
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cradle-to-cradle approach for successive upcycling of polyethylene to polymer electrolytes to organic acids†
Jerald Y. Q.
Teo‡
 a,
Ming Yan
Tan‡
a,
Dorsasadat
Safanama
a,
Sheau Wei
Chien
a,
Yixuan
Jiang
ad,
Lewis
Queh
b,
Tristan T. Y.
Tan
a,
Ning
Ding
a,
Derrick W. H.
Fam
*ac and
Jason Y. C.
Lim
*ad
a,
Ming Yan
Tan‡
a,
Dorsasadat
Safanama
a,
Sheau Wei
Chien
a,
Yixuan
Jiang
ad,
Lewis
Queh
b,
Tristan T. Y.
Tan
a,
Ning
Ding
a,
Derrick W. H.
Fam
*ac and
Jason Y. C.
Lim
*ad
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore. E-mail: jason_lim@imre.a-star.edu.sg; derrickfamwh@imre.a-star.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore
cSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Ave, Singapore 639798, Singapore
dDepartment of Materials Science and Engineering, National University of Singapore (NUS), 9 Engineering Drive 1, Singapore 117576, Singapore
First published on 9th July 2024
Abstract
Polymer electrolytes are poised to increasingly dominate energy storage devices such as lithium ion batteries (LiBs) in the near future. Although this has spurred development of new generations of polymer electrolytes derived from alternative non-petroleum feedstock by valorising societal waste, the solid electrolyte materials' end-of-life has thus far been largely overlooked. Inadvertently, their disposal contributes to the growing environmental and plastic waste pollution. To address this, we demonstrate herein a cradle-to-cradle upcycling approach for polymer electrolytes derived from polyethylene – the most abundant waste plastic produced annually. Polyethylene was first aerobically activated using N-hydroxyphthalimide organocatalysts, and subsequently modified to yield polyethylene-graft-polyester copolymers. Both components of the copolymers played synergistic roles to enable them to perform well as solid and gel polymer electrolytes with appreciable ionic conductivities. A prototype LiB cell formulated using the PE-derived copolymer as the gel polymer electrolyte showed high coulombic efficiency and excellent retention of charge/discharge capacity. Notably, we demonstrated the ease of chemically upcycling these post-use PE-polyester polymer electrolytes through organocatalytic oxidation to yield industrially relevant, short-chain dicarboxylic acids. Our method is tolerant to common additives such as lithium salts, even in significant quantities, thus enabling the polymer electrolytes to be directly upcycled without any additional pretreatment. Our approach of upcycling waste plastics into functional polymer electrolyte materials, which are in turn feedstock for further value creation at the end of their lives, provides a viable value chain for sustainable polymer electrolyte materials which can also be applicable to other functional materials derived from waste plastics.
 Jason Y. C. Lim | Jason Y. C. Lim is a Group Leader at the Institute of Materials Research and Engineering (IMRE), A*STAR in Singapore, and an adjunct faculty member in the Department of Materials Science and Engineering, National University of Singapore (NUS). He obtained his BSc degree from Imperial College London, and a DPhil from the University of Oxford, U.K. His current research centres on upcycling common plastics into diverse economically valuable functional materials, as well as sustainable catalytic methods to transform these plastics into industrially relevant chemicals. He is a recipient of the National Research Foundation Fellowship for his work on sustainable plastic upcycling. |
Introduction
With increasing demand for electrification and consumer electronics worldwide, the market for portable and rechargeable high-performance energy storage systems such as lithium ion batteries (LiBs) is expecting strong unabated growth for the foreseeable future. As an essential component of LiBs, electrolytes dictate the battery's electrochemical performance, stability and safety. In recent years, polymer electrolytes are gaining popularity over solvent-based liquid electrolytes due to their reduced risk of dendrite formation, which can cause short circuitry, overheating and thermal runaway.1,2 Thus, they can be expected to partially replace conventional liquid electrolytes in LiBs in the coming years.3 Although a number of petroleum-based polymers such as polyethylene oxides (PEOs) have been successfully applied in real-life LiBs used in electric vehicles,4 increasing global demand has driven attempts to develop more sustainable alternatives.5,6 Other than being produced from low-cost, renewable feedstock, the material's end-of-life is another important, but often overlooked, aspect of sustainable electrolyte design.7–9 Indeed, with increasing consumption of batteries, the need to develop strategies for post-lifespan treatment of waste battery materials will become increasingly urgent to reduce the environmental impact and recover the cost of raw materials (typically minerals).10 Although recycling batteries to recover precious metals has been the main focus of the industry, the increasing popularity of polymer-based solid state batteries presents a pressing need to uncover viable solutions for treating used polymer electrolytes. While natural polysaccharides (e.g. cellulose, chitosan, carrageenan) and their derivatives have been explored as sustainable polymer electrolytes,11–13 their crystallinity can be difficult to control and can give rise to poor ionic conductivities. This results in the need for creation of blends and composites13 which can later complicate end-of-life processing.14 Therefore, developing strategies for effective post-usage treatment of polymer electrolyte materials produced from sustainable sources that are also tolerant to different additives (e.g. lithium salt) present in the electrolyte formulation is of urgent necessity.Against this backdrop, the exploitation of low-cost, readily available societal waste as alternative feedstock for sustainable electrolyte production is highly desirable. Successful applications of polycarbonates synthesised from CO29 and poly(glycidols) from glycidol – a by-product of epichlorohydrin production15 – have been reported. Other than these feedstocks, plastic waste holds immense potential for upcycling into functional polymers. This exploits and transcends the inherent chemical and physical properties of plastics for more economically valuable applications,16–20 whilst simultaneously keeping them in sustainable material loops. Plastics containing cleavable linkages along their polymer backbones, such as bisphenol A polycarbonates21 and poly(ethylene terephthalate) (PET) polyesters, have been shown to be viable starting materials for polymer electrolytes in working cell prototypes.22 Notably for the latter, the rigid aromatic segments of PET were essential for achieving mechanically robust conductive polymer electrolyte films. However, upcycling of plastics containing saturated, chemically unreactive C–C polymer backbones, which constitute close to 60% of global plastic waste production,23 into viable polymer electrolyte materials has thus far remained elusive.
Herein, we report a proof-of-concept cradle-to-cradle approach where polymer electrolytes produced from low-cost, abundant polyethylene (PE) plastic can be converted post-use to aliphatic organic acid feedstock for diverse applications (Fig. 1). PE, the most abundant waste plastic produced,23 is post-synthetically chemically modified and upcycled into functional non-polyethylene oxide (PEO)-containing copolymers suitable as both solid and gel polymer electrolytes. To achieve a simple, readily accessible polymer design from PE, the non-polar PE main chain was grafted covalently with pendant polar polyester segments (PCL24/PLA) to form amphipathic branched copolymers. Other than potentially facilitating ionic transport,25,26 introduction of branching through the pendant polyester segments is expected to suppress polymer crystallisation,27 forming more amorphous regions that may facilitate ion transport. A prototype coin cell assembled shows promising electrochemical and cycling performance. Furthermore, we demonstrate the possibility of upcycling these PE-derived polymer electrolytes into valuable C4–C6 dicarboxylic acids that are useful for applications ranging from pharmaceutics to chemical precursors, thereby overcoming the difficulties of recycling copolymers which can contaminate recyclates. Notably, this process is unaffected by the significant quantities of LiTFSI present in the polymer electrolyte formulations. The simplicity of our PE-derived polymer electrolyte design, promising performance as a LiB prototype, and post-usage polymer end-of-life upcycling into useful diacids can offer new cradle-to-cradle opportunities for sustainable energy storage materials derived from waste plastics.
