
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical diversity of reagents that modify RNA 2′-OH in water: a review
Ryuta
Shioi
b and
Eric T.
Kool
 *a
*a
aDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: kool@stanford.edu
bDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA
First published on 12th September 2024
Abstract
Electrophilic water-soluble compounds have proven versatile in reacting selectively with 2′-OH groups in RNA, enabling structure mapping, probing, caging, labeling, crosslinking, and conjugation of RNAs in vitro and in living cells. While early work focused on one or two types of reagents with limited properties, recent studies have greatly diversified the structure, properties, and applications of these reagents. Here we review the scope of documented RNA hydroxyl-reactive species reported to date, with an eye to the effects of chemical structure on reactivity with RNA and other useful properties. Multiple forms of carbonyl electrophiles are now known to react at the 2′-OH, and recently, sulfonyl and aryl electrophiles have also been documented to form bonds there in high yields as well. In addition to electrophilicity, data also point to significant effects of reagent stability, steric bulk, and chirality on reaction yields and selectivity. Finally, we outline reagent properties and principles that define utility in applications with RNA, with an eye to the design of future reagents.
Introduction
The covalent reaction of small molecules with biomacromolecules is critically important both in basic and applied science. In the protein and proteomics fields, the development of reactions of small activated species with protein sidechains has revolutionized biology and biomedicine. It has made possible the fluorescence labelling of antibodies for biomedical imaging and quantitation,1,2 and has enabled the construction of medically important antibody-drug conjugates.3 Moreover, the use of simple chemical species reacting with protein sidechains throughout the cell has enabled the characterisation of interactions within the entire proteome.4,5The cellular RNA population, like the protein population, is complex and dynamic, and undergoes a multitude of biologically important interactions in the cell.5 We argue that RNA science and applications will likely benefit at least as much as protein studies do from similar bond-forming reactions with small molecules. Trace reactions that are structure-sensitive can be used to profile RNA folded structures and complexes in the cell.6–8 Higher-yielding reactions can be used to construct conjugates for labelling, separation, and enrichment applications.9–16 They can also be used to control folding and biological activity,13 and to stabilise the polymer against degradation.17 Finally, covalent reactions with RNA can be employed to profile the interactions of small molecules (such as drugs and other small ligands) with individual RNAs or throughout the whole transcriptome, by providing a covalent trap of local binding interactions.18–20
It is becoming increasingly clear that the 2′-OH group offers an extraordinarily versatile chemical handle on RNA (Fig. 1). To be sure, other reaction sites exist on this biopolymer; for example, researchers frequently perform aqueous covalent reactions with nucleobases in RNA. Aldehydes such as glyoxal react with exocyclic amines of some of the bases (particularly guanine),10,23 and alkylating species (such as dimethylsulfate or nitrogen mustard) react with nucleophilic nitrogens on adenine and cytosine.24,25 In addition, diazirines can be used as carbene precursors, leading to reactions with RNA bases (primarily guanine) under UV irradiation.26,27 While useful, those reactions are limited to a subpopulation of nucleobases in RNA, and are nonselective between DNA and RNA. In contrast, the 2′-OH group, absent from DNA, occurs at nearly every position of all RNAs in the cell. This provides an immense opportunity to interrogate and modify RNA at every position along the biopolymer. In addition, reagents targeted to the 2′-OH group have been shown to be highly tuneable, and the resulting adducts can be designed to be reversible,13,28 leaving no scar behind afterward.
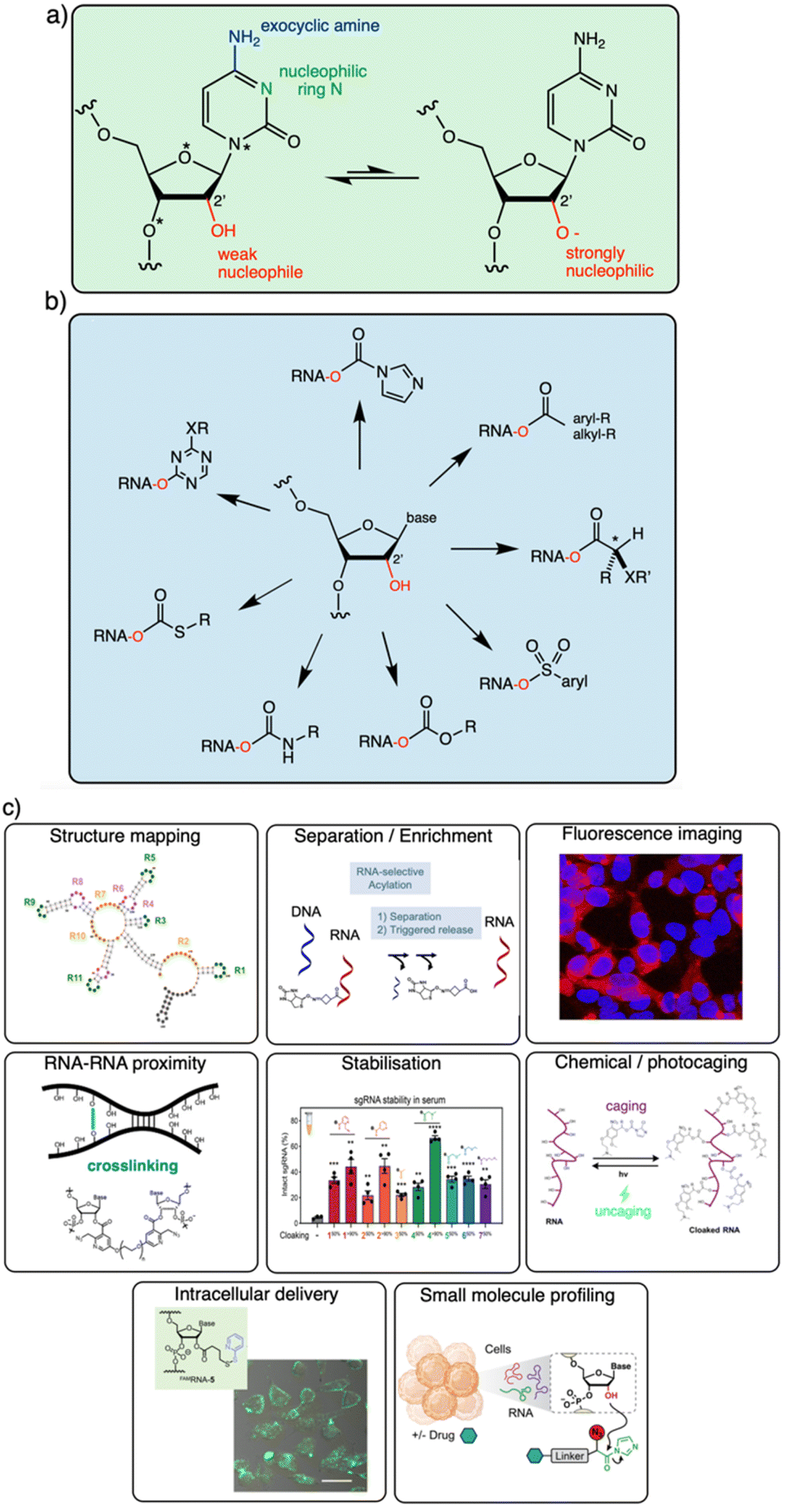 | ||
| Fig. 1 Diverse reactivity of the 2′-OH group in RNA. (a) Structure of cytidine nucleotide in RNA, showing commonly nucleophilic atoms, including N-3 ring nitrogen, exocyclic 4-amine group on the base, and 2′-OH group on the ribose sugar. This review focuses on reagents that are selective for the latter nucleophile on all four nucleotides. Adenosine, guanosine and uridine nucleotides also have similar (although not identical) 2′-OH reactivity. Proximal electronegative atoms (*) in the sugar and base inductively increase the acidity of the 2′-OH, which likely reacts with electrophiles in the rare but rapidly equilibrating anionic form (right).21,22 (b) Diversity of established 2′-OH group adduct structures achieved to date, ranging from carbonyl esters (both alkyl and aryl), to sulfonyl esters, carbonates, thiocarbonates, carbamates, and aryl ethers. Adducts can be flat or bulky, achiral or chiral, and can have alkyl or aryl substitution along with a variety of functional groups (X, R). (c) Overview of several biological applications of 2′-OH modification of RNA. | ||
The goal of this brief overview is to summarise the range of organic reagents that have been documented to react selectively with RNA at 2′-OH groups, with a view to structural variations, reactivity, and other relevant properties (Fig. 1). This is meant to outline for the reader the current state of the art in chemistry for RNA modification, and therefore focuses chiefly on high-yield preparatively useful reactions. Biological applications of 2′-OH reactions are rapidly expanding (see Fig. 1c for several) and have been reviewed recently.29,30 In addition, because excellent reviews already exist on the use of isatoic anhydride and acylimidazole reagents in trace-yield reactions for RNA structure mapping,29–31 we largely bypass that topic here with only brief mentions, and refer the reader to those sources.32–34
Mechanism of reaction
Successful reagent designs for bond-forming reactions often benefit from mechanistic knowledge of the reaction under study. For esterification of sugars, activated acyl species (e.g. acyl chlorides and anhydrides) have long been preparatively useful reagents. Such reactions are typically carried out in the presence of weak bases in organic solvents, excluding water that would compete for the reagent, and the reactions are usually selective for less sterically encumbered primary hydroxyls. In contrast, for RNA in water, it is observed that certain aqueous-soluble acyl species can react in high yields at the 2′-OH groups despite the competition with water itself (see below).7 It is notable that the reaction can be selective for this secondary hydroxyl over even the primary 5′-OH group of RNA strands, which is the reverse of the selectivity that is usually observed during esterification of typical alcohols.This begs the question of what makes the 2′-OH group in RNA specially reactive. McGinnis et al. investigated variable furanose conformations and possible general base catalysis of acylation by nearby basic groups on nucleobases to explain “hyper-reactive” hydroxyls.35 A recent study concluded that inductive effects of the heteroatoms near the 2′-OH group render it much more reactive, likely due to the increased concentration of the anionic form at neutral pH.21 The early data support the notion that acylation occurs by trapping the rare but rapidly equilibrating transient anionic form of the 2′-OH group. We suggest that in some respects, RNA 2′-OH is analogous to the hydroxyl of trichloroethanol, as both have three highly electronegative atoms at similar distances from the reacting alcohol. The pKa of RNA 2′-OH has been measured to be ∼12.5,36 very nearly the same as that of trichloroethanol.22 To be sure, the unusual 2′-OH reactivity is more complex than inductive effects alone, however, and local folding of RNA, sugar and chain conformation, electrostatic and steric effects, and solvation all likely play important roles. More work is needed to better understand these effects.
Isatoic anhydrides
The isatoic anhydrides as a class of ribose-reactive reagents (Fig. 2) were studied early on by Hiratsuka, who documented reactions of mononucleotides with the simple isatoic anhydrides NAIM (1) and 1M7 (2).37,38 Weeks made the important observation that reactions of these reagents also could be performed with RNAs in trace yields selectively at single-stranded regions of the polymer over duplex regions, and subsequently developed these reagents into powerful tools for analysis of folded structure, probing local sites of reaction by primer extension with a reverse transcriptase enzyme, which tends to stop at sites of acyl adducts.39 The general methodology (selective hydroxyl acylation analysed by primer extension, SHAPE) is widely used for RNA structure mapping, and has been reviewed elsewhere.34,40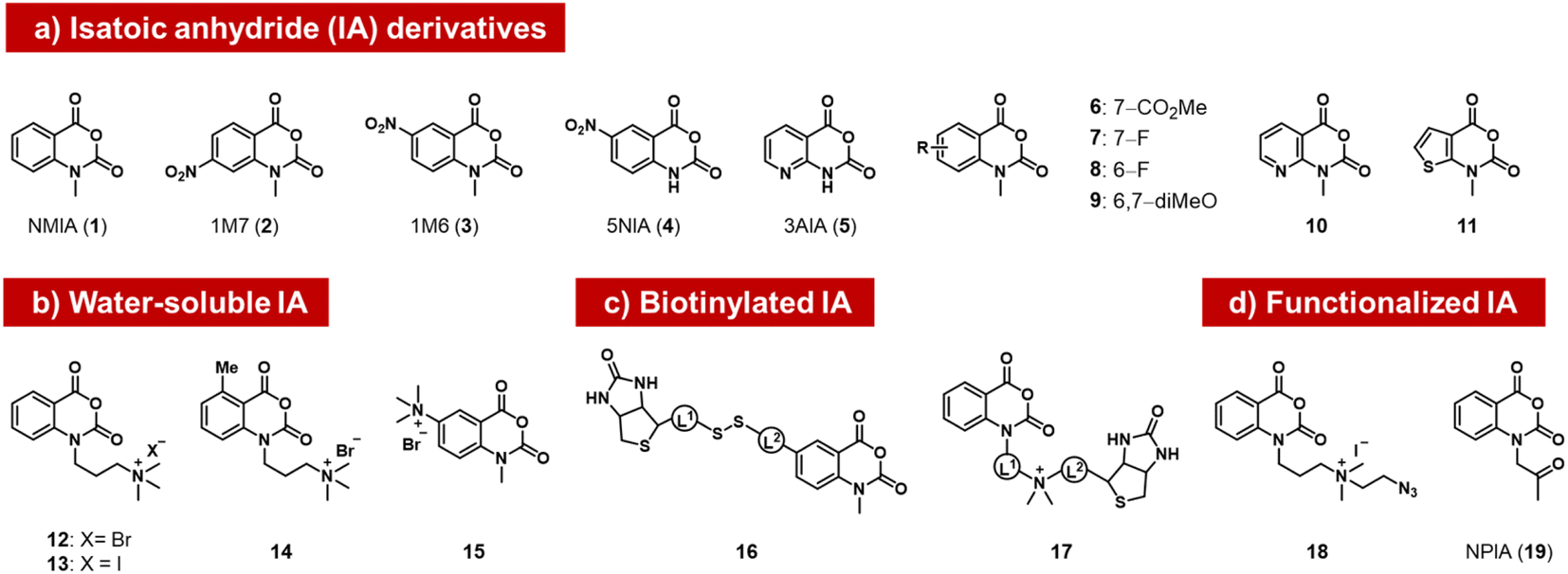 | ||
| Fig. 2 Isatoic anhydrides (IAs) documented as acylating species for RNA, designed and employed for mapping of RNA structure, as well as for labeling RNA with biotin, azide, or ketone groups. | ||
Subsequently, researchers tested structural variants of the isatoic anhydrides with an aim to expanding their properties. Isatoic anhydrides in these studies are typically prepared in two steps from 2-aminobenzoic acids, using triphosgene to form the anhydride, followed by an N-alkylation step to add substituents.41 Early reagents 1–5 can be used to generate trace-level reactions for RNA mapping, but were not documented to react in high yields.42–44 Possible limiting factors are low aqueous solubility and short half-life in water, which rapidly diminishes concentrations. Half-lives of isatoic anhydrides typically range from a few seconds to 14 minutes depending on functional groups and substitution patterns (6–11).45
Modified derivatives, such as biotinylated isatoic anhydrides (e.g.16) have also been reported and were employed to separate RNA from DNA in mixtures of the two, demonstrating higher yields of at least one adduct per strand.14,45 More recent studies by Fessler and coworkers have reported isatoic anhydrides with appended solubilising groups (e.g. 12–15),46,47 including azide-functionalised reagent 18, and this has resulted in reactions with favourable preparative-level reactions with RNA (although numeric yields were not reported). Analogous isatoic anhydride reagents with a cationic water-soluble linker and biotin were developed for quantification of protein and modification of RNA (16, 17).48 Finally, a ketone-linked isatoic anhydride reagent (19; NPIA) and its further conjugation with a hydrazide have been employed in a novel mapping method for RNA structure.49
Acylimidazoles
Acylimidazoles were among the earliest classes of reagents tested for reaction with RNA (20–84; Fig. 3). Acetylimidazole (20) and benzoylimidazole (34) were described several decades ago as acylating agents for RNA;50 researchers used radiolabeled reagents to quantify yields of 20–26% per tRNA strand. Acylimidazoles are conveniently prepared in one step from carboxylic acids via reaction with carbonyldiimidazole (CDI). Early studies of amino acids converted to acylimidazoles were carried out to test prebiotic chemistry hypotheses of the origins of amino acid-charged tRNAs.51,52 Reaction yields of up to ∼10% for alanine reagent (49) and other amino acid species (e.g.50–53) were documented with dinucleotides, and a few reactions were performed with homopolymer RNAs. It was later noted that having a free primary amine group on amino acids led to decomposition, likely due to polymerisation.53 In related work aimed at elucidating the prebiotic origins of tRNA aminoacylation, adenosine-5′-phosphorylimidazolide (54) was utilized as an aminoacyl acceptor, enabling the transfer of DL-serine to adenosine-5′-(O-methyl phosphate) with a preferential transfer of the D-isomer54 (Fig. 3c). | ||
| Fig. 3 Acylimidazoles reported to react at 2′-OH groups in RNA. (a) Acylimidazole derivatives of alkanoic acids; (b) acylimidazoles derived from aromatic carboxylic acids; (c) chiral acylimidazole species that have the potential to react stereoselectively with RNA (stereocenters not shown explicitly; each species (except 54) exists as a pair of enantiomers, which were tested separately). (d) Acylimidazole reagents with reductively-cleavable disulfide groups. | ||
Moving beyond prebiotic chemistry, Spitale et al. surveyed potential acyl reactants for RNA, and benzoylimidazole 35 gave promising levels of reaction.7 Reaction with CDI in anhydrous DMSO created a 1–2 M stock solution of the acylimidazole (along with an equivalent of the byproduct imidazole), and the stock reagent was added to the aqueous RNA solution to yield final concentrations in the 100–400 mM range. To increase solubility, the phenyl group of 35 was replaced with heterocycles pyridine (36; NAI) or furan (37; FAI). The electron-poor pyridine renders NAI more reactive with RNA but gives it a shorter lifespan toward hydrolysis in water (t1/2 = 33 min). FAI, with its electron-rich pi system, survives longer in water (73 min t1/2) but gives lower yields in preparative reactions with RNA.55 Both reagents are cell permeable and were subsequently used in mapping RNA structures in intact cells.7,55,56
An azide-modified form of NAI (38; NAI-N3) has shown utility both for in-cell mapping of the transcriptome12 and for high-yield modification of RNAs.13 Post-reaction, the azide group can be appended with biotin (via strained alkyne cycloaddition) to enrich reacted RNAs from unreacted ones, conferring improved signal over background.12 Similar azide-enabled modification for structure mapping was demonstrated with the furoyl species 39 as well.57 In tests of the possibility of superstoichiometric reactions, it was subsequently shown that 38 can modify over 80% of 2′-hydroxyls of an RNA strand, documented by MALDI-TOF mass spectrometry.13 The high-yield reaction was carried out in pH 7 buffer with 100 mM reagent (2–4 h). Little or no reaction with DNA was observed, demonstrating selectivity for 2′-OH on RNA. Polyacylation of ribozymes and fluorescent RNAs with 38 (also referred to as “cloaking”) was used to temporarily block folding and interactions, and the azide group could serve as a trigger for self-aminolysis of the ester after azide reduction by a reducing phosphine (see below). This enabled its use in chemical caging of RNAs.13 The same reagent was subsequently employed to modify CRISPR single guide RNA for control of CRISPR-Cas9 gene editing in two independent laboratories.58,59 The compound NAI-N3 (38) was also applied more recently in strategies developed for local preparative installation at sequence-programmed sites in RNAs.60,61
The concept of chemical caging of RNA via polyacylation of 2′-OH has subsequently received a good deal of attention, frequently using acylimidazole species. Examples (in addition to 38) include a trans-cyclooctene reagent (32)44 that is reversibly triggered by an amino-tetrazine, and a cyclobutanone adduct (167; see Fig. 6) that is reversed by histamine,62 and a biotin-substituted acylimidazole (33) that associates with streptavidin to block RNA activity.63
A survey of acylimidazoles reacting with RNA (see 21, 22) showed that multiple such species can react at superstoichiometric levels with RNA in water.63 In addition to the original aryl compounds, acylimidazole derivatives of alkyl carboxylates in general have also been found to react well. Park et al. reported the use of linear-chain species based on alkanoic acids or similar compounds with ethylene glycol linkages. These were substituted at the terminal position with azide groups for further reaction and conjugation.44
Marinus et al. reported tests of several aryl acylimidazole species (40–46) and investigated their capability as structure mapping reagents at trace yields.43 No data were reported regarding their ability to react at stoichiometric levels, but compound 45 (2AI) was reported as a promising mapping reagent for RNAs in intact cells, with especially good signals in bacteria.43 Chen et al. recently reported a structurally related fluorine-substituted pyridyl reagent (48) as an NMR reporter for RNA structure.64
Xiao et al. recently reported a survey of alkyl and aryl acylimidazoles with varying substitution and steric size, investigating their yields and selectivity for reaction among widely varied RNA structures.65 Compounds 25–31 and 47 were studied, and a wide range of reactivities was observed. Because both RNA structure and reagent structure affect local reactivity, a sequencing-based study of their reactivity with a library of 43 RNAs was performed. As was seen for previous compounds used in structure mapping, the new compounds showed a preference for loop nucleotides over those in duplex (stem) structure. More sterically bulky compounds (e.g., comparing 36, 46, 47) were shown to yield more localized signals in small loops. Following up on this for practical application, an azide-substituted compound (25) was prepared in one step from commercial precursor, and applied for local reaction at a single nucleotide in an RNA target, achieving preparative-scale fluorescent labeling.65
Since RNA is chiral, this offers the possibility of asymmetry in reactions with chiral electrophiles. In this vein, Shioi et al. investigated the effects of chirality on high-yield acylation of RNAs by simple acylimidazoles derived from amino acids and alkoxy acids (55–65).66 Early surveys showed that enantiomers of acylimidazole derivatives of methoxypropionic acid (58) and N,N-dimethylalanine (55) reacted with significant (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–4:1) diastereoselectivity. Following up on this, the authors also prepared an azide-substituted variant of the species ((R)-56) and employed it in site-localized high-yield labelling of RNA. One surprising observation from the study was the unusually high reactivity of (R)-N,N-dimethylaminoalanine acylimidazole (55), which reacted with RNA to give superstoichiometric yields at unusually low concentrations of 100 μM, 100 to 1000 times lower than standard concentrations of most other reagents, while simply omitting the α-methyl group (compound 49) lowers acylation efficiency by over 10-fold. A more recent study of chiral acylimidazoles with greater steric bulk (66–76), documented up to >99:1 diastereoselectivity for compound 76, the most highly stereoselective RNA acylating agent to date.53
:1–4:1) diastereoselectivity. Following up on this, the authors also prepared an azide-substituted variant of the species ((R)-56) and employed it in site-localized high-yield labelling of RNA. One surprising observation from the study was the unusually high reactivity of (R)-N,N-dimethylaminoalanine acylimidazole (55), which reacted with RNA to give superstoichiometric yields at unusually low concentrations of 100 μM, 100 to 1000 times lower than standard concentrations of most other reagents, while simply omitting the α-methyl group (compound 49) lowers acylation efficiency by over 10-fold. A more recent study of chiral acylimidazoles with greater steric bulk (66–76), documented up to >99:1 diastereoselectivity for compound 76, the most highly stereoselective RNA acylating agent to date.53
RNA in general is highly labile to nuclease enzymes, and longer RNA strands are also strongly susceptible to thermal degradation.67,68 The mechanism of cleavage of RNAs (both enzymatic and thermal) usually involves attack of the 2′-OH group on the neighbouring phosphodiester. Given this fact, Fang et al. tested superstoichiometric acylation at 2′-OH as a strategy to protect RNA from degradation.17 However, acylation can also block hybridisation and translation of RNAs,61 so for useful application in cells, acyl esters should be reversible. In this light, Fang et al. studied a small set of simple acylimidazole compounds (20, 23, 24, 26, 38, 46) for their ability to react in superstoichiometric yields with messenger RNAs (mRNAs) and protect them from thermal and enzymatic digestion. Most acyl groups were found to provide significant protection, and dimethylglycine in particular stabilised RNAs thermally at the elevated temperature of 37 °C for six days. It was subsequently found that small nucleophilic reagents in solution (such as Tris or DABCO) could promote removal of some of the ester adducts from the RNA, and interestingly, evidence suggested that dimethylglycine was spontaneously lost from RNAs in cells, enabling the recovery of protein expression.
