
Donor–acceptor hetero[6]radialene-based three-dimensional covalent organic frameworks for organic pollutant adsorption, photocatalytic degradation, and hydrogen production activity†
Jing Han
Wang
a,
Ahmed E.
Hassan
b,
Ahmed M.
Elewa
 c and
Ahmed F. M.
EL-Mahdy
*a
c and
Ahmed F. M.
EL-Mahdy
*a
aDepartment of Materials and Optoelectronic Science, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan. E-mail: ahmedelmahdy@mail.nsysu.edu.tw
bDepartment of Chemistry, Faculty of Science, Al-Azhar University, Nasr City 11884, Cairo, Egypt
cNuclear Chemistry Department, Hot Laboratories Center, Atomic Energy Authority, Cairo 13759, Egypt
First published on 2nd May 2024
Abstract
Covalent organic frameworks (COFs) are promising photocatalysts for treating organic pollutants and solar-to-hydrogen conversion; however, their suboptimal energy band structure and rapid charge recombination limit their photocatalytic performance. Herein, we synthesized two donor–acceptor hetero[6]radialene-based TFP 3D COFs, TFP-Py and TFP-BF, with a unique [3,4]-linked ffc topological structure for the adsorptive and photocatalytic degradation of the rhodamine B (RhB) dye and photocatalytic hydrogen production from water. The acceptor is hetero[6]radialene and the donors are planar pyrene (Py) and twisted bifluorenylidene (BF). TFP 3D COFs surpass previously reported COFs in removing RhB from water in 10 min with a maximum adsorption capacity (Qm) of 840 mg g−1. Interestingly, the incorporation of the twisted BF structure into the hetero[6]radialene-based 3D COF backbone improved the band gap structure, charge transport, and photoinduced electron and hole isolation. The TFP-BF 3D COF degrades organic dye (RhB) with 1.4-fold greater catalytic activity (1.5 × 10−2 min−1) than the planar pyrene donor-derived TFP-Py 3D COF (1.1 × 10−2 min−1). It demonstrates a hydrogen evolution (HER) rate of 21.04 mmol g−1 h−1, obviating the need for a Pt cocatalyst, the highest COF-specific capacity ever recorded. This study illuminates how electron donor topology and hetero[6]radialene 3D structure affect photocatalysis.
1 Introduction
Directly turning solar power into chemical fuels through the production of hydrogen from water using sunlight is considered a potential way to build a renewable and environmentally friendly energy system because of the ecologically benign characteristics and extremely dense energy content of hydrogen.1 The apparent ease of using powdered, distributed photocatalysts or thin catalyst films is deceptive; no available catalyst meets all three criteria (low cost, high stability, and high solar-to-hydrogen efficiency).2 Several inorganic semiconductors have been investigated for hydrogen evolution since the first report of TiO2 as a photocatalyst.3,4 However, due to their large bandgaps, most inorganic photocatalysts can only absorb light within narrow wavelength ranges.5,6 Therefore, organic photocatalysts have recently been explored owing to their desirable properties such as the capacity to absorb several photons and the appropriate flexibility to transfer charged particles.7,8 In 1985, it was discovered that poly(p-phenylene) could perform as a photocatalyst for hydrogen evolution; however, its activity is low and the reaction takes place only in the presence of ultraviolet irradiation.9 Since then, further examples of visible and visible-ultraviolet organic photocatalysts for hydrogen production have been reported, such as carbon nitrides (CNs),10 poly(azomethine)s,11 covalent triazine-based frameworks (CTFs),12,13 linear conjugated polymers,14 and conjugated microporous polymers (CMPs).15–17 Because of its non-hazardous, cheap, and thermally/chemically very stable nature, CN is an especially attractive organic photocatalyst for hydrogen production.18,19 Nevertheless, poor electrical conductivity, inability to absorb illumination with a wavelength >460 nm, high recombination rate of charge carriers at high temperatures, and insolubility in most solvents all restrict its usefulness.20 In addition, long-range order is absent in organic photocatalysts, including CNs, conjugated polymers, and CTFs (they are semicrystalline or amorphous).21 The movement of photoactive charges to the catalyst surface may be impeded by this disorder.22 On the other hand, numerous inorganic synthetic materials have been explored as photocatalysts for organic dye photodegradation, including metal sulfides and oxides. However, their limited surface areas and growing toxicity have greatly constrained their usefulness.23,24 Hence, organic photocatalytic materials have been the subject of current studies as promising new technical developments for remediating organic dye pollution in water.A covalent organic framework (COF) is a type of crystalline organic polymer, which is stretched in two or three dimensions (2D or 3D) through π–π stacking and covalent contact to produce a long-range ordered structure.25 The unique porosity, excellent stability, and structural changeability of COFs can be accomplished by incorporating numerous construction components into arrangements using different covalent-bond-forming processes.26–29 COFs have sparked enormous scientific curiosity for applications in a variety of domains since the pioneering study reported by Yaghi and colleagues a decade ago.26–29 Active sites on the outer layer of the backbone and quite good porosity contribute to their functionality in a wide variety of areas, including selective separation,30 energy storage,31–33 chemical capture,34,35 optoelectronics,36 catalysis,37–40 sensors,41,42 and heterogeneous catalysis.43 From a purely structural standpoint, most 2D COFs feature stacking structures with one-way channels.43,44 Regrettably, 3D COFs have not been explored nearly as much, with only tetrahedral-geometry-based nets having been described.45–47 This is likely due to the need for more suitable building units and the challenge of crystallizing such structures. However, the multiple open sites and unique confinement effects that may be found in 3D COFs provide several options for extending the possible uses of COFs.48–50 As a result, there is a strong need for the directed synthesis of 3D COFs with unique topologies and functionalities.
3D COFs provide visible optical absorption and charge movement throughout the entire polymeric framework, both of which are necessary for the photocatalysis process.51,52 To further increase catalytic activity, the ordered porosity of 3D COFs allows for the quick passage of hydrogen into the catalytic site.53,54 Regrettably, high exciton binding energies and low charge carrier mobilities are two downsides of using COFs as photocatalysts, leading to poor photocatalytic performance.55 Furthermore, there are significant obstacles to the development and manufacture of COF substances as an individual category of photoelectric effective platforms with excellent functionality due to the necessity for careful regulation of the inherent semiconductor characteristics of the skeleton.56,57 Nevertheless, these problems are alleviated once donor–acceptor (D–A) structure COFs are established. Developing a D–A-based COF would be extremely beneficial since it can exhibit the intramolecular charge transfer phenomena caused by visible light absorption and is suitable for metal-free photocatalysis.57–59 Moreover, including a D–A moiety in a polymer network facilitates effective charge separation by decreasing the exciton binding affinity, leading to efficient catalysis.57–59 A few COFs have been examined for their photocatalytic hydrogen evolution capability thus far. They have typically featured limited numbers of donor and acceptor units—namely tris(4-aminophenyl)amine, pyrene, porphyrin, tetraphenylethylene, benzobisthiazole, and benzotrithiophene as electron donors, and tetraphenylethylene, pyrazine, triazine, thiadiazole, thiazolo[5,4-d]thiazole, and phloroglucinol units as electron acceptors.60–65 In addition, only a report has been mentioned using 3D COF as a photocatalyst to degrade organic pollutants.66 Hence, creating environmentally friendly donor and acceptor 3D COFs as semiconductor materials for the adsorption and photocatalytic degradation of natural dyes in aqueous solutions and the production of hydrogen from water remains a formidable undertaking.
To develop functional 3D COF photocatalysts, the justification of the selection of covalent bonds and photoactive predecessors is essential. Therefore, we are intrigued by discovering innovative 3D COFs based on β-keto-enamines that feature D–A type structures, prolonged conjugation skeletons, and stronger water contact to enhance the photocatalytic activity. A trigeminal unit known as 1,3,5-triformylphloroglucinol (TFP-3OHCHO) can link with arylamine to achieve an enol-imine resonance configuration. The isomerization reaction of TFP-3OHCHO is conducive to the carbonyl formation (Fig. 1), resulting in the [6] radialene structure, which is an electron-deficient skeleton.67 In addition, the transformation of enol-imine into the β-keto-enamine resonance structure of the TFP-3OHCHO unit strongly improves the chemical stability and photoelectric properties through hydrogen-bond formation. Pyrene (Py) and bifluorenylidene (BF) units, which contain electron-rich arylamines are used as electron donors. The pyrene unit has a planar structure that can enhance stability, along with a long π-conjugated structure that could bring about photogenerated separation and transportation of charge carriers, gaining favorable photoelectric activity.68,69 9,9′-Bifluorenylidene is a suitable non-fullerene-type electron donor presenting a dimerized fluorene structure, twisted π-conjugated structure, and appealing charge transport and electron-donating properties (Fig. 1).70 To the best of our knowledge, no previous reports on the synthesis of three-dimensional (3D) COFs based on β-keto-enamine have been presented. Additionally, their use in photocatalytic processes has not been studied. In the present study, the TFP-3OHCHO acceptor was coupled with two electron donors of (4,4′,4′′,4′′′-pyrene-1,3,6,8-tetrayl) tetraaniline (PyTA-4NH2) and 4,4′,4′′,4′′′-([9,9′-bifluorenylidene]-3,3′,6,6′-tetrayl)tetraaniline (BFTB-4NH2) to produce two donor–acceptor β-keto-enamine-based TFP 3D COFs, namely TFP-Py and TFP-BF 3D COFs, for the adsorptive and photocatalytic degradation of organic chemical dyes, as well as photocatalytic production of H2 from water (Fig. 2a).
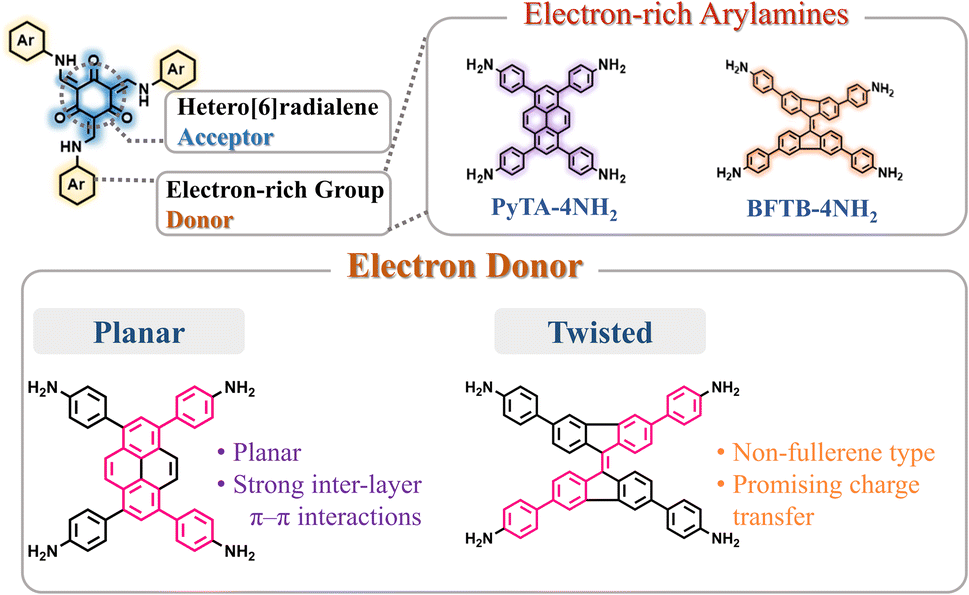 | ||
| Fig. 1 Schematic representation of our principle for the design of the donor–acceptor structure with the hetero[6]radialene core as the acceptor, and planar pyrene and twisted bifluorenylidene cores as donors. | ||
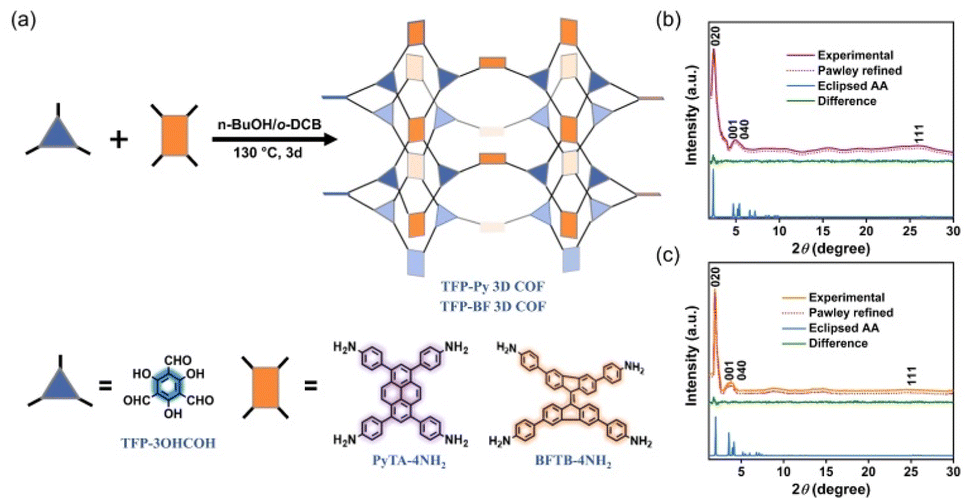 | ||
| Fig. 2 (a) Synthesis of the donor–acceptor hetero[6]radialene-based TFP 3D COFs, exhibiting a unique [3,4]-linked ffc topological structure. PXRD patterns of (b) TFP-Py and (c) TFP-BF 3D COFs: experimental patterns (violet and orange), simulated Pawley refined patterns (red), their difference (green), and simulated patterns obtained from the A–A (blue) stacking models. | ||
The results revealed that the photocatalytic activity of the derived 3D COF is strongly influenced by the configuration of the electron donor. TFP-BF 3D COF obtained from the twisted 9,9′-bifluorenylidene donor demonstrates significant catalytic activity in the degradation of organic dye (RhB), with a reaction rate constant (1.5 × 10−2 min−1) that is 1.4-fold larger than that of TFP-Py 3D COF obtained from the planar pyrene donor (1.1 × 10−2 min−1). Interestingly, photocatalytic hydrogen evolution rate (HER) measurements under UV-vis light revealed that the TFP-BF 3D COF has a value of 21.04 mmol h−1 g−1, significantly higher than that of the TFP-Py 3D COF (4.96 mmol h−1 g−1). This study sheds light on how the geometry of the electron donor and the three-dimensional structure of β-keto-enamines affect photocatalysis.