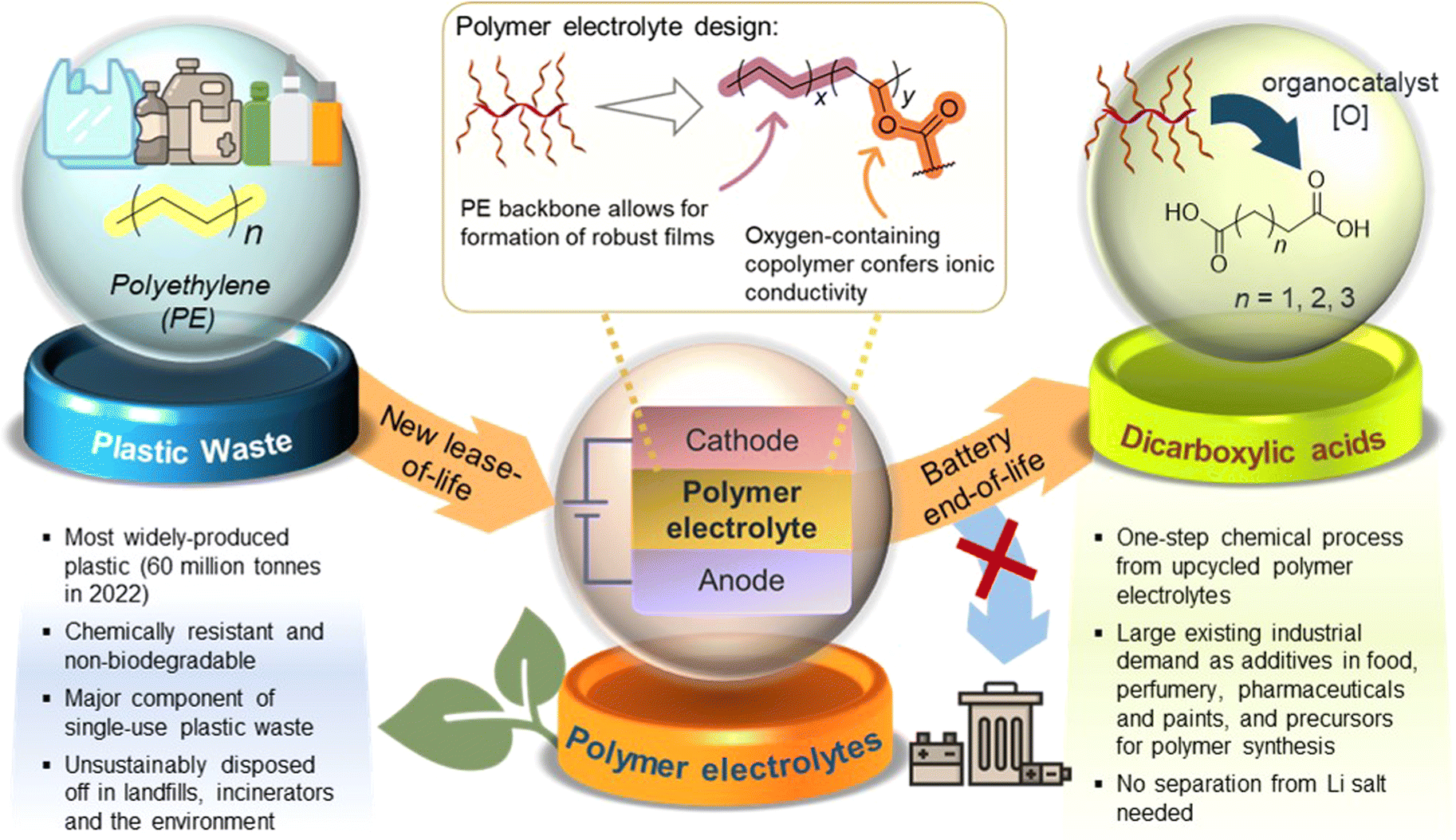 | ||
| Fig. 1 The cradle-to-cradle concept of upcycling post-use polyethylene to graft copolymers which can be used as polymer electrolytes for energy storage, and thereafter further valorising the end-of-life polymer electrolytes into industrially relevant dicarboxylic acids. | ||
Results and discussion
Synthesis and characterisation of PE-derived polymer electrolytes
To synthesise our targeted PE-derived polymer electrolytes, the C–H activation of PE to introduce reactive functional groups onto the polymer structure is necessary (Fig. 2). In this regard, oxygenated groups such as alcohols are particularly useful, as they can be initiating sites for ring-opening polymerisation (ROP) of cyclic lactones, allowing convenient grafting of polycaprolactone (PCL) as reported by Hartwig,24 as well as polylactic acid (PLA) segments onto the main PE polymer chain that can facilitate ionic transport.25 For PE activation, usage of O2 as an oxidant is preferred28 to avoid generation of additional waste from organic oxidants. As a result, N-hydroxyphthalimide (NHPI) organocatalysts were chosen for their efficacy in PE oxidation,29 and to avoid usage of transition metal catalysts which can degrade battery performance if incompletely removed.30 First introduced by Ishii and co-workers,31 NHPIs are attractive organocatalysts for the aerobic oxidation of hydrocarbons because of their low-toxicity, high activities under mild conditions,32 and ease of synthesis using green solvents such as acetic acid.33,34 NHPIs are precursors of the phthalimide N-oxyl (PINO) radical, which is the key catalytic species responsible for hydrogen abstraction from C–H bonds to promote the O2-mediated oxidation process.35 By varying the substituents on NHPI, which affects the hydrogen abstraction activation energy of the PINO radicals,36 the extents of activation and selectivity for PE oxidation may also be modulated.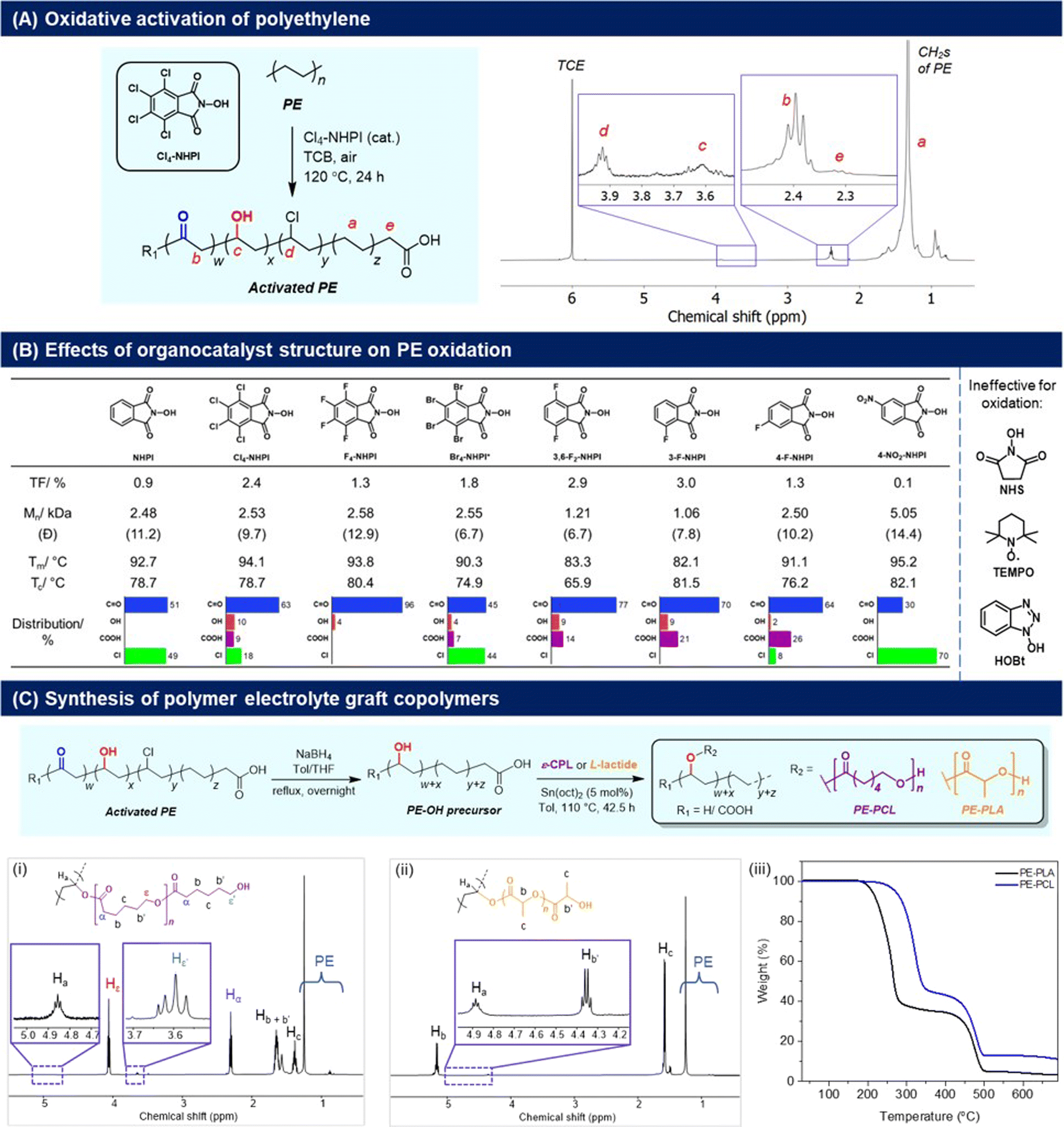 | ||
| Fig. 2 (A) Cl4-NHPI-catalysed PE aerobic oxidation and 1H NMR spectrum (C2D2Cl4, 80 °C) of the oxidised polymer; (B) effects of different NHPI-derived organocatalysts on PE oxidation, with TF values determined by 1H NMR analyses (Fig. S7–16†). Values of Mn and polydispersity (Đ) are determined by high-temperature GPC analysis in TCB (Fig. S5†), with PE starting material having Mn = 6870 and Đ = 12.7. Melting (Tm) and crystallisation temperatures (Tc) were determined by DSC analyses (2nd heating cycle), with Tm and Tc of PE starting material = 94.0 °C and 78.5 °C respectively; (C) conversion of the activated PE to PE-PCL and PE-PLA graft-copolymers through successive reduction and ring-opening polymerisation, with (i and ii) 1H NMR spectra (CDCl3) and (iii) TGA measurements of the copolymers shown. | ||
The aerobic oxidation reaction with PE resins was first performed in the presence of 2.5 mol% of Cl4-NHPI in 1,2,4-trichlorobenzene (TCB) at 120 °C in air. Product characterisation by 1H NMR spectroscopy (C2D2Cl4, 80 °C) reveals a signal at δ = 2.40 ppm corresponding to the α-protons of carbonyl groups on PE (Fig. 2A).37 Discernible signals at δ = 3.92 ppm (–CH–Cl–), 3.61 ppm (–CH–OH–), and 2.32 ppm [HO–C(O)–CH2–] also indicate some degree of chloro, hydroxyl and carboxylic acid functionalisation respectively. Chloro-substitution may be attributed to radical abstraction from the TCB solvent.38 Based on the relative integration of each of the aforementioned signals to that of the unreacted methylene groups of PE (0.7–1.8 ppm), the total degree of functionalisation (TF) is calculated to be 2.4% of PE's repeating units, with predominant carbonyl selectivity (63%) (Fig. S1†). The presence of carbonyls is corroborated by 13C NMR analysis, whereby resonances at δ = 211.3 and 42.9 ppm correspond to the carbonyl and α-methylene carbons respectively (Fig. S2†). The FTIR spectrum (Fig. S3†) showed a broad absorption peak at 1714 cm−1 from the overlapping C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of ketones and carboxylic acids, and alcohols were discerned by the broad O–H absorption band at 3294 cm−1 and diagnostic C–O stretch at 1101 cm−1. PE functionalisation was further verified by XPS analysis (Fig. S4†), where peak fitting of the high-resolution C 1s and O 1s spectra revealed component peaks corresponding to C–O/C–OH, CO and O–CO chemical states.39 High-temperature GPC analyses (Fig. 2B and S5†) revealed that some chain scission occurred during PE oxidation, with the termini of the oligomers likely containing carboxylic acid groups. Control experiments (see Section S2.1†) revealed that replacing air (21% O2) with a pure O2 atmosphere gave similar TF with a greater extent of chain scission, whilst the absence of Cl4-NHPI or O2 elicited no discernible oxidation. Additionally, the necessity of solvent was demonstrated as a very low degree of functionalisation (TF = 0.3) with poor selectivity was obtained when the reaction was repeated with neat molten PE at the same temperature (Fig. S6†). Compared to the use of 1,1,2,2-tetrachloroethane (TCE) that we have previously reported,29 TCB elicited greater selectivity for carbonyl installation on PE, albeit at the expense of TF.
O stretch of ketones and carboxylic acids, and alcohols were discerned by the broad O–H absorption band at 3294 cm−1 and diagnostic C–O stretch at 1101 cm−1. PE functionalisation was further verified by XPS analysis (Fig. S4†), where peak fitting of the high-resolution C 1s and O 1s spectra revealed component peaks corresponding to C–O/C–OH, CO and O–CO chemical states.39 High-temperature GPC analyses (Fig. 2B and S5†) revealed that some chain scission occurred during PE oxidation, with the termini of the oligomers likely containing carboxylic acid groups. Control experiments (see Section S2.1†) revealed that replacing air (21% O2) with a pure O2 atmosphere gave similar TF with a greater extent of chain scission, whilst the absence of Cl4-NHPI or O2 elicited no discernible oxidation. Additionally, the necessity of solvent was demonstrated as a very low degree of functionalisation (TF = 0.3) with poor selectivity was obtained when the reaction was repeated with neat molten PE at the same temperature (Fig. S6†). Compared to the use of 1,1,2,2-tetrachloroethane (TCE) that we have previously reported,29 TCB elicited greater selectivity for carbonyl installation on PE, albeit at the expense of TF.
A family of NHPI derivatives33,34 were then probed to identify the most suitable organocatalyst for PE oxidation (Fig. 2B). All catalysts attempted were soluble at 2.5 mol% loading in TCB for PE oxidation. As shown in Fig. 2B, carbonyl functionalisation on PE was dominant for most NHPI derivatives, with varying relative proportions of alcohol, acid and chloro groups. Compared to Cl4-NHPI, unsubstituted NHPI resulted in lower TF, though only carbonyl and chloro groups were observed by 1H NMR analysis (Fig. S7†). For the tetrahalo-substituted NHPIs, the TF seen for Cl4-NHPI exceeds that of F4-NHPI, which mirrors earlier findings of more electron-deficient PINO radicals being more reactive towards hydrogen abstraction by increasing their NO–H bond dissociation energies.40 F4-NHPI is also notable for giving the highest carbonyl selectivity amongst all organocatalysts tested. Even with a lower solubility of 0.5 mol% in TCB, Br4-NHPI could elicit a higher TF of 1.8 compared with F4-NHPI, indicative of its high reactivity for PE oxidation. Amongst the different fluorinated-NHPI derivatives, 3-F-NHPI and 3,6-F2-NHPI gave the highest TFs. Changing the position of fluorine substitution caused a drastic reduction in TF as seen for 4-F-NHPI, clearly demonstrating the important influence of positional isomerism on NHPI electronics. Despite different aromatic substituents significantly influencing the rate of PINO radical decay,41 it is likely that electronic effects on PE hydrogen abstraction dominate, as Cl4- and 3-F-substitutions give higher TF than their F4- and 4-F-counterparts, respectively, despite the former's more rapid decay.41 Notably, no C–H oxidation was observed in the presence of N-hydroxysuccinimide (NHS), TEMPO and hydroxybenzotriazole (HOBt) (Fig. S14–16†), in spite of their good solubility at 2.5 mol%, showing the necessity of the NHPI motif for appreciable aerobic PE oxidation to occur. High-temperature GPC analyses revealed that generally the more active NHPI catalysts that elicited greater degrees of functionalisation also resulted in more pronounced PE chain scissions.
Considering its commercial availability and the ability to achieve one of the highest TFs while inducing a lower extent of chain cleavage compared to 3,6-F2-NHPI and 3-F-NHPI, Cl4-NHPI was chosen as the catalyst of choice. After commercially available HDPE resins were oxidised with Cl4-NHPI, reduction with NaBH4 afforded PE with predominantly hydroxyl functionalisation. The hydroxylated PE (PE-OH) was subsequently subjected to ring-opening polymerisation (ROP) with ε-caprolactone and L-lactide to afford PE-PCL (78 wt% PCL) and PE-PLA (76 wt% PLA) copolymers respectively (Fig. 2C, further characterisation in Fig. S20†). The formation of these polymers was evident from the appearance of the resonances at 4.86 ppm (CDCl3) for PE-PCL and 4.88 ppm (CDCl3) for PE-PLA, corresponding to Cα–H on the PE segment bonded directly to the grafted polyesters as previously reported by Hartwig and co-workers (PE-PCL).24 Additionally, both copolymers were soluble in chloroform, unlike the PE-OH precursor.24 FTIR analysis of the PE-PCL copolymer also showed characteristic absorbance peaks corresponding to PCL which were absent in the PE-OH precursor (Fig. S17†). It is notable that real-life HDPE waste plastic bags could be similarly oxidised by Cl4-NHPI under the same reactions to afford predominant carbonyl functionalisation (Fig. S22†), thereby suggesting the possibility of converting real-life waste PE to functional polymer electrolytes.
The thermal properties of the PE-graft-copolymers were then investigated by TGA and DSC analyses. From the TGA measurements, an onset of decomposition above 200 °C was observed for both PE-PCL and PE-PLA (Fig. 2C), which is well above typical battery operating temperatures. The first thermal degradation peak, centred around 320 °C and 260 °C for PE-PCL and PE-PLA respectively (based on derivative thermogravimetry, Fig. S23†), corresponds to decomposition of the polyester segments. In both cases, thermal decomposition of the PE segment only commenced above 400 °C, which was similar to that of the PE-OH precursor (Fig. S24†). DSC measurements (Fig. S25A†) revealed two distinct endothermic peaks for PE-PCL centred at 48 °C and 113 °C, which correspond to the melting temperature (Tm) of the PCL and PE segments respectively,24 thereby suggesting the likelihood of microphase separation between both copolymer segments.42 In contrast, PE-PLA demonstrated greater homogeneity as indicated by its single well-defined Tm at 108 °C (Fig. S25B†). Although distinct Tg values were not observed by DSC measurements for the PE-PCL copolymer, we expect values of <−40 °C based on typical Tg values of <−50 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 and <−100 °C for PCL and HDPE44 respectively. Both PE-polyester copolymers were semicrystalline, and their percentages of crystallinity were determined to be 11.7% and 19.6% for PE-PCL and PE-PLA respectively through DSC analyses (Fig. S25†). Their semicrystalline nature was corroborated through XRD analyses, showing the presence of crystalline peaks arising from both PE and polyester segments (PE-PCL: 2θ = 21.2° and 23.5° arising from either HDPE45 or PCL;46 PE-PLA: 2θ = 21.2° from HDPE, and 16.4° and 18.7° from PLLA47) (Fig. S26†).
43 and <−100 °C for PCL and HDPE44 respectively. Both PE-polyester copolymers were semicrystalline, and their percentages of crystallinity were determined to be 11.7% and 19.6% for PE-PCL and PE-PLA respectively through DSC analyses (Fig. S25†). Their semicrystalline nature was corroborated through XRD analyses, showing the presence of crystalline peaks arising from both PE and polyester segments (PE-PCL: 2θ = 21.2° and 23.5° arising from either HDPE45 or PCL;46 PE-PLA: 2θ = 21.2° from HDPE, and 16.4° and 18.7° from PLLA47) (Fig. S26†).
Performance of polymer electrolytes
The applications of the PE-polyester copolymers as solid polymer electrolytes (SPEs) were first investigated. To ensure homogeneity in the resulting SPE films, the polymers were first mixed with appropriate quantities of LiTFSI salt in anhydrous 2-methyltetrahydrofuran, a bioderived green solvent,48 in an argon-filled glovebox. Removal of the solvent, followed by hot-pressing and drying in vacuo afforded the target films which were subjected to ionic conductivity measurements between 20 and 80 °C (see Section S1† for detailed experimental procedures). Unlike the resulting SPE films of PE-PCL and PE-PLA which were free-standing and easy to handle for ionic conductivity determination, those produced using PCL alone (similar molecular weight as the grafted chains on PE-PCL) with the same weight percentage loading of LiTFSI were found to be unsuitable for accurate electrochemical evaluation. Low loading of LiTFSI ([PCL]:[LiTFSI] = 12:1) formed highly brittle films which disintegrated easily (Fig. S27B†). However, doubling the LiTFSI loading ([PCL]:[LiTFSI] = 6:1) resulted in soft and tacky mixtures that could not be formed into films due to the plasticising effect. This highlighted the importance of the PE segment in improving the films' mechanical integrity.
As shown in Fig. 3A(i), the ionic conductivities of PE-PCL followed typical Arrhenius-like behaviour, with higher temperatures bringing about greater conductivity. Notably, the conductivities (Table 1) were comparable to those of SPEs previously reported from upcycled bisphenol A polycarbonates and PET,21,22 with higher LiTFSI loadings giving similar ionic conductivities measured (Table 1, entries 1 and 2). In comparison, the ionic conductivity of PE-PLA was found to be at least an order-of-magnitude lower than that of PE-PCL, even with a higher loading of LiTFSI (entry 3). Similarly, poor ionic conductivity was observed with the PE-OH precursor containing an identical weight loading of LiTFSI (entry 4), showing that the presence of PCL in the PE-PCL copolymer was essential for appreciable ionic conductivity. Indeed, solution phase 1H NMR studies of PE-PCL in a saturated 1,1,2,2-tetrachloroethane-d2 (TCE-d2) solution of LiTFSI showed downfield perturbations of only the PCL proton resonances and not those of PE, indicating the preferential interaction of Li+ with the oxygenated groups of PCL [Fig. 3A(ii)]. Consistent with the known plasticising effect of LiTFSI,49 DSC measurements revealed that its presence reduced the crystallinity of PE-PCL from 11.7% to ∼7% (Fig. S25†). This was further corroborated by XRD measurements of the PE-PCL-LiTFSI film which showed clear peak broadening (Fig. S26†). Conductivity was likely attributed to ion hopping between ester coordination sites on amorphous PCL phases, with added contributions from enhanced segmental chain motion.50 Using the Arrhenius equation:
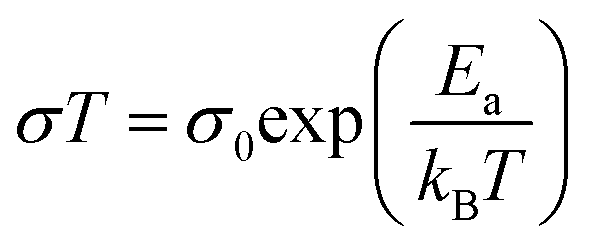
 | ||
| Fig. 3 (A) (i) Effects of higher LiTFSI loadings on ionic conductivities of PE-PCL SPEs; (ii) stacked expanded 1H NMR spectra of PE and PCL segments of PE-PCL in TCE-d2 (top, without LiTFSI) and in LiTFSI-saturated TCE-d2. (B) (i) Effects of different quantities of LiTFSI and EMI-TFSI ionic liquid on PE-PCL GPEs at different temperatures; (ii) photo of a free-standing PE-PCL GPE film; (iii) SEM cross-sectional morphology of PE-PCL GPEs after EMI-TFSI and LiTFSI removal at 10000× magnification. (C) (i) Galvanostatic charge–discharge curves of the Li/PE-PCL GPE/LFP cell at 60 °C (0.1C); (ii) cycling performance and coulombic efficiency of the Li/PE-PCL GPE/LFP cell over 120 cycles (60 °C, 0.1C); (iii) rate performance of the Li/PE-PCL GPE/LFP cell at various discharge rates at 60 °C. | ||
| S/N | Polymer | Film composition (molar ratio) | Ionic conductivity at 20 °C (S cm−1) | Ionic conductivity at 60 °C (S cm−1) | E a (eV) | R 2 |
|---|---|---|---|---|---|---|
| a Determined from the Arrhenius equation. b Goodness-of-fit determined from Fig. 3A(i) and B(i). c Not determined due to low ionic conductivity observed. d See Fig. S28 for temperature-dependence of ionic conductivity. | ||||||
| Performance as solid polymer electrolytes (SPEs) | ||||||
| 1 | PE-PCL | [PCL]:[LiTFSI] = 12:1 |
2.0 × 10−6 | 1.9 × 10−5 | 0.49 | 0.999 |
| 2 | PE-PCL | [PCL]:[LiTFSI] = 6:1 |
1.1 × 10−6 | 1.6 × 10−5 | 0.56 | 0.997 |
| 3 | PE-PLA | [PLA]:[LiTFSI] = 3:1 |
<10−7 | <10−7 | -c | -c |
| 4 | PE-OH | Same wt% loading as entry 1 | <10−7 | <10−7 | -c | -c |
|
||||||
| Performance as gel polymer electrolytes (GPEs) | ||||||
| 5 | PE-PCL | [PCL]:[LiTFSI] = 12:1 |
8.1 × 10−6 | 6.0 × 10−5 | 0.44 | 0.999 |
| 22 wt% EMITFSI w.r.t. polymer | ||||||
| 6 | PE-PCL | [PCL]:[LiTFSI] = 12:1 |
4.7 × 10−5 | 2.3 × 10−4 | 0.35 | 0.994 |
| 42 wt% EMITFSI w.r.t. polymer | ||||||
| 7 | PE-PCL | [PCL]:[LiTFSI] = 6:1 |
3.8 × 10−5 | 1.8 × 10−4 | 0.34 | 0.996 |
| 40 wt% EMITFSI w.r.t. polymer | ||||||
| 8 | PE-OH | 50 wt% LITFSI w.r.t. polymer | 1.1 × 10−7 | 2.4 × 10−7 | -c | -c |
| 20 wt% EMI-TFSI w.r.t. polymer | ||||||
| 9 | PE-PLAd | [PLA]:[LiTFSI] = 3:1 |
2.7 × 10−7 | 1.2 × 10−5 | 0.79 | 0.998 |
| 40 wt% EMITFSI w.r.t. polymer | ||||||
Compared with SPEs, GPEs offer improved ionic conductivities by the incorporation of a liquid into the solid polymer matrix without compromising on electrochemical stability and mechanical properties.54,55 To investigate the performance of the PE-polyesters as GPEs, the copolymers and LiTFSI were cast into free-standing, non-leaky GPE films containing varying quantities of EMI-TFSI ionic liquid (Fig. S27C†). Notably, free-standing homogeneous GPE films of PE-PCL containing LiTFSI and EMI-TFSI could be fabricated (Fig. 3B(ii)). In contrast, a physical blend of pure PE and PCL (similar molecular weight as that used in the PE-PCL graft copolymer), together with LiTFSI and EMI-TFSI in the same ratios as that of the PE-PCL copolymer film, could only form a paste under identical fabrication conditions (Fig. S27D†). This further demonstrates that the PE-PCL copolymer provided superior mechanical properties for film formation, even in the presence of the highly plasticising EMI-TFSI ionic liquid.
As expected, the presence of EMI-TFSI resulted in an increased ionic conductivity of PE-PCL GPEs at identical loadings of LiTFSI (Table 1, entries 1 and 5), which was augmented at higher temperatures [Fig. 3B(i)]. Notably, with 22 wt% EMI-TFSI loading, similar Ea values (via the Arrhenius model) to the SPE were obtained (entry 1). This indicates that ionic conduction was likely still dominated by ion-hopping or polymer chain segmental motion akin to SPEs. However, further increasing the EMI-TFSI loading from 22 wt% to 42 wt% w.r.t. polymer afforded more than an order-of-magnitude enhancement in ionic conductivity, with significant reduction in Ea (entry 6). These findings suggest a change in the dominant mechanism of ionic conductivity in the presence of sufficient EMI-TFSI loading. This likely involves conduction through the liquid phases percolating the PE-PCL polymer matrix enabled by the micropores (see Fig. 3B(ii) for SEM image). Doubling the LiTFSI loading (entry 7) did not result in further enhancement of ionic conductivity or Ea for ion conduction. Compared to PE-PCL, GPEs formed from the PE-OH precursor and PE-PLA polymer show poorer ionic conductivity (entries 8 and 9). For the latter, analysis of the film cross-section by SEM (Fig. S29†) showed smaller micropores compared to those of the aforementioned PE-PCL, which possibly result in poorer liquid phase percolation and, thus, poorer ionic conductivity.
Considering the superior performance of the PE-PCL GPE, we first assessed its electrochemical stability to determine its suitability for incorporation into prototype Li-ion cells. Linear sweep voltammetry (LSV) (Li/PE-PCL GPE/stainless steel, 1 mV s−1) shows the stability of the GPE within the potential window of up to 4.5 V versus Li/Li+, which is well above the upper voltage limit of LiFePO4 (LFP)-based cells (Fig. S30†). Additionally, cyclic voltammetry (CV) was performed at a scanning rate of 1 mV s−1 in the voltage range of −0.5 to 2 V for the Li/PE-PCL GPE/SS cell (Fig. S31A†) and in the voltage range of 2 to 4 V for the Li/PE-PCL GPE/LFP cell (Fig. S31B†) at 1 mV s−1. The CV plots show the typical Li plating and stripping behaviour on the cathodic and anodic scans with no other major peaks observed. Similarly, the only peaks observed for the LFP-based cells correspond to oxidation and reduction peaks of Fe2+/Fe3+, further confirming the stability of the cell.
Encouraged by these findings, the performance of the Li/PE-PCL GPE/LFP cell was further investigated. Galvanostatic charge–discharge cycles performed between 2.5 and 4 V at 0.1C (60 °C) showed flat charge and discharge profiles which indicated good electrochemical reversibility [Fig. 3C(i)]. According to the cycling performance as seen in Fig. 3C(ii), the Li/PE-PCL/LFP cell was able to maintain a very high coulombic efficiency of 100% and initial discharge capacity of 160 mA h g−1, close to the theoretical capacity of LFP at 170 mA h g−1. The cell maintains its high capacity over 120 cycles, with a capacity retention of >80%. Next, the rate performance of the cell was investigated [Fig. 3C(iii)]. As the cycling rate was increased from 0.1 to 0.3C, an excellent retention of capacity (2% capacity fade) was observed. Although higher cycling rates resulted in greater decreases of capacity to 60 mA h g−1 at a high rate of 2C, initial capacity could be recovered when the cycling rate returned to 0.1C. These experiments demonstrated the electrochemical stability of the PE-PCL polymer, and that no significant polymer degradation had occurred through repeated cycling.
Chemical upcycling of end-of-life electrolyte materials
With increasing functional applications of polymer electrolyte materials in energy storage devices, treatment of these polymers at their end-of-life is of the essence to avoid further contribution to the global problem of plastic waste. The polymer electrolyte recycling process can be further complicated by the presence of additives such as Li salt and liquid phase (for GPEs), which can contaminate waste streams and reduce the quality of recyclates. Although existing polyelectrolyte materials such as polyethylene glycol can be biodegraded,56 composite and copolymeric electrolyte materials can pose challenges for recycling and upcycling. For instance, inherent differences in reactivity between different copolymer segments often render a recycling/upcycling process suitable for one segment but unsuitable for another. For the PE-PCL copolymer used herein, while the PCL segment can theoretically be chemically recycled through catalytic reformation of the ε-caprolactone monomer,57 the PE segment will be chemically unaffected in this process. To circumvent this problem, we designed an oxidation process capable of upcycling both PE and PCL segments at the same time, whilst being tolerant to additives such as LiTFSI. This enables the formulated polymer electrolytes (e.g. as SPEs) to be directly used for oxidative upcycling without additional pretreatment.Building on our successes in utilising N-hydroxyl organocatalysts for oxidative conversion of polystyrene to benzoic acids,34 PE-PCL was reacted with HNO3 in the presence of the Cl4-NHPI catalyst in a green solvent, acetic acid, for 24 hours. Under such conditions, N-oxy radicals can be generated from the Cl4-NHPI precursor, which can abstract hydrogen atoms from the PE-PCL polymer.34 The resulting radicals can combine with O2 or nitrogen-containing species from HNO3 decomposition to form oxo-radicals on the polymer chain, ultimately propagating chain scission to form dicarboxylic acids.
While a range of dicarboxylic acids were formed during the oxidation, possessing chain lengths between C4–C11 determined via ESI-MS analysis (Fig. S33 and S34†), analysis of the product mixture by 1H NMR gave unequivocal evidence of succinic (C4), glutaric (C5) and adipic acid (C6) as the main products formed (Fig. S32†). Fig. 4 shows the amounts of each acid formed with different combinations of polymers and reagents, determined through quantitative 1H NMR analysis with 1,2-dichloroethane as the internal standard. By itself, Cl4-NHPI-catalysed oxidation of PE-OH afforded glutaric acid as the main product, with smaller quantities of succinic and adipic acid formed. This preference for C4 and C5 products can be expected as depolymerisation likely occurs through a zip depolymerisation reaction involving cyclic transition states via backbiting mechanisms.58 Under identical catalysed conditions, PCL produces mainly adipic acid, likely from tandem acid hydrolysis of the polyester followed by oxidation in situ. Under these conditions, shorter-chain succinic and glutaric acid were also formed, possibly via decarboxylation to form terminal alkyl radicals which could undergo further oxidation.59 Similar product distributions were observed when the PCL oxidation reaction was repeated in the presence of 15 wt% LiTFSI. For the PE-PCL copolymers, the absence of Cl4-NHPI in the reaction led to mainly adipic acid formation. However, addition of Cl4-NHPI resulted in production of succinic and glutaric acid, reflective of the catalyst's ability to enhance oxidation efficacy.34 The importance of the Cl4-NHPI catalyst was also apparent when solids could still be observed in the catalyst-free reaction after 24 h. These solids, as shown by 1H NMR analysis (Fig. S35†), were found to be mainly unreacted PE fragments. This indicates that only the PCL segment could be broken down in the absence of the catalyst, accounting for the predominant adipic acid formation. In contrast, the catalyst's presence led to the formation of clear homogeneous solutions after 24 h, which demonstrates that PE oxidative degradation had occurred. Although addition of LiTFSI afforded slightly higher yields of glutaric and adipic acid, the differences are small despite the greater stoichiometric excess of LiTFSI compared to the catalyst, suggesting only minor influences on reactivity. Indeed, these short-chain dicarboxylic acids are important industrially as they are used as intermediates for production of pharmaceuticals, adhesives, coatings, food additives, and polymers such as Nylon-6,6 from adipic acid, amongst others, with multi-billion-dollar immediate addressable markets expected to undergo rapid growth.60–62
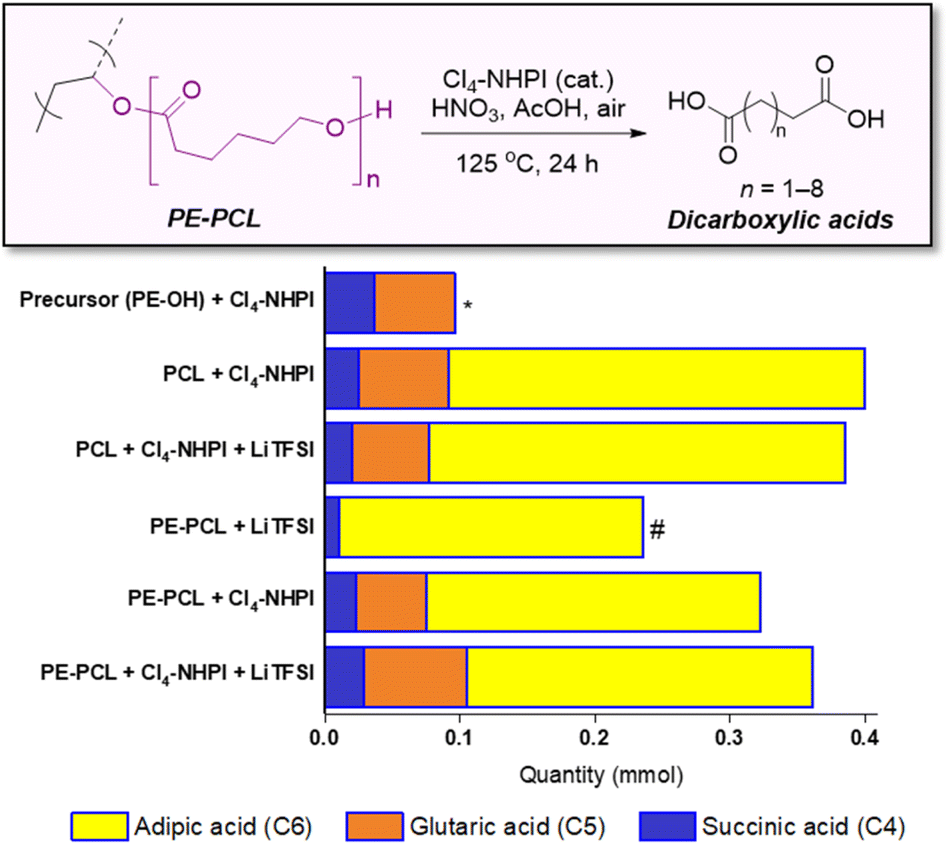 | ||
| Fig. 4 Different quantities of succinic, glutaric and adipic acid formed from oxidative upcycling of PE-PCL, PE-OH and PCL catalysed by Cl4-NHPI with or without LiTFSI. (* although adipic acid could be detected by ESI-MS, it was not present in sufficient quantities in the 1H NMR spectrum of the PE-OH oxidation reaction for accurate quantification; # solids, mainly PE, remained at the end of the reaction, hence the products are primarily from degradation of the PCL segment). | ||
Conclusions and outlook
In conclusion, we have demonstrated a cradle-to-cradle approach for upcycling polyethylene into PE-polyester polymer electrolytes, which can serve as feedstock for production of industrially relevant C4–C6 short-chain dicarboxylic acids after their end-of-life. Cl4-NHPI served as a convenient multi-purpose organocatalyst capable of first activating the unreactive PE polymer by installing reactive oxygenated groups on it, as well as for subsequent oxidation of PE-PCL polymer electrolytes to diacids. SPEs and GPEs formulated using the PE-PCL copolymer showed superior ionic conductivities compared with the analogous PE-PLA copolymer. Both PE and PCL components played synergistic roles which contributed to its success as an electrolyte material: while the PCL segment was necessary for ionic conductivity, the PE segment enhances film-forming ability which enabled formulations of self-standing, convenient-to-handle conductive films, despite some extent of oxidative PE chain cleavage resulting during PE functionalisation. Prototype Li/PE-PCL GPE/LFP cells showed excellent cycling performance, maintaining high electrochemical reversibility and stability throughout the cycling duration. Thereafter, oxidative upcycling of PE-PCL formed diacids from both copolymer segments simultaneously with greater selectivity for adipic acid and good tolerance for LiTFSI additives.The repurposing of low-value, high-volume waste plastics into polymer electrolyte materials is an emerging strategy for production of sustainable energy storage materials. For plastic upcycling to be economically feasible, the demand of the product should be sufficiently large and commensurate with the scale of plastic production. In this regard, the rapidly growing global demand for polymer electrolytes may offer a substantial market for upcycled PE-based electrolyte materials. As feedstock, sizeable streams of post-industrial PE waste from manufacturing processes may be plausible feedstocks, due to their well-defined compositions and high purity.63 This contrasts with municipal PE waste which is often heavily contaminated, thus necessitating extensive sorting and cleaning before it can be suitable for chemical upcycling. The use of TCB as a solvent for PE oxidative activation is unavoidable due to the excellent solvent resistance of PE. Furthermore, higher degrees of functionalisation could be achieved in TCB than in the absence of solvents. Nonetheless, TCB can conceivably be recovered and reused after the reaction, and can be possibly replaced by less hazardous bio-derived alternatives17 in future developments. With regard to the post-use fates of polymer electrolytes, our upcycling approach complements alternative strategies such as usage of dynamic covalent polymers which can extend the useful lifespan of these polymers by enabling repeated recyclability and reprocessibility.64 Despite this, inevitable material degradation by electrochemical processes through long-duration usage may ultimately compromise on recyclability.3 Our strategy of transforming used polymer electrolytes into small molecule diacids, on the other hand, offers new opportunities to keep the products from waste polymers in a circular materials economy for wholly different applications. Indeed, such a cradle-to-cradle upcycling approach will also be applicable to other polymeric materials from functional plastic upcycling at their end-of-lives, including antimicrobials, polymer coatings, and construction and packaging materials.65
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
J. Y. Q. Teo, M. Y. Tan, D. W. H. Fam and J. Y. C. Lim are listed as inventors on a patent application based on the results presented herein. The other authors have no conflicts to declare.Acknowledgements
Dr Debbie Seng is acknowledged for valuable assistance with XPS analysis. J. Y. C. Lim is grateful to the NRF Fellowship (Award number: NRF-NRFF15-2023-0007) for generous financial support; D. W. H. Fam acknowledges the AME Programmatic Grant (grant number A20H3b0140) on structural power for portable and electrified transportation.Notes and references
- B. Ramasubramanian, M. V. Reddy, K. Zaghib, M. Armand and S. Ramakrishna, Nanomaterials, 2021, 11, 2476 CrossRef CAS
.
- X.-Q. Xu, X.-B. Cheng, F.-N. Jiang, S.-J. Yang, D. Ren, P. Shi, H. Hsu, H. Yuan, J.-Q. Huang, M. Ouyang and Q. Zhang, SusMat, 2022, 2, 435–444 CrossRef CAS
- M. Ahuis, S. Doose, D. Vogt, P. Michalowski, S. Zellmer and A. Kwade, Nat. Energy, 2024, 9, 373–385 CrossRef CAS
- X. Judez, G. G. Eshetu, C. Li, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, Joule, 2018, 2, 2208–2224 CrossRef CAS
- J. C. Barbosa, R. Gonçalves, C. M. Costa and S. Lanceros-Méndez, ACS Omega, 2022, 7, 14457–14464 CrossRef CAS PubMed
- M. Y. Tan, D. Safanama, S. S. Goh, J. Y. C. Lim, C.-H. Lee, J. C. C. Yeo, W. Thitsartarn, M. Srinivasan and D. W. H. Fam, Chem.–Asian J., 2022, 17, e202200784 CrossRef CAS
- W. Gu, F. Li, T. Liu, S. Gong, Q. Gao, J. Li and Z. Fang, Adv. Sci., 2022, 9, 2103623 CrossRef CAS PubMed
- R. Zhang, X. Shi, O. C. Esan and L. An, Global chall., 2022, 6, 2200050 CrossRef
- H. Yeo, G. L. Gregory, H. Gao, K. Yiamsawat, G. J. Rees, T. McGuire, M. Pasta, P. G. Bruce and C. K. Williams, Chem. Sci., 2024, 15, 2371–2379 RSC
- Z. J. Baum, R. E. Bird, X. Yu and J. Ma, ACS Energy Lett., 2022, 7, 712–719 CrossRef CAS
- J. Hu, Z. Wang, Y. Si, C. Hong, C. Zhao, Y. Xing, W. Ling, Y. Wang, L. Feng and W. Feng, J. Mater. Chem. A, 2023, 11, 878–890 RSC
- M. Rayung, M. M. Aung, S. C. Azhar, L. C. Abdullah, M. S. Su’ait, A. Ahmad and S. N. Jamil, Materials, 2020, 13(4), 838 CrossRef CAS PubMed
- R. T. Abdulwahid, S. B. Aziz and M. F. Z. Kadir, Mater. Today Sustain., 2023, 23, 100472 CrossRef
- A. Dorigato, Adv. Ind. Eng. Polym. Res., 2021, 4, 53–69 CAS
- G. Piana, M. Ricciardi, F. Bella, R. Cucciniello, A. Proto and C. Gerbaldi, Chem. Eng. J., 2020, 382, 122934 CrossRef CAS
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS
- B.-N. T. Nguyen and J. Y. C. Lim, Trends Chem., 2024, 6, 100–114 CrossRef CAS
- J. Y. Q. Teo, A. Ong, T. T. Y. Tan, X. Li, X. J. Loh and J. Y. C. Lim, Green Chem., 2022, 24, 6086–6099 RSC
- J. B. Williamson, S. E. Lewis, R. R. Johnson
Iii, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed
- N. R. Ciccia, J. X. Shi, S. Pal, M. Hua, K. G. Malollari, C. Lizandara-Pueyo, E. Risto, M. Ernst, B. A. Helms, P. B. Messersmith and J. F. Hartwig, Science, 2023, 381, 1433–1440 CrossRef CAS PubMed
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo-Martínez, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC
- M. Y. Tan, L. Goh, D. Safanama, W. W. Loh, N. Ding, S. W. Chien, S. S. Goh, W. Thitsartarn, J. Y. C. Lim and D. W. H. Fam, J. Mater. Chem. A, 2022, 10, 24468–24474 RSC
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef
- A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895–903 CrossRef CAS PubMed
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Nat. Rev. Mater., 2019, 4, 312–330 CrossRef CAS
- L. Meabe, S. R. Peña, M. Martinez-Ibañez, Y. Zhang, E. Lobato, H. Manzano, M. Armand, J. Carrasco and H. Zhang, J. Phys. Chem. C, 2020, 124, 17981–17991 CrossRef CAS
- D. Li, L. Zhou, X. Wang, L. He and X. Yang, Materials, 2019, 12, 1746 CrossRef CAS PubMed
- J. Y. Q. Teo, C. W. S. Yeung, T. T. Y. Tan, W. W. Loh, X. J. Loh and J. Y. C. Lim, Green Chem., 2022, 24, 6287–6294 RSC
- C. W. S. Yeung, M. H. Periayah, J. Y. Q. Teo, E. T. L. Goh, P. L. Chee, W. W. Loh, X. J. Loh, R. Lakshminarayanan and J. Y. C. Lim, Macromolecules, 2023, 56, 815–823 CrossRef CAS
- Y. K. Lee, J. Power Sources, 2021, 484, 229270 CrossRef CAS
- Y. Ishii, K. Nakayama, M. Takeno, S. Sakaguchi, T. Iwahama and Y. Nishiyama, J. Org. Chem., 1995, 60, 3934–3935 CrossRef CAS
- L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296–1310 CrossRef CAS PubMed
- Q. Zhang, C. Chen, J. Xu, F. Wang, J. Gao and C. Xia, Adv. Synth. Catal., 2011, 353, 226–230 CrossRef CAS
- A. Ong, J. Y. Q. Teo, Z. Feng, T. T. Y. Tan and J. Y. C. Lim, ACS Sustainable Chem. Eng., 2023, 11, 12514–12522 CrossRef CAS
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS
- Y. Sun, W. Zhang, X. Hu and H. Li, J. Phys. Chem. B, 2010, 114, 8032 CrossRef CAS
- L. Chen, K. G. Malollari, A. Uliana, D. Sanchez, P. B. Messersmith and J. F. Hartwig, Chem, 2021, 7, 137–145 CAS
- A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895–903 CrossRef CAS
- T. K. Glaser, O. Plohl, A. Vesel, U. Ajdnik, N. P. Ulrih, M. K. Hrnčič, U. Bren and L. Fras Zemljič, Materials, 2019, 12, 2118 CrossRef CAS
- C. Annunziatini, M. F. Gerini, O. Lanzalunga and M. Lucarini, J. Org. Chem., 2004, 69, 3431–3438 CrossRef CAS
- O. Kushch, I. Hordieieva, K. Novikova, Y. Litvinov, M. Kompanets, A. Shendrik and I. Opeida, J. Org. Chem., 2020, 85, 7112–7124 CrossRef CAS
- E.-S. Park, M.-N. Kim and J.-S. Yoon, J. Polym. Sci. B Polym. Phys., 2002, 40, 2561–2569 CrossRef CAS
- C. Sångeland, G. Hernández, D. Brandell, R. Younesi, M. Hahlin and J. Mindemark, ACS Appl. Mater. Interfaces, 2022, 14, 28716–28728 CrossRef
- MP, Tg, and Structure of Common Polymers, https://resources.perkinelmer.com/corporate/cmsresources/images/44-74863tch_mptgandstructureofcommonpolymers.pdf, accessed 20 Mar 2024, 2024.
- G. Madhu, H. Bhunia, P. K. Bajpai and V. Chaudhary, J. Polym. Eng., 2014, 34, 813–821 CAS
- R. Longo, M. Catauro, A. Sorrentino and L. Guadagno, J. Therm. Anal. Calorim., 2022, 147, 5391–5399 CrossRef CAS
- K. Tashiro, N. Kouno, H. Wang and H. Tsuji, Macromolecules, 2017, 50, 8048–8065 CrossRef CAS
- H. H. Khoo, L. L. Wong, J. Tan, V. Isoni and P. Sharratt, Resour. Conserv. Recycl., 2015, 95, 174–182 CrossRef
- Y. Cui, J. Zhang, P. Wang, X. Zhang, J. Zheng, Q. Sun, J. Feng and Y. Zhu, Electrochim. Acta, 2012, 74, 194–200 CrossRef CAS
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Devices., 2018, 3, 1–17 Search PubMed
- H. Xu, Q. Zhu, Y. Zhao, Z. Du, B. Li and S. Yang, Adv. Mater., 2023, 35, 2212111 CrossRef CAS PubMed
- H. Tang, M. Zhu, J. Wang, T. Liu, H. He, S. Chen and X. Yao, Next Mater., 2024, 4, 100173 CrossRef
- R. Bakar, S. Darvishi, T. Li, M. Han, U. Aydemir, S. Nizamoglu, K. Hong and E. Senses, ACS Appl. Polym. Mater., 2022, 4, 179–190 CrossRef CAS
- J. Y. Song, Y. Y. Wang and C. C. Wan, J. Power Sources, 1999, 77, 183–197 CrossRef CAS
- J. Castillo, A. Santiago, X. Judez, I. Garbayo, J. A. Coca Clemente, M. C. Morant-Miñana, A. Villaverde, J. A. González-Marcos, H. Zhang, M. Armand and C. Li, Chem. Mater., 2021, 33, 8812–8821 CrossRef CAS
-
F. Kawai, in Biopolymers Online, 2005, DOI:10.1002/3527600035.bpol9012
- J. Su, G. Xu, B. Dong, R. Yang, H. Sun and Q. Wang, Polym. Chem., 2022, 13, 5897–5904 RSC
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef
- M. Gardette, A. Perthue, J.-L. Gardette, T. Janecska, E. Földes, B. Pukánszky and S. Therias, Polym. Degrad. Stab., 2013, 98, 2383–2390 CrossRef CAS
-
R. K. Saxena, S. Saran, J. Isar and R. Kaushik, in Current Developments in Biotechnology and Bioengineering, ed. A. Pandey, S. Negi and C. R. Soccol, Elsevier, 2017, pp. 601–630, DOI:10.1016/B978-0-444-63662-1.00027-0
- Glutaric acid market, https://www.futuremarketinsights.com/reports/glutaric-acid-during-market, accessed 21 Mar 2024, 2024.
- Adipic Acid Market, by Application (Nylon 6,6 Fiber, Nylon 6,6 Resin, Polyurethanes, and Adipate Esters), by End-use Industry (Automotive, Electrical & Electronics, Home Appliances, Textiles, and FMCG), and by Region (North America, Latin America, Asia Pacific, Europe, and Middle East & Africa)–Size, Share, Outlook, and Opportunity Analysis, 2022–2030, https://www.coherentmarketinsights.com/market-insight/adipic-acid-market-318, accessed 21 Mar 2024, 2024.
- K. Kleinhans, R. Demets, J. Dewulf, K. Ragaert and S. De Meester, Curr. Opin. Chem. Eng., 2021, 32, 100680 CrossRef
- Y. Lin, Y. Chen, Z. Yu, Z. Huang, J.-C. Lai, J. B. H. Tok, Y. Cui and Z. Bao, Chem. Mater., 2022, 34, 2393–2399 CrossRef CAS
- O. Guselnikova, O. Semyonov, E. Sviridova, R. Gulyaev, A. Gorbunova, D. Kogolev, A. Trelin, Y. Yamauchi, R. Boukherroub and P. Postnikov, Chem. Soc. Rev., 2023, 52, 4755–4832 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02178a |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |