
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule detection of a terrylenediimide-based near-infrared emitter†
Suvarna
Sujilkumar
 ,
Philip Daniel
Maret
,
Kavya
Vinod
,
Athira T.
John
and
Mahesh
Hariharan
*
,
Philip Daniel
Maret
,
Kavya
Vinod
,
Athira T.
John
and
Mahesh
Hariharan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala P.O., Vithura, Thiruvananthapuram, Kerala, India 695551. E-mail: mahesh@iisertvm.ac.in
First published on 24th September 2024
Abstract
Environment-sensitive fluorescent agents with near-infrared (NIR) emission are in great demand owing to their applications in biomedical and quantum technologies. We report a novel NIR absorbing (λAbsmax = 734 nm) and emitting (λFlmax = 814 nm) terrylenediimide (TDI) based donor–acceptor chromophore (TDI-TPA4), exhibiting polarity-sensitive single-photon emission. By virtue of the charge transfer (CT) character, ensemble level measurements revealed solvatochromism and NIR emission (ϕFl = 26.2%), overcoming the energy gap law. The CT nature of the excited states is further validated by state-of-the-art fragment-based excited state theoretical analysis. To mimic the polarity conditions at the single-molecule level, TDI-TPA4 was immobilized in polystyrene (PS; low polar) and poly(vinyl alcohol) (PVA; high polar) matrices, which enables tuning of the energy levels of the locally excited state and charge-separated (CS) state. Minimal blinking and prolonged survival time of the TDI-TPA4 molecule in the PS matrix, in contrast to the PVA matrix, possibly confirms the implication of the energy gap law and polarity sensitivity of TDI-TPA4. The existence of the CT state in nonpolar and CS state in polar solvents was confirmed by transient absorption measurements in the femtosecond regime. The current work sheds light on the design principle for NIR single-photon emitting organic chromophores for deep tissue imaging and probing the nanoscale heterogeneity.
Introduction
Near-infrared single-photon emitters are an emerging class of compounds for quantum information science, including computing, sensing, and communication.1–5 Single-molecule level scrutiny of organic near-infrared (NIR) emitting molecules has recently gained much attention.6–9 The efficiency of the NIR organic emitters is limited by the energy gap law; the intrinsic forbidden nature of emission from states with low energy gap results in negligible fluorescence quantum yield.10–13 The donor–acceptor systems with charge transfer (CT) character14 are better alternatives to circumvent the limitations possessed by conventional π-conjugated NIR molecules by narrowing the energy gap for NIR emission.15–18 The emission from the CT state is limited due to the forbidden nature of electronic transition as a result of spatial separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).19,20 However, hybridizing the local excited (LE) state with the CT state results in a favored transition dipole moment, subsequently leading to NIR emission.21–23 Exciton delocalization in the aggregates is one of the strategies implemented in Pt(II) complexes and bay-alkylated quaterrylene to successfully overcome the energy gap law for enhanced NIR fluorescence quantum yield at the ensemble level.24,25 Recently, Erker and Basché validated the energy gap law for NIR single-photon emitters by investigating the dielectric dependence of dibenzoterrylene (λFlmax = 741 nm) at the ensemble and single-molecule levels.26 In comparison to the previously investigated NIR single-photon emitters (Table S1†), probing the dielectric dependence of CT-mediated NIR emitting chromophores at the single-molecule level remains elusive. Polarity dependence of fluorescence at the nanoscale level can be probed using chromophores with emissive CT in the visible range, which was first demonstrated by Higgins and co-workers27 based on the model developed by Marcus.28,29 Monitoring the intrinsic NIR fluorescence fluctuations at the single-molecule level in different dielectric environments is beneficial for probing temporal inhomogeneity and acquiring autofluorescence-free contrasting images of (bio)specimens.30–38By virtue of the limited blinking and extended survival time before photobleaching,39 terrylenediimide (TDI) is known to be a suitable candidate for membrane labeling40 and single-molecule investigations.41–44 Our longstanding interest in monitoring CT excitons in rylenecarboximide derivatives at the ensemble45–47 and single-molecule levels48,49 motivated us to explore higher analogs, such as TDI-functionalized chromophores.50–56 Herein, we designed and synthesized a NIR absorbing and emitting donor–acceptor chromophore based on TDI (acceptor) with triphenylamine moiety (TPA) as a donor (TDI-TPA4) and explored its potential as a polarity-sensitive NIR single-photon source (Scheme 1). The striking contrast of fluorescence intensity time traces in different dielectric environments was demonstrated by single-molecule thin film measurements, and femtosecond excited state dynamics investigations corroborated the photoinduced processes at the ensemble level.
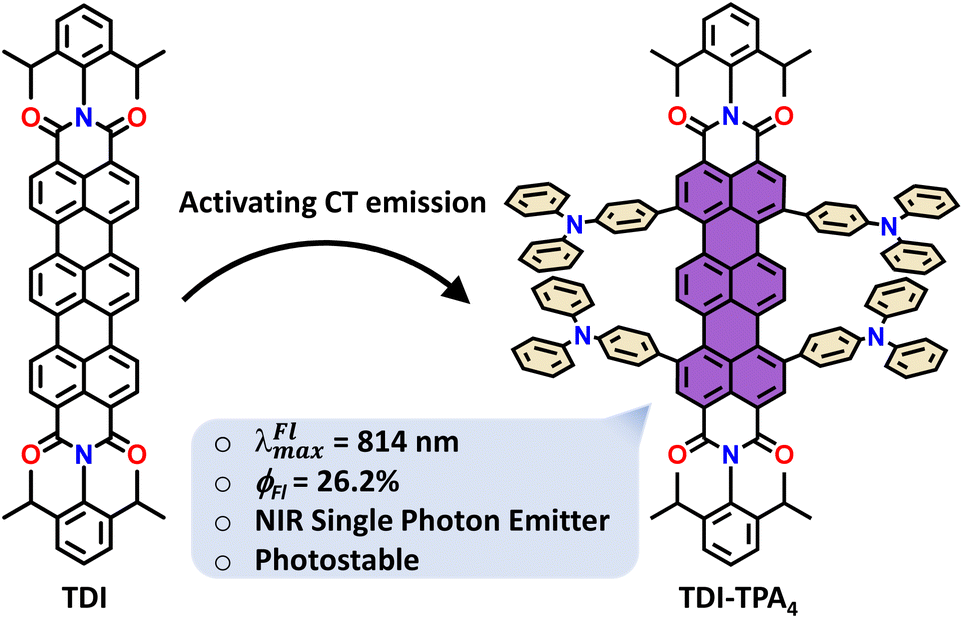 | ||
| Scheme 1 Design strategy and chemical structures of TDI and TDI-TPA4. | ||
Results and discussion
Synthesis
Novel TDI-TPA4 was synthesized via a Suzuki cross-coupling reaction between tetrabromo-terrylenediimide (TDI-Br4) and 4-(diphenylamino)phenylboronic acid (Schemes S1 and S2†). TDI and TDI-Br4 were synthesized and characterized as per the previously reported procedures (Scheme S2 and Fig. S1–S8†).50 The Suzuki coupling reaction between TDI-Br4 and 4-(diphenylamino)phenylboronic acid was carried out under inert conditions in the presence of potassium carbonate (K2CO3), dioxane, and a catalytic amount of tetrakis(triphenylphosphine)palladium(0) followed by heating of the reaction mixture at 85 °C. TDI-TPA4 was isolated and purified using column chromatography (ethyl acetate/hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) to yield purple-colored TDI-TPA4 with a 40% yield (Scheme S2†). The desired compound was successfully characterized by 1H and 13C NMR, IR spectroscopy, and HRMS mass spectrometry (see Fig. S9–S13†).
:10) to yield purple-colored TDI-TPA4 with a 40% yield (Scheme S2†). The desired compound was successfully characterized by 1H and 13C NMR, IR spectroscopy, and HRMS mass spectrometry (see Fig. S9–S13†).