More recently, Guo et al. designed a series of acylating reagents that introduce disulfide-containing adducts into RNA at superstoichiometric levels. The adducts were designed to be removed by intracellular reducing stimuli (77–84; Fig. 3d). Compound 81 showed enhancement of RNA cellular uptake and distribution without apparent lysosomal entrapment.69
Imidazole carbamates
While imidazole carbamates (Fig. 4) have been known as electrophiles for protein sidechains for some time,70 no such studies had been performed with RNA until relatively recently. Velema et al. first described the development of an acylating agent derived from an ortho-nitrobenzyl alcohol, with the aim of constructing a photocaging agent for 2′-OH groups.28 Tests with a simple imidazole carbamate revealed it to be too unreactive, giving little measurable reaction with RNA after 4 h. However, with survey of carbamate leaving groups led to replacement of the imidazole with 2-chloroimidazole (1.5 h t1/2 in water) increasing its leaving group ability, and the corresponding reagents (85, 86, which also contain a solubilizing dimethylamino group) were able to react with RNA at superstoichiometric levels. The resulting adducts had carbonate linkages that were stable in the dark. This enabled the use of light to switch on biochemical and biological activities of RNA in vitro and in live human cells.28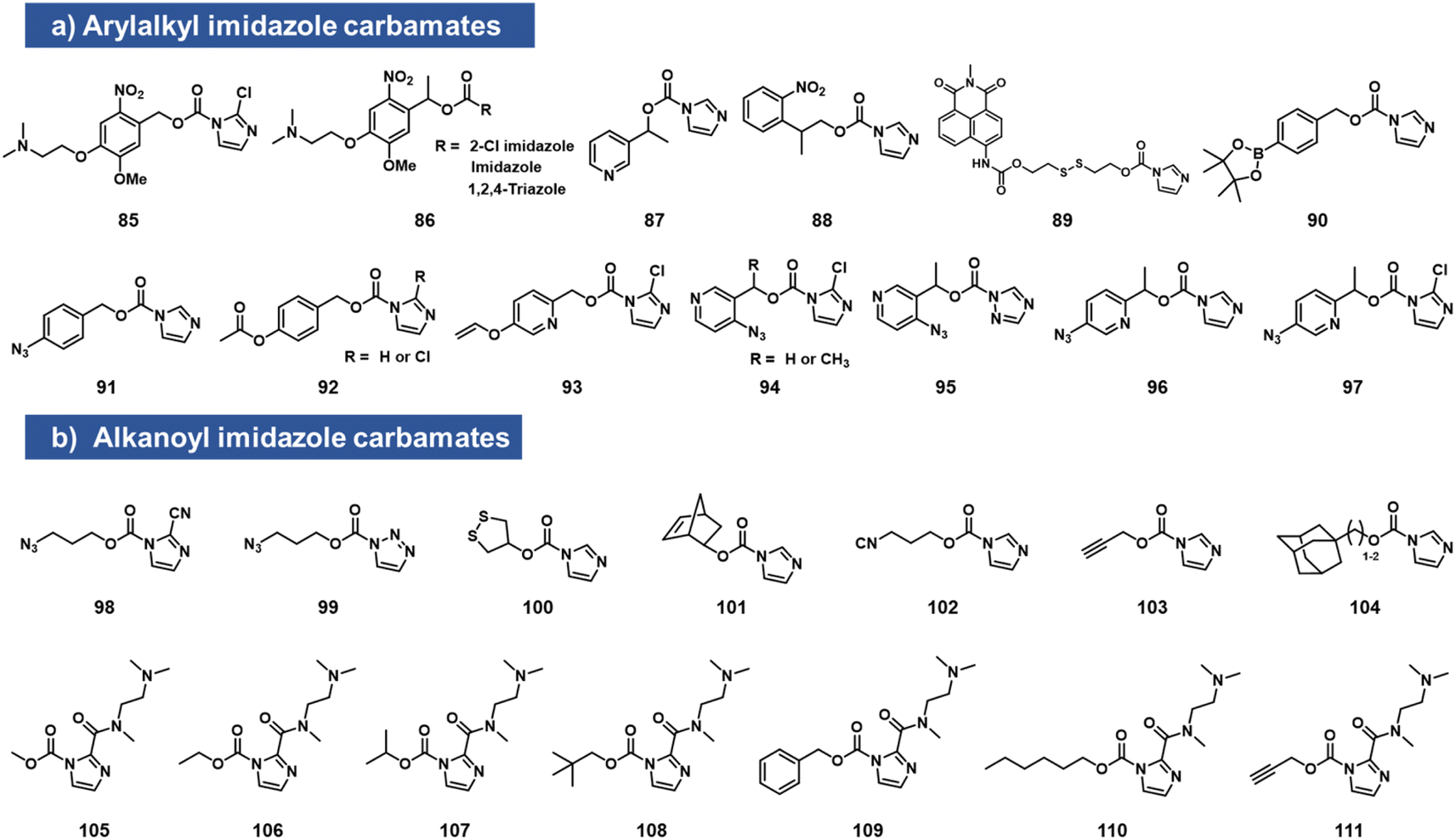 | ||
| Fig. 4 Imidazolecarbamate species reported to react with 2′-OH groups in RNA. To increase reactivity, the imidazole leaving group is often replaced with more active ones, either directly (as in 2-chloroimidazole or 2-carboxamido substituents), or by addition of a nucleophilic catalyst such as DMAP in solution. (a) Compounds with aromatic scaffolds; (b) aliphatic-based reactants. | ||
Simple imidazolecarbamates (98, 99) can in some contexts provide useful yields with RNA. Reagents (R)/(S) 87 were shown to react with a short RNA in ∼20% yields.66 A different photocleavable reagent (88) was recently reported by Liu et al., using DMAP catalysis to increase reactivity, and applied to control CRISPR-CAS12a activity with UV irradiation.71 The CRISPR system has been a test bed for multiple RNA caging approaches using imidazolecarbamate reagents, often assisted in the acylation reaction by DMAP catalysis. Lin et al. employed imidazole carbamates and triazole carbamates of photocleavable reagents in the presence of DMAP for modifying CRISPR crRNA, confirming successfully conjugation of RNA via PAGE gels.72 Similarly, Lei et al. used DMAP catalysis to modify CRISPR RNAs with imidazolecarbamates containing disulfide groups (89, 100) that could be reversed by addition of dithiothreitol.73 Other chemical caging strategies employed with imidazolecarbamates include a peroxide cleavage design (90),74 a norbornene–tetrazine strategy (101),75 an isonitrile–tetrazine cycloaddition (102),76 mesoporous metal–organic frameworks (103),77 an adamantane recognition system (104),78 Pd-catalyzed azide reduction (91),79 and enzyme-mediated deacylation (92).80 Employing the chloroimidazole leaving group, Park et al. reported studies of imidazolecarbamate reagents that use quinone methide mechanisms for triggering removal of acyl esters on RNA.81 Compounds 93–97 were shown to react with RNA at superstoichiometric levels. The compound adducts on RNA could be reversed with varying rates by addition of water-soluble phosphines. The most successful compound was 97, which could be added to RNA at a level of several adducts per strand, and then could be removed in ca. 1 h by addition of TPPMS phosphine (0.5 mM) or TCO-OH (5 mM).
Velema et al. described the use of a dialkylamino-substituted imidazole leaving group to activate other imidazole carbamate species.82 This elevated the water solubility of otherwise poorly soluble species (105–111), and made it possible to decorate an RNA strand with several hydrophobic groups, which remarkably rendered the RNA soluble in organic solvents. In addition, the same soluble imidazole was used to emplace several methylcarbonate groups on an RNA strand using reagent 105.82,83
Other acyl species
While the above carbonyl electrophiles are the best documented classes of reactants for RNA, many more sp2 carbon electrophiles exist, and there are a few reports of tests of their reactivity with RNA. The earliest reports of RNA acylation of transfer RNAs utilized acetic anhydride (112, Fig. 5) in DMF, giving yields of up to 70–92% of one adduct per tRNA.84,85 More recently, Spitale et al. tested several acyl species including other anhydrides, NHS esters, and acyl cyanides before arriving at the higher-yielding acylimidazoles (113–119).7,86 The NHS ester and some of the more hydrophobic anhydrides gave little or no reaction, while more hydrophilic anhydrides gave low levels of apparent acylation. Acyl cyanides were apparently too reactive, giving very short lifespans in water. In addition to this, in patent applications, Goldsborough envisioned the RNA reactions of species such as acyl chlorides, acyl bromides, and anhydrides with RNA, although yields were not given.87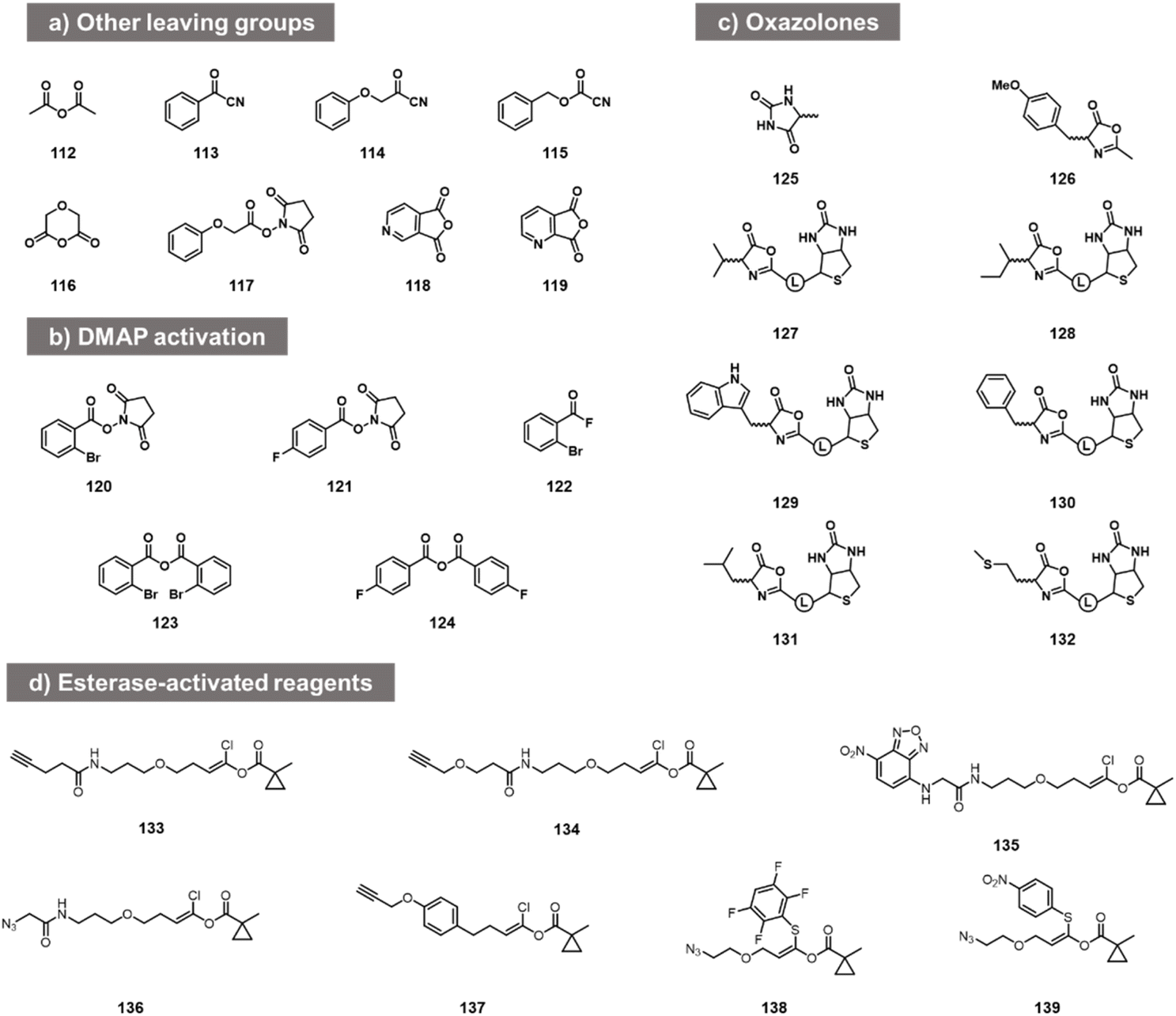 | ||
| Fig. 5 Other acyl reagent types reported to react at RNA 2′-OH. (a) Examples of anhydrides, acyl cyanides, and NHS esters tested for RNA acylation; (b) acylating agents that react with RNA in the presence of added DMAP catalyst; (c) oxazolone electrophiles employed for modification of nucleosides and of RNA 2′-OH; (d) esterase-activated acyl species. | ||
More recent studies with some of these acyl species (notably NHS esters 120, 121, acyl fluorides 122 and anhydrides 123, 124) showed that preparative-level yields in reaction with RNA can occur in the presence of the transacylation catalyst DMAP, observing substantial modification with RNA from the HIV-1 R region,88 although the quantitative yields were not provided. This catalyst strategy likely serves both to increase reactivity and solubility, and provides enhanced versatility by enabling the use of multiple precursor types.88
Other acyl species have also been studied recently. Oxazolones were described as a novel class of activated acyl species for reaction with the 3′- terminus of RNA at 3′- and 2′-OH. Liu et al. described reagents (125, 126; Fig. 5c) and documented reaction yields up to 30% with adenosine derivatives.89 Similarly, biotinylated oxazolones (127–132) were tested with ribozymes, and stoichiometric reactions were observed in gel shift assays (quantitative yields of acylation were not reported).90 Finally, chlorovinyl and related esters 133–139 have been reported by Pani et al. recently (Fig. 5d);91 these are designed to be activated by intracellular esterases to generate reactive acyl species (e.g. acyl chlorides) in cellulo. Tests with RNA alone showed reaction and labelling, although stoichiometric yields were not measured.
Bifunctional electrophiles
Bifunctional electrophiles (Fig. 6) offer the possibility of reaction with RNA and subsequent second reaction with another nucleophile. A recent study reported that carbonyldiimidazole 153 reacts rapidly with both RNA and DNA to provide imidazolecarbonyl adducts in superstoichiometric yields.92 The fact that DNA adduct yields were substantial (although below those of RNA) suggests strongly that a substantial fraction of the acylation occurs at exocyclic amine groups of the nucleobases, along with some reaction at 2′-OH in RNA, likely due to the very high reactivity of CDI. To attenuate reactivity, the authors replaced one of the imidazole groups with a series of phenolic leaving groups (154–162). They identified reagent 160 as an RNA-selective reagent for modification at 2′-OH. When incubated at 200 mM with RNA at pH 7.5 buffer supplemented with imidazole, the reagent provided high selectivity for RNA over DNA, resulting in several imidazolecarbonyl adducts. These are transiently stable in water (half-life on the order of 1 h), but offer the strategy of conjugation with nucleophiles (amines and thiols). Reaction with primary amines and thiols proceeded rapidly and in high yields, producing stable carbamate and thiocarbonate linkages and conjugates.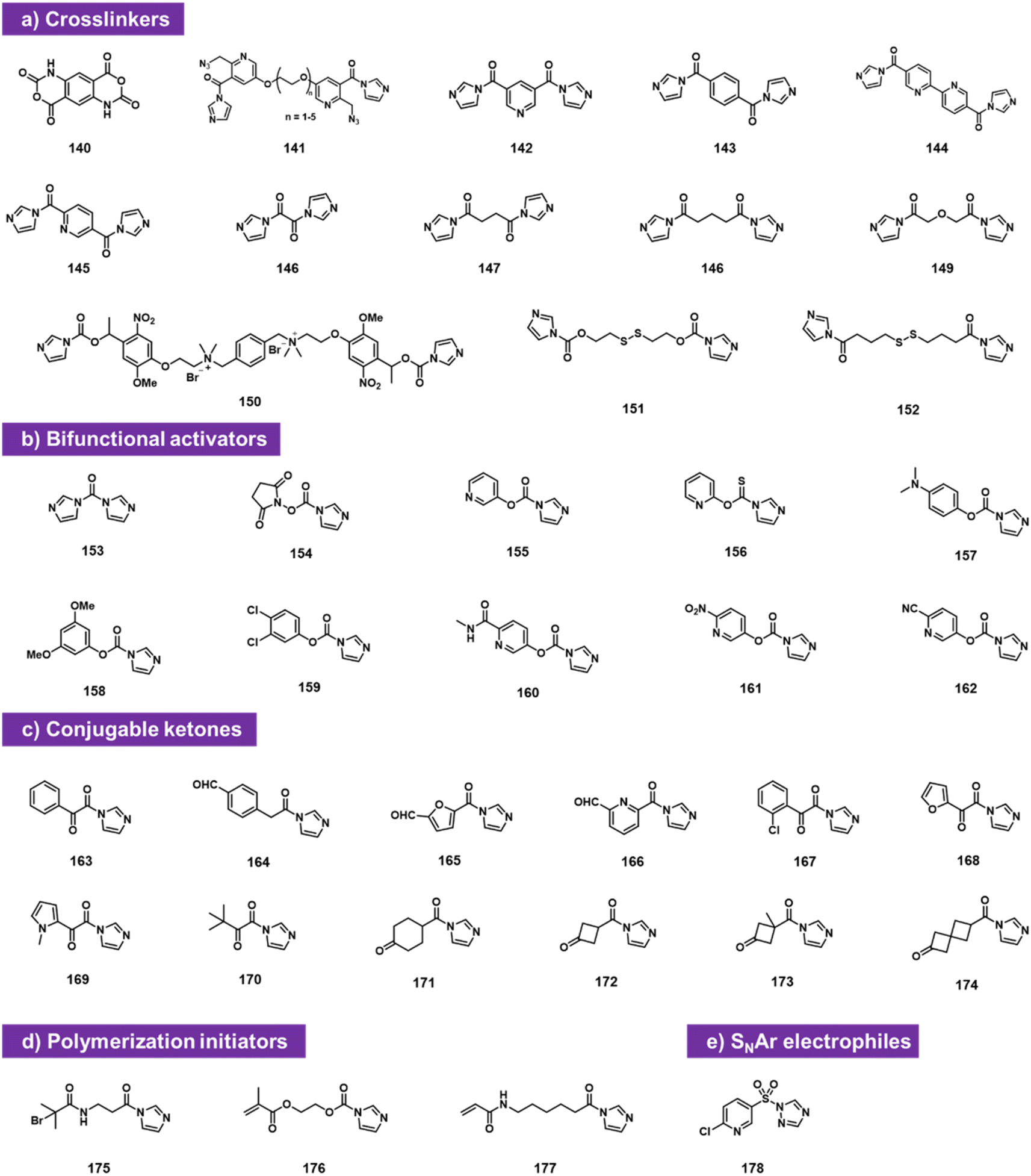 | ||
| Fig. 6 Doubly electrophilic reagents that react with aqueous RNA 2′-OH groups. Reagents are used as (a) RNA–RNA crosslinkers; (b) as activators that install electrophilic carbonyl groups at the 2′-O position of RNA; (c) as reagents for installing reactive ketones; (d) as initiators for polymerisation; and (e) as activating agents for conjugation via SNAr chemistry. | ||
Also falling into this category are bis-electrophiles designed for RNA–RNA crosslinking. Doubly reactive bis-acylimidazoles and bis-isatoic anhydrides have been studied for this purpose, and applied to analysing RNA–RNA contacts and distances. Named probe types include TBIA (140), BINARI (141), DPI (142), SHARC (143–149),93–96 and several others (150–152).72,73 Also studied as doubly reactive species for RNA are reagents that acylate RNA and then initiate polymerisation,97,98 for application in constructing RNA-polymer hybrids. Finally, sulfonyl triazole reagent 178 can sulfonylate RNA and then serve as an SNAr substrate for RNA conjugation with thiols.99
A commonly useful electrophile for bioconjugation is the carbonyl group of ketones. Multiple laboratories have reported the installation of ketones into RNA via isatoic anhydride (19)49 or acylimidazole reactivity (163–174).62 The ketones can then be reacted with aminooxy-substituted molecules (examples include biotin or fluorescent labels), resulting in oxime conjugates. Among the more recent ketone species, cyclobutanone compound 172 proved to be both highly reactive to RNA and readily conjugable with hydrazines; it also was selectively removed from RNA with histamine (Fig. 6c).62
Sulfonyl species
Small activated sulfonyl species have come under intense study in the recent decade for aqueous reactions with protein sidechains. For example, sulfonyl fluoride and sulfonyl triazole species have sufficient aqueous stability to survive in water for a substantial length of time, and remain reactive enough to modify hydroxy, amine, and sulphur-containing sidechains in proteins (Fig. 7).100,101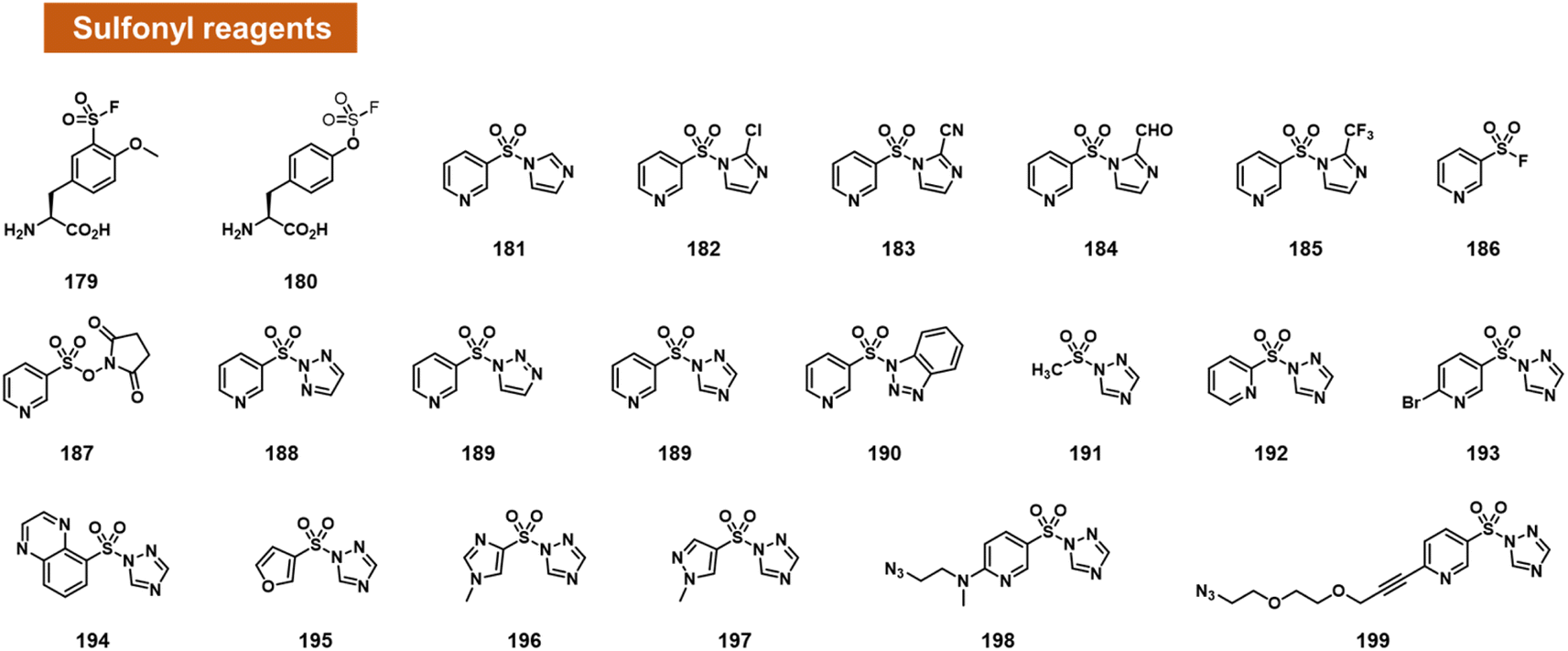 | ||
| Fig. 7 Activated small-molecule sulfonyl reagents that react at RNA 2′-OH groups. Among the leaving groups studied to date, 1,2,4-triazole appears to provide the highest RNA reactivity (e.g.189, 199). | ||
Based on this precedent, Sun et al. recently reported the engineering of proteins with reactive sulfonyl fluoride and fluorosulfate groups on aromatic amino acids (Fig. 7).102 These modified proteins were used to trap interactions with RNA in the cell, by reaction with nearby 2′-OH groups of RNAs bound to the protein. As controls to document the proposed reactions, the authors reacted two fluorosulfonyl substituted amino acids with mononucleotides, and observed up to 16% yield.
Chatterjee et al. tested a range of small-molecule sulfonyl species reacting at 2′-OH groups in RNA (Fig. 7).99 The authors surveyed different leaving groups for a pyridylsulfonyl (see 181–199), demonstrating highest yields (superstoichiometric in some cases) for sulfonyltriazole species. Little or no reaction was observed with DNA of the same sequence, indicating selectivity for 2′-OH. A test of alkyl sulfonyl species (191) revealed generally little reaction with RNA, possibly because of lower sulfonyl electrophilicity. With extended reaction times (typically 24 h), aryl sulfonyltriazoles could react with RNA with 100% or greater conversions, enabling labelling of RNA by use of an azide-substituted scaffold (199).
Sulfonyltriazoles can be synthesised conveniently from available sulfonyl chlorides in one step. Although slower to react with RNA than, for example, acylimidazoles, one beneficial property of the sulfonyl-1,2,4-triazoles in the study is their stability; they survive standard silica column chromatography and have attractive stability for storage. An example was found to last for t1/2 = 1.5 hours in water.99
Arylation at 2′-OH via SNAr reactivity
Chatterjee et al. recently documented the surprising finding that RNA 2′-OH groups could react efficiently with aryl electrophiles via nucleophilic aromatic substitution (SNAr; see Fig. 8).103 They observed moderate but positive reactivity of heterocyclic species such as chloropyrimidines (200–212). However, replacing halogen leaving groups with trialkylamines such as N-methylmorpholine (NMM) or trimethylamine (TMM) (see compounds 213–221) resulted in cationic ammonium salts that are much more reactive with RNA, producing >100% conversions with single-stranded RNA within ∼3 h. NMM salts of triazine have been employed previously for activating carboxylic acids for amide coupling,104 but were not known to be reactive with alcohols; notably, one RNA-reactive NMM triazine was shown to be stable in pure methanol for a day. Indeed, a NMM triazine salt was shown to be remarkably stable in water (half-life of >10 days), while it reacts with RNA to high conversions in as little as 2–3 hours. Testing the stability of a triazine ether adduct in RNA, the authors found no degradation after a week in buffer at room temperature. An additional feature of the triazine scaffold is its ease of multi-derivatisation; a dichlorotriazine was functionalised in one step with an azide-substituted amine, and then in a second step, reacted with NMM to produce a reagent (221) that can be employed to label RNA in high yields.103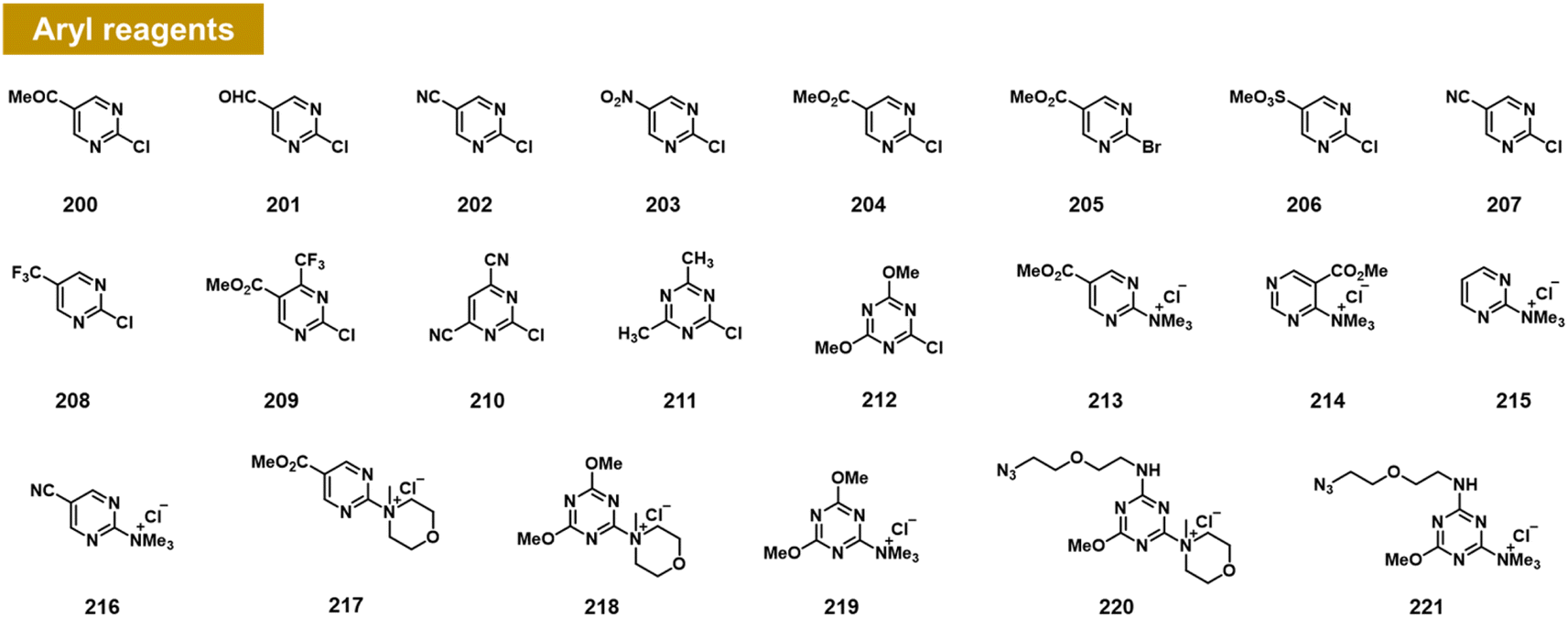 | ||
| Fig. 8 Aryl compounds found to react with RNA 2′-OH groups in preparative yields via the SNAr mechanism. Pyrimidines and triazines were found to react in low to moderate yields with chloride leaving groups; however, alkylammonium groups (213–221) greatly increase RNA reactivity and aqueous solubility. Distinct from previous reagents (Fig. 2–7), these produce an ether linkage with the biopolymer. | ||
DNA/RNA-catalysed reactions
A different approach to functionalising the 2′-OH group of RNA in water has been developed by Höbartner, who has made use of designer catalytic DNAs (DNAzymes) and RNAs (ribozymes) to transfer nucleotide analogues to the 2′-OH position of RNAs. Early work in the Silverman lab reported the discovery of the 10DM24 DNAzyme, which was shown to transfer nucleotides to the 2′-OH of internal adenosines in RNA strand.105 Büttner et al. subsequently made use of this DNAzyme, varying the sequence of the “arms” on either side of the active site, to direct the labelling of RNAs via GTP analogues (222–230; Fig. 9).106 Yields of >70% were reported, and a number of labelled GTP analogues (with labels at the 2′-O position) are commercially available for application. The same lab subsequently discovered a ribozyme that catalyses similar transfers of a different nucleotide, ATP, to adenosines in RNAs (231–240).107 In this case, labels are incorporated at the N6 position of ATP. More recently, Maghami et al. developed a ribozyme that transfers tenofovir-triphosphate analogues to RNA, which enables dual orthogonal labeling of two sites in RNAs (241–245).108 Taken together, the methods offer the favourable ability to direct modification to RNA in a sequence-directed way, with the only apparent limitation of adenosine as the specific labelled site.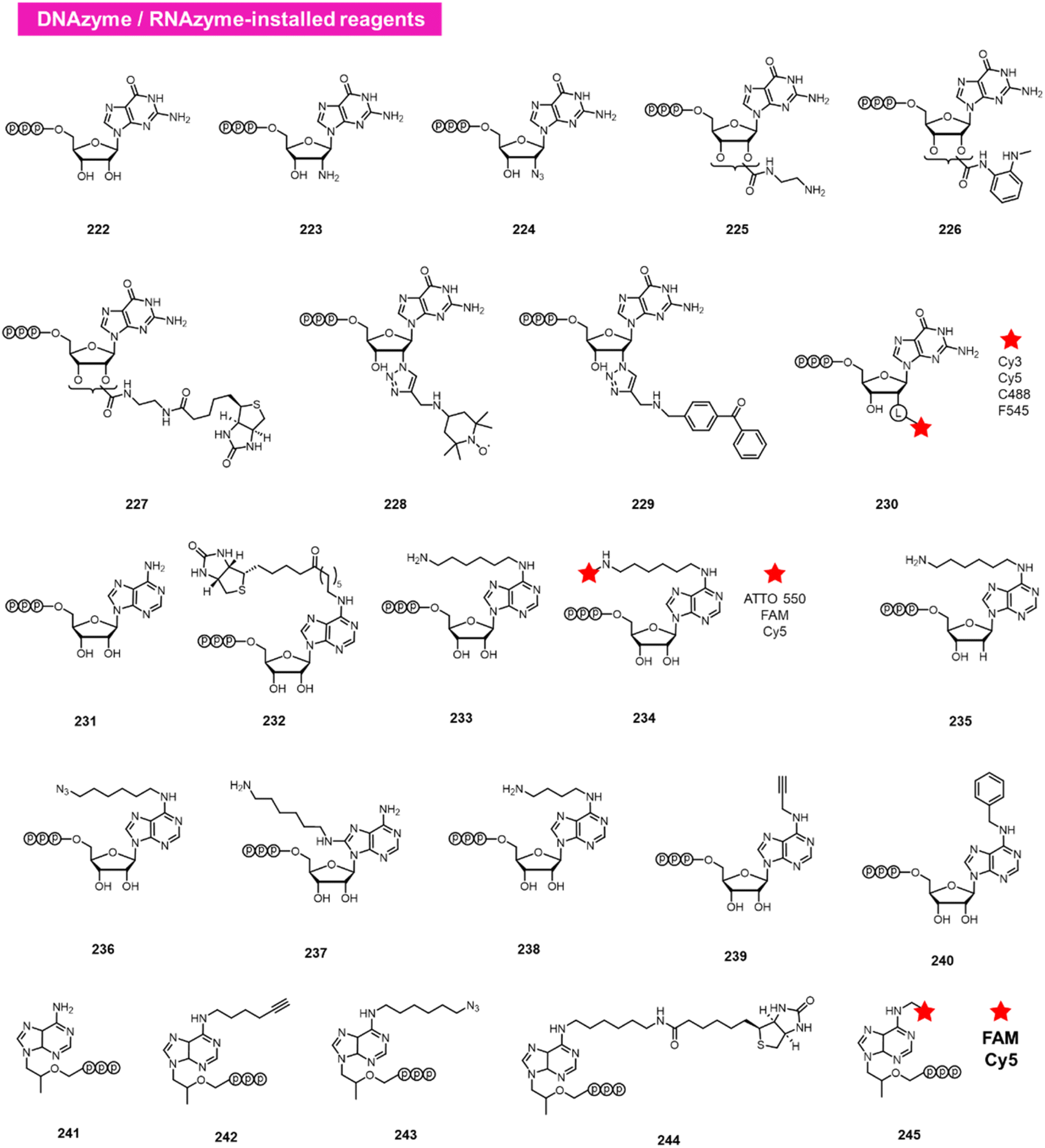 | ||
