
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coupling the high-temperature Fischer–Tropsch synthesis and the skeletal isomerization reaction at optimal operation conditions in the Power-to-Fuels process route for the production of sustainable aviation gasoline
Dorela
Dhamo
 *,
Jannis
Kühn
,
Simon
Lüttin
,
Michael
Rubin
and
Roland
Dittmeyer
*,
Jannis
Kühn
,
Simon
Lüttin
,
Michael
Rubin
and
Roland
Dittmeyer
Institute for Micro Process Engineering (IMVT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 10th April 2024
Abstract
In this study we presented a concept process route for the production of sustainable aviation gasoline and investigated experimentally the high-temperature Fischer–Tropsch (HTFT) synthesis and the first upgrading step, the skeletal isomerization reaction, both independently and coupled. The influence of the side products of the HTFT synthesis on the stability of the isomerization catalyst and the product distribution represents the main challenges of the coupled operation. While the individual steps of Power-to-Fuels processes are well investigated, there are not many studies on their coupled operation. In this work, the operating conditions of both reactions were firstly optimized independently from each other. Targeting the maximisation of the C3–C5 olefin fraction, especially butene, preferably at high CO conversion, the optimal operating parameters for HTFT synthesis were 300 °C, 20 bar(g), H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO = 2 and high GHSV. Targeting high isobutene selectivity at high 1-butene conversion, the optimal operating parameters for the isomerization reaction were 400 °C and atmospheric pressure. To investigate the coupling of both reactions, the gasous HTFT products together with the unconverted feed were separated from the liquid and solid products and fed into the isomerization reactor. The feed was diluted with N2 at 35.7 vol% and 8 vol% and the temperature of the isomerization reaction was increased from 300 °C to 400 °C. The coupling was run in steady-state at 400 °C and both 35.7 vol% and 8 vol% N2 dilution up to 65 h, while an operation temperature of 300 °C led to catalyst deactivation within the first hours.
:CO = 2 and high GHSV. Targeting high isobutene selectivity at high 1-butene conversion, the optimal operating parameters for the isomerization reaction were 400 °C and atmospheric pressure. To investigate the coupling of both reactions, the gasous HTFT products together with the unconverted feed were separated from the liquid and solid products and fed into the isomerization reactor. The feed was diluted with N2 at 35.7 vol% and 8 vol% and the temperature of the isomerization reaction was increased from 300 °C to 400 °C. The coupling was run in steady-state at 400 °C and both 35.7 vol% and 8 vol% N2 dilution up to 65 h, while an operation temperature of 300 °C led to catalyst deactivation within the first hours.
Introduction
In order to limit climate change, all individual sectors need to drastically reduce their greenhouse gas emissions, eventually achieving net-zero emissions by the middle of the century. As one of the main sectors responsible for CO2 emissions, the transport sector has to undergo a profound transformation by using environmentally friendly alternatives.1,2 While there are already quite established solutions as electrification of road traffic, there is still no applicable alternative for the maritime sector and air traffic. For example, airplanes require an energy carrier with a high volumetric and gravimetric energy density, which as of now cannot be provided by batteries (direct electrification).3 However, via the so-called Power-to-Fuels concepts, chemical energy carriers – so-called E-Fuels – can be synthesized from renewable electrical energy. Here, different products are feasible: besides gaseous products (hydrogen in the most straight forward scheme), liquid fuels such as methanol or hydrocarbons can be synthesized from green hydrogen (“Power-to”) and a CO2-source. When using renewable electricity for hydrogen generation and CO2 of non-fossil origin (eventually captured from ambient air via Direct Air Capture (DAC)), those E-Fuels can be considered as CO2 neutral. In the context of the transportation sector, E-Fuels via the Fischer–Tropsch route are very promising, as these synthetic hydrocarbons from the physico-chemical point of view can be tailored to match the properties of the fossil-based gasoline, diesel and kerosene and, thus, can readily be implemented in the existing infrastructure and substitute fossil fuels.4Power-to-Fuels pathway towards CO2-neutral aviation gasoline
This work focuses on the production of sustainable aviation gasoline over the Power-to-Fuels route. Aviation gasoline is an aviation fuel used in piston engine aircraft, therefore high-octane branched hydrocarbons (C7–C9) are required to prevent engine knocking and premature detonation. The main component is alkylate, which is a mixture of different isooctanes, branched chains with eight carbon atoms. With branching being a key property to achieve high octane numbers and knowing that unsaturated short hydrocarbons can be easier isomerized, high-temperature Fischer–Tropsch is more suitable than low-temperature Fischer–Tropsch for Syncrude production targeting aviation gasoline.5,6 The concept process route for the sustainable production of branched hydrocarbons (C7–C9) from CO2, water and renewable electricity is shown in Fig. 1. There are several technologies using CO2 feedstock as a carbon source, which can be extracted from ambient air (DAC), from concentrated gas streams of point sources like from biogas plants, or from industrial off-gases such as from cement or steel industry. Although, due to the higher efficiency and lower energy demand for CO2 capture, concentrated sources are more interesting in the first place, sustainability aspects should be taken into account: regarding CO2 balance, fossil carbon containing sources would still lead to net positive emissions, therefore, only renewable CO2 sources are suitable to produce CO2-neutral fuels.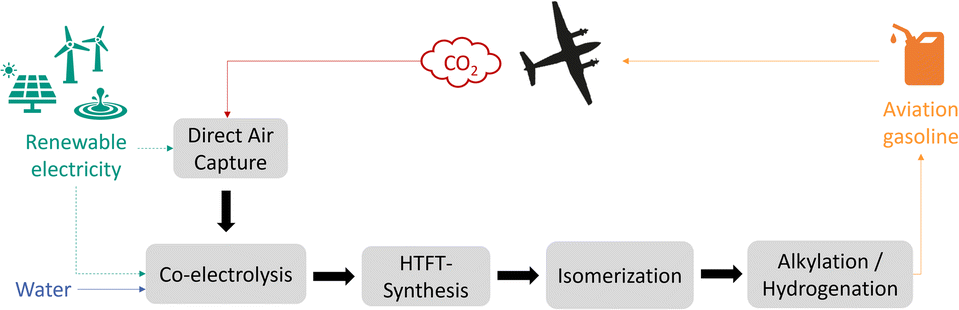 | ||
| Fig. 1 Illustration of a Power-to-Fuels approach to CO2 neutral production of aviation gasoline. | ||
Besides the carbon source, green hydrogen is needed in the Power-to-Fuels approach. The commonly used and efficient technology is water electrolysis either in liquid state (AEL, PEM, AEM) or in evaporated state (SOEC).7–9 Syngas can then be generated by a combination of water electrolysis and a reverse water–gas shift (RWGS) step.10 Alternatively, another promising technology is the high-temperature co-electrolysis (co-SOEC), in which both carbon dioxide and water are simultaneously reduced into carbon monoxide and hydrogen.11
Fischer–Tropsch synthesis
In the FT synthesis, carbon monoxide and hydrogen are converted to hydrocarbons. Targeting the production of short chain unsaturated molecules in the range of C3–C5 olefins, the suitable reaction is the HTFT synthesis. FT synthesis is an exothermal reaction (−154.1 kJ kmolCO−1)12 that produces mainly unbranched saturated alkanes (see eqn (1)), and unsaturated alkenes (see eqn (2)) of different carbon number n (chain length).| nCO + (2n + 1)H2 → CnH2n+2 + nH2O | (1) |
| nCO + (2n)H2 → CnH2n + nH2O | (2) |
The FT product distribution is commonly described in terms of carbon number distribution via the Anderson–Schulz–Flory (ASF) equation.18 According to ASF theory, the chain propagation probability does not depend on the carbon number. Later, from the 1980s on, deviations of the experimental results from the ASF model threw doubt on this theory, and new hypotheses, like dual site, particle size and promotor theories, were made trying to explain the mechanism of the FT synthesis.12,19–21
Besides the dependency on catalyst composition and size, there is a correlation between the operating conditions and the chain propagation probability. Studies by Ji et al. demonstrated an improvement in the light alkene selectivity with a Fe–Mn catalyst at relatively high H2:CO ratio (2.74) and high temperature (340 °C), while the flow rate did not show any impact.22 The study demonstrated that higher temperatures led to lower chain propagation probability. The work of Davis23 approved the temperature dependency and showed no significant dependency of the selectivity on the pressure and only a slight dependency of the light alkene selectivity on the H2:CO ratio. Furthermore, Davis considered the effect of high temperature and low H2:CO ratio on the water–gas shift (WGS) side reaction, leading to higher CO2 selectivity.
The state-of-the-art Fischer–Tropsch synthesis applied can be subdivided into two routes: (a) the low-temperature Fischer–Tropsch (LTFT) synthesis is operated at 220–250 °C on a cobalt- or iron-based catalysts, and produces saturated longer hydrocarbon chains, showing higher chain propagation probability. The high-temperature Fischer–Tropsch synthesis (HTFT) (b) is operated at 270–350 °C on an iron-based catalyst and produces rather short unsaturated hydrocarbons, showing lower chain propagation probability, which are required in the proposed PtL route.
The HTFT products do not meet the current fuel requirements; therefore, they need to be upgraded. The gaseous products, containing short chain hydrocarbons, and the unconverted feed are separated from the oil, wax and water phase and led to the upgrading unit. The first process step of the upgrading unit is the skeletal isomerization of the short chain hydrocarbons from the FT reaction, with the aim to increase the fraction of isobutene.
Skeletal isomerization of C4–C5 olefins
In the skeletal isomerization step, the structural change of aliphatic molecules takes place without hydrogenation (see eqn (3)). The reaction is acid-catalysed, slightly exothermic (−5 to 20 kJ mol−1) and thermodynamically limited. Beside branched molecules, double bond isomers are also formed.24,25 Skeletal isomerization is commonly used for the isomerization of n-butene, n-pentene and n-hexene. Longer chains are difficult to isomerize since the cracking is more favoured.26,27| n-CnH2n ↔ i-CnH2n | (3) |
There are different suitable catalysts for skeletal isomerization. On one hand, mesoporous and amorphous silicon and alumina catalysts show good activity and isobutene selectivity. They are usually operated at high temperatures up to 450 °C and atmospheric pressure. However, they have a relatively short life cycle due to carbon displacement on the catalyst, which makes them unsuitable for a commercial operation. Microporous catalysts, on the other hand, such as zeolites, show high isobutane selectivity even at lower temperatures around 350 °C and have a longer life cycle. The performance of these catalysts depends on their structure and acid strength. For the skeletal isomerization of n-butene, ferrierite catalysts are the most suitable. Ferrierites, and in general zeolites, consist of crosslinked SiO4 or AlO4 tetrahedra. The Si/Al ratio defines the acid strength.13,28–32
Referring to the structure, ferrierites are formed by 10-MR ring channels crossing 8-MR ring channels, containing ten and eight tetrahedra respectively. Skeletal isomerization occurs in the 10-MR channels while 8-MR channels allow byproducts to escape and thus improve the performance of the catalyst.13,31
The skeletal isomerization of hydrocarbons with five or more carbon atoms proceeds through a monomolecular mechanism. A carbocation is formed by the protonation of the double bond, which at the end leads to either a double bond- or a skeletal isomer. Besides the monomolecular mechanisms, the presence of byproducts, like propene or pentene during the skeletal isomerization of n-butene shows the presence of another mechanism: the bimolecular mechanism. According to this mechanism, two n-butenes are first dimerized, then isomerized and finally split into an isobutene and an n-butene molecule. During the first hours of operation, the byproduct fraction is quite high, suggesting a bimolecular mechanism. With increasing time-on-stream and therefore increased carbon deposits on the catalyst surface, the byproduct fraction decreases, which rather suggests the monomolecular mechanism or higher cracking during the bimolecular mechanism. Further carbon deposits on the catalyst surface lead to deactivation. Studies suggest a two-stage deposition mechanism. First, the 8-MR channels become clogged and slower filling occurs via the site-coverage mechanism, and afterwards, the 10-MR channels. The deactivation rate depends on the acid strength, reaction conditions, time-on-stream, and educt composition.33–38
Temperatures reported in the literature are 300 °C or higher. High temperatures lead to higher isobutene selectivity, but also favour coking and thus shorten the life of the catalysts. Besides temperature, the partial pressure of n-butene in the channels increases the formation of longer chains through the bimolecular mechanism, leading to pore blocking. Therefore, the skeletal isomerization of n-butene is usually run at atmospheric pressure.38,39
The second product upgrading step is alkylation. Due to the high olefinic feed, indirect alkylation is more suitable for the production of 2,2,4-trimethylpentene (isooctene). The alkylation reaction requires the presence of an acid catalyst, such as acidic resin or solid phosphoric acid. The products undergo a hydrogenation step to the final paraffinic fuel.14,15 Alternatively, aliphatic alkylation can be used to achieve a highly branched paraffinic product, where an olefin is alkylated with a branched paraffin.16,17 Therefore, the branched molecules from the skeletal isomerization reaction undergo a hydrogenation step before entering the alkylation reactor.
Remaining H2, CO, CO2 and light HTFT products found in the gas phase conceptionally can be recycled into the co-SOEC. This increases the carbon utilization and eventually the efficiency of the whole process chain. The effect of recycling steps on the product distribution should also be taken into account. To increase the overall process efficiency, the dissipated heat can be used in other process steps, where heat is required. Besides that, adjusting the operating conditions of each step, to maximize the target fraction also leads to higher end product yield.
While each process step has been well studied and established over the last decades, their coupling as a process chain remains to be investigated and optimized. One of the main concerns is how the side products from the Fischer–Tropsch synthesis as well as the unconverted feed influence the activity and stability of the upgrading steps. Referring to the risk of pore blocking through longer hydrocarbons, the coupling of the HTFT synthesis with the isomerization reactor might be challenging and needs further experimental investigations. In this publication we raise and answer the following research questions:
(I) What are the optimal operating conditions of the HTFT synthesis and of the skeletal isomerization reaction in the frame of the proposed process route?
(II) Do the side products of the HTFT synthesis cause a deactivation of the ferrierite catalyst?
(III) Do the side products of the HTFT synthesis influence the product distribution of skeletal isomerization reaction?
(IV) What effect do temperature and feed dilution have concerning the stability of the isomerization catalyst?
Experimental
Iron-based HTFT catalyst
In this work, a commercial iron-based catalyst was used (Riogen Inc., 0379-RG-FTS725). In detail, Fe is supported on SiO2 with copper and potassium being present as promoters. The exact catalyst composition and further detailed characterisation are not in the scope of this work.The catalyst was received as cylindrical extrudate (3 mm diameter, 4 mm height), crushed and sieved into particles in the range of 100–200 μm. Prior to the actual HTFT reaction, the catalyst was reduced in situ following the reduction procedure given by the catalyst supplier (9 vol% H2 in N2, GHSV = 6.61 × 104 h−1, at 350 °C, atmospheric pressure for approx. three hours).
Ferrierite catalyst
A ferrierite (HSZ-720NHA/01) from TOSOH with a crystal size ≤1 μm was used for the skeletal isomerization reaction. The SiO2/Al2O3 molar ratio was 17.6 and the pore size 0.48 nm.The catalyst was delivered as a powder with a particle size of approx. 6 μm. This particle size would cause a big pressure drop in the reactor therefore, first tablets were pressed, then crushed and finally sieved in a particle fraction between 100 and 200 μm. According to the catalyst supplier, there was no catalyst pre-treatment needed prior to the reaction starting.
Experimental setup
For the experimental investigation of both HTFT synthesis and skeletal isomerization reaction, two in-house manufactured fixed bed annular gap reactors were used with the catalyst placed in the gap (stainless steel; inner tube: Di,inner = 1.40 mm, Di,outer = 3.18 mm; outer tube Do,inner = 8.48 mm, Do,outer = 12.70 mm; i.e. width annular gap sag = 2.65 mm; total length L = 41 cm, length of the reaction zone LR = 8 cm – HTFT reactor and LR = 4 cm – isomerization reactor), see the illustration in Fig. 2. Temperature was regulated via three individual heating zones implemented via electrical heating of accordant aluminium blocks. The reactors, including the aluminium heating blocks, were insulated to minimize heat losses. The temperatures of the outer blocks were set to Ttop = Tbottom = TR + 3 °C in order to compensate heat losses and, thus, to achieve the desired reaction temperature TR.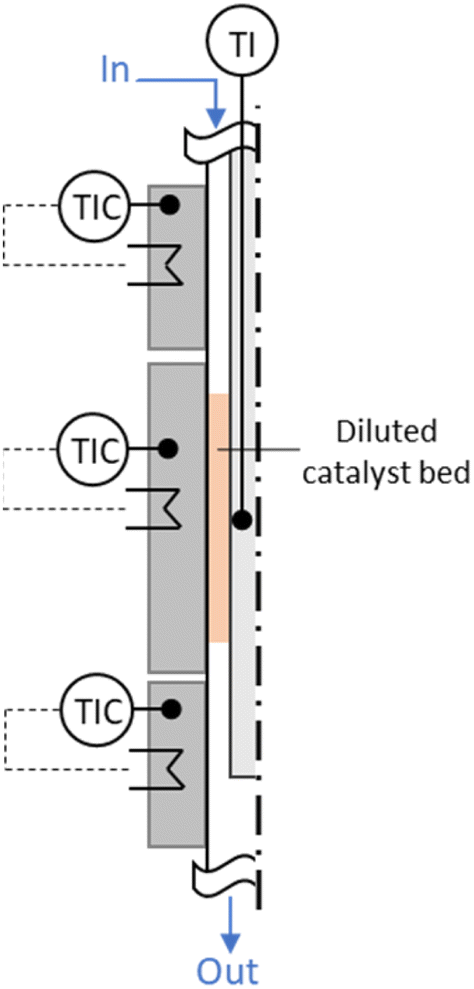 | ||
| Fig. 2 Annular gap reactor with three heating zones. The catalyst bed is diluted with SiC and placed in the gap in the second heating zone. | ||
Via mass flow controllers (Brooks Instrument) the reactants CO, H2 and N2 as internal standard (8 vol%) were preheated to 250 °C before being fed into the annular gap (HTFT reactor) and got further preheated to TR before entering the reaction zone. In general, due to the low amount of catalyst (0.7 g of the iron-based HTFT catalyst), the heat of reaction can be assumed to dissipate (also to the feed gas), and hotspot formation in the catalyst bed was limited by SiC dilution (3.0 g SiC (Thermo Fischer Scientific Inc., 100–200 μm)). Besides, the actual temperature in the reaction zone was determined via a moveable thermocouple. In steady state experiments at TR = 300 °C target temperature, the max. deviation along the reaction zone was less than 2 °C. The reaction products exited the reactor likewise at the bottom towards the downstream separation. The products from the HTFT reaction were solid, liquid, or gaseous at room temperature and, thus, need to be separated for further offline analysis. Product separation was achieved by two separation steps: in the so-called hot trap (140 °C) the wax phase (mostly > C17) is condensed while the shorter-chain hydrocarbons (mostly ≤ C17), unconverted reactants and the by-product water remain in the gas phase. In the subsequent cold trap (4 °C), the oil phase (mostly C6–17) and water were condensed. Gaseous products (mostly < C6) as well as the unconverted reactants were preheated up to 370 °C and enter the isomerization reactor. Similar to the HTFT reactor, the feed got further preheated to the reaction temperature in the first heating zone in the annular gap reactor before entering the reaction zone. The reaction zone was filled with 0.6 g of the ferrierite catalyst (100–200 μm) diluted with 1.2 g SiC. The products exited the isomerization reactor and were analysed online by a gas chromatograph (Agilent 6890N, equipped with a molecular sieve 5A column, a HP-Plot/Q column, a thermal conductivity detector (TCD) and a flame ionization detector (FID)). The compositions of the wax phase and the oil phase from the HTFT synthesis were determined via offline gas chromatography (wax phase: Agilent 7890B, oil phase (separated manually from water before): Agilent 7820A).