Optical properties
The electronic absorption spectra of the TDI-TPA4 and TDI were recorded in hexane (HEX, ε = 1.88, C = 5–10 μM) (Fig. 1). The absorption spectrum of TDI-TPA4 spans through the visible range and extends to the NIR window (400–850 nm) with absorption bands at 734, 597, and 522 nm with a molar extinction coefficient εmax = 3.54 × 104 M−1 cm−1 (at 734 nm) corresponding to the S0 to S1 transition (Table 1 and Fig. S14, S15†). TDI exhibited three distinct vibronic progressions with an absorption maximum at 634 nm corresponding to the S0 to S1 electronic transition. The fluorescence emission spectrum of TDI-TPA4 consists of a NIR band with a maximum at 814 nm when excited at 591 nm in HEX (Fig. 1). The remarkable red-shift in the fluorescence emission spectrum of TDI-TPA4 as compared to TDI (ΔλFlmax = 173 nm or 3315.16 cm−1) corroborate the existence of charge transfer between the four TPA moieties and the TDI core. The solvent-dependent absorption measurements of TDI-TPA4 depicted a red-shift (ΔλAbsmax = 20 nm) with increasing solvent polarity from HEX to ACN (Table 1 and Fig. S14–S17†). The solvent-dependent fluorescence spectra of TDI-TPA4 (C = 5–10 μM) depict a bathochromic shift along with significant fluorescence quenching upon increasing polarity from HEX (ε = 1.88, ϕFl = 26.2%), THF (ε = 7.58, ϕFl = 17.5%) to ACN (ε = 37.50, ϕFl = 10.2%) (Fig. S18† and Table 1). The decreased fluorescence quantum yield of TDI-TPA4 in hexane compared to TDI (ϕFl = 90%)50 is indicative of other nonradiative decay channels upon photoexcitation. The substantial Stokes shift of TDI-TPA4 in the polar solvents (ΔṽStokes = 227 nm or 3097.90 cm−1 in THF and ΔṽStokes = 231 nm or 3110.16 cm−1 in ACN) when compared to TDI-TPA4 in HEX (ΔṽStokes = 80 nm or 1338.77 cm−1) results from the lowering of the energy gap, leading to the reduced fluorescence quantum yield, which is in accordance with the energy gap law. The emission spectra recorded by exciting at 734, 597, and 522 nm depicted intact emission maximum at λFlmax = 814 nm (Fig. S19†). The excitation spectra collected at the respective emission maximum of TDI-TPA4 in HEX, THF, and ACN resemble the absorption spectra, validating the emissive nature of the CT state (Fig. S20†).47 Time-correlated single photon counting (TCSPC) measurement of TDI-TPA4 was performed in HEX (λex = 640 nm and monitored at 814 nm), suggesting the existence of a single emissive component with a fluorescence lifetime τFl = 1.37 ns (Fig. S21†). The concentration-dependent and temperature-dependent absorption measurements of TDI-TPA4 were performed in THF to understand the possibility of aggregate formation. The absorption spectra of TDI-TPA4 were recorded for different concentrations (C = 0.01 mM to 0.20 mM) and exhibited constant I0/I1 ratios, ruling out the possibility of aggregation (Fig. S22 and Table S2†). Monitoring the temperature-dependent absorption spectra of TDI-TPA4 (C = 5–10 μM) from 10 °C to 60 °C depicted negligible changes in the spectral feature, further strengthening the monomeric nature of TDI-TPA4 in the solution state (Fig. S23†). | ||
| Fig. 1 Normalized UV-vis absorption and fluorescence emission spectra of TDI and TDI-TPA4 in hexane. | ||
| Solvent | λ Absmax (nm) | λ Flmax (nm) | ε max × 104 (M−1 cm−1) | ϕ Fl (%) ± 5 | τ (ps) | ΔGCS (eV) | ΔGCR (eV) | k r × 109 (s−1) | k nr × 109 (s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a From global fits of fsTA spectra. ΔGCS and ΔGCR from Rehm–Weller analysis. kr and knr are calculated as per the reported procedure from the literature.26,57 | |||||||||
| HEX (ε = 1.88) | 734 | 814 | 3.54 | 26.2 | 1210.0 | 0.20 | −1.77 | 0.216 | 0.6 |
| THF (ε = 7.58) | 750 | 977 | 3.33 | 17.5 | 1.5 | −0.21 | −1.36 | 116.6 | 550.0 |
| ACN (ε = 37.50) | 754 | 985 | 3.13 | 10.2 | 0.5 | −0.33 | −1.24 | 204.0 | 1796.0 |
Theoretical investigations
The ground state optimization of the TDI-TPA4 was performed using density functional theory (DFT) in vacuum using the Gaussian 16 program.58 2,6-Diisopropylphenyl group at the imide position has been replaced with a methyl group to reduce computational cost. For ground state and first singlet excited state optimization, a range-separated hybrid exchange-correlation functional CAM-B3LYP (using the Coulomb-attenuating method) in combination with a def2-SVP basis set was used to account for the charge transfer excitations in TDI-TPA4. The optimized structure of TDI and TDI-TPA4 revealed a considerable contortion in the TDI core with a dihedral angle of 18.8° and 18.6° as a result of bulky TPA substitution in the bay region (Fig. 2a, b and S24†). The propeller-like TPA moieties are oriented at angles 115.3° and 115.1° with respect to the ortho position of the TDI core to decrease steric repulsion (Fig. S24 and S25†). Time-dependent DFT was employed to compute the vertical excitation energies and oscillator strengths at the CAM-B3LYP/def2-SVP level of theory. The singlet excited states S1 (2.105 eV), S2 (2.885 eV), and S3 (2.886 eV) are identified as bright states with considerable oscillator strength in TDI-TPA4 (Table S3†). Further investigation into the character of the excited states was carried out using fragment-based excited-state analysis developed by Plasser, implemented in TheoDORE.59,60 Becke, 3-parameter, Lee–Yang–Parr functional with D3, an empirical correction for dispersion forces and def2-SVP basis set was employed to perform the calculations. The degree of excitation energy delocalization in various fragments is determined by the participation ratio (PR). TDI core and four TPA