IX and XII along with their docking images. | ||
Kurt et al. (2019) synthesized coumarin-sulfonamide derivatives and evaluated their inhibition of hCA-IX and XII. Compound 211 was the most effective inhibitor of hCA-IX, with a Ki = 45.5 nM. It also demonstrated selective anti-proliferative activity, with IC50 = 17.01 μM in HT-29 cells and 118.73 μM in HEK293T cells. Compound 211 inhibited hCA-IX and XII expression in HT-29 cells, indicating its potential as a selective therapeutic agent for colon cancer by targeting these isoforms (Fig. 76).163
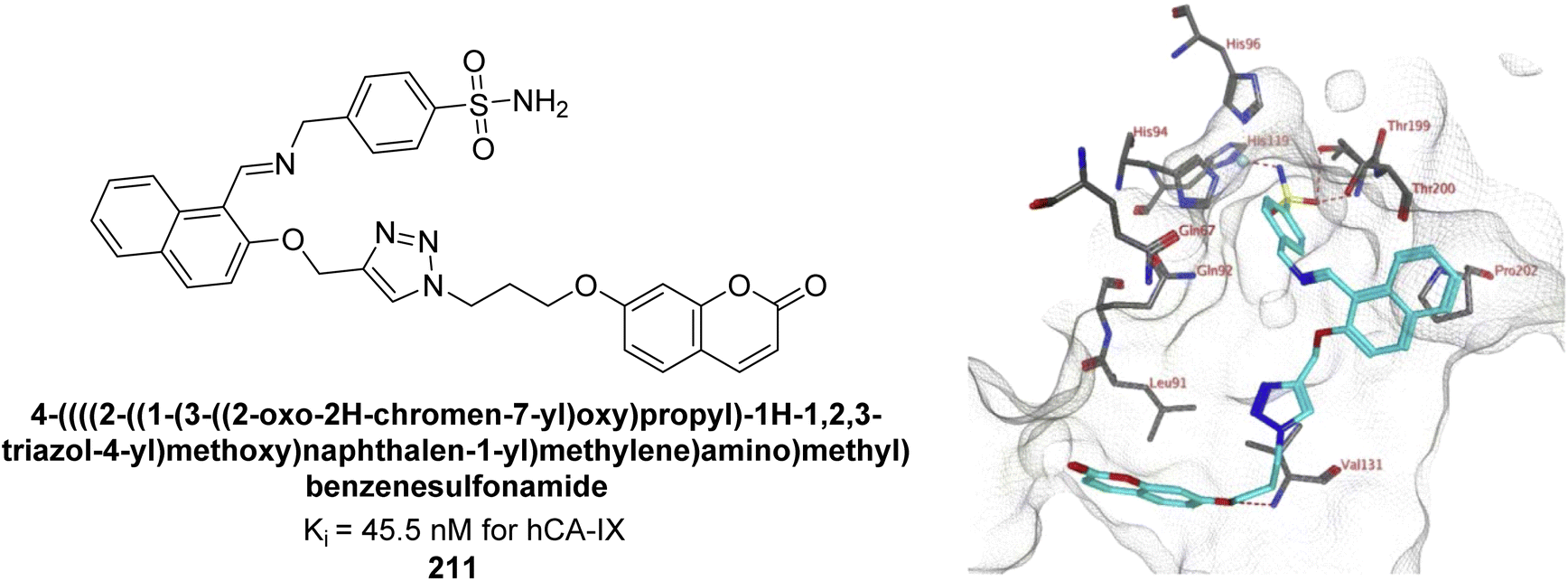 | ||
| Fig. 76 Chemical structure of compound 211; its Ki value against hCA-IX along with docking image of compound 211. | ||
Bayindir et al. (2019) synthesized novel N-substituted rhodanine derivatives using a green, one-pot method and tested their inhibitory effects on hCA-I and II. The Ki values ranged from 43.55–89.44 nM for hCA-I and from 16.97–64.57 nM for hCA-II. Compound 214 showed the strongest inhibition of hCA-I with a Ki = 43.55 nM, outperforming the standard drug AAZ (Ki = 110.11 nM). Compounds 212 and 213 also demonstrated significant inhibition of hCA-II, with Ki values better than AAZ (Ki = 116.97 nM) (Fig. 77).164
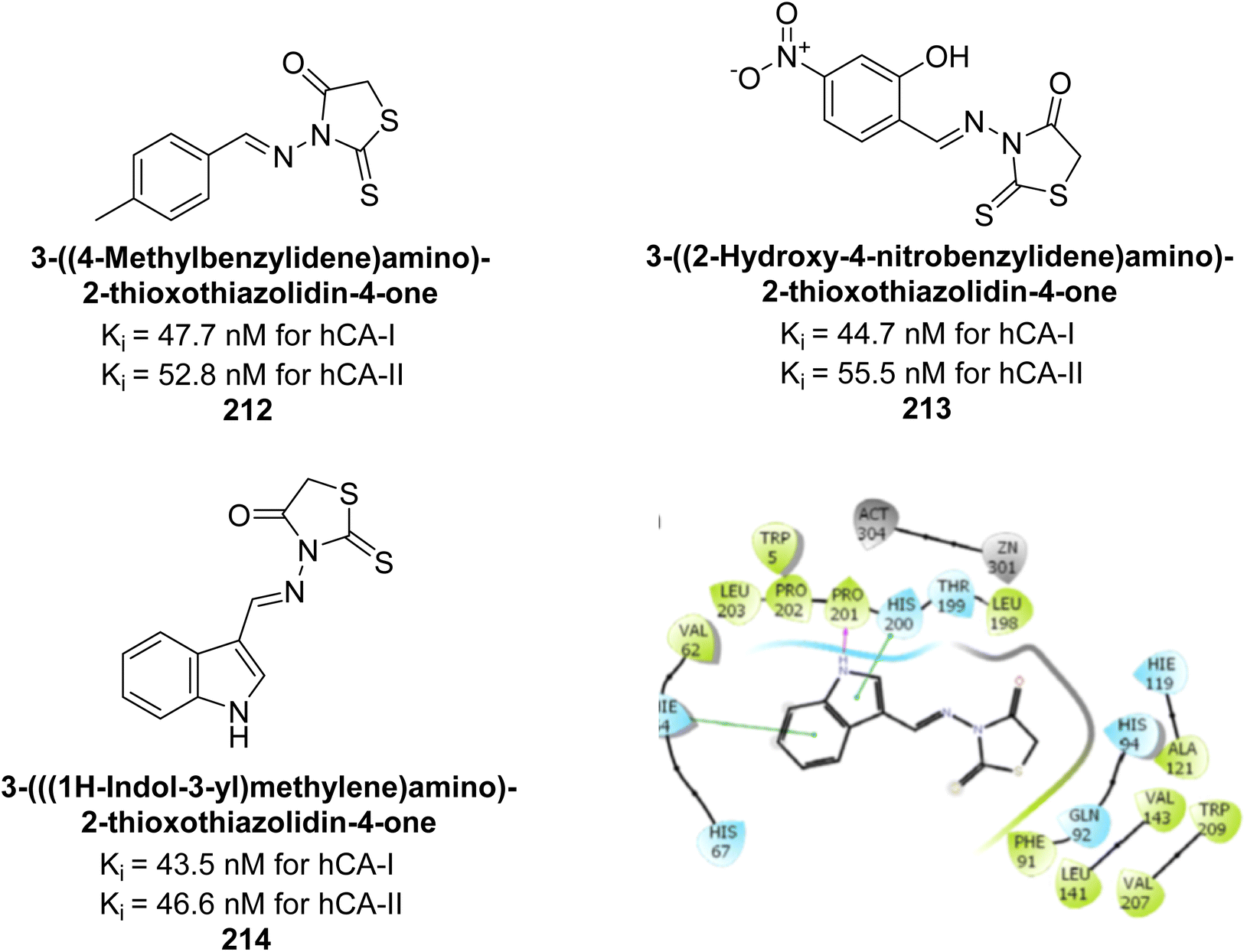 | ||
| Fig. 77 Chemical structures of compounds 212, 213 and 214; their Ki values against hCA-I and II along with docking image of compound 214. | ||
Supuran et al. (2019) synthesized imidazolone-based benzenesulfonamides. These compounds were evaluated as inhibitors of hCA-I, II, IX and XII. Compounds 215, 216, and 217 showed notable selectivity for hCA-IX and XII over hCA-I and II, with selectivity indices ranging from 6.2–19.4 for hCA-IX and 3.3–8 for hCA-XII. Among these, compound 216 was the most potent inhibitor of hCA-IX, with a Ki = 23 nM, outperforming the standard drug AAZ, which has a Ki = 25 nM (Fig. 78). Moreover, a molecular docking study was conducted to understand the binding interactions of the synthesized imidazolones within the active sites of CA-IX, using PDB structure 5FL4. This analysis helped rationalize the observed inhibitory activities of the compounds.165
 | ||
| Fig. 78 Chemical structures of compounds 215, 216 and 217; their Ki values against hCA-IX and XII along with docking image of compound 217. | ||
Akocak et al. (2020) synthesized 1,3-diaryltriazene-substituted sulfathiazole derivatives and evaluated for their inhibitory effects on hCA isoforms I and II. The Ki ranged from 450.37 to 1094.34 nM for hCA-I and 504.37 to 1205.36 nM for hCA-II. Among these, compound 218 demonstrated particularly potent inhibition against hCA-I and hCA-II with Ki values of 450.37 and 504.37, respectively (Fig. 79). Additionally, the binding interactions of the compound 218 were characterized by several key stabilizations. The benzenesulfonamide compound formed a strong hydrogen bond with Gln92 (2.15 Å) and engaged in π–π interactions with His94 and Phe131. The sulfonyl group created hydrogen bonds with Thr199 (1.90 Å) and Thr200 (2.74 Å), while the –SO2 group also bonded with Trp279 (2.24 Å). Hydrophobic interactions were observed between the thiazole, benzene, and fluorobenzene moieties and residues Tyr70, Trp84, Trp279, Phe330, Tyr121. These interactions suggest that these compounds have potential as lead candidates for developing selective inhibitors of metabolic enzymes.166
 | ||
| Fig. 79 Chemical structure of compound 218; its Ki values against hCA-I and II along with docking images of compound 218. | ||
Eldehna et al. (2020) synthesized thiazolo-benzimidazole and tested against hCA-I, II, and IX. Remarkably, compounds 219, 220, and 221 exhibited strong inhibition of hCA-IX with Ki values of 6.2, 9.7, and 5.5 nM, respectively, and showed good selectivity for hCA-IX over hCA-I and II. Docking studies revealed that the thiazolo-benzimidazole scaffold effectively mimics the phenyl moiety, establishing hydrophobic interactions and hydrogen bonds within the hCA-IX active site (Fig. 80).167
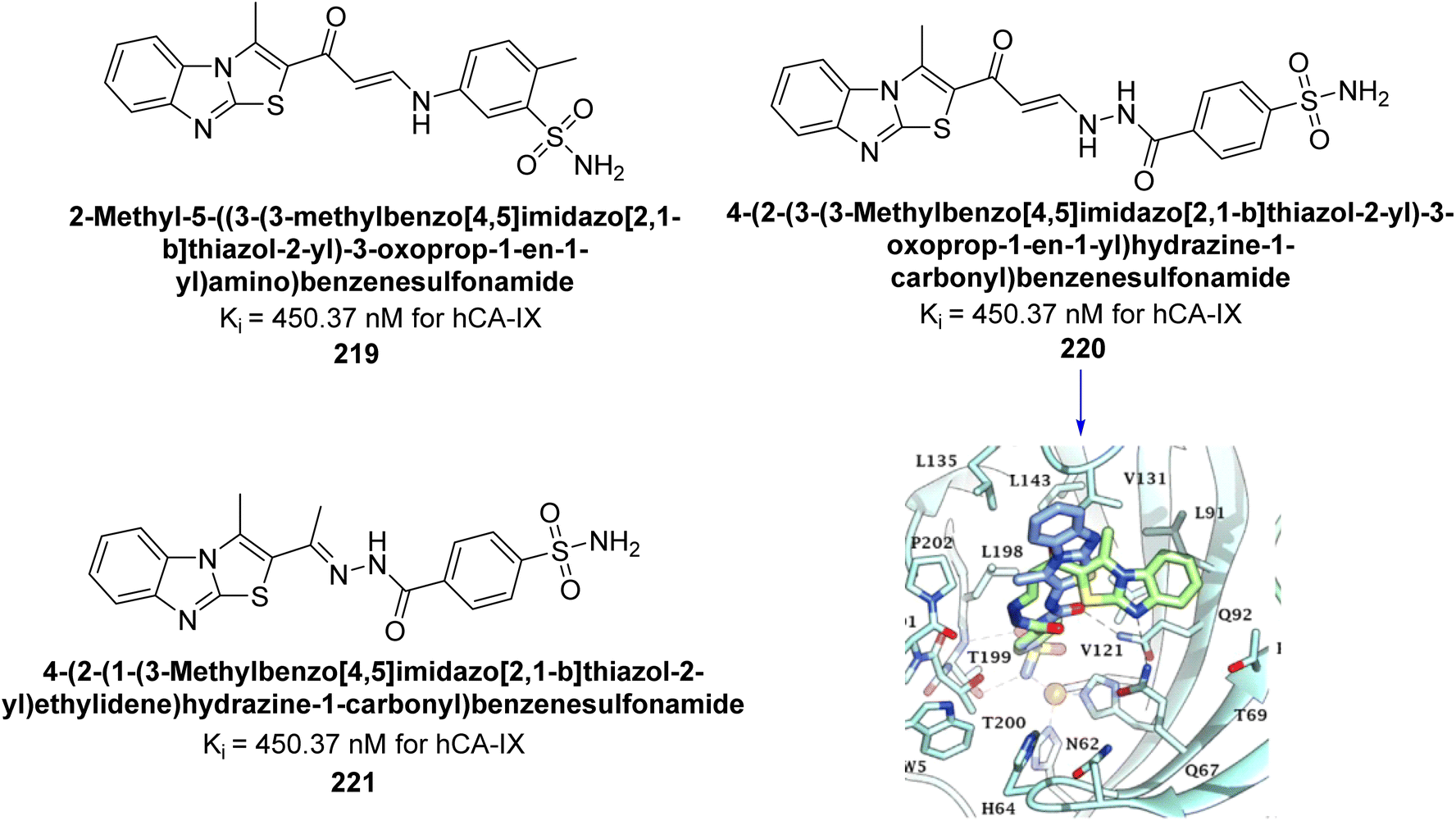 | ||
| Fig. 80 Chemical structures of compounds 219, 220 and 221; their Ki values against hCA-IX along with docking image of compound 220. | ||
Sever et al. (2020) synthesized a series of new thiazolyl-pyrazolines and evaluated for their efficacy using in vitro for hCA-I and II. Compound 222, featuring a cyanophenyl moiety at the 4th position of the thiazole scaffold, was the most potent, with a Ki = 13.04 nM. The synthesized compounds exhibited greater potency against hCA-II than AAZ (Ki = 98.28 nM). Particularly, compound 223, with a chlorophenyl group at the 4th position of the thiazole scaffold, was the most potent inhibitor of hCA-II, with a Ki = 7.47 nM. Compound 222, featuring a cyano group, interacted with Tyr204 through a π–π interaction and formed a hydrogen bond with Tyr20 (2.16 Å). Compound 223 showed an oxygen in the morpholine ring forming a hydrogen bond with Lys170 (2.12 Å). Additionally, face-to-face stacking interactions were observed between His64, His94, and Phe13 with benzene rings, while hydrophobic interactions involved residues Trp5, Val135, Tyr7, Ala65, Pro201, Leu198, Pro202, and Leu204 (Fig. 81).168
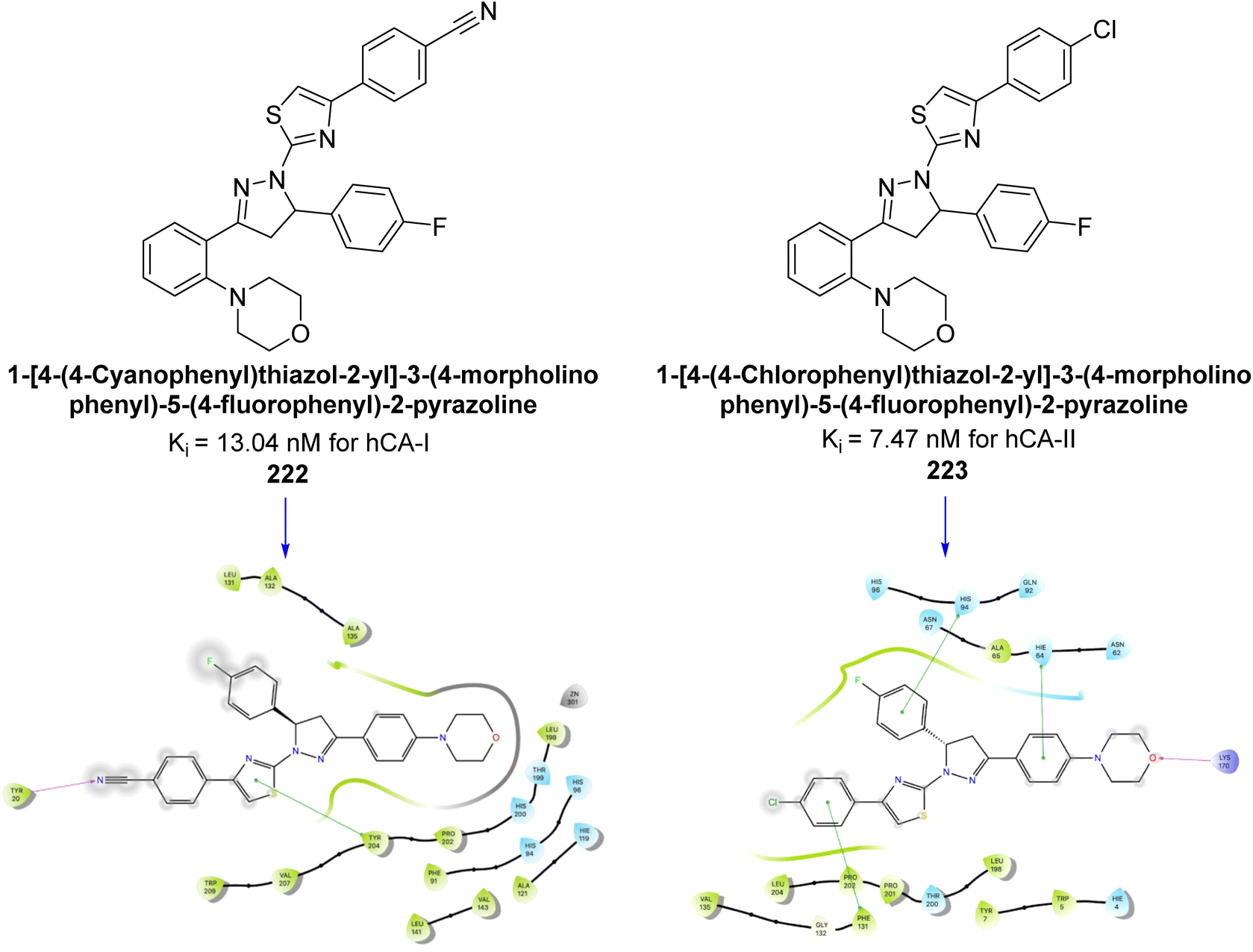 | ||
| Fig. 81 Chemical structures of compounds 222 and 223; their Ki values against hCA-I and II along with their docking images. | ||
Arslan et al. (2020) synthesized a series of novel sulfonamide-based ketenes and evaluated for their inhibitory effects on hCA-I and II. The compounds 224, 225 and 226 which contain halogen substitutions, showed the most potent inhibition, with Ki values in the low nanomolar range (9.01, 7.41 and 7.37 nM, respectively). Additionally, compounds 227 and 228 selectively inhibited hCA-I, while 229 and 230 selectively inhibited hCA-II (Fig. 82). Molecular docking studies were conducted to further investigate the interactions between the ligands and their respective receptors (Fig. 82).169
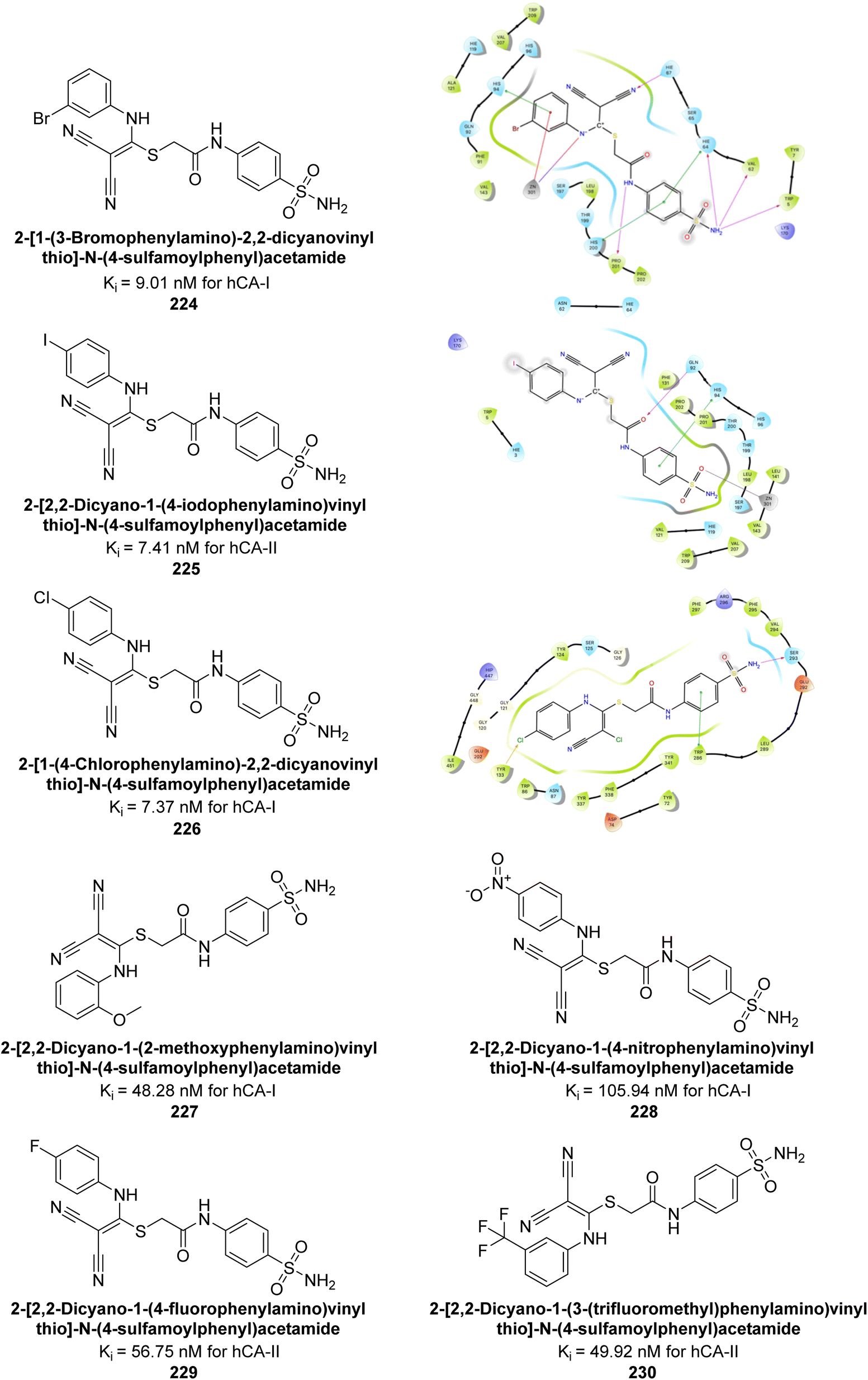 | ||
| Fig. 82 Chemical structures of compounds 224–230; their Ki values against hCA-I and II along with docking images of compounds 224, 225 and 226. | ||
Supuran et al. (2020) synthesized various triazolopyrimidine-based and triazole-based benzenesulfonamides. Especially, sulfonamides 231, 232 and 233 exhibited strong inhibition of hCA-IX and XII. Among them, pyrimidine-based sulfonamide 234 and triazole-based sulfonamide 233 demonstrated high selectivity for hCA-IX (Fig. 83). The strong inhibitory activity of compound 234 against hCA-IX is due to its effective interactions with a hydrophobic pocket formed by Val20, Trp5, Pro201, and Pro202. In compound 233, the phenylureido tail binds to a different pocket in hCA-II than in hCA-IX and XII. This variation is attributed to the Phe131 residue in hCA-II, which sterically blocks ligand binding in the region where it interacts with Gln92 and Leu91 in hCA-IX, and Thr91 and Lys67 in hCA-XII (Fig. 83).170
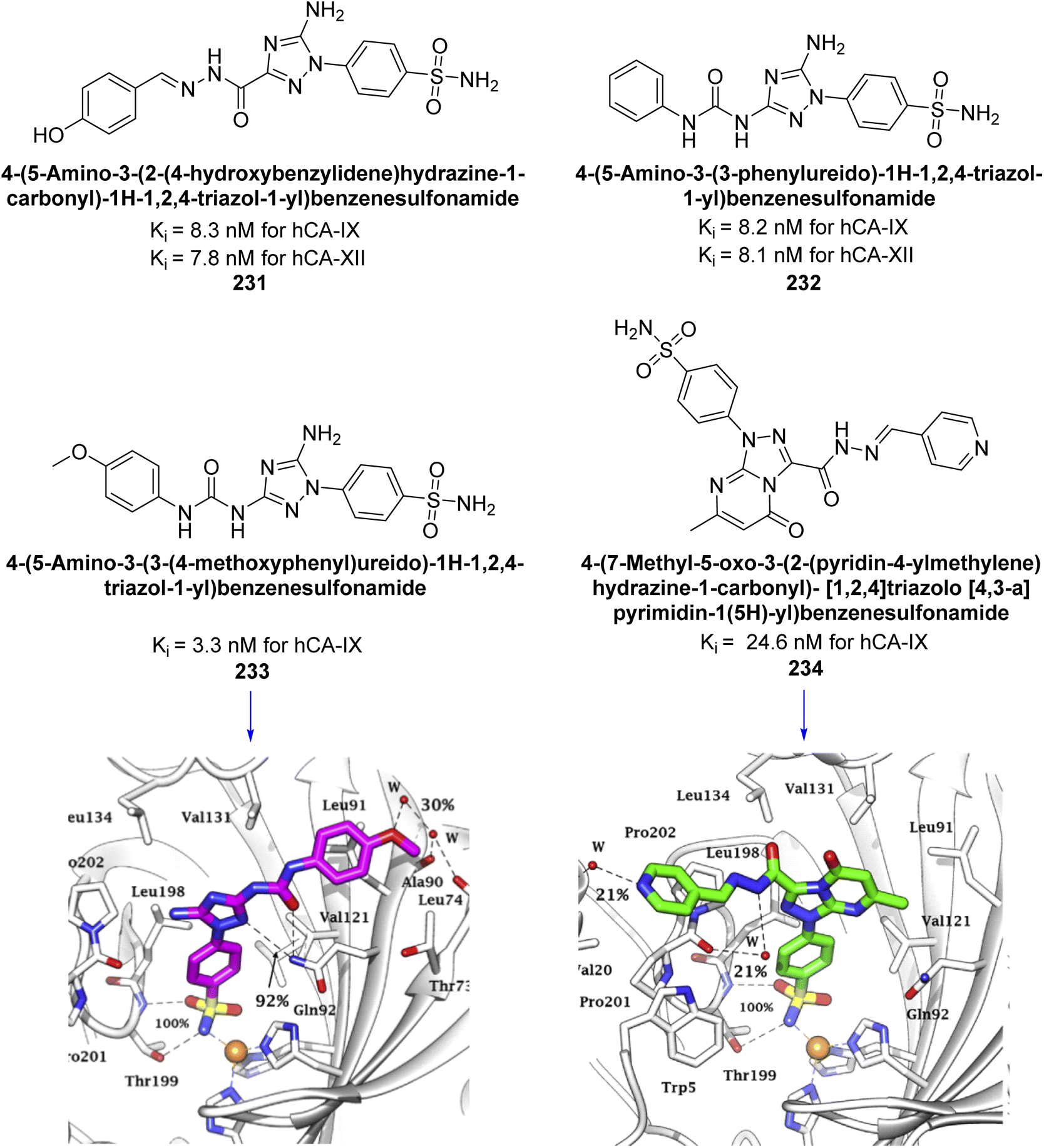 | ||
| Fig. 83 Chemical structures of compounds 231–234; their Ki values against hCA-IX and XII along with docking images of compounds 233 and 234. | ||
Shafiq et al. (2020) synthesized quinazolinone derivatives and assessed their inhibitory effects on bCA-II and hCA-II. The compounds exhibited moderate to significant inhibition, with IC50 values ranging from 8.9 to 67.3 μM for bCA-II and 14.0 to 59.6 μM for hCA-II. SAR analysis suggested that a nitro group at the R position enhances activity. The most active compound, 235, acted as a competitive inhibitor of both bCA-II and hCA-II, with Ki values of 13.0 μM and 14.25 μM, respectively. Compounds 236 and 237 showed higher selectivity for hCA-II, confirmed by molecular docking studies (Fig. 84).171
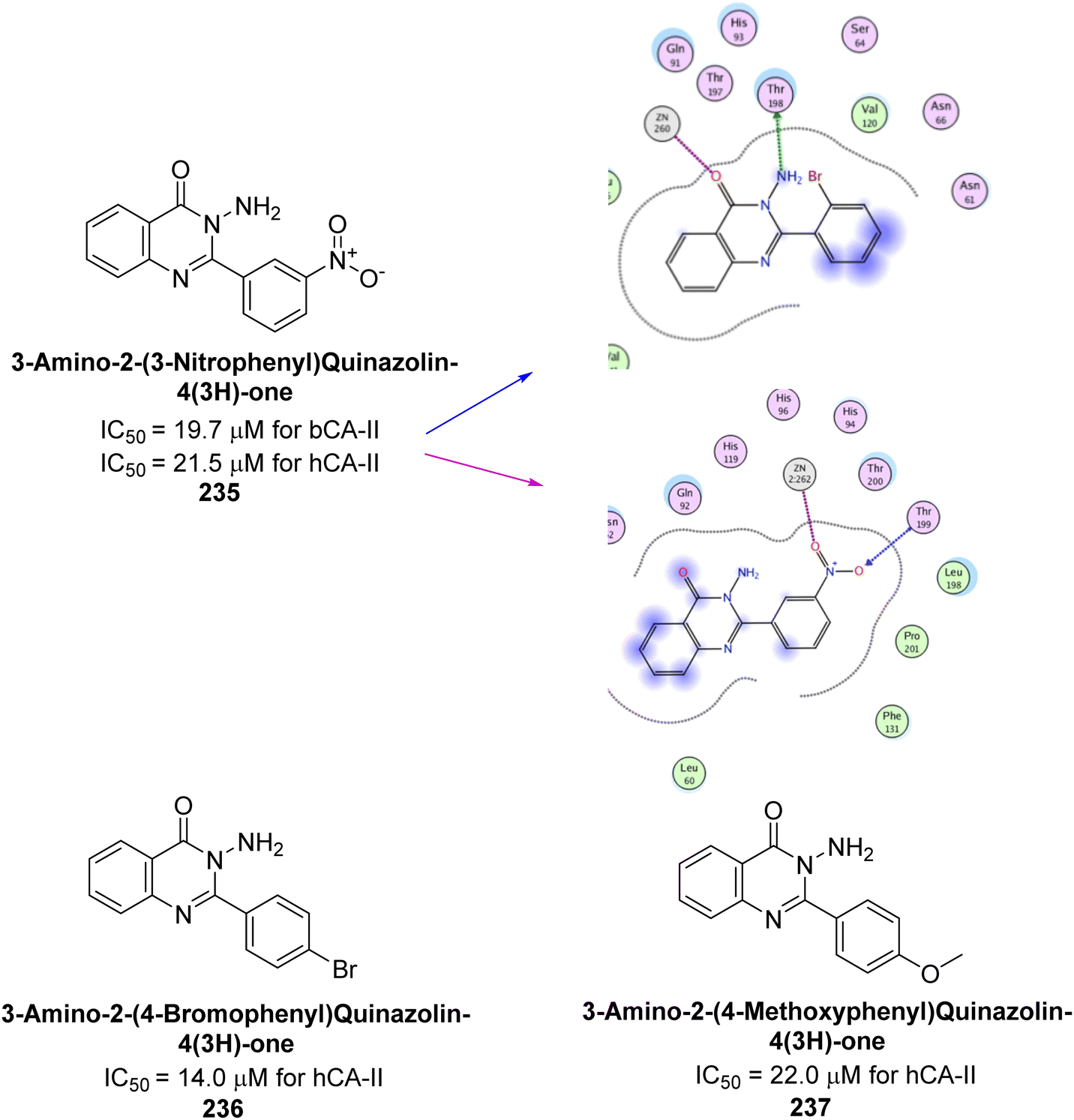 | ||
| Fig. 84 Chemical structures of compounds 235–237; their IC50 values against bCA-II and hCA-II along with docking image of compound 235. | ||
Saeed et al. (2020) synthesized a series of 2-substituted benzoxazoles and evaluated for their hCA-II inhibitory activity. Among them, compounds 238 and 239 demonstrated the highest inhibitory potential, with IC50 values of 0.00564 μM and 0.00596 μM, respectively. These values indicate a significantly stronger inhibitory effect compared to the standard AAZ, which has an IC50 value of 0.997 μM. Compound 238 confirmed an optimal binding conformation within the hCA-II binding pocket, primarily through hydrogen bonds with residues His63 and Pro199, which were more significant than hydrophobic interactions in the docking complex analysis (Fig. 85). Preliminary findings indicate that compound 238 exhibits favorable drug-like properties and may hold potential for further biomedical applications.172
 | ||
| Fig. 85 Chemical structures of compounds 238 and 239; their IC50 values against hCA-II along with docking image of compound 238. | ||
George et al. (2020) synthesized benzenesulfonamide derivatives and assessed against hCA-XII. The derivatives, specifically compounds 240 and 241, demonstrated the highest activity against hCA-XII, with Ki values of 21.5 nM and 22.3 nM, respectively. These compounds featured unsubstituted benzoyl and methoxybenzoyl groups, which contributed to their potent inhibitory effects. Moreover, compound 240 exhibited a binding mode similar to that of AAZ, interacting with key sites including Zn2+, Thr199, Thr200, and Leu198 through metal coordination and hydrogen bonding (Fig. 86).173
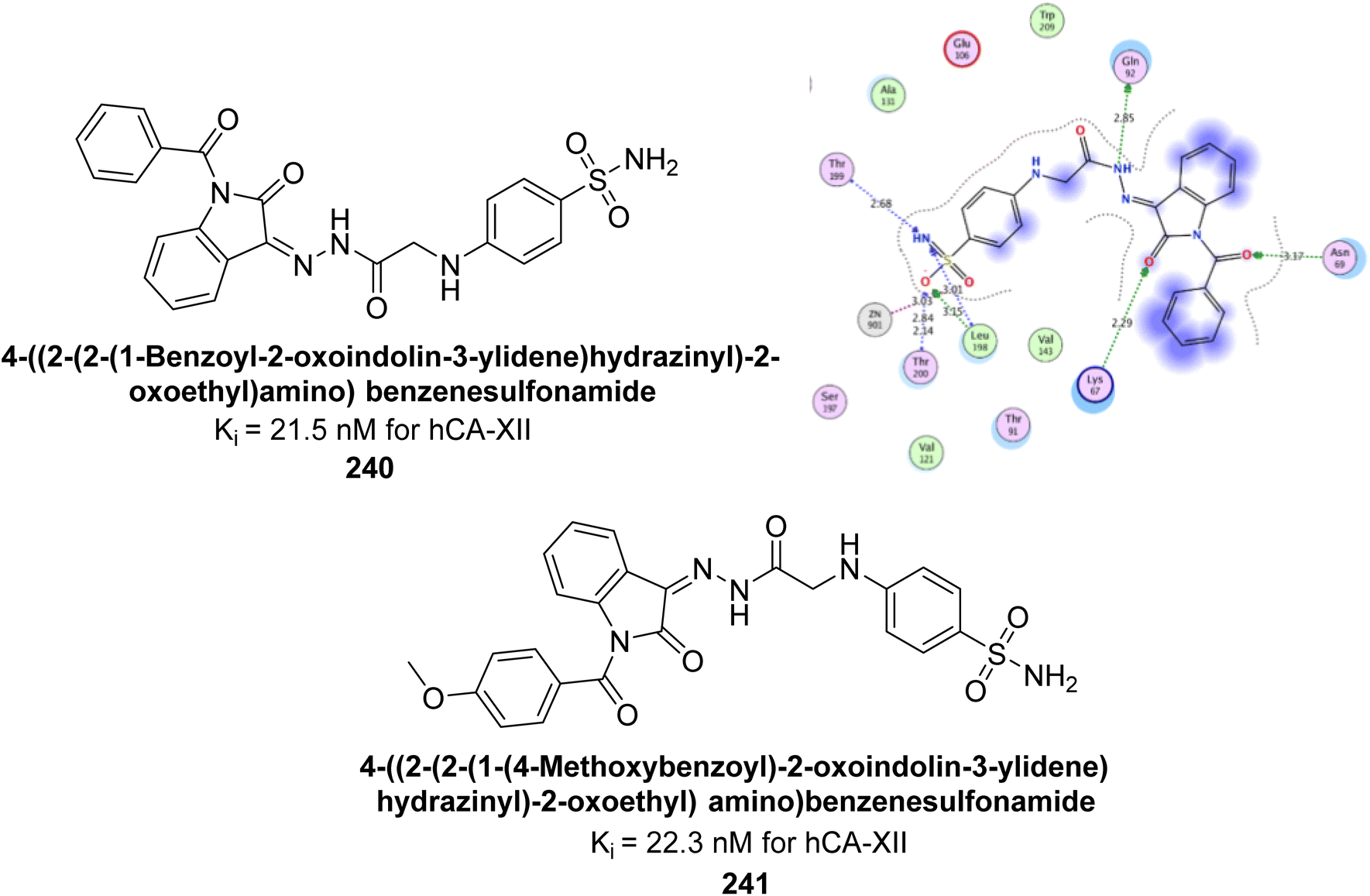 | ||
| Fig. 86 Chemical structures of compounds 240 and 241; their Ki values against hCA-XII along with docking image of compound 240. | ||
Supuran et al. (2020) synthesized coumarin-linked 1,2,4-oxadiazoles and evaluated their inhibitory activity against hCA-IX and XII. Compounds 242, 243, 244, and 245 displayed potent inhibition of hCA-XII, with compound 242 showing the strongest inhibition (Ki = 1 nM). Compound 245 was the most effective against hCA-IX (Ki = 23.6 nM). These results highlight compound 242 as a promising lead for hCA-XII inhibitors and compound 245 for dual hCA-IX/XII inhibition. Molecular docking studies were conducted to explore the binding interactions of the potent compounds 242 and 243 within the hCA-XII active site (Fig. 87).174
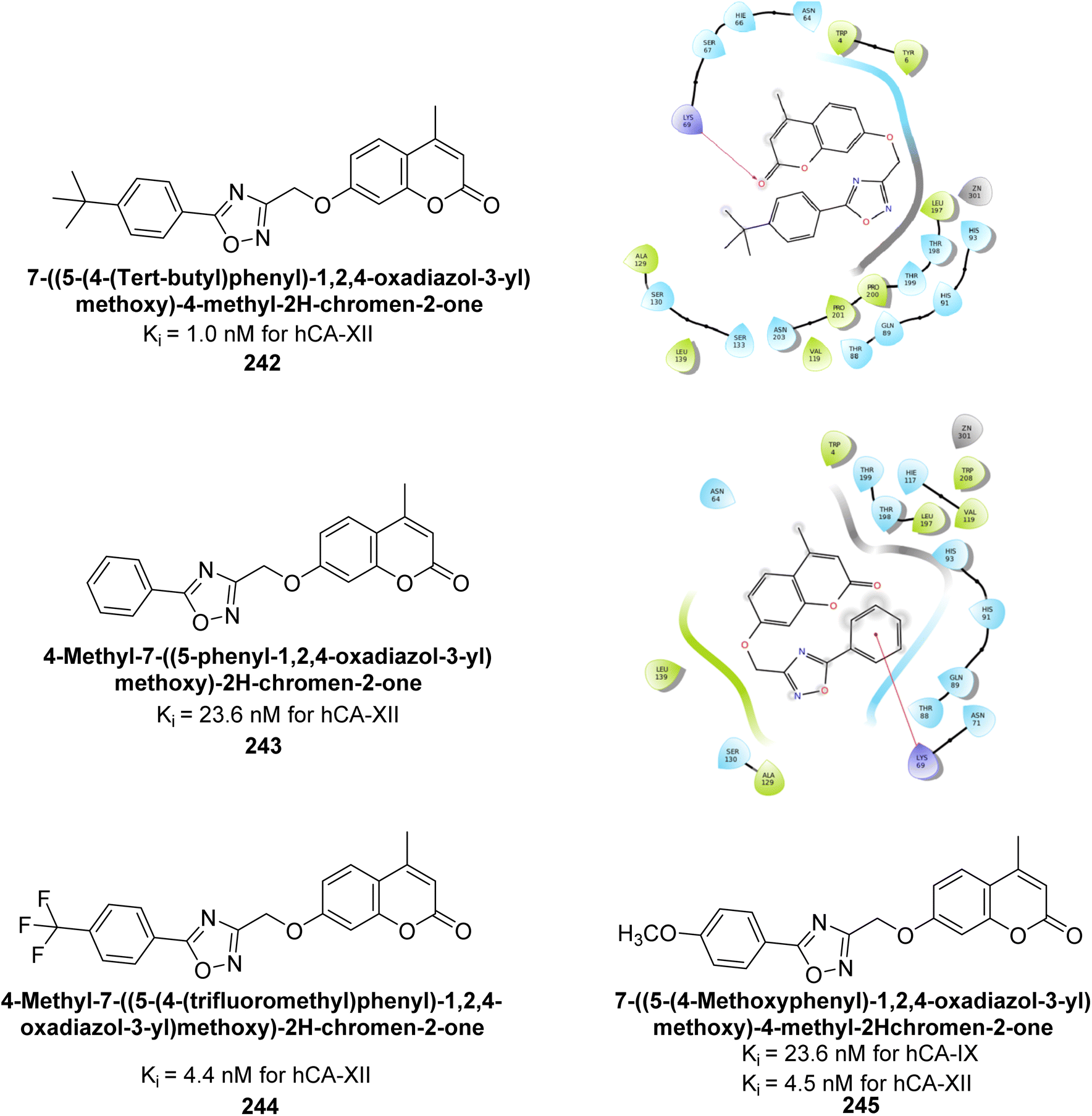 | ||
| Fig. 87 Chemical structures of compounds 242–245; their Ki values against hCA-IX and XII along with docking images of compounds 242 and 243. | ||
George et al. (2020) synthesized 2-oxindole benzenesulfonamide conjugates with various linkers and tested their inhibitory activity against hCA-I, II, IX, and XII. Several compounds demonstrated distinguished activity, particularly towards hCA-I, II, and IX, surpassing the standard AAZ. Particularly, compound 246 showed the strongest inhibition against hCA-II and IX, with Ki values of 3.0 and 13.9 nM, respectively. Molecular docking studies of compound 246 revealed binding modes similar to AAZ, aligning with its potent inhibition (Fig. 88).175
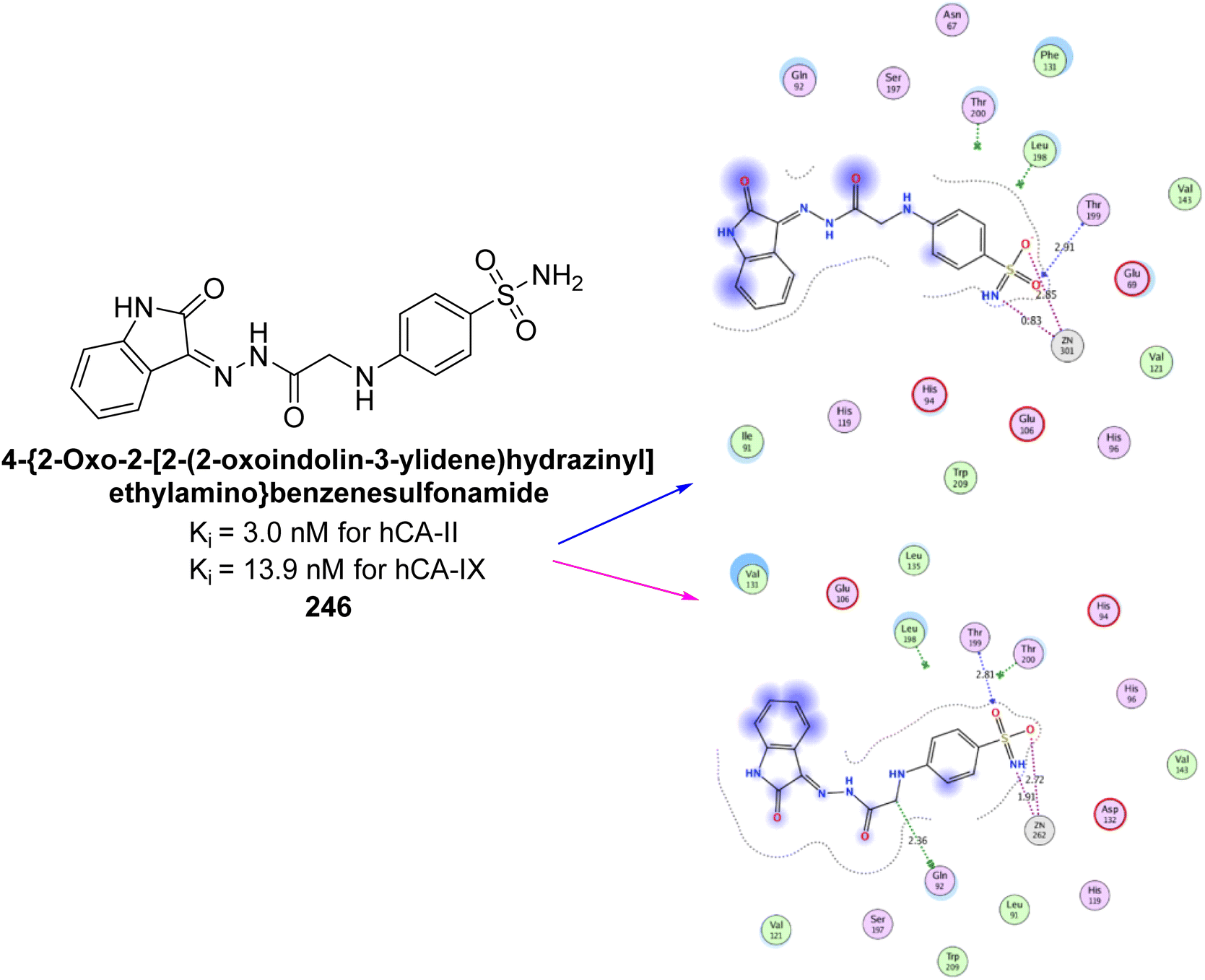 | ||
| Fig. 88 Chemical structure of compound 246; its Ki values against hCA-II and IX along with docking images of compound 246. | ||
Supuran et al. (2021) designed indolyl chalcone-linked sulfonamides and evaluated their inhibition of hCA-I, II, IX, and XIII isoforms. Compounds 247 (2.3 nM), 248 (2.4 nM), 249 (3.6 nM), and 250 (7.0 nM) showed superior potency against hCA-II compared to the standard AAZ (12.1 nM) (Fig. 89). Additionally, several compounds exhibited inhibition of hCA-XIII and hCA-IX within the 50–100 nM range. Compounds 247–250 are considered promising leads for further development as potent hCA-II inhibitors, targeting ocular disorders and cancer through structural optimization.176
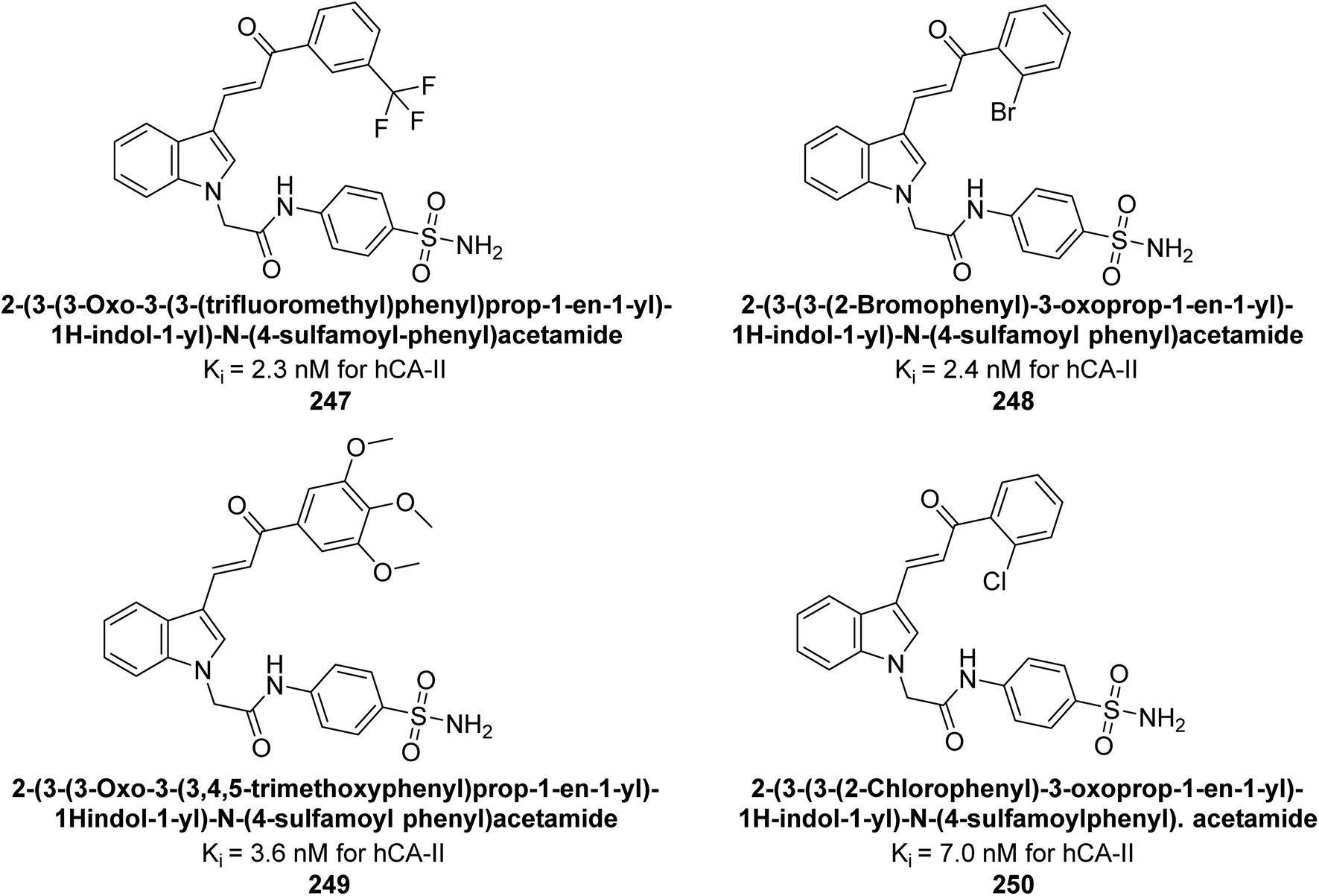 | ||
| Fig. 89 Chemical structures of compounds 247–250; their Ki values against hCA-II. | ||
Supuran et al. (2021) synthesized triazole-sulfonamide pyrimidine derivatives via click chemistry. These compounds demonstrated strong inhibitory activity against hCA-II and moderate to excellent activity against hCA-IX and XII. Remarkably, compounds 251 (Ki = 9.4 nM), 252 (Ki = 1.8 nM), and 253 (Ki = 0.82 nM) exhibited high selectivity for hCA-XII. Compound 254 showed potent inhibition of both hCA-IX (Ki = 2.9 nM) and hCA-XII (Ki = 0.82 nM). Molecular docking confirmed favorable interactions of these compounds with active site residues of hCA-XII and IX. The docking study indicates that the benzenesulfonamide moiety fits precisely into the active catalytic pocket near the zinc ion of CA (Fig. 90). These derivatives show promising results as selective anticancer agents targeting CA.177
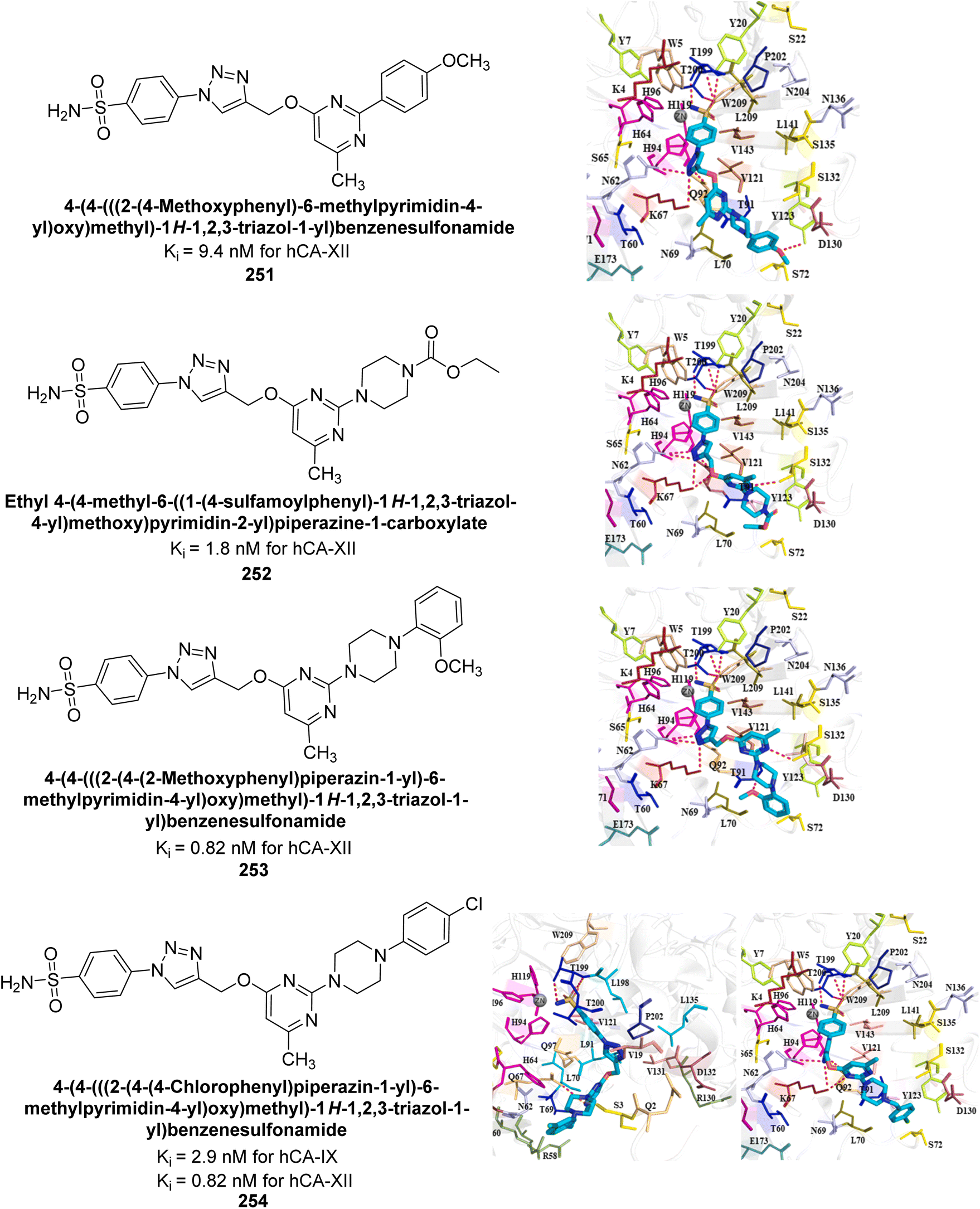 | ||
| Fig. 90 Chemical structures of compounds 251–254; their Ki values against hCA-IX and XII along with docking images of compounds 251–254. | ||
Arifuddin et al. (2021) synthesized benzenesulfonamide-linked 1,3,4-oxadiazole hybrids and evaluated their inhibitory effects on hCA-I and XIII. Many compounds exhibited strong inhibition, surpassing the standard drug AAZ against hCA-XIII. Compound 255 (75.8 nM) was three times more potent than AAZ (250.0 nM) against hCA-I. Additionally, compounds 255 (15.4 nM), 256 (16.2 nM), 257 (16.4 nM), and 258 (17.0 nM) outperformed AAZ (17.0 nM) against hCA-XIII. The presence of EWGs (–Cl, –NO2, –CN) contributed to enhanced activity, making these compounds promising leads for selective hCA-XIII inhibitors (Fig. 91).178
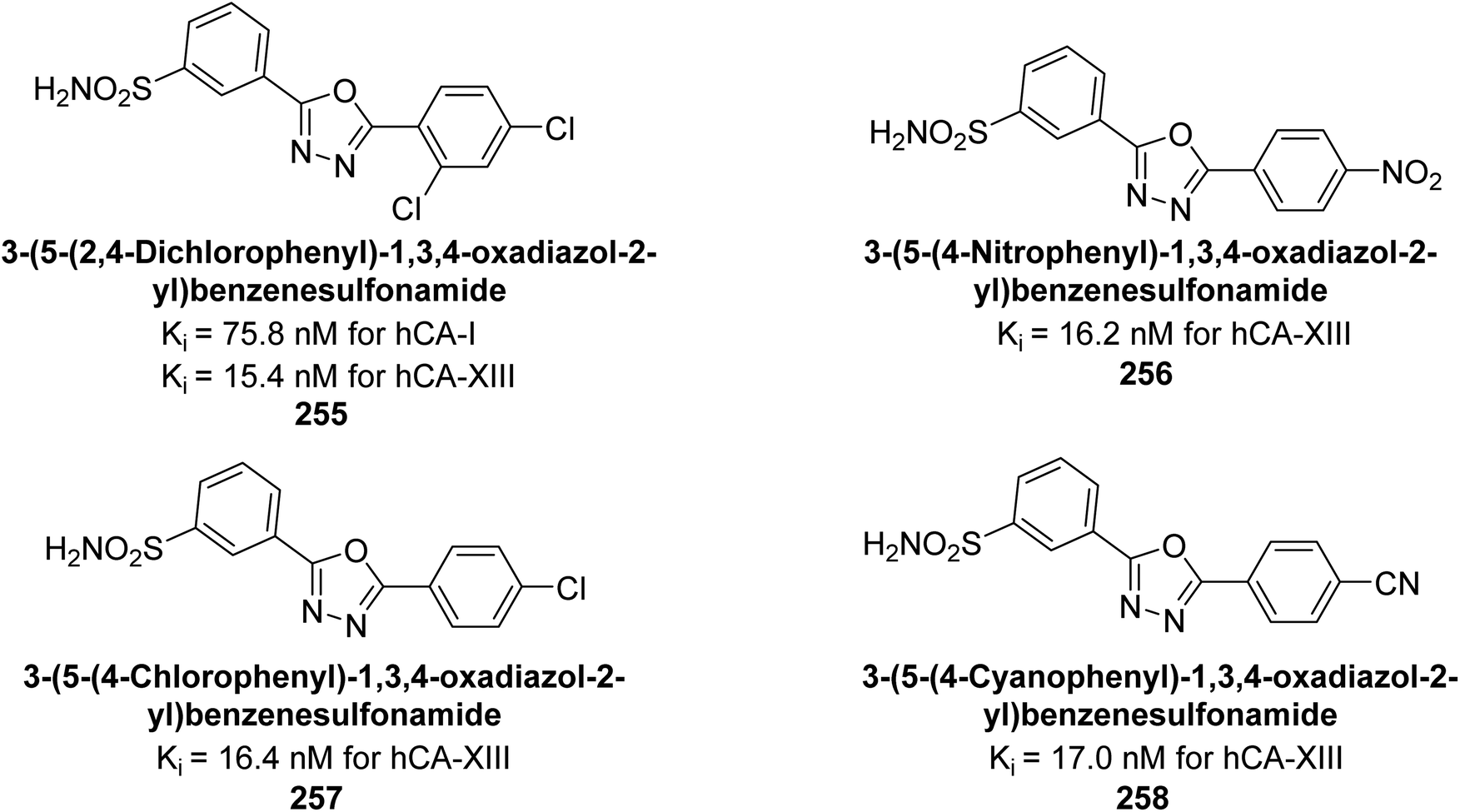 | ||
| Fig. 91 Chemical structures of compounds 255–258; their Ki values against hCA-I & XIII. | ||
Gitto et al. (2021) designed and synthesized a series of benzenesulfonamides and assessed against selected druggable hCA isoforms. Remarkably, compound 259 demonstrated remarkable affinity for hCA-VII (Ki: 4.3 nM) and exhibited good selectivity over the widely distributed hCA-I, outperforming TPM (Fig. 92).179
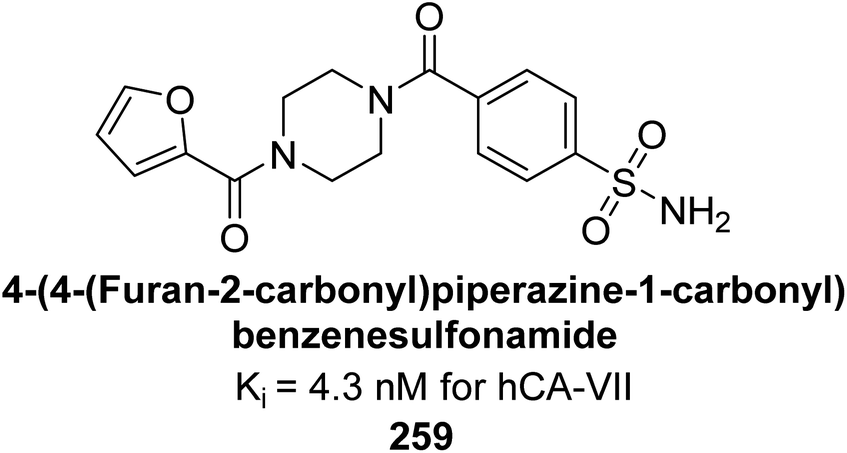 | ||
| Fig. 92 Chemical structure of compound 259; its Ki values against hCA-VII. | ||
Ashraf et al. (2021) synthesized sulfonamide derivatives with thiadiazole and evaluated their bCA-II inhibitory activity. Derivative 260 was the most potent, with an IC50 of 0.60 μM, outperforming AAZ (IC50 = 0.984 μM). Kinetic analysis revealed mixed-type inhibition for 260, with Ki and  values of 2.91 μM and 3.88 μM, respectively, indicating a preference for competitive binding. Docking studies showed hydrogen bonding between 260 and His3 and Trp4, along with π–π stacking with Trp4. Compound 260 also exhibited anticancer activity, inhibiting 40% of MCF-7 cell growth at 125 μM (Fig. 93). These findings suggest that derivative 260 could serve as a lead compound for designing more potent CA inhibitors.180
values of 2.91 μM and 3.88 μM, respectively, indicating a preference for competitive binding. Docking studies showed hydrogen bonding between 260 and His3 and Trp4, along with π–π stacking with Trp4. Compound 260 also exhibited anticancer activity, inhibiting 40% of MCF-7 cell growth at 125 μM (Fig. 93). These findings suggest that derivative 260 could serve as a lead compound for designing more potent CA inhibitors.180
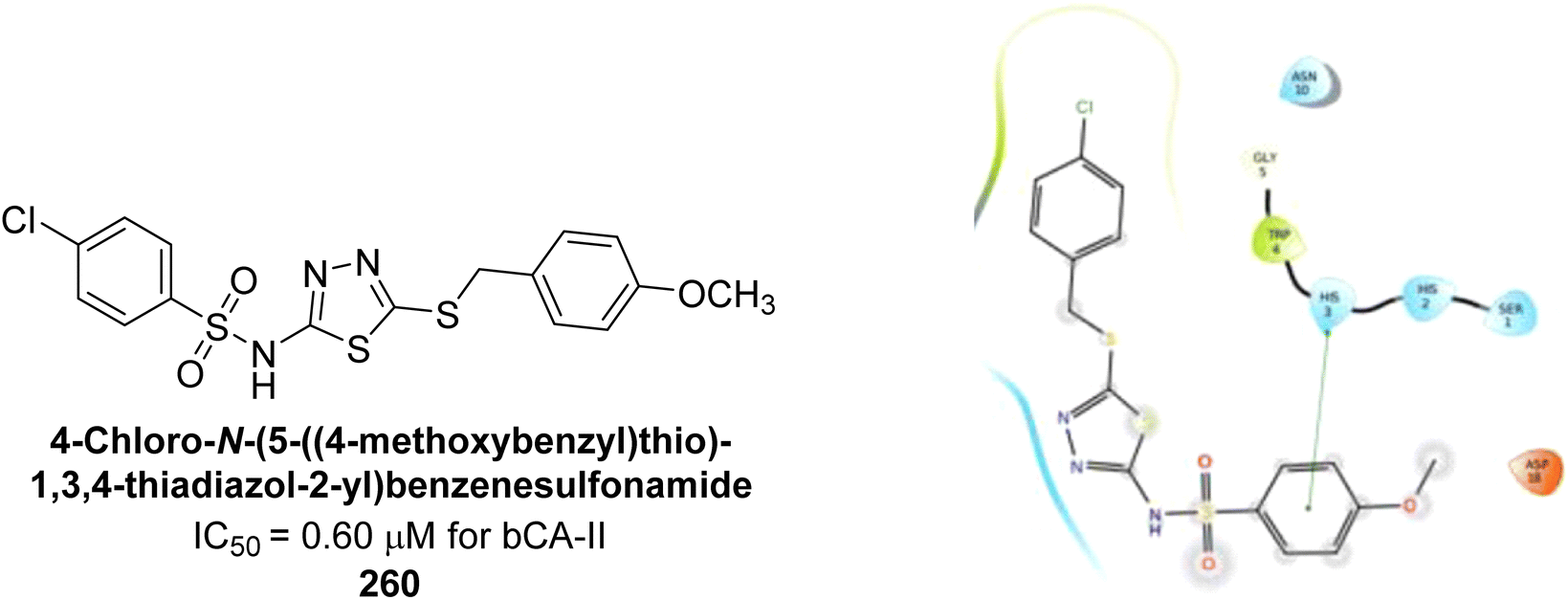 | ||
| Fig. 93 Chemical structure of compound 260; its Ki values against bCA-II and its docking image. | ||
Gul et al. (2021) synthesized pyrazole-based benzenesulfonamides and evaluated their inhibitory activity against hCA isoforms. Compound 261 exhibited high selectivity for hCA-IX (Ki = 7.0 nM), with 127 times greater selectivity over hCA-I, while compound 262 (Ki = 10.6 nM) showed 47 times higher selectivity for hCA-IX over hCA-II compared to AAZ. Compound 261 formed two hydrogen bonds in the hCA-IX complex versus one in hCA-I, with a shorter zinc coordination bond in hCA-IX. Compound 262's sulfonamide moiety coordinated with the Zn ion, forming hydrogen bonds with Thr199, Thr200, Gln92, and Ala129, contributing to its strong binding affinity for hCA-IX (Fig. 94).181
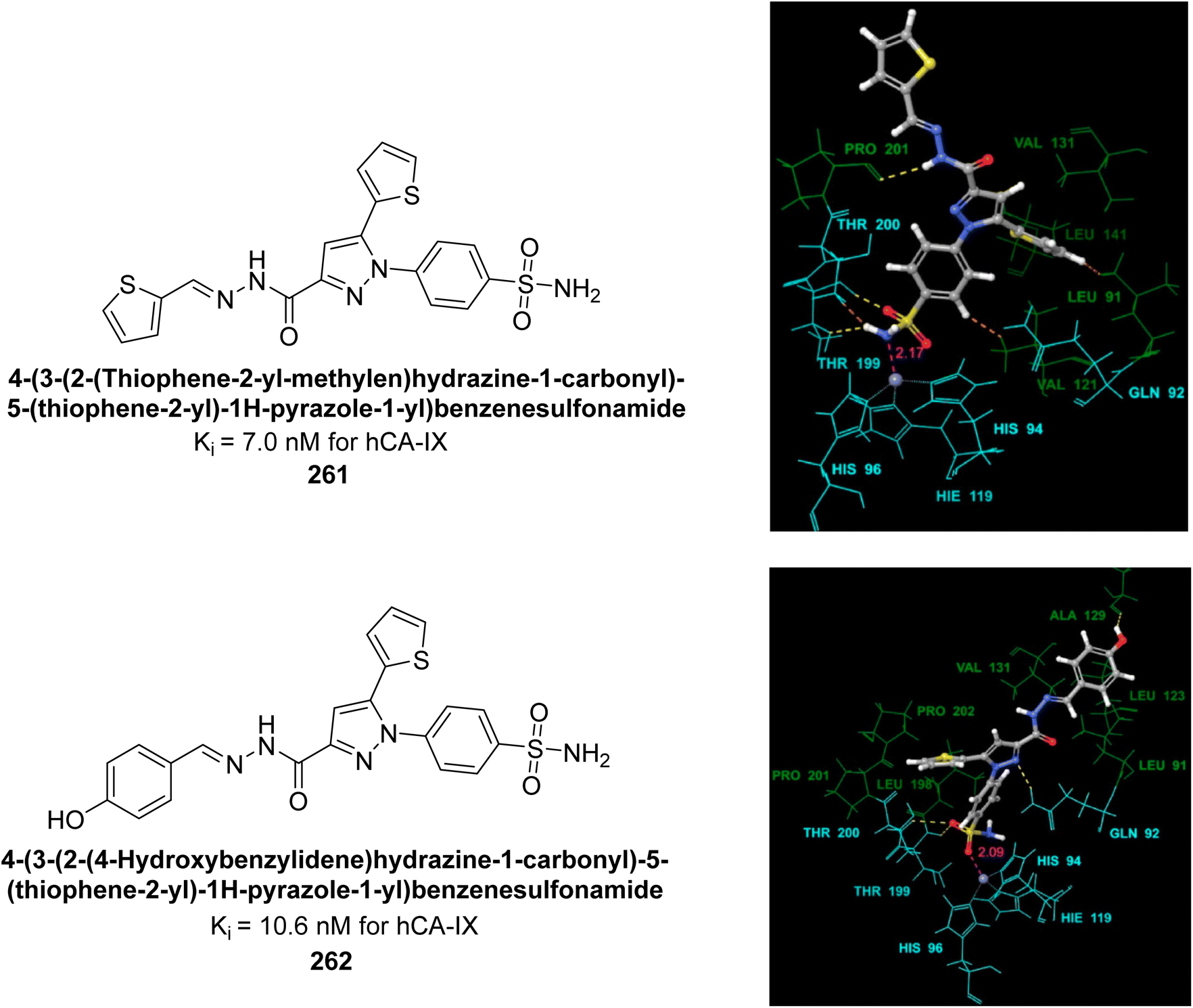 | ||
| Fig. 94 Chemical structures of compounds 261 and 262; their Ki values against hCA-IX along with docking images. | ||
Supuran et al. (2021) designed and prepared benzenesulfonamide and benzoic acid derivatives using a tail/dual tail approach to enhance potency and selectivity as CAIs. Benzenesulfonamide derivatives demonstrated moderate to potent inhibitory activity, particularly towards hCA-II, with compound 263 showing a Ki of 7.6 nM. In contrast, the benzoic acid analogues exhibited no significant activity, except for compounds 264, 265 and 266 which showed weak activity (Fig. 95).182
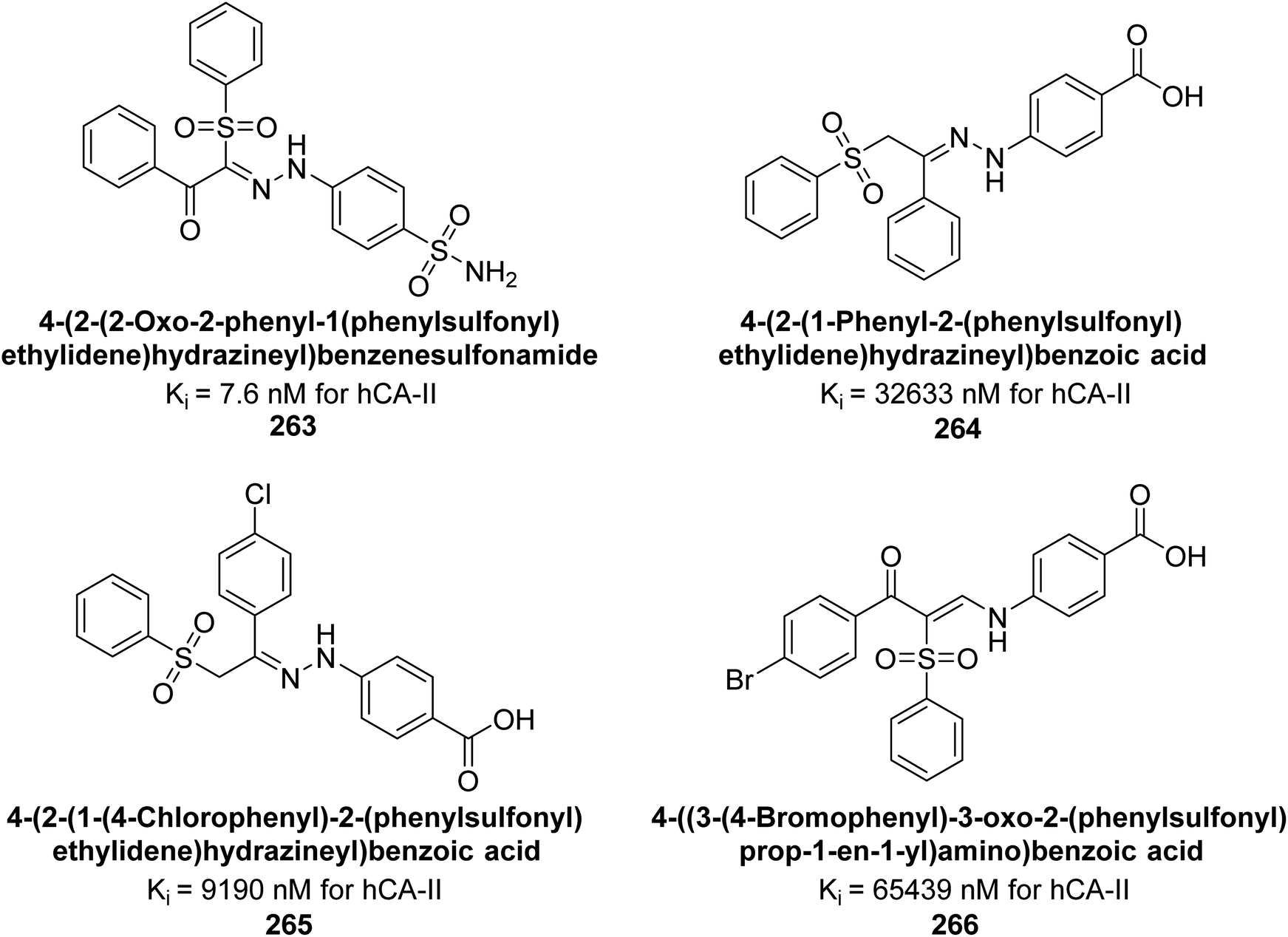 | ||
| Fig. 95 Chemical structures of compounds 263–266; their Ki values against hCA-II. | ||
Hassan et al. (2021) synthesized aryl thiazolone-benzenesulfonamides and evaluated their inhibitory effects on hCA-IX. Particularly, three sulfonamide derivatives, 267, 268 and 269 demonstrated excellent inhibition of hCA-IX with IC50 values ranging from 10.93 to 25.06 nM, and significantly less inhibition of hCA-II with IC50 values from 1.55 to 3.92 μM, indicating remarkable selectivity for hCA-IX over hCA-II. Docking results of active analogues 267–269 indicated precise fit within the active site, forming coordination and hydrogen bonds. The remarkable potency of these compounds against hCA-IX is likely due to their similar predicted binding patterns (Fig. 96).183
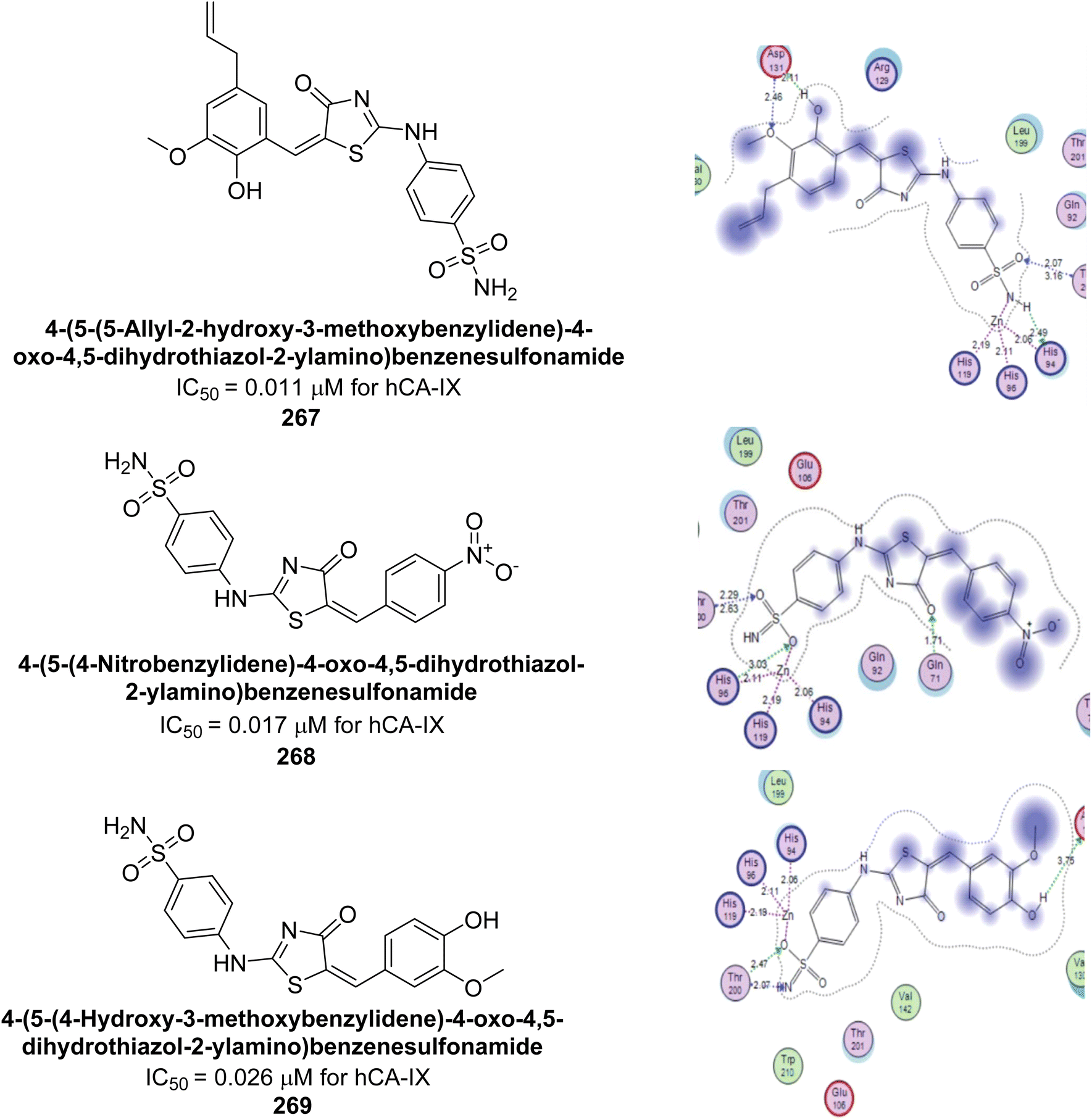 | ||
| Fig. 96 Chemical structures of compounds 267–269; their IC50 values against hCA-IX along with docking images. | ||
Saadiq et al. (2021) reported the synthesis of benzimidazole-derived N-acylhydrazones and their in vitro evaluation as potent bCA-II inhibitors. Among the target compounds, several exhibited higher inhibition than the standard AAZ (IC50: 18.6 μM), including compound 270 (IC50: 13.3 μM), compound 271 (IC50: 17.2 μM), compound 272 (IC50: 14.6 μM), and compound 273 (IC50: 14.5 μM). Molecular docking of the most active compounds confirmed their binding interactions with the enzyme's active site, corroborating the experimental results (Fig. 97).184
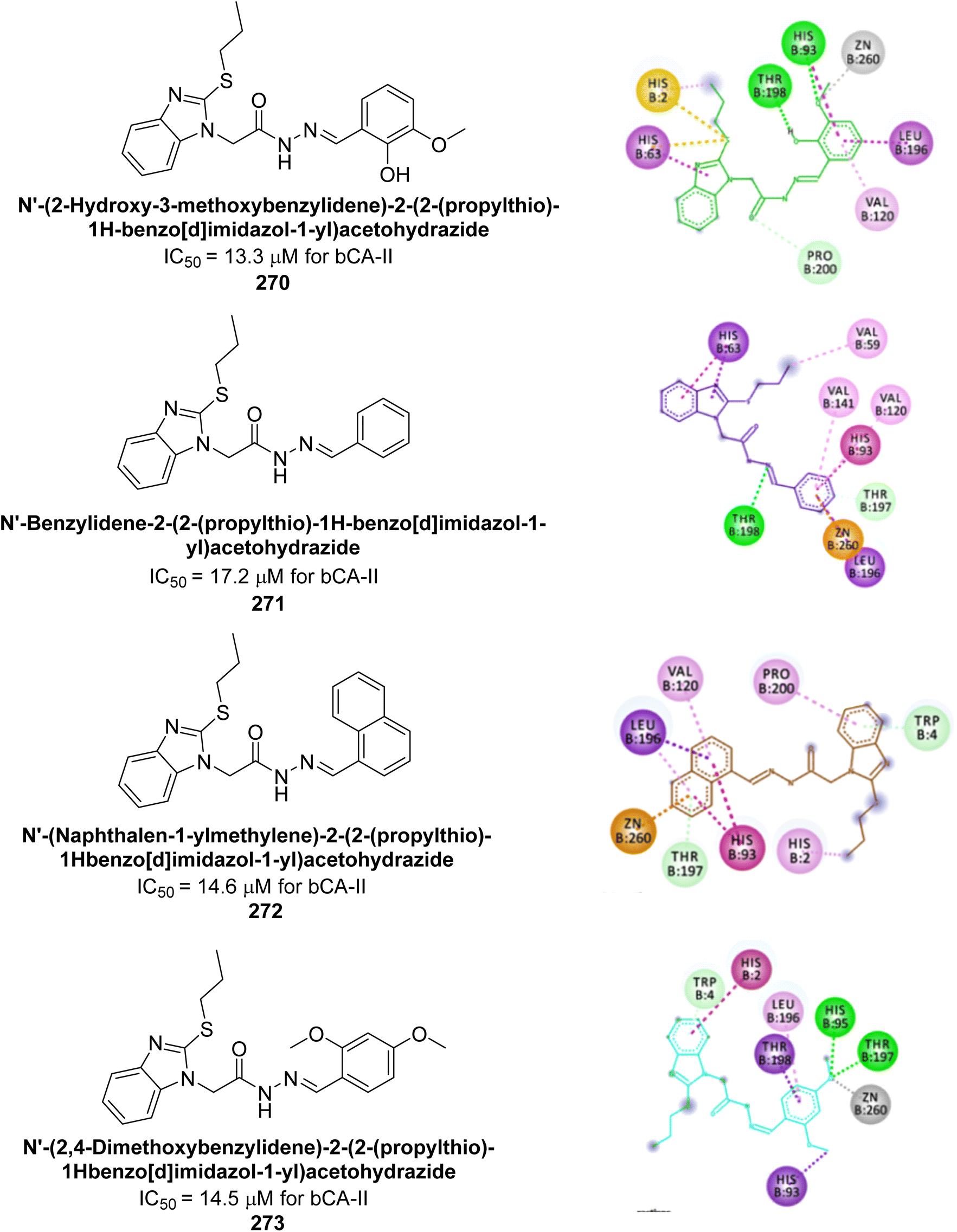 | ||
| Fig. 97 Chemical structures of compounds 270–273; their IC50 values against bCA-II along with docking images. | ||
Eldehna et al. (2021) developed new CA inhibitors using natural piperine as a tail moiety. Among the synthesized piperine-sulfonamides, the p-regioisomers 274 and 275 exhibited the strongest inhibitory activity, with nanomolar potency against hCA-II (Ki = 93.4 and 88.6 nM), hCA-IX (Ki = 38.7 and 68.2 nM), and hCA-XII (Ki = 57.5 and 45.6 nM) (Fig. 98).185
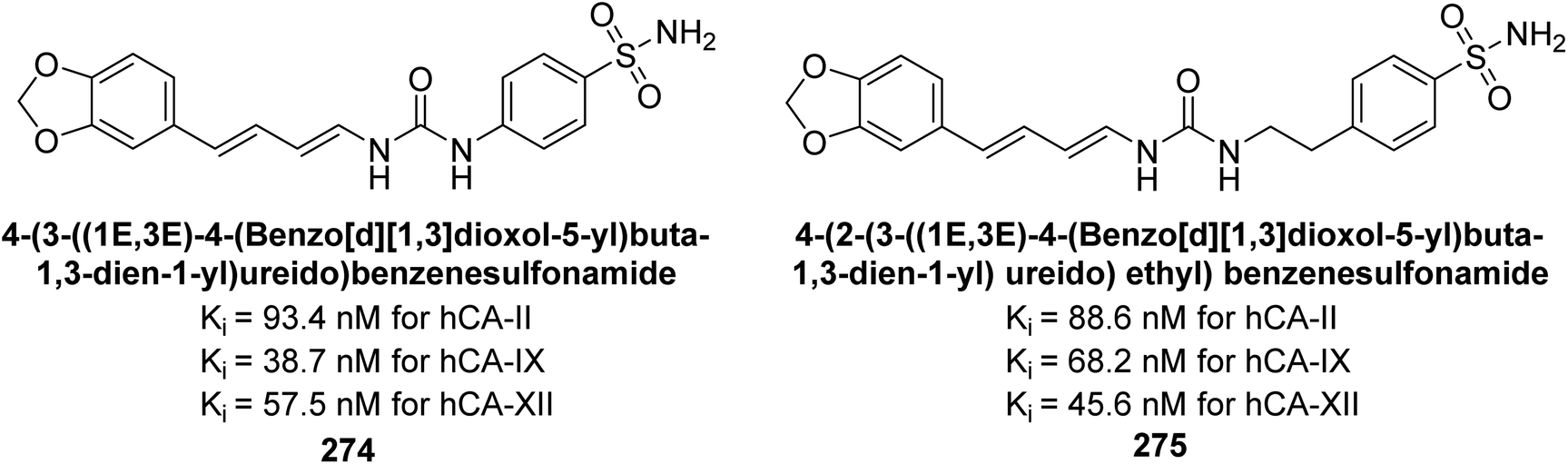 | ||
| Fig. 98 Chemical structures of compounds 274 and 275; their Ki values against hCA-II, XI and XII. | ||
Zaib et al. (2022) synthesized indole-picolinamide hybrids and found that compound 276 is a highly potent inhibitor of bCA-II, with an IC50 of 0.0440 μM, which is 22 times more effective than AAZ (IC50 = 0.9618 μM). The enhanced activity of 276 is attributed to its phenyl carbamate moiety. Substituting the carbamate linker with other linkers while keeping the phenyl ring unchanged resulted in reduced activity. Molecular docking studies confirmed that compound 276 interacts with His118, Thr197, His93, and His95 in the active site, exhibiting π-donor hydrogen, π–π T-shaped interactions, and π-alkyl interactions, as well as conventional hydrogen bonds (Fig. 99). These findings suggest that compound 276 could be a promising lead for developing new CA-II inhibitors.186
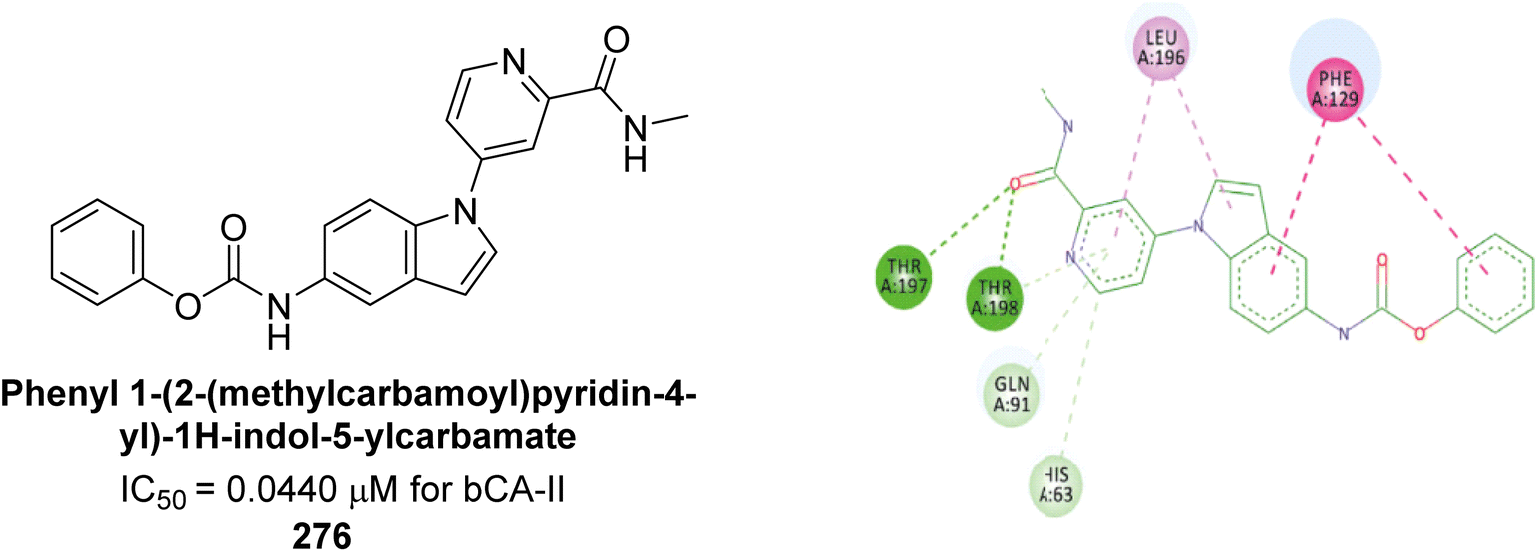 | ||
| Fig. 99 Chemical structure of compound 276; its IC50 value against bCA-II and its docking image. | ||
Supuran et al. (2022) developed and evaluated a series of quinoline/pyridine-linked indole-3-sulfonamide derivatives for their inhibitory effects on hCA-IX and XII. Among these, compound 277 showed prominent inhibition of hCA-IX with a Ki of 1.57 μM. Similar to the standard AAZ, compound 277 formed hydrogen bonds with Thr199 and engaged in hydrophobic interactions with several active site residues, including Asn62, Ser65, and Leu135. These interactions stabilize compound 277 within the hCA-IX active site, making these derivatives promising candidates for selective hCA-IX inhibitors in cancer therapy (Fig. 100).187
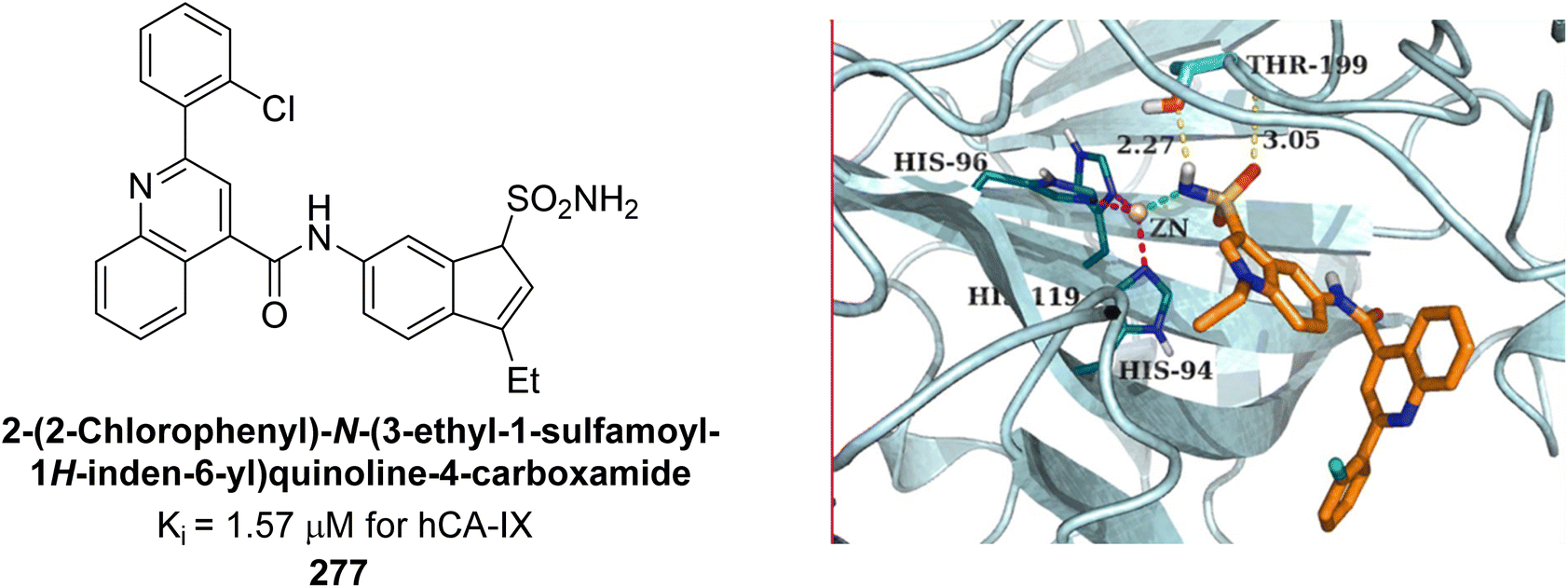 | ||
| Fig. 100 Chemical structure of compound 277; its Ki value against hCA-IX along with docking image. | ||
Mohsen et al. (2022) developed a series of 2-thiopyrimidine benzenesulfonamides utilizing a dual-tail design. This approach combines a benzenesulfonamide moiety for Zn2+ binding with a 2,4-disubstituted thiopyrimidine tail. Among the compounds, 278 was the most potent with a Ki of 1.72 nM and demonstrated high selectivity for hCA-II over hCA-IX and XII, with selectivity indices of 50 and 5.26, respectively. Compounds 279 and 280 also showed strong inhibition of hCA-IX (Ki = 7.4 and 7.0 nM, respectively) and better potency against hCA-XII (Ki = 4.67 nM) compared to the standard AAZ (Ki = 25 nM for hCA-IX and 5.7 nM for hCA-XII). Molecular docking revealed that the sulfonamide moiety interacts with the Zn2+ ion in the CA active site, while the dual-tail extension engages with surrounding residues, enhancing the compounds' potency and selectivity (Fig. 101).188
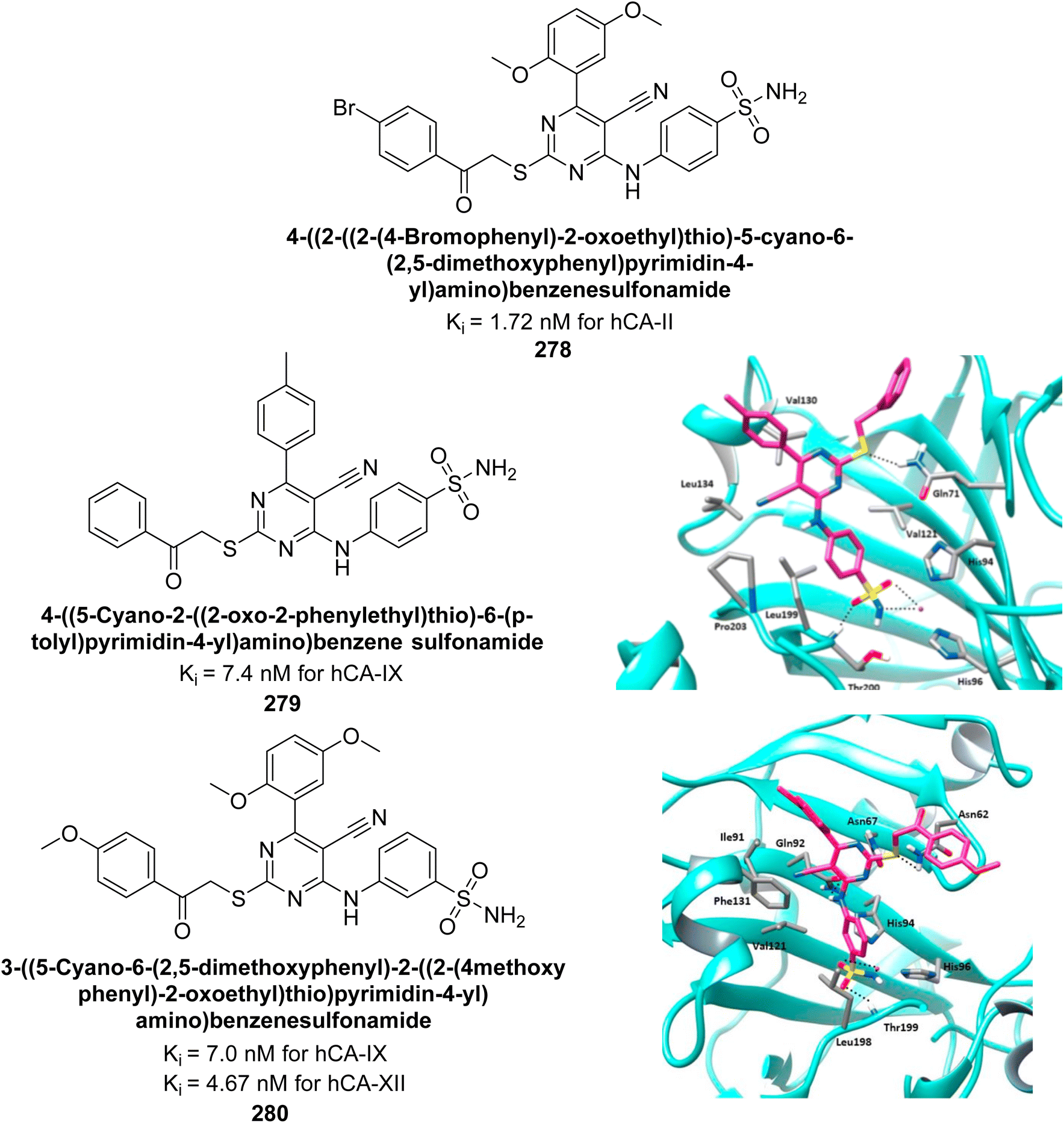 | ||
| Fig. 101 Chemical structures of compounds 278–280; their Ki values against hCA-IX and XII along with docking images. | ||
Sharma et al. (2022) synthesized arylthiazolylhydrazono-1,2,3-triazoles incorporating sulfanilamide and metanilamide moieties using a tail-approach. Additionally, metanilamide analogues were better inhibitors of the hCA-IV and IX. Compounds 281, 282, 283, and 284 potently inhibited hCA-II with Ki values of 18.1, 14.1, 14.9, and 17.8 nM, respectively, and 281–283 exhibited selectivity for hCA-II over hCA-I, IV, and IX isoforms by more than 15-fold. Compounds 282 and 285, significant inhibition profiles against hCA-IX (Ki range of 0.113 μM-0.318 μM). Remarkably, 284 was the only compound in the series that effectively inhibited all the tested isoforms (Table 13). These findings can be leveraged to design novel inhibitors that incorporate both sulfanilamide and metanilamide moieties, offering enhanced potency and selectivity.189
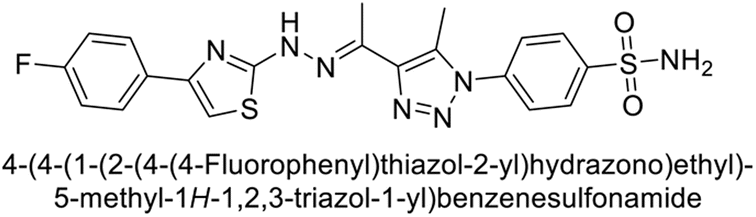
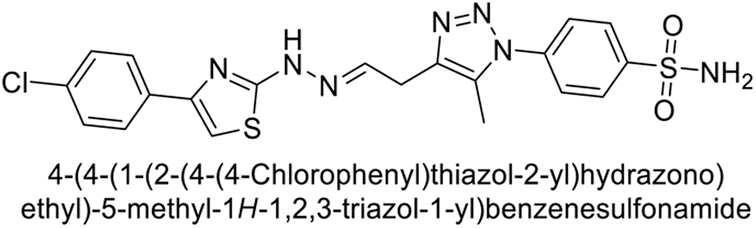
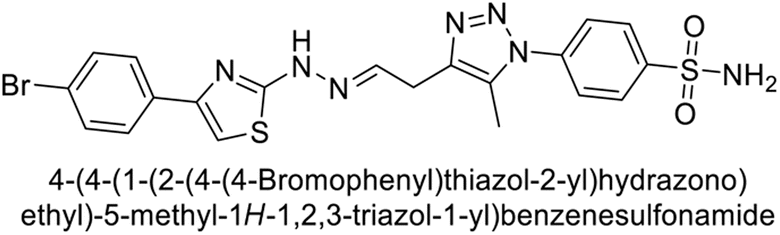
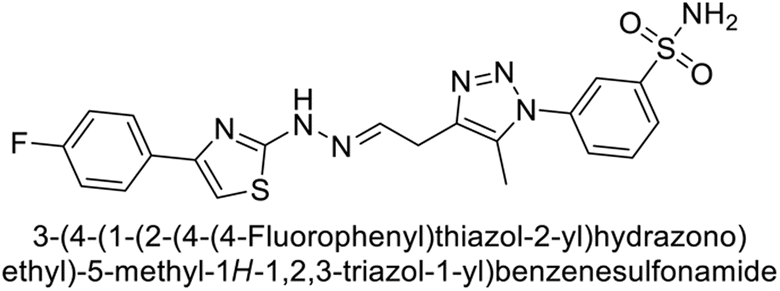
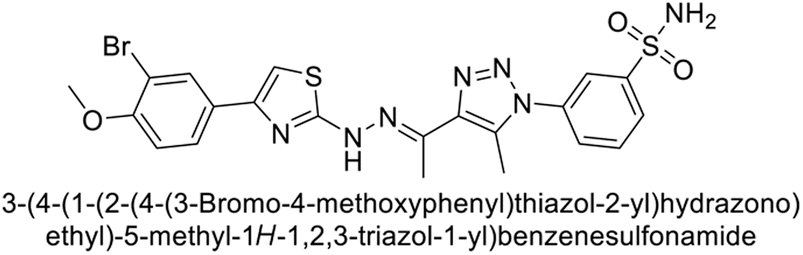
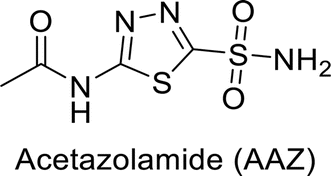
Tawfik et al. (2022) designed featuring a benzenesulfonamide moiety for zinc binding, with hydrazone to interact with hydrophilic and hydrophobic regions of the active site, respectively. All synthesized compounds demonstrated inhibition of hCA-IX, with IC50 values ranging from 13.3 to 259 nM. Particularly, derivatives 286, 287, 288, 289 and 290 showed high efficacy against the hCA-IX isoform, with Ki of 13.3, 22.6, 25.8, 26.9, and 27.2 nM, respectively (Fig. 102).190
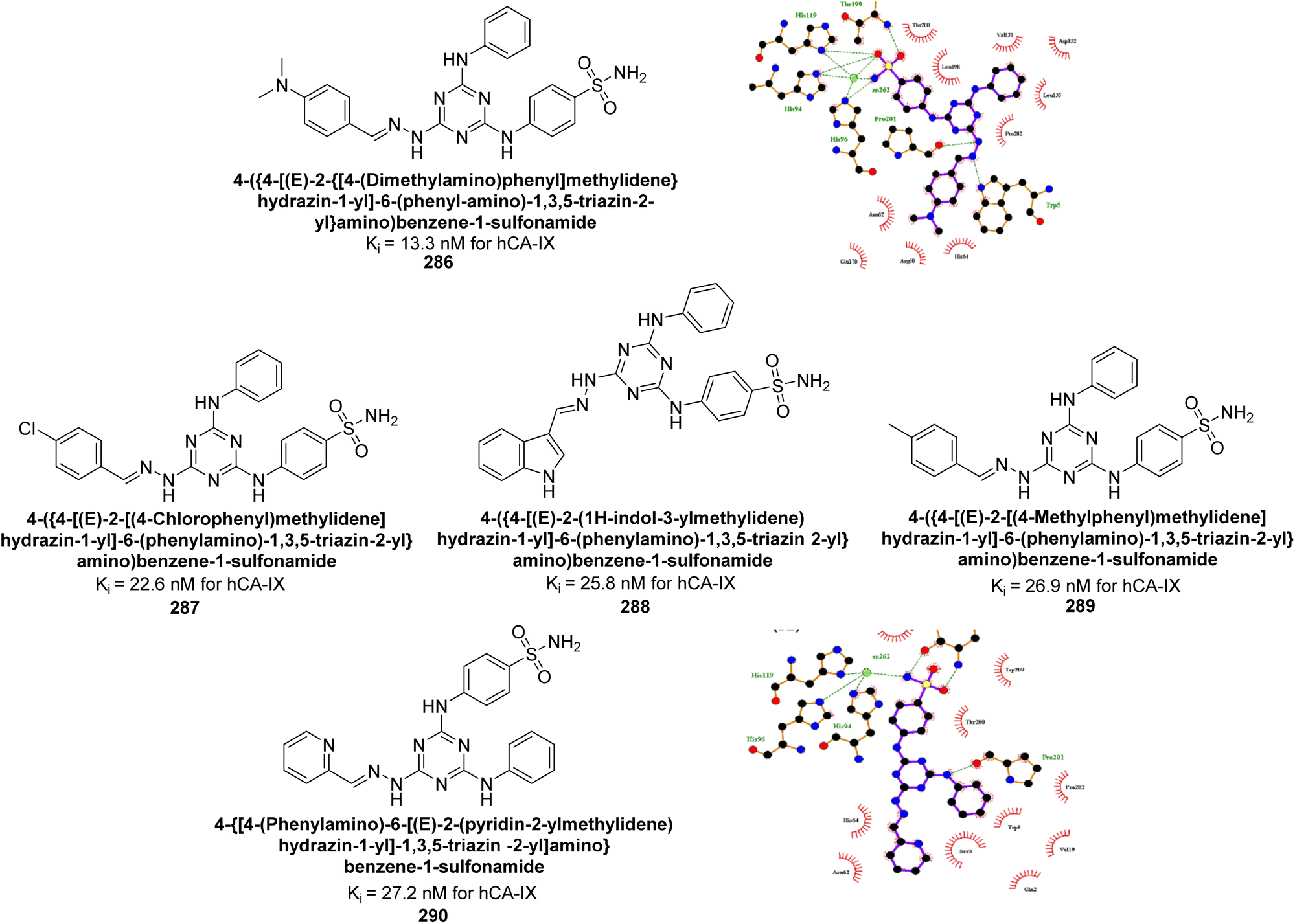 | ||
| Fig. 102 Chemical structures of compounds 286–290; their Ki values against hCA-IX along with docking images of 286 and 290. | ||
Carradori et al. (2022) synthesized rhodamine-linked benzenesulfonamides and evaluated for their inhibitory activity against hCA-I, II, IX, and XII. All synthesized compounds exhibited good to excellent inhibition in the nanomolar range, attributed to the sulfonamide moiety's zinc-binding properties. The most potent inhibitors of hCA-I were 291 (Ki = 22.4 nM) and 292 (Ki = 35.8 nM), significantly outperforming the standard drug AAZ (Ki = 250.0 nM) (Fig. 103). Both EWGs and EDGs on the aromatic ring significantly enhanced inhibition. The findings suggest that the newly synthesized benzenesulfonamide-linked rhodanine derivatives are promising candidates for developing isoform-selective inhibitors targeting CA isoforms.191
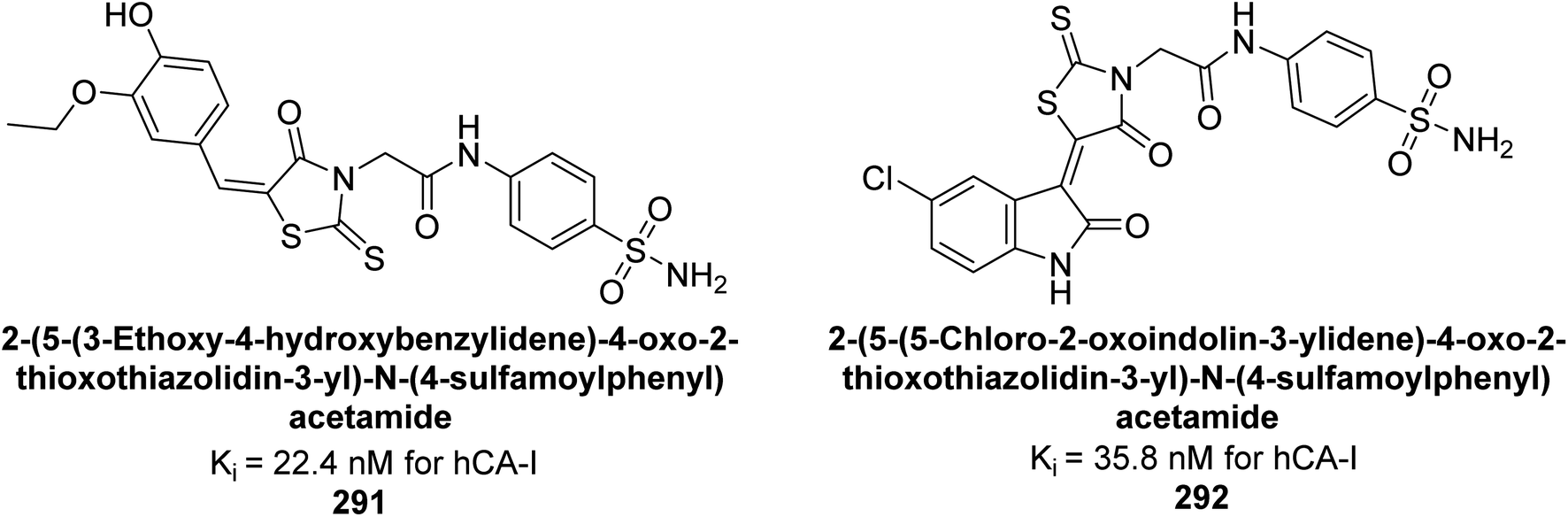 | ||
| Fig. 103 Chemical structures of compounds 291 and 292; their Ki values against hCA-I. | ||
Supuran et al. (2022) synthesized isatin N-phenylacetamide-based sulfonamides and evaluated for their inhibitory activity against hCA isoforms. Among these, derivative 293 exhibited the most potent inhibition against hCA-I and hCA-II, with Ki values of 45.10 nM and 5.87 nM, respectively, surpassing the standard inhibitor AAZ. Additionally, 293 demonstrated significant inhibition of hCA-XII, with a Ki value of 7.91 nM, comparable to AAZ's Ki of 5.70 nM. Docking studies of the most active compound, 293 predicted their binding patterns and affinities to the active sites of all the investigated isoforms (Fig. 104).192
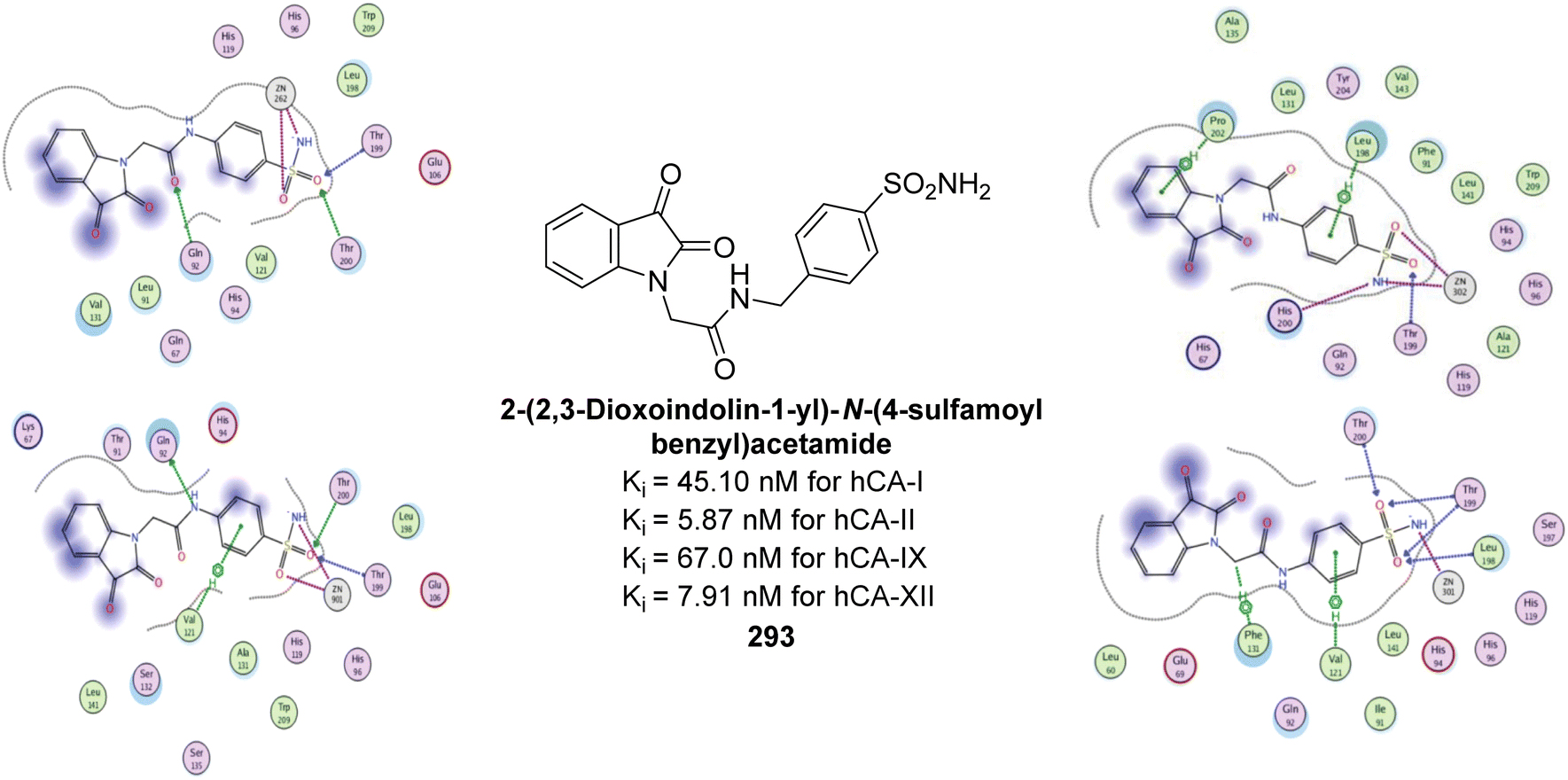 | ||
| Fig. 104 Chemical structure of compound 293; its Ki values against hCA-I, II, IX and XII along with its docking images. | ||
Thacker et al. (2022) synthesized coumarin-linked thiazole derivatives and assessed their inhibition of hCA-IX and XII. Remarkably, compound 294 showed the highest inhibition of hCA-XII with a Ki value of 91.1 nM (Fig. 105). The hydrolyzed form of compound 294 also demonstrated strong interactions and favorable docking scores with both isoforms, indicating its potential as a template for developing selective and potent hCA-XII inhibitors.193
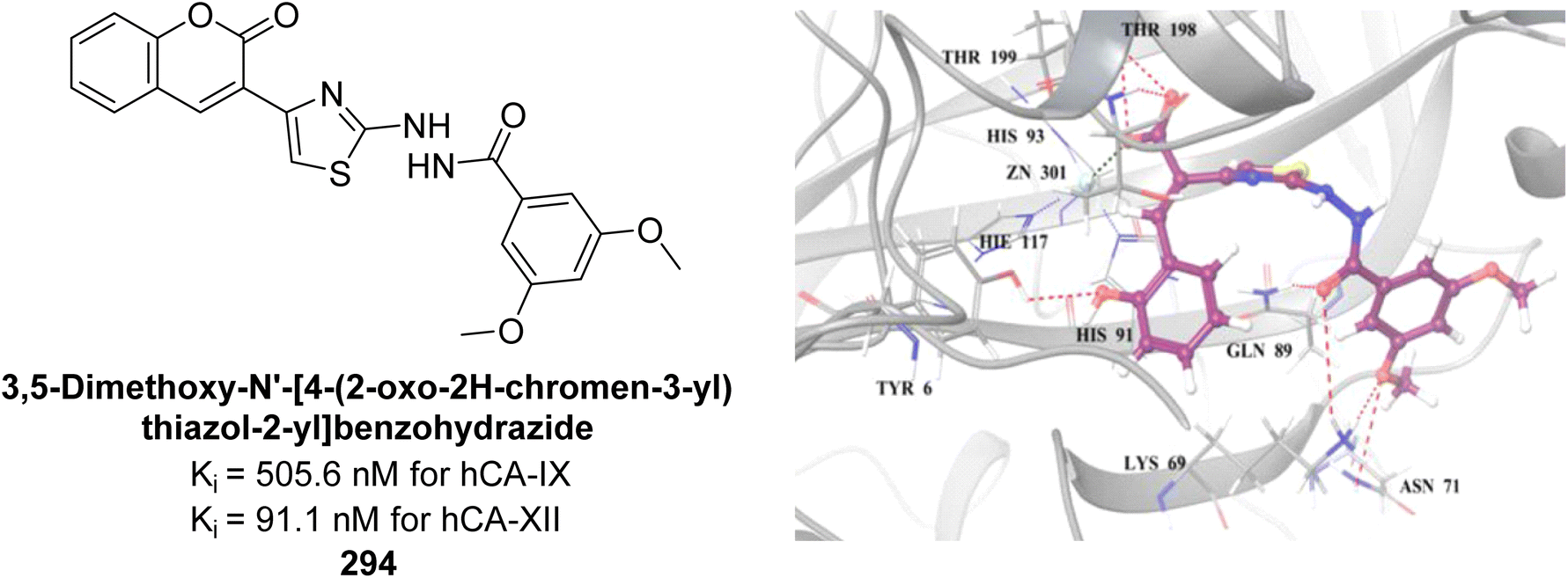 | ||
| Fig. 105 Chemical structure of compound 294; its Ki values against hCA-IX and XII along with its docking image against hCA-XII. | ||
Mohsen et al. (2022) synthesized 2-thioquinazoline-benzenesulfonamide hybrids and evaluated them as hCA inhibitors. Compounds 295 and 296 showed the highest potency across four hCA isoforms, with Ki values of 0.09 and 0.05 μM for hCA-II, 0.32 and 0.47 μM for hCA-IX, and 0.58 and 0.46 μM for hCA-XII, respectively. Compound 296 displayed distinguished selectivity for hCA-II. Molecular docking studies revealed that the benzenesulfonamide moiety binds deeply within the hCA-II active site, interacting with the Zn2+ ion and key residues Thr199 and Thr200, while the 2-substituted thioquinazoline moiety interacts with the hydrophilic region of the binding site (Fig. 106).194
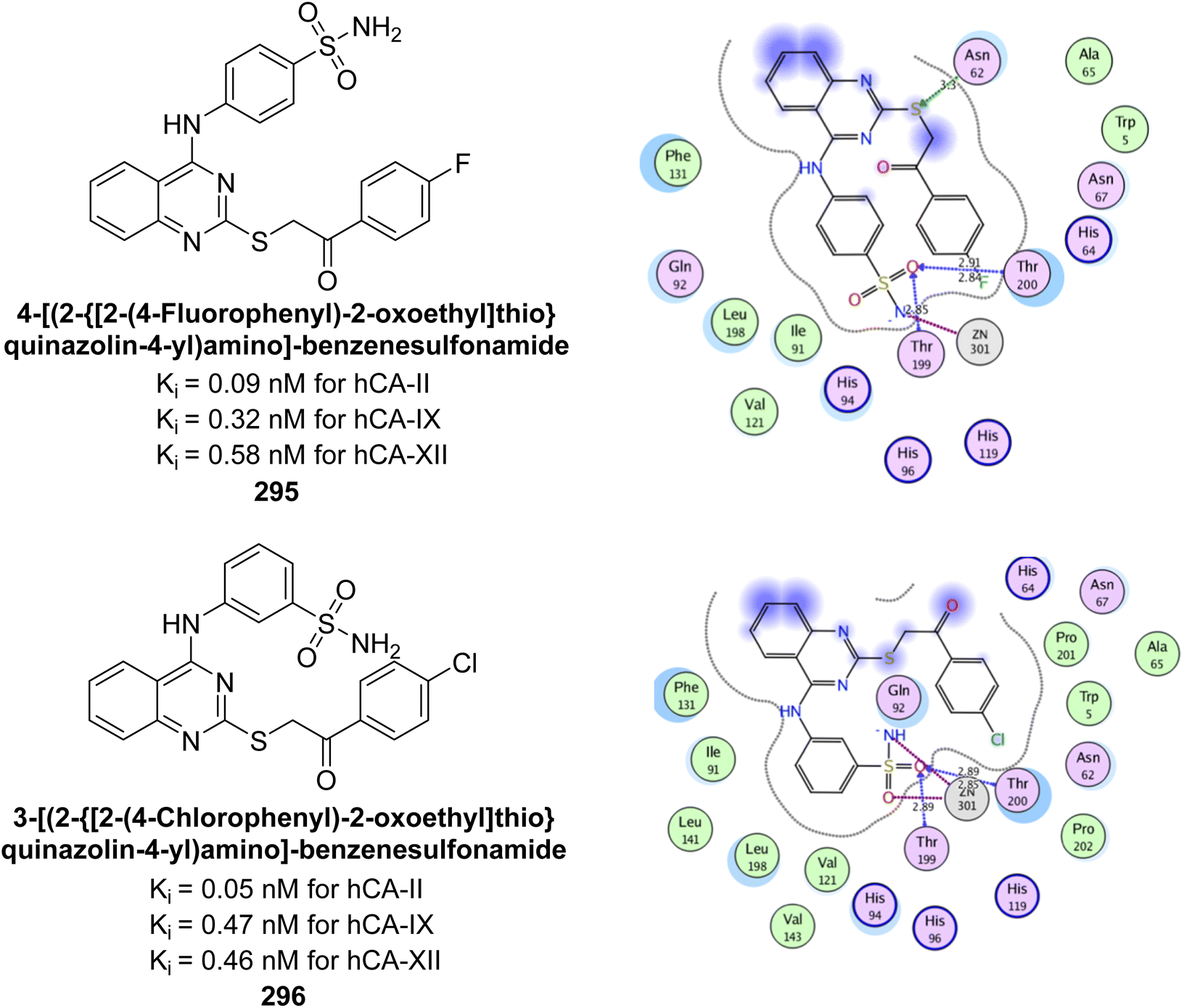 | ||
| Fig. 106 Chemical structures of compounds 295 and 296; their Ki values against hCA-II, IX and XII along with docking images. | ||
Winum et al. (2022) designed and synthesized a series of novel N-triazolo-benzenesulfonamides-1,5-benzodiazepines using click chemistry. The synthesized compounds showed nanomolar affinity for various hCA isoforms, including hCA-I, II, IV, VII, IX, and XII. Remarkably, derivative 297 demonstrated exceptional inhibitory activity across all tested isoforms, outperforming AAZ and showing superior multivalent effects compared to previously reported divalent CA inhibitors (Fig. 107).195
 | ||
| Fig. 107 Chemical structure of compound 297; its Ki values against hCA-I, II, IV, VII, IX, and XII. | ||
Arslan et al. (2023) designed and synthesized a series of benzenesulfonamides featuring a 1,2,3-triazole moiety as inhibitors of α-hCAs using a tail approach. Among the synthesized derivatives, the naphthyl (298, Ki = 68.6 nM) and methyl (299, Ki = 56.3 nM) derivatives showed distinguished selectivity towards hCA-IX, while the propyl (300, Ki = 95.6 nM) and pentyl (301, Ki = 51.1 nM) derivatives were selective for hCA-XII. Derivative 302 exhibited strong inhibitory effects against hCA-I (Ki = 47.8 nM), hCA-IX and XII (Ki values of 195.9 and 116.9 nM, respectively), as compared to the reference drug AAZ with Ki values of 451.8, 437.2, and 338.9 nM, respectively. Additionally, derivative 303 demonstrated higher potency against hCA-II (Ki = 33.2 nM) compared to AAZ (Ki = 327.3 nM) (Fig. 108). The study also utilized molecular docking techniques to explore the binding interactions of the synthesized compounds within the active sites of hCAs.196
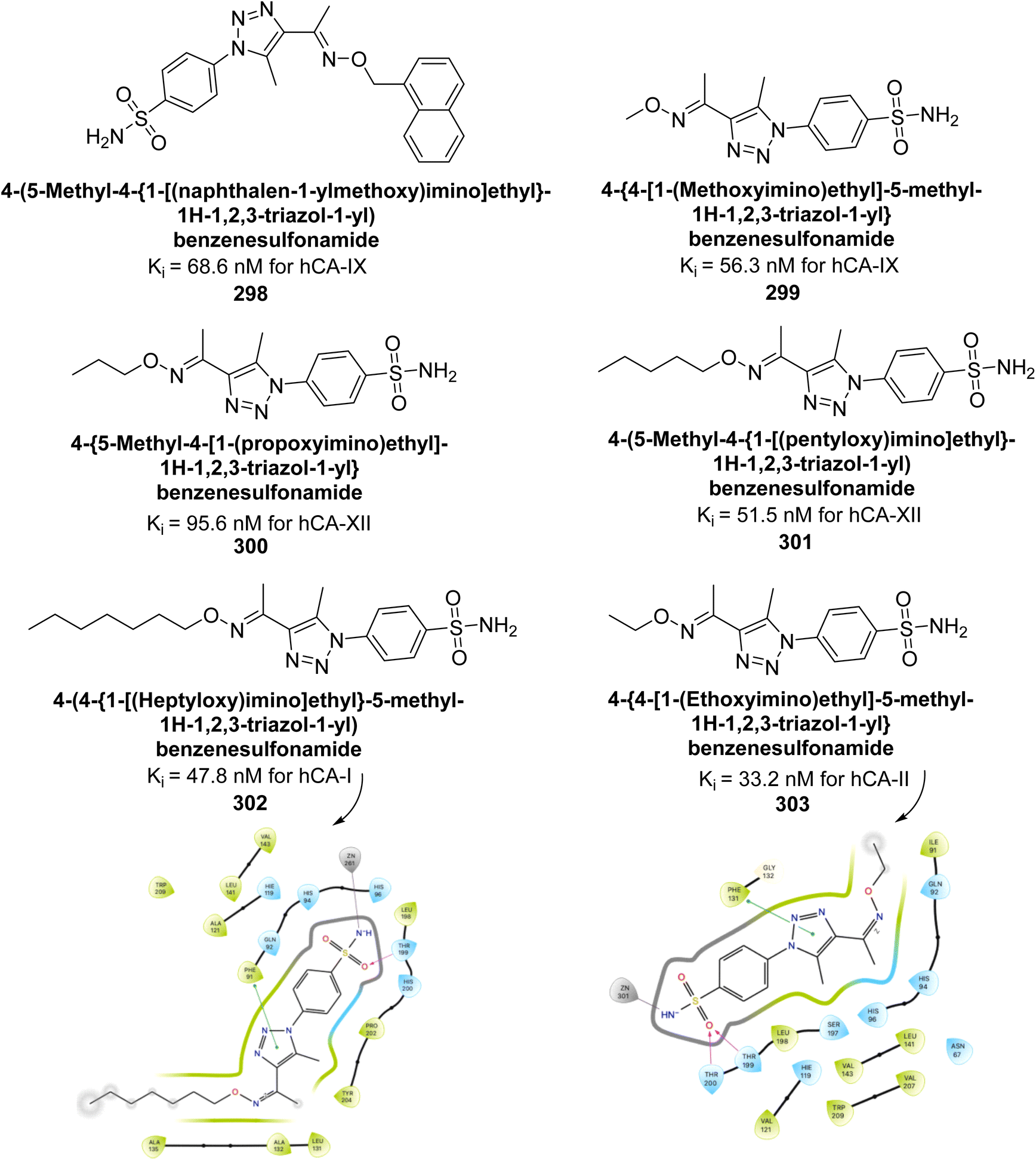 | ||
| Fig. 108 Chemical structures of compounds 298–303; their Ki values against hCA-I, II, IX and XII along with docking images of 302 and 303. | ||
Supuran et al. (2023) designed and synthesized pyrazole-based benzenesulfonamide with inhibitory activities against CA enzyme. Remarkably, hCA-IX and XII isoforms were potently inhibited by these compounds, exhibiting Ki values in the nanomolar range, between 13.0-82.1 nM for hCA-IX and 5.8–62.0 nM for hCA-XII. The study also utilized molecular docking techniques to explore the binding interactions of the synthesized compounds 304 and 305 within the active sites of hCA (Fig. 109).197
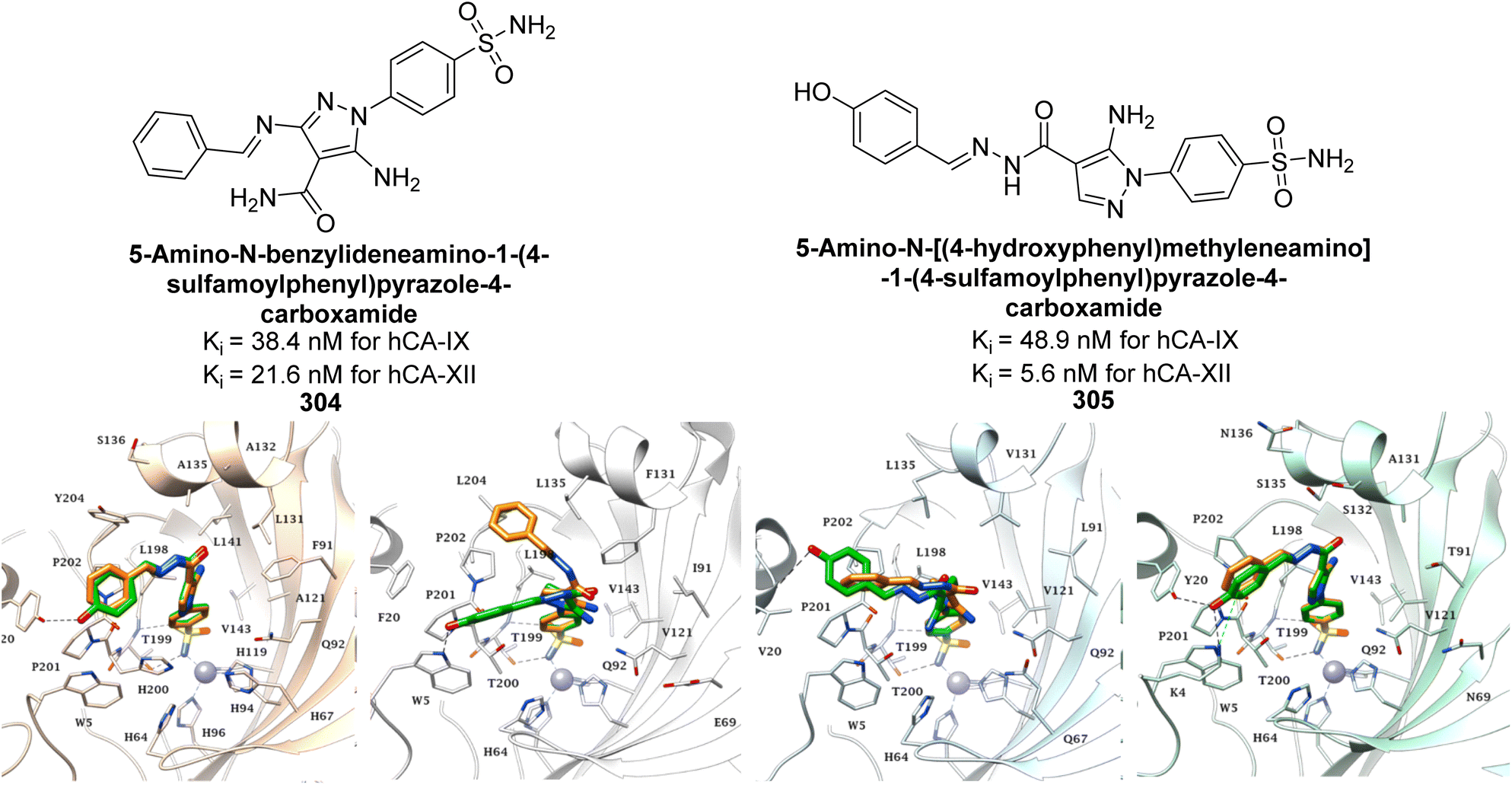 | ||
| Fig. 109 Chemical structures of compounds 304 and 305; their Ki values against hCA-I, II, IX and XII along with their docking images (predicted binding modes of compounds 304 (orange) and 305 (green)). | ||
Türkeş et al. (2023) synthesized benzenesulfonamide derivatives and evaluated against CA isoforms. Among these, the 2-iodophenyl 306 and 2-naphthyl 307 derivatives showed notable selectivity for hCA-IX and XII over hCA-I, while the phenyl 308 and 2,6-dimethylphenyl 309 analogs were more selective for hCA-IX and XII over hCA-II. Compound 308 exhibited strong inhibition of hCA-IX (Ki = 18.29 nM), outperforming AAZ (Ki = 437.20 nM), and compound 307 showed superior potency against hCA-XII (Ki = 9.22 nM) compared to AAZ (Ki = 338.90 nM). Molecular docking indicated that the sulfonamide moiety binds well to the hCA active sites, interacting with the Zn2+ ion, while the tail extension engages in hydrophilic and hydrophobic interactions with surrounding amino acids, affecting the compounds' potency and selectivity (Fig. 110).198
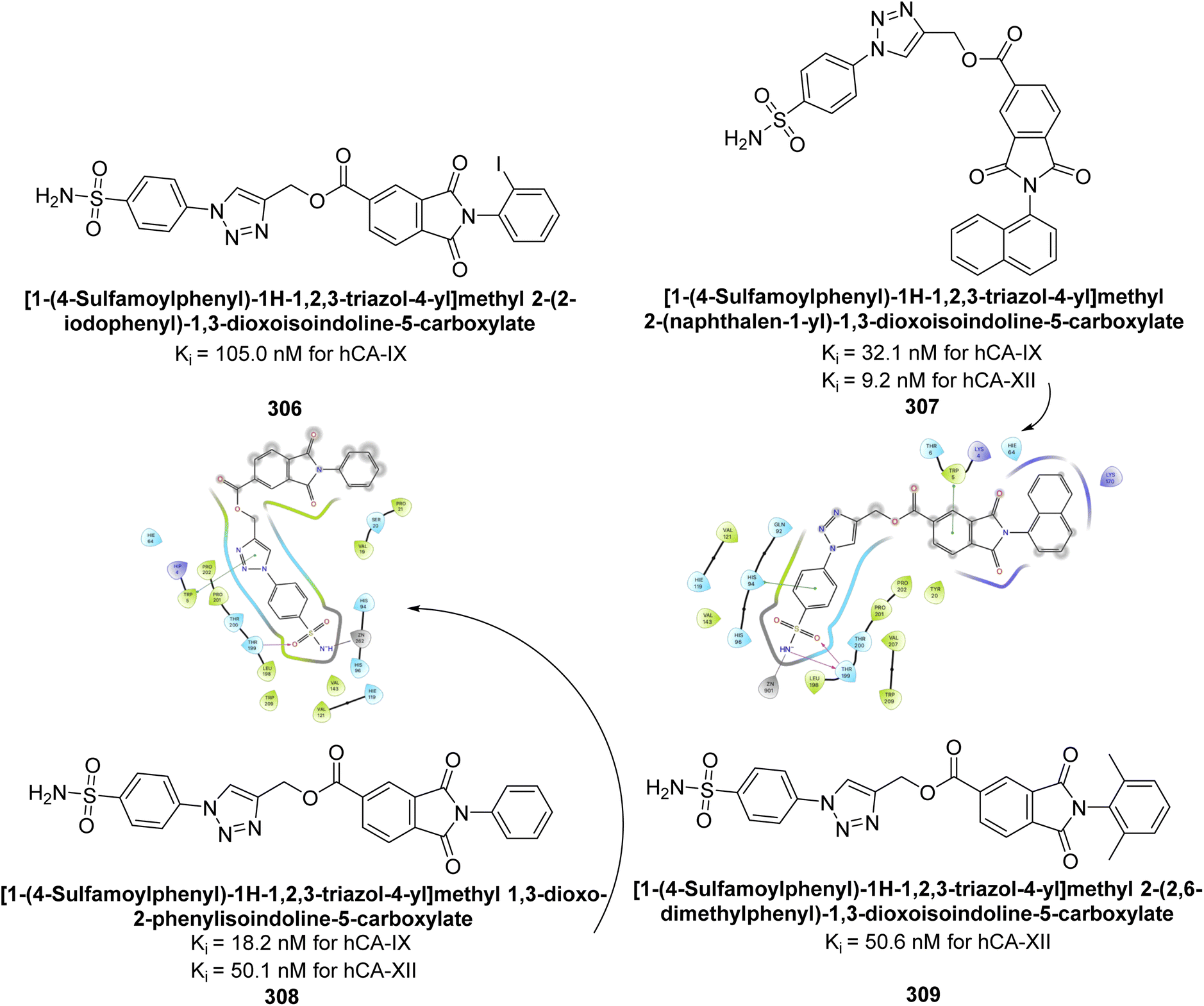 | ||
| Fig. 110 Chemical structures of compounds 306–309; their Ki values against hCA-I, IX and XII along with their docking images. | ||
Supuran et al. (2023) evaluated the inhibitory effects of quinazoline-based hydroxyl Schiff base derivatives against hCA isoforms. These quinazolines showed effective inhibition of hCA-I with Ki values ranging from 87.6 to 692.3 nM, comparable to or better than AAZ. For hCA-II, the inhibitors had Ki values between 16.9 and 29.7 nM, similar to AAZ. The compounds were particularly effective against hCA-IX, with Ki values from 8.9 to 88.3 nM, outperforming AAZ, and demonstrated strong inhibition of hCA-XII, with Ki values ranging from 5.4 to 19.5 nM. Molecular docking studies indicated that compound 310 binds strongly to hCA-IX and XII, highlighting its potential as a lead compound for developing hCA inhibitors (Fig. 111).199
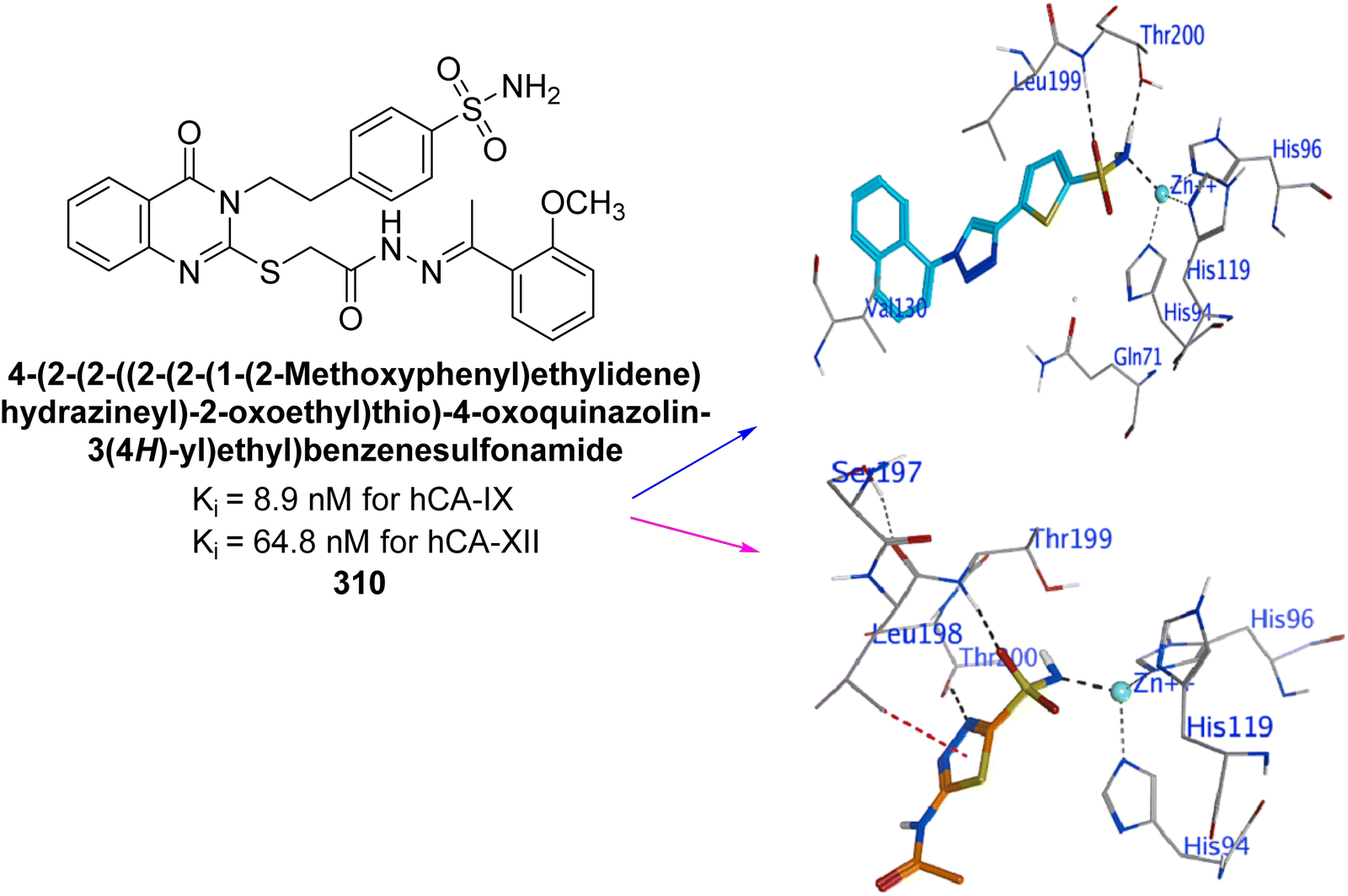 | ||
| Fig. 111 Chemical structure of compound 310; its Ki values against hCA-IX and XII along with its docking images. | ||
Kalay et al. (2023) developed secondary sulfonamide derivatives with a benzothiazole scaffold and tested their inhibitory activity against hCA-I and II. The compounds exhibited Ki values from 0.052 to 0.971 μM for hCA-I and 0.025 to 0.682 μM for hCA-II. Remarkably, several of these compounds demonstrated more potent inhibition than the standard drug, AAZ, with compounds 311 and 312 showing the highest efficacy against hCA-I and II, respectively. In compound 311 (Ki = 0.112 μM), the presence of an EDG (amide group) in its tail was found to enhance the inhibitory activity. Furthermore, compound 312 (Ki = 0.052 μM) demonstrated significantly higher potency, being five times more active than the reference drug (AAZ, Ki = 0.25 μM) (Fig. 112).200
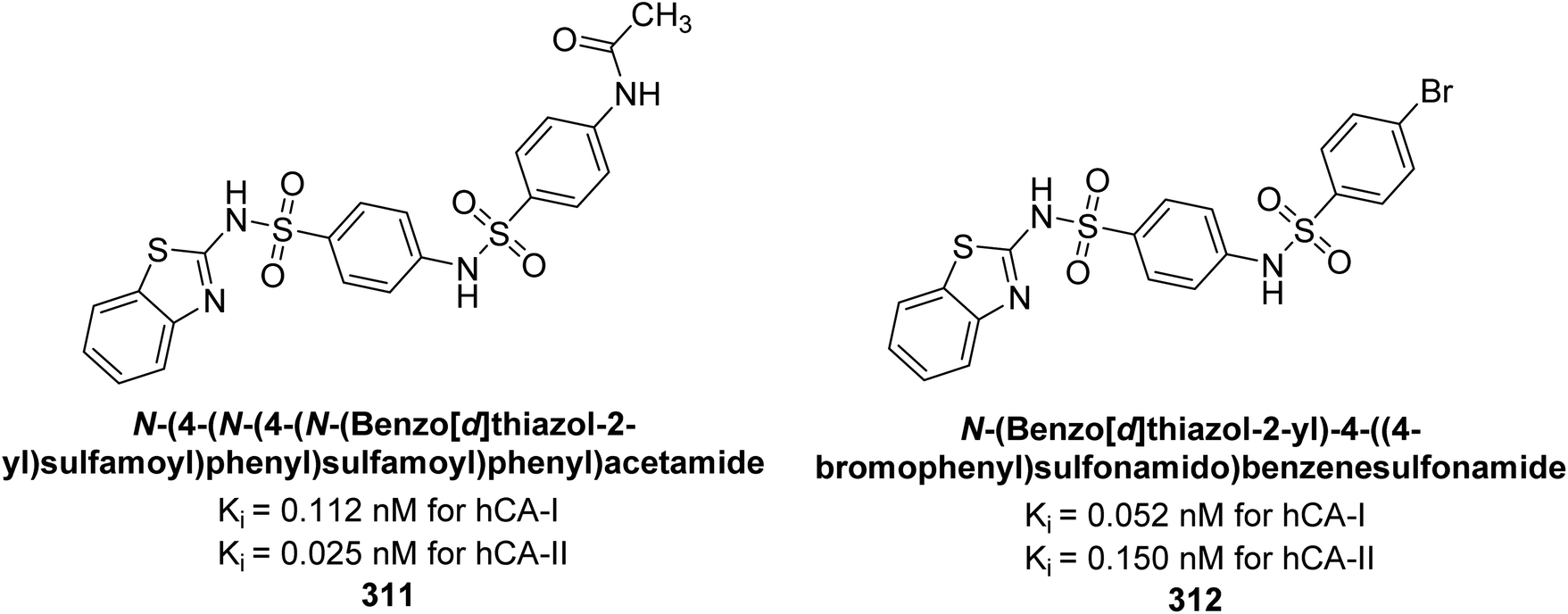 | ||
| Fig. 112 Chemical structures of compounds 311 and 312; their Ki values against hCA-I and II. | ||
Bostancı et al. (2023) synthesized thiadiazole and hydrazone derivatives and assessed their inhibition of hCA-I, II, and IX. Compound 313 notably inhibited hCA-I with an IC50 = 29.74 μM, while compound 314 was a potent inhibitor of hCA-II with an IC50 = 23.18 μM. The o-fluoro substituent on the phenyl ring enhanced inhibition of hCA-II, while an o-methoxy group increased activity against hCA-I and showed anticancer potential. Molecular docking studies were performed for compounds 313 and 314 to investigate their biological interactions (Fig. 113). These compounds hold potential as promising lead candidates for further exploration against the CA target.201
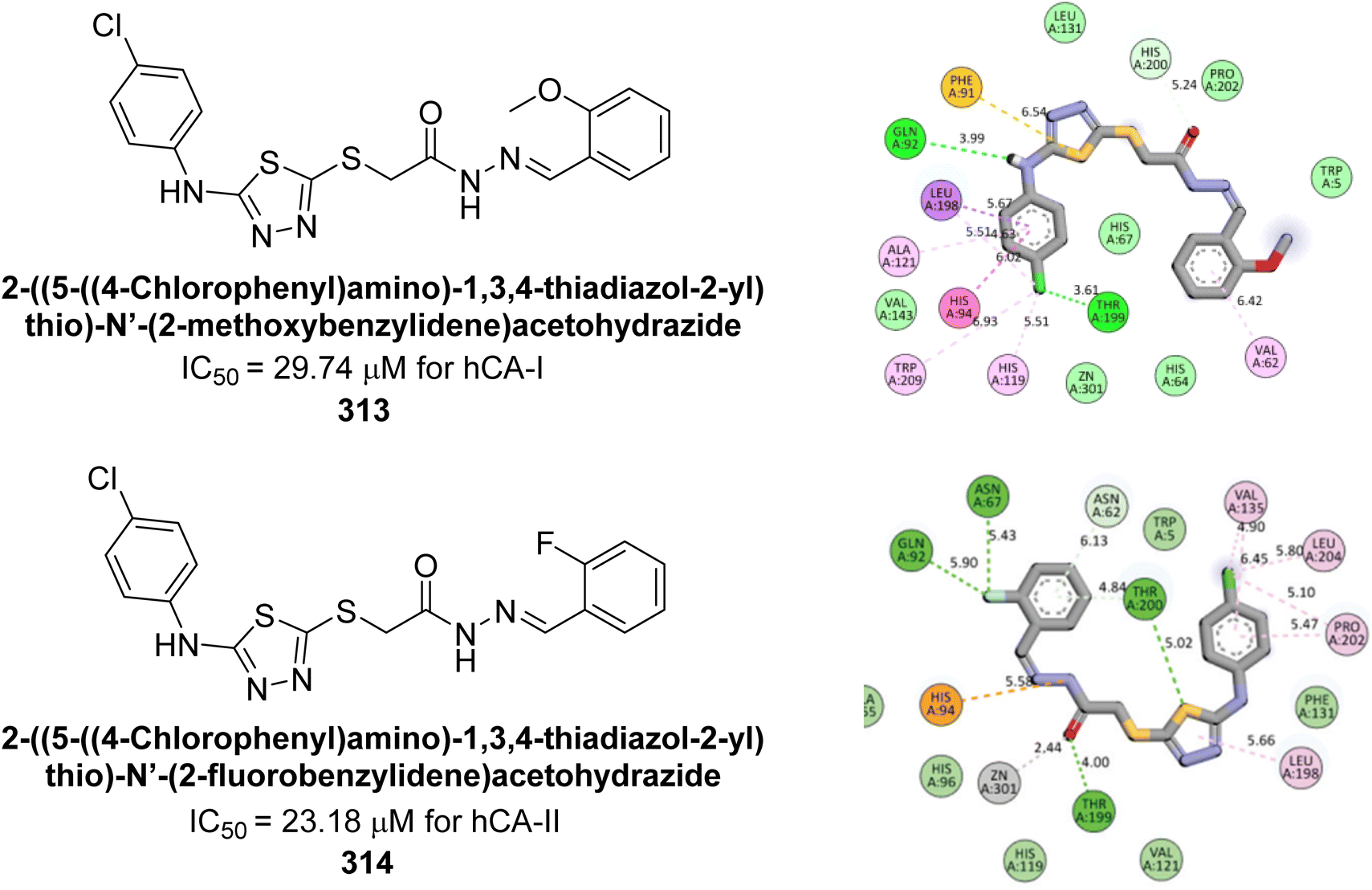 | ||
| Fig. 113 Chemical structures of compounds 313 and 314; their Ki values against hCA-I and II along with their docking images. | ||
Metwally et al. (2023) synthesized a series of thiopyrano[2,3-d]thiazoles linked to pyrazole moieties as potential anticancer agents through molecular hybridization. Remarkably, compound 315 exhibited an IC50 of 0.067 μM against hCA-IX, comparable to AAZ (0.059 μM). Similarly, compound 316 showed an IC50 of 0.123 μM against hCA-XII, close to AAZ's IC50 of 0.083 μM. Molecular docking studies further confirmed that compounds 315 and 316 effectively occupied the active sites of hCA-IX and XII through various interactions (Fig. 114).202
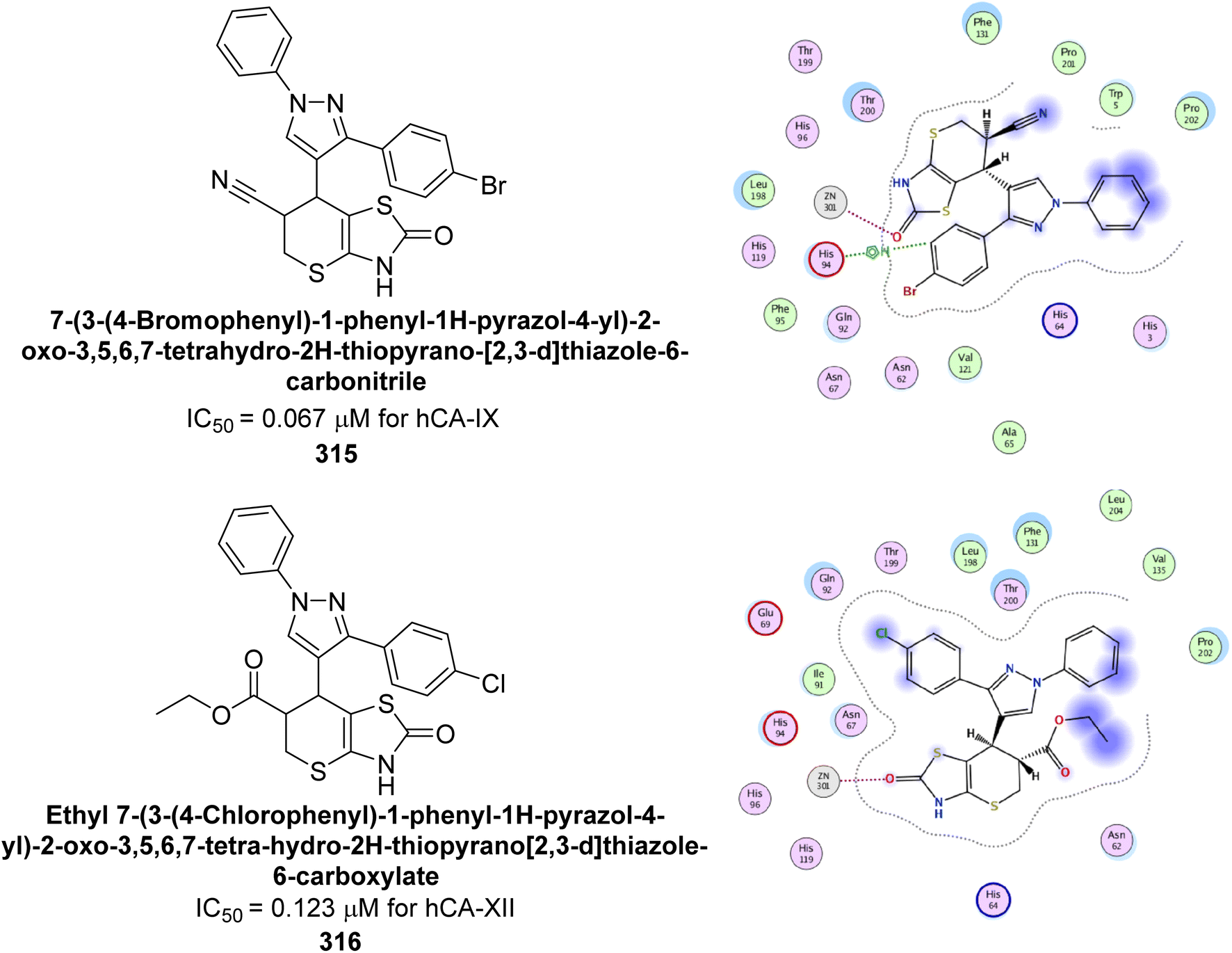 | ||
| Fig. 114 Chemical structures of compounds 315 and 316; their Ki values against hCA-IX and XII along with their docking images. | ||
Supuran et al. (2023) synthesized pyrimidines with modifications at the 7-position and evaluated their inhibition of hCA-IX and XII. The derivatives exhibited strong inhibition, with Ki values ranging from 5 to 96 nM for hCA-IX and 4 to 72 nM for hCA-XII. Especially, compounds 317, 318, 319, and 320 showed potent inhibition of hCA-IX (Ki = 5.1, 8.6, 4.7, and 5.1 nM), while compounds 321 and 322 were effective against hCA-XII (Ki = 8.8, 5.4, 4.3, and 9.0 nM). Preliminary cytotoxicity tests indicated these compounds are not toxic to human non-tumor cells. The results suggest that small structural modifications at the 7-position of the triazolopyrimidine scaffold can enhance both inhibitory activity and isoform selectivity (Fig. 115).203
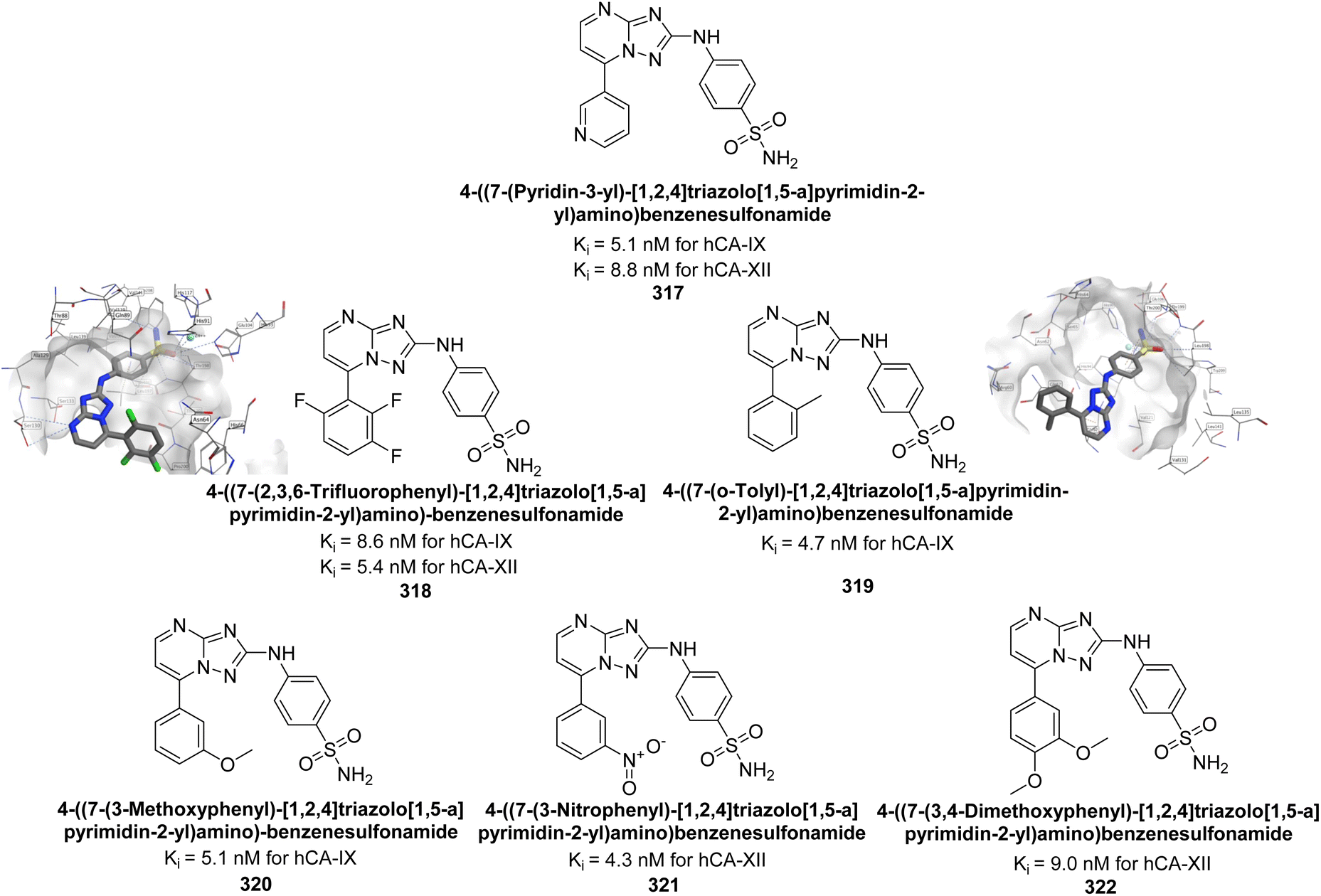 | ||
| Fig. 115 Chemical structures of compounds 317–322; their Ki values against hCA-IX and XII along with docking images of 318 and 319. | ||
Ünver et al. (2023) synthesized imidazole-hydrazone Schiff base derivatives and evaluated their inhibitory effects against hCA-I and II. Compound 323 exhibited the highest inhibition of hCA-I, with an IC50 of 0.94 μM. Compounds 324, 325, and 326 showed notable inhibition of hCA-II, with IC50 values of 3.3, 4.4, and 5.6 μM, respectively (Fig. 116). Thus, compound 323 emerges as a potential lead for developing selective cytotoxic agents targeting hCA-I, while compound 324 holds promising for targeting hCA-II.204
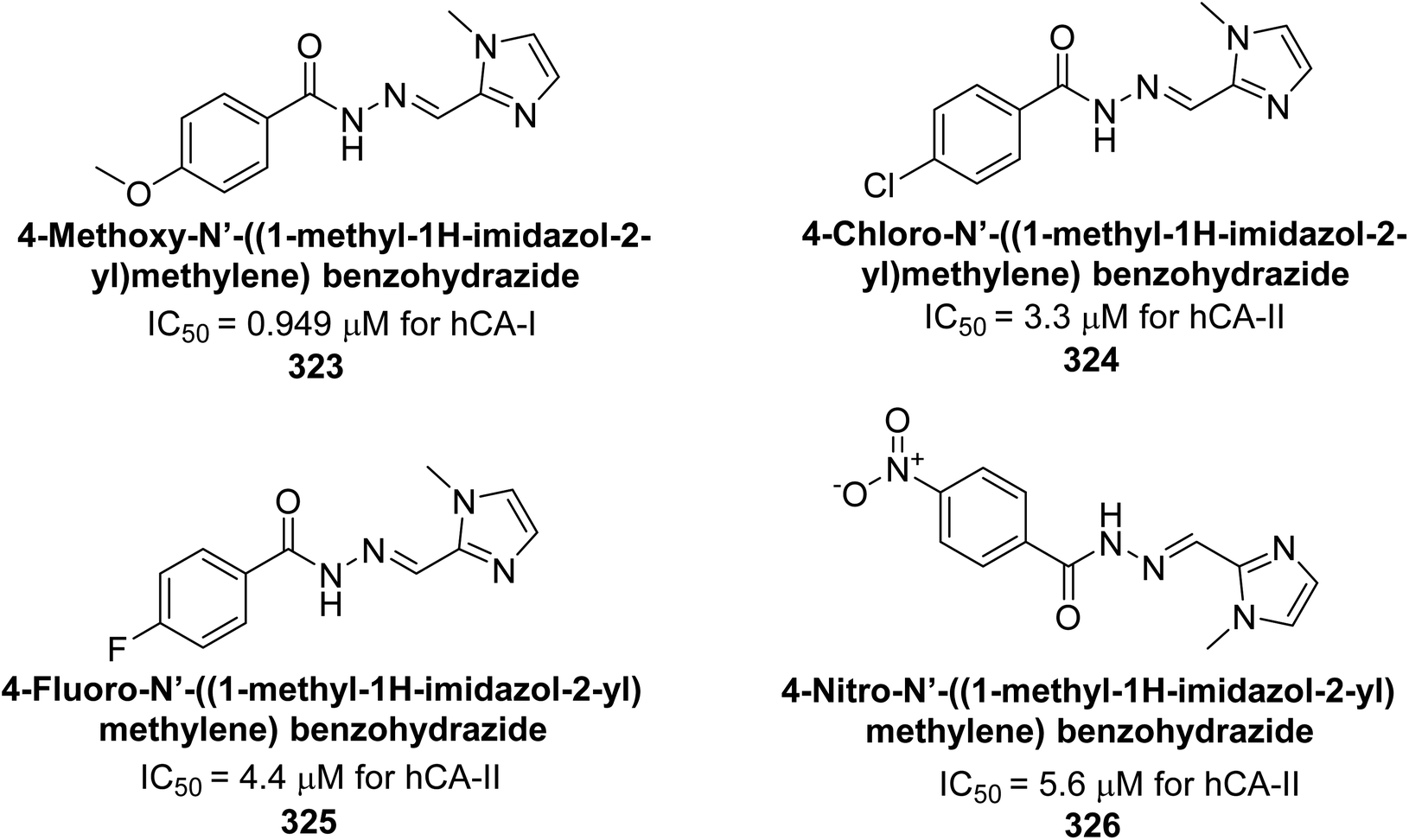 | ||
| Fig. 116 Chemical structures of compounds 323–326; their Ki values against hCA-I and II. | ||
Çevik et al. (2023) synthesized a series of benzimidazole-1,3,4-triazole derivatives with the objective of developing novel heterocyclic hybrids as potent enzyme inhibitors. Among them, compound 327 emerged as the most potent inhibitor of hCA-I, with an IC50 of 1.288 μM, while compound 328 was the most effective against hCA-II, with an IC50 of 1.532 μM. Enzyme inhibition kinetics revealed that all compounds act as non-competitive inhibitors. Molecular docking studies on compounds 327 and 328 confirmed their binding interactions within the enzyme's active site, corroborating the experimental results (Fig. 117).205
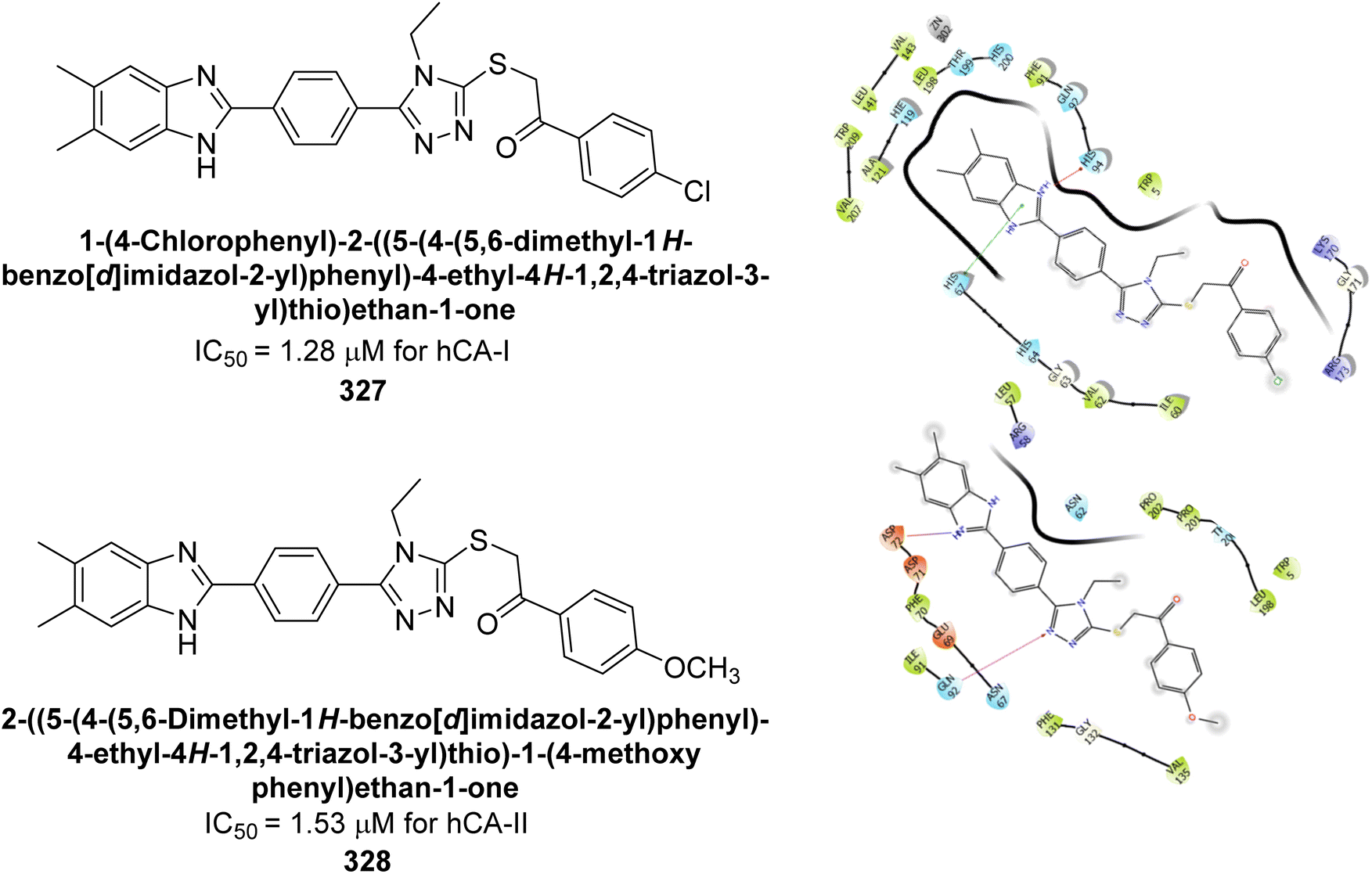 | ||
| Fig. 117 Chemical structures of compounds 327 and 328; their Ki values against hCA-I and II along with their docking images. | ||
Kucukoglu et al. (2023) designed and synthesized a series of oxadiazoles synthesized and evaluated for their inhibitory activity against hCA-I and II. Many of the compounds demonstrated superior potency compared to AAZ. Remarkably, compounds 329 and 330 exhibited the strongest inhibition of hCA-I, with IC50 values of 0.68 and 0.96 μM, respectively. Additionally, compounds 329, 330, 331 and 332 showed significant inhibitory effects against hCA-II, with IC50 values of 0.65, 0.40, 0.40, and 0.71 μM, respectively. Molecular docking analysis indicated that compounds 329 and 330 engage in π–π stacking interactions with Phe91 in the active site of hCA-I. For hCA-II, these compounds formed π–π stacking interactions with His94 and π-cation interactions with Phe131 (Fig. 118). The alkylamino groups of both compounds played an essential role in their binding to hCA-II.206
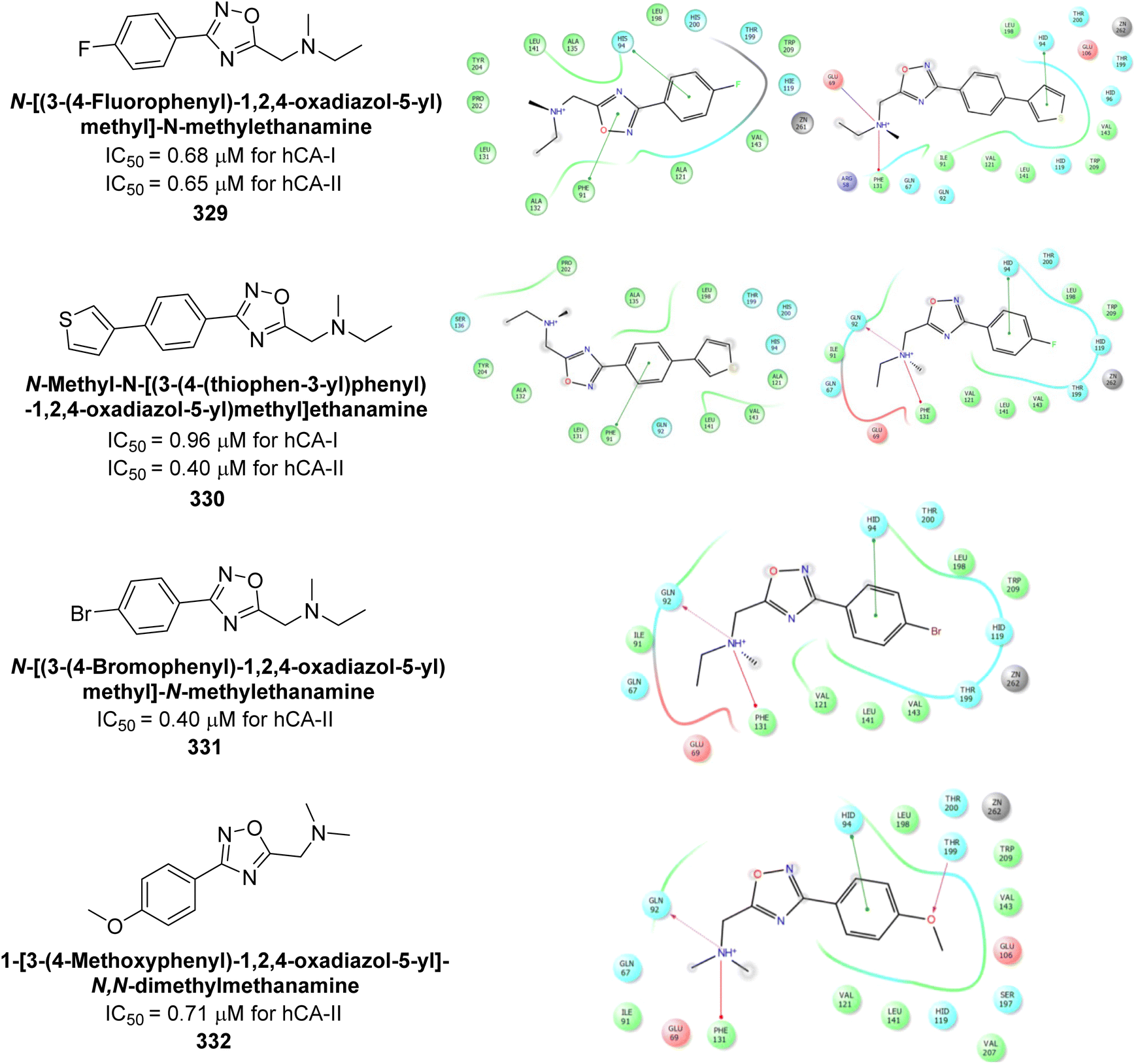 | ||
| Fig. 118 Chemical structures of compounds 329–332; their IC50 values against hCA-I and II along with their docking images. | ||
Supuran et al. (2023) synthesized a series of biotin-conjugated sulfonamide derivatives to evaluate their potential as inhibitors of hCA-IX and XII. Particularly, in compound 333 inclusion of a 4-fluorophenyl (4-F-C6H4) group, resulted in high selectivity for hypoxia-induced hCA-XII, with a Ki value of 4.5 nM. Additionally, the 2-naphthyl derivative 334 emerged as the most potent inhibitor of hCA-IX, with a Ki of 6.2 nM—four times more effective than AAZ (Ki = 25 nM)—and exhibited excellent selectivity (Fig. 119).207
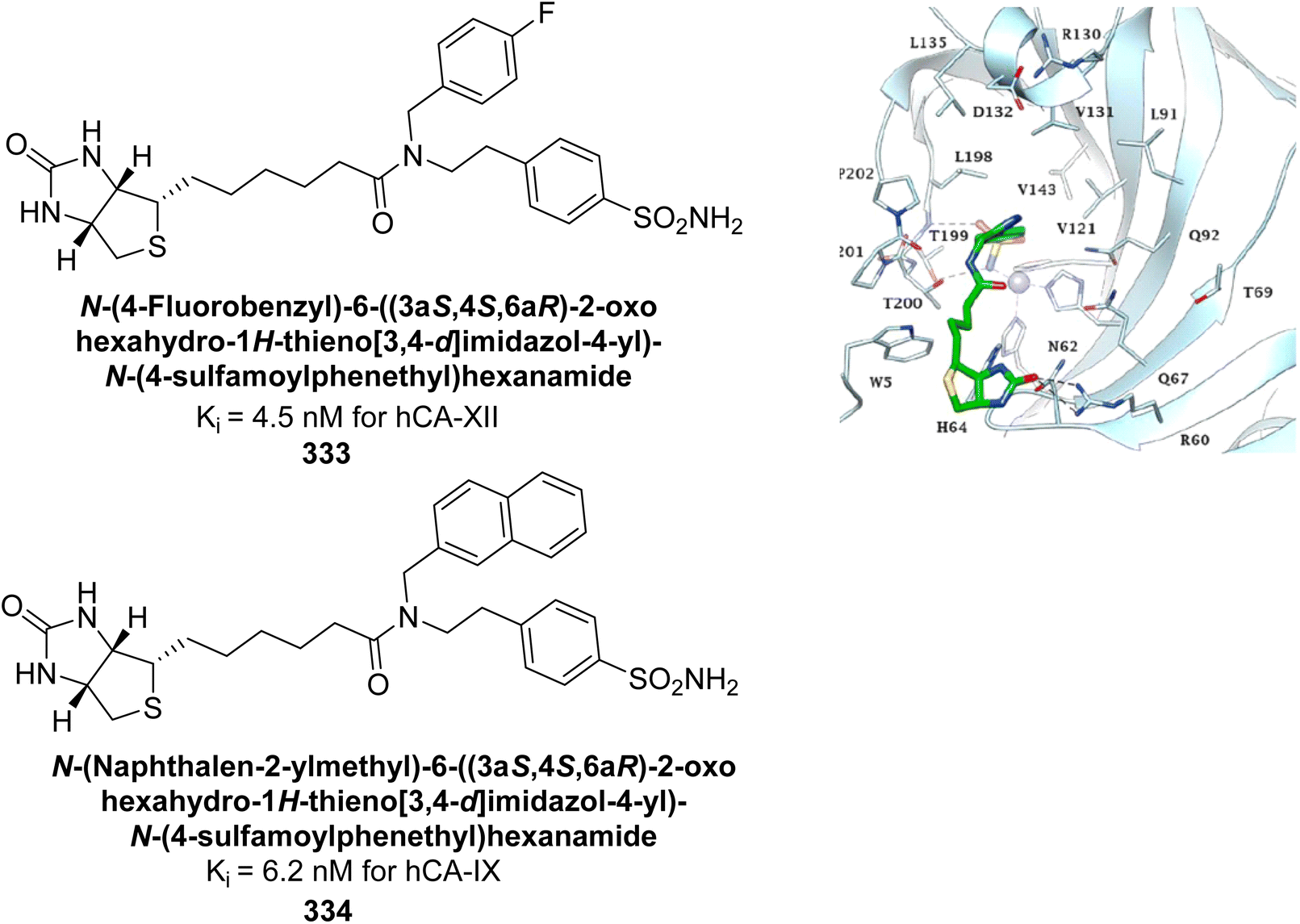 | ||
| Fig. 119 Chemical structures of compounds 333 and 334; their Ki values against hCA-IX and XII along with docking image of 333. | ||
Taslimi et al. (2023) synthesized a series of new coumarin-dihydropyridine derivatives using a molecular hybridization approach, combining key pharmacophore features. The synthesized derivatives were subsequently evaluated for their inhibitory activity against two key hCA isoforms, hCA-I and II. In vitro assays revealed that compounds 335, 336, and 337 were the most effective inhibitors of the studied hCA isoforms (Fig. 120). Compound 336, belonging to the diethyl series and featuring a 4-fluorobenzyl substituent, was identified as the most selective inhibitor of hCA-I and II.208
 | ||
| Fig. 120 Chemical structures of compounds 335–337; their IC50 values against hCA-I and II. | ||
Supuran et al. (2023) designed and synthesized furyl sulfonamides and tested for their inhibitory activity against hCA-I, II and IV. The findings indicated that compounds 338, 339, 340, and 341 demonstrated strong inhibitory activity against the hCA-I and II isoforms, while compound 342 showed notable activity against the hCA-IV and IX isoforms. Additionally, molecular docking studies were conducted to explore potential interactions of these compounds with the active site of each isoform (Fig. 121). The most potent inhibitors exhibited favorable bioavailability and drug-likeness properties.209
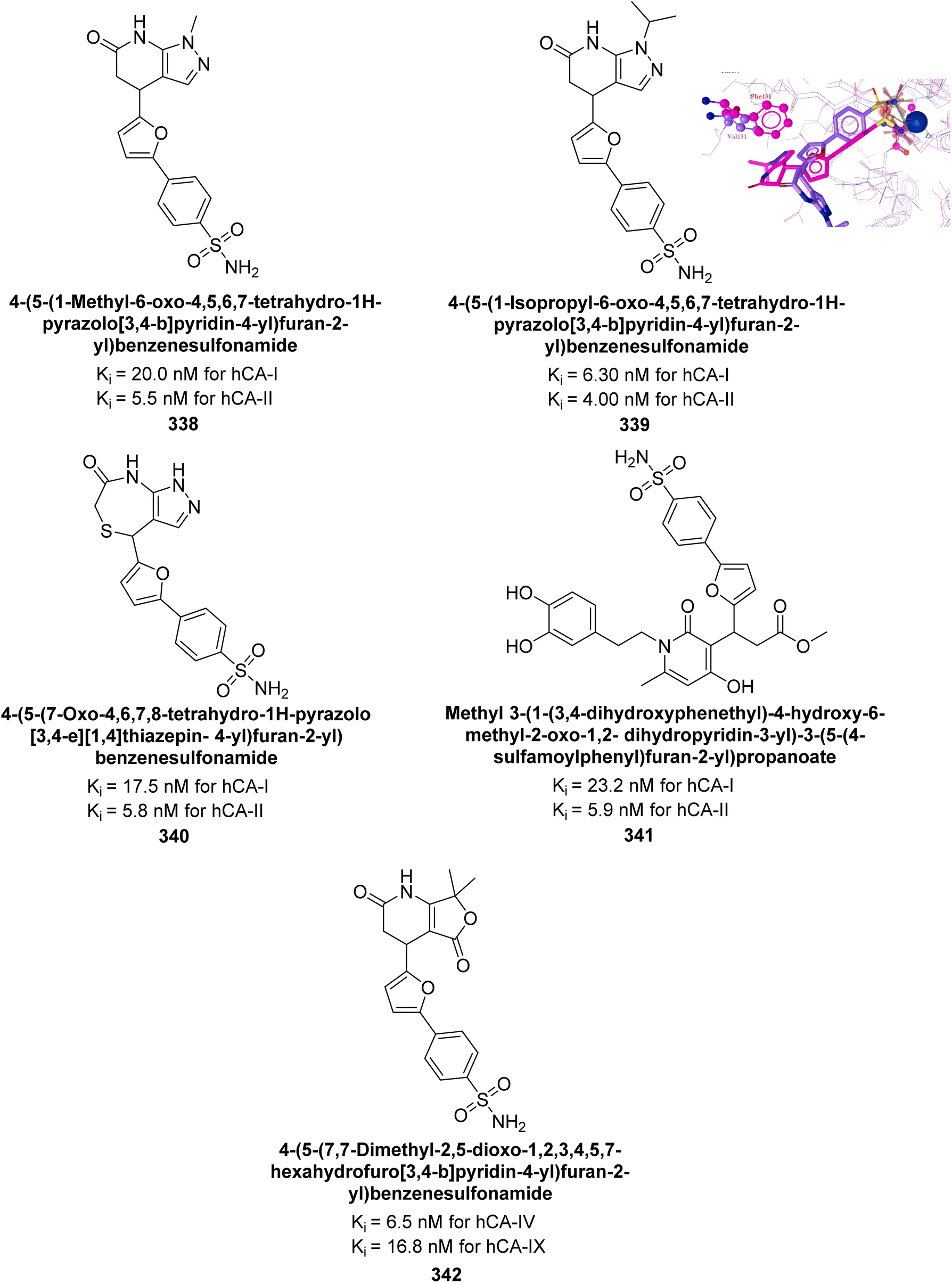 | ||
| Fig. 121 Chemical structures of compounds 338–342; their Ki values against hCA-I, II and IV; docking image of compound 339. | ||
Trippier et al. (2023) synthesized sulfonamides featuring a coumarin scaffold and evaluated as potent and selective CA inhibitors. Especially, compound 343 (Ki values: 21 nM for hCA-IX, and 5 nM for hCA-XII) was identified as a promising inhibitor of hCA-IX and XII, warranting further investigation (Fig. 122).210
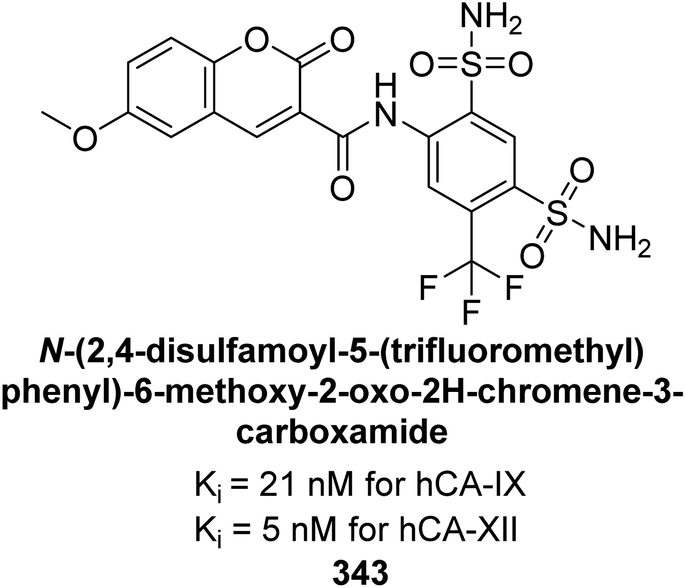 | ||
| Fig. 122 Chemical structure of compound 343; its Ki values against hCA-IX and XII. | ||
Metwaly et al. (2024) designed and synthesized a series of novel substituted thiophene derivatives and their corresponding thiophene-pyrazole analogues incorporating a benzenesulfonamide moiety. The inhibitory effects of these synthesized analogues on hCA-IX and XII, were evaluated, with AAZ (IC50 = 0.065 μM for hCA-IX; 0.083 μM for hCA-XII) used as a reference compound. Compounds 344 (IC50 = 0.092 μM) and 345 (IC50 = 0.069 μM) demonstrated significant inhibitory activity against hCA-IX, while compounds 346 (IC50 = 0.173 μM) and 347 (IC50 = 0.146 μM) exhibited distinguished activity against hCA-XII (Fig. 123). The newly synthesized thiophene analogues were analyzed through molecular docking studies against the hCA-XII crystal structure to investigate their binding interactions.211
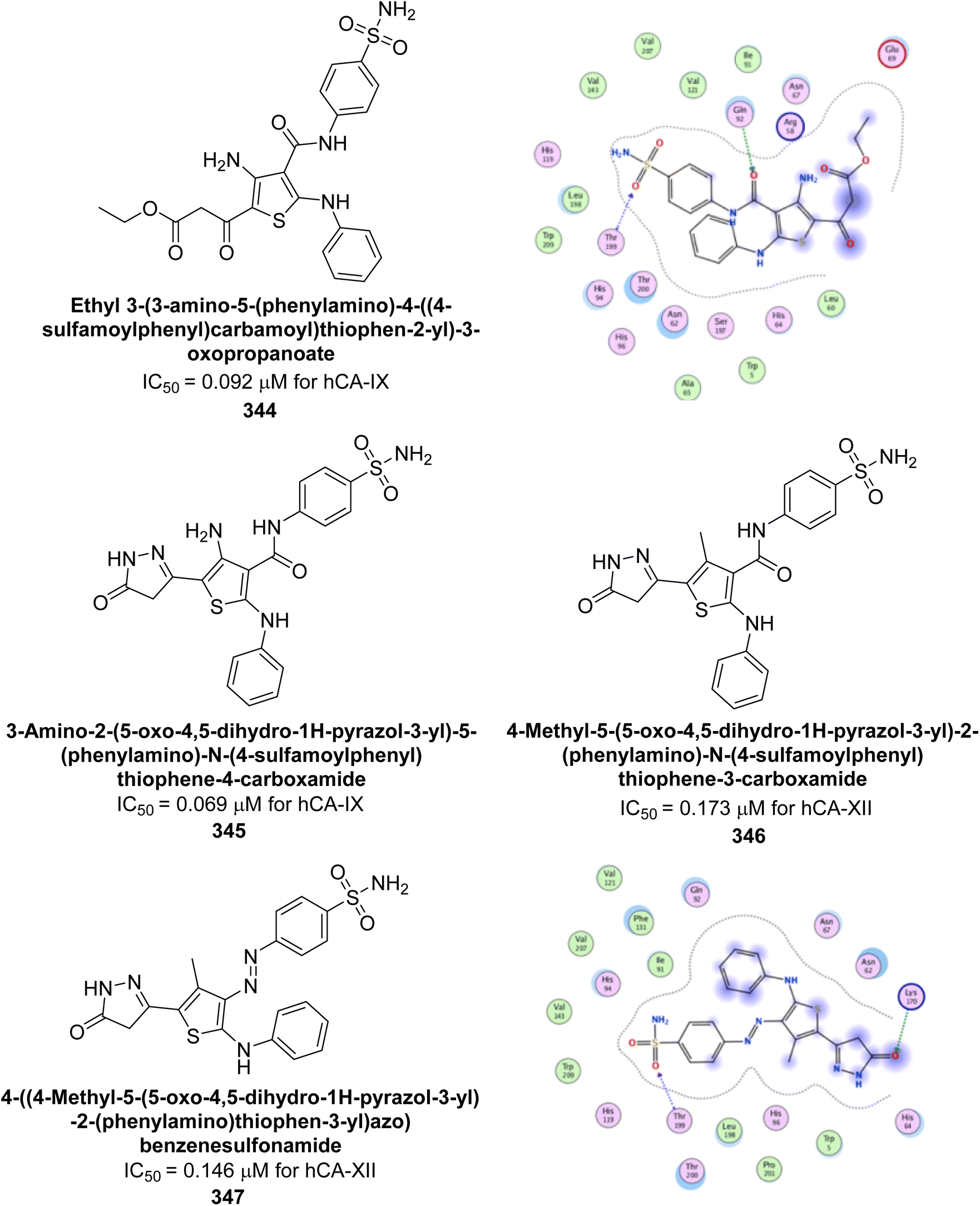 | ||
| Fig. 123 Chemical structures of compounds 344–347; their IC50 values against hCA-IX and XII along with docking images of 344 and 347. | ||
El-Messery et al. (2024) developed a series of coumarin sulfonate and sulfamate derivatives and evaluated their selectivity and inhibitory activity against hCA-II, IX, and XII, with AAZ as a reference. Derivatives with aryl or fluoroaryl sulfone groups favored cancer-related isoforms hCA-IX and XII. Markedly, the p-fluorine-substituted derivative 348 showed strong activity against hCA-IX (IC50 = 0.62 μM), while the benzene sulfone derivative 349 was selective for hCA-XII (IC50 = 0.56 μM). Coumarin derivatives with ethyl sulfone groups 350, 351, 352 were highly selective for hCA-II. Molecular docking studies indicated that these compounds interact with the hCA active site via various mechanisms (Fig. 124).212
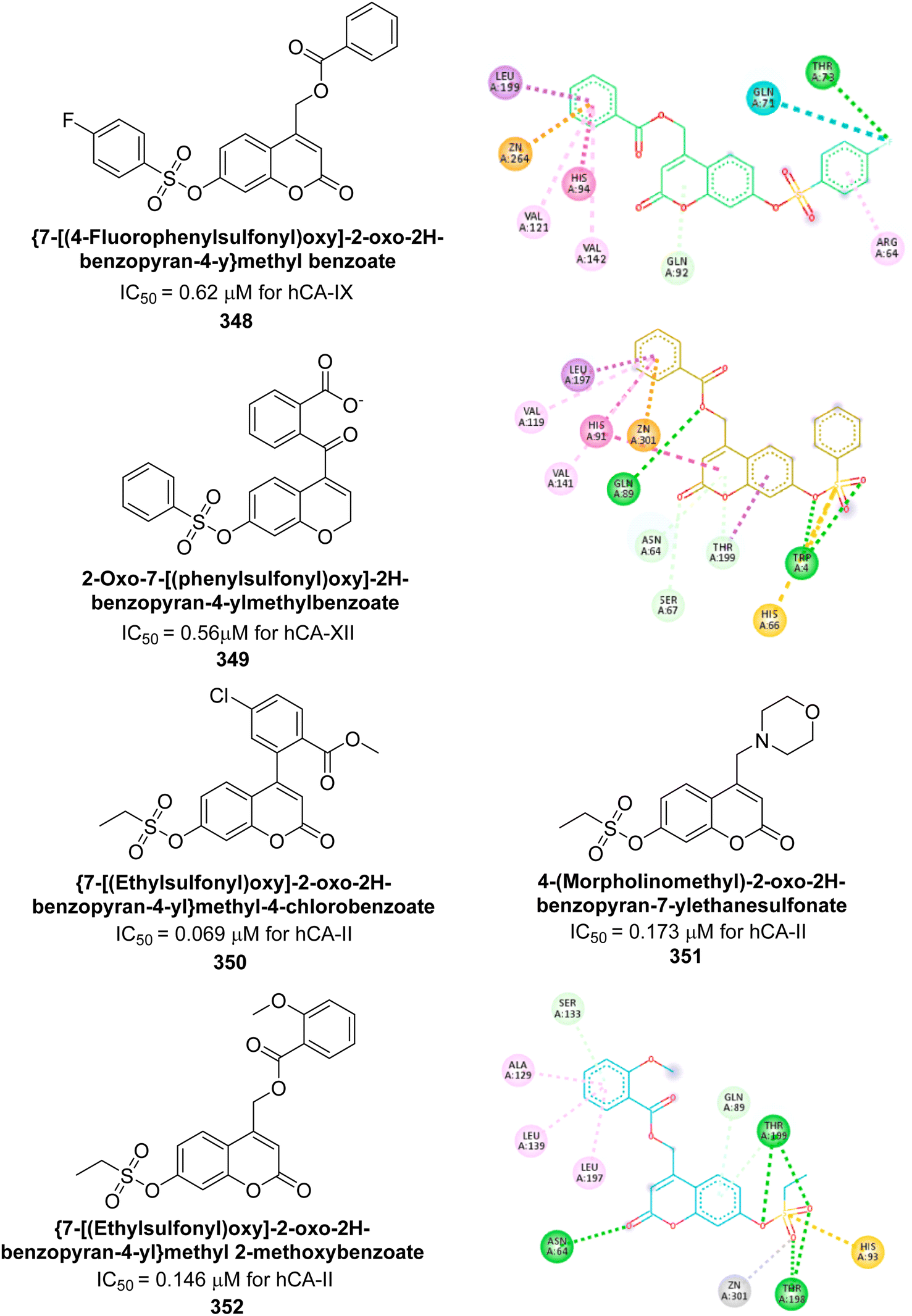 | ||
| Fig. 124 Chemical structures of compounds 348–352; their IC50 values against hCA-II, IX and XII along with their docking images. | ||
Çevik et al. (2024) developed a series of benzimidazole-triazole derivatives and evaluated their inhibitory effects on hCA-I and II. The compounds showed IC50 values between 1.158 μM and 3.48 μM. Remarkably, compound 353 demonstrated the highest activity, with IC50 values of 1.288 μM for hCA-I and 1.619 μM for hCA-II. Kinetic studies indicated that these compounds act as non-competitive inhibitors. Molecular docking of compound 353 confirmed its interactions with the enzyme's active site, aligning with the experimental data (Fig. 125).213
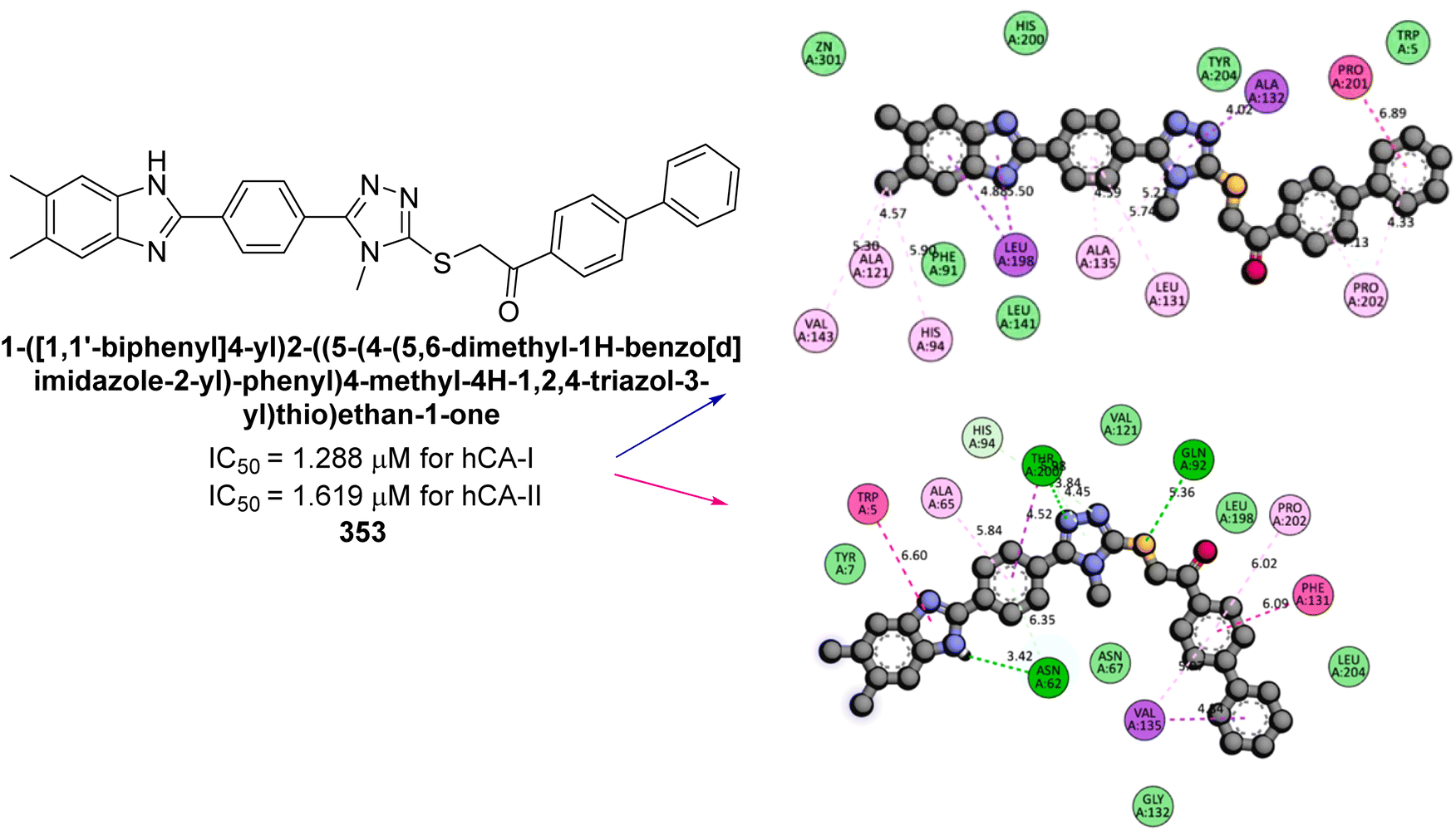 | ||
| Fig. 125 Chemical structure of compound 353; its IC50 values against hCA-I and II along with its docking images. | ||
Nural et al. (2024) developed naphthoquinone-thiazole hybrids and evaluated their inhibitory effects on hCA-I and II. The compounds exhibited significant inhibition, with Ki values ranging from 67.86 to 161.60 nM for hCA-I and 55.27 to 87.48 nM for hCA-II. The most potent compound, 354, had Ki values of 67.86 nM for hCA-I and 56.8 nM for hCA-II. Molecular docking studies showed that in hCA-I, the naphthalene, thiazole, and difluorobenzene rings engaged in π–π stacking with residues His67, His94 and Phe91. For hCA-II, hydrogen bonds were formed with carbonyl groups at distances of 1.86, 2.11, and 2.15 Å, and π–π stacking occurred between His64 and the difluorobenzene ring (Fig. 126). These findings suggest that the compounds align well with the active sites of these enzymes.214
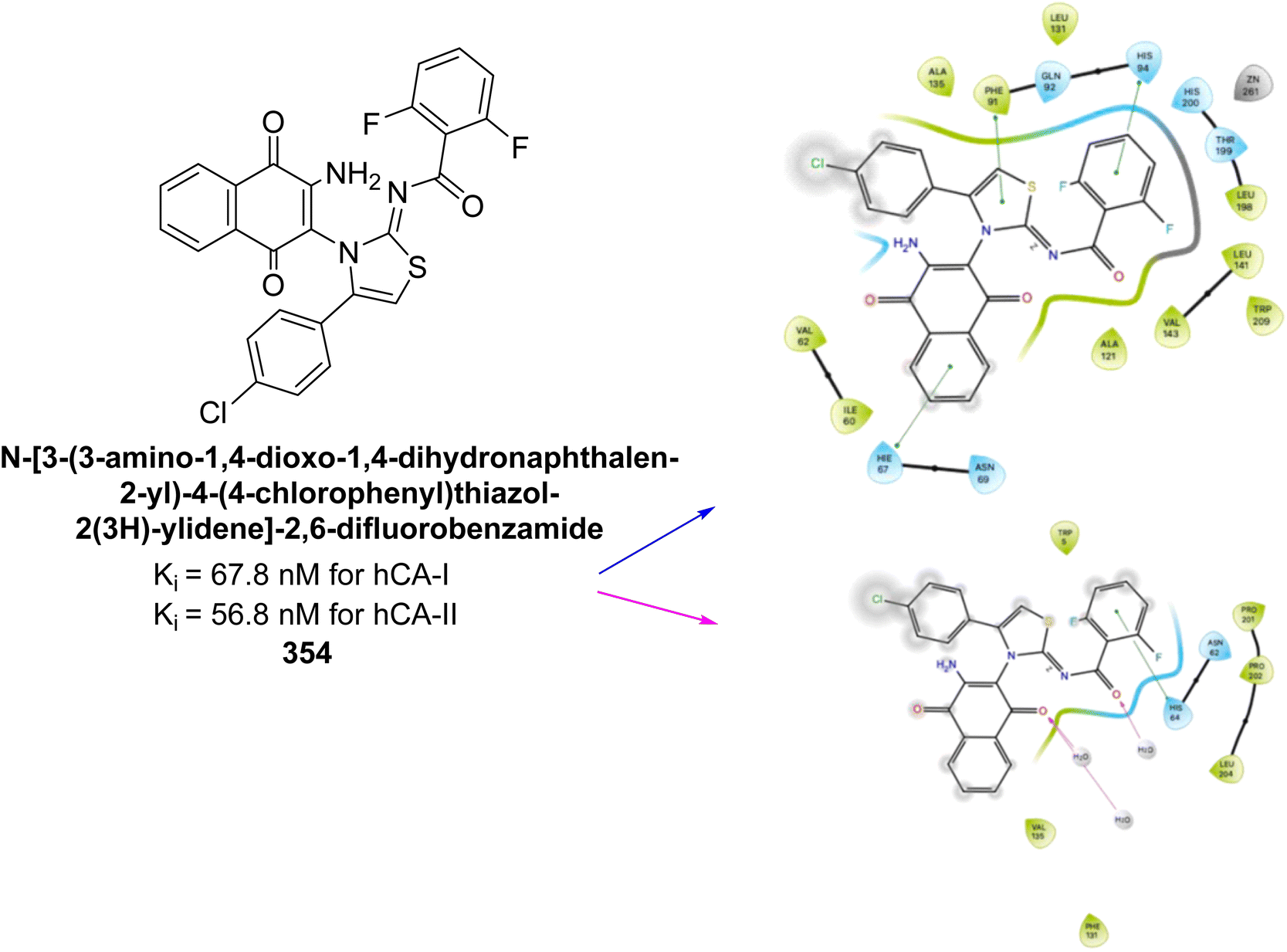 | ||
| Fig. 126 Chemical structure of compound 354; its Ki values against hCA-I and II along with its docking images. | ||
Mert et al. (2024) developed a series of novel pyrazole dicarboxylic acid derivatives. These derivatives were evaluated for their inhibitory activity against hCA-I and II, showing high potency. The compounds demonstrated significant inhibition, with Ki values ranging from 11.27 to 50.31 nM for hCA-I and 9.97 to 36.22 nM for hCA-II. Compound 355 (Ki = 11.27 nM) demonstrated exceptional activity against hCA-I. In contrast, compound 356 (Ki = 9.97 nM) exhibited excellent activity specifically against hCA-II. Molecular docking studies confirmed that these compounds effectively inhibit the target enzymes (Fig. 127).215
 | ||
| Fig. 127 Chemical structures of compounds 355 and 356; their Ki values against hCA-I and II along with their docking images. | ||
Supuran et al. (2024) synthesized coumarin analogues, incorporating a thiazole moiety and evaluated against hCAs. Particularly, compound 357, featuring a 4-methoxy substitution, exhibited significant activity with a Ki of 90.9 nM against the hCA-XII isoform (Fig. 128). Significantly, all compounds showed selective inhibition against the hCA-IX and XII isoforms, with Ki values below 1000 nM. These findings suggest that the newly synthesized coumarin–thiazole hybrids have the potential to serve as selective lead candidates targeting hCA-IX and XII over hCA-I and II.216
 | ||
| Fig. 128 Chemical structure of compound 357; its Ki value against hCA-XII. | ||
Lamie et al. (2024) designed and synthesized a series of 1,2,3-triazole benzenesulfonamide derivatives to target hCA-IX and XII isoforms. Most of the synthesized compounds demonstrated significant inhibitory activity against these isoforms. Remarkably, compounds 358, 359 and 360 exhibited exceptional inhibition of hCA-IX, with Ki values ranging from 0.03 to 0.06 μM, surpassing the potency of AAZ. Additionally, compounds 361 and 362 effectively inhibited the hCA-XII isoform, both with a Ki value of 0.02 μM, comparable to AAZ. ADME analysis indicated that compounds 358, 360, 361 and 362 complied with Lipinski's rule of five, suggesting they may be orally active, whereas compound 359 displayed lower oral bioavailability than AAZ. Molecular docking studies further elucidated the binding modes and stability of the target compounds within the active sites of hCA-IX and XII, particularly for compounds 360 and 362 (Fig. 129).217
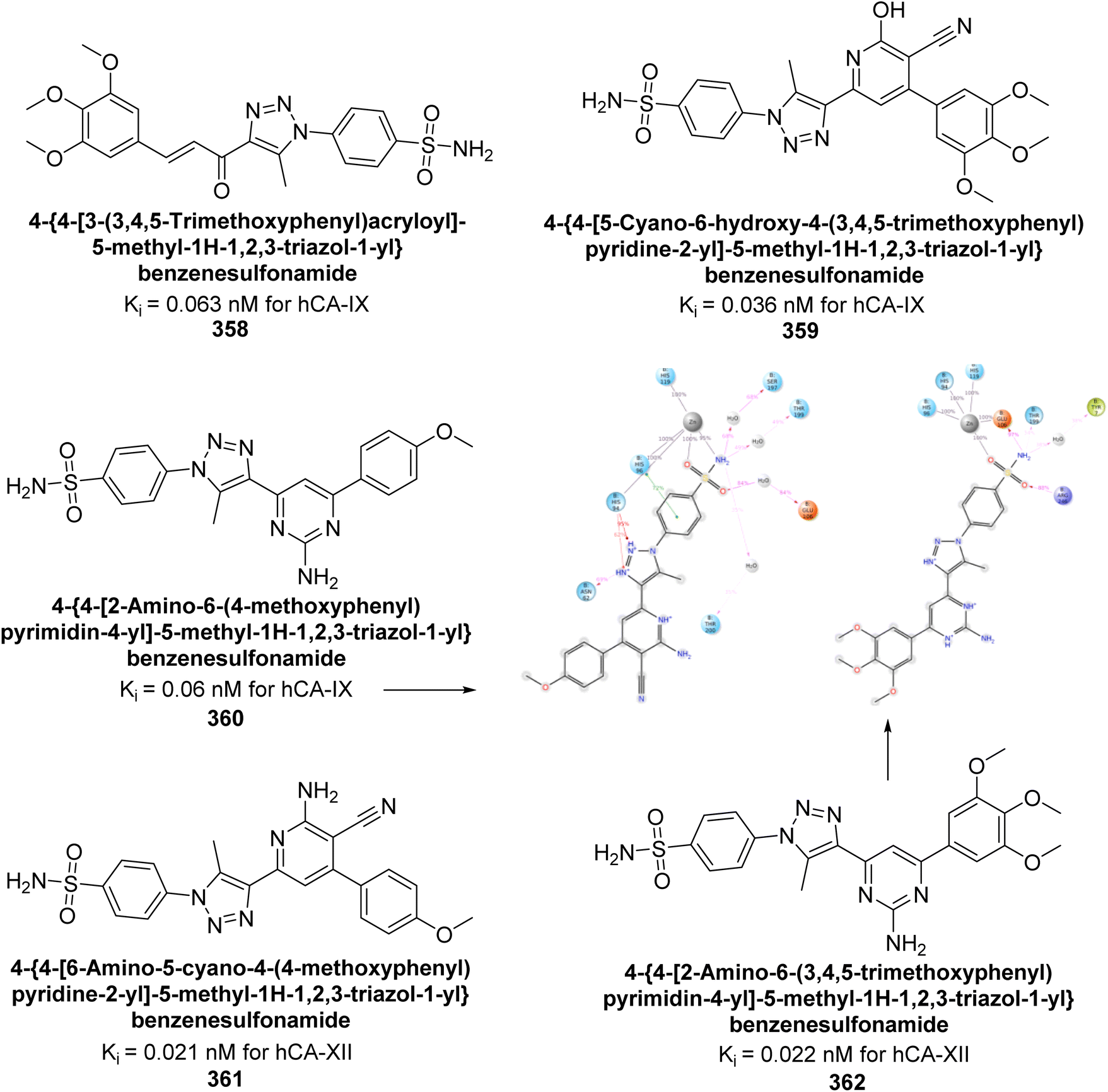 | ||
| Fig. 129 Chemical structures of compounds 358–362; their Ki values against hCA-XI and XII along with docking images of 360 and 362. | ||
Abouzid et al. (2024) synthesized a series of novel compounds incorporating the privileged thienopyrimidine scaffold. Among them, compounds 363, 364, 365 and 366 demonstrated the highest inhibition across all four isoforms. Particularly, compound 366 exhibited potent inhibitory activity against hCA-II, IX and XII, with Ki values of 8.6, 13.8, and 19 nM, respectively, comparing favorably to AAZ, which has Ki values of 12, 25, and 5.7 nM, respectively. Additionally, compound 365 showed significant activity against hCA-IX, with a Ki of 16.1 nM. Molecular docking studies were performed to predict the binding modes of these synthesized compounds within the active sites of the four CAIs. The results revealed that compounds 363–366 effectively mimicked the binding mode of AAZ, coordinating with the Zn2+ ion and interacting with critical amino acids Thr199, His96 and His94 essential for enzymatic activity (Fig. 130).218
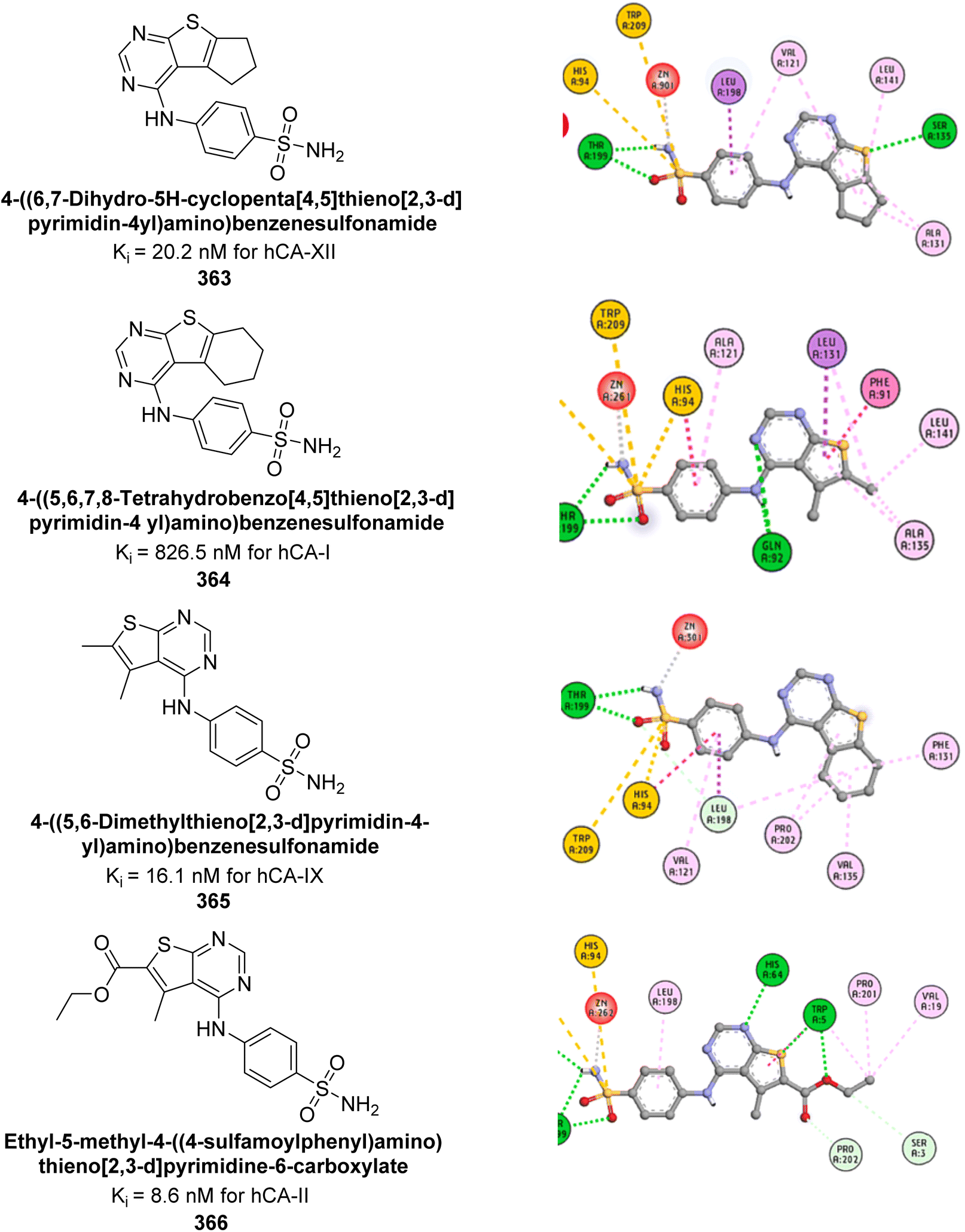 | ||
| Fig. 130 Chemical structures of compounds 363–366; their Ki values against hCA-I, II, XI and XII along with their docking images. | ||
Angeli et al. (2024) designed and synthesized dihydro-pyrrol-2-one compounds containing dual sulfonamide groups. Specifically, the ubiquitously expressed cytosolic hCA-I was inhibited with Ki values ranging from 3.9 (for compound 367) to 870.9 nM, while hCA-II also showed significant inhibition. Importantly, all compounds demonstrated effective inhibition of the hCA-IX (Ki = 1.9 [for compound 368] – 211.2 nM) and hCA-XII, with inhibition in the low nanomolar range (Fig. 131).219
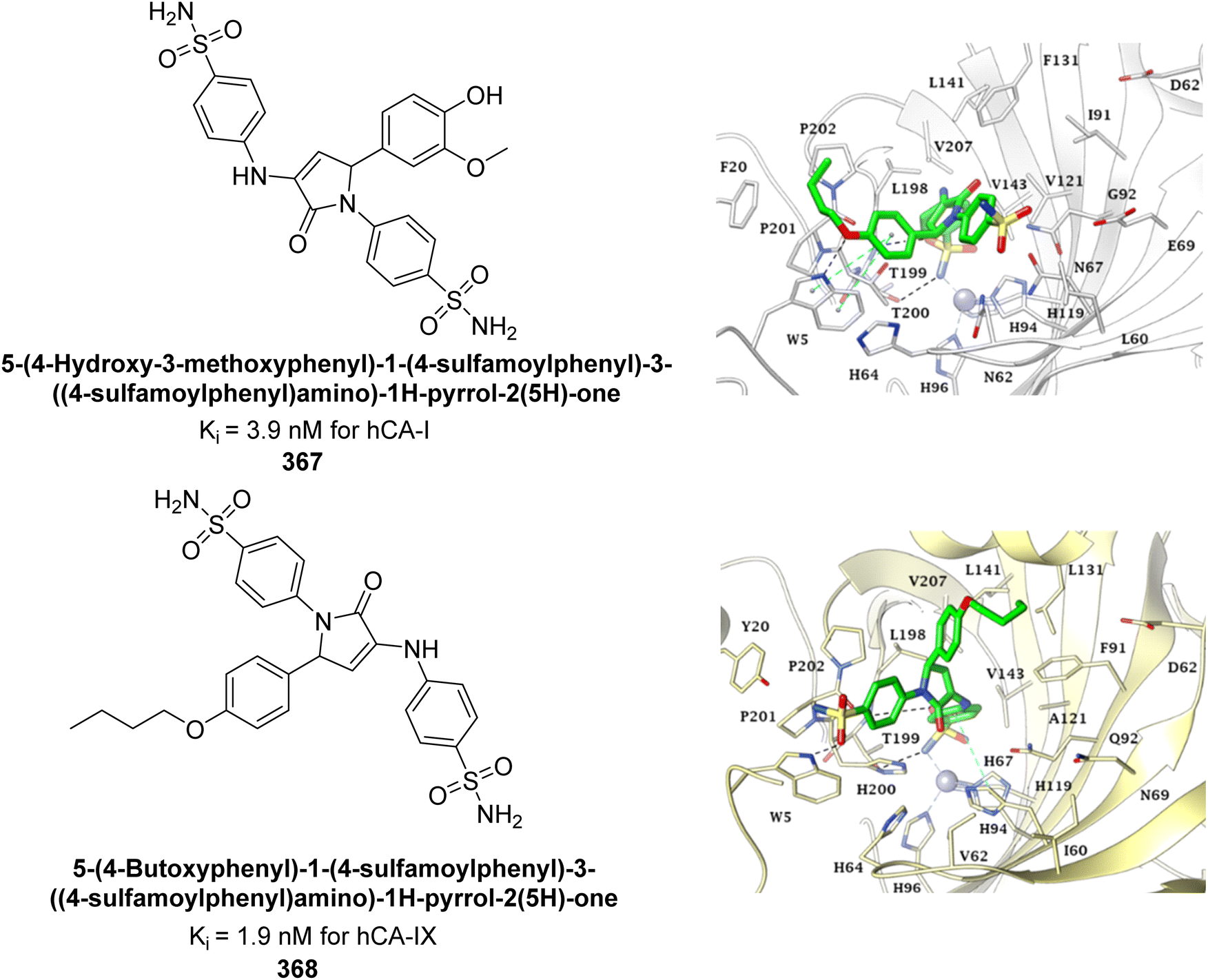 | ||
| Fig. 131 Chemical structures of compounds 367 and 368; their Ki values against hCA-I and IX along with their docking images. | ||
Supuran et al. (2024) developed rhodanine-linked enamine-carbohydrazide derivatives and evaluated their effectiveness as inhibitors of mycobacterial carbonic anhydrase (mtCA). The results demonstrate their efficacy, with significant selectivity toward the mtCA-2 enzyme. Although these compounds exhibited moderate activity against hCA isoforms, their distinguished selectivity highlights their potential as antitubercular agents. Among the series, compound 369 emerged as the most potent, with a Ki value of 9.5 μM against mtCA-2. Molecular docking studies indicated that compounds 369 and 370 interact with the Zn2+ ion through metal coordination, similar to classical CAIs (Fig. 132).220
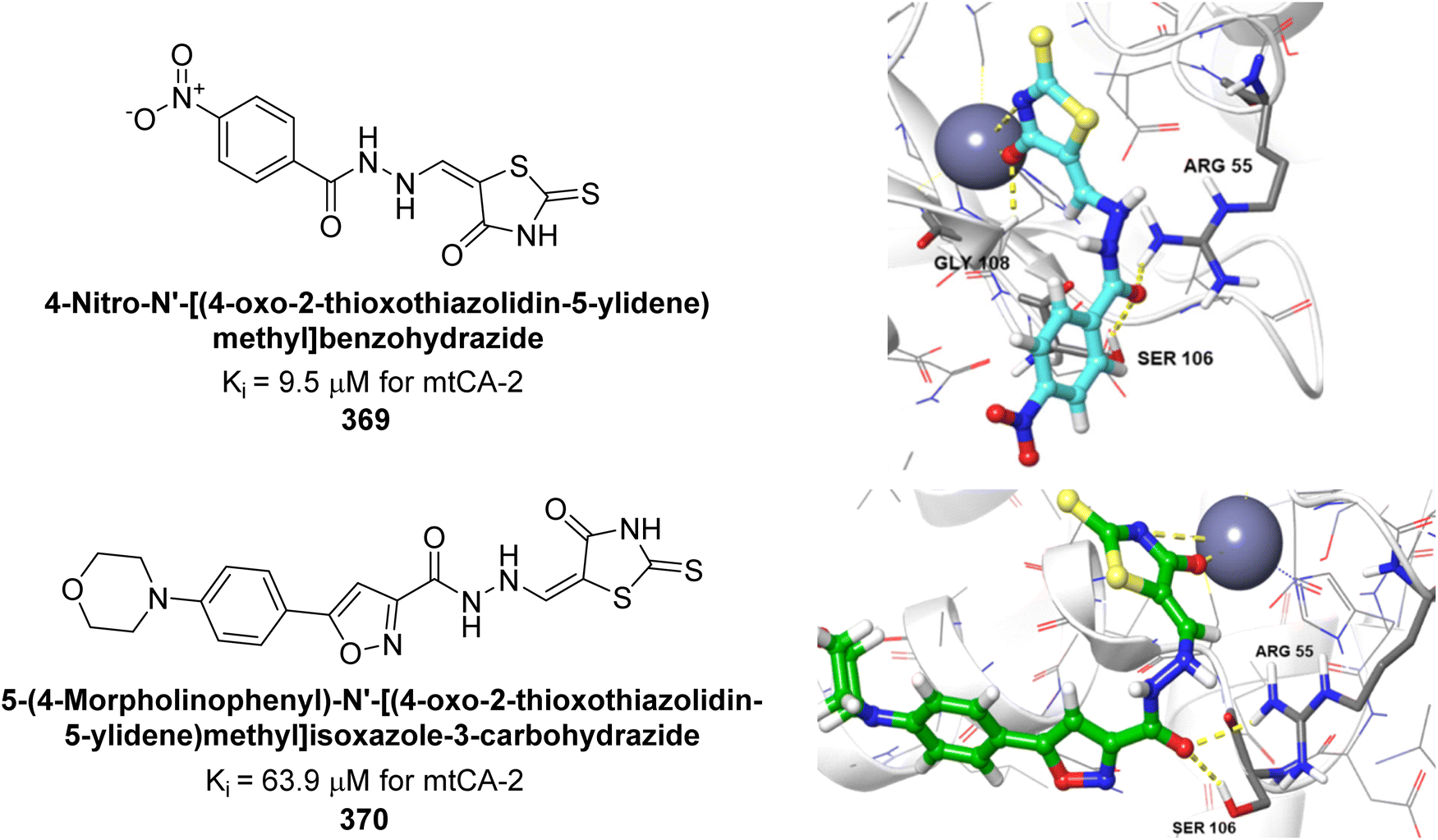 | ||
| Fig. 132 Chemical structures of compounds 369 and 370; their Ki values against mtCA-2 along with their docking images. | ||
Žalubovskis et al. (2024) synthesized sulfonamide-incorporated bis(α-aminophosphonates) and evaluated against hCA-I, II, VII, IX, and XIII. The compounds showed notable activity and selectivity, particularly towards hCA-IX. Compound 371, with a Ki of 15.1 nM, demonstrated high selectivity for hCA-IX. Additionally, these sulfonamides effectively inhibited hCA-XIII, with compound 372 showing the strongest inhibition (Ki = 17.2 nM), comparable to the well-known inhibitor AAZ but with greater selectivity (Fig. 133). We anticipate that this preliminary research will stimulate further exploration into the development of novel phosphorus-containing sulfonamide CAIs. Despite their promising inhibitory activities and selectivity, these compounds have been largely overlooked by researchers in this field.221
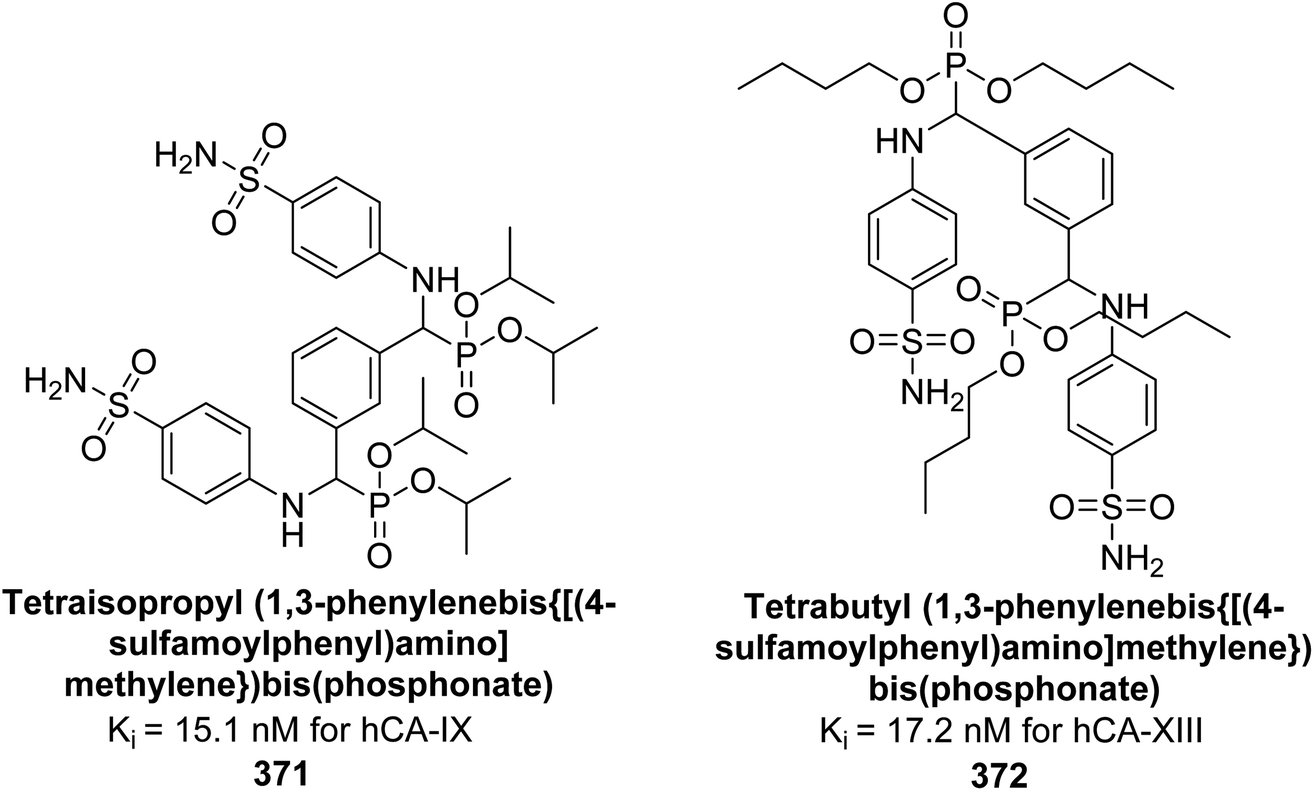 | ||
| Fig. 133 Chemical structures of compounds 371 and 372; their Ki values against hCA-IX and XIII. | ||
Arifuddin et al. (2024) synthesized isatin-linked benzenesulfonamides and evaluated for their inhibitory potency against hCA-I, II, IX, and XII. Among the most potent were compounds 373 (435.8 nM) and 374 (956.4 nM) against hCA-I; compounds 375 (60.5 nM), 376 (95.6 nM), 377 (92.1 nM), and 378 (75.4 nM) against hCA-IX; and compound 379 (84.5 nM) against hCA-XII (Fig. 134). These derivatives showed greater selectivity towards hCA-IX over hCA-I, II, and XII, indicating their potential for further development as isoform-selective hCA-IX inhibitors through additional structural modifications.222
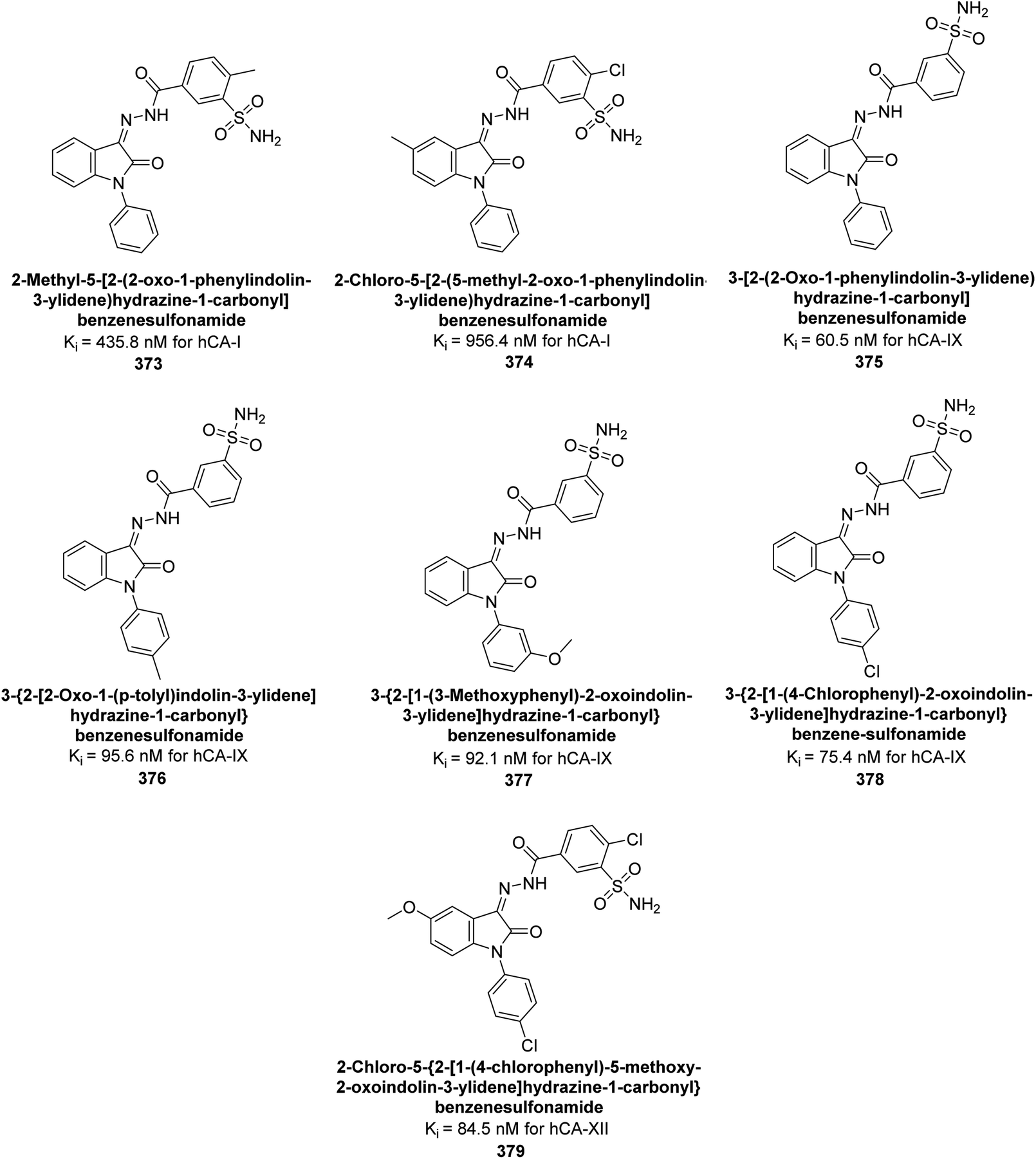 | ||
| Fig. 134 Chemical structures of compounds 373–379; their Ki values against hCA-I, IX and XII. | ||
Göksu et al. (2024) designed and synthesized a series of benzene sulfonamides and assessed for their in vitro and in silico inhibitory activities against hCA-I and II. These compounds demonstrated significant inhibition of hCAs, with Ki ranging from 39.20 nM to 131.54 nM for hCA-I, and 50.96 nM to 147.94 nM for hCA-II. Compounds 380 and 381 proved to be potent inhibitors for hCA-I and II, respectively. The leucinate group was particularly effective against hCA-I, while the N-butyl group showed greater efficacy for inhibiting hCA-II (Fig. 135).223
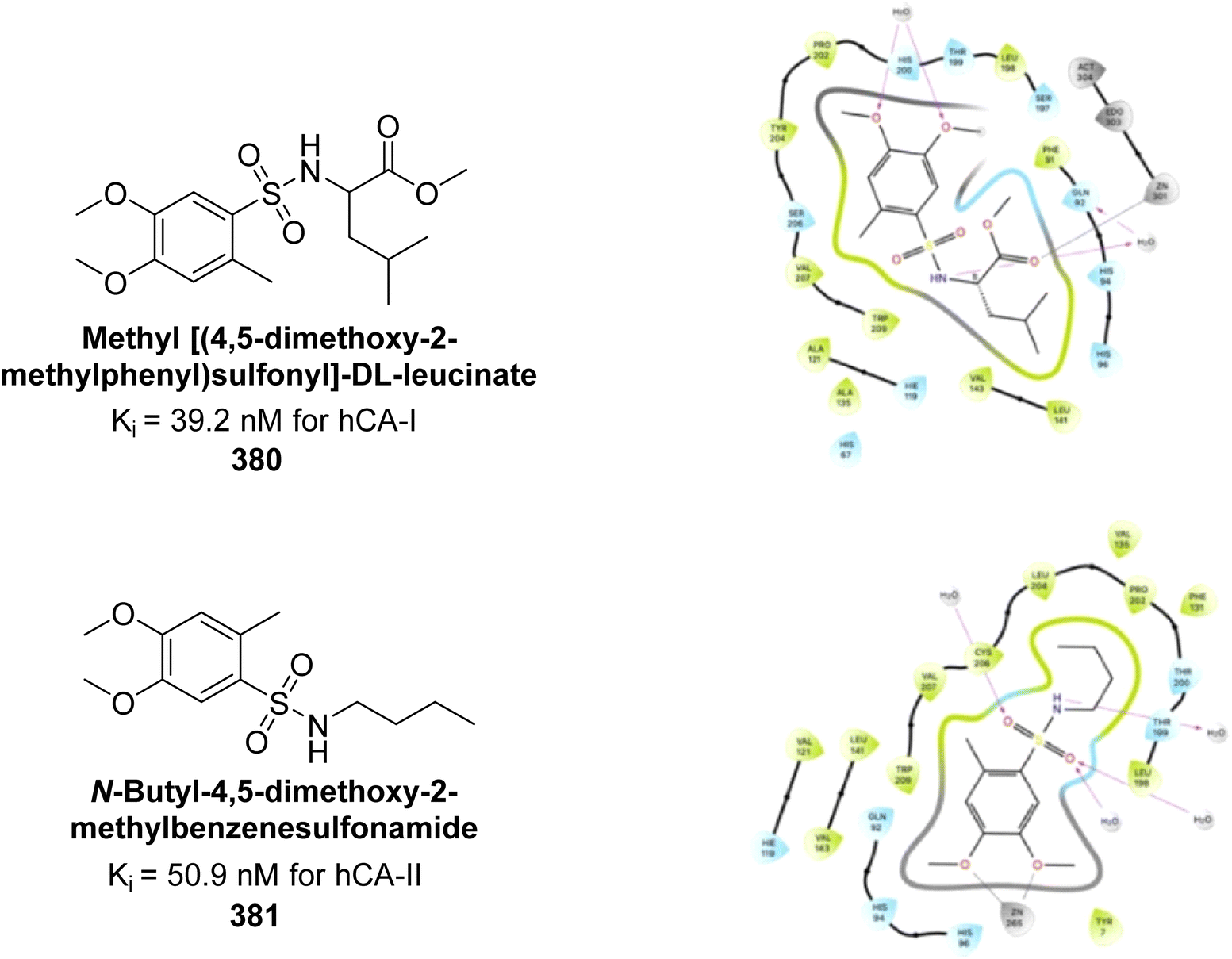 | ||
| Fig. 135 Chemical structures of compounds 380 and 381; their Ki values against hCA-I and II along with their docking images. | ||
Moi et al. (2024) synthesized a series of piperidine-4-carboxamides and evaluated against hCA isoforms. Among the compounds, benzenesulfonamido carboxamides 382 and 383 emerged as the most potent in the piperazine- and benzylamine-based series, respectively. Shifting the 4-methyl group to the 2-position led to an approximately 10-fold decrease in activity against the target isoform, as demonstrated by compound 383, which exhibited a Ki of 8.4 nM. Molecular docking studies revealed that compound 382 exhibited high selectivity for the tumor-associated hCA-IX and XII isoforms due to its ability to form favorable interactions within their active sites (Fig. 136). These initial findings highlighted the potential of piperidine-4-carboxamides as promising candidates for the continued development of therapies targeting cancer cell lines that express CA.224
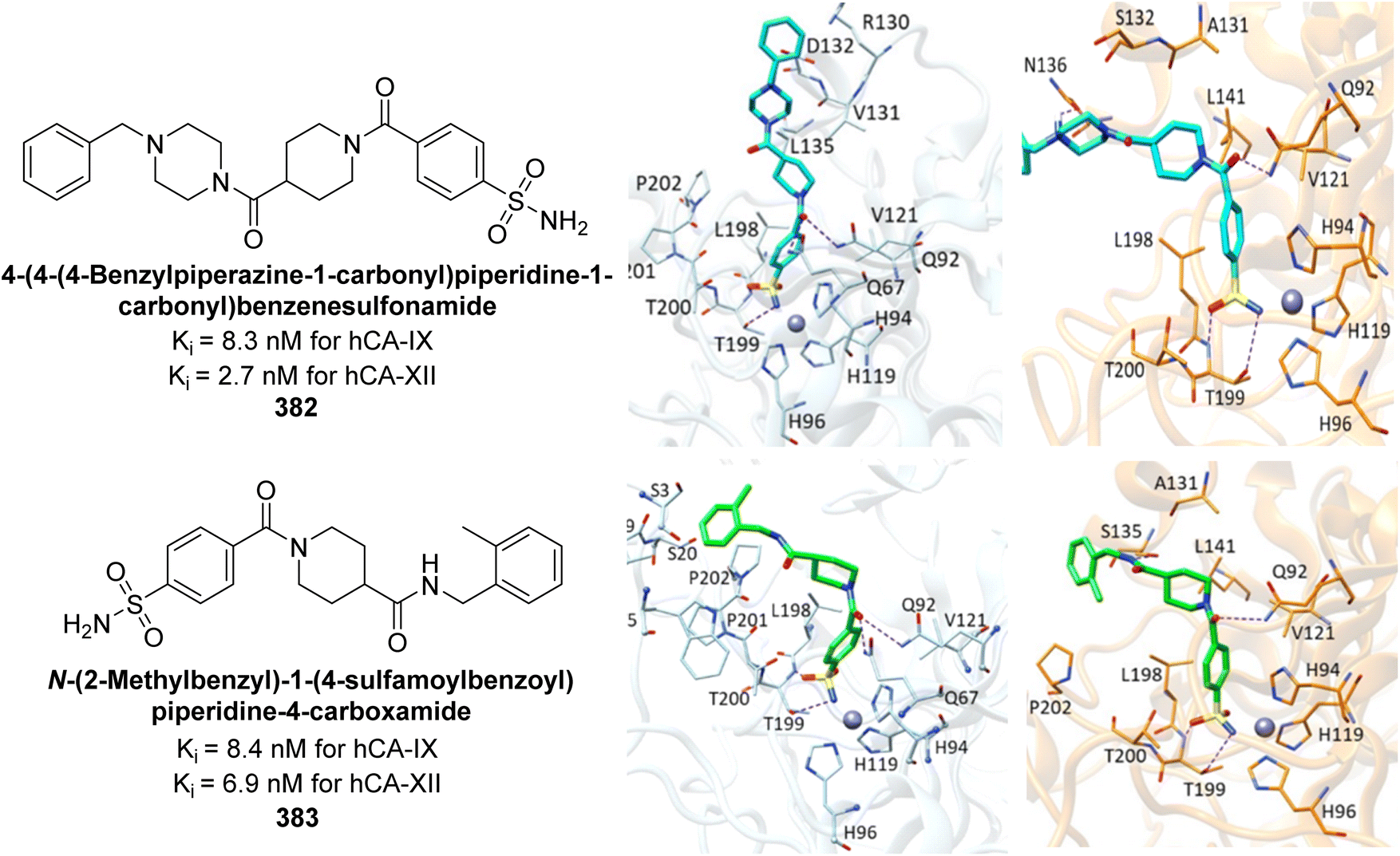 | ||
| Fig. 136 Chemical structures of compounds 382 and 383; their Ki values against hCA-IX and XII along with their docking images. | ||
Mert et al. (2024) designed and synthesized a series of novel pyrazole-dicarboxamides. The inhibition potency of the newly synthesized molecules against the hCA-I and II isoforms was evaluated, revealing Ki values ranging from 0.024 to 0.496 μM for hCA-I and 0.006 to 5.441 μM for hCA-II. Remarkably, compounds 384 and 386 exhibited nanomolar inhibition against hCA-II, demonstrating high selectivity for this isozyme. Molecular docking studies were conducted with the most active compounds, 385 and 386, alongside the reference inhibitor AAZ, to elucidate their binding mechanisms with hCA-I and II. These compounds showed superior interactions with the isozymes compared to AAZ (Fig. 137). Consequently, these sulfonamide derivatives, demonstrating superior enzyme inhibition capabilities compared to the reference compound AAZ, hold promise as precursor molecules for further studies aimed at enzyme inhibition.225
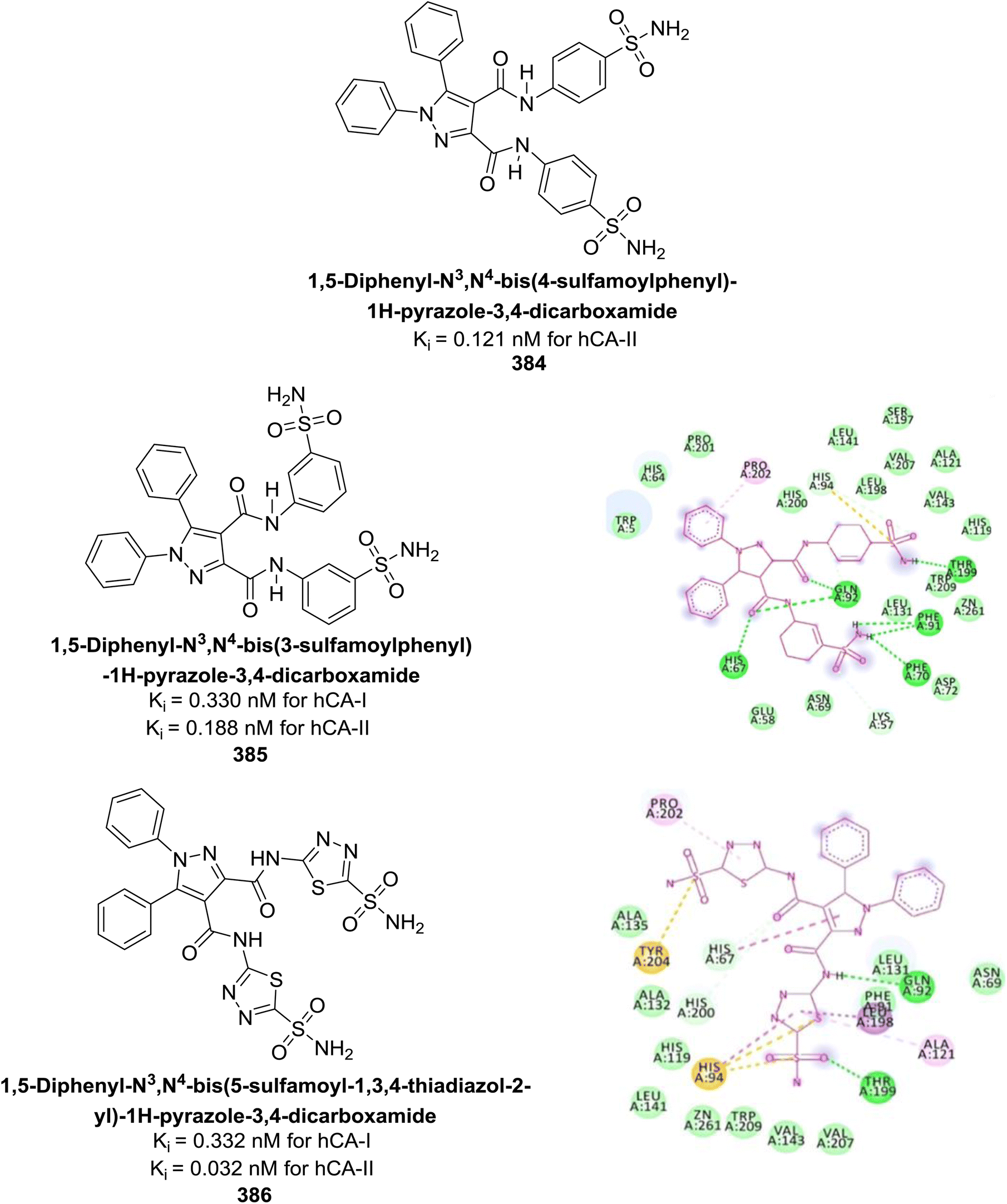 | ||
| Fig. 137 Chemical structures of compounds 384–386; their Ki values against hCA-I and II along with docking images of 385 and 386. | ||
Bagnoli et al. (2024) synthesized a diverse range of 3-selenylindoles through an eco-friendly method utilizing Oxone® as the oxidant with catalytic iodine. Additionally, several novel 3-selenylbenzenesulfonamide indoles were evaluated against hCAs. Particularly, 3-selenylbenzenesulfonamide indoles 387, 388 and 389 exhibited strong and selective inhibition of hCA-II, while compounds 390 and 391 displayed selective inhibition of hCA-IX (Fig. 138). These results suggest that 3-selenylbenzenesulfonamide indoles hold promise as lead candidates for the development of more potent and selective hCA inhibitors.226
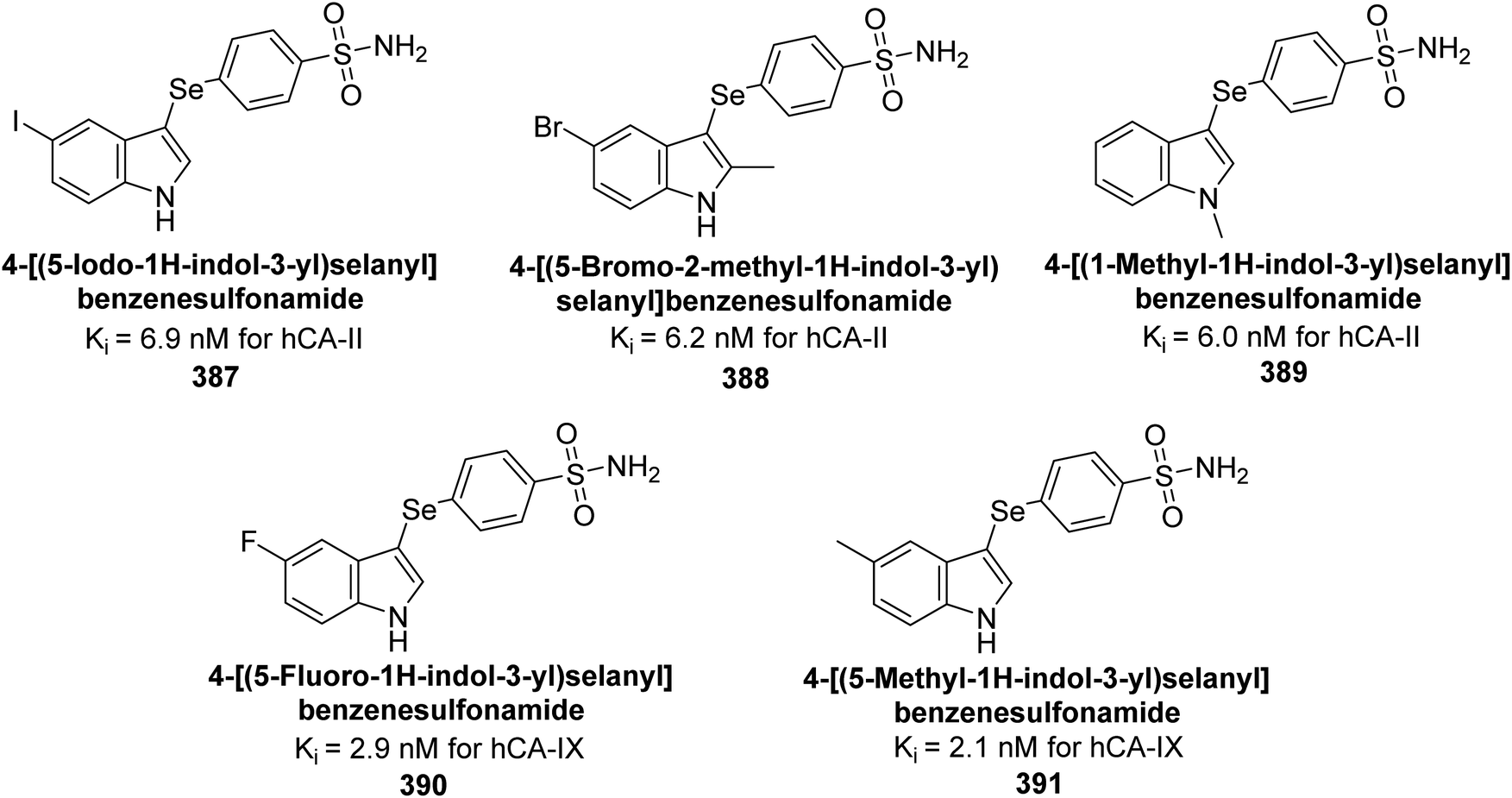 | ||
| Fig. 138 Chemical structures of compounds 387–391; their Ki values against hCA-II and IX. | ||
Durgun et al. (2024) synthesized a novel pyrazole carboxamide derivatives and evaluated their inhibitory effects on hCA-I and II. The compounds exhibited potential as inhibitors, with Ki values ranging from 10.69 nM to 70.87 nM for hCA-I, and from 20.01 nM to 56.63 nM for hCA-II. Especially, compounds 392 and 393 showed competitive inhibition of hCA-I, while the other compounds demonstrated non-competitive inhibition. Additionally, compound 392 displayed competitive inhibition against hCA-II, forming an H-bond with the Thr200 residue and a water molecule within the active site of hCA-II, while Phe131 interacted through π–π stacking (Fig. 139). These docking scores and binding poses suggest that the compounds have significant potential as inhibitors of hCAs, and may serve as alternative therapeutic agents for the treatment of glaucoma.227
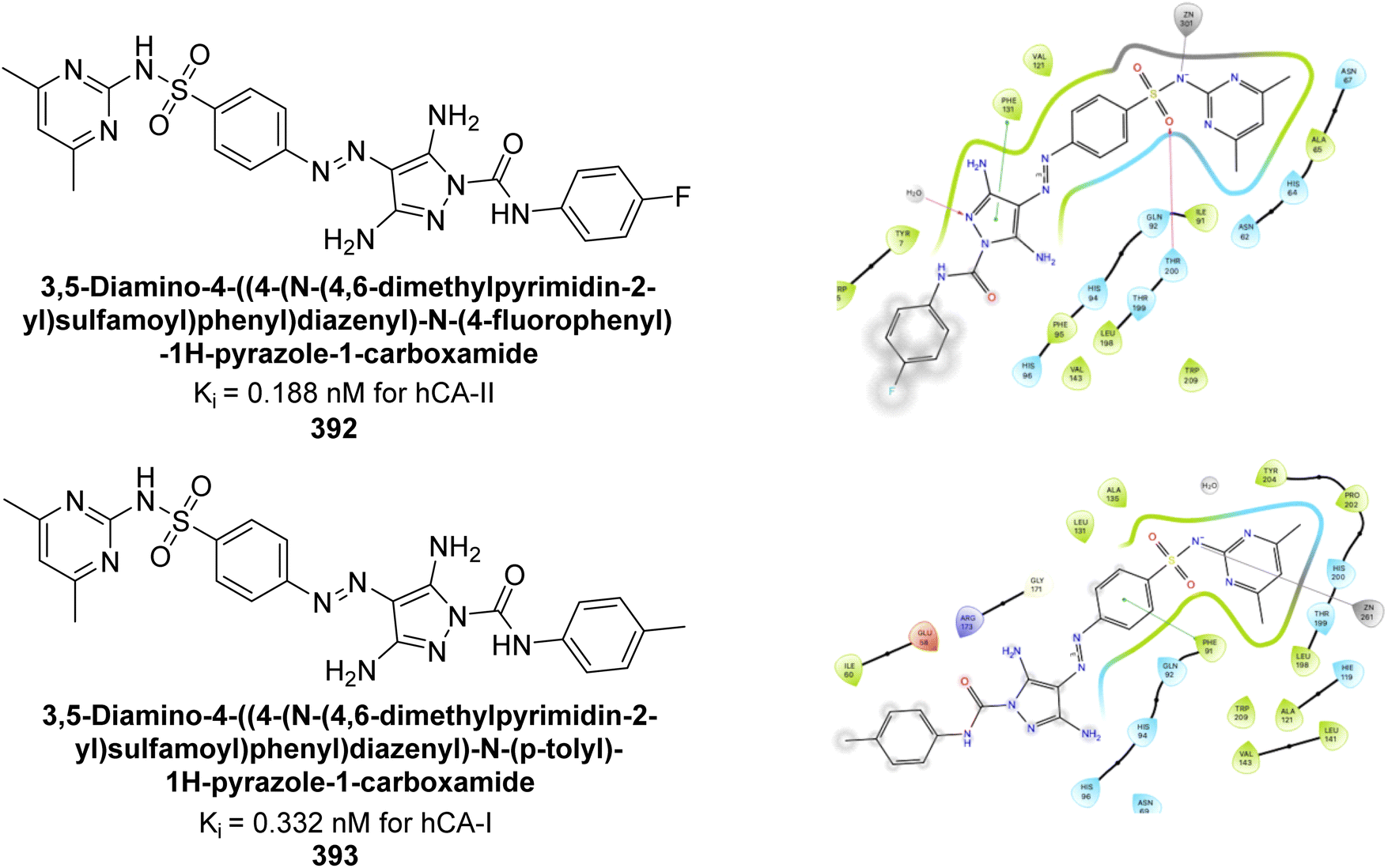 | ||
| Fig. 139 Chemical structures of compounds 392 and 393; their Ki values against hCA-I and II along with their docking images. | ||
Angeli et al. (2024) synthesized a novel phthalazine derivatives and assessed for their inhibitory activity against hCA-II and IX. Markedly, several compounds displayed high selectivity, with a selectivity index reaching up to 13.8. The presence of a benzamide group at the 4′ position of the phthalazine ring was found to enhance hCA-IX inhibitory activity, similar to its effect on hCA-II inhibition. Replacing the benzamide group with a 4-hydroxybenzene moiety resulted in compound 394, which was slightly less active than compound 395. Among the compounds tested, 394 exhibited notable activity with a Ki value of 4.6 nM. Molecular docking studies were conducted to elucidate the interactions of these compounds within the active sites of hCA-II and IX isoforms (Fig. 140). These findings suggest that the developed phthalazine derivatives may offer valuable alternatives in anticancer therapy by addressing key challenges such as resistance development and lack of selectivity, which often lead to acute or chronic toxicity.228
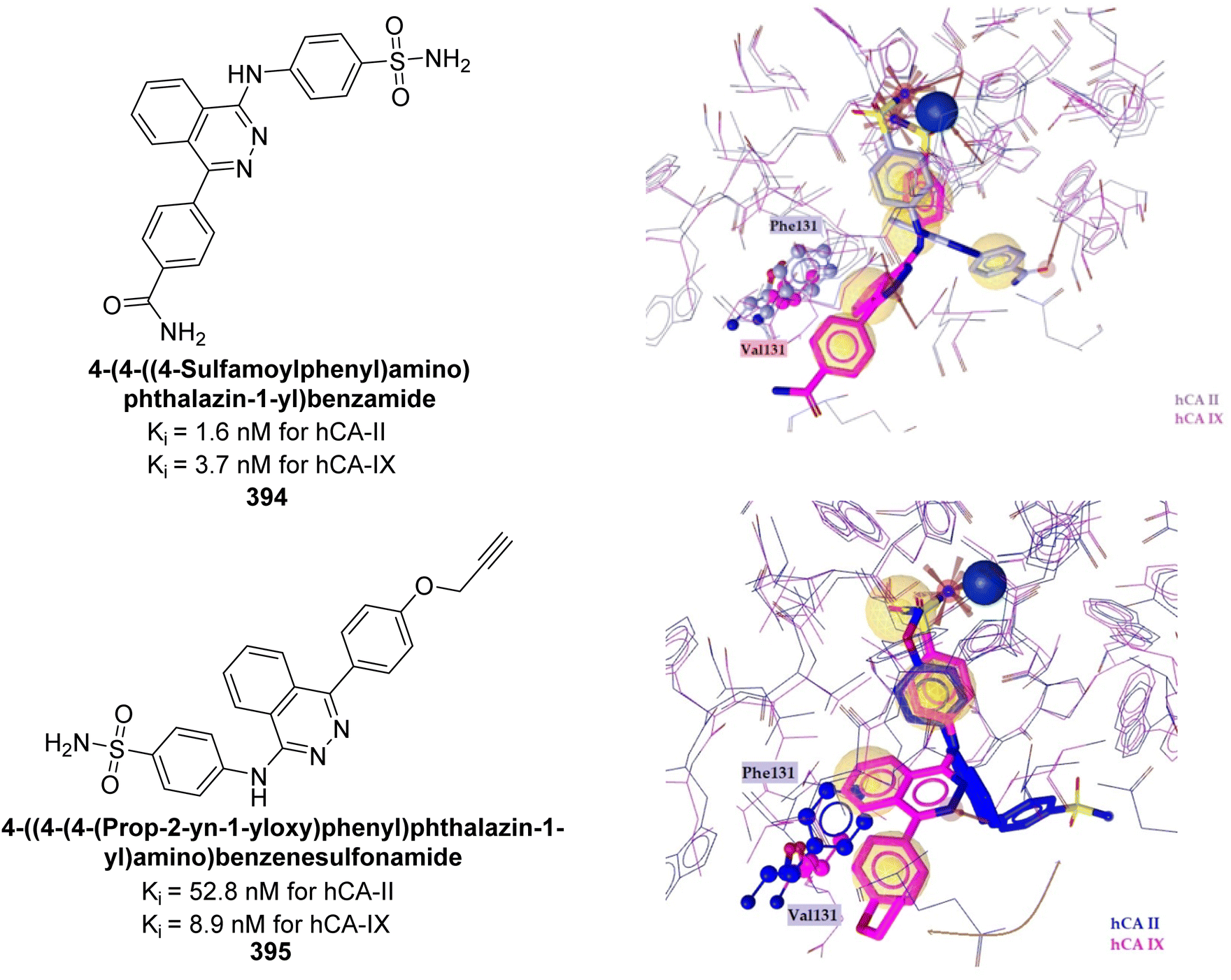 | ||
| Fig. 140 Chemical structures of compounds 394 and 395; their Ki values against hCA-II and IX along with their docking images. | ||
Aslan et al. (2024) designed and synthesized a series of Biginelli products incorporating the benzenesulfonamide moiety known for inhibiting CA enzymes. The introduction of a carboxamide group at the 5-position of the 6-phenyl-tetrahydropyrimidine ring, as in compound 396, resulted in significant and selective inhibition of hCA-IX and XII isoforms, with Ki values of 6.6 and 8.9 nM, respectively. Remarkably, compound 397 exhibited 1.7-fold higher potency against hCA-XII compared to hCA-IX, with Ki values of 5.3 and 8.9 nM, respectively (Fig. 141).229
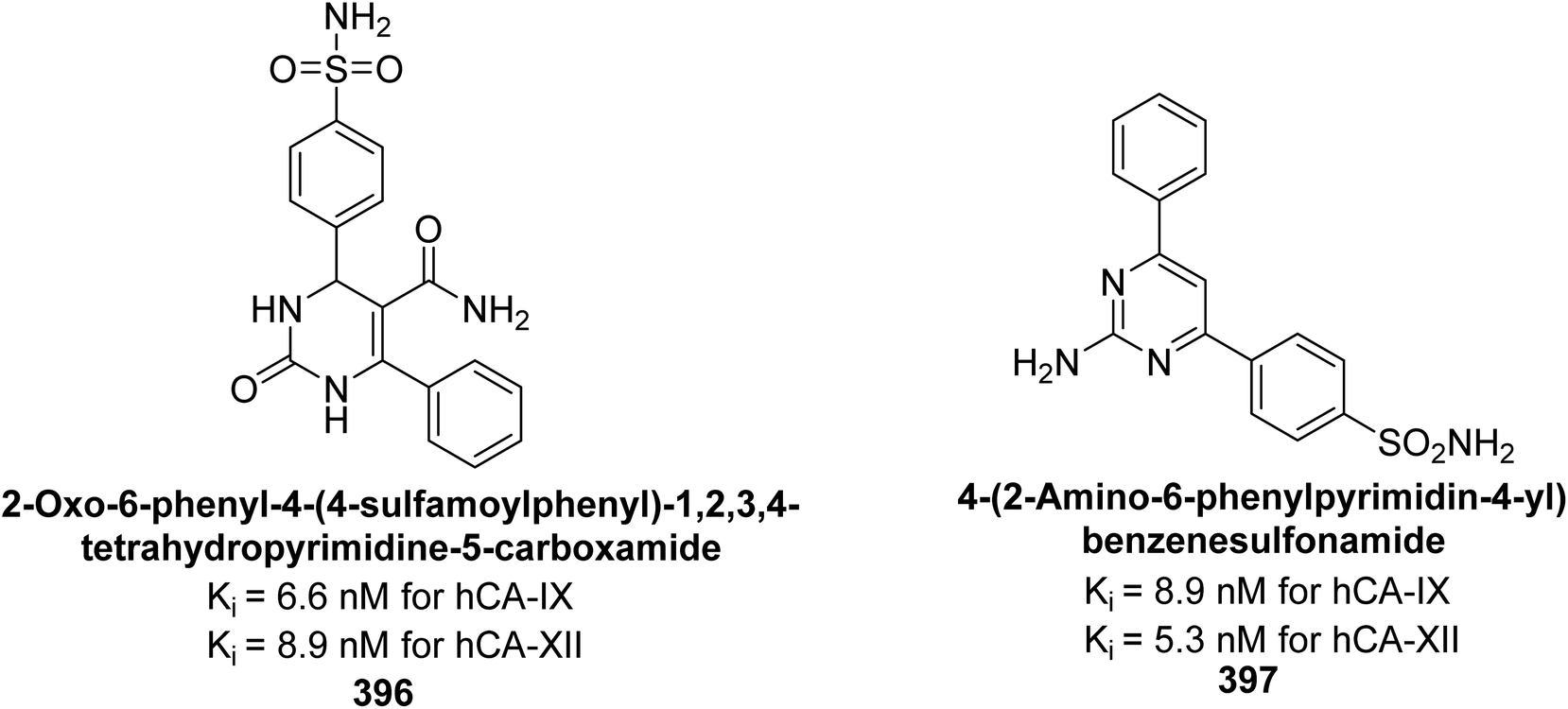 | ||
| Fig. 141 Chemical structures of compounds 396 and 397; their Ki values against hCA-IX and XII. | ||
Nafie et al. (2024) designed and synthesized a series of mono- and bis-1,2,3-triazoles and evaluated against hCA-IX. Compounds 398, 399 and 400 demonstrated potent inhibition of hCA-IX, with IC50 values of 0.113, 0.134, and 0.214 μM. Molecular modeling studies revealed that compound 398, in particular, shows a strong binding affinity for hCA-IX, which is a key target in PC-3 prostate cancer cells (Fig. 142). Future studies should investigate the anticancer activity of compound 398 in vivo to assess its efficacy and safety in experimental animal models, with the aim of clinical development.230
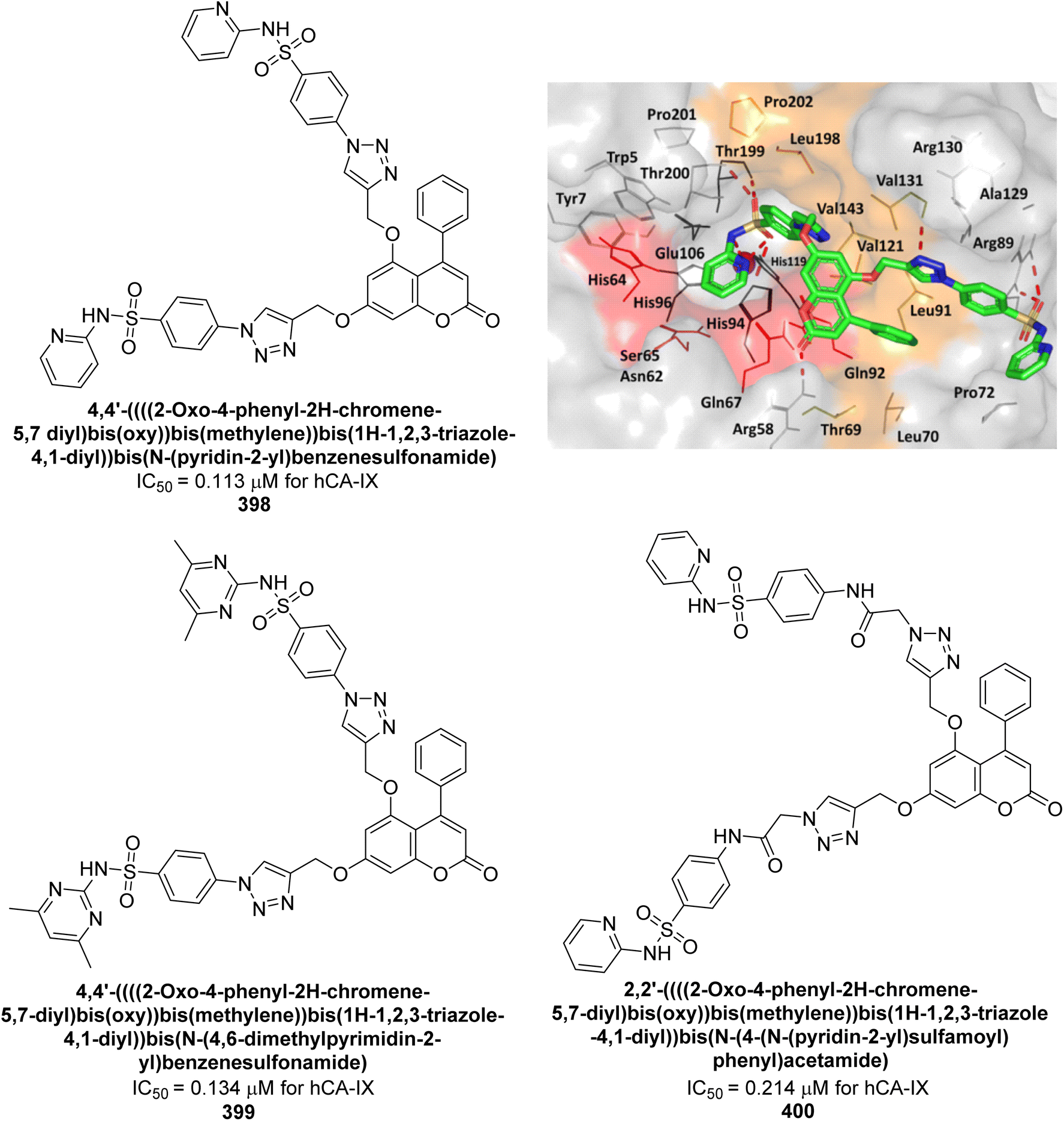 | ||
| Fig. 142 Chemical structures of compounds 398–400; their IC50 values against hCA-IX and docking image of 398. | ||
Kasımoğulları et al. (2024) synthesized a series of pyrazole-carboxamides and evaluated the inhibitory effects of these compounds on hCA-I and II, revealing Ki values ranging from 0.063 to 3.368 μM for hCA-I and from 0.007 to 4.235 μM for hCA-II. Compound 401 (IC50 = 0.420 μM for hCA-I & 0.310 μM for hCA-II) displayed non-competitive behavior. Compound 401, which contains a primary –SO2NH2 group in the para position, is the most effective esterase inhibitor for both hCA-I and II. The compound 401 exhibited superior binding interactions relative to AAZ (Fig. 143).231
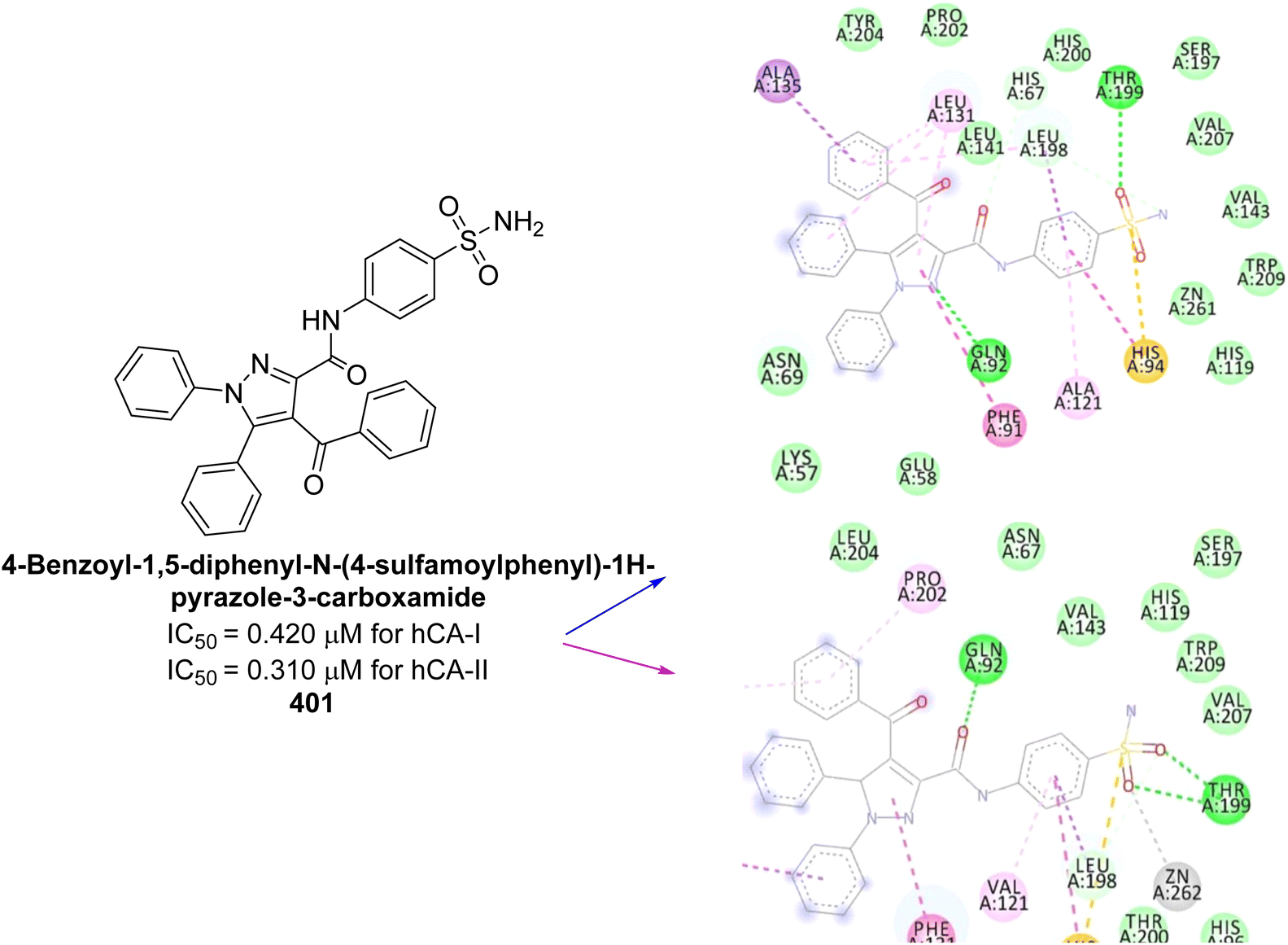 | ||
| Fig. 143 Chemical structure of compound 401; its IC50 values against hCA-I and II, and its docking images. | ||
6. Comparative analysis of the different heterocyclic scaffolds as CAIs against various isoforms based on IC50/Ki values
The tabular data presents a comparative analysis of various heterocyclic scaffolds as CAIs based on their IC50 and Ki values against different CA isoforms (Table 14). This includes scaffolds such as pyrimidine, triazole, thiazole, phthalazine, imidazole, oxadiazole, indole, isatin, chalcone, coumarin, rhodanine, quinolines, and sulfonamides. Each scaffold's inhibitory performance is evaluated, showcasing its potency and selectivity towards different CA isoforms. The data serves to highlight the SAR and potential of each scaffold in CA inhibition for therapeutic applications.| Compound no. | Chemical structure and IUPAC name | Isoform targeted, IC50 (μM)/Ki values (nM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| hCA-I | hCA-II | hCA-IV | hCA-VII | hCA-IX | hCA-XII | bCA-II | |||
| 1 |  |
346 | 61.4 | — | — | 24.8 | 34.2 | — | 84 |
| 2 |  |
438 | 58.5 | — | — | 23.1 | 27.8 | — | 84 |
| 3 |  |
541 | 76.3 | — | — | 48.6 | 39.1 | — | 84 |
| 4 | 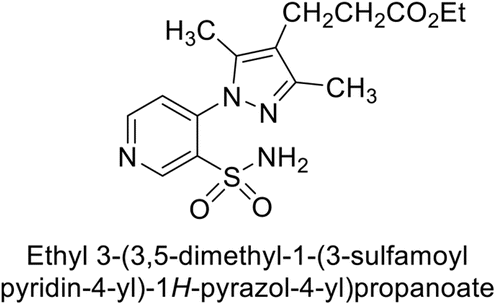 |
397 | 134 | — | — | 19.5 | 22.6 | — | 84 |
| 5 | 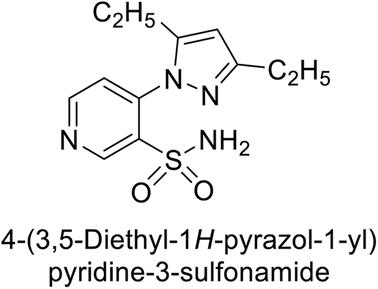 |
278 | 87.1 | — | — | 31.7 | 16.8 | — | 84 |
| 6 | 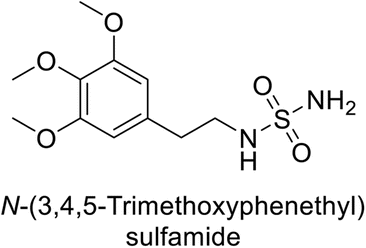 |
1.82 | 2.03 | — | — | 0.221 | 1.52 | — | 85 |
| 7 | 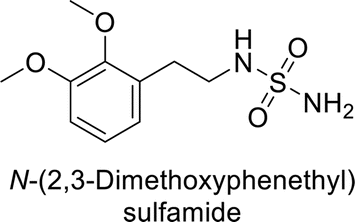 |
0.25 | 2.95 | — | — | >50 | 0.047 | — | 85 |
| 8 | 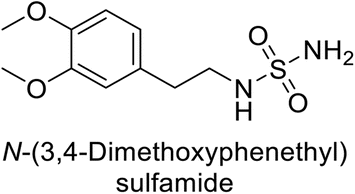 |
0.28 | 2.40 | — | — | 0.092 | 0.044 | — | 85 |
| 9 | 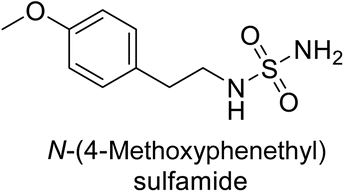 |
0.061 | 1.47 | — | — | 0.376 | 0.021 | — | 85 |
| 10 | 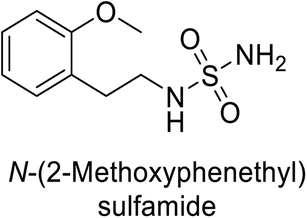 |
0.25 | 2.04 | — | — | 0.041 | 0.416 | — | 85 |
| 11 | 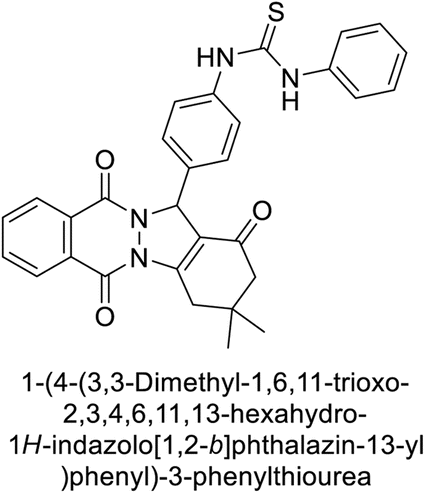 |
IC50 = 6.40 | IC50 = 6.13 | — | — | — | — | — | 86 |
| 12 | 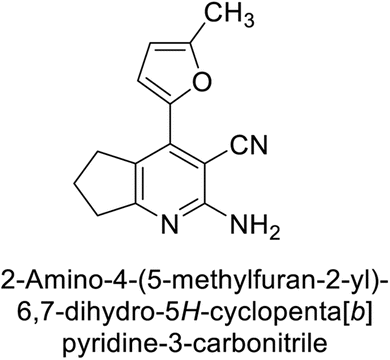 |
23.8 | 41 | — | — | — | — | — | 87 |
| 13 | 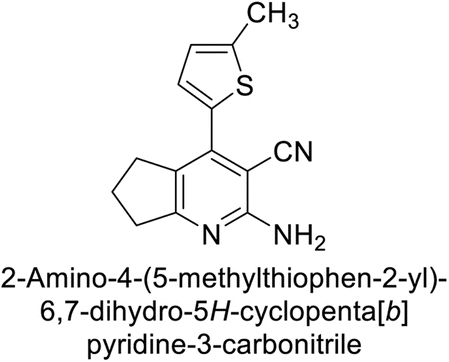 |
31 | 182 | — | — | — | — | — | 87 |
| 14 |  |
— | 23 | — | — | — | 124 | — | 88 |
| 15 |  |
— | 63 | — | — | — | 24 | — | 88 |
| 16 | 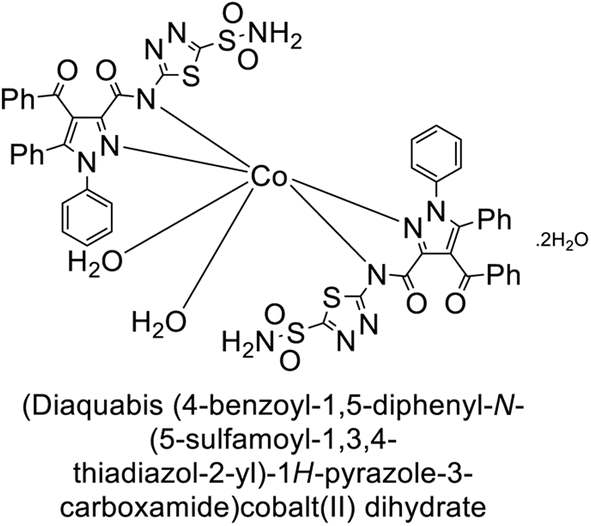 |
IC50 = 0.058 | IC50 = 0.110 | — | — | — | — | — | 89 |
| 17 | 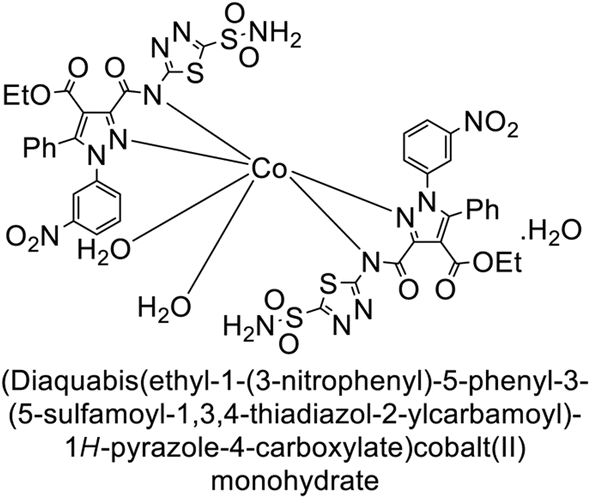 |
IC50 = 0.297 | IC50 = 0.052 | — | — | — | — | — | 89 |
| 18 | 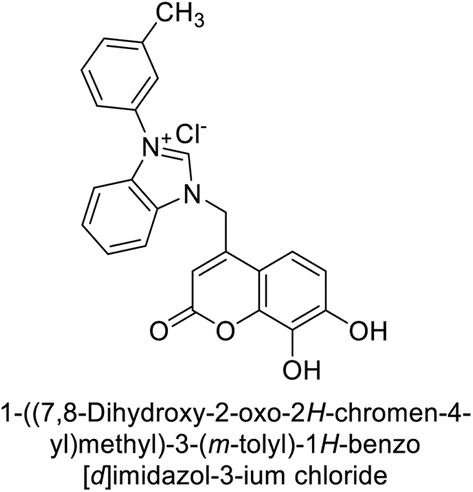 |
IC50 = 22.1 | IC50 = 28.9 | — | — | — | — | — | 90 |
| 19 | 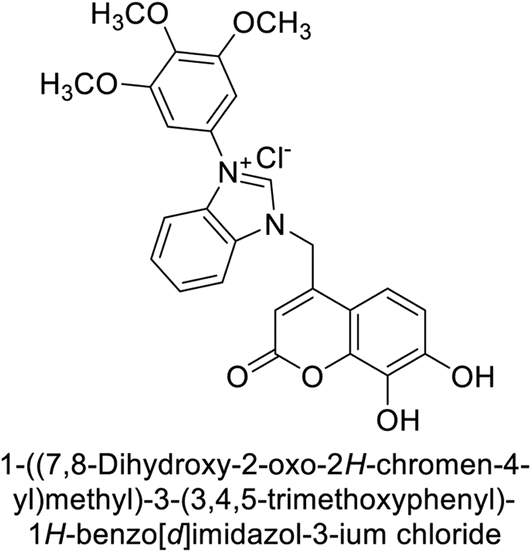 |
IC50 = 33.1 | IC50 = 23.3 | — | — | — | — | — | 90 |
| 20 | 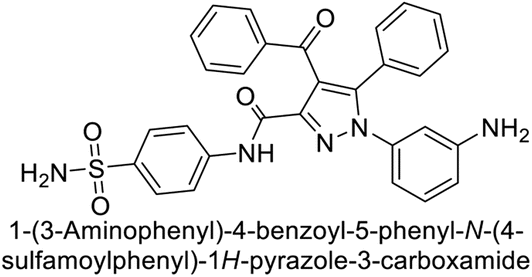 |
0.108 | 0.055 | — | — | — | — | — | 91 |
| 21 | 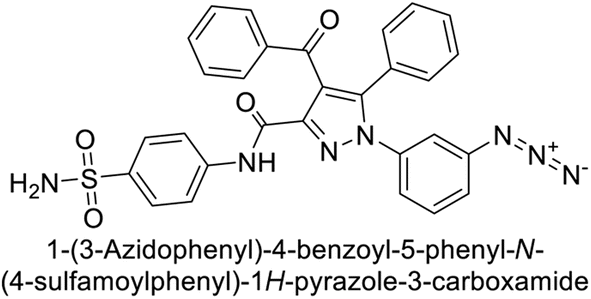 |
0.129 | 0.064 | — | — | — | — | — | 91 |
| 22 | 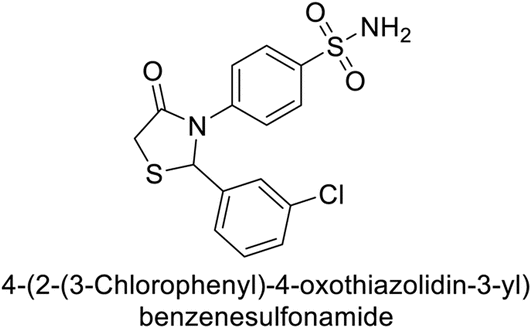 |
— | — | — | — | 2.2 | — | — | 92 |
| 23 | 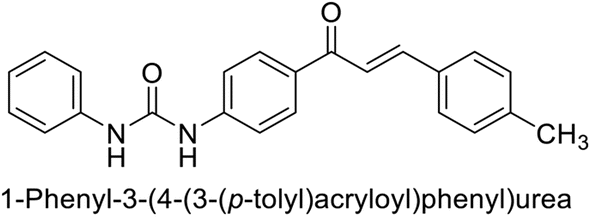 |
IC50 = 25.4 | IC50 = 27.2 | — | — | — | — | — | 93 |
| 24 | 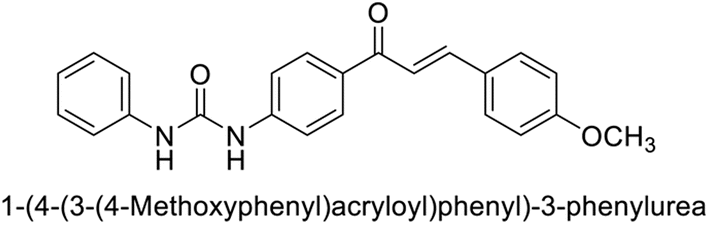 |
IC50 = 23.1 | IC50 = 19.4 | — | — | — | — | — | 93 |
| 25 | 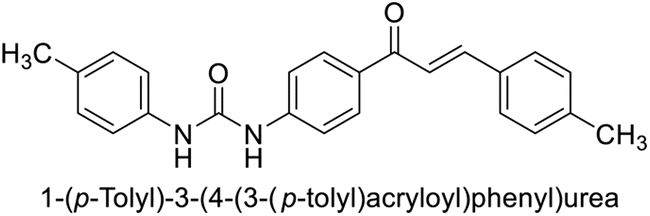 |
IC50 = 27.9 | IC50 = 14.4 | — | — | — | — | — | 93 |
| 26 |  |
— | — | — | — | 0.48 | — | — | 94 |
| 27 | 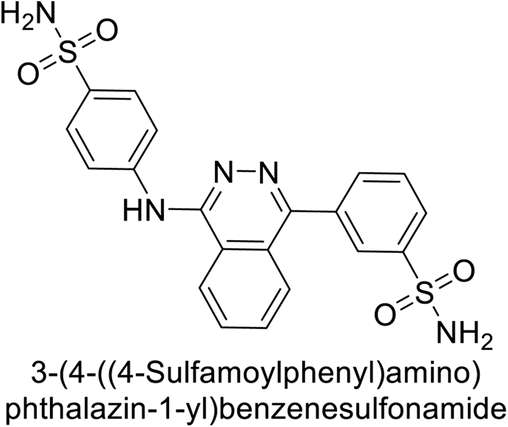 |
— | — | — | — | 18.5 | — | — | 94 |
| 28 | 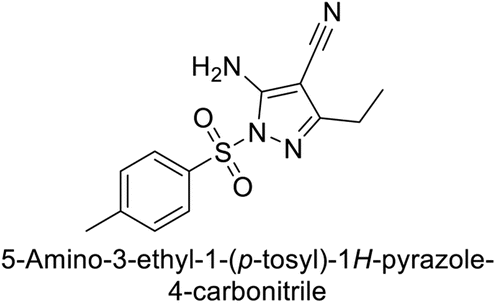 |
— | — | — | — | 0.47 | 0.15 | — | 95 |
| 29 | 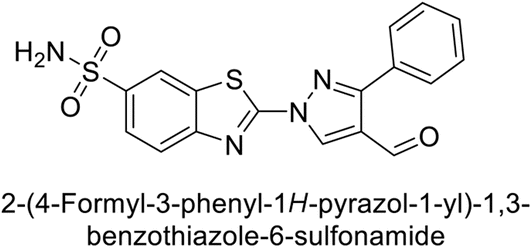 |
8806 | 10.9 | — | — | 1747 | 214 | — | 96 |
| 30 | 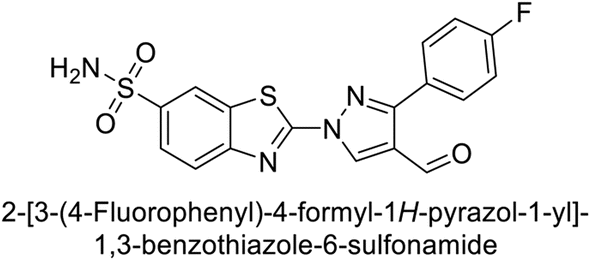 |
15690 |
10.4 | — | — | 1022 | >10000 |
— | 96 |
| 31 | 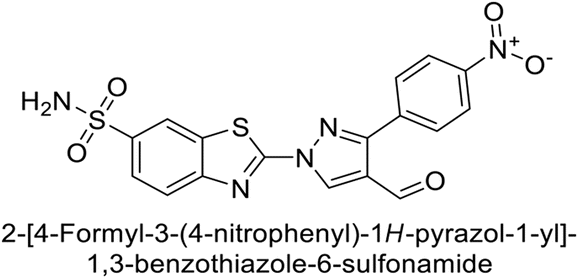 |
2331 | 3.8 | — | — | 2109 | 74.5 | — | 96 |
| 32 | 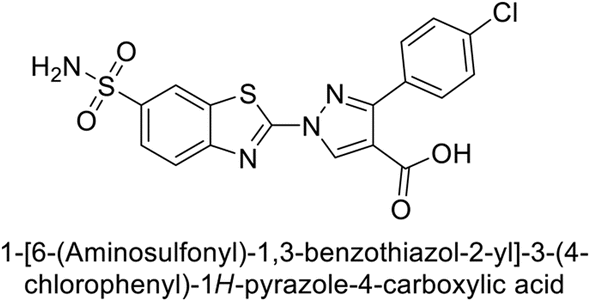 |
407 | 7.3 | — | — | 2.8 | 5.5 | — | 96 |
| 33 | 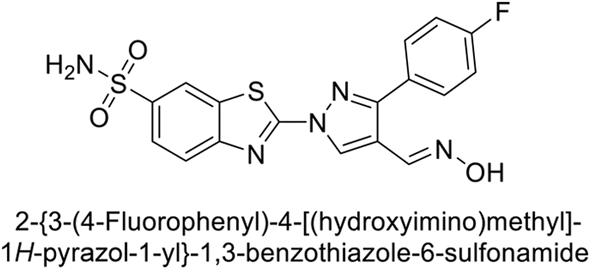 |
7161 | 8.5 | — | — | 224 | 83.8 | — | 96 |
| 34 | 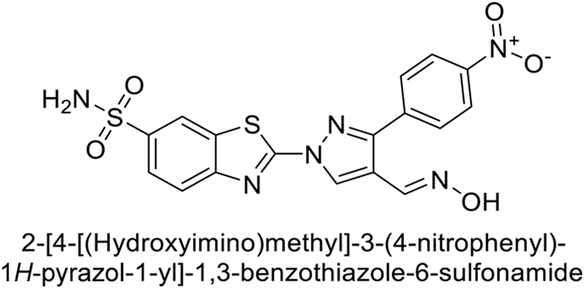 |
23102 |
11.6 | — | — | 119 | 96.8 | — | 96 |
| 35 | 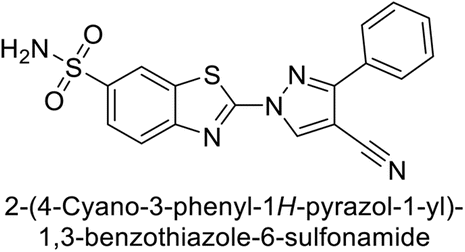 |
9819 | 139 | — | — | 81.6 | 6.6 | — | 96 |
| 36 | 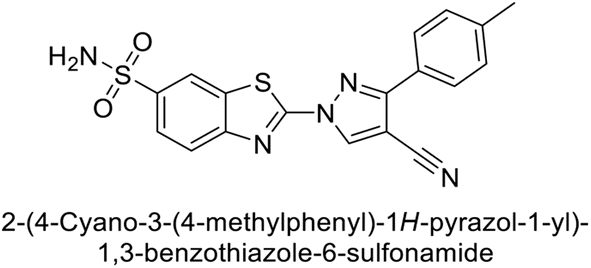 |
>50000 |
48.1 | — | — | 186 | 7.2 | — | 96 |
| 37 | 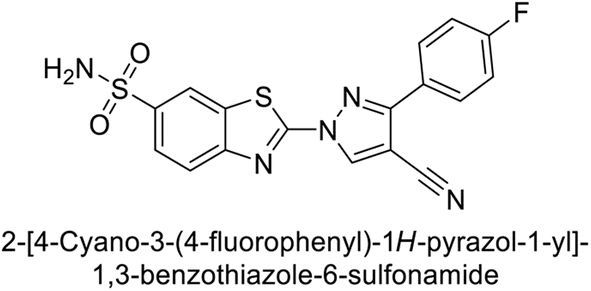 |
12108 |
27 | — | — | 739 | 7.4 | — | 96 |
| 38 | 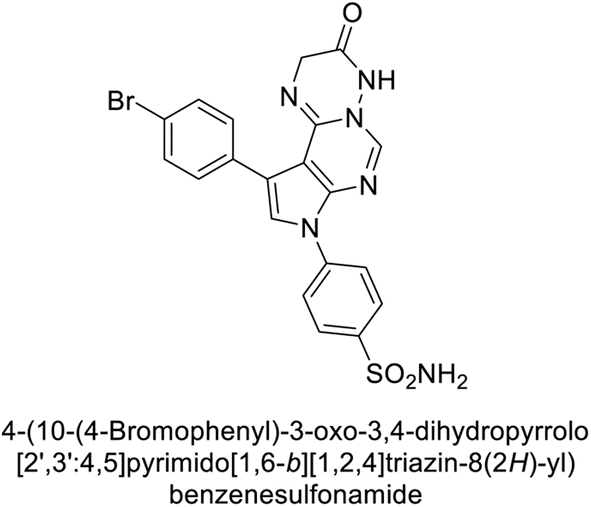 |
94.1 | 4.9 | — | — | 11.7 | 0.76 | — | 97 |
| 39 | 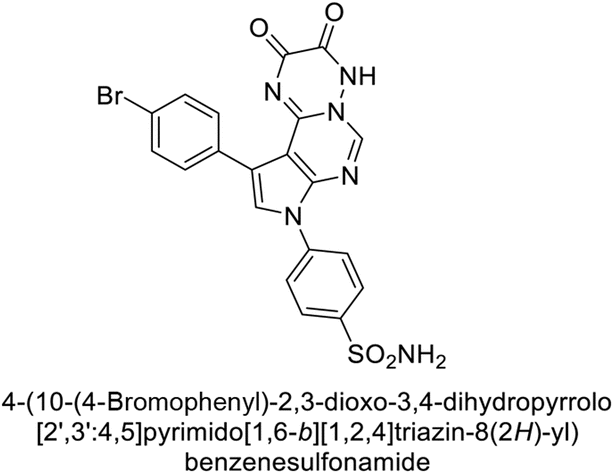 |
69.1 | 64.3 | — | — | 325 | 0.912 | — | 97 |
| 40 | 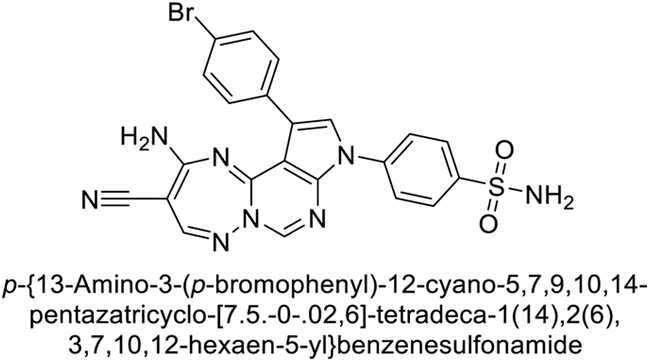 |
80.4 | 5.2 | — | — | 3.1 | 0.9 | — | 97 |
| 41 | 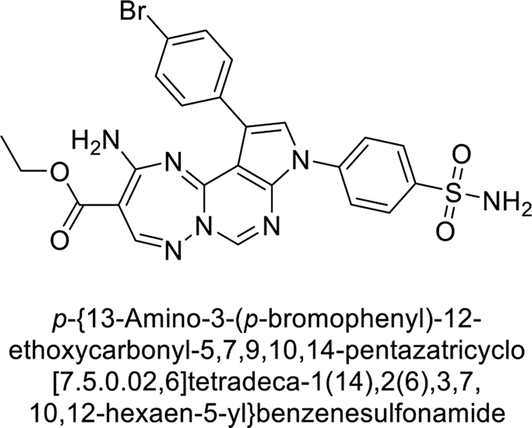 |
126 | 7.3 | — | — | 3 | 0.83 | — | 97 |
| 42 | 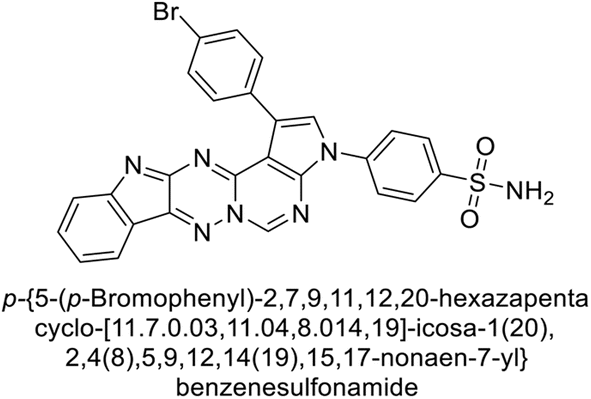 |
498 | 8.5 | — | — | 2.7 | 0.72 | — | 97 |
| 43 | 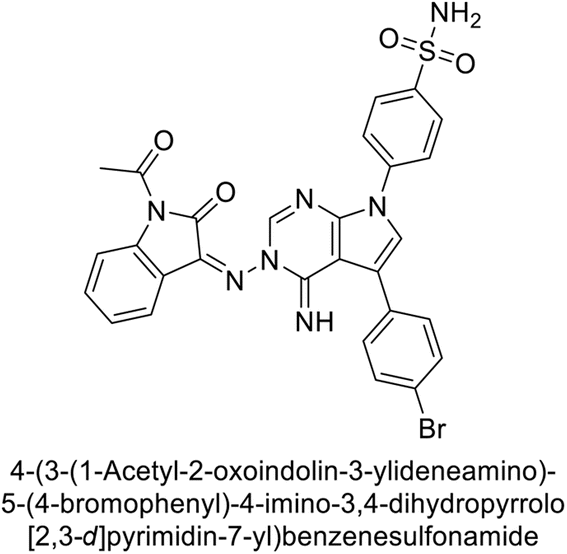 |
25.1 | 5.6 | — | — | 3.1 | 0.81 | — | 97 |
| 44 | 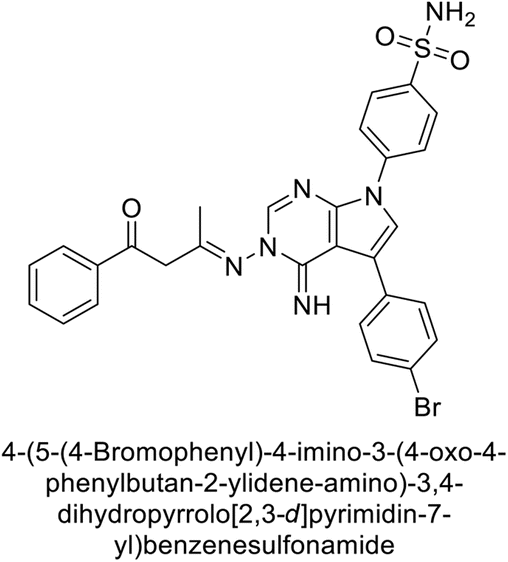 |
16.3 | 3.9 | — | — | 2.8 | 0.94 | — | 97 |
| 45 |  |
270 | 8 | — | — | 43.8 | 7.9 | — | 98 |
| 46 | 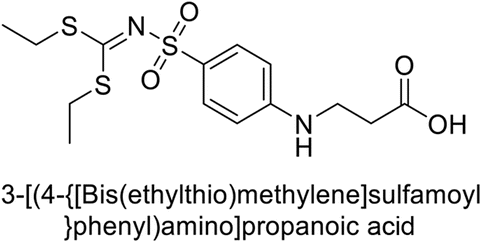 |
— | 40 | — | — | — | >200 | — | 99 |
| 47 |  |
— | 4.0 | — | — | — | 1.85 | — | 99 |
| 48 |  |
923 | 61.3 | — | — | 5.9 | 4.3 | — | 100 |
| 49 |  |
887 | 60.1 | — | — | 6.2 | 4.0 | — | 100 |
| 50 | 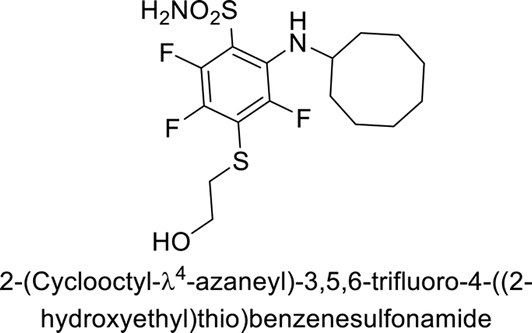 |
— | — | — | — | — | 1.1 | — | 101 |
| 51 | 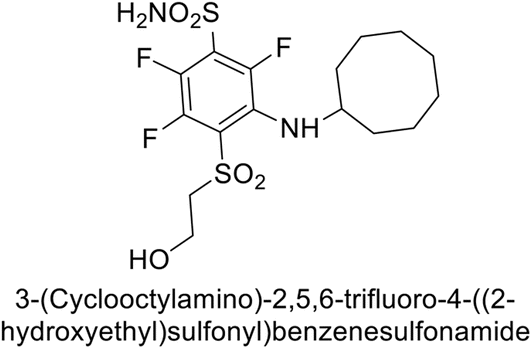 |
— | — | — | — | — | 0.05 | — | 101 |
| 52 |  |
5.3 | 5.0 | — | — | 8.3 | 7.2 | — | 102 |
| 53 |  |
6.6 | 5.1 | — | — | 5.8 | 6.5 | — | 102 |
| 54 | 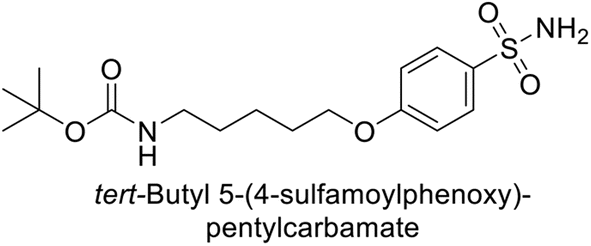 |
7.9 | 5.5 | — | — | 7.1 | 5.7 | — | 102 |
| 55 | 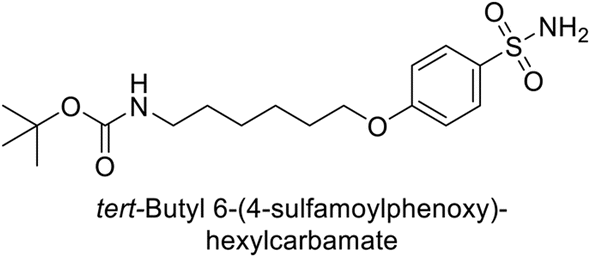 |
6.1 | 5.2 | — | — | 6.9 | 6.4 | — | 102 |
| 56 | 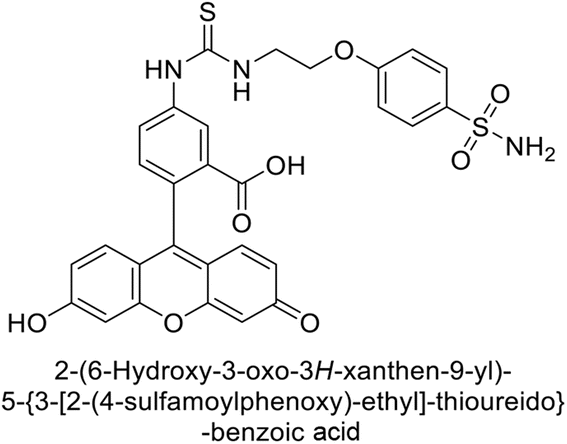 |
18.0 | 5.0 | — | — | 8.5 | 8.6 | — | 102 |
| 57 | 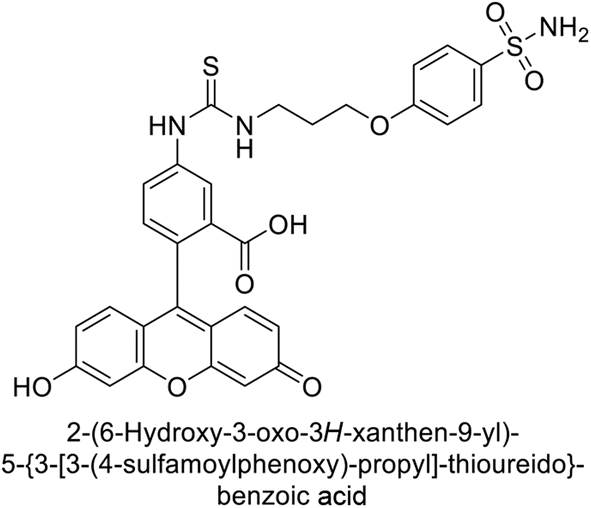 |
17.1 | 4.6 | — | — | 7.2 | 5.7 | — | 102 |
| 58 | 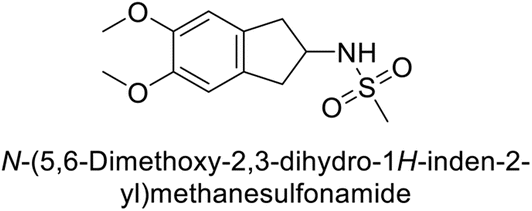 |
46 | 372 | — | — | — | — | — | 103 |
| 59 | 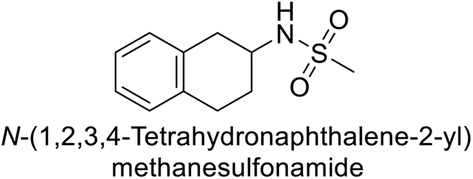 |
228 | 94 | — | — | — | — | — | 103 |
| 60 | 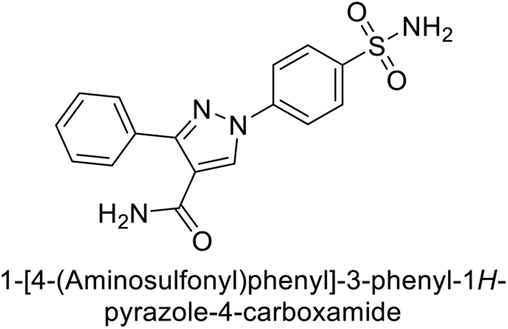 |
— | — | — | — | 4.3 | 2.7 | — | 104 |
| 61 | 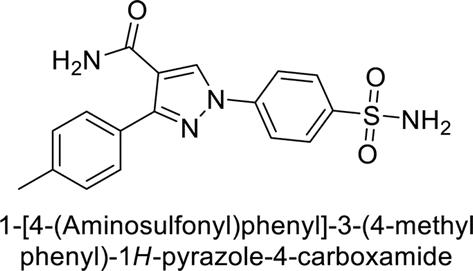 |
— | — | — | — | 4.2 | 3.1 | — | 104 |
| 62 | 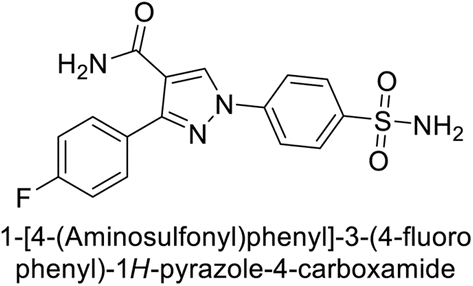 |
— | — | — | — | 5.0 | 4.5 | — | 104 |
| 63 | 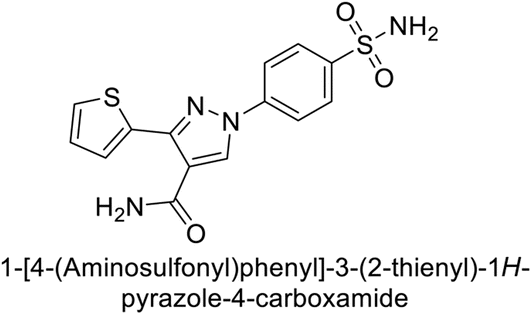 |
— | — | — | — | 5.2 | 4.7 | — | 104 |
| 64 | 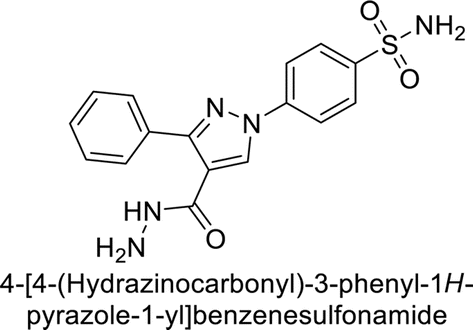 |
— | — | — | — | 2.3 | 1.9 | — | 104 |
| 65 | 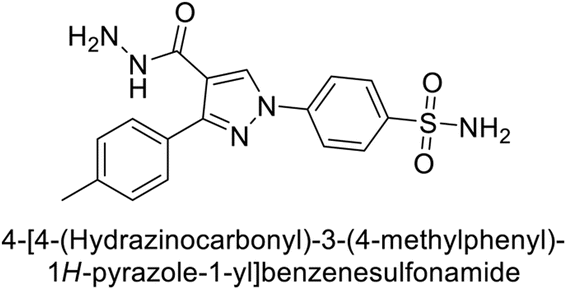 |
— | — | — | — | 2.8 | 12.9 | — | 104 |
| 66 | 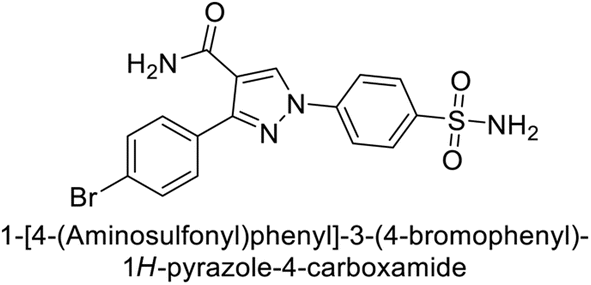 |
— | — | — | — | 2.0 | 10.4 | — | 104 |
| 67 | 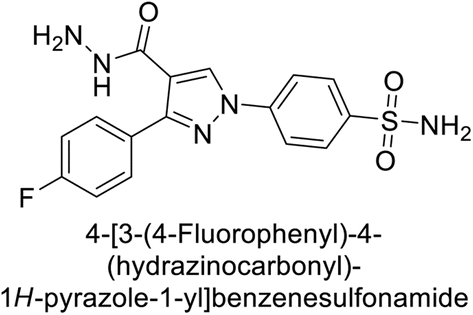 |
— | — | — | — | 2.1 | 3.9 | — | 104 |
| 68 | 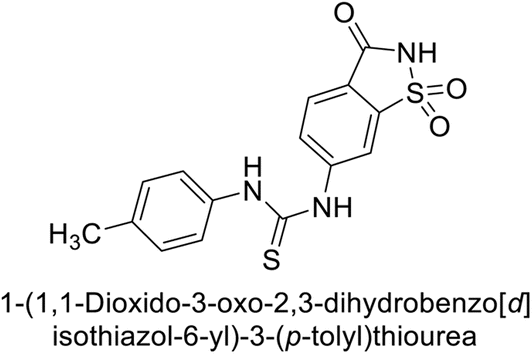 |
IC50 = 13.67 | IC50 = 11.54 | — | — | — | — | — | 105 |
| 69 | 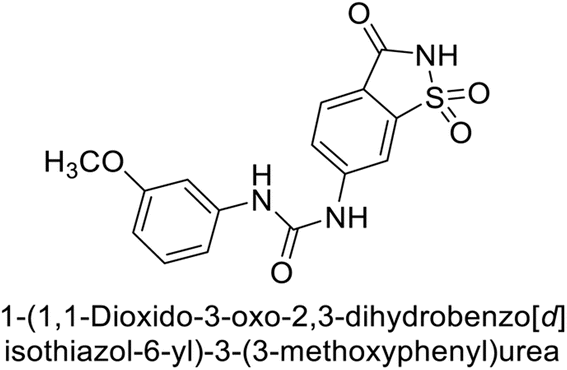 |
IC50 = 41.67 | IC50 = 6.54 | — | — | — | — | — | 105 |
| 70 | 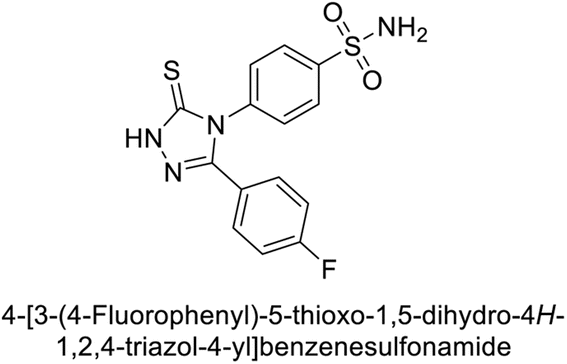 |
— | — | — | — | 2.8 | 4.9 | — | 106 |
| 71 | 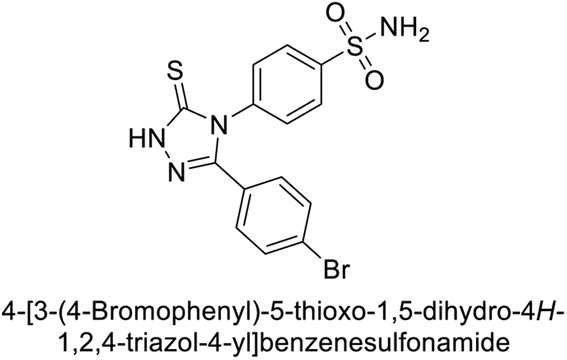 |
— | — | — | — | 7.6 | 1.3 | — | 106 |
| 72 | 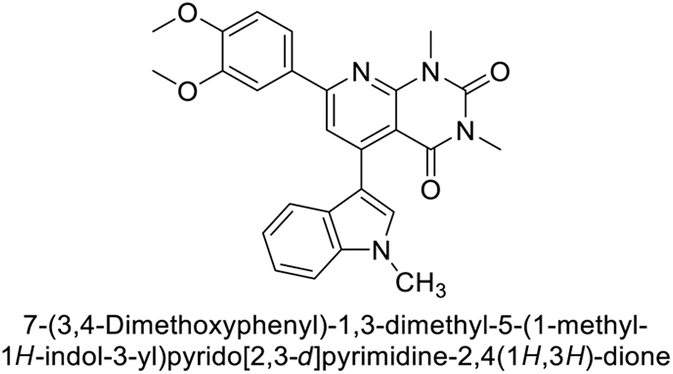 |
IC50 = 6.79 | IC50 = 8.06 | — | — | — | — | — | 107 |
| 73 | 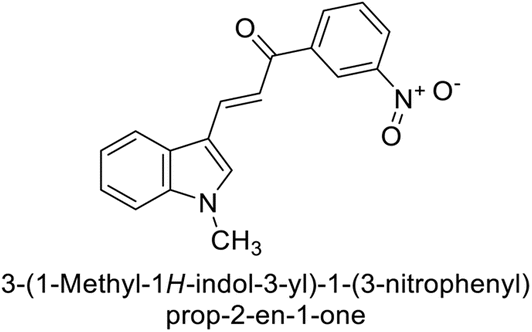 |
IC50 = 8.38 | IC50 = 7.22 | — | — | — | — | — | 107 |
| 74 | 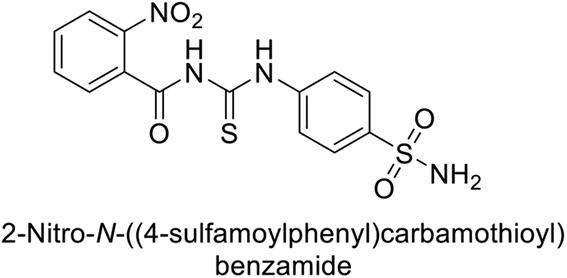 |
— | — | — | — | — | — | IC50 = 0.261 | 108 |
| 75 | 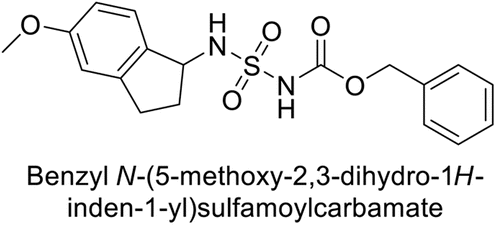 |
153 | 306 | — | — | — | — | — | 109 |
| 76 |  |
262 | 117 | — | — | — | — | — | 109 |
| 77 | 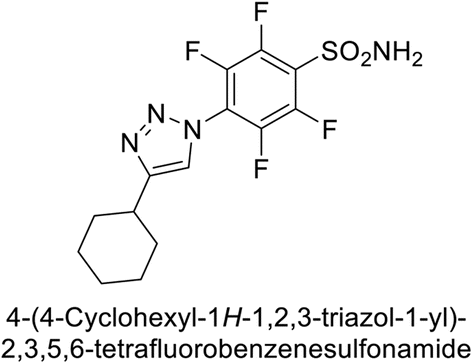 |
— | — | — | — | 1.5 | 0.8 | — | 110 |
| 78 | 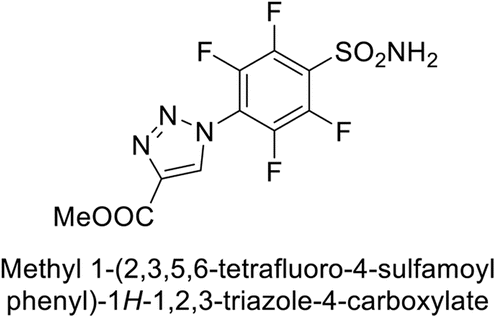 |
— | — | — | — | 115 | 58.9 | — | 110 |
| 79 | 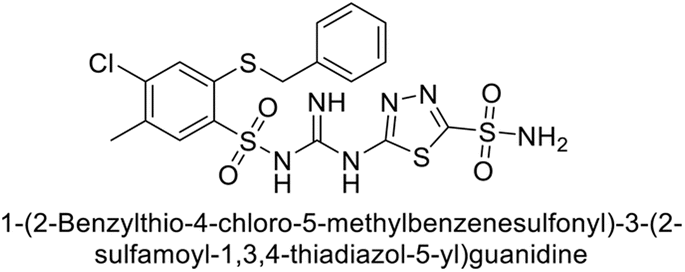 |
— | — | — | — | 4.7 | 0.96 | — | 111 |
| 80 |  |
IC50 = 0.05 | — | — | — | — | — | — | 112 |
| 81 |  |
IC50 = 2.12 | IC50 = 6.41 | — | — | — | — | — | 113 |
| 82 | 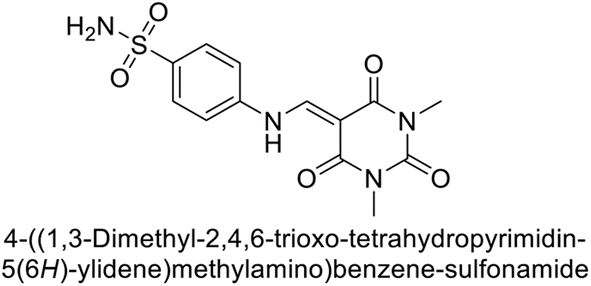 |
IC50 = 5.20 | IC50 = 2.52 | — | — | — | — | — | 113 |
| 83 | 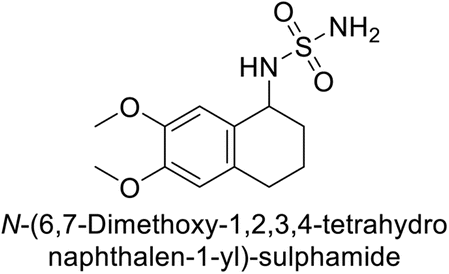 |
0.71 | 0.69 | — | — | — | — | — | 114 |
| 84 |  |
1.56 | 0.39 | — | — | — | — | — | 114 |
| 85 | 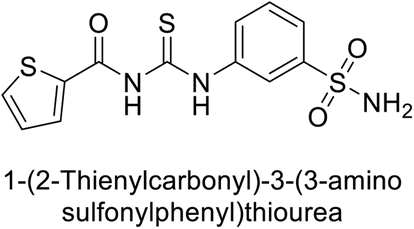 |
— | — | — | — | — | — | IC50 = 0.052 | 115 |
| 86 | 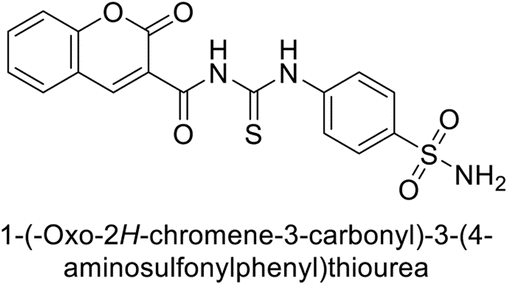 |
— | — | — | — | — | — | IC50 = 0.011 | 115 |
| 87 |  |
718 | 8.7 | — | — | 2.8 | 6.3 | — | 116 |
| 88 |  |
3025 | 10.5 | — | — | 4.8 | 2.7 | — | 116 |
| 89 |  |
2340 | 44 | — | — | 7.0 | 2.8 | — | 116 |
| 90 | 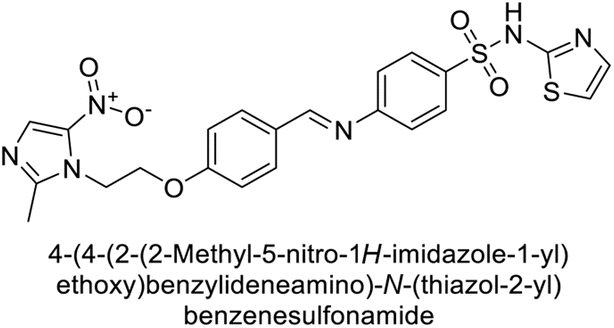 |
— | 16 | — | — | 93 | — | — | 117 |
| 91 | 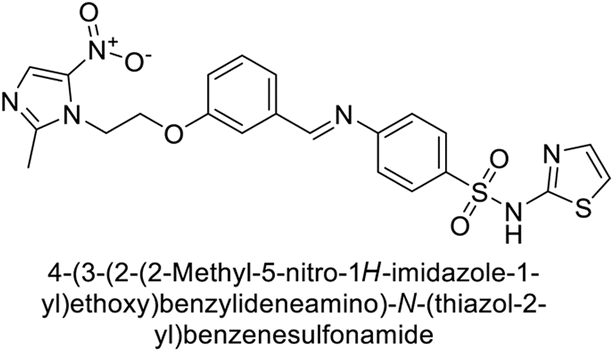 |
— | 89 | — | — | 38 | — | — | 117 |
| 92 | 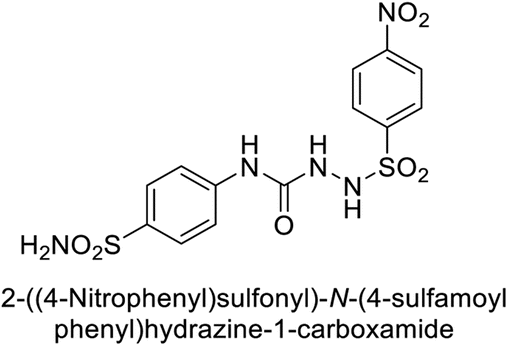 |
— | 6.0 | — | — | 0.79 | — | — | 118 |
| 93 | 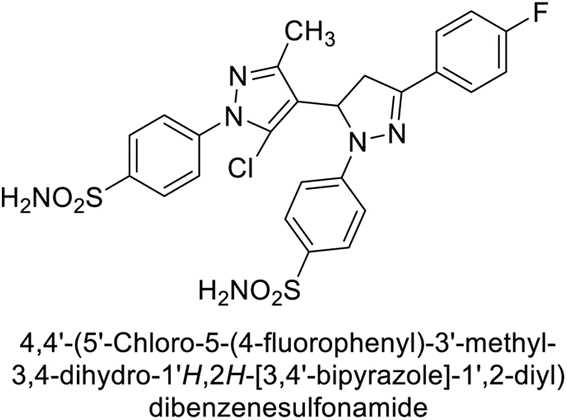 |
1.4 | — | — | 3.0 | — | — | — | 119 |
| 94 | 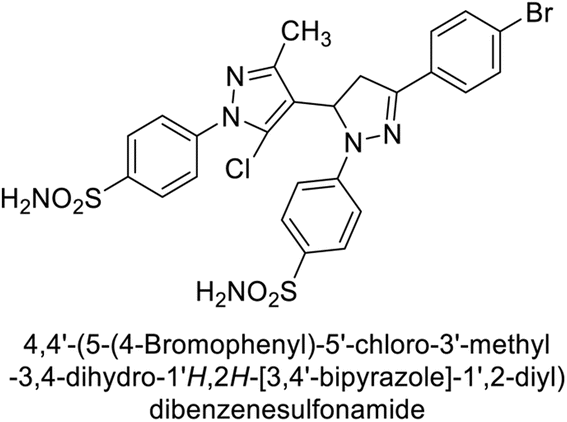 |
5.5 | — | — | 0.47 | — | — | — | 119 |
| 95 | 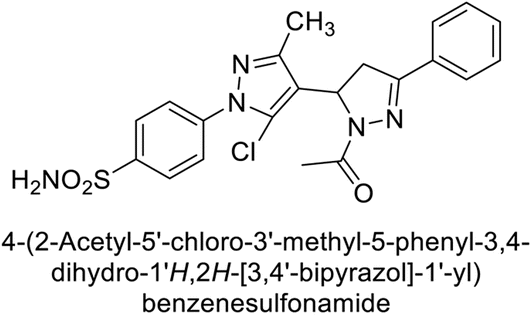 |
0.17 | — | — | 0.58 | — | — | — | 119 |
| 96 | 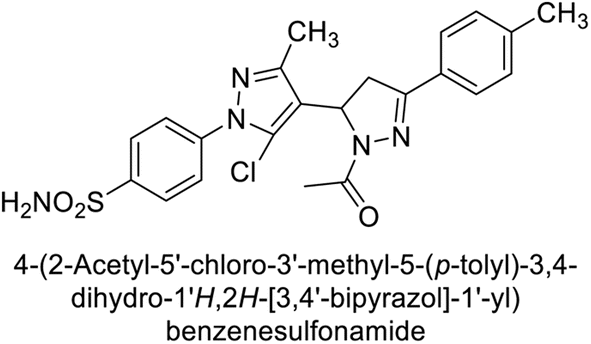 |
0.54 | — | — | 4.0 | — | — | — | 119 |
| 97 | 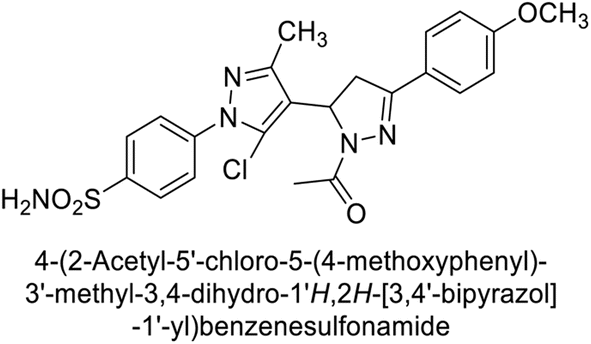 |
1.1 | — | — | 0.54 | — | — | — | 119 |
| 98 | 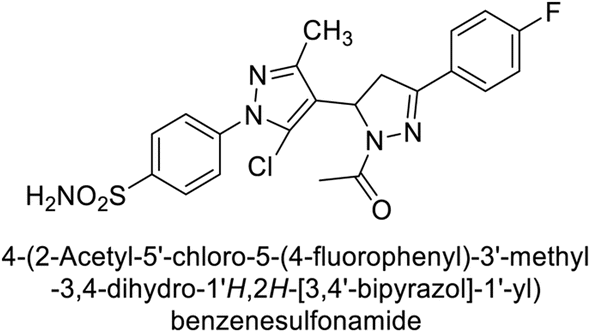 |
1.7 | — | — | 0.57 | — | — | — | 119 |
| 99 | 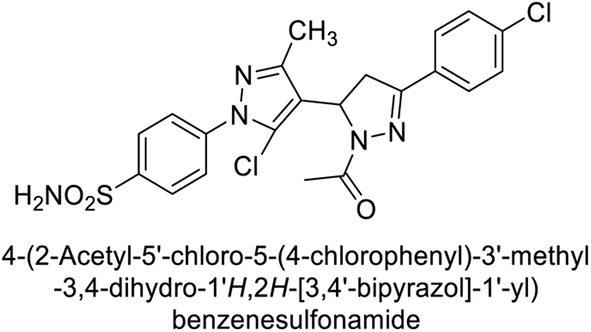 |
0.26 | — | — | 3.9 | — | — | — | 119 |
| 100 | 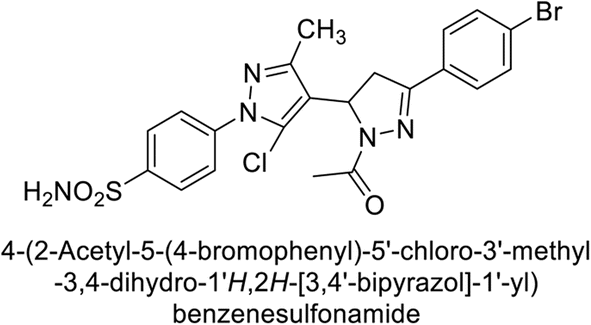 |
0.30 | — | — | 5.1 | — | — | — | 119 |
| 101 | 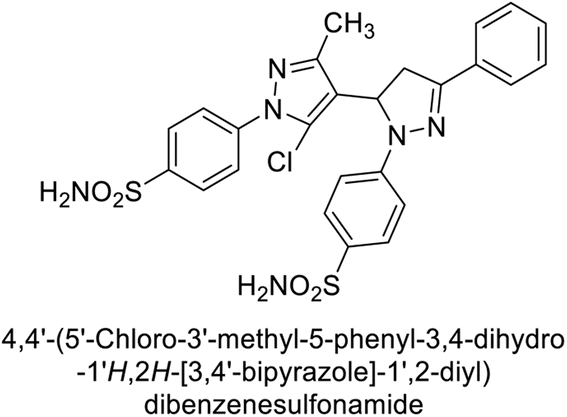 |
10.9 | — | — | 9.4 | — | — | — | 119 |
| 102 | 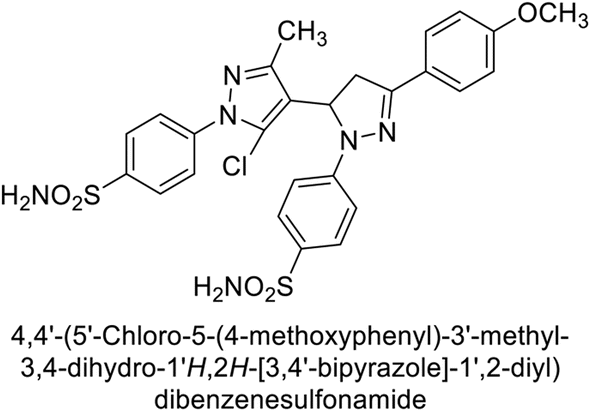 |
1.5 | — | — | 5.9 | — | — | — | 119 |
| 103 | 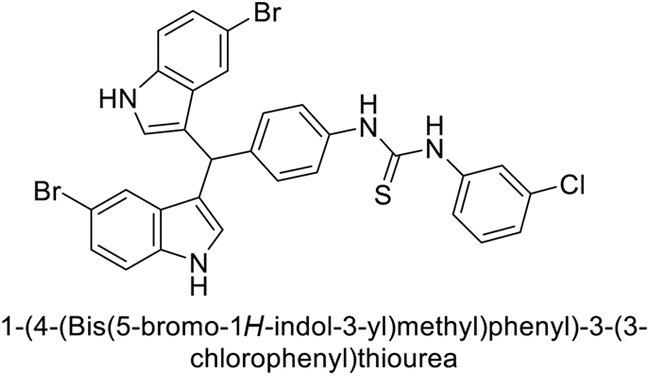 |
— | IC50 = 42.9 | — | — | — | — | — | 120 |
| 104 |  |
— | — | — | — | 3.8 | — | — | 121 |
| 105 |  |
— | — | — | — | 6.1 | — | — | 121 |
| 106 | 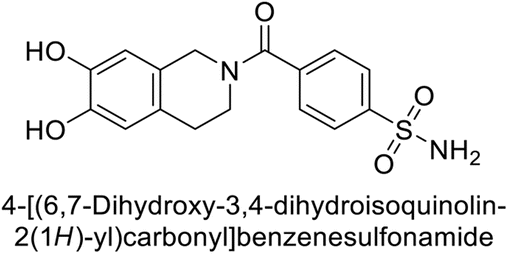 |
— | — | — | — | 5.01 | — | — | 121 |
| 107 | 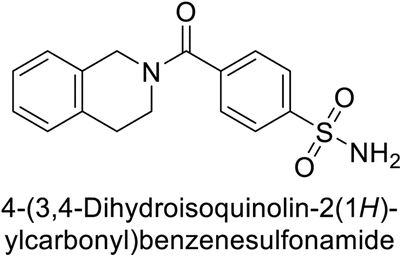 |
— | — | — | — | 7.5 | — | — | 121 |
| 108 | 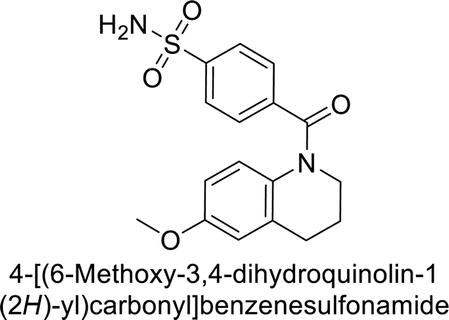 |
— | — | — | — | 6.5 | — | — | 121 |
| 109 | 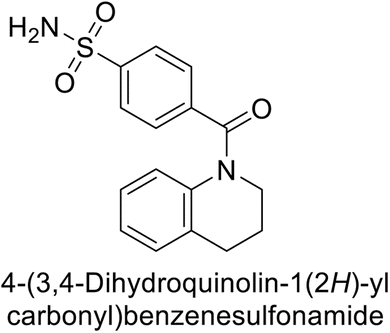 |
— | — | — | — | 9.4 | — | — | 121 |
| 110 | 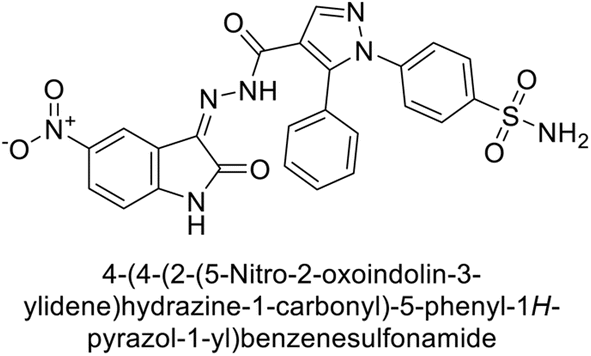 |
— | — | — | — | — | 3.7 | — | 122 |
| 111 | 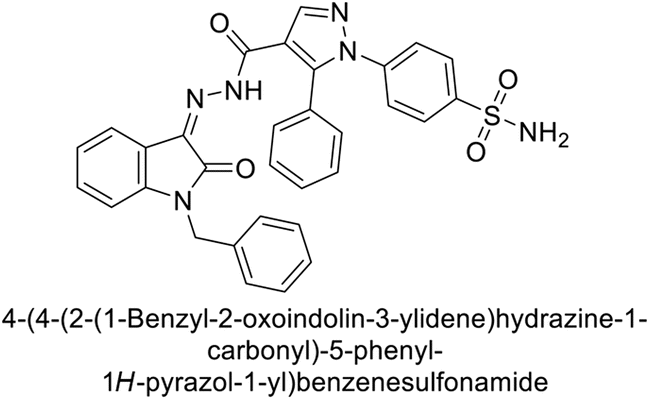 |
— | — | — | — | — | 6.5 | — | 122 |
| 112 | 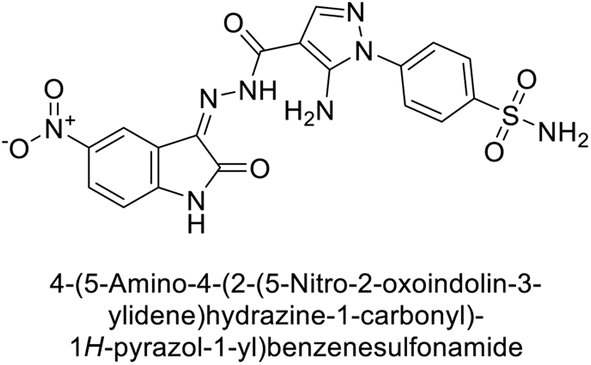 |
— | — | — | — | — | 5.4 | — | 122 |
| 113 | 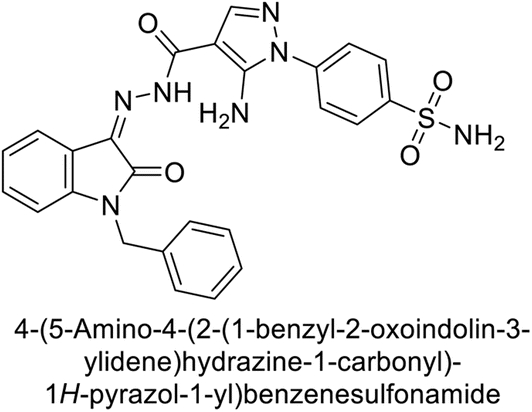 |
— | — | — | — | — | 7.2 | — | 122 |
| 114 | 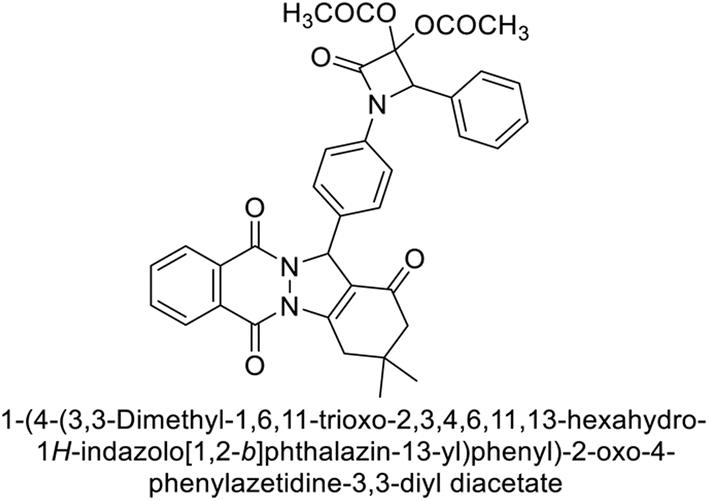 |
IC50 = 6.97 | IC50 = 8.48 | — | — | — | — | — | 123 |
| 115 |  |
95.2 | 6.2 | — | — | 90.4 | 92.3 | — | 124 |
| 116 |  |
71.2 | 4.0 | — | — | 103 | 63.1 | — | 124 |
| 117 |  |
77.6 | 4.2 | — | — | 213 | 42.3 | — | 124 |
| 118 | 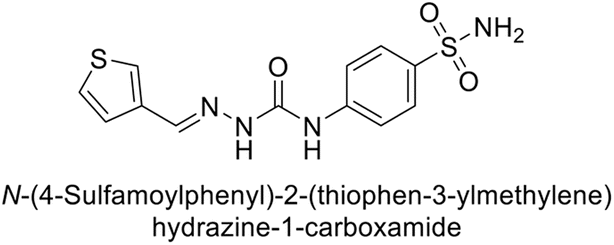 |
79.3 | 4.6 | — | — | 395 | 23.8 | — | 124 |
| 119 | 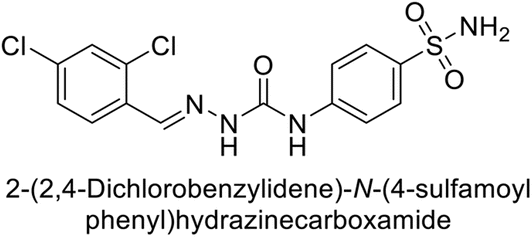 |
6530 | 75.0 | — | — | 98.1 | 90.7 | — | 124 |
| 120 | 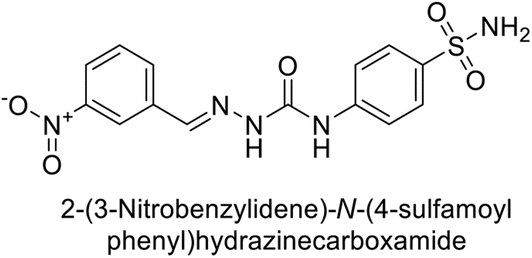 |
4930 | 51.6 | — | — | 415 | 83.9 | — | 124 |
| 121 | 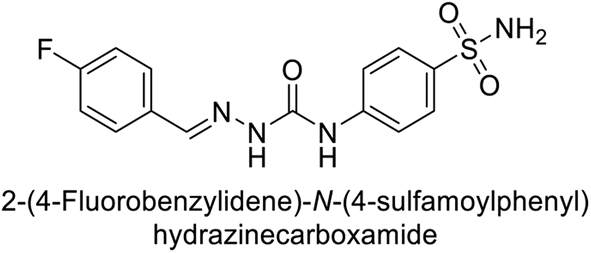 |
675 | 5.6 | — | — | 27.3 | 95.8 | — | 124 |
| 122 | 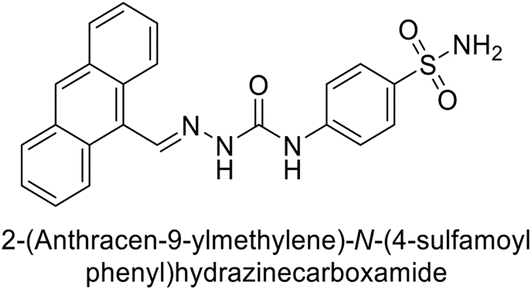 |
1640 | 19.5 | — | — | 381 | 21.5 | — | 124 |
| 123 |  |
— | — | — | — | — | — | IC50 = 61 | 125 |
| 124 |  |
— | — | — | — | — | — | IC50 = 108 | 125 |
| 125 |  |
— | — | — | — | — | — | IC50 = 191 | 125 |
| 126 | 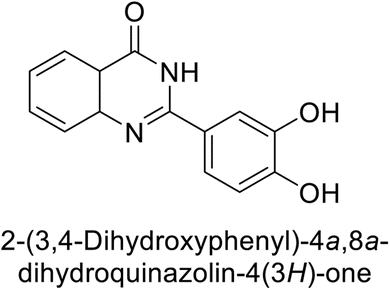 |
— | — | — | — | — | — | IC50 = 20 | 125 |
| 127 | 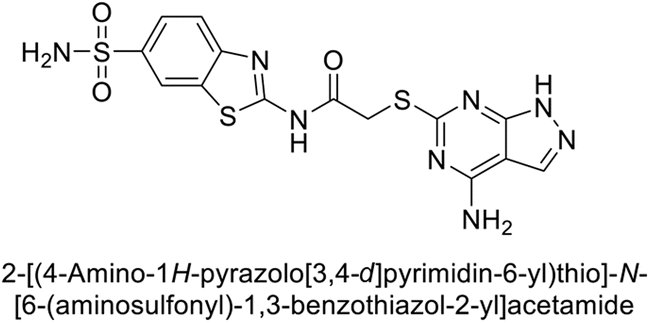 |
— | — | — | 9.5 | — | — | — | 126 |
| 128 | 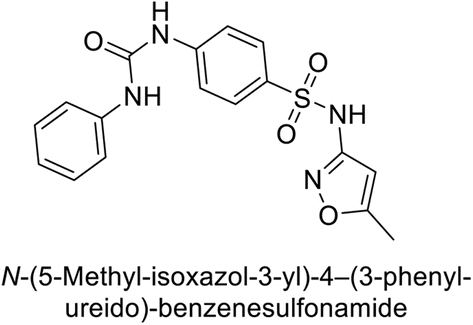 |
73 | — | — | 85 | — | — | — | 127 |
| 129 | 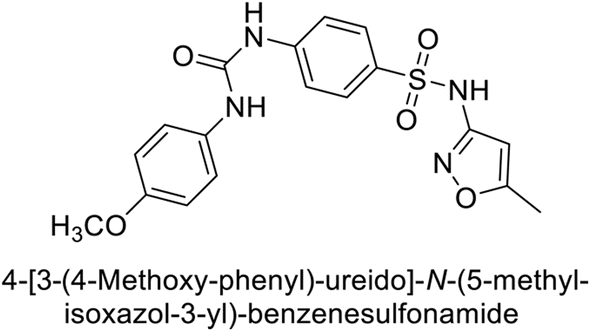 |
— | 96 | — | — | — | — | — | 127 |
| 130 | 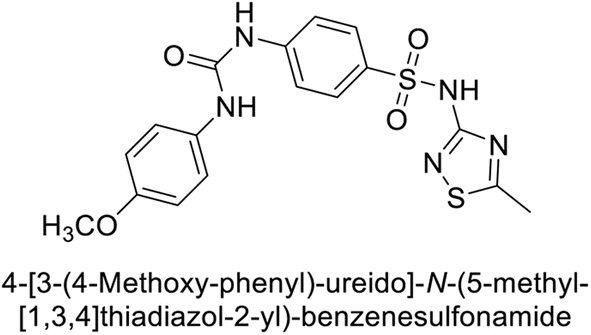 |
— | 87 | — | — | — | — | — | 127 |
| 131 | 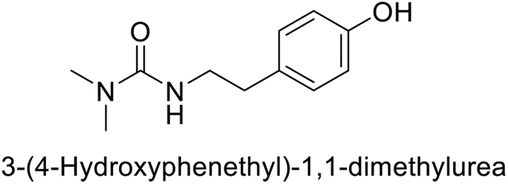 |
— | 0.307 | — | — | — | — | — | 128 |
| 132 | 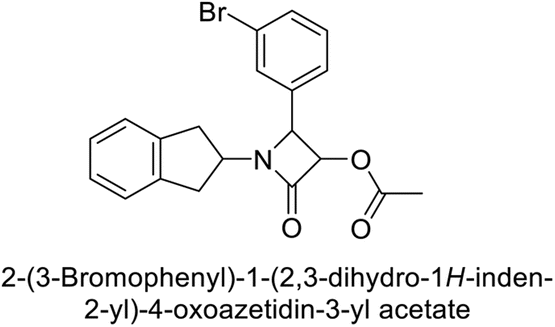 |
0.35 | 3.39 | — | — | — | — | — | 129 |
| 133 | 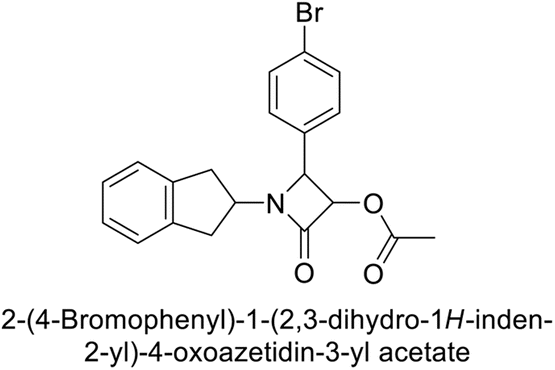 |
0.97 | 0.93 | — | — | — | — | — | 129 |
| 134 | 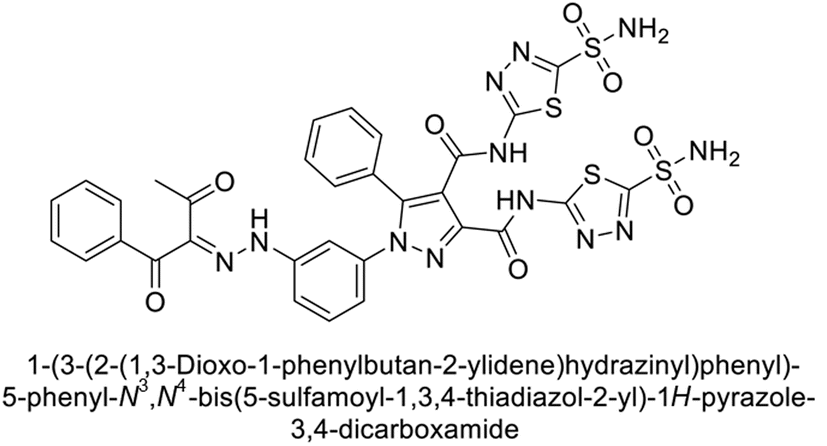 |
0.119 | — | — | — | — | — | — | 130 |
| 135 |  |
— | 0.084 | — | — | — | — | — | 130 |
| 136 |  |
47.40 | 76.06 | — | — | — | — | — | 131 |
| 137 |  |
77.68 | 30.63 | — | — | — | — | — | 131 |
| 138 | 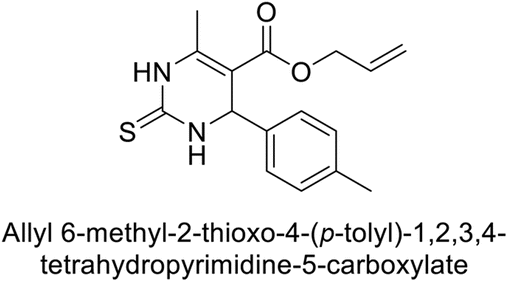 |
218.5 | 155.4 | — | — | — | — | — | 132 |
| 139 |  |
0.85 | — | — | — | — | — | — | 133 |
| 140 |  |
— | — | — | — | 29.2 | — | — | 133 |
| 141 |  |
26.5 | 18.9 | — | — | — | — | — | 134 |
| 142 | 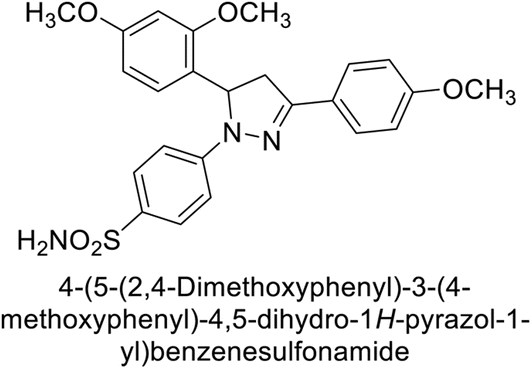 |
39.7 | 20.5 | — | — | — | — | — | 134 |
| 143 | 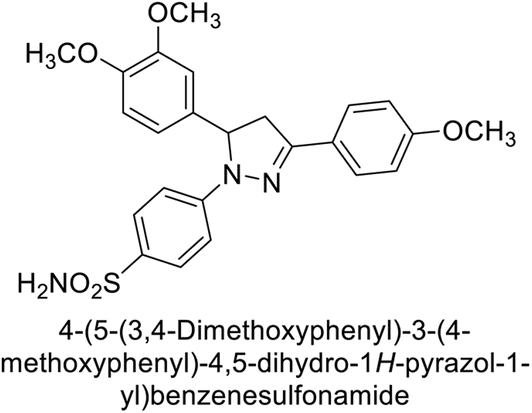 |
55.5 | 28.8 | — | — | — | — | — | 134 |
| 144 | 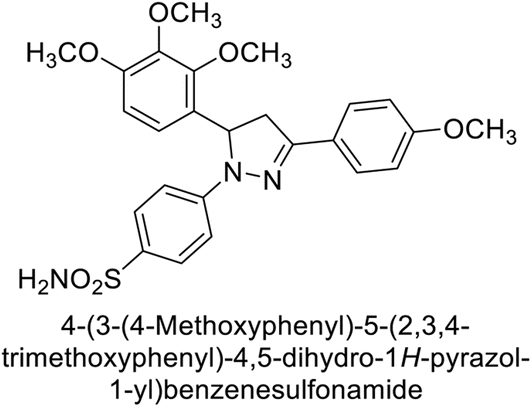 |
31.4 | 19.9 | — | — | — | — | — | 134 |
| 145 | 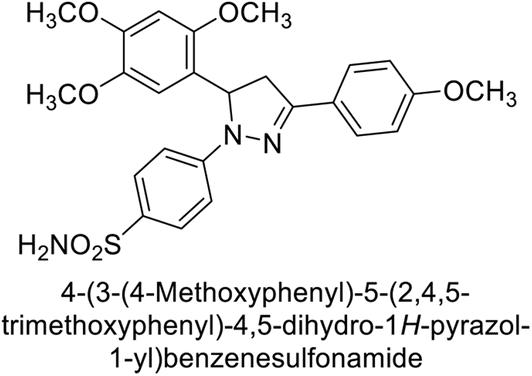 |
29.9 | 25.9 | — | — | — | — | — | 134 |
| 146 | 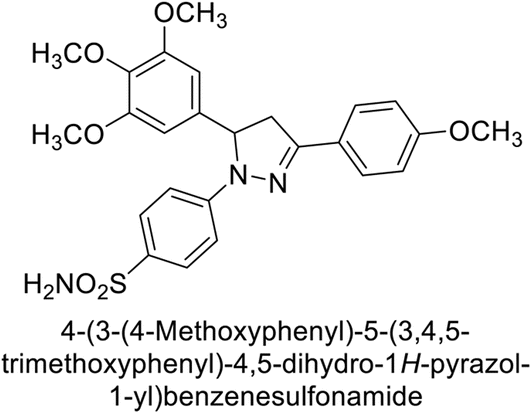 |
34.1 | 20.2 | — | — | — | — | — | 134 |
| 147 | 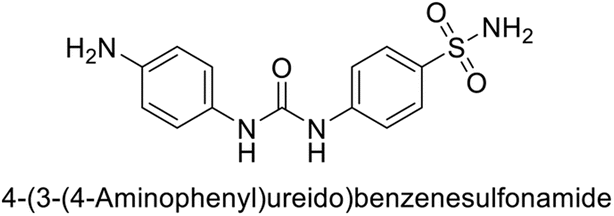 |
— | — | — | — | — | 0.71 | — | 135 |
| 148 | 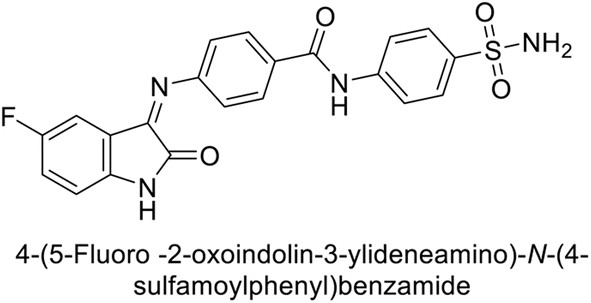 |
— | — | — | — | — | 0.69 | — | 135 |
| 149 | 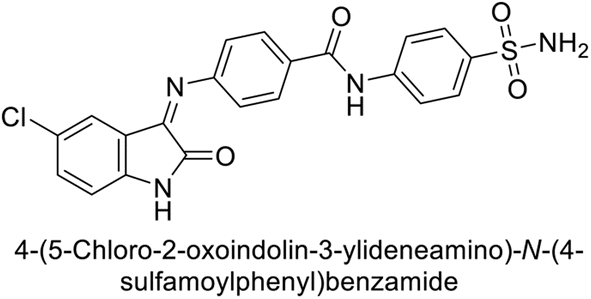 |
— | — | — | — | — | 0.54 | — | 135 |
| 150 | 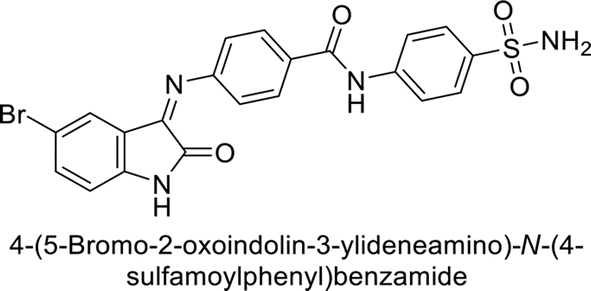 |
— | — | — | — | — | 0.58 | — | 135 |
| 151 | 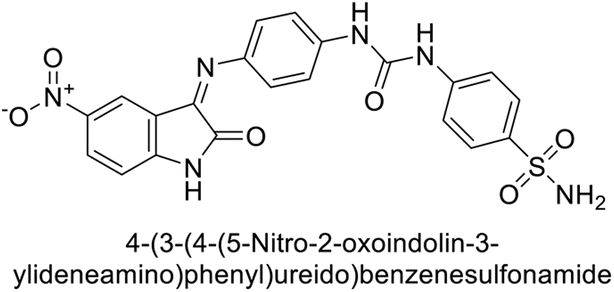 |
— | — | — | — | — | 0.67 | — | 135 |
| 152 | 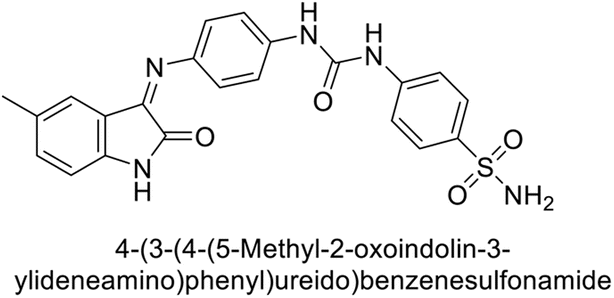 |
— | — | — | — | — | 0.64 | — | 135 |
| 153 | 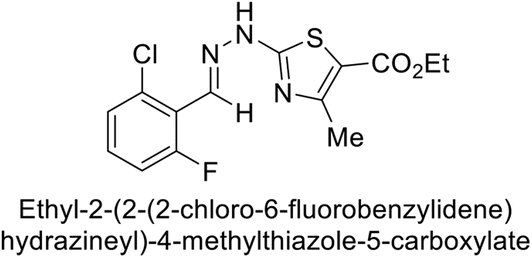 |
— | — | — | — | — | — | 1.26 | 136 |
| 154 | 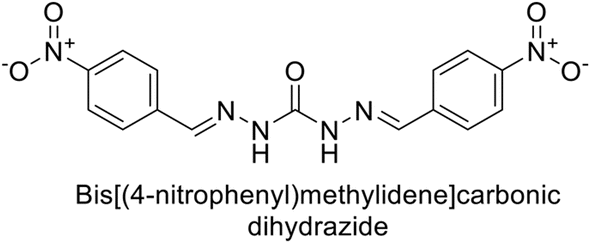 |
— | — | — | — | — | — | IC50 = 1.33 | 137 |
| 155 | 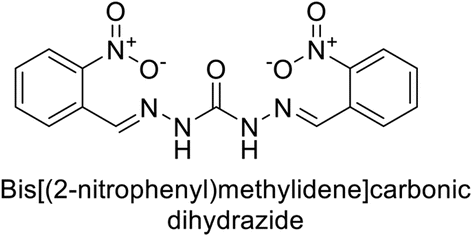 |
— | — | — | — | — | — | IC50 = 1.85 | 137 |
| 156 | 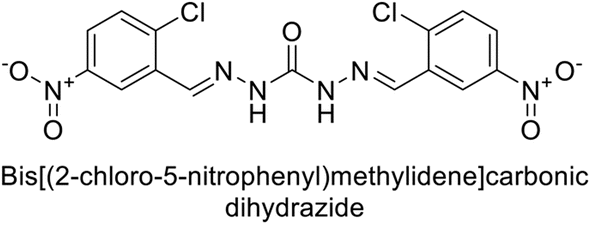 |
— | — | — | — | — | — | IC50 = 1.37 | 137 |
| 157 | 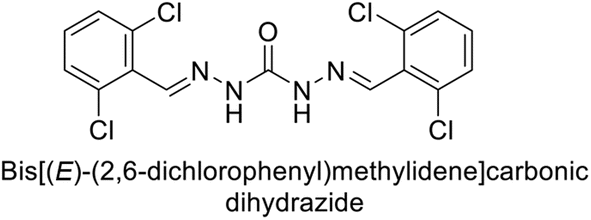 |
— | — | — | — | — | — | IC50 = 1.46 | 137 |
| 158 | 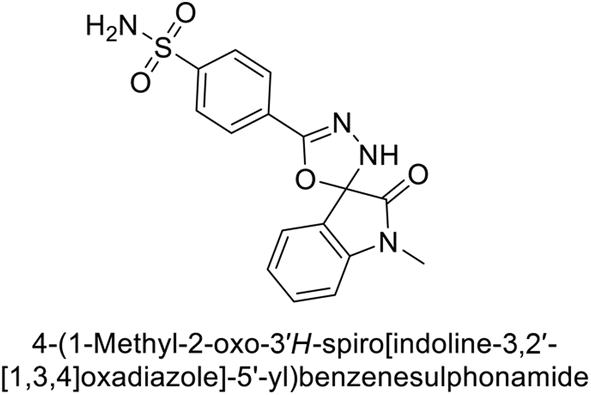 |
— | 0.042 | — | — | — | — | — | 138 |
| 159 | 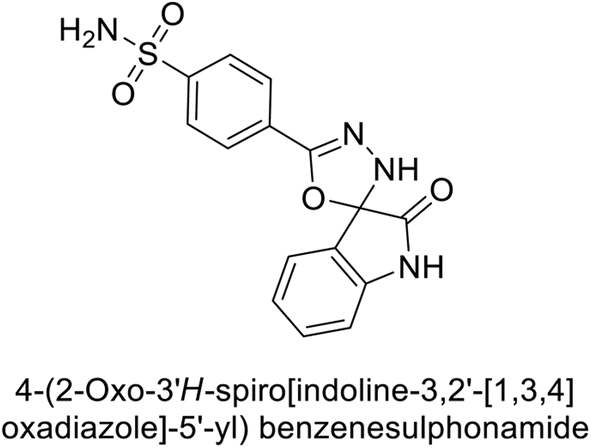 |
— | 0.151 | — | — | — | — | — | 138 |
| 160 | 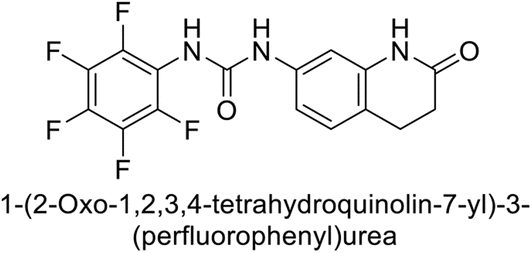 |
— | — | — | — | 243.6 | — | — | 139 |
| 161 |  |
— | — | — | — | 292.8 | — | — | 139 |
| 162 | 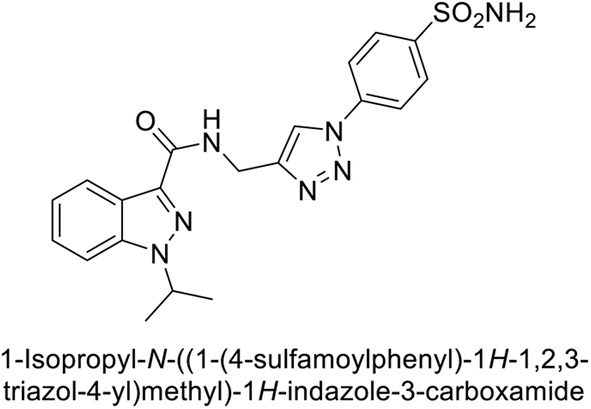 |
— | — | — | — | 1.8 | — | — | 140 |
| 163 | 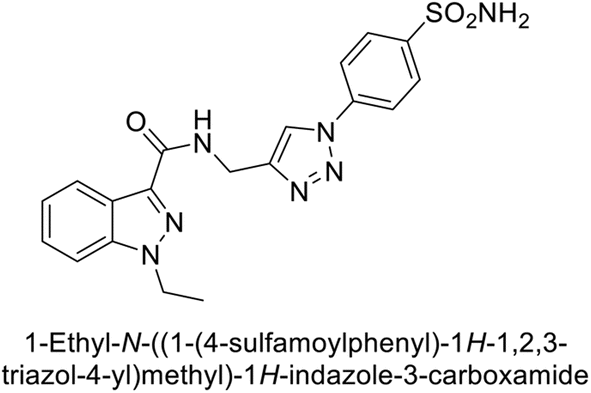 |
— | — | — | — | 2.3 | — | — | 140 |
| 164 | 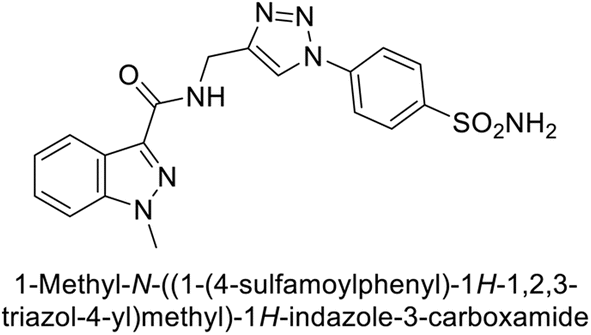 |
— | — | — | — | 2.0 | — | — | 140 |
| 165 |  |
0.89 | — | — | — | — | — | — | 141 |
| 166 | 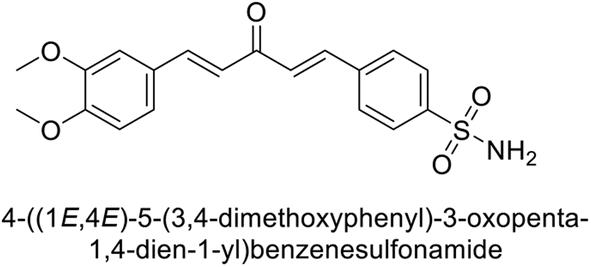 |
0.75 | — | — | — | — | — | — | 141 |
| 167 | 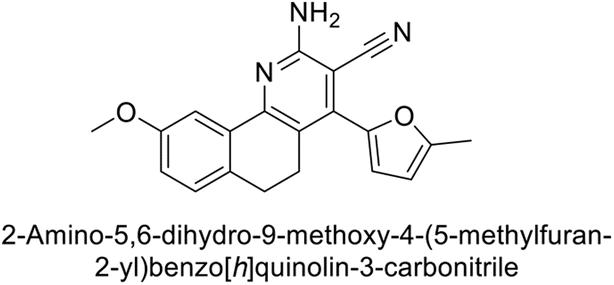 |
2.84 | 4.56 | — | — | — | — | — | 142 |
| 168 | 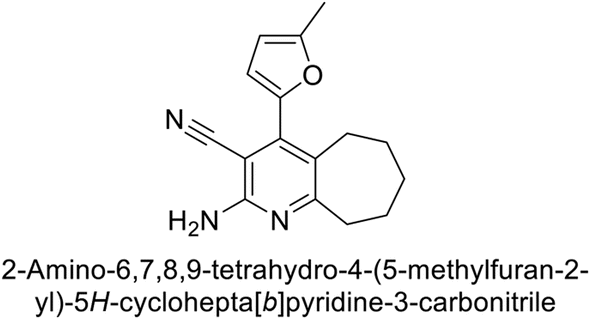 |
53.9 | 2.56 | — | — | — | — | — | 142 |
| 169 | 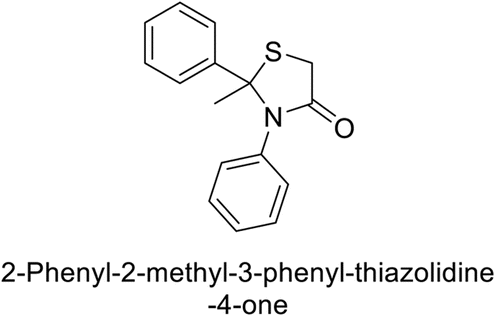 |
23.76 | — | — | — | — | — | — | 143 |
| 170 | 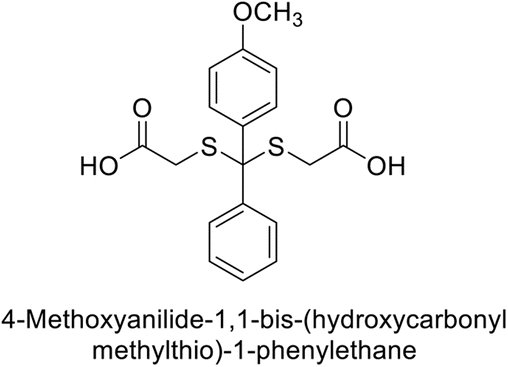 |
— | 58.92 | — | — | — | — | — | 143 |
| 171 |  |
23.27 | — | — | — | — | — | — | 144 |
| 172 | 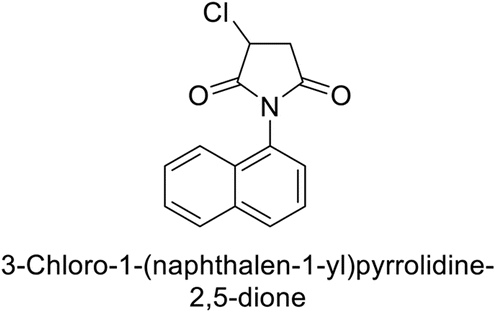 |
— | 10.64 | — | — | — | — | — | 144 |
| 173 |  |
— | — | — | — | 107.9 | — | — | 145 |
| 174 | 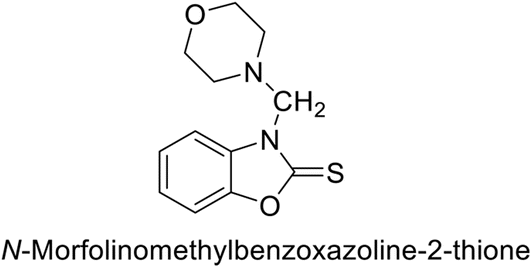 |
— | 58 | — | — | — | — | — | 146 |
| 175 | 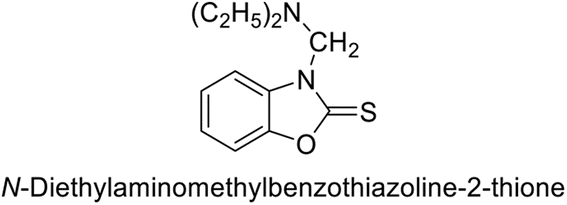 |
— | 70 | — | — | — | — | — | 146 |
| 176 | 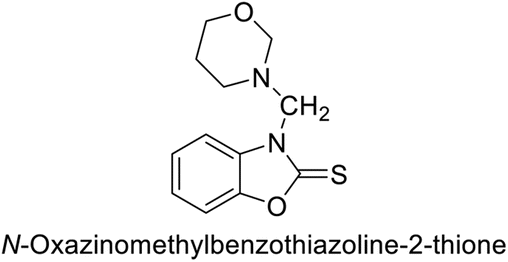 |
— | 76 | — | — | — | — | — | 146 |
| 177 | 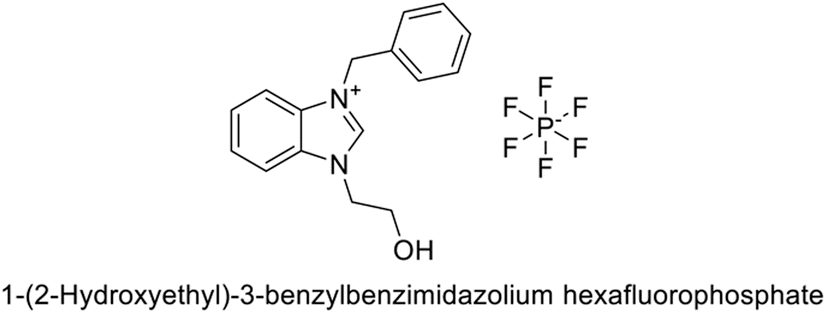 |
13.90 | 12.82 | — | — | — | — | — | 147 |
| 178 | 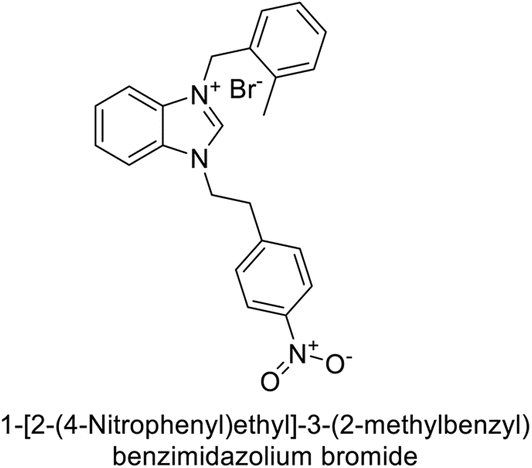 |
17.33 | 45.31 | — | — | — | — | — | 148 |
| 179 | 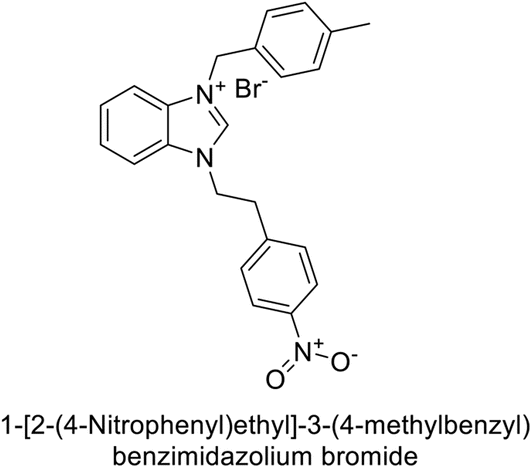 |
54.48 | 33.98 | — | — | — | — | — | 148 |
| 180 |  |
0.99 | — | — | — | — | — | 0.71 | 149 |
| 181 |  |
— | 0.67 | — | — | — | — | — | 149 |
| 182 | 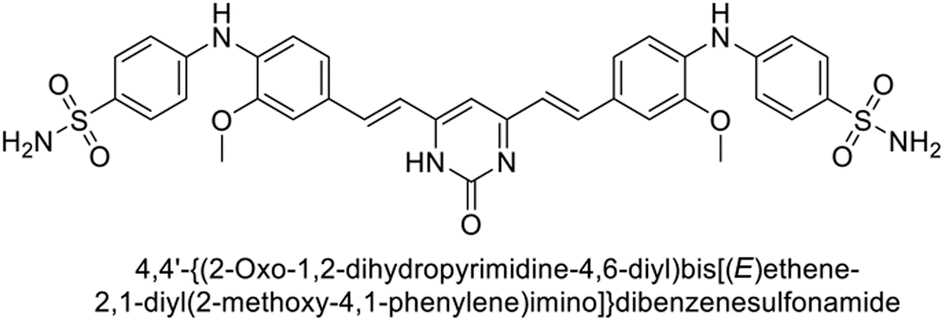 |
— | — | — | — | — | — | 0.71 | 149 |
| 183 | 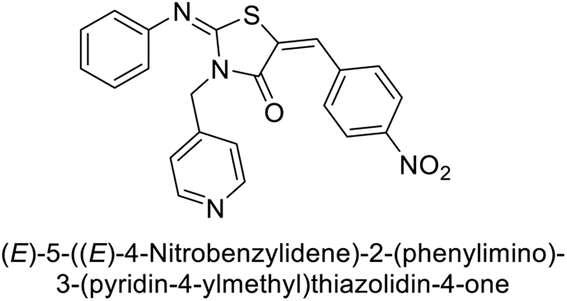 |
— | — | — | — | 1.61 | — | — | 150 |
| 184 | 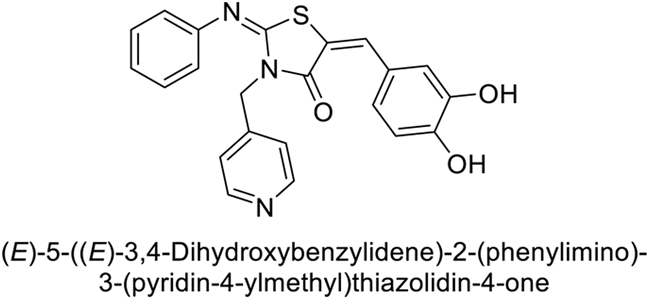 |
— | — | — | — | 1.84 | — | — | 150 |
| 185 | 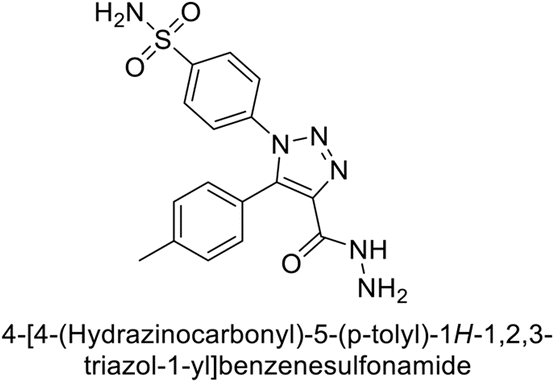 |
27.2 | — | — | — | — | — | — | 151 |
| 186 | 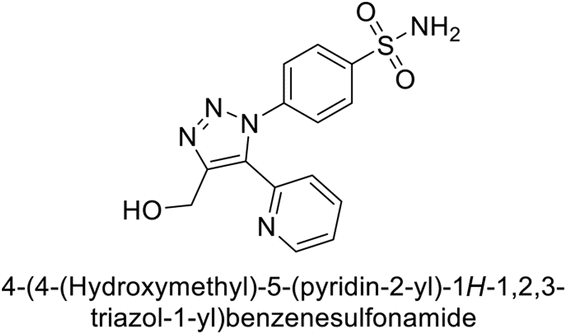 |
— | — | — | — | 14.3 | — | — | 151 |
| 187 | 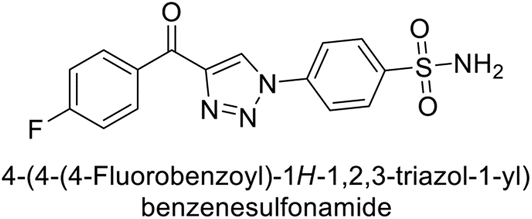 |
— | — | 52.4 | — | — | — | — | 152 |
| 188 |  |
— | — | — | — | 46.4 | — | — | 153 |
| 189 |  |
— | — | — | — | 42.6 | — | — | 153 |
| 190 | 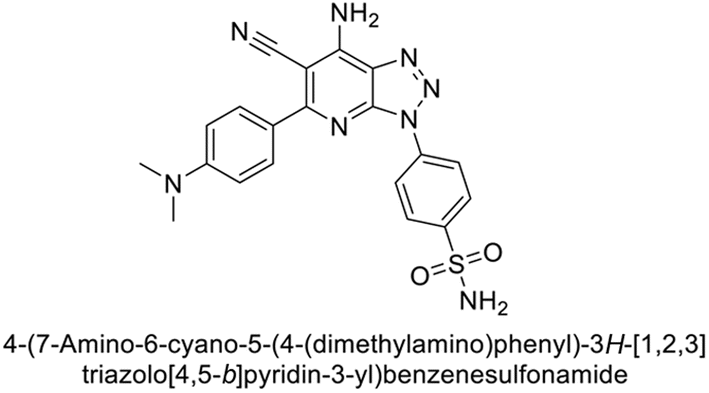 |
— | — | — | — | 35.1 | — | — | 153 |
| 191 | 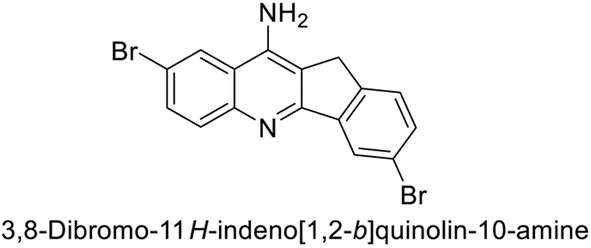 |
120.9 | 267.5 | — | — | — | — | — | 154 |
| 192 | 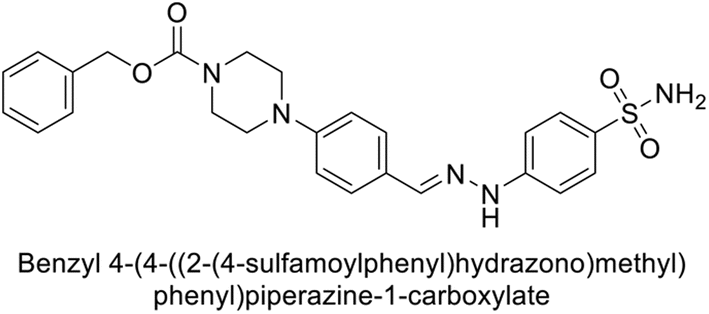 |
919.6 | 93.9 | — | 11.4 | 1631.4 | — | — | 155 |
| 193 | 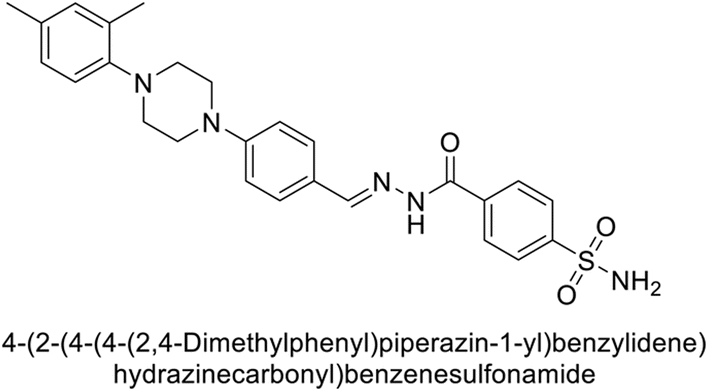 |
321.8 | 30.8 | — | 6.2 | 1788.6 | — | — | 155 |
| 194 | 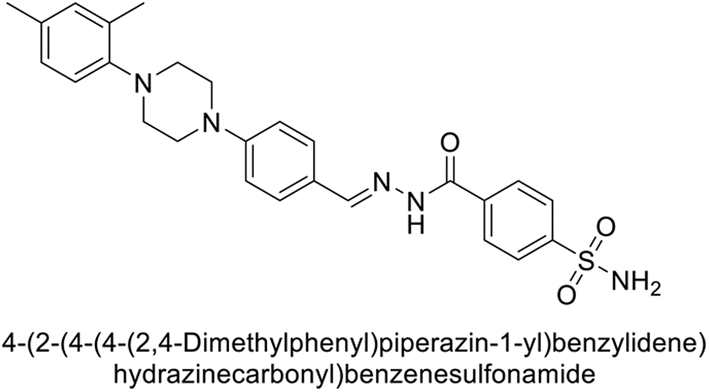 |
>10000 |
9385.9 | — | 17.8 | 1660.9 | — | — | 155 |
| 195 | 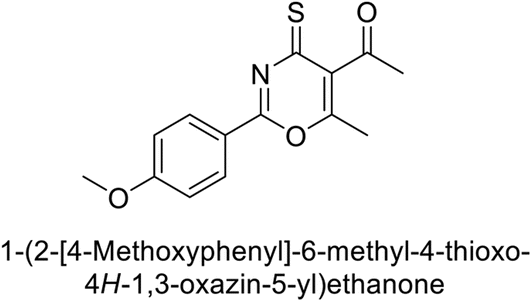 |
— | — | — | — | — | — | 0.144 | 156 |
| 196 |  |
— | 0.1676 | — | — | — | — | — | 157 |
| 197 |  |
— | 0.2880 | — | — | — | — | — | 157 |
| 198 | 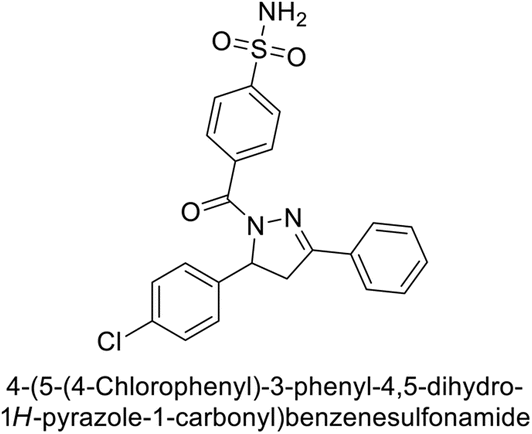 |
195.0 | — | — | — | 7.9 | 34.0 | — | 158 |
| 199 | 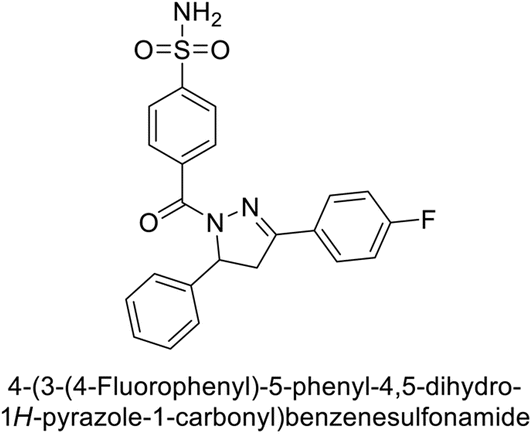 |
102.9 | — | — | — | 23.1 | 10.1 | — | 158 |
| 200 | 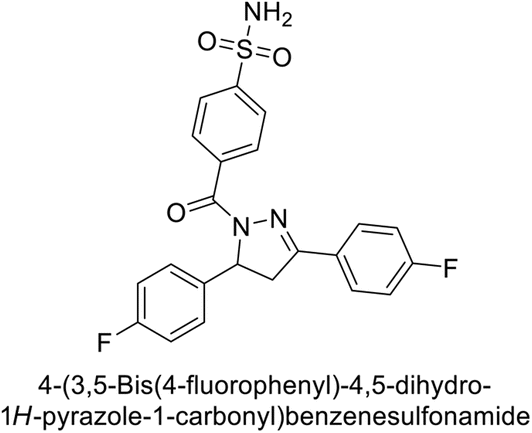 |
140.7 | — | — | — | 5.5 | 7.1 | — | 158 |
| 201 | 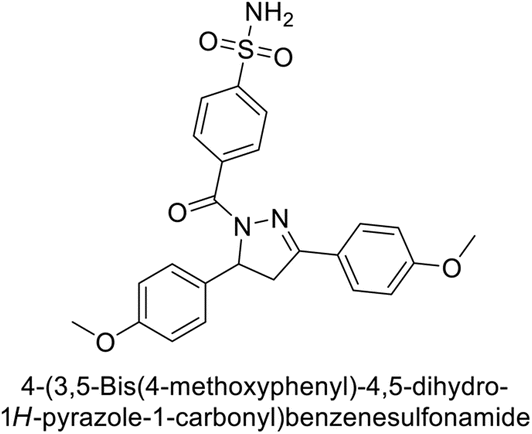 |
875.9 | — | — | — | 37.0 | 18.8 | — | 158 |
| 202 | 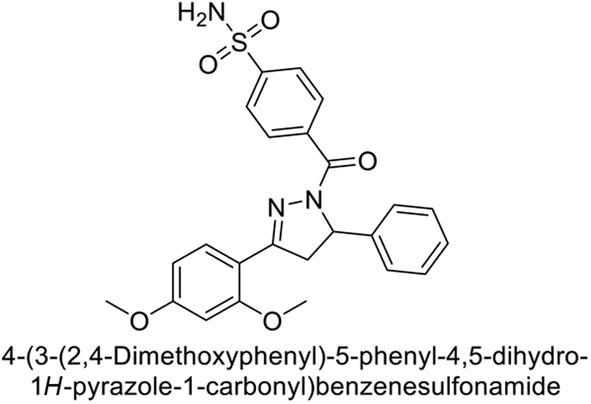 |
807.8 | — | — | — | 32.9 | 9.7 | — | 158 |
| 203 | 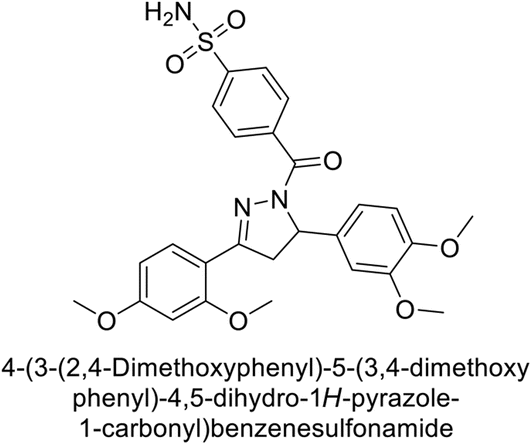 |
2961.3 | — | — | — | 36.3 | 28.9 | — | 158 |
| 204 | 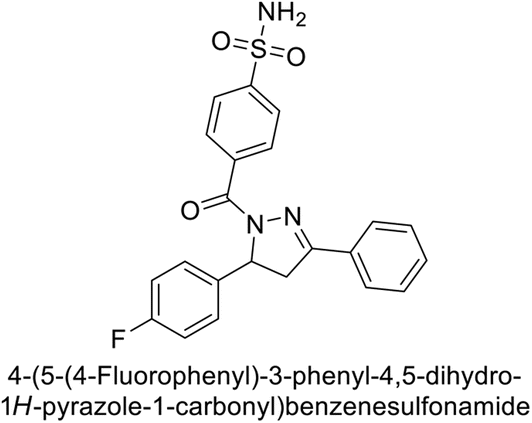 |
87.8 | — | — | — | 44.5 | 9.4 | — | 158 |
| 205 | 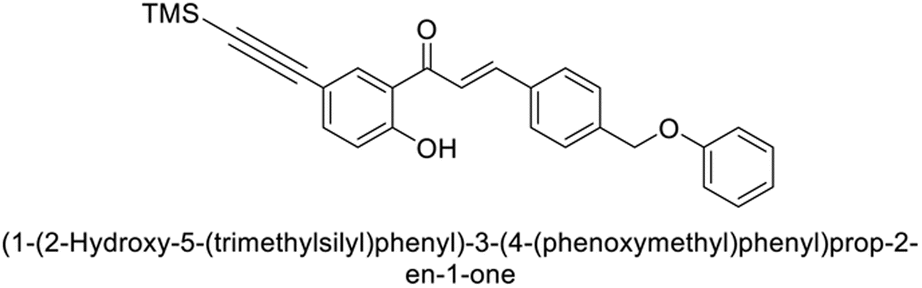 |
— | — | — | — | — | — | IC50 = 0.054 | 159 |
| 206 | 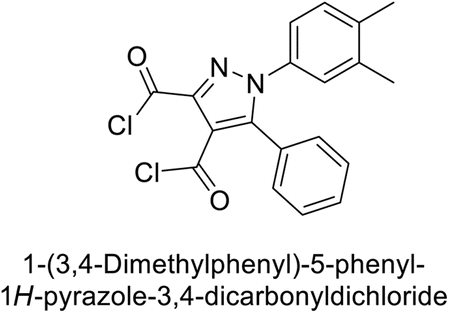 |
1.03 | 1.82 | — | — | — | — | — | 160 |
| 207 | 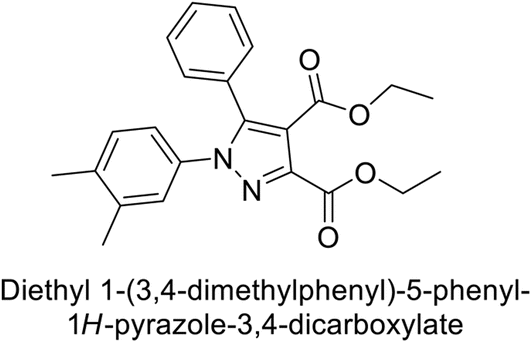 |
6.58 | 8.93 | — | — | — | — | — | 160 |
| 208 |  |
— | — | — | — | 0.2 | 0.2 | — | 161 |
| 209 |  |
— | — | — | — | 144.6 | — | — | 162 |
| 210 | 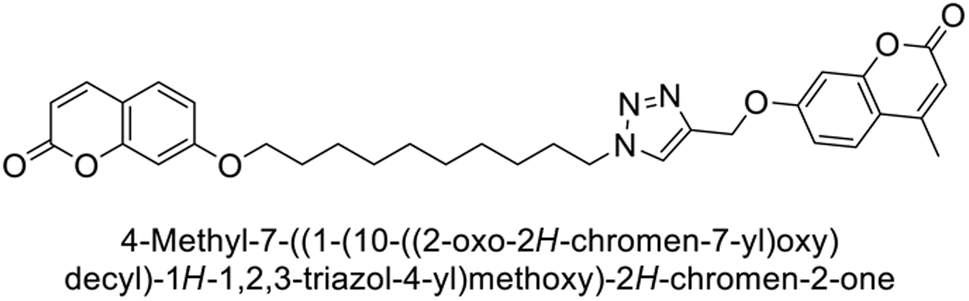 |
— | — | — | — | 71.5 | — | — | 162 |
| 211 |  |
— | — | — | — | 45.5 | — | — | 163 |
| 212 |  |
47.7 | 52.8 | — | — | — | — | — | 164 |
| 213 |  |
44.7 | 55.5 | — | — | — | — | — | 164 |
| 214 | 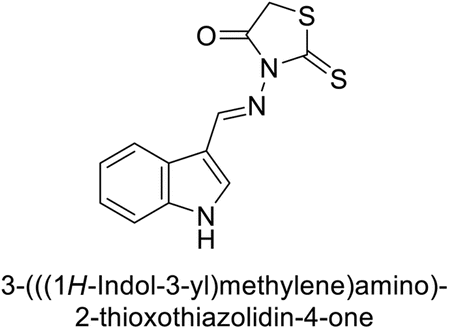 |
43.5 | 46.6 | — | — | — | — | — | 164 |
| 215 | 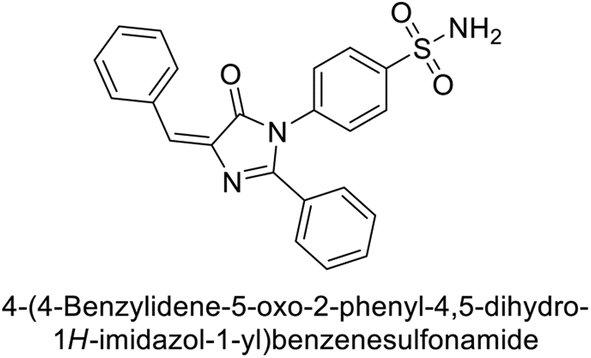 |
— | — | — | — | 58.7 | 76.5 | — | 165 |
| 216 | 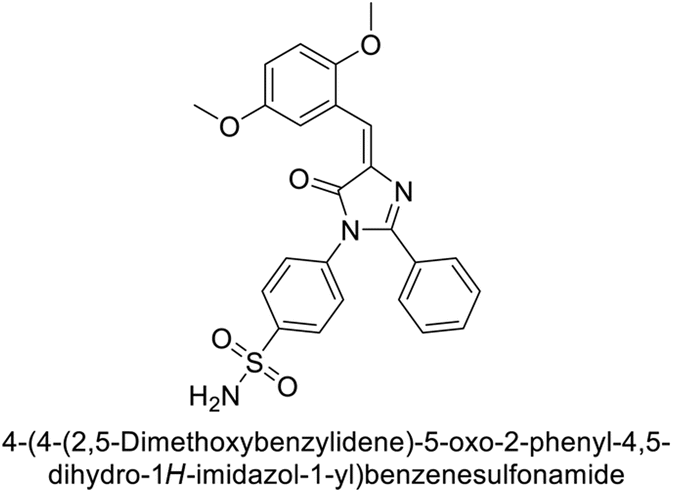 |
— | — | — | — | 23.0 | 54.8 | — | 165 |
| 217 | 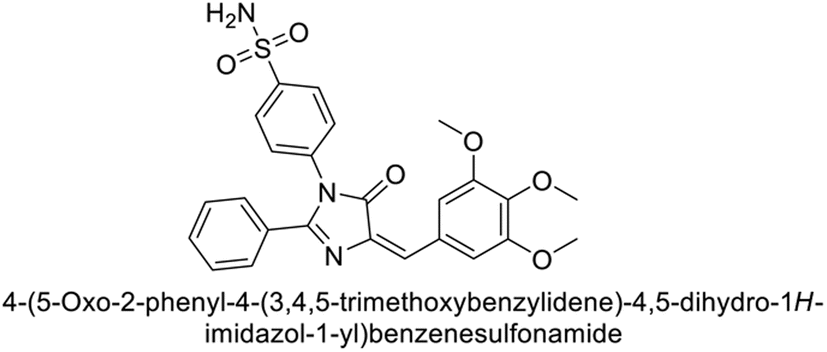 |
— | — | — | — | 68.2 | 179.6 | — | 165 |
| 218 | 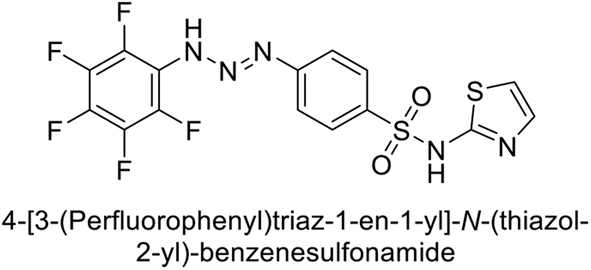 |
450.37 | 504.37 | — | — | — | — | — | 166 |
| 219 | 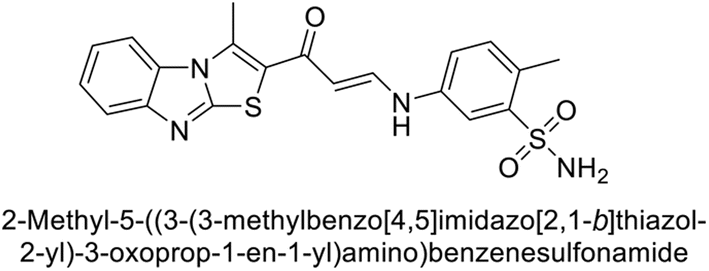 |
— | — | — | — | 450.37 | — | — | 167 |
| 220 | 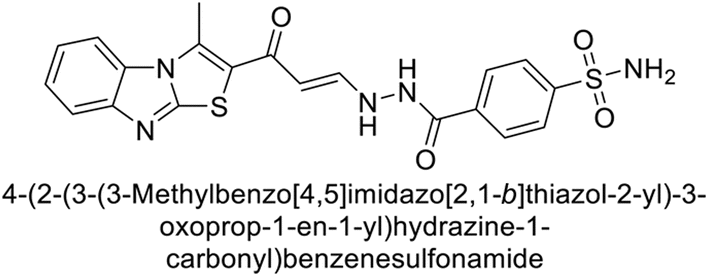 |
— | — | — | — | 450.37 | — | — | 167 |
| 221 | 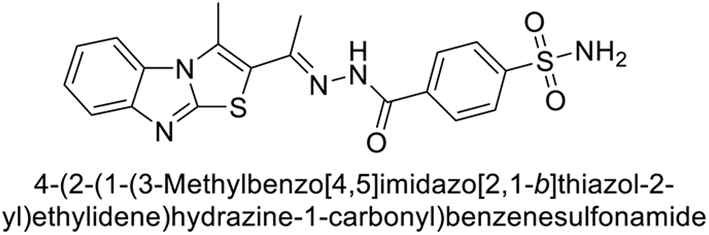 |
— | — | — | — | 450.37 | — | — | 167 |
| 222 | 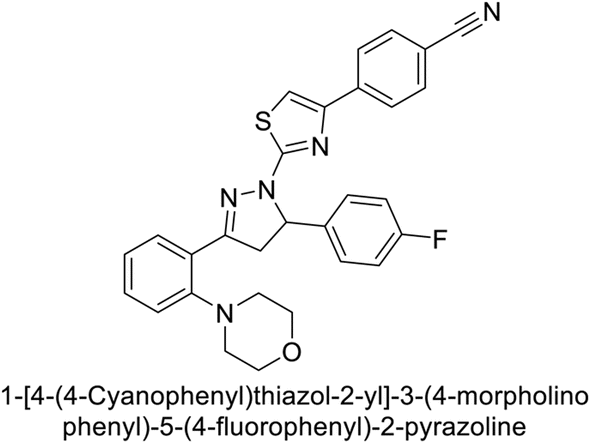 |
13.04 | — | — | — | — | — | — | 168 |
| 223 | 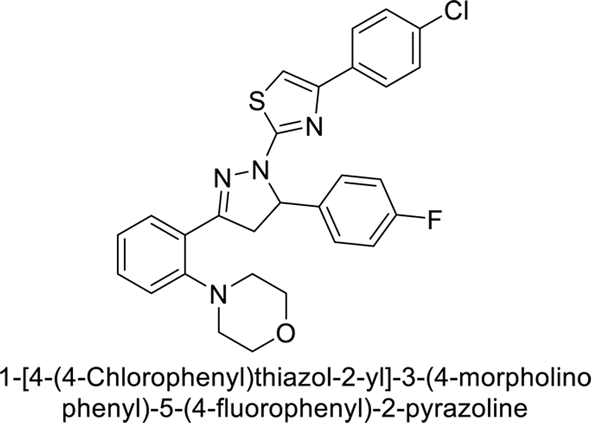 |
— | 7.47 | — | — | — | — | — | 168 |
| 224 | 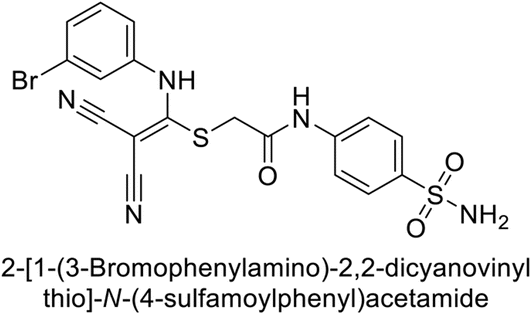 |
9.01 | — | — | — | — | — | — | 169 |
| 225 | 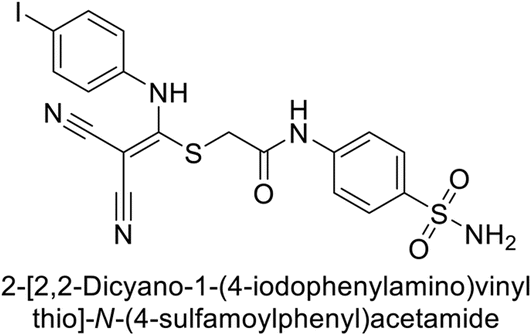 |
— | 7.41 | — | — | — | — | — | 169 |
| 226 | 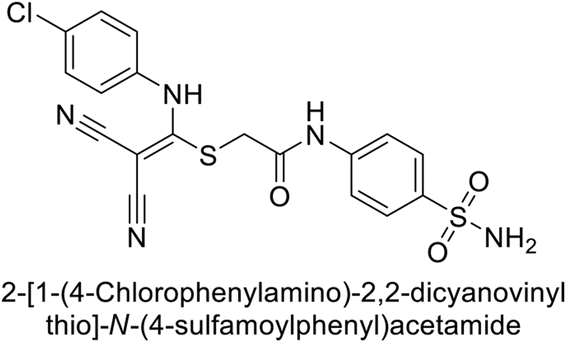 |
7.37 | — | — | — | — | — | — | 169 |
| 227 | 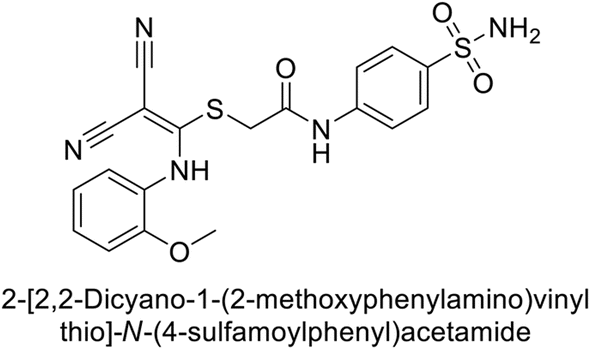 |
48.28 | — | — | — | — | — | — | 169 |
| 228 | 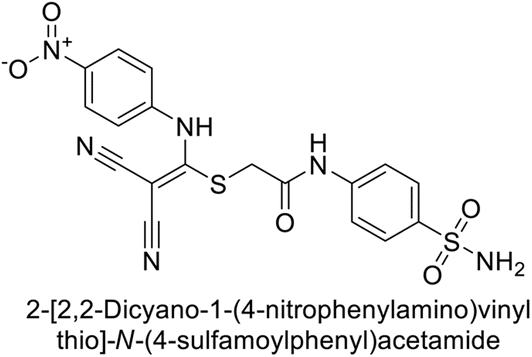 |
105.94 | — | — | — | — | — | — | 169 |
| 229 | 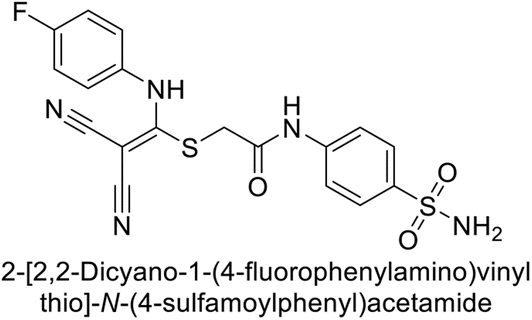 |
— | 56.67 | — | — | — | — | — | 169 |
| 230 | 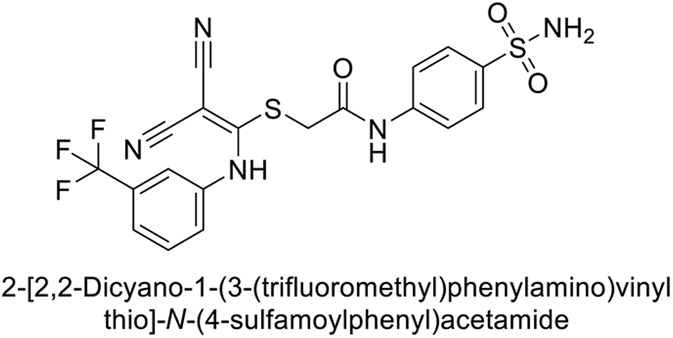 |
— | 49.92 | — | — | — | — | — | 169 |
| 231 | 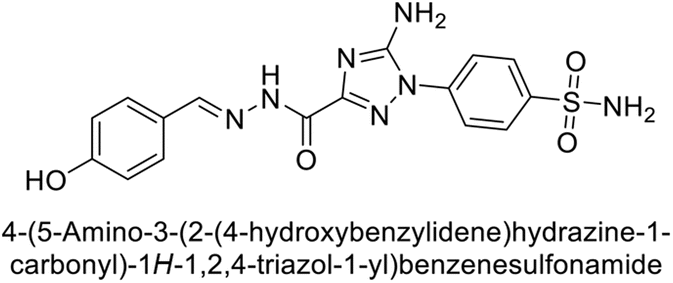 |
— | — | — | — | 8.3 | 7.8 | — | 170 |
| 232 | 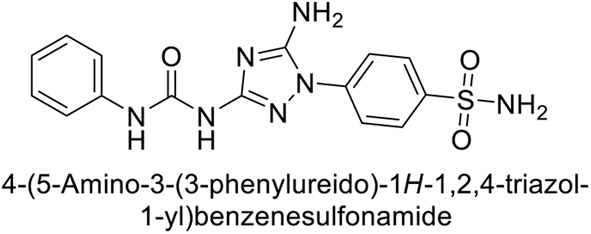 |
— | — | — | — | 8.2 | 8.1 | — | 170 |
| 233 | 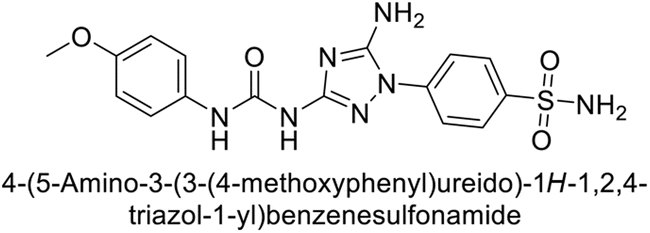 |
— | — | — | — | 3.3 | — | — | 170 |
| 234 | 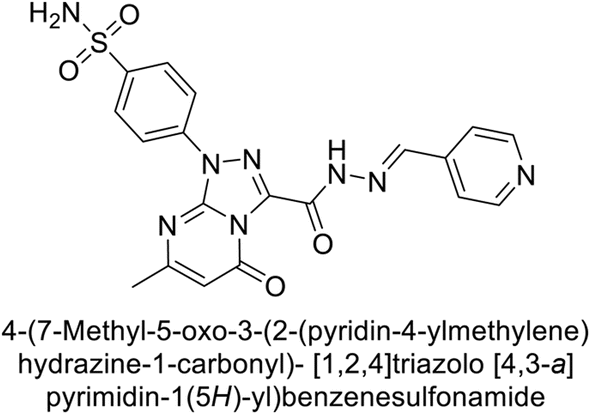 |
— | — | — | — | 24.6 | — | — | 170 |
| 235 | 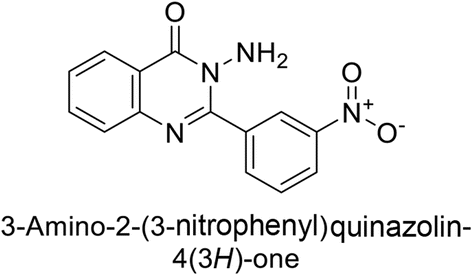 |
— | IC50 = 21.5 | — | — | — | — | IC50 = 19.7 | 171 |
| 236 | 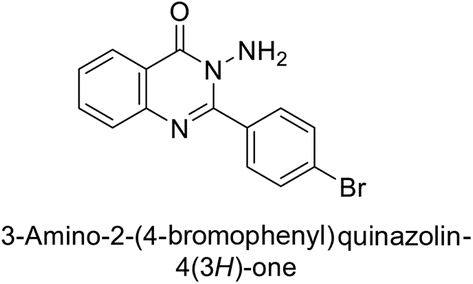 |
— | IC50 = 14.0 | — | — | — | — | — | 171 |
| 237 | 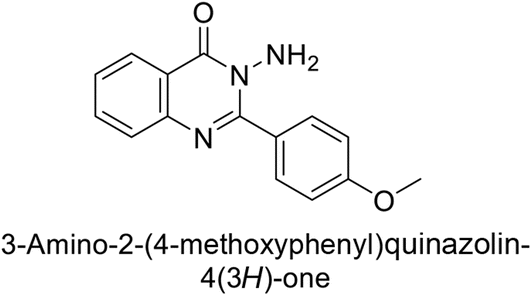 |
— | IC50 = 22.0 | — | — | — | — | — | 171 |
| 238 | 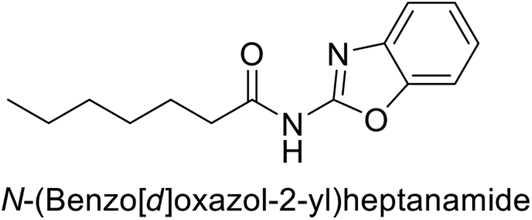 |
— | IC50 = 0.00564 | — | — | — | — | — | 172 |
| 239 | 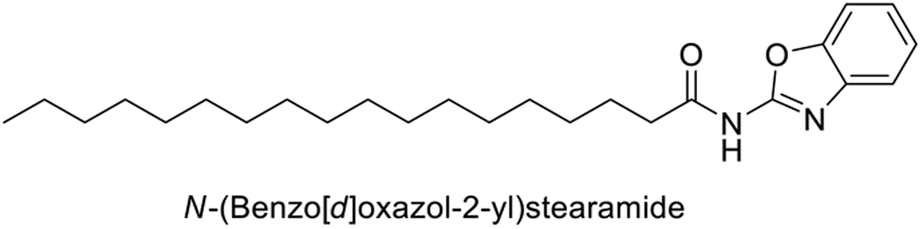 |
— | IC50 = 0.00596 | — | — | — | — | — | 172 |
| 240 | 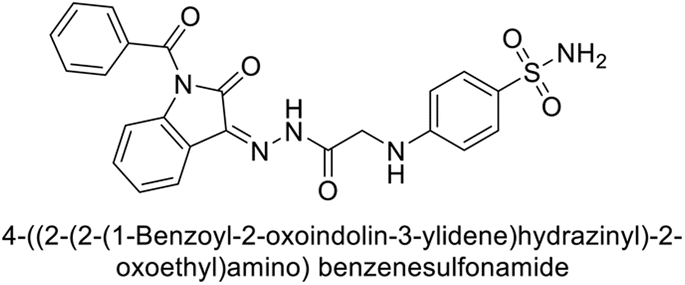 |
— | — | — | — | — | 21.5 | — | 173 |
| 241 | 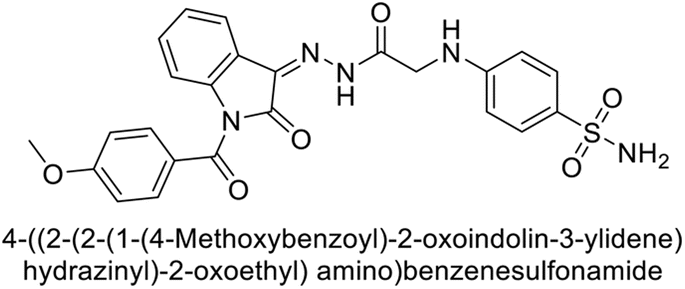 |
— | — | — | — | — | 22.3 | — | 173 |
| 242 | 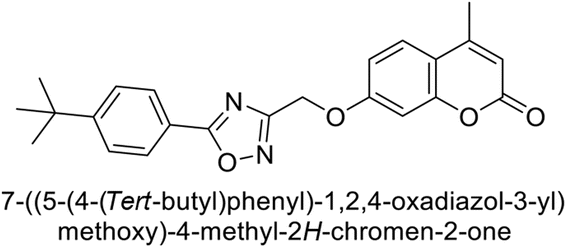 |
— | — | — | — | — | 1.0 | — | 174 |
| 243 | 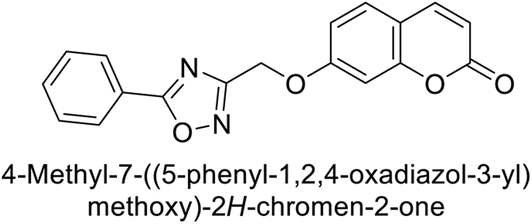 |
— | — | — | — | — | 23.6 | — | 174 |
| 244 | 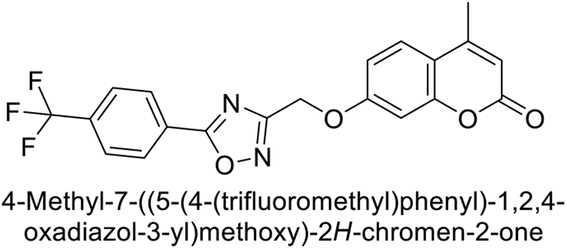 |
— | — | — | — | — | 4.4 | — | 174 |
| 245 | 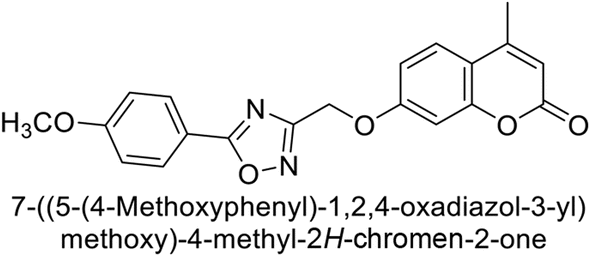 |
— | — | — | — | 23.6 | 4.5 | — | 174 |
| 246 | 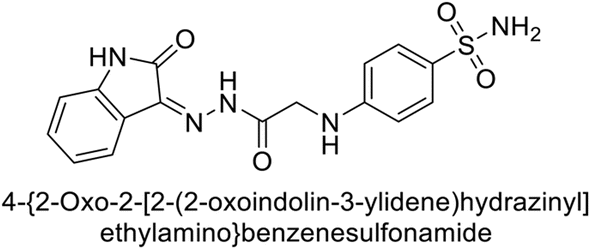 |
— | 3.0 | — | — | 13.9 | — | — | 175 |
| 247 | 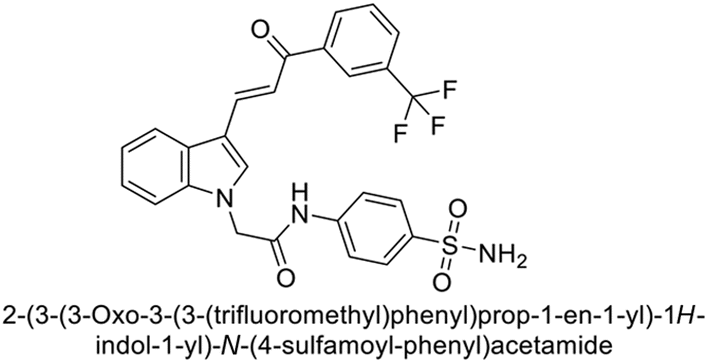 |
— | 2.3 | — | — | — | — | — | 176 |
| 248 | 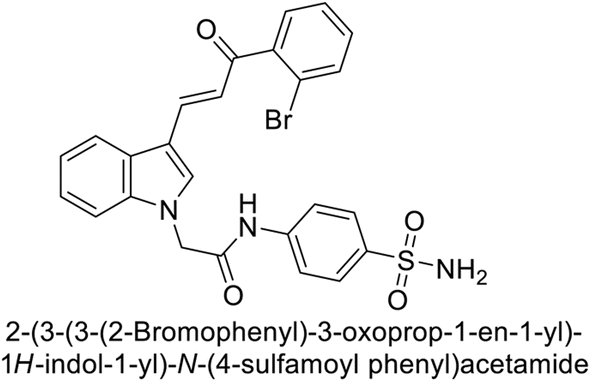 |
— | 2.4 | — | — | — | — | — | 176 |
| 249 | 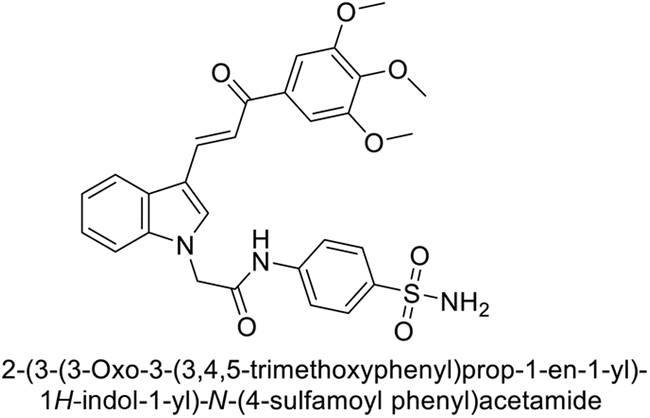 |
— | 3.6 | — | — | — | — | — | 176 |
| 250 | 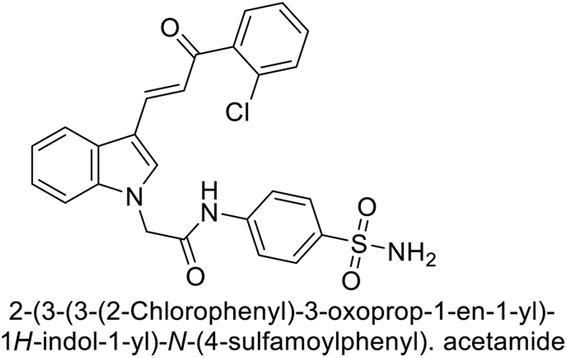 |
— | 7.0 | — | — | — | — | — | 176 |
| 251 | 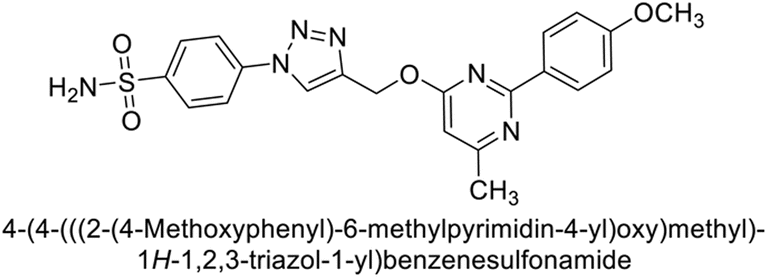 |
— | — | — | — | — | 9.4 | — | 177 |
| 252 | 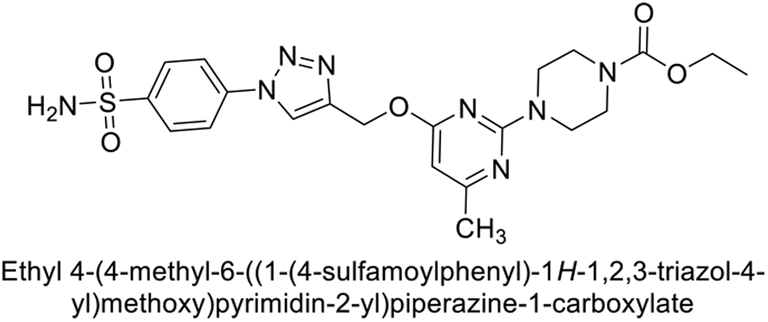 |
— | — | — | — | — | 1.8 | — | 177 |
| 253 | 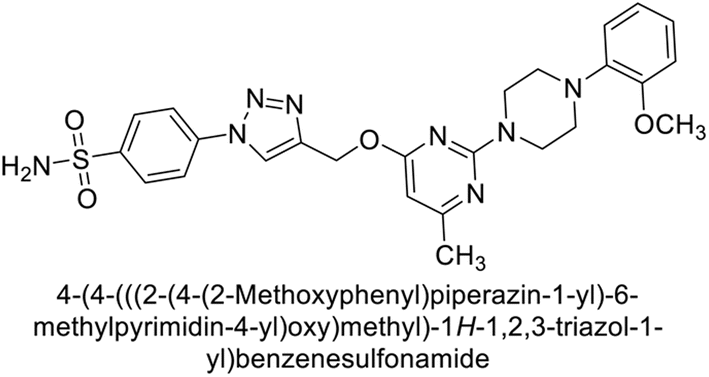 |
— | — | — | — | — | 0.82 | — | 177 |
| 254 | 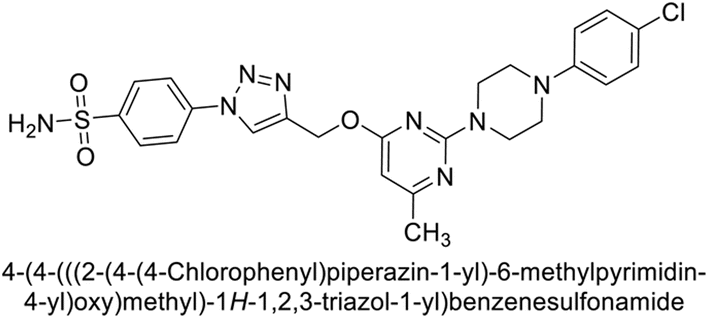 |
— | — | — | — | 2.9 | 0.82 | — | 177 |
| 255 | 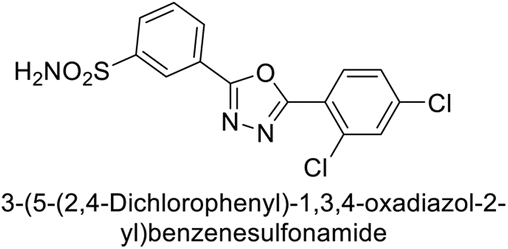 |
75.8 | — | — | — | — | 15.4 | — | 178 |
| 256 | 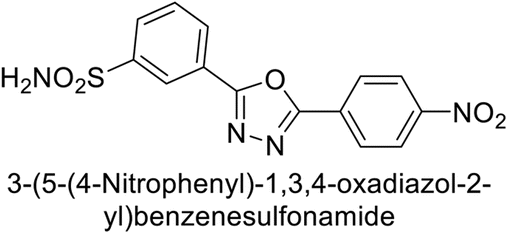 |
— | — | — | — | — | 16.2 | — | 178 |
| 257 | 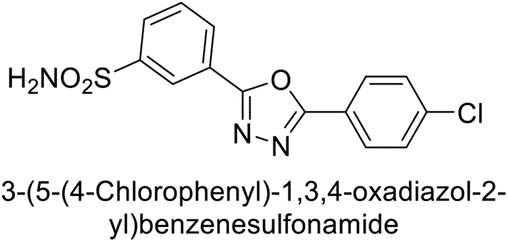 |
— | — | — | — | — | 16.4 | — | 178 |
| 258 | 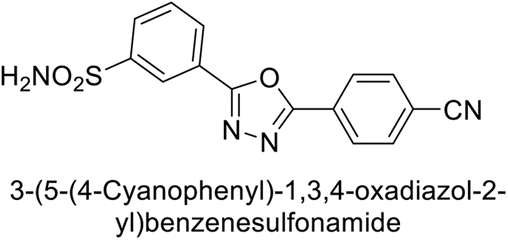 |
— | — | — | — | — | 17.0 | — | 178 |
| 259 | 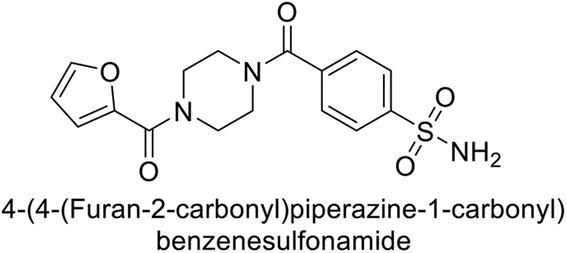 |
— | — | 4.3 | — | — | — | — | 179 |
| 260 | 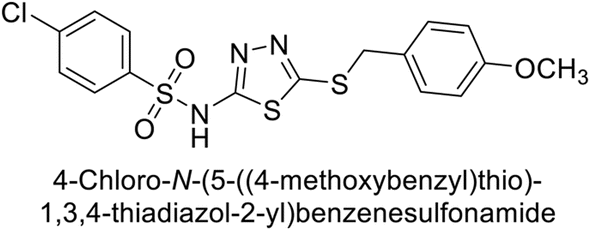 |
— | — | — | — | — | — | IC50 = 0.60 | 180 |
| 261 | 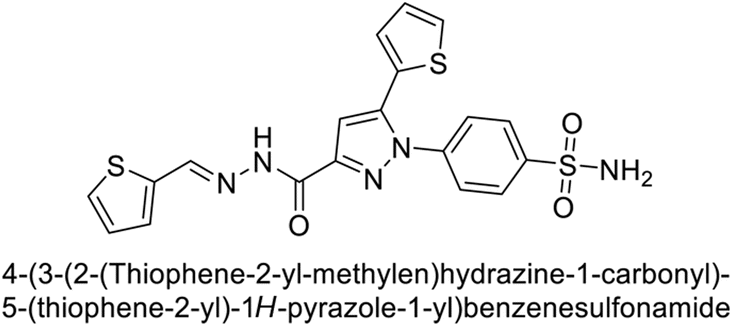 |
— | — | — | — | 7.0 | — | — | 181 |
| 262 | 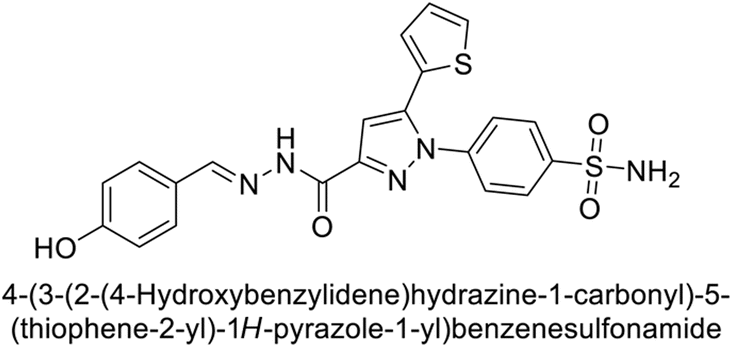 |
— | — | — | — | 10.6 | — | — | 181 |
| 263 | 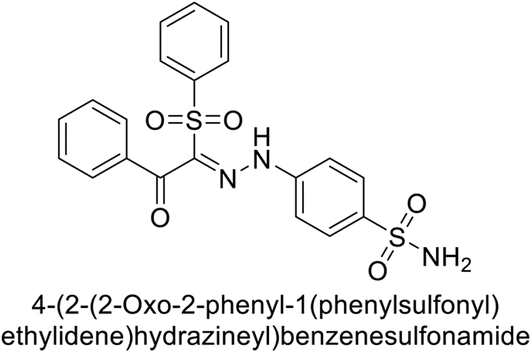 |
— | 7.6 | — | — | — | — | — | 182 |
| 264 | 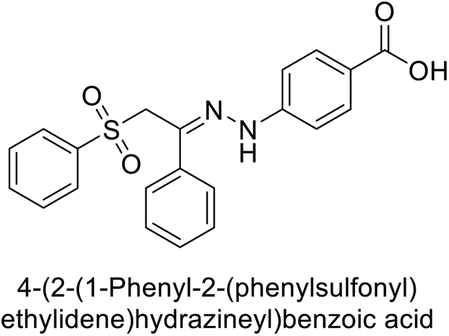 |
— | 32633 |
— | — | — | — | — | 182 |
| 265 | 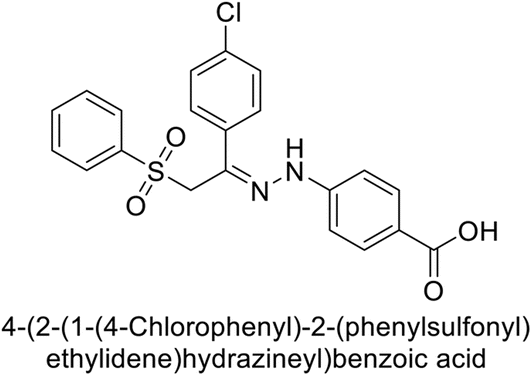 |
— | 9190 | — | — | — | — | — | 182 |
| 266 | 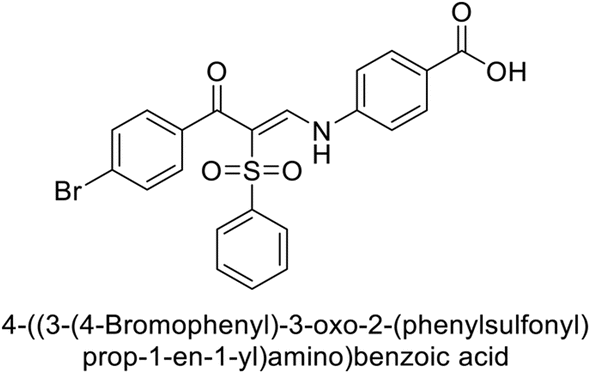 |
— | 65349 |
— | — | — | — | — | 182 |
| 267 | 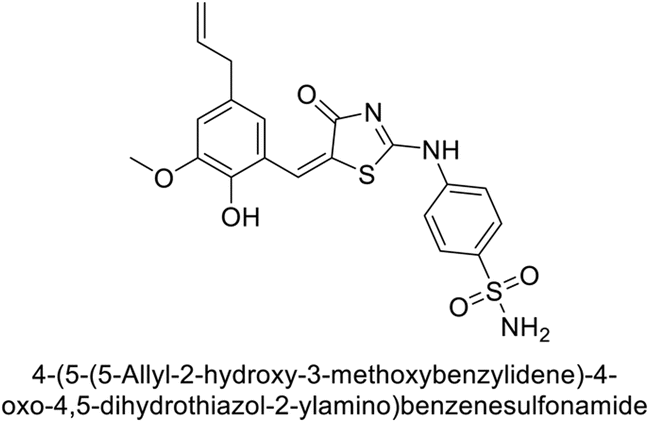 |
— | — | — | — | IC50 = 0.011 | — | — | 183 |
| 268 | 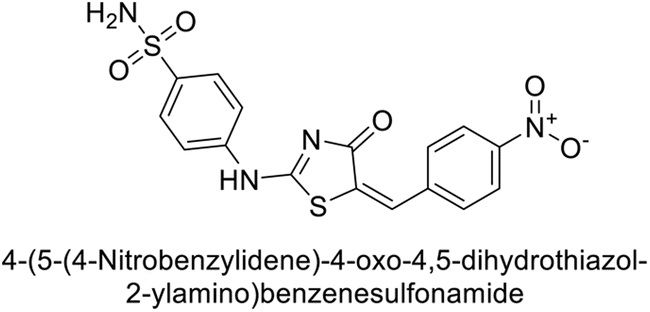 |
— | — | — | — | IC50 = 0.017 | — | — | 183 |
| 269 | 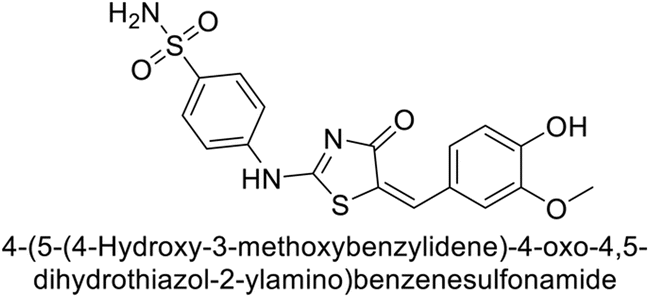 |
— | — | — | — | IC50 = 0.026 | — | — | 183 |
| 270 | 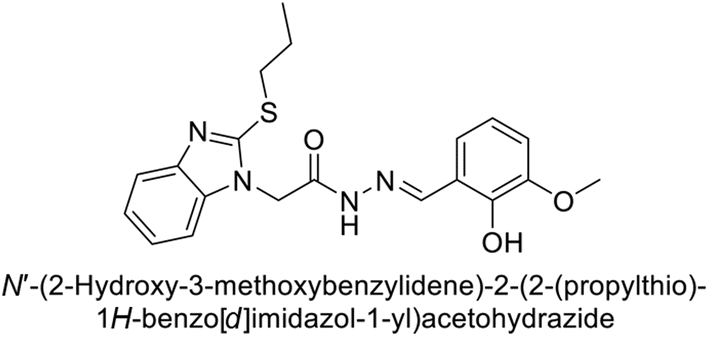 |
— | — | — | — | — | — | IC50 = 13.3 | 184 |
| 271 | 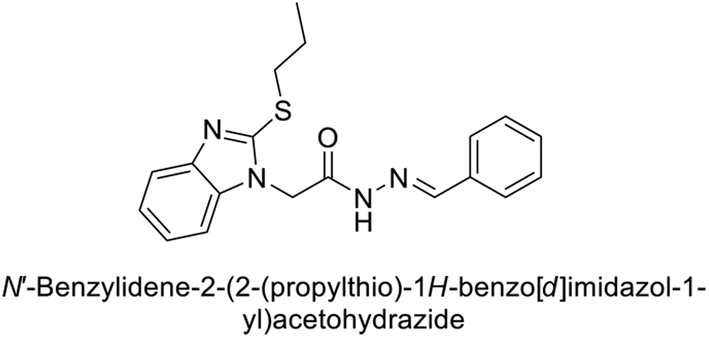 |
— | — | — | — | — | — | IC50 = 17.2 | 184 |
| 272 | 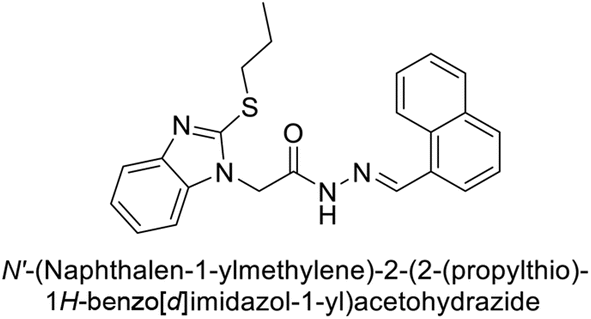 |
— | — | — | — | — | — | IC50 = 14.6 | 184 |
| 273 | 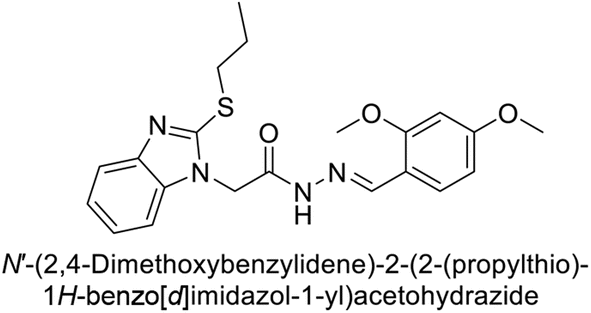 |
— | — | — | — | — | — | IC50 = 14.5 | 184 |
| 274 | 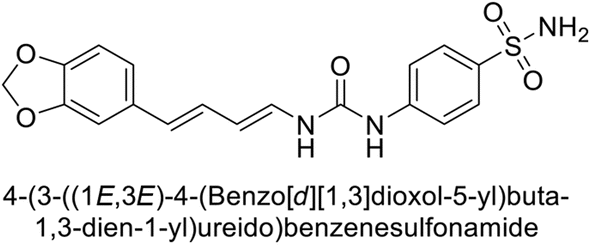 |
— | 93.4 | — | — | 38.7 | 57.5 | — | 185 |
| 275 | 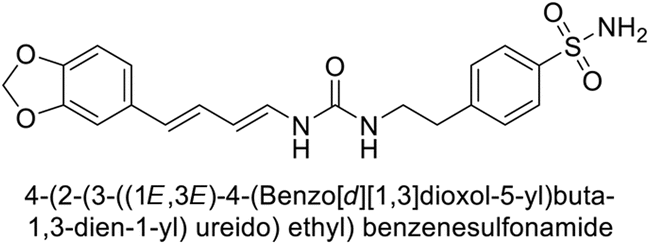 |
— | 88.6 | — | — | 68.2 | 45.6 | — | 185 |
| 276 |  |
— | — | — | — | — | — | IC50 = 0.0440 | 186 |
| 277 |  |
— | — | — | — | 1.57 | — | — | 187 |
| 278 | 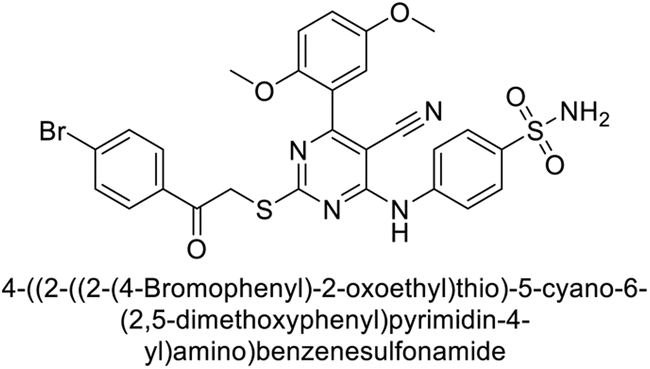 |
— | 1.72 | — | — | — | — | — | 188 |
| 279 | 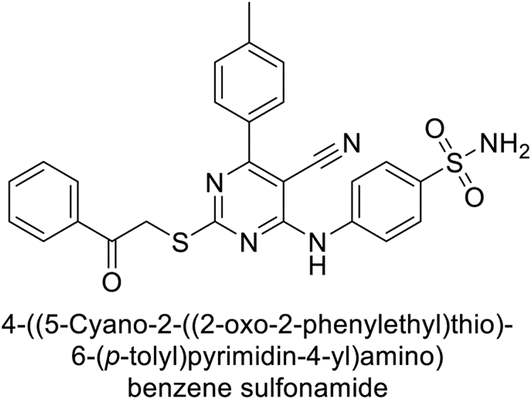 |
— | — | — | — | 7.4 | — | — | 188 |
| 280 | 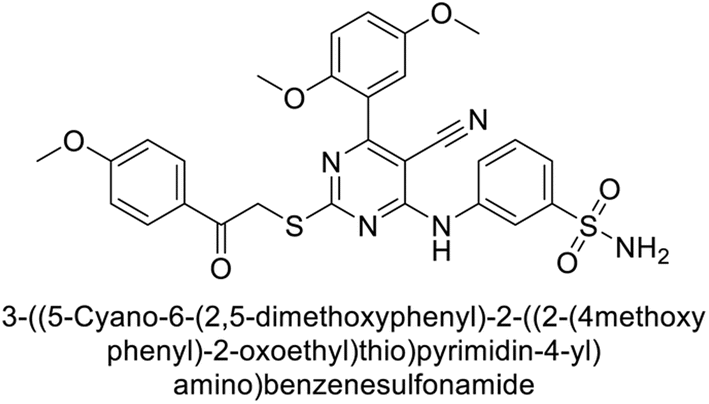 |
— | — | — | — | 7.0 | 4.67 | — | 188 |
| 281 | 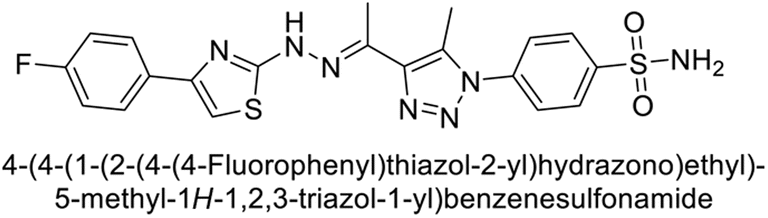 |
— | 18.1 | 5416 | — | 279.8 | — | — | 189 |
| 282 | 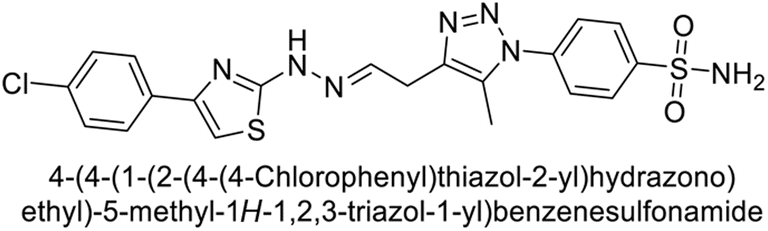 |
— | 14.1 | 8871 | — | 7893 | — | — | 189 |
| 283 | 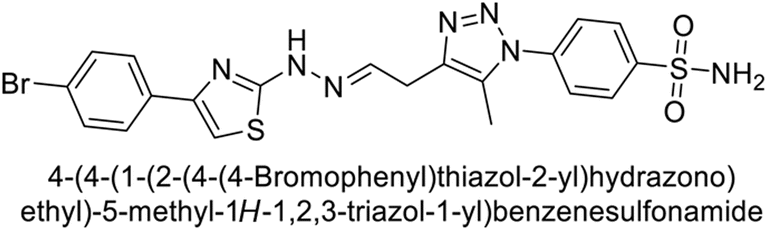 |
— | 14.9 | 8717 | — | 270.2 | — | — | 189 |
| 284 | 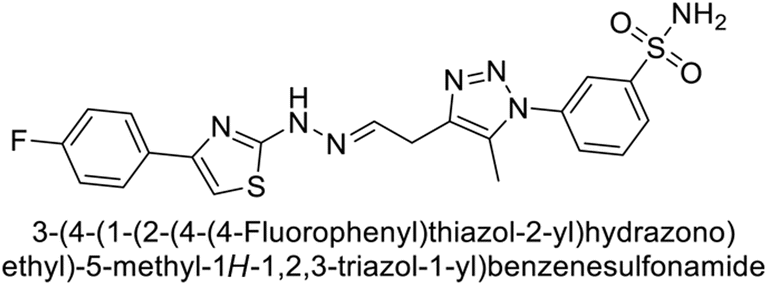 |
— | 17.8 | 88 | — | 113 | — | — | 189 |
| 285 | 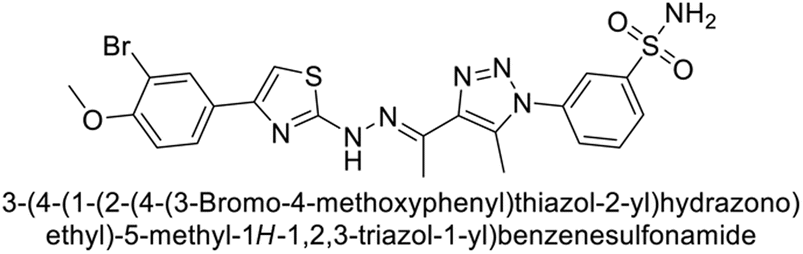 |
— | 810.4 | 5667 | — | 856 | — | — | 189 |
| 286 | 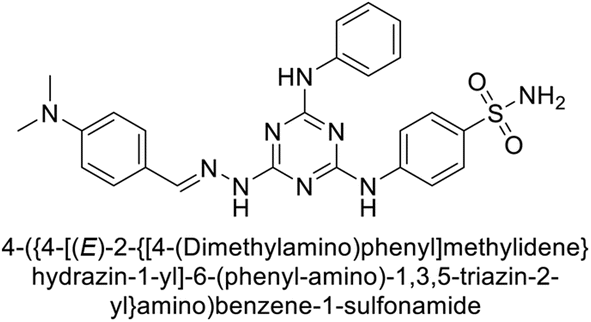 |
— | — | — | — | 13.3 | — | — | 190 |
| 287 | 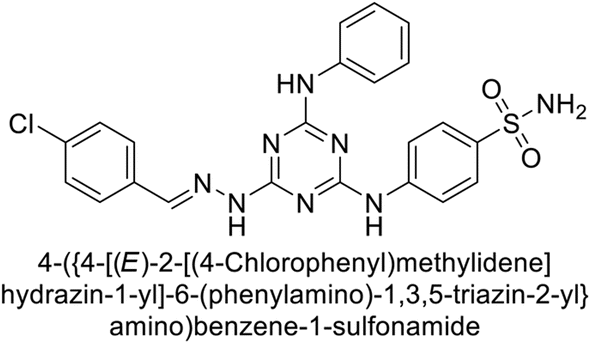 |
— | — | — | — | 22.6 | — | — | 190 |
| 288 | 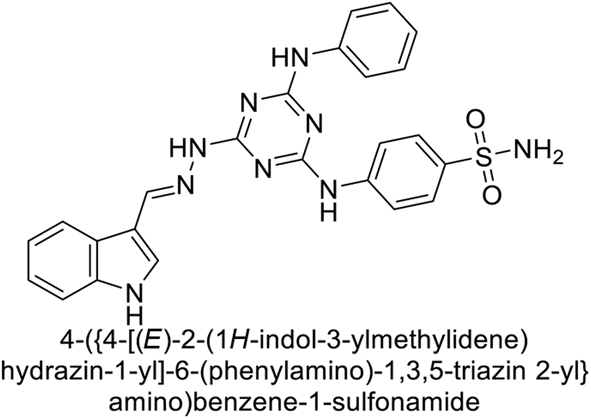 |
— | — | — | — | 25.8 | — | — | 190 |
| 289 | 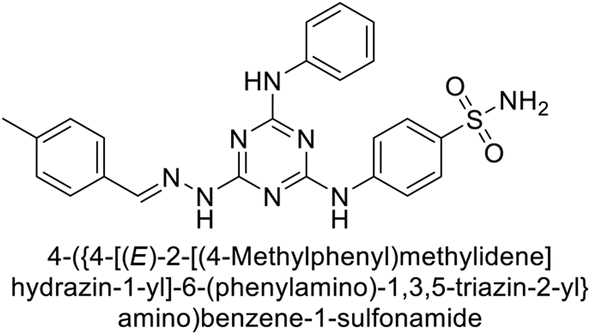 |
— | — | — | — | 26.9 | — | — | 190 |
| 290 | 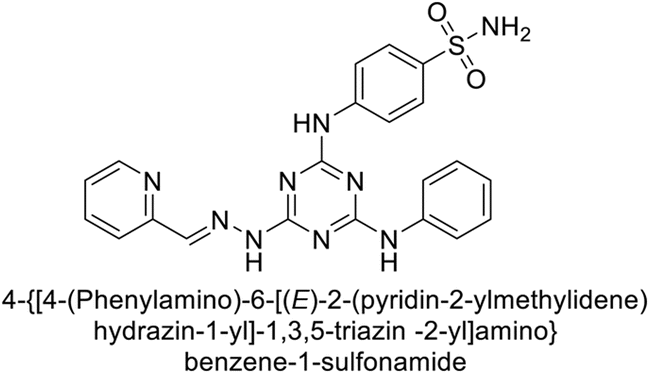 |
— | — | — | — | 27.2 | — | — | 190 |
| 291 | 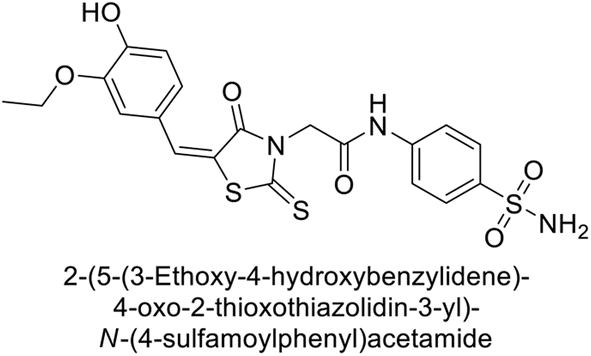 |
22.4 | — | — | — | — | — | — | 191 |
| 292 | 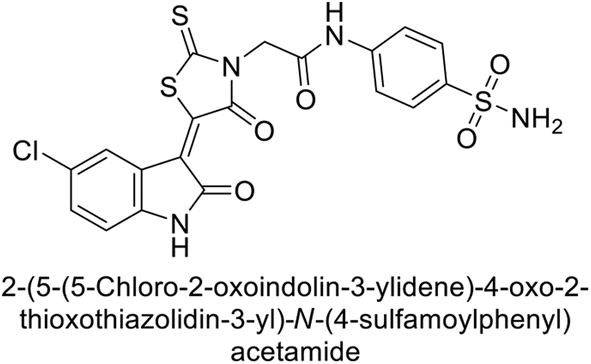 |
35.8 | — | — | — | — | — | — | 191 |
| 293 | 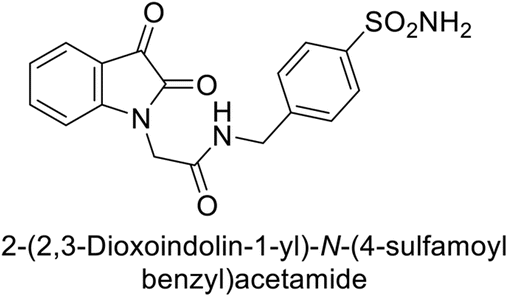 |
45.10 | 5.87 | — | — | 67.0 | 7.91 | — | 192 |
| 294 | 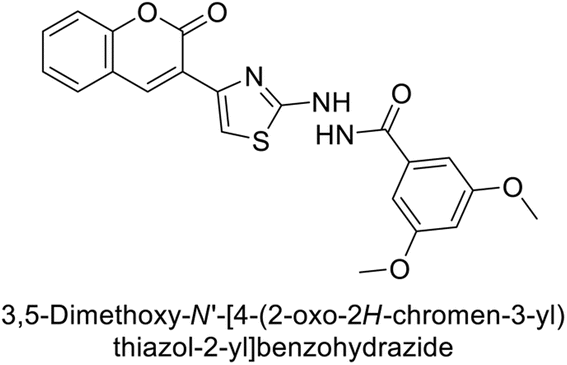 |
— | — | — | — | 505.6 | 91.1 | — | 193 |
| 295 | 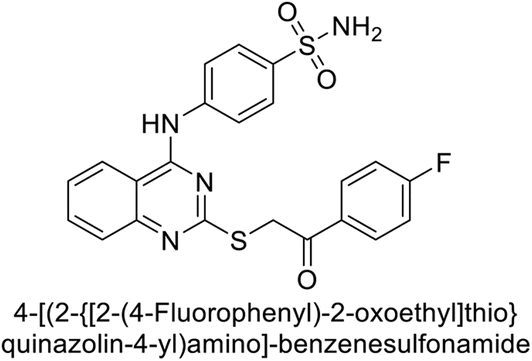 |
— | 0.09 | — | — | 0.32 | 0.58 | — | 194 |
| 296 | 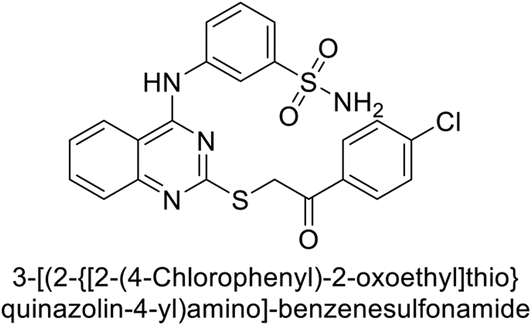 |
— | 0.05 | — | — | 0.47 | 0.46 | — | 194 |
| 297 | 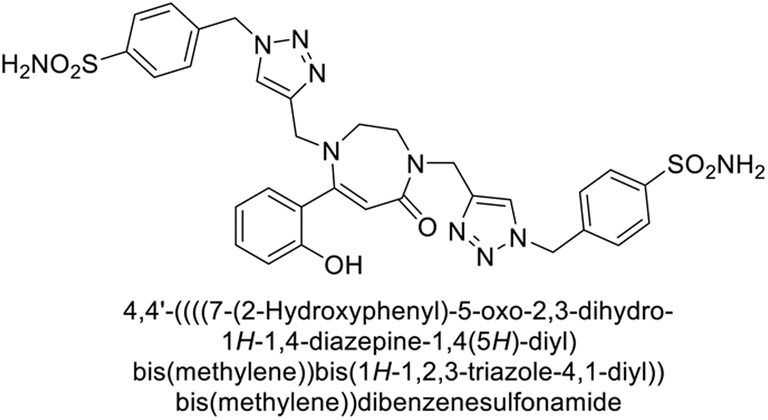 |
78.2 | 2.8 | 1013 | 8.9 | 13.2 | 6.5 | — | 195 |
| 298 | 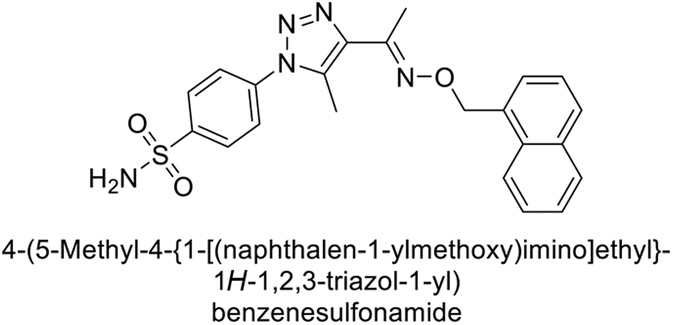 |
— | — | — | — | 68.6 | — | — | 196 |
| 299 | 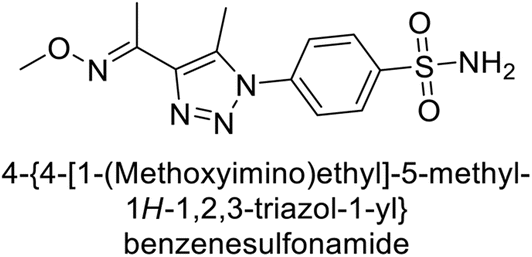 |
— | — | — | — | 56.3 | — | — | 196 |
| 300 | 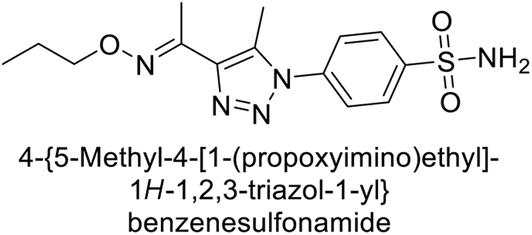 |
— | — | — | — | — | 95.6 | — | 196 |
| 301 | 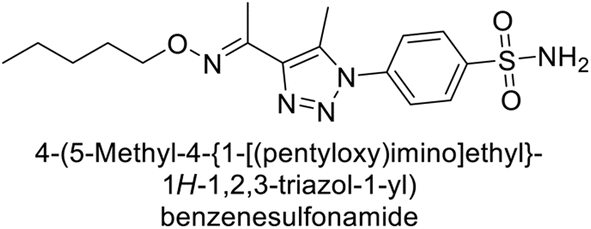 |
— | — | — | — | — | 51.5 | — | 196 |
| 302 | 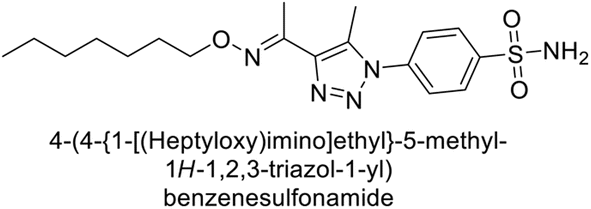 |
47.8 | — | — | — | — | — | — | 196 |
| 303 | 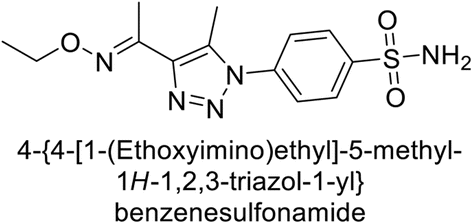 |
— | 33.2 | — | — | — | — | — | 196 |
| 304 | 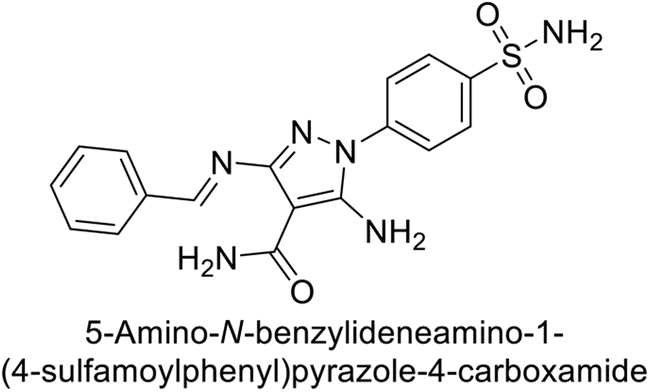 |
— | — | — | — | 38.4 | 21.6 | — | 197 |
| 305 | 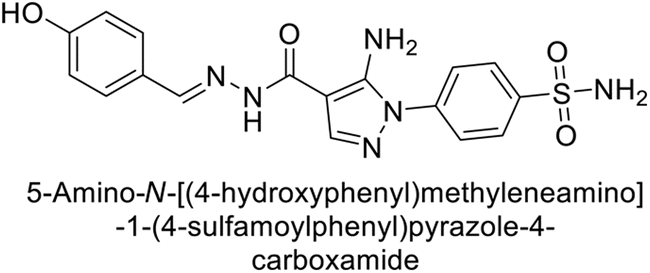 |
— | — | — | — | 48.9 | 5.6 | — | 197 |
| 306 | 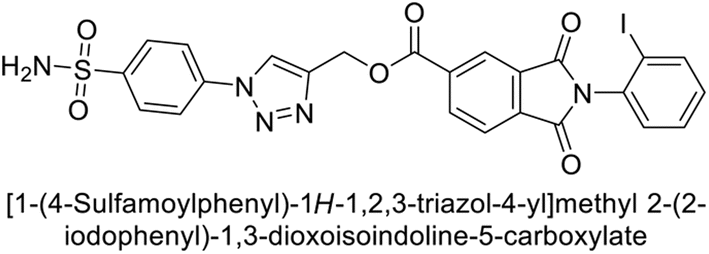 |
— | — | — | — | 105.0 | — | — | 198 |
| 307 | 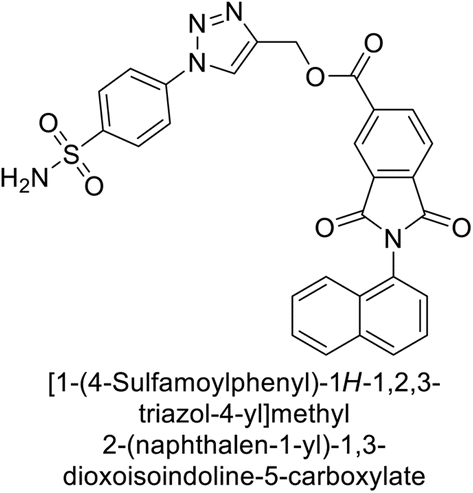 |
— | — | — | — | 32.1 | 9.2 | — | 198 |
| 308 | 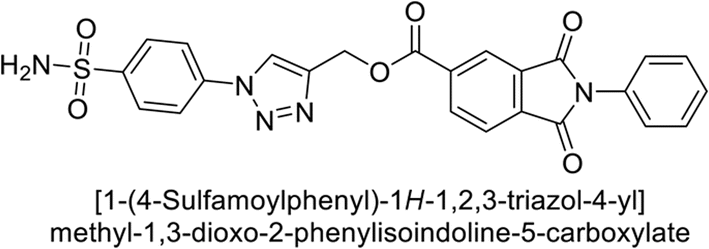 |
— | — | — | — | 18.2 | 50.1 | — | 198 |
| 309 | 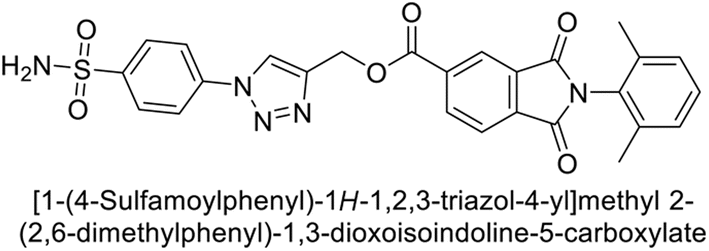 |
— | — | — | — | — | 50.6 | — | 198 |
| 310 | 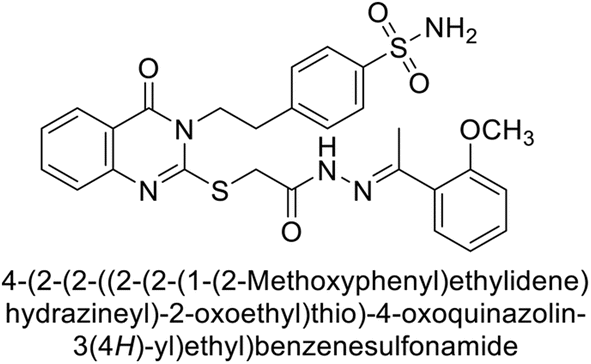 |
— | — | — | — | 8.9 | 64.8 | — | 199 |
| 311 | 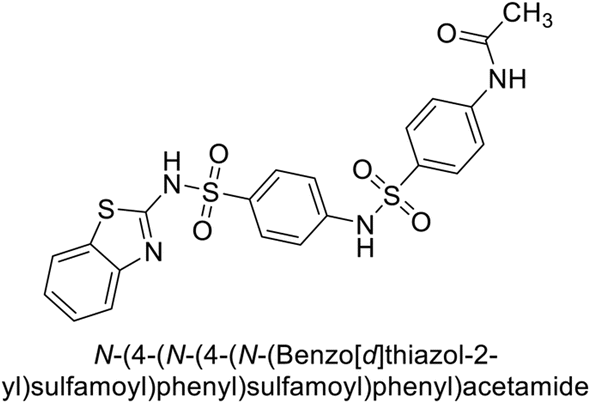 |
0.112 | 0.025 | — | — | — | — | — | 200 |
| 312 | 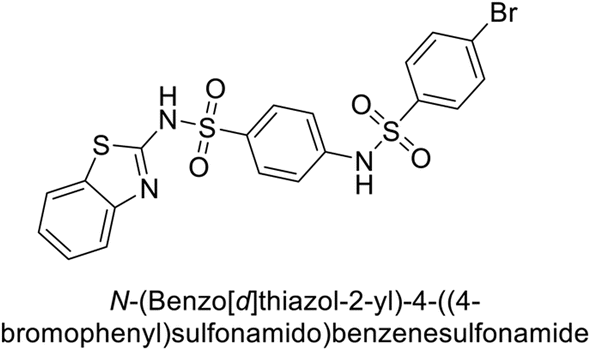 |
0.052 | 0.150 | — | — | — | — | — | 200 |
| 313 | 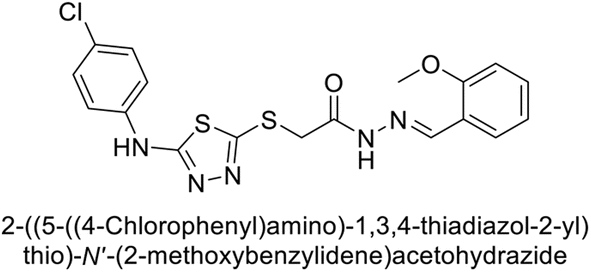 |
IC50 = 29.74 | — | — | — | — | — | — | 201 |
| 314 | 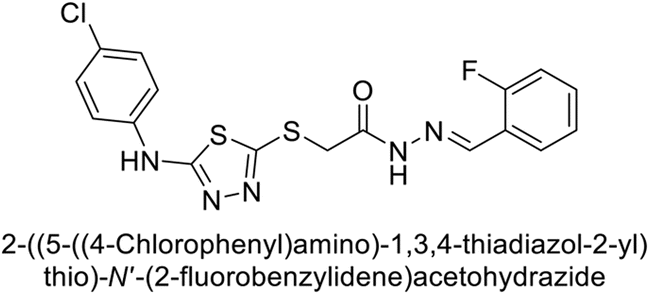 |
— | IC50 = 23.18 | — | — | — | — | — | 201 |
| 315 | 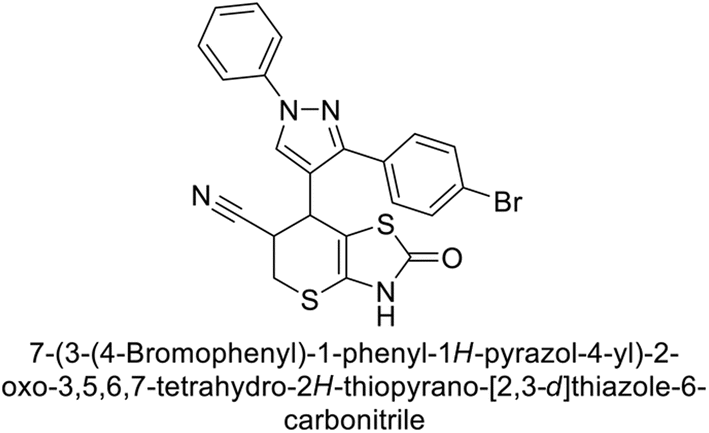 |
— | — | — | — | IC50 = 0.067 | — | — | 202 |
| 316 | 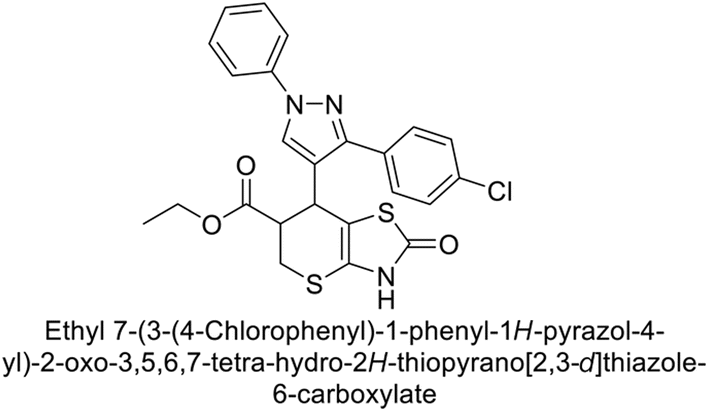 |
— | — | — | — | — | IC50 = 0.123 | — | 202 |
| 317 | 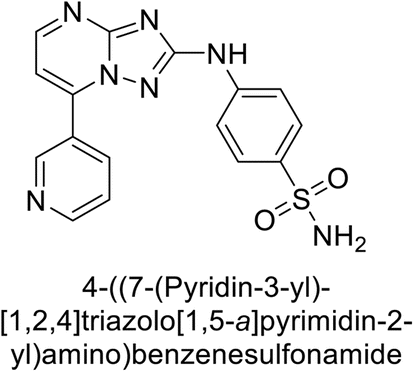 |
— | — | — | — | 5.1 | 8.8 | — | 203 |
| 318 | 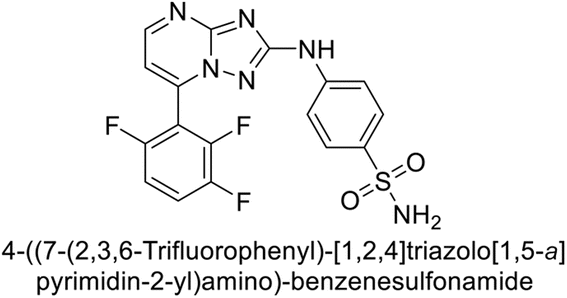 |
— | — | — | — | 8.6 | 5.4 | — | 203 |
| 319 | 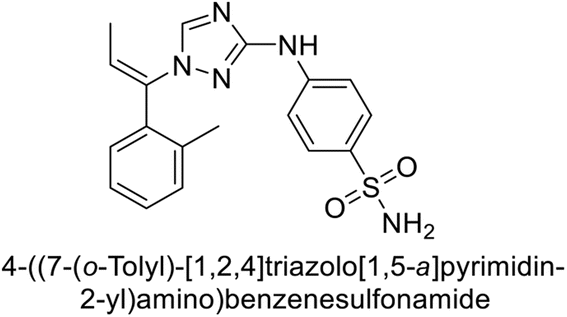 |
— | — | — | — | 4.7 | — | — | 203 |
| 320 | 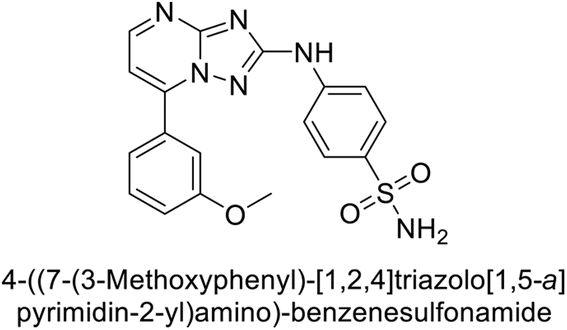 |
— | — | — | — | 5.1 | — | — | 203 |
| 321 | 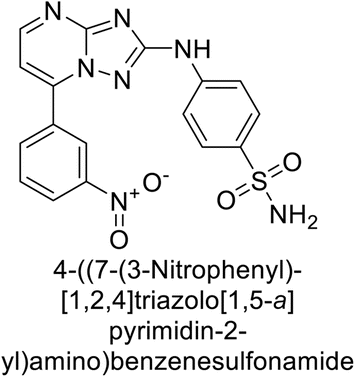 |
— | — | — | — | — | 4.3 | — | 203 |
| 322 | 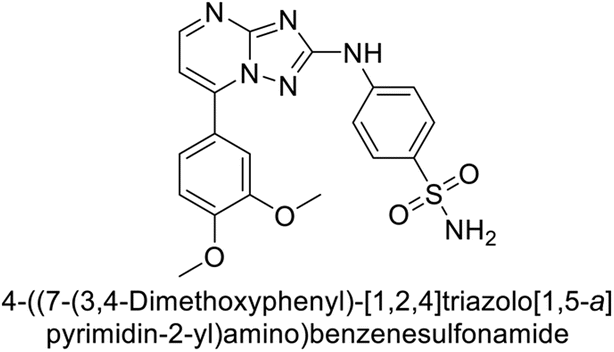 |
— | — | — | — | — | 9.0 | — | 203 |
| 323 | 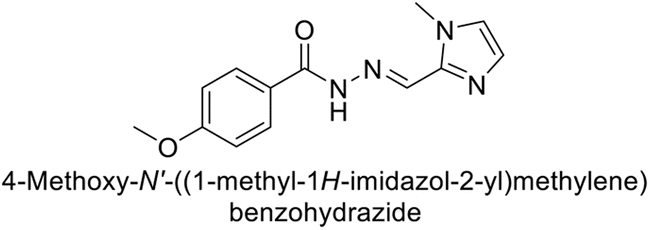 |
0.949 | — | — | — | — | — | — | 204 |
| 324 | 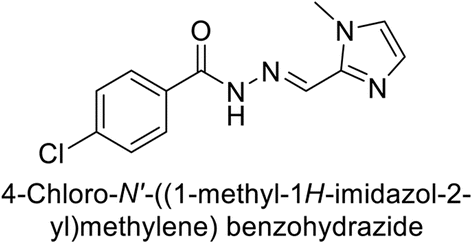 |
— | IC50 = 3.3 | — | — | — | — | — | 204 |
| 325 | 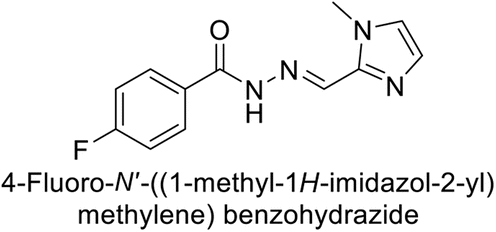 |
— | IC50 = 4.4 | — | — | — | — | — | 204 |
| 326 | 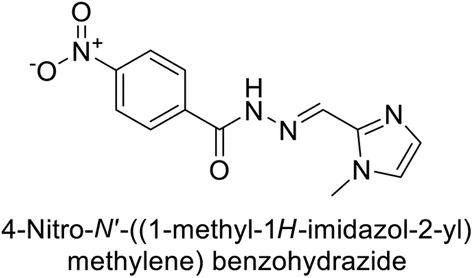 |
— | IC50 = 5.6 | — | — | — | — | — | 204 |
| 327 | 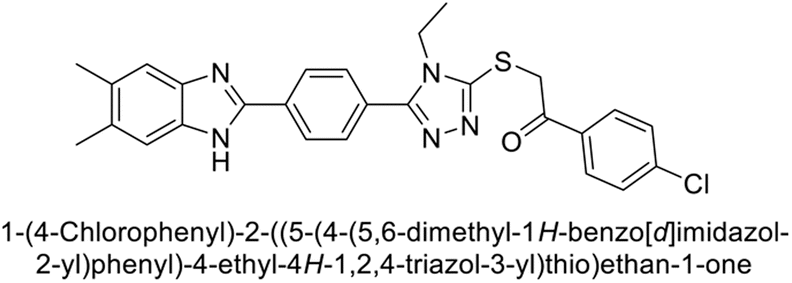 |
IC50 = 1.28 | — | — | — | — | — | — | 205 |
| 328 | 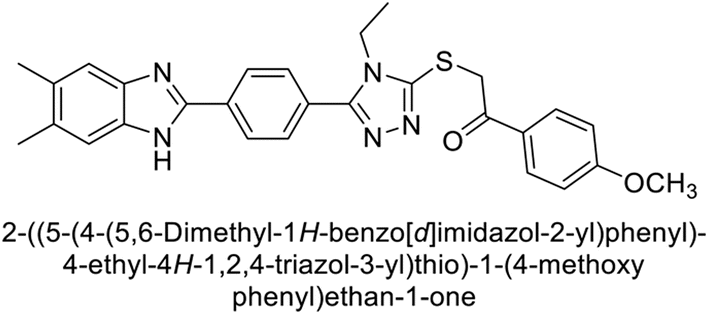 |
— | IC50 = 1.53 | — | — | — | — | — | 205 |
| 329 | 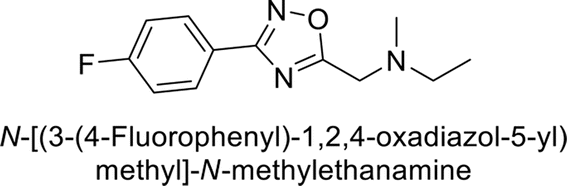 |
IC50 = 0.68 | IC50 = 0.65 | — | — | — | — | — | 206 |
| 330 |  |
IC50 = 0.96 | IC50 = 0.40 | — | — | — | — | — | 206 |
| 331 | 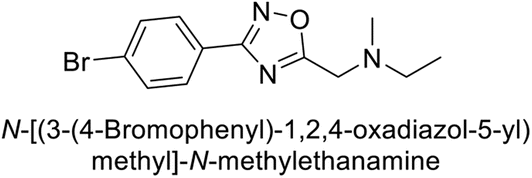 |
— | IC50 = 0.40 | — | — | — | — | — | 206 |
| 332 | 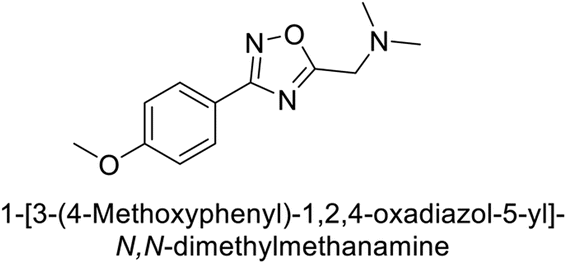 |
— | IC50 = 0.71 | — | — | — | — | — | 206 |
| 333 | 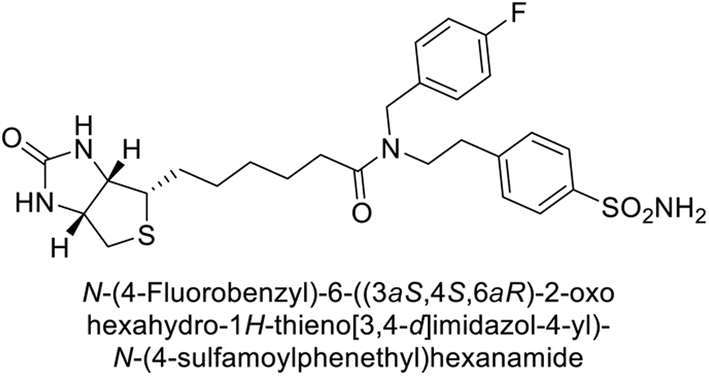 |
— | — | — | — | — | 4.5 | — | 207 |
| 334 | 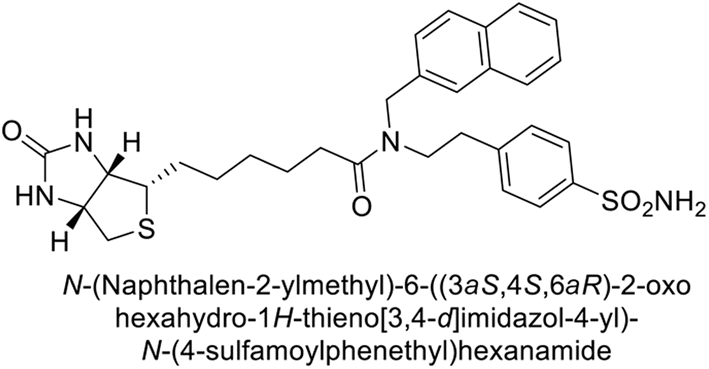 |
— | — | — | — | 6.2 | — | — | 207 |
| 335 | 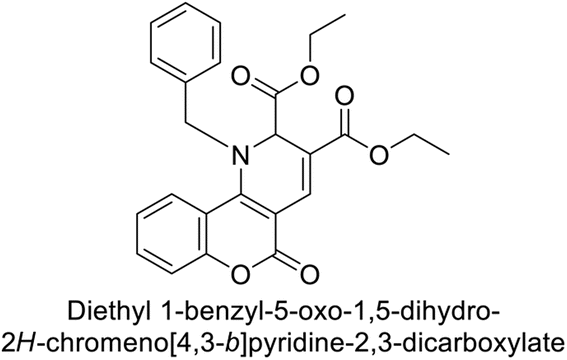 |
IC50 = 1.47 | IC50 = 1.58 | — | — | — | — | — | 208 |
| 336 | 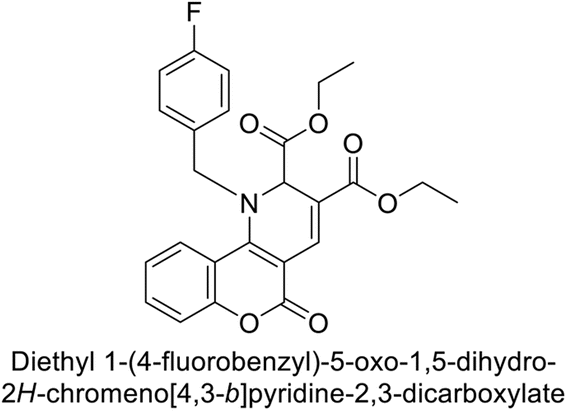 |
IC50 = 0.88 | IC50 = 1.10 | — | — | — | — | — | 208 |
| 337 | 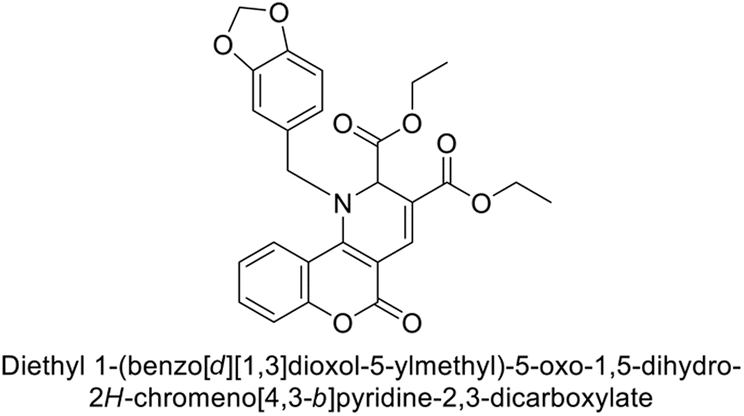 |
IC50 = 1.33 | IC50 = 1.53 | — | — | — | — | — | 208 |
| 338 | 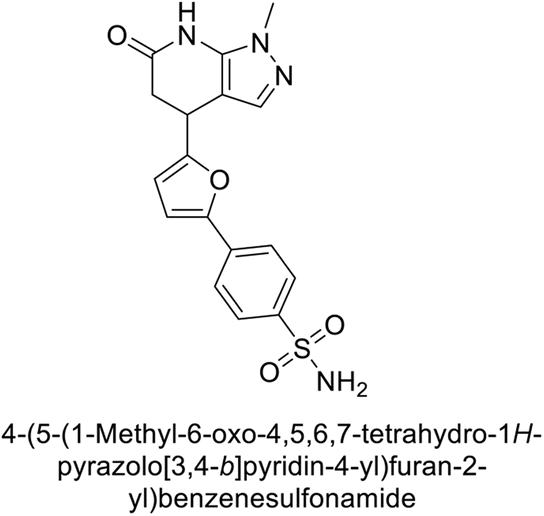 |
20.0 | 5.5 | — | — | — | — | — | 209 |
| 339 | 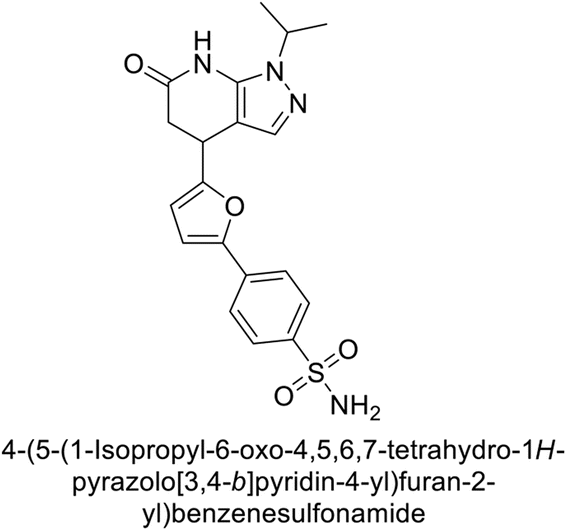 |
6.30 | 4.00 | — | — | — | — | — | 209 |
| 340 | 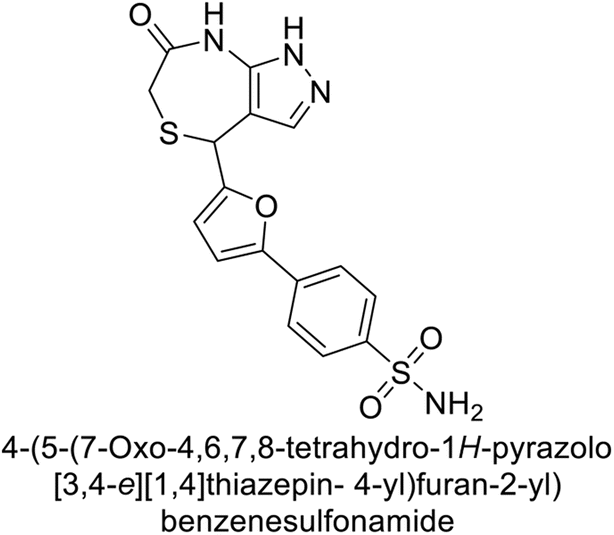 |
17.5 | 5.8 | — | — | — | — | — | 209 |
| 341 | 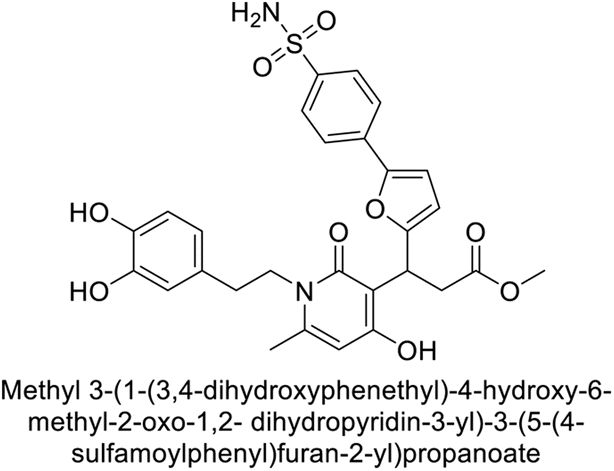 |
23.2 | 5.9 | — | — | — | — | — | 209 |
| 342 | 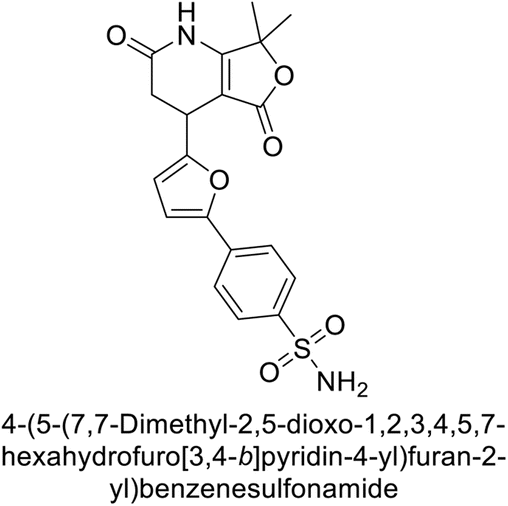 |
— | — | 6.5 | 16.8 | — | — | — | 209 |
| 343 | 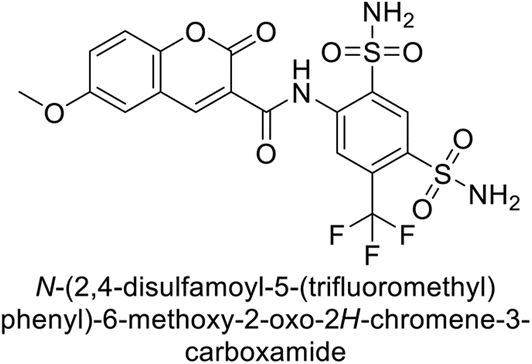 |
— | — | — | — | 21 | 5 | — | 210 |
| 344 |  |
— | — | — | — | 0.092 | — | — | 211 |
| 345 |  |
— | — | — | — | 0.069 | — | — | 211 |
| 346 |  |
— | — | — | — | — | IC50 = 0.173 | — | 211 |
| 347 | 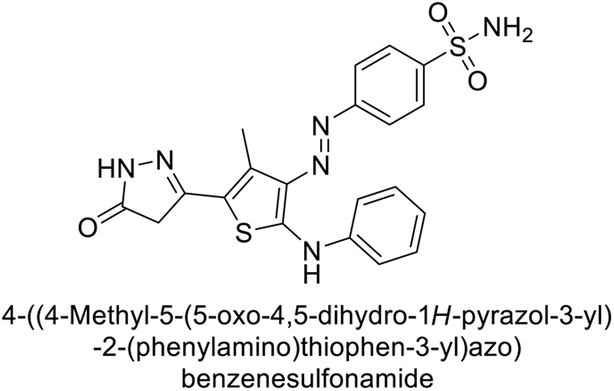 |
— | — | — | — | — | IC50 = 0.146 | — | 211 |
| 348 | 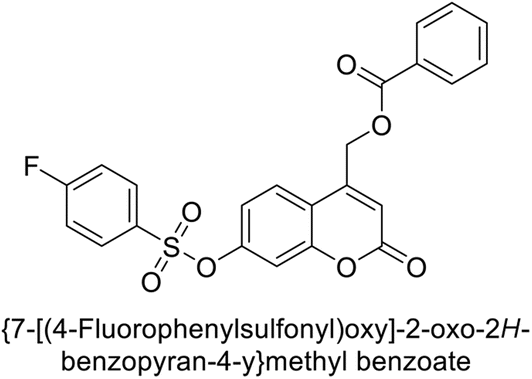 |
— | IC50 = 0.62 | — | — | — | — | — | 212 |
| 349 | 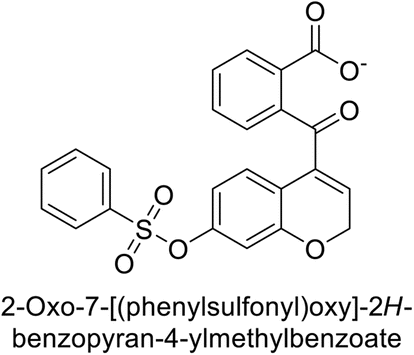 |
— | — | — | — | — | IC50 = 0.56 | — | 212 |
| 350 | 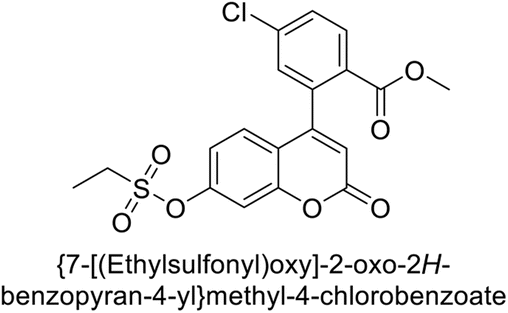 |
— | IC50 = 0.069 | — | — | — | — | — | 212 |
| 351 | 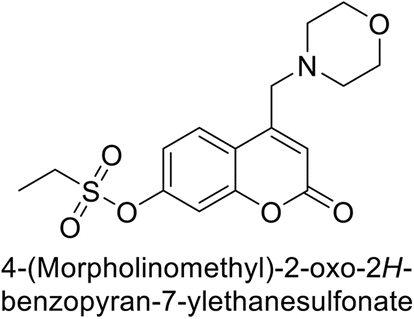 |
— | IC50 = 0.173 | — | — | — | — | — | 212 |
| 352 | 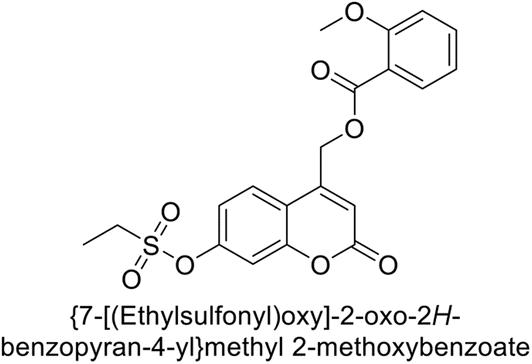 |
— | IC50 = 0.146 | — | — | — | — | — | 212 |
| 353 | 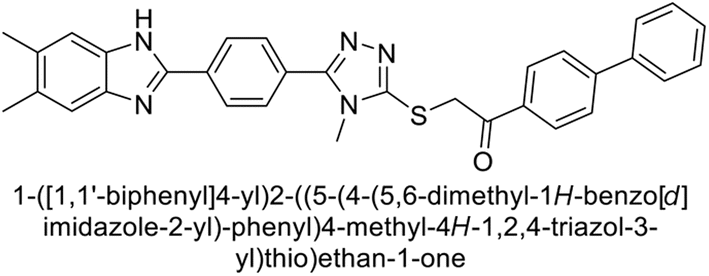 |
1.288 | 1.699 | — | — | — | — | — | 213 |
| 354 | 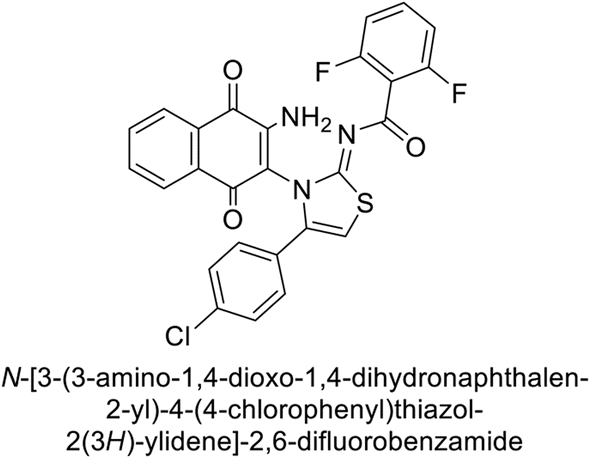 |
67.8 | 56.8 | — | — | — | — | — | 214 |
| 355 | 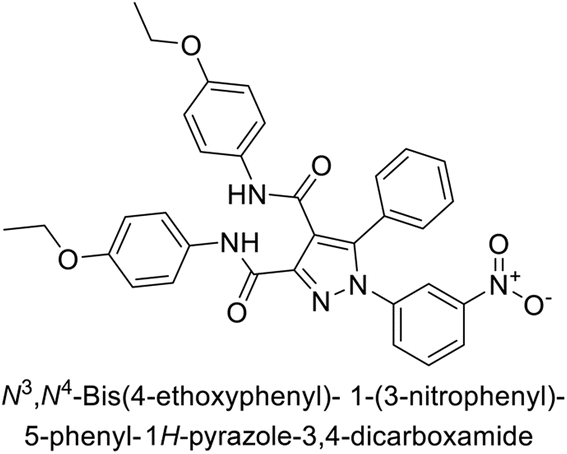 |
11.27 | — | — | — | — | — | — | 215 |
| 356 |  |
— | 9.97 | — | — | — | — | — | 215 |
| 357 |  |
— | — | — | — | — | 90.9 | — | 216 |
| 358 |  |
— | — | — | — | 0.063 | — | — | 217 |
| 359 | 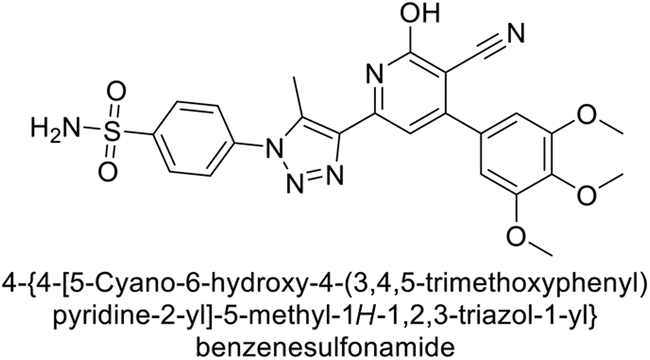 |
— | — | — | — | 0.036 | — | — | 217 |
| 360 | 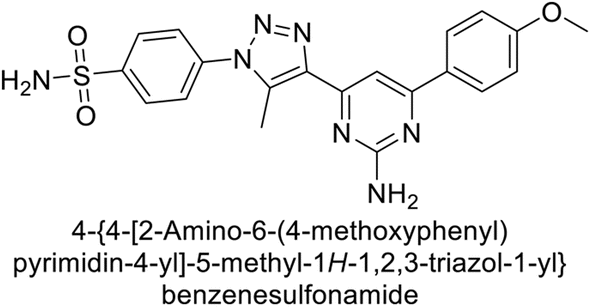 |
— | — | — | — | 0.06 | — | — | 217 |
| 361 | 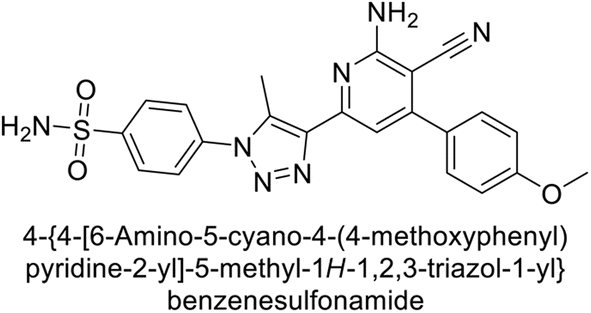 |
— | — | — | — | — | 0.021 | — | 217 |
| 362 | 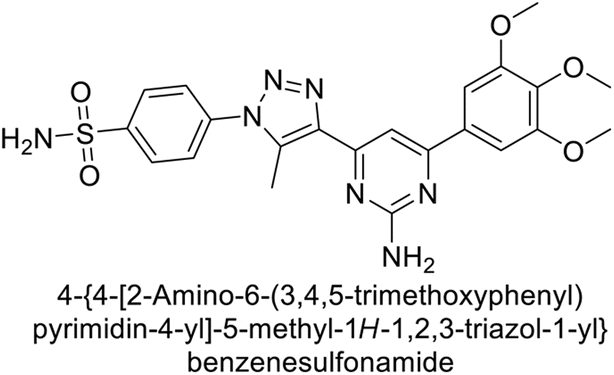 |
— | — | — | — | — | 0.022 | — | 217 |
| 363 | 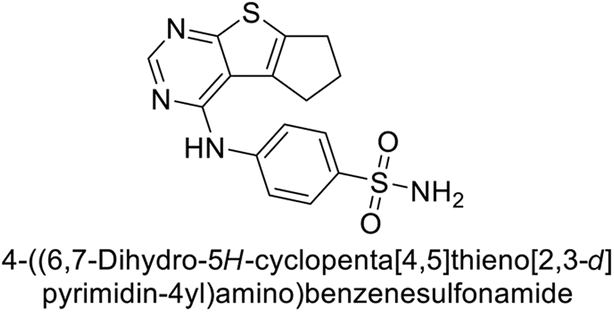 |
— | — | — | — | — | 20.2 | — | 218 |
| 364 | 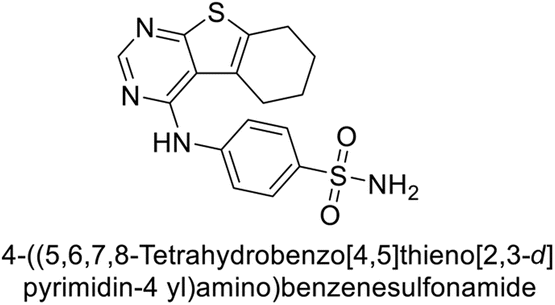 |
826.5 | — | — | — | — | — | — | 218 |
| 365 | 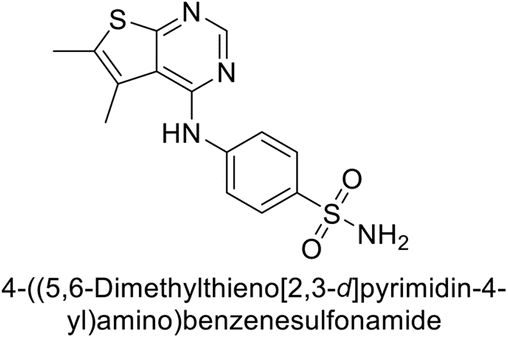 |
— | — | — | — | 16.9 | — | — | 218 |
| 366 | 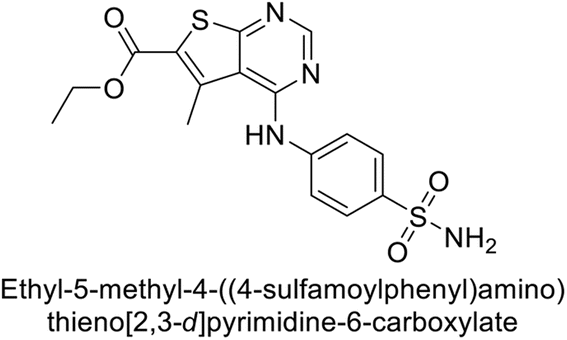 |
— | 8.6 | — | — | — | — | — | 218 |
| 367 | 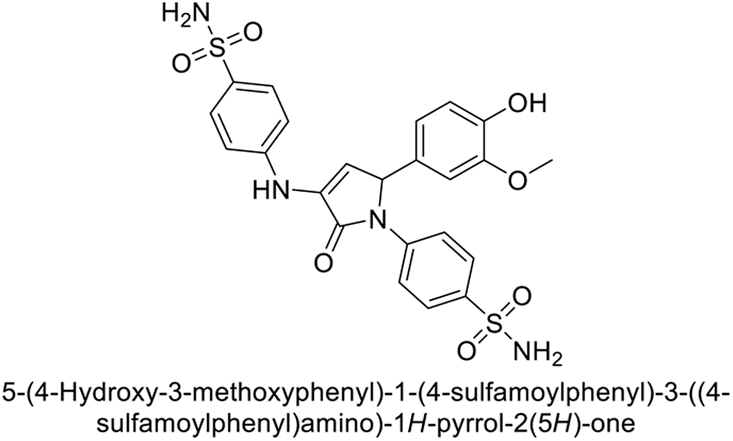 |
3.9 | — | — | — | — | — | — | 219 |
| 368 | 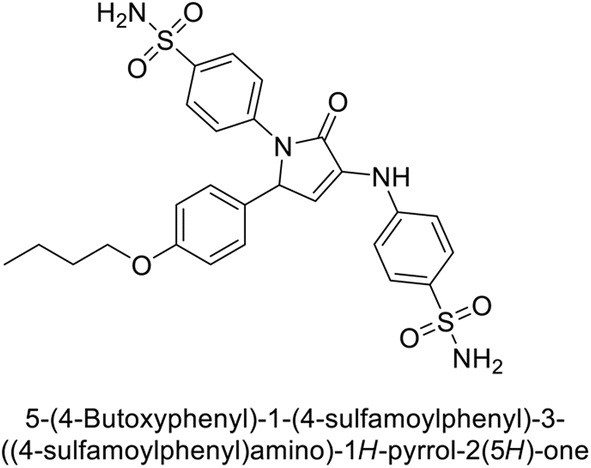 |
— | — | — | — | 1.9 | — | — | 219 |
| 369 | 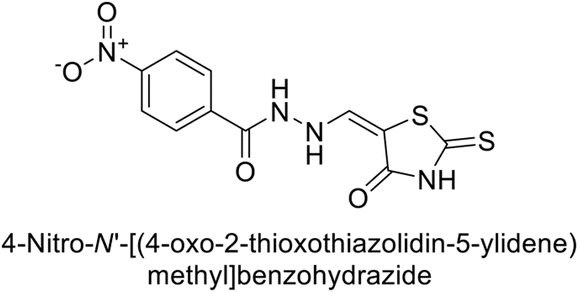 |
— | 9.5 | — | — | — | — | — | 220 |
| 370 | 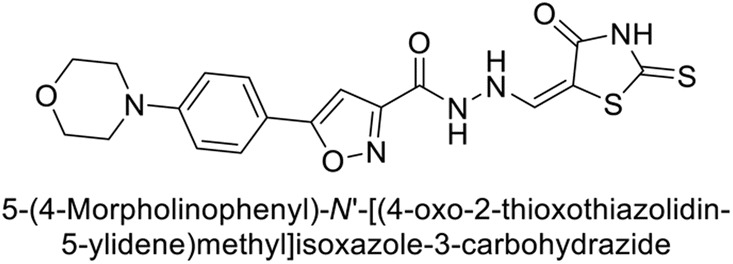 |
— | 63.9 | — | — | — | — | — | 220 |
| 371 |  |
— | — | — | — | 15.1 | — | — | 221 |
| 372 |  |
— | — | — | — | — | 17.2 | — | 221 |
| 373 |  |
435.8 | — | — | — | — | — | — | 222 |
| 374 |  |
956.4 | — | — | — | — | — | — | 222 |
| 375 | 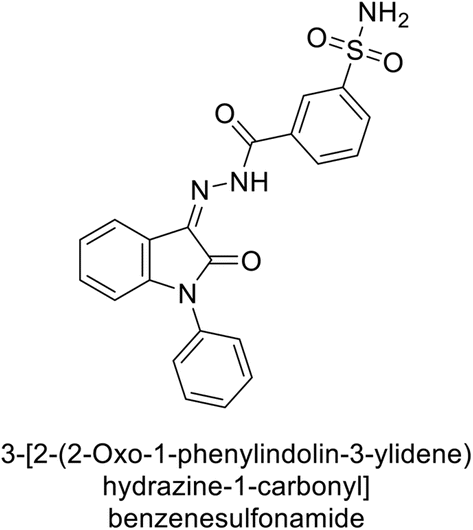 |
— | — | — | — | 60.5 | — | — | 222 |
| 376 | 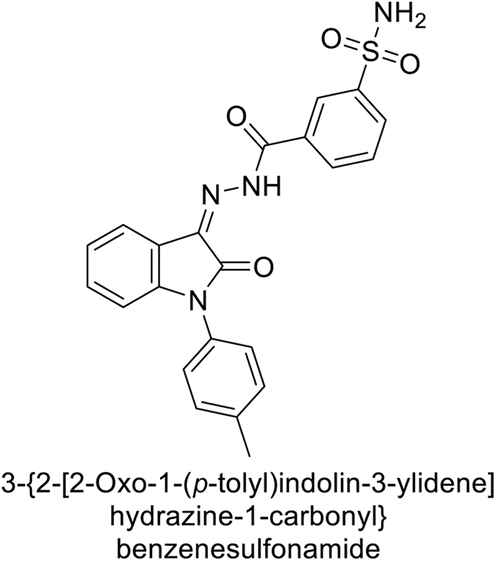 |
— | — | — | — | 95.6 | — | — | 222 |
| 377 | 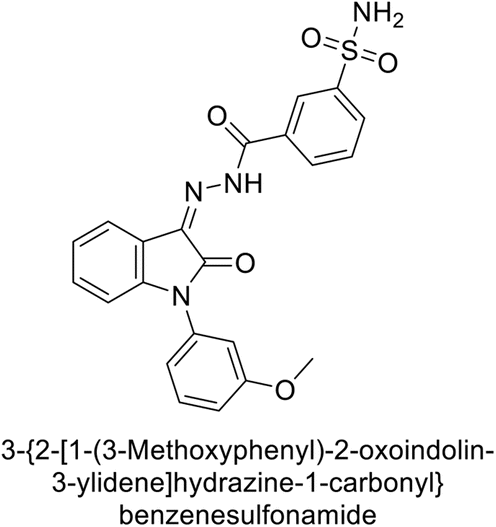 |
— | — | — | — | 92.1 | — | — | 222 |
| 378 | 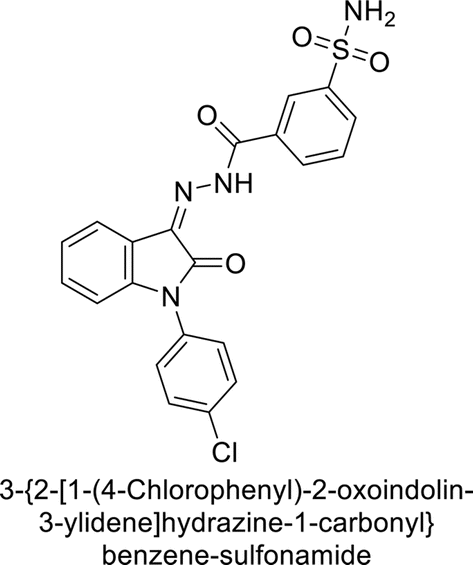 |
— | — | — | — | 75.4 | — | — | 222 |
| 379 | 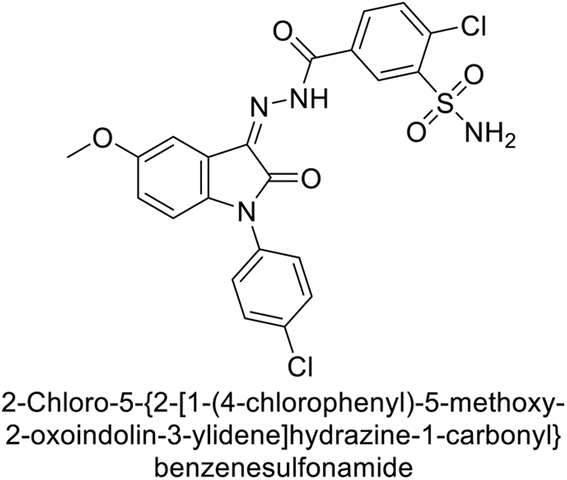 |
— | — | — | — | — | 84.5 | — | 222 |
| 380 | 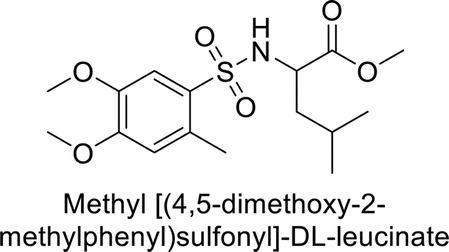 |
— | 39.2 | — | — | — | — | — | 223 |
| 381 | 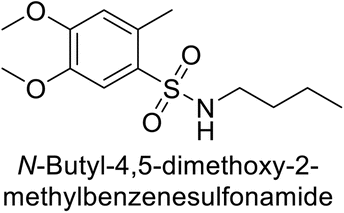 |
— | 50.9 | — | — | — | — | — | 223 |
| 382 | 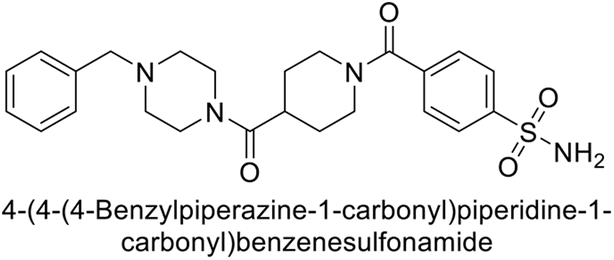 |
— | — | — | — | 8.3 | 2.7 | — | 224 |
| 383 | 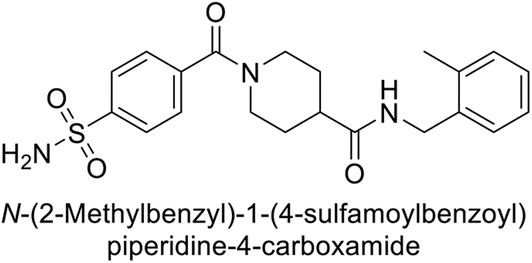 |
— | — | — | — | 8.4 | 6.9 | — | 224 |
| 384 |  |
— | 0.121 | — | — | — | — | — | 225 |
| 385 |  |
0.330 | 0.188 | — | — | — | — | — | 225 |
| 386 |  |
0.332 | 0.032 | — | — | — | — | — | 225 |
| 387 | 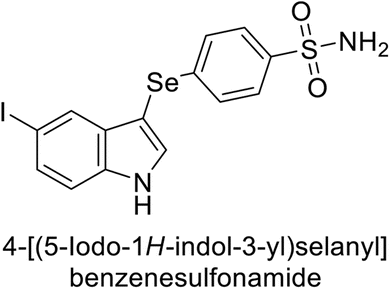 |
— | 6.9 | — | — | — | — | — | 226 |
| 388 | 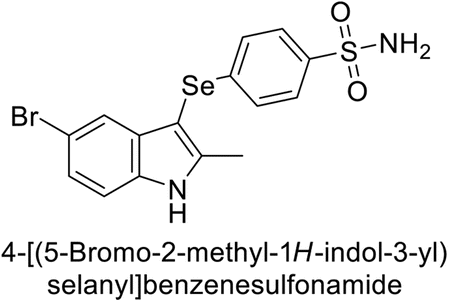 |
— | 6.2 | — | — | — | — | — | 226 |
| 389 | 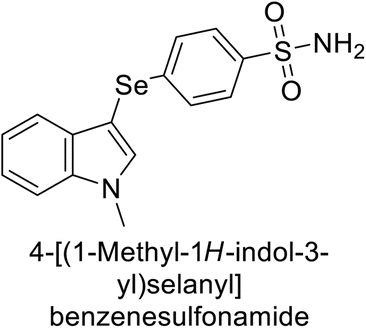 |
— | 6.0 | — | — | — | — | — | 226 |
| 390 | 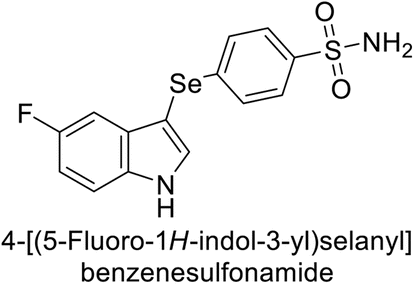 |
— | — | — | — | 2.9 | — | — | 226 |
| 391 | 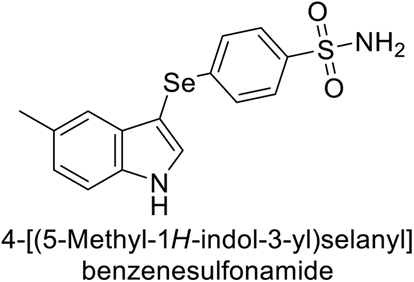 |
— | — | — | — | 2.1 | — | — | 226 |
| 392 | 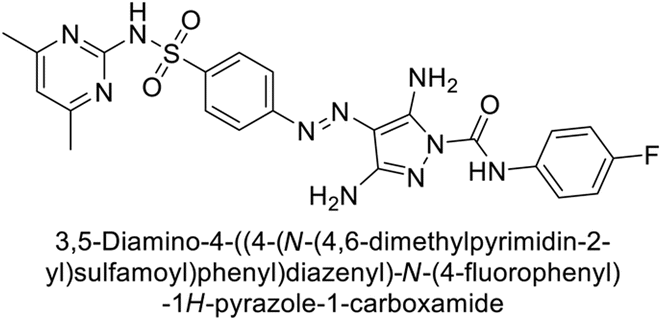 |
— | 0.188 | — | — | — | — | — | 227 |
| 393 | 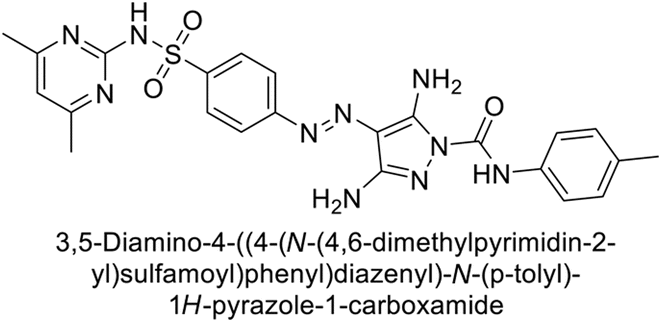 |
0.332 | — | — | — | — | — | — | 227 |
| 394 | 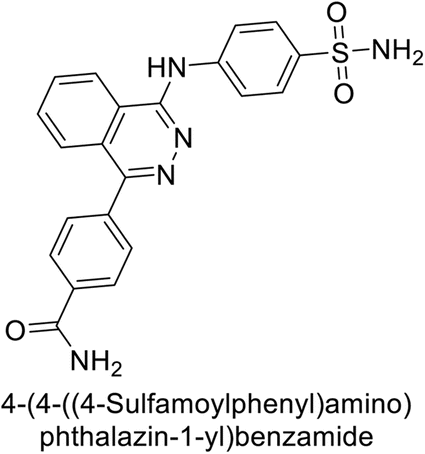 |
— | 1.6 | — | — | 3.7 | — | — | 228 |
| 395 | 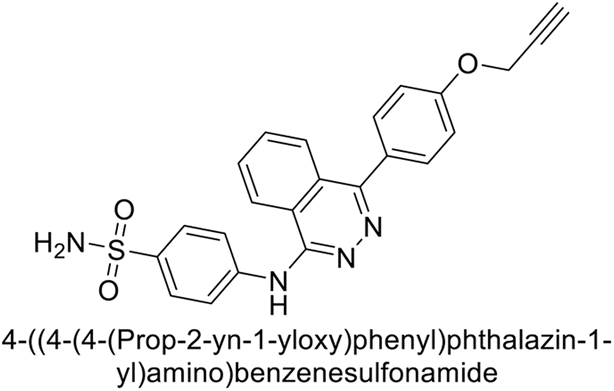 |
— | 52.8 | — | — | 8.9 | — | — | 228 |
| 396 | 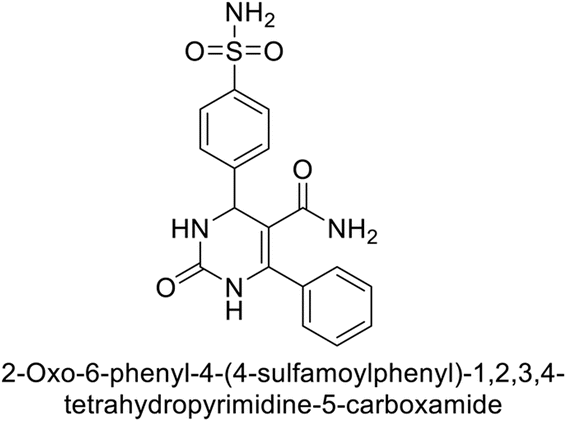 |
— | — | — | — | 6.6 | 8.9 | — | 229 |
| 397 | 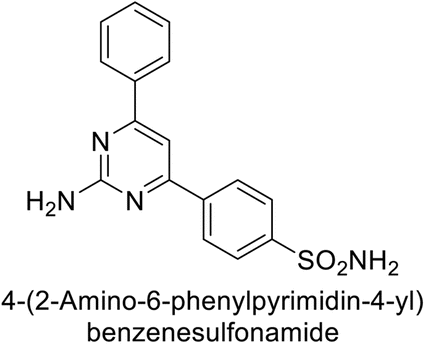 |
— | — | — | — | 8.9 | 5.3 | — | 229 |
| 398 | 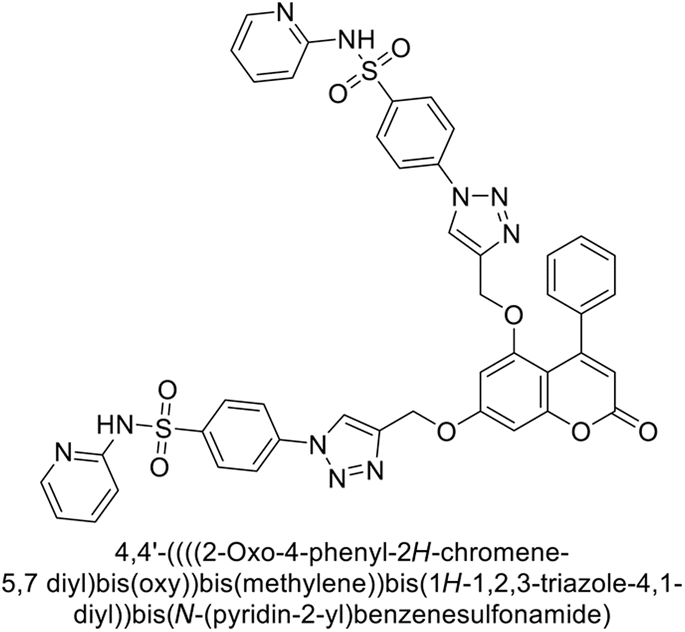 |
— | — | — | — | IC50 = 0.113 | — | — | 230 |
| 399 | 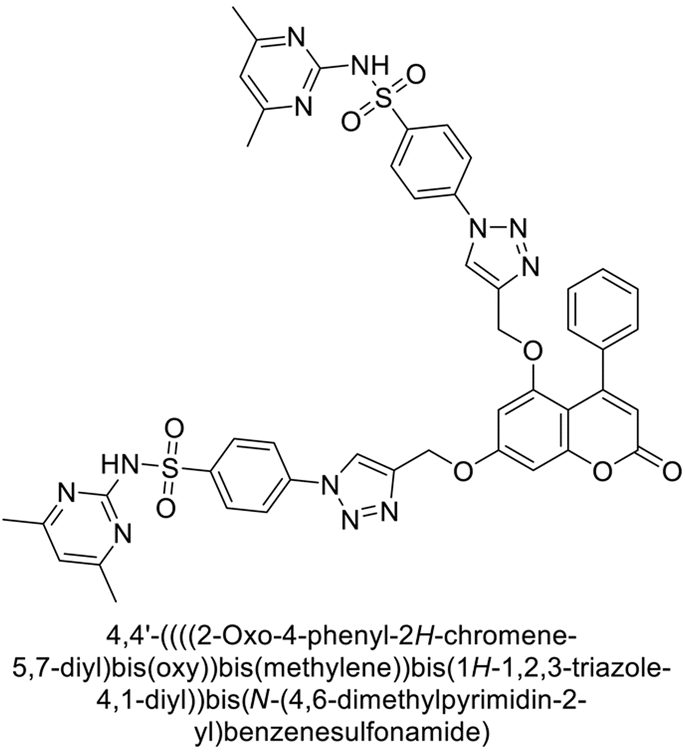 |
— | — | — | — | IC50 = 0.134 | — | — | 230 |
| 400 | 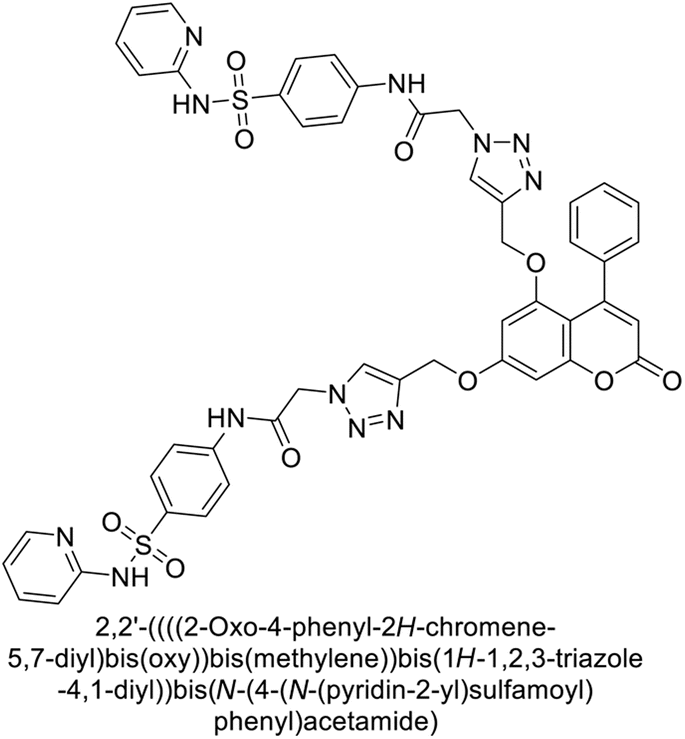 |
— | — | — | — | IC50 = 0.214 | — | — | 230 |
| 401 | 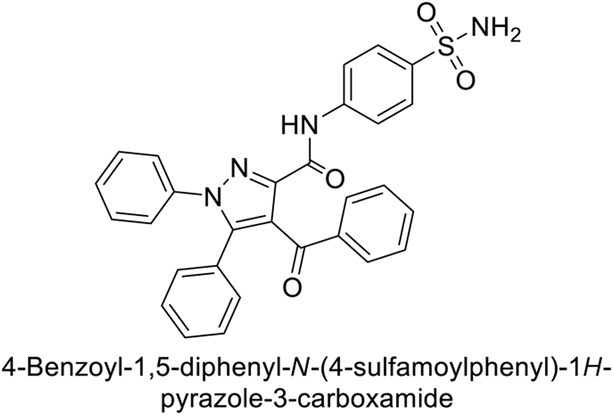 |
IC50 = 0.420 | IC50 = 0.310 | — | — | — | — | — | 231 |
Among the various heterocyclic scaffolds discussed, sulfonamide stand out as the most effective CAIs. Their established ability to coordinate directly with the zinc ion in the enzyme's active site contributes to their potent inhibitory activity, as reflected in low IC50/Ki values across multiple CA isoforms. Sulfonamides benefit from a well-defined structure–activity relationship, allowing for strategic modifications to enhance both potency and selectivity. While other scaffolds such as phthalazine, pyrimidine, triazole, and thiazole also exhibit promising inhibitory activities, their variations in binding affinity and effectiveness often depend on specific substituents and structural characteristics.
7. Structure–activity relationship based on different heterocyclic scaffolds
The SAR of different heterocyclic scaffolds-based CAIs can be extensively explored by considering the effects of various electron-donating (EDGs) and electron-withdrawing (EWGs) groups attached to the pyrazoline core. These substituents significantly impact the inhibitor's binding affinity, potency, and selectivity for different hCA isoforms. The interactions of these compounds with the active site of the enzyme can be further elucidated through molecular docking studies (Fig. 144).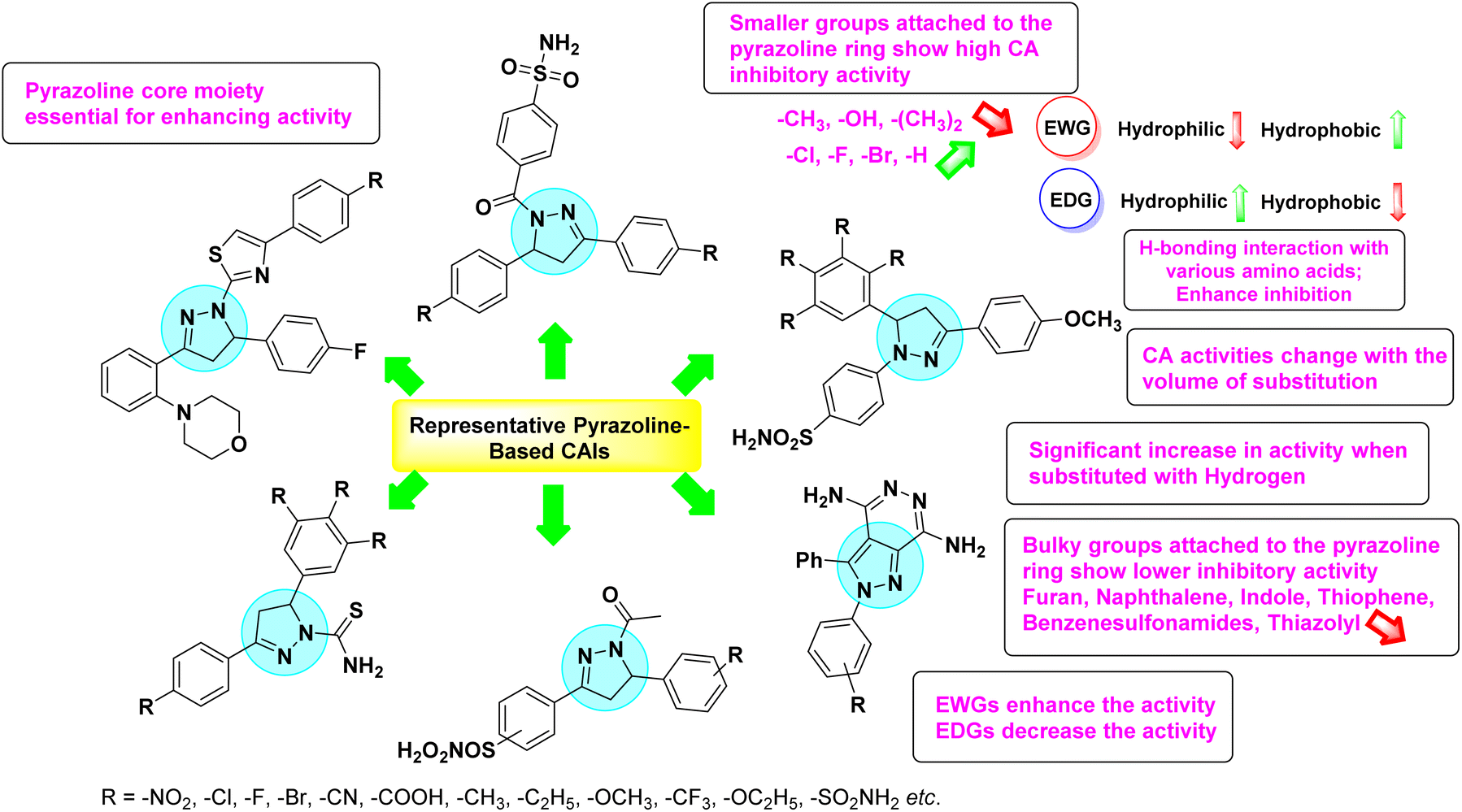 | ||
| Fig. 144 SAR of pyrazoline-based CAIs. | ||
7.1. Pyrazoline-based CAIs
The presence of hydroxyl or amino groups increases the potential for hydrogen bonding with amino acids like Thr199 and His94 in hCA-II or His64 in hCA-I. Such groups often enhance the selectivity and potency of CAIs, as they can establish strong hydrogen bonds with the active site. In molecular docking simulations, hydroxyl groups frequently participate in forming stable hydrogen bonds with key active site residues, improving the inhibitor's binding stability. These interactions also contribute to the overall efficacy of the compounds.
The carboxyl and sulfonamide groups are known to significantly improve binding affinity due to their ability to engage in both hydrogen bonding and ionic interactions with residues in the enzyme's active site. Sulfonamide derivatives are especially potent due to their ability to chelate the zinc ion, a key mechanism in CA inhibition. Molecular docking studies typically show that sulfonamide groups form strong coordinate bonds with the zinc ion, mimicking the interaction of classical CA inhibitors like AAZ. The sulfonamide group's interaction with the zinc ion is crucial for effective inhibition, especially for tumor-associated isoforms such as hCA-IX and XII.
7.1.3.1. Interaction with zinc ion. Most pyrazoline-based CAIs rely on functional groups such as sulfonamides or carboxylates to interact directly with the zinc ion in the active site. Docking studies frequently show that the sulfonamide moiety binds to the zinc ion via one of its oxygen atoms, displacing a water molecule and effectively inhibiting the enzyme's catalytic activity.
Compounds with carboxamide groups also demonstrate effective inhibition, with docking simulations revealing coordination between the carboxyl group and the zinc ion, enhancing the compound's inhibitory potency.
7.1.3.2. Hydrophobic interactions. Docking simulations illustrate that the hydrophobic interactions between EDGs like alkyl or alkoxy chains and hydrophobic residues (such as Val121, Leu198, and Phe131 in hCA-II) stabilize the pyrazoline scaffold within the active site. These interactions are particularly important for enhancing the selectivity of the compounds toward specific hCA isoforms. Pyrazoline derivatives with bulky substituents such as phenyl groups tend to occupy hydrophobic pockets within the enzyme, promoting favorable interactions that enhance the inhibitor's binding affinity and potency.
7.1.3.3. Hydrogen bonding. Hydrogen bonds formed between the inhibitor and active site residues are critical for stabilizing the inhibitor–enzyme complex. Pyrazoline CAIs with hydroxyl, amino, or carboxyl groups often form hydrogen bonds with residues such as Thr199, Glu106, or His94 in hCA-II. These bonds are observed in docking studies to significantly increase binding affinity. The positioning of electron-donating or withdrawing groups in relation to the hydrogen bonding residues in the enzyme is a determining factor in the compound's inhibitory efficiency.
7.2. Triazole and thiazole-based CAIs
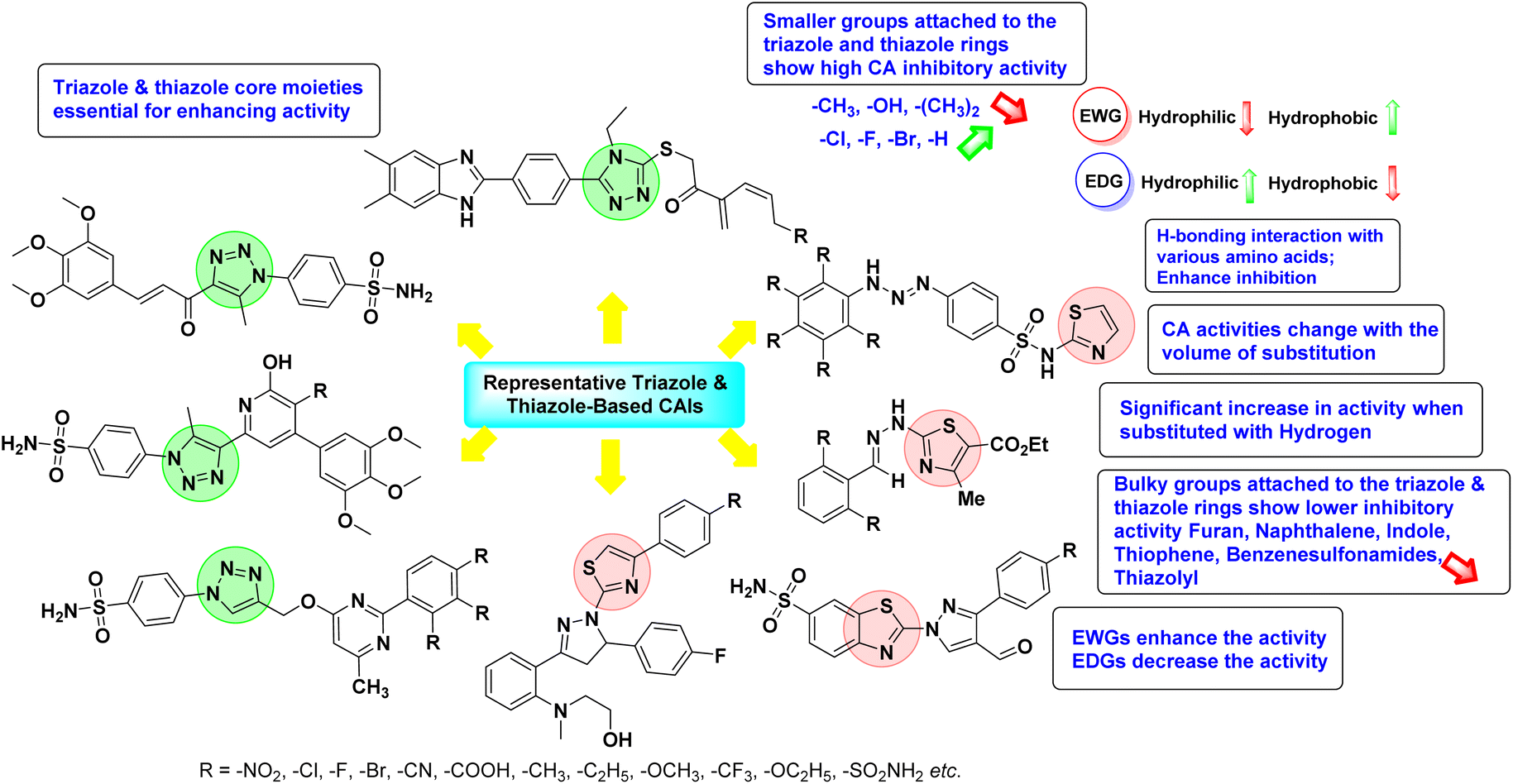 | ||
| Fig. 145 SAR of triazole and thiazole-based CAIs. | ||
7.2.1.1. Electron-donating groups. Alkyl groups such as methyl (–CH3) or ethyl (–C2H5) attached to the triazole ring tend to increase the electron density of the ring, enhancing hydrophobic interactions with the enzyme's active site. These interactions typically involve nonpolar residues in the enzyme, such as Val121 and Leu198 in hCA-II. Docking studies often show that these EDGs stabilize the triazole ring within the hydrophobic pocket of the enzyme, improving the binding affinity and selectivity of the inhibitor, particularly for isoforms like hCA-I and hCA-II.
The presence of hydroxyl or amino groups introduces the possibility of forming hydrogen bonds with key active site residues, such as Thr199 and His94 in hCA-II. These interactions are critical for enhancing the binding affinity and specificity of the inhibitor. Molecular docking simulations often reveal that these groups engage in strong hydrogen bonds, leading to enhanced stabilization of the inhibitor within the active site. This interaction is particularly beneficial for improving selectivity towards certain hCA isoforms.
7.2.1.2. Electron-withdrawing groups. Halogens, particularly fluorine (–F) and chlorine (–Cl), when attached to the triazole ring, decrease the electron density, which can strengthen interactions with the zinc ion at the enzyme's active site. These substituents also contribute to enhanced van der Waals interactions with hydrophobic residues. Docking studies often show that halogen atoms participate in halogen bonding with residues such as Thr199 or zinc-bound water molecules, which stabilizes the inhibitor and increases its potency against isoforms like hCA-IX and hCA-XII.
Strong EWGs like nitro (–NO2) and cyano (–CN) enhance the affinity of triazole-based CAIs for the zinc ion by creating a more favorable electronic environment for interaction with the metal center. Docking studies demonstrate that these groups often form electrostatic interactions with the zinc ion or polar residues near the active site, such as His64 or His94. This leads to stronger inhibition, particularly against tumor-associated hCA isoforms, where the zinc ion plays a crucial role in enzymatic activity.
7.2.2.1. Electron-donating groups. The addition of alkyl groups (methyl and isopropyl) to the thiazole ring increases hydrophobic character, which can enhance the interaction of the inhibitor with nonpolar residues in the enzyme's active site. Docking studies often show that alkyl-substituted thiazoles engage in van der Waals interactions with hydrophobic residues such as Leu198 and Val121 in hCA-II, contributing to better stabilization within the active site.
These groups hydroxyl (–OH) and alkoxy (–OR) can form hydrogen bonds with polar residues in the active site, enhancing the binding affinity of the inhibitor. The presence of a hydroxyl group on the thiazole ring may allow interaction with residues such as Thr199 or Glu106 in hCA-II, enhancing specificity. Docking simulations suggest that these groups stabilize the inhibitor through hydrogen bonding and may also participate in water-mediated interactions with the zinc ion, leading to improved inhibitory potency.
7.2.2.2. Electron-withdrawing groups. Halogen atoms (–Cl, –Br), when attached to the thiazole ring, can participate in both halogen bonding and electrostatic interactions with residues near the zinc ion. This is particularly true for fluorine, which can increase binding affinity by forming stable interactions with polarizable residues or the zinc ion. Molecular docking studies frequently reveal that halogenated thiazoles exhibit stronger binding affinities due to these interactions, particularly with hCA-IX and XII, where halogen atoms can enhance the inhibitor's potency by stabilizing the compound in the active site.
The introduction of nitro or sulfonamide groups enhances the binding affinity of thiazole-based CAIs by facilitating strong interactions with the zinc ion in the active site. The sulfonamide group, in particular, is well-known for its ability to chelate the zinc ion, which is a key mechanism of action for many CAIs. Docking studies show that these EWGs enhance the inhibitory activity by forming coordinate bonds with the zinc ion and additional hydrogen bonds with surrounding amino acid residues. This is especially effective against hCA-IX and XII isoforms, where such interactions are critical for potent inhibition.
7.2.3.1. Interaction with zinc ion. Both triazole and thiazole derivatives, particularly those with EWGs like sulfonamide or nitro, demonstrate strong interactions with the zinc ion. The binding mechanism typically involves the coordination of these groups with the zinc ion, leading to effective inhibition of enzymatic activity.
7.2.3.2. Hydrophobic and van der Waals interactions. The hydrophobic nature of alkyl-substituted triazoles and thiazoles allows these inhibitors to fit snugly within the enzyme's hydrophobic pocket, enhancing binding affinity. Docking simulations reveal that these interactions are crucial for stabilizing the inhibitor within the active site, particularly in isoforms like hCA-I and hCA-II.
7.2.3.3. Hydrogen bonding and π–π stacking. Triazoles and thiazoles with hydroxyl, amino, or carboxyl groups are often involved in hydrogen bonding with key residues such as Thr199, His94, and Glu106. These interactions are crucial for enhancing the binding affinity and selectivity of the inhibitors, as demonstrated by docking studies. π–π Stacking interactions, particularly with aromatic residues within the active site, also play a significant role in stabilizing the inhibitor, contributing to its overall potency.
7.3. Pyrimidine-based CAIs
Pyrimidine rings are commonly employed in CAI design due to their aromatic nature and nitrogen atoms, which can engage in various interactions with the active site of hCA isoforms. The substitution patterns on the pyrimidine ring play a crucial role in determining the activity and selectivity of the inhibitors (Fig. 146).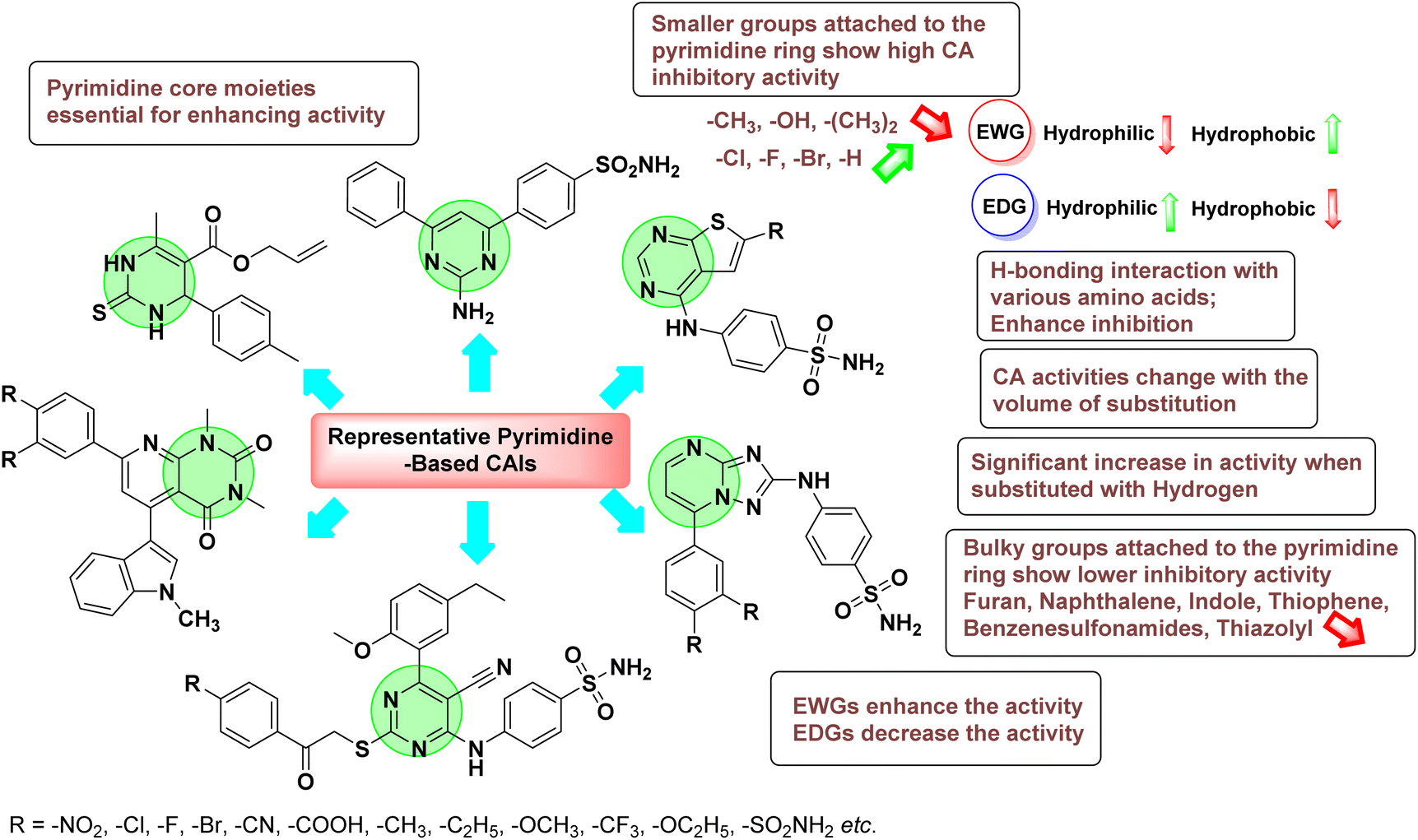 | ||
| Fig. 146 SAR of pyrimidine-based CAIs. | ||
The introduction of amino or hydroxyl groups on the pyrimidine ring allows for the formation of hydrogen bonds with key active site residues, such as Thr199, His94, and His64. These interactions are crucial for enhancing the binding affinity and selectivity of the inhibitor. Docking simulations reveal that these groups can participate in hydrogen bonding with the enzyme, leading to better stabilization of the inhibitor within the active site. This is particularly important for selective inhibition of hCA isoforms like hCA-IX and XII.
Strong EWGs like –NO2 and –CN groups enhance the pyrimidine ring's ability to interact with the zinc ion by creating a more favorable electronic environment. These interactions are essential for the potent inhibition of hCA isoforms. Docking studies demonstrate that these EWGs often participate in electrostatic interactions with the zinc ion or with polar residues near the active site, leading to stronger inhibition. This is particularly effective in isoforms like hCA-IX and XII, where the zinc ion is crucial for enzymatic activity.
7.3.3.1. Interaction with zinc ion. Pyrimidine derivatives, especially those with strong EWGs like sulfonamide or nitro groups, demonstrate robust interactions with the zinc ion. The binding mechanism often involves coordination with the zinc ion, which is a critical step in inhibiting the enzyme's activity.
7.3.3.2. Hydrophobic and van der Waals interactions. Alkyl and halogen-substituted pyrimidines tend to occupy hydrophobic pockets within the enzyme's active site, where they engage in van der Waals interactions. These interactions are crucial for stabilizing the inhibitor and enhancing binding affinity, particularly in hCA-I and II.
7.3.3.3. Hydrogen bonding and π–π stacking. Pyrimidine derivatives with hydroxyl, amino, or carboxyl groups often form hydrogen bonds with active site residues such as Thr199, His94, and Glu106. These interactions significantly enhance binding affinity and selectivity. π–π Stacking interactions, especially with aromatic residues in the active site, also contribute to the stabilization of the inhibitor, thus increasing its potency.
7.3.4.1. Substitutions at the 2-position. The 2-position of the pyrimidine ring is commonly substituted with groups like amines, which can interact with zinc-bound water molecules in the active site. This interaction enhances the inhibitor's binding affinity. Molecular docking studies reveal that amine groups at this position can form hydrogen bonds with key residues, improving selectivity for hCA-II over other isoforms.
7.3.4.2. Substitutions at the 4-position. The 4-position is often substituted with EWGs like sulfonamides, which are known to chelate the zinc ion effectively. This chelation is critical for inhibition, especially in tumor-associated isoforms like hCA-IX and XII. Docking simulations show that sulfonamide groups form stable interactions with the zinc ion, making them crucial for high inhibitory potency.
7.3.4.3. Substitutions at the 5-position. The 5-position is frequently modified with hydrophobic groups that engage in van der Waals interactions with the enzyme's active site. Alkyl or halogen substituents at this position can enhance the inhibitor's affinity by stabilizing the compound within the hydrophobic pocket. Molecular docking studies indicate that these interactions are essential for maintaining the inhibitor's orientation within the active site, contributing to its overall potency.
7.4. Phthalazine-based CAIs
Phthalazine derivatives have been explored as potential inhibitors of various hCA isoforms, with modifications at different positions of the scaffold offering insights into how electron-donating and EWGs affect activity and selectivity. The substitution pattern significantly influences the pharmacological profile of these compounds, as demonstrated by various molecular docking studies (Fig. 147).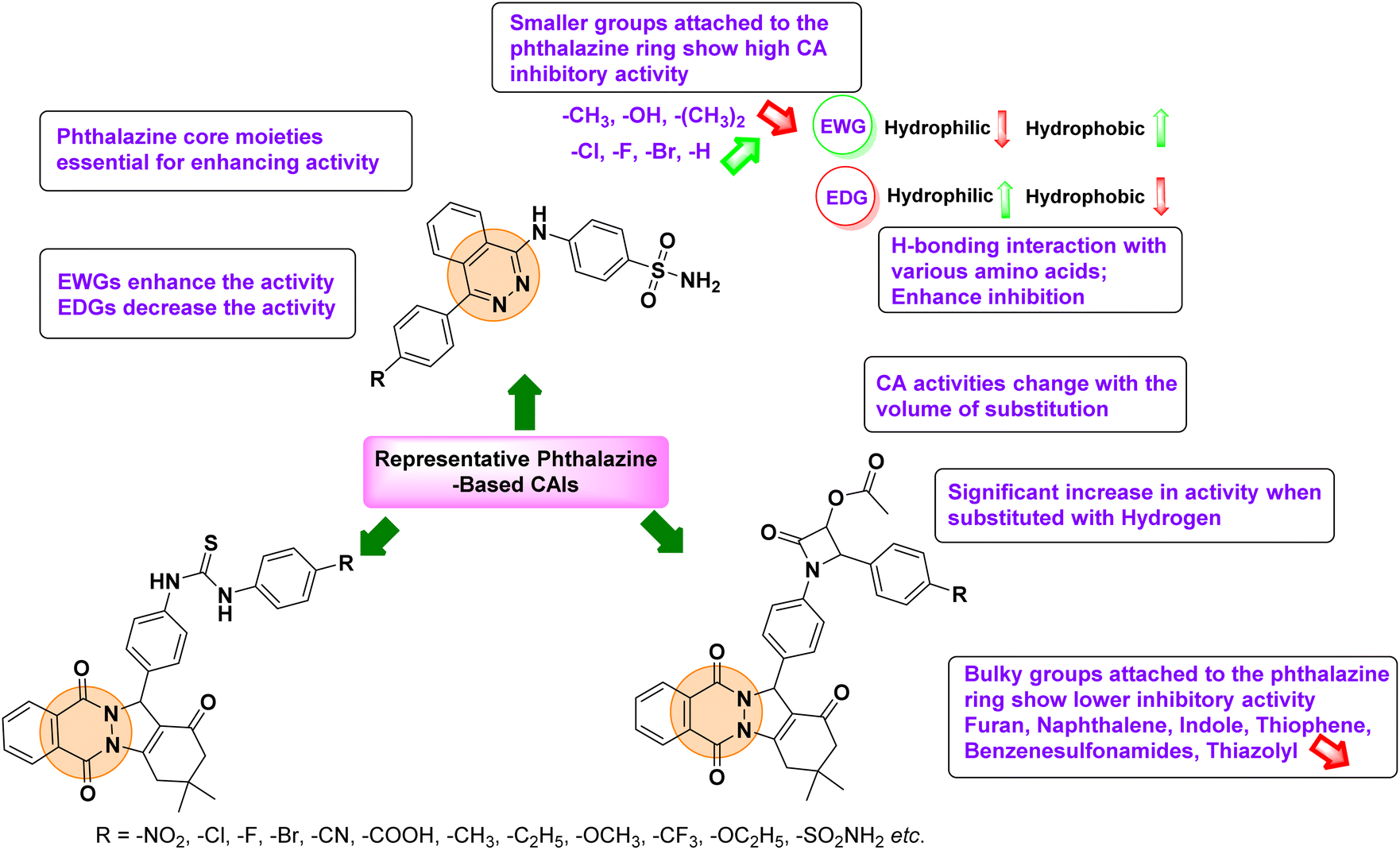 | ||
| Fig. 147 SAR of phthalazine-based CAIs. | ||
Alkyl substituents increase the electron density on the phthalazine ring, which can strengthen hydrophobic interactions with the non-polar residues in the enzyme's active site. These interactions help stabilize the inhibitor within the active site, enhancing its inhibitory activity. In docking simulations, alkyl-substituted phthalazines are observed to occupy hydrophobic pockets within the active site, contributing to stronger van der Waals interactions. This is particularly effective for inhibiting hCA-II, where such hydrophobic interactions play a significant role in enzyme inhibition.
Strong EWGs like nitro and cyano groups decrease the electron density of the phthalazine ring, making the compound more electrophilic and enhancing its ability to interact with the zinc ion at the core of the enzyme's active site. Molecular docking simulations show that phthalazine derivatives with nitro or cyano groups form robust interactions with the zinc ion, contributing to potent inhibition of hCA-IX and XII isoforms. These EWGs also engage in electrostatic interactions with nearby residues, further stabilizing the inhibitor within the enzyme's active site.
7.4.3.1. Interaction with zinc ion. The zinc ion located at the active site of carbonic anhydrases is critical for catalysis. Phthalazine-based CAIs, particularly those with strong EWGs like sulfonamides, nitro, or cyano groups, exhibit strong coordination with the zinc ion. This interaction is essential for inhibiting the enzyme's activity, as it disrupts the catalytic cycle. Docking studies show that sulfonamide-substituted phthalazines, for instance, coordinate directly with the zinc ion, leading to a significant reduction in enzymatic activity. This is especially effective for isoforms like hCA-IX and XII, which are targeted in anticancer therapies.
7.4.3.2. Hydrophobic interactions. Phthalazine derivatives with alkyl or halogen substituents often engage in hydrophobic interactions with nonpolar residues within the enzyme's active site, such as Val121, Leu198, and Phe131. These interactions help stabilize the inhibitor within the active site, enhancing its inhibitory potency. Docking studies demonstrate that these hydrophobic interactions are particularly important for hCA-II inhibition, where the enzyme's active site contains a number of hydrophobic pockets that can accommodate alkyl or halogenated substituents.
7.4.3.3. Hydrogen bonding and π–π stacking. Phthalazine derivatives with amino or hydroxyl groups often participate in hydrogen bonding with key active site residues, such as Thr199, Glu106, and His64. These interactions significantly enhance the binding affinity and selectivity of the inhibitors, especially for isoforms like hCA-II and IX. π–π Stacking interactions between the aromatic phthalazine ring and aromatic residues within the enzyme's active site, such as Phe131 and Trp209, also contribute to the stabilization of the inhibitor. Molecular docking studies frequently highlight these interactions as critical for the potent inhibition of hCA isoforms.
7.4.4.1. Substitutions at the 2-position. Substituents at the 2-position of the phthalazine ring, such as alkyl or halogen groups, can significantly influence the inhibitor's interaction with hydrophobic pockets in the enzyme's active site. These groups enhance the hydrophobic interactions, leading to increased binding affinity and inhibition potency, particularly for hCA-II.
7.4.4.2. Substitutions at the 4-position. EWGs such as sulfonamides or nitro groups at the 4-position can enhance the interaction with the zinc ion in the enzyme's active site. These groups are essential for inhibiting hCA-IX and XII, as they engage in strong electrostatic interactions with the zinc ion, which is critical for the enzyme's catalytic activity.
7.4.4.3. Substitutions at the 6-position. The 6-position of the phthalazine ring is often substituted with hydrophilic groups, such as amino or hydroxyl groups, which can form hydrogen bonds with polar residues within the enzyme's active site. These interactions increase the binding affinity and selectivity for hCA-II, which has several polar residues near the active site.
7.5. Imidazole and oxadiazole-based CAIs
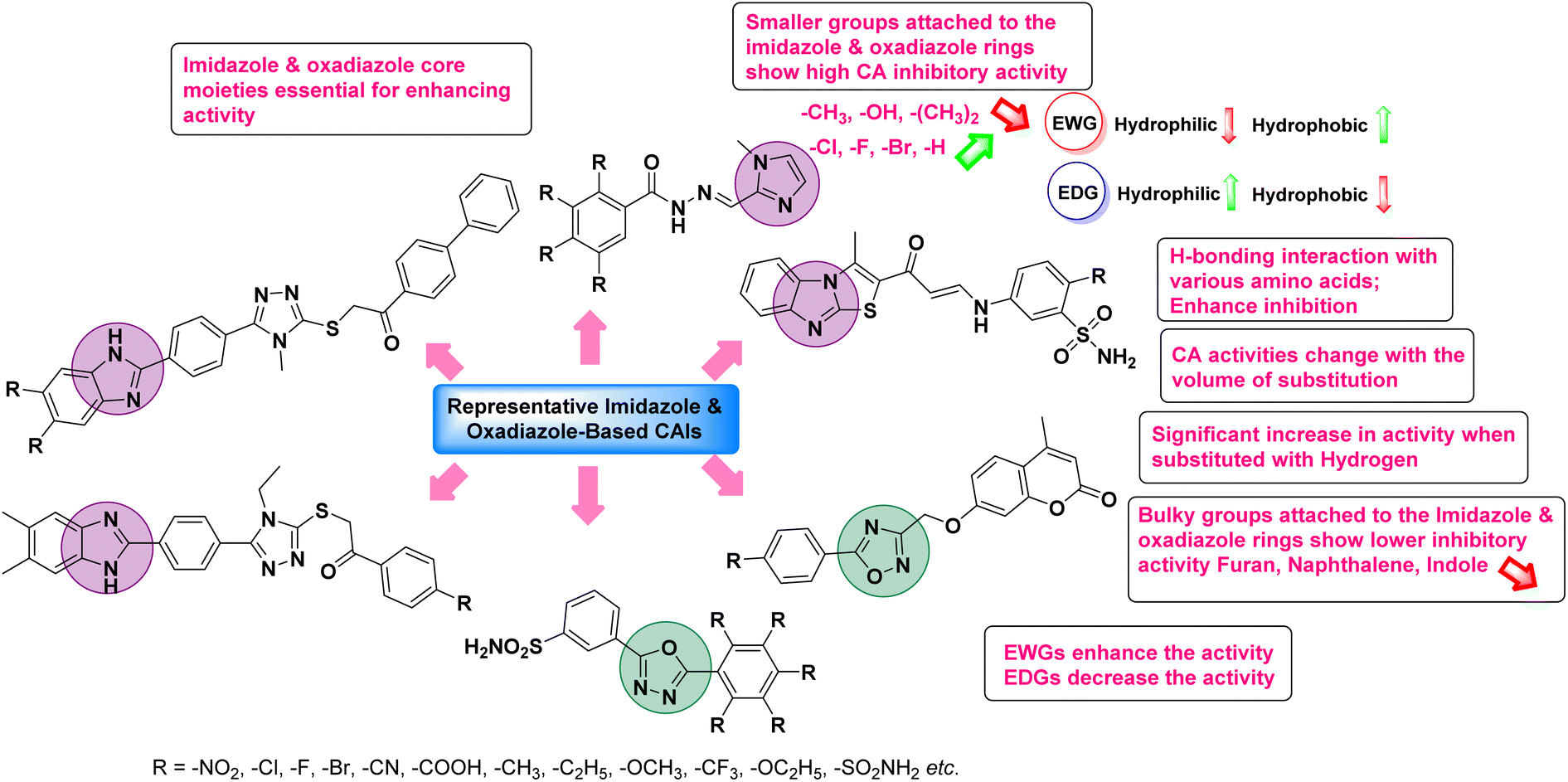 | ||
| Fig. 148 SAR of imidazole and oxadiazole-based CAIs. | ||
7.5.1.1. Electron-donating groups. Amino groups are strong electron donors and can enhance the nucleophilicity of the imidazole ring. When substituted at various positions on the imidazole ring, amino groups can engage in hydrogen bonding with the enzyme's active site residues, such as Thr199 and His94. Molecular docking studies often show that imidazole derivatives with amino groups exhibit strong interactions with the active site, improving binding affinity and selectivity for isoforms like hCA-II. The amino group can form a hydrogen bond with the carbonyl group of Thr199, stabilizing the inhibitor.
Hydroxyl groups are also electron-donating and can form hydrogen bonds with key residues in the enzyme's active site. This interaction can increase the inhibitor's binding affinity. Hydroxyl-substituted imidazoles are effective in forming hydrogen bonds with residues like Glu106 and His64. Docking studies reveal that these interactions contribute to the improved inhibition of isoforms such as hCA-I and IX.
7.5.1.2. Electron-withdrawing groups. Nitro group is strong EWG that reduce the electron density on the imidazole ring. This can enhance the interaction of the ring with the zinc ion in the enzyme's active site. Nitro-substituted imidazoles often show strong binding affinity due to their ability to interact electrostatically with the zinc ion and other polar residues. Docking studies highlight their effective inhibition of isoforms like hCA-IX, where the nitro group stabilizes the binding through electrostatic interactions with the zinc ion.
Chloro group is mild EWG that can alter the electronic environment of the imidazole ring, influencing its interaction with the enzyme. Chlorinated imidazoles can increase the hydrophobic interactions with non-polar residues in the enzyme's active site. Molecular docking studies show that these compounds bind effectively to isoforms like hCA-II, where the chloro group enhances the hydrophobic interactions.
7.5.2.1. Binding interactions. Imidazole-based CAIs often coordinate with the zinc ion at the active site, a crucial interaction for inhibition. EDGs like amino and hydroxyl groups can enhance this coordination through hydrogen bonding with nearby residues. EWGs like nitro group strengthen the binding through electrostatic interactions with the zinc ion and other polar residues.
7.5.2.2. Binding affinity. Docking studies reveal that imidazoles with EDGs generally exhibit higher binding affinity due to their ability to form hydrogen bonds and stabilize the inhibitor in the active site. EWGs improve binding through electrostatic and hydrophobic interactions.
7.5.3.1. Electron-donating groups. Amino groups enhance the electron density on the oxadiazole ring, improving its nucleophilicity and ability to interact with the enzyme's active site. Compounds with amino substitutions often show increased inhibition due to their ability to form hydrogen bonds with active site residues such as Thr199 and Glu106. Molecular docking studies indicate that amino-substituted oxadiazoles bind effectively to isoforms like hCA-I and hCA-IX.
Methoxy groups are electron-donating and can increase the electron density on the oxadiazole ring. They can also participate in hydrogen bonding. Methoxy groups enhance the binding of oxadiazoles by forming hydrogen bonds with polar residues in the enzyme's active site. Docking studies show that these compounds have improved inhibition profiles for isoforms like hCA-II.
7.5.3.2. Electron-withdrawing groups. Nitro groups decrease the electron density on the oxadiazole ring and increase its electrophilicity. This enhances interactions with the enzyme's active site. Nitro-substituted oxadiazoles exhibit strong binding affinity due to their electrostatic interactions with the zinc ion. Docking studies demonstrate that these compounds are potent inhibitors of isoforms such as hCA-IX and XII.
Cyano groups are strong EWGs that can stabilize the oxadiazole ring through electrostatic interactions. Cyano-substituted oxadiazoles bind effectively to the enzyme's active site through electrostatic interactions with the zinc ion. Molecular docking studies reveal their high potency against isoforms like hCA-XII.
7.5.4.1. Binding interactions. Oxadiazole-based CAIs often interact with the zinc ion in the enzyme's active site. EDGs like amino and methoxy groups improve binding through hydrogen bonding and increased electron density. EWGs like nitro and cyano groups enhance binding through electrostatic interactions with the zinc ion and other active site residues.
7.5.4.2. Binding affinity. Docking studies show that oxadiazole derivatives with EDGs generally exhibit higher binding affinity due to favorable hydrogen bonding interactions. EWGs improve binding through electrostatic interactions and stabilization of the enzyme–inhibitor complex.
7.6. Indole and isatin-based CAIs
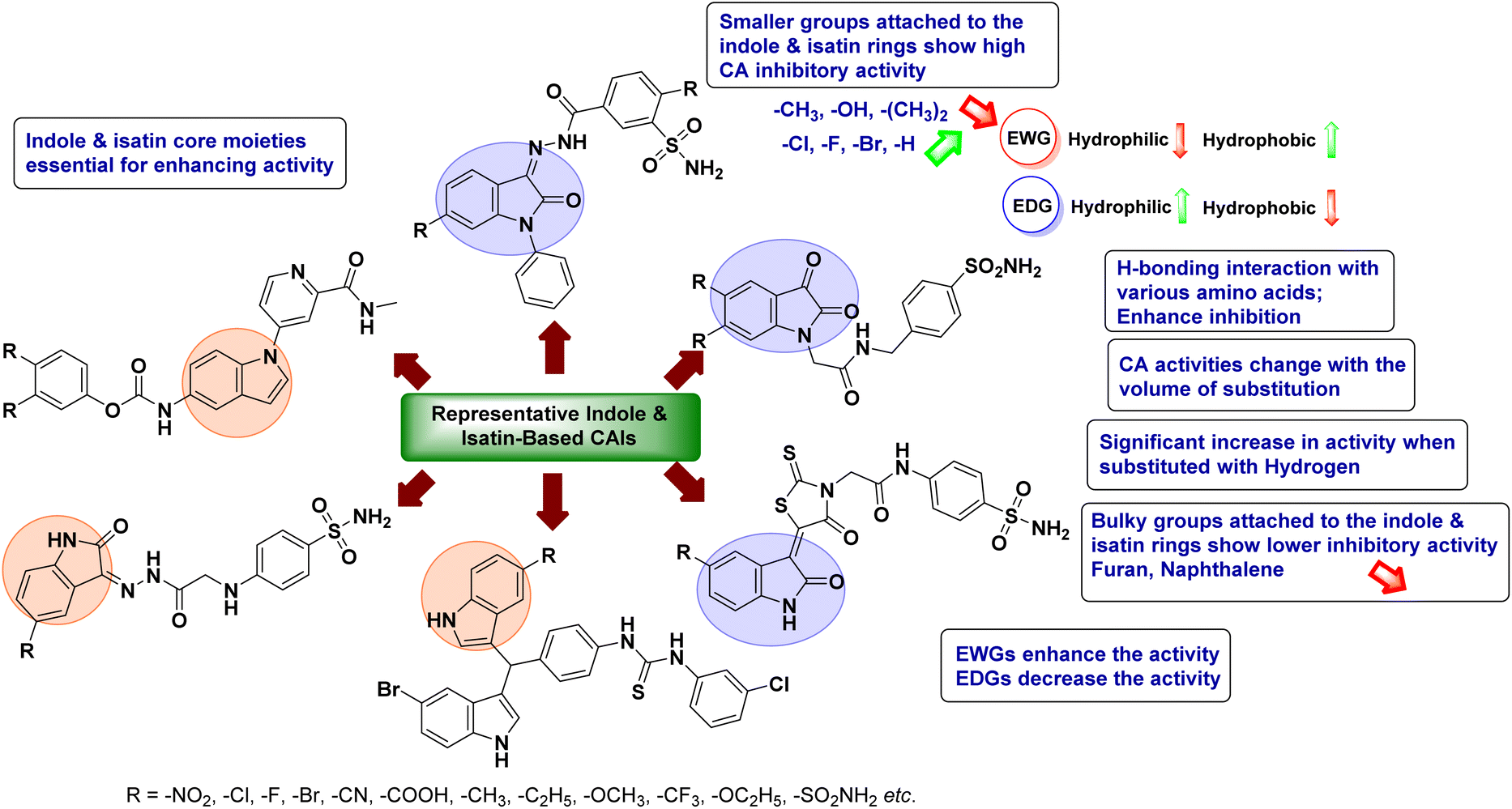 | ||
| Fig. 149 SAR of indole and isatin-based CAIs. | ||
7.6.1.1. Electron-donating groups. When –OH and –OCH3 groups positioned at the C5 or C6 of the indole ring, these EDGs enhance the electron density on the indole nitrogen, increasing its ability to form hydrogen bonds with amino acid residues in the active site of carbonic anhydrase enzymes. For instance, hydroxyl groups can form hydrogen bonds with residues such as Thr199 and Glu106, contributing to stronger binding affinity and improved inhibitory activity. Methoxy groups also enhance the interaction through van der Waals forces, increasing the overall stability of the enzyme–inhibitor complex.
The introduction of an amino group, particularly at the C2 or C3 position, enhances the inhibitor's ability to form hydrogen bonds, which can significantly improve the binding affinity towards hCA-I and II. Additionally, the amino group can participate in electrostatic interactions, further stabilizing the complex within the active site.
7.6.1.2. Electron-withdrawing groups. EWGs like nitro groups and halogens, when placed at the C5 or C7 positions, reduce the electron density on the indole ring, thereby enhancing π–π stacking interactions with aromatic residues in the enzyme's active site, such as Phe131 and Trp209. The nitro group, in particular, can participate in strong dipole interactions, enhancing the overall binding affinity.
Substituting carbonyl groups at C3 or C5 positions can increase the affinity of the inhibitor for the enzyme by forming additional hydrogen bonds with key residues in the active site, such as His94 and His119. This interaction is critical for the stabilization of the inhibitor within the enzyme's active site, leading to improved inhibitory potency.
7.6.1.3. Molecular docking studies. Molecular docking studies of indole-based CAIs typically show that inhibitors with EDGs exhibit enhanced hydrogen bonding interactions with active site residues, leading to a higher binding affinity. In contrast, inhibitors with EWGs tend to rely more on electrostatic interactions and π–π stacking, which also contribute to high binding affinity, albeit through a different mechanism. Docking studies have highlighted that indole derivatives with appropriately positioned EDGs and EWGs can achieve a fine balance between binding affinity and selectivity for different hCA isoforms, particularly hCA-IX and XII.
7.6.2.1. Electron-donating groups. When –OH and –OCH3 groups introduced at the N1 position or the aromatic ring, these EDGs can increase the electron density on the isatin scaffold, thereby enhancing the interaction with zinc ions and nearby amino acid residues in the active site. Hydroxyl groups often form hydrogen bonds with key residues such as Thr200 and His119, which are critical for potent inhibition.
Alkyl substitutions at the N1 or C5 position of the isatin scaffold can improve hydrophobic interactions with non-polar regions of the enzyme's active site, such as the hydrophobic pocket near Val121, thereby enhancing binding affinity and inhibitory potency.
7.6.2.2. Electron-withdrawing groups. Substitution of halogens or nitro groups at the C5 or C7 position can enhance the binding affinity through increased dipole–dipole interactions and the stabilization of the enzyme–inhibitor complex. These EWGs can also increase the binding strength by facilitating π–π stacking interactions with aromatic residues such as Phe131 and Trp209.
The –COOH and –CN groups, when introduced at the C3 or C7 positions, can increase the inhibitor's ability to coordinate with the zinc ion in the active site. The carboxyl group, in particular, can form salt bridges with positively charged residues, such as Lys67, enhancing the inhibitory effect.
7.6.2.3. Molecular docking studies. Docking studies of isatin-based CAIs have demonstrated that the presence of EWGs significantly enhances the binding affinity by stabilizing the inhibitor within the enzyme's active site. These studies reveal that isatin derivatives with appropriately placed EWGs tend to have a higher selectivity for hCA-IX and XII due to their stronger interactions with zinc ion and nearby residues. Conversely, derivatives with EDGs often show enhanced hydrogen bonding capabilities, contributing to higher overall binding affinity but with a potential reduction in selectivity.
7.7. Chalcone and coumarin-based CAIs
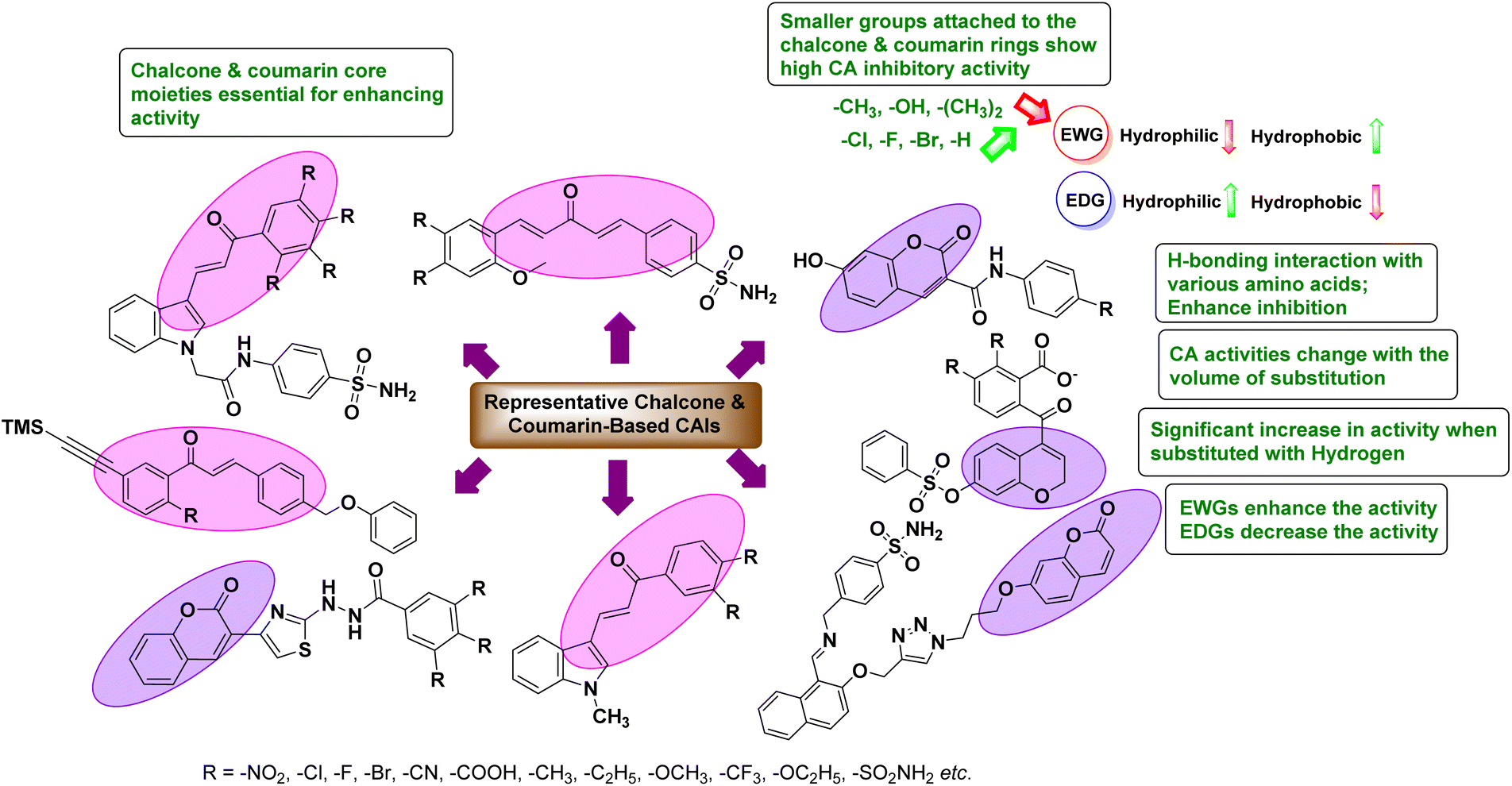 | ||
| Fig. 150 SAR of chalcone and coumarin-based CAIs. | ||
7.7.1.1. Electron-donating groups. When –OH and –OCH3 groups are attached to the A or B ring of the chalcone, especially at the ortho or para positions, they enhance hydrogen bonding interactions with amino acid residues in the active site of CA enzymes. Hydroxyl groups increase the polar character of the molecule, allowing strong hydrogen bonds with residues like Thr199 and His94 in the hCA-IX or XII isoforms, improving binding affinity.
The presence of amino groups enhances the electrostatic interactions and hydrogen bonding with CA residues. For example, amino-substituted chalcones have shown increased binding affinity towards hCA-IX due to stronger interactions with the zinc-bound water molecule and surrounding residues.
7.7.1.2. Electron-withdrawing groups. Halogen substitutions, particularly at the para position of the B ring, tend to enhance inhibitory activity through halogen bonding and hydrophobic interactions with residues like Val121 and Leu198 in the active site. Halogen groups also contribute to the stabilization of the π–π stacking interactions with aromatic amino acids like Phe131.
EWGs like nitro and cyano groups increase the binding affinity by forming dipole–dipole interactions with polar residues in the enzyme's active site. These groups enhance the inhibitor's interaction with residues near the zinc ion, promoting stronger binding and better inhibition.
7.7.1.3. Molecular docking studies. Molecular docking studies of chalcone-based CAIs reveal that the introduction of EDGs leads to enhanced hydrogen bonding with active site residues, resulting in improved binding affinity. Chalcones with EWGs, on the other hand, show better interaction with the hydrophobic regions and aromatic residues through halogen bonding and π–π stacking interactions, leading to effective enzyme inhibition. Chalcone derivatives generally show moderate selectivity towards hCA-IX and XII isoforms, with structural modifications fine-tuning the potency and selectivity.
7.7.2.1. Electron-donating groups. Substituting hydroxyl or alkoxy groups at positions 6, 7, or 8 on the coumarin ring significantly enhances the binding affinity through hydrogen bonding with residues at the entrance of the active site. These groups facilitate interactions with residues like Asn67 and His119, promoting strong and stable enzyme inhibition, particularly against hCA-IX and XII. The introduction of amino groups at positions 6 or 7 on the coumarin ring increases the inhibitor's polar character and enhances hydrogen bonding with residues near the enzyme's active site entrance. This modification generally leads to better inhibition of hCA isoforms, particularly membrane-bound isoforms such as hCA-IX.
7.7.2.2. Electron-withdrawing groups. Halogenation at positions 3 or 6 on the coumarin ring improves inhibitory activity by increasing the hydrophobic interactions with the enzyme's active site entrance. Halogenated coumarins interact with hydrophobic pockets within the enzyme, leading to improved binding stability. The EWGs (–COOH, –C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), when introduced at position 3, enhance the interaction with polar residues at the active site entrance, such as Arg64 and His200. The presence of carbonyl groups further strengthens the inhibitor's ability to stabilize within the active site, contributing to a stronger inhibition profile.
O), when introduced at position 3, enhance the interaction with polar residues at the active site entrance, such as Arg64 and His200. The presence of carbonyl groups further strengthens the inhibitor's ability to stabilize within the active site, contributing to a stronger inhibition profile.
7.7.2.3. Molecular docking studies. Molecular docking studies of coumarin-based CAIs reveal that EDGs enhance hydrogen bonding interactions with residues at the entrance of the active site, leading to higher binding affinity. EWGs improve the inhibitor's interaction with hydrophobic and polar regions of the enzyme, particularly with residues near the zinc ion. Coumarins exhibit moderate activity, and their effectiveness is highly dependent on the substituent pattern. Modifications with EDGs and EWGs can fine-tune the inhibitory potency, with most coumarins displaying higher selectivity for hCA-IX and XII.
7.8. Rhodanine-based CAIs
Rhodanine-based scaffolds have attracted considerable attention in medicinal chemistry, particularly for their role as CAIs. These molecules are characterized by a five-membered ring structure containing sulfur, nitrogen, and a ketone group, making them versatile in terms of functionalization and interactions with enzyme active sites (Fig. 151).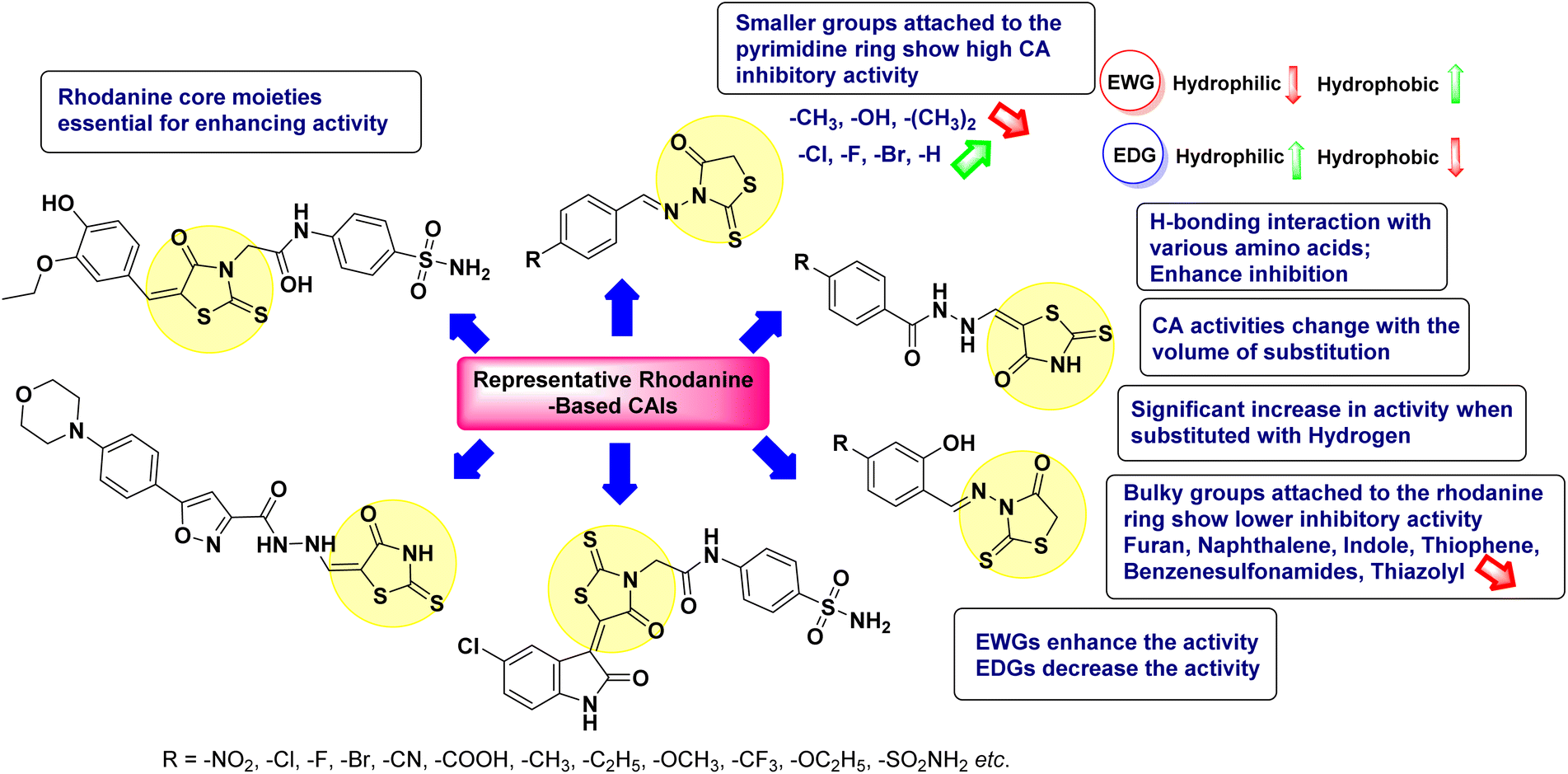 | ||
| Fig. 151 SAR of rhodanine-based CAIs. | ||
The presence of amino groups further enhances the electron density, allowing for strong interaction with amino acid residues in the active site, such as histidines and zinc ion coordination, crucial for CA inhibition.
Halogen substitution can significantly enhance binding affinity due to their size and ability to form halogen bonds. Fluorine, due to its small size and high electronegativity, often provides the best balance between binding affinity and selectivity for specific CA isoforms.
7.8.3.1. Zinc coordination. The sulfur atom in the rhodanine ring often coordinates with the zinc ion present in the active site of the CA enzyme, a critical interaction that is a hallmark of potent CAIs.
7.8.3.2. Hydrogen bonding. Functional groups like amines, hydroxyls, or carboxylates can form hydrogen bonds with key residues in the active site, such as Thr199, His94, and His119. These interactions are vital for high-affinity binding and selective inhibition of specific CA isoforms.
7.8.3.3. π–π Stacking and hydrophobic interactions. The rhodanine ring can participate in π–π stacking interactions with aromatic residues like Phe131 or hydrophobic interactions within the enzyme's active site, further stabilizing the inhibitor–enzyme complex.
7.9. Quinolines-based CAIs
Quinoline-based scaffolds are an important class of CAIs due to their ability to interact with various CA isoforms. The quinoline ring system, a fused bicyclic structure with a benzene ring fused to a pyridine, offers a versatile platform for chemical modifications that can significantly influence the inhibitory activity against CA enzymes (Fig. 152).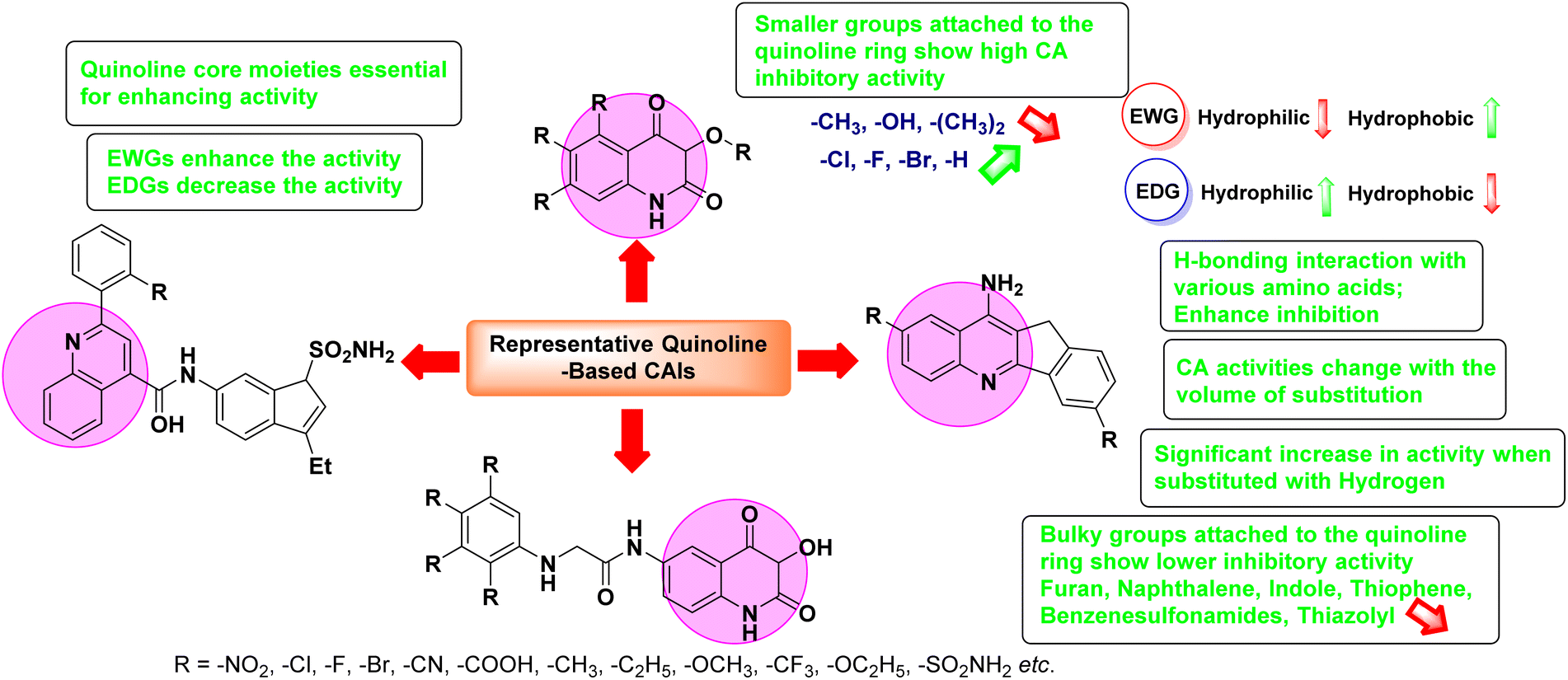 | ||
| Fig. 152 SAR of quinoline-based CAIs. | ||
The groups (OH, –OCH3) further increase the electron density, which can enhance hydrogen bonding with active site residues like Thr199, His94, and His119. The position of these substituents on the quinoline ring is crucial, as certain positions may favor stronger binding interactions, thus improving potency and selectivity.
Halogen atoms, especially fluorine, can be strategically placed on the quinoline ring to enhance binding affinity through halogen bonding and hydrophobic interactions. Fluorine, due to its small size and high electronegativity, often provides a good balance, leading to potent CA inhibition.
7.9.3.1. Zinc coordination. Although the quinoline scaffold itself does not directly coordinate with the zinc ion in the CA active site, functional groups attached to the scaffold can facilitate this interaction. For example, a sulfonamide group attached to the quinoline ring can coordinate with the zinc ion, which is a key interaction for potent CA inhibition.
7.9.3.2. Hydrogen bonding. Substituents such as hydroxyl or amino groups on the quinoline ring can form hydrogen bonds with key residues in the CA active site. These interactions are crucial for stabilizing the inhibitor within the enzyme's binding pocket, thereby enhancing selectivity and potency.
7.9.3.3. π–π Stacking and hydrophobic interactions. The planar structure of the quinoline ring allows for π–π stacking interactions with aromatic residues like Phe131, as well as hydrophobic interactions within the active site. These interactions contribute to the overall binding affinity and specificity of quinoline-based inhibitors.
7.10. Sulfonamides-based CAIs
Sulfonamides are a cornerstone class of CAIs due to their ability to effectively coordinate with the zinc ion in the active site of CA enzymes. The sulfonamide group (–SO2NH2) is essential for binding to the zinc ion and is a critical pharmacophore for CA inhibition. Modifying the surrounding structural elements, including electron-donating and EWGs, significantly influences their potency and selectivity across different CA isoforms (Fig. 153).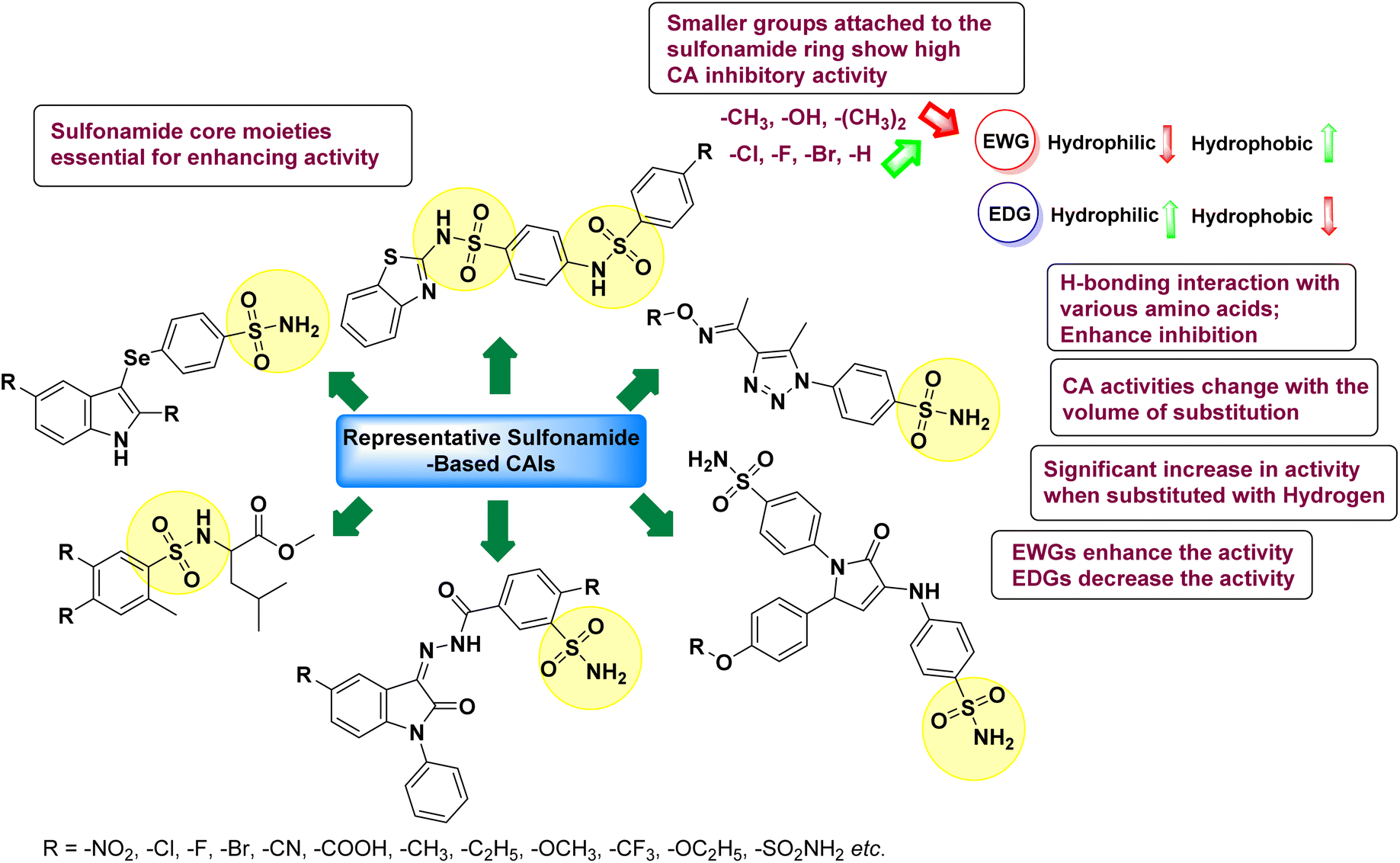 | ||
| Fig. 153 SAR of sulfonamide-based CAIs. | ||
These groups (–OH, –OCH3) can increase the electron density around the sulfonamide scaffold, which may enhance hydrogen bonding with key residues in the CA active site (e.g., Thr199 or Glu106). In addition, the hydroxyl group can form water-mediated interactions, further stabilizing the inhibitor in the enzyme pocket.
Halogen atoms (–Cl, –F, –Br), especially fluorine, when placed on the aryl ring of sulfonamides, tend to increase the potency of CA inhibition by enhancing both hydrophobic and electrostatic interactions. Fluorine is often favored due to its ability to enhance lipophilicity while maintaining a good balance between binding affinity and isoform selectivity.
7.10.3.1. Zinc ion coordination. The sulfonamide nitrogen forms a crucial interaction with the zinc ion in the CA active site. This coordination is the primary mechanism by which sulfonamides exert their inhibitory effect. The binding of the sulfonamide group displaces the water molecule coordinated to the zinc ion, thereby inhibiting the enzyme's catalytic activity.
7.10.3.2. Hydrogen bonding. The sulfonamide's nitrogen and oxygen atoms can form hydrogen bonds with surrounding active site residues, such as Thr199 and Glu106. The presence of EDGs on the sulfonamide scaffold can enhance this hydrogen bonding interactions, improving binding affinity and selectivity.
7.10.3.3. Electrostatic and hydrophobic interactions. Substituents on the sulfonamide scaffold, such as halogens or alkyl groups, can engage in hydrophobic interactions with non-polar residues lining the active site, such as Val121 and Phe131. Additionally, EWGs can improve electrostatic interactions with the enzyme's active site, further stabilizing the inhibitor.
7.11. Overall ranking based on CA inhibitory activity
Phthalazine and sulfonamides-based CAIs show the highest activity due to their strong binding interactions with the enzyme, especially against hCA-IX and hCA-I. Imidazole, oxadiazole, and triazole scaffolds also demonstrate significant potential, with their effectiveness stemming from their ability to engage in diverse interactions within the enzyme's active site. Other scaffolds like thiazole, pyrimidine, and indole/isatin offer moderate to high activity, influenced by their substitution patterns. Chalcone, coumarin, rhodanine, and quinolines show more variable effectiveness, highlighting the importance of functional group selection in optimizing inhibitory potential.In the context of CA inhibition, small molecules that form hydrogen bonds with the target protein typically interact with specific amino acid residues in or near the enzyme's active site. CA enzymes, particularly the human isoforms have a highly conserved active site containing a Zn2+ coordinated by three histidine residues. The small molecule inhibitors usually form key interactions around this zinc-binding region.
Here's a breakdown of important binding regions and how small molecules form H-bonds:
During a docking study, hydrogen bonds between the inhibitor and these residues or water molecules can contribute significantly to the binding affinity and specificity of the inhibitor toward the CA active site. Understanding these interactions helps in rationalizing the structure–activity relationship (SAR) of carbonic anhydrase inhibitors.
Moreover, the installation of EWGs or EDGs on various scaffolds plays a critical role in modulating CA inhibition. EWGs are often placed in positions that enhance hydrophobic interactions within the enzyme's active site, while EDGs are placed to improve hydrogen bonding or solubility. The exact placement depends on the scaffold and the target isoform, allowing for fine-tuning of inhibitor activity and selectivity.
8. Key positions for EWG/EDG introduction
8.1. Meta- or para-positions on aromatic rings or heterocyclic scaffolds
Substitutions at these positions generally have the most profound impact on the electronic and physicochemical properties of the molecule, influencing how the inhibitor interacts with both the hydrophobic and polar regions of CA.8.2. Nitrogen-containing heterocycles
In scaffolds like imidazole, thiazole, or triazole, substituents at key positions (such as C-2 or C-5) modulate the molecule's electron density, thereby affecting both zinc coordination and interactions with surrounding residues.8.3. Sulfonamide group modification
While the sulfonamide group itself is conserved for binding to the zinc ion, modifications at the periphery of this group can introduce EWGs or EDGs that affect the molecule's overall hydrophobicity or polarity.Each scaffold class has a unique SAR profile when it comes to CA inhibition. The effects of EWGs, EDGs, steric factors, and binding modes vary from one scaffold to another. While some general principles apply (e.g., EWGs increasing hydrophobicity and EDGs enhancing hydrogen bonding),13,232–235 the specific SAR for each scaffold has been discussed earlier w.r.t enzyme's active site, the targeted isoform, and the scaffold's binding interactions.
The SAR of CAIs reveals that both EDGs and EWGs significantly impact inhibitory activity. Scaffolds with strong EDGs tend to show enhanced binding through hydrogen bonding, while those with EWGs often improve binding through electrostatic interactions. Overall, phthalazine and sulfonamides-based CAIs exhibit the highest activity, with other scaffolds like imidazole, oxadiazole, and triazole also demonstrating significant potential (Fig. 154).
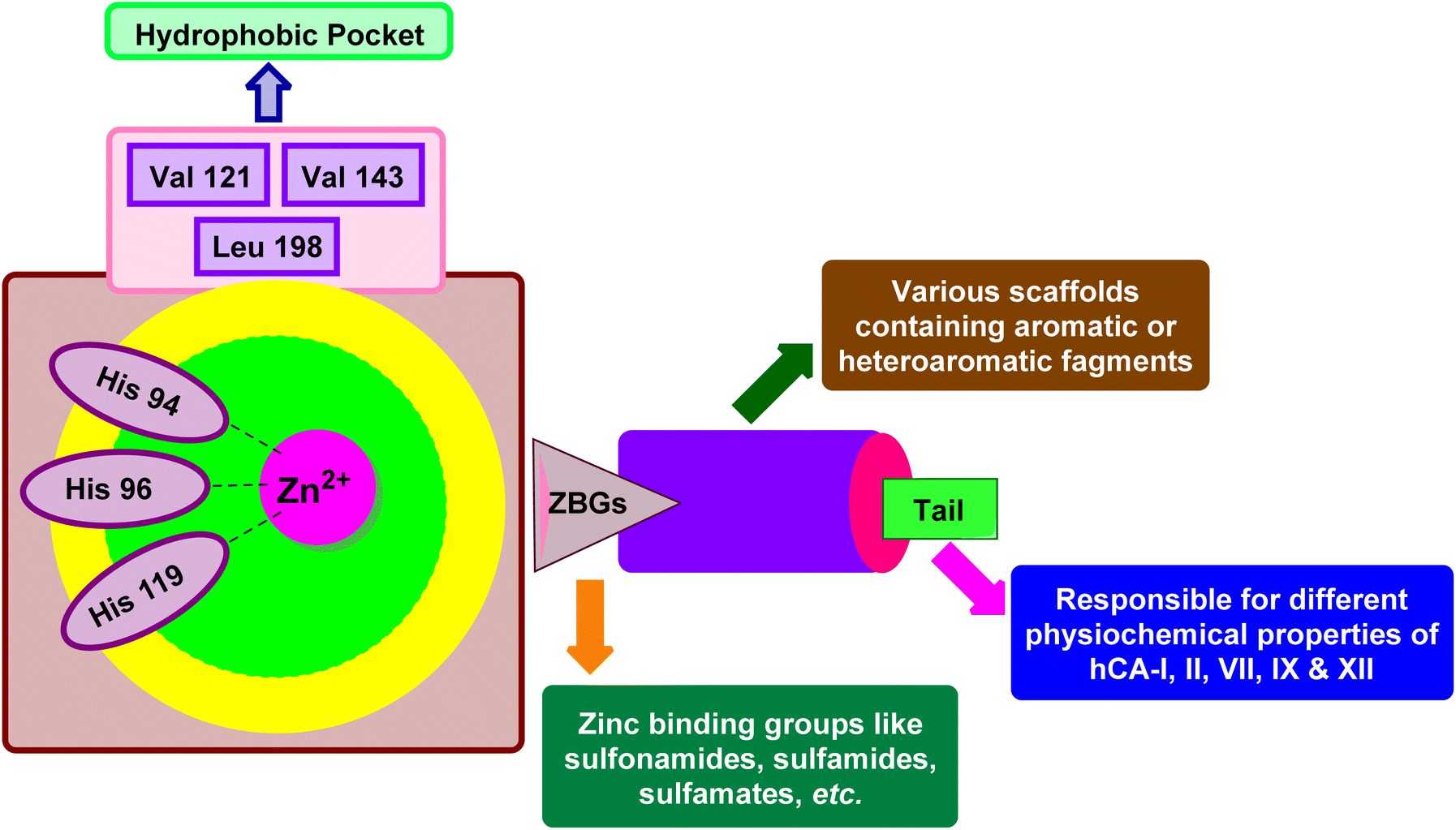 | ||
| Fig. 154 SAR of different heterocyclic scaffolds against hCA isoforms based on docking study. | ||
9. Conclusions
The inhibition of CA isoforms remains a validated therapeutic approach for treating various disorders such as glaucoma, cancer, retinopathy, hemolytic anemia, and edema, due to the enzymes' diverse cellular roles and physiological functions. Recent advancements in medicinal chemistry, particularly over the past eleven years (2013–2024), have led to the development of structurally diverse CAIs from synthetic compounds and rational design. These efforts have produced inhibitors with enhanced isoform selectivity, improved pharmacological profiles, and better physicochemical properties. Additionally, this review highlights the significant advancements in the development of CAIs across various chemical scaffolds, including phthalazine, sulfonamides, imidazole, oxadiazole, triazole, and others. Despite substantial progress, isoform selectivity remains a key challenge, limiting further clinical development of potent inhibitors due to the difficulty in balancing inhibitory activity and selectivity. A critical structural feature of potent CAIs, particularly against hCA-IX and XII, is the presence of a sulfonamide group, which coordinates with the zinc ion in the enzyme's active site and interacts with residues like His94 and His119. To address isoform selectivity, the “tail approach” has been extensively explored. This strategy involves attaching various molecular scaffolds to the core sulfonamide-bearing aromatic or heterocyclic ring to fine-tune binding interactions within the active site, enhancing both potency and selectivity. The SAR studies discussed emphasize the critical role of functional groups, particularly the balance between electron-donating and electron-withdrawing properties, in enhancing the inhibitory potency and selectivity of CAIs. Phthalazine and sulfonamide-based CAIs, in particular, have demonstrated exceptional efficacy, especially against cancer-related isoforms such as hCA-IX and hCA-XII. Targeting these isoforms (hCA-IX and XII) presents a promising strategy for cancer therapy. Benzenesulfonamides are commonly used in clinical applications. However, these compounds face two significant challenges: their lack of selectivity, as they tend to inhibit multiple isoforms indiscriminately, and their association with “sulfa allergies” in susceptible individuals. To address these issues, research has shifted towards exploring alternative scaffolds that offer both selectivity and safety. Coumarin and its derivatives have emerged as highly selective and potent inhibitors of hCA-IX and XII, and they have been combined with various pharmacophores, such as pyrazole, 1,2,3-triazole, 4-thiazolidinone, and thiourea, to enhance specificity and potency. The findings underscore the potential of CAIs as versatile therapeutic agents, with promising applications in oncology and other disease areas where carbonic anhydrases play a pivotal role.10. Future perspectives for CAIs
The promising results observed with various CAIs, including phthalazine, sulfonamides, imidazole, oxadiazole, and triazole-based scaffolds, underscore the potential for further advancements in this field. Based on the current findings and SAR, several future directions can be envisaged to enhance the development of CAIs (Table 15):| Research area | Key focus | Challenges |
|---|---|---|
| Isoform selectivity | Designing selective inhibitors for specific CA isoforms | Minimizing off-target effects and improving specificity |
| Resistance mechanisms | Addressing molecular mechanisms of CAI resistance | Developing novel scaffolds to overcome resistance |
| Pharmacokinetics and bioavailability | Optimizing ADME properties of CAIs | Ensuring adequate bioavailability and systemic exposure |
| Non-classical CAIs | Exploring novel heterocyclic frameworks | Discovering unique mechanisms with reduced side effects |
| Tumor-associated isoforms (CA-IX and CA-XII) | Targeting CA-IX and XII for cancer therapy | Minimizing inhibition of essential isoforms like CA-I, II |
| Combination therapies | Synergistic effects with other therapeutic agents | Optimizing dosing regimens and overcoming resistance |
| Clinical translation | Advancing promising candidates to clinical trials | Overcoming formulation, synthesis, and scalability issues |
(i) Enhancing isoform selectivity: one of the key challenges in CAI research is achieving selective inhibition of specific CA isoforms, particularly those implicated in disease. While significant progress has been made, future research should focus on designing inhibitors with greater selectivity to minimize off-target effects and improve therapeutic outcomes. Advanced computational modeling and structure-based drug design (SBDD) approaches could play a pivotal role in achieving this goal.
One of the primary challenges arises from the high degree of conservation in the active site among different carbonic anhydrase (CA) isoforms. The core structure of the active site is largely similar across these isoforms, which is essential for their catalytic function. Although individual isoforms may have variations in adjacent amino acid residues, these differences are often insufficient to create distinct binding pockets that could be targeted selectively by inhibitors. As a result, many potential inhibitors exhibit similar affinities for multiple isoforms, leading to off-target effects that diminish therapeutic efficacy.
Moreover, the structural plasticity of CA isoforms plays a significant role in this challenge. These enzymes can adopt various conformations depending on the presence of substrates, inhibitors, or changes in environmental conditions. Such conformational flexibility can result in unexpected binding interactions that are difficult to predict using static structural models. Consequently, inhibitors designed to target specific isoforms may inadvertently interact with others due to this dynamic nature, complicating the achievement of selectivity.
Additionally, the potential for allosteric regulation introduces another layer of complexity. While targeting allosteric sites offers a pathway to enhance isoform selectivity, the identification and characterization of these sites can be challenging. The subtle differences in the allosteric sites among isoforms may not always be apparent, making it difficult to design inhibitors that are both effective and selective.
Future research should focus on utilizing advanced techniques such as computational modeling, molecular dynamics simulations, and structural biology to gain deeper insights into the isoform-specific interactions of potential inhibitors. By addressing these challenges, the development of CAIs with improved selectivity and reduced off-target effects can become more attainable.
(ii) Overcoming resistance mechanisms: with long-term therapeutic use, resistance to CAIs may emerge, limiting their effectiveness. Investigating the underlying molecular mechanisms of resistance and developing CAIs that can overcome these barriers is an area that requires attention. This could involve exploring novel heterocyclic scaffolds or hybrid molecules that incorporate multiple mechanisms of action.
(iii) Optimizing pharmacokinetics and bioavailability: many potent CAIs suffer from suboptimal pharmacokinetics, leading to poor bioavailability or rapid metabolism. Future research should prioritize optimizing the physicochemical properties of CAIs to enhance their pharmacokinetic profiles, ensuring better systemic exposure and prolonged therapeutic efficacy. This includes improving absorption, distribution, metabolism, and excretion (ADME) characteristics of lead compounds.
(iv) Exploring non-classical CAIs: although sulfonamides remain the most widely studied CAIs, non-classical inhibitors based on different heterocyclic frameworks offer a promising avenue for future research. These scaffolds may provide unique mechanisms of action, potentially leading to the development of novel therapeutics with reduced side effects or improved efficacy.
(v) Targeting tumor-associated CA isoforms: CA-IX and CA-XII have been identified as key targets in cancer therapy due to their overexpression in hypoxic tumor environments. Future studies should focus on developing CAIs specifically targeting these isoforms while ensuring minimal inhibition of other physiologically essential isoforms such as CA-I and CA-II. This could enhance the therapeutic index and reduce toxicity in cancer treatments.
(vi) Exploring combination therapies: given the complexity of diseases involving carbonic anhydrases, there is growing interest in combining CAIs with other therapeutic agents to achieve synergistic effects. Research into combination therapies, particularly in oncology, may reveal new strategies for overcoming resistance or enhancing the efficacy of existing treatments.
(vii) Clinical translation of promising candidates: finally, while numerous CAIs have demonstrated efficacy in preclinical studies, the translation of these findings into clinical practice remains a challenge. Addressing formulation issues, scaling up synthesis, and conducting comprehensive clinical trials will be essential to bring new CAIs into therapeutic use.
(viii) Expanding therapeutic applications: exploring the potential of CAIs beyond cancer, such as in the treatment of glaucoma, epilepsy, and metabolic disorders, can broaden their clinical applications and improve patient outcomes.
One major challenge is the gap between preclinical efficacy and clinical outcomes. Preclinical studies often rely on in vitro assays or animal models that do not fully capture the complexity of human diseases, leading to discrepancies in therapeutic effects when tested in humans. This highlights the need for more advanced models that better mimic human pathophysiology, ensuring that findings from preclinical studies are more predictive of clinical success.
Another key challenge is optimizing the pharmacokinetic and pharmacodynamic profiles of CAIs for human use. Many compounds with excellent activity in vitro may suffer from poor bioavailability, rapid metabolism, or short half-lives in vivo, which reduces their efficacy. Developing formulations or delivery systems that improve the absorption, distribution, metabolism, and excretion (ADME) properties of CAIs is essential for translating preclinical candidates into viable clinical drugs.
Additionally, toxicity and off-target effects represent significant hurdles. While CAIs may selectively inhibit specific isoforms in controlled laboratory conditions, they may also inhibit non-target isoforms in clinical use, leading to adverse effects. Achieving isoform selectivity with minimal off-target activity is critical for improving the therapeutic index and minimizing side effects.
Lastly, scaling up the synthesis of promising CAIs from laboratory-scale to commercial-scale production can pose significant challenges. The complexity of certain synthetic pathways or the cost of raw materials may hinder large-scale manufacturing, impacting the overall feasibility of clinical development.
Addressing these challenges requires an integrated approach, combining advancements in drug design, formulation strategies, and more predictive preclinical models to facilitate the successful clinical translation of CAIs.
Data availability
All the data is provided in manuscript file.Conflicts of interest
Authors have no conflict of interests to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Research Project under grant number RGP2/255/45.References
- T. Qadir, A. Amin, P. K. Sharma, I. Jeelani and H. Abe, Open Med. Chem. J., 2022, 16, 1–34 CrossRef.
- E. Kabir and M. Uzzaman, Results Chem., 2022, 4, 100606 CrossRef.
- R. S. Jassas, N. Naeem, A. Sadiq, R. Mehmood, N. A. Alenazi, M. M. Al-Rooqi, E. U. Mughal, R. I. Alsantali and S. A. Ahmed, RSC Adv., 2023, 13, 16413–16452 RSC.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2022, 12, 19764–19855 RSC.
- P. N. Kalaria, S. C. Karad and D. K. Raval, Eur. J. Med. Chem., 2018, 158, 917–936 CrossRef.
- V. Srivastava, P. K. Singh, S. Tivari and P. P. Singh, Org. Chem. Front., 2022, 9, 1485–1507 RSC.
- M. H. A. Al-Jumaili, A. A. Hamad, H. E. Hashem, A. D. Hussein, M. J. Muhaidi, M. A. Ahmed, A. H. A. Albanaa, F. Siddique and E. A. Bakr, J. Mol. Struct., 2023, 1271, 133970 CrossRef.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- A. Husain and D. Madhesia, J. Enzyme Inhib. Med. Chem., 2012, 27, 773–783 CrossRef CAS PubMed.
- A. Al-Mulla, Der Pharma Chem., 2017, 9, 141–147 CAS.
- R. J. Obaid, E. U. Mughal, N. Naeem, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, Process Biochem., 2022, 120, 250–259 CrossRef CAS.
- P. Arora, V. Arora, H. Lamba and D. Wadhwa, Int. J. Pharm. Sci. Res., 2012, 3, 2947 Search PubMed.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2010, 20, 3467–3474 CrossRef CAS.
- C. T. Supuran and A. Scozzafava, Expert Opin. Ther. Pat., 2000, 10, 575–600 CrossRef CAS.
- J. Zhu, X. Jiang, X. Luo, R. Zhao, J. Li, H. Cai, X. Y. Ye, R. Bai and T. Xie, Drug Dev. Res., 2023, 84, 718–735 CrossRef CAS PubMed.
- C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27, 759–772 CrossRef CAS PubMed.
- W. Zuo, L. Zuo, X. Geng, Z. Li and L. Wang, Org. Chem. Front., 2023, 10, 6112–6116 RSC.
- R. Jing, Z. Jiang and X. Tang, Int. J. Mol. Sci., 2024, 25, 8638 CrossRef CAS.
- C. T. Supuran, Expert Opin. Ther. Pat., 2018, 28, 709–712 CrossRef CAS.
- A. Thiry, C. T. Supuran, B. Masereel and J.-M. Dogné, J. Med. Chem., 2008, 51, 3051–3056 CrossRef.
- S. Kumar, S. Rulhania, S. Jaswal and V. Monga, Eur. J. Med. Chem., 2021, 209, 112923 CrossRef PubMed.
- E. Masini, F. Carta, A. Scozzafava and C. T. Supuran, Expert Opin. Ther. Pat., 2013, 23, 705–716 CrossRef PubMed.
- G. De Simone and C. T. Supuran, Curr. Top. Med. Chem., 2007, 7, 879–884 CrossRef PubMed.
- A. Thiry, J.-M. Dogne, C. T. Supuran and B. Masereel, Curr. Top. Med. Chem., 2007, 7, 855–864 CrossRef PubMed.
- C. T Supuran, Future Med. Chem., 2021, 13, 1935–1937 CrossRef PubMed.
- C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 345–360 CrossRef PubMed.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef PubMed.
- C. T. Supuran, Expert Opin. Drug Discovery, 2017, 12, 61–88 CrossRef PubMed.
- C. T. Supuran and A. Scozzafava, Expert Opin. Ther. Pat., 2002, 12, 217–242 CrossRef.
- R. McKenna and C. T. Supuran, Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, 2013, pp. 291–323 Search PubMed.
- M. Aggarwal and R. McKenna, Expert Opin. Ther. Pat., 2012, 22, 903–915 CrossRef PubMed.
- K. M. Merz Jr, R. Hoffmann and M. J. Dewar, J. Am. Chem. Soc., 1989, 111, 5636–5649 CrossRef.
- C. T. Supuran, in Carbonic Anhydrase, CRC press, 2004, pp. 13–36 Search PubMed.
- C. T. Supuran, Clin. Sci., 2021, 135, 1233–1249 CrossRef PubMed.
- A. Angeli, F. Carta and C. T. Supuran, Catalysts, 2020, 10, 1008 CrossRef.
- R. J. DiMario, M. C. Machingura, G. L. Waldrop and J. V. Moroney, Plant Sci., 2018, 268, 11–17 CrossRef PubMed.
- M. Carter, Biol. Rev., 1972, 47, 465–513 CrossRef PubMed.
- A. Nocentini and C. T. Supuran, Carbonic Anhydrases, 2019, 3–16 CrossRef.
- L. Chen, Z. Jiang, L. Yang, Y. Fang, S. Lu, O. U. Akakuru, S. Huang, J. Li, S. Ma and A. Wu, Chin. J. Chem., 2023, 41, 199–206 CrossRef.
- C. Zhao, X. Tang, X. Chen and Z. Jiang, ACS Nano, 2024, 18, 17852–17868 CrossRef PubMed.
- P. Xu, C. Li, J. Yuan, Z. Bao and W. Liu, Front. Genet., 2024, 15, 1388015 CrossRef PubMed.
- M. I. Hassan, B. Shajee, A. Waheed, F. Ahmad and W. S. Sly, Bioorg. Med. Chem., 2013, 21, 1570–1582 CrossRef PubMed.
- K. Gilmour, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2010, 157, 193–197 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146–189 CrossRef PubMed.
- C. T. Supuran and A. Scozzafava, Bioorg. Med. Chem., 2007, 15, 4336–4350 CrossRef.
- C. T. Supuran and C. Capasso, Metabolites, 2017, 7, 56 CrossRef PubMed.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, X-Ray Crystallography of Carbonic Anhydrase Inhibitors and its Importance in Drug Design, Wiley, Hoboken, New Jersey, 2009 Search PubMed.
- C. T. Supuran, Biochem. J., 2016, 473, 2023–2032 CrossRef PubMed.
- L. Baranauskienė and D. Matulis, Carbonic Anhydrase as Drug Target: Thermodynamics and Structure of Inhibitor Binding, 2019, pp. 3–14 Search PubMed.
- M. Aggarwal, C. D. Boone, B. Kondeti and R. McKenna, J. Enzyme Inhib. Med. Chem., 2013, 28, 267–277 CrossRef.
- D. W. Christianson and C. A. Fierke, Acc. Chem. Res., 1996, 29, 331–339 CrossRef.
- C. T. Supuran, in Metalloenzymes, Elsevier, 2024, pp. 139–156 Search PubMed.
- J.-Y. Winum and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2015, 30, 321–324 CrossRef PubMed.
- S. Zamanova, A. M. Shabana, U. K. Mondal and M. A. Ilies, Expert Opin. Ther. Pat., 2019, 29, 509–533 CrossRef PubMed.
- S. Pastorekova, S. Parkkila and J. Zavada, Adv. Clin. Chem., 2006, 42, 167–216 Search PubMed.
- L. Ciccone, C. Cerri, S. Nencetti and E. Orlandini, Molecules, 2021, 26, 6380 CrossRef PubMed.
- C. T. Supuran, Expert Opin. Ther. Pat., 2003, 13, 1545–1550 CrossRef.
- C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2022, 37, 2478–2488 CrossRef PubMed.
- S. Bibi, T. Javed, F. Alam, A. Ali, S. Ali, M. Ullah, H. B. Asad, M. Hassham, F. Hasan and S. Muhammad, Pak. J. Pharm. Sci., 2019, 32, 709–720 Search PubMed.
- E. Berrino and F. Carta, in Carbonic Anhydrases, Elsevier, 2019, pp. 311–329 Search PubMed.
- J. Haapasalo, K. Nordfors, H. Haapasalo and S. Parkkila, Cancers, 2020, 12, 1723 CrossRef PubMed.
- E. Ruusuvuori and K. Kaila, Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, 2014, 271–290 Search PubMed.
- F. Carta and C. T. Supuran, Expert Opin. Ther. Pat., 2013, 23, 681–691 CrossRef PubMed.
- S. Bua, A. Nocentini and C. T. Supuran, in Carbonic Anhydrases, Elsevier, 2019, pp. 287–309 Search PubMed.
- M. Pandey, A. Singh, N. Agnihotri, R. Kumar, P. Saha, R. P. Pandey and A. Kumar, J. Res. Appl. Sci. Biotechnol., 2022, 1, 11–20 Search PubMed.
- C. T. Supuran, Expert Opin. Drug Metab. Toxicol., 2016, 12, 423–431 CrossRef.
- P. Challa and D. L. Epstein, in Chandler and Grant's Glaucoma, CRC Press, 2024, pp. 159–164 Search PubMed.
- G. Durand-Cavagna, R. Owen, L. Gordon, C. Peter and C. Boussiquet-Leroux, Toxicol. Sci., 1992, 18, 137–143 CrossRef CAS.
- L. Ambrosio, J. S. Williams, A. Gutierrez, E. A. Swanson, R. J. Munro, R. D. Ferguson, A. B. Fulton and J. D. Akula, Exp. Eye Res., 2021, 202, 108344 CrossRef CAS PubMed.
- A. Lafuma and G. Berdeaux, Curr. Med. Res. Opin., 2008, 24, 1519–1527 CrossRef CAS PubMed.
- N. A. Sharif, Exp. Eye Res., 2023, 232, 109444 CrossRef CAS PubMed.
- M. L. Shahsuvaryan, Med. Hypothesis, Discovery Innovation Ophthalmol., 2022, 11, 34 Search PubMed.
- M. F. Syed, A. Rehmani and M. Yang, Med. Clin., 2021, 105, 425–444 Search PubMed.
- F. Mincione, L. Menabuoni and C. T. Supuran, in Carbonic Anhydrase, CRC Press, 2004, pp. 255–266 Search PubMed.
- J. C. Clapham, J. R. Arch and M. Tadayyon, Pharmacol. Ther., 2001, 89, 81–121 CrossRef CAS.
- G. D. Simone, A. D. Fiore and C. T. Supuran, Curr. Pharm. Des., 2008, 14, 655–660 CrossRef PubMed.
- M. Y. Mboge, R. McKenna and S. C. Frost, Topics in Anti-Cancer Research, 2015, 5, 3 Search PubMed.
- S. G. Nerella, P. Singh, M. Arifuddin and C. T. Supuran, Expert Opin. Ther. Pat., 2022, 32, 833–847 CrossRef CAS PubMed.
- M. Y. Mboge, B. P. Mahon, R. McKenna and S. C. Frost, Metabolites, 2018, 8, 19 CrossRef PubMed.
- W. M. Dr. Pierce Jr, G. F. Nardin, M. F. Fuqua, E. Sabah-Maren and S. H. Stern, J. Bone Miner. Res., 1991, 6, 347–354 CrossRef.
- Z. Alkhayal, Z. Shinwari, A. Gaafar and A. Alaiya, Curr. Issues Mol. Biol., 2023, 45, 1373–1386 CrossRef.
- X. Chang, Y. Zheng, Q. Yang, L. Wang, J. Pan, Y. Xia, X. Yan and J. Han, Arthritis Res. Ther., 2012, 14, 1–14 CrossRef.
- M. A. Hayward and T. J. Caggiano, in Annual Reports in Medicinal Chemistry, Elsevier, 1987, vol. 22, pp. 169–178 Search PubMed.
- J. Sławiński, K. Szafrański, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2013, 69, 701–710 CrossRef.
- K. Aksu, M. Nar, M. Tanc, D. Vullo, İ. Gülçin, S. Göksu, F. Tümer and C. T. Supuran, Bioorg. Med. Chem., 2013, 21, 2925–2931 CrossRef.
- N. Berber, M. Arslan, E. Yavuz, C. Bilen and N. Gencer, Arthritis Res. Ther., 2013, 2013, 1–8 Search PubMed.
- S. Ayvaz, M. Çankaya, A. Atasever and A. Altuntas, J. Enzyme Inhib. Med. Chem., 2013, 28, 305–310 CrossRef.
- Z.-C. Wang, Y.-J. Qin, P.-F. Wang, Y.-A. Yang, Q. Wen, X. Zhang, H.-Y. Qiu, Y.-T. Duan, Y.-T. Wang, Y.-L. Sang and H.-L. Zhu, Eur. J. Med. Chem., 2013, 66, 1–11 CrossRef.
- N. Büyükkidan, B. Büyükkidan, M. Bülbül, R. Kasimoğullari, M. Serdar and S. Mert, J. Pharm. Pharmacol., 2013, 65, 363–369 CrossRef.
- M. O. Karatas, B. Alici, U. Cakir, E. Cetinkaya, D. Demir, A. Ergün, N. Gençer and O. Arslan, J. Enzyme Inhib. Med. Chem., 2013, 28, 299–304 CrossRef PubMed.
- E. Şen, Z. Alım, H. Duran, M. M. İşgör, Ş. Beydemir, R. Kasımoğulları and S. Ok, J. Enzyme Inhib. Med. Chem., 2013, 28, 328–336 CrossRef.
- S. K. Suthar, S. Bansal, S. Lohan, V. Modak, A. Chaudhary and A. Tiwari, Eur. J. Med. Chem., 2013, 66, 372–379 CrossRef PubMed.
- N. Gençer, Ç. Bilen, D. Demir, A. Atahan, M. Ceylan and M. Küçükislamoğlu, Artif. Cells, Nanomed., Biotechnol., 2013, 41, 384–388 CrossRef.
- R. Gao, S. Liao, C. Zhang, W. Zhu, L. Wang, J. Huang, Z. Zhao, H. Li, X. Qian and Y. Xu, Eur. J. Med. Chem., 2013, 62, 597–604 CrossRef.
- F. Allouche, F. Chabchoub, F. Carta and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2013, 28, 343–349 CrossRef PubMed.
- M. Ceruso, P. Khloya, C. T. Supuran and P. K. Sharma, Bioorg. Med. Chem., 2014, 22, 6945–6952 CrossRef PubMed.
- M. M. Ghorab, M. S. Alsaid, M. Ceruso, Y. M. Nissan and C. T. Supuran, Bioorg. Med. Chem., 2014, 22, 3684–3695 CrossRef PubMed.
- M. Mojzych, A. Bielawska, K. Bielawski, M. Ceruso and C. T. Supuran, Bioorg. Med. Chem., 2014, 22, 2643–2647 CrossRef PubMed.
- K. Rutkauskas, A. Zubrienė, I. Tumosienė, K. Kantminienė, M. Kažemėkaitė, A. Smirnov, J. Kazokaitė, V. Morkūnaitė, E. Čapkauskaitė, E. Manakova, S. Gražulis, Z.-J. Beresnevičius and D. Matulis, Molecules, 2014, 19, 17356–17380 CrossRef.
- J. Sławiński, Z. Brzozowski, B. Żołnowska, K. Szafrański, A. Pogorzelska, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2014, 84, 59–67 CrossRef.
- V. Dudutiene, J. Matuliene, A. Smirnov, D. D. Timm, A. Zubriene, L. Baranauskiene, V. Morkunaite, J. Smirnoviene, V. Michailoviene, V. Juozapaitiene and D. Matulis, J. Med. Chem., 2014, 57, 9435–9446 CrossRef.
- M. Bozdag, M. Pinard, F. Carta, E. Masini, A. Scozzafava, R. McKenna and C. T. Supuran, J. Med. Chem., 2014, 57, 9673–9686 CrossRef PubMed.
- Y. Akbaba, A. Akıncıoğlu, H. Göçer, S. Göksu, İ. Gülçin and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2014, 29, 35–42 CrossRef PubMed.
- P. Khloya, G. Celik, D. Vullo, C. T. Supuran and P. K. Sharma, Eur. J. Med. Chem., 2014, 76, 284–290 CrossRef PubMed.
- F. Sonmez, C. Bilen, S. Sumersan, N. Gençer, S. Isik, O. Arslan and M. Kucukislamoglu, J. Enzyme Inhib. Med. Chem., 2014, 29, 118–123 CrossRef PubMed.
- G. Celik, P. Khloya, D. Vullo, C. T. Supuran and P. K. Sharma, Bioorg. Med. Chem., 2014, 22, 1873–1882 CrossRef PubMed.
- H. Kuday, F. Sonmez, C. Bilen, E. Yavuz, N. Gençer and M. Kucukislamoglu, BioMed Res. Int., 2014, 1, 594879–594885 Search PubMed.
- A. Saeed, M. Al-Rashida, M. Hamayoun, A. Mumtaz and J. Iqbal, J. Enzyme Inhib. Med. Chem., 2014, 29, 901–905 CrossRef PubMed.
- Y. Akbaba, E. Bastem, F. Topal, İ. Gülçin, A. Maraş and S. Göksu, Arch. Pharm., 2014, 347, 950–957 CrossRef PubMed.
- N. Pala, L. Micheletto, M. Sechi, M. Aggarwal, F. Carta, R. McKenna and C. T. Supuran, ACS Med. Chem. Lett., 2014, 5, 927–930 CrossRef PubMed.
- B. Żołnowska, J. Sławiński, A. Pogorzelska, J. Chojnacki, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2014, 71, 135–147 CrossRef PubMed.
- F. Celik, M. Arslan, M. O. Kaya, E. Yavuz, N. Gençer and O. Arslan, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 58–62 CrossRef PubMed.
- T. Demirci, M. Arslan, Ç. Bilen, D. Demir, N. Gençer and O. Arslan, J. Enzyme Inhib. Med. Chem., 2014, 29, 132–136 CrossRef PubMed.
- A. Akıncıoğlu, M. Topal, İ. Gülçin and S. Göksu, Arch. Pharm., 2014, 347, 68–76 CrossRef PubMed.
- S. Zaib, A. Saeed, K. Stolte, U. Flörke, M. Shahid and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 140–150 CrossRef PubMed.
- J. Sławiński, A. Pogorzelska, B. Żołnowska, K. Brożewicz, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2014, 82, 47–55 CrossRef PubMed.
- Z.-C. Wang, Y.-T. Duan, H.-Y. Qiu, W.-Y. Huang, P.-F. Wang, X.-Q. Yan, S.-F. Zhang and H.-L. Zhu, RSC Adv., 2014, 4, 33029–33038 RSC.
- J. Pichake, P. S. Kharkar, M. Ceruso, C. T. Supuran and M. P. Toraskar, ACS Med. Chem. Lett., 2014, 5, 793–796 CrossRef PubMed.
- P. Khloya, M. Ceruso, S. Ram, C. T. Supuran and P. K. Sharma, Bioorg. Med. Chem. Lett., 2015, 25, 3208–3212 CrossRef PubMed.
- S. Imran, M. Taha, N. H. Ismail, S. Fayyaz, K. M. Khan and M. I. Choudhary, Bioorg. Chem., 2015, 62, 83–93 CrossRef PubMed.
- M. R. Buemi, L. De Luca, S. Ferro, E. Bruno, M. Ceruso, C. T. Supuran, K. Pospíšilová, J. Brynda, P. Řezáčová and R. Gitto, Eur. J. Med. Chem., 2015, 102, 223–232 CrossRef.
- H. S. Ibrahim, S. M. Abou-Seri, M. Tanc, M. M. Elaasser, H. A. Abdel-Aziz and C. T. Supuran, Eur. J. Med. Chem., 2015, 103, 583–593 CrossRef.
- N. Berber, M. Arslan, Ç. Bilen, Z. Sackes, N. Gençer and O. Arslan, Russ. J. Bioorg. Chem., 2015, 41, 414–420 CrossRef.
- N. Singasane, P. S. Kharkar, M. Ceruso, C. T. Supuran and M. P. Toraskar, J. Enzyme Inhib. Med. Chem., 2015, 30, 901–907 CrossRef PubMed.
- S. Muhammad Saad, M. Saleem, S. Perveen, M. Tanveer Alam, K. Mohammed Khan and M. Iqbal Choudhary, Med. Chem., 2015, 11, 336–341 CrossRef.
- D. A. Ibrahim, D. S. Lasheen, M. Y. Zaky, A. W. Ibrahim, D. Vullo, M. Ceruso, C. T. Supuran and D. A. Abou El Ella, Bioorg. Med. Chem., 2015, 23, 4989–4999 CrossRef.
- C. B. Mishra, S. Kumari, A. Angeli, S. M. Monti, M. Buonanno, A. Prakash, M. Tiwari and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 174–179 CrossRef.
- K. Aksu, B. Özgeriş, P. Taslimi, A. Naderi, İ. Gülçin and S. Göksu, Arch. Pharm., 2016, 349, 944–954 CrossRef.
- H. Genç, R. Kalin, Z. Köksal, N. Sadeghian, U. M. Kocyigit, M. Zengin, İ. Gülçin and H. Özdemir, Int. J. Mol. Sci., 2016, 17, 1736 CrossRef.
- S. Mert, Z. Alım, M. M. İşgör, Ş. Beydemir and R. Kasımoğulları, Bioorg. Chem., 2016, 68, 64–71 CrossRef PubMed.
- E. Garibov, P. Taslimi, A. Sujayev, Z. Bingol, S. Çetinkaya, İ. Gulçin, S. Beydemir, V. Farzaliyev, S. H. Alwasel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 1–9 CrossRef PubMed.
- A. Sujayev, L. Polat Kose, E. Garibov, İ. Gülçin, V. Farzaliev, S. H. Alwasel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 1192–1197 CrossRef.
- N. M. A. Gawad, N. H. Amin, M. T. Elsaadi, F. M. Mohamed, A. Angeli, V. De Luca, C. Capasso and C. T. Supuran, Bioorg. Med. Chem., 2016, 24, 3043–3051 CrossRef.
- K. Kucukoglu, F. Oral, T. Aydin, C. Yamali, O. Algul, H. Sakagami, I. Gulcin, C. T. Supuran and H. I. Gul, J. Enzyme Inhib. Med. Chem., 2016, 31, 20–24 CrossRef PubMed.
- W. M. Eldehna, M. Fares, M. Ceruso, H. A. Ghabbour, S. M. Abou-Seri, H. A. Abdel-Aziz, D. A. Abou El Ella and C. T. Supuran, Eur. J. Med. Chem., 2016, 110, 259–266 CrossRef PubMed.
- A. Saeed, S. U. Khan, P. A. Mahesar, P. A. Channar, G. Shabir and J. Iqbal, Biochem. Biophys. Res. Commun., 2017, 482, 176–181 CrossRef.
- S. Iqbal, M. Saleem, M. K. Azim, M. Taha, U. Salar, K. M. Khan, S. Perveen and M. I. Choudhary, Bioorg. Chem., 2017, 72, 89–101 CrossRef PubMed.
- Ö. Güleç, M. Arslan, N. Gencer, A. Ergun, C. Bilen and O. Arslan, Arch. Physiol. Biochem., 2017, 123, 306–312 CrossRef.
- M. Bozdag, S. Bua, S. M. Osman, Z. AlOthman and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 885–892 CrossRef PubMed.
- S. Angapelly, P. Sri Ramya, A. Angeli, C. T. Supuran and M. Arifuddin, ChemMedChem, 2017, 12, 1578–1584 CrossRef PubMed.
- P. S. Ramya, S. Angapelly, A. Angeli, C. S. Digwal, M. Arifuddin, B. N. Babu, C. T. Supuran and A. Kamal, J. Enzyme Inhib. Med. Chem., 2017, 32, 1274–1281 CrossRef.
- A. Altundas, B. Gül, M. Çankaya, A. Atasever and İ. Gülçin, J. Biochem. Mol. Toxicol., 2017, 31, e21998 CrossRef PubMed.
- P. Taslimi, S. Osmanova, İ. Gulçin, S. Sardarova, V. Farzaliyev, A. Sujayev, R. Kaya, F. Koc, S. Beydemir and S. H. Alwasel, J. Biochem. Mol. Toxicol., 2017, 31, e21931 CrossRef.
- K. Oktay, L. Polat Kose, K. Şendil, M. S. Gültekin and İ. Gülçın, Med. Chem. Res., 2017, 26, 1619–1627 CrossRef.
- B. Zengin Kurt, F. Sonmez, S. Durdagi, B. Aksoydan, R. Ekhteiari Salmas, A. Angeli, M. Kucukislamoglu and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 1042–1052 CrossRef.
- İ. Gulçin, M. Abbasova, P. Taslimi, Z. Huyut, L. Safarova, A. Sujayev, V. Farzaliyev, Ş. Beydemir, S. H. Alwasel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 1174–1182 CrossRef.
- F. Erdemir, D. B. Celepci, A. Aktaş, P. Taslimi, Y. Gök, H. Karabıyık and İ. Gülçin, J. Mol. Struct., 2018, 1155, 797–806 CrossRef.
- A. Behcet, T. Çağlılar, D. B. Celepci, A. Aktaş, P. Taslimi, Y. Gök, M. Aygün, R. Kaya and İ. Gülçin, J. Mol. Struct., 2018, 1170, 160–169 CrossRef.
- M. Ahmed, M. A. Qadir, A. Hameed, M. N. Arshad, A. M. Asiri and M. Muddassar, Bioorg. Chem., 2018, 76, 218–227 CrossRef PubMed.
- M. F. Ansari, D. Idrees, M. I. Hassan, K. Ahmad, F. Avecilla and A. Azam, Eur. J. Med. Chem., 2018, 144, 544–556 CrossRef.
- L. Vats, V. Sharma, A. Angeli, R. Kumar, C. T. Supuran and P. K. Sharma, Eur. J. Med. Chem., 2018, 150, 678–686 CrossRef.
- R. Kumar, L. Vats, S. Bua, C. T. Supuran and P. K. Sharma, Eur. J. Med. Chem., 2018, 155, 545–551 CrossRef.
- M. G. El-Gazzar, N. H. Nafie, A. Nocentini, M. M. Ghorab, H. I. Heiba and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2018, 33, 1565–1574 CrossRef.
- M. Ekiz, A. Tutar, S. Ökten, B. Bütün, Ü. M. Koçyiğit, P. Taslimi and G. Topçu, Arch. Pharm., 2018, 351, 1800167 CrossRef.
- C. B. Mishra, S. Kumari, A. Angeli, S. Bua, M. Buonanno, S. M. Monti, M. Tiwari and C. T. Supuran, Eur. J. Med. Chem., 2018, 156, 430–443 CrossRef.
- R. Qamar, A. Saeed, M. Saeed, Z. Ashraf, Q. Abbas, M. Hassan and F. Albericio, Drug Dev. Res., 2018, 79, 352–361 CrossRef.
- B. N. Sağlık, U. A. Cevik, D. Osmaniye, S. Levent, B. K. Çavuşoğlu, Y. Demir, S. Ilgın, Y. Özkay, A. S. Koparal and Ş. Beydemir, Bioorg. Chem., 2019, 91, 103153 CrossRef.
- A.-M. Alaa, A. S. El-Azab, S. Bua, A. Nocentini, M. A. A. El-Enin, M. M. Alanazi, N. A. AlSaif, M. M. Hefnawy and C. T. Supuran, Bioorg. Chem., 2019, 87, 425–431 CrossRef PubMed.
- J. Mahar, A. Saeed, K. D. Belfield, F. A. Larik, P. A. Channar, M. A. Kazi, Q. Abbas, M. Hassan, H. Raza and S.-Y. Seo, Bioorg. Chem., 2019, 84, 170–176 CrossRef PubMed.
- F. Turkan, A. Cetin, P. Taslimi, M. Karaman and İ. Gulçin, Bioorg. Chem., 2019, 86, 420–427 CrossRef PubMed.
- P. S. Thacker, M. Alvala, M. Arifuddin, A. Angeli and C. T. Supuran, Bioorg. Chem., 2019, 86, 386–392 CrossRef CAS PubMed.
- B. Z. Kurt, A. Dag, B. Doğan, S. Durdagi, A. Angeli, A. Nocentini, C. T. Supuran and F. Sonmez, Bioorg. Chem., 2019, 87, 838–850 CrossRef CAS.
- B. Z. Kurt, F. Sonmez, D. Ozturk, A. Akdemir, A. Angeli and C. T. Supuran, Eur. J. Med. Chem., 2019, 183, 111702 CrossRef.
- S. Bayindir, C. Caglayan, M. Karaman and İ. Gülcin, Bioorg. Chem., 2019, 90, 103096 CrossRef CAS PubMed.
- W. M. Eldehna, M. A. Abdelrahman, A. Nocentini, S. Bua, S. T. Al-Rashood, G. S. Hassan, A. Bonardi, A. A. Almehizia, H. M. Alkahtani, A. Alharbi and C. T. Supuran, Bioorg. Chem., 2019, 90, 103102 CrossRef CAS.
- M. Işık, S. Akocak, N. Lolak, P. Taslimi, C. Türkeş, İ. Gülçin, M. Durgun and Ş. Beydemir, Arch. Pharm., 2020, 353, 2000102 CrossRef.
- A. A. Alkhaldi, M. M. Al-Sanea, A. Nocentini, W. M. Eldehna, Z. M. Elsayed, A. Bonardi, M. F. Abo-Ashour, A. K. El-Damasy, M. S. Abdel-Maksoud and T. Al-Warhi, Eur. J. Med. Chem., 2020, 207, 112745 CrossRef CAS PubMed.
- B. Sever, C. Türkeş, M. D. Altıntop, Y. Demir and Ş. Beydemir, Int. J. Biol. Macromol., 2020, 163, 1970–1988 CrossRef CAS.
- Q. Istrefi, C. Türkeş, M. Arslan, Y. Demir, A. R. Nixha, Ş. Beydemir and Ö. İ. Küfrevioğlu, Arch. Pharm., 2020, 353, 1900383 CrossRef CAS.
- M. A. Said, W. M. Eldehna, A. Nocentini, A. Bonardi, S. H. Fahim, S. Bua, D. H. Soliman, H. A. Abdel-Aziz, P. Gratteri, S. M. Abou-Seri and C. T. Supuran, Eur. J. Med. Chem., 2020, 185, 111843 CrossRef CAS.
- A. Khan, M. Khan, S. A. Halim, Z. A. Khan, Z. Shafiq and A. Al-Harrasi, Front. Chem., 2020, 8, 598095 CrossRef PubMed.
- A. Saeed, P. A. Channar, M. Arshad, H. R. El-Seedi, Q. Abbas, M. Hassan, H. Raza and S. Y. Seo, J. Heterocycl. Chem., 2020, 57, 2831–2843 CrossRef.
- R. F. George, S. Bua, C. T. Supuran and F. M. Awadallah, Bioorg. Chem., 2020, 96, 103635 CrossRef PubMed.
- P. S. Thacker, A. Angeli, O. S. Argulwar, P. L. Tiwari, M. Arifuddin and C. T. Supuran, Bioorg. Chem., 2020, 98, 103739 CrossRef.
- R. F. George, M. F. Said, S. Bua and C. T. Supuran, Bioorg. Chem., 2020, 95, 103514 CrossRef.
- P. Singh, P. P. Yadav, B. Swain, P. S. Thacker, A. Angeli, C. T. Supuran and M. Arifuddin, Bioorg. Chem., 2021, 108, 104647 CrossRef PubMed.
- S. Manzoor, A. Petreni, M. K. Raza, C. T. Supuran and N. Hoda, Bioorg. Med. Chem. Lett., 2021, 48, 128249 CrossRef PubMed.
- B. Swain, P. Singh, A. Angeli, K. Aashritha, N. Nagesh, C. T. Supuran and M. Arifuddin, Bioorg. Med. Chem. Lett., 2021, 37, 127856 CrossRef PubMed.
- F. Mancuso, A. Di Fiore, L. De Luca, A. Angeli, G. De Simone, C. T. Supuran and R. Gitto, Bioorg. Med. Chem., 2021, 44, 116279 CrossRef.
- M. Abas, A. Bahadur, Z. Ashraf, S. Iqbal, M. S. R. Rajoka, S. Rashid, E. Jabeen, Z. Iqbal, Q. Abbas and A. Bais, J. Mol. Struct., 2021, 1246, 131145 CrossRef.
- C. Yamali, H. Sakagami, Y. Uesawa, K. Kurosaki, K. Satoh, Y. Masuda, S. Yokose, A. Ece, S. Bua, A. Angeli and H. I. Gul, Eur. J. Med. Chem., 2021, 217, 113351 CrossRef.
- A. H. Eldeeb, M. F. Abo-Ashour, A. Angeli, A. Bonardi, D. S. Lasheen, E. Z. Elrazaz, A. Nocentini, P. Gratteri, H. A. Abdel-Aziz and C. T. Supuran, Eur. J. Med. Chem., 2021, 221, 113486 CrossRef.
- M. T. Nemr, A. M. AboulMagd, H. M. Hassan, A. A. Hamed, M. I. Hamed and M. T. Elsaadi, RSC Adv., 2021, 11, 26241–26257 RSC.
- M. Saadiq, G. Uddin, A. Latif, M. Ali, N. Akbar, Ammara, S. Ali, M. Ahmad, M. Zahoor and A. Khan, ACS Omega, 2021, 7, 705–715 CrossRef.
- D. M. Elimam, A. A. Elgazar, A. Bonardi, M. Abdelfadil, A. Nocentini, R. A. El-Domany, H. A. Abdel-Aziz, F. A. Badria, C. T. Supuran and W. M. Eldehna, Eur. J. Med. Chem., 2021, 225, 113800 CrossRef.
- S. Zaib, I. Khan, H. S. Anbar, S.-O. Zaraei, R. M. Sbenati, H. T. Maryam, H. S. Shah and M. I. El-Gamal, J. Mol. Struct., 2022, 1262, 133048 CrossRef.
- P. Singh, N. S. Goud, B. Swain, S. K. Sahoo, A. Choli, A. Angeli, B. S. Kushwah, V. M. Yaddanapudi, C. T. Supuran and M. Arifuddin, Bioorg. Med. Chem. Lett., 2022, 70, 128809 CrossRef PubMed.
- H. T. Abdel-Mohsen, A. M. El Kerdawy, M. A. Omar, A. Petreni, R. M. Allam, H. I. El Diwani and C. T. Supuran, Eur. J. Med. Chem., 2022, 228, 114004 CrossRef PubMed.
- A. Kumar, K. Siwach, T. Rom, R. Kumar, A. Angeli, A. K. Paul, C. T. Supuran and P. K. Sharma, Bioorg. Chem., 2022, 123, 105764 CrossRef PubMed.
- H. O. Tawfik, A. Petreni, C. T. Supuran and M. H. El-Hamamsy, Eur. J. Med. Chem., 2022, 232, 114190 CrossRef PubMed.
- B. Swain, A. Khan, P. Singh, V. S. Marde, A. Angeli, K. K. Chinchilli, V. M. Yaddanapudi, S. Carradori, C. T. Supuran and M. Arifuddin, Molecules, 2022, 27, 8028 CrossRef PubMed.
- M. F. Said, R. F. George, A. Petreni, C. T. Supuran and N. M. Mohamed, J. Enzyme Inhib. Med. Chem., 2022, 37, 701–717 CrossRef PubMed.
- P. S. Thacker, V. Newaskar, A. Angeli, D. K. Sigalapalli, N. S. Goud, H. Chirra, A. B. Shaik, M. Arifuddin and C. T. Supuran, Arch. Pharm., 2022, 355, 2200232 CrossRef PubMed.
- H. T. Abdel-Mohsen, M. A. Omar, A. Petreni and C. T. Supuran, Arch. Pharm., 2022, 355, 2200180 CrossRef PubMed.
- C. Ismail, A. Nocentini, C. T. Supuran, J. Y. Winum and R. Gharbi, Arch. Pharm., 2022, 355, 2100405 CrossRef PubMed.
- A. Buza, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. R. Nixha and Ş. Beydemir, Int. J. Biol. Macromol., 2023, 239, 124232 CrossRef PubMed.
- M. A. Ragab, W. M. Eldehna, A. Nocentini, A. Bonardi, H. E. Okda, B. Elgendy, T. S. Ibrahim, M. M. Abd-Alhaseeb, P. Gratteri and C. T. Supuran, Eur. J. Med. Chem., 2023, 250, 115180 CrossRef CAS PubMed.
- C. Kakakhan, C. Türkeş, Ö. Güleç, Y. Demir, M. Arslan, G. Özkemahlı and Ş. Beydemir, Bioorg. Med. Chem., 2023, 77, 117111 CrossRef.
- A. S. El-Azab, A.-M. Alaa, S. Bua, A. Nocentini, A. H. Bakheit, H. M. Alkahtani, M. M. Hefnawy and C. T. Supuran, Saudi Pharm. J., 2023, 31, 101866 CrossRef PubMed.
- C. Öztürk, E. Kalay, S. Gerni, N. Balci, F. S. Tokali, O. N. Aslan and E. Polat, Biotechnol. Appl. Biochem., 2024, 71, 223–231 CrossRef PubMed.
- H. Bostancı, U. Çevik, R. Kapavarapu, Y. Güldiken, Z. Ş. Inan, Ö. Güler, T. Uysal, A. Uytun, F. Çetin and Y. Özkay, SAR QSAR Environ. Res., 2023, 34, 543–567 CrossRef.
- N. H. Metwally and E. A. El-Desoky, ACS Omega, 2023, 8, 5571–5592 CrossRef PubMed.
- R. Romagnoli, T. De Ventura, S. Manfredini, E. Baldini, C. T. Supuran, A. Nocentini, A. Brancale, C. Varricchio, R. Bortolozzi and L. Manfreda, J. Enzyme Inhib. Med. Chem., 2023, 38, 2270180 CrossRef.
- H. Ünver, U. Acar Cevik, H. E. Bostancı, O. Kaya and Ü. M. Kocyigit, ChemistrySelect, 2023, 8, e202301641 CrossRef.
- İ. Çelik, U. Acar Çevik, K. Küçükoğlu, H. Nadaroglu, H. E. Bostancı, A. Işık, Y. Özkay and Z. A. Kaplancıklı, Chem. Biol. Drug Des., 2024, 103, e14351 CrossRef.
- K. Kucukoglu, N. Faydali, D. Bul, H. Nadaroglu, B. Sever, M. D. Altıntop, B. Ozturk and I. Guzel, J. Mol. Struct., 2023, 1276, 134699 CrossRef CAS.
- P. Begines, A. Bonardi, A. Nocentini, P. Gratteri, S. Giovannuzzi, R. Ronca, C. Tavani, M. L. Massardi, Ó. López and C. T. Supuran, Bioorg. Med. Chem., 2023, 94, 117467 CrossRef CAS PubMed.
- N. A. Zahedi, M. Mohammadi-Khanaposhtani, P. Rezaei, M. Askarzadeh, M. Alikhani, M. Adib, M. Mahdavi, B. Larijani, S. Niakan, M. B. Tehrani and P. Taslimi, J. Mol. Struct., 2023, 1276, 134767 CrossRef CAS.
- A. Angeli, V. Kartsev, A. Petrou, B. Lichitsky, A. Komogortsev, A. Geronikaki and C. T. Supuran, Bioorg. Chem., 2023, 138, 106621 CrossRef CAS.
- B. I. Huwaimel, S. K. Jonnalagadda, S. Jonnalagadda, S. Kumari, A. Nocentini, C. T. Supuran and P. C. Trippier, Drug Dev. Res., 2023, 84, 681–702 CrossRef CAS PubMed.
- A. I. Alalawy, K. Alatawi, N. A. Alenazi, A. F. Qarah, O. M. Alatawi, R. B. Alnoman, A. Alharbi and N. M. El-Metwaly, J. Mol. Struct., 2024, 1295, 136609 CrossRef CAS.
- F. N. Takla, S. Zaib, W. A. Bayoumi, K. Shehzadi, S. M. El-Messery, S. Anjum, A. Faisal and M. N. Nasr, J. Mol. Struct., 2024, 1300, 137277 CrossRef CAS.
- U. A. Çevik, A. Işık, R. Kapavarapu, K. Küçükoğlu, H. Nadaroglu, H. E. Bostancı, Y. Özkay and Z. A. Kaplancıklı, J. Mol. Struct., 2024, 1295, 136770 CrossRef.
- C. Efeoglu, O. Selcuk, B. Demir, E. Sahin, H. Sari, C. Türkeş, Y. Demir, Y. Nural and Ş. Beydemir, J. Mol. Struct., 2024, 1301, 137365 CrossRef CAS.
- S. Mert, Y. Demir, Y. Sert, R. Kasımoğulları and İ. Gülçin, J. Mol. Struct., 2024, 139472 Search PubMed.
- P. Singh, S. G. Nerella, B. Swain, A. Angeli, Q. Ullah, C. T. Supuran and M. Arifuddin, Int. J. Biol. Macromol., 2024, 268, 131548 CrossRef PubMed.
- M. M. Abdelhakeem, M. M. Morcoss, D. A. Hanna and P. F. Lamie, Bioorg. Chem., 2024, 144, 107154 CrossRef PubMed.
- S. Higazy, N. Samir, A. El-Khouly, S. Giovannuzzi, P. Begines, H. M. Gaber, C. T. Supuran and K. A. Abouzid, Bioorg. Chem., 2024, 144, 107089 CrossRef PubMed.
- C. M. Al-Matarneh, M. Pinteala, A. Nicolescu, M. Silion, F. Mocci, R. Puf, A. Angeli, M. Ferraroni, C. T. Supuran and S. Zara, J. Med. Chem., 2024, 67, 3018–3038 CrossRef PubMed.
- S. Maddipatla, B. Bakchi, R. R. Gadhave, A. Ammara, S. Sau, B. Rani, S. Nanduri, N. P. Kalia, C. T. Supuran and V. M. Yaddanapudi, Arch. Pharm., 2024, e2400064 CrossRef PubMed.
- M. Sobati, M. Abdoli, A. Angeli, A. Bonardi, M. Ferraroni, C. T. Supuran and R. Žalubovskis, Arch. Pharm., 2024, e2400038 CrossRef PubMed.
- B. Swain, V. S. Marde, P. Singh, A. Angeli, A. Khan, V. M. Yaddanapudi, Q. Ullah, C. T. Supuran and M. Arifuddin, Arch. Pharm., 2024, 357, 2300718 CrossRef.
- R. El Ati, N. Öztaşkın, A. Çağan, A. Akıncıoğlu, Y. Demir, S. Göksu, R. Touzani and İ. Gülçin, Arch. Pharm., 2024, 357, 2300545 CrossRef PubMed.
- D. Moi, S. Vittorio, A. Angeli, C. T. Supuran and V. Onnis, ACS Med. Chem. Lett., 2024, 15, 470–477 CrossRef PubMed.
- Ş. Özkul, E. Tunca, S. Mert, A. Bayrakdar and R. Kasımoğulları, J. Biochem. Mol. Toxicol., 2024, 38, e23704 CrossRef.
- M. Palomba, A. Angeli, R. Galdini, A. Hughineata, G. Perin, E. J. Lenardao, F. Marini, C. Santi, C. Supuran and L. Bagnoli, Org. Biomol. Chem., 2024, 6532–6542 RSC.
- M. Durgun, S. Akocak, N. Lolak, F. Topal, Ü. M. Koçyiğit, C. Türkeş, M. Işık and Ş. Beydemir, Chem. Biodiversity, 2024, 21, e202301824 CrossRef.
- A. Angeli, A. Petrou, V. G. Kartsev, A. Zubenko, L. N. Divaeva, V. Chekrisheva, D. Iacopetta, M. S. Sinicropi, S. Sirakanyan and A. Geronikaki, ChemMedChem, 2024, e202400147 CrossRef.
- H. Aslan, G. Renzi, A. Angeli, I. D’Agostino, R. Ronca, M. L. Massardi, C. Tavani, S. Carradori, M. Ferraroni and P. Governa, RSC Med. Chem., 2024, 1929–1941 RSC.
- F. F. Albelwi, M. S. Nafie, N. R. Albujuq, W. Hourani, A. Aljuhani, K. M. Darwish, M. M. Tawfik, N. Rezki and M. R. Aouad, RSC Med. Chem., 2024, 15, 2440–2461 RSC.
- İ. Yetek, S. Mert, E. Tunca, A. Bayrakdar and R. Kasımoğulları, Mol. Diversity, 2024, 1–21 Search PubMed.
- C. T. Supuran, Curr. Pharm. Des., 2008, 14, 603–614 CrossRef.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421–4468 CrossRef PubMed.
- A. Maresca, C. Temperini, L. Pochet, B. Masereel, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2010, 53, 335–344 CrossRef PubMed.
- D. Vullo, M. Franchi, E. Gallori, J. Pastorek, A. Scozzafava, S. Pastorekova and C. T. Supuran, Bioorg. Med. Chem. Lett., 2003, 13, 1005–1009 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |