
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical trifluoromethylation of alkynes: the unique role of DMSO as a masking auxiliary†
Jihoon
Jang‡
,
Ho Seong
Hwang‡
,
Haeryeong
Jeong
and
Eun Jin
Cho
 *
*
Department of Chemistry, Chung-Ang University, 84 Heukseok-ro, Dongjak-gu, Seoul 06974, Republic of Korea. E-mail: ejcho@cau.ac.kr
First published on 4th November 2024
Abstract
Recent advancements in eco-friendly radical fluoroalkylation have substituted traditional two-electron-based reactions. However, the radical trifluoromethylation of terminal alkynes remains a significant challenge, primarily due to the high reactivity of alkenyl radical intermediates, which predominantly engage in reactions other than the desired elimination. In this work, we have developed an electrochemical trifluoromethylation method for terminal alkynes, facilitating the efficient formation of CF3-alkynes. The success of this method centers on the use of DMSO as a “masking auxiliary”, which effectively stabilizes the alkenyl radical intermediate, allowing the reaction to proceed smoothly under mild conditions. This approach is supported by extensive experimental and computational studies, which elucidate the unique mechanism and expand the potential applications of radical trifluoromethylation across chemical synthesis.
Introduction
Recent advancements in eco-friendly radical chemistry, particularly through photochemistry1 and electrochemistry,1a,2 have led to the development of sustainable methods for generating radical intermediates using environmentally benign photons and electric energy, respectively. Among these methods, radical fluoroalkylations have emerged as particularly efficient due to their mild reaction conditions, offering a sustainable alternative to traditional fluoroalkylation processes.3 Fluoroalkyl groups, especially the trifluoromethyl group, are crucial in enhancing the physical and biological properties of compounds, including metabolic stability, binding selectivity, bioavailability, and lipophilicity.4 There has been significant progress in the radical trifluoromethylation of (hetero)aromatics, alkenes, and alkynes, which serve as substitutes and complements to traditional two-electron based processes.3 However, the radical trifluoromethylation of terminal alkynes (Csp–H) to form CF3-alkynes remains a notable challenge.5,6 Despite recent advances in photoredox-mediated trifluoromethylation of terminal alkynes by our group7 and the Yang group,8 these methods do not directly utilize terminal alkynes. Instead, they depend on in situ generated alkynyl-iodide as the actual substrate (Scheme 1a). Direct terminal Csp–H functionalization using CF3 radicals has necessitated the pre-functionalization of alkynes. | ||
| Scheme 1 Alkynyl trifluoromethylations. | ||
Several challenges arise in the radical alkynyl trifluoromethylation of aromatic alkynes. Selectivity issues can occur between the aromatic and alkyne moieties (Scheme 1b(i)).9 More critically, the high reactivity of the alkenyl radical intermediate complicates the formation of alkynyl-CF3 compounds (Scheme 1b(ii)). Due to its low oxidation potential, the vinyl radical is prone to oxidation into its cationic form, making it susceptible to nucleophilic attack.9,10 Additionally, the alkenyl radical intermediate can be captured easily by hydrogen, halogens, or pi-systems, leading to the formation of other trifluoromethylated products,11 not CF3-alkynes.
Herein, an unprecedented radical electrochemical approach is presented for the direct synthesis of alkynyl-CF3 compounds from terminal alkynes, effectively overcoming these challenges (Scheme 1c). To address the issues mentioned above, DMSO was employed as a “masking auxiliary”, leveraging its ability to stabilize the free alkenyl radical intermediate. Although DMSO is typically limited in electrochemistry due to its redox active character,12 this characteristic was utilized to enable the crucial final step of regenerating the alkyne moiety.
Results and discussion
Electrochemical trifluoromethylation of alkynes
The investigation was initiated using phenylacetylene (1a) as a model substrate, with NaSO2CF3 (2) as the CF3 radical source in DMSO (Table 1). The reaction was conducted under constant voltage electrolysis in an undivided cell, utilizing tetrabutylammonium hexafluorophosphate (TBAPF6) as the electrolyte, and graphite [C(+)/C(−)] as both the working and counter electrodes. The cell potential was optimized within the range of 3.5 V to 5.0 V, ultimately determining that 4.4 V was optimal, yielding 3a in 79% yield. However, when the cell voltage exceeded 4.4 V, the formation of the reduced byproduct, ethylbenzene, increased (see Fig. S1† in the ESI for detailed experimental data†). Despite the inherent electrochemical challenges, DMSO facilitated notably high yields, suggesting a unique role in this transformation. In contrast, other solvents, such as MeCN and DMF, resulted in lower yields (entries 2 and 3). Substituting TBAPF6 with other tetra-n-butylammonium salts, such as TBABF4 and TBANO3, produced comparable yields (entries 4 and 5), whereas the use of inorganic electrolytes like KPF6 led to a reduced yield (entry 6). Zn(SO2CF3)2 can also serve as a CF3 radical source in place of NaSO2CF3, albeit with a slightly lower yield (entry 7). Further optimization of electrolyte concentration, ranging from 0.02 M to 0.06 M, did not increase the reactivity (entries 11–13). As a control, no reaction occurred in the absence of electrical input, supporting the necessity of the electrochemical redox process (entry 14).|
|
||
|---|---|---|
| Entry | Variations | Yieldb (%) |
| a 0.3 mmol scale. b Yields were determined by 19F NMR spectroscopy using α,α,α-trifluorotoluene as an internal standard. | ||
| 1 | None | 79 |
| 2 | MeCN instead of DMSO | 9 |
| 3 | DMF instead of DMSO | 46 |
| 4 | TBABF4 instead of TBAPF6 | 72 |
| 5 | TBANO3 instead of TBAPF6 | 77 |
| 6 | KPF6 instead of TBAPF6 | 35 |
| 7 | Zn(SO2CF3)2 instead of NaSO2CF3 | 60 |
| 8 | NaSO2CF3 1.0 equiv. | 32 |
| 9 | NaSO2CF3 2.0 equiv. | 76 |
| 10 | NaSO2CF3 3.0 equiv. | 46 |
| 11 | TBAPF6 (0.02 M) | 44 |
| 12 | TBAPF6 (0.04 M) | 70 |
| 13 | TBAPF6 (0.06 M) | 76 |
| 14 | No electricity | N.R |
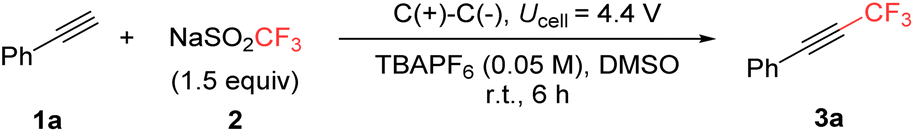
Mechanistic investigations of electrochemical trifluoromethylation of alkynes
To gain deeper insight into the reaction mechanism, particularly the role of DMSO, a series of mechanistic investigations were conducted. Initially, cyclic voltammetry experiments were performed (Scheme 2a). In MeCN with TBAPF6 as the electrolyte, NaSO2CF3 (2, black), DMSO (red), and phenylacetylene (1a, blue) exhibited irreversible oxidation peaks at 0.94 V, 1.74 V, and 2.08 V vs. Ag/AgCl, respectively. Notably, the oxidation peak at 2.08 V disappeared when NaSO2CF32 and phenylacetylene 1a were combined (purple), and a new oxidation peak emerged at 1.48 V, attributed to the oxidation of the alkenyl-CF3 radical (ii). Interestingly, upon the addition of DMSO (light brown), the oxidation peak corresponding to the alkenyl-CF3 radical (ii) vanished, while the oxidation peak for 1a reappeared. Additionally, a reduction peak for SO2 at −0.98 V vs. Ag/AgCl was observed, which was only detected in the presence of DMSO (green and light brown).13 These observations, along with previous report,13 suggest that in the presence of DMSO, the SO2CF3 radical decomposes into CF3 radical and SO2 more slowly than in its absence, indicating DMSO's involvement in this electrochemical trifluoromethylation. | ||
| Scheme 2 Mechanistic investigations. | ||
Further investigation revealed that under optimized conditions (Ucell = 4.4 V), the working electrode cell potential in a three-electrode setup did not exceed 1.22 V vs. Ag/AgCl (Scheme 2b). According to the cyclic voltammetry data in Scheme 2a, only the trifluoromethylsulfinate (–SO2CF3) could be oxidized under these conditions. This suggests that the oxidation of the free alkenyl radical intermediate (ii) (1.48 V) does not occur during the transformation, indicating the presence of a more reactive intermediate. This hypothesis was supported by experiments involving the radical quencher butylated hydroxy toluene (BHT). The reaction was inhibited, confirming that the process is radical-mediated (Scheme 2c). GC-MS analysis detected a BHT-CF3 adduct, while no BHT-alkenyl-CF3 adduct was observed.14 This suggests that the free alkenyl-CF3 radical (ii) is short-lived and, rather than undergoing oxidation, is converted to another intermediate that plays a key role in this electrochemical redox process.
