
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of N-perfluoro-tert-butyl secondary amines from N-trifluoromethyl secondary amines†
Leibing
Wang
 a,
Zhongyu
Feng
a,
Zhen
Luo
a,
Zihao
Guo
*a,
Jieping
Wang
*a and
Wenbin
Yi
*ab
a,
Zhongyu
Feng
a,
Zhen
Luo
a,
Zihao
Guo
*a,
Jieping
Wang
*a and
Wenbin
Yi
*ab
aSchool of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: zihao_guo@njust.edu.cn; jieping.wang@njust.edu.cn; yiwb@njust.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 6th November 2024
Abstract
N-Perfluoro-tert-butyl (N-PFtB) secondary amines, harboring a unique 19F-reporting moiety linked directly to nitrogen, are highly attractive due to their diverse potential applications. However, their mild and facile synthesis remains a significant challenge. Herein, we present a safe and efficient strategy for the direct synthesis of N-perfluoro-tert-butyl secondary amines from readily available N-trifluoromethyl secondary amines. Experiments and theoretical calculations demonstrate that this novel protocol encompasses three main processes: the elimination of hydrogen fluoride from the N-trifluoromethyl precursor, consecutive addition–elimination conversion of difluoromethyl imine (R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) to hexafluoropropyl imine (R–NC(CF3)2), and final addition of R–NC(CF3)2 with the in situ generated fluoroform (HCF3). Key advantages of this reaction include the utilization of a single trifluoromethyl source and the N-trifluoromethyl starting material itself as the hydrogen source. Notably, the elimination of hydrogen fluoride, facilitated by CsF, is critical for the success of this approach. This method is compatible with a broad range of functional groups, including heterocyclic compounds. 19F MRI experiments suggest promising prospects for PFtB-labeled secondary amines as 19F MRI contrast agents.
CF2) to hexafluoropropyl imine (R–NC(CF3)2), and final addition of R–NC(CF3)2 with the in situ generated fluoroform (HCF3). Key advantages of this reaction include the utilization of a single trifluoromethyl source and the N-trifluoromethyl starting material itself as the hydrogen source. Notably, the elimination of hydrogen fluoride, facilitated by CsF, is critical for the success of this approach. This method is compatible with a broad range of functional groups, including heterocyclic compounds. 19F MRI experiments suggest promising prospects for PFtB-labeled secondary amines as 19F MRI contrast agents.
Introduction
Fluorine chemistry has seen significant advancements in recent years, with applications emerging in biomedicine1,2 and materials science.3,4 Synthetic methods for mono-, di-, and trifluoromethylation are now well-established and highly sophisticated.5 Alongside these developments, short-chain perfluoroalkylation methodologies have garnered increasing attention due to the unique properties of perfluoroalkyl-containing compounds,6–10 which hold considerable promise in therapeutic and diagnostic applications.11–13 Perfluoro-tert-butyl (PFtB) is particularly valuable in this context, playing a crucial role in 19F-labeled NMR9,14 and MRI probes,15–17 as well as 19F MRI contrast agents.18,19 PFtB offers a distinct and singular 19F signal, providing significant advantages in imaging. Not only does 19F sensitivity rival that of 1H, but its signal originates solely from the imaging agent, eliminating interference from background signals. Evidently, the growing demand for perfluorinated compounds in these fields underscores the importance of methodological studies for PFtB group synthesis.Given the ubiquitous role of nitrogen in drug molecules, compounds combining nitrogen with the PFtB group, particularly N-PFtB secondary amines, are highly attractive and hold significant promise. However, the lack of a general synthesis method has resulted in limited reports of such compounds to date. The existing route developed by Petrov involves obtaining R–NHC(CF3)3 from the hexafluoropropyl imine intermediate (R–NC(CF3)2) following addition and hydrochloric acid treatment.20 This intermediate is typically derived from conventional reactions involving gaseous hexafluoroacetone with aromatic primary amines or isocyanates (Fig. 1A).21,22 However, these procedures suffer from some drawbacks, including the use of highly toxic gases, multiple steps, and demanding operations,23–25 which significantly hinder the exploration and application of these compounds. Therefore, overcoming these challenges and developing a general strategy for the safe and efficient construction of N-PFtB secondary amines is critical.