| Fig. 9 DNAzyme and RNAzyme-directed reactions have been employed to modify RNA 2′-OH. Reagents 222–245 are conjugated to adenosines in RNA with loss of pyrophosphate leaving group. | ||
Functional groups and conversions
Reactions carried out at 2′-OH not only serve to block or alter RNA interactions, but also can be employed to introduce functional groups into RNA, incorporating new reactivity and properties into the biopolymer (Fig. 1b and 10). As mentioned above, adducts with azide groups placed strategically near an acyl linkage to RNA can be reduced with phosphines, resulting in cyclisation and removal of the adduct.13,44 However, placement more remote from the acyl group enables azides to be reduced to produce amines as stable groups in RNA.44 An example of this is acylimidazole 21 (n = 3), which can be used to emplace a primary amine into RNA. Of course, azide groups can also be reacted with alkynes, and this strategy has been employed to conjugate fluorescent labels or biotin groups into RNA via azide or alkyne-substituted reagents.12,60 The Velema and Zhou laboratories have applied carbamates with alkyne groups to RNA,77,82 and Pani has also leveraged esterase-triggered reagents including an alkyne.91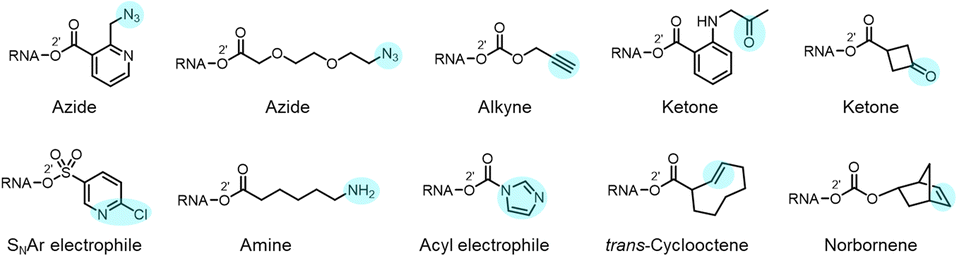 | ||
| Fig. 10 Examples of diverse functional groups that have been incorporated into RNA via reaction at 2′-OH groups. In RNA, these functional groups (highlighted in colour) enable further modification via cycloaddition and nucleophilic addition chemistries. | ||
Other functional groups have also been introduced into RNA at 2′-OH via acylation or sulfonylation chemistry. As mentioned above, bis-electrophiles (140–152) can react with RNA, introducing a second active acyl that can trap other nearby RNAs in a crosslinking strategy.93–96 Imidazole carbamate species have been used to introduce photocaging groups into RNA.28,71 Ketones can be introduced via isatoic anhydride or acylimidazole reagents.49,62 As mentioned above, a doubly reactive sulfonyltriazole reagent (178) has been employed to introduce a 2-chloropyridyl group into RNA, which undergoes subsequent high-yield SNAr chemistry with thiols (Fig. 10).103
General principles for design of reagents
While studies of reactions at 2′-OH in RNA are still in their early stages as compared with the large extant body of work on protein derivatisation, the data to date allow an initial assessment of some of the relevant properties that determine the most effective designs of reagents.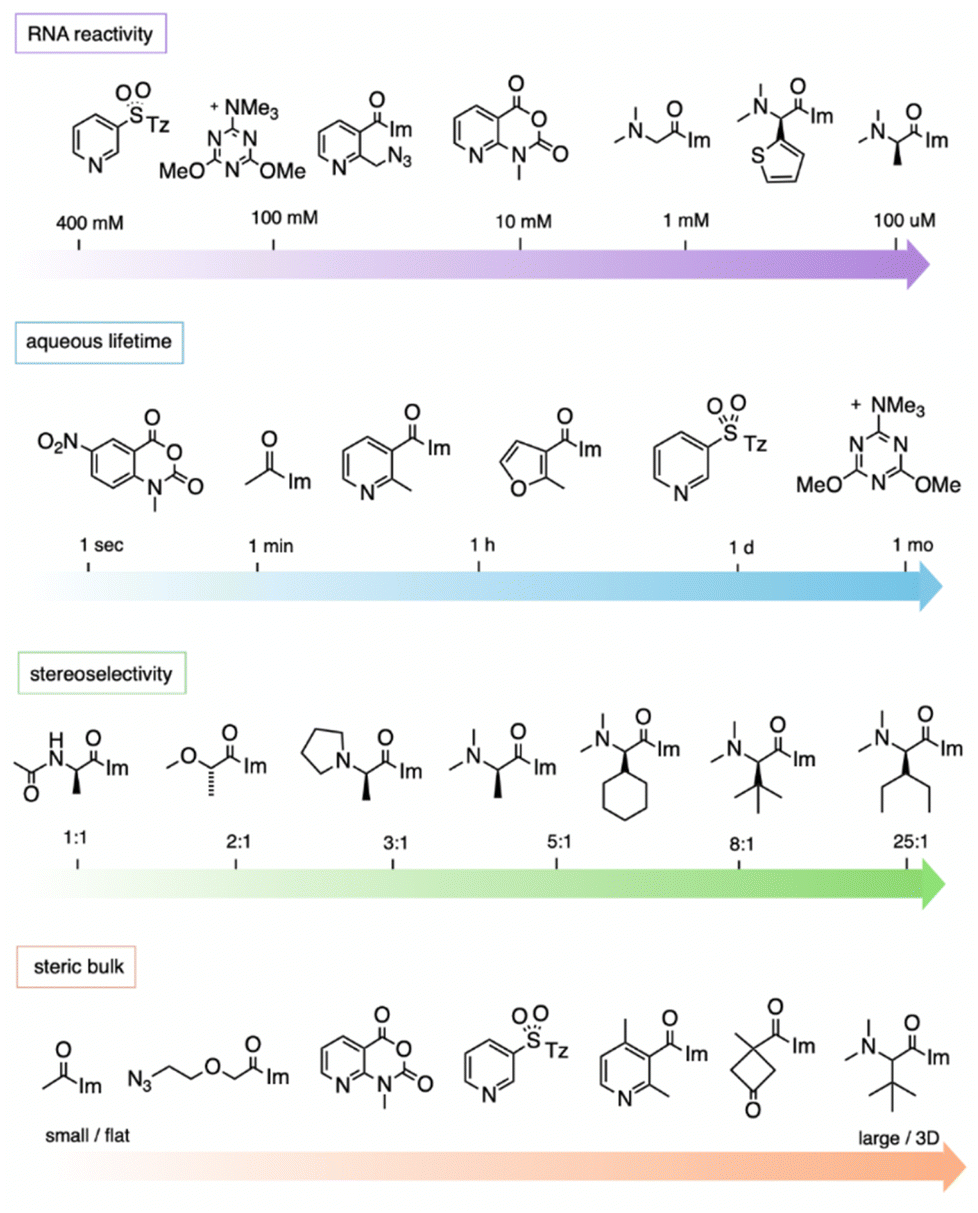 | ||
| Fig. 11 General trends of reagent structures and properties for reaction at 2′-OH groups in RNA, showing representative examples. “RNA reactivity” (top row) shows common concentrations required to yield >1 adduct per RNA strand with example reagents shown; the most highly RNA-reactive reagents can polyacylate RNA at 100 μM reagent. “Aqueous lifetime” illustrates the wide range of reported half-lives of electrophilic 2′-OH-reactive species in water. “Stereoselectivity” depicts diastereoselectivity in reactions of the enantiomers of chiral compounds with single-stranded RNA (favoured enantiomer shown). Stereoselectivity can be higher in folded RNAs. “Steric bulk” shows size of substituents near the reactive electrophilic atom (C or S). “Im”: imidazole; “Tz”: 1,2,4-triazole. | ||
It should be noted that the measurement of aqueous half-lives for electrophiles is illuminating but not entirely predictive of RNA reactivity. For example, (R)-dimethylalanine acylimidazole reagent 55 has a half-life only a few minutes shorter than that of NAI-N3 (another acylimidazole), but the former reacts with RNA in superstoichiometric levels at concentrations 100-fold lower than the latter.66 More dramatically, recent arylation reagents have demonstrated greatly extended aqueous lifetimes; for example, compound 218 has a t1/2 of 10.7 days in D2O, while it reacts at high yields with RNA in 2–3 hours.103 Given the large disparity of water vs. RNA reactivity of this latter compound, these arylating agents are the most RNA-selective reagents yet identified, with a reported ∼108-fold kinetic preference for 2′-OH. The results demonstrate that it is possible to increase selectivity for RNA 2′-OH over water, but it is currently difficult to predict which species will exhibit the highest selectivities.
In some applications, reversibility of the adduct on RNA is desirable. For example, in caging RNA, a triggering mechanism for easy reversibility is necessary. This has been achieved by incorporating light-cleavable groups as mentioned above,28,71 or chemically cleavable groups incorporating reducible azide groups.13,58,79,81 A second application is the use of acyl groups for protection of RNA from hydrolysis; the presence of the groups inhibits RNA degradation, but eventually the groups may need to be removed so as not to interfere with biological activity. With this in mind, Fang et al. described the use of non-basic nucleophiles to promote the loss of acyl esters, and also documented loss of the groups (such as the aforementioned dimethylglycine, 57) inside cells, restoring translation of the RNA.17 Multiple labs have successfully designed groups (38) that can be removed in cells by phosphines added to the growth medium, enabling control of RNA-mediated activities.58,59 Finally, Guo et al. recently employed disulfide acyl adducts (81, 82) that are reversed by reduction both outside and inside cells.69
Unpaired nucleotides in RNA are generally several-fold (generally 2–8-fold) more reactive than those in duplex (paired) structure. Loops are highly variable in their reactivity. Early studies of ribosomal RNAs by Weeks showed that not all unpaired nucleotides react equally with acylating agents. A study of NAIM revealed that there were “hyper-reactive” positions in certain loops of this context.22 In the study, this was attributed to possible general base catalysis by nearby nitrogens on bases. A more recent study by Xiao et al. carried out a quantitative survey of the effects of different loop and unpaired contexts on reactivity to the common reagents 1M7 and NAI-N3.109 A library of RNAs with systematically varied loop sizes and types, along with three single-stranded RNAs, was profiled at every position for reactivity, with data obtained by deep sequencing. The results showed, surprisingly, that very small loops were the most reactive in the library, while increasing loop sizes tended to decrease reactivity. This runs counter to the notion that increased RNA flexibility might favour reaction. Certain loop types displayed the highest reactivity; for example, 1–3 nt bulge loops and small asymmetric internal loops were among the most reactive motifs in the library, and proved to be 50-fold to 4000-fold more reactive than purely single-stranded RNA of the same sequence, likely by exposing specific 2′-OH groups in relatively rigid way.
From a practical standpoint, highly selective RNA loop reactivity can be important as a strategy for reacting at specific 2′-OH groups in a larger RNA. Xiao et al. have used complementary DNAs to induce small reactive loops at designed sites in RNAs, enabling sequence-specific modification (e.g. for fluorescence labelling without interfering with biological activity).61
Conclusions and future prospects
The early research in this field has pointed to high promise of applications of 2′-OH reactivity in RNA, and the chemical and structural diversity of reagents that undergo this reaction has rapidly increased in the last few years. Reagents are useful for mapping RNA structure in vitro and in intact cells; for labelling and functionalising RNA; for controlling RNA function; and for protecting RNA against hydrolytic degradation. To date, the most well-studied classes of reagents for trace-level reactions are the isatoic anhydrides and the acylimidazoles. For reagents that yield stoichiometric and superstoichiometric levels of reaction, a few isatoic anhydrides are known, and numerous acylimidazole species, from both alkyl and aryl scaffolds, have shown much utility and structural diversity.In addition to those two classes of reactive species, imidazolecarbamates, although often less reactive than acylimidazoles, have shown increasing utility in high-yield reactions with RNA, especially with the help of better imidazole leaving groups and nucleophilic catalysis. Aryl sulfonyltriazoles and other sulfonyl species can react, although research on this class of reactants remains in its early stages. Also at early stages is the study of chiral compounds, and the study of SNAr arylation. More work on these classes of reagents is needed to better understand optimal reagent designs and their sensitivity to varied RNA folded sequences and structures. In addition, other active acyl, aryl, and sulfonyl species beyond those tested with RNA to date are known in the literature, and studies of many of those species with RNA are merited. For new reagent species, issues of ease of preparation, solubility and half-life in water, RNA reactivity and RNA structure selectivity should all be kept in mind.
Given the growing versatility of 2′-OH-reactive reagents, it seems certain that further development of specialised reagents is coming in the near future, and new applications are likely to be reported as well. One interesting challenge is the development of new strategies for reacting at specific 2′-OH groups in an RNA strand (as opposed to random reaction); although useful inroads have been made,44,102 further research toward this goal seems likely.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
R. S. and E. T. K. conceived and wrote the manuscript.Conflicts of interest
Both authors are inventors on patent applications involving 2′-modification of RNA.Acknowledgements
We thank the U.S. National Institutes of Health for support of RNA studies (GM145357), and R. S. thanks the Kao Corporation for support.References
- K.-L. Wu, C. Yu, C. Lee, C. Zuo, Z. T. Ball and H. Xiao, Bioconjugate Chem., 2021, 32, 1947–1959 CrossRef PubMed.
- L. Qian, X. Lin, X. Gao, R. U. Khan, J.-Y. Liao, S. Du, J. Ge, S. Zeng and S. Q. Yao, Chem. Rev., 2023, 123, 7782–7853 CrossRef PubMed.
- K. Tsuchikama, Y. Anami, S. Y. Y. Ha and C. M. Yamazaki, Nat. Rev. Clin. Oncol., 2024, 21, 203–223 CrossRef PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Cell, 2017, 168, 527–541 CrossRef PubMed.
- M.-L. Änkö and K. M. Neugebauer, Trends Biochem. Sci., 2012, 37, 255–262 CrossRef PubMed.
- M. Zubradt, P. Gupta, S. Persad, A. M. Lambowitz, J. S. Weissman and S. Rouskin, Nat. Methods, 2017, 14, 75–82 CrossRef PubMed.
- R. C. Spitale, P. Crisalli, R. A. Flynn, E. A. Torre, E. T. Kool and H. Y. Chang, Nat. Chem. Biol., 2013, 9, 18–20 CrossRef PubMed.
- J. L. McGinnis, Q. Liu, C. A. Lavender, A. Devaraj, S. P. McClory, K. Fredrick and K. M. Weeks, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 2425–2430 CrossRef PubMed.
- T. Li, C. Cheng and J. Liu, ChemBioChem, 2024, e202400220 CrossRef PubMed.
- X. Weng, J. Gong, Y. Chen, T. Wu, F. Wang, S. Yang, Y. Yuan, G. Luo, K. Chen, L. Hu, H. Ma, P. Wang, Q. C. Zhang, X. Zhou and C. He, Nat. Chem. Biol., 2020, 16, 489–492 CrossRef PubMed.
- J. Hunt, Biochem. J., 1965, 95, 541–551 CrossRef PubMed.
- R. C. Spitale, R. A. Flynn, Q. C. Zhang, P. Crisalli, B. Lee, J.-W. Jung, H. Y. Kuchelmeister, P. J. Batista, E. A. Torre, E. T. Kool and H. Y. Chang, Nature, 2015, 519, 486–490 CrossRef PubMed.
- A. Kadina, A. M. Kietrys and E. T. Kool, Angew. Chem., Int. Ed., 2018, 57, 3059–3063 CrossRef PubMed.
- S. Ursuegui, N. Chivot, S. Moutin, A. Burr, C. Fossey, T. Cailly, A. Laayoun, F. Fabis and A. Laurent, Chem. Commun., 2014, 50, 5748–5751 RSC.
- D. Zhang, C. Y. Zhou, K. N. Busby, S. C. Alexander and N. K. Devaraj, Angew. Chem., Int. Ed., 2018, 57, 2822–2826 CrossRef PubMed.
- C. P. M. Scheitl, M. Ghaem Maghami, A.-K. Lenz and C. Höbartner, Nature, 2020, 587, 663–667 CrossRef PubMed.
- L. Fang, L. Xiao, Y. W. Jun, Y. Onishi and E. T. Kool, Nat. Chem., 2023, 15, 1296–1305 CrossRef PubMed.
- S. P. Velagapudi, Y. Li and M. D. Disney, Bioorg. Med. Chem. Lett., 2019, 29, 1532–1536 CrossRef PubMed.
- S. Balaratnam, C. Rhodes, D. D. Bume, C. Connelly, C. C. Lai, J. A. Kelley, K. Yazdani, P. J. Homan, D. Incarnato, T. Numata and J. S. Schneekloth Jr, Nat. Commun., 2021, 12, 5856 CrossRef PubMed.
- L. Fang, W. A. Velema, Y. Lee, L. Xiao, M. G. Mohsen, A. M. Kietrys and E. T. Kool, Nat. Chem., 2023, 15, 1374–1383 CrossRef PubMed.
- B. Jash and E. T. Kool, Chem. Commun., 2022, 58, 3693–3696 RSC.
- P. Ballinger and F. A. Long, J. Am. Chem. Soc., 1960, 82, 795–798 CrossRef.
- D. Mitchell, L. E. Ritchey, H. Park, P. Babitzke, S. M. Assmann and P. C. Bevilacqua, RNA, 2018, 24, 114–124 CrossRef.
- D. A. Peattie, Proc. Natl. Acad. Sci. U.S.A., 1979, 76, 1760–1764 CrossRef PubMed.
- L. Guan and M. D. Disney, Angew. Chem., Int. Ed., 2013, 52, 10010–10013 CrossRef PubMed.
- C. A. Weidmann, A. M. Mustoe, P. B. Jariwala, J. M. Calabrese and K. M. Weeks, Nat. Biotechnol., 2021, 39, 347–356 CrossRef.
- C. C. Smith, M. Hollenstein and C. J. Leumann, RSC Adv., 2014, 4, 48228–48235 RSC.
- W. A. Velema, A. M. Kietrys and E. T. Kool, J. Am. Chem. Soc., 2018, 140, 3491–3495 CrossRef PubMed.
- L. Xiao, L. Fang and E. T. Kool, Cell Chem. Biol., 2024, 31, 110–124 CrossRef PubMed.
- M. J. Smola, G. M. Rice, S. Busan, N. A. Siegfried and K. M. Weeks, Nat. Protoc., 2015, 10, 1643–1669 CrossRef PubMed.
- W. A. Velema and E. T. Kool, Nat. Rev. Chem, 2019, 4, 22–37 CrossRef PubMed.
- S. A. Mortimer, C. Trapnell, S. Aviran, L. Pachter and J. B. Lucks, Curr. Protoc. Chem. Biol., 2012, 4, 275–297 CrossRef.
- K. M. Kutchko and A. Laederach, Wiley Interdiscip. Rev.: RNA, 2017, 8, e1374 CrossRef.
- K. M. Weeks, Acc. Chem. Res., 2021, 54, 2502–2517 CrossRef PubMed.
- J. L. McGinnis, J. A. Dunkle, J. H. D. Cate and K. M. Weeks, J. Am. Chem. Soc., 2012, 134, 6617–6624 CrossRef PubMed.
- I. Velikyan, S. Acharya, A. Trifonova, A. Földesi and J. Chattopadhyaya, J. Am. Chem. Soc., 2001, 123, 2893–2894 CrossRef.
- T. Hiratsuka, J. Biol. Chem., 1982, 257, 13354–13358 CrossRef.
- T. Hiratsuka, Biochim. Biophys. Acta, 1983, 742, 496–508 CrossRef.
- E. J. Merino, K. A. Wilkinson, J. L. Coughlan and K. M. Weeks, J. Am. Chem. Soc., 2005, 127, 4223–4231 CrossRef CAS PubMed.
- K. M. Weeks, Biopolymers, 2015, 103, 438–448 CrossRef CAS.
- J. Liu, Y. Shi, Z. Tian, F. Li, Z. Hao, W. Wen, L. Zhang, Y. Wang, Y. Li and Z. Fan, J. Agric. Food Chem., 2022, 70, 5197–5206 CrossRef CAS PubMed.
- S. Busan, C. A. Weidmann, A. Sengupta and K. M. Weeks, Biochemistry, 2019, 58, 2655–2664 CrossRef CAS.
- T. Marinus, A. B. Fessler, C. A. Ogle and D. Incarnato, Nucleic Acids Res., 2021, 49, e34 CrossRef CAS.
- H. S. Park, A. M. Kietrys and E. T. Kool, Chem. Commun., 2019, 55, 5135–5138 RSC.
- S. Ursuegui, R. Yougnia, S. Moutin, A. Burr, C. Fossey, T. Cailly, A. Laayoun, A. Laurent and F. Fabis, Org. Biomol. Chem., 2015, 13, 3625–3632 RSC.
- A. B. Fessler, A. Dey, C. B. Garmon, D. S. Finis, N.-A. Saleh, A. J. Fowler, D. S. Jones, K. Chakrabarti and C. A. Ogle, Bioconjugate Chem., 2018, 29, 3196–3202 CrossRef CAS PubMed.
- A. B. Fessler, A. Dey, D. S. Finis, A. J. Fowler, K. Chakrabarti and C. A. Ogle, Bioconjugate Chem., 2020, 31, 884–888 CrossRef CAS.
- A. B. Fessler, A. J. Fowler and C. A. Ogle, Bioconjugate Chem., 2021, 32, 904–908 CrossRef CAS.
- L. D. Poulsen, L. J. Kielpinski, S. R. Salama, A. Krogh and J. Vinther, RNA, 2015, 21, 1042–1052 CrossRef CAS.
- R. J. Cedergren, N. Beauchemin and J. Toupin, Biochemistry, 1973, 12, 4566–4570 CrossRef CAS.
- A. T. Profy and D. A. Usher, J. Mol. Evol., 1984, 20, 147–156 CrossRef CAS PubMed.
- D. A. Usher and M. C. Needels, Adv. Space Res., 1984, 4, 163–166 CrossRef CAS PubMed.
- R. Shioi, S. Chatterjee, L. Xiao, W. Zhong and E. T. Kool, J. Org. Chem., 2024, 89, 8055–8063 CrossRef CAS.
- A. L. Weber, J. Mol. Evol., 1987, 25, 7–11 CrossRef CAS.
- R. C. Spitale, R. A. Flynn, E. A. Torre, E. T. Kool and H. Y. Chang, Wiley Interdiscip. Rev.: RNA, 2014, 5, 867–881 CrossRef CAS.
- B. Lee, R. A. Flynn, A. Kadina, J. K. Guo, E. T. Kool and H. Y. Chang, RNA, 2017, 23, 169–174 CrossRef.
- D. Chan, S. Beasley, Y. Zhen and R. C. Spitale, Bioorg. Med. Chem. Lett., 2018, 28, 601–605 CrossRef.
- S.-R. Wang, L.-Y. Wu, H.-Y. Huang, W. Xiong, J. Liu, L. Wei, P. Yin, T. Tian and X. Zhou, Nat. Commun., 2020, 11, 91 CrossRef PubMed.
- M. Habibian, C. McKinlay, T. R. Blake, A. M. Kietrys, R. M. Waymouth, P. A. Wender and E. T. Kool, Chem. Sci., 2020, 11, 1011–1016 RSC.
- L. Xiao, M. Habibian and E. T. Kool, J. Am. Chem. Soc., 2020, 142, 16357–16363 CrossRef PubMed.
- L. Xiao, Y. W. Jun and E. T. Kool, Angew. Chem., Int. Ed., 2021, 60, 26798–26805 CrossRef CAS PubMed.
- R. Shioi, L. Xiao, L. Fang and E. T. Kool, Chem. Commun., 2024, 60, 232–235 RSC.
- W. Shen, W. Xiong, Q. Qi, X. Liu, Z. Xie, Y. Zhang, J. Hou, T. Tian and X. Zhou, Chin. J. Chem., 2024, 42, 1387–1393 CrossRef CAS.
- X. Chen, Y. Guo and R. Wang, Bioorg. Med. Chem. Lett., 2024, 109, 129847 CrossRef CAS PubMed.
- L. Xiao, L. Fang, S. Chatterjee and E. T. Kool, J. Am. Chem. Soc., 2023, 145, 143–151 CrossRef CAS.
- R. Shioi, L. Xiao, S. Chatterjee and E. T. Kool, Chem. Sci., 2023, 14, 13235–13243 RSC.
- S. Ohta, S. Maul, A. J. Sinskey and S. R. Tannenbaum, Appl. Microbiol., 1971, 22, 415–421 CrossRef CAS.
- H. K. Wayment-Steele, D. S. Kim, C. A. Choe, J. J. Nicol, R. Wellington-Oguri, A. M. Watkins, R. A. Parra Sperberg, P.-S. Huang, E. Participants and R. Das, Nucleic Acids Res., 2021, 49, 10604–10617 CrossRef CAS.
- J. Guo, S. Chen, Y. Onishi, Q. Shi, Y. Song, H. Mei, L. Chen, E. T. Kool and R. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202402178 CrossRef CAS.
- M. R. Mortensen, N. L. Nielsen, J. Palmfeldt and K. V. Gothelf, Org. Biomol. Chem., 2019, 17, 1379–1383 RSC.
- P. Liu, Y. Lin, X. Zhuo, J. Zeng, B. Chen, Z. Zou, G. Liu, E. Xiong and R. Yang, Angew. Chem., Int. Ed., 2024, 63, e202401486 CrossRef CAS.
- C.-L. Lin, W.-D. Chen, L. Liu and L. Cheng, Chem. Commun., 2024, 60, 5197–5200 RSC.
- H. Lei, W. Xiong, M. Li, Q. Qi, X. Liu, S. Wang, T. Tian and X. Zhou, Bioorg. Med. Chem., 2024, 112, 117878 CrossRef CAS.
- H. Ji, W. Xiong, K. Zhang, T. Tian and X. Zhou, Chem.–Asian J., 2022, 17, e202200214 CrossRef CAS.
- Q. Qi, Y. Zhang, W. Xiong, X. Liu, S. Cui, X. Ye, K. Zhang, T. Tian and X. Zhou, Chem. Sci., 2022, 13, 12577–12587 RSC.
- H. Ji, W. Xiong, S. Guo, S. Wang, X. Xing, T. Tian and X. Zhou, ACS Chem. Biol., 2023, 18, 1829–1837 CrossRef CAS PubMed.
- J. Liu, X. Liu, Q. Liu, J. Cao, X. Lv, S. Wang, T. Tian, X. Zhou and H. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302649 CrossRef CAS.
- W. Xiong, X. Liu, Q. Qi, H. Ji, F. Liu, C. Zhong, S. Liu, T. Tian and X. Zhou, Nucleic Acids Res., 2022, 50, 1241–1255 CrossRef CAS PubMed.
- F. Fu, W. Xiong, X. Xu, Y. Liu, M. Li, Q. Qi, X. Liu, Y. Zhang, T. Tian and X. Zhou, Chin. J. Chem., 2024 DOI:10.1002/cjoc.202400254.
- K. Liu and A. M. Kietrys, bioRxiv, 2024, preprint, ASAP, DOI: DOI:10.1002/cjoc.202400254.
- H. S. Park, B. Jash, L. Xiao, Y. W. Jun and E. T. Kool, Org. Biomol. Chem., 2021, 19, 8367–8376 RSC.
- W. A. Velema and E. T. Kool, Org. Lett., 2018, 20, 6587–6590 CrossRef PubMed.
- M. Habibian, W. A. Velema, A. M. Kietrys, Y. Onishi and E. T. Kool, Org. Lett., 2019, 21, 5413–5416 CrossRef PubMed.
- O. S. Yu and A. B. Yu, FEBS Lett., 1990, 270, 111–114 CrossRef PubMed.
- D. G. Knorre and G. G. Shamovsky, Biochim. Biophys. Acta, 1967, 142, 555–558 CrossRef PubMed.
- S. A. Mortimer and K. M. Weeks, J. Am. Chem. Soc., 2008, 130, 16178–16180 CrossRef CAS PubMed.
- S. Goldsborough, US Patent, US20030039985A1, 2003 Search PubMed.
- L. Nodin, O. Noël, F. Chaminade, O. Maskri, V. Barbier, O. David, P. Fossé and J. Xie, Bioorg. Med. Chem. Lett., 2015, 25, 566–570 CrossRef CAS.
- Z. Liu, C. Hanson, G. Ajram, L. Boiteau, J.-C. Rossi, G. Danger and R. Pascal, Synlett, 2016, 28, 73–77 CrossRef.
- E. Janzen, Y. Shen, A. Vazquez-Salazar, Z. Liu, C. Blanco, J. Kenchel and I. Chen, Nat. Commun., 2022, 13, 3631 CrossRef CAS PubMed.
- S. Pani, T. Qiu, K. Kentala, S.-A. Azizi and B. C. Dickinson, Nat. Chem., 2024, 16, 717–726 Search PubMed.
- R. Shioi, L. Xiao and E. T. Kool, Bioconjugate Chem., 2024, 35, 43–50 CrossRef CAS.
- W. A. Velema, H. S. Park, A. Kadina, L. Orbai and E. T. Kool, Angew. Chem., Int. Ed., 2020, 59, 22017–22022 CrossRef CAS.
- H. Lei, T. Zeng, X. Ye, R. Fan, W. Xiong, T. Tian and X. Zhou, Advanced Science, 2023, 10, 2206433 CrossRef CAS.
- T. W. Christy, C. A. Giannetti, G. Houlihan, M. J. Smola, G. M. Rice, J. Wang, N. V. Dokholyan, A. Laederach, P. Holliger and K. M. Weeks, Biochemistry, 2021, 60, 1971–1982 CrossRef CAS PubMed.
- R. Van Damme, K. Li, M. Zhang, J. Bai, W. H. Lee, J. D. Yesselman, Z. Lu and W. A. Velema, Nat. Commun., 2022, 13, 911 CrossRef CAS PubMed.
- J. Jeong, X. Hu, H. Murata, G. Szczepaniak, M. Rachwalak, A. Kietrys, S. R. Das and K. Matyjaszewski, J. Am. Chem. Soc., 2023, 145, 14435–14445 CrossRef CAS PubMed.
- J. Jeong, S. Y. An, X. Hu, Y. Zhao, R. Yin, G. Szczepaniak, H. Murata, S. R. Das and K. Matyjaszewski, ACS Nano, 2023, 17, 21912–21922 CrossRef.
- S. Chatterjee, R. Shioi and E. T. Kool, ACS Cent. Sci., 2023, 9, 531–539 CrossRef CAS.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- J. T. Cruite, R. P. Nowak, K. A. Donovan, S. B. Ficarro, H. Huang, H. Liu, Y. Liu, J. A. Marto, R. J. Metivier, E. S. Fischer and L. H. Jones, ACS Med. Chem. Lett., 2023, 14, 1576–1581 CrossRef CAS.
- W. Sun, N. Wang, H. Liu, B. Yu, L. Jin, X. Ren, Y. Shen and L. Wang, Nat. Chem., 2023, 15, 21–32 CrossRef CAS.
- S. Chatterjee, L. Xiao, W. Zhong, S. Feng and E. T. Kool, Angew. Chem., Int. Ed., 2024, 63, e202403496 CrossRef.
- M. Kunishima, C. Kawachi, K. Hioki, K. Terao and S. Tani, Tetrahedron, 2001, 57, 1551–1558 CrossRef.
- C. Höbartner and S. K. Silverman, Angew. Chem., Int. Ed., 2007, 46, 7420–7424 CrossRef PubMed.
- L. Büttner, F. Javadi-Zarnaghi and C. Höbartner, J. Am. Chem. Soc., 2014, 136, 8131–8137 CrossRef.
- M. Ghaem Maghami, C. P. M. Scheitl and C. Höbartner, J. Am. Chem. Soc., 2019, 141, 19546–19549 CrossRef.
- M. Ghaem Maghami, S. Dey, A. Lenz and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 9335–9339 CrossRef CAS PubMed.
- L. Xiao, L. Fang and E. T. Kool, Cell Chem. Biol., 2022, 29, 1341–1352 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |