
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMg2+-driven selection of natural phosphatidic acids in primitive membranes†
Krishnakavya
Thaipurayil Madanan
 a,
Yuhan
Li
a,
Valeria J.
Boide-Trujillo
a,
David A.
Russell
ab and
Claudia
Bonfio
*ab
a,
Yuhan
Li
a,
Valeria J.
Boide-Trujillo
a,
David A.
Russell
ab and
Claudia
Bonfio
*ab
aInstitut de Science et d’Ingénierie Supramoléculaires (ISIS), CNRS UMR 7006, University of Strasbourg, 8 Allée Gaspard Monge, 67000 Strasbourg, France. E-mail: cb2036@cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, Tennis Court Road, CB2 1GA Cambridge, UK
First published on 29th October 2024
Abstract
Biological membranes are composed exclusively of phospholipids comprising glycerol-1-phosphate or glycerol-3-phosphate. By contrast, primitive membranes would have likely been composed of heterogeneous mixtures of phospholipids, including non-natural analogues comprising glycerol-2-phosphate, as delivered by prebiotic synthesis. Thus, it is not clear how the selection of natural phospholipids could have come about. Here we show how differences in supramolecular properties, but not molecular properties, could have driven the selection of natural phosphatidic acids in primitive membranes. First, we demonstrate that at the molecular level it is unlikely that any prebiotic synthesis or hydrolysis pathway would have enabled the selection of natural phosphatidic acids. Second, we report that at the supramolecular level, natural phospholipids display a greater tendency to self-assemble in more packed and rigid membranes than non-natural analogues of the same chain length. Finally, taking advantage of these differences, we highlight that Mg2+, but not Na+, K+, Ca2+ or Zn2+, drives the selective precipitation of non-natural phosphatidic acids from heterogeneous mixtures obtained by prebiotic synthesis, leaving membranes proportionally enriched in natural phosphatidic acids. Our findings delineate a plausible pathway by which the transition towards biological membranes could have occurred under conditions compatible with prebiotic metal-driven processes, such as non-enzymatic RNA polymerization.
Introduction
Phospholipids are major constituents of biological membranes.1 Their emergence on early Earth would thus have marked a pivotal step on the path towards primitive cells.2 Prebiotic synthesis would have produced heterogeneous mixtures of isomeric phospholipids, but only the natural phospholipids comprising glycerol-1-phosphate (G1P) and glycerol-3-phosphate (G3P) were retained in biological membranes while their non-natural analogues comprising glycerol-2-phosphate (G2P) were presumably lost to chemical evolution. Still, it is not clear how these natural phospholipids were selected.Prebiotic chemists have sought to identify mechanisms by which many of the natural building blocks of life could have been selected. Yet, investigations have focused almost exclusively on nucleosides and peptides. Canonical RNA and DNA nucleosides could have arisen by selective prebiotic synthesis3–5 or because of their greater hydrolytic and photochemical stability relative to non-canonical isomers.6–8 Selection could also have resulted from the increased stability of duplexes comprising natural oligonucleotides, compared to non-natural analogues.9 Similarly, the canonical amino acids could have resulted from selective synthesis,10 the preferential incorporation of natural over non-natural amino acids into peptides,11 or by retention due to the enhanced foldability of oligopeptides containing natural amino acids compared to those containing non-natural analogues.12 The idea that selection processes could have operated at both the molecular and supramolecular levels is central to every scheme.
Two recent reports have begun to address the selection of natural phospholipids. In work on the emergence of amino alcohol bearing phospholipids, our group13 described a general prebiotic synthesis and hydrolysis scheme that is selective for natural phospholipid headgroups comprising G1P over those comprising G2P. Then, in work advancing the prebiotic chemistry of cyclophospholipids, Krishnamurthy and co-workers14 showed that liposomes formed of glycerol monodecanoate and phospholipid comprising G1P are more tolerant of pH changes and metal ions, such as those required for prebiotic RNA chemistry,15 than those formed of phospholipid comprising G2P.
These studies raise further questions, particularly in light of the systems chemistry approach in which mixtures of reactants and products must be considered and their properties compared. First, it is not clear if prebiotic synthesis and hydrolysis processes could have led to the selection of natural over non-natural diacyl phospholipids. Prebiotic acylation reactions of individual glycerol phosphates have been reported,16,17 but no direct comparison has been made of the reactivity of G1P and G2P with activated fatty acids, nor of the hydrolytic stability of the corresponding phospholipids. Second, it is not clear if differences in membrane properties could have led to the selection of natural phospholipids from heterogeneous mixtures containing both natural and non-natural forms. Hybrid liposomes often behave differently from those made of individual lipids and so their behavior is difficult to predict.
Here we explore processes operating at the molecular and supramolecular levels that could have led to the selection of natural phosphatidic acids from heterogeneous mixtures on early Earth. We show that hydrolytically stable natural and non-natural phosphatidic acids could have formed on prebiotic acylation of mixtures of glycerol phosphates under a variety of conditions, together with a range of amphiphilic coproducts. We then highlight that natural phosphatidic acids have a greater propensity to self-assemble, compared to their non-natural isomers, and that their liposomes display enhanced rigidity and stability towards changes in pH and metal ion concentrations. Finally, harnessing these differences, we demonstrate that on exposure to Mg2+, but not Na+, K+, Ca2+ or Zn2+, natural phosphatidic acids could have been selected from liposomes composed of heterogeneous mixtures. Overall, our findings offer a plausible rationale for the selection of natural phospholipids, taking advantage of differences in properties that manifest only at the supramolecular level.
Note: throughout our discussion we treat G1P and G3P as synonymous and for simplicity refer to them with the acronym G1P. The stereospecific numbering (sn) scheme for glycerophospholipids is used only where appropriate.
Results and discussion
Prebiotic synthesis delivers mixtures of natural and non-natural phosphatidic acids
To probe the acylation of glycerol phosphates in solution, each glycerol phosphate was dissolved in water and the pH of each solution was adjusted to 7.4. A solution of N-decanoyl imidazole (NDI) in acetonitrile was then added,16 and each homogeneous mixture was stirred for 8 days at room temperature (Fig. 1a, b and S1†). The progress of each reaction was followed by 31P NMR and by TLC.27 In the reaction of G1P, 3(2)-monodecanoyl-glycerol-1-phosphatidic acid (MDG1PA, 1.68 and 1.59 ppm) were obtained alongside 2,3-didecanoyl-glycerol-1-phosphatidic acid (DDG1PA, 0.89 ppm) in yields of 20% and 4%, respectively (Fig. 1a, b and S1†). By contrast, in the reaction of G2P, 1-monodecanoyl-glycerol-2-phosphatidic acid (MDG2PA, 1.05 ppm), and 1,3,-didecanoyl-glycerol-2-phosphatidic acid (DDG2PA, 0.47 ppm) were obtained in yields of 43% and 44%, respectively (Fig. 1a, b and S2†).
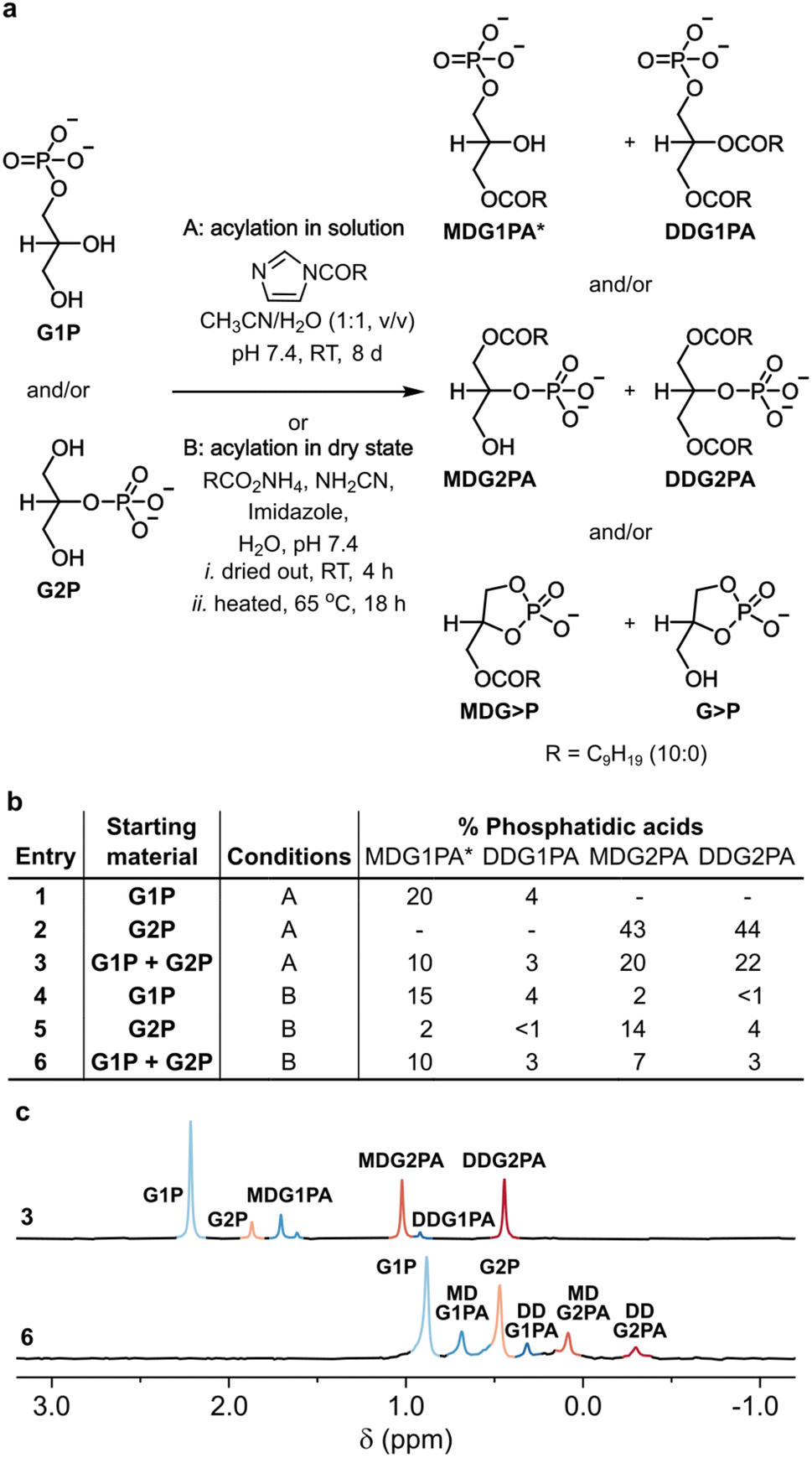 | ||
| Fig. 1 Prebiotic synthesis of phosphatidic acids. (a) Reactions of G1P (10 mM), G2P (10 mM), or both (5 mM each) with NDI (100 mM) in solution, or ammonium decanoate (100 mM), cyanamide (100 mM), and imidazole (100 mM) in the dry state, afford natural G1PAs and non-natural G2PAs. (b) Yields of G1PAs and G2PAs formed from G1P, G2P, or both. (c) 31P{1H} NMR spectra showing the signals for the G1PAs and G2PAs obtained in reactions described in entries 3 (pH ∼7) and 6 (pH ∼4 after extraction) of (b). Yields were determined by 31P NMR based on conversion of the respective glycerol phosphate (or sum total glycerol phosphates). The chemical shift of each phosphatidic acid is pH and concentration dependent. *The major positional isomer26 of MDG1PA is shown in (a) and combined yields of both isomers of MDG1PA are given in (b). See the ESI† for details. | ||
Since the acylation of G2P proceeds to a greater extent than the acylation of G1P, we wondered if, in a reaction starting from a mixture of glycerol phosphates, G2P would outcompete G1P for NDI and thus suppress the formation of G1PAs. However, in the event, acylation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of glycerol phosphates gave G1PAs and G2PAs in combined yields of 13% and 42%, respectively, or approximately half the yields obtained in experiments starting from G1P or G2P (Fig. 1a–c and S3†). This result shows that in solution G1P and G2P undergo acylation independently of one another. Nevertheless, as the yield of DDG2PA is approximately 10 times that of DDG1PA under our conditions, we infer DDG1PA would only have been produced as the major phosphatidic acid if the initial ratio of G1P:G2P exceeded 10:1. The literature on prebiotic phosphorylation of glycerol21–25 suggests that this requirement would rarely have been met, however, especially when hydrolysis of glycerol cyclic phosphate is taken into account.13,20,28
:1 mixture of glycerol phosphates gave G1PAs and G2PAs in combined yields of 13% and 42%, respectively, or approximately half the yields obtained in experiments starting from G1P or G2P (Fig. 1a–c and S3†). This result shows that in solution G1P and G2P undergo acylation independently of one another. Nevertheless, as the yield of DDG2PA is approximately 10 times that of DDG1PA under our conditions, we infer DDG1PA would only have been produced as the major phosphatidic acid if the initial ratio of G1P:G2P exceeded 10:1. The literature on prebiotic phosphorylation of glycerol21–25 suggests that this requirement would rarely have been met, however, especially when hydrolysis of glycerol cyclic phosphate is taken into account.13,20,28
The acylation of glycerol phosphates in solution occurs by a two-step phosphate-assisted mechanism in which intermolecular formation of a mixed anhydride intermediate precedes intramolecular transfer of the acyl group to a neighboring hydroxyl group (Fig. S4†).16 Acyl transfer to the proximal primary hydroxyl group of G2P is more efficient than acyl transfer to either the proximal secondary hydroxyl group or the distal primary hydroxyl group of G1P. Thus, given the preferential formation of G2PAs in these reactions in solution, we sought to determine whether acylation reactions in the dry state are subject to the same bias.
:4:1, v/v/v), and the formation of phosphatidic acids was verified by 31P NMR and TLC as before. In contrast to the results obtained in solution, a broader range of products was produced in the dry state. On acylation of G1P, MDG1PA and DDG1PA were obtained in yields of 15% and 4%, respectively, alongside G2P (2%), MDG2PA (2%), DDG2PA (1%), glycerol-1,2-cyclic phosphate (G>P, 16%), and 3-monodecanoyl glycerol-1,2-cyclic phosphate (MDG>P, 6%) (Fig. 1b and S5†). The positional isomers, G2P and MDG2PA probably form on hydrolysis of G>P and MDG>P, respectively,13,29 or by phosphoryl migration under the ultimately acidic conditions of the reaction.26 Similarly, the reaction of G2P afforded MDG2PA and DDG2PA in yields of 14% and 4%, respectively, along with the same array of products as obtained in the reaction starting from G1P (Fig. 1b and S6†). Finally, the reaction of a 1:1 mixture of G1P and G2P gave comparable amounts of G1PAs and G2PAs (combined yields of 13% and 10%, respectively) (Fig. 1a–c and S7†).
It is difficult to be certain of the mechanism of the acylation reaction in the dry state, but it is possible it involves direct attack of the acylating agent on the hydroxyl groups of each glycerol phosphate. This proposal would account for the comparable yields of phosphatidic acids obtained on acylation of each glycerol phosphate in the dry state. To test this idea, we subjected sn-glycero-3-phosphocholine (G3PC)30 to acylation in the dry state and in solution. This substrate was chosen as its phosphodiester group is not expected to participate in the reaction. On reaction of G3PC with ammonium decanoate, cyanamide, and imidazole in the dry state, 1(2)-monodecanoyl-sn-glycero-3-phosphocholine (MDG3PC, −0.20 and −0.38 ppm) and 1,2-didecanoyl-sn-glycero-3-phosphocholine (DDG3PC, −0.68 ppm) were obtained in a yields of 10% and 11%, respectively, together with glycero-2-phosphocholine (5%), and traces (<1%, combined) of MDG>P and G>P (Fig. S8†). By contrast, the reaction of G3PC with NDI in solution produced only MDG3PC in 4% yield (Fig. S9†). These results show that a free phosphate group is not required for acylation in the dry state, but is required for efficient acylation in solution.
Mechanism aside, our data show that dry state acylation reactions of G1P and G2P proceed without bias and afford G1PAs and G2PAs in yields proportional to the amount of each glycerol phosphate present. This result is significant as under certain conditions21,22G1P is the major product obtained on prebiotic phosphorylation of glycerol. Moreover, acylation in the dry state occurs under arguably more realistic early Earth conditions (the activated fatty acid is generated in situ and the reaction requires no organic solvent). However, even in a best-case scenario where only G1P was available, prebiotic acylation of glycerol phosphates in the dry state would have produced both natural G1PAs and non-natural G2PAs.
:1, v/v) or in the form of liposomes in carbonate buffer. The extent of hydrolysis of each lipid was greater in solution than in liposomes at every time point, as observed for other primitive amphiphiles,33 but differences between the extents of hydrolysis of DDG1PA and DDG2PA in solution and in liposomes were minimal (Fig. S10†). For example, after 11 days in solution 87% of DDG1PA and 87% of DDG2PA were hydrolyzed, while in liposomes 35% of DDG1PA and 34% of DDG2PA were hydrolyzed. Similar hydrolysis experiments were performed at pH 7 (imidazole buffer) and pH 4 (acetate buffer), but slow hydrolysis (data not shown) and precipitation of the lipid (see below) prevented us from observing any differences. These results suggest that, in contrast to the possible emergence of amino alcohol bearing phospholipids,13 it is not likely that the selection of natural phosphatidic acids on early Earth resulted from hydrolysis.
Natural and non-natural phosphatidic acids exhibit different supramolecular properties
As G1PAs and G2PAs form stable liposomes in aqueous buffers,14,16 we sought to identify whether differences in their respective supramolecular properties could have led to the selection of natural G1PAs. Microscopy observations showed that DDG1PA and DDG2PA form liposomes of similar size and lamellarity in phosphate buffer (50 mM, pH 7.5) (Fig. 2a).14,16 Yet, the biophysical properties of these membranes remain unknown. Previous NMR and neutron diffraction studies on 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPG3PC) and its isomer 1,3-dipalmitoyl-sn-glycero-2-phosphocholine (DPG2PC) showed that the two phospholipids differ in their gel-to-liquid transition temperature (41.5 °C for DPG3PCvs. 37.5 °C for DPG2PC), probably due to the different conformations of their glycerol backbones (perpendicular and parallel to the membrane surface for DPG3PC and DPG2PC, respectively).34 We thus investigated whether natural and non-natural phosphatidic acids also acquire a different spatial organization within their respective bilayer. Accordingly, liposomes prepared from DDG1PA and DDG2PA labelled with the fluorescent probe 1,6-diphenyl-1,3,5-hexatriene (DPH) were studied by fluorescence anisotropy. DPH is an apolar dye that localizes in the hydrophobic region of the lipid bilayer and so fluorescence anisotropy measurements provide information on the freedom of motion of the dye within the membrane, the viscosity of the environment in which the dye is embedded, and hence the fluidity of the membrane.35 Our data show that DDG1PA and DDG2PA form liposomes with membranes as fluid as those composed of unsaturated fatty acids, such as myristoleic acid (MA) (Fig. 2b), but that DDG1PA membranes are more rigid than DDG2PA membranes. Moreover, when DDG1PA and DDG2PA liposomes were labelled with Laurdan, an amphiphilic dye sensitive to lipid packing and order within the bilayer,36 generalized polarization studies showed that the DDG1PA membranes are more ordered and better packed than DDG2PA membranes, indicative of stronger hydrophobic interactions between the acyl chains (Fig. 2c and S11†). The fluorescence anisotropy and generalized polarization measurements were made at 25 °C, a temperature expected to be above the gel-to-fluid phase transition temperature of both lipids. These findings confirm that natural and non-natural phosphatidic acids acquire different conformations within their respective bilayers (Fig. 2d), as shown previously for long chain phosphatidylcholines.34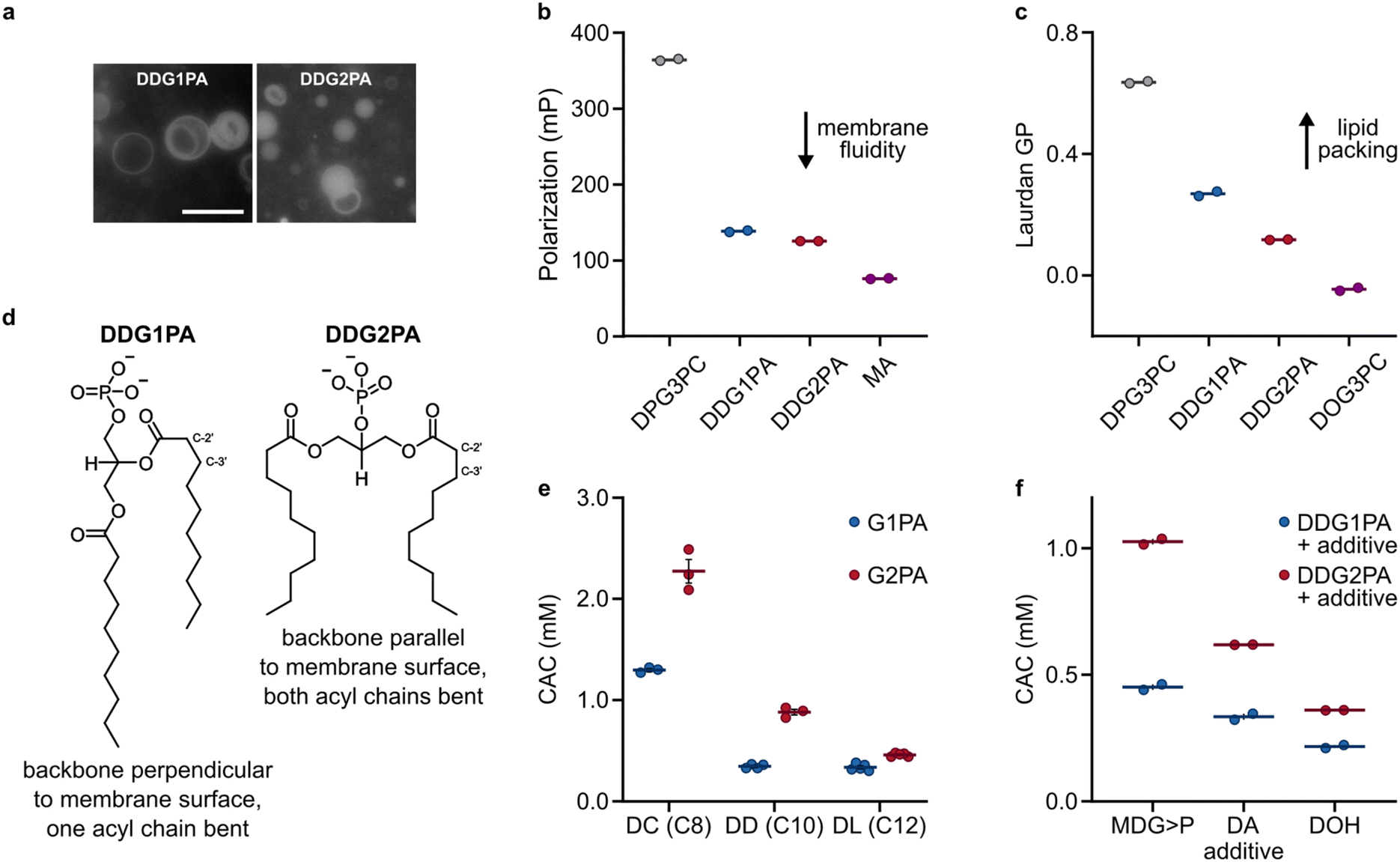 | ||
| Fig. 2 Self-assembly and supramolecular properties of G1PAs and G2PAs in pure or mixed systems. (a) Fluorescence microscopy images show that DDG1PA and DDG2PA (25 mM, containing 1 mol% NBD-PE) form liposomes of similar size and lamellarity. The scale bar represents 10 μm. (b) DPH fluorescence anisotropy (polarization) of DDG1PA and DDG2PA membranes compared with those of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPG3PC) and MA. Measurements were made at 25 °C. (c) Laurdan generalized polarization (GP) of DDG1PA and DDG2PA membranes compared with those of DPG3PC and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOG3PC). Measurements were made at 25 °C. (d) Structural and conformational differences between DDG1PA and DDG2PA in membranes. (e) CACs of G1PAs and G2PAs of different chain length. (f) CACs of mixtures of DDG1PA or DDG2PA and other C10-amphiphiles. Data show the mean ± SEM, n ≥ 2. See the ESI† for details. | ||
To determine whether natural phosphatidic acids exhibit a greater propensity to self-assemble compared to their non-natural analogues, we synthesized the C8-lipids, 2,3-dicapryloyl-glycerol-1-phosphatidic acid (DCG1PA) and 1,3-dicapryloyl-glycerol-2-phosphatidic acid (DCG2PA), and the C12-lipids, 2,3-dilauroyl-glycerol-1-phosphatidic acid (DLG1PA) and 1,3-dilauroyl-glycerol-2-phosphatidic acid (DLG2PA) (Fig. S12–S17†).31 With this series of isomeric phospholipids of different chain length in hand, we measured the critical aggregation concentration (CAC) of each species, i.e. the minimal concentration of lipid needed to form supramolecular aggregates (e.g. liposomes). Liposomes composed of G1PA or G2PA were prepared by lipid film rehydration in phosphate buffer at pH 7.5, and absorbance spectra were recorded at different lipid concentrations using the solvatochromic probe merocyanine 540, as described previously.33 Interestingly, we found that without exception the CAC of each natural G1PA is lower than that of each non-natural G2PA of the same chain length (Fig. 2e). This trend was conserved when G1PA and G2PA liposomes were prepared in other buffers or pure water at different pH values (Fig. S18†). Furthermore, the CACs of mixtures containing DDG1PA and other prebiotic amphiphiles, specifically decanoic acid (DA), decanol (DOH), and 3-monodecanoyl-glycerol-1,2-cyclic phosphate (MDG>P), are consistently lower than those of analogous mixtures containing DDG2PA (Fig. 2f). Finally, to see if CACs of natural phospholipids of other types are also lower than those of their non-natural isomers, we measured the CACs of 2,3-didecanoyl-rac-glycerol-1-phosphocholine (DDG1PC) and 1,3-didecanoyl-glycerol-2-phosphocholine (DDG2PC). These lipids were prepared from their respective phosphatidic acid precursors (Fig. S19 and S20†).37 In line with our observations for phosphatidic acids, we found that DDG1PC has a CAC approximately half that of DDG2PC (38 ± 5 μm vs. 71 ± 2 μm, respectively) (Fig. S21†). Taken together, these results suggest that the tendency of natural phospholipids to self-assemble at lower concentrations than non-natural analogues of the same chain length is conserved across different headgroup types.
Differences in supramolecular properties enable the selection of natural phosphatidic acids in primitive membranes
In a prebiotic scenario where G1P and G2P were acylated in the same environment, both natural G1PAs and non-natural G2PAs would have formed and co-assembled into mixed liposomes. The selection of natural phospholipids would therefore have necessitated the loss of the non-natural isomers from liposomes, either by dilution in the aqueous medium or by precipitation, as a result of their different supramolecular properties.:1, 5 mM each) was used to form liposomes, no precipitate was observed. Thus, the stabilizing effect of DDG1PA in mixed liposomes, previously inferred from CAC measurements (Fig. 2), probably results from its ability to maintain a hydrogen bonding network with DDG2PA headgroups at pH 4.38,39 Therefore, despite the contrasting pH tolerance of the individual lipids, our observations seemingly rule out a purely pH-driven selection process for natural phospholipids from heterogeneous mixtures.
To establish the relative Mg2+-tolerance of the isomeric phosphatidic acids, liposomes made of 5 mM DDG1PA or DDG2PA were assembled in 50 mM phosphate buffer and titrated with MgCl2. Our data show that DDG2PA liposomes are particularly sensitive to Mg2+ and precipitation of the lipid was quantified by 31P NMR (99% loss after addition of 5 mM Mg2+) (Fig. S25†). By contrast, DDG1PA liposomes are largely unaffected by Mg2+ (16% loss after addition of 5 mM Mg2+) (Fig. S26†). Similar observations were made by epifluorescence microscopy using a higher lipid concentration (25 mM), where precipitation of DDG2PA was observed at 7.5 mM MgCl2 (Fig. 3a). The concentration of Mg2+ required to induce precipitation is proportional to the concentration of the lipid. Analogously, DDG2PA liposomes were more susceptible to precipitation by CaCl2 and ZnCl2 than DDG1PA liposomes (Fig. S27–S30†). Finally, by comparison, neither lipid was susceptible to precipitation on addition of up to 100 mM NaCl or KCl, as expected for monovalent cations.43
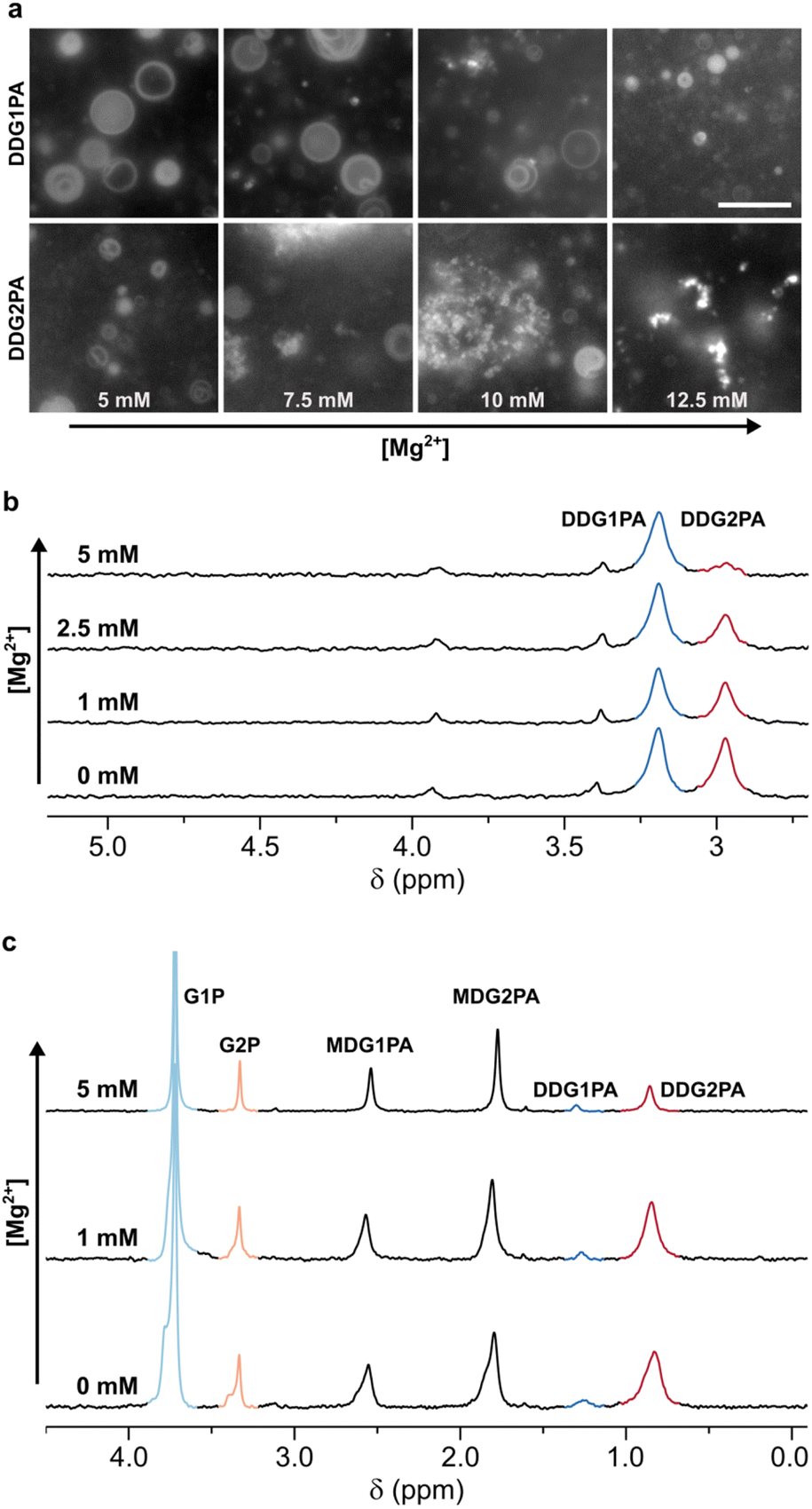 | ||
| Fig. 3 Mg2+-driven selection of natural G1PAs over non-natural G2PAs. (a) Microscopy images showing the relative Mg2+-tolerance of DDG1PA and DDG2PA liposomes (25 mM). The scale bar represents 10 μm. DDG1PA liposomes remain intact while DDG2PA liposomes disassemble following aggregation and precipitation of the lipid. The concentration of Mg2+ required to induce precipitation is proportional to the concentration of the lipid. (b) 31P{1H} NMR spectra showing the change in composition of mixed liposomes prepared from DDG1PA and DDG2PA (1:1, 5 mM each) on titration with Mg2+. (c) 31P{1H} NMR spectra showing the change in composition of mixed liposomes prepared from G1PAs and G2PAs obtained by prebiotic synthesis on titration with Mg2+. DDG2PA precipitates from mixed liposomes on titration with Mg2+, while DDG1PA remains in liposomes. See the ESI† for details. | ||
To probe the greater tolerance of DDG1PA over DDG2PA to Mg2+, we measured the change in membrane ζ-potential of liposomes made of DDG1PA or DDG2PA (5 mM each) on titration with MgCl2. In the absence of Mg2+, the ζ-potentials of DDG1PA and DDG2PA liposomes were −56 mV and −74 mV, respectively (Fig. S31†). The more negative surface potential of DDG2PA suggests that its phosphate group is less hydrated and hence forms weaker hydrogen bonding interactions in the headgroup space of the bilayer compared to DDG1PA. Further, it is possible that the phosphate group of DDG2PA is more exposed to the bulk medium than that of DDG1PA, as might be expected since DDG2PA forms less rigid and less well packed membranes than DDG1PA (Fig. 2c). On addition of Mg2+ to liposomes composed of each lipid, we observed a gradual increase in ζ-potential, demonstrating the electrostatic interactions of Mg2+ with the anionic headgroups and its charge shielding effect (Fig. S31†).43
Intrigued by the lower tolerance of DDG2PA over DDG1PA to divalent cations, we tested the stability of mixed liposomes made of DDG1PA and DDG2PA (1:1, 5 mM each, prepared in 50 mM phosphate buffer, pH 7.5) in the presence of 5 mM Mg2+. In the event, the sample remained turbid despite the immediate formation of a precipitate.
To determine the composition of the remaining liposomes, the precipitate was removed by filtration, the liposomes were dissolved by addition of 0.2 M Triton X-100,44 and the resulting solution was analyzed by 31P NMR (Fig. 3b and S32†). DDG1PA was found to be four times more abundant than DDG2PA in the filtrate, demonstrating that selective precipitation of the non-natural DDG2PA occurred on addition of Mg2+. This finding suggests that a Mg2+-driven selection process could have led to the enrichment of natural phosphatidic acids in mixed liposomes. Moreover, Mg2+ appears unique among the metal ions studied as selective precipitation of DDG2PA from mixed liposomes could not be effected by titration with Ca2+, Zn2+ (Fig. S33 and S34†), Na+ or K+ (data not shown).
The mechanism that underpins the Mg2+-induced selection from mixed membranes can be understood in terms of the electrostatic interactions between Mg2+ and the anionic surface of the membrane and the differential binding of the isomeric lipids. By analogy with the effect of Ca2+ on natural phospholipids,45 Mg2+ binding to phosphatidic acids would liberate water of hydration from the membrane, leading to tighter packing of the lipids and ultimately precipitation. Our data show that DDG2PA membranes have a more negative surface charge than those of DDG1PA and suggest that the headgroups of DDG2PA are less well hydrated and so more prone to disruption on dehydration. It is also possible that the binding affinity of Mg2+ for DDG2PA is greater than that for DDG1PA and the difference in binding affinities is enough to enable the selective precipitation of DDG2PA in the presence of DDG1PA, in contrast to the other metal ions tested. Whether the selective precipitation of DDG2PA from mixed membranes suggests that phase separation into isomeric lipid domains occurs prior to precipitation remains an intriguing possibility.
Finally, to demonstrate the plausibility of the Mg2+-driven selection process on lipid mixtures obtained by prebiotic synthesis, we prepared a mixture of G1PAs and G2PAs by acylation of G1P and G2P with NDI, as described above (Fig. 1). The mixture of phospholipids obtained after 8 days (MDG1PA, MDG2PA, DDG1PA, and DDG2PA) was lyophilized, then gently hydrated with water at pH 7.0 to give liposomes. The imidazole released on reaction of NDI served as the buffer. On titration with MgCl2, DDG2PA precipitated and the surviving liposomes became proportionally enriched in DDG1PA (31P NMR, Fig. 3c and Fig. S35–S37†). Interestingly, the lysophospholipids MDG1PA and MDG2PA were unaffected by Mg2+ up to a concentration of 5 mM, yet these species could undergo further acylation and thus Mg2+-induced precipitation.
Overall, these results highlight the unique role of Mg2+ in driving the selection of G1PAs from heterogeneous mixtures of phospholipids under conditions compatible with key metal-driven prebiotic processes like non-enzymatic RNA polymerisation.15
Conclusion
Due to the heterogeneity of prebiotic building blocks present on early Earth, efficient mechanisms would have been required for the selection of the now canonical biomolecules. In relation to the emergence of phospholipids, recent studies showed that the selection of natural phospholipid headgroups could have resulted from the preferential hydrolysis of their non-natural analogues13 and that liposomes composed of non-natural phosphatidic acids are poorly tolerant to pH changes and metal ions.14,16 However, no prior study addressed whether processes operating at the molecular level (prebiotic synthesis) or the supramolecular level (membrane properties) could have driven the selection of natural phospholipids from heterogeneous mixtures available on early Earth. Our work addresses these questions.First, we show that acylation of mixtures of glycerol phosphates in solution affords non-natural G2PAs in higher yields than natural G1PAs, but acylation in the dry state provides G1PAs and G2PAs in similar yields. These isomeric phospholipids are comparably stable to hydrolysis. Second, we highlight that natural G1PAs possess a greater propensity to self-assemble and to pack in ordered bilayers, compared to non-natural G2PAs, and that they form liposomes of enhanced rigidity and broader tolerance to pH and metal ions. Finally, we demonstrate that Mg2+ drives the selective precipitation of G2PAs and, thus, the accumulation of G1PAs in liposomes composed of mixtures of phospholipids obtained by prebiotic synthesis. The effect of Mg2+ is unique among the metal ions tested. Taken together, our results suggest that properties that manifest at the supramolecular level can be harnessed for the selection of natural phospholipids, while processes that operate at the molecular level (synthesis and hydrolysis) probably cannot.
In summary, our work outlines prebiotic synthesis and selection pathways that could have driven the transition towards biological phospholipid membranes on early Earth. The phosphatidic acids focused on here are minor constituents of biological membranes but serve as important biosynthetic intermediates in pathways leading to complex lipids, such as phosphatidylethanolamines and phosphatidylcholines. Since the transformation of G1PAs into both of these lipids under early Earth conditions has already been described,46,47 our study renders these foundational investigations all the more relevant. Importantly, the requirements of Mg2+-driven non-enzymatic RNA replication and the need for Mg2+-stable biological phospholipid liposomes can be harmonized. In fact, Mg2+ is essential in driving the selection of metal ion-resistant natural phospholipids. Beyond, the interactions between Mg2+ ions and membranes enriched in natural phosphatidic acids could have led to the co-localization of RNA on the membrane and enabled synergistic effects in prebiotic RNA chemistry48 as well as RNA-driven recognition and fusion of primitive liposomes.49
Data availability
Data are available upon reasonable request from the corresponding author.Author contributions
K. T. M., Y. L., V. J. B.-T. and D. A. R. carried out the experimental work. D. A. R. and C. B. conceived the project, directed the work and wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was funded by the Foundation Jean-Marie Lehn, the University of Strasbourg Institute for Advanced Study (USIAS) and the Agence Nationale de la Recherche (ITI SysChem within the Investissement d’Avenir program ANR-10-IDEX-0002, and AAP IdEx 2022 Attractivité to C. B.), and the CSC Graduate School, funded by the Agence Nationale de la Recherche (CSC-IGS ANR-17-EURE-0016 to K. T. M. and V. J. B. T.). The authors gratefully acknowledge support from Foundation Jean-Marie Lehn, the University of Strasbourg Institute for Advanced Study and the Agence Nationale de la Recherche. The authors thank colleagues at ISIS and lab members for stimulating discussions.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 2014 Search PubMed.
- J. C. Blain and J. W. Szostak, Annu. Rev. Biochem., 2014, 83, 615–640 CrossRef CAS PubMed.
- J. Xu, V. Chmela, N. J. Green, D. A. Russell, M. J. Janicki, R. W. Góra, R. Szabla, A. D. Bond and J. D. Sutherland, Nature, 2020, 582, 60–66 CrossRef CAS PubMed.
- J. Xu, N. J. Green, D. A. Russell, Z. Liu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 14482–14486 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- A. C. Rios and Y. Tor, Isr. J. Chem., 2013, 53, 469–483 CrossRef CAS PubMed.
- M. W. Powner and J. D. Sutherland, ChemBioChem, 2008, 9, 2386–2387 CrossRef CAS PubMed.
- R. A. Sanchez and L. E. Orgel, J. Mol. Biol., 1970, 47, 531–543 CrossRef CAS PubMed.
- S. Bhowmik and R. Krishnamurthy, Nat. Chem., 2019, 11, 1009–1018 CrossRef CAS PubMed.
- K. Ashe, C. Fernández-García, M. K. Corpinot, A. J. Coggins, D.-K. Bučar and M. W. Powner, Commun. Chem., 2019, 2, 1–7 CrossRef CAS.
- M. Frenkel-Pinter, J. W. Haynes, M. C, A. S. Petrov, B. T. Burcar, R. Krishnamurthy, N. V. Hud, L. J. Leman and L. D. Williams, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16338–16346 CrossRef CAS PubMed.
- M. Makarov, A. C. Sanchez Rocha, R. Krystufek, I. Cherepashuk, V. Dzmitruk, T. Charnavets, A. M. Faustino, M. Lebl, K. Fujishima, S. D. Fried and K. Hlouchova, J. Am. Chem. Soc., 2023, 145, 5320–5329 CrossRef CAS PubMed.
- M. Aleksandrova, F. Rahmatova, D. A. Russell and C. Bonfio, J. Am. Chem. Soc., 2023, 145, 25614–25620 CrossRef CAS PubMed.
- S. Pulletikurti, K. S. Veena, M. Yadav, A. A. Deniz and R. Krishnamurthy, Chem, 2024, 10, 1839–1867 CAS.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122–125 CrossRef CAS PubMed.
- C. Bonfio, C. Caumes, C. D. Duffy, B. H. Patel, C. Percivalle, M. Tsanakopoulou and J. D. Sutherland, J. Am. Chem. Soc., 2019, 141, 3934–3939 CrossRef CAS PubMed.
- D. E. Epps, E. Sherwood, J. Eichberg and J. Oró, J. Mol. Evol., 1978, 11, 279–292 CrossRef CAS PubMed.
- D. Fayolle, E. Altamura, A. D'Onofrio, W. Madanamothoo, B. Fenet, F. Mavelli, R. Buchet, P. Stano, M. Fiore and P. Strazewski, Sci. Rep., 2017, 7, 18106 CrossRef PubMed.
- W. R. Hargreaves, S. J. Mulvihill and D. W. Deamer, Nature, 1977, 266, 78–80 CrossRef CAS PubMed.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- D. J. Ritson and J. D. Sutherland, Nat. Chem., 2023, 15, 1470–1477 CrossRef CAS PubMed.
- D. E. Epps, D. W. Nooner, J. Eichberg, E. Sherwood and J. Oró, J. Mol. Evol., 1979, 14, 235–241 CrossRef CAS PubMed.
- C. Gibard, S. Bhowmik, M. Karki, E.-K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10, 212–217 CrossRef CAS PubMed.
- O. R. Maguire, I. B. A. Smokers and W. T. S. Huck, Nat. Commun., 2021, 12, 5517 CrossRef CAS PubMed.
- B. Burcar, M. Pasek, M. Gull, B. J. Cafferty, F. Velasco, N. V. Hud and C. Menor-Salv, Angew. Chem., Int. Ed., 2016, 55, 13249–13253 CrossRef CAS PubMed.
- A. Plueckthun and E. A. Dennis, Biochemistry, 1982, 21, 1743–1750 CrossRef CAS PubMed.
- E. K. Ryu and M. MacCoss, J. Lipid Res., 1979, 20, 561–563 CrossRef CAS.
- L. Kugel and M. Halmann, J. Am. Chem. Soc., 1967, 89, 4125–4128 CrossRef CAS.
- V. E. Ortuno, S. Pulletikurti, K. S. Veena and R. Krishnamurthy, Chem. Commun., 2022, 58, 6231–6234 RSC.
- H. Brockerhoff and M. Yurkowsky, Can. J. Biochem., 1965, 43, 1777 CrossRef CAS PubMed.
- C. M. Gupta, R. Radhakrishnan and H. G. Khorana, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4315–4319 CrossRef CAS PubMed.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS PubMed.
- C. Bonfio, D. A. Russell, N. J. Green, A. Mariani and J. D. Sutherland, Chem. Sci., 2020, 11, 10688–10697 RSC.
- G. Buldt and G. H. de Haas, J. Mol. Biol., 1982, 158, 55–71 CrossRef CAS PubMed.
- B. J. Litman and Y. Barenholz, in Methods in Enzymology, Academic Press, 1982, vol. 81, pp. 678–685 Search PubMed.
- T. Parasassi, G. De Stasio, G. Ravagnan, R. M. Rusch and E. Gratton, Biophys. J., 1991, 60, 179–189 CrossRef CAS PubMed.
- G. S. Harbison and R. G. Griffin, J. Lipid Res., 1984, 25, 1140–1142 CrossRef CAS.
- A. Blume and H. Eibl, Biochim. Biophys. Acta, 1979, 558, 13–21 CrossRef CAS PubMed.
- H. Eibl and A. Blume, Biochim. Biophys. Acta, 1979, 553, 476–488 CrossRef CAS PubMed.
- P. Dalai, P. Ustriyana and N. Sahai, Geochim. Cosmochim. Acta, 2018, 223, 216–228 CrossRef CAS.
- C. Bonfio, E. Godino, M. Corsini, F. Fabrizi de Biani, G. Guella and S. S. Mansy, Nat. Catal., 2018, 1, 616–623 CrossRef CAS.
- K. B. Muchowska, S. J. Varma and J. Moran, Nature, 2019, 569, 104–107 CrossRef CAS PubMed.
- H. Träuble and H. Eibl, Proc. Natl. Acad. Sci. U.S.A., 1974, 71, 214–219 CrossRef PubMed.
- E. London and G. W. Feigenson, J. Lipid Res., 1979, 20, 408–412 CrossRef CAS.
- H. Hauser, Chem. Phys. Lipids, 1991, 57, 309–325 CrossRef CAS PubMed.
- M. Rao, J. Eichberg and J. Oró, J. Mol. Evol., 1982, 18, 196–202 CrossRef CAS PubMed.
- M. Rao, J. Eichberg and J. Oró, J. Mol. Evol., 1987, 25, 1–6 CrossRef CAS PubMed.
- T. Czerniak and J. P. Saenz, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119235119 CrossRef PubMed.
- J. A. Peruzzi, M. L. Jacobs, T. Q. Vu, K. S. Wang and N. P. Kamat, Angew. Chem., Int. Ed., 2019, 58, 18683–18690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05362a |
| This journal is © The Royal Society of Chemistry 2024 |