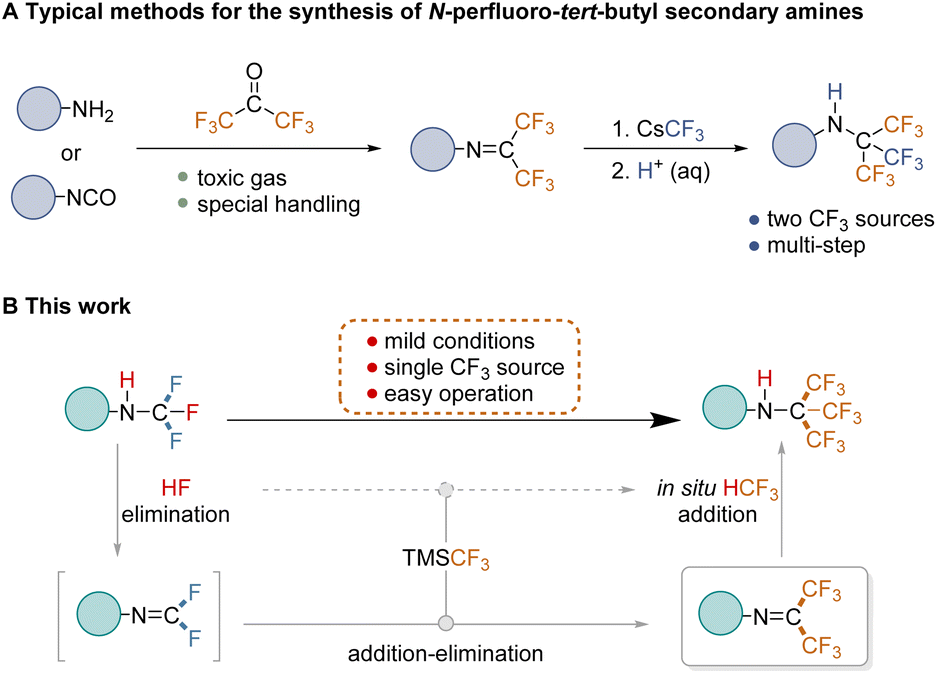 | ||
| Fig. 1 Previous work and reaction design. (A) Traditional approaches to synthesizing N-PFtB secondary amines; (B) our designed strategy for the construction of N-PFtB secondary amines. | ||
To the best of our knowledge, the conversion from difluoromethyl imine (R–NCF2) to hexafluoropropyl imine (R–NC(CF3)2) remains unreported. We propose this conversion as a significantly safer and more efficient approach for constructing multi-CF3 amines, eliminating the need for highly toxic gases. Our strategy leverages a readily available N-CF3 secondary amine as the starting material.26 This amine undergoes elimination of HF to generate the highly reactive difluoromethyl imine. Subsequently, in the presence of a CF3 source, this imine undergoes two consecutive addition–elimination processes to form hexafluoropropyl imine. Finally, the target N-PFtB secondary amine is obtained through the addition reaction of hexafluoropropyl imine with trifluoromethyl and hydrogen (Fig. 1B). Experimental and theoretical calculations support the notion that the eliminated HF reacts further with TMSCF3, in situ producing HCF3 which subsequently participates in the addition reaction with hexafluoropropyl imine. The key advantages of this novel strategy lie in its reliance on a single CF3 source and the starting material N-CF3 secondary amine as the hydrogen source, enabling a one-step reaction under mild conditions.
Results and discussion
To evaluate the proposed reaction pathway, we employed 4-cyanophenyl N-trifluoromethyl secondary amine as a model substrate. The substrate was treated with 8.0 equivalents of TMSCF3 and 1.0 equivalent of KF initiator in THF at room temperature for 6 hours. The desired product (compound 1) was isolated in a 33% yield (Table 1, entry 1). Subsequent attempts to improve the yield by individually or concurrently increasing the amounts of TMSCF3 or KF resulted in only modest improvement. Similarly, the addition of CuCl exhibited a negligible effect on the yield (entries 2–6). Notably, the absence of KF or its substitution with NaF or TBAF (tetrabutylammonium fluoride) completely ablated product formation (entries 7–9). Further investigation revealed that cesium fluoride (CsF) was crucial for this reaction system. When 8.0 equivalents of TMSCF3 were combined with 1.0 equivalent of CsF in the presence of the substrate, the yield remarkably increased to 65% (entry 10). Employing 1.5 equivalents of CsF led to a further enhancement in product yield (entry 11). However, increasing the CsF amount to 3.0 equivalents resulted in an inhibitory effect (increase in unknown by-products, entry 12). Likewise, decreasing the CsF quantity to 0.5 equivalents yielded a slightly lower product formation (generation of a dimer, entry 13) (see ESI, p. S4†). These observations collectively suggest that the liberated F− anion, after being displaced by CF3− during the reaction, participates again by reacting with TMSCF3 to release another CF3−. Among the tested solvents, THF provided the most favorable outcome (entries 14–15). Finally, the optimal reaction conditions were established by finetuning the ratio of TMSCF3 and CsF (entry 16).|
|
||||
|---|---|---|---|---|
| Entry | TMSCF3 (equiv.) | Additive (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: N-CF3 secondary amine (0.8 mmol, 1.0 equiv.), solvent (4.0 mL). b Isolated yields. | ||||
| 1 | 8 | KF (1) | THF | 33 |
| 2 | 8 | KF (3) | THF | 38 |
| 3 | 8 | KF (5), CuCl (1.0) | THF | 54 |
| 4 | 8 | KF (3), CuCl (1.0) | THF | 47 |
| 5 | 10 | KF (5) | THF | 43 |
| 6 | 10 | KF (3) | THF | 42 |
| 7 | 8 | — | THF | 0 |
| 8 | 8 | NaF (3.0) | THF | 0 |
| 9 | 8 | TBAF (3.0) | THF | 0 |
| 10 | 8 | CsF (1.0) | THF | 65 |
| 11 | 8 | CsF (1.5) | THF | 73 |
| 12 | 8 | CsF (3.0) | THF | 67 |
| 13 | 8 | CsF (0.5) | THF | 60 |
| 14 | 8 | CsF (1.5) | DCM | 23 |
| 15 | 8 | CsF (1.5) | MeCN | 41 |
| 16 | 6 | CsF (1.5) | THF | 76 |
| 17 | 10 | CsF (3.0) | THF | 57 |

To elucidate the reaction mechanism, we initially employed the N-CH3 analog of the starting substrate under standard conditions (Fig. 2A). This experiment aimed to preliminarily determine the reaction pathway. Notably, no signal corresponding to the anticipated product was detected by GC-MS or 19F NMR analysis. This observation suggests that the N–H bond in the N-CF3 secondary amine plays a critical role in the reaction, and a simple conversion of the three C–F bonds to C–CF3 bonds is not the operative mechanism. Secondly, the presence of a fluoroform (HCF3)27 signal was identified in the 19F NMR spectrum after 30 minutes of reaction time (Fig. 2B), although a very small amount of HCF3 is formed in the absence of substrate (see ESI, p. S7†). This finding implies the potential involvement of HF elimination during the reaction. Finally, we investigated the reaction under conditions employing a catalytic amount of CsF (0.2 equivalents) and 2.0 equivalents of TMSCF3 (Fig. 2C). Using GC-MS analysis, we observed a mixture of mono-, di-, and tri-substituted trifluoromethylated products (see ESI, p. S9†).
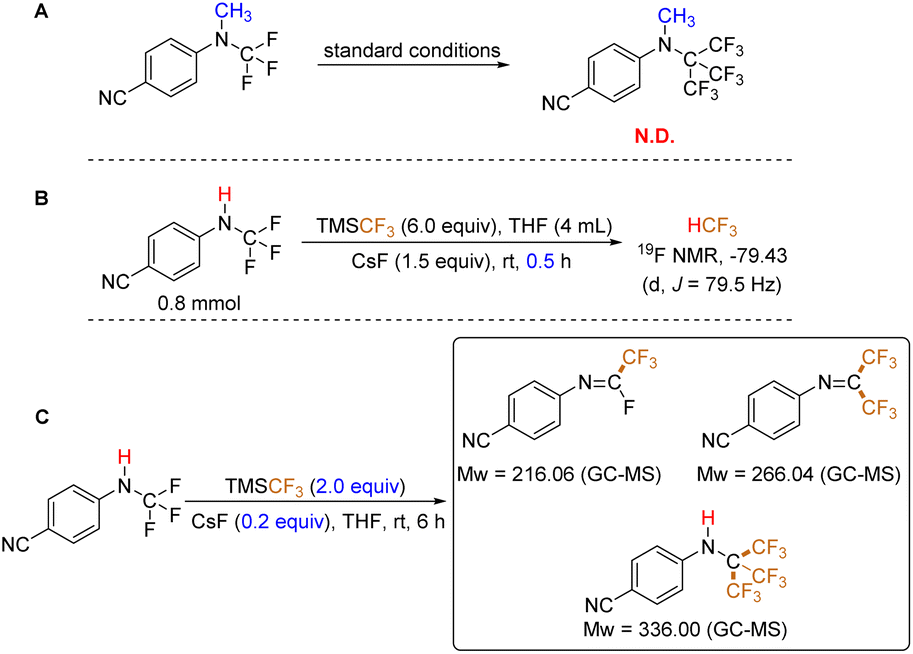 | ||
| Fig. 2 Preliminary mechanistic investigations. | ||
Density Functional Theory (DFT) calculations were employed to investigate the reaction mechanism (detailed in the ESI, p. S12†). The mechanism can be broadly divided into three key processes: firstly, CsF facilitates the removal of hydrogen fluoride (HF), forming CsHF2; secondly, two sequential addition–elimination processes occur, transforming C–F bonds into C–CF3 bonds; and finally, the resulting intermediate reacts with the in situ generated fluoroform (HCF3) at the NC double bond, restoring the hydrogen atom to its original position. The reaction energy barrier diagram is presented in Fig. 3. The DFT calculations reveal that CsF promotes the initial HF elimination process by forming intermediate IM-1 (−12.2 kcal mol−1) with a significantly lower energy barrier compared to direct HF elimination (kinetic barrier  , thermodynamic barrier
, thermodynamic barrier  ). Additionally, CsF exhibits a more favorable free energy change (
). Additionally, CsF exhibits a more favorable free energy change ( ) for intermediate formation compared to that of the generation of the intermediate IM-1via KF (
) for intermediate formation compared to that of the generation of the intermediate IM-1via KF ( ), underscoring its superior performance in the reaction. CsF reacts with TMSCF3 to form CsCF3, which then attacks IM-1 and undergoes an addition reaction to form IM-2 (−51.8 kcal mol−1). Subsequently, IM-2 undergoes β-fluoride elimination, losing one molecule of CsF to give IM-3 (−49.3 kcal mol−1), introducing the first CF3 group. Double trifluoromethyl-substituted IM-5 (−82.8 kcal mol−1) is formed after two consecutive processes. Notably, the CsHF2 formed during the generation of IM-1 reacts with TMSCF3 to afford HCF3 (
), underscoring its superior performance in the reaction. CsF reacts with TMSCF3 to form CsCF3, which then attacks IM-1 and undergoes an addition reaction to form IM-2 (−51.8 kcal mol−1). Subsequently, IM-2 undergoes β-fluoride elimination, losing one molecule of CsF to give IM-3 (−49.3 kcal mol−1), introducing the first CF3 group. Double trifluoromethyl-substituted IM-5 (−82.8 kcal mol−1) is formed after two consecutive processes. Notably, the CsHF2 formed during the generation of IM-1 reacts with TMSCF3 to afford HCF3 (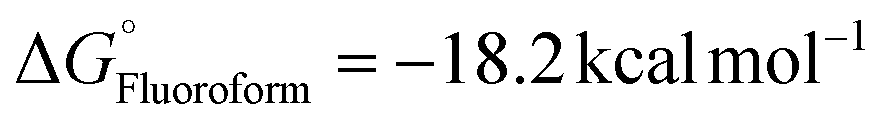 ), contributing to the exothermic free energy change in the reaction. Finally, the addition reaction of IM-5 with HCF3 produces the target product 1. Importantly, the calculations show that directly eliminating one HF using two CsF equivalents is energetically unfavorable.
), contributing to the exothermic free energy change in the reaction. Finally, the addition reaction of IM-5 with HCF3 produces the target product 1. Importantly, the calculations show that directly eliminating one HF using two CsF equivalents is energetically unfavorable.
 | ||
| Fig. 3 DFT study on the reaction mechanism. | ||
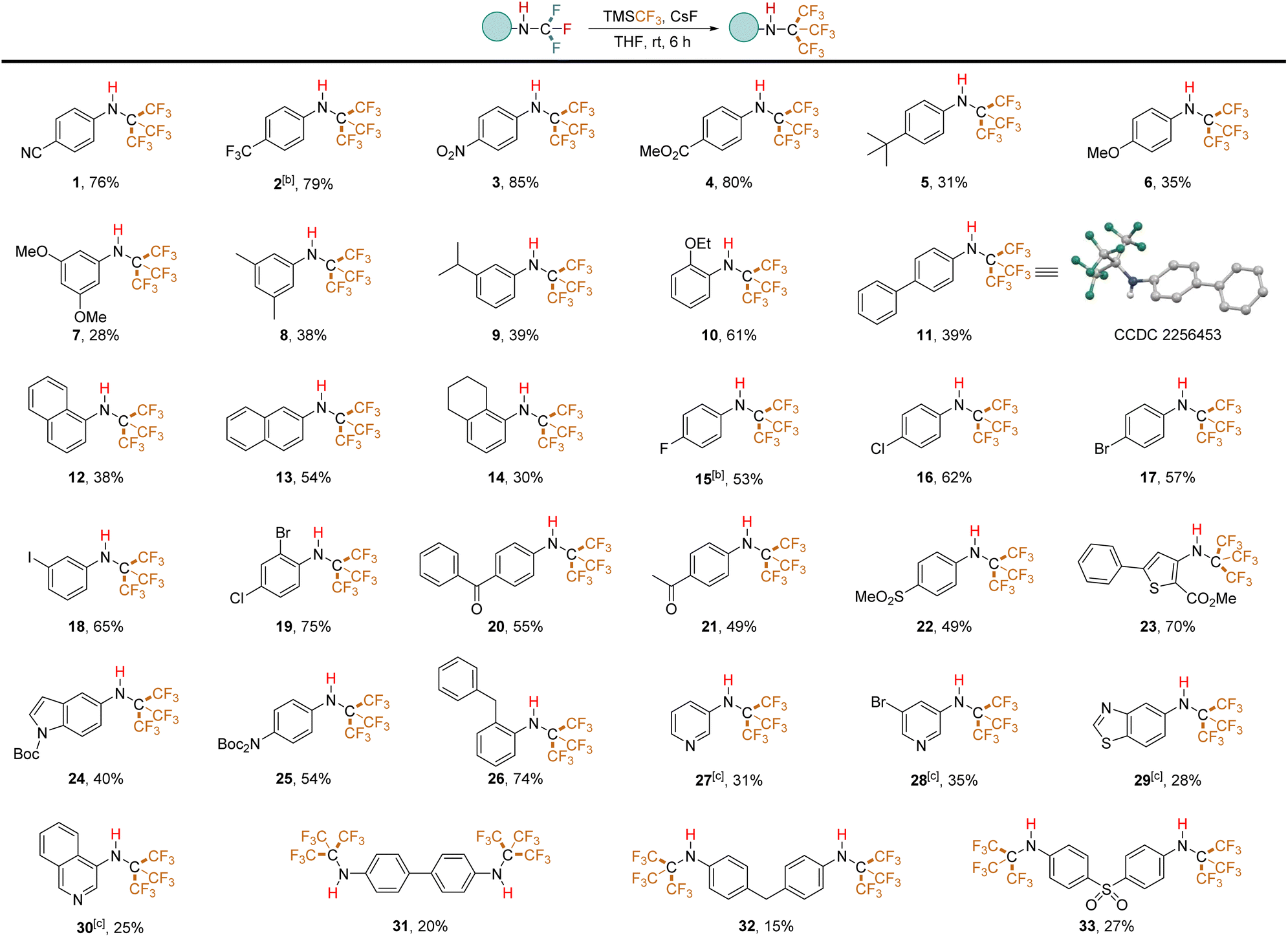
We evaluated the applicability of this method to various substrates under the optimized conditions (Table 2). The results reveal a clear influence of the electronic nature of the aryl ring on reaction efficiency. N-Trifluoromethyl secondary amines bearing electron-withdrawing groups, similar to the template substrate, yielded the corresponding trifluoromethylated products in good yields (e.g., trifluoromethyl (2), nitro (3), ester (4)). Conversely, electron-rich aromatic rings exhibited lower tolerance. Substrates with tert-butyl (5), methoxy (6, 7), methyl (8), and isopropyl (9) groups afforded N-PFtB secondary amines in lower to moderate yields. Unexpectedly, the ortho-ethoxy-substituted compound (10) produced the resulting product in a 61% yield. The naphthalene ring was also compatible, although α-naphthalene (12) displayed lower reactivity compared to β-naphthalene (13), while tetrahydronaphthalene (14) showed reactivity akin to that of α-naphthalene, possibly due to steric hindrance from the PFtB group. Notably, the presence of fluorine (15), chlorine (16), bromine (17), and iodine (18) substituents on the benzene ring did not affect the reaction. These halogens remained intact, as confirmed by the absence of additional trifluoromethyl signals in GC-MS and 19F NMR spectra. This compatibility is advantageous for downstream derivatization reactions. It's important to note that highly fluorinated molecules have a nonpolar character and an extremely low polarizability, inducing only weak intra- and intermolecular interactions.28 Consequently, substrates 2 and 15, which contain fluorine and trifluoromethyl groups on the benzene ring, exhibit lower boiling points compared to the other substrates, making it difficult to isolate compounds 2 and 15 under reduced pressure for extended periods. Moderate yields were obtained for ketone substrates (20, 21) and the sulfone substrate (22). Perfluoro-tert-butylation of thiophene (23) and protected nitrogen compounds (24, 25) also proceeded successfully. However, pyridine and thiazole starting materials did not convert to PFtB amines under standard conditions, potentially due to the instability of their N-CF3 secondary amine precursors in the solvent-free environment. Optimized conditions were developed for a one-pot conversion of isothiocyanates to N-PFtB pyridine derivatives (27, 28) in THF (details in ESI, p. S27†). Similarly, trifluoromethylated thiazole (29) and isoquinoline (30) precursors, synthesized in MeCN but THF instead of Et2O in the purification procedure, were successfully transformed to corresponding compounds. The synthesis of bis-NHC(CF3)3-substituted substrates (31–33) yielded low product quantities. Notably, the standard protocol is not suitable for aliphatic substrates. The structure of N-PFtB secondary amine 11 was confirmed by X-ray crystallographic analysis. Compound 1 exhibited stability under acidic (aqueous HCl, pH 0, 36 hours) conditions, with no degradation observed by 1H NMR (phenyl region). However, minimal decomposition was detected under basic conditions (aqueous NaOH, pH 14, 36 hours) (details in ESI, p. S42†). The predicted Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value of compound 1 (LogP = 4.85) is significantly higher than its N-tert-butyl counterpart (LogP = 2.27), indicating a substantial increase in lipophilicity due to the CF3 groups.29
P value of compound 1 (LogP = 4.85) is significantly higher than its N-tert-butyl counterpart (LogP = 2.27), indicating a substantial increase in lipophilicity due to the CF3 groups.29
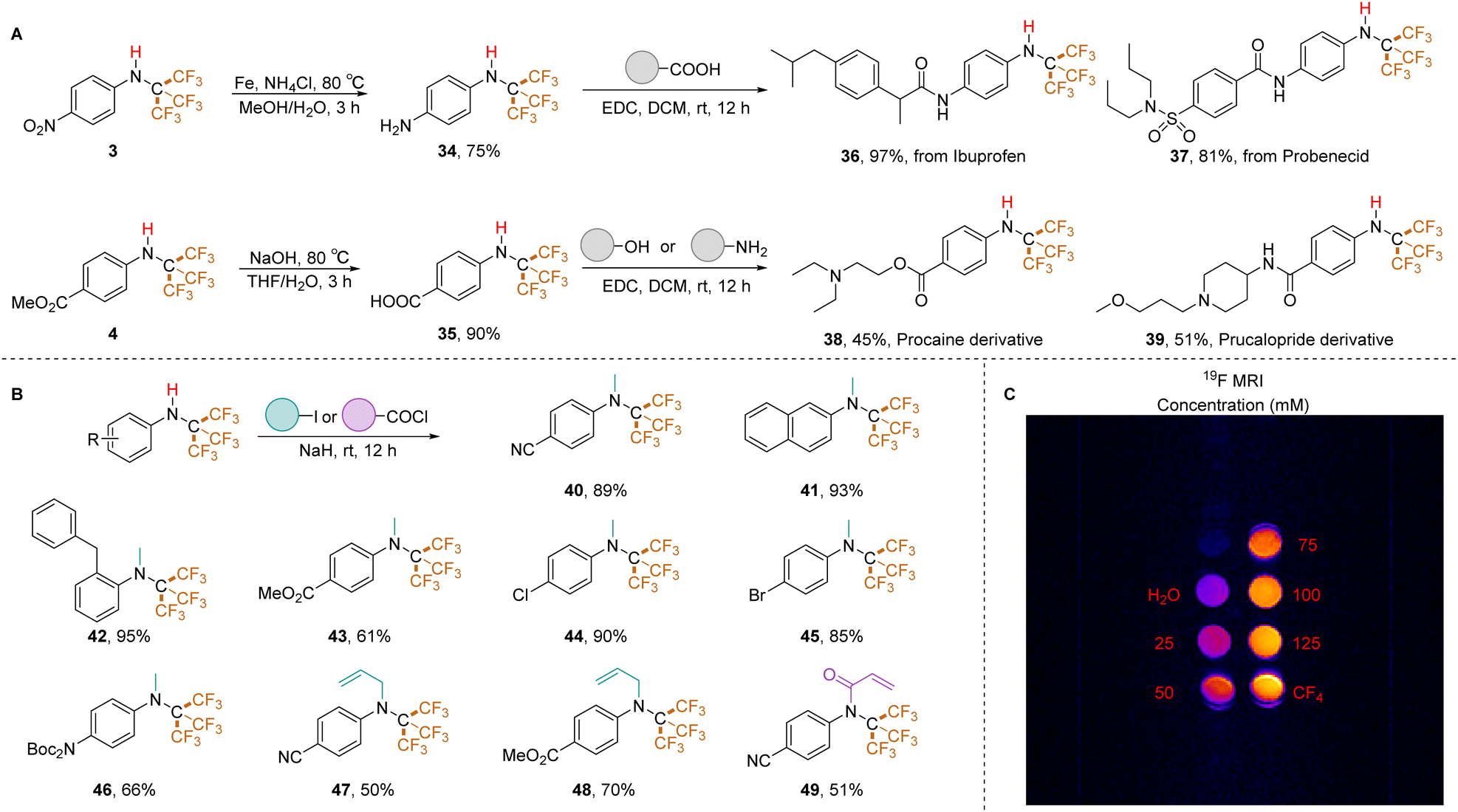
To assess the utility of this method for incorporating PFtB groups into drug-like molecules, we targeted ibuprofen (antipyretic/analgesic) and probenecid (antigout). Since N-CF3 secondary amines bearing an unprotected NH2 group were not accessible using our previously reported method,26 it is more feasible to obtain such an active precursor (34) by reducing a NO2 group containing N-PFtB secondary amine (3). Following the reduction of compound 3 and subsequent condensation, the PFtB group was successfully introduced into these drugs with excellent yields (ibuprofen: 36, 97%; probenecid: 37, 81%). Notably, the PFtB group remained intact during ester deprotection. Furthermore, N-PFtB derivatives of procaine (local anesthetic, 38) and prucalopride (constipation treatment, 39) were obtained via smooth condensation with the amino or hydroxyl groups (Table 3A). These examples demonstrate the compatibility of N-PFtB secondary amines with various derivatization reactions. Capitalizing on the stability of the N–H bond, we achieved N-methylation under strong basic conditions (NaH) using multiple substrates. Naphthalene (41), cyano (40), benzyl (42), ester (43), halogen (44, 45), and Boc-protected amino (46) substituted N-PFtB amines yielded the corresponding methylated products in high yields. Interestingly, allyl iodide reacted with compounds 1 and 4 to afford products 47 and 48 (Table 3B), while iodoethane, iodopropane, and iodobutane were unreactive. Additionally, the acylation (49) reaction of N–H bond in N-PFtB secondary amine was successfully performed. Finally, we explored the potential of PFtB-labeled molecules for 19F MRI applications using compound 36. Compared to negative (H2O) and positive (CF4) controls, compound 36 exhibited clear imaging sensitivity at various concentrations (25, 50, 75, 100, and 125 mM). Notably, a concentration of 75 mM displayed a high level of discrimination (Table 3C). These findings suggest promising prospects for PFtB-labeled secondary amines as 19F MRI contrast agents.
| a Derivatization and applications of N-PFtB secondary amines. (A) Synthesis of drug-like molecules (for details, see ESI). (B) Amine (0.3 mmol, 1.0 equiv.), iodide (5.0 equiv.), NaH (10.0 equiv.), THF, r.t., 12 h. (C) MR imaging of 19F at ultra-high field. |
|---|
|
Conclusions
In summary, this study describes a novel and efficient one-step method for the synthesis of aromatic N-PFtB secondary amines from readily available N-CF3 secondary amine precursors. The reaction involves the strategic elimination of HF from starting materials, rapid conversion of C–F bonds to C-CF3 groups, and incorporation of the in situ generated fluoroform. This method is compatible with a broad range of functional groups, including complex compounds. Experimental results and DFT calculations confirm the involvement of intermediates in the reaction process. This approach not only offers new insights into the reactivity of N-CF3 secondary amines but also provides a valuable pathway for the development of N-PFtB-containing drugs, imaging probes, and contrast agents.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. Y. and L. W. conceived the research. L. W. and J. W. carried out all the experiments and data analysis. Z. F. and Z. L. carried out synthesis of various starting materials. Z. G. performed DFT calculations. L. W., J. W. and W. Y. wrote the manuscript with input from all authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (22378205, 22078161), Fundamental Research Funds for the Central Universities (30922010403), Priority Academic Program Development of Jiangsu Higher Education Institutions, the Center for Advanced Materials and Technology in Nanjing University of Science and Technology for financial support.Notes and references
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- A. Vitale, R. Bongiovanni and B. Ameduri, Chem. Rev., 2015, 115, 8835–8866 CrossRef CAS PubMed.
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2014, 115, 826–870 CrossRef.
- J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS.
- C. L. Tong, X. H. Xu and F. L. Qing, Angew. Chem., Int. Ed., 2021, 60, 22915–22924 CrossRef CAS PubMed.
- Q. Wang, Q. Tao, H. Dong, C. Ni, X. Xie and J. Hu, Angew. Chem., Int. Ed., 2021, 60, 27318–27323 CrossRef CAS.
- Z. Wei, L. Wen, K. Zhu, Q. Wang, Y. Zhao and J. Hu, J. Am. Chem. Soc., 2022, 144, 22281–22288 CrossRef CAS PubMed.
- H. Meng, L. Wen, Z. Xu, Y. Li, J. Hao and Y. Zhao, Org. Lett., 2019, 21, 5206–5210 CrossRef CAS.
- C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Chem. Rev., 2022, 122, 167–208 CrossRef CAS.
- E. Hequet, C. Henoumont, R. N. Muller and S. Laurent, Future Med. Chem., 2019, 11, 1157–1175 CrossRef CAS PubMed.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, Chem. Rev., 2014, 115, 1106–1129 CrossRef.
- K. Tanabe, H. Harada, M. Narazaki, K. Tanaka, K. Inafuku, H. Komatsu, T. Ito, H. Yamada, Y. Chujo, T. Matsuda, M. Hiraoka and S. Nishimoto, J. Am. Chem. Soc., 2009, 131, 15982–15983 CrossRef CAS PubMed.
- A. Li, X. Luo, D. Chen, L. Li, H. Lin and J. Gao, Anal. Chem., 2023, 95, 70–82 CrossRef CAS.
- B. L. Bona, O. Koshkina, C. Chirizzi, V. Dichiarante, P. Metrangolo and F. Baldelli Bombelli, Acc. Mater. Res., 2022, 4, 71–85 CrossRef.
- B. Meng, S. L. Grage, O. Babii, M. Takamiya, N. MacKinnon, T. Schober, I. Hutskalov, O. Nassar, S. Afonin, S. Koniev, I. V. Komarov, J. G. Korvink, U. Strähle and A. S. Ulrich, Small, 2022, 18, 2107308 CrossRef CAS PubMed.
- K. Akazawa, F. Sugihara, T. Nakamura, H. Matsushita, H. Mukai, R. Akimoto, M. Minoshima, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed., 2018, 57, 16742–16747 CrossRef CAS.
- N. G. Taylor, S. H. Chung, A. L. Kwansa, R. R. Johnson, A. J. Teator, N. J. B. Milliken, K. M. Koshlap, Y. G. Yingling, Y. Z. Lee and F. A. Leibfarth, Chem. Eur. J., 2020, 26, 9982–9990 CrossRef CAS PubMed.
- V. A. Petrov, Tetrahedron Lett., 2000, 41, 6959–6963 CrossRef CAS.
- W. J. Middleton and C. G. Krespan, J. Org. Chem., 1965, 30, 1398–1402 CrossRef CAS.
- R. J. D. Pasquale, J. Fluorine Chem., 1976, 8, 311–322 CrossRef.
- Y. Guo, K. Fujiwara and K. Uneyama, Org. Lett., 2006, 8, 827–829 CrossRef CAS PubMed.
- J. F. Kögel, L. H. Finger, N. Frank and J. Sundermeyer, Inorg. Chem., 2014, 53, 3839–3846 CrossRef.
- Y. L. Yagupolskii and K. I. Petko, J. Fluorine Chem., 2015, 176, 9–13 CrossRef CAS.
- L. Wang, J. Wang, S. Ye, B. Jiang, Z. Guo, Y. Mumtaz and W. Yi, Angew. Chem., Int. Ed., 2022, 61, e202212115 CrossRef CAS.
- J. X. Xiang, Y. Ouyang, X. H. Xu and F. L. Qing, Angew. Chem., Int. Ed., 2019, 58, 10320–10324 CrossRef CAS.
- J. P. Bégué and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, Inc., Hoboken, 2008 Search PubMed.
- T. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. Zhang, Y. Li and R. Wang, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details of the experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, X-ray crystallographic data for 11. CCDC 2256453. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06335j |
| This journal is © The Royal Society of Chemistry 2024 |