The isomerization reactor could also be run independently of the HTFT reactor. Another MFC dosed the feed from a gas bottle through a parallel gas line to the reactor entry.
Experimental procedure
The aim of this work was to investigate the coupling of the two reaction steps at their optimal operating parameters. The experiments were divided into three parts: determining the optimal parameters of the HTFT synthesis, determining the optimal operating parameters of the skeletal isomerization reaction, and investigating the coupling of the two process steps.:CO ≤ 2 and 1.07 × 104 h−1 ≤ GHSV ≤ 3.22 × 104 h−1. One of the parameter combinations (300 °C, 20 bar(g), H2:CO = 2 and 2.58 × 104 h−1), was chosen as a central reference point and was applied every 100–300 h TOS, to check for potential deactivation of the catalyst.
According to eqn (1) and (2), HTFT volumetrically is a non-constant reaction scheme. Therefore, CO conversion and product selectivity were calculated based on GC data corrected by the internal standard as follows:
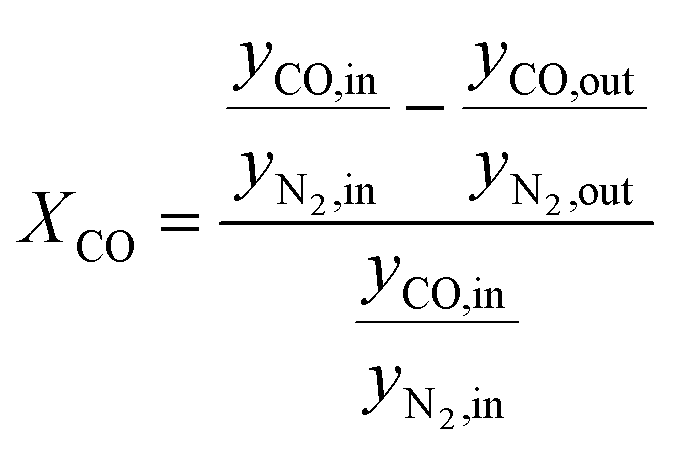 | (4) |
 | (5) |
In general, the carbon material balance was closed with deviations of ±15%. As both gas feed dosing system (mass flow controllers) and analytics (gas chromatography) were reliably calibrated, these deviations can be attributed to the low amount of overall product which results in residues in the separators which cannot be completely prevented. Besides, due to solubility reasons, cross-contamination of the wax phase by product water (hot trap) and components of the wax-phase in the oil phase (cold trap) cannot be avoided. From a practical point of view, manual separation of the product collected in the cold-trap (oil fraction and water) further induced some deviations in the carbon material balance.
The 1-butene conversion and the product selectivity were calculated with the following equations:
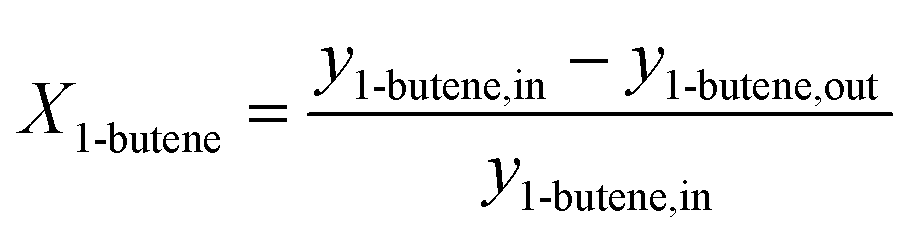 | (6) |
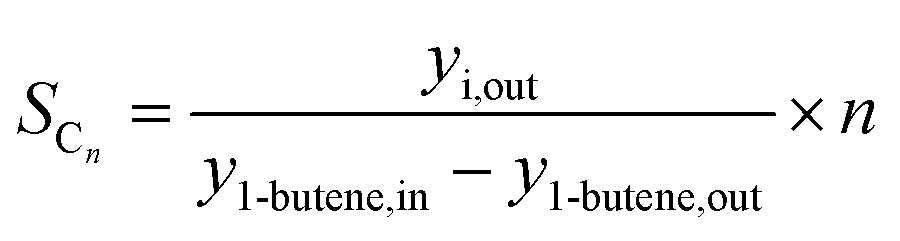 | (7) |
Besides the product distribution, the stability of the ferrierite was investigated in two ways. Firstly, the isobutene selectivity during the coupling over time was observed. If the isobutene selectivity (see eqn (5)) decreases over time, the catalyst is deactivating. Secondly, the isobutene selectivity, decoupled from HTFT calculated viaeqn (7), was checked before and after the coupling with the 1 vol% 1-butene in N2 gas bottle. If there is no significant difference to be noticed, then the coupling did not affect the ferrierite catalyst.
Results and discussion
We started the reaction at 300 °C, 20 bar(g) and H2:CO ratio of 2.5. Stable HTFT operation was reached after approx. 150 h. The reaction was considered to have reached a steady state when the relative change of at least the last three measurements was less than 5%.
Optimal operating parameters – HTFT synthesis
The aim of these experiments was to maximise the selectivity of C3–C5 olefins, preferably butene, at high CO conversions. The temperature was varied from 290 °C to 320 °C, pressure increased from 15 bar(g) to 25 bar(g), H2:CO ratio from 1.7 to 2.5 and the GHSV from 1.07 × 104 h−1 to 3.22 × 104 h−1. The best results were achieved at 300 °C, 20 bar(g), H2:CO = 2 and high GHSV, see following sections. There was no significant deactivation noticed after more than 430 h on stream.
The activity increase from 290 °C to 300 °C can be explained by the increase of the reaction rate with temperature. Above 300 °C, the exothermic adsorption might be a limiting step. The endothermic desorption step is favoured at higher temperatures, leading to higher selectivity of methane and C2 hydrocarbons and lower selectivity of C3–C5 hydrocarbons. Another explanation for the slightly higher increase of the CO conversion can be found in the formulation of the kinetic term for the iron based Fischer–Tropsch reaction rate from Ledakowicz et al.40 Being the main inhibitors, higher H2O, and CO2 partial pressures slow down the reaction rate.
The increased CO and H2 partial pressures due to the higher pressure in the reactor lead to an enhanced surfaced coverage and a higher reaction rate and therefore higher conversion. In addition, the higher partial pressures support the chain growth leading to longer chains.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :CO effect.
First, the H2:CO ratio was increased from 2 to 2.5. Due to the higher CO partial pressure, the experiments at the H2:CO ratio of 2 took place at a higher conversion of 46.6%, compared to 37.6% CO conversion at a H2:CO ratio of 2.5. The hydrocarbon chain length decreased, while increasing the H2:CO ratio. The C3–C5 olefin selectivity was 2.2% higher at a H2:CO ratio of 2.5 compared to a ratio of 2. In addition, increasing the H2:CO ratio led to a slightly lower olefin to paraffin ratio, from 3.0 to 2.8. The higher H2 partial pressure supports the hydrogenation of the adsorbed hydrocarbons and leads therefore to lower olefin to paraffin ratios.
:CO effect.
First, the H2:CO ratio was increased from 2 to 2.5. Due to the higher CO partial pressure, the experiments at the H2:CO ratio of 2 took place at a higher conversion of 46.6%, compared to 37.6% CO conversion at a H2:CO ratio of 2.5. The hydrocarbon chain length decreased, while increasing the H2:CO ratio. The C3–C5 olefin selectivity was 2.2% higher at a H2:CO ratio of 2.5 compared to a ratio of 2. In addition, increasing the H2:CO ratio led to a slightly lower olefin to paraffin ratio, from 3.0 to 2.8. The higher H2 partial pressure supports the hydrogenation of the adsorbed hydrocarbons and leads therefore to lower olefin to paraffin ratios.
After decreasing the H2:CO ratio to 1.7 and running the central reaction operating point again, we noticed that the catalyst was deactivated, and the CO conversion decreased from approx. 30.1% to 14.6%. The catalyst was reduced and the experiment at the central operating point was repeated, which resulted in an increase of the CO conversion up to 23.7%, still lower than the initial one. Catalyst deactivation is a complex phenomenon. Deactivation pathways can be described based on pore blocking, sintering or carbon deposition.26,42 Pore blocking occurs through heavy hydrocarbons and takes place mostly at lower temperatures or with cobalt catalysts as a result of shifting the product distribution towards longer chains.43 Considering that deactivation was observed after changing the H2:CO ratio to 1.7, deposits of carbonaceous compounds on the catalyst active sites might be the deactivation reason. This so-called “soft coke” can be removed by hydrogenation, or it forms “hard coke” which cannot be removed by hydrogenation anymore and requires a combustion step. The increase in conversion after the hydrogenation indicates that the soft coke was partly hydrogenated. Nevertheless, the catalyst could not reach the initial activity, which also indicated hard coke formation.
:CO ratio of 2. Tripling the GHSV from 1.07 × 104 to 3.22 × 104 h−1 at 300 °C decreased the conversion from 56.4% to 33.1%, due to the shorter residence time of the feed in the reactor. Increasing the GHSV led to higher selectivity of C3–C5 olefins and slightly higher butene selectivity, due to the decreasing chain growth probability. The lower residence time supressed the readsorption of the olefins. Here, one must find a compromise between high olefins selectivity and high conversion, in order to maximise the yield. Higher yields, but lower CO conversions lead to the necessity to recycle. Recycling a part of the unconverted reactants, as well as the gaseous FT products, might lead to a hydrogenation of the olefin fraction and thus a decrease of the target products amount. Therefore, the impact of a recycling step should be further investigated in order to determine an optimal GHSV for the overall process.
Optimal operating parameters – skeletal isomerization reaction
The aim of these experiments was to maximise the isobutene selectivity at high 1-butene conversion. Temperature was varied from 300 °C to 400 °C and pressure was increased from 1 bar to 5 bar. The feed composition was 1 vol% 1-butene in N2. The best results were achieved at 400 °C and atmospheric pressure, see following sections.In comparison to the conversion, the temperature had a considerable effect on product selectivity. At a volume flow of 12 l h−1 of 1 vol% 1-butene in N2 and a temperature increase from 300 °C to 400 °C, the isobutene selectivity doubled. Even with lower feed volume flows of 6 l h−1, there was a significantly higher isobutene selectivity from 32% at 300 °C to approximately 45% at 400 °C. The increase in temperature simultaneously led to lower cis- and trans-butene proportions in the product stream. The double bond isomerization takes place at lower temperatures, while the skeletal isomerization is not favoured at these temperatures, which leads to a high fraction of 2-butenes. If the temperature increases, both 1-butenes and 2-butenes react to form isobutenes.
After increasing the pressure to 5 bar and running the central reaction operating point again, we noticed that the catalyst was deactivated, and the isobutene selectivity decreased from approx. 38.5% to 29%. The increased pressure supports the bimolecular mechanism and consequently the formation of byproducts. The bimolecular mechanism consists of two reaction steps: dimerization and cracking. The dimerization step can result in the formation of long-chain products that no longer crack and block the pores of the ferrierite. This then leads to a decrease in isobutene selectivity.
Coupling the gaseous products of the HTFT synthesis with skeletal isomerization reaction
By coupling the HTFT synthesis with the skeletal isomerization reaction this study revealed, that the side products of the HTFT synthesis affected the stable operation of the isomerization reaction at low temperatures. In addition, the side products of the HTFT synthesis further reacted in the isomerization process step, changing the product distribution of the isomerization reaction. The isobutene selectivity increased by approx. 2.2% to 3.1% depending on the temperature and degree of dilution, which corresponds to a selectivity up to 6 times higher than before the coupling. In addition, 1-pentene decreased and 2-pentene and isopentene were formed. The isopentene selectivity increased from approx. 0.08% to 2–3% depending on temperature and dilution. Double bond isomerization took place increasing the fraction of both 2-butene and 2-pentene. Besides reactions effecting the C4–C5 hydrocarbons, there were significant fraction changes in the C2–C3 and C6–C7 chains. This can be explained either by dimerization to longer hydrocarbons or by a cracking step toward shorter molecules.The influence of the temperature and dilution on product distribution and catalyst stability are explained in detail in the following sections.
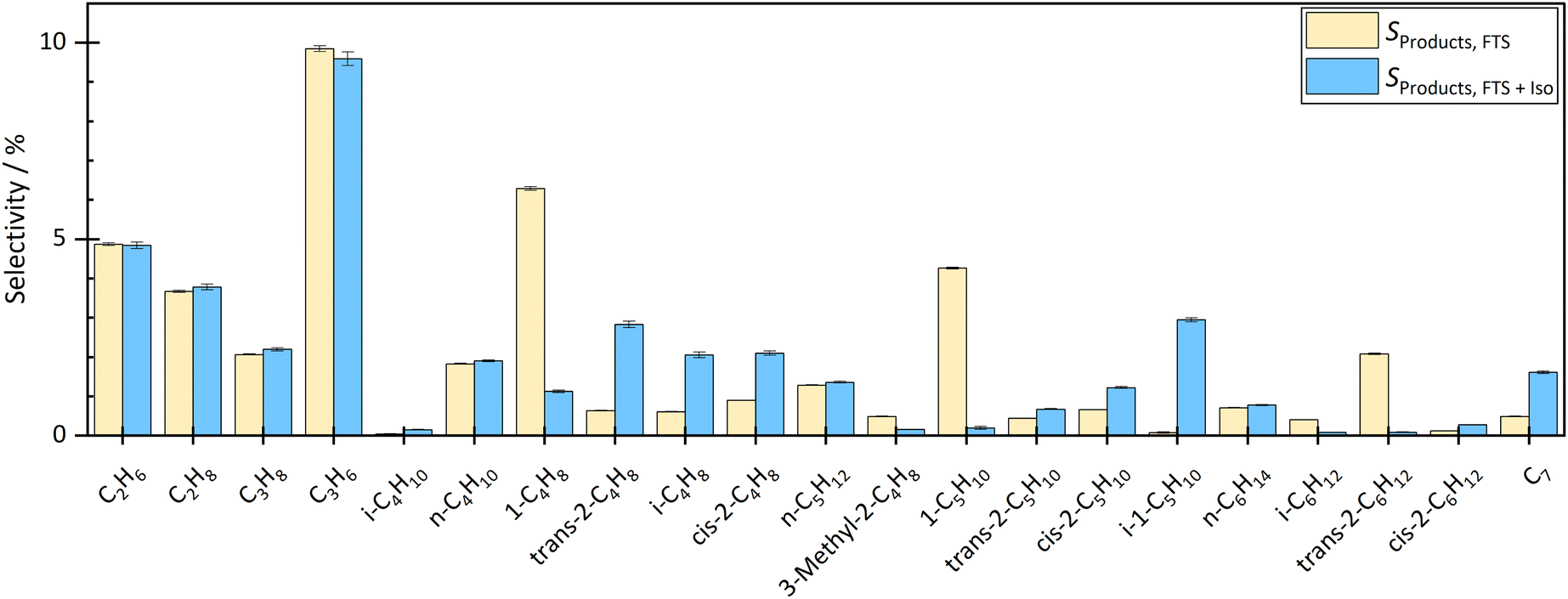 | ||
| Fig. 3 Comparison of product distribution before coupling and during coupling. The gas volume flow was VGas = 280 ml min−1. The N2 dilution in the feed was 35.7 vol%. The CO conversion of the HTFT was at T = 300 °C and p = 20 bar(g) with XCO = 35%. The temperature of the isomerization reactor during the coupling was T = 300 °C. The selectivity of methane remained unchanged at approximately SCH4 = 17%. | ||
 | ||
| Fig. 4 Comparison of product distribution before coupling and during coupling. The gas volume flow was VGas = 280 ml min−1. The N2 dilution in the feed was 35.7 vol%. The CO conversion of the HTFT was at T = 300 °C and p = 20 bar(g) with XCO = 37%. The temperature of the isomerization reactor during the coupling was T = 400 °C. The selectivity of methane remained unchanged at approximately SCH4 = 17%. | ||
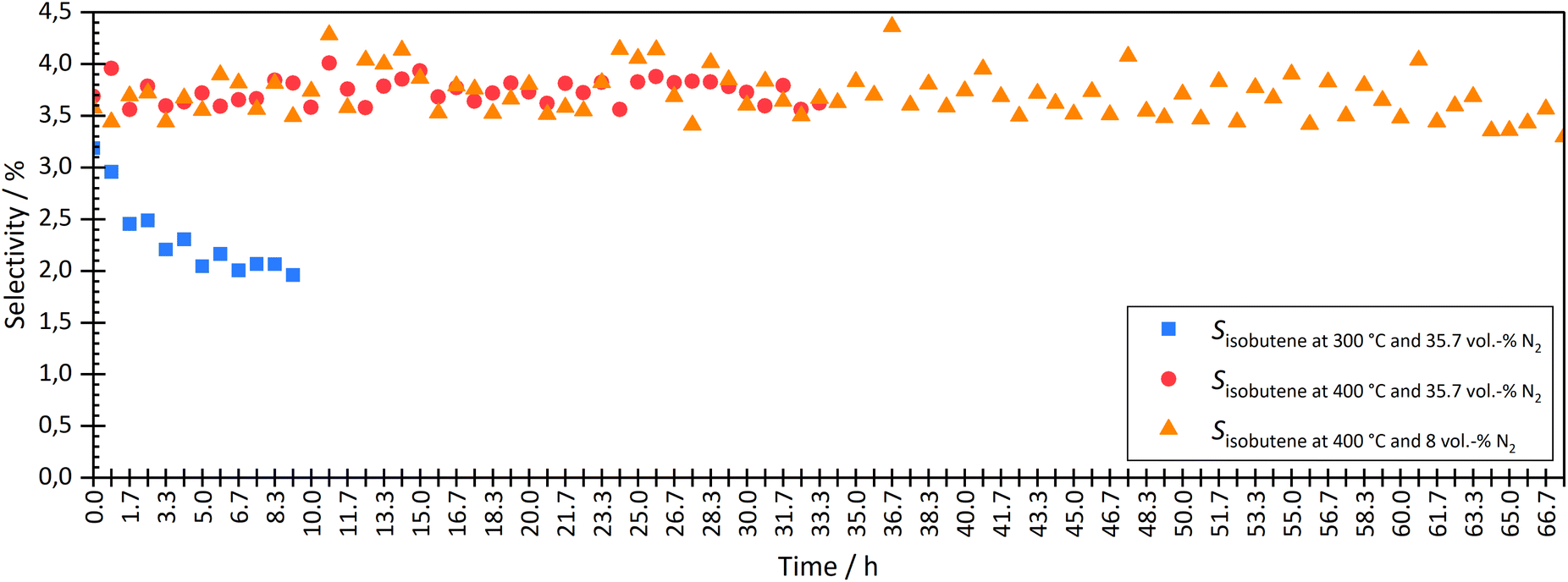 | ||
| Fig. 5 Comparison the isobutene selectivity over time during coupling at different temperatures (300 °C and 400 °C) and two different N2 dilutions (35.7 vol% and 8 vol%). | ||
At a temperature of the isomerization reactor of 300 °C, the isobutene selectivity decreased from approx. 2.8% to 2.0% within the first 10 hours. This indicated catalyst deactivation. The consumption of trans-hexene, without affecting the selectivity of short-chain molecules, indicated the formation of long-chain or cyclic molecules, which could not be measured with the given analytic system. At lower temperatures, the dimerization step of the bimolecular mechanism is more favoured than the cracking, which leads to the formation of longer chains and therefore pore blocking and catalyst deactivation over time on stream.
The experiment was carried out stably at 400 °C. At a high degree of dilution of approximately 35.7 vol% N2, the isobutene selectivity was on average approx. 3.7% ± 0.2% over a period of approximately 33 hours. There were no signs of deactivation. Even at a lower dilution level of 8 vol% N2, no deactivation was seen in the first 33 h. The average isobutene selectivity was also 3.7% ± 0.4%. The coupling continued until approximately 67 h. The average isobutene selectivity decreased by 0.1% between 33 h and 67 h. A slight but not significant deactivation was seen. Experiments with longer operating times are necessary to investigate the deactivation rate but were not carried out in this work.
Checking the catalyst decoupled from HTFT synthesis confirmed the results above. After coupling at 300 °C isomerization temperature and 35.7 vol% N2 dilution, isobutene selectivity decreased by 6.5%. These results also showed that coupling led to deactivation of the ferrierite catalyst. After coupling at 400 °C isomerization temperature and 35.7 vol% N2 dilution, no significant difference was seen in isobutene selectivity before and after coupling. Even at lower dilutions (8 vol% N2), no deactivation was seen. The selectivity difference of both isobutene and 2-butene before and after coupling was less than 0.5%.
Conclusions
The focus of this study was to investigate the coupling of the HTFT synthesis and the skeletal isomerization step in the proposed Power-to-Fuels route for the production of sustainable aviation gasoline. It is shown, that a stable operation of the coupled HTFT synthesis and the isomerization step is possible at 400 °C independently of the feed dilution. First, the operating parameters for each reaction were optimized. Varying the operating parameters of the HTFT synthesis, a high C3–C5 olefin selectivity, at preferably high CO conversion, was achieved at 300 °C, 20 bar(g) and H2:CO ratio of 2. The catalyst deactivated at H2:CO ratio lower than 2. An improvement of the catalyst stability at H2:CO ratios below 2 is necessary, since it would lead to higher C3–C5 olefin selectivity and production rates. Choosing the right GHSV is an engineering question which can be answered considering the overall process. Even if low GHSV leads to higher selectivity, the respective low production rate makes the process inefficient. Higher GHSV in the process, and as a result, higher production rate but also lower conversions, demands recycling units. The impact of such an operation mode on the secondary reactions, such as olefin hydrogenation, should be further investigated. Varying the operating parameters of the skeletal isomerization step, a high isobutene selectivity was reached at 400 °C and atmospheric pressure. Operating at a five-time higher pressure led to a decrease of the isobutene selectivity over time, most likely due to pore blocking. Higher pressures and therefore higher butene partial pressure support the bimolecular mechanism and form longer hydrocarbon chains, which then block the ferrierite channels. Coupling both reactions was investigated at two temperatures and two dilution rates. A steady-state operation coupled with a 35.7 vol% N2 dilution and 300 °C operating temperature of the isomerization reactor was not possible. Increasing the temperature of the isomerization reactor enabled a stable operation for at least 33 h. Decreasing the N2 dilution to 8 vol% did not affect the ferrierite stability significantly. After 67 h operation, the isobutene selectivity was 0.1% lower. Checking the catalyst decoupled from HTFT synthesis before and after the coupling confirmed these results. This study serves as a starting point for investigations of further process steps in the proposed Power-to-Fuels route for the production of sustainable aviation gasoline. While we showed the feasibility of a stable coupled operation of HTFT synthesis and the isomerization reaction, the influence of the side products on further steps like the indirect alkylation and the hydrotreatment has to be investigated experimentally in future studies.
Conflicts of interest
The authors declare no financial and/or personal relationship that might influence this work.Acknowledgements
The authors would like to thank Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH for funding this work as part of the project “ProQR – climate-neutral alternative fuels”, commissioned by Bundesministerium für Umwelt, Naturschutz, nukleare Sicherheit und Verbraucherschutz (BMUV).References
-
International Energy Agency, Global Energy Review 2021, 2021 Search PubMed
.
-
European Commission, The European Green Deal, 2019 Search PubMed
-
A. Schwab, A. Thomas, J. Bennett, E. Robertson and S. Cary, Electrification of Aircraft: Challenges, Barriers, and Potential Impacts, National Renewable Energy Laboratory, 2021 Search PubMed
- S. Drünert, U. Neuling, T. Zitscher and M. Kaltschmitt, Power-to-liquid fuels for aviation – processes, resources and supply potential under German conditions, Appl. Energy, 2020, 277, 115578 CrossRef
- B. I. Kamara and J. Coetzee, Overview of High-Temperature Fischer–Tropsch Gasoline and Diesel Quality, Energy Fuels, 2009, 23, 2242–2247 CrossRef CAS
- A. P. Steynberg, R. L. Espinoza, B. Jager and A. C. Vosloo, High temperature Fischer–Tropsch synthesis in commercial practice, Appl. Catal., A, 1999, 186, 41–54 CrossRef CAS
- M. F. Ahmad Kamaroddin, N. Sabli, T. A. Tuan Abdullah, S. I. Siajam, L. C. Abdullah, A. Abdul Jalil and A. Ahmad, Membrane-Based Electrolysis for Hydrogen Production: A Review, Membranes, 2021, 11(11), 810 CrossRef CAS PubMed
- J. Chi and H. Yu, Water electrolysis based on renewable energy for hydrogen production, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, Solid-state water electrolysis with an alkaline membrane, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed
-
Comparison of power-to-liquid processes using PEM electrolysis with RWGS and SOEC co-electrolysis, ed. D. H. König, R.-U. Dietrich and A. Wörner, 2015 Search PubMed
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, A review of high temperature co-electrolysis of H2O and CO2 to produce sustainable fuels using solid oxide electrolysis cells (SOECs): advanced materials and technology, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC
-
A. De Klerk and P. M. Maitlis, Greener Fischer-Tropsch processes for fuels and feedstocks, Wiley-VCH, Weinheim, 2013 Search PubMed
- W.-Q. Xu, Y.-G. Yin, S. L. Suib, J. C. Edwards and C.-L. OYoung, n-Butene Skeletal Isomerization to Isobutylene on Shape Selective Catalysts: Ferrierite/ZSM-35, J. Phys. Chem., 1995, 99, 9443–9451 CrossRef CAS
- A. De Klerk and P. L. De Vaal, Alkylate Technology Selection for Fischer–Tropsch Syncrude Refining, Ind. Eng. Chem. Res., 2008, 47, 6870–6877 CrossRef CAS
-
S. A. Treese, P. R. Pujadó and D. S. J. Jones, Handbook of Petroleum Processing, Springer International Publishing, Cham, 2015 Search PubMed
- S. I. Hommeltoft, Isobutane alkylation: recent developments and future perspectives, Appl. Catal., A, 2001, 221, 421–428 CrossRef CAS
- A. Corma and A. Martínez, Chemistry, Catalysts, and Processes for Isoparaffin–Olefin Alkylation: Actual Situation and Future Trends, Catal. Rev., 1993, 35, 483–570 CrossRef CAS
- R. A. Friedel and R. B. Anderson, Composition of Synthetic Liquid Fuels. I. Product Distribution and Analysis of C 5—C 8 Paraffin Isomers from Cobalt Catalyst 1, J. Am. Chem. Soc., 1950, 72, 1212–1215 CrossRef CAS
- Z. Gholami, F. Gholami, Z. Tišler, J. Hubáček, M. Tomas, M. Bačiak and M. Vakili, Production of Light Olefins via Fischer-Tropsch Process Using Iron-Based Catalysts: A Review, Catalysts, 2022, 12, 174 CrossRef CAS
- S. Lee, J.-C. Seo, H.-J. Chun, S. Yang, E. Sim, J. Lee and Y. T. Kim, Selective olefin production on silica based iron catalysts in Fischer–Tropsch synthesis, Catal. Sci. Technol., 2022, 12, 5814–5828 RSC
- D. B. Bukur, D. Mukesh and S. A. Patel, Promoter effects on precipitated iron catalysts for
Fischer-Tropsch synthesis, Ind. Eng. Chem. Res., 1990, 29, 194–204 CrossRef CAS
- Y.-Y. Ji, H.-W. Xiang, J.-L. Yang, Y.-Y. Xu, Y.-W. Li and B. Zhong, Effect of reaction conditions on the product distribution during Fischer–Tropsch synthesis over an industrial Fe-Mn catalyst, Appl. Catal., A, 2001, 214, 77–86 CrossRef CAS
- B. H. Davis, Fischer-Tropsch synthesis: relationship between iron catalyst composition and process variables, Catal. Today, 2003, 84, 83–98 CrossRef CAS
-
L. Domokos, Skeletal isomerization of n-butene over medium pore zeolites, 2000 Search PubMed
- C. W. Montgomery, J. H. McAteer and N. W. Franke, Catalytic Isomerization of n-Butane and Isobutane, J. Am. Chem. Soc., 1937, 59, 1768–1769 CrossRef CAS
-
A. De Klerk, Fischer-Tropsch Refining, Wiley-VCH, Weinheim, 1st edn, 2012 Search PubMed
-
J. B. Wise and D. Powers, in Environmental Catalysis, ed. J. N. Armor, American Chemical Society, Washington, DC, 1994, pp. 273–285 Search PubMed
- Y. Khitev, I. I. Ivanova, Y. Kolyagin and O. A. Ponomareva, Skeletal isomerization of 1-butene over micro/mesoporous materials based on FER zeolite, Appl. Catal., A, 2012, 441–442, 124–135 CrossRef CAS
-
R. Millini and G. Bellussi, in Zeolites in Catalysis, ed. J. Cejka, R. E. Morris and P. Nachtigall, Royal Society of Chemistry, Cambridge, 2017, pp. 1–36 Search PubMed
- M. Moliner, C. Martínez and A. Corma, Multipore zeolites: synthesis and catalytic applications, Angew. Chem., Int. Ed. Engl., 2015, 54, 3560–3579 CrossRef CAS PubMed
-
R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, Chemistry of Zeolites and Related Porous Materials, Wiley, 2007 Search PubMed
- J. T. F. Degnan, Applications of zeolites in petroleum refining, Top. Catal., 2000, 13, 349–356 CrossRef
- J. Houžvička and V. Ponec, Skeletal Isomerization of Butene: On the Role of the Bimolecular Mechanism, Ind. Eng. Chem. Res., 1997, 36, 1424–1430 CrossRef
- G. Seo, H. S. Jeong, D.-L. Jang, D. L. Cho and S. B. Hong, The role of carbonaceous deposits in the skeletal isomerization of 1-butene over ferrierite zeolites, Catal. Lett., 1996, 41, 189–194 CrossRef CAS
- L. Domokos, L. Lefferts, K. Seshan and J. Lercher, Isomerization of Linear Butenes to Iso-Butene over Medium Pore Zeolites, J. Catal., 2001, 197, 68–80 CrossRef CAS
- D. Jo, S. B. Hong and M. A. Camblor, Monomolecular Skeletal Isomerization of 1-Butene over Selective Zeolite Catalysts, ACS Catal., 2015, 5, 2270–2274 CrossRef CAS
- R. Byggningsbacka, N. Kumar and L.-E. Lindfors, Kinetic Model for Skeletal Isomerization of n-Butene over ZSM-22, Ind. Eng. Chem. Res., 1999, 38, 2896–2901 CrossRef CAS
- A. C. Butler and C. P. Nicolaides, Catalytic skeletal isomerization of linear butenes to isobutene, Catal. Today, 1993, 18, 443–471 CrossRef
- Z. X. Cheng and V. Ponec, On the problems of the mechanism of the skeletal isomerization of n-butene, Catal. Lett., 1994, 27, 113–117 CrossRef CAS
- S. Ledakowicz, H. Nettelhoff, R. Kokuun and W. D. Deckwer, Kinetics of the Fischer-Tropsch synthesis in the slurry phase on a potassium promoted iron catalyst, Ind. Eng. Chem. Process Des. Dev., 1985, 24, 1043–1049 CrossRef CAS
- J. Gorimbo, A. Muleja, X. Liu and D. Hildebrandt, Fischer–Tropsch synthesis: product distribution, operating conditions, iron catalyst deactivation and catalyst speciation, Int. J. Ind. Chem., 2018, 9, 317–333 CrossRef CAS
- N. E. Tsakoumis, M. Rønning, Ø. Borg, E. Rytter and A. Holmen, Deactivation of cobalt based Fischer–Tropsch catalysts: a review, Catal. Today, 2010, 154, 162–182 CrossRef CAS
- M. K. Niemel and A. O. I. Krause, The
long-term performance of Co/SiO2 catalysts in CO hydrogenation, Catal. Lett., 1996, 42, 161–166 CrossRef
| This journal is © The Royal Society of Chemistry 2024 |