2 Results and discussion
2.1. Material synthesis and characterization
The 3D COFs were specifically developed by connecting triangular and rectangular precursors using a [3 + 4] condensation process. Fig. 2a displays our syntheses of the two donor–acceptor β-keto-enamine-based TFP 3D COFs, namely, TFP-Py and TFP-BF 3D COFs, through the solvothermal condensations of the trialdehyde TFP-3OHCHO (Scheme S1 and Fig. S1−3†) with the tetramines PyTA-4NH2 and BFTB-4NH2 (Schemes S2−S4 and Fig. S4−S9†), respectively, in a mixture of o-DBC/n-butanol/6 M acetic acid (5 mL/5 mL/0.1 mL) at 120 °C for three days. The two 3D COFs are insoluble in common solvents such as N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), MeCN, ethanol (EtOH), acetone, dichloromethane (DCM), THF, etc. We employed powder X-ray diffraction (PXRD) using Cu Ka radiation for assessing the crystallinity of our 3D COFs (Fig. 2b and c). The experimental PXRD patterns of the TFP-Py 3D COF (Fig. 2b, violet curve) and the TFP-BF 3D COF (Fig. 2c, orange curve) featured strong peaks at 2θ = 2.46° and 1.91°, respectively, which we assigned to lattices with (020) reflection facets. For TFP-Py 3D COF, the minor overlapped peaks at 2θ of 4.66° and 5.15° were assigned to the (001) and (040) reflection planes, respectively. We attributed the small and broad peak observed at a 2θ value of 26.03° to the reflection originating from the (111) plane. For the TFP-BF 3D COF, the overlapped peaks corresponding to the (001) and (040) reflection planes appeared at 2θ of 3.40° and 4.20°, respectively, with another broad peak at 25.10°, which can be attributed to the (111) plane. These PXRD patterns proved that our new TFP-Py and TFP-BF 3D COFs are microcrystalline architectures and are highly organised. The frameworks of TFP-Py and TFP-BF 3D COFs were modeled using Material Studio, which suggested that both COFs adopt an ffc topology. The Pawley XRD refinements of the TFP-Py and TFP-BF 3D COFs (Fig. 2b and c, red curves) were carried out, and very little discrepancies between the results and experimental PXRD patterns showed that they were consistent (Fig. 2b and c, green curves). To obtain additional information about their unit cell parameters, we simulated ffc nets of the TFP-Py and TFP-BF 3D COFs. The simulated XRD patterns (Fig. 2b and c, blue curves) were highly consistent with the experimental ones. In addition, the refinement results yielded unit cell parameters nearly equivalent to the predictions with good agreement factors (a = 36.9148 Å, b = 19.2861 Å, c = 39.9578 Å, α = 76.58°, β = 76.58°, γ = 91.34° for TFP-Py 3D COF; a = 46.6438 Å, b = 25.6726 Å, c = 31.070 Å, α = 90.00°, β = 90.00°, γ = 93.10° for TFP-BF 3D COF) (Tables S1 and S2†). These results matched the previously reported 3D COFs with ffc topology.57 Fig. S10 and S11† depict the FTIR spectra of TFP-Py and TFP-BF 3D COFs and compare their monomers to highlight the growth and decay of the peak. TFP-3OHCHO exhibited two distinct signals for CH = O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C units at 1651 and 1428 cm−1 in addition to a strong, wide absorption for phenolic –OH groups at 3460 cm−1. FTIR spectra of the tetramines PyTA-4NH2 and BFTB-4NH2 monomers featured signals for amino NH2 units at 3452–3341 cm−1, C–H stretching at 3122–3023 cm−1, aromatic CC stretching at 1618–1516 cm−1, and C–N units at 1277 cm−1 (Fig. S10 and S11†). The FTIR spectra of the TFP-Py and TFP-BF 3D COFs lacked any discernible signals corresponding to the phenolic –OH groups of TFP-3OHCHO or the amino –NH2 groups of the unbound tetramine monomers. This observation confirms that these functional groups have been fully utilized and are no longer present in the COF structures. The presence of TFP-BF 3D COF in its keto-enamine form was verified through the observation of broad peaks at 3432–3428 cm−1 corresponding to the N–H functional groups, 1615–1614 cm−1 corresponding to the CO functional groups, 1459–1450 cm−1 corresponding to the conjugated CC functional groups, and 1294–1290 cm−1 corresponding to the C–N functional groups. The synthesized 3D COFs in our study were observed to exist in the keto-enamine tautomeric form. This structural arrangement is upheld by several robust hydrogen bonds formed between the carbonyl (CO) groups and the amino (N–H) groups.71
C units at 1651 and 1428 cm−1 in addition to a strong, wide absorption for phenolic –OH groups at 3460 cm−1. FTIR spectra of the tetramines PyTA-4NH2 and BFTB-4NH2 monomers featured signals for amino NH2 units at 3452–3341 cm−1, C–H stretching at 3122–3023 cm−1, aromatic CC stretching at 1618–1516 cm−1, and C–N units at 1277 cm−1 (Fig. S10 and S11†). The FTIR spectra of the TFP-Py and TFP-BF 3D COFs lacked any discernible signals corresponding to the phenolic –OH groups of TFP-3OHCHO or the amino –NH2 groups of the unbound tetramine monomers. This observation confirms that these functional groups have been fully utilized and are no longer present in the COF structures. The presence of TFP-BF 3D COF in its keto-enamine form was verified through the observation of broad peaks at 3432–3428 cm−1 corresponding to the N–H functional groups, 1615–1614 cm−1 corresponding to the CO functional groups, 1459–1450 cm−1 corresponding to the conjugated CC functional groups, and 1294–1290 cm−1 corresponding to the C–N functional groups. The synthesized 3D COFs in our study were observed to exist in the keto-enamine tautomeric form. This structural arrangement is upheld by several robust hydrogen bonds formed between the carbonyl (CO) groups and the amino (N–H) groups.71
It has been reported that the keto-enamine salicylideneanilines72 and β-keto-enamine-based COFs73 have strong intramolecular hydrogen bonds between the carbonyl (CO) groups and the amino (N–H) groups. To confirm the existence of the intramolecular hydrogen bonds between the carbonyl and amino groups in our β-keto-enamine-based TFP 3D COFs, we prepared the model compound (TFP-An) (Scheme S5†) and observed the chemical shift positions of the amino (N–H) groups in chloroform and chloroform/DMSO solvents. Due to its exceptional ability to accept hydrogen bonds, DMSO possesses a greater propensity to cleave intramolecular hydrogen bonds; however, this comes at the expense of a more intense interaction between DMSO and the substrate.74 The amino (N–H) group of TFP-An underwent a downfield shift in Fig. S12† when 30% DMSO-d6 was added, confirming the rupture of the intramolecular hydrogen bond at the expense of the formation of strong intermolecular hydrogen bonds with the DMSO solvent.
Fig. 3a and b display the solid-state (ss) 13C NMR spectra of our TFP 3D COFs that were spinning at a 13C cross-polarization magic angle. The 13C NMR spectrum of TFP-3OHCHO revealed a signal at 191.98 ppm corresponding to the CH = O carbon, and a signal at 173.63 ppm corresponding to the C–OH carbon atoms (Fig. S3†). The latter signal disappeared following the solvothermal condensations with the tetramine monomers, forming TFP 3D COFs. Signals at 190.24–180.74 ppm for CO units could be seen in the ss 13C NMR spectra of TFP-Py and TFP-BF 3D COFs (Fig. 3b). In addition, enamine (C–NH) and α-enamine (CC) signals both appeared in the ranges of 155.06–144.02 and 111.16–104.13 ppm, respectively. The ss13C NMR spectra demonstrated the proper formation of β-keto-enamine-linked 3D COFs (Fig. 3b).
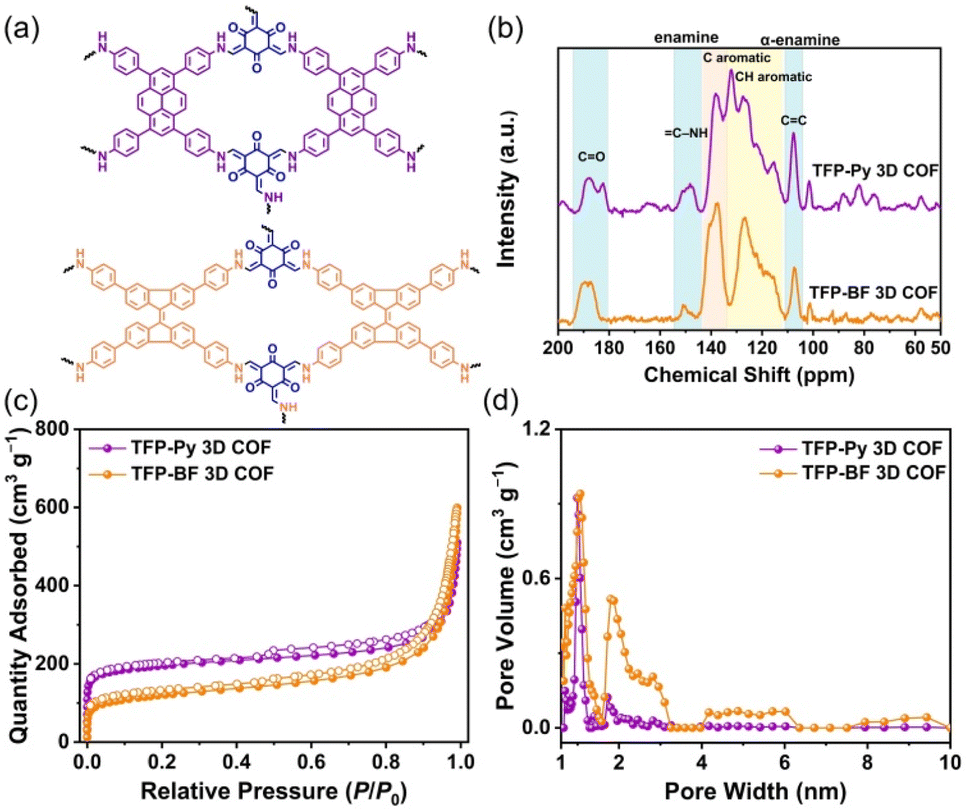 | ||
| Fig. 3 (a) Chemical structures of TFP-Py and TFP-BF 3D COFs. (b) 13C ssNMR of TFP-Py and TFP-BF 3D COFs. (c) Nitrogen sorption isotherms at 77 K of TFP-Py and TFP-BF 3D COFs. (d) Pore size distributions of TFP-Py and TFP-BF 3D COFs. | ||
Thermal gravimetric analysis (TGA) was performed to identify the thermal stability of our β-keto-enamine-linked 3D COFs. With a 10 °C per minute heating rate, the TGA investigations were conducted under a nitrogen environment where the temperature runs from 100 to 800 °C (Fig. S13†). The TFP-Py 3D COF obtained from the planar pyrene monomer exhibited higher thermal stability with a decomposition temperature of 10 weight percent (Td10) of around 427.52 °C and a char yield of 67.07%. In contrast, the TFP-BF 3D COF, which was synthesized using the twisted 9,9′-bifluorenylidene donor monomer, had reduced thermal stability. The Td10 value for this COF was measured to be 365.16 °C, demonstrating a char yield of 62.77% (Fig. S13 and Table S3†). The thermal stability of the resulting TFP 3D COF was shown to be considerably influenced by the planarity of the constructing linkers. A higher degree of planarity in the monomer led to increased thermal stability in the TFP 3D COF structure. Considering the importance of pore size and surface area for photocatalytic materials, we used N2 sorption isothermal studies at 77 K to examine the porosity characteristics of the TFP-Py and TFP-BF 3D COFs. The nitrogen sorption isotherms of both 3D COFs featured a type I isotherm with quick N2 absorption at low pressure (P/P0 < 0.05), confirming the microporous natures of our 3D COFs (Fig. 3c). The BET model was used to examine TFP 3D COF pore diameters and surface areas. The two TFP-Py and TFP-BF 3D COFs showed high BET surface areas, which were evaluated to be 731 and 447 m2 g−1, respectively, accompanied by pore volumes of 0.94 and 0.99 cm3 g−1, respectively (Table S4†). It was found that the planarity of the building blocks had a significant impact on the BET surface area of the COF, with a positive correlation between the two. Thus, the TFP-Py 3D COF obtained from the planar pyrene monomer exhibited a much higher BET surface area than TFP-BF 3D COF produced from the twisted 9,9′-bifluorenylidene. The hierarchical porous nature of these TFP 3D COFs was corroborated by pore size distribution profiles created by the nonlocal density functional theory (NLDFT). The results displayed that TFP-Py 3D COF exhibited pore sizes of 0.97/1.73 nm, while TFP-BF 3D COF was 1.01/1.82 (Fig. 3d and Table S4†). This agrees with the theoretical pore sizes indicated by the crystal structures; the bigger pore acts as two smaller ones, with sizes that are closer to the small pore in the frameworks (Fig. S14†).
X-ray photoelectron spectroscopy (XPS) examinations investigated the compositions and orbital spaces of the C, N, and O species in these catalysts. Fig. S15† reveals three peaks at 284 and 399 eV for architectural carbons and nitrogens, and 532 eV for adsorbed water and oxygen. We fitted the XPS profiles for the C 1s, N 1s, and O 1s orbitals to learn more about the TPP-CTFs' N and O species. Fig. S16 and Table S5† reveal that TFP-Py 3D COF had three primary peaks in the C 1s spectrum, corresponding to C–N, CC, and CO at 283.58, 284.42, and 286.40 eV, respectively. The N 1s spectrum displayed C–N at 399.47 eV, while the peaks at 531.08 and 533.15 eV were attributed to CO and H2O, respectively, in the O 1s spectrum. The peaks at 399.47 eV and 531.08 eV for C–N and CO were attributed to β-keto-enamine-linkage formed by a Schiff-base condensation reaction. Further, TFP-BF 3D COF showed three C 1s spectra peaks: C–N, CC, and CO at 283.58, 284.34, and 286.47 eV, respectively. The N 1s spectrum of N 1s showed C–N at 399.84 eV, whereas O 1s showed CO and H2O at 531.45 and 533.02 eV (Fig. S16 and Table S5†). The theoretical element ratio was calculated based on the atomic percentages of the respective elements constituting the donor and acceptor, while the experimental element ratio was calculated from the peak area of each species after using sensitivity factor correction in the XPS survey of TFP 3D COFs and the two ratios are listed in Table S6.† According to the literature, the sensitivity factors involving C 1s, N 1s, and O 1s had the following values: 0.296, 0.477, and 0.711.75
We explored the morphologies and microstructures of these TFP 3D COFs using transmission electron microscopy (TEM) and field-emission scanning electron microscopy (FE-SEM) (Fig. 4a–n). FE-SEM imaging revealed the micrometer-sized rod-like and twisted fiber-like morphologies of the TFP-Py and TFP-BF 3D COFs, respectively (Fig. 4a and h). The TEM images confirmed the formation of the rod-like morphology of TFP-Py 3D COF with an average diameter of 50–70 nm and a length of several micrometers (Fig. 4b and c), as well as the formation of the twisted fiber-like morphology of TFP-BF 3D COF with diameter and length of several nanometers (Fig. 4i and j). In addition, the TEM images of TFP-Py and TFP-BF 3D COFs revealed well-ordered nanochannel arrangements, indicating the high quality and excellent crystallinity of both TFP 3D COFs (Fig. 4d and k). The lattice fringe spacing is 1.73 and 1.95 nm, which we assigned to the (040) reflection plane of TFP-Py and TFP-BF 3D COFs (Fig. 4d and k), respectively, in agreement with the PXRD patterns.76 The crystalline structures of both TFP 3D COFs were also verified by the selected area electron diffraction (SAED) patterns (Fig. 4d and k). The TEM images of TFP-Py and TFP-BF 3D COFs were further analyzed using the fast.
 | ||
| Fig. 4 FE-SEM images of (a) TFP-Py and (h) TFP-BF 3D COFs. HR-TEM images of TFP-Py and TFP-BF COFs recorded at (b and i) high-magnification, with a scale bar of 100 nm, and (c–e, j–l) low-magnification, with (c and j) a scale bar of 20 nm and (d, e, k and l) a scale bar of 2 nm; inset: the SAED pattern. The FFT image from the corresponding yellow square using the image J software of (f) TFP-Py and (m) TFP-BF 3D COFs. The simulated FFT diagrams of a yellow selected square of (g) TFP-Py and (n) TFP-BF 3D COFs. | ||
Fourier transform (FFT) method of the Image J software. The simulated FFT diagrams of selected areas of TFP-Py and TFP-BF 3D COFs exhibited lattice fringe spacings of 1.77 nm for TFP-Py 3D COF (Fig. 4e–g) and 2.27 nm for TFP-BF 3D COF (Fig. 4l–n), corresponding to the (040) reflection plane. Therefore, the d040 spacing matched the PXRD and TEM analyses.
2.2. Photophysical and electrochemical properties
The photophysical and electrochemical properties of the two D–A TFP 3D COFs were characterized by ultraviolet-visible diffuse reflectance spectra (UV-vis DRS), photoluminescence (PL) emission spectra, photocurrent measurements, and electrochemical impedance spectroscopy (EIS) using the solid-state COF powder. Fig. 5a reveals broad light absorption intensities extending to 800 nm for both TFP 3D COFs, with absorption onsets at wavelengths of 523 nm for TFP-Py 3D COF and 607 nm for TFP-BF 3D COF, demonstrating their strong visible-light-capture capability. This property also implies that such TFP-Py and TFP-BF 3D COFs could substantially absorb visible radiation for photocatalytic activity. The red-shifting of the absorption signal of TFP-BF 3D COF, relative to that of TFP-Py 3D COF, was due to the twisted structure of the bifluorenylidene unit.77 Furthermore, the optical band gap was calculated to be 1.80 eV for TFP-BF 3D COF by the Tauc plot, which is narrower than TFP-Py 3D COF (Eg = 1.95 eV) (Fig. 5b). Thus, changing the donor unit structure allowed us to fine-tune Eg values. In addition, the TFP-BF 3D COF had a shorter bandgap than TFP-Py 3D COF, indicating that it could effectively enhance the photocatalytic activity (Table 1). The highest occupied molecular orbitals (HOMOs) of these TFP 3D COFs were identified by ultraviolet photoelectron spectroscopy (UPS) (Fig. S17 and S18†). The lowest unoccupied molecular orbitals (LUMOs) levels were obtained by subtracting the band gaps from the HOMO energy levels (Table S7,†Fig. 5c). For TFP-Py 3D COF, the HOMO and LUMO energy levels were −4.89 and −2.94 eV, while those for TFP-BF 3D COF were −4.99 and −3.19 eV, respectively (Fig. 5c). The higher negative value of the LUMO position for the TFP-BF 3D COF as compared to that for the TFP-Py 3D COF suggests that TFP-BF 3D COF has a stronger ability to reduce protons to hydrogen gas. The fundamental stage of the optical photocatalytic processes is photo-induced electron–hole pair production, separation, and migration. Therefore, photoluminescence (PL) spectra were employed to assess how well the charge carrier separation and trapping were carried out. In the field of PL spectroscopy, the emission efficiency is tightly connected to the probability of radiative (electron–hole recombination) and nonradiative (charge-carrier separation) transitions.78 Therefore, the decrease in PL emission observed in TFP-BF 3D COF could be linked to a greater probability of nonradiative charge-carrier separation, while the higher emission in TFP-Py 3D COF indicates a more favorable radiative electron–hole recombination mechanism (Fig. 5d). The lower emission in TFP-BF 3D COF implied that a larger number of photoexcited electrons were likely participating in the photocatalytic activity rather than undergoing recombination with holes through fluorescence emission. The reduction in band gap can also be ascribed to the heightened photocatalytic activity resulting from alterations in the redox potential for photocatalytic activities, thereby enhancing charge transfer. In addition, the enhanced crystallinity would diminish the occurrence of defects and inhibit electron–hole recombination.79 Thus, the crystallinity and band gap appear to be more influential factors for photocatalytic activity, promoting enhanced charge transfer and decreased electron–hole recombination. These factors elucidated the significant decrease in PL intensity observed in TFP-BF 3D COFs. Furthermore, the inclusion of the BF moiety has been shown to effectively impede the process of charge carriers recombining through radiation, resulting in a significant enhancement of charge-carrier separation. Furthermore, these hypotheses were reinforced by analyzing the fluorescence lifetime decays of the TFP-Py and TFP-BF 3D COFs (Fig. S19†). The fluorescence lifetime of the TFP-Py 3D COF was 4.42 ns, whereas the fluorescence lifetime of the TFP-Py 3D COF was 6.31 ns, indicating the enhanced charge-carrier separation efficiency of the TFP-BF 3D COF.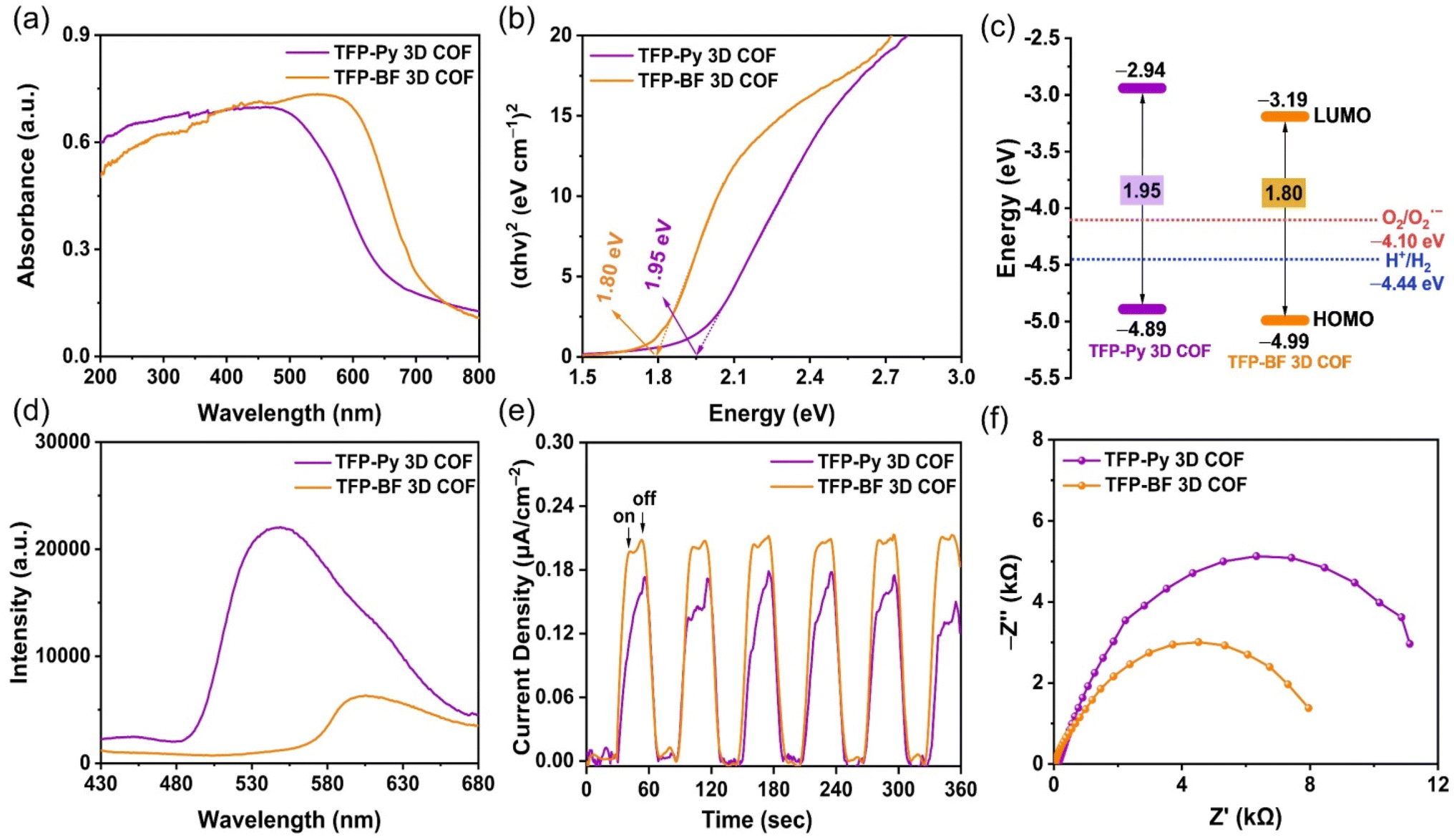 | ||
| Fig. 5 (a) UV-vis DRS spectra of TFP-Py and TFP-BF 3D COFs. (b) Tauc plots from the UV-vis spectra of TFP-Py and TFP-BF 3D COFs. (c) Energy level diagrams of TFP-Py and TFP-BF 3D COFs. (d) Photoluminescence spectra of TFP-Py and TFP-BF 3D COFs. (e) Periodic on/off photocurrent responses of TFP-Py and TFP-BF 3D COFs under irradiation with UV-vis light. (f) EIS spectra of TFP-Py and TFP-BF 3D COFs. | ||
| 3D COF | HOMO/LUMO (eV)a,b | Bandgap (eV)c | Pd (wt%) | HER (mmol g−1 h−1)d | AQY (%)e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 420 nm | 460 nm | 500 nm | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a HOMO measured using a photoelectron spectrometer. b LUMO = EHOMO − Eg. c Calculated from Tauc plots. d Conditions; the dosage of each photocatalyst was 2 mg using AC as the hole-scavenger under UV-vis light. e AQYs measured at 420, 460, and 500 nm. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TFP-Py | −4.89/−2.94 | 1.95 | 1.21 | 4.96 | 2.54 | 4.21 | 0.49 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TFP-BF | −4.99/−3.19 | 1.80 | 1.02 | 21.04 | 8.70 | 10.12 | 15.73 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This assertion was further substantiated by the results of the photocurrent test, which demonstrated that the TFP-BF 3D COF exhibited a higher photocurrent density as compared to the TFP-Py 3D COF when subjected to intermittent visible light irradiation (Fig. 5e). This observation suggested that a greater number of light-induced excitons can be generated throughout the photocatalytic process and that charge migration is more efficient in the TFP-BF 3D COF photocatalyst. The charge migration characteristic of the two D–A TFP 3D COFs was further investigated using electrochemical impedance spectroscopy (EIS). Fig. 5f and Fig. S20a† display the Nyquist plots of the TFP 3D COFs. By evaluating the Z′ axis intercept, we determined the intrinsic ohmic resistance (Rs) that represents the conductivity of TFP-Py and TFP-BF 3D COFs. The TFP-BF 3D COF revealed a lower Rs value of 27.5 Ω, whereas TFP-Py 3D COF revealed a higher Rs value of 138 Ω, implying that TFP-BF 3D COF has greater conductivity and more efficient charge transfer than TFP-Py 3D COF, which supports the results of the measurements of photoluminescence and photocurrent. The experimental Nyquist graphs (Fig. S20b, c and Table S8†) were fitted using a similar circuit, where Rs represents the ohmic resistance and Rct represents the charge transfer resistance. The TFP-Py 3D COF showed Rs and Rct of 138 and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 315 Ω, respectively, while the TFP-BF 3D COF showed Rs and Rct of 28 and 9062 Ω, respectively (Table S8†). The lower Rct value of the TFP-BF 3D COF compared to the TFP-Py 3D COF revealed that the TFP-BF 3D COF had greater charge transfer. The collective results confirm that our TFP-BF 3D COF has a commendable capacity to segregate and transport photogenerated electrons when subjected to illumination effectively.
315 Ω, respectively, while the TFP-BF 3D COF showed Rs and Rct of 28 and 9062 Ω, respectively (Table S8†). The lower Rct value of the TFP-BF 3D COF compared to the TFP-Py 3D COF revealed that the TFP-BF 3D COF had greater charge transfer. The collective results confirm that our TFP-BF 3D COF has a commendable capacity to segregate and transport photogenerated electrons when subjected to illumination effectively.
According to the aforementioned findings, the superior charge transfer and photocurrent density exhibited by TFP-BF 3D COF in comparison to TFP-Py 3D COF can be attributed to the following advantageous factors. First, the lower bandgap of the TFP-BF 3D COF facilitated a broader absorption of the solar spectrum and a more efficient generation of charge carriers. Second, the smaller diameter of the twisted fiber-like TFP-BF 3D COF (several nanometers), in comparison to the diameter of TFP-Py 3D COF (several micrometers), led to a decrease in the path that electrons and holes produced by light were required to travel to arrive at the sites of reactions on the outermost layer. Third, the more valuable electronic conductivity and small charge transfer obstructions of the TFP-BF 3D COF relative to the TFP-Py 3D COF enabled an ultrahigh charge transfer rate.
2.3. Dye adsorption: kinetics and reusability
The augmentation of N- and O-functionalized groups within the chemical structure of a conjugated polymer has been shown to significantly enhance its capacity for adsorbing organic dye molecules in aqueous environments. The active sites of these conjugated polymers have a significant charge density of nitrogen atoms, which enhances the bonding between the conjugated polymer and dye molecules. This bonding is sustained by several non-covalent interactions, as discussed in the study by Yuan et al.80 Additionally, it should be noted that the conjugated polymers possess high surface areas and porous architectures, which, therefore result in the significant adsorption of tiny dye molecules.81,82 Furthermore, it was hypothesized that the inclusion of pyrene and bifluorenylidene functional groups within the primary structures of our TFP 3D COFs would render them very favorable as organic adsorbents and photocatalysts for the degradation of dye molecules in aqueous environments. We employed a synthetic cationic pigment known as rhodamine B (RhB) as a standard guest molecule to evaluate the adsorption efficiencies of TFP 3D COFs. In our study, we assessed the efficacy of TFP-Py and TFP-BF 3D COFs in adsorbing RhB dye by analyzing the UV-vis spectra of aqueous solutions. We specifically focused on monitoring the changes in the maximum adsorption peak at a wavelength of 554 nm over different time intervals (ranging from 0 to 60 min) subsequent to the introduction of the COFs (Fig. 6).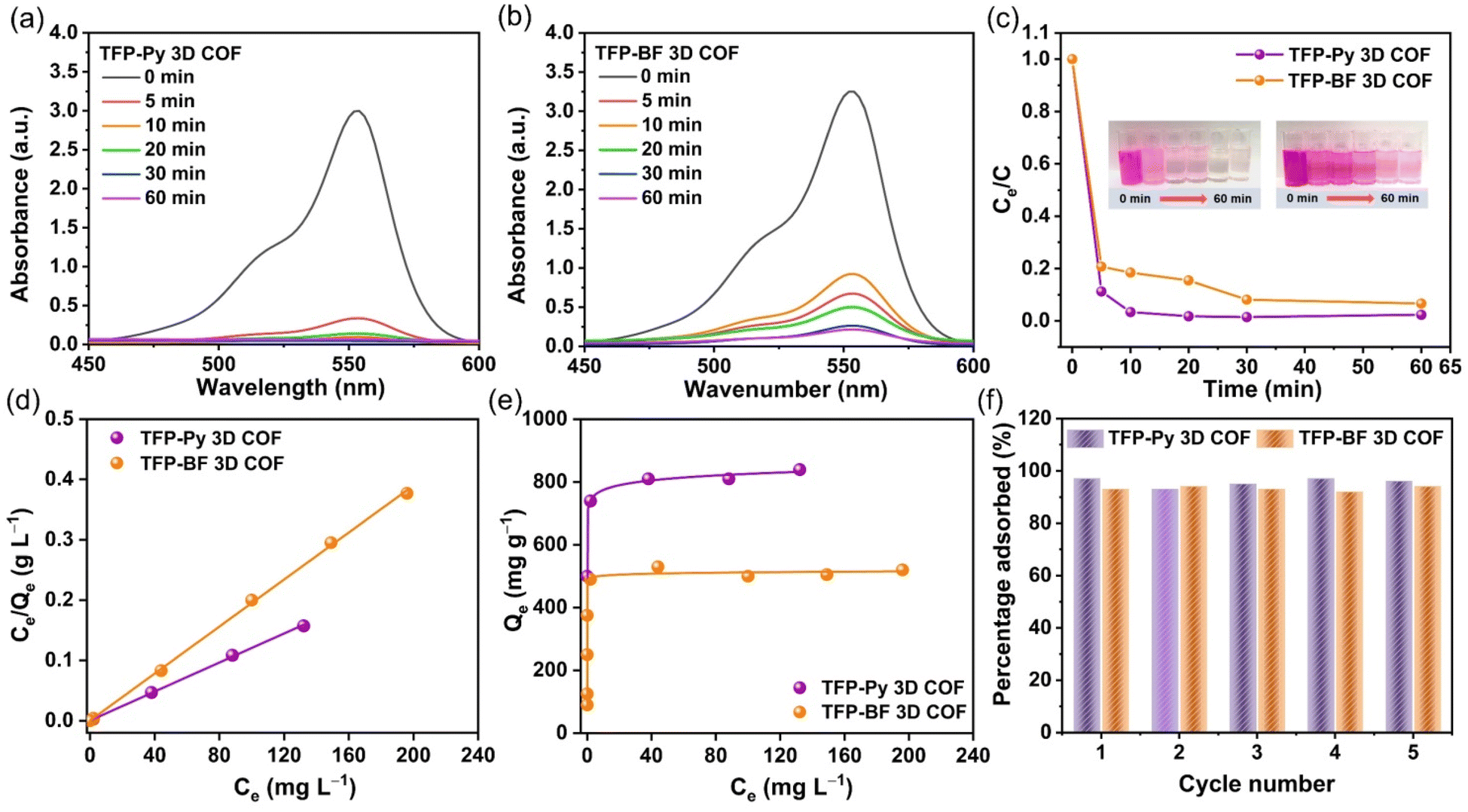 | ||
| Fig. 6 (a and b) UV-vis spectra of RhB solution (starting concentration: 18 mg L−1, 10 mL) at different times after the addition of (a) TFP-Py and (b) TFP-BF 3D COFs. (c) RhB adsorption rates from aqueous solutions (starting concentration: 18 mg L−1, 10 mL) evaluated over time on TFP-Py and TFP-BF 3D COFs. (Insets): corresponding photos. (d) Langmuir isothermal models and (e) adsorption isothermal curves for the adsorption of RhB onto TFP-Py and TFP-BF 3D COFs. (f) Reusability of TFP-Py and TFP-BF 3D COFs for the removal of RhB within 30 min. | ||
Fig. 6a and b demonstrate that the adsorption peak intensity at 554 nm reached complete adsorption within a time span of 10 min for TFP-Py 3D COF and 60 min for TFP-BF 3D COF when a minute quantity (4 mg) of TFP 3D COFs was introduced into a 10 mL aqueous solution containing RhB at a concentration of 18 mg L−1. Photos of the dye solution showed that after adding TFP-Py 3D COF within 10 minutes, the solution color changed from dark pink to almost colorless (illustration in Fig. 6c). The removal efficiencies of RhB reached approximately 98.56% and 92.10% within 30 min for TFP-Py and TFP-BF 3D COFs, respectively (Fig. 6c), demonstrating that our unique TFP 3D COFs had exceptional adsorption capacities for the removal of the organic dye from water. The interaction between the adsorbent and adsorbate when the adsorption process reaches an equilibrium state was examined by fitting the equilibrium adsorption data based on the Langmuir isothermal model.83Fig. 6d and Table S9† show that the Langmuir isothermal models of these TFP 3D COFs had good linear fits (Ce/Qe in relation to Ce), with correlation coefficients (RL2) of about 0.9995 for the TFP-Py 3D COF and 0.9994 for the TFP-BF 3DCOF. According to their Langmuir isotherm models, TFP-Py and TFP-BF 3D COFs had maximum adsorption capacities (Qm) of 840 and 520 mg g−1, respectively (Fig. 6e). The effectiveness of removal of RhB over TFP 3D COFs is influenced by four factors: N- and O-functionalized groups, porosity, π–π stacking interaction between the COFs and RhB, and surface area. As previously reported,80 the presence of N- and O-functionalized groups in the skeleton of adsorbents strongly improves their adsorption efficiency toward organic dyes. The high effectiveness of the TFP 3D COFs in removing dye can be related to the presence of N- and O-functionalized groups, specifically carbonyl CO and imino N–H groups, in their structures. A combination of electrostatic forces and hydrogen bonds allowed these groups to connect with RhB molecules. Because RhB is a cationic dye, it can be strongly adsorbed via electrostatic interactions between the positively charged groups on the RhB and the electron-rich oxygen and nitrogen atoms in the COF framework. In addition, the strong adsorption can be enhanced by hydrogen bonding between the C–O group of the RhB dye and the N–H group of the TFP 3D COFs. The N–H group of the TFP 3D COFs can form hydrogen bonds with the C–O group of the RhB dye, promoting strong adsorption. Regarding porosity, adsorbent materials with pore diameters smaller than 1.5 nm exhibit limited adsorption efficiency for dyes. Because most dye molecules are larger than 1 nm, they block the pores in these adsorbents.70 Alternatively, the adsorbent materials with pore diameters greater than 1.5 nm show a significant capacity for adsorbing dyes. The large pore diameters facilitated faster chemical transfer pathways and ample storage capacity, resulting in an increased adsorption capacity and rate. Hence, the TFP-Py and TFP-BF 3D COFs had large pore diameters of 1.73 and 1.82 nm, respectively, which exceeded the dimensions of the RhB molecule (1.59 × 1.18 × 0.56 nm). According to a previous report,84 an increase in the π–π stacking interaction between the dye and adsorbent material leads to an increase in the adsorption capacity of the dye. The high effectiveness of the TFP 3D COFs in eliminating dye from water can be related to the aromatic pyrene and bifluorenylidene units present in their structures. These units generate robust π–π stacking interactions with the aromatic rings in the dye molecules. The surface area of the adsorbent material is a crucial determinant of the adsorption efficacy towards organic dyes.70 Adsorbent materials with a large surface area show a significant capacity for adsorbing dyes. Hence, the remarkable effectiveness of the TFP 3D COFs in eliminating dyes from water can be related to the substantial surface area of TFP-Py (731 m2 g−1) and TFP-BF (447 m2 g−1) 3D COFs.
The surface area was the main factor that caused the TFP-Py 3D COF to have a higher dye removal efficiency than the TFP-BF 3D COFs. This is because both TFP-Py and TFP-BF 3D COFs have the same N- and O-functionalized groups, which are carbonyl CO and imino N–H groups, and they both have strong aromatic pyrene and bifluorenylidene units in their structures. The higher Qm value of the TFP-Py 3D COF may be explained by the greater surface area of TFP-Py 3D. These values of Qm are among the highest recently reported for COFs (Table S10†).
The Langmuir constant (KL), which is connected to the binding energy of RhB sorption, is related to the porosity of the adsorbent, indicating that the bigger the pore volume, the higher the KL.85 Table S9† reveals that the KL value of TFP-Py 3D COF with a 0.94 nm pore volume is 0.305 L mg−1; however, the KL value of TFP-BF 3D COF with a 0.99 nm pore volume is 0.712 L mg−1. Furthermore, we studied the adsorption mechanism between RhB dye and TFP 3D COFs by monitoring the changes in functional groups in the FTIR spectra of RhB and COFs before and after adsorption. Fig. S21 and S22† show that π–π interactions and hydrogen bonding are involved in the adsorption of RhB on the surface of COF. After RhB dye engagement, the aromatic CC bonds of TFP 3D COFs shifted to a higher wavenumber, demonstrating π–π stacking interactions between the TFP 3D COFs and the dye. It has been reported that increased electron density distribution around the aromatic ring leads to a higher bond strength, leading to a shift to a higher wavenumber.86 Therefore, the observed shift of the aromatic CC bond wavenumber in TFP-Py and TFP-BF 3D COFs upon RhB adsorption can be attributed to the increase in the electron density distribution within the COFs due to π–π stacking interactions. As the aromatic rings of the RhB molecule come close to the aromatic rings of the COFs, there can be a redistribution of electron density between them. This can lead to a slight alteration in the bond strength and electronic character of the CC bonds in the COFs, reflected in the observed shift in their wavenumber. On the other hand, the shifting of the N–H bond of the TFP 3D COFs to a lower wavenumber as well as the shifting of the C–O of the RhB dye to a higher wavenumber indicate the presence of hydrogen bonding between the N–H unit of the TFP 3D COFs and the C–O unit of the dye. We tested the recyclability of our TFP-Py and TFP-BF 3D COFs using cyclic adsorption/regeneration tests with the RhB solution. After five recovery cycles, the adsorption effectiveness of these COFs did not diminish noticeably (Fig. 6f), demonstrating the probable ability of our TFP-Py and TFP-BF 3D COFs to function as effective substances in eliminating organic contaminants from wastewater.
2.4. Photodegradation of RhB using 3D COF photocatalysts
The photocatalytic efficiencies of TFP-Py and TFP-BF 3D COFs were assessed for the degradation of RhB in aqueous solution under visible light exposure, considering their high surface areas, good porosities, and tight band gaps. In a control experiment, the UV-vis spectra of an aqueous solution containing RhB (10 mg L−1, 56 mL) were observed at different time intervals (ranging from 0 to 180 min) in the absence of any photocatalyst (Fig. S23†), confirming the challenging nature of degrading RhB dye without a TFP 3D COF photocatalyst under visible light conditions. The photodegradation performance of TFP 3D COFs (7 mg) was evaluated using a commonly employed aqueous solution of RhB (10 mg L−1, 56 mL). To attain adsorption–desorption equilibrium, the solution was subjected to continuous stirring in a light-free environment for 60 minutes prior to exposure to visible light. The TFP-BF 3D COF demonstrated superior photodegradation capabilities compared to the TFP-Py 3D COF. The photodegradation efficiencies of RhB on the TFP 3D COF were observed to be 60.40% for TFP-BF 3D COF and 48.40% for TFP-Py 3D COF after 60 minutes of visible light irradiation (>450 nm) (Fig. 7a–c). The TFP-Py and TFP-BF 3D COFs exhibited complete removal of RhB molecules within 150 min for TFP-BF 3D COF and 165 min for TFP-Py 3D COF, with photodegradation efficiencies of 96.60% and 95.90%, respectively (Fig. 7c and d). These results demonstrate that the newly developed TFP 3D COFs possess remarkable photocatalytic capabilities for degrading RhB under visible light conditions. The increased effectiveness of RhB photodegradation over the TFP-BF 3D COF, compared to the TFP-Py 3D COF, can be tentatively attributed to the smaller band gap and the closer energy level of the LUMO in TFP-BF 3D COF.87,88 The TFP-BF 3D COF exhibited a LUMO energy level of −3.19 eV, which closely approximated the oxygen reduction potential (O2/O2˙−, −4.10 eV) (Fig. 5c). The degradation efficiency of our newly developed photocatalyst TFP-BF 3D COF towards RhB is equivalent to, and in certain instances surpasses, the degradation efficiencies of several previously documented COFs and conjugated polymers when subjected to identical photocatalytic conditions (Table S11†). It is worth mentioning that the conjugated polymers mentioned before were synthesized using metal-catalyzed procedures. In contrast, our study describes the preparation of TFP-BF 3D COF using a straightforward and metal-free technique. The kinetic investigations of the photocatalytic degradation of TFP 3D COFs were conducted and analyzed using the Langmuir–Hinshelwood model (ln (C0/Ct) = kt), where C0 represents the initial concentration of the RhB aqueous solution, Ct represents the concentration of the RhB aqueous solution at time t (min) of the photodegradation reaction, k refers to the reaction rate constant, and t represents the duration (min) of UV-vis light irradiation. Fig. 7e confirms that the Langmuir–Hinshelwood models of TFP-Py and TFP-BF 3D COFs were fitted with the pseudo-first-order reaction kinetics during the degradation processes.89 This is evident from the observation of strong linear fits when plotting −ln (Co/Ct) against t with a correlation coefficient (R2) of about 0.9905 for the TFP-Py 3D COF and 0.9901 for the TFP-BF 3DCOF. The reaction rate constant of the TFP-BF 3DCOF was about 1.5 × 10−2 min−1, which is 1.4 times greater than that of TFP-Py 3D COF (approximately 1.1 × 10−2 min−1), confirming the high photocatalytic performance of TFP-BF 3D COF compared to TFP-Py 3D COF. In addition, the pseudo-first-order reaction kinetics of the photodegradation of RhB over TFP 3D COFs confirmed that the rate of the photodegradation reaction [RhB + H2O → degraded products (CO2 + H2O)] is dependent only on the concentration of RhB. The water concentration is sufficiently high, resulting in a negligible change in its concentration.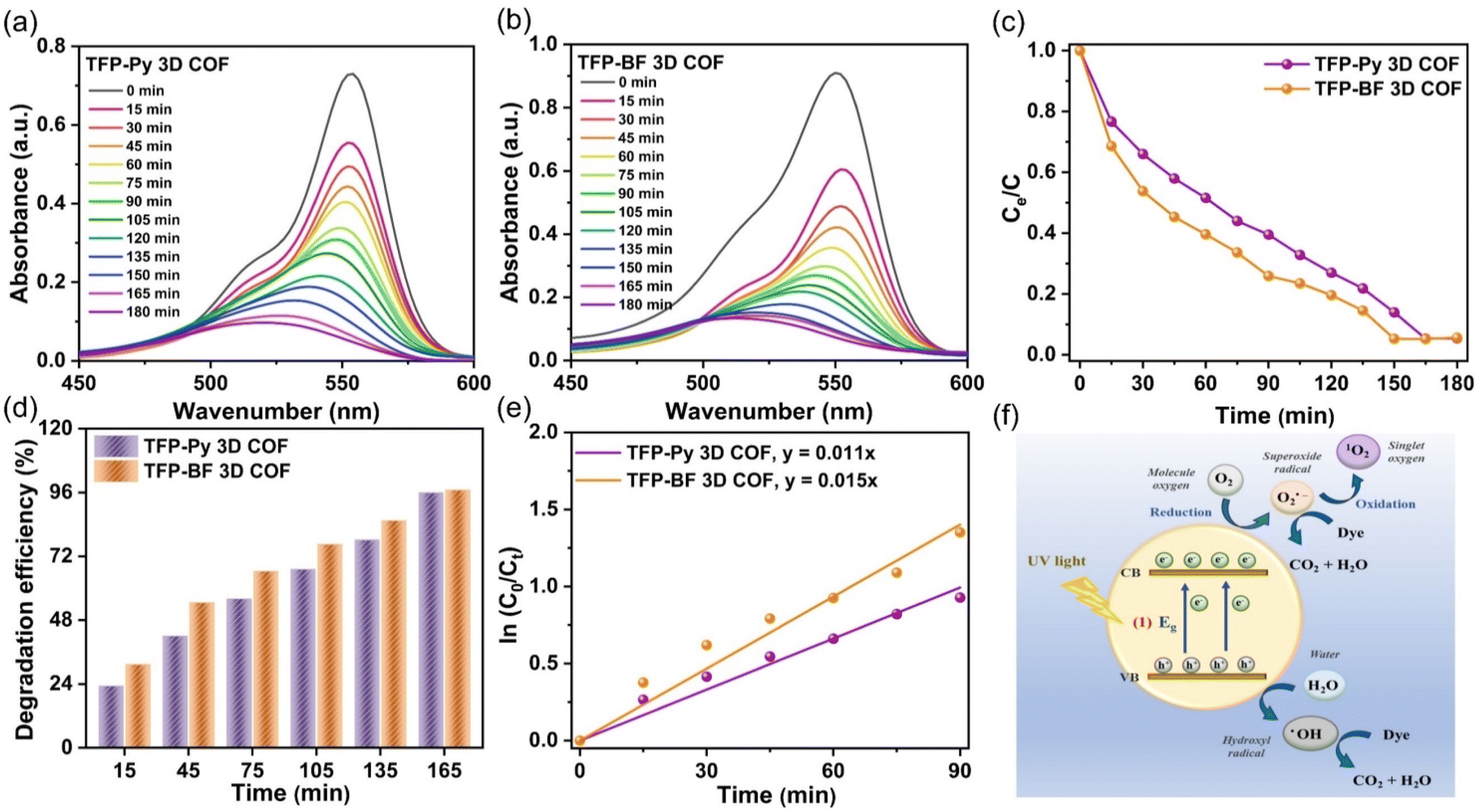 | ||
| Fig. 7 (a and b) UV-vis spectra revealing the photodegradation of RhB solution (starting concentration: 10 mg L−1, 56 mL) at different times after the addition of (a) TFP-Py and (b) TFP-BF 3D COFs. (c) Photocatalytic efficacies of TFP-Py and TFP-BF 3D COFs for the degradation of RhB solution under UV-vis light exposure for different periods. (d) Photodegradation efficiencies of TFP-Py and TFP-BF 3D COFs for degradation of RhB in water under UV-vis light irradiation at different times. (e) Pseudo-first-order kinetic curves for the photodegradation for RhB. (f) Schematic representation of photodegradation mechanism of RhB using TFP-Py and TFP-BF 3D COFs. | ||
The recycling and reuse of water treatment photocatalysts was essential for economic use and secondary pollution control. Therefore, the reusability of TFP-BF 3D COF was assessed in the degradation of RhB across ten cycles, with all conditions being kept identical. The TFP-BF 3D COF exhibited a sustained high level of photocatalytic activity, with a slight reduction of just 10% in performance (Fig. S24†), which can be attributed to the slight loss of catalyst powder during the recovery stage. Hence, it is plausible to utilize TFP-BF 3D COF repeatedly while maintaining a consistent level of photocatalytic performance. To investigate the stability of the TFP 3D COF photocatalysts, the TFP 3D COF powder was filtered after the ten cycles of RhB degradation, and PXRD and FE-SEM were measured. It was observed that the PXRD peaks of TFP-Py and TFP-BF 3D COFs showed no changes in intensity or location following the extended photodegradation cycles (Fig. S25a and b†). In addition, FE-SEM images revealed that both TFP-Py and TFP-BF 3D COFs maintained their micrometer-sized rod-like and twisted fiber-like morphologies after the lengthy photodegradation cycles (Fig. S25c and d†). These findings indicated the high stability of our TFP 3D COF photocatalysts.
We investigated RhB degradation over TFP 3D COF photocatalysts under visible light irradiation using TFP-BF 3D COF as a typical material. Photocatalytic degradation of organic pollutants is known to generate a variety of reactive species, including superoxide radicals (˙O2−), photogenerated holes (h+), singlet oxygen (1O2), and hydroxyl radicals (˙OH).90 This study employed p-benzoquinone (BQ), sodium azide (NaN3), isopropyl alcohol (IPA), and ethylenediaminetetraacetic acid disodium salt (EDTA-2Na) as scavengers for ˙O2−,1O2, ˙OH, and h+, respectively, aiming to prove the catalytic mechanism. Fig. S26† demonstrates that the lack of oxygen (O2) and the inclusion of BQ resulted in substantial decreases in activity. When comparing the effects of adding NaN3, or EDTA-2Na to the solution of RhB, it was observed that these additions resulted in minimal degradation efficiency reductions. In addition, the inclusion of IPA resulted in a decrease in the photocatalysis of about 25%. This study provides empirical evidence that TFP-BF 3D COF demonstrates remarkable photocatalytic characteristics, specifically in the generation of ˙O2− and to a lesser extent ˙OH (Fig. 7f). The ˙O2− species is generated as a result of the interaction between electrons and dissolved O2 inside an aqueous environment.91 Furthermore, when exposed to visible light, the observed high photocurrent density of TFP-BF 3D COF (Fig. 5e) aligns with its capacity to produce photoinduced electrons. These electrons then engage in a reaction with O2 molecules, resulting in the generation of superoxide radicals.
2.5. Photocatalytic hydrogen evolution using 3D COF photocatalysts
The shape and crystalline structure of photocatalysts affect their photo-driven hydrogen evolution activity. These parameters affect the band gap arrangement, electron migration, charge separation, photogenerated carrier movement, and photochemical reactions at the photocatalyst/electrolyte contact.92 The rod and twisted fiber-like morphologies provide a larger surface area in comparison to alternative morphologies, such as spherical-like ones, thereby enhancing the availability of active sites. Furthermore, the reduced distance that photogenerated electrons and holes need to travel to reach the reaction sites on the surface, in the form of rod and fiber-like structures, promotes efficient separation of the photogenerated electron/hole pairings.93 Therefore, our highly crystalline rod-like TFP-Py 3D COF and twisted fiber-like TFP-BF 3D COF are expected to enhance the performance of the photocatalytic hydrogen evolution from water.We examined the photocatalytic capabilities of the as-synthesized TFP-Py and TFP-BF 3D COF materials for H2 production from water at a temperature of 25 °C. The photocatalytic reactions were carried out using an Xe lamp as a source of ultraviolet and visible light, with a total illumination time of 4 hours. In a Pyrex reactor with appropriate sacrificial electron donors (SEDs), these TFP 3D COFs were dispersed in an H2O/DMF (9:1, v/v%) co-solvent to produce a photocatalytic system. A gas chromatograph (GC) was used to monitor and record the generated gas from the system once every hour. Several parameters were examined, encompassing the SED, source of light, and quantity of photocatalyst. We examined the impact of sacrificial agents, namely ascorbic acid (AC), triethylamine (TEA), and triethanolamine (TEOA), on the hydrogen evolution rate (HER) when a Pt cocatalyst is not present under ultraviolet and visible-light irradiation. It was observed that the 1.0 M AC solution exhibited the most significant photocatalytic HER when utilized with TFP-Py and TFP-BF 3D COFs (Fig. S27–S29†). Consequently, AC was selected for subsequent investigations. The enhanced HER observed in the TFP 3D COF photocatalysts can be attributed to the protonation of the N–H functional groups facilitated by the presence of an acidic catalyst (AC). Based on the available literature,94,95 the process of protonation in COF leads to an enhancement in its hydrophilicity and a reduction in its bandgap. Consequently, this alteration enables COF to exhibit a significant light absorption capacity. To verify these findings, we prepared AC-treated TFP 3D COFs by immersing the as-synthesized TFP 3D COFs in a 1 M AC aqueous solution for 5 minutes. The AC-treated TFP 3D COFs were then filtered and dried at room temperature. Subsequently, the water contact angle and UV-vis DRS measurements were determined. Fig. S30† shows that the water contact angle of the TFP-Py 3D COF dropped from 48.9° to 40.3°, and for the TFP-BF 3D COF, the contact angle decreased from 25.0° to 19.0°. This indicates that the hydrophilicity of the TFP 3D COFs was improved in the presence of AC. On the other hand, Fig. S31† shows that the absorption onsets of the as-synthesized TFP-Py and TFP-BF 3D COFs were observed at wavelengths of 523 nm and 607 nm, respectively. Upon protonation, these absorption onsets shifted to longer wavelengths, specifically 557 nm for the AC-treated TFP-Py 3D COF and 627 nm for the AC-treated TFP-BF 3D COF. The observed redshift cannot be ascribed to the absorption of the introduced AC, since it is a transparent substance that does not absorb visible light. The band gaps determined from Tauc plots for the as-synthesized TFP-Py and TFP-BF 3D COFs were 1.95 and 1.80 eV, respectively. These values decrease noticeably to 1.88 and 1.74 eV, respectively, after treatment with AC. Fig. S32 and S33† reveal no alteration in the absorption onset wavelengths for the TFP-Py and TFP-BF 3D COFs following treatment with 30% TEOA and 30% TEA aqueous solutions. This suggests that the bandgaps of the COFs remain unchanged under TEOA and TEA conditions.
The investigation also encompassed an examination of the illumination source, utilizing both visible and complete (ultraviolet and visible) illumination sources. Fig. S34 and S35† demonstrate that the photocatalytic HERs obtained under ultraviolet and visible illumination conditions are nearly identical to those achieved just under visible illumination. The absorption of the TFP-Py and TFP-BF 3D COFs in the visible range can be attributed to this phenomenon, as depicted in Fig. 5a. To enhance experimental convenience, ultraviolet and visible illumination were employed for subsequent investigations, obviating the necessity for a visible filter. The influence of the photocatalyst quantity on the photocatalytic efficiency has been demonstrated to be significant, mostly due to the phenomenon of saturation light absorption that occurs when an appropriate quantity of photocatalyst is introduced.96–98 Therefore, an investigation was conducted to determine the impact of the photocatalyst quantity on the photocatalytic HER by varying the mass within the range of 2.0 to 5.0 mg (Fig. S36–S38†). The greatest achieved performance was observed when using 2.0 mg of TFP-Py and TFP-BF 3D COF photocatalysts (Fig. 8a and S36–S38†). Consequently, a photocatalyst mass of 2.0 mg was chosen for subsequent investigations. Under the optimized parameters, the HERs for the TFP-Py and TFP-BF 3D COFs without the inclusion of co-catalysts were measured to be 4.96 and 21.04 mmol g−1 h−1, respectively (Fig. 8a and Video 1†). To assess the specific spectrum impacts on the H2 evolution activity of the TFP 3D COF photoabsorbers, we identified the apparent quantum yields (AQYs) by using three distinct bandpass filters (at 420, 460, and 500 nm). Fig. 8b and Table 1 show that the AQYs for the most active TFP-BF 3D COF were 8.70, 10.12, and 15.73% at light wavelengths of 420, 450, and 600 nm, respectively. These values were higher than the majority of COF-based photocatalysts previously reported. The AQY values were 2.54, 4.21, and 0.49% at light wavelengths of 420, 450, and 600 nm, respectively, for the TFP-Py 3D COF (Fig. 8b and Table 1). Moreover, the inclusion of a co-catalyst plays a pivotal role in the advancement of hydrogen production. The presence of heterojunctions between the co-catalyst and the photocatalyst greatly enhances the electron transfer from the photocatalyst to the co-catalyst in the photocatalytic reactions.99 Hence, the platinum (Pt) co-catalyst was immobilized on the TFP 3D COFs, and its immobilization exerted a notable impact on the HER (Fig. 8c, S39 and S40†). The hydrogen evolution rate (HER) of the TFP-BF 3D COF grew progressively from 21.07 to 30.72 mmol g−1 h−1 as the quantity of platinum (Pt) added rose from 0.0 to 1.0 wt% (Fig. 8c). After further augmenting the Pt content to 3 and 5 wt%, the HER of the TFP-BF 3D COF exhibited a decrease in performance, resulting in HER rates of 11.92 and 4.21 mmol g−1 h−1, respectively (Fig. 8c). This phenomenon may be attributed to an excessive amount of platinum impeding the transmission of light and obscuring the reactive sites on the surface of the TFP-BF 3D COF.
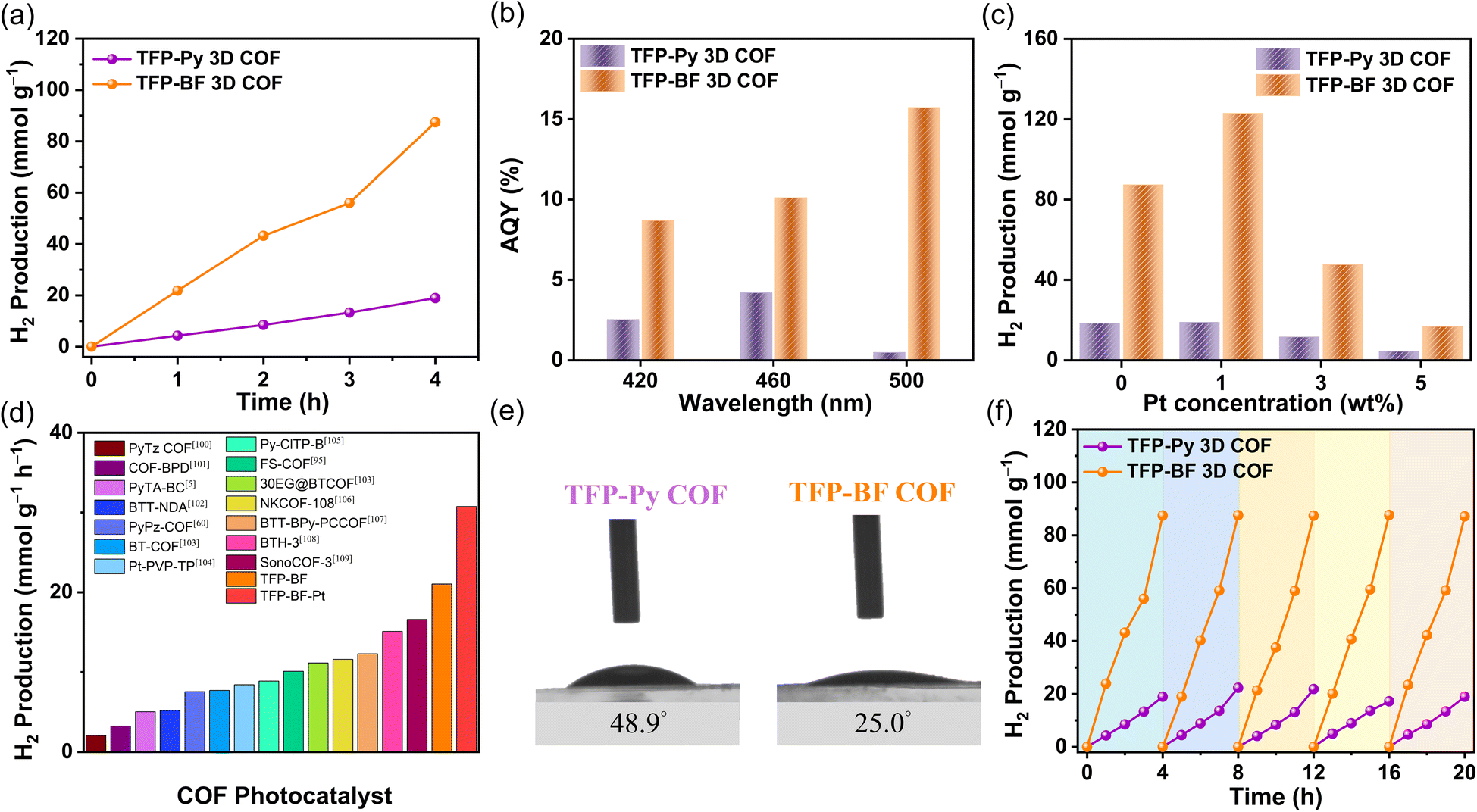 | ||
| Fig. 8 (a) Hydrogen evolution for the TFP-Py and TFP-BF 3D COFs under UV-vis light. (b) AQY of TFP-Py and TFP-BF 3D COFs at various incident light wavelengths. (c) Hydrogen evolution for the TFP-Py and TFP-BF 3D COFs with different concentrations of Pt co-catalyst under UV-vis light. (d) Comparison of hydrogen evolution for the TFP-Py and TFP-BF 3D COFs with previous COF materials. (e) Water contact angles for the TFP-Py and TFP-BF 3D COFs. (f) Stability test for the TFP-Py and TFP-BF 3D under UV-vis light. | ||
Overall, our TFP-BF 3D COF photocatalyst demonstrated remarkable photocatalytic performance for water reduction to generate H2, both with and without the inclusion of additional co-catalysts. The TFP-BF 3D COF has the highest photocatalytic HER efficiency among numerous COF-based photocatalysts including thiazolo[5,4-d]thiazole-based COF (PyTz-COF, 2.07 mmol g−1 h−1),100 vinylene-bipyridine-based COF (COF-BPDA, 3.23 mmol g−1 h−1),101 bicarbazole-based COF (PyTA-BC COF, 5.03 mmol g−1 h−1),5 benzotrithiophene-based COF (BTT-NDA COF, 5.22 mmol g−1 h−1),102 pyrazine-based COF (PyPz COF, 7.54 mmol g−1 h−1),60 benzo-2,1,3-thiadiazole-based COF (BT-COF, 7.70 mmol g−1 h−1),103 platinum-2,4,6-triformylphloroglucinol-based COF (Pt-PVP-TP, 8.42 mmol g−1 h−1),104 chloride-benzothiadiazole-based (Py-ClTP-B, 8.87 mmol g−1 h−1),105 sulfone (FS)-based COF (10.10 mmol g−1 h−1),95 pyrazine-polyethylene glycol-based COF (30 EG@BTCOF, 11.14 mmol g−1 h−1),103 benzothiazole-based COF (NKCOF-108, 11.60 mmol g−1 h−1),106 benzotrithiophene-bipyridine-based COF (BTT-BPy-PCCOF, 12.30 mmol g−1 h−1),107 sp2-benzobisthiazole-based COF (BTH-3 COF, 15.10 mmol g−1 h−1),108 sound-based COF (SonoCOF-3, 16.60 mmol g−1 h−1)109 (Fig. 8d and Table S12†). The contact angles exhibited by water droplets on the surfaces of TFP-Py and TFP-BF 3D COFs showed a gradual decrease from 48.9° to 25.0°, indicating an increase in hydrophilicity (Fig. 8e). Strong hydrophilicity, in combination with a small water contact angle, has been demonstrated to promote interactions between the photocatalytic reaction solution and the polymer surface, resulting in increased photocatalytic activity.110 According to the aforementioned findings, the superior photocatalytic activity exhibited by TFP-BF 3D COF in comparison to TFP-Py 3D COF can be attributed to the following advantageous factors. First, the lower bandgap of the TFP-BF 3D COF facilitated a higher production of electron–hole pairs, which resulted in greater photocatalytic activity. Second, the TFP-BF 3D COF exhibited a heightened negative LUMO position, hence augmenting the proton reduction capacity towards hydrogen gas. Third, the lower PL intensity of the TFP-BF 3D COF relative to the TFP-Py 3D COF induced more light-induced electrons, which are more likely to participate in the photocatalytic reaction rather than undergoing recombination with holes. Fourth, the TFP-BF 3D COF demonstrated a greater photocurrent density compared to the TFP-Py 3D COF. This suggests that a larger number of light-induced excitons could be generated during the photocatalytic process and that charge migration in the TFP-BF 3D COF photocatalyst was more efficient. Fifth, the TFP-BF 3D COF exhibited enhanced electronic conductivity and charge transfer resistance compared to the TFP-Py 3D COF, resulting in a significantly higher charge transfer rate. Lastly, the pronounced hydrophilic nature of the TFP-BF 3D COF facilitated its affinity for water, leading to an augmentation in its photocatalytic performance. The results of the control photocatalytic experiments demonstrated that the presence of a TFP 3D COF photocatalyst was necessary for the detection of H2 in water. In the absence of the COF photocatalyst, no H2 was observed (Fig. S41 and 42†). This suggests that both the COF photocatalyst and the sacrificial agent were crucial components in facilitating the photocatalytic generation of H2. The assessment of the photocatalytic potential of TFP-Py and TFP-BF 3D COFs for the generation of hydrogen through water splitting was conducted through iterative experiments under AM 1.5G conditions. Fig. 8f illustrates that the consistent photocatalytic performance of TFP-BF 3D COF was sustained throughout five cycles, albeit with a marginal decline of approximately 5% in photocatalytic activity. After the four cycles of photocatalytic H2 generation, the TFP 3D COF photocatalyst powders were filtered and PXRD and FE-SEM were evaluated to determine their stability. The PXRD peaks of TFP-Py and TFP-BF 3D COFs did not change intensity or location during extended photocatalytic H2 production cycles (Fig. S43a and b†). After the four photocatalytic H2 generation cycles, the FE-SEM images showed that TFP-Py and TFP-BF 3D COFs retained their micrometer-sized rod-like and twisted fiber-like morphologies, respectively (Fig. S43c and d†). These findings indicate the high stability of our TFP 3D COF photocatalysts.
2.6. DFT calculations
We performed density functional theory (DFT) calculations to investigate the electronic structure, chemical reactivity, and adsorption properties of the synthesized TFP-Py and TFP-BF COFs. In this study, the generalized gradient approximation111 from the DFT112 was utilized by the DMol3 module.113 Using the BLYP exchange-correlation functional114 and the double numerical with the polarization (DNP) basis set in DMol3, all geometry optimization calculations were executed. The analysis of the electronic structure and the frontier molecular orbitals (HOMO and LUMO) plays a crucial role in understanding photocatalytic behavior. Table S13† summarizes the calculated HOMO and LUMO energies. Both COFs exhibited similar HOMO energies (−4.83 eV for TFP-Py and −4.84 eV for TFP-BF), indicating a comparable ability to donate electrons. However, the LUMO energy of TFP-BF (−3.56 eV) is lower than that of TFP-Py (−3.27 eV), resulting in a smaller band gap (ΔE) of 1.28 eV for TFP-BF compared to 1.56 eV for TFP-Py. The smaller bandgap due to the twisted BF units allows for a broader absorption of the solar spectrum and more efficient generation of charge carriers. As illustrated in Fig. 9a, the TFP-BF and TFP-Py exhibit HOMO levels localized on the donor units (pyrene in TFP-Py and bifluorenylidene in TFP-BF), while LUMO levels spread across the molecule, with some concentration on the TFP acceptor unit. However, the twisted structure of the bifluorenylidene donor in TFP-BF might more effectively influence the orbital overlap and delocalization compared to the planar pyrene donor in TFP-Py. As a result, we anticipate that TFP-BF will have a greater capacity for separating e−/h+ pairs and a greater photocatalytic efficiency. Further insights into the electronic characteristics are provided by the global reactivity parameters (Table S13†), which were calculated using reactivity indices derived from HOMO and LUMO energy Eigenvalues.115 These parameters offer information on the susceptibility of the COFs to gain or lose electrons. Electron affinity (A), which reflects the ease of accepting electrons, is slightly higher for TFP-BF (3.56 eV) compared to TFP-Py (3.27 eV). The ionization potential (I), representing the energy required to remove an electron, remains similar for both frameworks. Consequently, the chemical hardness (η), a measure of resistance to electron transfer, is lower for TFP-BF (0.64 eV) compared to TFP-Py (0.78 eV). This indicates that TFP-BF might be more reactive towards both electron donation and acceptance processes. The softness (S), the inverse of hardness, follows the opposite trend, with TFP-BF being softer than TFP-Py. The electronegativity (χ) and electrophilicity (ω) indices also show a slightly higher propensity for electron-accepting ability in TFP-BF compared to TFP-Py. The chemical potential (μ) reflects the average tendency to gain or lose electrons. Both COFs have negative chemical potentials, indicating their inclination to gain electrons. Moving on, the electrostatic potential (ESP) maps of TFP-Py and TFP-BF suggest a comparable electronic foundation for both COFs, with subtle variations arising from the specific geometries of their donor units (Fig. S44†). Red regions (negative ESP) represent areas with high electron density, likely due to the electronegative N and O atoms. Conversely, blue regions (positive ESP) indicate areas with low electron density, potentially surrounding H atoms and the center of aromatic rings. The potential increases in the order red < orange < yellow < green < blue. Overall, the DFT results suggest that TFP-BF, with its narrower band gap, higher electron affinity, and lower chemical hardness, might have a greater propensity for efficient charge separation/transfer compared to TFP-Py. This could translate to better photocatalytic performance.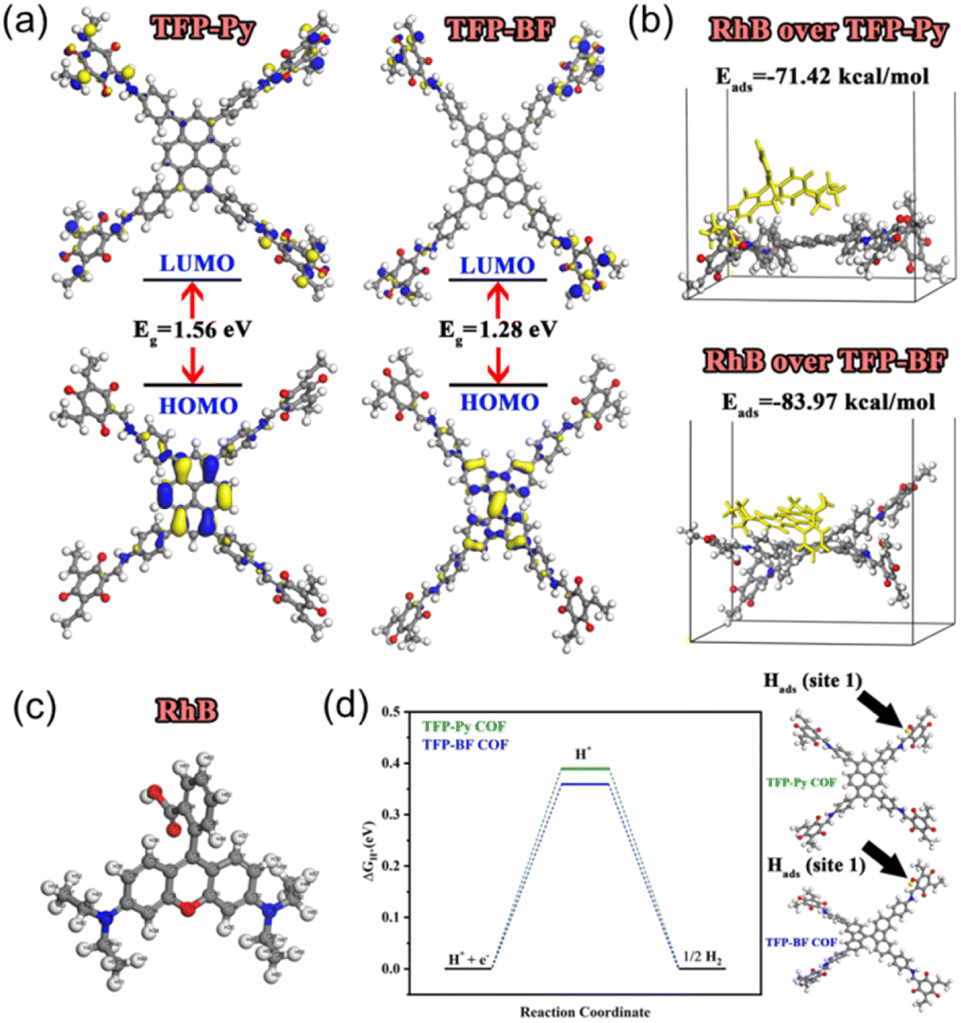 | ||
| Fig. 9 (a) Frontier molecular orbitals and HOMO–LUMO bandgap energies of as-prepared COFs. (b) Adsorption energies of RhB dye over as-prepared COFs. (c) Optimized structure of RhB dye. (d) Calculated free energy diagram (eV) for H2 evolution on the as-prepared COFs. | ||
The adsorption of organic dye molecules is another important aspect of photocatalysis. The adsorption energies of the RhB dye on the COF molecules are presented in Fig. 9b. Both TFP-Py and TFP-BF display strong binding affinities towards RhB, through π–π stacking and electrostatic interactions, with adsorption energies of −71.42 kcal mol−1 and −83.97 kcal mol−1, respectively. The significantly higher negative adsorption energy for TFP-BF suggests a much stronger interaction with the RhB dye compared to TFP-Py. This strong adsorption is expected to be beneficial for the photocatalytic degradation of RhB. The Fukui index, particularly the Fukui for radical attack (f0), is a valuable tool for understanding the mechanism of RhB degradation over COFs. Identifying the preferred sites for a radical attack on the RhB molecule helps elucidate the initial steps of the degradation process. Table S14† displays the Fukui index of the RhB dye, as determined by DFT. The electron density was low at the C(9) and C(10) atoms, while electrons were more localized on the other atoms. As a result of electrostatic attraction, the pollutants were more likely to come into touch with the COFs' surfaces. The atoms with larger condensed Fukui index (f0) values, such as N4, N5, C6, C18, and C19 of RhB (Table S14† and Fig. 9c), were suggested to be easily attacked by reactive species like hydroxyl radicals (OH˙) or superoxide radicals (O2˙−) to form small fragments, which were then mineralized into H2O and CO2.
Understanding the adsorption ability of hydrogen-active species is essential for achieving efficient photocatalytic H2 generation. Therefore, the Gibbs free energy changes (ΔG) for hydrogen adsorption by oxygen atoms (site 1) and nitrogen atoms (site 2) within the COF molecules were calculated and presented in Table S15.† The values are relatively low (around 0.4 eV for site 1 and 2.1 eV for site 2), suggesting that the process is closer to spontaneity. However, the ΔG values are lower for site 1 compared to site 2 for both COFs due to the higher electronegativity of oxygen atoms.116 Interestingly, TFP-BF exhibited slightly lower ΔG values compared to TFP-Py (Fig. 9d), implying a potentially better affinity for hydrogen adsorption. This could be related to the electronic properties of the COFs. TFP-BF has a higher electron affinity and softness compared to TFP-Py, potentially leading to a slightly more favorable interaction with the hydrogen molecule's electron density.
The DFT calculations revealed that the unique molecular structure (twisted structure) of TFP-BF, which possesses a narrower band gap, higher electron affinity, and softer character, may promote efficient charge separation and transport pathways compared to TFP-Py, resulting in distinct electronic behaviors and enhanced photocatalytic performance. While TFP-BF COF exhibits good hydrogen adsorption compared to TFP-Py COF, it also demonstrates a stronger binding affinity for the RhB dye. The Fukui index analysis pinpointed potential reactive sites on the RhB molecule for degradation by the COFs. These findings suggest that TFP-BF might be a more promising candidate for photocatalytic applications such as RhB dye degradation and H2 evolution.
3 Conclusions
In summary, two donor–acceptor hetero[6]radialene-based TFP 3D COFs–TFP-Py and TFP-BF 3D COFs–having a rare [3,4]-connected ffc topology were developed and synthesized through the coupling of the hetero[6]radialene (TFP-3OHCHO) acceptor with planar PyTA-4NH2 and twisted BFTB-4NH2 donors, respectively. The results obtained from the PXRD, BET, and TGA analyses demonstrated that the TFP 3D COFs exhibited exceptional crystallinity, significant thermal stability with a char yield of up to 67.07%, and a substantial surface area of up to 731 m2 g−1. Our TFP 3D COFs exhibited exceptional efficacy in the adsorption process of the organic dye RhB from aqueous solutions within 10 minutes and with a remarkable maximum adsorption capacity (Qm) of up to 840 mg g−1. Their performance is equivalent to, or potentially surpasses, that of various other COFs and conjugated polymers that have been previously documented in similar adsorption environments. The integration of a twisted BF structure into the core of the hetero[6]radialene-based 3D COF resulted in a highly favorable band gap configuration, effective charge transfer, and successful segregation of photoinduced holes and electrons. The TFP-BF 3D COF showed a significant degree of effectiveness in the process of photocatalytic degradation of RhB. It exhibited an impressive efficiency level of up to 96.60% with a reaction rate constant reaching as high as 1.5 × 10−2 min−1. The TFP-BF 3D COF exhibited a significant hydrogen evolution rate (HER) of 21.04 mmol g−1 h−1 under both UV and visible light irradiation, without requiring a Pt-cocatalyst. Furthermore, this COF demonstrated outstanding reusability. Notably, this HER value is the highest specific capacity achieved thus far among reported COFs. This study presents novel approaches for the accurate pre-design of photofunctional frameworks to enhance solar energy conversion and usage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported financially by the National Science and Technology Council, Taiwan, under contract NSTC 111-2221-E-110-003 and 112-2221-E-110-005-MY3.Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. S. Gopinath and N. Nalajala, J. Mater. Chem. A, 2021, 9, 1353–1371 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- A. F. M. EL-Mahdy, A. M. Elewa, S.-W. Huang, H.-H. Chou and S.-W. Kuo, Adv. Opt. Mater., 2020, 8, 2070074 CrossRef CAS.
- D. Friedmann, A. F. Lee, K. Wilson, R. Jalili and R. A. Caruso, J. Mater. Chem. A, 2019, 7, 10858–10878 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. D. Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. A. Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, Chem. Commun., 1985, 8, 474–475 RSC.
- R. Sharma, M. Almáši, S. P. Nehra, V. S. Rao, P. Panchal, D. R. Paul, I. P. Jain and A. Sharma, Renewable Sustainable Energy Rev., 2022, 168, 112776 CrossRef CAS.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14337–14341 CrossRef.
- Z. A. Lan, M. Wu, Z. Fang, Y. Zhang, X. Chen, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201482 CrossRef CAS PubMed.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan and A. Kudo, Angew. Chem., Int. Ed., 2022, 134, e202201299 CrossRef.
- Y. Yan, X. Yu, C. Shao, Y. Hu, W. Huang and Y. Li, Adv. Funct. Mater., 2023, 33, 2304604 CrossRef CAS.
- M. G. Kotp, C. L. Chang and A. F. M. EL-Mahdy, J. Water Process Eng., 2023, 53, 103675 CrossRef.
- M. G. Kotp, A. M. Elewa, A. F. M. EL-Mahdy, H. H. Chou and S. W. Kuo, ACS Appl. Energy Mater., 2021, 4, 13140–13151 CrossRef CAS.
- B. Kumru, V. Molinari, M. Hilgart, F. Rummel, M. Schäffler and B. V. K. J. Schmidt, Polym. Chem., 2019, 10, 3647–3656 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298–2321 RSC.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- B. Wang, G. M. Biesold, M. Zhang and Z. Lin, Chem. Soc. Rev., 2021, 50, 6914–6949 RSC.
- M. Arif, U. Fatima, A. Rauf, Z. H. Farooqi, M. Javed, M. Faizan and S. Zaman, Catalysts, 2023, 13, 231 CrossRef CAS.
- B. Cui and G. Fu, Nanoscale, 2022, 14, 1679–1699 RSC.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 6328 CrossRef PubMed.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- W. T. Chung, I. M. Mekhemer, M. G. Mohamed, A. M. Elewa, A. F. M. EL-Mahdy, H. H. Chou, S. W. Kuo and K. C. W. Wu, Coord. Chem. Rev., 2023, 483, 215066 CrossRef CAS.
- X. Wang, J. Yang, X. Shi, Z. Zhang, C. Yin and Y. Wang, Small, 2022, 18, 2107108 CrossRef CAS PubMed.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- H. Peng, S. Huang, V. Montes-García, D. Pakulski, H. Guo, F. Richard, X. Zhuang, P. Samorì and A. Ciesielski, Angew. Chem., 2023, 135, e202216136 CrossRef.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- S. Di, Q. Wu, C. Shi and S. Zhu, ACS Appl. Mater. Interfaces, 2023, 15, 1827–1842 CrossRef CAS PubMed.
- L. R. Ahmed, L. Gilmanova, C. T. Pan, S. Kaskel and A. F. M. EL-Mahdy, ACS Appl. Polym. Mater., 2022, 4, 9132–9143 CrossRef CAS.
- H. C. Ma, J. Zou, X. T. Li, G. J. Chen and Y. B. Dong, Chem.–Eur. J., 2020, 26, 13754–13770 CrossRef CAS PubMed.
- Q. Cao, L. L. Zhang, C. Zhou, J. H. He, A. Marcomini and J. M. Lu, Appl. Catal., B, 2021, 294, 120238 CrossRef CAS.
- Q. Sun, Y. Tang, B. Aguila, S. Wang, F. S. Xiao, P. K. Thallapally, A. M. Al-Enizi, A. Nafady and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 8670–8675 CrossRef CAS PubMed.
- S. Ruidas, A. Chowdhury, A. Ghosh, A. Ghosh, S. Mondal, A. D. Wonanke, M. Addicoat, A. K. Das, A. Modak and A. Bhaumik, Langmuir, 2023, 39, 4071–4081 CrossRef CAS PubMed.
- Z. Li, K. Geng, T. He, K. T. Tan, N. Huang, Q. Jiang, Y. Nagao and D. Jiang, Angew. Chem., Int. Ed., 2021, 60, 19419–19427 CrossRef CAS PubMed.
- Y. J. Li, W. R. Cui, Q. Q. Jiang, Q. Wu, R. P. Liang, Q. X. Luo and J. D. Qiu, Nat. Commun., 2021, 12, 4735 CrossRef CAS PubMed.
- J. Zhao, G. Guo, D. Wang, H. Liu, Z. Zhang, L. Sun, N. Ding, Z. Li and Y. Zhao, Green Chem., 2023, 25, 3103–3110 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- T. Y. Yu, Q. Niu, Y. Chen, M. Lu, M. Zhang, J. W. Shi, J. Liu, Y. Yan, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2023, 145, 8860–8870 CrossRef CAS PubMed.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- Y. R. Chen, X. Wang, W. Xu, K. Liu, W. B. Qiu, Y. Wu, Y. Li and W. R. Cui, Chem. Eng. J., 2023, 459, 141633 CrossRef CAS.
- J. Yang, F. Kang, X. Wang and Q. Zhang, Mater. Horiz., 2022, 9, 121–146 RSC.
- M. Wu, Z. Shan, J. Wang, T. Liu and G. Zhang, Chem. Eng. J., 2023, 454, 140121 CrossRef CAS.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng and A. Thomas, Nat. Rev. Methods Primers, 2023, 3, 1–19 CrossRef CAS.
- A. T. Partho, M. Tahir and B. Tahir, Int. J. Hydrogen Energy, 2022, 47, 34323–34375 CrossRef CAS.
- G. B. Wang, K. H. Xie, H. P. Xu, Y. J. Wang, F. Zhao, Y. Geng and Y. B. Dong, Coord. Chem. Rev., 2022, 472, 214774 CrossRef CAS.
- B. Zhang, H. Mao, R. Matheu, J. A. Reimer, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11420–11424 CrossRef CAS PubMed.
- A. Dey, F. A. Rahimi, S. Barman, A. Hazra and T. K. Maji, J. Mater. Chem. A, 2023, 11, 13615–13622 RSC.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S. W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CAS.
- S. Barman, A. Singh, F. A. Rahimi and T. K. Maji, J. Am. Chem. Soc., 2021, 143, 16284–16292 CrossRef CAS PubMed.
- F. D. Wang, L. J. Yang, X. X. Wang, Y. Rong, L. B. Yang, C. X. Zhang, F. Y. Yan and Q. L. Wang, Small, 2023, 19, 2207421 CrossRef CAS PubMed.
- C. Cui, X. Zhao, X. Su, N. Xi, X. Wang, X. Yu, X. L. Zhang, H. Liu and Y. Sang, Adv. Funct. Mater., 2022, 32, 2208962 CrossRef CAS.
- G. B. Wang, H. P. Xu, K. H. Xie, J. L. Kan, J. Fan, Y. J. Wang, Y. Geng and Y. B. Dong, J. Mater. Chem. A, 2023, 11, 4007–4012 RSC.
- H. Yu, J. Zhang, X. Yan, C. Wu, X. Zhu, B. Li, T. Li, Q. Guo, J. Gao, M. Hu and J. Yang, J. Mater. Chem. A, 2022, 10, 11010–11018 RSC.
- W. Huang, Y. Hu, Z. Qin, Y. Ji, X. Zhao, Y. Wu, Q. He, Y. Li, C. Zhang, J. Lu and Y. Li, Natl. Sci. Rev., 2023, 10, nwac171 CrossRef CAS PubMed.
- Q. Liao, W. Xu, X. Huang, C. Ke, Q. Zhang, K. Xi and J. Xie, Sci. China: Chem., 2020, 63, 707–714 CrossRef CAS.
- X. L. Wang, Y. Y. Sun, Y. Xiao, X. X. Chen, X. C. Huang and H. L. Zhou, Molecules, 2022, 27, 8002 CrossRef CAS PubMed.
- W. K. An, S. J. Zheng, X. Xu, L. J. Liu, J. S. Ren, L. Fan, Z. K. Yang, Y. Ren and C. Xu, Appl. Catal., B, 2022, 316, 121630 CrossRef CAS.
- H. Qiao, L. Yang, X. Yang, J. Wang, Y. Chen, L. Zhang, W. Sun, L. Zhai and L. Mi, Chem.–Eur. J., 2022, 28, e202200600 CrossRef CAS PubMed.
- S. Ruidas, B. Mohanty, P. Bhanja, E. Erakulan, R. Thapa, P. Das, A. Chowdhury, S. K. Mandal, B. K. Jena and A. Bhaumik, ChemSusChem, 2021, 14, 5057–5064 CrossRef CAS PubMed.
- A. F. M. EL-Mahdy, M. B. Zakaria, H.-X. Wang, T. Chen, Y. Yamauchi and S.-W. Kuo, J. Mater. Chem. A, 2020, 8, 25148–25155 RSC.
- J. H. Wang, T. A. Gaber, S. W. Kuo and A. F. M. EL-Mahdy, Polymers, 2023, 15, 1685 CrossRef CAS PubMed.
- J. H. Chong, M. Sauer, B. O. Patrick and M. J. MacLachlan, Org. Lett., 2003, 5, 3823–3826 CrossRef PubMed.
- L. Zhang, Q. H. Zhu, Y. R. Zhou, S. L. Wang, J. Fu, J. Y. Liu, G. H. Zhang, L. Ma, G. Tao, G. H. Tao and L. He, Nat. Commun., 2023, 14, 8181 CrossRef CAS PubMed.
- P. Dhanishta, P. S. S. kumar, S. K. Mishra and N. Suryaprakash, RSC Adv., 2018, 8, 11230–11240 RSC.
- M. Mohai, Surf. Interface Anal., 2004, 36, 828–832 CrossRef CAS.
- L. Peng, J. Sun, J. Huang, C. Song, Q. Wang, L. Wang, H. Yan, M. Ji, D. Wei, Y. Liu and D. Wei, Chem. Mater., 2022, 34, 2886–2895 CrossRef CAS.
- A. Takai, D. J. Freas, T. Suzuki, M. Sugimoto, J. Labuta, R. Haruki, R. Kumai, S. I. Adachi, H. Sakai, T. Hasobe and Y. Matsushita, Org. Chem. Front., 2017, 4, 650–657 RSC.
- C. Wang, F. Zheng, L. Zhang, J. Yang and P. Dong, Appl. Surf. Sci., 2023, 640, 158383 CrossRef CAS.
- A. M Elewa, A. F. M. EL-Mahdy, A. E. Hassan, Z. Wen, J. Jayakumar, T. L. Lee, L. Y. Ting, I. M. A. Mekhemer, T. F. Huang, M. H. Elsayed, C. L. Chang, W. C. Lin and H. H. Chou, J. Mater. Chem. A, 2022, 10, 12378–12390 RSC.
- Y. Yuan, H. Huang, L. Chen and Y. Chen, Macromolecules, 2017, 50(13), 4993–5003 CrossRef CAS.
- A. F. Saber and A. F. M. EL-Mahdy, New J. Chem., 2021, 45, 21834–21843 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons and S. Qiu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- S. Singh, S. Kaushal, J. Kaur, G. Kaur, S. K. Mittal and P. P. Singh, Chemosphere, 2021, 272, 129648 CrossRef CAS PubMed.
- A. F. M. EL-Mahdy, T. E. Liu and S. W. Kuo, J. Hazard. Mater., 2020, 391, 122163 CrossRef CAS PubMed.
- Z. M. Saigl and A. M. Ahmed, Indones. J. Chem., 2021, 1, 212–224 Search PubMed.
- J. Meurer, J. Hniopek, J. Ahner, M. Schmitt, J. Popp, S. Zechel, K. Peneva and M. D. Hager, Beilstein J. Org. Chem., 2021, 17, 2496–2504 CrossRef CAS PubMed.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- J. H. Wang, C. L. Chang, Z. W. Zhang and A. F. M. EL-Mahdy, Polym. Chem., 2022, 13, 5300–5308 RSC.
- S. X. Gan, C. Jia, Q. Y. Qi and X. Zhao, Chem. Sci., 2022, 13, 1009–1015 RSC.
- L. Cai, Y. Li, Y. Li, H. Wang, Y. Yu, Y. Liu and Q. Duan, J. Hazard. Mater., 2018, 348, 47–55 CrossRef CAS PubMed.
- W. Li, Z. Wang, Y. Li, J. B. Ghasemi, J. Li and G. Zhang, J. Hazard. Mater., 2022, 424, 127595 CrossRef CAS PubMed.
- J. Yu, Y. Yu, P. Zhou, W. Xiao and B. Cheng, Appl. Catal., B, 2014, 156–157, 184–191 CrossRef CAS.
- A. M. Elewa, M. H. Elsayed, A. F. M. EL-Mahdy, C. L. Chang, L. Y. Ting, W. C. Lin, C. Y. Lu and H. H. Chou, Appl. Catal., B, 2021, 285, 119802 CrossRef CAS.
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. V. D. Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- M. Qureshi and K. Takanabe, Chem. Mater., 2017, 29, 158–167 CrossRef CAS.
- P. Ravi, D. K. Kumaravel, D. Subramanian, D. Thoondyaiah, V. N. Rao, S. M. Venkatakrishnan and M. Sathish, ACS Appl. Energy Mater., 2021, 4, 13983–13996 CrossRef CAS.
- T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- S. Bi, F. Meng, D. Wu and F. Zhang, J. Am. Chem. Soc., 2022, 144, 3653–3659 CrossRef CAS PubMed.
- J. P. Jeon, Y. J. Kim, S. H. Joo, H. J. Noh, S. K. Kwak and J. B. Baek, Angew. Chem., Int. Ed., 2023, 62, e202217416 CrossRef CAS PubMed.
- T. Zhou, L. Wang, X. Huang, J. Unruangsri, H. Zhang, R. Wang, Q. Song, Q. Yang, W. Li, C. Wang, K. Takahashi, H. Xu and J. Guo, Nat. Commun., 2021, 12, 3934 CrossRef CAS PubMed.
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang, J. Hofkens, M. B. J. Roeffaers and J. Long, Angew. Chem., Int. Ed., 2019, 58, 18290 CrossRef CAS PubMed.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902 CrossRef CAS PubMed.
- Z. Zhao, Y. Zheng, C. Wang, S. Zhang, J. Song, Y. Li, S. Ma, P. Cheng, Z. Zhang and Y. Chen, ACS Catal., 2021, 11, 2098–2107 CrossRef CAS.
- L. Dai, A. Dong, X. Meng, H. Liu, Y. Li, P. Li and B. Wang, Angew. Chem., Int. Ed., 2023, 135, e202300224 CrossRef.
- Y. Wang, W. Hao, H. Liu, R. Chen, Q. Pan, Z. Li and Y. Zhao, Nat. Commun., 2022, 13, 100 CrossRef CAS PubMed.
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87–95 CrossRef.
- C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J. X. Jiang, Adv. Mater., 2021, 33, 2008498 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. M. Dreizler and E. K. Gross, Density Functional Theory: An Approach to the Quantum Many-Body Problem, Springer Science & Business Media, 2012 Search PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. E. Hassan, A. M. Elewa, M. S. Hussien, A. F. M. El-Mahdy, I. M. Mekhemer, I. S. Yahia, T. A. Mohamed, H. H. Chou and Z. Wen, J. Colloid Interface Sci., 2024, 653, 1650–1661 CrossRef CAS PubMed.
- L. Hao, R. Shen, C. Huang, Z. Liang, N. Li, P. Zhang, X. Li, C. Qin and X. Li, Appl. Catal., B, 2023, 330, 122581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07691a |
| This journal is © The Royal Society of Chemistry 2024 |