groups were categorized as two fragments for the fragment analysis of the TDI-TPA4 (Fig. S26†). The nature of the excited state is represented by the charge transfer (CT) value where CT > 0.8 denotes the CT state, and CT = 0 depicts the Frenkel exciton state (FE). A hole–electron correlation plot is used to pictorially represent the nature of the excited states. The S1 state of TDI-TPA4 is identified as a partial CT state (hybridized feature of CT and LE) with a CT value of 0.64 (PR = 1.46). Conversely, transitions S0 → S2, S0 → S3, and S0 → S4 possessed CT values of 0.91, 0.89, and 0.90, respectively, corresponding to a strong CT character (Fig. 2c). In the hole–electron isosurface of the S2 state, the electron is confined to the TDI core while the hole is confined to the TPA fragments, establishing the CT nature of TDI-TPA4 (Fig. 2d).61 To obtain further insights into the electron density distribution within the TDI-TPA4 molecule, the natural transition orbital (NTO) isosurface analysis for S1, S2, S3, and S4 excited states was carried out. For the S1 state, the electron density is distributed on both the TDI core and TPA fragment, and the lowest unoccupied NTO is localized on the TDI core, corroborating the hybrid nature of the CT and LE. The predominant CT character of higher S2, S3, and S4 excited states is further substantiated by the spatially disjoint electron density distribution of the highest occupied NTO on electron donating TPA fragment and the lowest unoccupied NTO on the TDI core (Fig. S27†).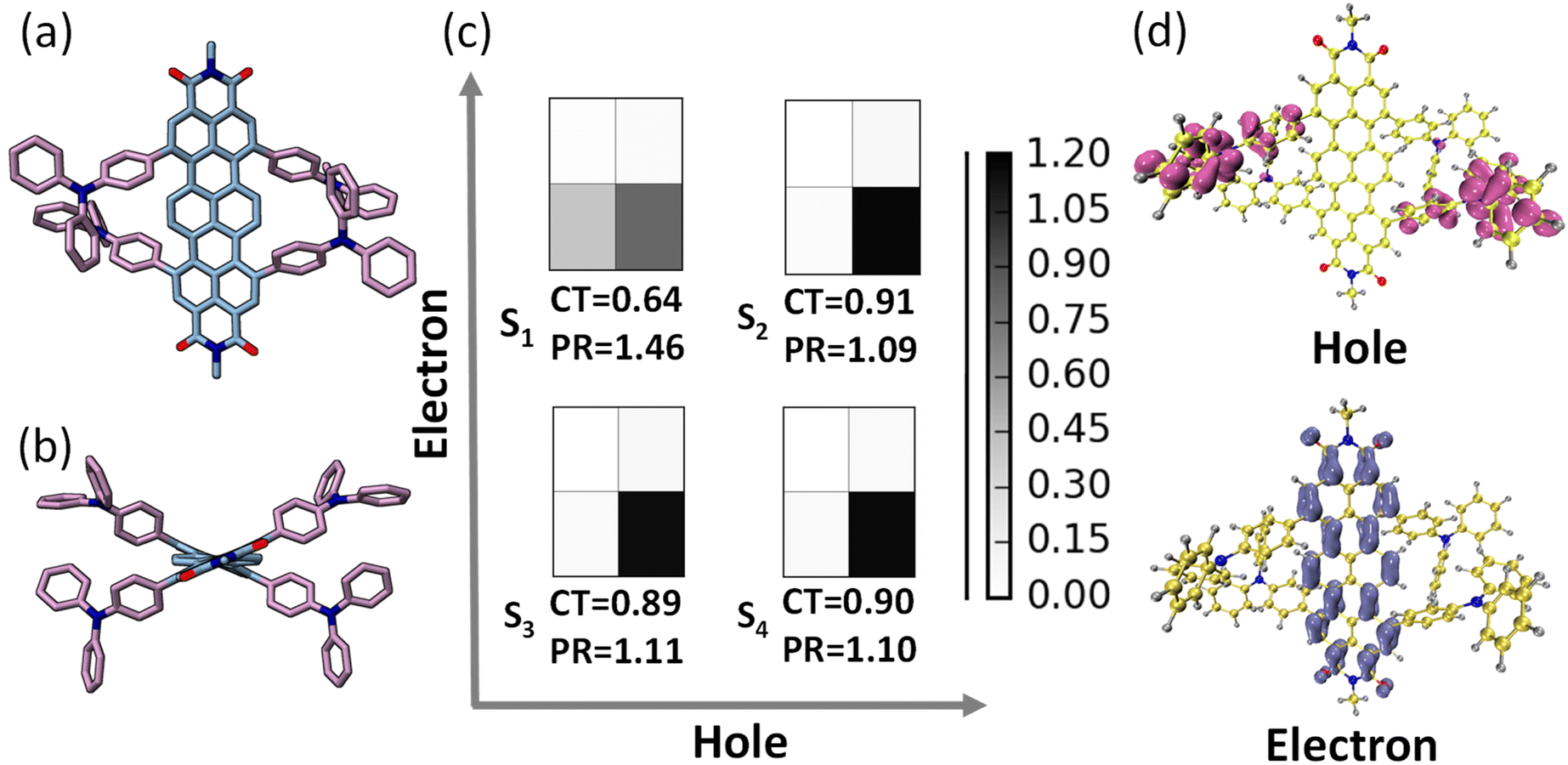 | ||
| Fig. 2 Ground state optimized geometry of TDI-TPA4 shown in (a) top view and (b) side view at CAM-B3LYP/def2-SVP level of theory (2,6-diisopropylphenyl group has been replaced with methyl group to reduce computational cost. Hydrogen atoms are omitted for clarity). (c) Hole–electron correlation plots showing the nature for the S1, S2, S3, and S4 excited states. (d) Corresponding hole–electron isosurface distribution of S2 state (isovalue = 0.001). The CT and PR values are shown to define the nature of excitations. | ||
Single-molecule fluorescence measurements
Exploitation of CT-based organic chromophores as a single-molecule source remained challenging due to quenching of local excited state fluorescence via electron transfer.62 Considerable efforts have been reported in the literature to monitor single-molecule electron transfer of rylenediimide dendrimers in the visible range.63–67 The fluorescence lifetime imaging (FLIM, Fig. 3a and e) and fluorescence intensity trajectories (FITs) were measured using the MicroTime 200 (MT200) time-resolved fluorescence microscope from PicoQuant. The inverted microscope (Olympus IX83) is equipped with a piezo-scanning stage (P-733.2CD, PI). The sample was excited using a 512 nm pulsed laser (LDH-D-C-512, PicoQuant) with a repetition rate of 20 MHz and scanned with a dwell time of 0.4 ms. To acquire the single-molecule emission in the NIR region, the intensity traces were recorded using an SPCM-AQRH single-photon avalanche detector (SPAD) having spectral range 400–1100 nm with 70% maximum detection efficiency at 700 nm and 180 nm diameter of active area.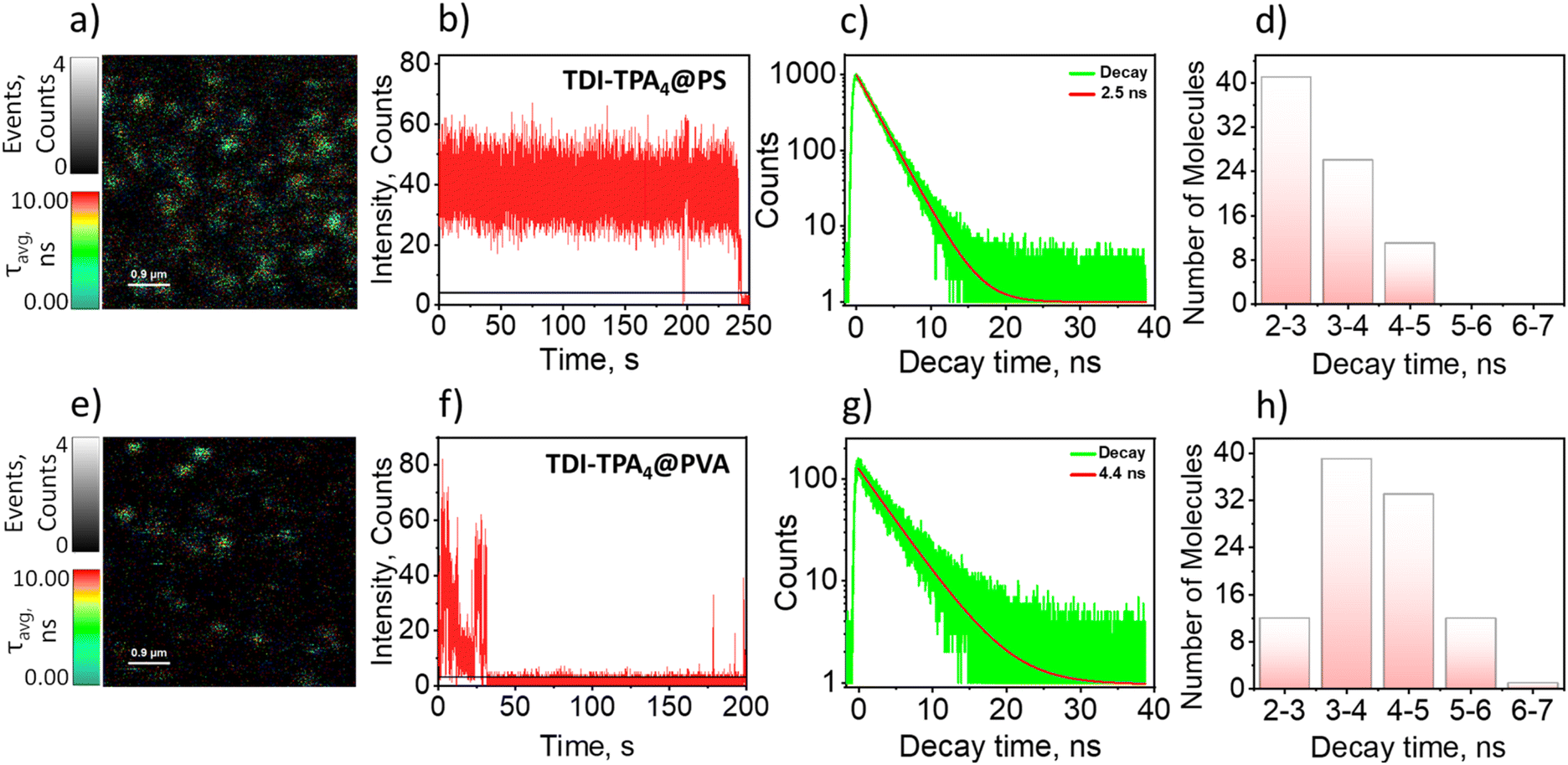 | ||
| Fig. 3 Representative fluorescence lifetime images with average lifetime (τavg) and events scale, of TDI-TPA4 in (a) PS and (e) PVA matrices. Fluorescence intensity trajectories (FITs) of TDI-TPA4 in (b) PS and (f) PVA matrices. Representative single-molecule fluorescence decay measurements of TDI-TPA4 in (c) PS and (g) PVA matrices. Fluorescence lifetime distribution plot of TDI-TPA4 in (d) PS and (h) PVA matrices. | ||
Single-molecule fluorescence (SMF) measurements of TDI-TPA4 were carried out in two different dielectric polymer media such as polystyrene (PS, ε = 2.6) and poly(vinyl alcohol) (PVA, ε = 7.8) nonpolar and polar analogs, respectively, akin to the ensemble measurements.64 FLIM images represent discrete TDI-TPA4 molecules in the PS and PVA matrices (Fig. 3a and e). FITs depict the real-time fluorescence fluctuations of single molecules in their local environment.68 The FITs under investigation include molecules showing blinking (reversible switching between on/bright and off/dark states) or single-step photobleaching (irreversible switch to off state). In the PS matrix, TDI-TPA4 depicted (81 single molecules) prolonged photon count and seldom blinking in agreement with the photophysical process occurring in the low dielectric ensemble measurements (Fig. 3b). The single-molecule fluorescence lifetime distribution of TDI-TPA4 in PS showed monoexponential lifetime distribution (Fig. 3c) peaking at ∼2–3 ns (Fig. 3d). Frequent blinking and low fluorescence emission were observed for TDI-TPA4 single molecules in the polar PVA matrix (119 single molecules). The contrasting blinking dynamics in matrices of distinct dielectric constants corroborated photoinduced charge separation is the major contributor to the dark states in TDI-TPA4. The feasibility of charge separation being less favored in low polar PS matrix accounts for the high photostability and relatively high rate of radiative process in TDI-TPA4 compared to PVA.67 Whereas charge separation is facilitated as an additional nonradiative pathway in the PVA matrix, corroborating the frequent blinking and relatively low fluorescence emission in TDI-TPA4 (Fig. 3f). The single-molecule fluorescence lifetime of TDI-TPA4 in the PVA matrix is spread over the 2–6 ns range (Fig. 3g and h). The broad distribution of lifetime implies the existence of a wide range of rate constants for electron transfer in the PVA matrix possibly due to the rigidity and residual low polar local environment offered by toluene molecules in PVA layer (see Section 1.10 in ESI†).27,69,70 An erratic blinking and irreversible photobleaching is reported as a consequence of the stabilization of the CS state in the polar nano-environment.71 The excited state stabilization results in the lowering of transition energy at the single-molecule domain, leading to the opening up of new nonradiative channels implied by the energy gap law.
Antibunching experiments of TDI-TPA4 in PS and PVA matrices were carried out using an 800 nm short-pass filter. A doublet peak was observed at zero delay point, which is characteristic of the afterglow (fluorescence from one detector initiating the other) from the silicon detector (Fig. S28†).72 Since there is a considerable contribution from afterglow above 700 nm, reliable antibunching data were not acquired. However, the contribution from the afterglow photons in the FIT measurements is buried in the background signal (Fig. S28†). The single-molecule investigation of the present work exclusively contains spatially separated single molecules that undergo blinking or single-step photobleaching. (Representative FITs; Fig. S29 and S30†).
Cyclic voltammetry and Rehm–Weller analysis
Having established the existence of bright and dark states of TDI-TPA4 in nonpolar and polar matrices, respectively at the single-molecule domain, we evaluated the thermodynamic feasibility of electron transfer at the ensemble level using cyclic voltammetry (CV) and differential pulse voltammetry (DPV). CV and DPV measurements were performed against Ag/Ag+ in dry dichloromethane (DCM) under a nitrogen atmosphere to examine the redox properties of TDI-TPA4 using ferrocene/ferrocenium (Fc/Fc+) couple as a reference (Fig. S31†). Two distinct reversible reduction waves (E1(red) = −1.01 V and E2(red) = −1.14 V) and three reversible oxidation waves (E1(ox) = 0.44 V, E2(ox) = 0.59 V and E3(ox) = 0.82 V) of TDI-TPA4 confirms profound electronic communication between the TDI core and TPA moiety. Rehm–Weller formalism provides the feasibility associated with the electron transfer process in the donor–acceptor system in terms of Gibbs free energy changes for charge separation (ΔGCS).73 The positive ΔGCS ≈ 0.20 eV indicates the infeasibility of charge separation of TDI-TPA4 in HEX. The ΔGCS ≈ −0.21 eV in THF and ΔGCS ≈ −0.33 eV in ACN suggests the feasibility of photoinduced electron transfer in medium and high polar solvents, respectively. The electrochemical energy gap (Eg) is estimated to be 1.45 eV from experimentally observed LUMO and HOMO energy levels (Table S4†).Femtosecond transient absorption measurements
Solvent-dependent transient absorption measurements provide insights into the excited state dynamics of TDI-TPA4 in different dielectric environments. Femtosecond transient absorption spectra (fsTA) of TDI-TPA4 were recorded in HEX (ε = 1.88), THF (ε = 7.58), and ACN (ε = 37.50) having optical density 0.2–0.3 using ∼100 fs laser pulse exciting at 520 nm (Fig. 4). A Spectra-Physics Mai Tai SP mode-locked laser (800 nm, 86 MHz) was used as a seed for Spectra-Physics Spitfire Ace regenerative amplifier (1 kHz, 5.5 mJ). Using TOPAS, a portion of the amplified 800 nm output beam was converted into a 520 nm pump pulse. The magic angle polarization (∼54.7°) between pump and probe pulses was used to ensure an isotropic signal of the sample. Singular value decomposition (SVD) and global analysis of the ΔA versus time-based wavelength-based three-dimensional map of the fsTA spectra were carried out to extract the kinetic components responsible for the total spectra. The fsTA spectra of TDI-TPA4 in HEX displayed negative ground state bleach (GSB) and stimulated emission (SE) band from ∼552–850 nm accompanied by a positive excited state absorption (ESA) band ranging from ∼876–1400 nm (Fig. 4a). The one-component sequential kinetic model A → GS (GS = ground state) was employed to acquire the evolution-associated spectra (EAS) of TDI-TPA4 in HEX. The deconvolution of the fsTA spectra revealed a single principal component with a decay constant of 1.21 ns in HEX, attributed to a CT state that matches the fluorescence lifetime obtained from TCSPC measurement. The inaccessibility of the CS state from the CT state in HEX is probably due to the large energy barrier for charge separation from the CT state, which is evident from the positive ΔGCS ≈ 0.20 eV calculated from the Rehm–Weller analysis.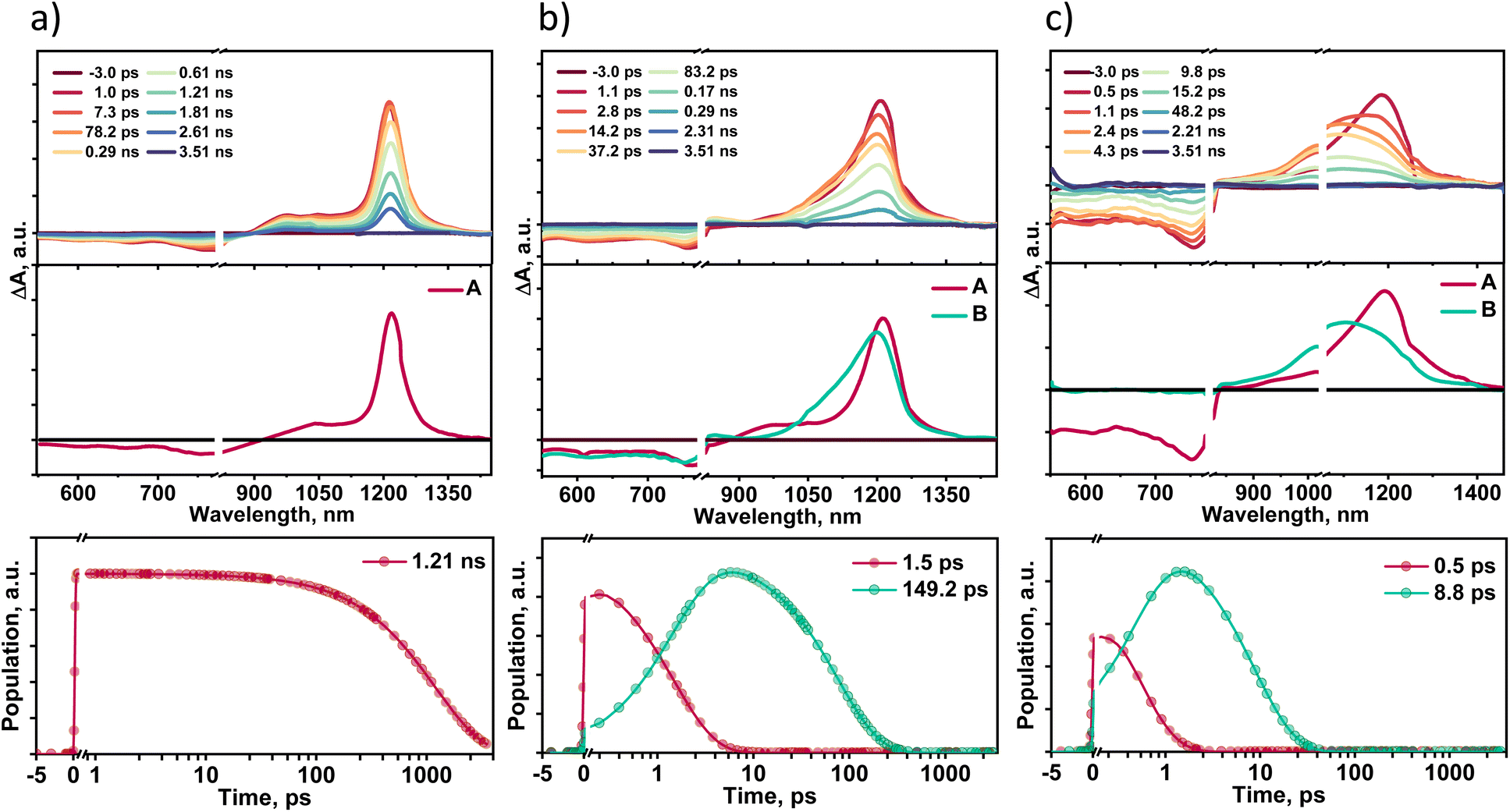 | ||
| Fig. 4 (Top row) Femtosecond transient absorption spectra of TDI-TPA4 in (a) HEX, (b) THF, and (c) ACN showing the excited state dynamics upon photoexcitation (λex = 520 nm). (Middle row) Species-associated spectra reconstructed from global analysis of TDI-TPA4 with A → GS model for HEX (left), the A → B → GS model for THF (center), and A → B → GS model for ACN (right). (Bottom row) Relative population profiles of the excited states in TDI-TPA4 in HEX, THF, and ACN. | ||
Transient absorption spectra recorded for TDI-TPA4 in a moderately polar solvent THF exhibited a negative band ∼550–827 nm corresponding to GSB and SE. The positive ESA consists of two bands: a low intense band (∼829–906 nm) and a broad intense absorption band ranging from ∼934–1396 nm (Fig. 4b). A two-component sequential kinetic model A → B → GS was employed to elucidate the excited state process in THF. Upon deconvolution, fsTA spectra result in two principal components centered at 1215 nm (A) and 1196 nm (B), which can be attributed to a CT state and a CS state, respectively. The negative ΔGCS ≈ −0.21 eV calculated from the Rehm–Weller analysis validates the feasibility of populating the CS state in THF.74 The CT state decays with a time constant of 1.5 ps, leading to the CS state, which subsequently decays to the ground state within 149.2 ps. The 3D plot of fsTA confirms the evolution of a new species in the later time delay in the ∼829–906 nm region, which resembles the spectroscopic signature of chemically oxidized absorption of TPA reported in the literature (Fig. S32 and S33†).74 The chemical oxidation experiments were carried out for the TPA in toluene and ACN using antimony(V) chloride, further confirming the characteristic absorption feature of the TPA radical cation in this wavelength regime (Fig. S34†).
The transient absorption spectra of TDI-TPA4 were recorded in ACN to probe excited state dynamics in a high dielectric environment. The negative GSB and SE featured in the ∼550–835 nm region, and the positive ESA band spanned from the ∼836–1445 nm region. The two-component sequential kinetic model A → B → GS is used to comprehend the photoinduced processes of TDI-TPA4 in ACN. The first component (A) at 1195 nm undergoes ultrafast decay to the second component (B) with a rate constant kA→B = (0.5 ps)−1. The formation of the second component at 1130 nm occurs in the later time delay, which further relaxes to the ground state with the rate constant of kB→GS = (8.8 ps)−1. The second band at 1130 nm matches with the chemically reduced anion formed from TDI-TPA4 in the ACN upon treatment with bis(cyclopentadienyl)cobalt(II) as a reducing agent (Fig. S35†). The A and B components can be attributed to CT and CS states, respectively, which are in agreement with the feasibility of charge separation ΔGCS ≈ −0.33 eV in ACN. The spectroelectrochemistry measurements further reinforced the evolution and spectral signature of TDI-TPA4 radical anion by applying electric potential. Electrochemical reduction of light-irradiated TDI-TPA4 solution in DCM exhibited an emergence of a new band at ∼983 nm upon applying the 3.5 V potential, which confirms the existence of the TDI-TPA4 radical anion (Fig. S36†). The fluorescence time constant (τ), ΔGCS, driving force for charge recombination (ΔGCR) and rate constants (k) of TDI-TPA4 in different solvents are summarized in Table 1. The remarkable increase in the nonradiative rate with the decrease in the transition energy validates the energy gap law in TDI-TPA4 at the ensemble level. The selected kinetic traces superimposed with the global analysis fitted curves at different wavelengths of fsTA in HEX, THF, and ACN are shown to display the fitting quality in Fig. S37.† To evaluate the possibility of triplet state formation upon charge recombination proposed by El-Sayed,75 nanosecond transient absorption spectroscopy (nsTA) measurements were performed in HEX, THF, and ACN by exciting the sample at 532 nm. The absence of a long-lived species with a positive absorption feature (τT > 8 ns) in the nsTA spectra further confirms the rapid charge recombination in TDI-TPA4 to a singlet state in THF and ACN (Fig. S38†).
The fsTA thin film measurements of TDI-TPA4 in PS and PVA polymer matrices were carried out to comprehend the excited state dynamics in low polar and high polar polymer environments upon photoexciting at 520 nm. The samples for thin film experiments were prepared by drop-casting the TDI-TPA4 and polymer solution on a cleaned round quartz cuvette. The thin film thickness of TDI-TPA4 in PS and PVA matrices were measured to be ∼430 ± 5 nm and 470 ± 5 nm respectively, using profilometry (KLA Tencor D600 stylus surface profiler; Fig. S39†). In the low polar PS matrix, the TDI-TPA4 exhibited negative GSB at ∼561–771 nm accompanied by ESA at ∼1046–1349 nm region (Fig. S40†). The wavelength range of the GSB and ESA of TDI-TPA4 in the PS matrix resembles the spectral signatures of TDI-TPA4 in HEX (Fig. 4a). The deconvolution of the fsTA spectra of TDI-TPA4 in PS thin film was carried out using a sequential kinetic model A → GS having a time constant of τA→GS = 0.87 ns, which is attributed to the CT state. A faster kinetic decay of TDI-TPA4 was observed in the PS thin films compared to the fsTA spectra of TDI-TPA4 in the HEX solution. The accelerated decay kinetics of TDI-TPA4 in PS compared to HEX is possibly due to the higher dielectric environment offered by PS. The fsTA spectra of TDI-TPA4 in the PVA matrix depict an ESA band centered at ∼1240 nm, which is characteristic of the ESA band observed in the polar solvents (THF and ACN). Deconvolution of the total spectra using A → B → GS sequential kinetic model resulted in two principal components with decay constants of 0.3 ps followed by 4.7 ps, which can be attributed to the CT and CS state of TDI-TPA4 in the PVA matrix, respectively (Fig. S40†). In thin film fsTA measurements, higher concentration of TDI-TPA4 (∼5 mM) reduces the interaction with residual toluene molecules per unit area. As a result, the influence of low polar environment offered by residual solvent molecules may not be detectable in the ensemble level transient absorption measurements. The thin film fsTA measurements of TDI-TPA4 in PVA matrix correspond to the excited state dynamics in the high dielectric environment, consistent with observations from high-polarity solution state fsTA measurements. This indeed affirms the capability of single-molecule techniques to probe unique events that are averaged out in ensemble level measurements.
A proposed Jablonski diagram illustrating the excited state dynamics of TDI-TPA4 in various dielectric environments is shown in Fig. 5. In the HEX or PS matrix, the deactivation from the CT state is identified as a major pathway as a result of a large energy barrier in achieving the CS state. In a moderately polar solvent THF and polar ACN or in a PVA matrix, the access to the CS state from the CT state is facilitated by the stabilization of the CT state in a higher dielectric environment.
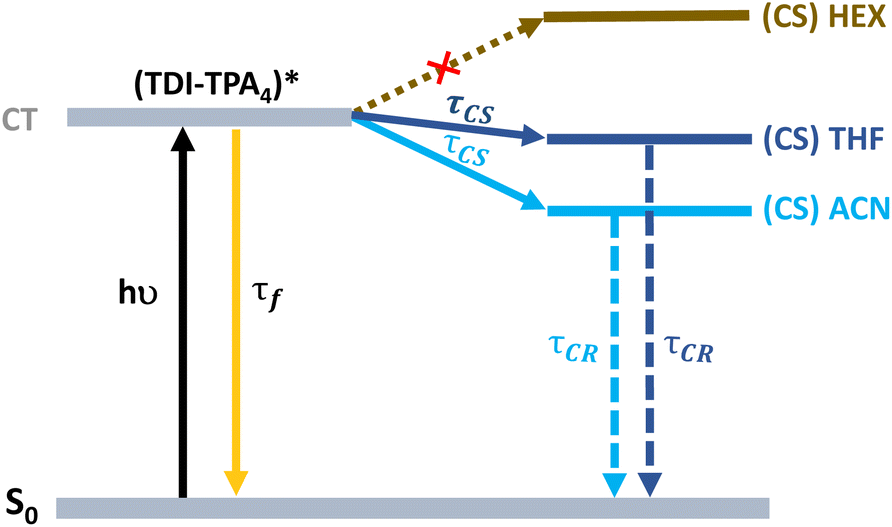 | ||
| Fig. 5 Jablonski diagram showing the excited state deactivation mechanism of TDI-TPA4 in HEX, THF, and ACN. | ||
Conclusions
In summary, a novel NIR absorbing and emitting TDI-TPA4 was explored at bulk and the single-molecule level. The solvatochromism and a significant red-shift in the emission spectrum exhibited by TDI-TPA4 with respect to TDI (ΔλFlmax = 173 nm) revealed CT character at the ensemble level and is supported by theoretical analysis. Quantum chemical calculations, including fragment-based excited state analysis of TDI-TPA4, suggested the nature of the first singlet excited state (S1) as a hybrid state of both CT and LE states and S2, S3, and S4 excited states as CT states. Single-molecule fluorescence measurements in PS and PVA unambiguously confirmed the dielectric dependence and implication of the energy gap law of the TDI-TPA4 to serve as a NIR emitting polarity probe in the nanoscale environment. In the low polar PS matrix, relatively high photostability and photon count were observed in comparison to the frequent blinking and photobleaching in the polar PVA matrix. The thermodynamic feasibility of charge separation of TDI-TPA4 in THF and ACN (ΔGCS ≈ −0.21 and ΔGCS ≈ −0.33 eV) is validated by Rehm–Weller formalism. The evolution of radical anion and cation of TDI-TPA4 in polar solvent is characterized by redox titration, spectroelectrochemistry, and femtosecond transient absorption measurements. Our findings manifest the potential of TDI-TPA4 donor–acceptor chromophore as a polarity-sensitive NIR single-photon emitter and pave the way for a wide range of futuristic applications.Data availability
All the experimental data were provided in the ESI.†Author contributions
The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. H. acknowledges the Nanomission project (DST/NM/TUE/EE01/2019) of the Department of Science and Technology (DST), Government of India, for financial support. S. S. acknowledges PMRF for the financial assistance. P. D. M. and K. V. thank IISER TVM and CSIR for financial assistance. We greatly acknowledge the support for high-performance computing time at the Padmanabha cluster, IISER Thiruvananthapuram, India. We kindly thank the CLIF facility at Kerala University for the FLS1000 spectrometer facility.Notes and references
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, The Rylene Colorant Family—Tailored Nanoemitters for Photonics Research and Applications, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed.
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson 3rd, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Exploiting chemistry and molecular systems for quantum information science, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS.
- F. Fuchs, B. Stender, M. Trupke, D. Simin, J. Pflaum, V. Dyakonov and G. V. Astakhov, Engineering near-infrared single-photon emitters with optically active spins in ultrapure silicon carbide, Nat. Commun., 2015, 6, 7578 CrossRef CAS PubMed.
- C. Toninelli, I. Gerhardt, A. S. Clark, A. Reserbat-Plantey, S. Götzinger, Z. Ristanović, M. Colautti, P. Lombardi, K. D. Major, I. Deperasińska, W. H. Pernice, F. H. L. Koppens, B. Kozankiewicz, A. Gourdon, V. Sandoghdar and M. Orrit, Single organic molecules for photonic quantum technologies, Nat. Mater., 2021, 20, 1615–1628 CrossRef CAS PubMed.
- M. B. Gaither-Ganim, S. A. Newlon, M. G. Anderson and B. Lee, Organic molecule single-photon sources, Oxford Open Mater. Sci., 2023, 3, itac017 CrossRef CAS.
- M. Musavinezhad, A. Shkarin, D. Rattenbacher, J. Renger, T. Utikal, S. Götzinger and V. Sandoghdar, Quantum Efficiency of Single Dibenzoterrylene Molecules in p-Dichlorobenzene at Cryogenic Temperatures, J. Phys. Chem. B, 2023, 127, 5353–5359 CrossRef CAS PubMed.
- C. Toninelli, K. Early, J. Bremi, A. Renn, S. Götzinger and V. Sandoghdar, Near-infrared single-photons from aligned molecules in ultrathin crystalline films at room temperature, Opt. Express, 2010, 18, 6577–6582 CrossRef CAS.
- X. Xu, Q. Yang, H. Zhao, S. Vasylevskyi, M. Bonn, X. Liu and A. Narita, Chiral Nanographene-Based Near-Infrared Fluorophore with Self-Blinking Properties, Adv. Funct. Mater., 2024, 34, 2308110 CrossRef CAS.
- L. E. McNamara, N. Liyanage, A. Peddapuram, J. S. Murphy, J. H. Delcamp and N. I. Hammer, Donor–Acceptor–Donor Thienopyrazines via Pd-Catalyzed C–H Activation as NIR Fluorescent Materials, J. Org. Chem., 2016, 81, 32–42 CrossRef CAS PubMed.
- R. Englman and J. Jortner, The energy gap law for radiationless transitions in large molecules, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, Application of the energy gap law to nonradiative, excited-state decay, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- S. J. Jang, A simple generalization of the energy gap law for nonradiative processes, J. Chem. Phys., 2021, 155, 164106 CrossRef CAS.
- R. Englman and J. Jortner, The energy gap law for non-radiative decay in large molecules, J. Lumin., 1970, 1–2, 134–142 CrossRef.
- H. Imahori, Y. Kobori and H. Kaji, Manipulation of Charge-Transfer States by Molecular Design: Perspective from “Dynamic Exciton”, Acc. Mater. Res., 2021, 2, 501–514 CrossRef CAS.
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Highly Efficient Near-Infrared Delayed Fluorescence Organic Light Emitting Diodes Using a Phenanthrene-Based Charge-Transfer Compound, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Near-Infrared-II Molecular Dyes for Cancer Imaging and Surgery, Adv. Mater., 2019, 31, 1900321 CrossRef.
- T. Shimada, T. Kaneko, Y. Notsuka, J. Kim, S. Mori, S. Shimizu, J. Kim, K. Kamada, D. Kim, Y. Yamaoka, H. Furuta and M. Ishida, Molecular Design for Stable Near-Infrared-II Two-Photon Excitation-Induced Photoacoustic Contrast Agents Based on Donor-Substituted BODIPYs, ACS Appl. Opt. Mater., 2024, 2, 211–219 CrossRef CAS.
- J. Xue, J. Xu, J. Ren, Q. Liang, Q. Ou, R. Wang, Z. Shuai and J. Qiao, Intermolecular charge-transfer aggregates enable high-efficiency near-infrared emissions by nonadiabatic coupling suppression, Sci. China: Chem., 2021, 64, 1786–1795 CrossRef CAS.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Influence of energy gap between charge-transfer and locally excited states on organic long persistence luminescence, Nat. Commun., 2020, 11, 191 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Multi-Resonance Deep-Red Emitters with Shallow Potential-Energy Surfaces to Surpass Energy-Gap Law**, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 CrossRef CAS PubMed.
- M.-P. Zhuo, X.-D. Wang and L.-S. Liao, Recent Progress of Novel Organic Near-Infrared-Emitting Materials, Small Sci., 2022, 2, 2200029 CrossRef CAS.
- Z. Lei and F. Zhang, Molecular Engineering of NIR-II Fluorophores for Improved Biomedical Detection, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef CAS PubMed.
- Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Hybridization and de-hybridization between the locally-excited (LE) state and the charge-transfer (CT) state: a combined experimental and theoretical study, Phys. Chem. Chem. Phys., 2016, 18, 24176–24184 RSC.
- A. Cravcenco, Y. Yu, F. Edhborg, J. F. Goebel, Z. Takacs, Y. Yang, B. Albinsson and K. Börjesson, Exciton Delocalization Counteracts the Energy Gap: A New Pathway toward NIR-Emissive Dyes, J. Am. Chem. Soc., 2021, 143, 19232–19239 CrossRef CAS PubMed.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Overcoming the energy gap law in near-infrared OLEDs by exciton–vibration decoupling, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- C. Erker and T. Basché, The Energy Gap Law at Work: Emission Yield and Rate Fluctuations of Single NIR Emitters, J. Am. Chem. Soc., 2022, 144, 14053–14056 CrossRef CAS PubMed.
- Y. Hou, A. M. Bardo, C. Martinez and D. A. Higgins, Characterization of Molecular Scale Environments in Polymer Films by Single Molecule Spectroscopy, J. Phys. Chem. B, 2000, 104, 212–219 CrossRef CAS.
- R. A. Marcus, Theory of charge-transfer spectra in frozen media, J. Phys. Chem., 1990, 94, 4963–4966 CrossRef CAS.
- R. A. Marcus, Chemical and electrochemical electron-transfer theory, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- L. Yang, M. Zhao, W. Chen, J. Zhu, W. Xu, Q. Li, K. Pu and Q. Miao, A Highly Bright Near-Infrared Afterglow Luminophore for Activatable Ultrasensitive In Vivo Imaging, Angew. Chem., Int. Ed., 2024, 63, e202313117 CrossRef CAS PubMed.
- J. Zhu, W. Chen, L. Yang, Y. Zhang, B. Cheng, W. Gu, Q. Li and Q. Miao, A Self-Sustaining Near-Infrared Afterglow Chemiluminophore for High-Contrast Activatable Imaging, Angew. Chem., Int. Ed., 2024, 63, e202318545 CrossRef CAS.
- Z.-H. Wu, X. Zhu, Q. Yang, Y. Zagranyarski, K. Mishra, H. Strickfaden, R. P. Wong, T. Basché, K. Koynov, M. Bonn, C. Li, X. Liu and K. Müllen, Near-Infrared Perylenecarboximide Fluorophores for Live-Cell Super-Resolution Imaging, J. Am. Chem. Soc., 2024, 146, 7135–7139 CrossRef CAS.
- X. Qin, X. Yang, L. Du and M. Li, Polarity-based fluorescence probes: properties and applications, Med. Chem., 2021, 12, 1826–1838 CAS.
- M. Xu, S. Liu, L. Zhang, W. Xie and X. Li, Near-infrared organic light-emitting materials, devices and applications, Mater. Chem. Front., 2023, 7, 4744–4767 RSC.
- Y.-J. Gong, X.-B. Zhang, G.-J. Mao, L. Su, H.-M. Meng, W. Tan, S. Feng and G. Zhang, A unique approach toward near-infrared fluorescent probes for bioimaging with remarkably enhanced contrast, Chem. Sci., 2016, 7, 2275–2285 RSC.
- F. Zhang and B. Z. Tang, Near-infrared luminescent probes for bioimaging and biosensing, Chem. Sci., 2021, 12, 3377–3378 RSC.
- R. Zhang, Y. Xu, Y. Zhang, H. S. Kim, A. Sharma, J. Gao, G. Yang, J. S. Kim and Y. Sun, Rational design of a multifunctional molecular dye for dual-modal NIR-II/photoacoustic imaging and photothermal therapy, Chem. Sci., 2019, 10, 8348–8353 RSC.
- X. Wang, J. Sun, W. Zhang, X. Ma, J. Lv and B. Tang, A near-infrared ratiometric fluorescent probe for rapid and highly sensitive imaging of endogenous hydrogen sulfide in living cells, Chem. Sci., 2013, 4, 2551–2556 RSC.
- M. Haase, C. G. Hübner, F. Nolde, K. Müllen and T. Basché, Photoblinking and photobleaching of rylene diimide dyes, Phys. Chem. Chem. Phys., 2011, 13, 1776–1785 RSC.
- C. Jung, B. K. Müller, D. C. Lamb, F. Nolde, K. Müllen and C. Bräuchle, A New Photostable Terrylene Diimide Dye for Applications in Single Molecule Studies and Membrane Labeling, J. Am. Chem. Soc., 2006, 128, 5283–5291 CrossRef CAS.
- H. Piwoński, A. Sokołowski and J. Waluk, In Search for the Best Environment for Single Molecule Studies: Photostability of Single Terrylenediimide Molecules in Various Polymer Matrices, J. Phys. Chem. Lett., 2015, 6, 2477–2482 CrossRef.
- K. Kennes, Y. Baeten, T. Vosch, W. Sempels, S. Yordanov, S. Stappert, L. Chen, K. Müllen, J. Hofkens, M. Van der Auweraer and E. Fron, Photophysical Investigation of Cyano-Substituted Terrylenediimide Derivatives, J. Phys. Chem. B, 2014, 118, 14662–14674 CrossRef CAS.
- S. Mais, J. Tittel, T. Basché, C. Bräuchle, W. Göhde, H. Fuchs, G. Müller and K. Müllen, Terrylenediimide: A Novel Fluorophore for Single-Molecule Spectroscopy and Microscopy from 1.4 K to Room Temperature, J. Phys. Chem. A, 1997, 101, 8435–8440 CrossRef CAS.
- P. Kukura, M. Celebrano, A. Renn and V. Sandoghdar, Single-Molecule Sensitivity in Optical Absorption at Room Temperature, J. Phys. Chem. Lett., 2010, 1, 3323–3327 CrossRef CAS.
- A. R. Mallia, P. S. Salini and M. Hariharan, Nonparallel Stacks of Donor and Acceptor Chromophores Evade Geminate Charge Recombination, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS.
- A. M. Philip, A. R. Mallia and M. Hariharan, Prolonged Charge Separated States in Twisted Stacks of All-Carbon Donor and Acceptor Chromophores, J. Phys. Chem. Lett., 2016, 7, 4751–4756 CrossRef CAS PubMed.
- A. R. Mallia, A. M. Philip, V. Bhat and M. Hariharan, Persistent Charge-Separated States in Self-Assembled Twisted Nonsymmetric Donor–Acceptor Triads, J. Phys. Chem. C, 2017, 121, 4765–4777 CrossRef CAS.
- P. D. Maret, D. Sasikumar, E. Sebastian and M. Hariharan, Symmetry-Breaking Charge Separation in a Chiral Bis(perylenediimide) Probed at Ensemble and Single-Molecule Levels, J. Phys. Chem. Lett., 2023, 14, 8667–8675 CrossRef CAS PubMed.
- A. Mazumder, K. Vinod, P. D. Maret, P. P. Das and M. Hariharan, Symmetry-Breaking Charge Separation Mediated Triplet Population in a Perylenediimide Trimer at the Single-Molecule Level, J. Phys. Chem. Lett., 2024, 15, 5896–5904 CrossRef CAS.
- F. Nolde, J. Qu, C. Kohl, N. G. Pschirer, E. Reuther and K. Müllen, Synthesis and Modification of Terrylenediimides as High-Performance Fluorescent Dyes, Chem.–Eur. J., 2005, 11, 3959–3967 CrossRef CAS PubMed.
- N. Liang, K. Sun, J. Feng, Y. Chen, D. Meng, W. Jiang, Y. Li, J. Hou and Z. Wang, Near-infrared electron acceptors based on terrylene diimides for organic solar cells, J. Mater. Chem. A, 2018, 6, 18808–18812 RSC.
- H. Wei, N. Jiang, N. Zhao, Y. Zhang and B. Gao, New Soluble Near-Infrared Dyes Based on Terrylene Diimides with Four Aromatic Heterocycles, Chin. J. Chem., 2014, 32, 356–360 CrossRef CAS.
- N.-H. Xie, C. Li, J.-X. Liu, W.-L. Gong, B. Z. Tang, G. Li and M.-Q. Zhu, The synthesis and aggregation-induced near-infrared emission of terrylenediimide–tetraphenylethene dyads, Chem. Commun., 2016, 52, 5808–5811 RSC.
- K. Singh Mehra, S. Jha, S. Bhandary, D. Mandal, R. Mishra and J. Sankar, Bridging the Bays, Both Ways: A Janus Butterfly-Shaped Intense NIR-Emitting Terrylene Diimide, Angew. Chem., Int. Ed., 2022, 61, e202205600 CrossRef CAS PubMed.
- J. Rühe, M. Rajeevan, K. Shoyama, R. S. Swathi and F. Würthner, A Terrylene Bisimide based Universal Host for Aromatic Guests to Derive Contact Surface-Dependent Dispersion Energies, Angew. Chem., Int. Ed., 2024, 63, e202318451 CrossRef PubMed.
- S. Jha, K. S. Mehra, M. Dey, S. S, D. Ghosh, P. K. Mondal, M. Polentarutti and J. Sankar, A nine-ring fused terrylene diimide exhibits switching between red TADF and near-IR room temperature phosphorescence, Chem. Sci., 2024, 15, 8974–8981 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Berlin, 3rd edn, 2006 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Plasser and H. Lischka, Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS.
- F. Plasser, TheoDORE: a toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS.
- A. Forde, V. M. Freixas, S. Fernandez-Alberti, A. J. Neukirch and S. Tretiak, Charge-Transfer Luminescence in a Molecular Donor–Acceptor Complex: Computational Insights, J. Phys. Chem. Lett., 2022, 13, 8755–8760 CrossRef CAS.
- S. Doose, H. Neuweiler and M. Sauer, Fluorescence Quenching by Photoinduced Electron Transfer: A Reporter for Conformational Dynamics of Macromolecules, ChemPhysChem, 2009, 10, 1389–1398 CrossRef CAS PubMed.
- T. D. Bell, A. Stefan, S. Masuo, T. Vosch, M. Lor, M. Cotlet, J. Hofkens, S. Bernhardt, K. Müllen, M. van der Auweraer, J. W. Verhoeven and F. C. De Schryver, Electron transfer at the single-molecule level in a triphenylamine-perylene imide molecule, ChemPhysChem, 2005, 6, 942–948 CrossRef CAS.
- R. Gronheid, A. Stefan, M. Cotlet, J. Hofkens, J. Qu, K. Müllen, M. Van der Auweraer, J. W. Verhoeven and F. C. De Schryver, Reversible Intramolecular Electron Transfer at the Single-Molecule Level, Angew. Chem., Int. Ed., 2003, 42, 4209–4214 CrossRef CAS.
- T. Tachikawa, S.-C. Cui, M. Fujitsuka and T. Majima, Interfacial electron transfer dynamics in dye-modified graphene oxide nanosheets studied by single-molecule fluorescence spectroscopy, Phys. Chem. Chem. Phys., 2012, 14, 4244–4249 RSC.
- E. Fron, R. Pilot, G. Schweitzer, J. Qu, A. Herrmann, K. Müllen, J. Hofkens, M. Van der Auweraer and F. C. De Schryver, Photoinduced electron-transfer in perylenediimide triphenylamine-based dendrimers: single photon timing and femtosecond transient absorption spectroscopy, Photochem. Photobiol. Sci., 2008, 7, 597–604 CrossRef CAS.
- M. Angeles Izquierdo, T. D. M. Bell, S. Habuchi, E. Fron, R. Pilot, T. Vosch, S. De Feyter, J. Verhoeven, J. Jacob, K. Müllen, J. Hofkens and F. C. De Schryver, Switching of the fluorescence emission of single molecules between the locally excited and charge transfer states, Chem. Phys. Lett., 2005, 401, 503–508 CrossRef CAS.
- G. S. Schlau-Cohen, S. Bockenhauer, Q. Wang and W. E. Moerner, Single-molecule spectroscopy of photosynthetic proteins in solution: exploration of structure–function relationships, Chem. Sci., 2014, 5, 2933–2939 RSC.
- M. Cotlet, S. Masuo, G. Luo, J. Hofkens, M. Van der Auweraer, J. Verhoeven, K. Müllen, X. S. Xie and F. De Schryver, Probing conformational dynamics in single donor-acceptor synthetic molecules by means of photoinduced reversible electron transfer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14343–14348 CrossRef CAS PubMed.
- D. Sluss, C. Bingham, M. Burr, E. D. Bott, E. A. Riley and P. J. Reid, Temperature-dependent fluorescence intermittency for single molecules of violamine R in poly(vinyl alcohol), J. Mater. Chem. B, 2009, 19, 7561–7566 RSC.
- M. W. Holman, R. Liu and D. M. Adams, Single-Molecule Spectroscopy of Interfacial Electron Transfer, J. Am. Chem. Soc., 2003, 125, 12649–12654 CrossRef CAS.
- K. J. Whitcomb, J. Q. Geisenhoff, D. P. Ryan, M. P. Gelfand and A. Van Orden, Photon Antibunching in Small Clusters of CdSe/ZnS Core/Shell Quantum Dots, J. Phys. Chem. B, 2015, 119, 9020–9028 CrossRef CAS PubMed.
- A. Osuka, S. Nakajma, K. Maruyama, N. Mataga and T. Asahi, Intramolecular Photoinduced Electron Transfer in Pyromellitimide-linked Porphyrins, Chem. Lett., 1991, 20, 1003–1006 CrossRef.
- J. Kong, W. Zhang, J.-Y. Shao, D. Huo, X. Niu, Y. Wan, D. Song, Y.-W. Zhong and A. Xia, Bridge-Length- and Solvent-Dependent Charge Separation and Recombination Processes in Donor-Bridge-Acceptor Molecules, J. Phys. Chem. B, 2021, 125, 13279–13290 CrossRef CAS PubMed.
- M. A. El-Sayed, Effect of spin orbit interactions on the dipolar nature of the radiative microwave zero-field transitions in aromatic molecules, J. Chem. Phys., 1974, 60, 4502–4507 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03861d |
| This journal is © The Royal Society of Chemistry 2024 |