
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent synthesis of stereogenic-at-boron fluorophores (BOSPYR) from boronic acids, salicylaldehydes, and 2-formylpyrrole hydrazones†
Ezgi
Bayer Kömüşdoğan
 a,
Sania
Batool
a,
Ertan
Şahin
b,
Erol
Yildirim
a,
Murat
Işık
*c and
Cihangir
Tanyeli
*a
a,
Sania
Batool
a,
Ertan
Şahin
b,
Erol
Yildirim
a,
Murat
Işık
*c and
Cihangir
Tanyeli
*a
aDepartment of Chemistry, Middle East Technical University, 06800 Ankara, Turkey. E-mail: tanyeli@metu.edu.tr
bDepartment of Chemistry, Atatürk University, Erzurum, 25240, Turkey
cDepartment of Food Engineering, Bingöl University, Bingöl, 12000, Turkey. E-mail: misik@bingol.edu.tr
First published on 5th December 2024
Abstract
This work describes one-step syntheses of various stereogenic-at-boron fluorochromes (BOSPYR) via multicomponent reactions involving readily accessible boronic acids, salicylaldehydes, and 2-formylpyrrole hydrazones. The dyes absorb and emit in the visible region of the electromagnetic radiation, and are characterized by large Stokes shifts (2850–4930 cm−1) with weak fluorescence emissions (Φfl: 1.5–9.1%). Notably, the dimmed fluorescence of BOSPYRs recovers upon transition to viscous media (21-fold for 1a). The representative compound 1a exhibits clear Cotton effects with dissymmetry factors of ca. |gabs| ∼ 1.9 × 10−3 in the visible region, indicating efficient asymmetry induction to the chromophore. The X-ray molecular structure of 1a shows that the chromophore deviates from planarity by 17.2°, which may contribute significantly to the inherent chirality of the fluorophore. A computational examination of excited states by time-dependent density functional theory (TD-DFT) identifies the emission mechanism as arising from a locally-excited (LE) state.
Fluorophores have become indispensable tools across various disciplines, including chemistry, biology, medicine, materials science, and numerous interdisciplinary areas.1 The element boron (B) plays a crucial role in modern fluorophore design thanks to forming stable tetravalent chelates, thereby restricting internal bond rotations in the π-skeleton of nascent fluorophores, enforcing planarity, and ensuring enhanced π-conjugation throughout the dye.2 This can be well exemplified by the archetypal boron dipyrromethene (BODIPY, Fig. 1) dyes3 or similar heterocyclic architectures, locked typically with a difluoro boron bridge.2 Despite their successful application as functional dyes in diverse fields, from probes/labels to photosensitizers for photodynamic action, the synthesis of such functional BODIPYs often involves intricate, long linear routes.4
 | ||
| Fig. 1 BODIPY dyes accessible through conventional synthesis vs. BASHY dyes and BOSPYR dyes (this work) through MCR approach. | ||
Unlike conventional synthesis approaches, multicomponent reactions (MCRs) offer a convergent synthesis approach, assembling often readily available starting materials in a single step. This has recently sparked growing interest in the development of functional organic dyes through MCRs.5,6 In contrast to the commonly used BF3, the group of Gois & Pischel has employed arylboronic acids as the boron source for the MCR-based development of intriguing BASHY fluorophores (Fig. 1),6 which exhibit photophysical properties highly comparable to BODIPYs.6–12 Notably, the sp3 hybridized boron atom in these dyes, accommodating four distinct ligands, creates asymmetry around it, imparting chirality to the dye.9
This remains a challenge in BODIPYs due to their highly symmetric nature (point group: C2v),13–15 particularly for those with chiral-at-boron arrangements.15 Therefore, currently, the majority of chiral-at-boron chromophores/fluorophores, although rare in number, generally rely on other miscellaneous heterocyclic systems with a tetravalent boron center.16 The visible light-absorbing BASHY fluorophores, as solely stereogenic-at-boron dyes, exhibited detectable chiroptical activities in electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) spectroscopy within the visible region.9 Similarly, although not a true MCR-type synthesis, research efforts by Hao & Jiao and co-workers employing arylboronic acids as the locking unit have resulted in a variety of readily accessible BOPBY, BOBHY, and BOSPY dyes, offering unique optical behaviors.17 Although these dyes also share stereogenic-at-boron characteristics similar to those of BASHY, no chiroptical investigation has been conducted yet.
Building on our previous interest in developing chiral dyes,15b in this communication, we describe the development of structurally diverse novel fluorophores (1a–h) through a tri-component reactions of commercially available salicylaldehyde derivatives (2a–c) and arylboronic acids (3a–c) with readily accessible 2-formylpyrrole hydrazones (4a–d) in a single step (Scheme 1). The stereogenic boron center in these compounds provides access to unique chiral-at-boron fluorophores (1a–h) through enantiomeric resolution on chiral HPLC column.
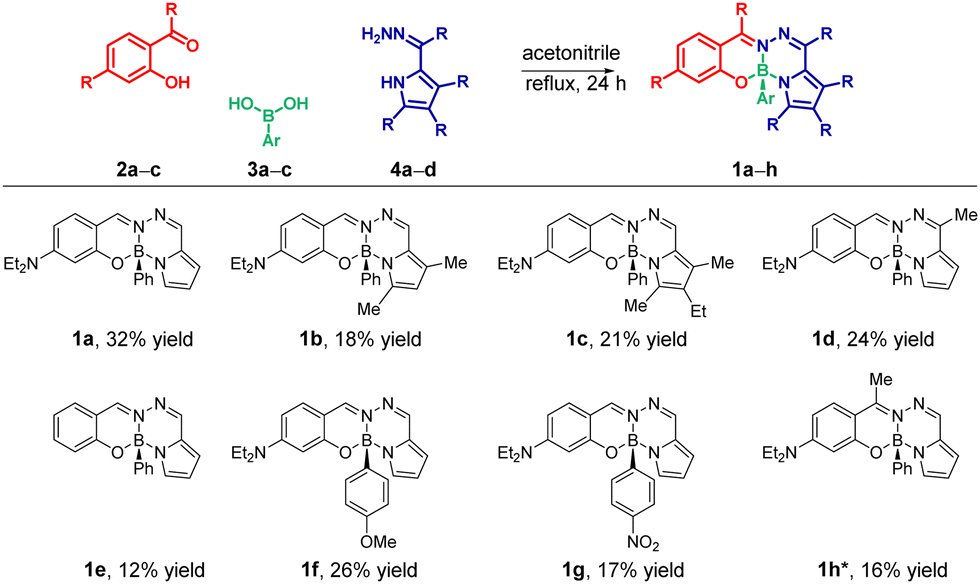 | ||
Scheme 1 Tri-component modular synthesis of BOSPYR dyes (1a–h): substrate scope. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The synthesis of compound 1h was achieved by employing the hydrazone of salicylaldehyde (2c) and the pyrrole-2-carboxaldehyde (see ESI† for the details). The synthesis of compound 1h was achieved by employing the hydrazone of salicylaldehyde (2c) and the pyrrole-2-carboxaldehyde (see ESI† for the details). | ||
This assembly system is designed with two key features in mind: ready availability of starting materials and (2) facile diversification potential through the use of substrates with diverse substitution patterns. Capitalizing on these advantages, we successfully synthesized compound 1a in a reasonable yield of 32% under optimized reaction condition (see ESI†). The reaction involved refluxing an equimolar mixture of 4-(diethylamino)salicylaldehyde (2a), phenylboronic acid (3a), and the hydrazone of pyrrole-2-carbaldehyde (4a) in acetonitrile for 24 hours (Scheme 1). The chemical structure of 1a was unequivocally confirmed by NMR spectroscopy, high-resolution mass spectrometry (HRMS), and single-crystal X-ray diffraction analysis (see ESI† and Fig. 3). Next, to explore the reaction's versatility, we performed the reaction with structural variations at each component: the pyrrole (1a–d), the salicyl-aldehyde/-ketone (1a, 1e, and 1h), and the arylboronic acid (1a, 1f, and 1g) units. The herein developed one-step, tri-assembly MCRs served as an enabling platform for efficient synthesis of fused tetracyclic boron-chelates 1a–h, despite the moderate yields (12–32%, Scheme 1), for which conventional synthetic approaches failed to give any trace of product (Scheme S5, ESI†).
The model dye 1a exhibited very low solvent-dependent absorbance and fluorescence—solvatochromism (Fig. S40, ESI†). It absorbs maximally at around 450 nm and emit at around 530, displaying large Stokes shifts (Δλ ∼ 3000 cm−1).18 Following the work by Hao and coworkers17c on the aggregation-induced emission (AIE) properties of structurally similar BOSPY dyes, we investigated the emission properties of our BOSPYR dyes, in water–DMSO mixtures (Fig. S41, ESI†). The fluorescence intensity exhibits negligible enhancement, gradually increasing up to 50% water fraction, followed by a sharp decrease at higher water fractions, likely due to aggregation or precipitation (Fig. S41, ESI†). To understand the dimmed fluorescence emissions of 1a in organic solvents, we recorded emission spectra in glycerol–ethanol mixtures with varying glycerol fractions (Fig. 2a). The fluorescence intensity increased by 21-fold when switching from pure ethanol to pure glycerol. The enhanced fluorescence observed in viscous glycerol medium suggests non-radiative vibrational relaxations as a key quenching mechanism for the dye 1a, making it a promising tool for studying biological processes associated with increased extracellular viscosity.19,20
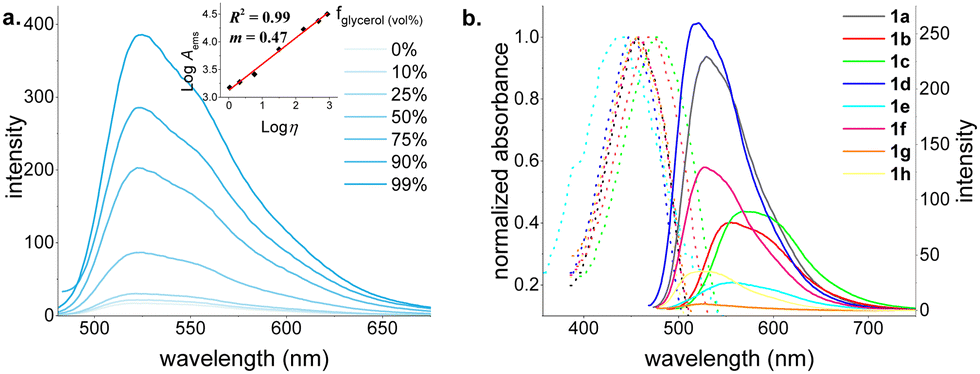 | ||
| Fig. 2 (a) Fluorescence emission spectra of 1a in glycerol–ethanol mixtures with varying glycerol fractions [fglycerol (vol%): 0–99%] at 25 °C. Integrated area ratio of fglycerol(0%)/fglycerol(99%) ∼ 1/21. (b) Normalized UV-vis absorption and fluorescence emission spectra of the BOSPYR dyes (1a–h, 2 μM) in DMSO. | ||
 | ||
| Fig. 3 (a) X-ray molecular structure of 1a with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level. (b) Dihedral angle between chelating mean planes defined by atoms N1–C10–C11–N3 and C13–C18–O1–C12; α = 17.2°. | ||
To understand the influence of structural variations on optical properties, we recorded absorption and fluorescence emission spectra for all BOSPYR dyes (1a–h) in DMSO (Fig. 2b and Table S1, ESI†). All dyes exhibit large Stokes shifts (2633–4930 cm−1) and weak emission (Φfl: 1.5–9.1%)21 with fluorescence lifetimes ranging from 0.47 to 2.00 ns, all characterized by a monoexponential fit. Alkyl substitution on the pyrrole unit (1a to 1c; entries 1–3, Table S1, ESI†) induces a bathochromic shift (530–573 nm). A more pronounced red-shift is observed for ethyl substitution at the pyrrole's 3-position (1c) compared to dimethylation (1b). Electrostatic potential surface calculations for compounds 1b and 1c (Fig. S51, ESI†) indicate that alkyl groups with mild electron-donating character decrease the energy required for reorganizing the delocalized π-network by increasing electron density on the pyrrole ring. This lowers the energy gap between ground and excited states, resulting in the observed bathochromic shift. Compared to 1a, compound 1d exhibits a small but noticeable blue shift in both absorption and emission maxima, accompanied by a 3-fold enhancement in quantum yield. Interestingly, removing the –NEt2 group on the salicylaldehyde unit (1avs.1e) leads to divergence of the absorption and emission maxima. While the absorption maximum of 1e exhibits a 20 nm blue shift, its emission maximum undergoes a 27 nm red shift, resulting in the largest Stokes shift of 4930 cm−1 (see Table S1, ESI†). This modification comes at the expense of a five-fold decrease in molar absorptivity and reduced chemical stability (Fig. S45, ESI†). The absence of the strong electron-donating –NEt2 group is believed to destabilize compound 1e, leading to lower electron density and reduced π-electron delocalization. Consequently, this results in a smaller transition dipole moment and hence a lower molar absorptivity compared to the others. Although thermally somewhat stable, it readily degrades upon exposure to mild acids and bases, and even undergoes solvolysis by DMSO (Fig. S46 and S47, ESI†). This is consistent with its reduced aromaticity revealed by DFT computation (vide infra). To explore the influence of the electronic nature of the pendant aryl unit, we synthesized dyes 1f and 1g simply by varying arylboronic acid substrate. As expected, the absorption and emission maxima of these dyes were nearly identical to those of the model dye 1a, consistent with electronic decoupling between the aryl unit and the chromophore. The minimal influence of electronic effects on the absorption and emission properties of the aryl unit introduced via the arylboronic acid module makes this position particularly attractive for a broad range of labeling applications.
To gain detailed structural insight, single crystals of 1a suitable for X-ray diffraction analysis were obtained by slow evaporation from methanol (Fig. 3 and Fig. S53, ESI†). The structure confirms the exact conformation of 1a, which crystallizes as a racemate in the centrosymmetric triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Both enantiomers occupy the asymmetric unit with opposite configurations at the boron center. Complexation around the boron atom leads to a four-fused ring system. The B–N chelate bonds within each molecule range from 1.515 to 1.588 Å. Additionally, B–O coordination bonds are observed with B1–O1 (1.482(3) Å) and B2–O2 (1.468(3) Å). The boron atoms exhibit slightly distorted tetrahedral geometries, displaced by 0.569 Å (B1) and 0.560 Å (B2) from the planes defined by the tri-coordinated O1/N1/N2 and O2/N5/N6 atoms, respectively. The dihedral angle between the mean planes of N1–C10–C11–N3 and C13–C18–O1–C12 is 17.2 degrees (Fig. 3).
space group. Both enantiomers occupy the asymmetric unit with opposite configurations at the boron center. Complexation around the boron atom leads to a four-fused ring system. The B–N chelate bonds within each molecule range from 1.515 to 1.588 Å. Additionally, B–O coordination bonds are observed with B1–O1 (1.482(3) Å) and B2–O2 (1.468(3) Å). The boron atoms exhibit slightly distorted tetrahedral geometries, displaced by 0.569 Å (B1) and 0.560 Å (B2) from the planes defined by the tri-coordinated O1/N1/N2 and O2/N5/N6 atoms, respectively. The dihedral angle between the mean planes of N1–C10–C11–N3 and C13–C18–O1–C12 is 17.2 degrees (Fig. 3).
The presence of a stereogenic boron atom in all BOSPYR dyes (1a–h) prompted us to investigate the chiroptical properties of enantiomerically pure 1a. (±)-1a was resolved via high-performance liquid chromatography (HPLC) using an analytical chiral column (Chiralcel® OD-H) under isocratic conditions (eluant: n-hexanes:2-propanol; 98:2). Gratifyingly, chiral HPLC separations were well achieved for the remaining racemic dyes (1b–h) as well (see ESI† for methods). The presence of two well-separated peaks with comparable integrated areas in the chiral HPLC chromatogram confirms the successful separation of enantiomers (Fig. S9, ESI†). The isolated enantiomers exhibited significant optical activity ([α]25D −2054.8° vs. +2082.2°, CHCl3), uncommon for small, point-chiral organic molecules.13–15 This mirror-image relationship in their specific rotations (almost equal but opposite in sign) is another strong indication of their enantiomeric nature. To assess the configurational stability of dye 1a, toluene solutions of (−)-1a were refluxed under open-air atmosphere for 1 hour. Gratifyingly, no epimerization was observed with chiral HPLC analysis (see Section S5.2 in ESI†), indicating high configurational stability. Compound (±)-1a demonstrated high stability towards light, acids (except acids like TFA), and bases (Fig. S43 and S44, ESI†).
The ECD spectra of (+)-1a and (−)-1a confirmed their relative configurations (Fig. S10, ESI†). The large specific rotations and distinct Cotton effects (|gabs| ∼ 1.9 × 10−3, λext 457 nm) suggest that the twisted planarity of the molecule, revealed by the X-ray structure, should contribute significantly to its pronounced chiroptical activity and inherent chirality.
To understand the excited state properties, computational studies were performed using DFT and TD-DFT methods. B3LYP-D3 provided the best agreement with experimental UV-vis spectra, accurately reproducing dihedral angle (17.3° vs. 17.2°) and λabs values (Fig. S49, ESI†). Vibrational frequency calculations and excited state geometry optimizations confirmed the thermal and photostability of most dyes. An exception was observed in molecule 1e, where the first triplet excited state exhibited ring deformations due to reduced aromaticity. The structure of 1e was also characterized by its low dipole moment and polarizability in the absence of the –NEt2 group, which explains the observed emission trends (Table S3, ESI†). The calculated UV-vis spectra for the first 30 singlet excited states agreed well with experimental data, including the red-shifted peaks observed in 1b and 1c (Fig. S49, ESI†). Analysis of natural transition orbitals for the first singlet excited state (S1) of 1a–1h (Fig. 4 and Table S4, Fig. S51, ESI†) revealed that the main absorption involves delocalized π-electrons across the chromophore, extending to the diethyl amine group upon excitation. This S0 → S1 excitation exhibits a locally excited (LE) state character, as evidenced by the redistribution of electron density within the conjugated ring structure, which accounts for the weak fluorescence (Fig. 4 and Fig. S51, S52, ESI†). This phenomenon is further corroborated by ground-state and excited-state atomic charge distribution analyses (Fig. S54, ESI†).22 Interestingly, theoretical calculations could not reproduce the experimental fluorescence spectra. Key computed parameters, including a singlet–triplet energy gap (ΔEST) of 0.75 eV, a reorganization energy (λS1/T1–S1/S1) of 0.2 eV, and spin–orbit coupling matrix elements (SOCME) for S1/T1 of 9.54 × 10−5 eV for 1a, suggest a low reverse intersystem crossing rate (kRISC) of 3.13 × 10−11. These findings rule out the possibility of these dyes acting as thermally activated delayed fluorescence (TADF) emitters. Additionally, the optimized geometry of the first excited state revealed a rotation of the B–Ph group, which may contribute to the observed enhancement of emission in viscous media (Fig. S48, ESI†).
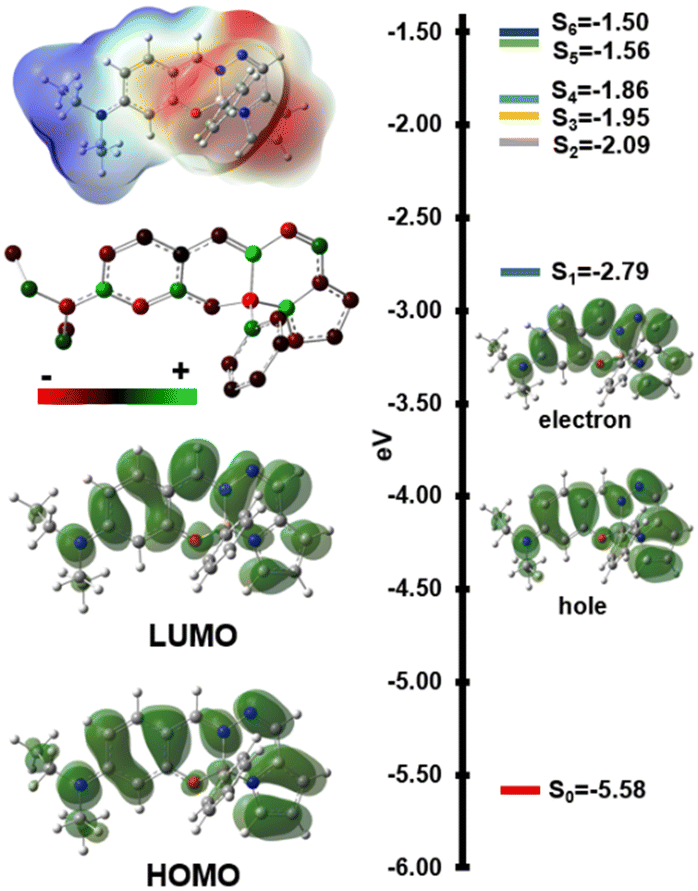 | ||
| Fig. 4 ESP, charge distribution, HOMO and LUMO surfaces, singlet energy levels and natural transition orbitals of S0 → S1 transition for 1a. | ||
The HOMO–LUMO energy gap decreased from 2.8 eV to 2.3 eV upon excited state geometry optimization, highlighting the influence of geometry relaxation on the emission spectra (e.g. large Δλ) and supporting the solvent-independent emissions observed experimentally (Fig. S40, ESI†). This relaxation also led to a good correlation between the electrochemical bandgap calculated through differential pulse voltammetry (DPV) experiments and the computed optical bandgap (2.27 eV vs. 2.30 eV, Fig. S47 and Table S2, ESI†).
In conclusion, this work presents a powerful approach for the synthesis of diverse stereogenic-at-boron BOSPYR fluorochromes through a straightforward one-step MCR. This method offers several advantages: it utilizes readily available starting materials, enables creation of fluorophores with an asymmetry at the boron atom, and yields visible light-absorbing dyes with large Stokes shifts and weak fluorescence that can be significantly enhanced in viscous media, making them promising viscosity imaging probes. Additionally, the inherent chirality of these fluorophores, arising from the bent conformation of the chromophore, evidenced by the large specific rotations and distinct Cotton effects of 1a, opens exciting possibilities for their development as chiroptical tools. The high configurational stability of B-stereocenter in these chelates further highlight them as reliable chiral fluorochromes. The combination of efficient synthesis, tunable properties, and inherent chirality positions these dyes as versatile, promising building blocks for advanced functional materials.
Data availability
Additional data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science + Business Media, New York, 3rd edn, 2006 CrossRef; (b) H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments, Wiley-VCH, 2003 Search PubMed.
- (a) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290 CrossRef PubMed; (b) H. Kim, A. Burghart, M. B. Welch, J. Reibenspies and K. Burgess, Chem. Commun., 1999, 1889 RSC; (c) W. Zhao and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 1677 CrossRef PubMed; (d) X. Li, G. Zhang and Q. Song, Chem. Commun., 2023, 59, 3812 RSC.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef PubMed; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed; (c) N. Boëns, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577 CrossRef CAS.
- (a) L. Levi and T. J. J. Müller, Chem. Soc. Rev., 2016, 45, 2825 RSC; (b) F. de Moliner, N. Kielland, R. Lavilla and M. Vendrell, Angew. Chem., Int. Ed., 2017, 56, 3758 CrossRef CAS PubMed; (c) L. Brandner and T. J. J. Müller, Front. Chem., 2023, 11, 1124209 CrossRef CAS PubMed; (d) A. Vázquez-Romero, N. Kielland, M. J. Arévalo, S. Preciado, R. J. Mellanby, Y. Feng, R. Lavilla and M. Vendrell, J. Am. Chem. Soc., 2013, 135, 16018 CrossRef PubMed.
- F. M. F. Santos and J. N. Rosa, et al. , Chem. – Eur. J., 2016, 22, 1631 CrossRef PubMed.
- P. M. S. D. Cal and F. Sieglitz, et al. , Chem. Commun., 2017, 53, 368 RSC.
- M. M. Alcaide and F. M. F. Santos, et al. , J. Org. Chem., 2017, 82, 7151 CrossRef CAS PubMed.
- V. G. Jiménez and F. M. F. Santos, et al. , J. Org. Chem., 2018, 83, 14057 CrossRef PubMed.
- F. M. F. Santos and Z. Domínguez, et al. , ChemPhotoChem, 2018, 2, 1038 Search PubMed.
- B. Zhang, S. Wang, J. Tan and X. Zhang, Dyes Pigm., 2018, 155, 186 Search PubMed.
- M. J. S. A. Silva and Y. Zhang, et al. , Bioconjugate Chem., 2023, 34, 2337 Search PubMed.
- H. Lu and J. Mack, et al. , Coord. Chem. Rev., 2016, 318, 1 CAS.
- (a) T. E. Wood and N. D. Dalgleish, et al. , J. Am. Chem. Soc., 2005, 127, 5740 CrossRef; (b) E. M. Sánchez-Carnerero and F. Moreno, et al. , Chem. Commun., 2013, 49, 11641 RSC; (c) S. Kolemen, Y. Cakmak, Z. Kostereli and E. U. Akkaya, Org. Lett., 2014, 16, 660 CrossRef PubMed; (d) E. M. Sánchez-Carnerero and F. Moreno, et al. , J. Am. Chem. Soc., 2014, 136, 3346 Search PubMed; (e) R. I. Lerrick and T. P. L. Winstanley, et al. , Chem. Commun., 2014, 50, 4714 RSC; (f) T. Bruhn and G. Pescitelli, et al. , Angew. Chem., Int. Ed., 2014, 53, 14592 CrossRef; (g) Y. Wu, S. Wang, Z. Li, Z. Shen and H. Lu, J. Mater. Chem. C, 2016, 4, 4668 RSC; (h) Y. Gobo, M. Yamamura, T. Nakamura and T. Nabeshima, Org. Lett., 2016, 18, 2719 CrossRef PubMed; (i) M. Saikawa, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 10727 RSC; (j) R. B. Alnoman and S. Rihn, et al. , Chem. – Eur. J., 2016, 22, 93 CrossRef PubMed; (k) R. Clarke and K. L. Ho, et al. , ChemPhotoChem, 2017, 1, 513 CrossRef CAS; (l) J. Jiménez and L. Cerdán, et al. , J. Phys. Chem. C, 2017, 121, 5287 CrossRef PubMed; (m) M. Toyoda, Y. Imai and T. Mori, J. Phys. Chem. Lett., 2017, 8, 42 CrossRef CAS; (n) Y. Gobo, R. Matsuoka, Y. Chiba, T. Nakamura and T. Nabeshima, Tetrahedron Lett., 2018, 59, 4149 CrossRef CAS; (o) C. Maeda, K. Nagahata, T. Shirakawa and T. Ema, Angew. Chem., Int. Ed., 2020, 59, 7813 CrossRef CAS PubMed; (p) H. Sakai and Y. Suzuki, et al. , J. Mater. Chem. C, 2023, 11, 2889 RSC.
- (a) A. Hafele, C. Zedde, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2010, 12, 1672 CrossRef PubMed; (b) M. Işık, E. Dündar, E. Şahin and C. Tanyeli, Chem. Commun., 2022, 58, 7188 RSC; (c) B. Zu, Y. Guo, L.-Q. Ren, Y. Li and C. He, Nat. Synth., 2023, 2, 564 CrossRef; (d) R. G. Clarke and J. Weatherston, et al. , ChemPhotoChem, 2023, 7, e202200194 CrossRef CAS; (e) C. Ray and E. Avellanal-Zaballa, et al. , Org. Chem. Front., 2023, 10, 5834 RSC.
- (a) N. Algoazy and J. G. Knight, et al. , Chem. – Eur. J., 2021, 27, 5246 CrossRef CAS PubMed; (b) B. Zu, Y. Guo and C. He, J. Am. Chem. Soc., 2021, 143, 16302 CrossRef CAS PubMed; (c) Y. Sun and C. Yu, et al. , Chem. Commun., 2023, 59, 13986 RSC; (d) G. Zhang and Z. Zhang, et al. , Nat. Commun., 2022, 13, 2624 CrossRef PubMed.
- (a) N. Chen and W. Zhang, et al. , Org. Lett., 2017, 19, 2026 CrossRef PubMed; (b) J. Wang and X. Fang, et al. , Org. Lett., 2021, 23, 4796 CrossRef PubMed; (c) H. Wang and X. Guo, et al. , Dyes Pigm., 2023, 210, 111013 CrossRef.
- A. Dhara and T. Sadhukhan, et al. , J. Am. Chem. Soc., 2020, 28, 12167 CrossRef PubMed.
- K. Bera and A. Kiepas, et al. , Nature, 2022, 611, 365 CrossRef PubMed.
- M. Paez-Perez and M. K. Kuimova, Angew. Chem., Int. Ed., 2024, 63, e202311233 CrossRef PubMed.
- Relative fluorescence quantum yields were calculated using quinine sulphate as the reference (Φfl = 0.60 in 0.5 M H2SO4). See: A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213 CrossRef.
- F. Weinhold and J. E. Carpenter, in The Structure of Small Molecules and Ions, ed. R. Naaman and Z. Vager, Plenum, 1988, pp. 227–236 DOI:10.1007/978-1-4684-7424-4.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details. CCDC 2344453. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03956d |
| This journal is © The Royal Society of Chemistry 2025 |