Based on our results, we propose a mechanism (Scheme 3a), supported by a series of further experimental and computational studies (Scheme 3b–f). The –SO2CF3 (2) undergoes single-electron oxidation, generating ·SO2CF3. This radical can be stabilized by DMSO, as confirmed by DFT calculations (uM062X/def2TZVP/PCM in DMSO), which showed a strong interaction between DMSO and the SO2CF3 radical (−15.69 kcal mol−1 and O–S distance 2.29 Å) (Scheme 3b).15 In the presence of phenylacetylene 1a, the CF3 radical adds to the alkyne moiety, and then reacts with DMSO. Crucially, DMSO functions as an electroauxiliary,16 effectively masking the free alkenyl radical intermediate. The involvement of DMSO was further supported by kinetic isotope effect (KIE) experiments (Scheme 3c and Fig. S3 in the ESI†). While the reaction with deuterium-exchanged terminal alkyne showed no significant KIE, the use of DMSO-d6 resulted in reduced reactivity, with a KIE value of 1.83, indicating DMSO's role in the process. Although the reaction did not proceed well in MeCN, the addition of another sulfoxide—4 equivalents of diphenylsulfoxide (DPSO)—improved the reaction yield, further supporting the involvement of sulfoxide in promoting the transformation (Scheme 3d). The rapid formation of an intermediate (B) between the alkenyl radical and DMSO leads to a masked species with an oxidation potential of 0.91 V vs. Ag/AgCl (uM062X/TZVP/PCM (DMSO)), which is easily oxidized under the applied cell conditions (<1.22 V). The presence of B was additionally confirmed by GC-MS and LC-MS analysis. The oxidation of B to B+ increases the acidity of the vinyl hydrogen atom, as demonstrated by the electrostatic potential map in Scheme 3e, where the ESP value shifts from 0.07 in B to 0.26 in B+, facilitating the key deprotonation step that results in the formation of the trifluoromethylated alkyne 3a. Further evidence supporting this deprotonation step was obtained from the reaction conducted in a divided cell (Scheme 3f and Fig. S4 in the ESI†). During the reaction, the anodic cell becomes increasingly acidic due to ongoing deprotonation. Additionally, performing the reaction in a divided cell resulted in a cleaner reaction profile, further validating the proposed mechanism.
 | ||
| Scheme 3 Proposed mechanism with supporting experimental evidence. | ||
Substrate scope
Next, under the optimized conditions, the substrate scope was explored by using various alkyne derivatives to synthesize the corresponding CF3-alkynes (Scheme 4). The reactions were successful across a broad range of phenylacetylene derivatives, regardless of the electron density or positional variation of the substituents. Both electron-donating (3ab–3al) and electron-withdrawing (3an–3ax) groups on the phenyl ring yielded the desired CF3-alkynes. Additionally, this method was effectively applied to extended conjugated systems, such as biphenyl (3am) and naphthalene (3b). Substituents at the ortho-, meta-, and para-positions of phenylacetylene were all compatible, demonstrating the versatility of the reaction conditions. Moreover, heterocyclic alkynes containing pyridine, indole, and quinoline moieties also proved to be suitable substrates (3c, 3d, 3e). The mild electrochemical conditions tolerated various functional groups, including reactive halides (3ao–3ar) and carbonyl substrates like aldehydes (3av), which are often incompatible with photoredox- or basic conditions. To further demonstrate the broad applicability of this methodology, its use was extended to the late-stage modification of pharmaceutical molecules. Structurally complex alkyne derivatives, such as those based on estrone and the anti-cancer drug erlotinib, were efficiently trifluoromethylated in a divided cell under standard conditions (3f and 3g). However, under the conditions, reactions involving aliphatic alkynes did not yield CF3-alkynes; instead, they produced CF3-alkenes, reduced alkanes, and some unidentified compounds.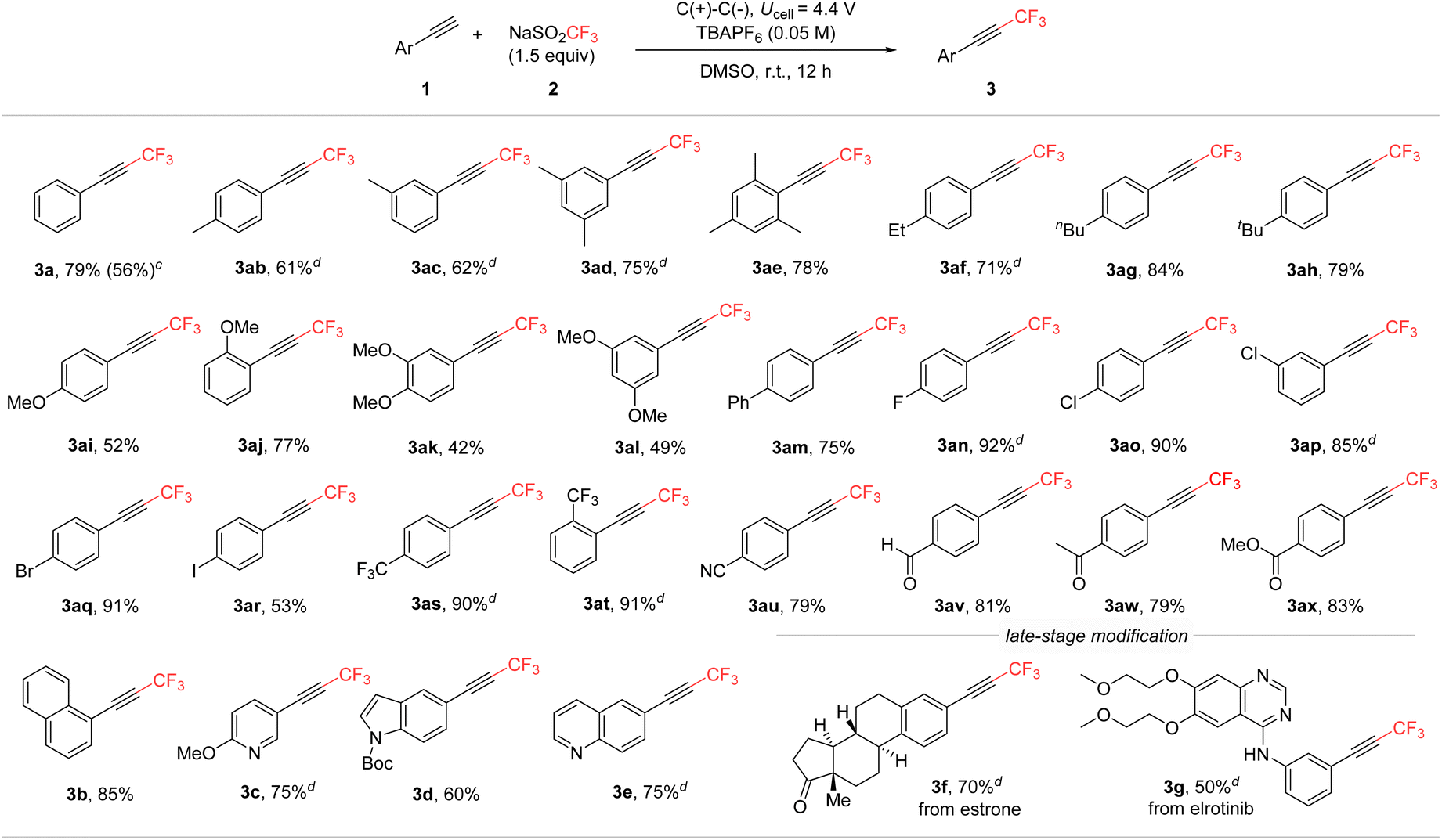 | ||
| Scheme 4 Substrate scopea,b; a0.3 mmol scale. bIsolated yields, c20 mmol scale with 136 h reaction time. dYields were determined by 19F NMR spectroscopy using α,α,α-trifluorotoluene as an internal standard. | ||
Conclusions
In conclusion, we successfully developed an electrochemical trifluoromethylation method for terminal alkynes, achieving the efficient formation of CF3-alkynes—a transformation that has posed significant challenges in radical chemistry. The success of this method is attributed to the strategic use of DMSO as an electroauxiliary, which effectively stabilizes the alkenyl radical intermediate, thereby enabling the reaction to proceed under mild conditions. Comprehensive experimental and computational studies strongly supported this approach, elucidating the unique mechanism and expanding the scope of radical trifluoromethylation for diverse applications in chemical synthesis.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
J. Jang, H. S. Hwang and H. Jeong performed synthetic experiments and mechanistic studies. E. J. Cho coordinated all of the experiments, analyses, and co-wrote the manuscript. All authors contributed to the discussion on the study and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Research Foundation of Korea (NRF-2020R1A2C2009636 and RS-2024-00409659) and the Ministry of Trade, Industry and Energy, Korea (Technology Innovation Program, RS-2023-00266039).References
- (a) R. Shaw, N. Sihag, H. Bhartiya and M. R. Yadav, Org. Chem. Front., 2024, 11, 954 RSC; (b) Y. Ouyang and F.-L. Qing, J. Org. Chem., 2024, 89, 2815 CrossRef CAS PubMed; (c) C. H. Ka, S. Kim and E. J. Cho, Chem. Rec., 2023, 23, e202300036 CrossRef; (d) F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615 CrossRef CAS PubMed; (e) E. H. Oh, H. J. Kim and S. B. Han, Synthesis, 2018, 50, 3346 CrossRef CAS; (f) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed; (g) E. J. Cho, Chem. Rec., 2016, 16, 47 CrossRef CAS.
- (a) S. Kim and H. Kim, J. Am. Chem. Soc., 2024, 146, 22498 CrossRef CAS; (b) Z. Zou, W. Zhang, Y. Wang and Y. Pan, Org. Chem. Front., 2021, 8, 2786 RSC; (c) R. P. Bhaskaran and B. P. Babu, Adv. Synth. Catal., 2020, 362, 5219 CrossRef CAS.
- (a) C. Urban, F. Cadoret, J.-C. Blazejewski and E. Magnier, Eur. J. Org Chem., 2011, 4862 CrossRef CAS; (b) T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156 CrossRef CAS.
- (a) A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhuan and H. A. Aisa, J. Chem. Inf. Model., 2020, 60, 6242 CrossRef CAS PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) D. Mandal, S. Maji, T. Pal, S. K. Sinha and D. Maiti, Chem. Commun., 2022, 58, 10442 RSC; (b) A. Hassanpour, M. R. P. Heravi, A. Ebadi, A. Hosseinian and E. Vessally, J. Fluorine Chem., 2021, 245, 109762 CrossRef CAS; (c) G. Li, C. Zhang, C. Song and Y. Ma, Beilstein J. Org. Chem., 2018, 14, 155 CrossRef CAS PubMed.
- (a) S. T. Shreiber and D. A. Vicic, Angew. Chem., Int. Ed., 2021, 60, 18162 CrossRef CAS; (b) J. Li, K. Liu, K. Zheng, C. Zheng, H. Xiao and S. Fan, J. Org. Chem., 2020, 85, 8723 CrossRef CAS PubMed; (c) H.-S. M. Siah and A. Fiksdahl, J. Fluorine Chem., 2017, 197, 24 CrossRef CAS; (d) L. He and G. C. Tsui, Org. Lett., 2016, 18, 2800 CrossRef CAS.
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS.
- (a) X. Shi, B. Yu, X. Zhou and Y. Yang, Chem. Commun., 2024, 60, 2532 RSC; (b) X. Shi, T. Song, Q. Li, X. Guo and Y. Yang, Org. Lett., 2022, 24, 8724 CrossRef CAS PubMed.
- W. Jud, C. O. Kappe and D. Cantillo, Org. Biomol. Chem., 2019, 17, 3529 RSC.
- (a) S. Tanaka, Y. Nakayama, Y. Konishi, T. Koike and M. Akita, Org. Lett., 2020, 22, 2801 CrossRef CAS PubMed; (b) W. Lee, Y. Lee, M. Yoo, S. B. Han and H. J. Kim, Org. Chem. Front., 2020, 7, 3209 RSC; (c) A.-L. Barthelemy, G. Dagousset and E. Magnier, Eur. J. Org Chem., 2020, 1429 CrossRef CAS; (d) Y. R. Malpani, B. K. Biswas, H. S. Han, Y.-S. Jung and S. B. Han, Org. Lett., 2018, 20, 1693 CrossRef CAS; (e) H. S. Han, Y. J. Lee, Y.-S. Jung and S. B. Han, Org. Lett., 2017, 19, 1962 CrossRef CAS PubMed; (f) R. Tomita, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2015, 54, 12923 CrossRef CAS PubMed.
- (a) F. Xiang, D. Wang, K. Xu and C.-C. Zeng, Org. Lett., 2024, 26, 411 CrossRef CAS PubMed; (b) X. Wang, W. Zhou, W. Xie, Q. Chen and J. Wu, Chin. Chem. Lett., 2023, 34, 107984 CrossRef CAS; (c) N. Petek, H. Brodnik, O. Reiser and B. Štefane, J. Org. Chem., 2023, 88, 6538 CrossRef CAS PubMed; (d) Z.-H. Yan, W.-C. Li, Y.-H. Wu, Q.-B. Yan, Z.-L. Wei and W.-W. Liao, Org. Chem. Front., 2022, 9, 5912 RSC; (e) H. Wang, S. Li, Y. Cui, M. Liu, X. Bu, H. Tian and X. Yang, New J. Chem., 2022, 46, 20412 RSC; (f) W. Zhang, Z. Zou, W. Zhao, S. Lu, Z. Wu, M. Huang, X. Wang, Y. Wang, Y. Liang, Y. Zhu and Y. Pan, Nat. Commun., 2020, 11, 2572 CrossRef CAS PubMed; (g) T. Shang, J. Zhang, Y. Zhang, F. Zhang, X.-S. Li and G. Zhu, Org. Lett., 2020, 22, 3667 CrossRef CAS PubMed.
- O. Hammerich, Organic Electrochemistry, Taylor & Francis Group, CRC Press, 2016 Search PubMed.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed.
- In our previous work using NaSO2CF3 and phenylacetylene in MeCN solvent, BHT-alkenyl-CF3 adduct was observed, see: J. Jang and E. J. Cho, Adv. Synth. Catal., 2024, 366, 3450 CrossRef CAS.
- Regarding two-center/three-electron bond structure, see: (a) S.-Q. Cai, K.-F. Zhang and X.-H. Cai, Curr. Org. Chem., 2022, 26, 91 CrossRef CAS; (b) R. Gleiter and G. Haberhauer, Coord. Chem. Rev., 2017, 344, 263 CrossRef CAS; (c) M. M. D. Pramanik and N. Rastogi, Chem. Commun., 2016, 52, 8557 RSC.
- J. Yoshida, K. Nishiwaki, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06780k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |