
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported ruthenium catalysts for the transformation of aqueous glycerol to hydrogen gas and lactic acid†
Ankit
Kumar
a,
Bhanu
Priya
a,
Rohit Kumar
Rai
b,
Parveen
Garg
c,
Uday
Deshpande
c and
Sanjay Kumar
Singh
 *a
*a
aCatalysis Group, Department of Chemistry, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, MP, India. E-mail: sksingh@iiti.ac.in
bKAUST Catalysis Center and Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia
cUGC-DAE Consortium for Scientific Research, Indore 452001, MP, India
First published on 6th November 2024
Abstract
Glycerol (GLY) is an attractive biobased platform chemical that produces valuable fine chemicals with a wide range of industrial applicability and has the potential to produce high-purity H2 gas. Herein, we established an efficient method for selective production of H2 gas and lactic acid (LA) from aqueous glycerol under mild reaction conditions (90–130 °C) over various supported ruthenium catalysts. Notably, we achieved a substantial yield of H2 gas (n(H2)/n(GLY) ratio of 1.4 with >99.9% H2 purity) and LA (86%) from glycerol over Ru nanoparticles immobilized over a La(OH)3 support (Ru/La(OH)3) in contrast to bare Ru nanoparticles where we observed a n(H2)/n(GLY) ratio of 1.6 with only 70% yield of LA as we reported previously. We could significantly boost the generation of both H2 gas and LA by tuning the reaction parameters, including reaction time, temperature, base, and water concentrations. Furthermore, the effect of various support materials such as Mg(OH)2, ZnO, ZrO2, and TiO2 was also tested for H2 production from GLY under optimized reaction conditions. The employment of various characterization techniques to understand the physicochemical properties of the synthesized supported Ru catalysts revealed that the choice of support material significantly influenced the catalytic activity towards the selective production of H2 and LA.
1. Introduction
In the recent past, there have been considerable efforts in exploring sustainable and environmentally benign energy resources to meet the ever-increasing global energy demand driven by population growth and industrialization.1,2 In this regard, hydrogen (H2), a sustainable energy carrier with high energy density (120 MJ kg−1), has garnered significant attention and is widely regarded as the most promising solution to address these energy crisis and climate change issues.3–6 The low volumetric energy density, high diffusivity, and flammability of hydrogen gas make it challenging to store or transport, one of the biggest hurdles to its extensive applications. Despite these hurdles, hydrogen gas is being produced at the industrial scale through various processes such as reforming of methane,7 aqueous phase reforming (APR),8 and steam reforming (SR) of non-renewable resources.9 Notably, these processes are very energy-intensive and emit greenhouse gases along with H2 gas. On the other hand, there are several liquid organic molecules such as methanol,10–13 ethanol,14–17 formic acid,18–21 formaldehyde,21–23 and other polyols24–27 showing high potential to store appreciably high volumetric and gravimetric content of H2, which can be released in the presence of a suitable catalyst. Advantageously, being liquid, these enable the easy storage and transportation of H2 gas.According to the Organization for Economic Co-operation and Development (OECD) and the Food and Agricultural Organization (FAO), global biodiesel production is projected to increase to 9.5 billion litres by 2030.28 The major by-product of the transesterification of triglycerides is glycerol (GLY), formed in abundance (10 wt% of biodiesel) during biodiesel production.29 Due to its non-toxicity and ability to be transformed into several valuable platform chemicals, including lactic acid (LA), 1,2-propanediol (PD), ethylene glycol (EG), glycolic acid (GA) and so on, glycerol (GLY) has emerged as a versatile bio-platform substrate.29 Furthermore, LA is one of the important products of GLY transformation, which finds vast application in industries. Notably, LA can be produced from GLY either by a catalytic oxidation process or an acceptor-less dehydrogenation process. Dehydrogenation of GLY to LA is considered as an attractive route for the simultaneous production of H2 gas along with LA. Hence, GLY, a waste product in biodiesel industries, can be explored as a promising resource for hydrogen production.
Catalytic transformation of glycerol via steam reforming (SR) or aqueous-phase reforming (APR) has been extensively explored for H2 production.30,31 Several catalysts such as Pt/SiO2,32 Ru/Y2O3,33 Ru/Al2O3,34–36 Ni/M (M = CeO2, MgO, TiO2),37 Ce-Sm-5Cu,38 Ni/La/Co/Al2O3,39 Co/MgO–La2O3,40 and others have been explored to produce H2 gas via an SR process albeit at high temperatures (>250 °C). On the other hand, the APR process works at relatively lower temperatures than the SR process. For the APR of GLY to H2 gas, a plethora of catalysts, including Ru/NaY (NaY – sodium zeolite),26 Ru-NMC-3 (NMC – N-doped mesoporous carbon),41 RuPt-NMC-3,41 PtMo/C,42 PtRe/C,43 Pt/Al2O3,44 Pt-KHT/28 (KHT – K-promoted hydrotalcite),45 Pt–Cu/Mg(Al)O,46 PtFe/γ-Al2O3,47 Ni/Al2O3–La2O3,48 and others have been investigated at 200–250 °C. Dumesic and co-workers achieved a breakthrough for the generation of H2 gas from biomass-derived compounds such as glucose, sorbitol, glycerol, ethylene glycol and methanol through APR over a Pt/Al2O3 catalyst.44 They discovered that when the reaction temperature and pressure were increased from 225 °C and 29 bar to 265 °C and 56 bar, the H2 selectivity decreased from 75% to 51%, while the selectivity for alkane products increased from 19% to 34% in the case of GLY. Although the reforming of GLY has resulted in the generation of H2 gas, the purity of H2 gas is compromised due to the co-production of other gases (CO, CO2, and CH4) following a series of uncontrolled pathways such as C3H8O3 → 4H2 + 3CO; CO + H2O → CO2 + H2; CO + 3H2 → CH4 + H2O; CO2 + 4H2 → CH4 + 2H2O.
Notably, the literature reveals that most of the catalytic transformations of aqueous GLY to H2 gas have been performed at high temperatures (200–900 °C), while LA production from GLY is performed at lower temperatures (<200 °C) (Table S1, ESI†). For instance, Raja et al. explored different metal loadings of Ru over a NaY support (2–5 wt% Ru–NaY) for the APR of GLY and achieved conv. up to 88% and 74% selectivity for H2 gas with contamination of other gases (24%) at 250 °C under a continuous flow of N2 gas (40 bar) over a 3 wt% Ru–NaY catalyst. They observed that the catalytic activity in APR of GLY was significantly improved by examining the structure–activity relationship between Ru and the NaY support, metal loading and dispersion of Ru metal.26 In a similar way, they also developed Ru-based catalysts (Ru-NMC-3 and RuPt-NMC-3) for APR of GLY and afforded 92% conversion with 88.5% H2 selectivity at 250 °C.37 On the other hand, researchers have also delved into the steam reforming of glycerol to generate hydrogen gas. For instance, Hirai et al. developed a Ru/Y2O3 catalyst and observed complete conversion of GLY with 90% H2 yield at 600 °C along with other gases (CO, CO2 and CH4).33 Adhikari et al. explored a Ru/Al2O3 catalyst for the SR of GLY at 900 °C and observed only 58% conversion of GLY with 42% yield of H2 gas.34 Vaidya et al. studied the kinetics of steam reforming of GLY over the Ru/Al2O3 catalyst in the temperature range of 350–500 °C. They performed a reaction at 500 °C for 2 h and obtained 50% GLY conversion with the H2 yield of 58.3% with co-production of CO (7.2%), CO2 (34.2%), and CH4 (0.3%).35 In a similar direction, Kousi et al. explored modified Ru/Al2O3 catalysts for GLY reforming and observed 93%, 85%, and 57% conversion of GLY over Ru/Al2O3, Ru/B2O3–Al2O3 and Ru/MgO–Al2O3 catalysts at 600 °C, respectively, with the H2 yield of ∼68% along with other gases (CO, CO2 and CH4).36 It is worth noting that a major drawback of steam reforming is the production of impure hydrogen, necessitating additional steps for purification. Even though the dehydrogenative method for converting GLY to LA is an appealing and sustainable approach to generate both H2 gas and LA, most of the research focused on the oxidative conversion of GLY to LA employing Cu, Ni, Au, or Pd-based catalysts.49–60 During the former process, excessive oxidation of glyceraldehyde produces glyceric acid, tartaric acid, and glycolic acid (GA) along with LA, which dramatically decreases the selectivity of the desired LA product (Schemes S1 and S2, ESI†).
In the literature, some non-noble metal-based catalysts such as Cu,49 Cu2O,50 CuO/ZrO2,51 CuO/CeO2,52 CuO/Al2O3,53 Cu–Cu2O@NC (NC – N-doped carbon),54 Co3O4/CeO2,55 CoCl2,56 and Ni/HAP (HAP – hydroxyapatite),57 have also been explored for the conversion of GLY to LA. Moreover, noble metal-based supported catalysts such as AuCu/CeO2,58 M/CeO2 (M = Au, Pt),59 Pd/HAP,61 M/ZnO62 (M = Pt, Pd, Rh, and Au), Pt/C,63 Pt/carbon material,64 Pt/L-Nb2O5,65 Pt/support (ZrO2, TiO2, C),66 Pt/ZrO2,67 Ru–Zn–Cu(I)/HAP,68 and Au/HAP/BN (BN – boron nitride),69 were primarily developed for the transformation of GLY to LA at higher temperature (>160 °C) under a gaseous atmosphere (He, N2 or ethylene). For instance, Wang et al. reported the hydrothermal conversion of GLY to LA over a Pd/HAP catalyst at 230 °C using NaOH (1.1 equiv.) and exhibited complete conversion of GLY with 95% selectivity of LA in 1.5 h.61 Similarly, Urbano et al. studied different noble metals (Pt, Pd, Rh Au) supported over ZnO under H2 or He pressure (20 bar) at 180 °C in an alkaline medium and achieved complete conversion of glycerol but with lower selectivity of LA (68%) over Rh/ZnO.62 Several catalysts documented in the literature require an inert atmosphere for achieving a high yield of lactic acid (LA) and primarily focus on the conversion of GLY only to LA, with limited reports on the generation of hydrogen gas.63–66 In the search for efficient catalytic systems for the production of H2 gas, Pescarmona et al. investigated a Pt/ZrO2 catalyst to facilitate a one-pot transfer hydrogenation reaction between glycerol and cyclohexene to produce LA (95%) and cyclohexane without additional H2 gas at 160 °C in 4.5 h under a N2 atmosphere (20 bar), which clearly indicates that GLY has the potential to be explored as a hydrogen storage material.67 Han et al. developed a Ru–Zn–Cu(I)/HAP catalyst for the transformation of GLY to LA at 140 °C and achieved >99% GLY conversion with the LA yield of 70.9% in 24 h. However, when they used a Ru/HAP catalyst, they achieved 63.7% yield of LA with complete conversion of GLY and 13.7% selectivity for methane gas in 12 h.68 A few Ru-based molecular catalysts have also been explored for the catalytic transformation of GLY to LA.70,71 Beller et al. reported a PNP-Ru catalyst and achieved 67% LA yield at 140 °C over 24 h using diglyme or NMP/water as a solvent with the evolution of CO and CO2 gases along with H2 gas.70 Similarly, Kumar et al. explored an NNN pincer Ru complex for the catalytic conversion of GLY to LA and achieved 90% yield of LA with complete conversion of GLY using ethanol as a solvent at 140 °C in 48 h. However, when the reaction was carried out using water as a solvent, they observed only a 40% yield of LA with 44% GLY conversion.71 Recently, we also reported selective hydrogen and LA production from GLY using NaOH over bare ruthenium nanoparticles in water, where we achieved complete conversion of GLY in 10 h with 1.6 equiv. H2 gas and 70% yield of LA at 110 °C.25 It is evident from the literature reports that most of the catalytic systems are active at high temperature and pressure conditions. Moreover, the purity of hydrogen gas is also one of the major issues as contaminants such as CO, CO2, and CH4 were also produced along with H2 gas.
In this regard, herein, we synthesized ruthenium nanoparticles immobilized over various supports (La(OH)3, Mg(OH)2, ZnO, ZrO2, and TiO2) and explored them for a sustainable, efficient, and low-temperature approach for the selective production of high-purity H2 gas along with high yield of LA from aqueous glycerol (GLY). Controlled experiments were performed to elucidate the plausible dehydrogenation pathway of GLY to H2 and LA. The developed supported ruthenium catalysts showed high activity for large scale generation of H2 gas from GLY with a productivity of 12 L H2/gRu/h at 130 °C. Additionally, the developed process was extended for H2 gas production from various other terminal diols such as ethylene glycol (EG), 1,3-propanediol (PDO), 1,4-butanediol (BDO), 1,5-pentanediol (PO) and 1,6-hexanediol (HDO).
2. Results and discussion
2.1. Synthesis, characterization, and screening of Ru catalysts for the transformation of GLY to H2 gas and SL
Supported metal catalysts have more advantages in terms of high dispersion, catalytic activity, stability, and reusability than unsupported metal catalysts.72 Metal hydroxides and metal oxides are an important class of support materials having notable properties such as high surface area and high thermal stability.73 Ru nanoparticles stabilized over different metal oxides, such as ZrO2, Nb2O5, CeO2, Al2O3, ZnO, etc., have been explored extensively for the transformation of GLY to lactic acid (LA).51–53,62,65 Therefore, at the outset, we synthesized various Ru nanoparticles immobilized over different basic (La(OH)3, Ru/Mg(OH)2), amphoteric (ZnO, ZrO2), and acidic (TiO2) supports using a wet-impregnation method followed by NaBH4 induced reduction. The P-XRD patterns of the synthesized Ru catalysts showed dominant peaks corresponding to the support material only, while no peaks corresponding to Ru were observed, suggesting that Ru nanoparticles are well dispersed over the support materials (Fig. S1, ESI†). The FE-SEM images of Ru catalysts showed no visible Ru nanoparticles due to their small particle size. However, EDX and elemental mapping evidenced the presence of Ru in all the supported catalysts (Fig. S2–S12, ESI†).We further screened these catalysts for the dehydrogenation of GLY in water (n(GLY)/n(H2O) of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) using NaOH (2.0 equiv.) at 130 °C (Table 1 and Fig. S13, S14, ESI†). Over the Ru/La(OH)3 catalyst having 9 wt% Ru loading, high catalytic activity was achieved with the complete conversion of GLY and evolution of 470 mL H2 gas (96% yield of H2, TON 152, and TOF 105 h−1) with 86% yield of sodium lactate (SL) in 4.5 h (Table 1, entry 1). Ru/Mg(OH)2, having Ru loading of 7.5 wt%, also showed appreciably good catalytic activity with the release of 402 mL of H2 gas (97% yield of H2, TON 146, and TOF 60 h−1) along with GLY conversion of 80% and 59% yield of SL in 4.5 h (Table 1, entry 2). Ru/ZnO having Ru loading of 8.2 wt% showed 70% conversion of GLY with 290 mL of H2 (92% yield of H2, TON 116, and TOF 40 h−1) and 61% yield of SL in 4.5 h (Table 1, entry 3). On the other hand, Ru/ZrO2 and Ru/TiO2, having metal loadings 8.3 and 8.6 wt%, showed 67% (TON 110 and TOF 59 h−1) and 62% conversion (TON 98 and TOF 62 h−1) with SL yield of 52% and 51%, respectively in 4.5 h (Table 1, entries 4 and 5).
:3) using NaOH (2.0 equiv.) at 130 °C (Table 1 and Fig. S13, S14, ESI†). Over the Ru/La(OH)3 catalyst having 9 wt% Ru loading, high catalytic activity was achieved with the complete conversion of GLY and evolution of 470 mL H2 gas (96% yield of H2, TON 152, and TOF 105 h−1) with 86% yield of sodium lactate (SL) in 4.5 h (Table 1, entry 1). Ru/Mg(OH)2, having Ru loading of 7.5 wt%, also showed appreciably good catalytic activity with the release of 402 mL of H2 gas (97% yield of H2, TON 146, and TOF 60 h−1) along with GLY conversion of 80% and 59% yield of SL in 4.5 h (Table 1, entry 2). Ru/ZnO having Ru loading of 8.2 wt% showed 70% conversion of GLY with 290 mL of H2 (92% yield of H2, TON 116, and TOF 40 h−1) and 61% yield of SL in 4.5 h (Table 1, entry 3). On the other hand, Ru/ZrO2 and Ru/TiO2, having metal loadings 8.3 and 8.6 wt%, showed 67% (TON 110 and TOF 59 h−1) and 62% conversion (TON 98 and TOF 62 h−1) with SL yield of 52% and 51%, respectively in 4.5 h (Table 1, entries 4 and 5).
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | H2 gasb (mL) | n(H2)/n(GLY) | Conv. (%) | Yield of productsc (% C) | CBd (%) | H2 yielde (%) | TON/TOFf (h−1) | |||
| SL (Sel.%) | SG | PD | SF | ||||||||
| a Reaction conditions: Ru/support (100 mg), GLY (13.68 mmol), NaOH (2.0 equiv.), water (41.04 mmol), 130 °C and 600 rpm. b Volume of gas was measured using the water displacement method. c Yield (at 4.5 h) was calculated by 1H NMR using sodium acetate as an internal standard. d CB is carbon balance based on the yield and conversion at 4.5 h. e H2 yield based on CB. f Turnover frequency was calculated based on the volume of H2 gas released in the initial 1 h; TOF = TON/t. SL (sodium lactate), SG (sodium glycolate), PD (1,2-propanediol), and SF (sodium formate). g In parentheses, gas evolved and conv.% in 7 h. h 8 h. i 8 h. j 7 h. k 6.5 h. n.r. (no reaction). | |||||||||||
| 1 | Ru/La(OH)3 | 470 | 1.4 | >99 | 86 (86) | — | — | 12 | 98 | 96 | 152/105 |
| 2 | Ru/Mg(OH)2 | 402 (522)g | 1.2 | 80 (>99)g | 59 (74) | 1 | — | 16 | 96 | 97 | 146/60 |
| 3 | Ru/ZnO | 290 (450)h | 0.9 | 70 (>99)h | 61 (87) | 1 | — | 6 | 98 | 92 | 116/40 |
| 4 | Ru/ZrO2 | 290 (490)i | 0.9 | 67 (>99)i | 52 (78) | 2 | — | 9 | 96 | 96 | 110/59 |
| 5 | Ru/TiO2 | 254 (340)j | 0.8 | 62 (88)j | 51 (82) | 2 | — | 6 | 97 | 92 | 98/62 |
| 6 | La(OH)3 | — | — | n.r. | — | — | — | — | — | — | — |
| 7 | Ru | 478 (534)k | 1.4 | 85 (>99)k | 63 (74) | — | — | 20 | 98 | 99 | —/82 |

Notably, the reaction could not proceed if only La(OH)3 was used (Table 1, entry 6), while with bare Ru nanoparticles, 85% GLY conversion with the evolution of 478 mL H2 gas (99% yield of H2, TOF 82 h−1) along with 63% SL and 20% sodium formate (SF) in 4.5 h was observed (Table 1, entry 7). The observed enhanced yield for SL (86%) over the Ru/La(OH)3 catalyst as compared to bare Ru nanoparticles (63% yield of SL) indicates the prominent effect of the support in tuning the catalytic activity. Previously also, we observed a lower SL yield (70%) over bare Ru nanoparticles using NaOH (2.0 equiv.) in 10 h at 110 °C with 1.6 equiv. of H2 gas.25 Moreover, the complete conversion of GLY to H2 gas and SL over Ru/Mg(OH)2, Ru/ZnO and Ru/ZrO2 catalysts took a longer time (7–8 h) at 130 °C (Table 1), as compared to the Ru/La(OH)3. The results indicated that Ru/La(OH)3 performed well over other supported ruthenium catalysts in terms of the high yield of H2 and SL. Though the metal loading, metal particle size, metal dispersion and metal active sites may influence the catalytic activity significantly, the higher catalytic activity of Ru/La(OH)3 could be primarily attributed to the strongly basic nature of the support with a higher surface area than the others. Notably, TEM imaging showed that all the supported ruthenium catalysts have analogous particle sizes ranging from 1.5 nm (for Ru/La(OH)3, Ru/ZnO and Ru/TiO2) to 1.8 nm (for Ru/ZrO2) (Fig. 1 and Table S2, ESI†). Moreover, Ru/La(OH)3, Ru/ZnO, and Ru/TiO2 displayed a metal dispersion of 87%, as compared to 76% for Ru/Mg(OH)2 and 72% for Ru/ZrO2. Notably, metal dispersion refers to the particle size of Ru nanoparticles, as estimated by TEM. However, to further distinguish the Ru particles from the La(OH)3 support, we performed HAADF-STEM and EDS mapping analysis (Fig. 1 and Fig. S3, ESI†). In the HAADF-STEM images, we can clearly see brighter Ru nanoparticles on the support. The difference between the ruthenium nanoparticles and the La(OH)3 support material was validated by comparing the TEM image of the Ru/La(OH)3 catalyst with the La(OH)3 support alone (Fig. S15, ESI†). The observed lattice parameter of 0.318 nm for both La(OH)3 and Ru/La(OH)3 further verifies the presence of the La(OH)3 support, which is consistent with the literature reports.74,75 Moreover, the SAED pattern of the La(OH)3 (Fig. S15, ESI†) depicts the polycrystalline nature of the La(OH)3 support.74,75 The EDS and elemental mapping further confirmed the elemental distribution of Ru and La within the sample. Furthermore, the Brunauer–Emmett–Teller (BET) surface area, as determined by N2 adsorption–desorption isotherms, of Ru/Mg(OH)2 and Ru/La(OH)3 are 172 m2 g−1 and 81 m2 g−1, respectively as compared to Ru/ZnO (30 m2 g−1), Ru/ZrO2 (30 m2 g−1), and Ru/TiO2 (27 m2 g−1) (Fig. S16 and Table S2, ESI†). Interestingly, Ru/La(OH)3 and Ru/TiO2 have analogous metal loading, metal particle size (1.5 nm), metal dispersion (87%), and surface metal active sites, but they exhibited contrasting catalytic performance, where complete conversion of GLY was achieved over Ru/La(OH)3 in 4.5 h, while Ru/TiO2 exhibited only 67% conversion under analogous conditions (Table 1, Fig. S13 and Table S2, ESI†). Furthermore, as revealed by CO2 TPD experiments, Ru/La(OH)3 and Ru/Mg(OH)2 exhibited CO2 desorption peaks at higher temperatures corresponding to moderate to strong basic sites (Fig. S17, ESI†). Generally, desorption peaks at low (<300 °C), medium (300–550 °C), and high temperature (>550 °C) can be ascribed to the presence of weak, moderate, and strong basic sites, respectively.76 In contrast, Ru/ZrO2 and Ru/TiO2 only exhibited a peak below 300 °C, indicating weak basic sites, while Ru/ZnO showed a peak with the lowest intensity, indicating very few basic sites (Fig. S17, ESI†).
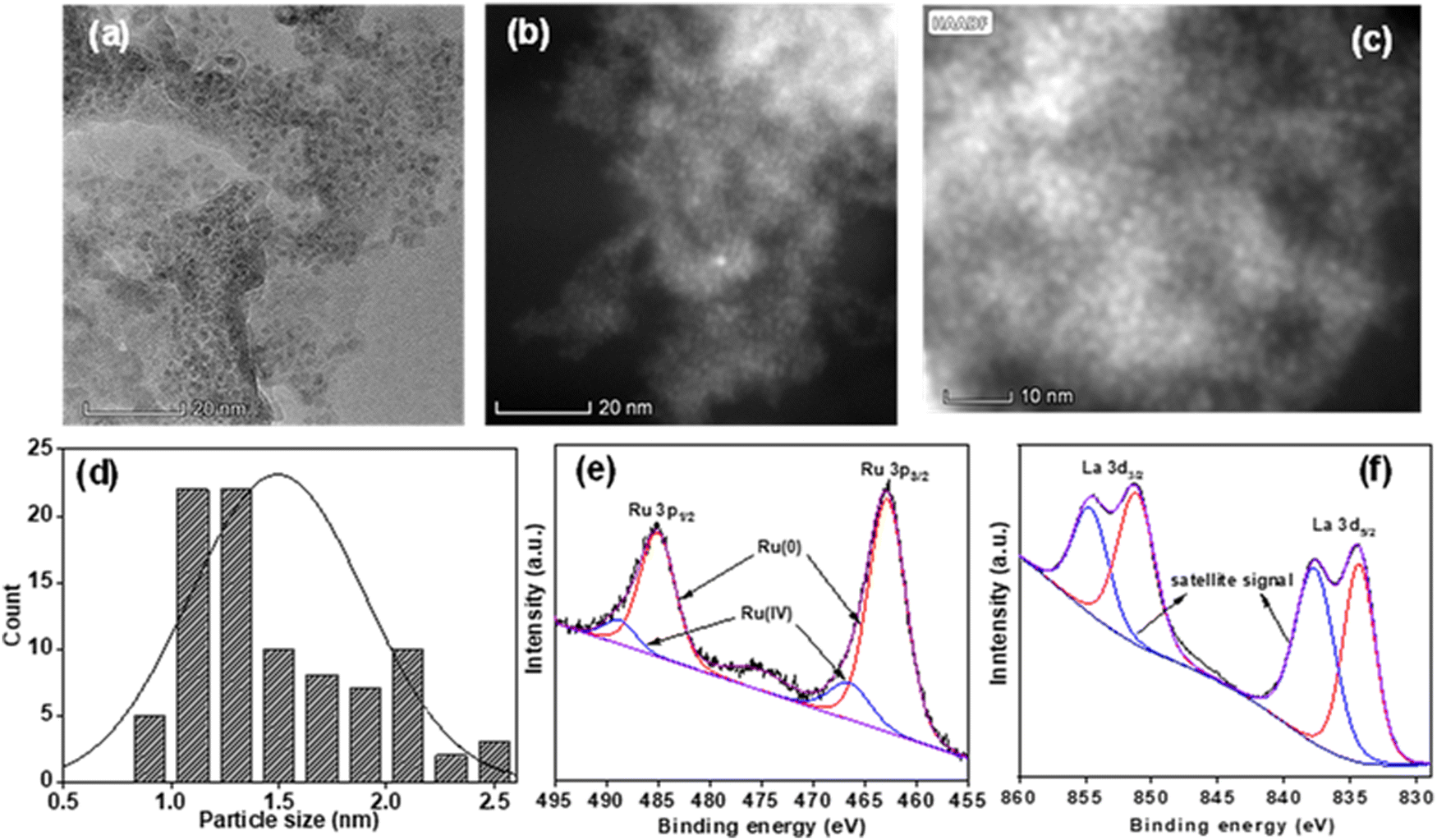 | ||
| Fig. 1 (a) HR-TEM, (b) and (c) HAADF-STEM images, and (d) particle size distribution curve. XPS spectra of the (e) Ru 3p region, and (f) La 3d region for the Ru/La(OH)3 catalyst. | ||
The wide scan XPS of the Ru/La(OH)3 catalyst also confirmed the presence of all elements, i.e., Ru, La, and O (Fig. S18, ESI†), which is consistent with the EDS data of the Ru/La(OH)3 catalyst (Fig. S2, ESI†). Deconvolution of the XPS data showed the peaks at binding energies of 462.9 eV and 485.2 eV for the Ru 3p3/2 and Ru 3p1/2, respectively, corresponding to the Ru(0) state (Fig. 1(e)). The observed binding energy values for Ru(0) in Ru/La(OH)3 are consistent with the available literature reported values for Ru(0) nanoparticles.77–81 Nevertheless, these binding energies exhibit a slight positive shift compared to those typically reported for bare Ru nanoparticles, suggesting that the Ru(0) is slightly electron-deficient in the sample Ru/La(OH)3. The observed change in binding energy can likely be attributed to the interaction between Ru nanoparticles and the La(OH)3 support.77–81 Accordingly, XPS peaks for Ru(0) 3d5/2 and Ru(0) 3d3/2 were found at 280.9 eV and 285.1 eV, respectively, where the peak corresponding to Ru 3d3/2 overlapped significantly with the peak of C 1s (284.6 eV) in the Ru/La(OH)3 catalyst (Fig. S19, ESI†). For La 3d, the deconvolution of peaks resulted in doublets at 834.6 eV and 851.3 eV, corresponding to La(III) 3d5/2 and La(III) 3d3/2, respectively. Additionally, the peaks at 837.9 eV and 854.6 eV are satellite peaks due to the shake-up processes (Fig. 1(f)). Spin–orbit coupling of 16.7 eV was observed, confirming the +3 oxidation state of La.82,83 In the O 1s spectra, two peaks centered at 530.4 eV and 532.5 eV were observed for oxygen bonded to La i.e. La–O, and adsorbed water molecules/hydroxyl groups present on the catalyst surface, respectively (Fig. S20, ESI†). Hence, metal–support interactions play a crucial role in achieving enhanced catalytic activity for GLY dehydrogenation over the Ru/La(OH)3 catalyst. As the first step in the catalytic GLY, dehydrogenation is the activation of the O–H bond via proton abstraction and alkoxide species formation over the catalyst surface. It can be facilitated by either a homogeneous base or the basic sites present on the catalyst.60,84 Advantageously, the observed Ru to La(OH)3 electronic interaction may facilitate the crucial hydride abstraction step forming glyceraldehyde and H2. Glyceraldehyde can then further undergo base-catalyzed reactions to produce lactate. However, an excess of the base can also catalyze various side reactions, such as C–C cleavage, leading to the production of formate and increasing the yield of H2 gas.84 Notably, Mg(OH)2 is sparingly soluble in aqueous solution at higher temperatures, further increasing the base concentration and, thereby, C–C cleavage reaction due to which, higher volume of H2 gas but poor lactate selectivity over the Ru/Mg(OH)2 catalyst were observed. In contrast, the weakly or poorly basic sites in Ru/ZnO, Ru/ZrO2, and Ru/TiO2 can be correlated with their lower catalytic activities. Moreover, the high selectivity (87%) towards SL in the case of Ru/ZnO could be attributed to the presence of Zn2+, which favoured the 1,2-hydride shift of pyruvaldehyde to SL compared to the in situ hydrogenation of pyruvaldehyde to PD (1,2-PDO).68 These results are consistent with previously reported systems for APR of glycerol.85
2.2. Catalytic transformation of GLY to H2 and SL over the Ru/La(OH)3 catalyst
Notably, the dehydrogenation of glycerol (GLY) was conducted without NaOH (with catalyst) and without catalyst (with NaOH) at 130 °C, where no conversion of GLY, in either case, was observed (Table 2, entries 1 and 2), suggesting the crucial role of base and the catalyst for the dehydrogenation of GLY. Reaction with neat GLY over the Ru/La(OH)3 showed 70% conversion of GLY with the release of 260 mL (92% yield of H2) [(n(H2)/n(GLY) ∼ 0.8)] of H2 in 8 h (Table 2, entry 3). GC-TCD of the released gas confirmed the presence of only H2 gas without any other gaseous contamination (CO, CO2, CH4 or alkanes), confirming the selective production of H2 from GLY (Fig. 2 and Fig. S21, ESI†).|
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | n(GLY)/n(H2O) | NaOH (equiv.) | t (h) | H2 gasb (mL) | n(H2)/n(GLY) | Conv. (%) | Yield of productsc (% C) | CBd (%) | H2 yielde (%) | TOFf (h−1) | |||
| SL (Sel.%) | SG | PD | SF | ||||||||||
| a Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (13.68 mmol), NaOH (27.36 mmol), water (0–20 equiv.), 130 °C, and 600 rpm. b Volume of gas was measured by the water displacement method. c Yield was calculated by 1H NMR using sodium acetate as an internal standard. d CB is carbon balance. e H2 yield based on CB. f Turnover frequency (TOF) was calculated based on the volume of H2 gas released in the initial 1 h; TOF = TON/t. g Reaction in the absence of a catalyst. The results reported are the average of at least two repeated reactions. SL (sodium lactate), SG (sodium glycolate), PD (1,2-propanediol), SF (sodium formate) and n.r. (no reaction). | |||||||||||||
| 1 | 1:3 |
— | 4.5 | — | — | n.r. | — | — | — | — | — | — | — |
| 2g | 1:3 |
2.0 | 4.5 | — | — | n.r. | — | — | — | — | — | — | — |
| 3 | 1:0 |
2.0 | 8 | 260 | 0.8 | 70 | 51 (73) | 3 | 3 | 9 | 96 | 92 | 33 |
| 4 | 1:1 |
2.0 | 6 | 446 | 1.3 | 95 | 77 (81) | <1 | <1 | 14 | 97 | 98 | 82 |
| 5 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :3 :3
|
2.0 | 4.5 | 470 | 1.4 | >99 | 86 (86) | — | — | 12 | 98 | 96 | 105 |
| 6 | 1:6 |
2.0 | 5.5 | 456 | 1.3 | 93 | 76 (82) | 3 | 1 | 10 | 97 | 92 | 97 |
| 7 | 1:10 |
2.0 | 6.5 | 386 | 1.1 | 81 | 66 (81) | 3 | 1 | 7 | 96 | 87 | 67 |
| 8 | 1:20 |
2.0 | 6.5 | 156 | 0.5 | 69 | 47 (68) | 9 | 1 | 3 | 91 | 88 | 38 |
| 9 | 1:3 |
0.5 | 3.5 | 138 | 0.4 | 52 | 37 (71) | 1 | 10 | 1 | 97 | 90 | 36 |
| 10 | 1:3 |
1.0 | 4 | 270 | 0.8 | 85 | 70 (82) | 2 | 4 | 5 | 96 | 92 | 68 |
| 11 | 1:3 |
1.5 | 5 | 356 | 1.0 | 93 | 79 (85) | 1 | 1 | 8 | 96 | 92 | 85 |
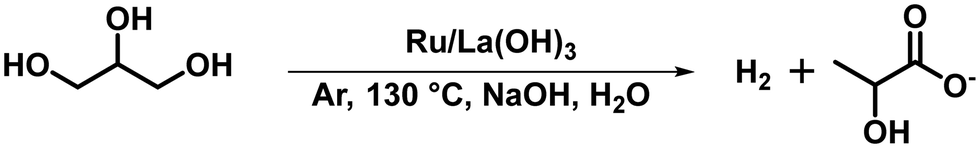
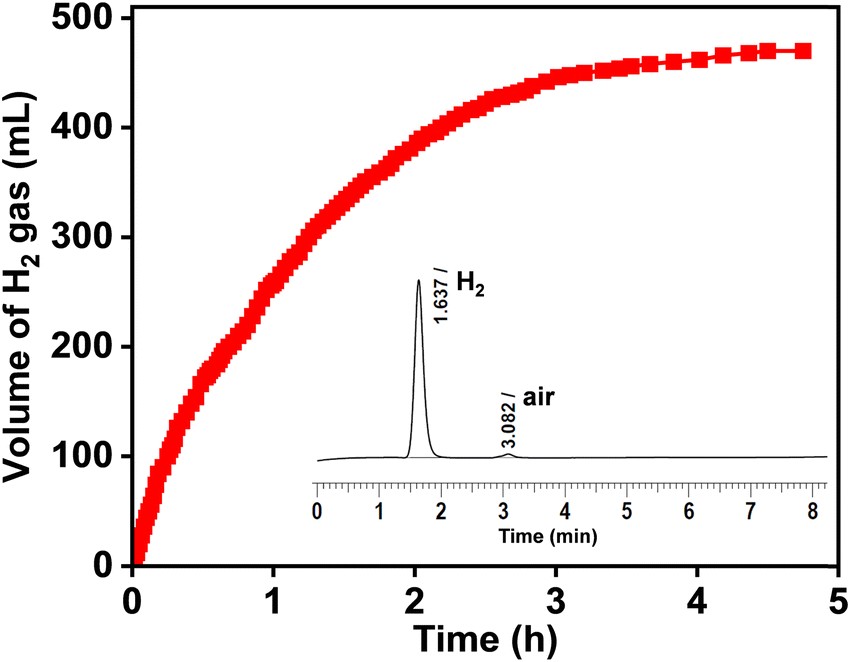 | ||
| Fig. 2 Time course plot for hydrogen gas production from GLY over the Ru/La(OH)3 catalyst. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (13.68 mmol), NaOH (27.36 mmol), water (41.04 mmol), 130 °C, and 600 rpm. | ||
The conversion of GLY was enhanced to be 95% when one equivalent of water was added to the reaction, releasing 446 mL (98% yield of H2) [(n(H2)/n(GLY) ∼ 1.3)] of gas in 6 h (Table 2 entry 4), indicating the crucial role of water in the H2 production from GLY. On further increase in the water content (GLY/H2O molar ratio of 1:3), enhanced conversion of GLY (>99%) was achieved with the release of 470 mL (96% yield of H2) [(n(H2)/n(GLY) ∼ 1.4)] of gas in 4.5 h with appreciably good yields of SL (86%) (Table 2, entry 5). Since water improves the solubility of NaOH, it increases the mass transfer of NaOH and decreases the viscosity of GLY and therefore, enhanced catalytic transformation of GLY to H2 and SL was achieved in the presence of water.86 However, upon the further increase in the water content, the amount of H2 gas was decreased to 456 mL (97% yield of H2) (93% GLY conversion) and 386 mL (87% yield of H2) (81% GLY conversion) for the n(GLY)/n(H2O) ratio of 1:6 and 1:10, respectively (Table 2, entries 6 and 7). For the more diluted aqueous solution of GLY (n(GLY)/n(H2O) ratio of 1:20), lower GLY conversion (69%) with the release of only 156 mL (88% yield of H2) of H2 gas was observed (Table 2, entry 8). Increasing the glycerol:water ratio to 1:6 or more, resulted in a decrease in the molar concentration of GLY in the reaction mixture. This dilution effect reduces the availability of GLY molecules at the catalyst active sites, thereby lowering the rate of H2 production and GLY conversion. Additionally, the higher water concentration may inhibit the adsorption of GLY over the catalyst surface, further contributing to decreased catalytic efficiency. These results indicated the crucial role of the n(GLY)/n(H2O) ratio in achieving a high yield of H2 gas and SL for the catalytic transformation of GLY over the Ru/La(OH)3 catalyst (Fig. S22, ESI†).
Moreover, the concentration of NaOH significantly influenced the efficiency of producing the selective H2 gas and SL (Table 2, entries 9–11, and Fig. S23, ESI†). Upon varying the NaOH concentration from 0.5 equiv. to 1.5 equiv., a significant enhancement in the volume of H2 gas as well as the conversion of GLY was observed at 130 °C. The results showed that when the reaction was carried out with 0.5 equiv. of NaOH, only 138 mL (0.4 equiv., 90% yield of H2) of H2 gas with GLY conversion of 52% in 3.5 h was observed (Table 2, entry 9). Furthermore, on increasing the NaOH concentration to 1.0 equiv., the volume of the gas increased to 270 mL (0.8 equiv., 92% yield of H2) with 85% conversion of GLY and 70% yield of SL in 4 h (Table 2, entry 10). The reaction conducted using 1.5 equiv. NaOH resulted in 356 mL (1.0 equiv., 92% yield of H2) of H2 gas with 93% GLY conversion (Table 2, entry 11). Moreover, other by-products such as SG and PD were formed in less amount as compared to SL and SF. These results clearly indicated that NaOH plays an important role in promoting the deprotonation of hydroxyl groups of GLY, thus enhancing the dehydrogenation of GLY to SL over the Ru/La(OH)3. Notably, a high yield of PD (10%) was observed while using 0.5 equiv. of NaOH, while with the high base amount (1.0–2.0 equiv.), less PD yield was observed, implying that further dehydrogenation of the produced PD occurred in the presence of base to produce H2 gas and SL.
Moreover, the promotional effect of different types of bases, such as NaOH, KOH, KOtBu, Na2CO3 and K2CO3, in the dehydrogenation of GLY was also investigated (Table S3, ESI†). We observed high catalytic activity for GLY dehydrogenation (n(H2)/n(GLY) ∼ 1.4), using NaOH with complete conversion of GLY as compared to KOH [(n(H2)/n(GLY) ∼ 0.9)] with 88% conversion of GLY under the optimum reaction conditions (Table S3, ESI†). Moreover, the rate of generation of H2 gas was sluggish with KOH (initial TOF 56 h−1) compared to that observed using NaOH (initial TOF 105 h−1). This is possible because of the larger ionic radius of the K+ ion than that of the Na+ ion, causing steric hindrance in the dehydrogenation of GLY.87 Notably, no conversion of GLY was observed for the reactions performed with Na2CO3 and K2CO3, while with KOtBu, only 72% conversion of GLY was achieved (Table S3 entries 3–5, ESI†). At 90 °C, 13.3 mmol of H2 gas (∼1.0 equiv.) was observed in 14 h (Table S3 entry 6, ESI†), while, at elevated temperatures (100–120 °C), higher yield of H2 gas was observed over the Ru/La(OH)3 catalyst with no significant change in the selectivity of SL (Table S3 entries 7–9, ESI†). Notably, GLY was completely converted for the reactions performed at ≥110 °C, where an increase in the initial TOF was also observed upon an increase in the reaction temperature (Fig. 3(a), Fig. S24 and Table S3, ESI†). Furthermore, the activation energy for the selective H2 generation from GLY over the Ru/La(OH)3 catalyst was calculated on the basis of gas evolved in the initial one hour (initial TOF), using the Arrhenius equation and was found to be 37.5 kJ mol−1 (Fig. S25, ESI†). Moreover, enhancement in hydrogen gas production from GLY was observed for the reactions performed with higher ruthenium loading (Table S3 entries 10–12, ESI†). The aforementioned findings demonstrated the vital role of base concentration, n(GLY)/n(H2O) ratio, and the reaction temperature for achieving high catalytic activity for producing H2 gas and SL from GLY. Hence, high catalytic activity for GLY dehydrogenation with a good yield of SL over the Ru/La(OH)3 catalyst was achieved using n(NaOH)/n(GLY) of 2.0 equiv. and n(GLY)/n(H2O) of 1:3 at 130 °C (Table 2 and entry 5).
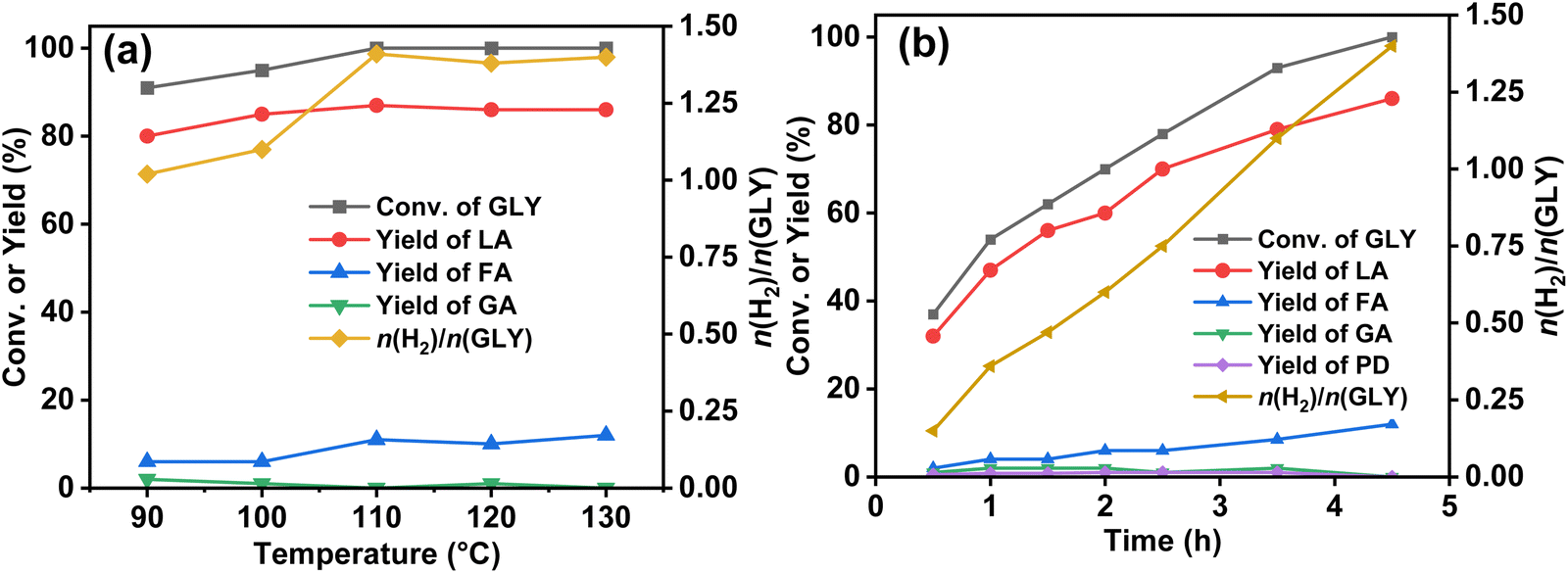 | ||
| Fig. 3 (a) Effect of temperature on the catalytic dehydrogenation of GLY over the Ru/La(OH)3 catalyst. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (13.68 mmol), NaOH (0.5–2.0 equiv.), water (41.04 mmol), 90 °C (14 h), 100 °C (12 h), 110 °C (9 h), 120 °C (6.5 h), 130 °C (4.5 h), and 600 rpm. (b) Time-dependent catalytic dehydrogenation of GLY over the Ru/La(OH)3 catalyst. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (13.68 mmol), NaOH (27.36 mmol), water (41.04 mmol), 130 °C, and 600 rpm. | ||
Furthermore, the time-dependent 1H NMR analysis of the catalytic reaction mixture at different time intervals during the dehydrogenation of GLY showed the continuous consumption of GLY with the increase in the yield of SL and SF. On the other hand, the yield of SG (<2%) and PD (<1%) was found to be constant throughout the reaction (Fig. 3(b)). Furthermore, to investigate the participation of these intermediates in the catalytic hydrogen production from GLY, several control experiments were performed using various substrates in the presence of NaOH (2.0 equiv.) (Scheme S3, ESI†). In agreement with the plausible reaction pathway, we observed the complete conversion of glyoxal to SG (yield 94%) using NaOH without catalyst (Scheme S3 and Fig. S26, ESI†). On the other hand, treating glyoxal with Ru/La(OH)3 catalyst under the optimized reaction conditions resulted in the formation of H2 gas (4.3 mmol), SG (yield 64%) and SF (yield 32%), suggesting that presumably sodium glycolate (SG) transformed to SF with the release of H2 over the Ru/La(OH)3 catalyst (Scheme 1(b), Scheme S3 and Fig. S27, ESI†). To investigate this further, performing a reaction with GA over the Ru/La(OH)3 catalyst in the presence of NaOH (2.0 equiv.) indeed resulted in the production of H2 gas (8.5 mmol) along with SF (yield 62%) (Scheme 1(c), Scheme S3 and Fig. S28, ESI†). Notably, further conversion of LA and formic acid (FA) over Ru/La(OH)3 was not observed during our investigation under the optimized reaction conditions (Scheme S3 and Fig. S29, S30, ESI†). Therefore, this experimental evidence clearly indicated that the selective catalytic dehydrogenation of GLY to H2 gas and SL followed the reaction pathway where initially, GLY dehydrogenated to glyceraldehyde with the release of one equivalent of H2 gas. Glyceraldehyde can further be transformed by two competing pathways. In the first pathway, glyceraldehyde is transformed into pyruvaldehyde through base-catalyzed dehydration, which can subsequently be converted into SL through the Cannizzaro reaction. Notably, pyruvaldehyde could also be hydrogenated using in situ generated H2 to generate PD.63 In another pathway, glyceraldehyde can be converted into formaldehyde and glycolaldehyde through a base-catalyzed retro-aldol reaction via C–C bond cleavage, where glycolaldehyde can further be dehydrogenated to SG via glyoxal, followed by the dehydrogenation of SG to SF (Scheme 1). Notably, there are multiple pathways (Schemes S1 and S2, ESI†) involved in the process of hydrogen gas generation during the catalytic conversion of GLY. Accordingly, the yield of H2 gas produced during the catalytic conversion of GLY over Ru/La(OH)3 at 130 °C was found to be in good agreement with the carbon balance (Table 2).
 | ||
| Scheme 1 (a) Plausible reaction pathways for hydrogen production from glycerol over the Ru/La(OH)3 catalyst. (b) Control experiment for the transformation of glyoxal over the Ru/La(OH)3 catalyst. (c) Control experiment for the transformation of glycolic acid over the Ru/La(OH)3 catalyst. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), glyoxal/glycolic acid (13.68 mmol), NaOH (27.36 mmol), water (41.04 mmol), 130 °C, and 600 rpm. | ||
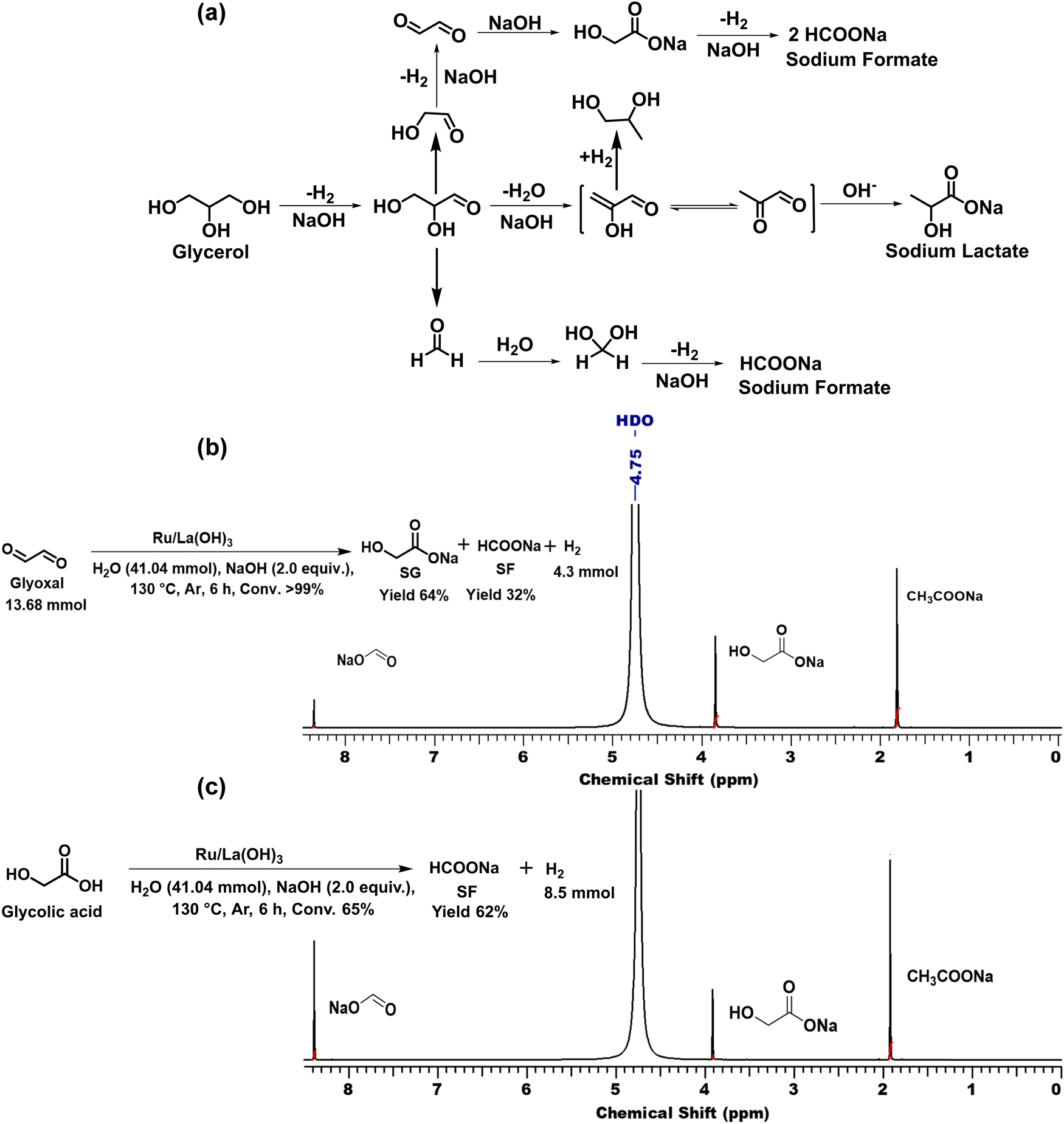
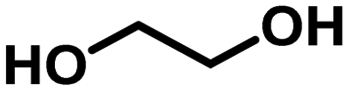
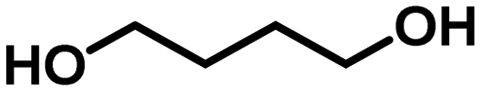
2.3. Scope of the Ru/La(OH)3 catalyst for H2 production from various terminal diols
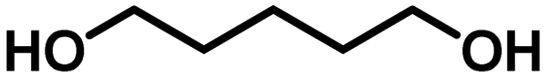
Encouraged by the high catalytic activity observed for the dehydrogenation of GLY, the Ru/La(OH)3 catalyst was also employed for the dehydrogenation of ethylene glycol (EG) and other terminal diols such as 1,3-propanediol (PDO), 1,4-butanediol (BDO), 1,5-pentanediol (PO) and 1,6-hexanediol (HDO), which were converted to sodium formate (SF) and other by-products due to C–C bond cleavage and/or aldol condensation. EG was dehydrogenated completely over the Ru/La(OH)3 in 8 h and produced 870 mL (35 mmol, 2.6 equiv.) of H2 gas (Table 3, entry 1). Notably, previously, we observed less gas release (2.3 equiv. H2 gas) with longer reaction time (9.5 h) for EG dehydrogenation over bare ruthenium nanoparticles in water, suggesting the important role of the support material.27 PO produced 570 mL (23 mmol) of H2 gas with >99% conversion (Table 3, entry 2) in 4 h. Furthermore, in the case of BDO, 67% conversion was achieved in 9 h with 390 mL (16 mmol) of H2 gas (Table 3, entry 3). However, for PDO, only 96 mL (4 mmol) of H2 gas evolved with 22% conversion of PDO in 7.5 h (Table 3, entry 4), whereas no evolution of H2 gas was observed for HDO (Table 3, entry 5). These results showed that the catalytic efficacy of Ru/La(OH)3 for the dehydrogenation reactions decreases with the increase in carbon chain length in terminal diols. However, in the case of long-chain diols, there is a decrease in the carbon balance due to the formation of some unidentified products via C–C bond cleavage or aldol-condensation reactions over the Ru/La(OH)3 catalyst. However, for other long-chain diols, the estimation of hydrogen yield based on carbon balance for these long-chain diols became very complex due to the involvement of several unidentified side reactions.88| Entry | Substrate | t (h) | H2 gasb (mL) | n(H2)/n(diol) | Conv. (%) | Yield of byproductsc (% C) | CBd (%) | |
|---|---|---|---|---|---|---|---|---|
| SF | Others | |||||||
| a Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), substrate (13.68 mmol), NaOH (27.36 mmol), water (41.04 mmol), 130 °C, and 600 rpm. b Volume of gas was measured by the water displacement method. c Yield was calculated by 1H NMR using sodium acetate as an internal standard. d CB is carbon balance. e H2 yield on the carbon basis. The results reported are the average of at least two repeated reactions. SF (sodium formate), SG (sodium glycolate), HPA (3-hydroxypropionate), SM (sodium malonate), SP (sodium propanoate), SS (sodium succinate), and n.r. (no reaction). | ||||||||
| 1 |
|
8 | 870 (84%)e | 2.6 | >99 | 52 | SG (32) | 84 |
| 2 |

|
4 | 570 | 1.7 | >99 | 55 | HPA (5) | 75 |
| SM (2) | ||||||||
| SP (13) | ||||||||
| 3 |
|
9 | 390 | 1.2 | 67 | 19 | SS (28) | 81 |
| 4 |
|
7.5 | 96 | 0.3 | 22 | 5 | — | 83 |
| 5 |
|
4.5 | — | — | n.r. | — | — | — |
2.4. Catalyst stability and recyclability
A mercury poisoning experiment was conducted to investigate the heterogeneity of the Ru/La(OH)3 catalyst, where the Ru/La(OH)3 catalyst was treated with an excess of elemental Hg(0) before proceeding to the catalytic reaction. A significant quenching of the reaction was observed, proving the heterogeneous nature of the catalyst (Fig. S31, ESI†). Furthermore, we performed a hot-filtration test for the Ru/La(OH)3 catalyst during hydrogen production from glycerol under the optimized reaction conditions. For this, we hot filtered the reaction mixture after 1.5 h to separate the catalyst and divide the reaction aliquots into two parts. The first part was analyzed by 1H NMR, which showed 62% conversion of GLY, while the second part continued to stir for another 3 h and was then analyzed by 1H NMR. The results showed no enhancement in the GLY conversion, which further confirmed the heterogeneous nature of the Ru/La(OH)3 catalyst. Moreover, large-scale H2 gas generation from GLY was conducted, where ∼2.2 L (90 mmol) of H2 gas was produced (TON 1016) in 18 h from 5 mL (68.4 mmol) of GLY with a productivity of 12 L H2/gRu/h and a yield of SL of 82% in the presence of NaOH (2.0 equiv.) at 130 °C (Fig. 4(a)). Moreover, large-scale dehydrogenation of EG was also performed, and we observed the release of ∼4.3 L H2 gas from 3.8 mL of EG in 42 h using NaOH (3.0 equiv.), and ∼3.7 L of H2 gas from 3.8 mL of EG in 37 h in the presence of 2.0 equiv. NaOH (Fig. S32, ESI†). The catalyst recyclability experiments were conducted at intermediate conversion (∼60%) over ten catalytic runs, revealing sustained catalytic activity through the initial six cycles (n(H2)/n(GLY) of 0.45). After the 6th cycle, a gradual decrease in conversion (by 2%) was observed from the 6th to 7th cycle and a further 6% loss in conversion [n(H2)/n(GLY) of 0.39] was observed after the 10th cycle (Fig. 4(b), Fig. S33b and Table S4, ESI†). On the other hand, catalyst recyclability experiments carried out for ten catalytic runs at the initial complete conversion of GLY showed that complete conversion of GLY was attained through the initial five cycles [(n(H2)/n(GLY)) of 1.4]. Though the activity decreased in subsequent catalytic runs, 88% GLY conversion [n(H2)/n(GLY) of ∼1.2] was achieved even after the 10th catalytic run (Fig. S33 and Table S5, ESI†). Though the selectivity of SL was maintained high for both intermediate and complete GLY conversion, the SL yield was significantly lower (∼54% yield) at intermediate GLY conversion as compared to higher SL yield (∼84%) obtained at complete GLY conversion, which is consistent with the respective n(H2)/n(GLY) at intermediate and complete conversion of glycerol (Scheme 1). It is evident that at intermediate conversion, along with SL, other by-products (SG, PD and SF) were also observed with a combined yield of ∼7% (Tables S4 and S5, ESI†). Notably, these intermediates were formed due to C–C bond cleavage of glyceraldehyde, which eventually transformed to SF during the complete conversion of GLY and hence would not influence the SL selectivity (Tables S4 and S5, ESI†).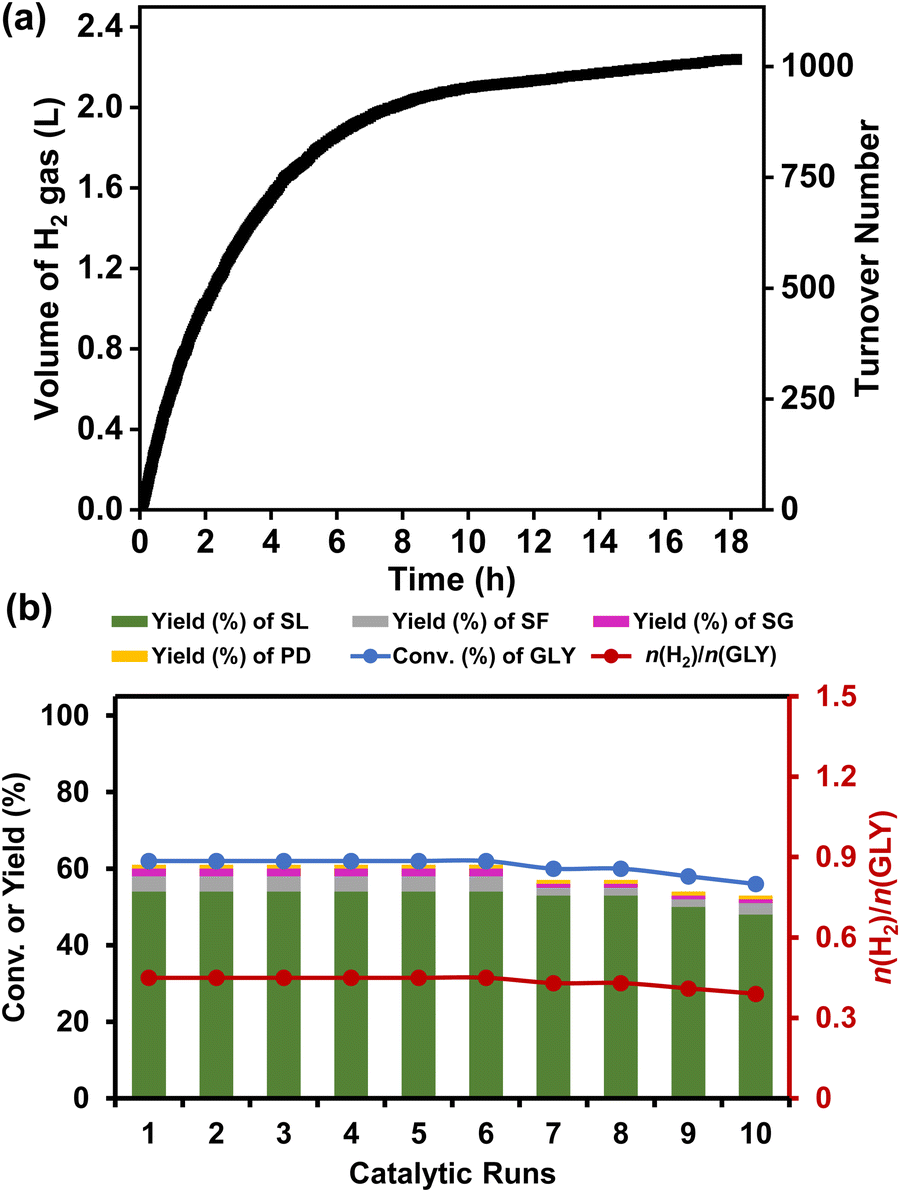 | ||
| Fig. 4 (a) Time course plot for the production of hydrogen gas from a large amount of glycerol (5 mL) over the Ru/La(OH)3 catalyst. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (68.4 mmol), NaOH (136.8 mmol), water (205.2 mmol), 130 °C, and 600 rpm. (b) Recyclability experiment for hydrogen production from glycerol over the Ru/La(OH)3 catalyst at an intermediate GLY conversion. Reaction conditions: Ru/La(OH)3 (100 mg, 9 wt% Ru), GLY (13.68 mmol), NaOH (27.36 mmol), water (41.04 mmol), 130 °C, 1.5 h, and 600 rpm. | ||
The observed loss in catalytic activity during the recyclability experiments can be attributed to the agglomeration of ruthenium nanoparticles and loss of the catalyst during the catalyst recovery process. The TEM analysis of the recovered catalyst after ten cycles also displayed a slight increase in particle size (∼3.6 nm) compared to the fresh Ru/La(OH)3 catalyst (particle size ∼1.5 nm) (Fig. S34, ESI†). ICP-AES analysis of the reaction aliquot after the 10th cycle showed no significant leaching of Ru (11 ppm) and La (0.18 ppm). Moreover, the ruthenium content in the Ru/La(OH)3 catalyst after the 10th catalytic run was also analyzed using ICP-AES and was found to be 7.5 wt%. Noticeably, the P-XRD pattern of the spent Ru/La(OH)3 catalyst was found to be analogous to the fresh Ru/La(OH)3 catalyst (Fig. S35, ESI†). These findings confirm the robustness and the long-term stability of the Ru/La(OH)3 catalyst, which is reflected in the observed enhanced catalytic activity of Ru/La(OH)3 for the dehydrogenation of GLY to H2 gas and SL. The developed Ru/La(OH)3 catalyst was also highly active for hydrogen production from crude glycerol under the optimized reaction conditions where we observed the complete dehydrogenation of crude glycerol into hydrogen gas (∼700 mL, 28.6 mmol), sodium lactate (11.83 mmol), and sodium formate (9.58 mmol) in 7 h (Fig. S36, ESI†).
3. Conclusion
Herein, we demonstrated a sustainable and efficient catalytic process for the selective and complete conversion of glycerol (GLY) to produce high-purity H2 gas (>99.9%) and sodium lactate (SL) over Ru/La(OH)3 catalyst in aq. NaOH solution at a lower temperature (90–130 °C). Our findings indicated the crucial role of base concentration, n(GLY)/n(H2O) ratio, reaction temperature, and different support materials in achieving complete conversion of GLY selectively to H2 and SL. We observed that Ru/La(OH)3 outperformed other supported ruthenium catalysts to achieve a high H2 yield (1.4 equiv. per mmol of GLY) along with a high yield of SL (86%) at 130 °C in the presence of NaOH (2.0 equiv.) in 4.5 h. Moreover, at 110 °C also, we could achieve a similar amount of H2 gas as well as a high yield of SL (87%) in 9 h using 2.0 equiv. NaOH. To validate the catalytic dehydrogenation pathway of GLY, several controlled experiments were carried out under the optimized reaction conditions. Advantageously, we could generate H2 gas on a large scale with high purity and selectivity (>99.9%) from aqueous GLY over the Ru/La(OH)3 catalyst, displaying the remarkably high long-term stability of the Ru/La(OH)3 catalyst with an efficiency of producing 220 L H2 per gRu with a productivity of 12 L H2 per gRu per h. This eliminates the need for additional purification, making it a cost-effective solution, a crucial factor for industrial applications. Moreover, the developed catalytic methodology was also equally effective for the large scale dehydrogenation of EG to yield ∼4.3 L H2 gas from 3.8 mL of EG in 42 h using NaOH (3.0 equiv.) at 130 °C. Therefore, the developed Ru/La(OH)3-based catalytic system provides new insights for a sustainable approach for exploring GLY, EG and other terminal diols as important substrates for selectively producing high-purity hydrogen gas in aqueous conditions at lower temperatures.4. Experimental section
4.1. Materials
RuCl3·3H2O (>99%), NaBH4 (98%), cetyltrimethylammonium bromide (CTAB) (98%), La(OH)3 (99.9%), TiO2 (99.5%), ZrO2 (99%), glycerol (99.5%), ethylene glycol (>99%), glyoxal solution (40 wt% in water), lactic acid (>90%), formic acid (>96%), and 1,6-hexanediol (HDO) (99%) were purchased from Sigma Aldrich, India. Glycolic acid (>98%) 1,2-propanediol (PD) (>98%), 1,3-propanediol (PO) (98%), 1,4-butanediol (BDO) (>99%), and 1,5-pentanediol (PDO) (>97%) were purchased from TCI analytics, India. MgO (99%) and ZnO (98%) were procured from S. D. Fine Chemical Limited, India. High-purity argon gas was procured from Sigma Gases, India. Crude glycerol was prepared by mixing using pure glycerol (80%), methanol (15%) and water (5%).89,90 Distilled water was used for performing all the experiments. All other chemical reagents and metal salts were available commercially and were used as received without any further purification.4.2. Catalyst impregnation
The wet impregnation method was used for the synthesis of supported Ru catalysts using the support La(OH)3, ZnO, Mg(OH)2, ZrO2, and TiO2. Typically, 90 mg of support was dispersed in 5 mL of distilled water in a 25 mL round bottom flask under sonication for 20 min. To this, ruthenium(III) chloride (0.1 mmol) and CTAB (0.050 g) were added, and the resulting mixture was heated at 60 °C for 2 h. The mixture was cooled to room temperature and then aqueous solution of NaBH4 (0.050 g in 5 mL water) was added dropwise under sonication to reduce Ru(III) to Ru(0). The obtained mixture was sonicated for 30 minutes, and then the precipitates were collected by centrifugation, washed several times with distilled water and ethanol, and dried under vacuum before use for the catalytic reactions. The actual loading of Ru in the Ru/La(OH)3 catalyst as calculated by ICP-AES measurement was found to be 9.0 wt% ± 0.2%, based on the analysis of three samples of Ru/La(OH)3 catalyst. The metal loading of other supported Ru catalysts is mentioned in Table S2 (ESI†).4.3. Catalyst characterization
Transmission electron microscopic (TEM) imaging and energy dispersive X-ray spectroscopy (EDS) mapping were performed on the FEI Titan Themis with an operating voltage of 300 kV. Samples for TEM were prepared by dispersing the solid samples in ethanol under sonication and dropping the suspension directly onto lacey carbon-coated copper grids. The Ru metal dispersion for all catalysts was determined through TEM analysis, assuming that the Ru nanoparticles are spherical and utilizing the formula provided in the ESI.† Scanning electron microscopic images and elemental mapping data were collected using a JOEL-7610F plus equipped with an EDS detector. Powder X-ray diffraction (P-XRD) measurements were performed using a Rigaku Smart Lab, Automated Multipurpose X-ray diffractometer with monochromatic Cu Kα radiation (λ = 0.154 nm). The XRD patterns were collected under 40 kV and 30 mA in the range 20 to 80°. The nitrogen physisorption isotherms were measured at −196 °C using a Quantachrome Autosorb iQ2 TPX automated gas sorption system. Samples were degassed at 200 °C for 8 h under high vacuum before analysis. X-Ray photoelectron Spectroscopy (XPS) was performed using a SPECS Surface Nano Analysis GmbH, Germany. All the XPS spectra were recorded using Al K-alpha (1486.61 eV) X-rays. The binding energy values were charge-corrected with respect to the C 1s signal (284.6 eV). Inductively coupled plasma atomic emission spectroscopic (ICP-AES) analyses were performed using ARCOS, a simultaneous spectrometer of SPECTRO analytical instruments. The catalyst was dissolved completely in aqua regia and then digested by heating the samples (catalyst in aqua regia) in an autoclave at 180 °C for 12 h. After cooling down, the samples were diluted with water and analysed by ICP-AES analysis. NMR spectra were recorded in deuterated solvents using a Bruker Ascend 500 and Bruker Avance 400 spectrometer (500 MHz and 400 MHz). The gas chromatography (GC) analyses were performed on a Shimadzu GC-2014 system using a shin carbon-ST packed column with a thermal conductivity detector (TCD) using argon as a carrier gas. Parameters were set for the program to detect H2, CO2, CO, and CH4 gas (detector temperature: 200 °C), and oven temperature program: 90 °C (hold time: 1 min), 90–200 °C (rate: 15 °C minute−1).4.4. Catalytic test
In a 25 mL round bottom flask fitted with a condenser and a water displacement set-up, a specified amount of the catalyst, glycerol (GLY), water and base were added. The reaction set-up was de-aerated by a repeated process of vacuuming and flushing with argon gas at least three times. The reaction mixture was stirred (600 rpm) in a pre-heated oil bath at 90–130 °C under an argon atmosphere. The amount of H2 gas evolved was quantified using a water displacement set-up, which was calibrated (thrice using water) as 4.0 ± 0.1 mL cm−1 (Fig. S37, ESI†). The composition of the evolved gas was analyzed by GC-TCD. After the reaction, the catalyst was recovered from the reaction mixture by centrifugation. The conversion of GLY and the yields of other co-products were determined on the carbon basis by 1H NMR of the reaction mixture using sodium acetate as an internal standard (Fig. S38, ESI†). Notably, acetate or any other undesired product was not observed during the NMR analysis (without internal standard) of the crude reaction mixture (Fig. S38, ESI†). LA was purified by acidifying (pH ∼ 1.5) the crude reaction mixture containing SL with 1 M HCl, and then the organic product (LA) was extracted using diethyl ether (5 × 10 mL). The organic fraction was dried over Na2SO4. The purified LA was obtained by removing the organic solvent under reduced pressure and was analyzed by NMR (1H and 13C) in D2O (Fig. S39 and S40, ESI†). The turnover number (TON) was calculated by considering the total metal-based ruthenium using the formula given below.| TON = n(H2)/n(catalyst) |
4.5. Heterogeneity test
Typically, an aqueous suspension of Ru/La(OH)3 catalyst (100 mg catalyst in 0.740 mL water) was stirred with a large excess of elemental mercury Hg(0) at room temperature for 3 h prior to the catalytic reaction. Then GLY (13.68 mmol, 1 mL) and NaOH (27.36 mmol, 1.095 g) were added to the above reaction vessel (25 mL round bottom flask) fitted with a condenser and gas burette. The water displacement set-up was then de-aerated by a repeated process of vacuum and flushing with argon gas at least three times. The reaction mixture was stirred at 130 °C in a pre-heated oil bath at a rpm of 600, and the progress of the reaction was monitored for the specified duration.4.6. Recyclability experiment
Catalytic reaction was performed as specified in Section 4.4. After the completion of the reaction, the catalyst was recovered from the reaction mixture by centrifugation, washed several times with distilled water and ethanol, dried overnight under vacuum and then reused for the next catalytic run.4.7. Large scale reaction
Typically, large scale reaction was performed using GLY (68.5 mmol) and water (205.5 mmol) in the presence of the Ru/La(OH)3 catalyst (100 mg) and NaOH (2.0 equiv.) under the optimized reaction conditions at 130 °C. Similarly, a large scale reaction for ethylene glycol (68.5 mmol) was also performed using water (205.5 mmol) and NaOH (2.0 and 3.0 equiv.) over the Ru/La(OH)3 catalyst (100 mg) at 130 °C. Large scale reaction was performed in a 50 mL round bottom flask using a reaction set-up as mentioned in Section 4.4.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from IIT Indore, CSIR (01(3045)/21/EMR-II) and SERB-DST (CRG/2021/000504). Sophisticated Instrumentation Centre (SIC) IIT Indore, DST-FIST 500 NMR facility of the Department of Chemistry, IIT Indore, Department of Metallurgy Engineering and Material Science (MEMS) IIT Indore, SAIF IIT Bombay, CIF IIT Gandhinagar, and UGC-DAE Consortium for Scientific Research, Indore are gratefully acknowledged for providing instrumentation facilities. A. K. and B. P. thank CSIR New Delhi and SERB, respectively, for their fellowships.References
- A. Midilli, M. Ay, I. Dincer and M. A. Rosen, Renewable Sustainable Energy Rev., 2005, 9, 255–271 CrossRef CAS.
- M. Balat, Int. J. Hydrogen Energy, 2008, 33, 4013–4029 CrossRef CAS.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2013, 38, 2579–2588 CrossRef CAS.
- S. Singh, S. Jain, V. PS, A. K. Tiwari, M. R. Nouni, J. K. Pandey and S. Goel, Renewable Sustainable Energy Rev., 2015, 51, 623–633 CrossRef CAS.
- L. Barreto, A. Makihira and K. Riahi, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS.
- F. Mueller-Langer, E. Tzimas, M. Kaltschmitt and S. Peteves, Int. J. Hydrogen Energy, 2007, 32, 3797–3810 CrossRef CAS.
- L. Chen, Z. Qi, S. Zhang, J. Su and G. A. Somorjai, Catalysts, 2020, 10, 858 CrossRef CAS.
- A. Fasolini, R. Cucciniello, E. Paone, F. Mauriello and T. Tabanelli, Catalysts, 2019, 9, 917 CrossRef CAS.
- D. Mei, V. D. Lebarbier, R. Xing, K. O. Albrecht and R. A. Dagle, ACS Catal., 2016, 6, 315–325 CrossRef CAS.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- M. K. Awasthi, R. K. Rai, S. Behrens and S. K. Singh, Catal. Sci. Technol., 2021, 11, 136–142 RSC.
- P. Sponholz, D. Mellmann, C. Cordes, P. G. Alsabeh, B. Li, Y. Li, M. Nielsen, H. Junge, P. Dixneuf and M. Beller, ChemSusChem, 2014, 7, 2419–2422 CrossRef CAS.
- Y. Q. Xia, Z. Chen, Y. Wang, F. Cheng, L. Qin and Z. Zheng, Organometallics, 2022, 41, 914–919 CrossRef.
- V. Arora, S. Dhole and A. Kumar, Catal. Sci. Technol., 2023, 13, 6699–6711 RSC.
- M. Kuwahara, M. Nishioka, M. Yoshida and K. I. Fujita, ChemCatChem, 2018, 10, 3636–3640 CrossRef CAS.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS.
- S. Patra and S. K. Singh, Inorg. Chem., 2020, 59, 4234–4243 CrossRef CAS PubMed.
- L. E. Heim, N. E. Schlörer, J. H. Choi and M. H. G. Prechtl, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- M. Trincado, V. Sinha, R. E. Rodriguez-Lugo, B. Pribanic, B. D. Bruin and H. Grützmacher, Nat. Commun., 2017, 8, 1–11 CrossRef PubMed.
- T. Van Haasterecht, T. W. Van Deelen, K. P. De Jong and J. H. Bitter, Catal. Sci. Technol., 2014, 4, 2353–2366 RSC.
- A. Kumar, M. K. Awasthi, B. Priya and S. K. Singh, ChemCatChem, 2022, 14, e202101951 CrossRef CAS.
- P. Gogoi, A. S. Nagpure, P. Kandasamy, C. V. V. Satyanarayana and T. Raja, Sustainable Energy Fuels, 2020, 4, 678–690 RSC.
- A. Kumar, B. Priya and S. K. Singh, ACS Sustainable Chem. Eng., 2023, 11, 3999–4008 CrossRef CAS.
- Agricultural Outlook 2021–2030. OECD-FAO Agricultural Outlook 2021–2030; 2021.
- A. A. Farias da Costas, A. de N. de Oliveira, R. Esposito, A. Auvigne, C. Len, R. Luque, R. C. R. Noronha and L. A. S. do. Nascimento, Sustainable Energy Fuels, 2023, 7, 1768–1792 RSC.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- C. A. Schwengber, H. J. Alves, R. A. Schaffner, F. A. da Silva, R. Sequinel, V. R. Bach and R. J. Ferracin, Renewable Sustainable Energy Rev., 2016, 58, 259–266 CrossRef CAS.
- F. Pompeo, G. F. Santori and N. N. Nichio, Catal. Today, 2011, 172, 183–188 CrossRef CAS.
- T. Hirai, N. Ikenaya, T. Miyake and T. Suzuki, Energy Fuels, 2005, 19, 1761–1762 CrossRef CAS.
- S. Adhikari, S. Fernando and A. Haryanto, Catal. Today, 2007, 129, 355–364 CrossRef CAS.
- R. Sundari and P. D. Vaidya, Energy Fuels, 2012, 26, 4195–4204 CrossRef CAS.
- K. Kousi, D. I. Kondarides, X. E. Verykios and C. Papadopoulou, Appl. Catal., A, 2017, 542, 201–211 CrossRef CAS.
- S. Adhikari, S. D. Fernando, S. D. F. To, R. M. Bricka, P. H. Steele and A. Haryanto, Energy Fuels, 2008, 22, 1220–1226 CrossRef CAS.
- K. Polychronopoulou, N. D. Charisiou, G. I. Siakavelas, A. A. Alkhoori, V. Sebastian, S. J. Hinder, M. A. Baker and M. A. Goula, Sustainable Energy Fuels, 2019, 3, 673–691 RSC.
- F. Ming, X. Qingli, Q. Wei, Z. Zhikai, Z. Suping and Y. Yongjie, Energy Sources, Part A, 2016, 38, 2128–2134 CrossRef.
- M. Surendar, D. Padmakar, N. Lingaiah, K. S. Rama Rao and P. S. Sai Prasad, Sustainable Energy Fuels, 2017, 1, 354–361 RSC.
- P. Gogoi, N. Kanna, P. Begum, R. C. Deka, C. V. V. Satyanarayana and T. Raja, ACS Catal., 2020, 10, 2489–2507 CrossRef CAS.
- P. J. Dietrich, R. J. Lobo-Lapidus, T. Wu, A. Sumer, M. C. Akatay, B. R. Fingland, N. Guo, J. A. Dumesic, C. L. Marshall, E. Stach, J. Jellinek, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Top. Catal., 2012, 55, 53–69 CrossRef CAS.
- D. L. King, L. Zhang, G. Xia, A. M. Karim, D. J. Heldebrant, X. Wang, T. Peterson and Y. Wang, Appl. Catal., B, 2010, 99, 206–213 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- C. Pendem, B. Sarkar, N. Siddiqui, L. N. S. Konathala, C. Baska and R. Bal, ACS Sustainable Chem. Eng., 2018, 6, 2122–2131 CrossRef CAS.
- D. A. Boga, R. Oord, A. M. Beale, Y. M. Chung, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 529–537 CrossRef CAS.
- Y. Guo, X. Liu and Y. Wang, Ind. Eng. Chem. Res., 2019, 58, 2749–2758 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Güemez, R. M. Navarro, M. C. S. Sánchez and J. L. G. Fierro, Top. Catal., 2008, 49, 46–58 CrossRef CAS.
- H. Yin, H. Yin, A. Wang, L. Shen, Y. Liu and Y. Zheng, J. Nanosci. Nanotechnol., 2017, 17, 1255–1266 CrossRef CAS PubMed.
- L. Shen, H. Yin, H. Yin, S. Liu and A. Wang, J. Nanosci. Nanotechnol., 2017, 17, 773–779 CrossRef.
- L. Shen, Z. Yu, D. Zhang, H. Yin, C. Wang and A. Wang, J. Chem. Technol. Biotechnol., 2019, 94, 204–215 CrossRef CAS.
- G. Y. Yang, Y. H. Ke, H. F. Ren, C. L. Liu, R. Z. Yang and W. S. Dong, Chem. Eng. J., 2016, 283, 759–767 CrossRef CAS.
- R. Palacio, S. Torres, S. Royer, A. S. Mamede, D. López and D. Hernández, Dalton Trans., 2018, 47, 4572–4582 RSC.
- D. Roy, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2011, 1, 548–551 CrossRef CAS.
- J. Zhang, X. Wu, J. Chen, X. Wang and Y. Mai, ChemCatChem, 2023, 15, e202201139 CrossRef CAS.
- H. Narjinari, S. Dhole and A. Kumar, Chem. – Eur. J., 2023, e202302686 Search PubMed.
- R. Palacio, S. Torres, D. Lopez and D. Hernandez, Catal. Today, 2018, 302, 196–202 CrossRef CAS.
- L. Qiu, H. Yin, H. Yin and A. Wang, J. Nanosci. Nanotechnol., 2017, 18, 4734–4745 CrossRef PubMed.
- R. Palacio, D. López and D. Hernández, J. Nanopart. Res., 2019, 21, 148 CrossRef.
- D. Akbulut and S. Ozkar, RSC Adv., 2022, 12, 18864–18883 RSC.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS.
- M. Checa, F. Auneau, J. Hidalgo-Carrillo, A. Marinas, J. M. Marinas, C. Pinel and F. J. Urbano, Catal. Today, 2012, 196, 91–100 CrossRef CAS.
- S. M. A. H. Siddiki, A. S. Touchy, K. Kon, T. Toyao and K. I. Shimizu, ChemCatChem, 2017, 9, 2816–2821 CrossRef CAS.
- W. Oberhauser, C. Evangelisti, C. Tiozzo, F. Vizza and R. Psaro, ACS Catal., 2016, 6, 1671–1674 CrossRef CAS.
- S. Feng, K. Takahashi, H. Miura and T. Shishido, Fuel Process. Technol., 2020, 197, 106202 CrossRef CAS.
- J. Ftouni, N. Villandier, F. Auneau, M. Besson, L. Djakovitch and C. Pinel, Catal. Today, 2015, 257, 267–273 CrossRef CAS.
- Z. Tang, P. Liu, H. Cao, S. Bals, H. J. Heeres and P. P. Pescarmona, ACS Catal., 2019, 9, 9953–9963 CrossRef CAS.
- Z. Jiang, Z. Zhang, T. Wu, P. Zhang, J. Song, C. Xie and B. Han, Chem. – Asian J., 2017, 12, 1598–1604 CrossRef CAS PubMed.
- G. Bharath, K. Rambabu, A. Hai, H. Taher and F. Banat, ACS Sustainable Chem. Eng., 2020, 8, 7278–7289 CrossRef CAS.
- Y. Li, M. Nielsen, B. Li, P. H. Dixneuf, H. Junge and M. Beller, Green Chem., 2015, 17, 193–198 RSC.
- M. Dutta, K. Das, S. J. Prathapa, H. K. Srivastava and A. Kumar, Chem. Commun., 2020, 56, 9886–9889 RSC.
- M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS.
- Z. Yuan, Z. Gao and B. Q. Xu, Chin. J. Catal., 2015, 36, 1543–1551 CrossRef CAS.
- J. G. Kang, Y. I. Kim, D. W. Cho and Y. Sohn, Mater. Sci. Semicond. Process., 2015, 40, 737–743 CrossRef CAS.
- S. R. Sanivarapu, J. B. Lawrence and G. Sreedhar, ACS Omega, 2018, 3, 6267–6278 CrossRef CAS PubMed.
- M. A. Aramendia, V. Borau, C. Jimenez, A. Marinas, J. M. Marinas, J. R. Ruiz and F. J. Urbano, J. Mol. Catal. A: Chem., 2004, 218, 81–90 CrossRef CAS.
- B. Sukhbaatar, W. Qing, J. Seo, S. Yoon and B. Yoo, Sci. Rep., 2024, 14, 5850 CrossRef CAS.
- J. R. Deka, D. Saikia, K. S. Hsia, H. M. Kao, Y. C. Yang and C. S. Chen, Catalysts, 2020, 10, 267 CrossRef CAS.
- Y. Zhang, H. Jiang, G. Li and M. Zhang, RSC Adv., 2016, 6, 16851 RSC.
- C. Antonetti, M. Oubenali, A. M. R. Galletti, P. Serp and G. Vannucci, Appl. Catal., A, 2012, 421, 99–107 CrossRef.
- Q. He, Y. Zhou, H. Shou, X. Wang, P. Zhang, W. Xu, S. Qiao, C. Wu, H. Liu, D. Liu, S. Chen, R. Long, Z. Qi, X. Wu and L. Song, Adv. Mater., 2022, 34, 2110604 CrossRef CAS.
- C. Fu, J. Wang, M. Yang, X. Su, J. Xu and B. Jiang, J. Non-Cryst. Solids, 2011, 357, 1172–1176 CrossRef CAS.
- X. Wang, N. Maeda and D. M. Meier, Catalysts, 2021, 11, 916 CrossRef CAS.
- H. Yan, S. Yao, S. Zhao, M. Liu, W. Zhang, X. Zhou, G. Zhang, X. Jin, Y. Liu, X. Feng, X. Chen, D. Chen and C. Yang, Chem. Eng. Sci., 2021, 230, 116191 CrossRef CAS.
- Y. Guo, M. U. Azmat, X. Liu, Y. Wang and G. Lu, Appl. Energy, 2012, 92, 218–223 CrossRef CAS.
- M. Finn, J. A. Ridenour, J. Heltzel, C. Cahill and A. V. Kostal, Organometallics, 2018, 37, 1400–1409 CrossRef CAS.
- G. Zhang, X. Jin, Y. Guan, B. Yin, X. Chen, Y. Liu, X. Feng, H. Shan and C. Yan, Ind. Eng. Chem. Res., 2019, 58, 14548–14558 CrossRef CAS.
- Y. Zhan, W. Hou, G. Li, Y. Shen, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2019, 7, 17559–17564 CrossRef CAS.
- F. Yang, M. A. Hanna and R. Sun, Biotechnol. Biofuels, 2012, 5, 13 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, V. L. Muldoon and S. Deng, Renewable Sustainable Energy Rev., 2022, 159, 112206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00213j |
| This journal is © The Royal Society of Chemistry 2025 |