
Theoretical study of p-block metal–nitrogen–carbon single-atom catalysts for heterogeneous Fenton-like reaction†
Chen
Zhou
a,
Haobin
Tan
a,
Shengbo
Wang
a,
Qiang
Liu
b,
Zhenhui
Xu
b,
Peng
Zhang
 *a and
Chun
Hu
*a
*a and
Chun
Hu
*a
aKey Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Institute of Environmental Research at Greater Bay, Guangzhou University, Guangzhou 510006, China. E-mail: pengzhang85@foxmail.com; huchun@gzhu.edu.cn
bHouma Special Industry Factory, Houma 043000, China
First published on 8th November 2024
Abstract
The heterogeneous Fenton-like reaction has been widely used in water purification and environmental remediation due to the highly reactive nature of hydroxyl radicals. Nevertheless, the intrinsic structure–activity relationship for heterogeneous Fenton-like reaction catalysts remains to be clarified. Metal/nitrogen/carbon (M/N/C) single-atom catalysts (SACs) provide an ideal opportunity to reveal the relationship between the structure and activity. In this work, the detailed catalytic mechanism and activity of H2O2 decomposition on p-block main-group metal/nitrogen/carbon (PM/N/C) catalysts were investigated systematically. A volcano relationship between the catalytic activity and the adsorption energies of reaction intermediates was found for H2O2 decomposition on PM/N/C SACs. PM-N2C2 and PM-C4 exhibit higher H2O2 decomposition activity than PM-N4, indicating that reducing the N/C ratio in the coordination environment can effectively adjust the catalytic activity. By altering the N/C coordination environment, it is possible to modify the p-band position of p-block main-group metal atoms in PM/N/C SACs, thereby enhancing the catalytic activity of H2O2 decomposition.
Environmental significanceThe heterogeneous Fenton process was considered as a promising strategy for the removal of organic pollutants from wastewater. Establishing the structure–activity relationship is crucial for the rational design of efficient heterogeneous Fenton catalysts. Metal/nitrogen/carbon (M/N/C) single-atom catalysts (SACs) provide an ideal opportunity to reveal the intrinsic structure–activity relationship. Herein, a volcano relationship was found between the catalytic activity of H2O2 decomposition and the adsorption strength of the reaction intermediates and the p-band center of p-block main-group metal/nitrogen/carbon (PM/N/C) catalysts. Furthermore, modifying the coordination environment can modulate the p-band position of PM atoms and enhance the catalytic activity of PM/N/C. This work can provide a valuable principle for the rational design of heterogeneous Fenton catalysts for efficient decontamination application. |
Introduction
With the rapid development of urbanization and industrialization, environmental pollution caused by the continuous release of toxic agents into water environments has become an overwhelming worldwide issue, particularly in developing and underdeveloped countries.1–3 The Fenton reaction, known as one of the most efficient advanced oxidation processes, is widely utilized for degrading persistent organic contaminants in wastewater treatment.4,5 Unlike the traditional homogeneous Fenton process, which involves the reaction of hydrogen peroxide (H2O2) with dissolved ferrous ions, the heterogeneous Fenton process offers significant advantages such as functionality across a wide range of pH values, minimal metal leaching, controlled iron sludge precipitation, and convenient catalyst recycling.6–8The hydroxyl radicals (·OH) generated from the catalytic decomposition of H2O2 on heterogeneous catalysts can oxidize various organic pollutant molecules with high activity and non-selectivity.9–11 Gao et al. found that sulfurized CoFe2O4 ensured effective H2O2 activation and sufficient generation of oxygen-containing radicals such as the hydroxyl radical and superoxide radical, which facilitated pollutant degradation.12 Cao et al. found that N-doped hierarchically porous carbon with embedded FeOx exhibited high activity and selectivity for H2O2 generation as well as effective H2O2 activation to ·OH.13 Up to now, most published research focused on H2O2 adsorption, activation and hydroxyl radical production.14–16 Although significant progress has been made, a comprehensive understanding of the detailed catalytic cycle of heterogeneous Fenton-like reactions remains to be clarified.17,18 Gaining a deep understanding of the physical and chemical mechanisms behind the decomposition reaction of H2O2 is important for the exploration of novel heterogeneous Fenton-like reaction catalysts.19,20 It is crucial to understand the intrinsic relationship between the catalytic activity and the surface atomic structure of heterogeneous Fenton-like reaction catalysts.21,22
Metal/nitrogen/carbon (M/N/C) single-atom catalysts (SACs) have attracted more and more attention as heterogeneous Fenton-like reaction catalysts because they hold great potential to provide unmatched high activity and selectivity for Fenton and Fenton-like reactions.23–27 Nevertheless, these investigations primarily focused on transition-metals as active centers, while p-block main-group metals (PM) have been less explored.28–32 Exploring diverse M/N/C configurations such as introducing extra metals or altering the coordination environment with different ligands is essential for modifying electronic and catalytic characteristics.33
The catalytic activity of transition-metal SACs benefit from the localized character of d orbitals, while that of PM atoms originates from the hybridized p orbitals.34–36 Although p-block main-group metal/nitrogen/carbon (PM/N/C) SACs are less reported for H2O2 decomposition, they show superior catalytic ability in electrocatalytic processes to achieve high activity and selectivity.37,38 Unlike SACs based on transition metals, main-group metal SACs possess several unique characteristics. Firstly, C atoms can easily stabilize central main-group metals with covalent bonds rather than coordinating with N atoms, which contributes to the structural stability of single metal atoms (SAs). Secondly, main-group metals usually have the inherent poor ability of hydrogen adsorption, which can improve the reaction selectivity. Finally, experimental and theoretical research studies indicate that adjusting the electronic configuration of s/p orbitals in main-group metal SACs to a particular state can enhance the absorption of reactive entities. Recently, Jiang et al. found that Sb/N/C consisting of Sb–N4 moieties anchored on N-doped carbon nanosheets could act as a CO2 reduction reaction catalyst to produce formate efficiently.39 Wu et al. demonstrated that Al and Ga can act as promising active centers in SACs toward the NO reduction reaction by the modulation of p-band filling of the PM atoms.40 These pioneering research studies have confirmed that the p electrons in PM atoms can also be activated through engineering PM/N/C SACs, resulting in promising catalytic activity.41,42 PM/N/C SACs, which featured atomically dispersed PM atoms with activating p electrons, could possess significant potential for heterogeneous catalytic reactions.43–45 However, the potential of PM/N/C SACs as heterogeneous Fenton-like reaction catalysts still needs to be explored. The intrinsic structure–activity relationship remains to be clarified, which is necessary for the rational design of PM SACs.46
In this work, the mechanism and catalytic activity of H2O2 decomposition on PM-N4, PM-N2C2, and PM-C4 moieties embedded in graphene were investigated based on density functional theory (DFT).47 It was found that these PM/N/C SACs exhibit excellent catalytic activity toward H2O2 decomposition by changing the N/C coordination environment and modulating the p-band filling of the PM centers.48 This work firmly demonstrates that PM atoms can serve as promising active centers in SACs toward H2O2 decomposition. This work not only elucidates the catalytic mechanism and reaction pathways of H2O2 decomposition, but also clarifies the intrinsic structure–activity relationship between PM/N/C SACs and their catalytic performance. Our work theoretically introduced PM atoms as active centers in SACs for H2O2 decomposition, which provides a potential new avenue for the rational design of heterogeneous Fenton-like catalysts, and sheds light on the further development of advanced oxidation process (AOP) catalysts for efficient decontamination application.
Results and discussion
Geometry structure and stability of PM/N/C SACs
Di-vacancy pyridine graphene, which can provide a range of coordination modes for active centers, has shown great stability and activity from the viewpoint of theoretical and experimental contexts.49 In this work, pyridine graphene's di-vacancy defect site was selected to support the dispersed PM atoms.50 In order to investigate the effect of the coordination number of N-atoms surrounding the metal atoms, three configurations (PM-N4, PM-N2C2, and PM-C4 shown in Fig. 1a) were considered to support the nine PM atoms (Al, Ga, In, Tl, Ge, Sn, Pb, Sb, Bi shown in Fig. 1b), resulting in 27 PM/N/C SACs.51 The full geometric optimization structures of the 27 PM/N/C SACs are illustrated in Fig. S1–S3 of the ESI.† Note that there are three allotropic structures for PM-N2C2 (Fig. S4†) and the most stable configuration with two para N atoms was considered. The corresponding energies of the three structures of PM-N2C2 are shown in Table S1 of the ESI.† The adsorption energies of the PM atoms in PM/N/C SACs and the cohesive energies of the nine PM atoms were calculated and summarized in Fig. 1c. The adsorption energy and cohesive energy decrease with the increase of the periodic number for PM atoms in the same group due to the decrease of electronegativity.52 In the three configurations, PM-N2C2 tends to show higher adsorption energy of the central metal atoms than those of PM-N4 and PM-C4, which suggests that PM-N2C2 is more stable. As depicted in Fig. 1c, the adsorption energies of PM in PM/N/C SACs (except TlC4 and BiN4) are higher than the cohesive energies of the corresponding bulk metals, indicating that the nine PM atoms can anchor stably on the majority of substrates except TlC4 and BiN4. In order to further investigate the thermodynamic stability, ab initio molecular dynamics (AIMD) simulation was conducted to evaluate the thermal stability of PM/N/C. As shown in Fig. S5,† the total energy oscillated within a fluctuation of less than 0.003 eV per atom and the geometric structure of InC4 was perfectly retained without obvious distortion up to 10 ps at 300 K, further confirming its high stability and experimental feasibility. Therefore, the studied PM/N/C SAC moieties are favorable for experimental realization. Both the Mulliken and Hirshfeld charge analyses show that the supported PM atom donates electrons to the substrate in all the PM/N/C SACs, as listed in Table S2.† The resultant positively charged metal atoms become active sites for catalytic reactions.53 | ||
| Fig. 1 (a) Schematic illustrations of PM-N4, PM-N2C2, and PM-C4 structures. (b) PM atoms considered in this work. (c) The adsorption energies between the PM atoms and the adjacent N and C atoms in PM-N4, PM-N2C2, and PM-C4, and the cohesive energies (Ecoh) of PM. | ||
Reaction pathways of H2O2 decomposition
Given that recently published work about H2O2 dissociation always focused on the dissociation of H2O2 and the generation of hydroxyl radicals, understanding the complete reaction cycle for H2O2 decomposition was necessary. The decomposition of H2O2 on PM/N/C is a complex process in which a variety of close-shell species (H2O2, H2O, O2, H2) and reaction intermediates (OH, OOH, H, O) can be present.54 Various elementary reactions were involved (Table S3†) based on those close-shell species and reaction intermediates, which include adsorption, O–O bond scission, dehydrogenation, hydrogen transfer, and desorption.19 Here, we further compiled possible pathways for H2O2 decomposition on PM/N/C (as shown in Fig. 2), including the OH*-assisted pathway (path I), direct dehydrogenation pathway (path II), O*-assisted pathway (path III), and O*–O* recombination pathway (path IV). A complete pathway for direct dehydrogenation (path II) was not shown for simplicity, because the O–H bond in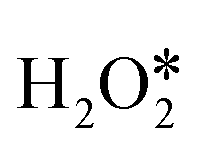 (* denotes the adsorbed state of the reactant molecule) is more difficult to be broken than the O–O bond (as shown in Table S4†). For the other three pathways, the first reaction step is the adsorption of H2O2 followed by the O–O bond scission to form two OH* or one O* and one H2O*. Then the second H2O2 can adsorb and undergo continuous hydrogen-transfer steps with OH* in path I or O* in path III. Moreover, the second
(* denotes the adsorbed state of the reactant molecule) is more difficult to be broken than the O–O bond (as shown in Table S4†). For the other three pathways, the first reaction step is the adsorption of H2O2 followed by the O–O bond scission to form two OH* or one O* and one H2O*. Then the second H2O2 can adsorb and undergo continuous hydrogen-transfer steps with OH* in path I or O* in path III. Moreover, the second 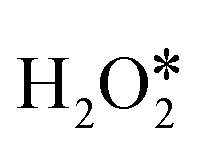 can directly decompose to form H2O* and O*, and O* can recombine with the O* formed in the first H2O2 dissociation step to form
can directly decompose to form H2O* and O*, and O* can recombine with the O* formed in the first H2O2 dissociation step to form 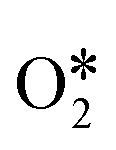 (path IV). At last, the final products O2 and H2O desorbed from the surface of the catalyst and the catalyst was refreshed. These pathways show a detailed and complete catalytic cycle for H2O2 decomposition, which will assist in developing efficient Fenton-like systems.
(path IV). At last, the final products O2 and H2O desorbed from the surface of the catalyst and the catalyst was refreshed. These pathways show a detailed and complete catalytic cycle for H2O2 decomposition, which will assist in developing efficient Fenton-like systems.
 | ||
| Fig. 2 Reaction pathways of H2O2 decomposition. | ||
Here, the reaction energies and barrier energies of all elementary reactions involved in the four pathways of H2O2 decomposition on the 27 PM SACs were calculated based on DFT calculations, which can help to establish the complete reaction energy diagram and then characterize the catalytic activity.55 By comparing the barrier energies of the rate-determining step in four pathways, the favourable reaction pathway can be determined.56
Reaction pathways and activity of H2O2 decomposition on the PM-N4 SACs
Based on the four pathways of H2O2 decomposition mentioned above, the detailed catalytic mechanism was explored. InN4 was first taken as an example (shown in Fig. 3). H2O2 adsorption is the first step in the catalytic cycle of H2O2 decomposition with an adsorption energy of −0.37 eV. The adsorbed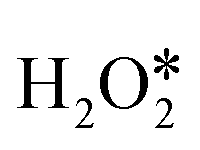 can be directly dehydrogenated to form H* and OOH* by the O–H bond scission, or dissociated to form two OH* or one O* atom and one H2O* by the O–O bond scission. As shown in Fig. 3c, direct dehydrogenation of
can be directly dehydrogenated to form H* and OOH* by the O–H bond scission, or dissociated to form two OH* or one O* atom and one H2O* by the O–O bond scission. As shown in Fig. 3c, direct dehydrogenation of 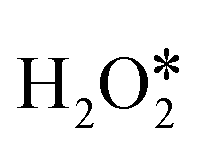 needs to conquer a high barrier energy of 1.11 eV, indicating the low efficiency of this process. For comparison, the decomposition of H2O2 to form two OH* or O* and H2O* via O–O bond scission only needs to overcome a small barrier energy of 0.08 eV and 0.11 eV, respectively, suggesting the high reaction rate of these two elementary steps. As the O–O bond scission is much more favourable than
needs to conquer a high barrier energy of 1.11 eV, indicating the low efficiency of this process. For comparison, the decomposition of H2O2 to form two OH* or O* and H2O* via O–O bond scission only needs to overcome a small barrier energy of 0.08 eV and 0.11 eV, respectively, suggesting the high reaction rate of these two elementary steps. As the O–O bond scission is much more favourable than 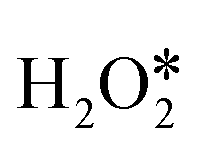 direct dehydrogenation due to the significantly lower energy barriers, one conclusion can be made, that is, the O–O bond breaking is the major pathway for the first adsorbed
direct dehydrogenation due to the significantly lower energy barriers, one conclusion can be made, that is, the O–O bond breaking is the major pathway for the first adsorbed 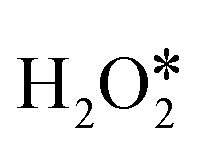 dissociation, excluding the possibility of
dissociation, excluding the possibility of  direct dehydrogenation (path II).57 Furthermore, the reaction energies of
direct dehydrogenation (path II).57 Furthermore, the reaction energies of  and
and  are −2.88 eV and −2.15 eV, respectively, indicating that H2O2 can split into two OH* or one O* and one H2O* easily on InN4. ·OH can generate at the transition states of H2O2 dissociation to form two OH* or O* and H2O* (Fig. 3c), indicating that ·OH can be easily formed on the InN4 surfaces. And then, the second H2O2 can adsorb and undergo a hydrogen transfer step to the adsorbed OH* in path I and O* in path III with energy barriers of 0.21 and 0 eV, respectively. Meanwhile, the adsorbed second
are −2.88 eV and −2.15 eV, respectively, indicating that H2O2 can split into two OH* or one O* and one H2O* easily on InN4. ·OH can generate at the transition states of H2O2 dissociation to form two OH* or O* and H2O* (Fig. 3c), indicating that ·OH can be easily formed on the InN4 surfaces. And then, the second H2O2 can adsorb and undergo a hydrogen transfer step to the adsorbed OH* in path I and O* in path III with energy barriers of 0.21 and 0 eV, respectively. Meanwhile, the adsorbed second  can directly decompose to form H2O* and O* with an energy barrier of 1.59 eV, which is much higher than that of hydrogen transfer steps as shown in Fig. 3d, indicating that the hydrogen transfer step for the second adsorbed
can directly decompose to form H2O* and O* with an energy barrier of 1.59 eV, which is much higher than that of hydrogen transfer steps as shown in Fig. 3d, indicating that the hydrogen transfer step for the second adsorbed 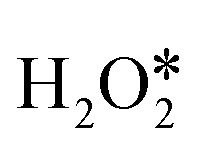 molecule is more energetically favorable than the direct dissociation step (path IV). Therefore, only path I and path III were discussed in the following part of this work.
molecule is more energetically favorable than the direct dissociation step (path IV). Therefore, only path I and path III were discussed in the following part of this work.
 | ||
Fig. 3 Reaction energies (a) and barrier energies (b) of elementary reactions along the four possible pathways for H2O2 decomposition on InN4. The energy diagrams of the first (c) and second (d) adsorbed  decomposition. decomposition. | ||
Due to the low efficiency of path II and path IV for H2O2 decomposition on InN4, the reaction energy diagrams of the preferable pathways (path I and path III) were identified from the viewpoint of thermodynamics (shown in Fig. 4). In path I, the second  adsorbed with an adsorption energy of −1.04 eV and then transfers an H atom to OH* to form H2O*, characterized by an energy barrier of 0.21 eV and a reaction energy of 0.05 eV. The formed H2O* was desorbed from the InN4 surface with a desorption energy of 0.55 eV. Additionally, the OOH* adsorbed on InN4 undergoes the second H-transfer step to the other OH* with a reaction energy and barrier energy of 0.70 and 0.71 eV, respectively. The final products H2O* and
adsorbed with an adsorption energy of −1.04 eV and then transfers an H atom to OH* to form H2O*, characterized by an energy barrier of 0.21 eV and a reaction energy of 0.05 eV. The formed H2O* was desorbed from the InN4 surface with a desorption energy of 0.55 eV. Additionally, the OOH* adsorbed on InN4 undergoes the second H-transfer step to the other OH* with a reaction energy and barrier energy of 0.70 and 0.71 eV, respectively. The final products H2O* and 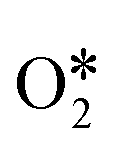 are sequentially released from the InN4 surface with a desorption energy of 0.44 eV and 0.60 eV, respectively. In path III, the first H2O2 dissociates to form O* and H2O*. Subsequently, H2O* desorbs from the InN4 surface with a desorption energy of 0.66 eV, and then the second H2O2 adsorbs onto the InN4 surface with an adsorption energy of −0.14 eV. The adsorbed second
are sequentially released from the InN4 surface with a desorption energy of 0.44 eV and 0.60 eV, respectively. In path III, the first H2O2 dissociates to form O* and H2O*. Subsequently, H2O* desorbs from the InN4 surface with a desorption energy of 0.66 eV, and then the second H2O2 adsorbs onto the InN4 surface with an adsorption energy of −0.14 eV. The adsorbed second 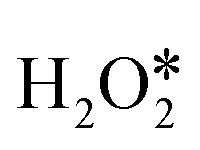 undergoes sequential H-transfer steps with O*. The barrier energies for the two sequential H-transfer steps were calculated as 0 and 0.71 eV, respectively, and the corresponding reaction energies were calculated to be −1.73 and 0.70 eV. At last, H2O* and
undergoes sequential H-transfer steps with O*. The barrier energies for the two sequential H-transfer steps were calculated as 0 and 0.71 eV, respectively, and the corresponding reaction energies were calculated to be −1.73 and 0.70 eV. At last, H2O* and  sequentially desorb from the InN4 surface with desorption energies of 0.44 eV and 0.60 eV, respectively. The catalytic cycle of H2O2 decomposition was limited by the elemental step with the largest energy barrier in a reaction pathway, which is the rate-determining step for H2O2 decomposition. The reaction pathway with a smaller energy barrier of the rate-determining step holds higher potential toward H2O2 decomposition. Based on the thermodynamic analysis, the rate-determining step of H2O2 decomposition on InN4 along path I and path III is identified to be
sequentially desorb from the InN4 surface with desorption energies of 0.44 eV and 0.60 eV, respectively. The catalytic cycle of H2O2 decomposition was limited by the elemental step with the largest energy barrier in a reaction pathway, which is the rate-determining step for H2O2 decomposition. The reaction pathway with a smaller energy barrier of the rate-determining step holds higher potential toward H2O2 decomposition. Based on the thermodynamic analysis, the rate-determining step of H2O2 decomposition on InN4 along path I and path III is identified to be  with a barrier energy of 0.71 eV, indicating that path I and path III can occur simultaneously.
with a barrier energy of 0.71 eV, indicating that path I and path III can occur simultaneously.
 | ||
| Fig. 4 The reaction energy diagram of H2O2 decomposition on InN4via path I and path III. | ||
As summarized in Table S4,† the reaction energies and barrier energies of the direct dehydrogenation of H2O2 (path II) were much larger than those of O–O bond scission (paths I and III) on all 27 PM SACs, indicating that the O–H bond was more difficult to break and path II was energetically unfavorable. Furthermore, when the active sites were covered by O* or OH*, the second H2O2 was hard to dissociate directly to form O* and H2O* (path IV) due to the large barrier energy, suggesting that path IV was disadvantaged. Therefore, only paths I and III were discussed in the following part.
Fig. S6–S8 in the ESI† summarized the decomposition process of H2O2 on the remaining eight PM-N4 (AlN4, GaN4, TlN4, GeN4, SnN4, PbN4, SbN4, BiN4) SACs. It was found that the rate-determining step of H2O2 dissociation on PM-N4 is different. The desorption of  is the rate-determining step for H2O2 decomposition on AlN4 and GaN4 with barrier energies of 1.80 and 1.37 eV, respectively. For InN4, BiN4, SbN4, GeN4, and SnN4, the rate-determining step is the reaction of OOH* with OH* to form the final products H2O* and
is the rate-determining step for H2O2 decomposition on AlN4 and GaN4 with barrier energies of 1.80 and 1.37 eV, respectively. For InN4, BiN4, SbN4, GeN4, and SnN4, the rate-determining step is the reaction of OOH* with OH* to form the final products H2O* and  with barrier energies of 0.71, 0.93, 0.82, 0.74, and 0.86 eV, respectively. For PbN4, the reaction of the adsorbed O* atom with the second
with barrier energies of 0.71, 0.93, 0.82, 0.74, and 0.86 eV, respectively. For PbN4, the reaction of the adsorbed O* atom with the second 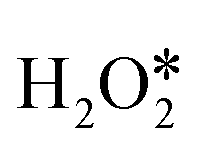 molecule to form OOH* and OH* is the rate-determining step with a barrier energy of 0.84 eV. For TlN4, the O–O bond scission of the initial H2O2 molecule to form two OH* is the rate-determining step with a barrier energy of 0.91 eV.
molecule to form OOH* and OH* is the rate-determining step with a barrier energy of 0.84 eV. For TlN4, the O–O bond scission of the initial H2O2 molecule to form two OH* is the rate-determining step with a barrier energy of 0.91 eV.
From the discussion above, it was known that OH* and O* are two critical reaction intermediates in path I and path III, and the moderate adsorption strength of OH* and O* can ensure relatively high reaction rates for the whole process of H2O2 dissociation. Interestingly, as shown in Fig. 5a, the adsorption energy of OH* exhibits a linear relationship versus that of O* on PM-N4 sites. Therefore, the adsorption energy of OH* can act as a valid descriptor to characterize the ability of PM-N4 SACs for H2O2 decomposition. A straightforward volcano relationship (Fig. 5b) was observed between the barrier energies of the rate-determining step and the adsorption energies of OH*. For AlN4 and GaN4 in the left leg of the volcano curve, the release of the final product  is the rate-determining step of H2O2 decomposition due to the strong adsorption strength of the reaction intermediates. In contrast, PM-N4 in the right leg displayed improved activity for H2O2 decomposition due to the moderate adsorption strength of reaction intermediates. InN4 exhibits the most excellent catalytic activity for H2O2 decomposition due to the favourable adsorption strength of reaction intermediates.
is the rate-determining step of H2O2 decomposition due to the strong adsorption strength of the reaction intermediates. In contrast, PM-N4 in the right leg displayed improved activity for H2O2 decomposition due to the moderate adsorption strength of reaction intermediates. InN4 exhibits the most excellent catalytic activity for H2O2 decomposition due to the favourable adsorption strength of reaction intermediates.
 | ||
| Fig. 5 (a) The linear relationship between the adsorption energies of OH* and O* on PM-N4. (b) The volcano relationship of the barrier energies of the rate-determining step involved in the H2O2 decomposition process against the adsorption energies of OH* on PM-N4. (c) The linear relationship between the adsorption energies of O* and OH* and the p-band centre (εp) of the PM atoms in PM-N4. (d) The volcano relationship of the barrier energies of the rate-determining step involved in the H2O2 decomposition process against the p-band centre (εp) of the PM atoms in PM-N4. | ||
The adsorption strength of the reaction intermediates depends on the electronic interaction between the reaction intermediates and the metal centers. The electronic structure was investigated to further understand the reaction mechanism of the PM-N4 SACs for the decomposition of H2O2.58,59 By examining the partial density of states (PDOS) of the p-band for PM atoms in PM-N4 (shown in Fig. 5c), a strong linear relationship was found between the adsorption energies of the reaction intermediates (O* and OH*) and the p-band centre (εp) of the PM atoms in PM-N4. The higher the energy level of the p-band center, the weaker the adsorption strength of the reaction intermediate, indicating that the p-band center can also act as a descriptor to build the volcano curve (Fig. 5d). The energy level of the p-band near the Fermi level is responsible for the binding strength. With a higher p-band location (higher εp position), the resonance state after reactant adsorption is closer to the Fermi level, which results in the filling of anti-bonding states of the p-band and then the weaker adsorption strength (Fig. 6a).60 According to orbital analysis, the bonding state of OH*/O* adsorption on the PM center depends on the coupling between the 2p states of oxygen and the p states of the PM atom. The PM–O interaction is mainly assigned to the head-to-head σ-bonds formed between pz orbitals (Fig. 6b). As shown in Fig. 6c, the pz-band of the central metal shifts to the left relative to the Fermi level as the central metals vary from Al to Tl. A downshift of the pz states leads to a downward shift of the antibonding states, indicating the formation of weaker chemical bonds. Moreover, the crystal orbital overlap population (COOP) was adopted to gain deeper insight into the electronic interaction between PM-N4 and O-containing adsorbates by revealing the corresponding bonding and antibonding states. As shown in Fig. 6d, the COOP value of O* adsorbed on AlN4, InN4, and TlN4 near the Fermi level gradually decreases, indicating more occupied antibonding states near the Fermi level on TlN4. This is consistent with the increase of the p-band center and the decrease of the adsorption strength of O*.
 | ||
| Fig. 6 (a) Schematic illustrations of the interactions between OH molecular orbitals and the p-band of the PM atom. (b) The pz–pz orbital hybridization between the central metal atom and O atom. (c) The PDOS of pz in central metal atoms of AlN4, GaN4, InN4, and TlN4. (d) The COOP of O* adsorbed on AlN4, InN4, and TlN4. | ||
Reaction pathways and activity of H2O2 decomposition on PM-N2C2 and PM-C4
Except for InN4 and GeN4, the barrier energies of rate-determining steps for the decomposition of H2O2 on PM-N4 always exceed 0.8 eV, which is challenging to achieve at room temperature. Therefore, the effect of the N/C coordination environment on the catalytic activity of H2O2 decomposition was further investigated. Tables S4 and S5† show the reaction energies and barrier energies of the direct dehydrogenation of the first adsorbed and the direct decomposition of the second adsorbed
and the direct decomposition of the second adsorbed 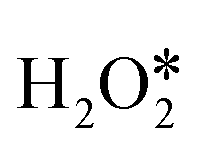 on PM-N2C2 and PM-C4. In path II and path IV, the efficiency is low because of the high barrier energies of the direct dehydrogenation of the first adsorbed
on PM-N2C2 and PM-C4. In path II and path IV, the efficiency is low because of the high barrier energies of the direct dehydrogenation of the first adsorbed  and direct decomposition of the second adsorbed
and direct decomposition of the second adsorbed  . The energy diagrams for H2O2 decomposition along path I and path III on PM-N2C2 and PM-C4 are shown in Fig. S9–S14.† A nearly linear relationship was also found between the adsorption energies of OH* and the adsorption energies of O* (Fig. 7a). The volcano relationship between the adsorption energies of OH* and the barrier energies of the rate-determining step for H2O2 decomposition on PM-N4, PM-N2C2, and PM-C4 is shown in Fig. 7b. The two volcano curves in Fig. 5b and 7b show that PM/N/C SACs exhibit optimal catalytic activity for H2O2 decomposition when the adsorption energy for OH* is around −3.34 eV.
. The energy diagrams for H2O2 decomposition along path I and path III on PM-N2C2 and PM-C4 are shown in Fig. S9–S14.† A nearly linear relationship was also found between the adsorption energies of OH* and the adsorption energies of O* (Fig. 7a). The volcano relationship between the adsorption energies of OH* and the barrier energies of the rate-determining step for H2O2 decomposition on PM-N4, PM-N2C2, and PM-C4 is shown in Fig. 7b. The two volcano curves in Fig. 5b and 7b show that PM/N/C SACs exhibit optimal catalytic activity for H2O2 decomposition when the adsorption energy for OH* is around −3.34 eV.
 | ||
| Fig. 7 (a) The linear relationship between the adsorption energies of OH* and O* on PM-N4, PM-N2C2, and PM-C4. (b) The volcano relationship of the barrier energies of the rate-determining step involved in the H2O2 decomposition against the adsorption energies of OH* on PM-N4, PM-N2C2, and PM-C4. (c) The linear relationship between the adsorption energies of OH* and the ICOOP. (d) The COOP of O* adsorbed on InN4, InN2C2, and InC4. | ||
It can be observed from Fig. 7b and Table S6† that the barrier energies of the rate-determining step on AlN2C2, AlC4, GaN2C2, and GaC4 are significantly reduced compared to AlN4 and GaN4 due to the weak adsorption of the reaction intermediates. For Al, In, Ge, Sn, and Sb, the barrier energies of H2O2 decomposition gradually decrease with the reduction of N atoms in the coordination environment of PM/N/C SACs. In all the three configurations (PM-N4, PM-N2C2, and PM-C4), In presented the best catalytic efficiency for H2O2 decomposition compared with the other eight PM atoms. InC4 presents the highest catalytic activity among the 27 PM SACs in this work, and the barrier energy of the rate-determining step for H2O2 decomposition on InC4 is 0.59 eV, which can be overcome easily at room temperature.61 Furthermore, metal Ge also shows significant catalytic efficiency for H2O2 decomposition in PM/N/C. Generally, PM-N2C2 and PM-C4 exhibit higher catalytic activity than PM-N4 for H2O2 decomposition. The barrier energies of the rate-determining steps for the decomposition of H2O2 on PM-N2C2 and PM-C4 were decreased to smaller than 0.80 eV, except for AlN2C2, AlC4, and PbC4, as shown in Table S6.† Based on the discussion above, we can conclude that changing the N/C coordination environments of the PM SACs can enhance the catalytic efficiency of H2O2 decomposition effectively.
The catalytic efficiency of H2O2 decomposition is governed by the adsorption strength of reaction intermediates, which in turn depends on the surface electronic states of catalysts. In order to gain further insight into the origin of the different catalytic activities of PM/N/C with different N/C coordination environments, the electronic structures of PM/N/C were considered. For a more quantitative comparison, the integrated COOP (ICOOP) was calculated between the PM atoms in PM/N/C and the absorbed OH*. As shown in Fig. 7c, a strong linear relation between the adsorption energies of OH* and the ICOOP value was found. Interestingly, the adsorption strength of intermediates (OH* and O*) on AlN4, AlN2C2, and AlC4 decreased with the decrease of the number of N atoms in the coordination environment, and the catalytic activity of H2O2 decomposition increased to near the apex of the volcano curve. The Mulliken charge value of the Al atom in AlN4, AlN2C2, and AlC4 gradually decreases (Table S2†), which implies that the electrostatic interaction between the reaction intermediates (OH* and O*) and Al atom is weakening. At the same time, the p-band center of the Al atom in AlN4, AlN2C2, and AlC4 shifts to the right (Fig. 8a), which results in the shifting of the 1π-adsorbate resonance state at the OH* molecule to the Fermi level (as shown in Fig. 9a), leading to the weaker adsorption strength. In contrast, the adsorption strength of intermediates (OH* and O*) on TlN4, TlN2C2, and TlC4 increased with the reduction of the number of N atoms in the coordination environment. The ICOOP of OH* adsorbed on TlN4, TlN2C2, and TlC4 gradually increased (Table S6†), indicating that the adsorption strength of OH* on TlN4, TlN2C2, and TlC4 gradually enhanced.62 Meanwhile, as the N atoms in the coordination environment decrease, the p-band center of the Tl atom in TlN4, TlN2C2, and TlC4 moves leftward (Fig. 8b), and the 1π resonance states after OH adsorption on Tl shift to the left, resulting in less occupied antibonding states near the Fermi level (Fig. 9b). Fig. 7b also reveals that InN4, InN2C2, and InC4 exhibit the best activity of H2O2 decomposition in PM-N4, PM-N2C2, and PM-C4, respectively, situated at the volcano curve's peak, exhibiting a steady rise in activity for H2O2 decomposition. However, the adsorption strength of intermediates (OH* and O*) on InN4, InN2C2, and InC4 doesn't change significantly, as demonstrated by the similar COOP value of O* adsorbed on InN4, InN2C2, and InC4 near the Fermi level (Fig. 7d). As shown in Fig. 9c, the hybridization between In-p states and O-p states becomes stronger as the number of N atom decreases.63 The px and py orbitals of the In atom in InN4, InN2C2 and InC4 were further investigated based on the PDOS (shown in Fig. 10). It was found that the px and py orbitals of the In atom shift to higher energy position relative to the Fermi level as the N/C coordination environment varies from InN4 to InN2C4 and InC4, which results in the upward shift of the antibonding states and stronger adsorption strength of OH*.
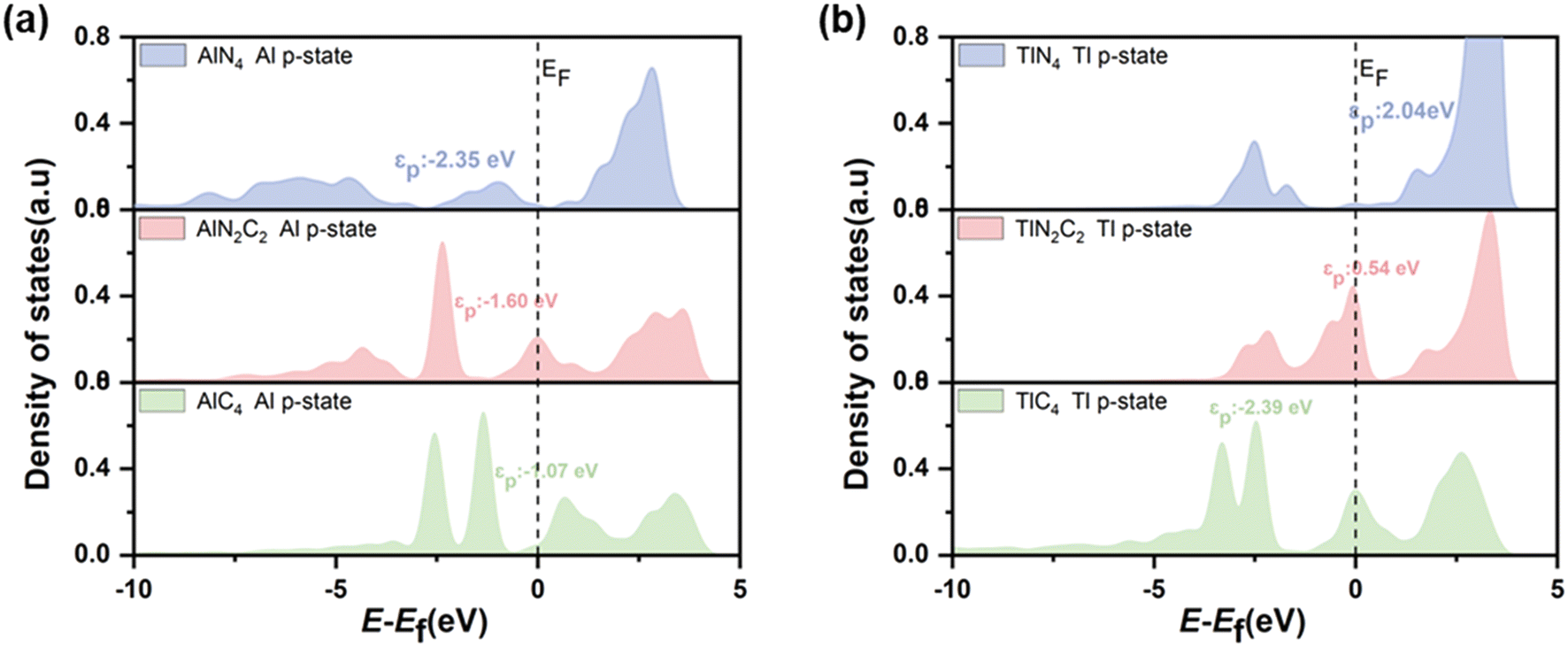 | ||
| Fig. 8 (a) The PDOS of the Al p-state in AlN4, AlN2C2, and AlC4. (b) The PDOS of the Tl p-state in TlN4, TlN2C2, and TlC4. | ||
 | ||
| Fig. 9 The PDOS for OH vacuum states and OH* adsorbed on the AlN4, AlN2C2, AlC4 (a), TlN4, TlN2C2, TlC4 (b) and InN4, InN2C2, InC4 (c). | ||
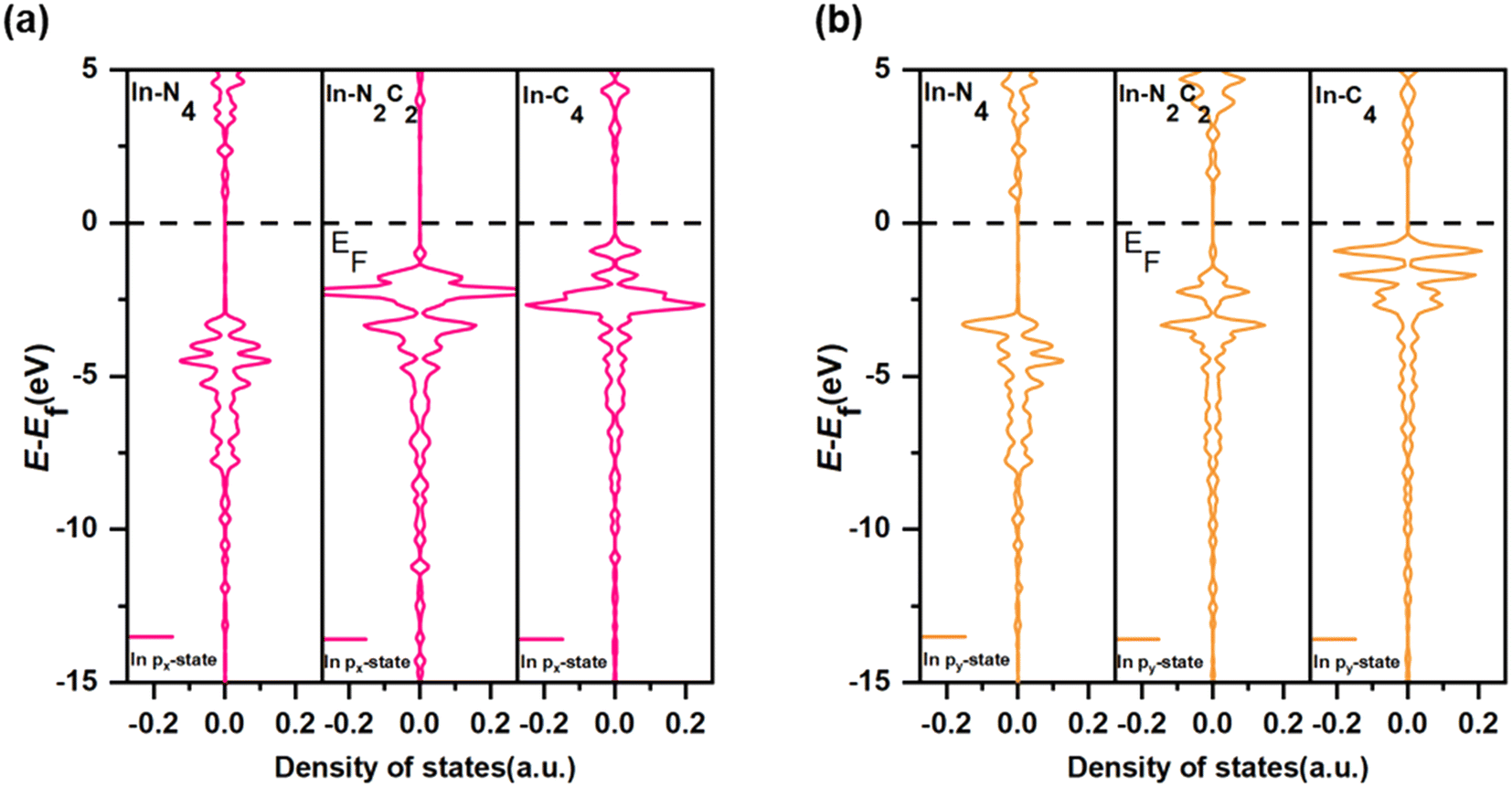 | ||
| Fig. 10 px (a) and py (b) orbitals of In atoms for OH* adsorbed InN4, InN2C2 and InC4. | ||
Conclusion
In this work, the catalytic cycles of H2O2 decomposition on PM/N/C SACs were systematically studied based on density functional theory calculation. Linear relationships were found between the adsorption energies of reaction intermediates and the p-band centre of the PM atoms in PM-N4, and a straightforward volcano plot was found between the catalytic activity and the adsorption strength of the reaction intermediates for H2O2 decomposition on PM-N4. Among the nine PM-N4 sites, InN4 and GeN4 exhibit excellent catalytic activity toward H2O2 decomposition with low activation energies of the rate-determining step (less than 0.8 eV). The coordination environment also plays an important role in the activity of PM/N/C SACs. PM-N2C2 and PM-C4 exhibit higher H2O2 decomposition activity than PM-N4 by altering the N/C coordination environment and then modifying the p-band position of PM atoms in PM/N/C SACs. This means that reducing the N/C ratio in the coordination environment of PM/N/C SACs can adjust the catalytic activity of H2O2 decomposition efficiently. In other words, PM metals possess the capability to serve as viable active centers in PM/N/C SACs for the Fenton-like reaction to form ·OH for pollutant degradation, which is important not only in environmental remediation but also in the biomedical field.Data availability
All data included in this study are available upon request by contact with the corresponding author.Author contributions
Chen Zhou: investigation, writing – original draft. Haobin Tan, Shengbo Wang: formal analysis. Peng Zhang: conceptualization, supervision, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Natural Science Foundation of Guangdong Province (2024A1515012765), the National Natural Science Foundation of China (52350005, 51838005, and 21403092), and the basic research projects jointly funded by Guangzhou City and Guangzhou University (202201020194).References
- Z. Chen, D. Yao, C. Chu and S. Mao, Photocatalytic H2O2 production Systems: Design strategies and environmental applications, Chem. Eng. J., 2023, 451, 138489 CrossRef CAS.
- D. J. Kim, Q. Zhu, K. Rigby, X. Wu, J. H. Kim and J.-H. Kim, A Protocol for Electrocatalyst Stability Evaluation: H2O2 Electrosynthesis for Industrial Wastewater Treatment, Environ. Sci. Technol., 2022, 56, 1365–1375 CrossRef CAS PubMed.
- T. Zhang, Y. Wen, Z. Pan, Y. Kuwahara, K. Mori, H. Yamashita, Y. Zhao and X. Qian, Overcoming Acidic H2O2/Fe(II/III) Redox-Induced Low H2O2 Utilization Efficiency by Carbon Quantum Dots Fenton-like Catalysis, Environ. Sci. Technol., 2022, 56, 2617–2625 CrossRef CAS PubMed.
- F. Wang, J. Xu, Z. Wang, Y. Lou, C. Pan and Y. Zhu, Unprecedentedly efficient mineralization performance of photocatalysis-self-Fenton system towards organic pollutants over oxygen-doped porous g-C3N4 nanosheets, Appl. Catal., B, 2022, 312, 121438 CrossRef CAS.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Turning main-group element magnesium into a highly active electrocatalyst for oxygen reduction reaction, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- K. Roos and P. E. M. Siegbahn, Activation of Dimanganese Class Ib Ribonucleotide Reductase by Hydrogen Peroxide: Mechanistic Insights from Density Functional Theory, Inorg. Chem., 2013, 52, 4173–4184 CrossRef CAS.
- M.-C. Kim, G.-H. Han, X. Xiao, J. Song, J. Hong, E. Jung, H.-K. Kim, J.-P. Ahn, S. S. Han, K.-Y. Lee and T. Yu, Anisotropic growth of Pt on Pd nanocube promotes direct synthesis of hydrogen peroxide, Appl. Surf. Sci., 2021, 562, 150031 CrossRef CAS.
- M. Malček, L. Bučinský, F. Teixeira and M. N. D. S. Cordeiro, Detection of simple inorganic and organic molecules over Cu-decorated circumcoronene: a combined DFT and QTAIM study, Phys. Chem. Chem. Phys., 2018, 20, 16021–16032 RSC.
- X. Liu, Y. Zhou, J. Zhang, L. Luo, Y. Yang, H. Huang, H. Peng, L. Tang and Y. Mu, Insight into electro-Fenton and photo-Fenton for the degradation of antibiotics: Mechanism study and research gaps, Chem. Eng. J., 2018, 347, 379–397 CrossRef CAS.
- Y. Zhu, Q. Xie, F. Deng, Z. Ni, Q. Lin, L. Cheng, X. Chen, R. Qiu and R. Zhu, The differences in heterogeneous Fenton catalytic performance and mechanism of various iron minerals and their influencing factors: A review, Sep. Purif. Technol., 2023, 325, 124702 CrossRef CAS.
- B. Jain, A. K. Singh, H. Kim, E. Lichtfouse and V. K. Sharma, Treatment of organic pollutants by homogeneous and heterogeneous Fenton reaction processes, Environ. Chem. Lett., 2018, 16, 947–967 CrossRef CAS.
- Y. Gao, W. Zhu, J. Liu, P. Lin, J. Zhang, T. Huang and K. Liu, Mesoporous sulfur-doped CoFe2O4 as a new Fenton catalyst for the highly efficient pollutants removal, Appl. Catal., B, 2021, 295, 120273 CrossRef CAS.
- P. Cao, X. Quan, K. Zhao, S. Chen, H. Yu and J. Niu, Selective electrochemical H2O2 generation and activation on a bifunctional catalyst for heterogeneous electro-Fenton catalysis, J. Hazard. Mater., 2020, 382, 121102 CrossRef CAS PubMed.
- A. Saravanan, V. C. Deivayanai, P. S. Kumar, G. Rangasamy, R. V. Hemavathy, T. Harshana, N. Gayathri and K. Alagumalai, A detailed review on advanced oxidation process in treatment of wastewater: Mechanism, challenges and future outlook, Chemosphere, 2022, 308, 136524 CrossRef CAS PubMed.
- X. Du, S. Wang, F. Ye and Z. Qingrui, Derivatives of metal-organic frameworks for heterogeneous Fenton-like processes: From preparation to performance and mechanisms in wastewater purification – A mini review, Environ. Res., 2022, 206, 112414 CrossRef CAS.
- Z.-Y. Guo, Y. Si, W.-Q. Xia, F. Wang, H.-Q. Liu, C. Yang, W.-J. Zhang and W.-W. Li, Electron delocalization triggers nonradical Fenton-like catalysis over spinel oxides, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201607119 CrossRef CAS.
- N. Li, X. He, J. Ye, H. Dai, W. Peng, Z. Cheng, B. Yan, G. Chen and S. Wang, H2O2 activation and contaminants removal in heterogeneous Fenton-like systems, J. Hazard. Mater., 2023, 458, 131926 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- A. Plauck, E. E. Stangland, J. A. Dumesic and M. Mavrikakis, Active sites and mechanisms for H2O2 decomposition over Pd catalysts, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14–16 CrossRef PubMed.
- A. S. Novikov, M. L. Kuznetsov, A. J. L. Pombeiro, N. A. Bokach and G. B. Shul'pin, Generation of HO· Radical from Hydrogen Peroxide Catalyzed by Aqua Complexes of the Group III Metals [M(H2O)n]3+ (M = Ga, In, Sc, Y, or La): A Theoretical Study, ACS Catal., 2013, 3, 1195–1208 CrossRef CAS.
- C. W. Yoon, K. F. Hirsekorn, M. L. Neidig, X. Yang and T. D. Tilley, Mechanism of the Decomposition of Aqueous Hydrogen Peroxide over Heterogeneous TiSBA15 and TS-1 Selective Oxidation Catalysts: Insights from Spectroscopic and Density Functional Theory Studies, ACS Catal., 2011, 1, 1665–1678 CrossRef CAS.
- Y. Wang and P. B. Balbuena, Potential Energy Surface Profile of the Oxygen Reduction Reaction on a Pt Cluster: Adsorption and Decomposition of OOH and H2O2, J. Chem. Theory Comput., 2005, 1, 935–943 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, A universal principle for a rational design of single-atom electrocatalysts, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen, H. Liu, Y. Li and G. Wang, High Durability of Fe–N–C Single-Atom Catalysts with Carbon Vacancies toward the Oxygen Reduction Reaction in Alkaline Media, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Organic wastewater treatment by a single-atom catalyst and electrolytically produced H2O2, Nat. Sustain., 2020, 4, 233–241 CrossRef.
- T. Wang, X. Cao, H. Qin, L. Shang, S. Zheng, F. Fang and L. Jiao, P-Block Atomically Dispersed Antimony Catalyst for Highly Efficient Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2021, 60, 21237–21241 CrossRef CAS.
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Single-Atom Fe Catalysts for Fenton-Like Reactions: Roles of Different N Species, Adv. Mater., 2022, 34, 2110653 CrossRef CAS PubMed.
- J. Liu, J. Zhu, H. Xu and D. Cheng, Rational Design of Heteroatom-Doped Fe–N–C Single-Atom Catalysts for Oxygen Reduction Reaction via Simple Descriptor, ACS Catal., 2024, 14, 6952–6964 CrossRef CAS.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Single atom electrocatalysts supported on graphene or graphene-like carbons, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- Y. Chen, F. Sun and Q. Tang, The active structure of p-block SnNC single-atom electrocatalysts for the oxygen reduction reaction, Phys. Chem. Chem. Phys., 2022, 24, 27302–27311 RSC.
- J. Hu, Y. Li, Y. Zou, L. Lin, B. Li and X. Li, Transition metal single-atom embedded on N-doped carbon as a catalyst for peroxymonosulfate activation: A DFT study, Chem. Eng. J., 2022, 437, 135428 CrossRef CAS.
- H. Qi, J. Yang, F. Liu, L. Zhang, J. Yang, X. Liu, L. Li, Y. Su, Y. Liu, R. Hao, A. Wang and T. Zhang, Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones, Nat. Commun., 2021, 12, 3295 CrossRef CAS.
- J. Guo, G. Song, X. Zhang and M. Zhou, Transition metal catalysts in the heterogeneous electro-Fenton process for organic wastewater treatment: a review, Environ. Sci.: Water Res. Technol., 2023, 9, 2429–2445 RSC.
- P. Zhang, H. Tan, Z. Wang, L. Lyu and C. Hu, Efficient H2O2 dissociation and formation on zinc chalcogenides: A density functional theory study, Appl. Surf. Sci., 2023, 616, 156495 CrossRef CAS.
- L. Li, C. Tang, H. Jin, K. Davey and S.-Z. Qiao, Main-group elements boost electrochemical nitrogen fixation, Chem, 2021, 7, 3232–3255 CAS.
- K. Chen, Y. Zhang, J. Xiang, X. Zhao, X. Li and K. Chu, p-Block Antimony Single-Atom Catalysts for Nitric Oxide Electroreduction to Ammonia, ACS Energy Lett., 2023, 8, 1281–1288 CrossRef CAS.
- A. Vojvodic, J. K. Nørskov and F. Abild-Pedersen, Electronic Structure Effects in Transition Metal Surface Chemistry, Top. Catal., 2014, 57, 25–32 CrossRef CAS.
- K. Chen, N. Zhang, F. Wang, J. Kang and K. Chu, Main-group indium single-atom catalysts for electrocatalytic NO reduction to NH3, J. Mater. Chem. A, 2023, 11, 6814–6819 RSC.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Discovery of main group single Sb–N4 active sites for CO2 electroreduction to formate with high efficiency, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- Q. Wu, B. Huang, Y. Dai, T. Heine and Y. Ma, Main-group metal elements as promising active centers for single-atom catalyst toward nitric oxide reduction reaction, npj 2D Mater. Appl., 2022, 6, 52 CrossRef CAS.
- B. Li, W. Gao and Q. Jiang, A universal picture for ejecting atoms on metallics, Acta Mater., 2022, 228, 117792 CrossRef CAS.
- P. Zhang, X. Hou, L. Liu, J. Mi and M. Dong, Two-Dimensional π-Conjugated Metal Bis(dithiolene) Complex Nanosheets as Selective Catalysts for Oxygen Reduction Reaction, J. Phys. Chem. C, 2015, 119, 28028–28037 CrossRef CAS.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, Single-Atom Catalysts for Electrochemical Water Splitting, ACS Energy Lett., 2018, 3, 1713–1721 CrossRef CAS.
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, Atomic (single, double, and triple atoms) catalysis: frontiers, opportunities, and challenges, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC.
- W. Gao, Y. Chen, B. Li, S.-P. Liu, X. Liu and Q. Jiang, Determining the adsorption energies of small molecules with the intrinsic properties of adsorbates and substrates, Nat. Commun., 2020, 11, 1196 CrossRef CAS.
- W. Wan, C. A. Triana, J. Lan, J. Li, C. S. Allen, Y. Zhao, M. Iannuzzi and G. R. Patzke, Bifunctional Single Atom Electrocatalysts: Coordination–Performance Correlations and Reaction Pathways, ACS Nano, 2020, 14, 13279–13293 CrossRef CAS.
- B. Li, W. Gao and Q. Jiang, Determinants of local chemical environments and magnetic moments of high-entropy alloys, Mater. Res. Lett., 2023, 11, 259–265 CrossRef CAS.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Theoretical Understandings of Graphene-based Metal Single-Atom Catalysts: Stability and Catalytic Performance, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS.
- Y. Zhang, C. Kang, W. Zhao, Y. Song, J. Zhu, H. Huo, Y. Ma, C. Du, P. Zuo, S. Lou and G. Yin, d-p Hybridization-Induced “Trapping–Coupling–Conversion” Enables High-Efficiency Nb Single-Atom Catalysis for Li–S Batteries, J. Am. Chem. Soc., 2023, 145, 1728–1739 CrossRef CAS.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Identifying and tailoring C–N coupling site for efficient urea synthesis over diatomic Fe–Ni catalyst, Nat. Commun., 2022, 13, 5337 CrossRef CAS.
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, N,O symmetric double coordination of an unsaturated Fe single-atom confined within a graphene framework for extraordinarily boosting oxygen reduction in Zn–air batteries, Energy Environ. Sci., 2023, 16, 2629–2636 RSC.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J.-F. Li, Z.-Q. Tian, W. Liu, A. Wang and T. Zhang, Potential-Driven Restructuring of Cu Single Atoms to Nanoparticles for Boosting the Electrochemical Reduction of Nitrate to Ammonia, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS.
- L. Luo, L. Fu, H. Liu, Y. Xu, J. Xing, C.-R. Chang, D.-Y. Yang and J. Tang, Synergy of Pd atoms and oxygen vacancies on In2O3 for methane conversion under visible light, Nat. Commun., 2022, 13, 2930 CrossRef CAS.
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, Mechanism of H2O2 Decomposition on Transition Metal Oxide Surfaces, J. Phys. Chem. C, 2012, 116, 9533–9543 CrossRef CAS.
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, Reactivity of metal oxide clusters with hydrogen peroxide and water – a DFT study evaluating the performance of different exchange–correlation functionals, Phys. Chem. Chem. Phys., 2013, 15, 5539 RSC.
- J. Li, A. Staykov, T. Ishihara and K. Yoshizawa, Theoretical Study of the Decomposition and Hydrogenation of H2O2 on Pd and Au@Pd Surfaces: Understanding toward High Selectivity of H2O2 Synthesis, J. Phys. Chem. C, 2011, 115, 7392–7398 CrossRef CAS.
- L. Wang, Q. Wang, F. Ren and Y. Wang, An unexpected interaction between a H2O2 molecule and anatase TiO2(101) surface, Appl. Surf. Sci., 2019, 493, 926–932 CrossRef CAS.
- C. Wang, J. Ye, L. Liang, X. Cui, L. Kong, N. Li, Z. Cheng, W. Peng, B. Yan and G. Chen, Application of MXene-based materials in Fenton-like systems for organic wastewater treatment: A review, Sci. Total Environ., 2023, 862, 160539 CrossRef CAS.
- Y. Pan, J. Cao, M. Xing and Y. Zhang, Current Mechanism of Peroxymonosulfate Activation by Cobalt-Based Heterogeneous Catalysts in Degrading Organic Compounds, ACS ES&T Eng., 2024, 4, 19–46 Search PubMed.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M.-X. Chen, E. Jin, J. Yang, H.-W. Liang, Y. Zhao, X. Feng, A. Narita and K. Müllen, Multilayer stabilization for fabricating high-loading single-atom catalysts, Nat. Commun., 2020, 11, 5892 CrossRef CAS PubMed.
- Y. Gu, B. J. Xi, H. Zhang, Y. C. Ma and S. L. Xiong, Activation of Main-Group Antimony Atomic Sites for Oxygen Reduction Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202202200 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Promises of Main Group Metal–Based Nanostructured Materials for Electrochemical CO2 Reduction to Formate, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- Z. Xue, X. Zhang, J. Qin and R. Liu, High-throughput identification of high activity and selectivity transition metal single-atom catalysts for nitrogen reduction, Nano Energy, 2021, 80, 105527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details; atomic structures; reaction energies; barrier energies; adsorption energies. See DOI: https://doi.org/10.1039/d4en00778f |
| This journal is © The Royal Society of Chemistry 2025 |