
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic nanoparticles: advances in fundamental investigations and catalytic applications
Hongxia Lin,
Yuxi Liu*,
Jiguang Deng ,
Lin Jing,
Zhiwei Wang,
Lu Wei,
Zhen Wei,
Zhiquan Hou,
Jinxiong Tao and
Hongxing Dai*
,
Lin Jing,
Zhiwei Wang,
Lu Wei,
Zhen Wei,
Zhiquan Hou,
Jinxiong Tao and
Hongxing Dai*
Beijing Key Laboratory for Green Catalysis and Separation, Key Laboratory of Beijing on Regional Air Pollution Control, Key Laboratory of Advanced Functional Materials, Education Ministry of China, Laboratory of Catalysis Chemistry and Nanoscience, Department of Chemical Engineering, Faculty of Environment and Life, Beijing University of Technology, Beijing 100124, China. E-mail: yxliu@bjut.edu.cn; hxdai@bjut.edu.cn
First published on 21st October 2024
Abstract
Bimetallic nanoparticles provide promising active sites for many reactions, and such materials can be synthesized with different spatial distributions, such as disordered alloys, core–shell structures, and Janus-type heterogeneous structures. Catalytic activity, selectivity, and stability of bimetallic nanoparticles can be modified by the geometric, electronic, multifunctional and mixing effects, as compared with single metals. Accurate control of bimetallic compositions and their distributions is crucial to obtain high-performance catalysts. The present review summarizes the recent advances in preparation methods and catalytic applications of supported bimetallic nanomaterials. In addition, representative case studies are also provided to investigate how bimetallic nanoparticles can be used as desired catalysts and how specific functional catalysts are designed for targeted reactions. The structure–performance relationships of supported bimetallic catalysts for a number of reactions are discussed to achieve a fundamental understanding. Synthetic strategies and perspectives for precise control of bimetallic active components and element distributions with distinctive nanostructures are proposed for potential industrial applications.
Environmental significanceThis review covers recent advances in the preparation methods and catalytic applications of bimetallic nanomaterials. The relationships between structural features and catalytic properties of different bimetals are discussed. Deactivation and regeneration of the catalysts are highlighted, which are essential for catalyst design and optimization. Typical reactions, such as catalytic oxidation, hydrogen evolution reaction, and carbon dioxide reduction, are provided to demonstrate the wide range of catalytic applications of bimetallic catalysts. The structural features and advantages of bimetallic nanoparticles are discussed in detail to provide an in-depth understanding of the role of the catalysts during the reaction processes. Furthermore, fundamental understanding and strategies to design specific bimetallic components and dual active sites are prospected for potential industrial applications. |
1. Introduction
Bimetallic nanoparticle catalysts exhibit better catalytic performance compared to their corresponding monometallic counterparts and have an expansive industrial application prospect. Just as the name implies, the two metallic elements are usually present as alloys or a mixture of particles. The unique synergistic effect between the two metal components provides the outstanding catalytic properties of bimetallic nanoparticles, which provides better activity, stability or selectivity.1,2 This review covers recent advances in the preparation methods and catalytic applications of bimetallic nanomaterials. The relationships between structural features and catalytic properties of different bimetals are discussed. Deactivation and regeneration of the catalysts are highlighted, which are essential for catalyst design and optimization. Typical reactions, such as catalytic oxidation, hydrogen evolution reaction, and carbon dioxide reduction, are provided to demonstrate the wide range of catalytic applications of bimetallic catalysts. The structural features and advantages of bimetallic nanoparticles are discussed in detail to provide an in-depth understanding of the role of the catalysts during the reaction processes. Furthermore, fundamental understanding and strategies to design specific bimetallic components and dual active sites are prospected for potential industrial applications. The synergistic effect in bimetallic catalysts may be ascribable to the complex structure from various atomic arrangements of two metallic elements. The existence form of bimetallic nanoparticles can be classified into random alloys, intermetallic compounds, biased structures or mixtures, which present special geometric structures and electronic properties. Bimetallic nanoparticles exhibit superior catalytic performance in different types of chemical reactions.3–10 Bimetallic nanoparticles possess the properties of both metals as well as other additional superior properties. The unique electronic and geometrical structures (bond lengths, tensions, etc.) of bimetallic nanoparticle surfaces after alloying provide multifunctional active sites.11 Meanwhile, the adsorption energies of reactants and intermediates on the bimetallic catalyst surface are changed, thus leading to better catalytic activities and selectivities of bimetallic catalysts. On the other hand, enhanced bimetallic contact sites designed by introducing a second metal to a noble substrate effectively improve the utilization efficiency of the noble metal.Metal additives in bimetallic catalysts enhance the activity, selectivity, and stability of a catalyst by the following four different effects:12 (i) the geometric structure effect: the addition of a second metal modifies the active site geometry of the substrate metal. The enhanced product selectivity as well as the initial activity of the bimetallic nanoparticle is ascribed to altered size of the metal active site by introducing the second element; (ii) the electronic structure effect: the addition of a second metal facilitates intermetallic electron transfer to optimize the electronic structure of active sites. The electron transfer between the two metals changes the surface electronic properties of the catalyst, which adjusts the adsorption energy and strength of the reaction substrate on the catalyst surface; (iii) the synergistic effect: the two metal species in bimetallic nanoparticles participate in chemical bonding to reaction intermediates or transition states. Bimetallic catalysts co-ordinate the properties of two metals to realize a bifunctional effect or the interaction between the two metals offers improved catalytic performance; and (iv) the stabilization effect: the addition of a second metal inhibits particle agglomeration or carbon deposition to obtain excellent stability. Also, excessive reduction or over-oxidation of the active sites is suppressed to realize high selectivity.
In this review article, we provide a comprehensive overview of the structural features and catalytic behaviors of bimetallic nanoparticle catalysts. The catalytic applications of bimetallic nanoparticles are summarized in the following three aspects: (i) the surface catalytic active sites of noble metals are exposed to promote metal availability and reduce consumption by introducing a second metal; (ii) the morphology and chemical components of bimetallic nanoparticles can be adjusted during the synthesis process. Target bimetallic nanoparticles can be constructed to regulate catalytic activity and selectivity according to the specific purposive reactions; and (iii) the synergistic interaction between bimetallic nanoparticles provides high intrinsic activity and selectivity.
This review covers recent advances in the preparation methods and catalytic applications of bimetallic nanomaterials. The synthesis approaches of bimetallic catalysts are described, and the relationship between structural features and catalytic properties of different bimetals (e.g., bimetallic nanoparticles and dual active sites) is discussed, which is a key issue in catalysis. In addition, deactivation and regeneration of the catalysts are highlighted, which are essential for catalyst design and optimization. Typical reactions, such as catalytic oxidation, the hydrogen evolution reaction, and carbon dioxide reduction, are provided to demonstrate the wide range of catalytic applications of bimetallic catalysts. The structural features and advantages of bimetallic nanoparticles are discussed in detail to provide an in-depth understanding of the role of the catalysts during the reaction processes. Furthermore, fundamental understanding and strategies to design specific bimetallic components and dual active sites are discussed for potential industrial applications.
2. Structural features of bimetallic nanoparticles
The structure of a bimetallic material determines its catalytic properties. Bimetallic nanocatalysts can be divided into two types based on the dispersion extent of the two metal atoms.13 Bimetallic nanoparticles exist as alloys and intermetallic compounds when metal atoms are highly dispersed. The segregation effect is a dominant factor in low dispersion, in which heterogeneous and core–shell structures are the main forms of bimetallic nanoparticles. On the one hand, the formation of bimetallic structures relies on regulating the synthetic method, i.e., using the addition sequence of metal precursors, the difference in electrode potentials, and regulating the reaction kinetics. On the other hand, the intrinsic properties of the two metals determine whether segregation happens to reduce the system energy and achieve the most stable state of dispersion.2.1. Geometric structures of bimetallic nanoparticles
Metal additives in bimetallic nanoparticles can effectively alter the size of the active site, which gives rise to the desirable design of catalytic activity and stability as well as product selectivity. Geometric effects are decorations on the metal surface where the second metal is partially deposited or completely incorporated on the substrate. The presence of different atoms in bimetallic nanoparticles also leads to the complexity of the spatial distribution of the two metal elements.15 The two metallic elements may form various types of geometric arrangements (Fig. 1), such as disordered alloys, core–shell structures, and Janus-type structures.14 The decrease in geometry size of bimetallic nanoparticles gives rise to an increase in surface atoms (i.e., the ratio of the number of surface atoms to the total number of atoms). Increased coordination unsaturation of surface atoms appears, accompanied by the significant improvement in catalytic properties. The coordination number of surface atoms plays a determining role in catalytic performance, and the adsorption of reactants and intermediates on surface atoms is influenced by neighbor atoms. It has been reported that the specific catalytic reaction may follow different reaction pathways when the reactants are adsorbed on different surface sites. The geometrical environment of a Pt-based catalyst surface can be dramatically changed by embedding a second metal in the surface lattice of Pt to form an alloy or intermetallic compound (Fig. 2).16 The second metal can isolate Pt atoms. The isolated Pt atoms can only adsorb on the top atom or molecule (Fig. 2d-Ia), leading to bridging adsorption between the two Pt atoms (Fig. 2d-Ib). And the collection of three atoms allows the reactant molecules to react in the hollow sites (Fig. 2d-Ic), which can effectively enhance the selectivity of the reaction.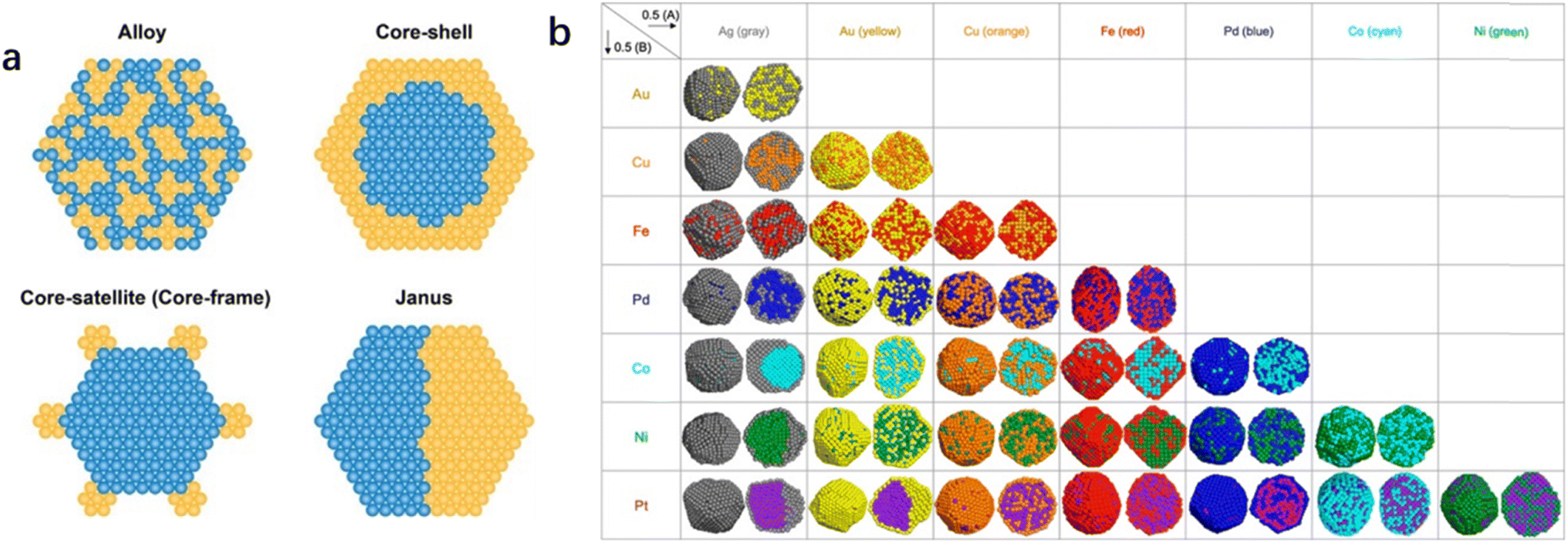 | ||
| Fig. 1 (a) Schematic illustration of several types of bimetallic nanoparticles with different spatial distributions of the two metal elements within the individual bimetallic nanoparticles,14 (Reproduced with permission from ref. 14 Copyright 2021, ACS) and (b) results of the combined simulations of molecular dynamics (MD) and Monte Carlo (MC) simulations for 0.5(A): 0.5(B) composition for 28 combinations of bimetallic nanoparticles.15 Reproduced with permission from ref. 15 Copyright 2021, Wiley. | ||
 | ||
| Fig. 2 (a) Overview of bimetallic systems and electrochemical reactions contained in this review. Bold elements appear in rows and columns to enable a concise scheme. RE stands for rare earth elements. “Other” reactions are electrochemical oxidation reactions of higher alcohols, BH4−, hydrazine, and methane and the oxygen evolution reaction (OER). Summarized optimization strategies: (b) ligand effects, (c) strain effects, (d) geometric effects, and (e) the bifunctional mechanism. (d) Geometric effects are divided into two parts. The first is the site isolation effect, accompanied by different adsorption possibilities of a molecule, that is, (Ia) in the hollow site, (Ib) on the bridge site or (Ic) on top of an atom. (II) The second is the equality of sites.16 Reproduced with permission from ref. 16 Copyright 2019, ACS. | ||
2.2. Electronic structures of bimetallic nanoparticles
The electronic structure of bimetallic nanoparticles is less sensitive to the particle size variation. The electronic effect of bimetallic nanoparticles controls the metal surface properties by modulating electronic states. Modulation of the electronic structure of the metal sites can give rise to synergistic effects and electron re-distribution between the bimetallic sites.17 This reconstruction facilitates the optimization of adsorption energy of the oxygen-containing species, which moderates adsorption intensity of the main intermediates at the diatomic site.18 Bimetallic sites can provide higher metal loadings and flexible active sites, which promote simultaneous adsorption of different reactants on the catalysts as well as different adsorption mechanisms. The electronic structure resulting from the pairing of two atoms also influences the adsorption mechanism. In summary, bimetallic site catalysts not only provide more adsorption sites, but also present different reaction paths and electronic structures, which greatly enhance the selectivity and stability. For example, bimetallic BiCu alloy nanosheets were synthesized to reconstruct the electronic structure for the carbon dioxide reduction reaction.19 The results indicated that the improved bimetallic catalytically active sites constructed by Cu alloying successfully enhanced the interfacial electron transfer in BiCu alloy nanosheets to achieve the ideal reaction kinetics. Density functional theory (DFT) calculations suggested that the Cu alloying contributed to the increased density of states near the Bi Fermi surface and the optimized adsorption of *OCHO intermediates at the Bi sites, leading to good electrocatalytic activity and superior operating stability.3. Synthesis of different types of bimetallic entities
3.1. Synthesis of bimetallic nanoparticles
Generally speaking, the standard reduction potential (E0, V vs. the standard hydrogen electrode (SHE)) of a metal determines the difficulty to reduce a metal ion. The more positive reduction potential indicates that the metal precursor is easier to reduce to the metallic element, which provides a reference for estimating the reduction speed of metal ions. However, when the ions (e.g., halide ions) and molecules (e.g., organic amines) with coordination ability are added to adjust the coordination environment, the coordination interaction decreases the reduction potential of the metal to an extent which relates to the coordination stability constant. During the reaction, the decrease in the metal reduction potential leads to a negative reduction rate, and the ligand which acts as a capping agent is adsorbed on the surface of the metal atom after the precursor is reduced. Hence, bimetallic nanoparticles with the desired structures can be predicted and designed by examining or modulating the reduction potentials of the two metal precursors. In general, similar reduction potentials can yield alloy-type structures, widely differing from the reduction potential generated deviatoric-like structures. For instance, the standard reduction electrode potentials for [PtCl4]2−/Pt, [PdCl4]2−/Pd, [RhCl6]3−/Rh, and Ru3+/Ru in acidic media are very close to each other, being +0.76, +0.59, +0.43, and +0.39 V vs. SHE (Table 1),20 respectively. Thus, the synthesis of bimetallic alloys (e.g., AuPd,21 AuPt,22 and PdPt23) has been widely reported. However, the reduction rate of the metal precursor, as one of the structural causative factors, is influenced by many factors, such as reduction potential, precursor concentration, and the selection of the reducing agent. The reducing agent is considered one of the crucial factors. For example, a strong reducing agent (i.e., sodium borohydride) should be able to rapidly reduce the metal precursors to generate a bimetallic alloy even in the presence of large reduction potential differences. When weak reducing agents (e.g., formaldehyde) are used, the reduction potential of the metal precursor becomes the dominant factor in the formation of the final structure. The interfacial energy between the two metals (phases) determines which kind of segregation type structure is formed. A low interfacial energy implies that the two metals have good affinity for each other and can be able to infiltrate the surface, hence tending to form core–shell structures. In contrast, a high interfacial energy brings about the island growth and generates bimetallic nanocrystals with heterogeneous structures. Dai's group did a lot of work in the preparation of double metal alloys (e.g., PdAu,21 PdPt,23 AuRu,24 PdRu,25 PtRu,26 and PdCo27) using the polyvinyl alcohol (PVA)-protected reduction strategy with NaBH4 as the reducing agent.| Half reaction | E0 (V vs. SHE) | Element electronegativity |
|---|---|---|
| [PtCl4]2− + 2e− → Pt + 4Cl− | +0.76 | 2.28 |
| [PdCl4]2− + 2e− → Pd + 4Cl− | +0.59 | 2.20 |
| [RhCl6]3− + 3e− → Rh + 6Cl− | +0.43 | 2.28 |
| Ru3+ + 3e− → Ru | +0.39 | 2.20 |
| Cu2+ + 2e− → Cu | +0.34 | 1.90 |
| Ni2+ + 2e− → Ni | 0.23 | 1.91 |
| Co2+ + 2e− → Co | −0.28 | 1.88 |
The multistep synthesis of bimetallic nanocrystals is typified by the crystal seeding and displacement methods, which feature the successful reduction of the two metal precursors to the metal in sequence.28,29 The crystal seeding method is the most common strategy for the synthesis of bimetallic nanocrystals with core–shell and heterostructures. The important factors to be taken into account in the crystal seed method include the stability of the crystal seed and the smooth deposition of the second metal to obtain ideal bimetallic nanocrystals. A suitable surfactant or stabilizer is added during the preparation process to ensure that the crystals are well dispersed in the solution, which can facilitate the uniform deposition of the second metal. The reduction speed of the second metal atoms should be slow and the concentrations of the precursors are not high to ensure that the initial deposition takes place on the surface of the seed. When the atomic deposition rate is dominant, the heterogeneous structures can be readily obtained (by the kinetic control); if the atomic diffusion speed is faster, the bimetallic nanoparticles with stable core–shell structures tend to be formed (by the thermodynamic control). To generate a stable core–shell and heterogeneous structure, the crystal structures of the two metals should be identical (the same atomic arrangements). Most of the noble metals (e.g., Rh, Pd, Ag, Ir, Pt, and Au) and common transition metals (e.g., Ni and Cu) meet such a requirement with a face-centered cubic structure as the most stable phase.30,31 Based on this rule, face-centered cubic structures can be constructed for metals with other crystal phases. For example, Xia et al. synthesized Pd–Ru core-skeleton octahedra using truncated octahedral Pd as the crystal seeds.32 The resultant Ru octahedral nanoframes replicated the face-centered cubic crystal structure of Pd seeds by the kinetic control (Fig. 3).
 | ||
| Fig. 3 Structural and compositional analyses of the Pd–Ru core–frame octahedra that were obtained by growing Ru on the corners and edges of Pd truncated octahedra as the seeds. (a) Typical TEM image showing the octahedral shape and good uniformity of the sample (inset shows the corresponding atomic model), and (b) line-scan EDX spectra of elemental Pd and Ru that were acquired from an individual octahedron (inset) along a corner-to-corner and an edge-to-edge direction as indicated by the white arrows. (c) High-resolution HAADF–STEM image of an individual octahedron orientated along 〈110〉 direction, and (d) schematic illustration showing the two steps involved in the synthesis of Ru NFs: (1) selective nucleation and growth of Ru on the corners and edges of Pd truncated octahedra, yielding Pd–Ru core–frame octahedra, and (2) formation of Ru octahedral NFs by etching away the Pd cores.32 Reproduced with permission from ref. 32 Copyright 2016, ACS. | ||
Liu's group fabricated mesoporous intermetallic (PtSn, PtPb, PtCd, PtZn, and PdSn) samples using a general stepwise concurrent template strategy (Fig. 4).33,34 In this approach, the intermediate concurrent template was first constructed to obtain a hybrid of mesoporous platinum or palladium and a KIT-6 template (meso-Pt/KIT-6 or meso-Pd/KIT-6), which could be transformed using the second precursor under reducing conditions. The second precursor could either be a second metal or a metalloid. meso-Pt/KIT-6 or meso-Pd/KIT-6 could be re-crystallized with the second metal precursor (e.g., SnCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33) at elevated temperatures to form atomically ordered intermetallic compounds, whereas the mesoporous SiO2 template (KIT-6 or SBA-15) provided a nano-restricted mesopore environment to grow the ordered mesoporous nanocrystals.35–38 The concurrent template method can be easily extended to design other mesoporous intermetallic nanoparticles with macroscopic morphologies, mesoscopic structures, and atomic intermetallic phases (ordering). The obtained mesoporous intermetallic compounds exhibited the best surface electronic states and chemisorption properties with controlled catalytic selectivity and excellent stability.
33) at elevated temperatures to form atomically ordered intermetallic compounds, whereas the mesoporous SiO2 template (KIT-6 or SBA-15) provided a nano-restricted mesopore environment to grow the ordered mesoporous nanocrystals.35–38 The concurrent template method can be easily extended to design other mesoporous intermetallic nanoparticles with macroscopic morphologies, mesoscopic structures, and atomic intermetallic phases (ordering). The obtained mesoporous intermetallic compounds exhibited the best surface electronic states and chemisorption properties with controlled catalytic selectivity and excellent stability.
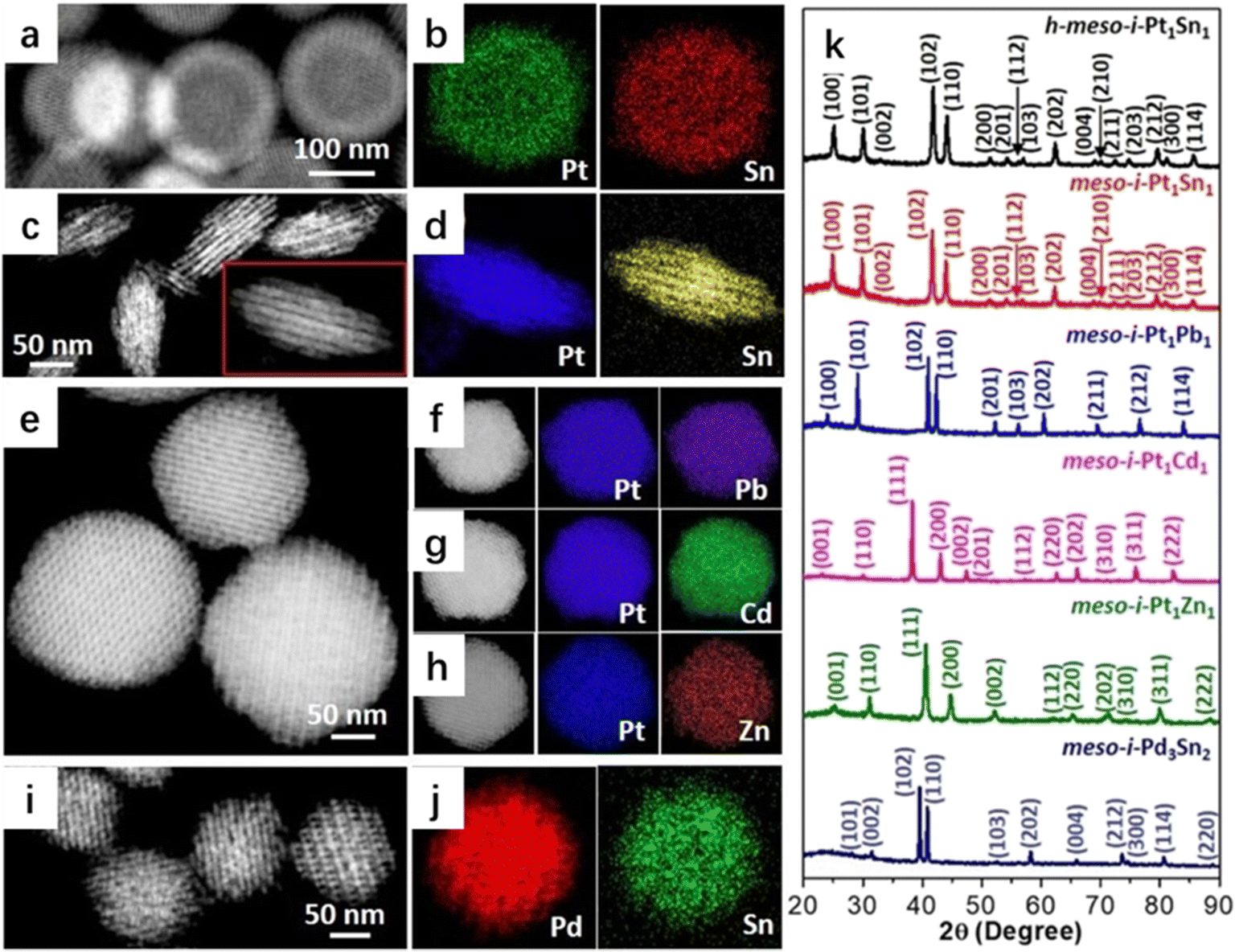 | ||
| Fig. 4 (a) HAADF–STEM image and (b) EDX mappings of h-meso-i-Pt1Sn1 nanoparticles, (c) HAADF–STEM image and (d) EDX mappings of meso-i-Pt1Sn1 nanobundles, (e) HAADF–STEM image of meso-i-Pt1Pb1 nanoparticles, (f–h) HAADF–STEM images and corresponding EDX mappings of (f) meso-i-Pt1Pb1, (g) meso-i-Pt1Cd1, and (h) meso-i-Pt1Zn1 nanoparticles, (i) HAADF–STEM image and (j) EDX mappings of meso-i-Pd3Sn2 nanoparticles, and (k) PXRD patterns of h-meso-i-Pt1Sn1 nanoparticles, meso-i-Pt1Sn1 nanobundles, meso-i-Pt1Pb1, meso-i-Pt1Cd1, meso-i-Pt1Zn1, and meso-i-Pd3Sn2 nanoparticles.33 Reproduced with permission from ref. 33 Copyright 2022, Wiley. | ||
The metal displacement method is another common strategy for the synthesis of bimetallic nanocrystals, which involves the replacement of a weakly active metallic element with a highly reactive metal as a sacrificial metal. The essential requirements are the difference in the metallic activity between the two metal ions. Active metals, such as Ag, Ni, Co, and Cu, are commonly used for synthesizing alloys or bimetallic nanocrystals with core–shell structures. The initial reduced metal atoms are attached on the surface of the sacrificial metal, and the bimetallic nanoparticles are formed through a complex process among atomic diffusion, migration, and mixing. For example, the PdAg, PtAg or AuAg alloys with different morphologies were constructed using Ag as a sacrificial metal.39,40 The addition of Na2PdCl4 or Na2PtCl4 allowed the well-dispersed silver nanocubes in water to be converted into Pd–Ag or Pt–Ag nanoboxes.39 The nanocages formed by the replacement of silver with palladium were composed of a Pd–Ag single-crystalline alloy, while the nanocages formed by the replacement of Ag with Pt were assembled with different platinum nanoparticles. The shells of bimetallic nanoparticles of the core–shell structure obtained by the metal displacement method were all alloys. Lee et al. prepared Ag@AgAu core–shell-structured nanoparticles with an alloy shell by the galvanic substitution reaction between HAuCl4 and Ag nanoparticles.41 Time-course studies by HRTEM and UV/Vis spectroscopy revealed the following sequence of events in the evolution of core–shell structures from Ag nanoparticle seeds: (i) oxidative dissolution of Ag atoms from the {111} facets of Ag nanoparticles and initial preferential deposition of Au atoms from AuCl4− reduction on the Ag{100} facets, followed by alloying with the Ag atoms below; (ii) subsequent Au atom deposition on the Ag{111} facets followed by Ag–Au alloying there; and (iii) re-arrangement of the surface atoms to form the final core–shell nanoparticles. In practice, researchers commonly etch and dissolve unreacted intermediates or by-products to remove impurities and form bimetallic nanocrystals with hollow or porous structures, hence improving the utilization efficiency and catalytic activity of the active metal.
3.2. Deactivation and regeneration of bimetallic catalysts
Catalyst deactivation is the loss of catalytic activity or selectivity during reaction processes, which is a major drawback in industrial applications. Resources are invested in catalyst replacement or regeneration to overcome deactivation. Deactivation is common for solid catalysts as well as bimetallic catalysts. Possible deactivation mechanisms of bimetallic catalysts include sintering of bimetallic particles, coking, leaching of metallic substances, segregation of the two metallic elements, phase transitions, and poisoning (Table 2 and Fig. 5).42 Deactivation processes are reported to stem from three general effects. Among these effects, modification of the active phase shows the largest diversity of origins that may lead to simultaneous blockage. Processes such as sintering or leaching lead to irreversible deactivation of a catalyst, but coke deposition can be reversed by calcination to remove the carbonaceous species covered on the catalyst surface. The study of structural transformation of bimetallic catalysts can guide the regeneration of bimetallic catalysts. The catalytic activity rejuvenation of bimetallic PtSn nanoparticles after coke deposition could be realized by satisfactory oxychlorination treatment to achieve effective Pt re-dispersion.43| Generalized mode | Definition |
|---|---|
| Depletion | Loss of the active phase into the reaction mixture via dissolution or sublimation |
| Promoter depletion | Loss of catalyst components other than the active phase (e.g., co-catalysts or other promoting additives) |
| Fragmentation | Loss of the active phase due to structural modifications leading to their detachment as whole entities |
| Segregation | Loss of the active phase due to physical separation of the catalyst from the reaction mixture with or without prior agglomeration |
| Species deposition | Deposition of the species from the reaction mixture leading to the coverage of the active sites |
| Aggregation | Agglomeration of catalyst components reducing the active site accessibility |
| Sintering | Size increase of the active phase reducing the number of accessible sites with possible modification of their nature |
| Poisoning | Strong adsorption of the species from the reaction mixture blocking active sites or unfavorably altering their properties |
| Relocation | Spatial reorganization of catalyst components reducing the active site accessibility or unfavorably altering their properties |
| Dispersion | Size reduction of the active phase unfavorably altering the properties of active sites |
| Structural collapse | Total loss of the active phase structure radically changing the nature of the active sites |
| Restructuring | Partial alteration of the active phase structure unfavorably altering the properties of the active sites |
| Skeletal modification | Localized change of the catalyst framework unfavorably altering the properties of the active sites |
| Phase change | Bulk compositional alteration of the active phase unfavorably altering the properties of the active sites |
 | ||
| Fig. 5 Generality of deactivation modes.42 Reproduced with permission from ref. 42 Copyright 2022, Springer Nature. | ||
Deactivation of bimetallic catalysts is a complex phenomenon caused by various reasons including carbon deposition, sintering, poisoning (e.g., adsorption of sulfur or other compounds), and coverage or loss of active components. Restoring catalytic performance of the bimetallic catalysts requires the selection of a suitable regeneration strategy based on the specific cause of deactivation. The following steps are usually involved:29,42,43 (i) thermal treatment: carbon or other organic deposits on the surface of the catalyst can be removed and catalytic activity of the catalyst can be restored by the appropriate high-temperature treatment; (ii) reduction treatment: for oxidized metal catalysts, the active sites of the metal can be restored by calcining in a reducing atmosphere (such as hydrogen or carbon monoxide) at an appropriate temperature; (iii) regeneration treatment: catalysts deactivated by sintering can be treated at high temperatures in a hydrogen atmosphere to restore their original surface area and pore structure. The high temperature can induce re-dispersion of the metal particles, and hydrogen helps to reduce the metal oxides to the metallic states; (iv) ion exchange treatment: bimetallic catalysts that contain exchangeable metal ions and deactivated or lost active components can be replaced by ion exchange processing; and (v) chemical cleaning: if the deactivation is caused by the adsorption of toxic or chemical compounds (e.g., sulphides, halogens, etc.) on bimetallic catalysts, they can be cleaned with a suitable chemical solvent to remove the above toxic or chemical substances.
4. Catalytic applications of bimetallic nanoparticles
Bimetallic nanoparticles possess several significant performance advantages over single metal nanoparticles: (i) enhanced activity: bimetallic nanoparticles can increase catalytic performance under milder conditions due to bimetallic synergistic effects. Electronic interactions between two different metals can adjust the surface electronic properties, thus reducing the activation energy of the reaction; (ii) improved selectivity: bimetallic nanoparticles can provide higher selectivities toward the desired products in certain reactions. The combination of bimetals may change the reaction paths or stabilize specific intermediates; (iii) developed stability: bimetallic nanoparticles exhibit better stability than monometallic nanoparticles, especially at high temperatures or under severe practical reaction conditions. The second metal may form a protective layer on the surface that inhibits corrosion or sintering of another active metal component; (iv) enhanced resistance to toxicity: adsorption of toxic substances (e.g., SO2, H2O or by-products) leads to deactivation of the active component. The second metal can increase desorption capacity and release the active sites by regulating the surface properties; (v) enhanced mass transfer: bimetallic nanoparticles show stronger mass transfer capability owing to the rougher surface or more active sites, which improves adsorption and diffusion of the reactants; and (vi) regulated optical and electronic properties: the optical and electronic properties of bimetallic nanoparticles can be modulated by changing the ratio and structure of the bimetallic components to meet specific application requirements.There are various explanations for the mechanism of the promotion of catalytic performance using bimetallic nanoparticles, which are summarized as follows:45,46 (i) the synergistic interaction between bimetals effectively increases resistance to poisoning; (ii) altering the spacing between the two metal elements to regulate the d-band center and improve the catalytic activity; (iii) inhibiting particle aggregation to enhance stability by adding a second metal; and (iv) surface roughness which is accountable for introducing the second metal to increase the active sites.
4.1. Methane combustion
Methane has a stable molecular structure with a low polarity and requires a high light-off temperature for catalytic oxidation. Methane combustion at low temperatures has always been one of the biggest challenges in the field of catalytic oxidation. Due to the high reaction temperature, even Pd-based catalysts often suffer from catalyst deactivation owing to carbon accumulation or nanoparticle sintering. Pd-based bimetallic nanoparticles have been widely used in methane oxidation due to unique structural and electronic effects. Pd-based bimetallic catalysts can reduce Pd consumption while improving the catalytic activity and stability. Numerous studies (Table 3) have shown that the introduction of a second metal into Pd can increase the surface active sites and adjust the adsorption strength and binding energy between the catalyst and the reactants or intermediates.47,50,53,55,56 For example, Zhao et al.47 adopted the KIT-6-templating strategy to generate mesoporous cubic Pd, Pt, and PdxPt (x = 0.43–8.52) for methane oxidation. It was found that Pd and Pt were well dispersed in the ordered mesoporous architecture of the PdxPt alloys. The redox properties of Pd showed a significant adjustment by Pt doping, and the Pt–Pd oxide (i.e., PdO–PtO2) exhibited better catalytic performance than the Pt0–Pd0 metallic material with a good thermal stability. The meso-Pd, meso-Pt, and meso-Pd2.41Pt showed a T50% and T90% (the temperatures required for achieving methane conversions of 50 and 90%) of 350, 372, and 303 and 368, 506, and 322 °C at a space velocity (SV) of 100000 mL g−1 h−1, respectively. The meso-Pd2.41Pt sample performed the best with the highest TOFPd+Pt at 280 °C and specific reaction rate at 280 °C of 0.59 × 10−3 s−1 and 4.46 μmol gcat−1 s−1 at 280 °C, respectively. The deactivation of the Pd2.41Pt catalyst caused by the introduction of carbon dioxide or water vapor was reversible, whereas the deactivation caused by the introduction of sulfur dioxide was irreversible. The authors thought that the well-ordered and developed mesoporous structure and good oxygen activation ability were accountable for the high catalytic activity.
| Catalyst | Preparation method | Noble metal particle size (nm) | Noble metal loading (wt%) | CH4 concentration (vol%) | SV (mL g−1 h−1) | T50% (°C) | T90% (°C) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Meso Pd2.41Pt | PVA-protected NaBH4 reduction strategy | 7.1 | — | 2.5 | 100000 |
303 | 322 | 47 |
| AuPd/meso-Co3O4 | PVA-protected NaBH4 reduction strategy | 3.5 | 1.94 | 2.5 | 20000 |
280 | 324 | 21 |
| PdPt/meso-Mn2O3 | PVA-protected NaBH4 reduction strategy | 2.5 | 1.42 | 2.5 | 20000 |
345 | 425 | 23 |
| AuRu/meso-Mn2O3 | PVA-protected NaBH4 reduction strategy | 2–5 | 0.97 | 2.5 | 20000 |
470 | 540 | 24 |
| AuPd/3DOM CoCr2O4 | PVA-protected NaBH4 reduction strategy | 3.3 | 1.93 | 2.5 | 20000 |
353 | 394 | 48 |
| AuPd/3DOM La0.6Sr0.4MnO3 | PVA-protected NaBH4 reduction strategy | 2.2 | 2.92 | 5.0 | 50000 |
314 | 336 | 49 |
| PdPt/3DOM LaMnAl11O19 | PVA-protected NaBH4 reduction strategy | 4.3 | 1.14 | 2.5 | 20000 |
487 | 549 | 50 |
| PdPt/MnOx/3DOM CoFe2O4 | PVA-protected NaBH4 reduction strategy | 3.0 | 2.10 | 2.5 | 20000 |
301 | 372 | 51 |
| AuPd/3DOM Mn2O3 | PVA-protected NaBH4 reduction strategy | 3.7 | 1.97 | 2.5 | 40000 |
393 | 440 | 52 |
| AuPd–Co/3DOM Mn2O3 | PVA-protected NaBH4 reduction strategy | 3.6 | 1.94 | 2.5 | 40000 |
365 | 442 | 52 |
| Au–Pd/3DOM Co3O4 | PVA-protected NaBH4 reduction strategy | 2.6 | 1.99 | 2.5 | 20000 |
337 | 379 | 53 |
| Au–Pd–CoO/3DOM Co3O4 | PVA-protected NaBH4 reduction strategy | 2.7 | 1.99 | 2.5 | 20000 |
312 | 341 | 53 |
| Au–Pd–CoO/3DOM Mn2O3 | PVA-protected NaBH4 reduction strategy | 3.1 | 1.97 | 2.5 | 20000 |
430 | 500 | 53 |
| Au–Pd–CoO/3DOM Al2O3 | PVA-protected NaBH4 reduction strategy | 3.0 | 1.95 | 2.5 | 20000 |
543 | 625 | 53 |
| CoPd/3DOM CeO2 | PVA-protected NaBH4 reduction strategy | 4.1 | 0.77 | 2.5 | 40000 |
430 | 480 | 27 |
| AuPd/Co3O4/3DOM MnCo2O4 | PVA-protected NaBH4 reduction strategy | 4.6 | 1.98 | 2.5 | 40000 |
340 | 408 | 54 |
Cargnello et al. developed a supramolecular approach to homogeneously deposit single units composed of a palladium (Pd) core and a ceria (CeO2) shell onto a modified hydrophobic alumina (Fig. 6).57 The commercial alumina substrate was highly hydrophilic, while the Pd@CeO2 active component was hydrophobic. The hydrophobic Pd@CeO2 structure presented agglomeration instead of adhering to alumina, resulting in irreversible catalyst deactivation. The authors aimed to develop an efficiency strategy to modulate special properties of the alumina surface to be hydrophobic and deposit the active component as a single unit on the surface of the substrate. The alumina surface was decorated with organosilane triethoxy(octyl)silane (TEOOS) to ensure that the surface was covered by the formed alkyl chain, leading to a larger adsorption capacity of the Pd@CeO2 structure without deactivation. The water droplets deposited on the hydrophobic alumina were repulsed instead of the favorable interaction with the OH groups of alumina, which further confirmed the adopted tactic. The as-prepared sample showed a remarkably higher methane oxidation activity and the supported catalyst remained intact without segregation or agglomeration during the calcination at 850 °C. The authors pointed out that the excellent methane complete oxidation activity was associated with the strong metal–support synergy interaction, well-defined ordered geometry mesopore structure, and outstanding thermal stability under the demanding conditions. Dai and co-workers systematically studied the correlation between noble metals and several supports for methane oxidation activity.21 It was observed that AuPd nanoparticles were highly distributed on the surface of meso-Co3O4 with an average size of 2.7–4.5 nm.21 The 2.94Au0.50Pd/meso-Co3O4 catalyst exhibited an excellent performance and possessed the highest specific reaction rate. Over 2.94Au0.50Pd/meso-Co3O4 at a SV of 20000 mL g−1 h−1, the T10% (the temperature required for achieving a methane conversion of 10%), T50%, and T90% were 230, 280, and 324 °C, respectively, with the lowest apparent activation energy and the highest TOFAuPd being achieved over this catalyst. Similarly, meso-Mn2O3-supported PdPt alloy (2.1–2.8 nm in size) materials with uniformly dispersed noble metals also showed good methane combustion performance.23 The 1.41Pd5.1Pt/meso-Mn2O3 sample gave the best methane oxidation performance with the highest TOFPdPt and best specific reaction rate at 400 °C. In addition, the as-prepared sample displayed an increased endurance performance in the presence of SO2, CO2 or water vapor by introducing an appropriate amount of Pt. Moreover, AuRu/meso-Mn2O3 was generated to possess good low-temperature reducibility, in which the 0.97AuRu/meso-Mn2O3 sample showed the highest catalytic activity.24 The same research group investigated three-dimensionally ordered macroporous (3DOM) Cr-based spinel-type oxide-supported gold–palladium alloy catalysts with an abundant adsorbed oxygen (Oads) species concentration and good low-temperature reducibility for methane combustion. 1.93AuPd1.95/3DOM CoCr2O4 achieved the highest methane conversion, with superior TOFAuPd at 320 °C of 0.271 × 10−3 s−1 and the highest specific reaction rate at 320 °C of 1.15 μmol gcat−1 s−1.48 The presence of SO2 in the reaction system over the 0.97AuRu/meso-Mn2O3 sample caused a permanent loss in activity, while the deactivation induced by moisture addition was reversible. The thermal stability and adsorbed oxygen species of the AuPd/3DOM La0.6Sr0.4MnO3 bimetallic catalysts were investigated.49 It was found that the 2.92AuPd/3DOM La0.6Sr0.4MnO3 sample possessed the best suitable Au/Pd molar ratio (1:2) for CH4 oxidation, which exhibited the highest TOFAuPd (10.2 × 10−3 s−1) and the lowest apparent activation energy (Ea = 72.4 kJ mol−1). The sizes of the bimetallic Au–Pd catalyst increased from 2.15 to 3.76 nm after its exposure to water, indicating that the presence of water vapor caused an irreversible deactivation. Hexaaluminates (AAl12O19, in which A is an alkali, alkaline earth or rare earth element) are regarded as excellent catalyst candidates due to good thermal stability (their sintering temperatures are above 1200 °C). The same research group also tested methane combustion activities of PdPt/3DOM LaMnAl11O19 with noble metal particle sizes of 3–5 nm,50 in which the 1.14Pd2.8Pt/3DOM LaMnAl11O19 sample performed the best. Ex situ X-ray photoelectron spectroscopy was used to investigate the formation of the active PdO species during the oxidation processes of Pd in 1.14Pd2.8Pt/3DOM LaMnAl11O19 at different temperatures. Doping with an appropriate amount of Pt to the Pd-based catalyst could improve the tolerance performance in the presence of H2O, CO2, and SO2 (which did not exert significant influence on catalytic stability). Recently, the same group has investigated ternary metal catalysts for methane combustion, which are combined with noble metal nanoparticles, transition metal oxides, and porous spinel-type mixed oxide. The as-prepared PdPt/MnOx/3DOM CoFe2O4 sample possessed the highest Oads species content and the best low-temperature reducibility with a PdPt particle size of 2.2–3.0 nm.51 The 1.81PdPt/6.7MnOx/3DOM CoFe2O4 sample displayed the highest activity with the lowest apparent activation energy, as well as the highest TOFNoble metal and TOFCoFe2O4 of 37.44 × 10−3 s−1 and 0.28 × 10−3 s−1, respectively.
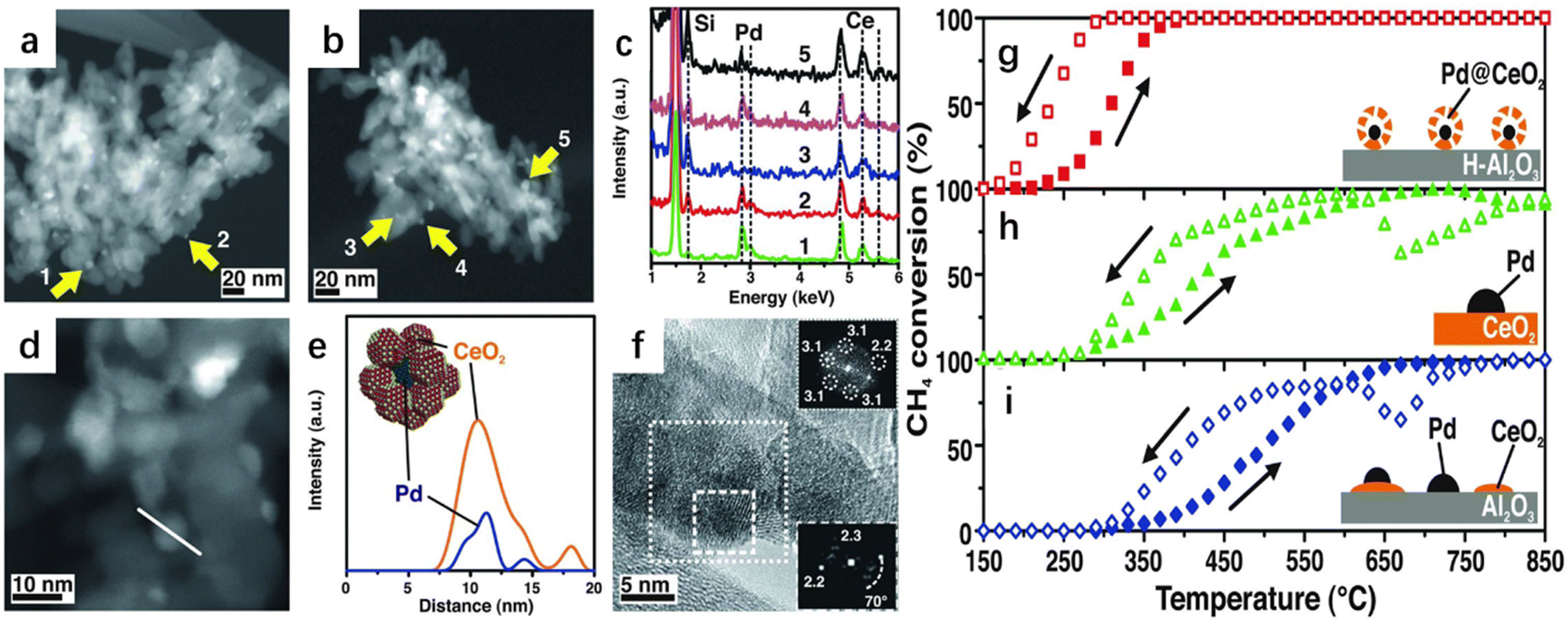 | ||
| Fig. 6 TEM investigations of Pd@CeO2 core shell structures dispersed on hydrophobic alumina. (a) HAADF-STEM image after calcining to 500 °C for 5 hours and (b) to 850 °C for 5 hours. (c) EDS spot analysis of the indicated particles. (d) High magnification HAADF-STEM image of the Pd@ CeO2/H–Al2O3 catalysts calcined to 500 °C and (e) the corresponding EDS line profile together with a model. (f) HRTEM image of a single Pd@CeO2 structure on the Pd@CeO2/H–Al2O3 catalysts calcined to 500 °C. Heating and cooling light-off curves of CH4 conversion against the temperature for the three catalyst formulations used. (g) Pd@CeO2/H–Al2O3 core–shell catalyst, (h) Pd/CeO2-IWI, and (i) Pd/CeO2/Al2O3-IMP.57 Reproduced with permission from ref. 57 Copyright 2012, Science. | ||
The modified polyvinyl alcohol (PVA)-protected reduction method was applied to synthesize transition metal (M = Mn, Cr, Fe, and Co)-doped Au–Pd–M nanoparticles. The Au–Pd–M nanoparticles with a diameter of 3.6–4.4 nm were uniformly loaded on the surface of 3DOM Mn2O352 or 3DOM Co3O453 via the gas bubble-assisted adsorption route. Among the Au–Pd–M nanoparticles/3DOM Mn2O3 samples, supported Au–Pd–0.21Co performed the best for methane oxidation due to its highest Pd2+ concentration; the supported Au–Pd–0.22Fe catalyst possessed the best ability for oxygen activation, resulting in the highest catalytic activity for o-xylene oxidation (T90% = 213 °C). The supported Au–Pd–yCoO nanocatalyst with a Co/Pd molar ratio of 0.40 performed the best for methane oxidation, whereas the Au–Pd–3.61CoO/3DOM Co3O4 catalyst showed the highest TOFPd at 280 °C and methane reaction rate at 280 °C. Furthermore, the supported Au–Pd–CoO catalysts still possessed good thermal stability for methane oxidation, which was associated with the strong interaction between the noble metal nanoparticles and the 3DOM support and the Pd–CoO interface stabilized by the Au species. The doping of CoO into Au–Pd nanoparticles enhanced the adsorption and activation ability of methane, in which CoO was the methane adsorption site and the Pd–CoO interface was the active center for methane oxidation. It was concluded that doping the transition metal into Au–Pd nanoparticles could modify the microstructure of Au–Pd to generate an interface between the noble metal and the transition oxide, hence enhancing the adsorption and activation ability of the reactants (O2, CH4, and/or o-xylene) and catalytic performance. Moreover, the doping of the transition metal led to a decrease in the amount of noble metals, significantly improving the utilization efficiency of noble metals. The PVA-protected two-step reduction strategy was adopted to fabricate the CoPd nanoparticles. The CoPd nanoparticles displayed a core–shell (core: Pd; shell: Co) structure, with an average size of 3.5–4.5 nm, and were well dispersed on the wall surface of 3DOM CeO2.27 The CoxPd/3DOM CeO2 samples were highly active and exhibited super thermal stability for methane oxidation, with the Co3.5Pd/3DOM CeO2 sample showing the highest catalytic activity and excellent thermal stability in the range of 400–800 °C. Based on the activity data and characterization results, it was concluded that the excellent catalytic activity of Co3.5Pd/3DOM CeO2 was related to its good O2 and CH4 adsorption abilities and unique Pd@Co core–shell structure. It is envisioned that fabricating an active catalyst with a unique structure can further reduce the use of noble metals, thus improving their utilization efficiency. By studying AuPd/Co3O4/3DOM MnCo2O4 catalysts for CH4 oxidation, Han et al.54 found that the high Oads species concentration and unique pore structure were responsible for the good activity.
4.2. VOC oxidation
The dual active site structure–effect relationship refers to the influence of the spatial distribution, interactions, and electronic properties of two different metal components, which play a crucial role in catalytic activity, selectivity, and stability. The structure–effect relationship of the dual active site is mainly reflected in the following aspects:44 (i) ligand effect: the introduction of a second metal can change the electronic state of the host metal (M), thus altering the coordination environment of the M atom. The number of the second metal atoms and the coordination mode can affect the electron density and energy level of the host M atoms, thus influencing the catalytic activity and selectivity; (ii) geometric effect: the relative position and spacing of the two metal atoms in a bimetallic site affects adsorption and activation of the reactant molecules. Specific interatomic distances can promote the formation and breaking of specific chemical bonds, thus changing the reaction paths; (iii) synergistic effect: synergistic interaction of bimetals in the active sites enhances the catalytic performance. The introduction of a second metal may promote dispersion of another metal or inhibit sintering under the reaction conditions; (iv) electronic effects: electronic interactions between two metals can change the electronic states, including electron donating and accepting abilities, which are critical for redox capacity and electron transfer processes; (v) lattice strain effect: the introduction of a second metal with mismatched lattice constants produces localized strains, which can change the electronic structure and surface energy of the host metal; and (vi) surface reconstruction: different surface structures of bimetallic catalysts, such as alloys, core–shell structure, and Janus or core frame, change catalytic activity and selectivity of the catalysts. Different surface structures can provide different active sites, thus influencing adsorption of the reactants and catalytic reaction paths.Noble metals (e.g., Pd and Pt) are the most efficient elements in the oxidation reaction due to their excellent C–H cleavage and C–C bond breakage abilities. Monometallic catalysts have only isolated active centers and the addition of a second metal provides intermetallic electron transformation. Therefore, synergistic combinations between bimetals re-configure the active sites through the modulation of the electron arrangement of monometallic or the increase of new active sites. As mentioned above, geometrical or electronic modifications with the addition of a second metal are the most effective strategies to develop catalysts with better performance and stability (Table 4). Exploring the optimal bimetallic structure and design for specific reactants is an important perspective in catalysis research. For instance, Xie et al.58 prepared 3DOM Co3O4 and its supported gold–palladium alloy (AuPd/3DOM Co3O4) nanocatalysts using a PVA-protected reduction method. The 3DOM Co3O4-supported Au–Pd samples performed much better than the supported single Au or Pd sample for toluene oxidation, with the 1.99AuPd/3DOM Co3O4 sample showing the best performance: the T10%, T50%, and T90% were 145, 164, and 168 °C at a SV of 40000 mL g−1 h−1, respectively. The AuPd/3DOM Co3O4 sample displayed a hydrothermal stability than the supported single Au or Pd sample. O2 could be conveniently activated on a gold nanoparticle cluster by formation of the hydroperoxyl species via the H-transfer reaction in the presence of water vapor. The Pd nanoparticle cluster possessed a good ability to adsorb water molecules, whereas the Au nanoparticle cluster showed a good capability to activate O2 molecules in the presence of water molecules. The formation rate of the active oxygen species was important for the development of a catalyst with high stability. Compared with the Pt/M catalyst, the Pt–Cu/M catalyst owned a stronger ability of VOC adsorption and gaseous oxygen activation by introducing additional sites.59 The Langmuir–Hinshelwood (adsorbed oxygen) and Mars–van Krevelen (lattice oxygen) mechanisms existed in toluene oxidation over the present Pt/M and Pt–Cu/M catalysts, respectively. The change in the involved active oxygen species during toluene oxidation resulted from the Pt–Cu alloy structure. In addition to the adsorption of O2, some of the active lattice oxygen species could also be replenished by migration of the bulk lattice oxygen in Pt–Cu/Mn2O3. The XPS results demonstrated that the Pt–Cu/Mn2O3 catalyst exhibited a stronger ability of activating O2 to Oads than the Pt/M catalyst. A part of the bulk lattice oxygen could be converted into the surface active Olatt to participate in the reaction. Introduction of Cu, as an additional activation site of O2, not only enhanced the adsorption capacity of precious metals for toluene/iso-hexane, but also increased the O2 activation ability. Wang et al. synthesized uniformly dispersed Au–Pd nanoparticles with sizes of 2.7–3.2 nm for toluene and o-xylene oxidation by the PVA-protected reduction approach.60 The T50% (T90%) for the oxidation of toluene and o-xylene over Au–Pd/Co3O4 were 171 (180) and 181 (187) °C at a SV of 40000 mL g−1 h−1, respectively. The redox ability and oxygen vacancy concentration of the catalyst certainly played an important role in the oxidative removal of VOCs. It was concluded that the enhanced low-temperature reducibility and surface oxygen species concentration accounted for the high catalytic activity of the Au–Pd/Co3O4 sample. Xia et al. prepared well-dispersed Au–Pd/α-MnO2 nanocatalysts via hydrothermal and PVA-protected reduction routes.61 It was found that the interaction between Au–Pd alloy nanoparticles and α-MnO2 nanotubes significantly improved the reactivity of lattice oxygen species. The interaction between the Au–Pd alloy nanoparticles and the α-MnO2 support made the Au and Pd species exist in a mixture of metallic (Au0 and Pd0) and ionic (Auδ+ and Pd2+) states. The T50% (T90%) for the oxidation of toluene and m-xylene over Au–Pd/α-MnO2 were 175 (185) and 210 (220) °C at a SV of 40000 mL g−1 h−1, respectively. The 0.91Au0.48Pd/α-MnO2 nanotube catalyst exhibited high catalytic stability as well as good tolerance to water vapor and CO2 in the oxidation of VOC mixtures. Au–Pd/Ce0.6Zr0.3Y0.1O2 with a noble metal particle size of 4.6–5.6 nm was prepared using cetyltrimethyl ammonium bromide (CTAB)-assisted hydrothermal and PVA-protected reduction methods.62 The 0.90Au–Pd/Ce0.6Zr0.3Y0.1O2 sample possessed lower apparent activation energies (37–43 kJ mol−1) than the Ce0.6Zr0.3Y0.1O2 sample (88 kJ mol−1) and hence performed the best for toluene oxidation: the T50% and T90% were 190 and 218 °C at a SV of 20000 mL g−1 h−1, respectively. The excellent catalytic performance of 0.90Au–Pd/Ce0.6Zr0.3Y0.1O2 was attributed to the highest adsorbed oxygen concentration, the best low-temperature reducibility, and the strong interaction between Au–Pd nanoparticles and Ce0.6Zr0.3Y0.1O2 nanorods. AuxMny/meso-Fe2O3 catalysts for benzene and toluene oxidation were prepared by KIT-6-templating and reduction strategies.63 The Au5Mn2/meso-Fe2O3 catalyst exhibited the best benzene conversion activity: T50% and T90% were 237 and 254 °C at a SV of 20000 mL g−1 h−1, with the TOFAu and specific reaction rates at 210 and 230 °C being 1.08 s−1 and 6.92 mmol gAu−1 s−1 and 2.29 s−1 and 11.58 mmol gAu−1 s−1, respectively. The benzene chemisorption mechanism and adsorbed oxygen species could greatly influence the oxidation of benzene. The Au in Au5Mn2/meso-Fe2O3 could obtain electrons from the benzene ring in the presence of benzene adsorption, resulting in stronger adsorption of benzene and thus increasing the catalytic activity for benzene oxidation. Such excellent performance was related to its well-dispersed AuMn nanoparticles, high adsorbed oxygen species concentration, good low-temperature reducibility, high benzene adsorption capacity, and strong benzene adsorption. Au–Pd/meso-Cr2O3 catalysts with uniformly dispersed noble metal nanoparticles at a size of 2.9–3.7 nm were fabricated by KIT-6-templating and PVA-protected reduction methods.64 The 1.95Au1Pd2/meso-Cr2O3 catalyst exhibited the best toluene oxidation activity: T50% and T90% were 145 and 165 °C at a SV of 20000 mL g−1 h−1, respectively, with the lowest apparent activation energy (31 kJ mol−1). The Pd nanoparticles possessed a good ability to adsorb water molecules, whereas the Au nanoparticles showed a good capability of activating O2 molecules in the presence of water. Therefore, the introduction of an appropriate amount of moisture to the feedstock could facilitate the easy activated adsorption of O2 molecules on the supported Au–Pd alloy sample, resulting in a positive effect on toluene oxidation over 1.95Au1Pd2/meso-Cr2O3. Using the hydrothermal and PVA-protected NaBH4 reduction strategies, highly active PdPty/V2O5–TiO2 catalysts with good moisture- and sulfur dioxide-resistant performance in toluene oxidation were prepared.65 The 0.46PdPty/V2O5–TiO2 catalyst exhibited the best toluene oxidation activity: T50% and T90% were 220 and 245 °C at a SV of 40000 mL g−1 h−1, respectively, with an apparent activation energy (Ea) = 45 kJ mol−1), specific reaction rate at 230 °C = 98.6 μmol gPt−1 s−1,
and turnover frequency (TOFNoble metal) at 230 °C = 142.2 × 10−3 s−1. No significant changes in toluene conversion were detected when 5.0 vol% H2O or 50 ppm SO2 was introduced to the reaction system. It was concluded that vanadium was the main site for SO2 adsorption, while PdO was the secondary site for SO2 adsorption, which protected the active Pt site from being poisoned by SO2, thus making the 0.46PdPty/V2O5–TiO2 catalyst show good sulfur dioxide resistance. The superior activity, excellent thermal stability, water resistance, and CO2 resistance of 0.46PdPty/V2O5–TiO2 were associated with its well-dispersed PdPt nanoparticles, high adsorbed oxygen species concentration, good redox ability, large toluene adsorption capacity, and strong interaction between PdPty and V2O5–TiO2. PtRu/3DOM Ce0.7Zr0.3O2 catalysts with uniformly dispersed noble metal nanoparticles at a size of 4.2–5.1 nm were prepared by polymethyl methacrylate (PMMA)-templating and ethylene glycol reduction methods.26 The 2.03PtRu/3DOM Ce0.7Zr0.3O2 catalyst exhibited the best performance in toluene oxidation: T50% and T90% were 163 and 194 °C at a SV of 40000 mL g−1 h−1, respectively, with the lowest apparent activation energy (45 kJ mol−1). However, 1.98PtRu@3DOM Ce0.7Zr0.3O2 possessed excellent thermal stability, while the thermal stability of 2.03PtRu/3DOM Ce0.7Zr0.3O2 decreased greatly after calcination at 800 °C for 5 h. After the high-temperature treatment, the average particle size of PtRu nanoparticles in 1.98PtRu@3DOM Ce0.7Zr0.3O2 increased slightly from 4.2 to 6.7 nm, whereas that of PtRu nanoparticles in 2.03PtRu/3DOM Ce0.7Zr0.3O2 increased significantly from 5.1 to 17.3 nm. The good performance of 1.98PtRu@3DOM Ce0.7Zr0.3O2 was associated with its highly dispersed partially embedded PtRu nanoparticles and good toluene and oxygen adsorption ability, and the formation of Pt–O–Ce-like bonds that possessed a strong interaction between PtRu nanoparticles and 3DOM Ce0.7Zr0.3O2 contributed to its better thermal stability.
| Catalyst | Preparation method | Mean noble metal particle size (nm) | Reaction conditions | T50% (°C) | T90% (°C) | Ref. |
|---|---|---|---|---|---|---|
| Au–Pd/3DOM Co3O4 | PVA-protected reduction method | 2.7 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
164 | 168 | 58 |
| Pt–Cu/Mn2O3 | Oleylamine method | 2.8 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
199 | 220 | 59 |
| Pt–Cu/Mn2O3 | Oleylamine method | 2.8 | 1000 ppm iso-hexane, SV = 40000 mL g−1 h−1 |
205 | 280 | 59 |
| Au–Pd/Co3O4 | PVA-protected reduction method | 2.7–3.2 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
171 | 180 | 60 |
| Au–Pd/Co3O4 | PVA-protected reduction method | 2.7–3.2 | 1000 ppm o-xylene, SV = 40000 mL g−1 h−1 |
181 | 187 | 60 |
| Au–Pd/α-MnO2 | PVA-protected reduction method | 2.5 ± 1.5 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
175 | 185 | 61 |
| Au–Pd/α-MnO2 | PVA-protected reduction method | 2.5 ± 1.5 | 1000 ppm m-xylene, SV = 40000 mL g−1 h−1 |
210 | 220 | 61 |
| Au–Pd/Ce0.6Zr0.3Y0.1O2 | PVA-protected reduction method | 4.6–5.6 | 1000 ppm toluene, SV = 20000 mL g−1 h−1 |
190 | 218 | 62 |
| AuMn/meso-Fe2O3 | Oleylamine co-reduction method | 3.5 | 1000 ppm toluene, SV = 20000 mL g−1 h−1 |
210 | 230 | 63 |
| AuMn/meso-Fe2O3 | Oleylamine co-reduction method | 3.5 | 1000 ppm benzene, SV = 20000 mL g−1 h−1 |
237 | 254 | 63 |
| Au–Pd/meso-Cr2O3 | PVA-protected reduction method | 2.9–3.7 | 1000 ppm toluene, SV = 20000 mL g−1 h−1 |
145 | 165 | 64 |
| PdPty/V2O5–TiO2 | PVA-protected reduction method | 3.1 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
220 | 245 | 65 |
| PtRu/3DOM Ce0.7Zr0.3O2 | Ethylene glycol reduction method | 3.0 | 1000 ppm toluene, SV = 40000 mL g−1 h−1 |
163 | 194 | 26 |
| Pd1Co1/Al2O3 | Oleic acid method | 4.1 | 1000 ppm benzene, SV = 40000 mL g−1 h−1 |
220 | 250 | 66 |
| PtW/Al2O3 | Solvothermal method | 4.5–8.8 | 1000 ppm benzene, SV = 40000 mL g−1 h−1 |
133 | 140 | 67 |
| AuPd/3DOM CeO2 | PVA-protected reduction method | 3–4 | 750 ppm trichloroethylene, SV = 20000 mL g−1 h−1 |
330 | 415 | 68 |
| RuPd/3DOM CeO2 | PVA-protected reduction method | 2.9–3.5 | 1000 ppm trichloroethylene, SV = 20000 mL g−1 h−1 |
237 | 298 | 25 |
| RuPt/3DOM CeO2 | PVA-protected reduction method | 2.9–3.5 | 1000 ppm trichloroethylene, SV = 20000 mL g−1 h−1 |
321 | 373 | 25 |
| RuAu/3DOM CeO2 | PVA-protected reduction method | 2.9–3.5 | 1000 ppm trichloroethylene, SV = 20000 mL g−1 h−1 |
290 | 349 | 25 |
| RuCo/meso-MgO | Cetyltrimethylammonium bromide-assisted method | 1.8 | 1000 ppm1,2-dichloroethane, SV = 20000 mL g−1 h−1 |
403 | 445 | 69 |
| RuCo/meso-Al2O3 | Cetyltrimethylammonium bromide-assisted method | 1.8 | 1000 ppm1,2-dichloroethane, SV = 20000 mL g−1 h−1 |
363 | 391 | 69 |
| RuCo/HZSM-5 | Cetyltrimethylammonium bromide -assisted method | 1.8 | 1000 ppm 1,2-dichloroethane, SV = 20000 mL g−1 h−1 |
238 | 281 | 69 |
Supported bimetallic single-atom palladium–cobalt catalysts with high catalytic efficiency and enhanced sulfur resistance in benzene oxidation (Fig. 7) were fabricated by a novel strategy.66 The PdCo/Al2O3 catalyst exhibited the best catalytic activity, and the gradual recovery of activity after introduction of 25 ppm SO2 for a long time was observed. By investigating the sulfur resistance in catalytic benzene oxidation, we found that the PdCo/Al2O3 catalyst showed a good sulfur resistance, and its activity for benzene oxidation experienced a process of first decline and then recovery after the introduction of SO2. The results of surface compositions and adsorption–desorption behaviors of PdCo/Al2O3 showed that there was no competitive adsorption between benzene and oxygen on the surface of the catalyst. It could be inferred that its excellent catalytic activity was due to the dual active sites composed of Pd and Co double single atoms. The adsorption–desorption behaviors of the catalysts before and after sulfur resistance were investigated, and the enhanced sulfur resistance was mainly attributed to its good active site regeneration ability, which was due to the rapid decomposition of the sulfite or sulfate species formed on the surface of single-atom dispersed Pd and Co. In addition, the results of in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and other characterization studies revealed that benzene oxidation over PdCo/Al2O3 might take place according to the sequence of benzene → cyclohexadiene → phenol → quinone → maleate → acetic acid → CO2 and H2O. PtM (M = W, Mo) bimetallic nanoparticles and commercial γ-Al2O3-supported PtM catalysts were prepared by the solvothermal method.67 The shape of the PtW(Mo) nanoparticles was regular, while Pt nanoparticles displayed an inhomogeneous rod-like morphology. Compared to the as-obtained metal nanoparticles, the sizes of the metal nanoparticles in the supported samples had no obvious changes, and they were well distributed on the support surface. The PtW/Al2O3 catalyst exhibited the best catalytic activity for benzene oxidation: T50% and T90% were 133 and 140 °C at a SV of 40000 mL g−1 h−1, respectively. The presence of WOx or MoOx clusters in the bimetallic samples accelerated the reaction. Electrons were transferred from the WOx or MoOx to Pt, as evidenced by the increase in the W6+/W5+ or Mo6+/Mo5+ molar ratio after the increase in W or Mo doping in the bimetallic system (Ptσ+ + W5+ → Ptσ+ + W6+), which made the Pt more electronegative. Concurrently, O2 could be activated by picking up the electrons from the surface Pt to give electrophilic oxygen site (O−, O2− or O22−) species, which was verified by the oxygen adspecies concentrations on the catalyst surface. In situ DRIFTS studies revealed that the transformation from phenol to benzoquinone was the key step in the oxidation process of benzene. The supported bimetallic catalysts exhibited stronger benzene adsorption ability than the supported Pt catalyst. Therefore, it was reasonably concluded that the key role of the MOx was adsorption and activation of VOC molecules. 3DOM CeO2-supported Au–Pd alloys were synthesized using PMMA-templating and PVA-protected reduction methods.68 It was found that the AuPd/3DOM CeO2 samples displayed a good-quality 3DOM architecture, and the noble metal nanoparticles with a size of 3–4 nm were uniformly dispersed on the skeleton surface of 3DOM CeO2. Among all of the samples, 2.85AuPd1.87/3DOM CeO2 performed the best for the oxidation of trichloroethylene (TCE): T50% and T90% were 330 and 415 °C at a SV of 20000 mL g−1 h−1, respectively. Furthermore, the 2.85AuPd1.87/3DOM CeO2 sample possessed the lowest apparent activation energy (33 kJ mol−1). In addition, the synergistic effect of Au and Pd in 2.85AuPd1.87/3DOM CeO2 also contributed to the superior thermal stability and water and Cl resistance. The alloying of Au and Pd improved the adsorption and activation of oxygen molecules and enhanced the interaction between the AuPd alloy nanoparticles and the support, resulting in a significant increase in catalytic activity and resistance to chlorine poisoning. It was concluded that the excellent catalytic performance for TCE oxidation of 2.85AuPd1.87/3DOM CeO2 was associated with the highly dispersed AuPd1.87 alloy nanoparticles, high adsorbed oxygen species concentration, good low-temperature reducibility, and strong interaction between AuPd nanoparticles and 3DOM CeO2 as well as the high-quality 3DOM structure and high surface acidity.
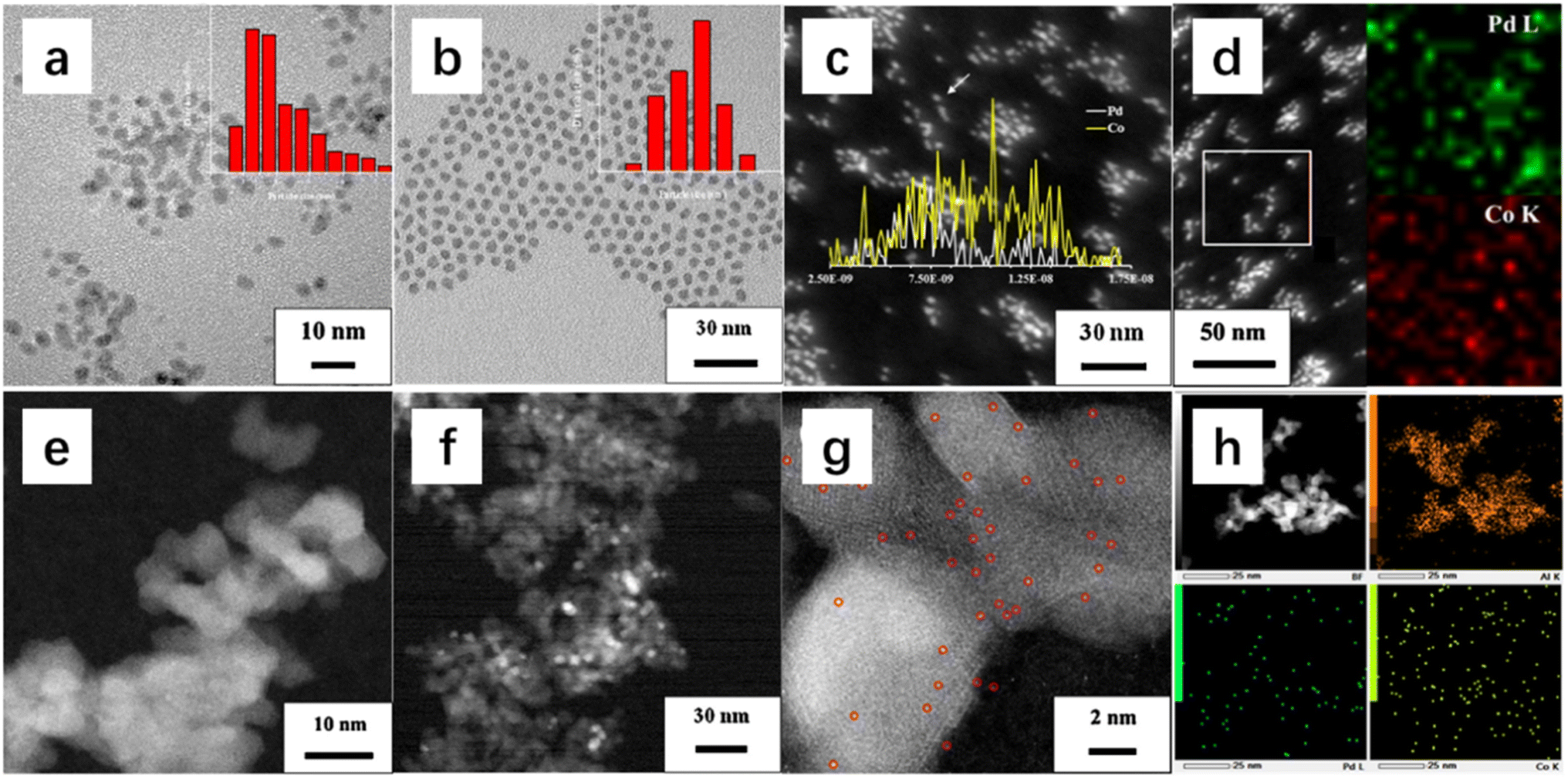 | ||
| Fig. 7 TEM images and particle-size distributions (inside) of (A) PdCo nanoparticles and (B) Pd nanoparticles, (C) STEM image and EDX line scan (inside) of PdCo nanoparticles, (D) EDX elemental mapping of PdCo nanoparticles, STEM images of (E) Pd1Co1/Al2O3 and (F) Pd/Al2O3, aberration-corrected STEM images of (G) Pd1Co1/Al2O3, and EDX elemental mapping of (H) Pd1Co1/Al2O3.66 Reproduced with permission from ref. 66 Copyright 2021, Elsevier. | ||
Ruthenium-based materials are promising catalysts for the removal of chlorinated VOCs (CVOCs) due to their good ability in cleaving the C–Cl bonds and decreasing the Cl deposition. However, the roles of noble bimetals alloyed with Ru have not been clarified clearly. Bimetallic RuM (M = Au, Pd or Pt) alloys supported on 3DOM CeO2 were fabricated by PMMA-templating and PVA-protected reduction methods (Fig. 8).25 The Ru, Pd, and RuyM nanoparticles with an average size of 2.9–3.5 nm were uniformly dispersed on the skeleton surface of 3DOM CeO2. The 0.93Ru2.87Pd/3DOM CeO2 sample showed the highest catalytic activity and the lowest apparent activation energy (34 kJ mol−1) in TCE oxidation, with the T50% and T90% being 237 and 298 °C at SV = 20000 mL g−1 h−1, respectively. Simultaneously, 0.93Ru2.87Pd/3DOM CeO2 exhibited the highest TCE oxidation rate at 250 °C (30.3 μmol gNoble metal−1 s−1) and the highest TOFNoble metal at 250 °C (3.1 × 10−3 s−1). 0.93Ru2.87Pd/3DOM CeO2 also possessed a better hydrothermal stability than 0.85Ru/3DOM CeO2 after the hydrothermal ageing treatment at 750 °C. Effects of H2O, CO2, and HCl on catalytic activity of 0.93Ru2.87Pd/3DOM CeO2 were also examined. The presence of Ru in the samples was favorable for generation of HCl and Cl2 and reduction of the by-products in TCE oxidation, and tetrachloroethylene was the main by-product. The possible catalytic mechanism over 0.93Ru2.87Pd/3DOM CeO2 was also proposed. The outstanding catalytic efficiency of 0.93Ru2.87Pd/3DOM CeO2 could be assigned to the high adsorbed oxygen species, good low-temperature reducibility, strong TCE adsorption and activation ability, and formation of an intimate nanointerface between RuPd nanoparticles and 3DOM CeO2. The effect of the nature of porous supports (mesoporous Al2O3 (meso-Al2O3), mesoporous MgO (meso-MgO), and microporous HZSM-5) on catalytic performance of the bimetallic RuCo nanoparticles was investigated for the oxidation of 1,2-dichloroethane (1,2-DCE).69 The order in activity was RuCo/HZSM-5 (T50% = 238 °C and T90% = 281 °C) > Ru/HZSM-5 (T50% = 260 °C and T90% = 308 °C) > Co/HZSM-5 (T50% = 285 °C and T90% = 329 °C) > RuCo/meso-Al2O3 (T50% = 363 °C and T90% = 391 °C) > RuCo/meso-MgO (T50% = 403 °C and T90% = 445 °C), with the more acidic RuCo/HZSM-5 showing the highest catalytic activity. The excellent oxidation performance was related to the abundant acidic sites, porous structure, and redox centers on the surface of HZSM-5. The large surface area and pore structure of the catalyst facilitated the adsorption of 1,2-DCE molecules, and then 1,2-DCE could remove HCl at the acidic sites to obtain C2H3Cl. The loading of RuCo nanoparticles as a strong oxidation center facilitated the deep oxidation of the intermediate products, thus effectively inhibiting the formation of the toxic by-product C2H3Cl and reducing secondary contamination. The X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) results confirmed that the RuO2 species were dominantly present in the bimetallic sample, and doping of the Co species could increase the length of the Ru–O bond. Over the RuCo/HZSM-5 sample, the partial deactivation induced by water vapor or HCl addition was reversible, while that induced by SO2 introduction was irreversible. According to the characterization results, the oxidation of 1,2-DCE over RuCo/meso-Al2O3, RuCo/meso-MgO or RuCo/HZSM-5 might take place concurrently via the Langmuir–Hinshelwood and Mars–van Krevelen mechanisms. The synergistic promotion of the redox, the surface acid, and the strong interaction of the highly dispersed RuCo nanoparticles with HZSM-5 improved the oxidation performance of the catalyst.
 | ||
| Fig. 8 (a, b, e, f, i, j, and m–p) TEM and (c, g, and k) HAADF–STEM images, EDX elemental mappings, and (d, h, and l) EDX line scan profiles of (a and b) 3DOM CeO2, (c and d) 0.93Ru2.87Pd/3DOM CeO2, (e–h) 0.91Ru2.85Au/3DOM CeO2, (i–l) 0.90Ru2.77Pt/3DOM CeO2, (m and n) 0.85Ru/3DOM CeO2, and (o and p) 0.88Pd/3DOM CeO2.25 Reproduced with permission from ref. 25 Copyright 2020, RSC. | ||
4.3. Others
The addition of another metal to a platinum catalyst to exert a bimetallic synergistic effect is an effective way to significantly improve the catalytic performance. When the metal composition changes from single to double elements, however, the complexity of the active sites increases. For example, Bao et al. reported Pt–Fe/SiO2 catalysts with coordinatively unsaturated metal sites and lower valent states for the preferential oxidation of CO.70 The excellent performance catalyst exhibited a structure consisting of a Pt-rich core with the ferrous species encapsulated on the surface. The oxygen molecules were adsorbed at the interface between the coordinatively unsaturated Fe2+ and the Pt atoms, which were dissociated into active oxygen atoms to participate in the reaction. By the way, the Pt–FeO2 interface confinement stabilized the ligand-unsaturated ferrous species against further oxidation to Fe2O3 to achieve a relative stability. The ultra-low-temperature and high-performance Fe–Pt/SiO2 catalysts reported by Lu et al. demonstrated that the Fe1(OH)–Pt interface was the catalyst active site in CO-selective catalytic oxidation,71 which was in good agreement with the above result. Unlike the active and stable Fe2+ site in the Fe–Pt/SiO2 catalyst reported by Bao et al.,70 the Fe2+ species was oxidized to the Fe3+ species in the gas feed, and the Fe1(OH)x clusters were anchored on the Pt nanoparticles to form the active Fe1(OH)–Pt interface reported by Lu et al.71 Density functional theory calculations indicated that the Fe1(OH)x–Pt single interfacial sites could readily react with CO and facilitate oxygen activation to realize 100% selective CO elimination. Platinum-based bimetallic nanoparticles were regarded as the most efficient catalysts for CO oxidation. Construction of Pt–metal (hydr)oxide interface active sites was highly efficient for CO oxidation at low temperatures. Another type of the active site was the Pt–M alloy structure, in which platinum was directly bonded to the second metal atom. The Pt–Co/SiO2 and Au–Co/SiO2 catalysts were subjected to different redox behaviors and applications in CO oxidation.72 The Co oxide species were completely reduced to metallic Co0 atoms and formed an alloy structure with Pt atoms below 300 °C, which showed a superior catalytic activity for CO oxidation.72 However, the Co species in the reduced Au–Co/SiO2 samples were rapidly oxidized at room temperature in an O2 atmosphere, since the Co atoms were mostly located at the sample surface. The result indicated that a suitable reduction treatment could form a high density of the interface sites between the surface CoO structure and the noble metal to enhance the reactivity. Wei et al. reported that the Pt–Co(OH)/SiO2 bimetallic catalysts were treated with a reduction pre-treatment to form the Pt–Co bonds directly, which acted as the active site in CO oxidation at low temperatures.73 Meanwhile, Co atoms were oxidized to CoOx in the catalytic reaction process, and the synergistic interaction between the Pt–CoOx interfaces became a new active site. Two-dimensional planar Pt sub-nanoclusters with a single atomic layer were loaded on the SiO2 surface using the atomic layer deposition (ALD) method, and CoOx was deposited to decorate the Pt atoms. The resulting Pt–CoOx/SiO2 catalysts exhibited extremely high catalytic performance in the selective oxidation of CO, achieving a 100% CO conversion and 100% selectivity in the range of 25–140 °C.74 The Pt clusters and CoOx species formed a Pt–CoOx interface through O atoms after the high-temperature reduction, and the structure of the Pt–CoOx sub-nanoclusters remained stable during the preferential oxidation of CO in H2. The results of the DRIFTS and XPS characterization showed that the charge transfer from Pt to CoOx in the sub-nanoclusters of Pt–CoOx reduced the charge feedback from Pt to CO molecules, which weakened the adsorption strength of CO at the Pt site and effectively enhanced its catalytic performance.Alloying 3d transition metals with Pt was an effective strategy to improve the catalytic activity for the oxygen reduction reaction (ORR). Table 5 gives an activity overview of the various bimetallic catalysts for certain reactions reported in the literature. The challenge was insufficient catalyst durability due to the rapid leaching of transition metal elements. To overcome the defect and achieve enhanced durability, Huang et al. reported a novel catalytic structure based on PtGa ultrathin alloy nanowires (NWs) with the unconventional strong p–d hybridization interactions.76 The ORR mass activity and specific activity of PtGa NWs increased by 10.5 and 12.1 times, respectively, compared with those of the commercial Pt catalyst. The mass activity of the PtGa NWs decreased by only 15.8% after 30000 endurance tests, while a 79.6% reduction happened over the commercial Pt catalyst. It was found that the strong p–d hybridization interaction between Ga and Pt was responsible for the superior ORR performance via synergistically upgrading the surface electronic structure and improving the oxidation resistance of Pt as well as suppressing the leaching of lattice Ga. Conventional alloy catalysts lack an accurate control over the local atomic structure of the active site. Mueller et al. investigated the active site synergistic effect of bimetallic Pd–Au electrocatalysts by coating the surface of Au nanoparticles with different doses of palladium (Fig. 9).77 It was found that different contents of the Pd-modified gold nanoparticles induced changes in the Pd ensemble size and corresponding adsorption properties. Density functional theory calculations revealed that the modification of Pd sites in the Pd@Au electrocatalysts effectively reduced the CO2 activation energy barrier. Moreover, the *CO intermediates combined with the bimetallic surface were less toxic than the pure Pd, and the synergistic effect between the Pd@Au electrocatalysts was accountable for the high activity for the electroreduction of CO2 to CO.
| Catalyst | Preparation method | Particle size (nm) | Reaction | Reaction conditions | Catalytic performance | Ref. |
|---|---|---|---|---|---|---|
| Pt–Fe/SiO2 | Chemical deposition | 2–4 | The preferential oxidation of CO in excess H2 | 1 vol% CO, 0.5% O2, H2 balance. SV = 36000 mL g−1 h−1 |
Pt–Fe/SiO2 catalysts showed almost 100% CO conversion and 100% CO selectivity at room temperature | 70 |
| Pt–Co/SiO2 | Two-step NaBH4 reduction method | 3.8 | CO oxidation in excess O2 | 1 vol% CO, 0.5 vol% O2, He balance; SV = 75000 mL g−1 h−1 |
Complete CO conversion at room temperature was observed when the catalyst was reduced at 100 °C | 72 |
| Au–Co/SiO2 | Two-step NaBH4 reduction method | 2.5 | CO oxidation in excess O2 | 1 vol% CO, 0.5 vol% O2, He balance; SV = 75000 mL g−1 h−1 |
Complete CO conversion at room temperature was observed when the catalyst was reduced at 500 and 600 °C | 72 |
| Au–Rh | Oleylamine co-reduction method | 8.5 | Hydrogen evolution reaction | 10 μL Au–Rh/C was added in a mixture of water, isopropanol and Nafion (5%) at a volumetric ratio of 4:1:0.0025 to make the working electrode |
Au75Rh25 core–shell decahedra exhibited an overpotential of 64.1 mV at a current density of 10 mA cm−2 and mass activity of 10.76 mA μgRh−1 | 75 |
| PtGa | Oleylamine co-reduction method | 1.2 ± 0.4 | Oxygen reduction reaction | O2-saturated 0.1 M HClO4 solutions at a sweep rate of 10 mV s−1 and a rotation rate of 1600 rpm at room temperature | Pt4.31Ga NWs/C showed the highest specific and mass activities of 3.28 mA cm−2 and 1.89 A mgPt−1 at 0.9 VRHE | 76 |
| Pd–Au | Chemical deposition | 5.3 | CO2 reduction | A solution of 0.1 mol L−1 KHCO3 was used as electrolyte. CO2 was bubbled at a constant rate of 20 sccm and purged for 10 minutes prior to each measurement | Pd5@Au95 delivered a CO2 reduction current density of up to 5.8 mA cmgeo−2 at −0.8 V | 77 |
| AuAg | Nitric acid leaching to obtain nanoporous AgAu alloys | Oxidative coupling of methanol | Oxygen (0.2 Torr) and methanol (0.1 Torr) were introduced into the vacuum chamber via leak valves | AgAu alloys producing methyl formate selectively without deactivation at 150 °C | 4 | |
| Au–Pt | Oleylamine co-reduction method | 16 | Oxygen reduction reaction | Ar-purged 0.1 mol L−1 HClO4 solutions at a sweep rate of 50 mV s−1 between 0.05 and 0.1 V at room temperature | AuPt1.03 star-shaped decahedra achieved the highest mass activity (0.94 mA μgPt−1) and area activity (1.09 mA cmPt−1) | 78 |
| PtRu | Oleylamine co-reduction method | 12.7 | Methanol electro oxidation | CV curves were recorded in an Ar-purged 0.5 mol L−1 H2SO4/0.5 mol L−1 CH3OH solution at a sweep rate 50 mV s−1 in the range of 0.2 V to 1.2 V vs. RHE | Pt7Ru bimetallic icosahedra achieved a specific activity of 0.76 mA cm−2 and mass activity of 74.43 mA mgPt−1 | 79 |
 | ||
| Fig. 9 Synthesis and HAADF−STEM images of the Pd@Au nanoparticles. (a) Illustration of the synthetic scheme for the Pd@Au nanoparticles with control over the dose of Pd, and (b–d) STEM images and EELS-based element maps for (b) Pd2@Au98, (c) Pd5@Au95, (d) Pd10@Au90, and (e) Pd20@Au80, where Au and Pd atoms are represented by red and green pixels, respectively.77 Reproduced with permission from ref. 77 Copyright 2019, ACS. | ||
Activation pretreatments were commonly used to adjust the surface compositions and structures of bimetallic alloy materials. Friend et al. investigated the transformations of nanoporous Ag0.03Au0.97 alloys by the O3 activation induced treatment and subsequent adjustments under steady-state CH3OH oxidation conditions.4 The activation treatment produced enriched AgO and Au2O3 in the near-surface region of Ag. The reduction of generated oxides happened in the O2/CH3OH mixture to produce CO2 and highly Ag-enriched surface alloys. However, bulk agglomerate diffusion led to sintering and Ag re-distribution at high temperatures, and the selectivity for the catalytic generation of methyl formate was reduced. The results revealed that the material properties which determined the catalytic activity were dynamic, and the substable form of the catalysts might account for the catalytic performance.
The ultimate goal of catalyst design was to optimize catalytic performance by revealing the relationship between the structure and activity. However, it remains a great challenge to distinguish the role of electronic and geometrical effects (ensemble effects) for bimetallic core–shell electrocatalysts. Yang et al. reported an efficient strategy to study the local epitaxial growth of Rh atoms on Au decahedra to investigate the electronic and geometrical effects.78 A series of Au–Rh core–shell star-shaped structured decahedra with different compositions were prepared using the seed growth method. Hollow Ru nanoframe-structured nanocrystals were generated by the selective etching of Au nanocrystal cores with I3−/I− ions as the etchant. The Au75Rh25 core–shell star-shaped decahedra exhibited the best catalytic activity for the hydrogen evolution reaction (HER) with an overpotential of 64.1 mV at a current density of 10 mA cm−1. In comparison with the commercial Rh/C catalyst, the Au–Rh decahedra showed better decomposition efficiency and selectivity. The results of density functional theory calculations indicated that geometric effect (ensemble effect) was the dominant factor in accelerating the kinetics of the HER, as the exposure of Au atoms on the surface weakened the strong adsorption of the intermediates on the Rh surface. The Au–Pt core–shell structured star decahedra (Fig. 10) were fabricated using a one-step thermal synthesis method with oleylamine as solvent and a reducing agent for the oxygen reduction reaction (ORR) in fuel cells.75 The introduction of amine groups reduced the surface energy of Pt, thus achieving the epitaxial growth of Pt nanocrystals on the Au core and regulating the thickness of the Pt epitaxial layer at the atomic scale. Significantly, Br− ions could stabilize the ORR favored Pt {111} crystalline surface to a large extent, thus preventing the formation of the spherical nanoparticles. The AuPt1.03 star decahedron exhibited the highest mass activity (0.94 mA μgPt−1) and area activity (1.09 mA cmPt−2), which was 6.7 and 5 times higher than that of the commercial Pt/C catalyst, respectively. The decahedron of Au–Pt core–shell with a thicker Pt shell layer effectively enhanced the catalytic activity and durability of the composite structure due to the synergetic effect and twin structure, with essentially no loss of activity for the ORR after 30000 cycles of testing. The Pt–Cu alloy octahedra with different compositions were prepared using the solvothermal method for methanol oxidation.80 Cl− ions in the copper chloride reactant were found to selectively modulate the underpotential deposition (UPD) of copper atoms on the Pt crystalline surface, which was crucial to the formation of Pt–Cu octahedral nanocrystals. Element-specific growth trajectories of Pt–Cu nanostructures coupled with compositional variations and geometrical morphologies were observed: the PtxCu1−x polydendrimer started from Pt-rich crystalline species (x > 0.6) and evolved into a Pt–Cu alloying phase (x ≈ 0.5), which in turn formed Pt-rich dendrites (when x > 0.8). Due to the synergistic interaction between Pt and Cu bimetallic multipod nanostructures, the alloy octahedron exhibited resistance to CO toxicity and a superior catalytic activity to the commercial Pt/C catalyst. The PtRu bimetallic particles have been known to be promising commercial catalysts for methanol oxidation. Shape-controlled synthesis of PtRu bimetallic nanocrystals, especially for {100} (e.g., cubic) or {111} cuts (e.g., icosahedral) exposed to the catalytic Platonic structure limits the development of catalytic properties. Lin et al. reported a facile method for the synthesis of Ru-decorated Pt bimetallic cubes and icosahedra in mixed solvents for methanol oxidation.79 It was found that the formation of cubes might be due to the selective adsorption of CO from the catalytic decomposition of N,N-dimethylformamide (DMF) by the Ru salt precursor on Pt {100} in solvents which contained N,N-dimethylformamide and oleylamine. The synthesis became a two-phase interfacial reaction due to the large difference in solvent polarity and slowed down the reaction kinetics by introducing hexadecane to the above solvent, which facilitated the formation of the icosahedra instead of a cube structure. Ru-modified Pt icosahedra displayed better catalytic performance in terms of specific activity and mass activity than the cubes for methanol oxidation, due to a combination of twin-induced strain and facet effects. The Ru-decorated Pt bimetallic icosahedra showed a specific activity of 0.76 mA cm−2 and a mass activity of 74.43 mA mgPt−1, which were 6.7 and 2.2 times higher than those of the PtRu nanoparticles, respectively.
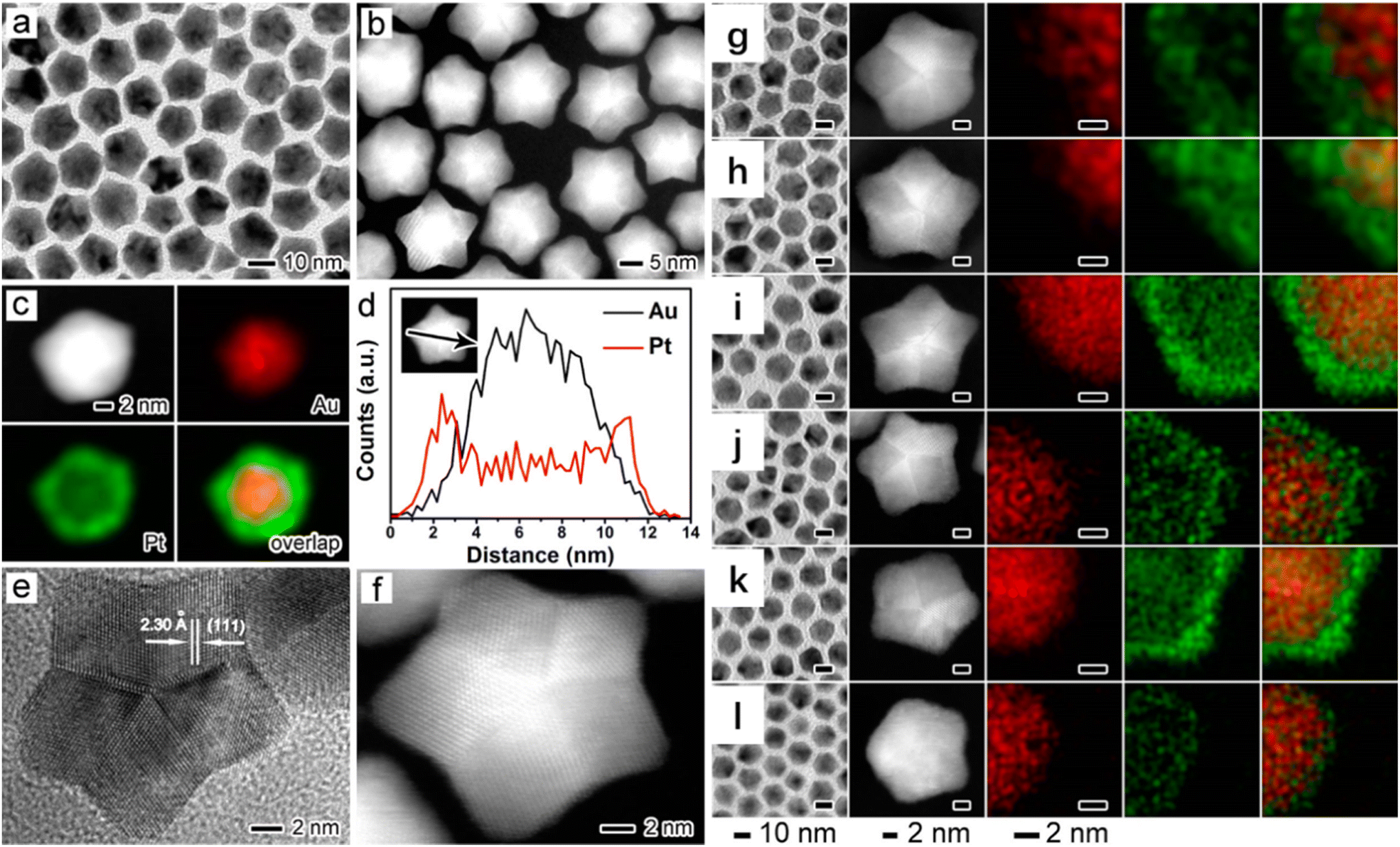 | ||
| Fig. 10 Morphological, structural, and compositional characterization studies of Au–Pt star-shaped decahedra prepared using the standard procedure: (a) TEM image, (b) HAADF-STEM image, (c) EDX mapping, (d) line-scan analysis, (e) HRTEM image, and (f) HAADF-STEM image at a higher magnification. The insets of a and b show the magnified TEM and HAADF-STEM images of a single star-shaped decahedron, respectively. The red and green colors in (c) correspond to Au and Pt elements, respectively.75 TEM and HAADF-STEM-EDX mapping images of the Au–Pt nanocrystals prepared using the standard procedure, except for the different molar ratios of Au to Pt salt precursors: (a) 1.0:1.0, (b) 1.0:0.9, (c) 1.0:0.8, (d) 1.0:0.7, (e) 1.0:0.6, and (f) 1,0:0.5.75 Reproduced with permission from ref. 75 Copyright 2015, ACS. | ||
There are water, sulfur dioxide or other organic compounds in practical industrial exhaust gases. The presence of water and reactant molecules exhibits competitive adsorption, which causes the partial deactivation of catalysts. Simultaneously, the adsorbed water molecules generate a large number of hydroxyl groups and occupy the exposed surface active sites, which inhibit generation of the surface active oxygen species. It was reported that the bimetallic PdW/TiO2 catalysts showed better water resistance than monometallic Pd/TiO2.81 After the introduction of 1.0 vol% H2O, benzene conversions over PdW/TiO2 remained almost unchanged while those over Pd/TiO2 were decreased by 20%. The in situ FTIR results showed that the absorption intensity of OH species on the Pd/TiO2 catalyst was significantly stronger than that over the bimetallic PdW/TiO2 catalyst. The absorption bands of OH species on PdW/TiO2 almost disappeared after water was cut off. In contrast, the adsorbed OH species on the Pd/TiO2 catalyst were desorbed slowly. The results indicated that the bimetallic PdW/TiO2 catalyst inhibited the formation of surface hydroxyl groups and accelerated the desorption of water. The adsorbed SO2 molecules on the catalyst surface always occupy the active sites, which result in poisoning of the catalyst. The catalyst deactivation caused by inducing SO2 is ascribed to the formation of difficultly decomposed sulfate species. Pd/ZrO2 and PtPd/ZrO2 catalysts were prepared to investigate their SO2-resistant performance.3 Methane conversions over PtPd/ZrO2 were decreased slightly after the introduction of 100 ppm SO2, and catalytic activities were returned to the initial levels when SO2 was cut off. However, the introduction of 100 ppm SO2 to the Pd/ZrO2 catalyst resulted in a methane conversion drop by 45% (indicating an irreversible deactivation). The XPS results demonstrated that a new component appeared on the S 2p spectrum of the PdPt/ZrO2 catalyst compared with the Pd/ZrO2 catalyst, which could be attributed to the adsorption of SO2 at the Pt site. The introduction of Pt facilitated the preferential adsorption of SO2 at the Pt site, thereby protecting the PdO active phase from being poisoned by SO2.
The adsorption and desorption behaviors of VOC molecules and intermediates on the surface of the catalysts determine catalytic performance of the bimetallic nanoparticles. For example, PtFe/MnO2 nanoparticles were fabricated for toluene and isohexane oxidation.82 The VOCs-TPD results revealed that the Pt/MnO2 catalyst showed a large toluene adsorption capacity, while the PtFe/MnO2 catalyst was more favorable for isohexane adsorption. The conversion of toluene over Pt/MnO2 was decreased by 50% after 6 h of the reaction, whereas that over PtFe/MnO2 remained stable after 20 h of the reaction. The PtFe/MnO2 catalyst exhibited stronger reducing properties and a higher lattice oxygen activity, which changed the toluene oxidation intermediate pathways.
Based on the above discussion, the introduction of a second metal modifies the surface properties of a bimetallic catalyst so as to obtain multifunctional active sites, which optimize adsorption behaviors of the reactant molecules and reaction pathways. Future work should focus on designing and synthesizing bimetallic catalysts with high activities and stability in the presence of H2O, SO2 or other organic compounds.
As mentioned above, bimetallic catalysts show excellent activity and stability in many reactions. Catalytic performance of bimetallic catalysts can be adjusted for the reactions of different reactants by changing the metal nature, metal composition or support nature. Bimetallic catalysts exhibit better thermal stability and resistance to sintering. The bimetallic sites optimize the reaction paths and adsorption properties of the intermediates, exhibiting better resistance to toxicity. However, reaction mechanisms over bimetallic catalysts may be more complex than those over their monometallic counterparts. The exact function of the dual active sites may be unclear, which makes it more difficult to optimize the catalytic performance. Preparation of bimetallic catalysts with uniform distributions and desired particle sizes requires complex steps, which may limit large-scale production of such kinds of bimetallic catalysts for industrial applications. Bimetallic catalysts require a more complex regeneration process after deactivation since the interaction of the two metals may influence their regeneration efficiencies. Overall, bimetallic nanoparticles offer many advantages in catalytic oxidation reactions. The catalytic performance and application values of bimetallic catalysts can be maximized by controlling their chemical compositions, structures, and preparation methods.
5. Conclusion and perspectives
Surface composition and elemental arrangement play a crucial role in determining activity and selectivity of a bimetallic catalyst. Understanding the factors that regulate the structure optimization is important for both engineering the surface electronic properties of bimetallic nanoparticles and achieving the desired catalytic reactivity. This review article provides an overview of the preparation methods, characterization, and catalytic applications of bimetallic nanoparticles. Bimetallic nanoparticles offer enriched catalytic properties, which is ascribed to the specific interactions between the two metals, the tunable compositions, and the reconfigurable electronic arrangements. Generally speaking, an improved chemical engineering method is designed to control the sizes, morphologies, chemical components, and coordination environments of bimetallic nanoparticles. The significant surface reactivity factors of bimetallic nanoparticles are investigated and summarized, including solid-state diffusion, intrinsic surface energy, surface reactant adsorption, and surface chemical reactivity between reactants and metal atoms. Diverse bimetallic nanostructures with individualized active sites present different structure–reactivity relationships for the specific reactions. It is essential to elucidate the different predominant roles and reaction pathways of the active sites in two-metal catalysts.The improved catalytic performance of bimetallic catalysts is associated with the following four effects: the geometric structure effect, electronic structure effect, synergistic effect, and stabilization effect. Modification in the coordination number, atomic arrangement, atomic size, and local atomic charge of reactive metals provides the corresponding multifunction and synergistic effect in heterogeneous catalysis, such as dual adsorption/activation of reactants, decoking, and enhanced redox ability. The unique geometrical structures and electronic properties of bimetallic nanoparticles overcome various bottlenecks in terms of conversion efficiency and product selectivity. It is hopeful that the insights summarized in this review article will contribute to further studies on bimetallic nanoparticles.
Industrial exhaust gases have complex chemical compositions and are usually mixed with H2O, SO2 or other organic compounds under practical conditions. In general, the presence of other components introduces competitive adsorption, which induces the decrease in catalytic activity. Bimetallic catalysts exhibit high activities toward targeted VOC oxidation and reduce the generation of harmful by-products. High-performance bimetallic catalysts without noble metals are economically viable and promising for large-scale preparation. Future work should pay much attention to designing efficient and economical bimetallic catalysts for industrial applications.
Data availability
This thesis is a review and the data in the text are derived from the references. Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFB3504101 and 2022YFB3506200), National Natural Science Foundation of China (22322601 and 22306008), National Natural Science Committee of China-Liaoning Provincial People's Government Joint Fund (U1908204), R&D Program of Beijing Municipal Education Commission (KZ202210005011), Postdoctoral Science Foundation of China (2022M720315), and Postdoctoral Research Foundation of Beijing (2023ZZ-139).References
- Y. Nakaya and S. Furukawa, Catalysis of alloys: classification, principles, and design for a variety of materials and reactions, Chem. Rev., 2022, 123(9), 5859–5947 CrossRef.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Dual-metal atom electrocatalysts: theory, synthesis, characterization, and applications, Adv. Energy Mater., 2022, 12(3), 2102715 CrossRef CAS.
- X. F. Zhang, Y. X. Liu, J. G. Deng, L. Jing, L. K. Wu and H. X. Dai, Catalytic performance and SO2 resistance of zirconia-supported platinum-palladium bimetallic nanoparticles for methane combustion, Catal. Today, 2022, 402(15), 138–148 CrossRef CAS.
- B. Zugic, M. A. van Spronsen, C. Heine, M. M. Montemore, Y. Y. Li, D. N. Zakharov, S. Karakalos, B. A. J. Lechner, E. Crumlin, M. M. Biener, A. I. Frenkel, J. Biener, E. A. Stach, M. B. Salmeron, E. Kaxiras, R. J. Madix and C. M. Friend, Evolution of steady-state material properties during catalysis: oxidative coupling of methanol over nanoporous Ag0.03Au0.97, J. Catal., 2019, 380, 366–374 CrossRef CAS.
- K. Kusada, D. S. Wu, Y. Nanba, M. Koyama, T. Yamamoto, X. Q. Tran, T. Toriyama, S. Matsumura, A. Ito, K. Sato, K. Nagaoka, O. Seo, C. Song, Y. N. Chen, N. Palina, L. S. R. Kumara, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Highly stable and active solid-solution-alloy three-way catalyst by utilizing configurational-entropy effect, Adv. Mater., 2021, 33(16), 2005206 CrossRef CAS.
- H. Arandiyan, Y. Wang, J. Scott, S. Mesgari, H. X. Dai and R. Amal, In situ exsolution of bimetallic Rh-Ni nanoalloys: a highly efficient catalyst for CO2 methanation, ACS Appl. Mater. Interfaces, 2018, 10(19), 16352–16357 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts, Science, 2006, 311(5759), 362–365 CrossRef CAS.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Direct synthesis of hydrogen peroxide from H2 and O2 using Pd and Au catalysts, Phys. Chem. Chem. Phys., 2003, 5(9), 1917–1923 RSC.
- W. R. Schwartz and L. D. Pfefferle, Combustion of methane over palladium-based catalysts: support interactions, J. Phys. Chem. C, 2012, 116(15), 8571–8578 CrossRef CAS.
- J. S. Tian, R. Kong, Z. Wang, L. Fang, T. Y. He, D. Jiang, H. G. Peng, T. L. Sun, Y. H. Zhu and Y. Wang, Enhancing methane combustion activity by modulating the local environment of Pd single atoms in Pd1/CeO2 catalysts, ACS Catal., 2024, 14(1), 183–191 CrossRef CAS.
- X. H. Huo, G. L. Li, X. Wang and W. B. Zhang, Bimetallic catalysis in stereo divergent synthesis, Angew. Chem., 2022, 134(45), e202210086 CrossRef.
- T. T. Li, N. Ji, Z. C. Jia, X. Y. Diao, Z. J. Wang, Q. L. Liu, C. F. Song and X. B. Lu, Effects of metal promoters in bimetallic catalysts in hydrogenolysis of lignin derivatives into value-added chemicals, ChemCatChem, 2020, 12(21), 5288–5302 CrossRef CAS.
- S. Ashraf, Y. Y. Liu, H. J. Wei, R. F. Shen, H. H. Zhang, X. L. Wu, S. Mehdi, T. Liu and B. J. Li, Bimetallic nanoalloy catalysts for green energy production: advances in synthesis routes and characterization techniques, Small, 2023, 19(43), 2303031 CrossRef CAS.
- N. Eom, M. E. Messing, J. Johansson and K. Deppert, General trends in core–shell preferences for bimetallic nanoparticles, ACS Nano, 2021, 15(5), 8883–8895 CrossRef CAS PubMed.
- J. C. Qiu, Q. N. Nguyen, Z. H. Lyu, Q. X. Wang and Y. N. Xia, Bimetallic Janus nanocrystals: syntheses and applications, Adv. Mater., 2022, 34(1), 2102591 CrossRef CAS PubMed.
- L. Rößner and M. Armbrüster, Electrochemical energy conversion on intermetallic compounds: a review, ACS Catal., 2019, 9(3), 2018–2062 CrossRef.
- H. N. Shi, J. H. Li, H. Z. Wang, J. G. Hou, K. Y. Li and X. W. Guo, Chlorine tailored p-d blocks dual-metal atomic catalyst for efficient photocatalytic CO2 reduction, Appl. Catal., B, 2023, 322, 122139 CrossRef CAS.
- Z. H. Xu, W. Zuo, Y. Y. Yu, J. Y. Liu, G. Z. Cheng and P. P. Zhao, Surface reconstruction facilitated by fluorine migration and bimetallic center in NiCo bimetallic fluoride toward oxygen evolution reaction, Adv. Sci., 2024, 11(6), 2306758 CrossRef CAS.
- W. B. Wu, J. Y. Zhu, Y. Tong, S. F. Xiang and P. Z. Chen, Electronic structural engineering of bimetallic Bi-Cu alloying nanosheet for highly-efficient CO2 electroreduction and Zn-CO2 batteries, Nano Res., 2024, 17, 3684–3692 CrossRef CAS.
- Y. Q. Kang, O. Cretu, J. Kikkawa, K. Kimoto, H. Nara, A. S. Nugraha, H. Kawamoto, M. Eguchi, T. Liao, Z. Q. Sun, T. Asahi and Y. Yamauchi, Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites, Nat. Commun., 2023, 14, 4182 CrossRef CAS PubMed.
- Z. X. Wu, J. G. Deng, Y. X. Liu, S. H. Xie, Y. Jiang, X. T. Zhao, J. Yang, H. Arandiyan, G. S. Guo and H. X. Dai, Three-dimensionally ordered mesoporous Co3O4-supported Au-Pd alloy nanoparticles: high-performance catalysts for methane combustion, J. Catal., 2015, 332, 13–24 CrossRef CAS.
- H. F. Wang, G. H. Lin, X. Q. Li, W. S. Lu and Z. C. Peng, Self-standing hollow porous AuPt nanospheres and their enhanced electrocatalytic performance, J. Colloid Interface Sci., 2019, 554, 396–403 CrossRef CAS PubMed.
- P. Xu, Z. X. Wu, J. G. Deng, Y. X. Liu, S. H. Xie, G. S. Guo and H. X. Dai, Catalytic performance enhancement by alloying Pd with Pt on ordered mesoporous manganese oxide for methane combustion, Chin. J. Catal., 2017, 38(1), 92–105 CrossRef CAS.
- Z. Han, J. Y. Fang, S. H. Xie, J. G. Deng, Y. X. Liu and H. X. Dai, AuRu/meso-Mn2O3: a highly active and stable catalyst for methane combustion, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 359, 012022 Search PubMed.
- X. Zhang, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, X. H. Yu, Z. Han, K. F. Zhang and H. X. Dai, 3DOM CeO2-supported RuyM (M = Au, Pd, Pt) alloy nanoparticles with improved catalytic activity and chlorine-tolerance in trichloroethylene oxidation, Catal. Sci. Technol., 2020, 10(11), 3755–3770 RSC.
- W. B. Pei, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, K. F. Zhang, Z. Q. Hou, Z. Han, A. Rastegarpanah and H. X. Dai, PtRu nanoparticles partially embedded in the 3DOM Ce0.7Zr0.3O2 skeleton: Active and stable catalysts for toluene combustion, J. Catal., 2020, 385, 274–288 CrossRef CAS.
- S. H. Xie, Y. X. Liu, J. G. Deng, X. T. Zhao, J. Yang, K. F. Zhang, Z. Han and H. X. Dai, Three-dimensionally ordered macroporous CeO2-supported Pd@Co nanoparticles: highly active catalysts for methane oxidation, J. Catal., 2016, 342, 17–26 CrossRef CAS.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. N. Xia, Bimetallic nanocrystals: syntheses, properties, and applications, Chem. Rev., 2016, 116(18), 10414–10472 CrossRef CAS PubMed.
- C. L. Chen, M. R. Sun, K. X. Wang and Y. J. Li, Dual-metal single-atomic catalyst: the challenge in synthesis, characterization, and mechanistic investigation for electrocatalysis, SmartMat, 2022, 3(4), 533–564 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys: from theory to applications of alloy clusters and nanoparticles, Chem. Rev., 2008, 108(3), 845–910 CrossRef CAS PubMed.
- D. Skachkov, C. Venkateswara Rao and Y. Ishikawa, Combined first-principles molecular dynamics/density functional theory study of ammonia electrooxidation on Pt (100) electrode, J. Phys. Chem. C, 2013, 117(48), 25451–25466 CrossRef CAS.
- H. H. Ye, Q. X. Wang, M. Catalano, N. Lu, J. Vermeylen, M. J. Kim, Y. Z. Liu, Y. G. Sun and X. H. Xia, Ru nanoframes with an fcc structure and enhanced catalytic properties, Nano Lett., 2016, 16(4), 2812–2817 CrossRef CAS PubMed.
- H. Lv, H. Y. Qin, K. Ariga, Y. Yamauchi and B. Liu, A general concurrent template strategy for ordered mesoporous intermetallic nanoparticles with controllable catalytic performance, Angew. Chem., 2022, 134(17), e202116179 CrossRef.
- H. Lv, Y. Z. Wang, L. Z. Sun, Y. Yamauchi and B. Liu, A general protocol for precise syntheses of ordered mesoporous intermetallic nanoparticles, Nat. Protoc., 2023, 18, 3126–3154 CrossRef CAS.
- Y. Z. Wang, H. Lv, L. Z. Sun, F. R. Jia and B. Liu, Ordered mesoporous intermetallic trimetals for efficient and pH-universal hydrogen evolution electrocatalysis, Adv. Energy Mater., 2022, 12(30), 2201478 CrossRef CAS.
- Y. Z. Wang, X. Y. Zhang, H. J. He, J. J. Chen and B. Liu, Ordered mesoporous high-entropy intermetallics for efficient oxygen reduction electrocatalysis, Adv. Energy Mater., 2024, 14(8), 2303923 CrossRef CAS.
- L. Z. Sun, H. Lv, J. Feng, O. Guselnikova, Y. Z. Wang, Y. Yamauchi and B. Liu, Noble-metal-based hollow mesoporous nanoparticles: synthesis strategies and applications, Adv. Mater., 2022, 34(31), 2201954 CrossRef CAS.
- H. J. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, Shape- and size-controlled synthesis in hard templates: sophisticated chemical reduction for mesoporous monocrystalline platinum nanoparticles, J. Am. Chem. Soc., 2011, 133(37), 14526–14529 CrossRef CAS PubMed.
- J. Y. Chen, B. Wiley, J. McLellan, Y. J. Xiong, Z.-Y. Li and Y. N. Xia, Optical properties of Pd-Ag and Pt-Ag nanoboxes synthesized via galvanic replacement reactions, Nano Lett., 2005, 5(10), 2058–2062 CrossRef CAS PubMed.
- X. M. Lu, H.-Y. Tuan, J. Y. Chen, Z.-Y. Li, B. A. Korgel and Y. N. Xia, Mechanistic studies on the galvanic replacement reaction between multiply twinned particles of Ag and HAuCl4 in an organic medium, J. Am. Chem. Soc., 2007, 129(6), 1733–1742 CrossRef CAS PubMed.
- Q. B. Zhang, J. P. Xie, J. Y. Lee, J. X. Zhang and C. Boothroyd, Synthesis of Ag@AgAu metal core/alloy shell bimetallic nanoparticles with tunable shell compositions by a galvanic replacement reaction, Small, 2008, 4(8), 1067–1071 CrossRef CAS PubMed.
- A. J. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Pérez-Ramírez, Unifying views on catalyst deactivation, Nat. Catal., 2022, 5, 854–866 CrossRef.
- G. H. Kim, K.-D. Jung, W.-I. Kim, B.-H. Um, C.-H. Shin, K. Oh and H. L. Koh, Effect of oxychlorination treatment on the regeneration of Pt-Sn/Al2O3 catalyst for propane dehydrogenation, Res. Chem. Intermed., 2016, 42, 351–365 CrossRef CAS.
- F. Tao and Y. T. Li, A new type of catalysts: catalysts of singly dispersed bimetallic sites, Trends Chem., 2023, 5(6), 486–499 CrossRef CAS.
- P. Buchwalter, J. Rose and P. Braunstein, Multimetallic catalysis based on heterometallic complexes and clusters, Chem. Rev., 2015, 115(1), 28–126 CrossRef CAS.
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction, Chem. Soc. Rev., 2010, 39(6), 2184–2202 RSC.
- X. T. Zhao, Y. X. Liu, J. G. Deng, P. Xu, J. Yang, K. F. Zhang, Z. Han and H. X. Dai, Mesoporous PdxPt alloys: high-performance catalysts for methane combustion, Mol. Catal., 2017, 442, 191–201 CrossRef CAS.
- Z. W. Wang, J. G. Deng, Y. X. Liu, H. G. Yang, S. H. Xie, Z. X. Wu and H. X. Dai, Three-dimensionally ordered macroporous CoCr2O4-supported Au-Pd alloy nanoparticles: highly active catalysts for methane combustion, Catal. Today, 2017, 281, 467–476 CrossRef CAS.
- Y. Wang, H. Arandiyan, J. Scott, M. Akia, H. X. Dai, J. G. Deng, K.-F. Aguey-Zinsou and R. Amal, High performance Au-Pd supported on 3D hybrid strontium-substituted lanthanum manganite perovskite catalyst for methane combustion, ACS Catal., 2016, 6(10), 6935–6947 CrossRef CAS.
- P. Xu, X. Zhang, X. T. Zhao, J. Yang, Z. Q. Hou, L. Bai, H. Q. Chang, Y. X. Liu, J. G. Deng, G. S. Guo, H. X. Dai and C.-T. Au, Preparation, characterization, and catalytic performance of PdPt/3DOM LaMnAl11O19 for the combustion of methane, Appl. Catal., A, 2018, 562, 284–293 CrossRef CAS.
- X. Y. Li, Y. X. Liu, J. G. Deng, S. H. Xie, X. T. Zhao, Y. Zhang, K. F. Zhang, H. Arandiyan, G. S. Guo and H. X. Dai, Enhanced catalytic performance for methane combustion of 3DOM CoFe2O4 by co-loading MnOx and Pd-Pt alloy nanoparticles, Appl. Surf. Sci., 2017, 403, 590–600 CrossRef CAS.
- S. H. Xie, Y. X. Liu, J. G. Deng, X. T. Zhao, J. Yang, K. F. Zhang, Z. Han, H. Arandiyan and H. X. Dai, Effect of transition metal doping on the catalytic performance of Au-Pd/3DOM Mn2O3 for the oxidation of methane and o-xylene, Appl. Catal., B, 2017, 206, 221–232 CrossRef CAS.
- S. H. Xie, Y. X. Liu, J. G. Deng, S. M. Zang, Z. H. Zhang, H. Arandiyan and H. X. Dai, Efficient removal of methane over cobalt-monoxide-doped AuPd nanocatalysts, Environ. Sci. Technol., 2017, 51(4), 2271–2279 CrossRef CAS.
- Z. Han, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, Y. X. Zhang, K. F. Zhang, X. Zhang, Z. Q. Hou, W. B. Pei and H. X. Dai, AuPd/Co3O4/3DOM MnCo2O4: highly active catalysts for methane combustion, Catal. Today, 2021, 376, 134–143 CrossRef CAS.
- Z. Q. Hou, L. Y. Dai, J. G. Deng, G. F. Zhao, L. Jing, Y. S. Wang, X. H. Yu, R. Y. Gao, X. R. Tian, H. X. Dai, D. S. Wang and Y. X. Liu, Electronically engineering water resistance in methane combustion with an atomically dispersed tungsten on PdO catalyst, Angew. Chem., 2022, 134(27), e202201655 CrossRef.
- H. G. Peng, C. Rao, N. Zhang, X. Wang, W. M. Liu, W. T. Mao, L. Han, P. F. Zhang and S. Dai, Confined ultrathin Pd-Ce nanowires with outstanding moisture and SO2 tolerance in methane combustion, Angew. Chem., 2018, 130(29), 8953–8957 CrossRef.
- M. Cargnello, J. J. D. Jaén, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gámez, R. J. Gorte and P. Fornasiero, exceptional activity for methane combustion over modular Pd@CeO2 subunits on functionalized Al2O3, Science, 2012, 337(6095), 713–717 CrossRef CAS.
- S. H. Xie, J. G. Deng, S. M. Zang, H. G. Yang, G. S. Guo, H. Arandiyan and H. X. Dai, Au-Pd/3DOM Co3O4: highly active and stable nanocatalysts for toluene oxidation, J. Catal., 2015, 322, 38–48 CrossRef CAS.
- Y. Feng, L. Wei, Z. W. Wang, Y. X. Liu, H. X. Dai, C. Wang, H.-C. Hsi, E. H. Duan, Y. Peng and J. G. Deng, Boosting catalytic stability for VOCs removal by constructing PtCu alloy structure with superior oxygen activation behavior, J. Hazard. Mater., 2022, 439, 129612 CrossRef CAS PubMed.
- Z. W. Wang, Y. X. Liu, T. Yang, J. G. Deng, S. H. Xie and H. X. Dai, Catalytic performance of cobalt oxide-supported gold-palladium nanocatalysts for the removal of toluene and o-xylene, Chin. J. Catal., 2017, 38(2), 207–216 CrossRef CAS.
- Y. S. Xia, L. Xia, Y. X. Liu, T. Yang, J. G. Deng and H. X. Dai, Concurrent catalytic removal of typical volatile organic compound mixtures over Au-Pd/α-MnO2 nanotubes, J. Environ. Sci., 2018, 64, 276–288 Search PubMed.
- W. Tan, J. G. Deng, S. H. Xie, H. G. Yang, Y. Jiang, G. S. Guo and H. X. Dai, Ce0.6Zr0.3Y0.1O2 nanorod supported gold and palladium alloy nanoparticles: high-performance catalysts for toluene oxidation, Nanoscale, 2015, 7(18), 8510–8523 RSC.
- J. Wang, L. Y. Dai, J. G. Deng, Y. X. Liu, L. Jing, W. B. Pei, Z. Q. Hou, X. Zhang, X. H. Yu and H. X. Dai, Experimental and density functional theory investigations on the oxidation of typical aromatics over the intermetallic compounds-derived AuMn/meso-Fe2O3 catalysts, J. Catal., 2022, 405, 273–287 CrossRef CAS.
- Z. X. Wu, J. G. Deng, S. H. Xie, H. G. Yang, X. T. Zhao, K. F. Zhang, H. X. Lin, H. X. Dai and G. S. Guo, Mesoporous Cr2O3-supported Au-Pd nanoparticles: high-performance catalysts for the oxidation of toluene, Microporous Mesoporous Mater., 2016, 224, 311–322 CrossRef CAS.
- J. J. Sun, Y. X. Liu, J. G. Deng, L. Jing, M. M. Bao, Q. P. Sun, L. L. Li, L. K. Wu, X. Q. Hao and H. X. Dai, PdPty/V2O5-TiO2: highly active catalysts with good moisture- and sulfur dioxide-resistant performance in toluene oxidation, Catalysts, 2022, 12, 1302 CrossRef CAS.
- Z. Q. Hou, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, W. B. Pei, R. Y. Gao, Y. Feng and H. X. Dai, Highly efficient and enhanced sulfur resistance supported bimetallic single-atom palladium-cobalt catalysts for benzene oxidation, Appl. Catal., B, 2021, 285, 119844 CrossRef CAS.
- K. Y. Zhang, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, K. F. Zhang, Z. Q. Hou, X. Zhang, J. Wang, Y. Feng, Y. X. Zhang and H. X. Dai, Insights into the active sites of chlorine-resistant Pt-based bimetallic catalysts for benzene oxidation, Appl. Catal., B, 2020, 279, 119372 CrossRef CAS.
- X. Zhang, Y. X. Liu, J. G. Deng, X. H. Yu, Z. Han, K. F. Zhang and H. X. Dai, Alloying of gold with palladium: an effective strategy to improve catalytic stability and chlorine-tolerance of the 3DOM CeO2-supported catalysts in trichloroethylene combustion, Appl. Catal., B, 2019, 257, 117879 CrossRef CAS.
- X. Zhang, L. Y. Dai, Y. X. Liu, J. G. Deng, L. Jing, Z. W. Wang, W. B. Pei, X. H. Yu, J. Wang and H. X. Dai, Effect of support nature on catalytic activity of the bimetallic RuCo nanoparticles for the oxidative removal of 1,2-dichloroethane, Appl. Catal., B, 2021, 285, 119804 Search PubMed.
- Q. Fu, W. X. Li, Y. X. Yao, H. Y. Liu, H. Y. Su, D. Ma, X. K. Gu, L. M. Chen, Z. Wang, H. Zhang, B. Wang and X. H. Bao, Interface-confined ferrous centers for catalytic oxidation, Science, 2010, 328, 1141–1144 CrossRef CAS PubMed.
- L. N. Cao, W. Liu, Q. Q. Luo, R. T. Yin, B. Wang, J. Weissenrieder, M. Soldemo, H. Yan, Y. Lin, Z. H. Sun, C. Ma, W. H. Zhang, S. Chen, H. W. Wang, Q. Q. Guan, T. Yao, S. Q. Wei, J. L. Yang and J. L. Lu, Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in H2, Nature, 2019, 565, 631–635 CrossRef CAS PubMed.
- X. J. Xu, Q. Fu, M. M. Wei, X. Wu and X. H. Bao, Comparative studies of redox behaviors of Pt-Co/SiO2 and Au-Co/SiO2 catalysts and their activities in CO oxidation, Catal. Sci. Technol., 2014, 4, 3151–3158 RSC.
- C. H. Wu, C. Liu, D. Su, H. L. Xin, H. T. Fang, B. Eren, S. Zhang, C. B. Murray and M. B. Salmeron, Bimetallic synergy in cobalt-palladium nanocatalysts for CO oxidation, Nat. Catal., 2018, 2, 78–85 CrossRef.
- L. Huang, X. Y. Song, Y. Lin, C. Y. Liu, W. X. He, S. Y. Wang, Z. X. Long and Z. H. Sun, In situ observations of the structural dynamics of platinum-cobalt-hydroxide nanocatalysts under CO oxidation, Nanoscale, 2020, 12, 3273–3283 RSC.
- T. Bian, H. Zhang, Y. Y. Jiang, C. H. Jin, J. B. Wu, H. Yang and D. R. Yang, Epitaxial growth of twinned Au-Pt core-shell star-shaped decahedra as highly durable electrocatalysts, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS.
- L. Gao, X. X. Li, Z. Y. Yao, H. J. Bai, Y. F. Lu, C. Ma, S. F. Lu, Z. M. Peng, J. L. Yang, A. L. Pan and H. W. Huang, Unconventional p-d hybridization interaction in PtGa ultrathin nanowires boosts oxygen reduction electrocatalysis, J. Am. Chem. Soc., 2019, 141, 18083–18090 CrossRef CAS.
- Y. X. Wang, L. Cao, N. J. Libretto, X. Li, C. Y. Li, Y. L. Wan, C. N. He, J. S. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, Ensemble effect in bimetallic electrocatalysts for CO2 reduction, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS.
- T. Bian, B. B. Xiao, B. Sun, L. Huang, S. Su, Y. Jiang, J. K. Xiao, A. H. Yuan, H. Zhang and D. R. Yang, Local epitaxial growth of Au-Rh core-shell star-shaped decahedra: a case for studying electronic and ensemble effects in hydrogen evolution reaction, Appl. Catal., B, 2020, 263, 118255 CrossRef CAS.
- Z. Q. Lin, W. L. Chen, Y. Jiang, T. Bian, H. Zhang, J. B. Wu, Y. Wang and D. R. Yang, Facile synthesis of Ru-decorated Pt cubes and icosahedra as highly active electrocatalysts for methanol oxidation, Nanoscale, 2016, 8, 12812–12818 RSC.
- Y. Y. Jiang, T. Bian, F. Lin, H. Zhang, C. H. Jin, Z. Y. Li, D. R. Yang and Z. Zhang, Revealing the elemental-specific growth dynamics of Pt-Cu multipods by scanning transmission electron microscopy and chemical mapping, J. Mater. Chem. A, 2015, 3, 21284–21289 RSC.
- Y. J. Liang, Y. X. Liu, J. G. Deng, K. F. Zhang, Z. Q. Hou, X. T. Zhao, X. Zhang, K. Y. Zhang, R. J. Wei and H. X. Dai, Coupled palladium–tungsten bimetallic nanosheets/TiO2 hybrids with enhanced catalytic activity and stability for the oxidative removal of benzene, Environ. Sci. Technol., 2019, 53, 5926–5935 CrossRef CAS.
- Y. Feng, L. Wei, Y. X. Liu, H. X. Dai, Z. X. Zhao and J. G. Deng, Rapid supplement of active oxygen by constructing Pt-Fe alloy structure to improve catalytic stability for furniture paints industry VOCs removal, Sep. Purif. Technol., 2023, 324, 124621 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |